User login
Battling physician burnout delivers monetary benefits for health care organizations
The financial impact of physician burnout can provide a guide to help organizations address the problem, according to a special communication published online in JAMA Internal Medicine.
Approximately half of U.S. physicians experience some degree of professional burnout, but health care organizations have done little to respond, wrote Tait Shanafelt, MD, of Stanford (Calif.) University, and colleagues (JAMA Intern Med. 2017 Sep 25. doi: 10.1001/jamainternmed.2017.4340).
The researchers cited “a lack of awareness regarding the economic costs of physician burnout” and a lack of confidence that anything can be done as key factors in why the organizational response to burnout has been so limited.
The business end of physician burnout includes the costs associated with physician turnover and decreased productivity, as well as “financial risks to the organization’s long-term viability due to the relationship between burnout and lower quality of care, decreased patient satisfaction, and problems with patient safety,” the researchers said.
Organizations can combat physician burnout, the authors noted, and the effort is financially worthwhile, as well as morally and ethically important. “Burnout is primarily a system-level problem driven by excess job demands and inadequate resources and support, not an individual problem triggered by personal limitations,” they wrote.
The researchers developed a model outlining “Typical Steps in an Organization’s Journey Toward Expertise in Physician Well-being.” The steps begin with strategies that have a minor impact, including awareness of the problem of physician burnout and a focus on individual interventions such as mindfulness training, exercise, and nutrition.
However, the “transformative” changes in an organization include developing a “culture of wellness,” strategic investment in physician well-being, the presence of a “chief well-being officer” at the executive level, and accountability for physician wellness shared among organizational leaders.
The cost of such strategies varies among institutions, and the researchers provided worksheets to calculate the organizational cost of burnout and the return on investment if intervention steps are taken.
For example, an organization employing 450 physicians could potentially spend $1 million per year on an intervention to reduce physician burnout from 50% to 40%. The intervention investment could yield organizational cost savings of $1.125 million per year, with a 12.5% return on investment, as well as the potential financial benefits of improved patient satisfaction and quality of care.
“Understanding the business case to reduce burnout and promote engagement, as well as overcoming the misperception that nothing meaningful can be done, are key steps for organizations to begin to take action,” the researchers concluded.
“Improvement is possible, investment is justified, and return on investment measurable,” they said.
Dr. Shanafelt is a coinventor of the Physician Well-Being Index, Medical Student Well-Being Index, and Well-Being Index; copyright and licensing rights for these tools belong to the Mayo Clinic, and Dr. Shanafelt receives part of the royalties. The researchers had no other financial conflicts to disclose.
The financial impact of physician burnout can provide a guide to help organizations address the problem, according to a special communication published online in JAMA Internal Medicine.
Approximately half of U.S. physicians experience some degree of professional burnout, but health care organizations have done little to respond, wrote Tait Shanafelt, MD, of Stanford (Calif.) University, and colleagues (JAMA Intern Med. 2017 Sep 25. doi: 10.1001/jamainternmed.2017.4340).
The researchers cited “a lack of awareness regarding the economic costs of physician burnout” and a lack of confidence that anything can be done as key factors in why the organizational response to burnout has been so limited.
The business end of physician burnout includes the costs associated with physician turnover and decreased productivity, as well as “financial risks to the organization’s long-term viability due to the relationship between burnout and lower quality of care, decreased patient satisfaction, and problems with patient safety,” the researchers said.
Organizations can combat physician burnout, the authors noted, and the effort is financially worthwhile, as well as morally and ethically important. “Burnout is primarily a system-level problem driven by excess job demands and inadequate resources and support, not an individual problem triggered by personal limitations,” they wrote.
The researchers developed a model outlining “Typical Steps in an Organization’s Journey Toward Expertise in Physician Well-being.” The steps begin with strategies that have a minor impact, including awareness of the problem of physician burnout and a focus on individual interventions such as mindfulness training, exercise, and nutrition.
However, the “transformative” changes in an organization include developing a “culture of wellness,” strategic investment in physician well-being, the presence of a “chief well-being officer” at the executive level, and accountability for physician wellness shared among organizational leaders.
The cost of such strategies varies among institutions, and the researchers provided worksheets to calculate the organizational cost of burnout and the return on investment if intervention steps are taken.
For example, an organization employing 450 physicians could potentially spend $1 million per year on an intervention to reduce physician burnout from 50% to 40%. The intervention investment could yield organizational cost savings of $1.125 million per year, with a 12.5% return on investment, as well as the potential financial benefits of improved patient satisfaction and quality of care.
“Understanding the business case to reduce burnout and promote engagement, as well as overcoming the misperception that nothing meaningful can be done, are key steps for organizations to begin to take action,” the researchers concluded.
“Improvement is possible, investment is justified, and return on investment measurable,” they said.
Dr. Shanafelt is a coinventor of the Physician Well-Being Index, Medical Student Well-Being Index, and Well-Being Index; copyright and licensing rights for these tools belong to the Mayo Clinic, and Dr. Shanafelt receives part of the royalties. The researchers had no other financial conflicts to disclose.
The financial impact of physician burnout can provide a guide to help organizations address the problem, according to a special communication published online in JAMA Internal Medicine.
Approximately half of U.S. physicians experience some degree of professional burnout, but health care organizations have done little to respond, wrote Tait Shanafelt, MD, of Stanford (Calif.) University, and colleagues (JAMA Intern Med. 2017 Sep 25. doi: 10.1001/jamainternmed.2017.4340).
The researchers cited “a lack of awareness regarding the economic costs of physician burnout” and a lack of confidence that anything can be done as key factors in why the organizational response to burnout has been so limited.
The business end of physician burnout includes the costs associated with physician turnover and decreased productivity, as well as “financial risks to the organization’s long-term viability due to the relationship between burnout and lower quality of care, decreased patient satisfaction, and problems with patient safety,” the researchers said.
Organizations can combat physician burnout, the authors noted, and the effort is financially worthwhile, as well as morally and ethically important. “Burnout is primarily a system-level problem driven by excess job demands and inadequate resources and support, not an individual problem triggered by personal limitations,” they wrote.
The researchers developed a model outlining “Typical Steps in an Organization’s Journey Toward Expertise in Physician Well-being.” The steps begin with strategies that have a minor impact, including awareness of the problem of physician burnout and a focus on individual interventions such as mindfulness training, exercise, and nutrition.
However, the “transformative” changes in an organization include developing a “culture of wellness,” strategic investment in physician well-being, the presence of a “chief well-being officer” at the executive level, and accountability for physician wellness shared among organizational leaders.
The cost of such strategies varies among institutions, and the researchers provided worksheets to calculate the organizational cost of burnout and the return on investment if intervention steps are taken.
For example, an organization employing 450 physicians could potentially spend $1 million per year on an intervention to reduce physician burnout from 50% to 40%. The intervention investment could yield organizational cost savings of $1.125 million per year, with a 12.5% return on investment, as well as the potential financial benefits of improved patient satisfaction and quality of care.
“Understanding the business case to reduce burnout and promote engagement, as well as overcoming the misperception that nothing meaningful can be done, are key steps for organizations to begin to take action,” the researchers concluded.
“Improvement is possible, investment is justified, and return on investment measurable,” they said.
Dr. Shanafelt is a coinventor of the Physician Well-Being Index, Medical Student Well-Being Index, and Well-Being Index; copyright and licensing rights for these tools belong to the Mayo Clinic, and Dr. Shanafelt receives part of the royalties. The researchers had no other financial conflicts to disclose.
FROM JAMA INTERNAL MEDICINE
Early cognitive impairment associated with later Parkinson’s disease
Adults with early cognitive impairment are at greater risk for developing parkinsonism than those without cognitive impairment, based on data from 7,386 adults participating in the ongoing Rotterdam Study. The findings were published online Sept. 25 in JAMA Neurology.
“Between 15% and 43% of patients with newly diagnosed Parkinson disease (PD) are cognitively impaired,” wrote Sirwan K. L. Darweesh, MD, of Erasmus MC University Medical Center, Rotterdam, the Netherlands, and his colleagues. However, data on the predictive value of cognitive impairment for parkinsonism has not been well studied, they wrote (JAMA Neurol. 2017. doi: 10.1001/jamaneurol.2017.2248).
Over approximately 8 years’ follow-up, 1% of the participants were diagnosed with incident parkinsonism.
“Poor global cognition at baseline was associated with a higher risk of incident parkinsonism” with a hazard ratio of 1.79, the researchers said.
“To enable translation of our findings to clinical practice, we present likelihood ratios (LRs) for the baseline presence of isolated or combined cognitive dysfunction and subtle motor features for incident parkinsonism during follow-up,” they noted.
Approximately half of participants diagnosed with incident parkinsonism during the study period had subtle motor features, cognitive dysfunction, or both, at baseline. Baseline cognitive impairment alone showed a likelihood ratio of 1.76 for development of parkinsonism, but the likelihood ratio was greater when both cognitive impairment and subtle motor findings were present (2.66).
“In individuals who received a diagnosis of both incident dementia and incident parkinsonism, baseline cognitive dysfunction was not associated with incident dementia,” the researchers noted.
The researchers determined that the most likely explanation for the association between cognitive decline and increased Parkinson’s risk was that “low baseline cognitive scores may indicate ongoing cognitive decline in prediagnostic patients who probably will develop parkinsonism, most of whom have prediagnostic PD,” they said.
The study findings were limited by several factors including the potential misclassification of parkinsonism diagnosis, the researchers noted. However, the association between poor cognitive function and the risk of parkinsonism and probably Parkinson’s disease remained for the executive, attention, cognitive speed, and memory domains of cognition, they said. “Our findings suggest that poor cognitive functioning can be considered a prodromal sign of PD,” they concluded.
This study was supported in part by Stichting ParkinsonFonds. The researchers had no financial conflicts to disclose.
The long-term nature of the Rotterdam Study makes it an excellent source for examining the association between poor cognition and parkinsonism, wrote Ethan G. Brown, MD, and Caroline M. Tanner, MD, in an accompanying editorial.
“This study reiterates the presence of cognitive impairment very early in PD, emphasizing the need for therapeutic trials to target this symptom as an outcome. Although only some patients with cognitive impairment progress to PD, the study provides some clues on how to distinguish those most at risk. Progression to parkinsonism was more likely with baseline impairment of several individual cognitive tests, but only changes in semantic fluency predicted probable PD. Semantic fluency has been previously found to be specific for progression of cognitive impairment in PD, and this study again suggests the importance of this cognitive test early on,” they wrote.
“Yet the presence of cognitive impairment so early also gives rise to questions about the underlying pathology of PD progression. A commonly cited mechanism for progression of PD involves prion-like spread of synuclein pathology up through the dorsal nucleus of the vagus and substantia nigra. This spread presumably causes the autonomic, sleep, and motor dysfunction common in PD and supposedly leads to cognitive impairment only once Lewy bodies enter the neocortex. The current evidence that cognitive impairment can be evident in the prodromal stage challenges the universality of the model of vagal spread,” they noted.
However, recognizing the role of cognitive impairment as an early sign of PD can help clinicians plan screening and care, they said.
“This recognition can allow physicians to screen for falls or other nonmotor aspects of PD in these cases and provide early treatment for these symptoms. Physicians may recommend interventions, such as physical activity, that are helpful for motor and cognitive changes in PD,” they added (JAMA Neurol. 2017. doi: 10.1001/jamaneurol.2017.1474).
Dr. Brown and Dr. Tanner are affiliated with the Movement Disorders and Neuromodulation Center in the department of neurology at the University of California, San Francisco. Dr. Brown disclosed compensation for serving on the Fellowship Advisory Board for AbbVie. Dr. Tanner disclosed grants from a variety of nonprofit sources, as well as compensation for serving on Data Monitoring Committees for Biotie Therapeutics, Voyager Therapeutics, and Intec Pharma. Dr. Tanner also disclosed personal consulting fees from Neurocrine Biosciences, Adamas Therapeutics, and PhotoPharmics.
The long-term nature of the Rotterdam Study makes it an excellent source for examining the association between poor cognition and parkinsonism, wrote Ethan G. Brown, MD, and Caroline M. Tanner, MD, in an accompanying editorial.
“This study reiterates the presence of cognitive impairment very early in PD, emphasizing the need for therapeutic trials to target this symptom as an outcome. Although only some patients with cognitive impairment progress to PD, the study provides some clues on how to distinguish those most at risk. Progression to parkinsonism was more likely with baseline impairment of several individual cognitive tests, but only changes in semantic fluency predicted probable PD. Semantic fluency has been previously found to be specific for progression of cognitive impairment in PD, and this study again suggests the importance of this cognitive test early on,” they wrote.
“Yet the presence of cognitive impairment so early also gives rise to questions about the underlying pathology of PD progression. A commonly cited mechanism for progression of PD involves prion-like spread of synuclein pathology up through the dorsal nucleus of the vagus and substantia nigra. This spread presumably causes the autonomic, sleep, and motor dysfunction common in PD and supposedly leads to cognitive impairment only once Lewy bodies enter the neocortex. The current evidence that cognitive impairment can be evident in the prodromal stage challenges the universality of the model of vagal spread,” they noted.
However, recognizing the role of cognitive impairment as an early sign of PD can help clinicians plan screening and care, they said.
“This recognition can allow physicians to screen for falls or other nonmotor aspects of PD in these cases and provide early treatment for these symptoms. Physicians may recommend interventions, such as physical activity, that are helpful for motor and cognitive changes in PD,” they added (JAMA Neurol. 2017. doi: 10.1001/jamaneurol.2017.1474).
Dr. Brown and Dr. Tanner are affiliated with the Movement Disorders and Neuromodulation Center in the department of neurology at the University of California, San Francisco. Dr. Brown disclosed compensation for serving on the Fellowship Advisory Board for AbbVie. Dr. Tanner disclosed grants from a variety of nonprofit sources, as well as compensation for serving on Data Monitoring Committees for Biotie Therapeutics, Voyager Therapeutics, and Intec Pharma. Dr. Tanner also disclosed personal consulting fees from Neurocrine Biosciences, Adamas Therapeutics, and PhotoPharmics.
The long-term nature of the Rotterdam Study makes it an excellent source for examining the association between poor cognition and parkinsonism, wrote Ethan G. Brown, MD, and Caroline M. Tanner, MD, in an accompanying editorial.
“This study reiterates the presence of cognitive impairment very early in PD, emphasizing the need for therapeutic trials to target this symptom as an outcome. Although only some patients with cognitive impairment progress to PD, the study provides some clues on how to distinguish those most at risk. Progression to parkinsonism was more likely with baseline impairment of several individual cognitive tests, but only changes in semantic fluency predicted probable PD. Semantic fluency has been previously found to be specific for progression of cognitive impairment in PD, and this study again suggests the importance of this cognitive test early on,” they wrote.
“Yet the presence of cognitive impairment so early also gives rise to questions about the underlying pathology of PD progression. A commonly cited mechanism for progression of PD involves prion-like spread of synuclein pathology up through the dorsal nucleus of the vagus and substantia nigra. This spread presumably causes the autonomic, sleep, and motor dysfunction common in PD and supposedly leads to cognitive impairment only once Lewy bodies enter the neocortex. The current evidence that cognitive impairment can be evident in the prodromal stage challenges the universality of the model of vagal spread,” they noted.
However, recognizing the role of cognitive impairment as an early sign of PD can help clinicians plan screening and care, they said.
“This recognition can allow physicians to screen for falls or other nonmotor aspects of PD in these cases and provide early treatment for these symptoms. Physicians may recommend interventions, such as physical activity, that are helpful for motor and cognitive changes in PD,” they added (JAMA Neurol. 2017. doi: 10.1001/jamaneurol.2017.1474).
Dr. Brown and Dr. Tanner are affiliated with the Movement Disorders and Neuromodulation Center in the department of neurology at the University of California, San Francisco. Dr. Brown disclosed compensation for serving on the Fellowship Advisory Board for AbbVie. Dr. Tanner disclosed grants from a variety of nonprofit sources, as well as compensation for serving on Data Monitoring Committees for Biotie Therapeutics, Voyager Therapeutics, and Intec Pharma. Dr. Tanner also disclosed personal consulting fees from Neurocrine Biosciences, Adamas Therapeutics, and PhotoPharmics.
Adults with early cognitive impairment are at greater risk for developing parkinsonism than those without cognitive impairment, based on data from 7,386 adults participating in the ongoing Rotterdam Study. The findings were published online Sept. 25 in JAMA Neurology.
“Between 15% and 43% of patients with newly diagnosed Parkinson disease (PD) are cognitively impaired,” wrote Sirwan K. L. Darweesh, MD, of Erasmus MC University Medical Center, Rotterdam, the Netherlands, and his colleagues. However, data on the predictive value of cognitive impairment for parkinsonism has not been well studied, they wrote (JAMA Neurol. 2017. doi: 10.1001/jamaneurol.2017.2248).
Over approximately 8 years’ follow-up, 1% of the participants were diagnosed with incident parkinsonism.
“Poor global cognition at baseline was associated with a higher risk of incident parkinsonism” with a hazard ratio of 1.79, the researchers said.
“To enable translation of our findings to clinical practice, we present likelihood ratios (LRs) for the baseline presence of isolated or combined cognitive dysfunction and subtle motor features for incident parkinsonism during follow-up,” they noted.
Approximately half of participants diagnosed with incident parkinsonism during the study period had subtle motor features, cognitive dysfunction, or both, at baseline. Baseline cognitive impairment alone showed a likelihood ratio of 1.76 for development of parkinsonism, but the likelihood ratio was greater when both cognitive impairment and subtle motor findings were present (2.66).
“In individuals who received a diagnosis of both incident dementia and incident parkinsonism, baseline cognitive dysfunction was not associated with incident dementia,” the researchers noted.
The researchers determined that the most likely explanation for the association between cognitive decline and increased Parkinson’s risk was that “low baseline cognitive scores may indicate ongoing cognitive decline in prediagnostic patients who probably will develop parkinsonism, most of whom have prediagnostic PD,” they said.
The study findings were limited by several factors including the potential misclassification of parkinsonism diagnosis, the researchers noted. However, the association between poor cognitive function and the risk of parkinsonism and probably Parkinson’s disease remained for the executive, attention, cognitive speed, and memory domains of cognition, they said. “Our findings suggest that poor cognitive functioning can be considered a prodromal sign of PD,” they concluded.
This study was supported in part by Stichting ParkinsonFonds. The researchers had no financial conflicts to disclose.
Adults with early cognitive impairment are at greater risk for developing parkinsonism than those without cognitive impairment, based on data from 7,386 adults participating in the ongoing Rotterdam Study. The findings were published online Sept. 25 in JAMA Neurology.
“Between 15% and 43% of patients with newly diagnosed Parkinson disease (PD) are cognitively impaired,” wrote Sirwan K. L. Darweesh, MD, of Erasmus MC University Medical Center, Rotterdam, the Netherlands, and his colleagues. However, data on the predictive value of cognitive impairment for parkinsonism has not been well studied, they wrote (JAMA Neurol. 2017. doi: 10.1001/jamaneurol.2017.2248).
Over approximately 8 years’ follow-up, 1% of the participants were diagnosed with incident parkinsonism.
“Poor global cognition at baseline was associated with a higher risk of incident parkinsonism” with a hazard ratio of 1.79, the researchers said.
“To enable translation of our findings to clinical practice, we present likelihood ratios (LRs) for the baseline presence of isolated or combined cognitive dysfunction and subtle motor features for incident parkinsonism during follow-up,” they noted.
Approximately half of participants diagnosed with incident parkinsonism during the study period had subtle motor features, cognitive dysfunction, or both, at baseline. Baseline cognitive impairment alone showed a likelihood ratio of 1.76 for development of parkinsonism, but the likelihood ratio was greater when both cognitive impairment and subtle motor findings were present (2.66).
“In individuals who received a diagnosis of both incident dementia and incident parkinsonism, baseline cognitive dysfunction was not associated with incident dementia,” the researchers noted.
The researchers determined that the most likely explanation for the association between cognitive decline and increased Parkinson’s risk was that “low baseline cognitive scores may indicate ongoing cognitive decline in prediagnostic patients who probably will develop parkinsonism, most of whom have prediagnostic PD,” they said.
The study findings were limited by several factors including the potential misclassification of parkinsonism diagnosis, the researchers noted. However, the association between poor cognitive function and the risk of parkinsonism and probably Parkinson’s disease remained for the executive, attention, cognitive speed, and memory domains of cognition, they said. “Our findings suggest that poor cognitive functioning can be considered a prodromal sign of PD,” they concluded.
This study was supported in part by Stichting ParkinsonFonds. The researchers had no financial conflicts to disclose.
FROM JAMA NEUROLOGY
Key clinical point: Mild cognitive impairment may appear early in adults who go on to develop Parkinson’s disease.
Major finding: Poor global cognition at baseline was associated with a greater risk of incident parkinsonism (hazard ratio, 1.79) over approximately 8 years.
Data source: The data come from 7,386 adults in the population-based Rotterdam Study.
Disclosures: This study was supported in part by Stichting ParkinsonFonds. The researchers had no financial conflicts to disclose.
No reduction in preterm bronchopulmonary dysplasia with inhaled NO
Inhaled nitric oxide (NO) therapy does not appear to achieve reduction in the incidence of bronchopulmonary dysplasia in preterm infants, according to data published online Sept. 25 in JAMA Pediatrics.
Shabih U. Hasan, MD, from the Cumming School of Medicine at the University of Calgary, and his coauthors wrote that inhaled nitric oxide is currently approved for the treatment of hypoxic respiratory failure in infants with pulmonary hypertension. Animal studies have prompted interest in its potential to prevent bronchopulmonary dysplasia in preterm infants, but randomized trials so far have shown mixed results (JAMA Pediatr. 2017 Sep 25. doi: 10.1001/jamapediatrics.2017.2618).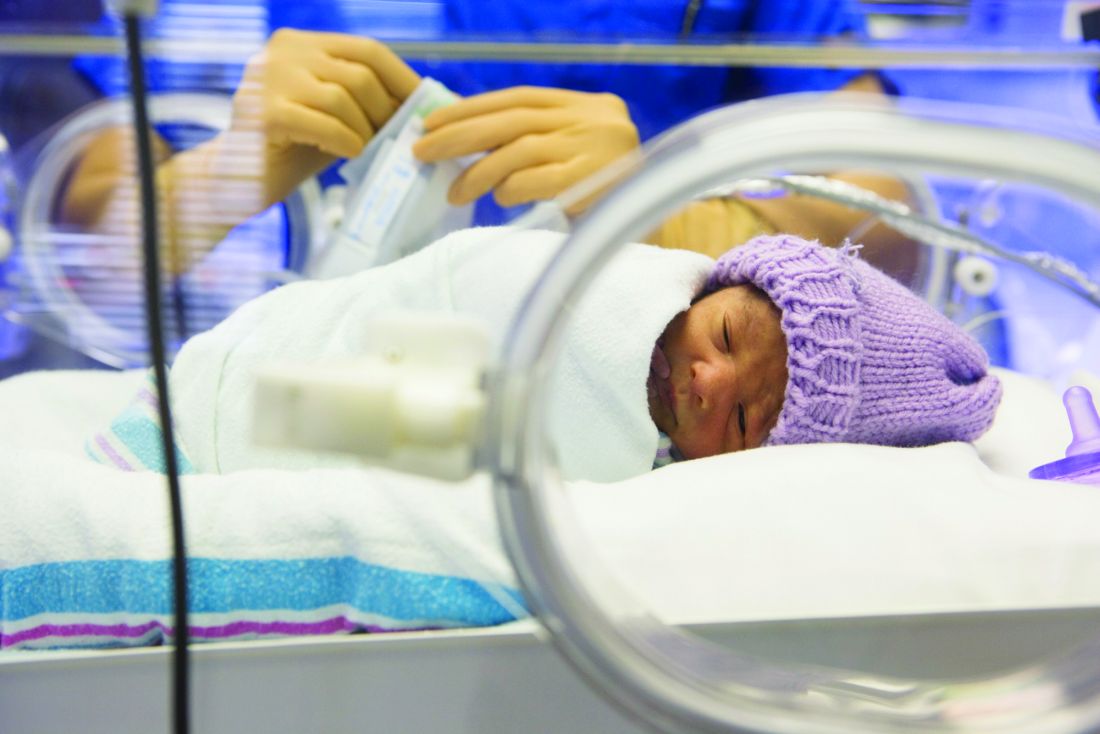
The dosage selected was higher, and the treatment was given for a longer period and initiated later than in some previous studies, which the authors hypothesized might improve outcomes.
However, there was no significant difference between the placebo and inhaled NO groups in the primary outcome of survival to 36 weeks postmenstrual age without bronchopulmonary dysplasia (31.5% vs. 34.9%).
Similarly, the rate of severe bronchopulmonary dysplasia was similar for placebo and inhaled nitric oxide (26.6% vs. 20.5%), as was the rate of postnatal corticosteroid use (41.0% vs. 41.5%), mean days of positive pressure respiratory support (55 vs. 54), mean days of oxygen therapy (88 vs. 91) and mean days of hospitalization (105 vs. 108).
The subgroup analysis revealed that characteristics such as birth weight, gestational age, sex, postnatal age at study entry, maternal race or mode of respiratory support also did not influence the outcomes.
While the rates of severe bronchopulmonary dysplasia were similar between the placebo and inhaled nitric oxide groups, the inhaled NO group had a larger number of infants whose mothers were white and a higher rate of rupture of membranes for more than 7 days, compared with the placebo group.
The two groups had similar incidence of prematurity complications, such as sepsis, patent ductus arteriosus, necrotizing enterocolitis, retinopathy, intraventricular hemorrhage, and pulmonary air leak.
There were also no significant differences in neurodevelopmental or respiratory outcomes at 18-24 months postmenstrual age.
The authors commented that they had hoped their results would be similar to the earlier NO CLD trial, which hinted at a substantial increase in survival without bronchopulmonary dysplasia, compared with placebo in infants aged 7-14 days at the start of treatment.
“The NO CLD trial was not powered to assess the primary outcome in the subgroup enrolled between ages 7 and 14 days, whereas our study was powered specifically for that purpose and included twice as many infants in each treatment arm,” the authors wrote.
Despite this, and a lack of any obvious differences between the study populations, the authors could not identify a reason for the lack of efficacy seen in their own study, compared with this earlier study.
The authors noted that their findings of a lack of benefit from prophylactic but delayed NO on bronchopulmonary dysplasia were consistent with previous meta-analyses, and with a consensus statement from the National Institutes of Health.
The study was sponsored by Mallinckrodt Pharmaceuticals. Four authors declared honorarium, speaking engagements, advisory positions or consultancies with Mallinckrodt Pharmaceuticals. No other conflicts of interest were declared.
Inhaled nitric oxide (NO) therapy does not appear to achieve reduction in the incidence of bronchopulmonary dysplasia in preterm infants, according to data published online Sept. 25 in JAMA Pediatrics.
Shabih U. Hasan, MD, from the Cumming School of Medicine at the University of Calgary, and his coauthors wrote that inhaled nitric oxide is currently approved for the treatment of hypoxic respiratory failure in infants with pulmonary hypertension. Animal studies have prompted interest in its potential to prevent bronchopulmonary dysplasia in preterm infants, but randomized trials so far have shown mixed results (JAMA Pediatr. 2017 Sep 25. doi: 10.1001/jamapediatrics.2017.2618).
The dosage selected was higher, and the treatment was given for a longer period and initiated later than in some previous studies, which the authors hypothesized might improve outcomes.
However, there was no significant difference between the placebo and inhaled NO groups in the primary outcome of survival to 36 weeks postmenstrual age without bronchopulmonary dysplasia (31.5% vs. 34.9%).
Similarly, the rate of severe bronchopulmonary dysplasia was similar for placebo and inhaled nitric oxide (26.6% vs. 20.5%), as was the rate of postnatal corticosteroid use (41.0% vs. 41.5%), mean days of positive pressure respiratory support (55 vs. 54), mean days of oxygen therapy (88 vs. 91) and mean days of hospitalization (105 vs. 108).
The subgroup analysis revealed that characteristics such as birth weight, gestational age, sex, postnatal age at study entry, maternal race or mode of respiratory support also did not influence the outcomes.
While the rates of severe bronchopulmonary dysplasia were similar between the placebo and inhaled nitric oxide groups, the inhaled NO group had a larger number of infants whose mothers were white and a higher rate of rupture of membranes for more than 7 days, compared with the placebo group.
The two groups had similar incidence of prematurity complications, such as sepsis, patent ductus arteriosus, necrotizing enterocolitis, retinopathy, intraventricular hemorrhage, and pulmonary air leak.
There were also no significant differences in neurodevelopmental or respiratory outcomes at 18-24 months postmenstrual age.
The authors commented that they had hoped their results would be similar to the earlier NO CLD trial, which hinted at a substantial increase in survival without bronchopulmonary dysplasia, compared with placebo in infants aged 7-14 days at the start of treatment.
“The NO CLD trial was not powered to assess the primary outcome in the subgroup enrolled between ages 7 and 14 days, whereas our study was powered specifically for that purpose and included twice as many infants in each treatment arm,” the authors wrote.
Despite this, and a lack of any obvious differences between the study populations, the authors could not identify a reason for the lack of efficacy seen in their own study, compared with this earlier study.
The authors noted that their findings of a lack of benefit from prophylactic but delayed NO on bronchopulmonary dysplasia were consistent with previous meta-analyses, and with a consensus statement from the National Institutes of Health.
The study was sponsored by Mallinckrodt Pharmaceuticals. Four authors declared honorarium, speaking engagements, advisory positions or consultancies with Mallinckrodt Pharmaceuticals. No other conflicts of interest were declared.
Inhaled nitric oxide (NO) therapy does not appear to achieve reduction in the incidence of bronchopulmonary dysplasia in preterm infants, according to data published online Sept. 25 in JAMA Pediatrics.
Shabih U. Hasan, MD, from the Cumming School of Medicine at the University of Calgary, and his coauthors wrote that inhaled nitric oxide is currently approved for the treatment of hypoxic respiratory failure in infants with pulmonary hypertension. Animal studies have prompted interest in its potential to prevent bronchopulmonary dysplasia in preterm infants, but randomized trials so far have shown mixed results (JAMA Pediatr. 2017 Sep 25. doi: 10.1001/jamapediatrics.2017.2618).
The dosage selected was higher, and the treatment was given for a longer period and initiated later than in some previous studies, which the authors hypothesized might improve outcomes.
However, there was no significant difference between the placebo and inhaled NO groups in the primary outcome of survival to 36 weeks postmenstrual age without bronchopulmonary dysplasia (31.5% vs. 34.9%).
Similarly, the rate of severe bronchopulmonary dysplasia was similar for placebo and inhaled nitric oxide (26.6% vs. 20.5%), as was the rate of postnatal corticosteroid use (41.0% vs. 41.5%), mean days of positive pressure respiratory support (55 vs. 54), mean days of oxygen therapy (88 vs. 91) and mean days of hospitalization (105 vs. 108).
The subgroup analysis revealed that characteristics such as birth weight, gestational age, sex, postnatal age at study entry, maternal race or mode of respiratory support also did not influence the outcomes.
While the rates of severe bronchopulmonary dysplasia were similar between the placebo and inhaled nitric oxide groups, the inhaled NO group had a larger number of infants whose mothers were white and a higher rate of rupture of membranes for more than 7 days, compared with the placebo group.
The two groups had similar incidence of prematurity complications, such as sepsis, patent ductus arteriosus, necrotizing enterocolitis, retinopathy, intraventricular hemorrhage, and pulmonary air leak.
There were also no significant differences in neurodevelopmental or respiratory outcomes at 18-24 months postmenstrual age.
The authors commented that they had hoped their results would be similar to the earlier NO CLD trial, which hinted at a substantial increase in survival without bronchopulmonary dysplasia, compared with placebo in infants aged 7-14 days at the start of treatment.
“The NO CLD trial was not powered to assess the primary outcome in the subgroup enrolled between ages 7 and 14 days, whereas our study was powered specifically for that purpose and included twice as many infants in each treatment arm,” the authors wrote.
Despite this, and a lack of any obvious differences between the study populations, the authors could not identify a reason for the lack of efficacy seen in their own study, compared with this earlier study.
The authors noted that their findings of a lack of benefit from prophylactic but delayed NO on bronchopulmonary dysplasia were consistent with previous meta-analyses, and with a consensus statement from the National Institutes of Health.
The study was sponsored by Mallinckrodt Pharmaceuticals. Four authors declared honorarium, speaking engagements, advisory positions or consultancies with Mallinckrodt Pharmaceuticals. No other conflicts of interest were declared.
FROM JAMA PEDIATRICS
Key clinical point: Treatment with inhaled nitric oxide does not reduce the incidence of bronchopulmonary dysplasia in preterm infants.
Major finding: The incidence of bronchopulmonary dysplasia in preterm infants was not reduced with inhaled nitric oxide therapy.
Data source: Prospective randomized placebo-controlled trial in 451 preterm infants of less than 30 weeks gestation.
Disclosures: The study was sponsored by Mallinckrodt Pharmaceuticals. Four authors declared honorarium, speakers fees, advisory positions, or consultancies with Mallinckrodt Pharmaceuticals. No other conflicts of interest were declared.
Primary care deficient in cancer survivor care
Even advanced primary care practices are not providing comprehensive cancer survivorship care, with deficiencies in how cancer survivors are categorized, how they’re transitioned to primary care, and in the information systems used in their care, according to a new study published online September 25 in JAMA Internal Medicine.
The analysis came from data gathered by investigators at Rutgers Robert Wood Johnson Medical School, New Brunswick, N.J., who performed case studies on 12 advanced primary care centers across a variety of practice types and geographic settings. The centers were chosen using a national registry of “workforce innovators” compiled by the Robert Wood Johnson Foundation in 2011 and 2012. All but three of the centers were designated patient-centered medical homes (JAMA Intern Med. 2017. doi: 10.1001/jamainternmed.2017.4747).
Researchers noted the tremendous importance of primary care to cancer survivors. Only about a third of cancer survivors continue to be seen by a cancer specialist 5 years after their diagnosis, but 75% are seen in primary care. The importance of preventive screening, surveillance for recurrence, interventions for long-term effects, and care coordination between specialty and primary care were noted in an Institute of Medicine report in 2006.
Researchers found that the primary care clinicians don’t treat cancer survivors as a distinct population; they get limited information or follow-up guidance on cancer care; and information systems aren’t good at supporting survivorship care.
“Codifying survivorship as a distinct clinical category that belongs on problem lists with payment-linked – fee, value-based, or capitated – care services is a critical first step toward bringing comprehensive cancer survivorship services to primary care,” Dr. Rubinstein said.
Researchers described what they called “cancer exceptionalism,” in which a cancer diagnosis follows a different clinical norm and patients are referred to oncology and then become disengaged with primary care.
On transition of care, one primary care physician told an interviewer that it seems that patients’ cancer treatment “kind of happens in a black box” and that they feel “a little intimidated” in providing the needed follow-up care.
Another said that while a patient’s cancer history could be seen “at a glance” in old paper charts, their electronic health record requires searching multiple screens and “sometimes it’s a needle in a haystack.”
“Despite the push from national organizations to enhance cancer survivorship care capacity in primary care,” Dr. Rubinstein said, “findings from this study suggest that cancer survivorship care does not integrate easily into advanced primary care.”
The researchers reported no conflicts of interest.
Even advanced primary care practices are not providing comprehensive cancer survivorship care, with deficiencies in how cancer survivors are categorized, how they’re transitioned to primary care, and in the information systems used in their care, according to a new study published online September 25 in JAMA Internal Medicine.
The analysis came from data gathered by investigators at Rutgers Robert Wood Johnson Medical School, New Brunswick, N.J., who performed case studies on 12 advanced primary care centers across a variety of practice types and geographic settings. The centers were chosen using a national registry of “workforce innovators” compiled by the Robert Wood Johnson Foundation in 2011 and 2012. All but three of the centers were designated patient-centered medical homes (JAMA Intern Med. 2017. doi: 10.1001/jamainternmed.2017.4747).
Researchers noted the tremendous importance of primary care to cancer survivors. Only about a third of cancer survivors continue to be seen by a cancer specialist 5 years after their diagnosis, but 75% are seen in primary care. The importance of preventive screening, surveillance for recurrence, interventions for long-term effects, and care coordination between specialty and primary care were noted in an Institute of Medicine report in 2006.
Researchers found that the primary care clinicians don’t treat cancer survivors as a distinct population; they get limited information or follow-up guidance on cancer care; and information systems aren’t good at supporting survivorship care.
“Codifying survivorship as a distinct clinical category that belongs on problem lists with payment-linked – fee, value-based, or capitated – care services is a critical first step toward bringing comprehensive cancer survivorship services to primary care,” Dr. Rubinstein said.
Researchers described what they called “cancer exceptionalism,” in which a cancer diagnosis follows a different clinical norm and patients are referred to oncology and then become disengaged with primary care.
On transition of care, one primary care physician told an interviewer that it seems that patients’ cancer treatment “kind of happens in a black box” and that they feel “a little intimidated” in providing the needed follow-up care.
Another said that while a patient’s cancer history could be seen “at a glance” in old paper charts, their electronic health record requires searching multiple screens and “sometimes it’s a needle in a haystack.”
“Despite the push from national organizations to enhance cancer survivorship care capacity in primary care,” Dr. Rubinstein said, “findings from this study suggest that cancer survivorship care does not integrate easily into advanced primary care.”
The researchers reported no conflicts of interest.
Even advanced primary care practices are not providing comprehensive cancer survivorship care, with deficiencies in how cancer survivors are categorized, how they’re transitioned to primary care, and in the information systems used in their care, according to a new study published online September 25 in JAMA Internal Medicine.
The analysis came from data gathered by investigators at Rutgers Robert Wood Johnson Medical School, New Brunswick, N.J., who performed case studies on 12 advanced primary care centers across a variety of practice types and geographic settings. The centers were chosen using a national registry of “workforce innovators” compiled by the Robert Wood Johnson Foundation in 2011 and 2012. All but three of the centers were designated patient-centered medical homes (JAMA Intern Med. 2017. doi: 10.1001/jamainternmed.2017.4747).
Researchers noted the tremendous importance of primary care to cancer survivors. Only about a third of cancer survivors continue to be seen by a cancer specialist 5 years after their diagnosis, but 75% are seen in primary care. The importance of preventive screening, surveillance for recurrence, interventions for long-term effects, and care coordination between specialty and primary care were noted in an Institute of Medicine report in 2006.
Researchers found that the primary care clinicians don’t treat cancer survivors as a distinct population; they get limited information or follow-up guidance on cancer care; and information systems aren’t good at supporting survivorship care.
“Codifying survivorship as a distinct clinical category that belongs on problem lists with payment-linked – fee, value-based, or capitated – care services is a critical first step toward bringing comprehensive cancer survivorship services to primary care,” Dr. Rubinstein said.
Researchers described what they called “cancer exceptionalism,” in which a cancer diagnosis follows a different clinical norm and patients are referred to oncology and then become disengaged with primary care.
On transition of care, one primary care physician told an interviewer that it seems that patients’ cancer treatment “kind of happens in a black box” and that they feel “a little intimidated” in providing the needed follow-up care.
Another said that while a patient’s cancer history could be seen “at a glance” in old paper charts, their electronic health record requires searching multiple screens and “sometimes it’s a needle in a haystack.”
“Despite the push from national organizations to enhance cancer survivorship care capacity in primary care,” Dr. Rubinstein said, “findings from this study suggest that cancer survivorship care does not integrate easily into advanced primary care.”
The researchers reported no conflicts of interest.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Advanced primary care practices are not providing comprehensive cancer survivorship care.
Major finding: Primary care clinicians don’t treat cancer survivors as a distinct population; they get limited information or follow-up guidance on cancer care; and information systems aren’t good at supporting survivorship care.
Data source: A comparative case study of 12 primary care practices compiled using a national registry of “workforce innovators.”
Disclosures: None reported.
Gods and Monsters
For the first time in history, four generations of physicians work side by side in the U.S. health care system. An expanding population, longer life expectancies, and later retirement ages all contribute to this phenomenon. Each of these generations has made significant contributions to modern surgery and how we practice it. For better and for worse.
Traditionalists, or the Greatest Generation, were true surgical pioneers. DeBakey, Cooley, Fogarty, their names now adorn everything from instruments to medical centers. They truly founded the modern system of surgery. Born between 1900 and 1945, Traditionalists were forged in the crucibles of the Great War and the Great Depression. Their core values were hard work, discipline, and sacrifice. A large number were combat veterans who valued conformity and adherence to the rules. Traditionalists set up our current hierarchical departments of surgery. Mirroring their values, they employed a military chain of command approach. Many traditionalists rose to positions of absolute power, and some were corrupted by this power. Gods became monsters. Abuse, both verbal and physical, came to be commonplace and accepted in the surgical work environment.
Born between 1946 and 1964, Baby Boomers were raised in the aftermath of a war none of them saw. More optimistic and idealistic than the Traditionalists, the Boomers valued success. Their goals became more individualistic. Chasing money, titles, and recognition, Boomers wanted to build a stellar career. Fifty-hour work weeks became 70, 80, or 90. Ambition led to wealth, dramatic successes, and remarkable careers. Their choices also led to divorce, drug abuse, and suicide. While burnout has become a modern concern, its roots are clearly tied to this era. Now serving as our deans and department chairs, the Boomers also made several notable contributions. Specific to our field, Boomers oversaw the development of vascular surgery as an independent specialty and the expansion of fellowship training programs. Coming of age in the 1960s, Boomers also led the integration of our field with the acceptance of both minorities and women.
When I first heard the term “Generation X” I thought “Dumb name, won’t last.” Not my best prediction. Born between 1965 and 1980, Generation X grew up during the home computer revolution. Quick to adopt new technologies, Gen Xers were far more adaptive to change than previous generations. Labeled as having short attention spans, most Gen Xers were task/goal oriented. While these attributes helped drive the endovascular revolution, they also may be the reason we have approximately 983 FDA-approved devices to treat SFA disease. Generation X entered surgical training eager to please the more senior Traditionalists and Boomers. This wouldn’t last. Children of divorce and latch-key kids, Generation Xers are eclectic, resourceful, and self-reliant. Most of all they value freedom. Watching their predecessors work themselves and others to near death, Generation X revolted. Uncapped duty hours, limitless call, and pyramidal residencies were all institutions in the 1980s, and they all fell. Generation X were portrayed as nihilistic slackers, but their true motivation was often distrust of institutions. Watching the Boomers descend into burnout, Xers tried to achieve a more reasonable work-life balance. Though they successfully fought for lessening the abuses of surgical training, few Gen Xers actually reaped the benefits. I vividly recall watching slack-jawed as an intern scrubbed out of a case to go home because he was post call. A Martian landing in the OR and offering to assist with the anastomosis would have brought no less amazement.
With their careers spanning the endovascular revolution, Generation X has seen perhaps the greatest era of transformation in our profession. Our competition is no longer general or cardiac surgery, but rather interventional radiology and interventional cardiology. Gen X is also the first generation to earn less than its predecessors. Throw in their obscene tuition payments and one can see how Gen Xers fell well short of the financial heights of the Traditionalists and Boomers. The Gen Xers are the masters of the work hard/play hard ethos. You will see them at VEITH entertaining their European colleagues at 3 a.m. and then running the 6 a.m. breakfast sessions. While the Boomers often seemed old by 40, Xers appear desperate to salvage their lost youth.
Born between 1981 and 2006, Millennials are already the most populous generation. Their chief attributes are confidence, sociability, and a realistic outlook. Knowing they can’t please everyone, they rarely try. They want work to be meaningful in and of itself. They also value teamwork over individual approaches. Millennials are civic minded and have a strong sense of volunteerism. Their parents often tried to shelter them from the evils of the world, and they were the first generation of children with schedules. Because of their upbringing, Millennials are far more likely to seek guidance than the independent-minded Gen Xers. Raised to believe their voice mattered, they are now often reviled for it. It is with some degree of awe that I watch our Millennial students brazenly march into the dean’s and chancellor’s office to discuss their “careers.” As a medical student I first saw my dean at graduation, and I certainly didn’t even know what a chancellor was. Generation X is often baffled by the self-interest Millennials exude. But we shouldn’t be, we have seen it before. Raised by Baby Boomers (The Me Generation), Millennials inherited their self-driven outlook. This is also the reason Boomers and Millennials struggle to work together. They are too alike. Boomers see Millennials as “snowflakes” who are scared of work and selfie obsessed. Millennials bristle at the authoritarian nature of Boomers.
For vascular surgery to advance as a field, we need to recruit, train, and mentor this new generation. If only there was some guide: “The Proper Care and Feeding of Millennials." As senior attendings, program directors, and section chiefs, Generation X must now serve as a bridge between two larger forces, the Boomers and their offspring, the Millennials. Of course, whatever generation you are from is the best, but we must confront our biases. It is easy to seek out the same personalities to be your trainees and partners. Don’t. This pool will shrink every year. Millennials are more self-aware of their capabilities and therefore of their limitations. We may become flustered by their need for hand-holding, but what if it is appropriate? Was all of the autonomy you were granted during training truly good for the patients? Graduated responsibility and roles that push their limits help Millennials grow. I know they don’t value punctuality or dress codes, but they are better team players and openly motivated by learning. I formed our integrated vascular residency with two positions per year specifically to foster the team building Millennials crave. Yes, this is the generation that got 8th-place trophies so you must constantly award progress. Fortunately, now that surgery is unencumbered by such things as massive salaries and status, Millennials enter our workforce with purer intentions.

We may want to make Millennials match our values, traits, and behaviors, but each generation has departed radically from the ethos of their predecessors. Let’s see what the kids can do.
Dr. Sheahan is a professor of surgery and Program Director, Vascular Surgery Residency and Fellowship Programs, Louisiana State University Health Sciences Center, School of Medicine, New Orleans. He is also the Deputy Medical Editor of Vascular Specialist.
For the first time in history, four generations of physicians work side by side in the U.S. health care system. An expanding population, longer life expectancies, and later retirement ages all contribute to this phenomenon. Each of these generations has made significant contributions to modern surgery and how we practice it. For better and for worse.
Traditionalists, or the Greatest Generation, were true surgical pioneers. DeBakey, Cooley, Fogarty, their names now adorn everything from instruments to medical centers. They truly founded the modern system of surgery. Born between 1900 and 1945, Traditionalists were forged in the crucibles of the Great War and the Great Depression. Their core values were hard work, discipline, and sacrifice. A large number were combat veterans who valued conformity and adherence to the rules. Traditionalists set up our current hierarchical departments of surgery. Mirroring their values, they employed a military chain of command approach. Many traditionalists rose to positions of absolute power, and some were corrupted by this power. Gods became monsters. Abuse, both verbal and physical, came to be commonplace and accepted in the surgical work environment.
Born between 1946 and 1964, Baby Boomers were raised in the aftermath of a war none of them saw. More optimistic and idealistic than the Traditionalists, the Boomers valued success. Their goals became more individualistic. Chasing money, titles, and recognition, Boomers wanted to build a stellar career. Fifty-hour work weeks became 70, 80, or 90. Ambition led to wealth, dramatic successes, and remarkable careers. Their choices also led to divorce, drug abuse, and suicide. While burnout has become a modern concern, its roots are clearly tied to this era. Now serving as our deans and department chairs, the Boomers also made several notable contributions. Specific to our field, Boomers oversaw the development of vascular surgery as an independent specialty and the expansion of fellowship training programs. Coming of age in the 1960s, Boomers also led the integration of our field with the acceptance of both minorities and women.
When I first heard the term “Generation X” I thought “Dumb name, won’t last.” Not my best prediction. Born between 1965 and 1980, Generation X grew up during the home computer revolution. Quick to adopt new technologies, Gen Xers were far more adaptive to change than previous generations. Labeled as having short attention spans, most Gen Xers were task/goal oriented. While these attributes helped drive the endovascular revolution, they also may be the reason we have approximately 983 FDA-approved devices to treat SFA disease. Generation X entered surgical training eager to please the more senior Traditionalists and Boomers. This wouldn’t last. Children of divorce and latch-key kids, Generation Xers are eclectic, resourceful, and self-reliant. Most of all they value freedom. Watching their predecessors work themselves and others to near death, Generation X revolted. Uncapped duty hours, limitless call, and pyramidal residencies were all institutions in the 1980s, and they all fell. Generation X were portrayed as nihilistic slackers, but their true motivation was often distrust of institutions. Watching the Boomers descend into burnout, Xers tried to achieve a more reasonable work-life balance. Though they successfully fought for lessening the abuses of surgical training, few Gen Xers actually reaped the benefits. I vividly recall watching slack-jawed as an intern scrubbed out of a case to go home because he was post call. A Martian landing in the OR and offering to assist with the anastomosis would have brought no less amazement.
With their careers spanning the endovascular revolution, Generation X has seen perhaps the greatest era of transformation in our profession. Our competition is no longer general or cardiac surgery, but rather interventional radiology and interventional cardiology. Gen X is also the first generation to earn less than its predecessors. Throw in their obscene tuition payments and one can see how Gen Xers fell well short of the financial heights of the Traditionalists and Boomers. The Gen Xers are the masters of the work hard/play hard ethos. You will see them at VEITH entertaining their European colleagues at 3 a.m. and then running the 6 a.m. breakfast sessions. While the Boomers often seemed old by 40, Xers appear desperate to salvage their lost youth.
Born between 1981 and 2006, Millennials are already the most populous generation. Their chief attributes are confidence, sociability, and a realistic outlook. Knowing they can’t please everyone, they rarely try. They want work to be meaningful in and of itself. They also value teamwork over individual approaches. Millennials are civic minded and have a strong sense of volunteerism. Their parents often tried to shelter them from the evils of the world, and they were the first generation of children with schedules. Because of their upbringing, Millennials are far more likely to seek guidance than the independent-minded Gen Xers. Raised to believe their voice mattered, they are now often reviled for it. It is with some degree of awe that I watch our Millennial students brazenly march into the dean’s and chancellor’s office to discuss their “careers.” As a medical student I first saw my dean at graduation, and I certainly didn’t even know what a chancellor was. Generation X is often baffled by the self-interest Millennials exude. But we shouldn’t be, we have seen it before. Raised by Baby Boomers (The Me Generation), Millennials inherited their self-driven outlook. This is also the reason Boomers and Millennials struggle to work together. They are too alike. Boomers see Millennials as “snowflakes” who are scared of work and selfie obsessed. Millennials bristle at the authoritarian nature of Boomers.
For vascular surgery to advance as a field, we need to recruit, train, and mentor this new generation. If only there was some guide: “The Proper Care and Feeding of Millennials." As senior attendings, program directors, and section chiefs, Generation X must now serve as a bridge between two larger forces, the Boomers and their offspring, the Millennials. Of course, whatever generation you are from is the best, but we must confront our biases. It is easy to seek out the same personalities to be your trainees and partners. Don’t. This pool will shrink every year. Millennials are more self-aware of their capabilities and therefore of their limitations. We may become flustered by their need for hand-holding, but what if it is appropriate? Was all of the autonomy you were granted during training truly good for the patients? Graduated responsibility and roles that push their limits help Millennials grow. I know they don’t value punctuality or dress codes, but they are better team players and openly motivated by learning. I formed our integrated vascular residency with two positions per year specifically to foster the team building Millennials crave. Yes, this is the generation that got 8th-place trophies so you must constantly award progress. Fortunately, now that surgery is unencumbered by such things as massive salaries and status, Millennials enter our workforce with purer intentions.
We may want to make Millennials match our values, traits, and behaviors, but each generation has departed radically from the ethos of their predecessors. Let’s see what the kids can do.
Dr. Sheahan is a professor of surgery and Program Director, Vascular Surgery Residency and Fellowship Programs, Louisiana State University Health Sciences Center, School of Medicine, New Orleans. He is also the Deputy Medical Editor of Vascular Specialist.
For the first time in history, four generations of physicians work side by side in the U.S. health care system. An expanding population, longer life expectancies, and later retirement ages all contribute to this phenomenon. Each of these generations has made significant contributions to modern surgery and how we practice it. For better and for worse.
Traditionalists, or the Greatest Generation, were true surgical pioneers. DeBakey, Cooley, Fogarty, their names now adorn everything from instruments to medical centers. They truly founded the modern system of surgery. Born between 1900 and 1945, Traditionalists were forged in the crucibles of the Great War and the Great Depression. Their core values were hard work, discipline, and sacrifice. A large number were combat veterans who valued conformity and adherence to the rules. Traditionalists set up our current hierarchical departments of surgery. Mirroring their values, they employed a military chain of command approach. Many traditionalists rose to positions of absolute power, and some were corrupted by this power. Gods became monsters. Abuse, both verbal and physical, came to be commonplace and accepted in the surgical work environment.
Born between 1946 and 1964, Baby Boomers were raised in the aftermath of a war none of them saw. More optimistic and idealistic than the Traditionalists, the Boomers valued success. Their goals became more individualistic. Chasing money, titles, and recognition, Boomers wanted to build a stellar career. Fifty-hour work weeks became 70, 80, or 90. Ambition led to wealth, dramatic successes, and remarkable careers. Their choices also led to divorce, drug abuse, and suicide. While burnout has become a modern concern, its roots are clearly tied to this era. Now serving as our deans and department chairs, the Boomers also made several notable contributions. Specific to our field, Boomers oversaw the development of vascular surgery as an independent specialty and the expansion of fellowship training programs. Coming of age in the 1960s, Boomers also led the integration of our field with the acceptance of both minorities and women.
When I first heard the term “Generation X” I thought “Dumb name, won’t last.” Not my best prediction. Born between 1965 and 1980, Generation X grew up during the home computer revolution. Quick to adopt new technologies, Gen Xers were far more adaptive to change than previous generations. Labeled as having short attention spans, most Gen Xers were task/goal oriented. While these attributes helped drive the endovascular revolution, they also may be the reason we have approximately 983 FDA-approved devices to treat SFA disease. Generation X entered surgical training eager to please the more senior Traditionalists and Boomers. This wouldn’t last. Children of divorce and latch-key kids, Generation Xers are eclectic, resourceful, and self-reliant. Most of all they value freedom. Watching their predecessors work themselves and others to near death, Generation X revolted. Uncapped duty hours, limitless call, and pyramidal residencies were all institutions in the 1980s, and they all fell. Generation X were portrayed as nihilistic slackers, but their true motivation was often distrust of institutions. Watching the Boomers descend into burnout, Xers tried to achieve a more reasonable work-life balance. Though they successfully fought for lessening the abuses of surgical training, few Gen Xers actually reaped the benefits. I vividly recall watching slack-jawed as an intern scrubbed out of a case to go home because he was post call. A Martian landing in the OR and offering to assist with the anastomosis would have brought no less amazement.
With their careers spanning the endovascular revolution, Generation X has seen perhaps the greatest era of transformation in our profession. Our competition is no longer general or cardiac surgery, but rather interventional radiology and interventional cardiology. Gen X is also the first generation to earn less than its predecessors. Throw in their obscene tuition payments and one can see how Gen Xers fell well short of the financial heights of the Traditionalists and Boomers. The Gen Xers are the masters of the work hard/play hard ethos. You will see them at VEITH entertaining their European colleagues at 3 a.m. and then running the 6 a.m. breakfast sessions. While the Boomers often seemed old by 40, Xers appear desperate to salvage their lost youth.
Born between 1981 and 2006, Millennials are already the most populous generation. Their chief attributes are confidence, sociability, and a realistic outlook. Knowing they can’t please everyone, they rarely try. They want work to be meaningful in and of itself. They also value teamwork over individual approaches. Millennials are civic minded and have a strong sense of volunteerism. Their parents often tried to shelter them from the evils of the world, and they were the first generation of children with schedules. Because of their upbringing, Millennials are far more likely to seek guidance than the independent-minded Gen Xers. Raised to believe their voice mattered, they are now often reviled for it. It is with some degree of awe that I watch our Millennial students brazenly march into the dean’s and chancellor’s office to discuss their “careers.” As a medical student I first saw my dean at graduation, and I certainly didn’t even know what a chancellor was. Generation X is often baffled by the self-interest Millennials exude. But we shouldn’t be, we have seen it before. Raised by Baby Boomers (The Me Generation), Millennials inherited their self-driven outlook. This is also the reason Boomers and Millennials struggle to work together. They are too alike. Boomers see Millennials as “snowflakes” who are scared of work and selfie obsessed. Millennials bristle at the authoritarian nature of Boomers.
For vascular surgery to advance as a field, we need to recruit, train, and mentor this new generation. If only there was some guide: “The Proper Care and Feeding of Millennials." As senior attendings, program directors, and section chiefs, Generation X must now serve as a bridge between two larger forces, the Boomers and their offspring, the Millennials. Of course, whatever generation you are from is the best, but we must confront our biases. It is easy to seek out the same personalities to be your trainees and partners. Don’t. This pool will shrink every year. Millennials are more self-aware of their capabilities and therefore of their limitations. We may become flustered by their need for hand-holding, but what if it is appropriate? Was all of the autonomy you were granted during training truly good for the patients? Graduated responsibility and roles that push their limits help Millennials grow. I know they don’t value punctuality or dress codes, but they are better team players and openly motivated by learning. I formed our integrated vascular residency with two positions per year specifically to foster the team building Millennials crave. Yes, this is the generation that got 8th-place trophies so you must constantly award progress. Fortunately, now that surgery is unencumbered by such things as massive salaries and status, Millennials enter our workforce with purer intentions.
We may want to make Millennials match our values, traits, and behaviors, but each generation has departed radically from the ethos of their predecessors. Let’s see what the kids can do.
Dr. Sheahan is a professor of surgery and Program Director, Vascular Surgery Residency and Fellowship Programs, Louisiana State University Health Sciences Center, School of Medicine, New Orleans. He is also the Deputy Medical Editor of Vascular Specialist.
JVS, JVS-VL Editors Seek Members' Help for Reviews, Meta-analyses
The staffs of the Journal of Vascular Surgery and Journal of Vascular Surgery: Venous & Lymphatic Disorders are interested in creating a list of SVS members who have interest, expertise and resources to perform high-quality systematic reviews and meta-analyses.
If interested, please submit your name, institution and topics consistent with your area of expertise. Members may reach out directly via email to Dr. Cynthia Shortell, assistant editor of reviews for JVS-VL, and Dr. Ron Fairman, assistant editor of reviews for JVS.
The staffs of the Journal of Vascular Surgery and Journal of Vascular Surgery: Venous & Lymphatic Disorders are interested in creating a list of SVS members who have interest, expertise and resources to perform high-quality systematic reviews and meta-analyses.
If interested, please submit your name, institution and topics consistent with your area of expertise. Members may reach out directly via email to Dr. Cynthia Shortell, assistant editor of reviews for JVS-VL, and Dr. Ron Fairman, assistant editor of reviews for JVS.
The staffs of the Journal of Vascular Surgery and Journal of Vascular Surgery: Venous & Lymphatic Disorders are interested in creating a list of SVS members who have interest, expertise and resources to perform high-quality systematic reviews and meta-analyses.
If interested, please submit your name, institution and topics consistent with your area of expertise. Members may reach out directly via email to Dr. Cynthia Shortell, assistant editor of reviews for JVS-VL, and Dr. Ron Fairman, assistant editor of reviews for JVS.
PAD Resources for SVS Members
September is Peripheral Artery Disease Awareness Month. To help SVS members educate patients and to spread awareness about vascular surgeons, we have prepared several things you can share.
1. An infographic for patients and their families. We urge you to print it and post around the office or your institution.
2. A quick resource web page for patients, offering patients a PAD video playlist and links to articles and information on PAD.
3. The latest PAD research information for physicians, along with clinical practice guideline links. If you have contacts among primary care physicians or other referrers, please feel free to send them this link.
4. Two press releases on PAD, to share with your communications people, public relations departments and/or patients
• Don't Fall for These 6 Internet Myths About Statins
• Often misdiagnosed, PAD can be mild or deadly
September is Peripheral Artery Disease Awareness Month. To help SVS members educate patients and to spread awareness about vascular surgeons, we have prepared several things you can share.
1. An infographic for patients and their families. We urge you to print it and post around the office or your institution.
2. A quick resource web page for patients, offering patients a PAD video playlist and links to articles and information on PAD.
3. The latest PAD research information for physicians, along with clinical practice guideline links. If you have contacts among primary care physicians or other referrers, please feel free to send them this link.
4. Two press releases on PAD, to share with your communications people, public relations departments and/or patients
• Don't Fall for These 6 Internet Myths About Statins
• Often misdiagnosed, PAD can be mild or deadly
September is Peripheral Artery Disease Awareness Month. To help SVS members educate patients and to spread awareness about vascular surgeons, we have prepared several things you can share.
1. An infographic for patients and their families. We urge you to print it and post around the office or your institution.
2. A quick resource web page for patients, offering patients a PAD video playlist and links to articles and information on PAD.
3. The latest PAD research information for physicians, along with clinical practice guideline links. If you have contacts among primary care physicians or other referrers, please feel free to send them this link.
4. Two press releases on PAD, to share with your communications people, public relations departments and/or patients
• Don't Fall for These 6 Internet Myths About Statins
• Often misdiagnosed, PAD can be mild or deadly
Docs, insurers condemn latest ‘repeal and replace’ plan
Medical societies and insurers are voicing their opposition to legislation that would alter provisions of the Affordable Care Act and fundamentally change how Medicaid is funded.
The bill, introduced by Sen. Lindsey Graham (R-S.C.), Sen. Bill Cassidy (R-La.), Sen. Dean Heller (R-Nev.), and Sen. Ron Johnson (R-Wis.), features a number of provisions long sought by the GOP, including the repeal of the individual and employer mandates, repeal of individual tax credits as of 2020, and repeal of the medical device tax. The bill also would promote the use of health savings accounts and turn Medicaid funding into a block grant program, allowing states to implement policies such as work requirements.
James L. Madara, MD, CEO of the American Medical Association, told congressional leaders in a Sept. 19 letter that the bill would violate the precept of “first do no harm” and results in millions of Americans losing their health coverage. Additionally, it would destabilize health insurance markets and decrease access to affordable coverage.
“We are also concerned that the proposal would convert the Medicaid program into a system that limits federal support to care for needy patients to an insufficient predetermined formula based on per capita caps,” Dr. Madara continued. “Per capita caps fail to take into account unanticipated costs of new medical innovations or the fiscal impact of public health epidemics, such as the crisis of opioid abuse currently ravaging our nation. In addition, the amendment does not take steps toward coverage and access for all Americans, and while insurers are still required to offer coverage to patients with preexisting conditions, allowing states to get waivers to vary premiums based on health status would allow insurers to charge unaffordable premiums based on those preexisting conditions. Also, waivers of essential health benefits will mean patients may not have access to coverage for services pertinent to treating their conditions.”
The American Congress of Obstetricians and Gynecologists called the bill an “assault on women’s health.” The bill would end guaranteed insurance coverage of maternity care and women’s health preventive services, including cancer screenings and contraception, ACOG president Haywood Brown, MD, said in a statement.
Dr. Brown added that the bill “jeopardizes access to care for women with high-risk and expensive pregnancies, such as those with Zika virus, opioid use disorder, and preeclampsia. It further obstructs safety net patients’ access to care by forbidding Planned Parenthood’s participation in the Medicaid program.”
AGA is also concerned that there are no guarantees that states have to provide essential benefits, patients that gained coverage through the ACA would lose that coverage, and most importantly, patients with pre-existing conditions have no guarantee that they will continue to receive affordable coverage.
Doctors aren’t the only ones objecting to the GOP legislation. America’s Health Insurance Plans president and CEO Marilyn Tavenner said in a Sept. 20 letter to Congress that the bill would further destabilize the individual health insurance market.
The bill’s road to passage is far from certain. Once again, the GOP is aiming to use the budget reconciliation process to pass this legislation, which means it needs only a simple majority to pass (a minimum of 50 votes with Vice President Mike Pence offering the tiebreaker if the bill cannot get 51 votes). But even getting to 50 votes is going to be a challenge as the last attempt to pass similar repeal and replace language failed when Sen. Susan Collins (R-Maine), Sen. Lisa Murkowski (R-Alaska), and Sen. John McCain (R-Ariz.) voted that package down. Given the similar features, Sen. Collins and Sen. Murkowski may still oppose the bill, while Sen. Rand Paul (R-Ky.) has been vocal about his displeasure with the bill and other GOP senators are getting pressure from their state governors to oppose the bill.
The Senate Finance Committee has scheduled a Sept. 25 hearing to consider the bill, but as of press time, no witnesses have been announced, and the bill likely will not follow the regular order of allowing for amendments by committee members prior to its introduction on the Senate floor later that week.
Based on current budget rules, the bill must be passed by Sept. 30 in order for the budget reconciliation process to be used and to allow for passage with a simple majority. If the Senate is able to pass the bill, House Speaker Paul Ryan (R-Wisc.) has said he will bring it up in the House. President Trump has indicated he will sign it into law if it reaches his desk.
Medical societies and insurers are voicing their opposition to legislation that would alter provisions of the Affordable Care Act and fundamentally change how Medicaid is funded.
The bill, introduced by Sen. Lindsey Graham (R-S.C.), Sen. Bill Cassidy (R-La.), Sen. Dean Heller (R-Nev.), and Sen. Ron Johnson (R-Wis.), features a number of provisions long sought by the GOP, including the repeal of the individual and employer mandates, repeal of individual tax credits as of 2020, and repeal of the medical device tax. The bill also would promote the use of health savings accounts and turn Medicaid funding into a block grant program, allowing states to implement policies such as work requirements.
James L. Madara, MD, CEO of the American Medical Association, told congressional leaders in a Sept. 19 letter that the bill would violate the precept of “first do no harm” and results in millions of Americans losing their health coverage. Additionally, it would destabilize health insurance markets and decrease access to affordable coverage.
“We are also concerned that the proposal would convert the Medicaid program into a system that limits federal support to care for needy patients to an insufficient predetermined formula based on per capita caps,” Dr. Madara continued. “Per capita caps fail to take into account unanticipated costs of new medical innovations or the fiscal impact of public health epidemics, such as the crisis of opioid abuse currently ravaging our nation. In addition, the amendment does not take steps toward coverage and access for all Americans, and while insurers are still required to offer coverage to patients with preexisting conditions, allowing states to get waivers to vary premiums based on health status would allow insurers to charge unaffordable premiums based on those preexisting conditions. Also, waivers of essential health benefits will mean patients may not have access to coverage for services pertinent to treating their conditions.”
The American Congress of Obstetricians and Gynecologists called the bill an “assault on women’s health.” The bill would end guaranteed insurance coverage of maternity care and women’s health preventive services, including cancer screenings and contraception, ACOG president Haywood Brown, MD, said in a statement.
Dr. Brown added that the bill “jeopardizes access to care for women with high-risk and expensive pregnancies, such as those with Zika virus, opioid use disorder, and preeclampsia. It further obstructs safety net patients’ access to care by forbidding Planned Parenthood’s participation in the Medicaid program.”
AGA is also concerned that there are no guarantees that states have to provide essential benefits, patients that gained coverage through the ACA would lose that coverage, and most importantly, patients with pre-existing conditions have no guarantee that they will continue to receive affordable coverage.
Doctors aren’t the only ones objecting to the GOP legislation. America’s Health Insurance Plans president and CEO Marilyn Tavenner said in a Sept. 20 letter to Congress that the bill would further destabilize the individual health insurance market.
The bill’s road to passage is far from certain. Once again, the GOP is aiming to use the budget reconciliation process to pass this legislation, which means it needs only a simple majority to pass (a minimum of 50 votes with Vice President Mike Pence offering the tiebreaker if the bill cannot get 51 votes). But even getting to 50 votes is going to be a challenge as the last attempt to pass similar repeal and replace language failed when Sen. Susan Collins (R-Maine), Sen. Lisa Murkowski (R-Alaska), and Sen. John McCain (R-Ariz.) voted that package down. Given the similar features, Sen. Collins and Sen. Murkowski may still oppose the bill, while Sen. Rand Paul (R-Ky.) has been vocal about his displeasure with the bill and other GOP senators are getting pressure from their state governors to oppose the bill.
The Senate Finance Committee has scheduled a Sept. 25 hearing to consider the bill, but as of press time, no witnesses have been announced, and the bill likely will not follow the regular order of allowing for amendments by committee members prior to its introduction on the Senate floor later that week.
Based on current budget rules, the bill must be passed by Sept. 30 in order for the budget reconciliation process to be used and to allow for passage with a simple majority. If the Senate is able to pass the bill, House Speaker Paul Ryan (R-Wisc.) has said he will bring it up in the House. President Trump has indicated he will sign it into law if it reaches his desk.
Medical societies and insurers are voicing their opposition to legislation that would alter provisions of the Affordable Care Act and fundamentally change how Medicaid is funded.
The bill, introduced by Sen. Lindsey Graham (R-S.C.), Sen. Bill Cassidy (R-La.), Sen. Dean Heller (R-Nev.), and Sen. Ron Johnson (R-Wis.), features a number of provisions long sought by the GOP, including the repeal of the individual and employer mandates, repeal of individual tax credits as of 2020, and repeal of the medical device tax. The bill also would promote the use of health savings accounts and turn Medicaid funding into a block grant program, allowing states to implement policies such as work requirements.
James L. Madara, MD, CEO of the American Medical Association, told congressional leaders in a Sept. 19 letter that the bill would violate the precept of “first do no harm” and results in millions of Americans losing their health coverage. Additionally, it would destabilize health insurance markets and decrease access to affordable coverage.
“We are also concerned that the proposal would convert the Medicaid program into a system that limits federal support to care for needy patients to an insufficient predetermined formula based on per capita caps,” Dr. Madara continued. “Per capita caps fail to take into account unanticipated costs of new medical innovations or the fiscal impact of public health epidemics, such as the crisis of opioid abuse currently ravaging our nation. In addition, the amendment does not take steps toward coverage and access for all Americans, and while insurers are still required to offer coverage to patients with preexisting conditions, allowing states to get waivers to vary premiums based on health status would allow insurers to charge unaffordable premiums based on those preexisting conditions. Also, waivers of essential health benefits will mean patients may not have access to coverage for services pertinent to treating their conditions.”
The American Congress of Obstetricians and Gynecologists called the bill an “assault on women’s health.” The bill would end guaranteed insurance coverage of maternity care and women’s health preventive services, including cancer screenings and contraception, ACOG president Haywood Brown, MD, said in a statement.
Dr. Brown added that the bill “jeopardizes access to care for women with high-risk and expensive pregnancies, such as those with Zika virus, opioid use disorder, and preeclampsia. It further obstructs safety net patients’ access to care by forbidding Planned Parenthood’s participation in the Medicaid program.”
AGA is also concerned that there are no guarantees that states have to provide essential benefits, patients that gained coverage through the ACA would lose that coverage, and most importantly, patients with pre-existing conditions have no guarantee that they will continue to receive affordable coverage.
Doctors aren’t the only ones objecting to the GOP legislation. America’s Health Insurance Plans president and CEO Marilyn Tavenner said in a Sept. 20 letter to Congress that the bill would further destabilize the individual health insurance market.
The bill’s road to passage is far from certain. Once again, the GOP is aiming to use the budget reconciliation process to pass this legislation, which means it needs only a simple majority to pass (a minimum of 50 votes with Vice President Mike Pence offering the tiebreaker if the bill cannot get 51 votes). But even getting to 50 votes is going to be a challenge as the last attempt to pass similar repeal and replace language failed when Sen. Susan Collins (R-Maine), Sen. Lisa Murkowski (R-Alaska), and Sen. John McCain (R-Ariz.) voted that package down. Given the similar features, Sen. Collins and Sen. Murkowski may still oppose the bill, while Sen. Rand Paul (R-Ky.) has been vocal about his displeasure with the bill and other GOP senators are getting pressure from their state governors to oppose the bill.
The Senate Finance Committee has scheduled a Sept. 25 hearing to consider the bill, but as of press time, no witnesses have been announced, and the bill likely will not follow the regular order of allowing for amendments by committee members prior to its introduction on the Senate floor later that week.
Based on current budget rules, the bill must be passed by Sept. 30 in order for the budget reconciliation process to be used and to allow for passage with a simple majority. If the Senate is able to pass the bill, House Speaker Paul Ryan (R-Wisc.) has said he will bring it up in the House. President Trump has indicated he will sign it into law if it reaches his desk.
Comments sought on VTE Guidelines
The American Society of Hematology (ASH) is seeking feedback from SVS members by Oct. 2 on its draft recommendations for ASH guidelines on VTE in the context of pregnancy and Heparin-Induced Thrombocytopenia.
The guidelines have been posted for comment. Members can review the comment page here and download the draft recommendations. The page includes a link to the online survey where members can provide their comments.
The American Society of Hematology (ASH) is seeking feedback from SVS members by Oct. 2 on its draft recommendations for ASH guidelines on VTE in the context of pregnancy and Heparin-Induced Thrombocytopenia.
The guidelines have been posted for comment. Members can review the comment page here and download the draft recommendations. The page includes a link to the online survey where members can provide their comments.
The American Society of Hematology (ASH) is seeking feedback from SVS members by Oct. 2 on its draft recommendations for ASH guidelines on VTE in the context of pregnancy and Heparin-Induced Thrombocytopenia.
The guidelines have been posted for comment. Members can review the comment page here and download the draft recommendations. The page includes a link to the online survey where members can provide their comments.
How is MACRA Data Gathering Going? Final 2017 90-day Reporting Period Begins Oct. 2
Payment adjustments for Medicare reimbursement come in 2019 -- but the adjustments are based on data being collected now, in 2017. If you have not begun collecting data as yet, you MUST begin no later than Oct. 2, 2017!
You will be required to send in your performance data no later than March 31, 2018. If your practice is participating in a CMS-approved Alternative Payment Model (APM), MIPS participation is not required. For 2017 there is currently no APM specific to vascular surgery. The SVS has formed a Task Force to develop a vascular APM.
The first payment adjustments based on performance go into effect on Jan. 1, 2019. Those members who do not participate in 2017 will receive a 4 percent penalty from Medicare.
SVS has held four webinars recently helping members learn what they need to know for the changes. View the materials here.
Payment adjustments for Medicare reimbursement come in 2019 -- but the adjustments are based on data being collected now, in 2017. If you have not begun collecting data as yet, you MUST begin no later than Oct. 2, 2017!
You will be required to send in your performance data no later than March 31, 2018. If your practice is participating in a CMS-approved Alternative Payment Model (APM), MIPS participation is not required. For 2017 there is currently no APM specific to vascular surgery. The SVS has formed a Task Force to develop a vascular APM.
The first payment adjustments based on performance go into effect on Jan. 1, 2019. Those members who do not participate in 2017 will receive a 4 percent penalty from Medicare.
SVS has held four webinars recently helping members learn what they need to know for the changes. View the materials here.
Payment adjustments for Medicare reimbursement come in 2019 -- but the adjustments are based on data being collected now, in 2017. If you have not begun collecting data as yet, you MUST begin no later than Oct. 2, 2017!
You will be required to send in your performance data no later than March 31, 2018. If your practice is participating in a CMS-approved Alternative Payment Model (APM), MIPS participation is not required. For 2017 there is currently no APM specific to vascular surgery. The SVS has formed a Task Force to develop a vascular APM.
The first payment adjustments based on performance go into effect on Jan. 1, 2019. Those members who do not participate in 2017 will receive a 4 percent penalty from Medicare.
SVS has held four webinars recently helping members learn what they need to know for the changes. View the materials here.