User login
What’s Eating You? Head Lice (Pediculus humanus capitis)
The head louse (Pediculus humanus capitis) is a blood-sucking arthropod of the suborder Anoplura. Lice are obligate human parasites that have infested humans since antiquity. Pediculosis capitis is an infestation of the scalp by head lice. It is estimated that 6 to 12 million individuals in the United States are affected with head lice per year.1 Resistance to topical chemical pediculicides is widespread, and new agents have been developed to address this gap in care.
Characteristics of Head Lice
The head louse is a tan-gray–colored, wingless insect measuring approximately 2- to 3-mm long with 3 body segments. It has 6 legs with claws used to grasp individual hairs, and it moves by crawling; it does not fly or jump.2,3 The head louse has an elongated abdomen and a small head with short antennae and anterior piercing mouthparts (Figure 1).4 Nits are transparent, flask-shaped, 0.5- to 0.8-mm egg cases found firmly cemented to the hair shafts approximately 1 to 4 mm above the level of the scalp (Figure 2).5 The head louse resides on scalp hair and feeds off the scalp itself. Both lice and nits can be present throughout the scalp but are most commonly found in the postauricular and occipital scalp.3,4
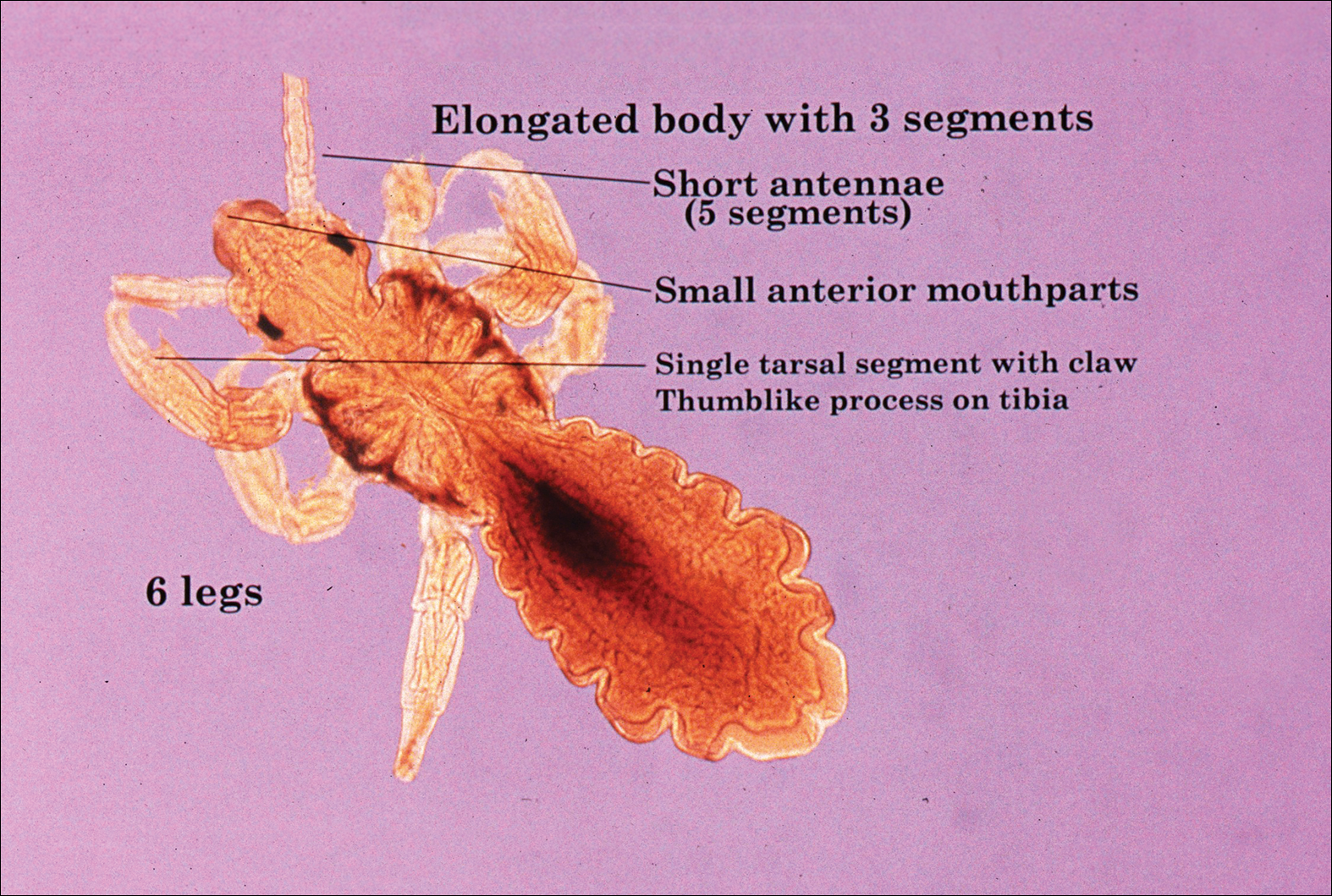

Female lice live approximately 30 days and lay 5 to 10 eggs per day. Eggs incubate individually in nits laid close to the scalp for 8 to 10 days before hatching.1,6 The newly hatched nymphs (also called instars) have multiple exoskeletons that are shed as they grow.7 Nymphs mature into adults in approximately 2 weeks, and the life cycle begins again.8 Head lice are obligate human parasites, feeding approximately every 4 to 6 hours on the blood of the host; however, they can survive up to 4 days without a blood meal on fomites if the climate and conditions are favorable.5,9
Epidemiology and Transmission
Head lice infestations commonly occur in children aged 3 to 11 years and are more prevalent in girls and women.1,10 Infestation rates are not reliably recorded, and few population-based studies have been performed; however, it is estimated that 6 to 12 million individuals are infested annually in the United States.1 Prevalence in some European populations has been estimated to range from 1% to 20%.11 A 2008 literature review found that worldwide prevalence varied across populations from 0.7% to 59%.10
Transmission occurs most frequently from direct head-to-head contact. One study found that transmission is most likely to occur when hairs are arranged in a parallel alignment and move slowly in relation to one another.12 Although controversial and probably less notable, transmission also may occur indirectly via fomites or the sharing of hairbrushes, hats, or other headgear.13,14 Classrooms are a common place for transmission.1 A 2009 study in Germany found an increase in health department consultations for head lice when schools reopened after vacations. The investigators also found that pediculicide sales peaked from mid-September through October, subsequent to schools reopening after the summer holiday.15 There is some evidence that overcrowded housing also can lead to increased incidence and transmission.16,17 There is no consistent correlation of infestation with socioeconomic status.1,17,18
Clinical Manifestations and Diagnosis
Clinically, patients with head lice present with scalp pruritus and sometimes posterior cervical or occipital lymphadenopathy. Pediculosis also can be asymptomatic. With the first exposure, symptoms may not develop for up to 4 to 6 weeks as the immune system develops sensitivity to the louse saliva.6 Bite reactions consisting of papules or wheals are related to immune sensitization.5 Louse feces and excoriations from scratching to relieve itch also may be present on examination. Secondary infection of excoriations also is possible.1
Diagnosis of an active infestation is made by identifying living lice. Because lice move quickly and can be difficult to detect, tightly attached nits on the hair shaft within 4 mm of the scalp are at least indicative of a historic infestation and can be suggestive of active infestation.1,19 Dermoscopy is a helpful tool in differentiating eggs containing nymphs from the empty cases of hatched lice and also from amorphous pseudonits (hair casts)(Figure 3).19,20 Wet combing improves the accuracy of diagnosing an active infection.21

Treatment
Effective treatment of head lice requires eradication of all living lice as well as louse eggs. Topically applied pyrethroids, including pyrethrin shampoos and mousses and permethrin lotion 1%, are considered the first-line therapy.8 Pyrethroids are over-the-counter treatments that act by interfering with sodium transport in the louse, causing depolarization of the neuromembranes and respiratory paralysis.22 Pyrethrins are natural compounds derived from the chrysanthemum plant; permethrin is a synthetic compound. Pyrethrins often are combined with piperonyl butoxide, an insecticide synergist that improves efficacy by inhibiting pyrethrin catabolism.23 Resistance to pyrethroids has become an increasingly important problem in the United States and worldwide.
Malathion lotion 0.5% is another therapeutic option for head lice. Malathion is a prescription organophosphate cholinesterase inhibitor that also causes respiratory paralysis of the louse and is one of the few treatments that is ovicidal.22 It was withdrawn from the market in 1995 due to its flammability and a theoretical risk of respiratory depression if ingested; however, it was reintroduced in 1999 and remains an effective treatment option with little resistance in the United States.24
Lindane 1% (shampoo and lotion), an organochloride compound that acts by causing neuronal hyperstimulation and eventual paralysis of lice, is no longer recommended due to its serious side effects, including central nervous system toxicity and increased risk of seizure.8,24
New US Food and Drug Administration–Approved Therapies
Newer topical treatments include benzyl alcohol lotion 5%, spinosad topical suspension 0.9%, ivermectin lotion 0.5%, and dimethicone-based products. Benzyl alcohol was approved by the US Food and Drug Administration (FDA) in 2009 and is available in the United States by prescription.25 Benzyl alcohol kills lice by asphyxiation. Phase 2 and 3 clinical trials showed significant treatment success 1 day posttreatment (fewer live lice than the vehicle alone; P=.004) and 2 weeks posttreatment (absence of live lice compared to the vehicle alone; P=.001).26
Spinosad was approved by the FDA in 2011 and is available in the United States by prescription.25 It contains the compounds spinosyn A and spinosyn D, which are naturally derived through fermentation by the soil bacterium Saccharopolyspora spinosa. It also contains benzyl alcohol. Spinosad paralyzes lice by disrupting neuronal activity and is at least partially ovicidal.27 Phase 3 clinical trials published in 2009 showed that spinosad was significantly more effective than permethrin in eradicating head lice (P<.001).28
Topical ivermectin was approved by the FDA in 2012 for prescription use.25 It acts on chloride ion channels, causing hyperpolarization of the muscle cells of lice and resulting in paralysis and death. Oral ivermectin (200 μg/kg) given once and repeated in 10 days is not FDA approved for the treatment of head lice but has shown some effectiveness and is sometimes used.8 A comparison study of topical versus oral ivermectin published in 2014 found that eradication was achieved in 88% (n=27) of topical ivermectin users after 1 treatment and 100% (n=31) after 2 treatments. Oral ivermectin produced cure rates of 45% (n=14) after 1 treatment and 97% (n=30) after 2 treatments. Both topical and oral ivermectin treatments are well tolerated.29
Physically Acting Preparations
Products with a physical mode of action are a new attractive option for treatment of pediculosis because the development of resistance is less likely. Studies of silicone-based fluids that physically occlude the respiratory system of the louse, such as dimethicone liquid gel 4%, have shown superiority over treatment with pyrethroids.30,31 Although the safety of dimethicone has been demonstrated, silicone-based treatments have not yet been widely adopted in the United States and are not currently used as a first-line treatment.32 However, use of such physically acting pediculicides may in time surpass traditional neurotoxic treatments due to their low susceptibility to resistance and good safety profile.33,34
Alternative Therapies
Nonchemical treatments for head lice that have shown variable success include wet combing, hot air treatments, and varying occlusive treatments. Physical removal via wet combing requires persistent repeated treatments over several weeks; for example, wet combing may be performed every 3 days for at least 2 weeks or until no head lice are detected on 4 consecutive occasions.35 Cure rates range from 38% to 75% with wet combing as a sole treatment of head lice.36 Because this treatment has minimal risks and no adverse side effects, it can be considered as an alternative treatment for some patients.
Hot air treatments also have been studied. A 2006 study showed that a hot air treatment device had the potential to eradicate head lice, most likely by desiccation. Specifically, 30 minutes of exposure to hot air (at 58.9°F, slightly cooler than a standard hair dryer) using the custom-built device resulted in 98% mortality of eggs and 80% mortality of hatched lice.37 Large randomized controlled trials of hot air treatments have not been performed.
Other alternative treatments include plant-derived oils. A laboratory study of essential oils found that spearmint, cassia, and clove showed pediculicidal activity similar to malathion with improved ovicidal activity.38 However, there is a potential for development of contact dermatitis from essential oils.
Complete Eradication of Head Lice
Removal of nits is an important component of effective lice eradication. Biochemical analysis has revealed that the nit sheath of the head louse is similar in composition to amyloid, rendering it difficult to design products that will unravel the nit sheath while leaving human hair undamaged.39 Because pediculicides are not necessarily ovicidal and complete physical nit removal is difficult to achieve, re-treatment in 7 to 10 days often is advisable to ensure that lice in all stages of the life cycle have been killed.4 Treatment of any secondary bacterial infection also is important. Although transmission of lice via fomites is less likely than from head-to-head contact, the cleaning of hats, hairbrushes, and linens is prudent. Diagnosing and treating infested close contacts also is essential to achieving eradication.4 Coordinated surveillance, education, and treatment efforts in high-risk communities can help detect asymptomatic cases and control local epidemics in a cost-effective manner.40 However, “no nit” policies at schools likely cause a net harm, as nit removal is difficult and children with nonviable nits are then excluded from the classroom.5
Treatment Resistance
Resistance to topical neurotoxic treatments is becoming increasingly common.41-43 Therefore, it is important to identify local patterns of resistance, if possible, when selecting a therapy for head lice. Improper usage, changes in pediculicide formulations and packaging, decreased product efficacy, and natural selection have all contributed to this rise in resistance.7 Additionally, due to protection from multiple exoskeletons and the natural molting process as they mature into adults, nymphs may only receive a sublethal dose when exposed to pediculicides, contributing further to resistance.7 Resistance to synthetic pyrethroids is most predominant, likely due to selection pressure because permethrin historically has been the most widely used insecticide for pediculosis. A 2014 study found that the frequency of sodium-channel insensitivity to pyrethroids, also known as knockdown resistance (or kdr), in US head louse populations collected over a 10-year period was 84.4% and approached 100% in some communities in recent years.44 This evidence strongly supports the use of alternative therapeutic categories to effectively eradicate head lice infestations.
Conclusion
Head lice infestation is common in children, and although it is not harmful to the host, it can be an irritating and symptomatic problem and can lead to notable distress, missed days of school, and secondary infections. Identifying active adult lice is the gold standard for diagnosis. Current recommended treatments include pyrethroids as the first-line therapy; however, resistance to these neurotoxic agents is becoming increasingly common. Alternative therapies such as newer neurotoxic agents or pediculicides with physical mechanisms of action (eg, dimethicone-based products) should be considered, particularly in regions where resistance is known to be high. Education about head lice, proper use of treatment, and coordinated diagnosis are necessary for effective management of this problem.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Centers for Disease Control and Prevention. Head lice. http://www.cdc.gov/parasites/lice/head/index.html. Updated September 24, 2013. Accessed November 9, 2017.
- Hurwitz S. Lice (pediculosis). In: Hurwitz S. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 2nd ed. Philadelphia, PA: WB Saunders Company; 1993:416-419.
- Elston DM. What’s eating you? Pediculus humanus (head louse and body louse). Cutis. 1999;63:259-264.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12.
- Frankowski BL, Weiner LB. Head lice. Pediatrics. 2002;110:638-643.
- Meinking TL. Clinical update on resistance and treatment of pediculosis capitis. Am J Manag Care. 2004;10(9 suppl):S264-S268.
- Devore CD, Schutze GE. Head lice. Pediatrics. 2015;135:E1355-E1365.
- Burkhart CN. Fomite transmission with head lice: a continuing controversy. Lancet. 2003;361:99-100.
- Falagas ME, Matthaiou DK, Rafailidis PI, et al. Worldwide prevalence of head lice. Emerg Infect Dis. 2008;14:1493-1494.
- Feldmeier H. Pediculosis capitis: new insights into epidemiology, diagnosis and treatment. Eur J Clin Microbiol Infect Dis. 2012;31:2105-2110.
- Canyon DV, Speare R, Muller R. Spatial and kinetic factors for the transfer of head lice (Pediculus capitis) between hairs. J Invest Dermatol. 2002;119:629-631.
- Burkhart CN, Burkhart CG. Fomite transmission in head lice. J Am Acad Dermatol. 2007;56:1044-1047.
- Canyon DV, Speare R. Indirect transmission of head lice via inanimate objects. Open Dermatol J. 2010;4:72-76.
- Bauer E, Jahnke C, Feldmeier H. Seasonal fluctuations of head lice infestation in Germany. Parasitol Res. 2009;104:677-681.
- Balcioglu IC, Kurt O, Limoncu ME, et al. Rural life, lower socioeconomic status and parasitic infections. Parasitol Int. 2007;56:129-133.
- Lesshafft H, Baier A, Guerra H, et al. Prevalence and risk factors associated with pediculosis capitis in an impoverished urban community in Lima, Peru. J Glob Infect Dis. 2013;5:138-143.
- Tagka A, Lambrou GI, Braoudaki M, et al. Socioeconomical factors associated with pediculosis (Phthiraptera: Pediculidae) in Athens, Greece. J Med Entomol. 2016;53:919-922.
- Di Stefani A, Hofmann-Wellenhof R, Zalaudek I. Dermoscopy for diagnosis and treatment monitoring of pediculosis capitis. J Am Acad Dermatol. 2006;54:909-911.
- Bakos RM, Bakos L. Dermoscopy for diagnosis of pediculosis capitis. J Am Acad Dermatol. 2007;57:727-728.
- Jahnke C, Bauer E, Hengge UR, et al. Accuracy of diagnosis of pediculosis capitis: visual inspection vs wet combing. Arch Dermatol. 2009;145:309-313.
- Elston DM. Drugs used in the treatment of pediculosis. J Drugs Dermatol. 2005;4:207-211.
- National Pesticide Information Center. Piperonyl butoxide (general fact sheet). http://npic.orst.edu/factsheets/pbogen.pdf/. Accessed November 13, 2017.
- Diamantis SA, Morrell DS, Burkhart CN. Treatment of head lice. Dermatol Ther. 2009;22:273-278.
- United States Food and Drug Administration. Treating and preventing head lice. http://www.fda.gov/forconsumers/consumerupdates/ucm171730.htm. Published July 13, 2010. Updated November 8, 2017. Accessed November 13, 2017.
- Meinking TL, Villar ME, Vicaria M, et al. The clinical trials supporting benzyl alcohol lotion 5% (UlesfiaTM): a safe and effective topical treatment for head lice (Pediculosis Humanus Capitis). Pediatr Dermatol. 2010;27:19-24.
- McCormack PL. Spinosad in pediculosis capitis. Am J Clin Dermatol. 2011;12:349-353.
- Stough D, Shellabarger S, Quiring J, et al. Efficacy and safety of spinosad and permethrin creme rinses for pediculosis capitis (head lice). Pediatrics. 2009;124:E389-E395.
- Ahmad HM, Abdel-Azim ES, Abdel-Aziz RT. Assessment of topical versus oral ivermectin as a treatment for head lice. Dermatol Ther. 2014;27:307-310.
- Heukelbach J, Pilger D, Oliveira FA, et al. A highly efficacious pediculicide based on dimethicone: randomized observer blinded comparative trial. BMC Infect Dis. 2008;8:115.
- Burgess IF, Brunton ER, Burgess NA. Single application of 4% dimethicone liquid gel versus two applications of 1% permethrin creme rinse for treatment of head louse infestation: a randomised controlled trial. BMC Dermatol. 2013;13:5.
- Ihde ES, Boscamp JR, Loh JM, et al. Safety and efficacy of a 100% dimethicone pediculocide in school-age children. BMC Pediatr. 2015;15:70.
- Heukelbach J, Oliveira FA, Richter J, et al. Dimethicone-based pediculicides: a physical approach to eradicate head lice. Open Dermatol J. 2010;4:77-81.
- Feldmeier H. Treatment of pediculosis capitis: a critical appraisal of the current literature. Am J Clin Dermatol. 2014;15:401-412.
- Glasziou P, Bennett J, Greenberg P, et al; Handbook Of Non Drug Intervention (HANDI) Project Team. Wet combing for the eradication of head lice. Aust Fam Physician. 2013;42:129-130.
- Tebruegge M, Runnacles J. Is wet combing effective in children with pediculosis capitis infestation? Arch Dis Child. 2007;92:818-820.
- Goates BM, Atkin JS, Wilding KG, et al. An effective nonchemical treatment for head lice: a lot of hot air. Pediatrics. 2006;118:1962-1970.
- Yones DA, Bakir HY, Bayoumi SA. Chemical composition and efficacy of some selected plant oils against Pediculus humanus capitis in vitro. Parasitol Res. 2016;115:3209-3218.
- Burkhart CN, Burkhart CG. Head lice: scientific assessment of the nit sheath with clinical ramifications and therapeutic options. J Am Acad Dermatol. 2005;53:129-133.
- Ibarra J, Fry F, Wickenden C, et al. The impact of well-developed preventative strategies on the eradication of head lice. Perspect Public Health. 2009;129:165-173.
- Mumcuoglu KY, Hemingway J, Miller J, et al. Permethrin resistance in the head louse pediculus humanus capitis from Israel. Med Vet Entomol. 1995;9:427-432.
- Meinking TL, Serrano L, Hard B, et al. Comparative in vitro pediculicidal efficacy of treatments in a resistant head lice population in the United States. Arch Dermatol. 2002;138:220-224.
- Hemingway J, Miller J, Mumcuoglu KY. Pyrethroid resistance mechanisms in the head louse Pediculus capitis from Israel: implications for control. Med Vet Entomol. 1999;13:89-96.
- Yoon KS, Previte DJ, Hodgdon HE, et al. Knockdown resistance allele frequencies in North American head louse (Anoplura: Pediculidae) populations. J Med Entomol. 2014;51:450-457.
The head louse (Pediculus humanus capitis) is a blood-sucking arthropod of the suborder Anoplura. Lice are obligate human parasites that have infested humans since antiquity. Pediculosis capitis is an infestation of the scalp by head lice. It is estimated that 6 to 12 million individuals in the United States are affected with head lice per year.1 Resistance to topical chemical pediculicides is widespread, and new agents have been developed to address this gap in care.
Characteristics of Head Lice
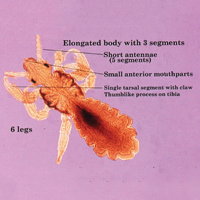
The head louse is a tan-gray–colored, wingless insect measuring approximately 2- to 3-mm long with 3 body segments. It has 6 legs with claws used to grasp individual hairs, and it moves by crawling; it does not fly or jump.2,3 The head louse has an elongated abdomen and a small head with short antennae and anterior piercing mouthparts (Figure 1).4 Nits are transparent, flask-shaped, 0.5- to 0.8-mm egg cases found firmly cemented to the hair shafts approximately 1 to 4 mm above the level of the scalp (Figure 2).5 The head louse resides on scalp hair and feeds off the scalp itself. Both lice and nits can be present throughout the scalp but are most commonly found in the postauricular and occipital scalp.3,4
Female lice live approximately 30 days and lay 5 to 10 eggs per day. Eggs incubate individually in nits laid close to the scalp for 8 to 10 days before hatching.1,6 The newly hatched nymphs (also called instars) have multiple exoskeletons that are shed as they grow.7 Nymphs mature into adults in approximately 2 weeks, and the life cycle begins again.8 Head lice are obligate human parasites, feeding approximately every 4 to 6 hours on the blood of the host; however, they can survive up to 4 days without a blood meal on fomites if the climate and conditions are favorable.5,9
Epidemiology and Transmission
Head lice infestations commonly occur in children aged 3 to 11 years and are more prevalent in girls and women.1,10 Infestation rates are not reliably recorded, and few population-based studies have been performed; however, it is estimated that 6 to 12 million individuals are infested annually in the United States.1 Prevalence in some European populations has been estimated to range from 1% to 20%.11 A 2008 literature review found that worldwide prevalence varied across populations from 0.7% to 59%.10
Transmission occurs most frequently from direct head-to-head contact. One study found that transmission is most likely to occur when hairs are arranged in a parallel alignment and move slowly in relation to one another.12 Although controversial and probably less notable, transmission also may occur indirectly via fomites or the sharing of hairbrushes, hats, or other headgear.13,14 Classrooms are a common place for transmission.1 A 2009 study in Germany found an increase in health department consultations for head lice when schools reopened after vacations. The investigators also found that pediculicide sales peaked from mid-September through October, subsequent to schools reopening after the summer holiday.15 There is some evidence that overcrowded housing also can lead to increased incidence and transmission.16,17 There is no consistent correlation of infestation with socioeconomic status.1,17,18
Clinical Manifestations and Diagnosis
Clinically, patients with head lice present with scalp pruritus and sometimes posterior cervical or occipital lymphadenopathy. Pediculosis also can be asymptomatic. With the first exposure, symptoms may not develop for up to 4 to 6 weeks as the immune system develops sensitivity to the louse saliva.6 Bite reactions consisting of papules or wheals are related to immune sensitization.5 Louse feces and excoriations from scratching to relieve itch also may be present on examination. Secondary infection of excoriations also is possible.1
Diagnosis of an active infestation is made by identifying living lice. Because lice move quickly and can be difficult to detect, tightly attached nits on the hair shaft within 4 mm of the scalp are at least indicative of a historic infestation and can be suggestive of active infestation.1,19 Dermoscopy is a helpful tool in differentiating eggs containing nymphs from the empty cases of hatched lice and also from amorphous pseudonits (hair casts)(Figure 3).19,20 Wet combing improves the accuracy of diagnosing an active infection.21
Treatment
Effective treatment of head lice requires eradication of all living lice as well as louse eggs. Topically applied pyrethroids, including pyrethrin shampoos and mousses and permethrin lotion 1%, are considered the first-line therapy.8 Pyrethroids are over-the-counter treatments that act by interfering with sodium transport in the louse, causing depolarization of the neuromembranes and respiratory paralysis.22 Pyrethrins are natural compounds derived from the chrysanthemum plant; permethrin is a synthetic compound. Pyrethrins often are combined with piperonyl butoxide, an insecticide synergist that improves efficacy by inhibiting pyrethrin catabolism.23 Resistance to pyrethroids has become an increasingly important problem in the United States and worldwide.
Malathion lotion 0.5% is another therapeutic option for head lice. Malathion is a prescription organophosphate cholinesterase inhibitor that also causes respiratory paralysis of the louse and is one of the few treatments that is ovicidal.22 It was withdrawn from the market in 1995 due to its flammability and a theoretical risk of respiratory depression if ingested; however, it was reintroduced in 1999 and remains an effective treatment option with little resistance in the United States.24
Lindane 1% (shampoo and lotion), an organochloride compound that acts by causing neuronal hyperstimulation and eventual paralysis of lice, is no longer recommended due to its serious side effects, including central nervous system toxicity and increased risk of seizure.8,24
New US Food and Drug Administration–Approved Therapies
Newer topical treatments include benzyl alcohol lotion 5%, spinosad topical suspension 0.9%, ivermectin lotion 0.5%, and dimethicone-based products. Benzyl alcohol was approved by the US Food and Drug Administration (FDA) in 2009 and is available in the United States by prescription.25 Benzyl alcohol kills lice by asphyxiation. Phase 2 and 3 clinical trials showed significant treatment success 1 day posttreatment (fewer live lice than the vehicle alone; P=.004) and 2 weeks posttreatment (absence of live lice compared to the vehicle alone; P=.001).26
Spinosad was approved by the FDA in 2011 and is available in the United States by prescription.25 It contains the compounds spinosyn A and spinosyn D, which are naturally derived through fermentation by the soil bacterium Saccharopolyspora spinosa. It also contains benzyl alcohol. Spinosad paralyzes lice by disrupting neuronal activity and is at least partially ovicidal.27 Phase 3 clinical trials published in 2009 showed that spinosad was significantly more effective than permethrin in eradicating head lice (P<.001).28
Topical ivermectin was approved by the FDA in 2012 for prescription use.25 It acts on chloride ion channels, causing hyperpolarization of the muscle cells of lice and resulting in paralysis and death. Oral ivermectin (200 μg/kg) given once and repeated in 10 days is not FDA approved for the treatment of head lice but has shown some effectiveness and is sometimes used.8 A comparison study of topical versus oral ivermectin published in 2014 found that eradication was achieved in 88% (n=27) of topical ivermectin users after 1 treatment and 100% (n=31) after 2 treatments. Oral ivermectin produced cure rates of 45% (n=14) after 1 treatment and 97% (n=30) after 2 treatments. Both topical and oral ivermectin treatments are well tolerated.29
Physically Acting Preparations
Products with a physical mode of action are a new attractive option for treatment of pediculosis because the development of resistance is less likely. Studies of silicone-based fluids that physically occlude the respiratory system of the louse, such as dimethicone liquid gel 4%, have shown superiority over treatment with pyrethroids.30,31 Although the safety of dimethicone has been demonstrated, silicone-based treatments have not yet been widely adopted in the United States and are not currently used as a first-line treatment.32 However, use of such physically acting pediculicides may in time surpass traditional neurotoxic treatments due to their low susceptibility to resistance and good safety profile.33,34
Alternative Therapies
Nonchemical treatments for head lice that have shown variable success include wet combing, hot air treatments, and varying occlusive treatments. Physical removal via wet combing requires persistent repeated treatments over several weeks; for example, wet combing may be performed every 3 days for at least 2 weeks or until no head lice are detected on 4 consecutive occasions.35 Cure rates range from 38% to 75% with wet combing as a sole treatment of head lice.36 Because this treatment has minimal risks and no adverse side effects, it can be considered as an alternative treatment for some patients.
Hot air treatments also have been studied. A 2006 study showed that a hot air treatment device had the potential to eradicate head lice, most likely by desiccation. Specifically, 30 minutes of exposure to hot air (at 58.9°F, slightly cooler than a standard hair dryer) using the custom-built device resulted in 98% mortality of eggs and 80% mortality of hatched lice.37 Large randomized controlled trials of hot air treatments have not been performed.
Other alternative treatments include plant-derived oils. A laboratory study of essential oils found that spearmint, cassia, and clove showed pediculicidal activity similar to malathion with improved ovicidal activity.38 However, there is a potential for development of contact dermatitis from essential oils.
Complete Eradication of Head Lice
Removal of nits is an important component of effective lice eradication. Biochemical analysis has revealed that the nit sheath of the head louse is similar in composition to amyloid, rendering it difficult to design products that will unravel the nit sheath while leaving human hair undamaged.39 Because pediculicides are not necessarily ovicidal and complete physical nit removal is difficult to achieve, re-treatment in 7 to 10 days often is advisable to ensure that lice in all stages of the life cycle have been killed.4 Treatment of any secondary bacterial infection also is important. Although transmission of lice via fomites is less likely than from head-to-head contact, the cleaning of hats, hairbrushes, and linens is prudent. Diagnosing and treating infested close contacts also is essential to achieving eradication.4 Coordinated surveillance, education, and treatment efforts in high-risk communities can help detect asymptomatic cases and control local epidemics in a cost-effective manner.40 However, “no nit” policies at schools likely cause a net harm, as nit removal is difficult and children with nonviable nits are then excluded from the classroom.5
Treatment Resistance
Resistance to topical neurotoxic treatments is becoming increasingly common.41-43 Therefore, it is important to identify local patterns of resistance, if possible, when selecting a therapy for head lice. Improper usage, changes in pediculicide formulations and packaging, decreased product efficacy, and natural selection have all contributed to this rise in resistance.7 Additionally, due to protection from multiple exoskeletons and the natural molting process as they mature into adults, nymphs may only receive a sublethal dose when exposed to pediculicides, contributing further to resistance.7 Resistance to synthetic pyrethroids is most predominant, likely due to selection pressure because permethrin historically has been the most widely used insecticide for pediculosis. A 2014 study found that the frequency of sodium-channel insensitivity to pyrethroids, also known as knockdown resistance (or kdr), in US head louse populations collected over a 10-year period was 84.4% and approached 100% in some communities in recent years.44 This evidence strongly supports the use of alternative therapeutic categories to effectively eradicate head lice infestations.
Conclusion
Head lice infestation is common in children, and although it is not harmful to the host, it can be an irritating and symptomatic problem and can lead to notable distress, missed days of school, and secondary infections. Identifying active adult lice is the gold standard for diagnosis. Current recommended treatments include pyrethroids as the first-line therapy; however, resistance to these neurotoxic agents is becoming increasingly common. Alternative therapies such as newer neurotoxic agents or pediculicides with physical mechanisms of action (eg, dimethicone-based products) should be considered, particularly in regions where resistance is known to be high. Education about head lice, proper use of treatment, and coordinated diagnosis are necessary for effective management of this problem.
The head louse (Pediculus humanus capitis) is a blood-sucking arthropod of the suborder Anoplura. Lice are obligate human parasites that have infested humans since antiquity. Pediculosis capitis is an infestation of the scalp by head lice. It is estimated that 6 to 12 million individuals in the United States are affected with head lice per year.1 Resistance to topical chemical pediculicides is widespread, and new agents have been developed to address this gap in care.
Characteristics of Head Lice
The head louse is a tan-gray–colored, wingless insect measuring approximately 2- to 3-mm long with 3 body segments. It has 6 legs with claws used to grasp individual hairs, and it moves by crawling; it does not fly or jump.2,3 The head louse has an elongated abdomen and a small head with short antennae and anterior piercing mouthparts (Figure 1).4 Nits are transparent, flask-shaped, 0.5- to 0.8-mm egg cases found firmly cemented to the hair shafts approximately 1 to 4 mm above the level of the scalp (Figure 2).5 The head louse resides on scalp hair and feeds off the scalp itself. Both lice and nits can be present throughout the scalp but are most commonly found in the postauricular and occipital scalp.3,4
Female lice live approximately 30 days and lay 5 to 10 eggs per day. Eggs incubate individually in nits laid close to the scalp for 8 to 10 days before hatching.1,6 The newly hatched nymphs (also called instars) have multiple exoskeletons that are shed as they grow.7 Nymphs mature into adults in approximately 2 weeks, and the life cycle begins again.8 Head lice are obligate human parasites, feeding approximately every 4 to 6 hours on the blood of the host; however, they can survive up to 4 days without a blood meal on fomites if the climate and conditions are favorable.5,9
Epidemiology and Transmission
Head lice infestations commonly occur in children aged 3 to 11 years and are more prevalent in girls and women.1,10 Infestation rates are not reliably recorded, and few population-based studies have been performed; however, it is estimated that 6 to 12 million individuals are infested annually in the United States.1 Prevalence in some European populations has been estimated to range from 1% to 20%.11 A 2008 literature review found that worldwide prevalence varied across populations from 0.7% to 59%.10
Transmission occurs most frequently from direct head-to-head contact. One study found that transmission is most likely to occur when hairs are arranged in a parallel alignment and move slowly in relation to one another.12 Although controversial and probably less notable, transmission also may occur indirectly via fomites or the sharing of hairbrushes, hats, or other headgear.13,14 Classrooms are a common place for transmission.1 A 2009 study in Germany found an increase in health department consultations for head lice when schools reopened after vacations. The investigators also found that pediculicide sales peaked from mid-September through October, subsequent to schools reopening after the summer holiday.15 There is some evidence that overcrowded housing also can lead to increased incidence and transmission.16,17 There is no consistent correlation of infestation with socioeconomic status.1,17,18
Clinical Manifestations and Diagnosis
Clinically, patients with head lice present with scalp pruritus and sometimes posterior cervical or occipital lymphadenopathy. Pediculosis also can be asymptomatic. With the first exposure, symptoms may not develop for up to 4 to 6 weeks as the immune system develops sensitivity to the louse saliva.6 Bite reactions consisting of papules or wheals are related to immune sensitization.5 Louse feces and excoriations from scratching to relieve itch also may be present on examination. Secondary infection of excoriations also is possible.1
Diagnosis of an active infestation is made by identifying living lice. Because lice move quickly and can be difficult to detect, tightly attached nits on the hair shaft within 4 mm of the scalp are at least indicative of a historic infestation and can be suggestive of active infestation.1,19 Dermoscopy is a helpful tool in differentiating eggs containing nymphs from the empty cases of hatched lice and also from amorphous pseudonits (hair casts)(Figure 3).19,20 Wet combing improves the accuracy of diagnosing an active infection.21
Treatment
Effective treatment of head lice requires eradication of all living lice as well as louse eggs. Topically applied pyrethroids, including pyrethrin shampoos and mousses and permethrin lotion 1%, are considered the first-line therapy.8 Pyrethroids are over-the-counter treatments that act by interfering with sodium transport in the louse, causing depolarization of the neuromembranes and respiratory paralysis.22 Pyrethrins are natural compounds derived from the chrysanthemum plant; permethrin is a synthetic compound. Pyrethrins often are combined with piperonyl butoxide, an insecticide synergist that improves efficacy by inhibiting pyrethrin catabolism.23 Resistance to pyrethroids has become an increasingly important problem in the United States and worldwide.
Malathion lotion 0.5% is another therapeutic option for head lice. Malathion is a prescription organophosphate cholinesterase inhibitor that also causes respiratory paralysis of the louse and is one of the few treatments that is ovicidal.22 It was withdrawn from the market in 1995 due to its flammability and a theoretical risk of respiratory depression if ingested; however, it was reintroduced in 1999 and remains an effective treatment option with little resistance in the United States.24
Lindane 1% (shampoo and lotion), an organochloride compound that acts by causing neuronal hyperstimulation and eventual paralysis of lice, is no longer recommended due to its serious side effects, including central nervous system toxicity and increased risk of seizure.8,24
New US Food and Drug Administration–Approved Therapies
Newer topical treatments include benzyl alcohol lotion 5%, spinosad topical suspension 0.9%, ivermectin lotion 0.5%, and dimethicone-based products. Benzyl alcohol was approved by the US Food and Drug Administration (FDA) in 2009 and is available in the United States by prescription.25 Benzyl alcohol kills lice by asphyxiation. Phase 2 and 3 clinical trials showed significant treatment success 1 day posttreatment (fewer live lice than the vehicle alone; P=.004) and 2 weeks posttreatment (absence of live lice compared to the vehicle alone; P=.001).26
Spinosad was approved by the FDA in 2011 and is available in the United States by prescription.25 It contains the compounds spinosyn A and spinosyn D, which are naturally derived through fermentation by the soil bacterium Saccharopolyspora spinosa. It also contains benzyl alcohol. Spinosad paralyzes lice by disrupting neuronal activity and is at least partially ovicidal.27 Phase 3 clinical trials published in 2009 showed that spinosad was significantly more effective than permethrin in eradicating head lice (P<.001).28
Topical ivermectin was approved by the FDA in 2012 for prescription use.25 It acts on chloride ion channels, causing hyperpolarization of the muscle cells of lice and resulting in paralysis and death. Oral ivermectin (200 μg/kg) given once and repeated in 10 days is not FDA approved for the treatment of head lice but has shown some effectiveness and is sometimes used.8 A comparison study of topical versus oral ivermectin published in 2014 found that eradication was achieved in 88% (n=27) of topical ivermectin users after 1 treatment and 100% (n=31) after 2 treatments. Oral ivermectin produced cure rates of 45% (n=14) after 1 treatment and 97% (n=30) after 2 treatments. Both topical and oral ivermectin treatments are well tolerated.29
Physically Acting Preparations
Products with a physical mode of action are a new attractive option for treatment of pediculosis because the development of resistance is less likely. Studies of silicone-based fluids that physically occlude the respiratory system of the louse, such as dimethicone liquid gel 4%, have shown superiority over treatment with pyrethroids.30,31 Although the safety of dimethicone has been demonstrated, silicone-based treatments have not yet been widely adopted in the United States and are not currently used as a first-line treatment.32 However, use of such physically acting pediculicides may in time surpass traditional neurotoxic treatments due to their low susceptibility to resistance and good safety profile.33,34
Alternative Therapies
Nonchemical treatments for head lice that have shown variable success include wet combing, hot air treatments, and varying occlusive treatments. Physical removal via wet combing requires persistent repeated treatments over several weeks; for example, wet combing may be performed every 3 days for at least 2 weeks or until no head lice are detected on 4 consecutive occasions.35 Cure rates range from 38% to 75% with wet combing as a sole treatment of head lice.36 Because this treatment has minimal risks and no adverse side effects, it can be considered as an alternative treatment for some patients.
Hot air treatments also have been studied. A 2006 study showed that a hot air treatment device had the potential to eradicate head lice, most likely by desiccation. Specifically, 30 minutes of exposure to hot air (at 58.9°F, slightly cooler than a standard hair dryer) using the custom-built device resulted in 98% mortality of eggs and 80% mortality of hatched lice.37 Large randomized controlled trials of hot air treatments have not been performed.
Other alternative treatments include plant-derived oils. A laboratory study of essential oils found that spearmint, cassia, and clove showed pediculicidal activity similar to malathion with improved ovicidal activity.38 However, there is a potential for development of contact dermatitis from essential oils.
Complete Eradication of Head Lice
Removal of nits is an important component of effective lice eradication. Biochemical analysis has revealed that the nit sheath of the head louse is similar in composition to amyloid, rendering it difficult to design products that will unravel the nit sheath while leaving human hair undamaged.39 Because pediculicides are not necessarily ovicidal and complete physical nit removal is difficult to achieve, re-treatment in 7 to 10 days often is advisable to ensure that lice in all stages of the life cycle have been killed.4 Treatment of any secondary bacterial infection also is important. Although transmission of lice via fomites is less likely than from head-to-head contact, the cleaning of hats, hairbrushes, and linens is prudent. Diagnosing and treating infested close contacts also is essential to achieving eradication.4 Coordinated surveillance, education, and treatment efforts in high-risk communities can help detect asymptomatic cases and control local epidemics in a cost-effective manner.40 However, “no nit” policies at schools likely cause a net harm, as nit removal is difficult and children with nonviable nits are then excluded from the classroom.5
Treatment Resistance
Resistance to topical neurotoxic treatments is becoming increasingly common.41-43 Therefore, it is important to identify local patterns of resistance, if possible, when selecting a therapy for head lice. Improper usage, changes in pediculicide formulations and packaging, decreased product efficacy, and natural selection have all contributed to this rise in resistance.7 Additionally, due to protection from multiple exoskeletons and the natural molting process as they mature into adults, nymphs may only receive a sublethal dose when exposed to pediculicides, contributing further to resistance.7 Resistance to synthetic pyrethroids is most predominant, likely due to selection pressure because permethrin historically has been the most widely used insecticide for pediculosis. A 2014 study found that the frequency of sodium-channel insensitivity to pyrethroids, also known as knockdown resistance (or kdr), in US head louse populations collected over a 10-year period was 84.4% and approached 100% in some communities in recent years.44 This evidence strongly supports the use of alternative therapeutic categories to effectively eradicate head lice infestations.
Conclusion
Head lice infestation is common in children, and although it is not harmful to the host, it can be an irritating and symptomatic problem and can lead to notable distress, missed days of school, and secondary infections. Identifying active adult lice is the gold standard for diagnosis. Current recommended treatments include pyrethroids as the first-line therapy; however, resistance to these neurotoxic agents is becoming increasingly common. Alternative therapies such as newer neurotoxic agents or pediculicides with physical mechanisms of action (eg, dimethicone-based products) should be considered, particularly in regions where resistance is known to be high. Education about head lice, proper use of treatment, and coordinated diagnosis are necessary for effective management of this problem.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Centers for Disease Control and Prevention. Head lice. http://www.cdc.gov/parasites/lice/head/index.html. Updated September 24, 2013. Accessed November 9, 2017.
- Hurwitz S. Lice (pediculosis). In: Hurwitz S. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 2nd ed. Philadelphia, PA: WB Saunders Company; 1993:416-419.
- Elston DM. What’s eating you? Pediculus humanus (head louse and body louse). Cutis. 1999;63:259-264.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12.
- Frankowski BL, Weiner LB. Head lice. Pediatrics. 2002;110:638-643.
- Meinking TL. Clinical update on resistance and treatment of pediculosis capitis. Am J Manag Care. 2004;10(9 suppl):S264-S268.
- Devore CD, Schutze GE. Head lice. Pediatrics. 2015;135:E1355-E1365.
- Burkhart CN. Fomite transmission with head lice: a continuing controversy. Lancet. 2003;361:99-100.
- Falagas ME, Matthaiou DK, Rafailidis PI, et al. Worldwide prevalence of head lice. Emerg Infect Dis. 2008;14:1493-1494.
- Feldmeier H. Pediculosis capitis: new insights into epidemiology, diagnosis and treatment. Eur J Clin Microbiol Infect Dis. 2012;31:2105-2110.
- Canyon DV, Speare R, Muller R. Spatial and kinetic factors for the transfer of head lice (Pediculus capitis) between hairs. J Invest Dermatol. 2002;119:629-631.
- Burkhart CN, Burkhart CG. Fomite transmission in head lice. J Am Acad Dermatol. 2007;56:1044-1047.
- Canyon DV, Speare R. Indirect transmission of head lice via inanimate objects. Open Dermatol J. 2010;4:72-76.
- Bauer E, Jahnke C, Feldmeier H. Seasonal fluctuations of head lice infestation in Germany. Parasitol Res. 2009;104:677-681.
- Balcioglu IC, Kurt O, Limoncu ME, et al. Rural life, lower socioeconomic status and parasitic infections. Parasitol Int. 2007;56:129-133.
- Lesshafft H, Baier A, Guerra H, et al. Prevalence and risk factors associated with pediculosis capitis in an impoverished urban community in Lima, Peru. J Glob Infect Dis. 2013;5:138-143.
- Tagka A, Lambrou GI, Braoudaki M, et al. Socioeconomical factors associated with pediculosis (Phthiraptera: Pediculidae) in Athens, Greece. J Med Entomol. 2016;53:919-922.
- Di Stefani A, Hofmann-Wellenhof R, Zalaudek I. Dermoscopy for diagnosis and treatment monitoring of pediculosis capitis. J Am Acad Dermatol. 2006;54:909-911.
- Bakos RM, Bakos L. Dermoscopy for diagnosis of pediculosis capitis. J Am Acad Dermatol. 2007;57:727-728.
- Jahnke C, Bauer E, Hengge UR, et al. Accuracy of diagnosis of pediculosis capitis: visual inspection vs wet combing. Arch Dermatol. 2009;145:309-313.
- Elston DM. Drugs used in the treatment of pediculosis. J Drugs Dermatol. 2005;4:207-211.
- National Pesticide Information Center. Piperonyl butoxide (general fact sheet). http://npic.orst.edu/factsheets/pbogen.pdf/. Accessed November 13, 2017.
- Diamantis SA, Morrell DS, Burkhart CN. Treatment of head lice. Dermatol Ther. 2009;22:273-278.
- United States Food and Drug Administration. Treating and preventing head lice. http://www.fda.gov/forconsumers/consumerupdates/ucm171730.htm. Published July 13, 2010. Updated November 8, 2017. Accessed November 13, 2017.
- Meinking TL, Villar ME, Vicaria M, et al. The clinical trials supporting benzyl alcohol lotion 5% (UlesfiaTM): a safe and effective topical treatment for head lice (Pediculosis Humanus Capitis). Pediatr Dermatol. 2010;27:19-24.
- McCormack PL. Spinosad in pediculosis capitis. Am J Clin Dermatol. 2011;12:349-353.
- Stough D, Shellabarger S, Quiring J, et al. Efficacy and safety of spinosad and permethrin creme rinses for pediculosis capitis (head lice). Pediatrics. 2009;124:E389-E395.
- Ahmad HM, Abdel-Azim ES, Abdel-Aziz RT. Assessment of topical versus oral ivermectin as a treatment for head lice. Dermatol Ther. 2014;27:307-310.
- Heukelbach J, Pilger D, Oliveira FA, et al. A highly efficacious pediculicide based on dimethicone: randomized observer blinded comparative trial. BMC Infect Dis. 2008;8:115.
- Burgess IF, Brunton ER, Burgess NA. Single application of 4% dimethicone liquid gel versus two applications of 1% permethrin creme rinse for treatment of head louse infestation: a randomised controlled trial. BMC Dermatol. 2013;13:5.
- Ihde ES, Boscamp JR, Loh JM, et al. Safety and efficacy of a 100% dimethicone pediculocide in school-age children. BMC Pediatr. 2015;15:70.
- Heukelbach J, Oliveira FA, Richter J, et al. Dimethicone-based pediculicides: a physical approach to eradicate head lice. Open Dermatol J. 2010;4:77-81.
- Feldmeier H. Treatment of pediculosis capitis: a critical appraisal of the current literature. Am J Clin Dermatol. 2014;15:401-412.
- Glasziou P, Bennett J, Greenberg P, et al; Handbook Of Non Drug Intervention (HANDI) Project Team. Wet combing for the eradication of head lice. Aust Fam Physician. 2013;42:129-130.
- Tebruegge M, Runnacles J. Is wet combing effective in children with pediculosis capitis infestation? Arch Dis Child. 2007;92:818-820.
- Goates BM, Atkin JS, Wilding KG, et al. An effective nonchemical treatment for head lice: a lot of hot air. Pediatrics. 2006;118:1962-1970.
- Yones DA, Bakir HY, Bayoumi SA. Chemical composition and efficacy of some selected plant oils against Pediculus humanus capitis in vitro. Parasitol Res. 2016;115:3209-3218.
- Burkhart CN, Burkhart CG. Head lice: scientific assessment of the nit sheath with clinical ramifications and therapeutic options. J Am Acad Dermatol. 2005;53:129-133.
- Ibarra J, Fry F, Wickenden C, et al. The impact of well-developed preventative strategies on the eradication of head lice. Perspect Public Health. 2009;129:165-173.
- Mumcuoglu KY, Hemingway J, Miller J, et al. Permethrin resistance in the head louse pediculus humanus capitis from Israel. Med Vet Entomol. 1995;9:427-432.
- Meinking TL, Serrano L, Hard B, et al. Comparative in vitro pediculicidal efficacy of treatments in a resistant head lice population in the United States. Arch Dermatol. 2002;138:220-224.
- Hemingway J, Miller J, Mumcuoglu KY. Pyrethroid resistance mechanisms in the head louse Pediculus capitis from Israel: implications for control. Med Vet Entomol. 1999;13:89-96.
- Yoon KS, Previte DJ, Hodgdon HE, et al. Knockdown resistance allele frequencies in North American head louse (Anoplura: Pediculidae) populations. J Med Entomol. 2014;51:450-457.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Centers for Disease Control and Prevention. Head lice. http://www.cdc.gov/parasites/lice/head/index.html. Updated September 24, 2013. Accessed November 9, 2017.
- Hurwitz S. Lice (pediculosis). In: Hurwitz S. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 2nd ed. Philadelphia, PA: WB Saunders Company; 1993:416-419.
- Elston DM. What’s eating you? Pediculus humanus (head louse and body louse). Cutis. 1999;63:259-264.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12.
- Frankowski BL, Weiner LB. Head lice. Pediatrics. 2002;110:638-643.
- Meinking TL. Clinical update on resistance and treatment of pediculosis capitis. Am J Manag Care. 2004;10(9 suppl):S264-S268.
- Devore CD, Schutze GE. Head lice. Pediatrics. 2015;135:E1355-E1365.
- Burkhart CN. Fomite transmission with head lice: a continuing controversy. Lancet. 2003;361:99-100.
- Falagas ME, Matthaiou DK, Rafailidis PI, et al. Worldwide prevalence of head lice. Emerg Infect Dis. 2008;14:1493-1494.
- Feldmeier H. Pediculosis capitis: new insights into epidemiology, diagnosis and treatment. Eur J Clin Microbiol Infect Dis. 2012;31:2105-2110.
- Canyon DV, Speare R, Muller R. Spatial and kinetic factors for the transfer of head lice (Pediculus capitis) between hairs. J Invest Dermatol. 2002;119:629-631.
- Burkhart CN, Burkhart CG. Fomite transmission in head lice. J Am Acad Dermatol. 2007;56:1044-1047.
- Canyon DV, Speare R. Indirect transmission of head lice via inanimate objects. Open Dermatol J. 2010;4:72-76.
- Bauer E, Jahnke C, Feldmeier H. Seasonal fluctuations of head lice infestation in Germany. Parasitol Res. 2009;104:677-681.
- Balcioglu IC, Kurt O, Limoncu ME, et al. Rural life, lower socioeconomic status and parasitic infections. Parasitol Int. 2007;56:129-133.
- Lesshafft H, Baier A, Guerra H, et al. Prevalence and risk factors associated with pediculosis capitis in an impoverished urban community in Lima, Peru. J Glob Infect Dis. 2013;5:138-143.
- Tagka A, Lambrou GI, Braoudaki M, et al. Socioeconomical factors associated with pediculosis (Phthiraptera: Pediculidae) in Athens, Greece. J Med Entomol. 2016;53:919-922.
- Di Stefani A, Hofmann-Wellenhof R, Zalaudek I. Dermoscopy for diagnosis and treatment monitoring of pediculosis capitis. J Am Acad Dermatol. 2006;54:909-911.
- Bakos RM, Bakos L. Dermoscopy for diagnosis of pediculosis capitis. J Am Acad Dermatol. 2007;57:727-728.
- Jahnke C, Bauer E, Hengge UR, et al. Accuracy of diagnosis of pediculosis capitis: visual inspection vs wet combing. Arch Dermatol. 2009;145:309-313.
- Elston DM. Drugs used in the treatment of pediculosis. J Drugs Dermatol. 2005;4:207-211.
- National Pesticide Information Center. Piperonyl butoxide (general fact sheet). http://npic.orst.edu/factsheets/pbogen.pdf/. Accessed November 13, 2017.
- Diamantis SA, Morrell DS, Burkhart CN. Treatment of head lice. Dermatol Ther. 2009;22:273-278.
- United States Food and Drug Administration. Treating and preventing head lice. http://www.fda.gov/forconsumers/consumerupdates/ucm171730.htm. Published July 13, 2010. Updated November 8, 2017. Accessed November 13, 2017.
- Meinking TL, Villar ME, Vicaria M, et al. The clinical trials supporting benzyl alcohol lotion 5% (UlesfiaTM): a safe and effective topical treatment for head lice (Pediculosis Humanus Capitis). Pediatr Dermatol. 2010;27:19-24.
- McCormack PL. Spinosad in pediculosis capitis. Am J Clin Dermatol. 2011;12:349-353.
- Stough D, Shellabarger S, Quiring J, et al. Efficacy and safety of spinosad and permethrin creme rinses for pediculosis capitis (head lice). Pediatrics. 2009;124:E389-E395.
- Ahmad HM, Abdel-Azim ES, Abdel-Aziz RT. Assessment of topical versus oral ivermectin as a treatment for head lice. Dermatol Ther. 2014;27:307-310.
- Heukelbach J, Pilger D, Oliveira FA, et al. A highly efficacious pediculicide based on dimethicone: randomized observer blinded comparative trial. BMC Infect Dis. 2008;8:115.
- Burgess IF, Brunton ER, Burgess NA. Single application of 4% dimethicone liquid gel versus two applications of 1% permethrin creme rinse for treatment of head louse infestation: a randomised controlled trial. BMC Dermatol. 2013;13:5.
- Ihde ES, Boscamp JR, Loh JM, et al. Safety and efficacy of a 100% dimethicone pediculocide in school-age children. BMC Pediatr. 2015;15:70.
- Heukelbach J, Oliveira FA, Richter J, et al. Dimethicone-based pediculicides: a physical approach to eradicate head lice. Open Dermatol J. 2010;4:77-81.
- Feldmeier H. Treatment of pediculosis capitis: a critical appraisal of the current literature. Am J Clin Dermatol. 2014;15:401-412.
- Glasziou P, Bennett J, Greenberg P, et al; Handbook Of Non Drug Intervention (HANDI) Project Team. Wet combing for the eradication of head lice. Aust Fam Physician. 2013;42:129-130.
- Tebruegge M, Runnacles J. Is wet combing effective in children with pediculosis capitis infestation? Arch Dis Child. 2007;92:818-820.
- Goates BM, Atkin JS, Wilding KG, et al. An effective nonchemical treatment for head lice: a lot of hot air. Pediatrics. 2006;118:1962-1970.
- Yones DA, Bakir HY, Bayoumi SA. Chemical composition and efficacy of some selected plant oils against Pediculus humanus capitis in vitro. Parasitol Res. 2016;115:3209-3218.
- Burkhart CN, Burkhart CG. Head lice: scientific assessment of the nit sheath with clinical ramifications and therapeutic options. J Am Acad Dermatol. 2005;53:129-133.
- Ibarra J, Fry F, Wickenden C, et al. The impact of well-developed preventative strategies on the eradication of head lice. Perspect Public Health. 2009;129:165-173.
- Mumcuoglu KY, Hemingway J, Miller J, et al. Permethrin resistance in the head louse pediculus humanus capitis from Israel. Med Vet Entomol. 1995;9:427-432.
- Meinking TL, Serrano L, Hard B, et al. Comparative in vitro pediculicidal efficacy of treatments in a resistant head lice population in the United States. Arch Dermatol. 2002;138:220-224.
- Hemingway J, Miller J, Mumcuoglu KY. Pyrethroid resistance mechanisms in the head louse Pediculus capitis from Israel: implications for control. Med Vet Entomol. 1999;13:89-96.
- Yoon KS, Previte DJ, Hodgdon HE, et al. Knockdown resistance allele frequencies in North American head louse (Anoplura: Pediculidae) populations. J Med Entomol. 2014;51:450-457.
Practice Points
- Transmission of head lice occurs most frequently from direct head-to-head contact; however, head lice can survive up to 4 days on fomites.
- Patients present with scalp pruritus and bite reactions (papules or wheals), but pediculosis can be asymptomatic, particularly with the first exposure before the immune system has developed sensitivity to the louse saliva.
- Topical pyrethroids are available over-the-counter and are considered first-line therapy; however, resistance to pyrethroids has become an important problem in the United States and worldwide.
- Newer topical treatments such as benzyl alcohol lotion 5%, spinosad topical suspension 0.9%, and ivermectin lotion 0.5% can be prescribed as alternative therapies, particularly if resistance to pyrethroids is a concern.
Levofloxacin prophylaxis in AML reduces febrile neutropenia admissions
In patients with acute myeloid leukemia (AML), giving levofloxacin for febrile neutropenia prophylaxis reduced hospital admissions due to this complication by about one-quarter, according to results of a retrospective cohort study.
“Multiple studies have demonstrated the use of an oral fluoroquinolone antibiotic to prevent febrile neutropenia readmission to hospital after receiving consolidation chemotherapy,” Samantha S. F. Lee, PharmD, a clinical pharmacist at St. Michael’s Hospital in Toronto, and her colleagues wrote. “However, this is the first study to demonstrate a positive outcome specific to the AML population.”
The results support recommendations made by the Infectious Diseases Society of America and the National Comprehensive Cancer Network related to treatment of AML after receiving consolidation chemotherapy.
For the study, the investigators retrospectively reviewed the charts of consecutive patients with AML treated at London Health Sciences Centre, a tertiary academic medical center, between 2006 and 2013. They compared outcomes between 50 patients who were prescribed prophylactic levofloxacin (Levaquin) after consolidation therapy and 50 patients who were not prescribed any antibiotics.
Overall, patients given levofloxacin had a lower rate of hospital readmission due to febrile neutropenia whether given the antibiotic after the first chemotherapy consolidation cycle (42% vs. 72%, P = .002) or after all cycles (51.4% vs. 67%, P = .023).
None of the patients in the levofloxacin group developed Clostridium difficile–associated diarrhea within 30 days from discharge after receiving the first cycle of consolidation chemotherapy, compared with one patient in the group not given the antibiotic (Support Care Cancer. 2017 Nov 23. doi: 10.1007/s00520-017-3976-1).
The use of levofloxacin did not significantly impact secondary outcomes, including total days of antibiotic treatment provided to febrile neutropenic patients, days to readmission for febrile neutropenia, or the rate of positive bacterial cultures returned in febrile neutropenic patients.
It was not possible to assess differences in the rate of fluoroquinolone resistance in positive bacterial cultures because fluoroquinolone susceptibilities were infrequently reported.
“This study provides evidence of a strong association between prophylaxis with oral levofloxacin and the rate of febrile neutropenia, but further study is required to evaluate the impact of fluoroquinolone use on antibiotic resistance and [Clostridium difficile–associated diarrhea] rates in this patient setting,” the investigators wrote.
The study was limited by the small sample size, which precluded accurate ascertainment of some outcomes, including safety outcomes, they noted. Additionally, adherence to oral therapy was unknown, and some data, such as fluoroquinolone resistance, were missing for patients admitted for febrile neutropenia to outside hospitals.
Dr. Lee reported having no relevant conflicts of interest.
In patients with acute myeloid leukemia (AML), giving levofloxacin for febrile neutropenia prophylaxis reduced hospital admissions due to this complication by about one-quarter, according to results of a retrospective cohort study.
“Multiple studies have demonstrated the use of an oral fluoroquinolone antibiotic to prevent febrile neutropenia readmission to hospital after receiving consolidation chemotherapy,” Samantha S. F. Lee, PharmD, a clinical pharmacist at St. Michael’s Hospital in Toronto, and her colleagues wrote. “However, this is the first study to demonstrate a positive outcome specific to the AML population.”
The results support recommendations made by the Infectious Diseases Society of America and the National Comprehensive Cancer Network related to treatment of AML after receiving consolidation chemotherapy.
For the study, the investigators retrospectively reviewed the charts of consecutive patients with AML treated at London Health Sciences Centre, a tertiary academic medical center, between 2006 and 2013. They compared outcomes between 50 patients who were prescribed prophylactic levofloxacin (Levaquin) after consolidation therapy and 50 patients who were not prescribed any antibiotics.
Overall, patients given levofloxacin had a lower rate of hospital readmission due to febrile neutropenia whether given the antibiotic after the first chemotherapy consolidation cycle (42% vs. 72%, P = .002) or after all cycles (51.4% vs. 67%, P = .023).
None of the patients in the levofloxacin group developed Clostridium difficile–associated diarrhea within 30 days from discharge after receiving the first cycle of consolidation chemotherapy, compared with one patient in the group not given the antibiotic (Support Care Cancer. 2017 Nov 23. doi: 10.1007/s00520-017-3976-1).
The use of levofloxacin did not significantly impact secondary outcomes, including total days of antibiotic treatment provided to febrile neutropenic patients, days to readmission for febrile neutropenia, or the rate of positive bacterial cultures returned in febrile neutropenic patients.
It was not possible to assess differences in the rate of fluoroquinolone resistance in positive bacterial cultures because fluoroquinolone susceptibilities were infrequently reported.
“This study provides evidence of a strong association between prophylaxis with oral levofloxacin and the rate of febrile neutropenia, but further study is required to evaluate the impact of fluoroquinolone use on antibiotic resistance and [Clostridium difficile–associated diarrhea] rates in this patient setting,” the investigators wrote.
The study was limited by the small sample size, which precluded accurate ascertainment of some outcomes, including safety outcomes, they noted. Additionally, adherence to oral therapy was unknown, and some data, such as fluoroquinolone resistance, were missing for patients admitted for febrile neutropenia to outside hospitals.
Dr. Lee reported having no relevant conflicts of interest.
In patients with acute myeloid leukemia (AML), giving levofloxacin for febrile neutropenia prophylaxis reduced hospital admissions due to this complication by about one-quarter, according to results of a retrospective cohort study.
“Multiple studies have demonstrated the use of an oral fluoroquinolone antibiotic to prevent febrile neutropenia readmission to hospital after receiving consolidation chemotherapy,” Samantha S. F. Lee, PharmD, a clinical pharmacist at St. Michael’s Hospital in Toronto, and her colleagues wrote. “However, this is the first study to demonstrate a positive outcome specific to the AML population.”
The results support recommendations made by the Infectious Diseases Society of America and the National Comprehensive Cancer Network related to treatment of AML after receiving consolidation chemotherapy.
For the study, the investigators retrospectively reviewed the charts of consecutive patients with AML treated at London Health Sciences Centre, a tertiary academic medical center, between 2006 and 2013. They compared outcomes between 50 patients who were prescribed prophylactic levofloxacin (Levaquin) after consolidation therapy and 50 patients who were not prescribed any antibiotics.
Overall, patients given levofloxacin had a lower rate of hospital readmission due to febrile neutropenia whether given the antibiotic after the first chemotherapy consolidation cycle (42% vs. 72%, P = .002) or after all cycles (51.4% vs. 67%, P = .023).
None of the patients in the levofloxacin group developed Clostridium difficile–associated diarrhea within 30 days from discharge after receiving the first cycle of consolidation chemotherapy, compared with one patient in the group not given the antibiotic (Support Care Cancer. 2017 Nov 23. doi: 10.1007/s00520-017-3976-1).
The use of levofloxacin did not significantly impact secondary outcomes, including total days of antibiotic treatment provided to febrile neutropenic patients, days to readmission for febrile neutropenia, or the rate of positive bacterial cultures returned in febrile neutropenic patients.
It was not possible to assess differences in the rate of fluoroquinolone resistance in positive bacterial cultures because fluoroquinolone susceptibilities were infrequently reported.
“This study provides evidence of a strong association between prophylaxis with oral levofloxacin and the rate of febrile neutropenia, but further study is required to evaluate the impact of fluoroquinolone use on antibiotic resistance and [Clostridium difficile–associated diarrhea] rates in this patient setting,” the investigators wrote.
The study was limited by the small sample size, which precluded accurate ascertainment of some outcomes, including safety outcomes, they noted. Additionally, adherence to oral therapy was unknown, and some data, such as fluoroquinolone resistance, were missing for patients admitted for febrile neutropenia to outside hospitals.
Dr. Lee reported having no relevant conflicts of interest.
FROM SUPPORTIVE CARE IN CANCER
Key clinical point:
Major finding: Patients given levofloxacin had lower rates of hospital readmission due to febrile neutropenia whether it was given after the first consolidation cycle (42% vs. 72%, P = .002) or after all cycles (51.4% vs. 67%, P = .023).
Data source: A single-center retrospective cohort study of 100 patients with AML, half of whom were prescribed levofloxacin after consolidation chemotherapy.
Disclosures: Dr. Lee reported having no relevant conflicts of interest.
Approach to Treatment of Medical and Cosmetic Facial Concerns in Skin of Color Patients
The approach to the treatment of common skin disorders and cosmetic concerns in patients with skin of color (SOC) requires the clinician to understand the biological differences, nuances, and special considerations that are unique to patients with darker skin types.1-3 This article addresses 4 common facial concerns in SOC patients—acne, rosacea, facial hyperpigmentation, and cosmetic enhancement—and provides treatment recommendations and management pearls to assist the clinician with optimal outcomes for SOC patients.
Acne in SOC Patients
Acne vulgaris is one of the most common conditions that dermatologists treat and is estimated to affect 40 to 50 million individuals in the United States.1 Many of these acne patients are individuals with SOC.2-4 A study of 2835 females (aged 10–70 years) conducted in 4 different cities—Los Angeles, California; London, United Kingdom; Akita, Japan; and Rome, Italy—demonstrated acne prevalence of 37% in blacks, 32% in Hispanics, 30% in Asians, 24% in whites, and 23% in Continental Indians.5 Blacks, Hispanics, and Continental Indians demonstrated equal prevalence with comedonal and inflammatory acne. Asians displayed more inflammatory acne lesions than comedones. In contrast, whites demonstrated more comedones than inflammatory acne. Dyspigmentation, postinflammatory hyperpigmentation (PIH), and atrophic scars were more common in black and Hispanic females than other ethnicities.5 This study illustrated that acne-induced PIH is a common sequela in SOC patients and is the main reason they seek treatment.6,7
The pathogenesis of acne is the same in all racial and ethnic groups: (1) follicular hyperkeratinization and the formation of a microcomedone caused by abnormal desquamation of the keratinocytes within the sebaceous follicle, (2) production of sebum by circulating androgens, (3) proliferation of Propionibacterium acnes, and (4) inflammation. Subclinical inflammation is present throughout all stages of acne, including normal-appearing skin, inflammatory lesions, comedones, and scarring, and may contribute to PIH in acne patients with SOC (Figure 1).8 A thorough history should be obtained from acne patients, including answers to the following questions7:
- What skin and hair care products do you use?
- Do you use sunscreen daily?
- What cosmetic products or makeup do you use?
- Do you use any ethnic skin care products, including skin lightening creams?
- Do you have a history of keloids?

It is important to ask these questions to assess if the SOC patient has developed pomade acne,9 acne cosmetica,10 or a potential risk of skin irritation from the use of skin care practices. It is best to take total control of the patient’s skin care regimen and discontinue use of toners, astringents, witch hazel, exfoliants, and rubbing alcohol, which may lead to skin dryness and irritation, particularly when combined with topical acne medications.
Treatment
Treatment of acne in SOC patients is similar to generally recommended treatments, with special considerations. Consider the following key points when treating acne in SOC patients:
- Treat acne early and aggressively to prevent or minimize subsequent PIH and acne scarring.
- Balance aggressive treatment with nonirritating topical skin care.
- Most importantly, target PIH in addition to acne and choose a regimen that limits skin irritation that might exacerbate existing PIH.7
Develop a maintenance program to control future breakouts. Topical agents can be used as monotherapy or in fixed combinations and may include benzoyl peroxide, antibiotics, dapsone, azelaic acid (AZA), and retinoids. Similar to white patients, topical retinoids remain a first-line treatment for acne in patients with SOC.11,12
Tolerability must be managed in SOC acne patients. Therapeutic maneuvers that can be instituted should include a discussion on using gentle skin care, initiating therapy with a retinoid applied every other night starting with a low concentration and gradually titrating up, and applying a moisturizer before or after applying acne medication. Oral therapies consist of antibiotics (doxycycline, minocycline), retinoids (isotretinoin), and hormonal modulators (oral contraceptives, spironolactone). Isotretinoin, recommended for patients with nodulocystic acne, may play a possible role in treating acne-induced PIH.13
Two common procedural therapies for acne include comedone extraction and intralesional corticosteroid injection. A 6- to 8-week course of a topical retinoid prior to comedonal extraction may facilitate the procedure and is recommended in SOC patients to help reduce cutaneous trauma and PIH.11 Inflammatory acne lesions can be treated with intralesional injection of triamcinolone acetonide 2.5 or 5.0 mg/mL, which usually reduces inflammation within 2 to 5 days.11
Treatment of acne-induced PIH includes sun protection, topical and oral medications, chemical peels, lasers, and energy devices. Treatment of hypertrophic scarring and keloids involves intralesional injection of triamcinolone acetonide 20, 30, or 40 mg/mL every 4 weeks until the lesion is flat.11
Superficial chemical peels can be used to treat acne and PIH in SOC patients,14 such as salicylic acid (20%–30%), glycolic acid (20%–70%), trichloroacetic acid (15%–30%), and Jessner peels.
Acne Scarring
Surgical approaches to acne scarring in patients with SOC include elliptical excision, punch excision, punch elevation, punch autografting, dermal grafting, dermal planning, subcutaneous incision (subcision), dermabrasion, microneedling, fillers, and laser skin resurfacing. The treatment of choice depends on the size, type, and depth of the scar and the clinician’s preference.
Lasers
Fractional photothermolysis has emerged as a treatment option for acne scars in SOC patients. This procedure produces microscopic columns of thermal injury in the epidermis and dermis, sparing the surrounding tissue and minimizing downtime and adverse events. Because fractional photothermolysis does not target melanin and produces limited epidermal injury, darker Fitzpatrick skin types (IV–VI) can be safely and effectively treated with this procedure.15
Rosacea in SOC Patients
Rosacea is a chronic inflammatory disorder that affects the vasculature and pilosebaceous units of the face. It commonly is seen in Fitzpatrick skin types I and II; however, rosacea can occur in all skin types (Figure 2). Triggers include emotional stress, extreme environmental temperatures, hot and spicy foods, red wine or alcohol, and topical irritants or allergens found in common cosmetic products.16
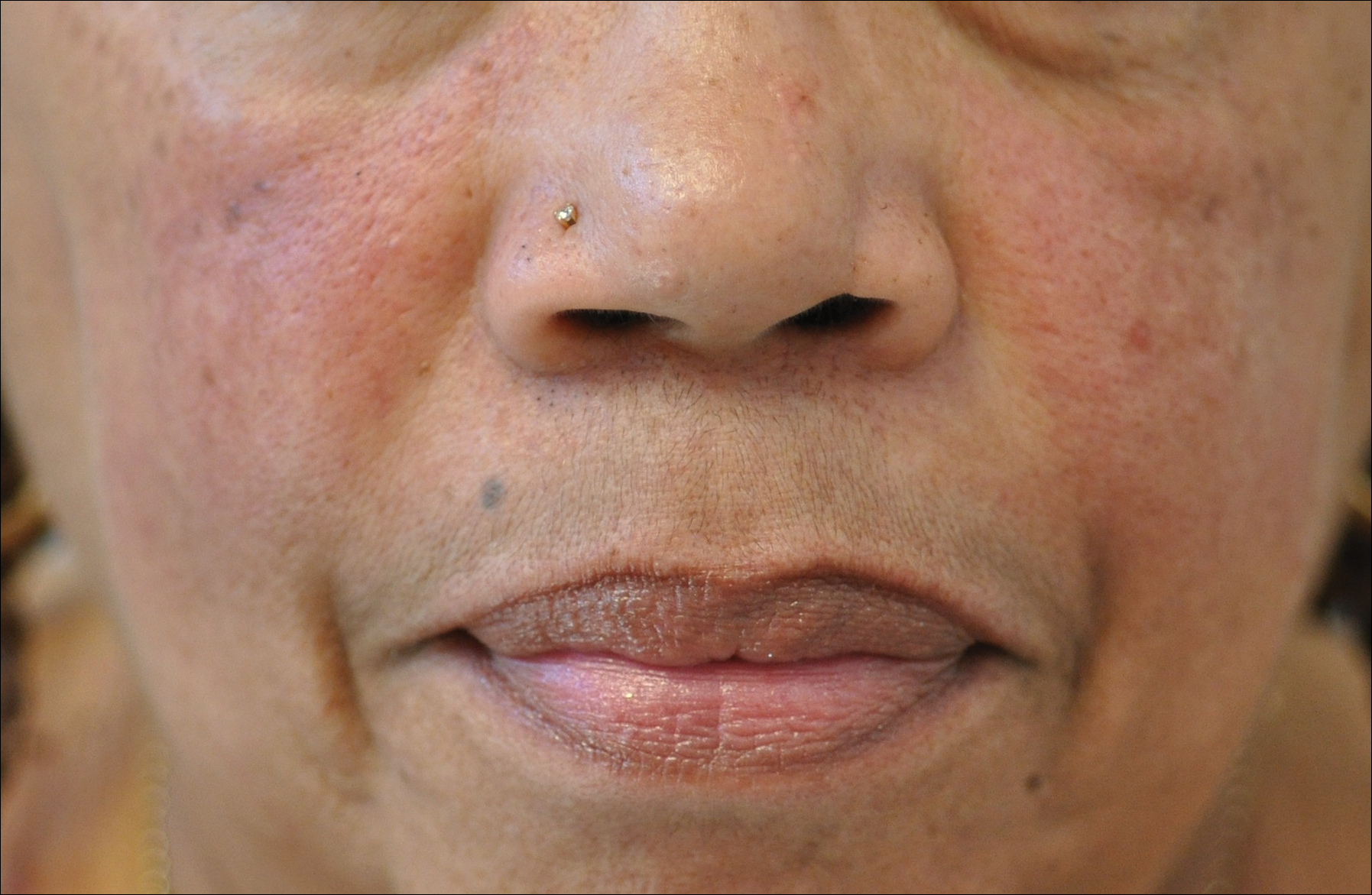
Data suggest that 4% of rosacea patients in the United States are of African, Latino, or Asian descent.11 National Ambulatory Medical Care Survey data revealed that of 31.5 million rosacea visits, 2% of patients were black, 2.3% were Asian or Pacific Islander, and 3.9% were Hispanic or Latino. In a 5-year longitudinal study of 2587 rosacea patients enrolled in Medicaid in North Carolina who were prescribed at least 1 topical treatment for rosacea, 16.27% were black and 10% were of a race other than white.17
Although the pathogenesis of rosacea is unclear, hypotheses include immune system abnormalities, neurogenic dysregulation, presence of microorganisms (eg, Demodex folliculorum), UV damage, and skin barrier dysfunction.18
The 4 major subtypes of rosacea are erythematotelangiectatic, papulopustular, phymatous, and ocular rosacea.16 Interestingly, rosacea in SOC patients may present with hypopigmentation surrounding the borders of the facial erythema. For phymatous rosacea, isotretinoin may reduce incipient rhinophyma but must be carefully monitored and pregnancy must be excluded. Surgical or laser therapy may be indicated to recontour the nose if severe.
There are several skin conditions that can present with facial erythema in patients with SOC, including seborrheic dermatitis, systemic lupus erythematosus, and contact dermatitis. It is important to note that the detection of facial erythema in darker skin types may be difficult; therefore, laboratory evaluation (antinuclear antibodies), patch testing, and skin biopsy should be considered if the clinical diagnosis is unclear.
Treatment
Treatment of rosacea in SOC patients does not differ from other racial groups. Common strategies include gentle skin care, sun protection (sun protection factor 30+), and barrier repair creams. Topical agents include metronidazole, AZA, sodium sulfacetamide/sulfur, ivermectin, and retinoids.16 Oral treatments include antibiotics in the tetracycline family (eg, subantimicrobial dose doxycycline) and isotretinoin.16 Persistent erythema associated with rosacea can be treated with brimonidine19 and oxymetazoline.20 Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea21; however, the latter is not recommended in Fitzpatrick skin types IV through VI.
Facial Hyperpigmentation in SOC Patients
Hyperpigmentation disorders can be divided into conditions that affect Fitzpatrick skin types I through III and IV though VI. Mottled hyperpigmentation (photodamage) and solar lentigines occur in patients with lighter skin types as compared to melasma, PIH, and age-related (UV-induced) hyperpigmentation, which occur more commonly in patients with darker skin types. Facial hyperpigmentation is a common concern in SOC patients. In a survey of cosmetic concerns of 100 women with SOC, hyperpigmentation or dark spots (86%) and blotchy uneven skin (80%) were the top concerns.22 In addition, facial hyperpigmentation has been shown to negatively impact quality of life.23
Postinflammatory hyperpigmentation occurs from a pathophysiological response to inflammation, cutaneous irritation or injury, and subsequent melanocyte lability. Postinflammatory hyperpigmentation is a common presenting concern in patients with SOC and is seen as a result of many inflammatory skin disorders (eg, acne, eczema) and dermatologic procedures (eg, adverse reaction to electrodesiccation, microdermabrasion, chemical peels, laser surgery).24
Melasma is an acquired idiopathic disorder of hyperpigmentation and often referred to as the mask of pregnancy (Figure 3). It occurs on sun-exposed areas of skin, mainly in women with Fitzpatrick skin types III through V. Associated factors or triggers include pregnancy, hormonal treatments, exposure to UV radiation, and medications.25 Hereditary factors play a role in more than 40% of cases.26
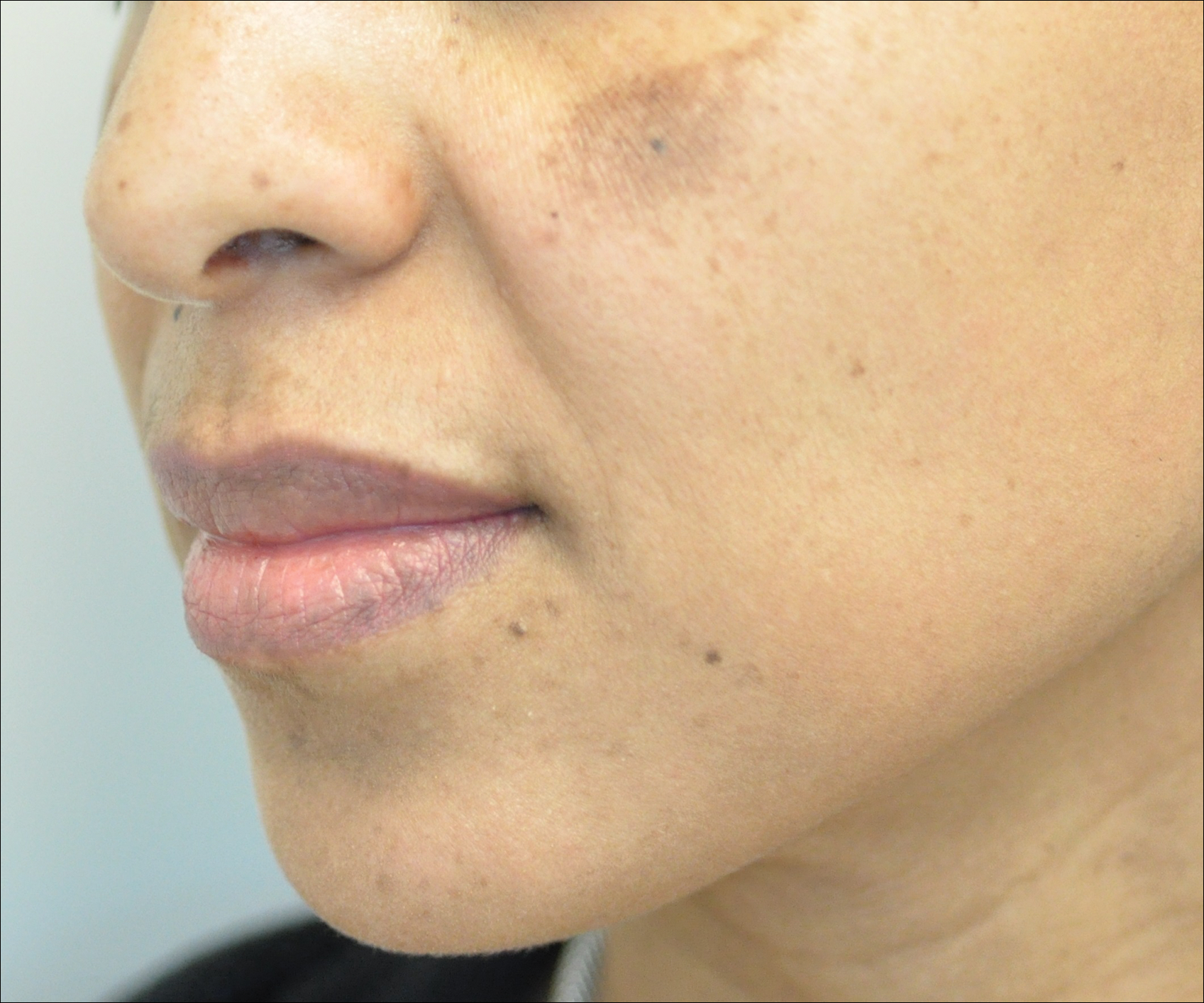
Other not-so-common facial dyschromias include contact dermatitis, acanthosis nigricans, exogenous ochronosis, lichen planus pigmentosus (associated with frontal fibrosing alopecia),27 drug-induced hyperpigmentation (associated with minocycline or diltiazem),28,29 and UV-induced (age-related) hyperpigmentation.
Treatment
The treatment of hyperpigmentation should provide the following: (1) protection from sun exposure; (2) inhibition of tyrosinase, the enzyme responsible for the conversion of tyrosine to melanin; (3) inhibition of melanosome transfer from the melanocyte to the keratinocyte; (4) removal of melanin from the epidermis through exfoliation; and (5) destruction or disruption of melanin in the dermis.30 Therapies for facial hyperpigmentation are listed in Table 1.

Topical therapies include prescription medications and nonprescription cosmeceuticals. Prescription medications include hydroquinone (HQ), topical retinoids, and AZA. Hydroquinone, a tyrosinase inhibitor, is the gold standard for skin lightening and often is used as a first-line therapy. It is used as a monotherapy (HQ 4%) or as a fixed combination with tretinoin 0.05% and fluocinolone 0.01%.31 Use caution with HQ in high concentrations (6% and higher) and low concentrations (2% [over-the-counter strength]) used long-term due to the potential risk of exogenous ochronosis.
Topical retinoids have been shown to be effective therapeutic agents for melasma and PIH. Tretinoin,32 tazarotene,33 and adapalene34 all have demonstrated efficacy for acne and acne-induced PIH in SOC patients. Patients must be monitored for the development of retinoid dermatitis and worsening of hyperpigmentation.
Azelaic acid is a naturally occurring dicarboxylic acid obtained from cultures of Malassezia furfur. Azelaic acid inhibits tyrosinase activity, DNA synthesis, and mitochondrial enzymes, thus blocking direct cytotoxic effects toward melanocytes. Azelaic acid is approved by the US Food and Drug Administration for acne in a 20% cream formulation and rosacea in 15% gel and foam formulations, and it is used off label for melasma and PIH.35
Oral tranexamic acid is currently used as a hemostatic agent due to its ability to inhibit the plasminogen-plasmin pathway. In melasma, it blocks the interaction between melanocytes and keratinocytes in the epidermis and modulates the vascular component of melasma in the dermis. In an open-label study, 561 Asian melasma patients were treated with oral tranexamic acid 250 mg twice daily for 4 months. Results demonstrated improvement in 90% of patients, and 7.1% reported adverse effects (eg, abdominal bloating and pain, nausea, vomiting, headache, tinnitus, numbness, menstrual irregularities).36 Coagulation screening should be monitored monthly, and any patient with a history of clotting abnormalities should be excluded from off-label treatment with oral tranexamic acid.
Nonprescription cosmeceuticals are available over-the-counter or are office dispensed.37 For optimal results, cosmeceutical agents for skin lightening are used in combination. Most of these combinations are HQ free and have additive benefits such as a multimodal skin lightening agent containing key ingredients that correct and prevent skin pigmentation via several pathways affecting melanogenesis.38 It is an excellent alternative to HQ for mottled and diffuse UV-induced hyperpigmentation and can be used for maintenance therapy in patients with melasma.
Photoprotection is an essential component of therapy for melasma and PIH, but there is a paucity of data on the benefits for SOC patients. Halder et al39 performed a randomized prospective study of 89 black and Hispanic patients who applied sunscreen with a sun protection factor of 30 or 60 daily for 8 weeks. Clinical grading, triplicate L*A*B chromameter, and clinical photography were taken at baseline and weeks 4 and 8. The results demonstrated skin lightening in both black and Hispanic patients and support the use of sunscreen in the prevention and management of dyschromia in SOC patients.39 Visible light also may play a role in melasma development, and thus use of sunscreens or makeup containing iron oxides are recommended.40
Procedural treatments for facial hyperpigmentation include microdermabrasion, chemical peels, lasers, energy-based devices, and microneedling. There are many types and formulations of chemical peeling agents available; however, superficial and medium-depth chemical peels are recommended for SOC patients (Table 2). Deep chemical peels are not recommended for SOC patients due to the potential increased risk for PIH and scarring.

Cosmetic Enhancement in SOC Patients
Cosmetic procedures are gaining popularity in the SOC population and account for more than 20% of cosmetic procedures in the United States.41 Facial cosmetic concerns in SOC include dyschromia, benign growths (dermatosis papulosa nigra), hyperkinetic facial lines, volume loss, and skin laxity.42 Key principles to consider when treating SOC patients are the impact of ethnicity on aging and facial structure, the patient’s desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient’s expectations for recommended therapies.
Aging in SOC Patients
Skin aging can be classified as intrinsic aging or extrinsic aging. Intrinsic aging is genetic and involves subsurface changes such as volume loss, muscle atrophy, and resorption of bony structure. Extrinsic aging (or photoaging) involves surface changes of the epidermis/dermis and manifests as mottled pigmentation, textural changes, and fine wrinkling. Due to the photoprotection of melanin (black skin=SPF 13.4), skin aging in SOC patients is delayed by 10 to 20 years.43 In addition, SOC patients have more reactive collagen and can benefit from noninvasive cosmetic procedures such as fillers and skin-tightening procedures.42
Cosmetic Treatments and Procedures
Dermatosis papulosa nigra (benign growths of skin that have a genetic predisposition)44 occur mainly on the face but can involve the entire body. Treatment modalities include electrodesiccation, cryotherapy, scissor excision, and laser surgery.45
Treatment of hyperkinetic facial lines with botulinum toxin type A is a safe and effective procedure in patients with SOC. Grimes and Shabazz46 performed a 4-month, randomized, double-blind study that evaluated the treatment of glabellar lines in women with Fitzpatrick skin types V and VI. The results demonstrated that the duration of effects was the same in the patients who received either 20 or 30 U of botulinum toxin type A.46 Dynamic rhytides (furrows and frown/scowl lines arising from laughing, frowning, or smiling) can be treated safely in patients with SOC using botulinum toxin type A off label for relaxation of the upper and lower hyperkinetic muscles that result in these unwanted signs of aging. Botulinum toxin type A often is used for etched-in crow’s-feet, which rarely are evident in SOC patients.47 Facial shaping also can be accomplished by injecting botulinum toxin type A in combination with soft-tissue dermal fillers.47
Although black individuals do not experience perioral rhytides at the frequency of white individuals, they experience a variety of other cosmetic issues related to skin sagging and sinking. Currently available hyaluronic acid (HA) fillers have been shown to be safe in patients with Fitzpatrick skin types IV through VI.48 Two studies evaluated fillers in patients with SOC, specifically HA49 and calcium hydroxylapatite,50 focused on treatment of the nasolabial folds and the potential risk for dyspigmentation and keloidal scarring. Taylor et al49 noted that the risk of hyperpigmentation was 6% to 9% for large- and small-particle HA, respectively, and was associated with the serial or multiple puncture injection technique. No hypertrophic or keloidal scarring occurred in both studies.49,50
Facial contouring applications with fillers include glabellar lines, temples, nasal bridge, tear troughs, malar and submalar areas, nasolabial folds, radial lines, lips, marionette lines, mental crease, and chin. Hyaluronic acid fillers also can be used for lip enhancement.47 Although white women are looking to increase the size of their lips, black women are seeking augmentation to restore their lip size to that of their youth. Black individuals do not experience the same frequency of perioral rhytides as white patients, but they experience a variety of other issues related to skin sagging and sinking. Unlike white women, enhancement of the vermilion border rarely is performed in black women due to development of rhytides, predominantly in the body of the lip below the vermilion border in response to volume loss in the upper lip while the lower lip usually maintains its same appearance.47
Facial enhancement utilizing poly-L-lactic acid can be used safely in SOC patients.51 Poly-L-lactic acid microparticles induce collagen formation, leading to dermal thickening over 3 to 6 months; however, multiple sessions are required to achieve optimal aesthetic results.
Patients with more reactive collagen can benefit from noninvasive cosmetic procedures such as skin-tightening procedures.52 Radiofrequency and microfocused ultrasound are cosmetic procedures used to provide skin tightening and facial lifting. They are safe and effective treatments for patients with Fitzpatrick skin types IV to VI.53 Histologically, there is less thinning of collagen bundles and elastic tissue in ethnic skin. Due to stimulation of collagen by these procedures, most SOC patients will experience a more enhanced response, requiring fewer treatment sessions than white individuals.
Conclusion
Medical and aesthetic facial concerns in SOC patients vary and can be a source of emotional and psychological distress that can negatively impact quality of life. The approach to the treatment of SOC patients should be a balance between tolerability and efficacy, considering the potential risk for PIH.
- White GM. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J Am Acad Dermatol. 1998;39(2 pt 3):S34-S37.
- Halder RM, Grimes PE, McLaurin CL, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasians, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
- Halder RM, Holmes YC, Bridgeman-Shah S, et al. A clinicohistologic study of acne vulgaris in black females (abstract). J Invest Dermatol. 1996;106:888.
- Plewig G, Fulton JE, Kligman AM. Pomade acne. Arch Dermatol. 1970;101:580-584.
- Kligman AM, Mills OH. Acne cosmetica. Arch Dermatol. 1972;106:893-897.
- Halder RM, Brooks HL, Callender VD. Acne in ethnic skin. Dermatol Clin. 2003;21:609-615.
- Callender VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Winhoven SM. Postinflammatory hyperpigmentation in an Asian patient. a dramatic response to oral isotretinoin (13-cis-retinoic acid). Br J Med. 2005;152:368-403.
- Sarkar R, Bansal S, Garg VK. Chemical peels for melasma in dark-skinned patients. J Cutan Aesthet Surg. 2012;5:247-253.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Culp B, Scheinfeld N. Rosacea: a review. P T. 2009;34:38-45.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014:20. pii:13030/qt1mv9r0ss.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Jackson JM, Knuckles M, Minni JP, et al. The role of brimonidine tartrate gel in the treatment of rosacea. Clin Cosmet Investig Dermatol. 2015;23:529-538.
- Patel NU, Shukla S, Zaki J, et al. Oxymetazoline hydrochloride cream for facial erythema associated with rosacea. Expert Rev Clin Pharmacol. 2017;10:104954.
- Weinkle AP, Doktor V, Emer J. Update on the management of rosacea. Clin Cosmet Investig Dermatol. 2015;8:159-177.
- Grimes PE. Skin and hair cosmetic issues in women of color. Dermatol Clin. 2000;19:659-665.
- Taylor A, Pawaskar M, Taylor SL, et al. Prevalence of pigmentary disorders and their impact on quality of life: a prospective cohort study. J Cosmet Dermatol. 2008;7:164-168.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Grimes PE. Melasma: etiologic and therapeutic considerations. Arch Dermatol. 1995;131:1453-1457.
- Handel AC, Miot LD, Miot HA. Melasma: a clinical and epidemiological review. An Bras Dermatol. 2014;89:771-782.
- Callender VD, Reid SD, Obayan O, et al. Diagnostic clues to frontal fibrosing alopecia in patients of African descent. J Clin Aesthet Dermatol. 2016;9:45-51.
- Narang T, Sawatkar GU, Kumaran MS, et al. Minocycline for recurrent and/or chronic erythema nodosum leprosum. JAMA Dermatol. 2015;151:1026-1028.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10.
- Pandya AG, Guevara IL. Disorders of hyperpigmentation. Dermatol Clin. 2000;18:91-98.
- Taylor SC, Torok H, Jones T, et al. Efficacy and safety of a new triple-combination agent for the treatment of facial melasma. Cutis. 2003;72:67-72.
- Bulengo-Ransby SM. Topical tretinoin (retinoic acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Jacyk WK. Adapalene in the treatment of African patients. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):37-42.
- Kircik LH. Efficacy and safety of azelaic acid (AzA) gel 15% in the treatment of postinflammatory hyperpigmentation and acne: a 16-week, baseline-controlled study. J Drugs Dermatol. 2011;10:586-590.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma. J Am Acad Dermatol. 2016;75:385-392.
- Kindred C, Okereke U, Callender VD. Skin-lightening agents: an overview of prescription, office-dispensed, and over-the-counter products. Cosmet Dermatol. 2013;26:18-26.
- Makino ET, Kadoya K, Sigler ML, et al. Development and clinical assessment of a comprehensive product for pigmentation control in multiple ethnic populations. J Drugs Dermatol. 2016;15:1562-1570.
- Halder R, Rodney I, Munhutu M, et al. Evaluation and effectiveness of a photoprotection composition (sunscreen) on subjects of skin of color. J Am Acad Dermatol. 2015;72(suppl 1):AB215.
- Castanedo-Cazares JP, Hernandez-Blanco D, Carlos-Ortega B, et al. Near-visible light and UV photoprotection in the treatment of melasma: a double-blind randomized trial. Photodermatol Photoimmunol Photomed. 2014;30:35-42.
- American Society for Aesthetic Plastic Surgery. 2016 Cosmetic Surgery National Data Bank Statistics. https://www.surgery.org/sites/default/files/ASAPS-Stats2016.pdf. Accessed November 15, 2017.
- Burgess CM. Soft tissue augmentation in skin of color: market growth, available fillers and successful techniques. J Drugs Dermatol. 2007;6:51-55.
- Davis EC, Callender VD. Aesthetic dermatology for aging ethnic skin. Dermatol Surg. 2011;37:901-917.
- Grimes PE, Arora S, Minus HR, et al. Dermatosis papulosa nigra. Cutis. 1983;32:385-386.
- Lupo M. Dermatosis papulosa nigra: treatment options. J Drugs Dermatol. 2007;6:29-30.
- Grimes PE, Shabazz D. A four-month randomized, double-blind evaluation of the efficacy of botulinum toxin type A for the treatment of glabellar lines in women with skin types V and VI. Dermatol Surg. 2009;35:429-435.
- Burgess CM, Awosika O. Ethnic and gender considerations in the use of facial injectables: African-American patients. Plast Reconstr Surg. 2015;136(5 suppl):28S-31S.
- Taylor SC, Kelly AP, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. New York, NY: McGraw-Hill Education; 2016.
- Taylor SC, Burgess CM, Callender VD. Safety of nonanimal stabilized hyaluronic acid dermal fillers in patients with skin of color: a randomized, evaluator-blinded comparative trial. Dermatol Surg. 2009;35(suppl 2):1653-1660.
- Marmur ES, Taylor SC, Grimes PE, et al. Six-month safety results of calcium hydroxylapatite for treatment of nasolabial folds in Fitzpatrick skin types IV to VI. Dermatol Surg. 2009;35(suppl 2):1641-1645.
- Hamilton TK, Burgess CM. Consideration for the use of injectable poly-L-lactic acid in people of color. J Drugs Dermatol. 2010;9:451-456.
- Fabi SG, Goldman MP. Retrospective evaluation of micro-focused ultrasound for lifting and tightening of the face and neck. Dermatol Surg. 2014;40:569-575.
- Harris MO, Sundaram HA. Safety of microfocused ultrasound with visualization in patients with Fitzpatrick skin phototypes III to VI. JAMA Facial Plast Surg. 2015;17:355-357.
The approach to the treatment of common skin disorders and cosmetic concerns in patients with skin of color (SOC) requires the clinician to understand the biological differences, nuances, and special considerations that are unique to patients with darker skin types.1-3 This article addresses 4 common facial concerns in SOC patients—acne, rosacea, facial hyperpigmentation, and cosmetic enhancement—and provides treatment recommendations and management pearls to assist the clinician with optimal outcomes for SOC patients.
Acne in SOC Patients
Acne vulgaris is one of the most common conditions that dermatologists treat and is estimated to affect 40 to 50 million individuals in the United States.1 Many of these acne patients are individuals with SOC.2-4 A study of 2835 females (aged 10–70 years) conducted in 4 different cities—Los Angeles, California; London, United Kingdom; Akita, Japan; and Rome, Italy—demonstrated acne prevalence of 37% in blacks, 32% in Hispanics, 30% in Asians, 24% in whites, and 23% in Continental Indians.5 Blacks, Hispanics, and Continental Indians demonstrated equal prevalence with comedonal and inflammatory acne. Asians displayed more inflammatory acne lesions than comedones. In contrast, whites demonstrated more comedones than inflammatory acne. Dyspigmentation, postinflammatory hyperpigmentation (PIH), and atrophic scars were more common in black and Hispanic females than other ethnicities.5 This study illustrated that acne-induced PIH is a common sequela in SOC patients and is the main reason they seek treatment.6,7
The pathogenesis of acne is the same in all racial and ethnic groups: (1) follicular hyperkeratinization and the formation of a microcomedone caused by abnormal desquamation of the keratinocytes within the sebaceous follicle, (2) production of sebum by circulating androgens, (3) proliferation of Propionibacterium acnes, and (4) inflammation. Subclinical inflammation is present throughout all stages of acne, including normal-appearing skin, inflammatory lesions, comedones, and scarring, and may contribute to PIH in acne patients with SOC (Figure 1).8 A thorough history should be obtained from acne patients, including answers to the following questions7:
- What skin and hair care products do you use?
- Do you use sunscreen daily?
- What cosmetic products or makeup do you use?
- Do you use any ethnic skin care products, including skin lightening creams?
- Do you have a history of keloids?
It is important to ask these questions to assess if the SOC patient has developed pomade acne,9 acne cosmetica,10 or a potential risk of skin irritation from the use of skin care practices. It is best to take total control of the patient’s skin care regimen and discontinue use of toners, astringents, witch hazel, exfoliants, and rubbing alcohol, which may lead to skin dryness and irritation, particularly when combined with topical acne medications.
Treatment
Treatment of acne in SOC patients is similar to generally recommended treatments, with special considerations. Consider the following key points when treating acne in SOC patients:
- Treat acne early and aggressively to prevent or minimize subsequent PIH and acne scarring.
- Balance aggressive treatment with nonirritating topical skin care.
- Most importantly, target PIH in addition to acne and choose a regimen that limits skin irritation that might exacerbate existing PIH.7
Develop a maintenance program to control future breakouts. Topical agents can be used as monotherapy or in fixed combinations and may include benzoyl peroxide, antibiotics, dapsone, azelaic acid (AZA), and retinoids. Similar to white patients, topical retinoids remain a first-line treatment for acne in patients with SOC.11,12
Tolerability must be managed in SOC acne patients. Therapeutic maneuvers that can be instituted should include a discussion on using gentle skin care, initiating therapy with a retinoid applied every other night starting with a low concentration and gradually titrating up, and applying a moisturizer before or after applying acne medication. Oral therapies consist of antibiotics (doxycycline, minocycline), retinoids (isotretinoin), and hormonal modulators (oral contraceptives, spironolactone). Isotretinoin, recommended for patients with nodulocystic acne, may play a possible role in treating acne-induced PIH.13
Two common procedural therapies for acne include comedone extraction and intralesional corticosteroid injection. A 6- to 8-week course of a topical retinoid prior to comedonal extraction may facilitate the procedure and is recommended in SOC patients to help reduce cutaneous trauma and PIH.11 Inflammatory acne lesions can be treated with intralesional injection of triamcinolone acetonide 2.5 or 5.0 mg/mL, which usually reduces inflammation within 2 to 5 days.11
Treatment of acne-induced PIH includes sun protection, topical and oral medications, chemical peels, lasers, and energy devices. Treatment of hypertrophic scarring and keloids involves intralesional injection of triamcinolone acetonide 20, 30, or 40 mg/mL every 4 weeks until the lesion is flat.11
Superficial chemical peels can be used to treat acne and PIH in SOC patients,14 such as salicylic acid (20%–30%), glycolic acid (20%–70%), trichloroacetic acid (15%–30%), and Jessner peels.
Acne Scarring
Surgical approaches to acne scarring in patients with SOC include elliptical excision, punch excision, punch elevation, punch autografting, dermal grafting, dermal planning, subcutaneous incision (subcision), dermabrasion, microneedling, fillers, and laser skin resurfacing. The treatment of choice depends on the size, type, and depth of the scar and the clinician’s preference.
Lasers
Fractional photothermolysis has emerged as a treatment option for acne scars in SOC patients. This procedure produces microscopic columns of thermal injury in the epidermis and dermis, sparing the surrounding tissue and minimizing downtime and adverse events. Because fractional photothermolysis does not target melanin and produces limited epidermal injury, darker Fitzpatrick skin types (IV–VI) can be safely and effectively treated with this procedure.15
Rosacea in SOC Patients
Rosacea is a chronic inflammatory disorder that affects the vasculature and pilosebaceous units of the face. It commonly is seen in Fitzpatrick skin types I and II; however, rosacea can occur in all skin types (Figure 2). Triggers include emotional stress, extreme environmental temperatures, hot and spicy foods, red wine or alcohol, and topical irritants or allergens found in common cosmetic products.16
Data suggest that 4% of rosacea patients in the United States are of African, Latino, or Asian descent.11 National Ambulatory Medical Care Survey data revealed that of 31.5 million rosacea visits, 2% of patients were black, 2.3% were Asian or Pacific Islander, and 3.9% were Hispanic or Latino. In a 5-year longitudinal study of 2587 rosacea patients enrolled in Medicaid in North Carolina who were prescribed at least 1 topical treatment for rosacea, 16.27% were black and 10% were of a race other than white.17
Although the pathogenesis of rosacea is unclear, hypotheses include immune system abnormalities, neurogenic dysregulation, presence of microorganisms (eg, Demodex folliculorum), UV damage, and skin barrier dysfunction.18
The 4 major subtypes of rosacea are erythematotelangiectatic, papulopustular, phymatous, and ocular rosacea.16 Interestingly, rosacea in SOC patients may present with hypopigmentation surrounding the borders of the facial erythema. For phymatous rosacea, isotretinoin may reduce incipient rhinophyma but must be carefully monitored and pregnancy must be excluded. Surgical or laser therapy may be indicated to recontour the nose if severe.
There are several skin conditions that can present with facial erythema in patients with SOC, including seborrheic dermatitis, systemic lupus erythematosus, and contact dermatitis. It is important to note that the detection of facial erythema in darker skin types may be difficult; therefore, laboratory evaluation (antinuclear antibodies), patch testing, and skin biopsy should be considered if the clinical diagnosis is unclear.
Treatment
Treatment of rosacea in SOC patients does not differ from other racial groups. Common strategies include gentle skin care, sun protection (sun protection factor 30+), and barrier repair creams. Topical agents include metronidazole, AZA, sodium sulfacetamide/sulfur, ivermectin, and retinoids.16 Oral treatments include antibiotics in the tetracycline family (eg, subantimicrobial dose doxycycline) and isotretinoin.16 Persistent erythema associated with rosacea can be treated with brimonidine19 and oxymetazoline.20 Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea21; however, the latter is not recommended in Fitzpatrick skin types IV through VI.
Facial Hyperpigmentation in SOC Patients
Hyperpigmentation disorders can be divided into conditions that affect Fitzpatrick skin types I through III and IV though VI. Mottled hyperpigmentation (photodamage) and solar lentigines occur in patients with lighter skin types as compared to melasma, PIH, and age-related (UV-induced) hyperpigmentation, which occur more commonly in patients with darker skin types. Facial hyperpigmentation is a common concern in SOC patients. In a survey of cosmetic concerns of 100 women with SOC, hyperpigmentation or dark spots (86%) and blotchy uneven skin (80%) were the top concerns.22 In addition, facial hyperpigmentation has been shown to negatively impact quality of life.23
Postinflammatory hyperpigmentation occurs from a pathophysiological response to inflammation, cutaneous irritation or injury, and subsequent melanocyte lability. Postinflammatory hyperpigmentation is a common presenting concern in patients with SOC and is seen as a result of many inflammatory skin disorders (eg, acne, eczema) and dermatologic procedures (eg, adverse reaction to electrodesiccation, microdermabrasion, chemical peels, laser surgery).24
Melasma is an acquired idiopathic disorder of hyperpigmentation and often referred to as the mask of pregnancy (Figure 3). It occurs on sun-exposed areas of skin, mainly in women with Fitzpatrick skin types III through V. Associated factors or triggers include pregnancy, hormonal treatments, exposure to UV radiation, and medications.25 Hereditary factors play a role in more than 40% of cases.26
Other not-so-common facial dyschromias include contact dermatitis, acanthosis nigricans, exogenous ochronosis, lichen planus pigmentosus (associated with frontal fibrosing alopecia),27 drug-induced hyperpigmentation (associated with minocycline or diltiazem),28,29 and UV-induced (age-related) hyperpigmentation.
Treatment
The treatment of hyperpigmentation should provide the following: (1) protection from sun exposure; (2) inhibition of tyrosinase, the enzyme responsible for the conversion of tyrosine to melanin; (3) inhibition of melanosome transfer from the melanocyte to the keratinocyte; (4) removal of melanin from the epidermis through exfoliation; and (5) destruction or disruption of melanin in the dermis.30 Therapies for facial hyperpigmentation are listed in Table 1.
Topical therapies include prescription medications and nonprescription cosmeceuticals. Prescription medications include hydroquinone (HQ), topical retinoids, and AZA. Hydroquinone, a tyrosinase inhibitor, is the gold standard for skin lightening and often is used as a first-line therapy. It is used as a monotherapy (HQ 4%) or as a fixed combination with tretinoin 0.05% and fluocinolone 0.01%.31 Use caution with HQ in high concentrations (6% and higher) and low concentrations (2% [over-the-counter strength]) used long-term due to the potential risk of exogenous ochronosis.
Topical retinoids have been shown to be effective therapeutic agents for melasma and PIH. Tretinoin,32 tazarotene,33 and adapalene34 all have demonstrated efficacy for acne and acne-induced PIH in SOC patients. Patients must be monitored for the development of retinoid dermatitis and worsening of hyperpigmentation.
Azelaic acid is a naturally occurring dicarboxylic acid obtained from cultures of Malassezia furfur. Azelaic acid inhibits tyrosinase activity, DNA synthesis, and mitochondrial enzymes, thus blocking direct cytotoxic effects toward melanocytes. Azelaic acid is approved by the US Food and Drug Administration for acne in a 20% cream formulation and rosacea in 15% gel and foam formulations, and it is used off label for melasma and PIH.35
Oral tranexamic acid is currently used as a hemostatic agent due to its ability to inhibit the plasminogen-plasmin pathway. In melasma, it blocks the interaction between melanocytes and keratinocytes in the epidermis and modulates the vascular component of melasma in the dermis. In an open-label study, 561 Asian melasma patients were treated with oral tranexamic acid 250 mg twice daily for 4 months. Results demonstrated improvement in 90% of patients, and 7.1% reported adverse effects (eg, abdominal bloating and pain, nausea, vomiting, headache, tinnitus, numbness, menstrual irregularities).36 Coagulation screening should be monitored monthly, and any patient with a history of clotting abnormalities should be excluded from off-label treatment with oral tranexamic acid.
Nonprescription cosmeceuticals are available over-the-counter or are office dispensed.37 For optimal results, cosmeceutical agents for skin lightening are used in combination. Most of these combinations are HQ free and have additive benefits such as a multimodal skin lightening agent containing key ingredients that correct and prevent skin pigmentation via several pathways affecting melanogenesis.38 It is an excellent alternative to HQ for mottled and diffuse UV-induced hyperpigmentation and can be used for maintenance therapy in patients with melasma.
Photoprotection is an essential component of therapy for melasma and PIH, but there is a paucity of data on the benefits for SOC patients. Halder et al39 performed a randomized prospective study of 89 black and Hispanic patients who applied sunscreen with a sun protection factor of 30 or 60 daily for 8 weeks. Clinical grading, triplicate L*A*B chromameter, and clinical photography were taken at baseline and weeks 4 and 8. The results demonstrated skin lightening in both black and Hispanic patients and support the use of sunscreen in the prevention and management of dyschromia in SOC patients.39 Visible light also may play a role in melasma development, and thus use of sunscreens or makeup containing iron oxides are recommended.40
Procedural treatments for facial hyperpigmentation include microdermabrasion, chemical peels, lasers, energy-based devices, and microneedling. There are many types and formulations of chemical peeling agents available; however, superficial and medium-depth chemical peels are recommended for SOC patients (Table 2). Deep chemical peels are not recommended for SOC patients due to the potential increased risk for PIH and scarring.
Cosmetic Enhancement in SOC Patients
Cosmetic procedures are gaining popularity in the SOC population and account for more than 20% of cosmetic procedures in the United States.41 Facial cosmetic concerns in SOC include dyschromia, benign growths (dermatosis papulosa nigra), hyperkinetic facial lines, volume loss, and skin laxity.42 Key principles to consider when treating SOC patients are the impact of ethnicity on aging and facial structure, the patient’s desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient’s expectations for recommended therapies.
Aging in SOC Patients
Skin aging can be classified as intrinsic aging or extrinsic aging. Intrinsic aging is genetic and involves subsurface changes such as volume loss, muscle atrophy, and resorption of bony structure. Extrinsic aging (or photoaging) involves surface changes of the epidermis/dermis and manifests as mottled pigmentation, textural changes, and fine wrinkling. Due to the photoprotection of melanin (black skin=SPF 13.4), skin aging in SOC patients is delayed by 10 to 20 years.43 In addition, SOC patients have more reactive collagen and can benefit from noninvasive cosmetic procedures such as fillers and skin-tightening procedures.42
Cosmetic Treatments and Procedures
Dermatosis papulosa nigra (benign growths of skin that have a genetic predisposition)44 occur mainly on the face but can involve the entire body. Treatment modalities include electrodesiccation, cryotherapy, scissor excision, and laser surgery.45
Treatment of hyperkinetic facial lines with botulinum toxin type A is a safe and effective procedure in patients with SOC. Grimes and Shabazz46 performed a 4-month, randomized, double-blind study that evaluated the treatment of glabellar lines in women with Fitzpatrick skin types V and VI. The results demonstrated that the duration of effects was the same in the patients who received either 20 or 30 U of botulinum toxin type A.46 Dynamic rhytides (furrows and frown/scowl lines arising from laughing, frowning, or smiling) can be treated safely in patients with SOC using botulinum toxin type A off label for relaxation of the upper and lower hyperkinetic muscles that result in these unwanted signs of aging. Botulinum toxin type A often is used for etched-in crow’s-feet, which rarely are evident in SOC patients.47 Facial shaping also can be accomplished by injecting botulinum toxin type A in combination with soft-tissue dermal fillers.47
Although black individuals do not experience perioral rhytides at the frequency of white individuals, they experience a variety of other cosmetic issues related to skin sagging and sinking. Currently available hyaluronic acid (HA) fillers have been shown to be safe in patients with Fitzpatrick skin types IV through VI.48 Two studies evaluated fillers in patients with SOC, specifically HA49 and calcium hydroxylapatite,50 focused on treatment of the nasolabial folds and the potential risk for dyspigmentation and keloidal scarring. Taylor et al49 noted that the risk of hyperpigmentation was 6% to 9% for large- and small-particle HA, respectively, and was associated with the serial or multiple puncture injection technique. No hypertrophic or keloidal scarring occurred in both studies.49,50
Facial contouring applications with fillers include glabellar lines, temples, nasal bridge, tear troughs, malar and submalar areas, nasolabial folds, radial lines, lips, marionette lines, mental crease, and chin. Hyaluronic acid fillers also can be used for lip enhancement.47 Although white women are looking to increase the size of their lips, black women are seeking augmentation to restore their lip size to that of their youth. Black individuals do not experience the same frequency of perioral rhytides as white patients, but they experience a variety of other issues related to skin sagging and sinking. Unlike white women, enhancement of the vermilion border rarely is performed in black women due to development of rhytides, predominantly in the body of the lip below the vermilion border in response to volume loss in the upper lip while the lower lip usually maintains its same appearance.47
Facial enhancement utilizing poly-L-lactic acid can be used safely in SOC patients.51 Poly-L-lactic acid microparticles induce collagen formation, leading to dermal thickening over 3 to 6 months; however, multiple sessions are required to achieve optimal aesthetic results.
Patients with more reactive collagen can benefit from noninvasive cosmetic procedures such as skin-tightening procedures.52 Radiofrequency and microfocused ultrasound are cosmetic procedures used to provide skin tightening and facial lifting. They are safe and effective treatments for patients with Fitzpatrick skin types IV to VI.53 Histologically, there is less thinning of collagen bundles and elastic tissue in ethnic skin. Due to stimulation of collagen by these procedures, most SOC patients will experience a more enhanced response, requiring fewer treatment sessions than white individuals.
Conclusion
Medical and aesthetic facial concerns in SOC patients vary and can be a source of emotional and psychological distress that can negatively impact quality of life. The approach to the treatment of SOC patients should be a balance between tolerability and efficacy, considering the potential risk for PIH.
The approach to the treatment of common skin disorders and cosmetic concerns in patients with skin of color (SOC) requires the clinician to understand the biological differences, nuances, and special considerations that are unique to patients with darker skin types.1-3 This article addresses 4 common facial concerns in SOC patients—acne, rosacea, facial hyperpigmentation, and cosmetic enhancement—and provides treatment recommendations and management pearls to assist the clinician with optimal outcomes for SOC patients.
Acne in SOC Patients
Acne vulgaris is one of the most common conditions that dermatologists treat and is estimated to affect 40 to 50 million individuals in the United States.1 Many of these acne patients are individuals with SOC.2-4 A study of 2835 females (aged 10–70 years) conducted in 4 different cities—Los Angeles, California; London, United Kingdom; Akita, Japan; and Rome, Italy—demonstrated acne prevalence of 37% in blacks, 32% in Hispanics, 30% in Asians, 24% in whites, and 23% in Continental Indians.5 Blacks, Hispanics, and Continental Indians demonstrated equal prevalence with comedonal and inflammatory acne. Asians displayed more inflammatory acne lesions than comedones. In contrast, whites demonstrated more comedones than inflammatory acne. Dyspigmentation, postinflammatory hyperpigmentation (PIH), and atrophic scars were more common in black and Hispanic females than other ethnicities.5 This study illustrated that acne-induced PIH is a common sequela in SOC patients and is the main reason they seek treatment.6,7
The pathogenesis of acne is the same in all racial and ethnic groups: (1) follicular hyperkeratinization and the formation of a microcomedone caused by abnormal desquamation of the keratinocytes within the sebaceous follicle, (2) production of sebum by circulating androgens, (3) proliferation of Propionibacterium acnes, and (4) inflammation. Subclinical inflammation is present throughout all stages of acne, including normal-appearing skin, inflammatory lesions, comedones, and scarring, and may contribute to PIH in acne patients with SOC (Figure 1).8 A thorough history should be obtained from acne patients, including answers to the following questions7:
- What skin and hair care products do you use?
- Do you use sunscreen daily?
- What cosmetic products or makeup do you use?
- Do you use any ethnic skin care products, including skin lightening creams?
- Do you have a history of keloids?
It is important to ask these questions to assess if the SOC patient has developed pomade acne,9 acne cosmetica,10 or a potential risk of skin irritation from the use of skin care practices. It is best to take total control of the patient’s skin care regimen and discontinue use of toners, astringents, witch hazel, exfoliants, and rubbing alcohol, which may lead to skin dryness and irritation, particularly when combined with topical acne medications.
Treatment
Treatment of acne in SOC patients is similar to generally recommended treatments, with special considerations. Consider the following key points when treating acne in SOC patients:
- Treat acne early and aggressively to prevent or minimize subsequent PIH and acne scarring.
- Balance aggressive treatment with nonirritating topical skin care.
- Most importantly, target PIH in addition to acne and choose a regimen that limits skin irritation that might exacerbate existing PIH.7
Develop a maintenance program to control future breakouts. Topical agents can be used as monotherapy or in fixed combinations and may include benzoyl peroxide, antibiotics, dapsone, azelaic acid (AZA), and retinoids. Similar to white patients, topical retinoids remain a first-line treatment for acne in patients with SOC.11,12
Tolerability must be managed in SOC acne patients. Therapeutic maneuvers that can be instituted should include a discussion on using gentle skin care, initiating therapy with a retinoid applied every other night starting with a low concentration and gradually titrating up, and applying a moisturizer before or after applying acne medication. Oral therapies consist of antibiotics (doxycycline, minocycline), retinoids (isotretinoin), and hormonal modulators (oral contraceptives, spironolactone). Isotretinoin, recommended for patients with nodulocystic acne, may play a possible role in treating acne-induced PIH.13
Two common procedural therapies for acne include comedone extraction and intralesional corticosteroid injection. A 6- to 8-week course of a topical retinoid prior to comedonal extraction may facilitate the procedure and is recommended in SOC patients to help reduce cutaneous trauma and PIH.11 Inflammatory acne lesions can be treated with intralesional injection of triamcinolone acetonide 2.5 or 5.0 mg/mL, which usually reduces inflammation within 2 to 5 days.11
Treatment of acne-induced PIH includes sun protection, topical and oral medications, chemical peels, lasers, and energy devices. Treatment of hypertrophic scarring and keloids involves intralesional injection of triamcinolone acetonide 20, 30, or 40 mg/mL every 4 weeks until the lesion is flat.11
Superficial chemical peels can be used to treat acne and PIH in SOC patients,14 such as salicylic acid (20%–30%), glycolic acid (20%–70%), trichloroacetic acid (15%–30%), and Jessner peels.
Acne Scarring
Surgical approaches to acne scarring in patients with SOC include elliptical excision, punch excision, punch elevation, punch autografting, dermal grafting, dermal planning, subcutaneous incision (subcision), dermabrasion, microneedling, fillers, and laser skin resurfacing. The treatment of choice depends on the size, type, and depth of the scar and the clinician’s preference.
Lasers
Fractional photothermolysis has emerged as a treatment option for acne scars in SOC patients. This procedure produces microscopic columns of thermal injury in the epidermis and dermis, sparing the surrounding tissue and minimizing downtime and adverse events. Because fractional photothermolysis does not target melanin and produces limited epidermal injury, darker Fitzpatrick skin types (IV–VI) can be safely and effectively treated with this procedure.15
Rosacea in SOC Patients
Rosacea is a chronic inflammatory disorder that affects the vasculature and pilosebaceous units of the face. It commonly is seen in Fitzpatrick skin types I and II; however, rosacea can occur in all skin types (Figure 2). Triggers include emotional stress, extreme environmental temperatures, hot and spicy foods, red wine or alcohol, and topical irritants or allergens found in common cosmetic products.16
Data suggest that 4% of rosacea patients in the United States are of African, Latino, or Asian descent.11 National Ambulatory Medical Care Survey data revealed that of 31.5 million rosacea visits, 2% of patients were black, 2.3% were Asian or Pacific Islander, and 3.9% were Hispanic or Latino. In a 5-year longitudinal study of 2587 rosacea patients enrolled in Medicaid in North Carolina who were prescribed at least 1 topical treatment for rosacea, 16.27% were black and 10% were of a race other than white.17
Although the pathogenesis of rosacea is unclear, hypotheses include immune system abnormalities, neurogenic dysregulation, presence of microorganisms (eg, Demodex folliculorum), UV damage, and skin barrier dysfunction.18
The 4 major subtypes of rosacea are erythematotelangiectatic, papulopustular, phymatous, and ocular rosacea.16 Interestingly, rosacea in SOC patients may present with hypopigmentation surrounding the borders of the facial erythema. For phymatous rosacea, isotretinoin may reduce incipient rhinophyma but must be carefully monitored and pregnancy must be excluded. Surgical or laser therapy may be indicated to recontour the nose if severe.
There are several skin conditions that can present with facial erythema in patients with SOC, including seborrheic dermatitis, systemic lupus erythematosus, and contact dermatitis. It is important to note that the detection of facial erythema in darker skin types may be difficult; therefore, laboratory evaluation (antinuclear antibodies), patch testing, and skin biopsy should be considered if the clinical diagnosis is unclear.
Treatment
Treatment of rosacea in SOC patients does not differ from other racial groups. Common strategies include gentle skin care, sun protection (sun protection factor 30+), and barrier repair creams. Topical agents include metronidazole, AZA, sodium sulfacetamide/sulfur, ivermectin, and retinoids.16 Oral treatments include antibiotics in the tetracycline family (eg, subantimicrobial dose doxycycline) and isotretinoin.16 Persistent erythema associated with rosacea can be treated with brimonidine19 and oxymetazoline.20 Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea21; however, the latter is not recommended in Fitzpatrick skin types IV through VI.
Facial Hyperpigmentation in SOC Patients
Hyperpigmentation disorders can be divided into conditions that affect Fitzpatrick skin types I through III and IV though VI. Mottled hyperpigmentation (photodamage) and solar lentigines occur in patients with lighter skin types as compared to melasma, PIH, and age-related (UV-induced) hyperpigmentation, which occur more commonly in patients with darker skin types. Facial hyperpigmentation is a common concern in SOC patients. In a survey of cosmetic concerns of 100 women with SOC, hyperpigmentation or dark spots (86%) and blotchy uneven skin (80%) were the top concerns.22 In addition, facial hyperpigmentation has been shown to negatively impact quality of life.23
Postinflammatory hyperpigmentation occurs from a pathophysiological response to inflammation, cutaneous irritation or injury, and subsequent melanocyte lability. Postinflammatory hyperpigmentation is a common presenting concern in patients with SOC and is seen as a result of many inflammatory skin disorders (eg, acne, eczema) and dermatologic procedures (eg, adverse reaction to electrodesiccation, microdermabrasion, chemical peels, laser surgery).24
Melasma is an acquired idiopathic disorder of hyperpigmentation and often referred to as the mask of pregnancy (Figure 3). It occurs on sun-exposed areas of skin, mainly in women with Fitzpatrick skin types III through V. Associated factors or triggers include pregnancy, hormonal treatments, exposure to UV radiation, and medications.25 Hereditary factors play a role in more than 40% of cases.26
Other not-so-common facial dyschromias include contact dermatitis, acanthosis nigricans, exogenous ochronosis, lichen planus pigmentosus (associated with frontal fibrosing alopecia),27 drug-induced hyperpigmentation (associated with minocycline or diltiazem),28,29 and UV-induced (age-related) hyperpigmentation.
Treatment
The treatment of hyperpigmentation should provide the following: (1) protection from sun exposure; (2) inhibition of tyrosinase, the enzyme responsible for the conversion of tyrosine to melanin; (3) inhibition of melanosome transfer from the melanocyte to the keratinocyte; (4) removal of melanin from the epidermis through exfoliation; and (5) destruction or disruption of melanin in the dermis.30 Therapies for facial hyperpigmentation are listed in Table 1.
Topical therapies include prescription medications and nonprescription cosmeceuticals. Prescription medications include hydroquinone (HQ), topical retinoids, and AZA. Hydroquinone, a tyrosinase inhibitor, is the gold standard for skin lightening and often is used as a first-line therapy. It is used as a monotherapy (HQ 4%) or as a fixed combination with tretinoin 0.05% and fluocinolone 0.01%.31 Use caution with HQ in high concentrations (6% and higher) and low concentrations (2% [over-the-counter strength]) used long-term due to the potential risk of exogenous ochronosis.
Topical retinoids have been shown to be effective therapeutic agents for melasma and PIH. Tretinoin,32 tazarotene,33 and adapalene34 all have demonstrated efficacy for acne and acne-induced PIH in SOC patients. Patients must be monitored for the development of retinoid dermatitis and worsening of hyperpigmentation.
Azelaic acid is a naturally occurring dicarboxylic acid obtained from cultures of Malassezia furfur. Azelaic acid inhibits tyrosinase activity, DNA synthesis, and mitochondrial enzymes, thus blocking direct cytotoxic effects toward melanocytes. Azelaic acid is approved by the US Food and Drug Administration for acne in a 20% cream formulation and rosacea in 15% gel and foam formulations, and it is used off label for melasma and PIH.35
Oral tranexamic acid is currently used as a hemostatic agent due to its ability to inhibit the plasminogen-plasmin pathway. In melasma, it blocks the interaction between melanocytes and keratinocytes in the epidermis and modulates the vascular component of melasma in the dermis. In an open-label study, 561 Asian melasma patients were treated with oral tranexamic acid 250 mg twice daily for 4 months. Results demonstrated improvement in 90% of patients, and 7.1% reported adverse effects (eg, abdominal bloating and pain, nausea, vomiting, headache, tinnitus, numbness, menstrual irregularities).36 Coagulation screening should be monitored monthly, and any patient with a history of clotting abnormalities should be excluded from off-label treatment with oral tranexamic acid.
Nonprescription cosmeceuticals are available over-the-counter or are office dispensed.37 For optimal results, cosmeceutical agents for skin lightening are used in combination. Most of these combinations are HQ free and have additive benefits such as a multimodal skin lightening agent containing key ingredients that correct and prevent skin pigmentation via several pathways affecting melanogenesis.38 It is an excellent alternative to HQ for mottled and diffuse UV-induced hyperpigmentation and can be used for maintenance therapy in patients with melasma.
Photoprotection is an essential component of therapy for melasma and PIH, but there is a paucity of data on the benefits for SOC patients. Halder et al39 performed a randomized prospective study of 89 black and Hispanic patients who applied sunscreen with a sun protection factor of 30 or 60 daily for 8 weeks. Clinical grading, triplicate L*A*B chromameter, and clinical photography were taken at baseline and weeks 4 and 8. The results demonstrated skin lightening in both black and Hispanic patients and support the use of sunscreen in the prevention and management of dyschromia in SOC patients.39 Visible light also may play a role in melasma development, and thus use of sunscreens or makeup containing iron oxides are recommended.40
Procedural treatments for facial hyperpigmentation include microdermabrasion, chemical peels, lasers, energy-based devices, and microneedling. There are many types and formulations of chemical peeling agents available; however, superficial and medium-depth chemical peels are recommended for SOC patients (Table 2). Deep chemical peels are not recommended for SOC patients due to the potential increased risk for PIH and scarring.
Cosmetic Enhancement in SOC Patients
Cosmetic procedures are gaining popularity in the SOC population and account for more than 20% of cosmetic procedures in the United States.41 Facial cosmetic concerns in SOC include dyschromia, benign growths (dermatosis papulosa nigra), hyperkinetic facial lines, volume loss, and skin laxity.42 Key principles to consider when treating SOC patients are the impact of ethnicity on aging and facial structure, the patient’s desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient’s expectations for recommended therapies.
Aging in SOC Patients
Skin aging can be classified as intrinsic aging or extrinsic aging. Intrinsic aging is genetic and involves subsurface changes such as volume loss, muscle atrophy, and resorption of bony structure. Extrinsic aging (or photoaging) involves surface changes of the epidermis/dermis and manifests as mottled pigmentation, textural changes, and fine wrinkling. Due to the photoprotection of melanin (black skin=SPF 13.4), skin aging in SOC patients is delayed by 10 to 20 years.43 In addition, SOC patients have more reactive collagen and can benefit from noninvasive cosmetic procedures such as fillers and skin-tightening procedures.42
Cosmetic Treatments and Procedures
Dermatosis papulosa nigra (benign growths of skin that have a genetic predisposition)44 occur mainly on the face but can involve the entire body. Treatment modalities include electrodesiccation, cryotherapy, scissor excision, and laser surgery.45
Treatment of hyperkinetic facial lines with botulinum toxin type A is a safe and effective procedure in patients with SOC. Grimes and Shabazz46 performed a 4-month, randomized, double-blind study that evaluated the treatment of glabellar lines in women with Fitzpatrick skin types V and VI. The results demonstrated that the duration of effects was the same in the patients who received either 20 or 30 U of botulinum toxin type A.46 Dynamic rhytides (furrows and frown/scowl lines arising from laughing, frowning, or smiling) can be treated safely in patients with SOC using botulinum toxin type A off label for relaxation of the upper and lower hyperkinetic muscles that result in these unwanted signs of aging. Botulinum toxin type A often is used for etched-in crow’s-feet, which rarely are evident in SOC patients.47 Facial shaping also can be accomplished by injecting botulinum toxin type A in combination with soft-tissue dermal fillers.47
Although black individuals do not experience perioral rhytides at the frequency of white individuals, they experience a variety of other cosmetic issues related to skin sagging and sinking. Currently available hyaluronic acid (HA) fillers have been shown to be safe in patients with Fitzpatrick skin types IV through VI.48 Two studies evaluated fillers in patients with SOC, specifically HA49 and calcium hydroxylapatite,50 focused on treatment of the nasolabial folds and the potential risk for dyspigmentation and keloidal scarring. Taylor et al49 noted that the risk of hyperpigmentation was 6% to 9% for large- and small-particle HA, respectively, and was associated with the serial or multiple puncture injection technique. No hypertrophic or keloidal scarring occurred in both studies.49,50
Facial contouring applications with fillers include glabellar lines, temples, nasal bridge, tear troughs, malar and submalar areas, nasolabial folds, radial lines, lips, marionette lines, mental crease, and chin. Hyaluronic acid fillers also can be used for lip enhancement.47 Although white women are looking to increase the size of their lips, black women are seeking augmentation to restore their lip size to that of their youth. Black individuals do not experience the same frequency of perioral rhytides as white patients, but they experience a variety of other issues related to skin sagging and sinking. Unlike white women, enhancement of the vermilion border rarely is performed in black women due to development of rhytides, predominantly in the body of the lip below the vermilion border in response to volume loss in the upper lip while the lower lip usually maintains its same appearance.47
Facial enhancement utilizing poly-L-lactic acid can be used safely in SOC patients.51 Poly-L-lactic acid microparticles induce collagen formation, leading to dermal thickening over 3 to 6 months; however, multiple sessions are required to achieve optimal aesthetic results.
Patients with more reactive collagen can benefit from noninvasive cosmetic procedures such as skin-tightening procedures.52 Radiofrequency and microfocused ultrasound are cosmetic procedures used to provide skin tightening and facial lifting. They are safe and effective treatments for patients with Fitzpatrick skin types IV to VI.53 Histologically, there is less thinning of collagen bundles and elastic tissue in ethnic skin. Due to stimulation of collagen by these procedures, most SOC patients will experience a more enhanced response, requiring fewer treatment sessions than white individuals.
Conclusion
Medical and aesthetic facial concerns in SOC patients vary and can be a source of emotional and psychological distress that can negatively impact quality of life. The approach to the treatment of SOC patients should be a balance between tolerability and efficacy, considering the potential risk for PIH.
- White GM. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J Am Acad Dermatol. 1998;39(2 pt 3):S34-S37.
- Halder RM, Grimes PE, McLaurin CL, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasians, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
- Halder RM, Holmes YC, Bridgeman-Shah S, et al. A clinicohistologic study of acne vulgaris in black females (abstract). J Invest Dermatol. 1996;106:888.
- Plewig G, Fulton JE, Kligman AM. Pomade acne. Arch Dermatol. 1970;101:580-584.
- Kligman AM, Mills OH. Acne cosmetica. Arch Dermatol. 1972;106:893-897.
- Halder RM, Brooks HL, Callender VD. Acne in ethnic skin. Dermatol Clin. 2003;21:609-615.
- Callender VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Winhoven SM. Postinflammatory hyperpigmentation in an Asian patient. a dramatic response to oral isotretinoin (13-cis-retinoic acid). Br J Med. 2005;152:368-403.
- Sarkar R, Bansal S, Garg VK. Chemical peels for melasma in dark-skinned patients. J Cutan Aesthet Surg. 2012;5:247-253.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Culp B, Scheinfeld N. Rosacea: a review. P T. 2009;34:38-45.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014:20. pii:13030/qt1mv9r0ss.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Jackson JM, Knuckles M, Minni JP, et al. The role of brimonidine tartrate gel in the treatment of rosacea. Clin Cosmet Investig Dermatol. 2015;23:529-538.
- Patel NU, Shukla S, Zaki J, et al. Oxymetazoline hydrochloride cream for facial erythema associated with rosacea. Expert Rev Clin Pharmacol. 2017;10:104954.
- Weinkle AP, Doktor V, Emer J. Update on the management of rosacea. Clin Cosmet Investig Dermatol. 2015;8:159-177.
- Grimes PE. Skin and hair cosmetic issues in women of color. Dermatol Clin. 2000;19:659-665.
- Taylor A, Pawaskar M, Taylor SL, et al. Prevalence of pigmentary disorders and their impact on quality of life: a prospective cohort study. J Cosmet Dermatol. 2008;7:164-168.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Grimes PE. Melasma: etiologic and therapeutic considerations. Arch Dermatol. 1995;131:1453-1457.
- Handel AC, Miot LD, Miot HA. Melasma: a clinical and epidemiological review. An Bras Dermatol. 2014;89:771-782.
- Callender VD, Reid SD, Obayan O, et al. Diagnostic clues to frontal fibrosing alopecia in patients of African descent. J Clin Aesthet Dermatol. 2016;9:45-51.
- Narang T, Sawatkar GU, Kumaran MS, et al. Minocycline for recurrent and/or chronic erythema nodosum leprosum. JAMA Dermatol. 2015;151:1026-1028.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10.
- Pandya AG, Guevara IL. Disorders of hyperpigmentation. Dermatol Clin. 2000;18:91-98.
- Taylor SC, Torok H, Jones T, et al. Efficacy and safety of a new triple-combination agent for the treatment of facial melasma. Cutis. 2003;72:67-72.
- Bulengo-Ransby SM. Topical tretinoin (retinoic acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Jacyk WK. Adapalene in the treatment of African patients. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):37-42.
- Kircik LH. Efficacy and safety of azelaic acid (AzA) gel 15% in the treatment of postinflammatory hyperpigmentation and acne: a 16-week, baseline-controlled study. J Drugs Dermatol. 2011;10:586-590.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma. J Am Acad Dermatol. 2016;75:385-392.
- Kindred C, Okereke U, Callender VD. Skin-lightening agents: an overview of prescription, office-dispensed, and over-the-counter products. Cosmet Dermatol. 2013;26:18-26.
- Makino ET, Kadoya K, Sigler ML, et al. Development and clinical assessment of a comprehensive product for pigmentation control in multiple ethnic populations. J Drugs Dermatol. 2016;15:1562-1570.
- Halder R, Rodney I, Munhutu M, et al. Evaluation and effectiveness of a photoprotection composition (sunscreen) on subjects of skin of color. J Am Acad Dermatol. 2015;72(suppl 1):AB215.
- Castanedo-Cazares JP, Hernandez-Blanco D, Carlos-Ortega B, et al. Near-visible light and UV photoprotection in the treatment of melasma: a double-blind randomized trial. Photodermatol Photoimmunol Photomed. 2014;30:35-42.
- American Society for Aesthetic Plastic Surgery. 2016 Cosmetic Surgery National Data Bank Statistics. https://www.surgery.org/sites/default/files/ASAPS-Stats2016.pdf. Accessed November 15, 2017.
- Burgess CM. Soft tissue augmentation in skin of color: market growth, available fillers and successful techniques. J Drugs Dermatol. 2007;6:51-55.
- Davis EC, Callender VD. Aesthetic dermatology for aging ethnic skin. Dermatol Surg. 2011;37:901-917.
- Grimes PE, Arora S, Minus HR, et al. Dermatosis papulosa nigra. Cutis. 1983;32:385-386.
- Lupo M. Dermatosis papulosa nigra: treatment options. J Drugs Dermatol. 2007;6:29-30.
- Grimes PE, Shabazz D. A four-month randomized, double-blind evaluation of the efficacy of botulinum toxin type A for the treatment of glabellar lines in women with skin types V and VI. Dermatol Surg. 2009;35:429-435.
- Burgess CM, Awosika O. Ethnic and gender considerations in the use of facial injectables: African-American patients. Plast Reconstr Surg. 2015;136(5 suppl):28S-31S.
- Taylor SC, Kelly AP, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. New York, NY: McGraw-Hill Education; 2016.
- Taylor SC, Burgess CM, Callender VD. Safety of nonanimal stabilized hyaluronic acid dermal fillers in patients with skin of color: a randomized, evaluator-blinded comparative trial. Dermatol Surg. 2009;35(suppl 2):1653-1660.
- Marmur ES, Taylor SC, Grimes PE, et al. Six-month safety results of calcium hydroxylapatite for treatment of nasolabial folds in Fitzpatrick skin types IV to VI. Dermatol Surg. 2009;35(suppl 2):1641-1645.
- Hamilton TK, Burgess CM. Consideration for the use of injectable poly-L-lactic acid in people of color. J Drugs Dermatol. 2010;9:451-456.
- Fabi SG, Goldman MP. Retrospective evaluation of micro-focused ultrasound for lifting and tightening of the face and neck. Dermatol Surg. 2014;40:569-575.
- Harris MO, Sundaram HA. Safety of microfocused ultrasound with visualization in patients with Fitzpatrick skin phototypes III to VI. JAMA Facial Plast Surg. 2015;17:355-357.
- White GM. Recent findings in the epidemiologic evidence, classification, and subtypes of acne vulgaris. J Am Acad Dermatol. 1998;39(2 pt 3):S34-S37.
- Halder RM, Grimes PE, McLaurin CL, et al. Incidence of common dermatoses in a predominantly black dermatologic practice. Cutis. 1983;32:388, 390.
- Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
- Davis SA, Narahari S, Feldman SR, et al. Top dermatologic conditions in patients of color: an analysis of nationally representative data. J Drugs Dermatol. 2012;11:466-473.
- Perkins AC, Cheng CE, Hillebrand GG, et al. Comparison of the epidemiology of acne vulgaris among Caucasians, Asian, Continental Indian and African American women. J Eur Acad Dermatol Venereol. 2011;25:1054-1060.
- Taylor SC, Cook-Bolden F, Rahman Z, et al. Acne vulgaris in skin of color. J Am Acad Dermatol. 2002;46(2 suppl):S98-S106.
- Davis EC, Callender VD. A review of acne in ethnic skin: pathogenesis, clinical manifestations, and management strategies. J Clin Aesthet Dermatol. 2010;3:24-38.
- Halder RM, Holmes YC, Bridgeman-Shah S, et al. A clinicohistologic study of acne vulgaris in black females (abstract). J Invest Dermatol. 1996;106:888.
- Plewig G, Fulton JE, Kligman AM. Pomade acne. Arch Dermatol. 1970;101:580-584.
- Kligman AM, Mills OH. Acne cosmetica. Arch Dermatol. 1972;106:893-897.
- Halder RM, Brooks HL, Callender VD. Acne in ethnic skin. Dermatol Clin. 2003;21:609-615.
- Callender VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Winhoven SM. Postinflammatory hyperpigmentation in an Asian patient. a dramatic response to oral isotretinoin (13-cis-retinoic acid). Br J Med. 2005;152:368-403.
- Sarkar R, Bansal S, Garg VK. Chemical peels for melasma in dark-skinned patients. J Cutan Aesthet Surg. 2012;5:247-253.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Culp B, Scheinfeld N. Rosacea: a review. P T. 2009;34:38-45.
- Al-Dabagh A, Davis SA, McMichael AJ, et al. Rosacea in skin of color: not a rare diagnosis. Dermatol Online J. 2014:20. pii:13030/qt1mv9r0ss.
- Del Rosso JQ. Advances in understanding and managing rosacea: part 1: connecting the dots between pathophysiological mechanisms and common clinical features of rosacea with emphasis on vascular changes and facial erythema. J Clin Aesthet Dermatol. 2012;5:16-25.
- Jackson JM, Knuckles M, Minni JP, et al. The role of brimonidine tartrate gel in the treatment of rosacea. Clin Cosmet Investig Dermatol. 2015;23:529-538.
- Patel NU, Shukla S, Zaki J, et al. Oxymetazoline hydrochloride cream for facial erythema associated with rosacea. Expert Rev Clin Pharmacol. 2017;10:104954.
- Weinkle AP, Doktor V, Emer J. Update on the management of rosacea. Clin Cosmet Investig Dermatol. 2015;8:159-177.
- Grimes PE. Skin and hair cosmetic issues in women of color. Dermatol Clin. 2000;19:659-665.
- Taylor A, Pawaskar M, Taylor SL, et al. Prevalence of pigmentary disorders and their impact on quality of life: a prospective cohort study. J Cosmet Dermatol. 2008;7:164-168.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Grimes PE. Melasma: etiologic and therapeutic considerations. Arch Dermatol. 1995;131:1453-1457.
- Handel AC, Miot LD, Miot HA. Melasma: a clinical and epidemiological review. An Bras Dermatol. 2014;89:771-782.
- Callender VD, Reid SD, Obayan O, et al. Diagnostic clues to frontal fibrosing alopecia in patients of African descent. J Clin Aesthet Dermatol. 2016;9:45-51.
- Narang T, Sawatkar GU, Kumaran MS, et al. Minocycline for recurrent and/or chronic erythema nodosum leprosum. JAMA Dermatol. 2015;151:1026-1028.
- Boyer M, Katta R, Markus R. Diltiazem-induced photodistributed hyperpigmentation. Dermatol Online J. 2003;9:10.
- Pandya AG, Guevara IL. Disorders of hyperpigmentation. Dermatol Clin. 2000;18:91-98.
- Taylor SC, Torok H, Jones T, et al. Efficacy and safety of a new triple-combination agent for the treatment of facial melasma. Cutis. 2003;72:67-72.
- Bulengo-Ransby SM. Topical tretinoin (retinoic acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Grimes P, Callender V. Tazarotene cream for postinflammatory hyperpigmentation and acne vulgaris in darker skin: a double-blind, randomized, vehicle-controlled study. Cutis. 2006;77:45-50.
- Jacyk WK. Adapalene in the treatment of African patients. J Eur Acad Dermatol Venereol. 2001;15(suppl 3):37-42.
- Kircik LH. Efficacy and safety of azelaic acid (AzA) gel 15% in the treatment of postinflammatory hyperpigmentation and acne: a 16-week, baseline-controlled study. J Drugs Dermatol. 2011;10:586-590.
- Lee HC, Thng TG, Goh CL. Oral tranexamic acid (TA) in the treatment of melasma. J Am Acad Dermatol. 2016;75:385-392.
- Kindred C, Okereke U, Callender VD. Skin-lightening agents: an overview of prescription, office-dispensed, and over-the-counter products. Cosmet Dermatol. 2013;26:18-26.
- Makino ET, Kadoya K, Sigler ML, et al. Development and clinical assessment of a comprehensive product for pigmentation control in multiple ethnic populations. J Drugs Dermatol. 2016;15:1562-1570.
- Halder R, Rodney I, Munhutu M, et al. Evaluation and effectiveness of a photoprotection composition (sunscreen) on subjects of skin of color. J Am Acad Dermatol. 2015;72(suppl 1):AB215.
- Castanedo-Cazares JP, Hernandez-Blanco D, Carlos-Ortega B, et al. Near-visible light and UV photoprotection in the treatment of melasma: a double-blind randomized trial. Photodermatol Photoimmunol Photomed. 2014;30:35-42.
- American Society for Aesthetic Plastic Surgery. 2016 Cosmetic Surgery National Data Bank Statistics. https://www.surgery.org/sites/default/files/ASAPS-Stats2016.pdf. Accessed November 15, 2017.
- Burgess CM. Soft tissue augmentation in skin of color: market growth, available fillers and successful techniques. J Drugs Dermatol. 2007;6:51-55.
- Davis EC, Callender VD. Aesthetic dermatology for aging ethnic skin. Dermatol Surg. 2011;37:901-917.
- Grimes PE, Arora S, Minus HR, et al. Dermatosis papulosa nigra. Cutis. 1983;32:385-386.
- Lupo M. Dermatosis papulosa nigra: treatment options. J Drugs Dermatol. 2007;6:29-30.
- Grimes PE, Shabazz D. A four-month randomized, double-blind evaluation of the efficacy of botulinum toxin type A for the treatment of glabellar lines in women with skin types V and VI. Dermatol Surg. 2009;35:429-435.
- Burgess CM, Awosika O. Ethnic and gender considerations in the use of facial injectables: African-American patients. Plast Reconstr Surg. 2015;136(5 suppl):28S-31S.
- Taylor SC, Kelly AP, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. New York, NY: McGraw-Hill Education; 2016.
- Taylor SC, Burgess CM, Callender VD. Safety of nonanimal stabilized hyaluronic acid dermal fillers in patients with skin of color: a randomized, evaluator-blinded comparative trial. Dermatol Surg. 2009;35(suppl 2):1653-1660.
- Marmur ES, Taylor SC, Grimes PE, et al. Six-month safety results of calcium hydroxylapatite for treatment of nasolabial folds in Fitzpatrick skin types IV to VI. Dermatol Surg. 2009;35(suppl 2):1641-1645.
- Hamilton TK, Burgess CM. Consideration for the use of injectable poly-L-lactic acid in people of color. J Drugs Dermatol. 2010;9:451-456.
- Fabi SG, Goldman MP. Retrospective evaluation of micro-focused ultrasound for lifting and tightening of the face and neck. Dermatol Surg. 2014;40:569-575.
- Harris MO, Sundaram HA. Safety of microfocused ultrasound with visualization in patients with Fitzpatrick skin phototypes III to VI. JAMA Facial Plast Surg. 2015;17:355-357.
Practice Points
- Treat acne in skin of color (SOC) patients early and aggressively to prevent or minimize subsequent postinflammatory hyperpigmentation (PIH) and acne scarring.
- Vascular lasers and intense pulsed light may be used to address the vascular components of rosacea; however, the latter is not recommended in Fitzpatrick skin types IV to VI.
- Hydroquinone is the gold standard for skin lightening and is often used as a first-line therapy for melasma and PIH.
- Photoprotection is an essential component of therapy for hyperpigmented skin disorders.
- Cosmetic procedures are gaining popularity in the SOC population. When treating SOC patients, consider the impact of ethnicity on aging and facial structure, the patient's desired cosmetic outcome, tissue reaction to anticipated treatments, and the patient's expectations for recommended therapies.
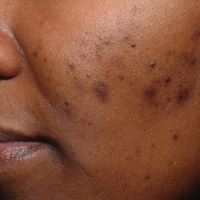
What is the optimal opioid prescription length after women’s health surgical procedures?
WHAT DOES THIS MEAN FOR PRACTICE?
- 7-day opioid prescriptions should be sufficient after common gyn procedures
- Monitor patients closely
- Transfer patients as soon as possible to non-opioid pain medication

Pediatric Periorificial Dermatitis
Perioral dermatitis is an acneform eruption presenting with erythematous papules, vesicles, and rarely pustules clustered around the orifices of the face. 1 Lesions may be found near the eyes, mouth, and nose but typically spare the vermilion border of the lips. 2 Nguyen and Eichenfield 3 preferred the term periorificial dermatitis (POD), which has since been adopted by others. 4 Patients may report pruritus, but there generally are no systemic symptoms unless patients have comorbid conditions such as atopic dermatitis. 5 Although this condition has been well examined in the literature on adults, data in the pediatric population are far more limited, consisting of case series and retrospective chart reviews. In 1979, Wilkinson et al 6 published a study of more than 200 patients with perioral dermatitis, but only 15 patients younger than 12 years were included.
Etiology
Although the exact pathogenesis of POD is unknown, a common denominator among many patients is prior exposure to topical corticosteroids.3,7-9 Periorificial dermatitis also has been linked to the use of systemic corticosteroids in pediatric patients.10 The exact relationship between steroid use and dermatitis is unknown; it may be related to a change in the flora of hair follicles and in particular an association with fusiform bacteria–rich conditions.11 Aside from steroid exposure, POD has been associated with the use of physical sunscreen in pediatric patients with dry skin,12 rosin in chewing gum,13 and inhaled corticosteroids in those with asthma.14 In one case, a 15-year-old adolescent girl developed POD and swelling of the lips after 2 years of playing a flute made of cocus wood.15,16
Epidemiology
Comorbidities and Family History
Goel et al17 (N=222) reported the following comorbidities associated with pediatric POD: atopic dermatitis (29.3%), asthma (14.9%), and allergies (9.9%). Steroid exposure was noted in 58.1% of patients.17 Similarly, Nguyen and Eichenfield3 (N=79) found that the most common comorbidities were atopic dermatitis (14%), keratosis pilaris (14%), viral infections (14%), acne (10%), and seborrheic dermatitis (10%). Family history of atopy was noted in 55% of patients and family history of rosacea was noted in 3%. In a case series of 11 pediatric patients, 3 (27%) had keratosis pilaris, 7 (64%) had a family history of atopy, and 2 (18%) had a family history of rosacea.8 Weston and Morelli9 found a much higher incidence of familial rosacea (20%) in 106 children with steroid rosacea.
Clinical Presentation
Periorificial dermatitis generally presents with small, pink- to flesh-colored papules in a perioral, periocular, and perinasal distribution. Although many patients are white, a particularly prominent variant has been noted in black children with papules that may be hyperpigmented.18 In a 2006 chart review in 79 pediatric POD patients aged 6 months to 18 years, Nguyen and Eichenfield3 reported that 92% (73/79) of patients presented for a facial rash with an average duration ranging from 2 weeks to 4 years.
Boeck et al19 described 7 pediatric patients with perioral dermatitis. Six (86%) patients had perioral lesions, and 6 (86%) had previously been treated with moderate- to high-potency topical corticosteroids. Skin prick tests were negative in 6 (86%) patients.19 In one case report, a 6-year-old boy did not present with the classic acneform lesions but rather sharply demarcated eczematous patches around the eyes, nose, and mouth. The rash began to fade after 2 weeks of using metronidazole gel 1%, and after 4 months he was only left with mild hyperpigmentation.4
Periorificial dermatitis was once thought to be a juvenile form of rosacea.5 In 1972, Savin et al8 described 11 pediatric patients with “rosacea-like” facial flushing, papules, pustules, and scaling over the cheeks, forehead, and chin. In some patients, the eyelids also were involved. At least 8 patients had been using potent topical corticosteroids and had noticed exacerbation of their skin lesions after stopping therapy.8
Variants of POD
Several other variants of POD have been described in pediatric patients including childhood granulomatous periorificial dermatitis (CGPD)(also known as facial Afro-Caribbean [childhood] eruption) and lupus miliaris disseminatus faciei. Childhood granulomatous periorificial dermatitis presents in prepubertal children as dome-shaped, red to yellow-brown, monomorphous papules around the eyes, nose, and mouth; there are no systemic findings.20,21 It occurs equally in males and females and is more commonly seen in dark-skinned patients. Childhood granulomatous periorificial dermatitis usually resolves within a few months to years but may be associated with blepharitis or conjunctivitis.20 Urbatsch et al20 analyzed extrafacial lesions in 8 patients (aged 2–12 years) with CGPD. Lesions were found on the trunk (38% [3/8]), neck (25% [2/8]), ears (25% [2/8]), extremities (50% [4/8]), labia majora (38% [3/8]), and abdomen (13% [1/8]). In addition, 2 (25% [2/8]) patients had blepharitis.20
Lupus miliaris disseminatus faciei, which occurs in adolescents and adults, commonly involves the eyelids and central areas of the face such as the nose and upper lips. Patients typically present with erythematous or flesh-colored papules.1
Diagnosis
Diagnosis of POD is made clinically based on the observation of papules (and sometimes pustules) around the orifices of the face, sparing the vermilion border, together with a lack of comedones.17 Laboratory tests are not useful.5 Biopsies rarely are performed, and the results mimic those of rosacea, demonstrating a perifollicular lymphohistiocytic infiltrate, epithelioid cells, and occasionally giant cells.5,22,23 Early papular lesions can show mild acanthosis, epidermal edema, and parakeratosis.23 Biopsies in patients with CGPD reveal noncaseating perifollicular granulomas.20
Treatment and Clinical Outcome
Although topical corticosteroids can improve facial lesions in pediatric POD, the eruption often rebounds when therapy is discontinued.1 One therapy frequently used in adults is oral tetracyclines; however, these agents must not be used in patients younger than 9 years due to potential dental staining.4 The standards are either topical metronidazole twice daily with clearance in 3 to 8 weeks or oral erythromycin.7
In the review conducted by Goel et al,17 treatment included azithromycin (44.6%), topical metronidazole (42.3%), sodium sulfacetamide lotion (35.6%), oral antibiotic monotherapy (15.3%), topical agent monotherapy (44.6%), and combined oral and topical agent therapy (40.1%). Of those patients who presented for a follow-up visit (59%), 72% of cases resolved and 10.7% showed some improvement. For those patients who returned for follow-up, the average duration until symptom resolution was approximately 4 months. The most common side effects were pigmentation changes (1.8%), worsening of symptoms (1.8%), gastrointestinal upset (0.9%), irritant dermatitis (0.9%), and xerosis (0.5%).17
Changes were made to the treatment plans for 16 patients, most often due to inadequate treatment response.17 Five patients treated with sodium sulfacetamide lotion also were started on oral azithromycin. Four patients treated with oral antibiotics were given a topical agent (metronidazole or sodium sulfacetamide lotion). Other modifications included replacing sodium sulfacetamide lotion with topical metronidazole and an oral antibiotic (azithromycin or doxycycline, n=3), adjusting the doses of oral or topical medications (n=2), adding tacrolimus (n=1), and replacing topical metronidazole with sodium sulfacetamide lotion (n=1). Of the patients who underwent a change in treatment plan, 5 experienced symptom recurrence, 4 had mild improvement, and 1 patient had no improvement. Six patients were lost to follow-up.17
In the study conducted by Nguyen and Eichenfield,3 follow-up visits occurred approximately 3 months after the first visit.
In the case series by Boeck et al,19 all patients were started on metronidazole gel 1% applied once daily for the first week, and then twice daily until the lesions resolved. All patients showed improvement after 4 to 6 weeks, and eventually the disease cleared between 3 and 6 months. All patients were still symptom free during a 2-year observation period.19
Manders and Lucky7 described 14 patients with POD (aged 9 months to 6.5 years). Eight patients used only metronidazole gel 0.75%, while 5 used the gel in combination with topical corticosteroids (21% [3/14]), oral erythromycin (7% [1/14]), or topical erythromycin (7% [1/14]); 1 patient remained on hydrocortisone 1% and cleared. Patients responded well within 1 to 8 weeks and were symptom free for up to 16 months. Mid- to high-potency steroids were discontinued in all patients.7
In some pediatric patients with CGPD, recovery occurs faster with the use of oral macrolides or tetracyclines, either alone or in combination with topical antibiotics or sulfur-based lotions.20 Extrafacial lesions associated with CGPD do not appear to negatively impact treatment response or duration of disease. In the review conducted by Urbatsch et al,20 7 of 8 (88%) CGPD patients with extrafacial lesions were treated with oral agents including erythromycin, hydroxychloroquine, cyclosporine, minocycline, and azithromycin. Most of these patients also were using topical agents such as triamcinolone acetonide, desonide, metronidazole, and erythromycin. The time to resolution ranged from several weeks to 6 months.20
Weston and Morelli9 described a treatment regimen for steroid rosacea. The study included data on 106 children (60 females, 46 males) who had been exposed to mostly class 7 low-potency agents. All patients were advised to immediately stop topical steroid therapy without gradual withdrawal and to begin oral erythromycin stearate 30 mg/kg daily in 2 doses per day for 4 weeks. Patients who were unable to tolerate erythromycin were advised to use topical clindamycin phosphate twice daily for 4 weeks (n=6). Eighty-six percent of patients showed resolution within 4 weeks, and 100% showed clearance by 8 weeks. Twenty-two percent of patients had clearance within 3 weeks. There was no difference in the duration until resolution for those who had used oral or topical antibiotics.9 A different study suggested that low-potency topical steroids can be used to control inflammation when weaning patients off of strong steroids.5
Differential Diagnosis
The differential diagnosis should include acne vulgaris, allergic contact dermatitis, irritant contact dermatitis, seborrheic dermatitis, impetigo, dermatophyte infection, rosacea, and angiofibromas.4
Acne vulgaris commonly is found in older adolescents, and unlike POD, it will present with open or closed comedones.2 In patients aged 1 to 7 years, acne is a reason to consider endocrine evaluation. Allergic contact dermatitis is extremely pruritic, and the lesions often are papulovesicular with active weeping or crusting. Patients with irritant contact dermatitis often report burning and pain, and papules and pustules typically are absent. A thorough history can help rule out allergic or irritant contact dermatitis. Seborrheic dermatitis presents with erythema and scaling of the scalp, eyebrows, and nasolabial folds; it tends to spare the perioral regions and also lacks papules.2 The lesions of impetigo typically have a yellow-brown exudate, which forms a honey-colored crust.24 Tinea faciei, unlike the other tinea infections, can have an extremely variable presentation. Lesions usually begin as scaly macules that develop raised borders with central hypopigmentation, but papules, vesicles, and crusts can be seen.25 Potassium hydroxide
Conclusion
Diagnosis of POD is clinical and rests upon the finding of erythematous papules on the face near the eyes, mouth, and nose. Extrafacial lesions also have been described, particularly in pediatric patients with CGPD. Many patients will report a history of atopic dermatitis and asthma. Therapy for POD includes both topical and systemic agents. For those with mild disease, topical metronidazole commonly is used. For patients requiring oral antibiotics, tetracyclines or macrolides can be prescribed based on the age of the patient. Many pediatric patients who begin with both oral and topical agents can later be maintained on topical therapy, sometimes with a low-dose oral antibiotic. Periorificial dermatitis has an excellent prognosis and most pediatric patients show marked improvement within weeks to months.
- Tempark T, Shwayder TA. Perioral dermatitis: a review of the condition with special attention to treatment options. Am J Clin Dermatol. 2014;15:101-113.
- McFarland SL, Polcari IC. Morphology-based diagnosis of acneiform eruptions. Pediatr Ann. 2015;44:E188-E193.
- Nguyen V, Eichenfield LF. Periorificial dermatitis in children and adolescents. J Am Acad Dermatol. 2006;55:781-785.
- Kihiczak GG, Cruz MA, Schwartz RA. Periorificial dermatitis in children: an update and description of a child with striking features. Int J Dermatol. 2009;48:304-306.
- Laude TA, Salvemini JN. Perioral dermatitis in children. Sem Cutan Med Surg. 1999;18:206-209.
- Wilkinson DS, Kirton V, Wilkinson JD. Perioral dermatitis: a 12-year review. Br J Dermatol. 1979;101:245-257.
- Manders SM, Lucky AW. Perioral dermatitis in childhood. J Am Acad Dermatol. 1992;27(5 pt 1):688-692.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Dermatol. 1972;87:425-429.
- Weston WL, Morelli JG. Steroid rosacea in prepubertal children. Arch Pediatr Adolesc Med. 2000;154:62-64.
- Clementson B, Smidt AC. Periorificial dermatitis due to systemic corticosteroids in children: report of two cases. Pediatr Dermatol. 2012;29:331-332.
- Takiwaki H, Tsuda H, Arase S, et al. Differences between intrafollicular microorganism profiles in perioral and seborrhoeic dermatitis. Clin Exp Dermatol. 2003;28:531-534.
- Abeck D, Geisenfelder B, Brandt O. Physical sunscreens with high sun protection factor may cause perioral dermatitis in children. J Dtsch Dermatol Ges. 2009;7:701-703.
- Satyawan I, Oranje AP, van Joost T. Perioral dermatitis in a child due to rosin in chewing gum. Contact Dermatitis. 1990;22:182-183.
- Dubus JC, Marguet C, Deschildre A, et al. Local side-effects of inhaled corticosteroids in asthmatic children: influence of drug, dose, age, and device. Allergy. 2001;56:944-948.
- Hausen BM, Bruhn G, Koenig WA. New hydroxyisoflavans as contact sensitizers in cocus wood Brya ebenus DC (Fabaceae). Contact Dermatitis. 1991;25:149-155.
- Dirschka T, Weber K, Tronnier H. Topical cosmetics and perioral dermatitis. J Dtsch Dermatol Ges. 2004;2:194-199.
- Goel NS, Burkhart CN, Morrell DS. Pediatric periorificial dermatitis: clinical course and treatment outcomes in 222 patients. Pediatr Dermatol. 2015;32:333-336.
- Cribier B, Lieber-Mbomeyo A, Lipsker D. Clinical and histological study of a case of facial Afro-Caribbean childhood eruption (FACE) [in French][published online July 23, 2008]. Ann Dermatol Venerol. 2008;135:663-667.
- Boeck K, Abeck D, Werfel S, et al. Perioral dermatitis in children—clinical presentation, pathogenesis-related factors and response to topical metronidazole. Dermatology. 1997;195:235-238.
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358.
- Kroshinsky D, Glick SA. Pediatric rosacea. Dermatol Ther. 2006;19:196-201.
- Ramelet AA, Delacrétaz J. Histopathologic study of perioral dermatitis [in French]. Dermatologica. 1981;163:361-369.
- Ljubojevi´c S, Lipozenci´c J, Turci´c P. Perioral dermatitis. Acta Dermatovenerol Croat. 2008;16:96-100.
- Nichols RL, Florman S. Clinical presentations of soft-tissue infections and surgical site infections. Clin Infect Dis. 2001;33(suppl 2):S84-S93.
- Lin RL, Szepietowski JC, Schwartz RA. Tinea faciei, an often deceptive facial eruption. Int J Dermatol. 2004;43:437-440.
Perioral dermatitis is an acneform eruption presenting with erythematous papules, vesicles, and rarely pustules clustered around the orifices of the face. 1 Lesions may be found near the eyes, mouth, and nose but typically spare the vermilion border of the lips. 2 Nguyen and Eichenfield 3 preferred the term periorificial dermatitis (POD), which has since been adopted by others. 4 Patients may report pruritus, but there generally are no systemic symptoms unless patients have comorbid conditions such as atopic dermatitis. 5 Although this condition has been well examined in the literature on adults, data in the pediatric population are far more limited, consisting of case series and retrospective chart reviews. In 1979, Wilkinson et al 6 published a study of more than 200 patients with perioral dermatitis, but only 15 patients younger than 12 years were included.
Etiology
Although the exact pathogenesis of POD is unknown, a common denominator among many patients is prior exposure to topical corticosteroids.3,7-9 Periorificial dermatitis also has been linked to the use of systemic corticosteroids in pediatric patients.10 The exact relationship between steroid use and dermatitis is unknown; it may be related to a change in the flora of hair follicles and in particular an association with fusiform bacteria–rich conditions.11 Aside from steroid exposure, POD has been associated with the use of physical sunscreen in pediatric patients with dry skin,12 rosin in chewing gum,13 and inhaled corticosteroids in those with asthma.14 In one case, a 15-year-old adolescent girl developed POD and swelling of the lips after 2 years of playing a flute made of cocus wood.15,16
Epidemiology
Comorbidities and Family History
Goel et al17 (N=222) reported the following comorbidities associated with pediatric POD: atopic dermatitis (29.3%), asthma (14.9%), and allergies (9.9%). Steroid exposure was noted in 58.1% of patients.17 Similarly, Nguyen and Eichenfield3 (N=79) found that the most common comorbidities were atopic dermatitis (14%), keratosis pilaris (14%), viral infections (14%), acne (10%), and seborrheic dermatitis (10%). Family history of atopy was noted in 55% of patients and family history of rosacea was noted in 3%. In a case series of 11 pediatric patients, 3 (27%) had keratosis pilaris, 7 (64%) had a family history of atopy, and 2 (18%) had a family history of rosacea.8 Weston and Morelli9 found a much higher incidence of familial rosacea (20%) in 106 children with steroid rosacea.
Clinical Presentation
Periorificial dermatitis generally presents with small, pink- to flesh-colored papules in a perioral, periocular, and perinasal distribution. Although many patients are white, a particularly prominent variant has been noted in black children with papules that may be hyperpigmented.18 In a 2006 chart review in 79 pediatric POD patients aged 6 months to 18 years, Nguyen and Eichenfield3 reported that 92% (73/79) of patients presented for a facial rash with an average duration ranging from 2 weeks to 4 years.
Boeck et al19 described 7 pediatric patients with perioral dermatitis. Six (86%) patients had perioral lesions, and 6 (86%) had previously been treated with moderate- to high-potency topical corticosteroids. Skin prick tests were negative in 6 (86%) patients.19 In one case report, a 6-year-old boy did not present with the classic acneform lesions but rather sharply demarcated eczematous patches around the eyes, nose, and mouth. The rash began to fade after 2 weeks of using metronidazole gel 1%, and after 4 months he was only left with mild hyperpigmentation.4
Periorificial dermatitis was once thought to be a juvenile form of rosacea.5 In 1972, Savin et al8 described 11 pediatric patients with “rosacea-like” facial flushing, papules, pustules, and scaling over the cheeks, forehead, and chin. In some patients, the eyelids also were involved. At least 8 patients had been using potent topical corticosteroids and had noticed exacerbation of their skin lesions after stopping therapy.8
Variants of POD
Several other variants of POD have been described in pediatric patients including childhood granulomatous periorificial dermatitis (CGPD)(also known as facial Afro-Caribbean [childhood] eruption) and lupus miliaris disseminatus faciei. Childhood granulomatous periorificial dermatitis presents in prepubertal children as dome-shaped, red to yellow-brown, monomorphous papules around the eyes, nose, and mouth; there are no systemic findings.20,21 It occurs equally in males and females and is more commonly seen in dark-skinned patients. Childhood granulomatous periorificial dermatitis usually resolves within a few months to years but may be associated with blepharitis or conjunctivitis.20 Urbatsch et al20 analyzed extrafacial lesions in 8 patients (aged 2–12 years) with CGPD. Lesions were found on the trunk (38% [3/8]), neck (25% [2/8]), ears (25% [2/8]), extremities (50% [4/8]), labia majora (38% [3/8]), and abdomen (13% [1/8]). In addition, 2 (25% [2/8]) patients had blepharitis.20
Lupus miliaris disseminatus faciei, which occurs in adolescents and adults, commonly involves the eyelids and central areas of the face such as the nose and upper lips. Patients typically present with erythematous or flesh-colored papules.1
Diagnosis
Diagnosis of POD is made clinically based on the observation of papules (and sometimes pustules) around the orifices of the face, sparing the vermilion border, together with a lack of comedones.17 Laboratory tests are not useful.5 Biopsies rarely are performed, and the results mimic those of rosacea, demonstrating a perifollicular lymphohistiocytic infiltrate, epithelioid cells, and occasionally giant cells.5,22,23 Early papular lesions can show mild acanthosis, epidermal edema, and parakeratosis.23 Biopsies in patients with CGPD reveal noncaseating perifollicular granulomas.20
Treatment and Clinical Outcome
Although topical corticosteroids can improve facial lesions in pediatric POD, the eruption often rebounds when therapy is discontinued.1 One therapy frequently used in adults is oral tetracyclines; however, these agents must not be used in patients younger than 9 years due to potential dental staining.4 The standards are either topical metronidazole twice daily with clearance in 3 to 8 weeks or oral erythromycin.7
In the review conducted by Goel et al,17 treatment included azithromycin (44.6%), topical metronidazole (42.3%), sodium sulfacetamide lotion (35.6%), oral antibiotic monotherapy (15.3%), topical agent monotherapy (44.6%), and combined oral and topical agent therapy (40.1%). Of those patients who presented for a follow-up visit (59%), 72% of cases resolved and 10.7% showed some improvement. For those patients who returned for follow-up, the average duration until symptom resolution was approximately 4 months. The most common side effects were pigmentation changes (1.8%), worsening of symptoms (1.8%), gastrointestinal upset (0.9%), irritant dermatitis (0.9%), and xerosis (0.5%).17
Changes were made to the treatment plans for 16 patients, most often due to inadequate treatment response.17 Five patients treated with sodium sulfacetamide lotion also were started on oral azithromycin. Four patients treated with oral antibiotics were given a topical agent (metronidazole or sodium sulfacetamide lotion). Other modifications included replacing sodium sulfacetamide lotion with topical metronidazole and an oral antibiotic (azithromycin or doxycycline, n=3), adjusting the doses of oral or topical medications (n=2), adding tacrolimus (n=1), and replacing topical metronidazole with sodium sulfacetamide lotion (n=1). Of the patients who underwent a change in treatment plan, 5 experienced symptom recurrence, 4 had mild improvement, and 1 patient had no improvement. Six patients were lost to follow-up.17
In the study conducted by Nguyen and Eichenfield,3 follow-up visits occurred approximately 3 months after the first visit.
In the case series by Boeck et al,19 all patients were started on metronidazole gel 1% applied once daily for the first week, and then twice daily until the lesions resolved. All patients showed improvement after 4 to 6 weeks, and eventually the disease cleared between 3 and 6 months. All patients were still symptom free during a 2-year observation period.19
Manders and Lucky7 described 14 patients with POD (aged 9 months to 6.5 years). Eight patients used only metronidazole gel 0.75%, while 5 used the gel in combination with topical corticosteroids (21% [3/14]), oral erythromycin (7% [1/14]), or topical erythromycin (7% [1/14]); 1 patient remained on hydrocortisone 1% and cleared. Patients responded well within 1 to 8 weeks and were symptom free for up to 16 months. Mid- to high-potency steroids were discontinued in all patients.7
In some pediatric patients with CGPD, recovery occurs faster with the use of oral macrolides or tetracyclines, either alone or in combination with topical antibiotics or sulfur-based lotions.20 Extrafacial lesions associated with CGPD do not appear to negatively impact treatment response or duration of disease. In the review conducted by Urbatsch et al,20 7 of 8 (88%) CGPD patients with extrafacial lesions were treated with oral agents including erythromycin, hydroxychloroquine, cyclosporine, minocycline, and azithromycin. Most of these patients also were using topical agents such as triamcinolone acetonide, desonide, metronidazole, and erythromycin. The time to resolution ranged from several weeks to 6 months.20
Weston and Morelli9 described a treatment regimen for steroid rosacea. The study included data on 106 children (60 females, 46 males) who had been exposed to mostly class 7 low-potency agents. All patients were advised to immediately stop topical steroid therapy without gradual withdrawal and to begin oral erythromycin stearate 30 mg/kg daily in 2 doses per day for 4 weeks. Patients who were unable to tolerate erythromycin were advised to use topical clindamycin phosphate twice daily for 4 weeks (n=6). Eighty-six percent of patients showed resolution within 4 weeks, and 100% showed clearance by 8 weeks. Twenty-two percent of patients had clearance within 3 weeks. There was no difference in the duration until resolution for those who had used oral or topical antibiotics.9 A different study suggested that low-potency topical steroids can be used to control inflammation when weaning patients off of strong steroids.5
Differential Diagnosis
The differential diagnosis should include acne vulgaris, allergic contact dermatitis, irritant contact dermatitis, seborrheic dermatitis, impetigo, dermatophyte infection, rosacea, and angiofibromas.4
Acne vulgaris commonly is found in older adolescents, and unlike POD, it will present with open or closed comedones.2 In patients aged 1 to 7 years, acne is a reason to consider endocrine evaluation. Allergic contact dermatitis is extremely pruritic, and the lesions often are papulovesicular with active weeping or crusting. Patients with irritant contact dermatitis often report burning and pain, and papules and pustules typically are absent. A thorough history can help rule out allergic or irritant contact dermatitis. Seborrheic dermatitis presents with erythema and scaling of the scalp, eyebrows, and nasolabial folds; it tends to spare the perioral regions and also lacks papules.2 The lesions of impetigo typically have a yellow-brown exudate, which forms a honey-colored crust.24 Tinea faciei, unlike the other tinea infections, can have an extremely variable presentation. Lesions usually begin as scaly macules that develop raised borders with central hypopigmentation, but papules, vesicles, and crusts can be seen.25 Potassium hydroxide
Conclusion
Diagnosis of POD is clinical and rests upon the finding of erythematous papules on the face near the eyes, mouth, and nose. Extrafacial lesions also have been described, particularly in pediatric patients with CGPD. Many patients will report a history of atopic dermatitis and asthma. Therapy for POD includes both topical and systemic agents. For those with mild disease, topical metronidazole commonly is used. For patients requiring oral antibiotics, tetracyclines or macrolides can be prescribed based on the age of the patient. Many pediatric patients who begin with both oral and topical agents can later be maintained on topical therapy, sometimes with a low-dose oral antibiotic. Periorificial dermatitis has an excellent prognosis and most pediatric patients show marked improvement within weeks to months.
Perioral dermatitis is an acneform eruption presenting with erythematous papules, vesicles, and rarely pustules clustered around the orifices of the face. 1 Lesions may be found near the eyes, mouth, and nose but typically spare the vermilion border of the lips. 2 Nguyen and Eichenfield 3 preferred the term periorificial dermatitis (POD), which has since been adopted by others. 4 Patients may report pruritus, but there generally are no systemic symptoms unless patients have comorbid conditions such as atopic dermatitis. 5 Although this condition has been well examined in the literature on adults, data in the pediatric population are far more limited, consisting of case series and retrospective chart reviews. In 1979, Wilkinson et al 6 published a study of more than 200 patients with perioral dermatitis, but only 15 patients younger than 12 years were included.
Etiology
Although the exact pathogenesis of POD is unknown, a common denominator among many patients is prior exposure to topical corticosteroids.3,7-9 Periorificial dermatitis also has been linked to the use of systemic corticosteroids in pediatric patients.10 The exact relationship between steroid use and dermatitis is unknown; it may be related to a change in the flora of hair follicles and in particular an association with fusiform bacteria–rich conditions.11 Aside from steroid exposure, POD has been associated with the use of physical sunscreen in pediatric patients with dry skin,12 rosin in chewing gum,13 and inhaled corticosteroids in those with asthma.14 In one case, a 15-year-old adolescent girl developed POD and swelling of the lips after 2 years of playing a flute made of cocus wood.15,16
Epidemiology
Comorbidities and Family History
Goel et al17 (N=222) reported the following comorbidities associated with pediatric POD: atopic dermatitis (29.3%), asthma (14.9%), and allergies (9.9%). Steroid exposure was noted in 58.1% of patients.17 Similarly, Nguyen and Eichenfield3 (N=79) found that the most common comorbidities were atopic dermatitis (14%), keratosis pilaris (14%), viral infections (14%), acne (10%), and seborrheic dermatitis (10%). Family history of atopy was noted in 55% of patients and family history of rosacea was noted in 3%. In a case series of 11 pediatric patients, 3 (27%) had keratosis pilaris, 7 (64%) had a family history of atopy, and 2 (18%) had a family history of rosacea.8 Weston and Morelli9 found a much higher incidence of familial rosacea (20%) in 106 children with steroid rosacea.
Clinical Presentation
Periorificial dermatitis generally presents with small, pink- to flesh-colored papules in a perioral, periocular, and perinasal distribution. Although many patients are white, a particularly prominent variant has been noted in black children with papules that may be hyperpigmented.18 In a 2006 chart review in 79 pediatric POD patients aged 6 months to 18 years, Nguyen and Eichenfield3 reported that 92% (73/79) of patients presented for a facial rash with an average duration ranging from 2 weeks to 4 years.
Boeck et al19 described 7 pediatric patients with perioral dermatitis. Six (86%) patients had perioral lesions, and 6 (86%) had previously been treated with moderate- to high-potency topical corticosteroids. Skin prick tests were negative in 6 (86%) patients.19 In one case report, a 6-year-old boy did not present with the classic acneform lesions but rather sharply demarcated eczematous patches around the eyes, nose, and mouth. The rash began to fade after 2 weeks of using metronidazole gel 1%, and after 4 months he was only left with mild hyperpigmentation.4
Periorificial dermatitis was once thought to be a juvenile form of rosacea.5 In 1972, Savin et al8 described 11 pediatric patients with “rosacea-like” facial flushing, papules, pustules, and scaling over the cheeks, forehead, and chin. In some patients, the eyelids also were involved. At least 8 patients had been using potent topical corticosteroids and had noticed exacerbation of their skin lesions after stopping therapy.8
Variants of POD
Several other variants of POD have been described in pediatric patients including childhood granulomatous periorificial dermatitis (CGPD)(also known as facial Afro-Caribbean [childhood] eruption) and lupus miliaris disseminatus faciei. Childhood granulomatous periorificial dermatitis presents in prepubertal children as dome-shaped, red to yellow-brown, monomorphous papules around the eyes, nose, and mouth; there are no systemic findings.20,21 It occurs equally in males and females and is more commonly seen in dark-skinned patients. Childhood granulomatous periorificial dermatitis usually resolves within a few months to years but may be associated with blepharitis or conjunctivitis.20 Urbatsch et al20 analyzed extrafacial lesions in 8 patients (aged 2–12 years) with CGPD. Lesions were found on the trunk (38% [3/8]), neck (25% [2/8]), ears (25% [2/8]), extremities (50% [4/8]), labia majora (38% [3/8]), and abdomen (13% [1/8]). In addition, 2 (25% [2/8]) patients had blepharitis.20
Lupus miliaris disseminatus faciei, which occurs in adolescents and adults, commonly involves the eyelids and central areas of the face such as the nose and upper lips. Patients typically present with erythematous or flesh-colored papules.1
Diagnosis
Diagnosis of POD is made clinically based on the observation of papules (and sometimes pustules) around the orifices of the face, sparing the vermilion border, together with a lack of comedones.17 Laboratory tests are not useful.5 Biopsies rarely are performed, and the results mimic those of rosacea, demonstrating a perifollicular lymphohistiocytic infiltrate, epithelioid cells, and occasionally giant cells.5,22,23 Early papular lesions can show mild acanthosis, epidermal edema, and parakeratosis.23 Biopsies in patients with CGPD reveal noncaseating perifollicular granulomas.20
Treatment and Clinical Outcome
Although topical corticosteroids can improve facial lesions in pediatric POD, the eruption often rebounds when therapy is discontinued.1 One therapy frequently used in adults is oral tetracyclines; however, these agents must not be used in patients younger than 9 years due to potential dental staining.4 The standards are either topical metronidazole twice daily with clearance in 3 to 8 weeks or oral erythromycin.7
In the review conducted by Goel et al,17 treatment included azithromycin (44.6%), topical metronidazole (42.3%), sodium sulfacetamide lotion (35.6%), oral antibiotic monotherapy (15.3%), topical agent monotherapy (44.6%), and combined oral and topical agent therapy (40.1%). Of those patients who presented for a follow-up visit (59%), 72% of cases resolved and 10.7% showed some improvement. For those patients who returned for follow-up, the average duration until symptom resolution was approximately 4 months. The most common side effects were pigmentation changes (1.8%), worsening of symptoms (1.8%), gastrointestinal upset (0.9%), irritant dermatitis (0.9%), and xerosis (0.5%).17
Changes were made to the treatment plans for 16 patients, most often due to inadequate treatment response.17 Five patients treated with sodium sulfacetamide lotion also were started on oral azithromycin. Four patients treated with oral antibiotics were given a topical agent (metronidazole or sodium sulfacetamide lotion). Other modifications included replacing sodium sulfacetamide lotion with topical metronidazole and an oral antibiotic (azithromycin or doxycycline, n=3), adjusting the doses of oral or topical medications (n=2), adding tacrolimus (n=1), and replacing topical metronidazole with sodium sulfacetamide lotion (n=1). Of the patients who underwent a change in treatment plan, 5 experienced symptom recurrence, 4 had mild improvement, and 1 patient had no improvement. Six patients were lost to follow-up.17
In the study conducted by Nguyen and Eichenfield,3 follow-up visits occurred approximately 3 months after the first visit.
In the case series by Boeck et al,19 all patients were started on metronidazole gel 1% applied once daily for the first week, and then twice daily until the lesions resolved. All patients showed improvement after 4 to 6 weeks, and eventually the disease cleared between 3 and 6 months. All patients were still symptom free during a 2-year observation period.19
Manders and Lucky7 described 14 patients with POD (aged 9 months to 6.5 years). Eight patients used only metronidazole gel 0.75%, while 5 used the gel in combination with topical corticosteroids (21% [3/14]), oral erythromycin (7% [1/14]), or topical erythromycin (7% [1/14]); 1 patient remained on hydrocortisone 1% and cleared. Patients responded well within 1 to 8 weeks and were symptom free for up to 16 months. Mid- to high-potency steroids were discontinued in all patients.7
In some pediatric patients with CGPD, recovery occurs faster with the use of oral macrolides or tetracyclines, either alone or in combination with topical antibiotics or sulfur-based lotions.20 Extrafacial lesions associated with CGPD do not appear to negatively impact treatment response or duration of disease. In the review conducted by Urbatsch et al,20 7 of 8 (88%) CGPD patients with extrafacial lesions were treated with oral agents including erythromycin, hydroxychloroquine, cyclosporine, minocycline, and azithromycin. Most of these patients also were using topical agents such as triamcinolone acetonide, desonide, metronidazole, and erythromycin. The time to resolution ranged from several weeks to 6 months.20
Weston and Morelli9 described a treatment regimen for steroid rosacea. The study included data on 106 children (60 females, 46 males) who had been exposed to mostly class 7 low-potency agents. All patients were advised to immediately stop topical steroid therapy without gradual withdrawal and to begin oral erythromycin stearate 30 mg/kg daily in 2 doses per day for 4 weeks. Patients who were unable to tolerate erythromycin were advised to use topical clindamycin phosphate twice daily for 4 weeks (n=6). Eighty-six percent of patients showed resolution within 4 weeks, and 100% showed clearance by 8 weeks. Twenty-two percent of patients had clearance within 3 weeks. There was no difference in the duration until resolution for those who had used oral or topical antibiotics.9 A different study suggested that low-potency topical steroids can be used to control inflammation when weaning patients off of strong steroids.5
Differential Diagnosis
The differential diagnosis should include acne vulgaris, allergic contact dermatitis, irritant contact dermatitis, seborrheic dermatitis, impetigo, dermatophyte infection, rosacea, and angiofibromas.4
Acne vulgaris commonly is found in older adolescents, and unlike POD, it will present with open or closed comedones.2 In patients aged 1 to 7 years, acne is a reason to consider endocrine evaluation. Allergic contact dermatitis is extremely pruritic, and the lesions often are papulovesicular with active weeping or crusting. Patients with irritant contact dermatitis often report burning and pain, and papules and pustules typically are absent. A thorough history can help rule out allergic or irritant contact dermatitis. Seborrheic dermatitis presents with erythema and scaling of the scalp, eyebrows, and nasolabial folds; it tends to spare the perioral regions and also lacks papules.2 The lesions of impetigo typically have a yellow-brown exudate, which forms a honey-colored crust.24 Tinea faciei, unlike the other tinea infections, can have an extremely variable presentation. Lesions usually begin as scaly macules that develop raised borders with central hypopigmentation, but papules, vesicles, and crusts can be seen.25 Potassium hydroxide
Conclusion
Diagnosis of POD is clinical and rests upon the finding of erythematous papules on the face near the eyes, mouth, and nose. Extrafacial lesions also have been described, particularly in pediatric patients with CGPD. Many patients will report a history of atopic dermatitis and asthma. Therapy for POD includes both topical and systemic agents. For those with mild disease, topical metronidazole commonly is used. For patients requiring oral antibiotics, tetracyclines or macrolides can be prescribed based on the age of the patient. Many pediatric patients who begin with both oral and topical agents can later be maintained on topical therapy, sometimes with a low-dose oral antibiotic. Periorificial dermatitis has an excellent prognosis and most pediatric patients show marked improvement within weeks to months.
- Tempark T, Shwayder TA. Perioral dermatitis: a review of the condition with special attention to treatment options. Am J Clin Dermatol. 2014;15:101-113.
- McFarland SL, Polcari IC. Morphology-based diagnosis of acneiform eruptions. Pediatr Ann. 2015;44:E188-E193.
- Nguyen V, Eichenfield LF. Periorificial dermatitis in children and adolescents. J Am Acad Dermatol. 2006;55:781-785.
- Kihiczak GG, Cruz MA, Schwartz RA. Periorificial dermatitis in children: an update and description of a child with striking features. Int J Dermatol. 2009;48:304-306.
- Laude TA, Salvemini JN. Perioral dermatitis in children. Sem Cutan Med Surg. 1999;18:206-209.
- Wilkinson DS, Kirton V, Wilkinson JD. Perioral dermatitis: a 12-year review. Br J Dermatol. 1979;101:245-257.
- Manders SM, Lucky AW. Perioral dermatitis in childhood. J Am Acad Dermatol. 1992;27(5 pt 1):688-692.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Dermatol. 1972;87:425-429.
- Weston WL, Morelli JG. Steroid rosacea in prepubertal children. Arch Pediatr Adolesc Med. 2000;154:62-64.
- Clementson B, Smidt AC. Periorificial dermatitis due to systemic corticosteroids in children: report of two cases. Pediatr Dermatol. 2012;29:331-332.
- Takiwaki H, Tsuda H, Arase S, et al. Differences between intrafollicular microorganism profiles in perioral and seborrhoeic dermatitis. Clin Exp Dermatol. 2003;28:531-534.
- Abeck D, Geisenfelder B, Brandt O. Physical sunscreens with high sun protection factor may cause perioral dermatitis in children. J Dtsch Dermatol Ges. 2009;7:701-703.
- Satyawan I, Oranje AP, van Joost T. Perioral dermatitis in a child due to rosin in chewing gum. Contact Dermatitis. 1990;22:182-183.
- Dubus JC, Marguet C, Deschildre A, et al. Local side-effects of inhaled corticosteroids in asthmatic children: influence of drug, dose, age, and device. Allergy. 2001;56:944-948.
- Hausen BM, Bruhn G, Koenig WA. New hydroxyisoflavans as contact sensitizers in cocus wood Brya ebenus DC (Fabaceae). Contact Dermatitis. 1991;25:149-155.
- Dirschka T, Weber K, Tronnier H. Topical cosmetics and perioral dermatitis. J Dtsch Dermatol Ges. 2004;2:194-199.
- Goel NS, Burkhart CN, Morrell DS. Pediatric periorificial dermatitis: clinical course and treatment outcomes in 222 patients. Pediatr Dermatol. 2015;32:333-336.
- Cribier B, Lieber-Mbomeyo A, Lipsker D. Clinical and histological study of a case of facial Afro-Caribbean childhood eruption (FACE) [in French][published online July 23, 2008]. Ann Dermatol Venerol. 2008;135:663-667.
- Boeck K, Abeck D, Werfel S, et al. Perioral dermatitis in children—clinical presentation, pathogenesis-related factors and response to topical metronidazole. Dermatology. 1997;195:235-238.
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358.
- Kroshinsky D, Glick SA. Pediatric rosacea. Dermatol Ther. 2006;19:196-201.
- Ramelet AA, Delacrétaz J. Histopathologic study of perioral dermatitis [in French]. Dermatologica. 1981;163:361-369.
- Ljubojevi´c S, Lipozenci´c J, Turci´c P. Perioral dermatitis. Acta Dermatovenerol Croat. 2008;16:96-100.
- Nichols RL, Florman S. Clinical presentations of soft-tissue infections and surgical site infections. Clin Infect Dis. 2001;33(suppl 2):S84-S93.
- Lin RL, Szepietowski JC, Schwartz RA. Tinea faciei, an often deceptive facial eruption. Int J Dermatol. 2004;43:437-440.
- Tempark T, Shwayder TA. Perioral dermatitis: a review of the condition with special attention to treatment options. Am J Clin Dermatol. 2014;15:101-113.
- McFarland SL, Polcari IC. Morphology-based diagnosis of acneiform eruptions. Pediatr Ann. 2015;44:E188-E193.
- Nguyen V, Eichenfield LF. Periorificial dermatitis in children and adolescents. J Am Acad Dermatol. 2006;55:781-785.
- Kihiczak GG, Cruz MA, Schwartz RA. Periorificial dermatitis in children: an update and description of a child with striking features. Int J Dermatol. 2009;48:304-306.
- Laude TA, Salvemini JN. Perioral dermatitis in children. Sem Cutan Med Surg. 1999;18:206-209.
- Wilkinson DS, Kirton V, Wilkinson JD. Perioral dermatitis: a 12-year review. Br J Dermatol. 1979;101:245-257.
- Manders SM, Lucky AW. Perioral dermatitis in childhood. J Am Acad Dermatol. 1992;27(5 pt 1):688-692.
- Savin JA, Alexander S, Marks R. A rosacea-like eruption of children. Br J Dermatol. 1972;87:425-429.
- Weston WL, Morelli JG. Steroid rosacea in prepubertal children. Arch Pediatr Adolesc Med. 2000;154:62-64.
- Clementson B, Smidt AC. Periorificial dermatitis due to systemic corticosteroids in children: report of two cases. Pediatr Dermatol. 2012;29:331-332.
- Takiwaki H, Tsuda H, Arase S, et al. Differences between intrafollicular microorganism profiles in perioral and seborrhoeic dermatitis. Clin Exp Dermatol. 2003;28:531-534.
- Abeck D, Geisenfelder B, Brandt O. Physical sunscreens with high sun protection factor may cause perioral dermatitis in children. J Dtsch Dermatol Ges. 2009;7:701-703.
- Satyawan I, Oranje AP, van Joost T. Perioral dermatitis in a child due to rosin in chewing gum. Contact Dermatitis. 1990;22:182-183.
- Dubus JC, Marguet C, Deschildre A, et al. Local side-effects of inhaled corticosteroids in asthmatic children: influence of drug, dose, age, and device. Allergy. 2001;56:944-948.
- Hausen BM, Bruhn G, Koenig WA. New hydroxyisoflavans as contact sensitizers in cocus wood Brya ebenus DC (Fabaceae). Contact Dermatitis. 1991;25:149-155.
- Dirschka T, Weber K, Tronnier H. Topical cosmetics and perioral dermatitis. J Dtsch Dermatol Ges. 2004;2:194-199.
- Goel NS, Burkhart CN, Morrell DS. Pediatric periorificial dermatitis: clinical course and treatment outcomes in 222 patients. Pediatr Dermatol. 2015;32:333-336.
- Cribier B, Lieber-Mbomeyo A, Lipsker D. Clinical and histological study of a case of facial Afro-Caribbean childhood eruption (FACE) [in French][published online July 23, 2008]. Ann Dermatol Venerol. 2008;135:663-667.
- Boeck K, Abeck D, Werfel S, et al. Perioral dermatitis in children—clinical presentation, pathogenesis-related factors and response to topical metronidazole. Dermatology. 1997;195:235-238.
- Urbatsch AJ, Frieden I, Williams ML, et al. Extrafacial and generalized granulomatous periorificial dermatitis. Arch Dermatol. 2002;138:1354-1358.
- Kroshinsky D, Glick SA. Pediatric rosacea. Dermatol Ther. 2006;19:196-201.
- Ramelet AA, Delacrétaz J. Histopathologic study of perioral dermatitis [in French]. Dermatologica. 1981;163:361-369.
- Ljubojevi´c S, Lipozenci´c J, Turci´c P. Perioral dermatitis. Acta Dermatovenerol Croat. 2008;16:96-100.
- Nichols RL, Florman S. Clinical presentations of soft-tissue infections and surgical site infections. Clin Infect Dis. 2001;33(suppl 2):S84-S93.
- Lin RL, Szepietowski JC, Schwartz RA. Tinea faciei, an often deceptive facial eruption. Int J Dermatol. 2004;43:437-440.
Practice Points
- Periorificial dermatitis (POD) affects young children and presents as flesh-colored papules around the mouth, nose, and even groin.
- Periorificial dermatitis has been associated with prior use of topical or inhaled steroids.
- Children with POD can be treated with oral erythromycin.
SABCS 2017: Top picks from Dr. William J. Gradishar
Oncology Practice Associate Editor William J. Gradishar, MD, reveals several anticipated studies from the 40th annual San Antonio Breast Cancer Symposium, set to begin on Wednesday, Dec. 6:

• GS1-01. Increasing the dose density of adjuvant chemotherapy by shortening intervals between courses or by sequential drug administration significantly reduces both disease recurrence and breast cancer mortality: an EBCTCG meta-analysis of 21,000 women in 16 randomized trials.
• GS4-02. Randomized comparison of adjuvant aromatase inhibitor exemestane plus ovarian function suppression vs. tamoxifen plus OFS in premenopausal women with hormone receptor–positive early breast cancer: Update of the combined TEXT and SOFT trials.
• GS1-03. Perioperative aromatase inhibitor treatment in determining or predicting long-term outcome in early breast cancer–the POETIC Trial.
• GS1-06. Extended adjuvant bisphosphonate treatment over 5 years in early breast cancer does not improve disease-free and overall survival, compared with 2 years of treatment: Phase III data from the SUCCESS A study.
• GS2-05. First-line ribociclib vs. placebo with goserelin and tamoxifen or a nonsteroidal aromatase inhibitor in premenopausal women with hormone receptor–positive, HER2-negative advanced breast cancer: Results from the randomized phase III MONALEESA-7 trial.
• GS2-06. Phase Ib/II study evaluating safety and efficacy of pembrolizumab and trastuzumab in patients with trastuzumab-resistant HER2-positive metastatic breast cancer: Results from the PANACEA (IBCSG 45-13/KEYNOTE-014) study.
• GS2-07. MANTA – A randomized phase 2 study of fulvestrant in combination with the dual mTOR inhibitor AZD2014 or everolimus or fulvestrant alone in estrogen receptor–positive advanced or metastatic breast cancer.
• GS3-01. A prospective, randomized, multicenter phase 3 trial of additional 2 versus additional 5 years of anastrozole after initial 5 years of adjuvant endocrine therapy – results from 3,484 postmenopausal women in the ABCSG-16 trial.
• GS3-02. Invasive disease-free survival and gene-expression signatures in CALGB (Alliance) 40601, a randomized phase III neoadjuvant trial of dual HER2 targeting with lapatinib added to chemotherapy plus trastuzumab.
• GS3-03. A phase III, multicenter, double-blind randomized trial of celecoxib versus placebo in primary breast cancer patients (REACT – Randomized European Celecoxib Trial).
• GS3-04. A randomized phase III study of adjuvant trastuzumab for a duration of 9 weeks versus 1 year, combined with adjuvant taxane-anthracycline chemotherapy, for early HER2-positive breast cancer (the SOLD study).
• GS3-06. Long-term follow-up of CALGB 40502/NCCTG N063H (Alliance): A randomized phase III trial of weekly paclitaxel, compared with weekly nanoparticle albumin-bound (nab)–paclitaxel or ixabepilone +/– bevacizumab as first-line therapy for locally recurrent or metastatic breast cancer.
• GS3-08. Pathological complete response predicts event-free and distant disease-free survival in the I-SPY2 TRIAL.
• GS4-01. Pooled analysis of five randomized trials investigating temporary ovarian suppression with gonadotropin-releasing hormone analogues during chemotherapy as a strategy to preserve ovarian function and fertility in premenopausal early breast cancer patients.
• GS4-02. Randomized comparison of adjuvant aromatase inhibitor exemestane plus ovarian function suppression vs. tamoxifen plus OFS in premenopausal women with hormone receptor–positive early breast cancer: Update of the combined TEXT and SOFT trials.
• GS4-03. Randomized comparison of adjuvant tamoxifen plus ovarian function suppression versus tamoxifen in premenopausal women with hormone receptor–positive early breast cancer: Update of the SOFT trial.
• GS4-04. Randomized blinded sham- and waitlist-controlled trial of acupuncture for joint symptoms related to aromatase inhibitors in women with early-stage breast cancer.
• GS5-03. Risk of arm morbidity after local therapy in young breast cancer survivors.
• GS5-05. Primary endocrine therapy for ER-positive ductal carcinoma in situ: CALGB 40903 (Alliance).
• GS6-01. Integration of clinical variables for the prediction of late distant recurrence in patients with estrogen receptor–positive breast cancer treated with 5 years of endocrine therapy.
• GS6-02. The benefit of abemaciclib in prognostic subgroups: An exploratory analysis of combined data from the MONARCH 2 and 3 studies.
• GS6-07. EMBRACA: A phase 3 trial comparing talazoparib, an oral PARP inhibitor, to physician’s choice of therapy in patients with advanced breast cancer and a germline BRCA mutation.
Dr. Gradishar is the Betsy Bramsen Professor of Breast Oncology, professor of medicine, and interim chief of the division of hematology/oncology, Northwestern University Feinberg School of Medicine, Chicago, and deputy director for the Clinical Network and director of the Maggie Daley Center for Women’s Cancer Care, Robert H. Lurie Comprehensive Cancer Center Network of Northwestern University, Chicago.
Oncology Practice Associate Editor William J. Gradishar, MD, reveals several anticipated studies from the 40th annual San Antonio Breast Cancer Symposium, set to begin on Wednesday, Dec. 6:
• GS1-01. Increasing the dose density of adjuvant chemotherapy by shortening intervals between courses or by sequential drug administration significantly reduces both disease recurrence and breast cancer mortality: an EBCTCG meta-analysis of 21,000 women in 16 randomized trials.
• GS4-02. Randomized comparison of adjuvant aromatase inhibitor exemestane plus ovarian function suppression vs. tamoxifen plus OFS in premenopausal women with hormone receptor–positive early breast cancer: Update of the combined TEXT and SOFT trials.
• GS1-03. Perioperative aromatase inhibitor treatment in determining or predicting long-term outcome in early breast cancer–the POETIC Trial.
• GS1-06. Extended adjuvant bisphosphonate treatment over 5 years in early breast cancer does not improve disease-free and overall survival, compared with 2 years of treatment: Phase III data from the SUCCESS A study.
• GS2-05. First-line ribociclib vs. placebo with goserelin and tamoxifen or a nonsteroidal aromatase inhibitor in premenopausal women with hormone receptor–positive, HER2-negative advanced breast cancer: Results from the randomized phase III MONALEESA-7 trial.
• GS2-06. Phase Ib/II study evaluating safety and efficacy of pembrolizumab and trastuzumab in patients with trastuzumab-resistant HER2-positive metastatic breast cancer: Results from the PANACEA (IBCSG 45-13/KEYNOTE-014) study.
• GS2-07. MANTA – A randomized phase 2 study of fulvestrant in combination with the dual mTOR inhibitor AZD2014 or everolimus or fulvestrant alone in estrogen receptor–positive advanced or metastatic breast cancer.
• GS3-01. A prospective, randomized, multicenter phase 3 trial of additional 2 versus additional 5 years of anastrozole after initial 5 years of adjuvant endocrine therapy – results from 3,484 postmenopausal women in the ABCSG-16 trial.
• GS3-02. Invasive disease-free survival and gene-expression signatures in CALGB (Alliance) 40601, a randomized phase III neoadjuvant trial of dual HER2 targeting with lapatinib added to chemotherapy plus trastuzumab.
• GS3-03. A phase III, multicenter, double-blind randomized trial of celecoxib versus placebo in primary breast cancer patients (REACT – Randomized European Celecoxib Trial).
• GS3-04. A randomized phase III study of adjuvant trastuzumab for a duration of 9 weeks versus 1 year, combined with adjuvant taxane-anthracycline chemotherapy, for early HER2-positive breast cancer (the SOLD study).
• GS3-06. Long-term follow-up of CALGB 40502/NCCTG N063H (Alliance): A randomized phase III trial of weekly paclitaxel, compared with weekly nanoparticle albumin-bound (nab)–paclitaxel or ixabepilone +/– bevacizumab as first-line therapy for locally recurrent or metastatic breast cancer.
• GS3-08. Pathological complete response predicts event-free and distant disease-free survival in the I-SPY2 TRIAL.
• GS4-01. Pooled analysis of five randomized trials investigating temporary ovarian suppression with gonadotropin-releasing hormone analogues during chemotherapy as a strategy to preserve ovarian function and fertility in premenopausal early breast cancer patients.
• GS4-02. Randomized comparison of adjuvant aromatase inhibitor exemestane plus ovarian function suppression vs. tamoxifen plus OFS in premenopausal women with hormone receptor–positive early breast cancer: Update of the combined TEXT and SOFT trials.
• GS4-03. Randomized comparison of adjuvant tamoxifen plus ovarian function suppression versus tamoxifen in premenopausal women with hormone receptor–positive early breast cancer: Update of the SOFT trial.
• GS4-04. Randomized blinded sham- and waitlist-controlled trial of acupuncture for joint symptoms related to aromatase inhibitors in women with early-stage breast cancer.
• GS5-03. Risk of arm morbidity after local therapy in young breast cancer survivors.
• GS5-05. Primary endocrine therapy for ER-positive ductal carcinoma in situ: CALGB 40903 (Alliance).
• GS6-01. Integration of clinical variables for the prediction of late distant recurrence in patients with estrogen receptor–positive breast cancer treated with 5 years of endocrine therapy.
• GS6-02. The benefit of abemaciclib in prognostic subgroups: An exploratory analysis of combined data from the MONARCH 2 and 3 studies.
• GS6-07. EMBRACA: A phase 3 trial comparing talazoparib, an oral PARP inhibitor, to physician’s choice of therapy in patients with advanced breast cancer and a germline BRCA mutation.
Dr. Gradishar is the Betsy Bramsen Professor of Breast Oncology, professor of medicine, and interim chief of the division of hematology/oncology, Northwestern University Feinberg School of Medicine, Chicago, and deputy director for the Clinical Network and director of the Maggie Daley Center for Women’s Cancer Care, Robert H. Lurie Comprehensive Cancer Center Network of Northwestern University, Chicago.
Oncology Practice Associate Editor William J. Gradishar, MD, reveals several anticipated studies from the 40th annual San Antonio Breast Cancer Symposium, set to begin on Wednesday, Dec. 6:
• GS1-01. Increasing the dose density of adjuvant chemotherapy by shortening intervals between courses or by sequential drug administration significantly reduces both disease recurrence and breast cancer mortality: an EBCTCG meta-analysis of 21,000 women in 16 randomized trials.
• GS4-02. Randomized comparison of adjuvant aromatase inhibitor exemestane plus ovarian function suppression vs. tamoxifen plus OFS in premenopausal women with hormone receptor–positive early breast cancer: Update of the combined TEXT and SOFT trials.
• GS1-03. Perioperative aromatase inhibitor treatment in determining or predicting long-term outcome in early breast cancer–the POETIC Trial.
• GS1-06. Extended adjuvant bisphosphonate treatment over 5 years in early breast cancer does not improve disease-free and overall survival, compared with 2 years of treatment: Phase III data from the SUCCESS A study.
• GS2-05. First-line ribociclib vs. placebo with goserelin and tamoxifen or a nonsteroidal aromatase inhibitor in premenopausal women with hormone receptor–positive, HER2-negative advanced breast cancer: Results from the randomized phase III MONALEESA-7 trial.
• GS2-06. Phase Ib/II study evaluating safety and efficacy of pembrolizumab and trastuzumab in patients with trastuzumab-resistant HER2-positive metastatic breast cancer: Results from the PANACEA (IBCSG 45-13/KEYNOTE-014) study.
• GS2-07. MANTA – A randomized phase 2 study of fulvestrant in combination with the dual mTOR inhibitor AZD2014 or everolimus or fulvestrant alone in estrogen receptor–positive advanced or metastatic breast cancer.
• GS3-01. A prospective, randomized, multicenter phase 3 trial of additional 2 versus additional 5 years of anastrozole after initial 5 years of adjuvant endocrine therapy – results from 3,484 postmenopausal women in the ABCSG-16 trial.
• GS3-02. Invasive disease-free survival and gene-expression signatures in CALGB (Alliance) 40601, a randomized phase III neoadjuvant trial of dual HER2 targeting with lapatinib added to chemotherapy plus trastuzumab.
• GS3-03. A phase III, multicenter, double-blind randomized trial of celecoxib versus placebo in primary breast cancer patients (REACT – Randomized European Celecoxib Trial).
• GS3-04. A randomized phase III study of adjuvant trastuzumab for a duration of 9 weeks versus 1 year, combined with adjuvant taxane-anthracycline chemotherapy, for early HER2-positive breast cancer (the SOLD study).
• GS3-06. Long-term follow-up of CALGB 40502/NCCTG N063H (Alliance): A randomized phase III trial of weekly paclitaxel, compared with weekly nanoparticle albumin-bound (nab)–paclitaxel or ixabepilone +/– bevacizumab as first-line therapy for locally recurrent or metastatic breast cancer.
• GS3-08. Pathological complete response predicts event-free and distant disease-free survival in the I-SPY2 TRIAL.
• GS4-01. Pooled analysis of five randomized trials investigating temporary ovarian suppression with gonadotropin-releasing hormone analogues during chemotherapy as a strategy to preserve ovarian function and fertility in premenopausal early breast cancer patients.
• GS4-02. Randomized comparison of adjuvant aromatase inhibitor exemestane plus ovarian function suppression vs. tamoxifen plus OFS in premenopausal women with hormone receptor–positive early breast cancer: Update of the combined TEXT and SOFT trials.
• GS4-03. Randomized comparison of adjuvant tamoxifen plus ovarian function suppression versus tamoxifen in premenopausal women with hormone receptor–positive early breast cancer: Update of the SOFT trial.
• GS4-04. Randomized blinded sham- and waitlist-controlled trial of acupuncture for joint symptoms related to aromatase inhibitors in women with early-stage breast cancer.
• GS5-03. Risk of arm morbidity after local therapy in young breast cancer survivors.
• GS5-05. Primary endocrine therapy for ER-positive ductal carcinoma in situ: CALGB 40903 (Alliance).
• GS6-01. Integration of clinical variables for the prediction of late distant recurrence in patients with estrogen receptor–positive breast cancer treated with 5 years of endocrine therapy.
• GS6-02. The benefit of abemaciclib in prognostic subgroups: An exploratory analysis of combined data from the MONARCH 2 and 3 studies.
• GS6-07. EMBRACA: A phase 3 trial comparing talazoparib, an oral PARP inhibitor, to physician’s choice of therapy in patients with advanced breast cancer and a germline BRCA mutation.
Dr. Gradishar is the Betsy Bramsen Professor of Breast Oncology, professor of medicine, and interim chief of the division of hematology/oncology, Northwestern University Feinberg School of Medicine, Chicago, and deputy director for the Clinical Network and director of the Maggie Daley Center for Women’s Cancer Care, Robert H. Lurie Comprehensive Cancer Center Network of Northwestern University, Chicago.
FROM SABCS 2017
Blisibimod shows mixed results for lupus in phase 3 trial
SAN DIEGO – While use of the investigational agent blisibimod did not meet the primary endpoint in a phase 3 trial in systemic lupus erythematosus (SLE), it was associated with steroid sparing, decreased urine protein, a trend toward decreased anti–double-stranded DNA antibodies, and significant decreases in anticardiolipin antibodies and immunoglobulin levels.
Those results were reported from CHABLIS-SC1 trial, a randomized (5:4), double-blind, placebo-controlled phase 3 study of 442 patients, and were presented by Joan T. Merrill, MD, at the annual meeting of the American College of Rheumatology. Blisibimod is a subcutaneously injected inhibitor of B-cell activating factor.

The SRI-6 primary endpoint at 52 weeks was met by 44% of patients on blisibimod, compared with 42% on placebo. But the SRI-6 endpoint widened when the most common features at entry were excluded (mucocutaneous and musculoskeletal), suggesting the possibility that blisibimod affects the more objective and potentially organ-threatening renal endpoint.
To be eligible for the trial, patients had to have a Safety of Estrogen in Lupus Erythematosus National Assessment Group–SLE Disease Activity Index (SELENA-SLEDAI) score of 10 or greater and be receiving steroids. The SRI-6 primary endpoint also required no worsening on the British Isles Lupus Assessment Group (BILAG) disease activity index or Physician Global Assessment. Key secondary endpoints included proteinuria and achievement of steroid taper.
Patients in the study were required to be receiving stable doses of prednisone or an equivalent steroid at less than or equal to 0.5 mg/kg or 40 mg daily for at least 28 days prior to randomization. Other permitted standard-of-care oral medications included methotrexate up to 25 mg weekly, azathioprine up to 300 mg daily, mycophenolate mofetil or sodium salt up to 3 mg daily, leflunomide up to 40 mg daily, hydroxychloroquine up to 400 mg daily, and nonsteroidal drugs within locally approved dose ranges.
Of the 442 patients enrolled, 245 received blisibimod, and 197 received placebo. They were well matched in demographics and baseline disease characteristics. “There were very few patients of African descent in this trial,” Dr. Merrill noted. “Also, about 30% of patients had some renal involvement. In fact, patients with stable, active renal disease were encouraged to participate in this trial.” The mean prednisone dose at entry was between 15 and 16 mg daily, and about 60% of patients were taking an antimalarial. An equal proportion of patients in both groups discontinued the study (22%). “A few more patients withdrew due to adverse events in the blisibimod group, and a few more patients withdrew due to lack of efficacy in the placebo group,” she said.
Of 135 patients with a baseline urine protein-to-creatinine ratio equal to or greater than 0.5, blisibimod treatment led to significantly greater improvement in proteinuria at several time points than did treatment with placebo, which also showed improvement in proteinuria.
Treatment with blisibimod also was associated with a reduction in anti–double-stranded DNA antibodies, as well as significant reductions in peripheral B-cell lineages, anticardiolipin antibodies, and immunoglobulins, and with significant increases in complement C3 and C4. “The expected pharmacodynamic markers were [also] met ... and more patients treated with blisibimod were able to achieve a prednisone milestone of reduction to less than or equal to 7.5 mg/day, compared with those in the placebo group,” she said. “This was statistically significant over time at multiple time points.”
Adverse events were balanced between treatment arms except for injection site reactions, which occurred in 7.3% of blisibimod-treated patients versus 2.6% of placebo patients. There were no major safety issues in the study.
Dr. Merrill hypothesized that the higher mean doses of corticosteroid at baseline could have contributed to the higher-than-usual placebo response rates and failure to meet the primary SRI-6 endpoint.
The study was supported by Anthera. Dr. Merrill disclosed that she has received research support from Anthera, Amgen, EMD Serono, GlaxoSmithKline. and Novartis.
SAN DIEGO – While use of the investigational agent blisibimod did not meet the primary endpoint in a phase 3 trial in systemic lupus erythematosus (SLE), it was associated with steroid sparing, decreased urine protein, a trend toward decreased anti–double-stranded DNA antibodies, and significant decreases in anticardiolipin antibodies and immunoglobulin levels.
Those results were reported from CHABLIS-SC1 trial, a randomized (5:4), double-blind, placebo-controlled phase 3 study of 442 patients, and were presented by Joan T. Merrill, MD, at the annual meeting of the American College of Rheumatology. Blisibimod is a subcutaneously injected inhibitor of B-cell activating factor.
The SRI-6 primary endpoint at 52 weeks was met by 44% of patients on blisibimod, compared with 42% on placebo. But the SRI-6 endpoint widened when the most common features at entry were excluded (mucocutaneous and musculoskeletal), suggesting the possibility that blisibimod affects the more objective and potentially organ-threatening renal endpoint.
To be eligible for the trial, patients had to have a Safety of Estrogen in Lupus Erythematosus National Assessment Group–SLE Disease Activity Index (SELENA-SLEDAI) score of 10 or greater and be receiving steroids. The SRI-6 primary endpoint also required no worsening on the British Isles Lupus Assessment Group (BILAG) disease activity index or Physician Global Assessment. Key secondary endpoints included proteinuria and achievement of steroid taper.
Patients in the study were required to be receiving stable doses of prednisone or an equivalent steroid at less than or equal to 0.5 mg/kg or 40 mg daily for at least 28 days prior to randomization. Other permitted standard-of-care oral medications included methotrexate up to 25 mg weekly, azathioprine up to 300 mg daily, mycophenolate mofetil or sodium salt up to 3 mg daily, leflunomide up to 40 mg daily, hydroxychloroquine up to 400 mg daily, and nonsteroidal drugs within locally approved dose ranges.
Of the 442 patients enrolled, 245 received blisibimod, and 197 received placebo. They were well matched in demographics and baseline disease characteristics. “There were very few patients of African descent in this trial,” Dr. Merrill noted. “Also, about 30% of patients had some renal involvement. In fact, patients with stable, active renal disease were encouraged to participate in this trial.” The mean prednisone dose at entry was between 15 and 16 mg daily, and about 60% of patients were taking an antimalarial. An equal proportion of patients in both groups discontinued the study (22%). “A few more patients withdrew due to adverse events in the blisibimod group, and a few more patients withdrew due to lack of efficacy in the placebo group,” she said.
Of 135 patients with a baseline urine protein-to-creatinine ratio equal to or greater than 0.5, blisibimod treatment led to significantly greater improvement in proteinuria at several time points than did treatment with placebo, which also showed improvement in proteinuria.
Treatment with blisibimod also was associated with a reduction in anti–double-stranded DNA antibodies, as well as significant reductions in peripheral B-cell lineages, anticardiolipin antibodies, and immunoglobulins, and with significant increases in complement C3 and C4. “The expected pharmacodynamic markers were [also] met ... and more patients treated with blisibimod were able to achieve a prednisone milestone of reduction to less than or equal to 7.5 mg/day, compared with those in the placebo group,” she said. “This was statistically significant over time at multiple time points.”
Adverse events were balanced between treatment arms except for injection site reactions, which occurred in 7.3% of blisibimod-treated patients versus 2.6% of placebo patients. There were no major safety issues in the study.
Dr. Merrill hypothesized that the higher mean doses of corticosteroid at baseline could have contributed to the higher-than-usual placebo response rates and failure to meet the primary SRI-6 endpoint.
The study was supported by Anthera. Dr. Merrill disclosed that she has received research support from Anthera, Amgen, EMD Serono, GlaxoSmithKline. and Novartis.
SAN DIEGO – While use of the investigational agent blisibimod did not meet the primary endpoint in a phase 3 trial in systemic lupus erythematosus (SLE), it was associated with steroid sparing, decreased urine protein, a trend toward decreased anti–double-stranded DNA antibodies, and significant decreases in anticardiolipin antibodies and immunoglobulin levels.
Those results were reported from CHABLIS-SC1 trial, a randomized (5:4), double-blind, placebo-controlled phase 3 study of 442 patients, and were presented by Joan T. Merrill, MD, at the annual meeting of the American College of Rheumatology. Blisibimod is a subcutaneously injected inhibitor of B-cell activating factor.
The SRI-6 primary endpoint at 52 weeks was met by 44% of patients on blisibimod, compared with 42% on placebo. But the SRI-6 endpoint widened when the most common features at entry were excluded (mucocutaneous and musculoskeletal), suggesting the possibility that blisibimod affects the more objective and potentially organ-threatening renal endpoint.
To be eligible for the trial, patients had to have a Safety of Estrogen in Lupus Erythematosus National Assessment Group–SLE Disease Activity Index (SELENA-SLEDAI) score of 10 or greater and be receiving steroids. The SRI-6 primary endpoint also required no worsening on the British Isles Lupus Assessment Group (BILAG) disease activity index or Physician Global Assessment. Key secondary endpoints included proteinuria and achievement of steroid taper.
Patients in the study were required to be receiving stable doses of prednisone or an equivalent steroid at less than or equal to 0.5 mg/kg or 40 mg daily for at least 28 days prior to randomization. Other permitted standard-of-care oral medications included methotrexate up to 25 mg weekly, azathioprine up to 300 mg daily, mycophenolate mofetil or sodium salt up to 3 mg daily, leflunomide up to 40 mg daily, hydroxychloroquine up to 400 mg daily, and nonsteroidal drugs within locally approved dose ranges.
Of the 442 patients enrolled, 245 received blisibimod, and 197 received placebo. They were well matched in demographics and baseline disease characteristics. “There were very few patients of African descent in this trial,” Dr. Merrill noted. “Also, about 30% of patients had some renal involvement. In fact, patients with stable, active renal disease were encouraged to participate in this trial.” The mean prednisone dose at entry was between 15 and 16 mg daily, and about 60% of patients were taking an antimalarial. An equal proportion of patients in both groups discontinued the study (22%). “A few more patients withdrew due to adverse events in the blisibimod group, and a few more patients withdrew due to lack of efficacy in the placebo group,” she said.
Of 135 patients with a baseline urine protein-to-creatinine ratio equal to or greater than 0.5, blisibimod treatment led to significantly greater improvement in proteinuria at several time points than did treatment with placebo, which also showed improvement in proteinuria.
Treatment with blisibimod also was associated with a reduction in anti–double-stranded DNA antibodies, as well as significant reductions in peripheral B-cell lineages, anticardiolipin antibodies, and immunoglobulins, and with significant increases in complement C3 and C4. “The expected pharmacodynamic markers were [also] met ... and more patients treated with blisibimod were able to achieve a prednisone milestone of reduction to less than or equal to 7.5 mg/day, compared with those in the placebo group,” she said. “This was statistically significant over time at multiple time points.”
Adverse events were balanced between treatment arms except for injection site reactions, which occurred in 7.3% of blisibimod-treated patients versus 2.6% of placebo patients. There were no major safety issues in the study.
Dr. Merrill hypothesized that the higher mean doses of corticosteroid at baseline could have contributed to the higher-than-usual placebo response rates and failure to meet the primary SRI-6 endpoint.
The study was supported by Anthera. Dr. Merrill disclosed that she has received research support from Anthera, Amgen, EMD Serono, GlaxoSmithKline. and Novartis.
AT ACR 2017
Key clinical point:
Major finding: The primary endpoint of CHABLIS-SC1 was not met, but expected pharmacodynamic markers were.
Study details: A phase 3 study in which 245 SLE patients received blisibimod and 197 received placebo.
Disclosures: The study was supported by Anthera Pharmaceuticals. Dr. Merrill disclosed that she has received research support from Anthera, Amgen, EMD Serono, GlaxoSmithKline, and Novartis.
Topical 5-Fluorouracil Made Easy?
What is the recent research behind 5-fluorouracil cream 5% combined with calcipotriol ointment 0.005% for actinic keratoses?
Cunningham et al published a randomized double-blind study in which 131 patients with actinic keratoses (AKs) were assigned to either 5-fluorouracil (5-FU) cream 5% combined with calcipotriol (calcipotriene) ointment 0.005% twice daily to the face, scalp, and arms for 4 days, or 5-FU 5% combined with petrolatum applied in the same fashion. There was an 87.8% versus 26.3% mean reduction in the number of AKs and less severe pain, crusting, and ulceration in the study cohort compared to the 5-FU plus petrolatum group.
The same study also investigated immune parameters in these patients and found that the study group preferentially displayed activated thymic stromal lymphopoietin and a CD4 T cell-mediated reaction, among other effects. In prior studies, thymic stromal lymphopoietin has been shown to be upregulated in barrier-defective skin, displays antitumor activity, and is enhanced by topical calcipotriol application based on its original indication for psoriasis.
How do these study results impact patient care?
In a perfect world, every patient could tolerate and afford chemopreventative measures such as 5-FU cream, apply it diffusely to sun-exposed skin, and experience no severe irritant reactions and/or social pariah status. We all know that this product is effective, and we all overprepare patients to use it, knowing that they will call our offices panicked and fearful that they are allergic to or are becoming infected by this cream.
Although further study clearly is needed to determine the optimal application amount, duration of use, and vehicle mix, this new compound utilizing 2 topicals that are familiar to us--5-FU cream approved for AKs and early squamous cell skin cancers and calcipotriol ointment (though available only in cream in the United States currently) for psoriasis--is an encouraging step. Home therapy for AKs and possibly early nonmelanoma skin cancers that is more tolerable, of shorter duration, and in turn more effective than the current options would lessen the burden of treating these lesions surgically or rescheduling 5-FU patients often for irritation reaction education.
How do patients respond to this regimen?
In my own anecdotal experience, this regimen has been well received by patients and often is covered by most insurances when written as 2 separate prescriptions (both in cream vehicle). They still report some irritation, but I prefer to utilize it segmentally instead of treating all sun-exposed areas at once (ie, treat one side of the face/scalp twice daily for 4 days, then the other, or even divide it into smaller segments once the prior segment has healed). This combination, in addition to, for example, adding nicotinamide 500 mg twice daily to a patient's skin cancer chemopreventative sequence, is in my opinion a novel but safe, effective, and well-tolerated field therapy recommendation.
American Academy of Dermatology Actinic Keratosis Overview
American Academy of Family Physicians Actinic Keratoses Information
Suggested Readings
- Cunningham TJ, Tabacchi M, Eliane JP, et al. Randomized trial of calcipotriol combined with 5-fluorouracil for skin cancer precursor immunotherapy. J Clin Invest. 2017;127:106-116.
- Demehri S, Turkoz A, Manivasagam S, et al. Elevated epidermal thymic stromal lymphopoietin levels establish an antitumor environment in the skin. Cancer Cell. 2012;22:494-505.
- Rosamilia LL. Three Cheers for B3? Cutis. July 7, 2015. http://www.mdedge.com/cutis/article/101102/nonmelanoma-skin-cancer/three-cheers-b3. Accessed November 20, 2017.
- Sato-Deguchi E, Imafuku S, Chou B, et al. Topical vitamin D(3) analogues induce thymic stromal lymphopoietin and cathelicidin in psoriatic skin lesions. Br J Dermatol. 2012;167:77-84.
What is the recent research behind 5-fluorouracil cream 5% combined with calcipotriol ointment 0.005% for actinic keratoses?
Cunningham et al published a randomized double-blind study in which 131 patients with actinic keratoses (AKs) were assigned to either 5-fluorouracil (5-FU) cream 5% combined with calcipotriol (calcipotriene) ointment 0.005% twice daily to the face, scalp, and arms for 4 days, or 5-FU 5% combined with petrolatum applied in the same fashion. There was an 87.8% versus 26.3% mean reduction in the number of AKs and less severe pain, crusting, and ulceration in the study cohort compared to the 5-FU plus petrolatum group.
The same study also investigated immune parameters in these patients and found that the study group preferentially displayed activated thymic stromal lymphopoietin and a CD4 T cell-mediated reaction, among other effects. In prior studies, thymic stromal lymphopoietin has been shown to be upregulated in barrier-defective skin, displays antitumor activity, and is enhanced by topical calcipotriol application based on its original indication for psoriasis.
How do these study results impact patient care?
In a perfect world, every patient could tolerate and afford chemopreventative measures such as 5-FU cream, apply it diffusely to sun-exposed skin, and experience no severe irritant reactions and/or social pariah status. We all know that this product is effective, and we all overprepare patients to use it, knowing that they will call our offices panicked and fearful that they are allergic to or are becoming infected by this cream.
Although further study clearly is needed to determine the optimal application amount, duration of use, and vehicle mix, this new compound utilizing 2 topicals that are familiar to us--5-FU cream approved for AKs and early squamous cell skin cancers and calcipotriol ointment (though available only in cream in the United States currently) for psoriasis--is an encouraging step. Home therapy for AKs and possibly early nonmelanoma skin cancers that is more tolerable, of shorter duration, and in turn more effective than the current options would lessen the burden of treating these lesions surgically or rescheduling 5-FU patients often for irritation reaction education.
How do patients respond to this regimen?
In my own anecdotal experience, this regimen has been well received by patients and often is covered by most insurances when written as 2 separate prescriptions (both in cream vehicle). They still report some irritation, but I prefer to utilize it segmentally instead of treating all sun-exposed areas at once (ie, treat one side of the face/scalp twice daily for 4 days, then the other, or even divide it into smaller segments once the prior segment has healed). This combination, in addition to, for example, adding nicotinamide 500 mg twice daily to a patient's skin cancer chemopreventative sequence, is in my opinion a novel but safe, effective, and well-tolerated field therapy recommendation.
American Academy of Dermatology Actinic Keratosis Overview
American Academy of Family Physicians Actinic Keratoses Information
Suggested Readings
- Cunningham TJ, Tabacchi M, Eliane JP, et al. Randomized trial of calcipotriol combined with 5-fluorouracil for skin cancer precursor immunotherapy. J Clin Invest. 2017;127:106-116.
- Demehri S, Turkoz A, Manivasagam S, et al. Elevated epidermal thymic stromal lymphopoietin levels establish an antitumor environment in the skin. Cancer Cell. 2012;22:494-505.
- Rosamilia LL. Three Cheers for B3? Cutis. July 7, 2015. http://www.mdedge.com/cutis/article/101102/nonmelanoma-skin-cancer/three-cheers-b3. Accessed November 20, 2017.
- Sato-Deguchi E, Imafuku S, Chou B, et al. Topical vitamin D(3) analogues induce thymic stromal lymphopoietin and cathelicidin in psoriatic skin lesions. Br J Dermatol. 2012;167:77-84.
What is the recent research behind 5-fluorouracil cream 5% combined with calcipotriol ointment 0.005% for actinic keratoses?
Cunningham et al published a randomized double-blind study in which 131 patients with actinic keratoses (AKs) were assigned to either 5-fluorouracil (5-FU) cream 5% combined with calcipotriol (calcipotriene) ointment 0.005% twice daily to the face, scalp, and arms for 4 days, or 5-FU 5% combined with petrolatum applied in the same fashion. There was an 87.8% versus 26.3% mean reduction in the number of AKs and less severe pain, crusting, and ulceration in the study cohort compared to the 5-FU plus petrolatum group.
The same study also investigated immune parameters in these patients and found that the study group preferentially displayed activated thymic stromal lymphopoietin and a CD4 T cell-mediated reaction, among other effects. In prior studies, thymic stromal lymphopoietin has been shown to be upregulated in barrier-defective skin, displays antitumor activity, and is enhanced by topical calcipotriol application based on its original indication for psoriasis.
How do these study results impact patient care?
In a perfect world, every patient could tolerate and afford chemopreventative measures such as 5-FU cream, apply it diffusely to sun-exposed skin, and experience no severe irritant reactions and/or social pariah status. We all know that this product is effective, and we all overprepare patients to use it, knowing that they will call our offices panicked and fearful that they are allergic to or are becoming infected by this cream.
Although further study clearly is needed to determine the optimal application amount, duration of use, and vehicle mix, this new compound utilizing 2 topicals that are familiar to us--5-FU cream approved for AKs and early squamous cell skin cancers and calcipotriol ointment (though available only in cream in the United States currently) for psoriasis--is an encouraging step. Home therapy for AKs and possibly early nonmelanoma skin cancers that is more tolerable, of shorter duration, and in turn more effective than the current options would lessen the burden of treating these lesions surgically or rescheduling 5-FU patients often for irritation reaction education.
How do patients respond to this regimen?
In my own anecdotal experience, this regimen has been well received by patients and often is covered by most insurances when written as 2 separate prescriptions (both in cream vehicle). They still report some irritation, but I prefer to utilize it segmentally instead of treating all sun-exposed areas at once (ie, treat one side of the face/scalp twice daily for 4 days, then the other, or even divide it into smaller segments once the prior segment has healed). This combination, in addition to, for example, adding nicotinamide 500 mg twice daily to a patient's skin cancer chemopreventative sequence, is in my opinion a novel but safe, effective, and well-tolerated field therapy recommendation.
American Academy of Dermatology Actinic Keratosis Overview
American Academy of Family Physicians Actinic Keratoses Information
Suggested Readings
- Cunningham TJ, Tabacchi M, Eliane JP, et al. Randomized trial of calcipotriol combined with 5-fluorouracil for skin cancer precursor immunotherapy. J Clin Invest. 2017;127:106-116.
- Demehri S, Turkoz A, Manivasagam S, et al. Elevated epidermal thymic stromal lymphopoietin levels establish an antitumor environment in the skin. Cancer Cell. 2012;22:494-505.
- Rosamilia LL. Three Cheers for B3? Cutis. July 7, 2015. http://www.mdedge.com/cutis/article/101102/nonmelanoma-skin-cancer/three-cheers-b3. Accessed November 20, 2017.
- Sato-Deguchi E, Imafuku S, Chou B, et al. Topical vitamin D(3) analogues induce thymic stromal lymphopoietin and cathelicidin in psoriatic skin lesions. Br J Dermatol. 2012;167:77-84.

Diversity in the Dermatology Workforce: 2017 Status Update
Physician diversity benefits patient care: Patients are more satisfied during race-concordant visits, report their physicians as more engaged and responsive to their needs, and experience notably longer visits.1,2 Nonwhite physicians (ie, races and ethnicities that are underrepresented in medicine [URM] with respect to the general population) are more likely to care for underserved communities. Furthermore, increased diversity in the learning environment supports preparedness of all trainees to serve diverse patients.3 For these reasons, a more diverse physician workforce can contribute to better access to care in all communities, thus addressing health disparities.1,4
Increasing diversity in the dermatology workforce has been identified as an emerging priority.5 Dermatology is one of the least diverse specialties,5 and the representation of URM dermatologists is lower compared to other medical specialties and the general US population. The proportion of specialty leaders from underrepresented backgrounds may be even smaller. The lack of diversity in academic dermatology has negative consequences for patients and communities. Increasing the diversity of resident trainees is the only way to improve the diversity gap within the dermatology workforce.6
Recent commentary on this topic has highlighted several priorities for addressing the dermatology diversity gap,6-11 including the following: (1) making diversity an explicit goal in dermatology; (2) ensuring early exposure to dermatology in medical school; (3) supporting mentorship programs for minority medical students; (4) increasing medical student diversity; (5) encouraging that all dermatology program directors and leaders train in implicit bias; and (6) reviewing residency admission criteria to ensure they are objective and equitable, not biased against any applicants.
The process of reviewing residency selection criteria has begun. In 2017, Chen and Shinkai7 called for our specialty to rethink the selection process. The authors argued that emphasis on test scores, grades, and publications systematically disadvantages underrepresented minorities and students from lower socioeconomic statuses. The authors proposed several solutions: (1) make diversity an explicit goal of the selection process, (2) shift away from test scores for all applicants, (3) change the interview format, (4) prioritize other competencies such as observation skills, and (5) recruit and retain faculty who support URM trainees.7
Several dermatology leadership groups have taken action to promote programs that aim to improve diversity within dermatology. The Dermatology Diversity Champions initiative includes 6 US dermatology residency programs that are committed to increasing diversity and collaborate to evaluate pilot approaches. The American Academy of Dermatology President’s Conference on Diversity in Dermatology in Chicago, Illinois, in August 2017, as well as the focus on diversity in residency training programs at the Annual Meeting of the Association of Professors of Dermatology in Chicago, Illinois, in October 2017, are strong indicators that our specialty as a whole is aware and eager to embrace diversity as a priority. The American Academy of Dermatology President’s Conference, which was comprised of representatives from many leadership organizations and interest groups within dermatology, identified 3 action items: (1) increase the pipeline of URM students into medical school, (2) increase interest in dermatology among URM medical students, and (3) increase URM representation in residency training programs.
There are many strengths, weaknesses, opportunities, and threats/barriers (SWOT) to attaining this goal. Current strengths include strong support from dermatology leaders and activities that build on existing mentorship and diversity efforts by leaders within our specialty. SWOT analysis highlights several key opportunities of this mission, including connecting with the House of Medicine in shared efforts to improve diversity, as well as increased understanding of skin of color, health disparities, and implicit bias among physicians. Although faculty development will require time and financial investment, it will lead to tremendous benefits and opportunities for all dermatologists, including URM physicians. Other weaknesses and threats/barriers are outlined in the Figure.
Final Thoughts
We are far from reaching our goal of a diverse dermatology workforce, and the road ahead is long. We have a start and we have momentum. We can move forward by spreading the word that all types of diversity are a priority for our specialty. Making a true difference will require commitment and sustained efforts. Dermatology can lead the way as all of American medicine strives to attain workforce diversity.
- Saha S. Taking diversity seriously: the merits of increasing minority representation in medicine. JAMA Intern Med. 2014;174:291-292.
- Cooper LA, Roter DL, Johnson RL, et al. Patient-centered communication, ratings of care, and concordance of patient and physician race. Ann Intern Med. 2003;139:907-915.
- Saha S, Guiton G, Wimmers PF, et al. Student body racial and ethnic composition and diversity-related outcomes in US medical schools. JAMA. 2008;300:1135-1145.
- Marrast LM, Zallman L, Woolhandler S, et al. Minority physicians’ role in the care of underserved patients: diversifying the physician workforce may be key in addressing health disparities. JAMA Intern Med. 2014;174:289-291.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587.
- Lester J, Wintroub B, Linos E. Disparities in academic dermatology. JAMA Dermatol. 2016;152:878-879.
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260.
- Granstein RD, Cornelius L, Shinkai K. Diversity in dermatology—a call for action. JAMA Dermatol. 2017;153:499-500.
- McKesey J, Berger TG, Lim HW, et al. Cultural competence for the 21st century dermatologist practicing in the United States. J Am Acad Dermatol. 2017;77:1159-1169.
- Van Voorhees AS, Enos CW. Diversity in dermatology residency programs. J Investig Dermatol Symp Proc. 2017;18:S46-S49.
- Imadojemu S, James WD. Increasing African American representation in dermatology. JAMA Dermatol. 2016;152:15-16.
Physician diversity benefits patient care: Patients are more satisfied during race-concordant visits, report their physicians as more engaged and responsive to their needs, and experience notably longer visits.1,2 Nonwhite physicians (ie, races and ethnicities that are underrepresented in medicine [URM] with respect to the general population) are more likely to care for underserved communities. Furthermore, increased diversity in the learning environment supports preparedness of all trainees to serve diverse patients.3 For these reasons, a more diverse physician workforce can contribute to better access to care in all communities, thus addressing health disparities.1,4
Increasing diversity in the dermatology workforce has been identified as an emerging priority.5 Dermatology is one of the least diverse specialties,5 and the representation of URM dermatologists is lower compared to other medical specialties and the general US population. The proportion of specialty leaders from underrepresented backgrounds may be even smaller. The lack of diversity in academic dermatology has negative consequences for patients and communities. Increasing the diversity of resident trainees is the only way to improve the diversity gap within the dermatology workforce.6
Recent commentary on this topic has highlighted several priorities for addressing the dermatology diversity gap,6-11 including the following: (1) making diversity an explicit goal in dermatology; (2) ensuring early exposure to dermatology in medical school; (3) supporting mentorship programs for minority medical students; (4) increasing medical student diversity; (5) encouraging that all dermatology program directors and leaders train in implicit bias; and (6) reviewing residency admission criteria to ensure they are objective and equitable, not biased against any applicants.
The process of reviewing residency selection criteria has begun. In 2017, Chen and Shinkai7 called for our specialty to rethink the selection process. The authors argued that emphasis on test scores, grades, and publications systematically disadvantages underrepresented minorities and students from lower socioeconomic statuses. The authors proposed several solutions: (1) make diversity an explicit goal of the selection process, (2) shift away from test scores for all applicants, (3) change the interview format, (4) prioritize other competencies such as observation skills, and (5) recruit and retain faculty who support URM trainees.7
Several dermatology leadership groups have taken action to promote programs that aim to improve diversity within dermatology. The Dermatology Diversity Champions initiative includes 6 US dermatology residency programs that are committed to increasing diversity and collaborate to evaluate pilot approaches. The American Academy of Dermatology President’s Conference on Diversity in Dermatology in Chicago, Illinois, in August 2017, as well as the focus on diversity in residency training programs at the Annual Meeting of the Association of Professors of Dermatology in Chicago, Illinois, in October 2017, are strong indicators that our specialty as a whole is aware and eager to embrace diversity as a priority. The American Academy of Dermatology President’s Conference, which was comprised of representatives from many leadership organizations and interest groups within dermatology, identified 3 action items: (1) increase the pipeline of URM students into medical school, (2) increase interest in dermatology among URM medical students, and (3) increase URM representation in residency training programs.
There are many strengths, weaknesses, opportunities, and threats/barriers (SWOT) to attaining this goal. Current strengths include strong support from dermatology leaders and activities that build on existing mentorship and diversity efforts by leaders within our specialty. SWOT analysis highlights several key opportunities of this mission, including connecting with the House of Medicine in shared efforts to improve diversity, as well as increased understanding of skin of color, health disparities, and implicit bias among physicians. Although faculty development will require time and financial investment, it will lead to tremendous benefits and opportunities for all dermatologists, including URM physicians. Other weaknesses and threats/barriers are outlined in the Figure.
Final Thoughts
We are far from reaching our goal of a diverse dermatology workforce, and the road ahead is long. We have a start and we have momentum. We can move forward by spreading the word that all types of diversity are a priority for our specialty. Making a true difference will require commitment and sustained efforts. Dermatology can lead the way as all of American medicine strives to attain workforce diversity.
Physician diversity benefits patient care: Patients are more satisfied during race-concordant visits, report their physicians as more engaged and responsive to their needs, and experience notably longer visits.1,2 Nonwhite physicians (ie, races and ethnicities that are underrepresented in medicine [URM] with respect to the general population) are more likely to care for underserved communities. Furthermore, increased diversity in the learning environment supports preparedness of all trainees to serve diverse patients.3 For these reasons, a more diverse physician workforce can contribute to better access to care in all communities, thus addressing health disparities.1,4
Increasing diversity in the dermatology workforce has been identified as an emerging priority.5 Dermatology is one of the least diverse specialties,5 and the representation of URM dermatologists is lower compared to other medical specialties and the general US population. The proportion of specialty leaders from underrepresented backgrounds may be even smaller. The lack of diversity in academic dermatology has negative consequences for patients and communities. Increasing the diversity of resident trainees is the only way to improve the diversity gap within the dermatology workforce.6
Recent commentary on this topic has highlighted several priorities for addressing the dermatology diversity gap,6-11 including the following: (1) making diversity an explicit goal in dermatology; (2) ensuring early exposure to dermatology in medical school; (3) supporting mentorship programs for minority medical students; (4) increasing medical student diversity; (5) encouraging that all dermatology program directors and leaders train in implicit bias; and (6) reviewing residency admission criteria to ensure they are objective and equitable, not biased against any applicants.
The process of reviewing residency selection criteria has begun. In 2017, Chen and Shinkai7 called for our specialty to rethink the selection process. The authors argued that emphasis on test scores, grades, and publications systematically disadvantages underrepresented minorities and students from lower socioeconomic statuses. The authors proposed several solutions: (1) make diversity an explicit goal of the selection process, (2) shift away from test scores for all applicants, (3) change the interview format, (4) prioritize other competencies such as observation skills, and (5) recruit and retain faculty who support URM trainees.7
Several dermatology leadership groups have taken action to promote programs that aim to improve diversity within dermatology. The Dermatology Diversity Champions initiative includes 6 US dermatology residency programs that are committed to increasing diversity and collaborate to evaluate pilot approaches. The American Academy of Dermatology President’s Conference on Diversity in Dermatology in Chicago, Illinois, in August 2017, as well as the focus on diversity in residency training programs at the Annual Meeting of the Association of Professors of Dermatology in Chicago, Illinois, in October 2017, are strong indicators that our specialty as a whole is aware and eager to embrace diversity as a priority. The American Academy of Dermatology President’s Conference, which was comprised of representatives from many leadership organizations and interest groups within dermatology, identified 3 action items: (1) increase the pipeline of URM students into medical school, (2) increase interest in dermatology among URM medical students, and (3) increase URM representation in residency training programs.
There are many strengths, weaknesses, opportunities, and threats/barriers (SWOT) to attaining this goal. Current strengths include strong support from dermatology leaders and activities that build on existing mentorship and diversity efforts by leaders within our specialty. SWOT analysis highlights several key opportunities of this mission, including connecting with the House of Medicine in shared efforts to improve diversity, as well as increased understanding of skin of color, health disparities, and implicit bias among physicians. Although faculty development will require time and financial investment, it will lead to tremendous benefits and opportunities for all dermatologists, including URM physicians. Other weaknesses and threats/barriers are outlined in the Figure.
Final Thoughts
We are far from reaching our goal of a diverse dermatology workforce, and the road ahead is long. We have a start and we have momentum. We can move forward by spreading the word that all types of diversity are a priority for our specialty. Making a true difference will require commitment and sustained efforts. Dermatology can lead the way as all of American medicine strives to attain workforce diversity.
- Saha S. Taking diversity seriously: the merits of increasing minority representation in medicine. JAMA Intern Med. 2014;174:291-292.
- Cooper LA, Roter DL, Johnson RL, et al. Patient-centered communication, ratings of care, and concordance of patient and physician race. Ann Intern Med. 2003;139:907-915.
- Saha S, Guiton G, Wimmers PF, et al. Student body racial and ethnic composition and diversity-related outcomes in US medical schools. JAMA. 2008;300:1135-1145.
- Marrast LM, Zallman L, Woolhandler S, et al. Minority physicians’ role in the care of underserved patients: diversifying the physician workforce may be key in addressing health disparities. JAMA Intern Med. 2014;174:289-291.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587.
- Lester J, Wintroub B, Linos E. Disparities in academic dermatology. JAMA Dermatol. 2016;152:878-879.
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260.
- Granstein RD, Cornelius L, Shinkai K. Diversity in dermatology—a call for action. JAMA Dermatol. 2017;153:499-500.
- McKesey J, Berger TG, Lim HW, et al. Cultural competence for the 21st century dermatologist practicing in the United States. J Am Acad Dermatol. 2017;77:1159-1169.
- Van Voorhees AS, Enos CW. Diversity in dermatology residency programs. J Investig Dermatol Symp Proc. 2017;18:S46-S49.
- Imadojemu S, James WD. Increasing African American representation in dermatology. JAMA Dermatol. 2016;152:15-16.
- Saha S. Taking diversity seriously: the merits of increasing minority representation in medicine. JAMA Intern Med. 2014;174:291-292.
- Cooper LA, Roter DL, Johnson RL, et al. Patient-centered communication, ratings of care, and concordance of patient and physician race. Ann Intern Med. 2003;139:907-915.
- Saha S, Guiton G, Wimmers PF, et al. Student body racial and ethnic composition and diversity-related outcomes in US medical schools. JAMA. 2008;300:1135-1145.
- Marrast LM, Zallman L, Woolhandler S, et al. Minority physicians’ role in the care of underserved patients: diversifying the physician workforce may be key in addressing health disparities. JAMA Intern Med. 2014;174:289-291.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587.
- Lester J, Wintroub B, Linos E. Disparities in academic dermatology. JAMA Dermatol. 2016;152:878-879.
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260.
- Granstein RD, Cornelius L, Shinkai K. Diversity in dermatology—a call for action. JAMA Dermatol. 2017;153:499-500.
- McKesey J, Berger TG, Lim HW, et al. Cultural competence for the 21st century dermatologist practicing in the United States. J Am Acad Dermatol. 2017;77:1159-1169.
- Van Voorhees AS, Enos CW. Diversity in dermatology residency programs. J Investig Dermatol Symp Proc. 2017;18:S46-S49.
- Imadojemu S, James WD. Increasing African American representation in dermatology. JAMA Dermatol. 2016;152:15-16.

The Effects of Sunscreen on Marine Environments
Coastal travel accounts for 80% of all tourism worldwide, a number that continues to grow. The number of travelers to the Mediterranean Sea alone is expected to rise to 350 million individuals per year within the next 20 years.1 As the number of tourists visiting the world’s oceans increases, the rate of sunscreen unintentionally washed into these marine environments also rises. One study estimated that approximately one-quarter of the sunscreen applied to the skin is washed off over a 20-minute period spent in the water.2 Four of the most common sunscreen agents—benzophenone-3 (BP-3),
Benzophenone-3
4-Methylbenzylidene Camphor
Environmental concerns have also been raised about another common chemical UV filter: 4-MBC, or enzacamene. In laboratory studies, 4-MBC has been shown to cause oxidative stress to Tetrahymena thermophila, an aquatic protozoan, which results in inhibited growth. At higher concentrations, damage to the cellular membrane was seen as soon as 4 hours after exposure.6 In embryonic zebrafish, elevated 4-MBC levels were correlated to improper nerve and muscular development, resulting in developmental defects.7 Another study demonstrated that 4-MBC was toxic to Mytilus galloprovincialis, known as the Mediterranean mussel, and Paracentrotus lividus, a species of sea urchin.8 Although these studies utilized highly controlled laboratory settings, further studies are needed to examine the effects of 4-MBC on these species at environmentally relevant concentrations.
Physical Sunscreens
Physical sunscreens, as compared to the chemical filters referenced above, use either zinc or titanium to protect the skin from the sun’s rays. Nanoparticles, in particular, are preferred because they do not leave a white film on the skin.9 Both titanium dioxide and zinc oxide nanoparticles have been found to inhibit the growth and photosynthesis of marine phytoplankton, the most abundant primary producers on Earth.10,11 These metal contaminants can be transferred to organisms of higher trophic levels, including zooplankton,12 and filter-feeding organisms, including marine abalone13 and the Mediterranean mussel.14 These nanoparticles have been shown to cause oxidative stress to these organisms, making them less fit to withstand environmental stressors. It is difficult to show their true impact, however, as it is challenging to accurately detect and quantify nanoparticle concentrations in vivo.15
Final Thoughts
- Marine problems: tourism & coastal development. World Wide Fund for Nature website. http://wwf.panda.org/about_our_earth/blue_planet/problems/tourism/. Published 2017. Accessed November 14, 2017.
- Danovaro R, Bongiorni L, Corinaldesi C, et al. Sunscreens cause coral bleaching by promoting viral infections. Environ Health Perspect. 2008;116:441-447.
- Downs C, Kramarsky-Winter E, Segal R, et al. Toxicopathological effects of the sunscreen UV filter, oxybenzone (benzophenone-3), on coral planulae and cultured primary cells and its environmental contamination in Hawaii and the US Virgin Islands. Arch Environ Contam Toxicol. 2016;70:265-288.
- Sánchez Rodríguez A, Rodrigo Sanz M, Betancort Rodríguez JR. Occurrence of eight UV filters in beaches of Gran Canaria (Canary Islands)[published online March 17, 2015]. Chemosphere. 2015;131:85-90.
- Bratkovics S, Sapozhnikova Y. Determination of seven commonly used organic UV filters in fresh and saline waters by liquid chromatography-tandem mass spectrometry. Analytical Methods. 2011;3:2943-2950.
- Gao L, Yuan T, Zhou C, et al. Effects of four commonly used UV filters on the growth, cell viability and oxidative stress responses of the Tetrahymena thermophila. Chemosphere. 2013;93:2507-2513.
- Li VW, Tsui MP, Chen X, et al. Effects of 4-methylbenzylidene camphor (4-MBC) on neuronal and muscular development in zebrafish (Danio rerio) embryos [published online February 18, 2016]. Environ Sci Pollut Res Int. 2016;23:8275-8285.
- Paredes E, Perez S, Rodil R, et al. Ecotoxicological evaluation of four UV filters using marine organisms from different trophic levels Isochrysis galbana, Mytilus galloprovincialis, Paracentrotus lividus, and Siriella armata. Chemosphere. 2014;104:44-50.
- Osterwalder U, Sohn M, Herzog B. Global state of sunscreens. Photodermatol Photoimmunol Photomed. 2014;30:62-80.
- Miller RJ, Bennett S, Keller AA, et al. TiO2 nanoparticles are phototoxic to marine phytoplankton. PloS One. 2012;7:E30321.
- Spisni E. Toxicity Assessment of Industrial- and Sunscreen-derived ZnO Nanoparticles [master’s thesis]. Coral Gables, FL: University of Miami Libraries Scholarly Repository; 2016. http://scholarlyrepository.miami.edu/cgi/viewcontent.cgi?article=1625&context=oa_theses. Accessed November 10, 2017.
- Jarvis TA, Miller RJ, Lenihan HS, et al. Toxicity of ZnO nanoparticles to the copepod Acartia tonsa, exposed through a phytoplankton diet [published online April 15, 2013]. Environ Toxicol Chem. 2013;32:1264-1269.
- Zhu X, Zhou J, Cai Z. The toxicity and oxidative stress of TiO2 nanoparticles in marine abalone (Haliotis diversicolor supertexta). Mar Pollut Bull. 2011;63:334-338.
- Barmo C, Ciacci C, Canonico B, et al. In vivo effects of n-TiO2 on digestive gland and immune function of the marine bivalve Mytilus galloprovincialis. Aquatic Toxicol. 2013;132:9-18.
- Sánchez-Quiles D, Tovar-Sánchez A. Are sunscreens a new environmental risk associated with coastal tourism? Environ Int. 2015;83:158-170.
- Xu S, Kwa M, Agarwal A, et al. Sunscreen product performance and other determinants of consumer preferences. JAMA Dermatol. 2016;152:920-927.
- Vesper I. Hawaii seeks to ban ‘reef-unfriendly’ sunscreen. Nature. February 3, 2017. https://www.nature.com/news/hawaii-seeks-to-ban-reef-unfriendly-sunscreen-1.21332. Accessed November 16, 2017.
Coastal travel accounts for 80% of all tourism worldwide, a number that continues to grow. The number of travelers to the Mediterranean Sea alone is expected to rise to 350 million individuals per year within the next 20 years.1 As the number of tourists visiting the world’s oceans increases, the rate of sunscreen unintentionally washed into these marine environments also rises. One study estimated that approximately one-quarter of the sunscreen applied to the skin is washed off over a 20-minute period spent in the water.2 Four of the most common sunscreen agents—benzophenone-3 (BP-3),
Benzophenone-3
4-Methylbenzylidene Camphor
Environmental concerns have also been raised about another common chemical UV filter: 4-MBC, or enzacamene. In laboratory studies, 4-MBC has been shown to cause oxidative stress to Tetrahymena thermophila, an aquatic protozoan, which results in inhibited growth. At higher concentrations, damage to the cellular membrane was seen as soon as 4 hours after exposure.6 In embryonic zebrafish, elevated 4-MBC levels were correlated to improper nerve and muscular development, resulting in developmental defects.7 Another study demonstrated that 4-MBC was toxic to Mytilus galloprovincialis, known as the Mediterranean mussel, and Paracentrotus lividus, a species of sea urchin.8 Although these studies utilized highly controlled laboratory settings, further studies are needed to examine the effects of 4-MBC on these species at environmentally relevant concentrations.
Physical Sunscreens
Physical sunscreens, as compared to the chemical filters referenced above, use either zinc or titanium to protect the skin from the sun’s rays. Nanoparticles, in particular, are preferred because they do not leave a white film on the skin.9 Both titanium dioxide and zinc oxide nanoparticles have been found to inhibit the growth and photosynthesis of marine phytoplankton, the most abundant primary producers on Earth.10,11 These metal contaminants can be transferred to organisms of higher trophic levels, including zooplankton,12 and filter-feeding organisms, including marine abalone13 and the Mediterranean mussel.14 These nanoparticles have been shown to cause oxidative stress to these organisms, making them less fit to withstand environmental stressors. It is difficult to show their true impact, however, as it is challenging to accurately detect and quantify nanoparticle concentrations in vivo.15
Final Thoughts
Coastal travel accounts for 80% of all tourism worldwide, a number that continues to grow. The number of travelers to the Mediterranean Sea alone is expected to rise to 350 million individuals per year within the next 20 years.1 As the number of tourists visiting the world’s oceans increases, the rate of sunscreen unintentionally washed into these marine environments also rises. One study estimated that approximately one-quarter of the sunscreen applied to the skin is washed off over a 20-minute period spent in the water.2 Four of the most common sunscreen agents—benzophenone-3 (BP-3),
Benzophenone-3
4-Methylbenzylidene Camphor
Environmental concerns have also been raised about another common chemical UV filter: 4-MBC, or enzacamene. In laboratory studies, 4-MBC has been shown to cause oxidative stress to Tetrahymena thermophila, an aquatic protozoan, which results in inhibited growth. At higher concentrations, damage to the cellular membrane was seen as soon as 4 hours after exposure.6 In embryonic zebrafish, elevated 4-MBC levels were correlated to improper nerve and muscular development, resulting in developmental defects.7 Another study demonstrated that 4-MBC was toxic to Mytilus galloprovincialis, known as the Mediterranean mussel, and Paracentrotus lividus, a species of sea urchin.8 Although these studies utilized highly controlled laboratory settings, further studies are needed to examine the effects of 4-MBC on these species at environmentally relevant concentrations.
Physical Sunscreens
Physical sunscreens, as compared to the chemical filters referenced above, use either zinc or titanium to protect the skin from the sun’s rays. Nanoparticles, in particular, are preferred because they do not leave a white film on the skin.9 Both titanium dioxide and zinc oxide nanoparticles have been found to inhibit the growth and photosynthesis of marine phytoplankton, the most abundant primary producers on Earth.10,11 These metal contaminants can be transferred to organisms of higher trophic levels, including zooplankton,12 and filter-feeding organisms, including marine abalone13 and the Mediterranean mussel.14 These nanoparticles have been shown to cause oxidative stress to these organisms, making them less fit to withstand environmental stressors. It is difficult to show their true impact, however, as it is challenging to accurately detect and quantify nanoparticle concentrations in vivo.15
Final Thoughts
- Marine problems: tourism & coastal development. World Wide Fund for Nature website. http://wwf.panda.org/about_our_earth/blue_planet/problems/tourism/. Published 2017. Accessed November 14, 2017.
- Danovaro R, Bongiorni L, Corinaldesi C, et al. Sunscreens cause coral bleaching by promoting viral infections. Environ Health Perspect. 2008;116:441-447.
- Downs C, Kramarsky-Winter E, Segal R, et al. Toxicopathological effects of the sunscreen UV filter, oxybenzone (benzophenone-3), on coral planulae and cultured primary cells and its environmental contamination in Hawaii and the US Virgin Islands. Arch Environ Contam Toxicol. 2016;70:265-288.
- Sánchez Rodríguez A, Rodrigo Sanz M, Betancort Rodríguez JR. Occurrence of eight UV filters in beaches of Gran Canaria (Canary Islands)[published online March 17, 2015]. Chemosphere. 2015;131:85-90.
- Bratkovics S, Sapozhnikova Y. Determination of seven commonly used organic UV filters in fresh and saline waters by liquid chromatography-tandem mass spectrometry. Analytical Methods. 2011;3:2943-2950.
- Gao L, Yuan T, Zhou C, et al. Effects of four commonly used UV filters on the growth, cell viability and oxidative stress responses of the Tetrahymena thermophila. Chemosphere. 2013;93:2507-2513.
- Li VW, Tsui MP, Chen X, et al. Effects of 4-methylbenzylidene camphor (4-MBC) on neuronal and muscular development in zebrafish (Danio rerio) embryos [published online February 18, 2016]. Environ Sci Pollut Res Int. 2016;23:8275-8285.
- Paredes E, Perez S, Rodil R, et al. Ecotoxicological evaluation of four UV filters using marine organisms from different trophic levels Isochrysis galbana, Mytilus galloprovincialis, Paracentrotus lividus, and Siriella armata. Chemosphere. 2014;104:44-50.
- Osterwalder U, Sohn M, Herzog B. Global state of sunscreens. Photodermatol Photoimmunol Photomed. 2014;30:62-80.
- Miller RJ, Bennett S, Keller AA, et al. TiO2 nanoparticles are phototoxic to marine phytoplankton. PloS One. 2012;7:E30321.
- Spisni E. Toxicity Assessment of Industrial- and Sunscreen-derived ZnO Nanoparticles [master’s thesis]. Coral Gables, FL: University of Miami Libraries Scholarly Repository; 2016. http://scholarlyrepository.miami.edu/cgi/viewcontent.cgi?article=1625&context=oa_theses. Accessed November 10, 2017.
- Jarvis TA, Miller RJ, Lenihan HS, et al. Toxicity of ZnO nanoparticles to the copepod Acartia tonsa, exposed through a phytoplankton diet [published online April 15, 2013]. Environ Toxicol Chem. 2013;32:1264-1269.
- Zhu X, Zhou J, Cai Z. The toxicity and oxidative stress of TiO2 nanoparticles in marine abalone (Haliotis diversicolor supertexta). Mar Pollut Bull. 2011;63:334-338.
- Barmo C, Ciacci C, Canonico B, et al. In vivo effects of n-TiO2 on digestive gland and immune function of the marine bivalve Mytilus galloprovincialis. Aquatic Toxicol. 2013;132:9-18.
- Sánchez-Quiles D, Tovar-Sánchez A. Are sunscreens a new environmental risk associated with coastal tourism? Environ Int. 2015;83:158-170.
- Xu S, Kwa M, Agarwal A, et al. Sunscreen product performance and other determinants of consumer preferences. JAMA Dermatol. 2016;152:920-927.
- Vesper I. Hawaii seeks to ban ‘reef-unfriendly’ sunscreen. Nature. February 3, 2017. https://www.nature.com/news/hawaii-seeks-to-ban-reef-unfriendly-sunscreen-1.21332. Accessed November 16, 2017.
- Marine problems: tourism & coastal development. World Wide Fund for Nature website. http://wwf.panda.org/about_our_earth/blue_planet/problems/tourism/. Published 2017. Accessed November 14, 2017.
- Danovaro R, Bongiorni L, Corinaldesi C, et al. Sunscreens cause coral bleaching by promoting viral infections. Environ Health Perspect. 2008;116:441-447.
- Downs C, Kramarsky-Winter E, Segal R, et al. Toxicopathological effects of the sunscreen UV filter, oxybenzone (benzophenone-3), on coral planulae and cultured primary cells and its environmental contamination in Hawaii and the US Virgin Islands. Arch Environ Contam Toxicol. 2016;70:265-288.
- Sánchez Rodríguez A, Rodrigo Sanz M, Betancort Rodríguez JR. Occurrence of eight UV filters in beaches of Gran Canaria (Canary Islands)[published online March 17, 2015]. Chemosphere. 2015;131:85-90.
- Bratkovics S, Sapozhnikova Y. Determination of seven commonly used organic UV filters in fresh and saline waters by liquid chromatography-tandem mass spectrometry. Analytical Methods. 2011;3:2943-2950.
- Gao L, Yuan T, Zhou C, et al. Effects of four commonly used UV filters on the growth, cell viability and oxidative stress responses of the Tetrahymena thermophila. Chemosphere. 2013;93:2507-2513.
- Li VW, Tsui MP, Chen X, et al. Effects of 4-methylbenzylidene camphor (4-MBC) on neuronal and muscular development in zebrafish (Danio rerio) embryos [published online February 18, 2016]. Environ Sci Pollut Res Int. 2016;23:8275-8285.
- Paredes E, Perez S, Rodil R, et al. Ecotoxicological evaluation of four UV filters using marine organisms from different trophic levels Isochrysis galbana, Mytilus galloprovincialis, Paracentrotus lividus, and Siriella armata. Chemosphere. 2014;104:44-50.
- Osterwalder U, Sohn M, Herzog B. Global state of sunscreens. Photodermatol Photoimmunol Photomed. 2014;30:62-80.
- Miller RJ, Bennett S, Keller AA, et al. TiO2 nanoparticles are phototoxic to marine phytoplankton. PloS One. 2012;7:E30321.
- Spisni E. Toxicity Assessment of Industrial- and Sunscreen-derived ZnO Nanoparticles [master’s thesis]. Coral Gables, FL: University of Miami Libraries Scholarly Repository; 2016. http://scholarlyrepository.miami.edu/cgi/viewcontent.cgi?article=1625&context=oa_theses. Accessed November 10, 2017.
- Jarvis TA, Miller RJ, Lenihan HS, et al. Toxicity of ZnO nanoparticles to the copepod Acartia tonsa, exposed through a phytoplankton diet [published online April 15, 2013]. Environ Toxicol Chem. 2013;32:1264-1269.
- Zhu X, Zhou J, Cai Z. The toxicity and oxidative stress of TiO2 nanoparticles in marine abalone (Haliotis diversicolor supertexta). Mar Pollut Bull. 2011;63:334-338.
- Barmo C, Ciacci C, Canonico B, et al. In vivo effects of n-TiO2 on digestive gland and immune function of the marine bivalve Mytilus galloprovincialis. Aquatic Toxicol. 2013;132:9-18.
- Sánchez-Quiles D, Tovar-Sánchez A. Are sunscreens a new environmental risk associated with coastal tourism? Environ Int. 2015;83:158-170.
- Xu S, Kwa M, Agarwal A, et al. Sunscreen product performance and other determinants of consumer preferences. JAMA Dermatol. 2016;152:920-927.
- Vesper I. Hawaii seeks to ban ‘reef-unfriendly’ sunscreen. Nature. February 3, 2017. https://www.nature.com/news/hawaii-seeks-to-ban-reef-unfriendly-sunscreen-1.21332. Accessed November 16, 2017.
