User login
-
Genetic Signatures May Predict CAR T Responders
“Our transcriptomic analysis of ZUMA-7 dataset identified novel gene expression signatures predictive of outcome with axi-cel,” the authors reported in research presented at the annual meeting of the American Association for Cancer Research earlier in April. “These gene expression signatures could support risk-stratification of LBCL patients.”
The results are from a subanalysis of the phase 3 ZUMA-7 trial in which patients with early relapsed or primary refractory LBCL were treated with axi-cel, administered as a one-time dose in the second-line setting.
Long-term results from the trial showed a 4-year overall survival of 54.6% with axi-cel versus 46.0% with the standard of care (P = .03), with a median rate of progression-free survival of 14.7 months with axi-cel versus 3.7 months in the standard-second-line treatment.
In the study, the authors noted that, “although the use of axi-cel resulted in long-term survival in more than half of treated patients, it is important to continue to strive to improve patient outcomes.”
Following up on that, senior author Simone Filosto, of Kite, a Gilead Company, of Santa Monica, California, and colleagues launched their analysis of the genetic profiles of those who did and did not have favorable responses, using data from the ZUMA-7 trial.
Using gene expression profiling with the IO-360 Nanostring gene expression panel of 769 genes, they evaluated pretreated LBCL tumor samples from 134 of the patients treated with axi-cel.
After multivariate adjustment, the results showed that those with a distinctive 6-transcript genetic expression signature, consisting of CD19, CD45RA, CCL22, KLRK1, SOX11, and SIGLEC5, had a significantly higher rate of event-free survival (hazard ratio [HR], 0.27; P = 1.82 x 10-8), as well as progression-free survival (HR, 0.27; P = 1.35 x 10-7) after treatment with axi-cel, compared with those who did not have the signature.
The authors speculated that “the 6-gene expression signature may capture lymphomas with abundant adhesion molecules, a relatively low inflammation, and abundant expression of the targeted antigen (CD19).”
Conversely, the analysis showed that increased levels of an unfavorable 17-transcript gene expression signature had a strong negative correlation with event-free survival (HR, 6.19; P = 1.51 x 10-13) and progression-free survival (HR, 7.58; P = 2.70 x 10-14).
The 17-transcript signature included CD45RO, BCL2, IL-18R1, TNFSF4 [OX40L], KLRB1 [CD161], KIR3DL2, ITGB8, DUSP5, GPC4, PSMB5, RPS6KB1, SERPINA9, NBN, GLUD1, ESR1, ARID1A, and SLC16A1.
“The 17-gene expression signature is consistent with a high level of immune infiltration and inflammation paralleled by the activation of immune-escape mechanisms, such as the upregulation of anti-apoptotic genes,” the authors explain.
Of note, the 17-gene expression signature was elevated among 18 patients who progressed after axi-cel treatment.
Importantly, the gene expression signatures were not associated with outcomes observed among patients receiving second-line standard of care in the ZUMA-7 trial. And the signatures also did not correspond with outcomes following first-line R-CHOP chemotherapy reported in two online datasets, indicating their predictive rather than prognostic value.
Commenting on the findings, Marco Ruella, MD, noted that “stratifying the [CAR T-treated] patients is extremely important given that only a subset of them, 30%-40%, will experience long-term remission.”
“In an ideal scenario, we would want to treat only the patients who would benefit from such a complex and expensive therapy,” underscored Dr. Ruella, assistant professor in the Division of Hematology/Oncology and the Center for Cellular Immunotherapies and Scientific Director of the Lymphoma Program at the Hospital of the University of Pennsylvania in Philadelphia.
A key caveat is that the results need more validation before they true gain clinical value, he noted.
“We need more data before we can use such a score in the clinic as we would need to be absolutely confident on the predictive value of such a score in additional confirmatory cohorts.”
Furthermore, caution is warranted in avoiding excluding any patients unnecessarily, he added.
“Only if there are approximately zero chances of response would we be able to exclude a patient from a treatment,” Dr. Ruella noted. “If the chance of long-term cure are minimal but still present, it might still make sense for the patient.”
Nevertheless, such findings advance the understanding of the therapy’s implication in a meaningful way, he said.
“I think this study [and similar others] are important studies that help us better understand the mechanisms of relapse,” he said.
“Translationally, we are getting closer to reaching a point where we can precisely predict outcomes and, perhaps in the future, select the patients that would benefit the most from these treatments.”
Dr. Filosto and other authors are employees of Kite, which manufactures axi-cel. Dr. Ruella treats patients with CAR T products that have been licensed to Novartis, Kite, and Vittoria Bio.
“Our transcriptomic analysis of ZUMA-7 dataset identified novel gene expression signatures predictive of outcome with axi-cel,” the authors reported in research presented at the annual meeting of the American Association for Cancer Research earlier in April. “These gene expression signatures could support risk-stratification of LBCL patients.”
The results are from a subanalysis of the phase 3 ZUMA-7 trial in which patients with early relapsed or primary refractory LBCL were treated with axi-cel, administered as a one-time dose in the second-line setting.
Long-term results from the trial showed a 4-year overall survival of 54.6% with axi-cel versus 46.0% with the standard of care (P = .03), with a median rate of progression-free survival of 14.7 months with axi-cel versus 3.7 months in the standard-second-line treatment.
In the study, the authors noted that, “although the use of axi-cel resulted in long-term survival in more than half of treated patients, it is important to continue to strive to improve patient outcomes.”
Following up on that, senior author Simone Filosto, of Kite, a Gilead Company, of Santa Monica, California, and colleagues launched their analysis of the genetic profiles of those who did and did not have favorable responses, using data from the ZUMA-7 trial.
Using gene expression profiling with the IO-360 Nanostring gene expression panel of 769 genes, they evaluated pretreated LBCL tumor samples from 134 of the patients treated with axi-cel.
After multivariate adjustment, the results showed that those with a distinctive 6-transcript genetic expression signature, consisting of CD19, CD45RA, CCL22, KLRK1, SOX11, and SIGLEC5, had a significantly higher rate of event-free survival (hazard ratio [HR], 0.27; P = 1.82 x 10-8), as well as progression-free survival (HR, 0.27; P = 1.35 x 10-7) after treatment with axi-cel, compared with those who did not have the signature.
The authors speculated that “the 6-gene expression signature may capture lymphomas with abundant adhesion molecules, a relatively low inflammation, and abundant expression of the targeted antigen (CD19).”
Conversely, the analysis showed that increased levels of an unfavorable 17-transcript gene expression signature had a strong negative correlation with event-free survival (HR, 6.19; P = 1.51 x 10-13) and progression-free survival (HR, 7.58; P = 2.70 x 10-14).
The 17-transcript signature included CD45RO, BCL2, IL-18R1, TNFSF4 [OX40L], KLRB1 [CD161], KIR3DL2, ITGB8, DUSP5, GPC4, PSMB5, RPS6KB1, SERPINA9, NBN, GLUD1, ESR1, ARID1A, and SLC16A1.
“The 17-gene expression signature is consistent with a high level of immune infiltration and inflammation paralleled by the activation of immune-escape mechanisms, such as the upregulation of anti-apoptotic genes,” the authors explain.
Of note, the 17-gene expression signature was elevated among 18 patients who progressed after axi-cel treatment.
Importantly, the gene expression signatures were not associated with outcomes observed among patients receiving second-line standard of care in the ZUMA-7 trial. And the signatures also did not correspond with outcomes following first-line R-CHOP chemotherapy reported in two online datasets, indicating their predictive rather than prognostic value.
Commenting on the findings, Marco Ruella, MD, noted that “stratifying the [CAR T-treated] patients is extremely important given that only a subset of them, 30%-40%, will experience long-term remission.”
“In an ideal scenario, we would want to treat only the patients who would benefit from such a complex and expensive therapy,” underscored Dr. Ruella, assistant professor in the Division of Hematology/Oncology and the Center for Cellular Immunotherapies and Scientific Director of the Lymphoma Program at the Hospital of the University of Pennsylvania in Philadelphia.
A key caveat is that the results need more validation before they true gain clinical value, he noted.
“We need more data before we can use such a score in the clinic as we would need to be absolutely confident on the predictive value of such a score in additional confirmatory cohorts.”
Furthermore, caution is warranted in avoiding excluding any patients unnecessarily, he added.
“Only if there are approximately zero chances of response would we be able to exclude a patient from a treatment,” Dr. Ruella noted. “If the chance of long-term cure are minimal but still present, it might still make sense for the patient.”
Nevertheless, such findings advance the understanding of the therapy’s implication in a meaningful way, he said.
“I think this study [and similar others] are important studies that help us better understand the mechanisms of relapse,” he said.
“Translationally, we are getting closer to reaching a point where we can precisely predict outcomes and, perhaps in the future, select the patients that would benefit the most from these treatments.”
Dr. Filosto and other authors are employees of Kite, which manufactures axi-cel. Dr. Ruella treats patients with CAR T products that have been licensed to Novartis, Kite, and Vittoria Bio.
“Our transcriptomic analysis of ZUMA-7 dataset identified novel gene expression signatures predictive of outcome with axi-cel,” the authors reported in research presented at the annual meeting of the American Association for Cancer Research earlier in April. “These gene expression signatures could support risk-stratification of LBCL patients.”
The results are from a subanalysis of the phase 3 ZUMA-7 trial in which patients with early relapsed or primary refractory LBCL were treated with axi-cel, administered as a one-time dose in the second-line setting.
Long-term results from the trial showed a 4-year overall survival of 54.6% with axi-cel versus 46.0% with the standard of care (P = .03), with a median rate of progression-free survival of 14.7 months with axi-cel versus 3.7 months in the standard-second-line treatment.
In the study, the authors noted that, “although the use of axi-cel resulted in long-term survival in more than half of treated patients, it is important to continue to strive to improve patient outcomes.”
Following up on that, senior author Simone Filosto, of Kite, a Gilead Company, of Santa Monica, California, and colleagues launched their analysis of the genetic profiles of those who did and did not have favorable responses, using data from the ZUMA-7 trial.
Using gene expression profiling with the IO-360 Nanostring gene expression panel of 769 genes, they evaluated pretreated LBCL tumor samples from 134 of the patients treated with axi-cel.
After multivariate adjustment, the results showed that those with a distinctive 6-transcript genetic expression signature, consisting of CD19, CD45RA, CCL22, KLRK1, SOX11, and SIGLEC5, had a significantly higher rate of event-free survival (hazard ratio [HR], 0.27; P = 1.82 x 10-8), as well as progression-free survival (HR, 0.27; P = 1.35 x 10-7) after treatment with axi-cel, compared with those who did not have the signature.
The authors speculated that “the 6-gene expression signature may capture lymphomas with abundant adhesion molecules, a relatively low inflammation, and abundant expression of the targeted antigen (CD19).”
Conversely, the analysis showed that increased levels of an unfavorable 17-transcript gene expression signature had a strong negative correlation with event-free survival (HR, 6.19; P = 1.51 x 10-13) and progression-free survival (HR, 7.58; P = 2.70 x 10-14).
The 17-transcript signature included CD45RO, BCL2, IL-18R1, TNFSF4 [OX40L], KLRB1 [CD161], KIR3DL2, ITGB8, DUSP5, GPC4, PSMB5, RPS6KB1, SERPINA9, NBN, GLUD1, ESR1, ARID1A, and SLC16A1.
“The 17-gene expression signature is consistent with a high level of immune infiltration and inflammation paralleled by the activation of immune-escape mechanisms, such as the upregulation of anti-apoptotic genes,” the authors explain.
Of note, the 17-gene expression signature was elevated among 18 patients who progressed after axi-cel treatment.
Importantly, the gene expression signatures were not associated with outcomes observed among patients receiving second-line standard of care in the ZUMA-7 trial. And the signatures also did not correspond with outcomes following first-line R-CHOP chemotherapy reported in two online datasets, indicating their predictive rather than prognostic value.
Commenting on the findings, Marco Ruella, MD, noted that “stratifying the [CAR T-treated] patients is extremely important given that only a subset of them, 30%-40%, will experience long-term remission.”
“In an ideal scenario, we would want to treat only the patients who would benefit from such a complex and expensive therapy,” underscored Dr. Ruella, assistant professor in the Division of Hematology/Oncology and the Center for Cellular Immunotherapies and Scientific Director of the Lymphoma Program at the Hospital of the University of Pennsylvania in Philadelphia.
A key caveat is that the results need more validation before they true gain clinical value, he noted.
“We need more data before we can use such a score in the clinic as we would need to be absolutely confident on the predictive value of such a score in additional confirmatory cohorts.”
Furthermore, caution is warranted in avoiding excluding any patients unnecessarily, he added.
“Only if there are approximately zero chances of response would we be able to exclude a patient from a treatment,” Dr. Ruella noted. “If the chance of long-term cure are minimal but still present, it might still make sense for the patient.”
Nevertheless, such findings advance the understanding of the therapy’s implication in a meaningful way, he said.
“I think this study [and similar others] are important studies that help us better understand the mechanisms of relapse,” he said.
“Translationally, we are getting closer to reaching a point where we can precisely predict outcomes and, perhaps in the future, select the patients that would benefit the most from these treatments.”
Dr. Filosto and other authors are employees of Kite, which manufactures axi-cel. Dr. Ruella treats patients with CAR T products that have been licensed to Novartis, Kite, and Vittoria Bio.
FROM AACR 2024
Federal Trade Commission Bans Noncompete Agreements, Urges More Protections for Healthcare Workers
But business groups have vowed to challenge the decision in court.
The proposed final rule passed on a 3-2 vote, with the dissenting commissioners disputing the FTC’s authority to broadly ban noncompetes.
Tensions around noncompetes have been building for years. In 2021, President Biden issued an executive order supporting measures to improve economic competition, in which he urged the FTC to consider its rulemaking authority to address noncompete clauses that unfairly limit workers’ mobility. In January 2023, per that directive, the agency proposed ending the restrictive covenants.
While the FTC estimates that the final rule will reduce healthcare costs by up to $194 billion over the next decade and increase worker earnings by $300 million annually, the ruling faces legal hurdles.
US Chamber of Commerce president and CEO Suzanne P. Clark said in a statement that the move is a “blatant power grab” that will undermine competitive business practices, adding that the Chamber will sue to block the measure.
The FTC received more than 26,000 comments on noncompetes during the public feedback period, with about 25,000 supporting the measure, said Benjamin Cady, JD, an FTC attorney.
Mr. Cady called the feedback “compelling,” citing instances of workers who were forced to commute long distances, uproot their families, or risk expensive litigation for wanting to pursue job opportunities.
For example, a comment from a physician working in Appalachia highlights the potential real-life implications of the agreements. “With hospital systems merging, providers with aggressive noncompetes must abandon the community that they serve if they [choose] to leave their employer. Healthcare providers feel trapped in their current employment situation, leading to significant burnout that can shorten their [career] longevity.”
Commissioner Alvaro Bedoya said physicians have had their lives upended by cumbersome noncompetes, often having to move out of state to practice. “A pandemic killed a million people in this country, and there are doctors who cannot work because of a noncompete,” he said.
It’s unclear whether physicians and others who work for nonprofit healthcare groups or hospitals will be covered by the new ban. FTC Commissioner Rebecca Slaughter acknowledged that the agency’s jurisdictional limitations mean that employees of “certain nonprofit organizations” may not benefit from the rule.
“We want to be transparent about the limitation and recognize there are workers, especially healthcare workers, who are bound by anticompetitive and unfair noncompete clauses, that our rule will struggle to reach,” she said. To cover nonprofit healthcare employees, Ms. Slaughter urged Congress to pass legislation banning noncompetes, such as the Workforce Mobility Act of 2021 and the Freedom to Compete Act of 2023.
The FTC final rule will take effect 120 days after it is published in the federal register, and new noncompete agreements will be banned as of this date. However, existing contracts for senior executives will remain in effect because these individuals are less likely to experience “acute harm” due to their ability to negotiate accordingly, said Mr. Cady.
States, AMA Take Aim at Noncompetes
Before the federal ban, several states had already passed legislation limiting the reach of noncompetes. According to a recent article in the Journal of the American College of Cardiology, 12 states prohibit noncompete clauses for physicians: Alabama, California, Colorado, Delaware, Massachusetts, Montana, New Hampshire, New Mexico, North Dakota, Oklahoma, Rhode Island, and South Dakota.
The remaining states allow noncompetes in some form, often excluding them for employees earning below a certain threshold. For example, in Oregon, noncompete agreements may apply to employees earning more than $113,241. Most states have provisions to adjust the threshold annually. The District of Columbia permits 2-year noncompetes for “medical specialists” earning over $250,000 annually.
Indiana employers can no longer enter into noncompete agreements with primary care providers. Other specialties may be subject to the clauses, except when the physician terminates the contract for cause or when an employer terminates the contract without cause.
Rachel Marcus, MD, a cardiologist in Washington, DC, found out how limiting her employment contract’s noncompete clause was when she wanted to leave a former position. Due to the restrictions, she told this news organization that she couldn’t work locally for a competitor for 2 years. The closest location she could seek employment without violating the agreement was Baltimore, approximately 40 miles away.
Dr. Marcus ultimately moved to another position within the same organization because of the company’s reputation for being “aggressive” in their enforcement actions.
Although the American Medical Association (AMA) does not support a total ban, its House of Delegates adopted policies last year to support the prohibition of noncompete contracts for physicians employed by for-profit or nonprofit hospitals, hospital systems, or staffing companies.
Challenges Await
The American Hospital Association, which opposed the proposed rule, called it “bad policy.” The decision “will likely be short-lived, with courts almost certain to stop it before it can do damage to hospitals’ ability to care for their patients and communities,” the association said in a statement.
To ease the transition to the new rule, the FTC also released a model language for employers to use when discussing the changes with their employees. “All employers need to do to comply with the rule is to stop enforcing existing noncompetes with workers other than senior executives and provide notice to such workers,” he said.
Dr. Marcus hopes the ban improves doctors’ lives. “Your employer is going to have to treat you better because they know that you can easily go across town to a place that has a higher salary, and your patient can go with you.”
A version of this article appeared on Medscape.com.
But business groups have vowed to challenge the decision in court.
The proposed final rule passed on a 3-2 vote, with the dissenting commissioners disputing the FTC’s authority to broadly ban noncompetes.
Tensions around noncompetes have been building for years. In 2021, President Biden issued an executive order supporting measures to improve economic competition, in which he urged the FTC to consider its rulemaking authority to address noncompete clauses that unfairly limit workers’ mobility. In January 2023, per that directive, the agency proposed ending the restrictive covenants.
While the FTC estimates that the final rule will reduce healthcare costs by up to $194 billion over the next decade and increase worker earnings by $300 million annually, the ruling faces legal hurdles.
US Chamber of Commerce president and CEO Suzanne P. Clark said in a statement that the move is a “blatant power grab” that will undermine competitive business practices, adding that the Chamber will sue to block the measure.
The FTC received more than 26,000 comments on noncompetes during the public feedback period, with about 25,000 supporting the measure, said Benjamin Cady, JD, an FTC attorney.
Mr. Cady called the feedback “compelling,” citing instances of workers who were forced to commute long distances, uproot their families, or risk expensive litigation for wanting to pursue job opportunities.
For example, a comment from a physician working in Appalachia highlights the potential real-life implications of the agreements. “With hospital systems merging, providers with aggressive noncompetes must abandon the community that they serve if they [choose] to leave their employer. Healthcare providers feel trapped in their current employment situation, leading to significant burnout that can shorten their [career] longevity.”
Commissioner Alvaro Bedoya said physicians have had their lives upended by cumbersome noncompetes, often having to move out of state to practice. “A pandemic killed a million people in this country, and there are doctors who cannot work because of a noncompete,” he said.
It’s unclear whether physicians and others who work for nonprofit healthcare groups or hospitals will be covered by the new ban. FTC Commissioner Rebecca Slaughter acknowledged that the agency’s jurisdictional limitations mean that employees of “certain nonprofit organizations” may not benefit from the rule.
“We want to be transparent about the limitation and recognize there are workers, especially healthcare workers, who are bound by anticompetitive and unfair noncompete clauses, that our rule will struggle to reach,” she said. To cover nonprofit healthcare employees, Ms. Slaughter urged Congress to pass legislation banning noncompetes, such as the Workforce Mobility Act of 2021 and the Freedom to Compete Act of 2023.
The FTC final rule will take effect 120 days after it is published in the federal register, and new noncompete agreements will be banned as of this date. However, existing contracts for senior executives will remain in effect because these individuals are less likely to experience “acute harm” due to their ability to negotiate accordingly, said Mr. Cady.
States, AMA Take Aim at Noncompetes
Before the federal ban, several states had already passed legislation limiting the reach of noncompetes. According to a recent article in the Journal of the American College of Cardiology, 12 states prohibit noncompete clauses for physicians: Alabama, California, Colorado, Delaware, Massachusetts, Montana, New Hampshire, New Mexico, North Dakota, Oklahoma, Rhode Island, and South Dakota.
The remaining states allow noncompetes in some form, often excluding them for employees earning below a certain threshold. For example, in Oregon, noncompete agreements may apply to employees earning more than $113,241. Most states have provisions to adjust the threshold annually. The District of Columbia permits 2-year noncompetes for “medical specialists” earning over $250,000 annually.
Indiana employers can no longer enter into noncompete agreements with primary care providers. Other specialties may be subject to the clauses, except when the physician terminates the contract for cause or when an employer terminates the contract without cause.
Rachel Marcus, MD, a cardiologist in Washington, DC, found out how limiting her employment contract’s noncompete clause was when she wanted to leave a former position. Due to the restrictions, she told this news organization that she couldn’t work locally for a competitor for 2 years. The closest location she could seek employment without violating the agreement was Baltimore, approximately 40 miles away.
Dr. Marcus ultimately moved to another position within the same organization because of the company’s reputation for being “aggressive” in their enforcement actions.
Although the American Medical Association (AMA) does not support a total ban, its House of Delegates adopted policies last year to support the prohibition of noncompete contracts for physicians employed by for-profit or nonprofit hospitals, hospital systems, or staffing companies.
Challenges Await
The American Hospital Association, which opposed the proposed rule, called it “bad policy.” The decision “will likely be short-lived, with courts almost certain to stop it before it can do damage to hospitals’ ability to care for their patients and communities,” the association said in a statement.
To ease the transition to the new rule, the FTC also released a model language for employers to use when discussing the changes with their employees. “All employers need to do to comply with the rule is to stop enforcing existing noncompetes with workers other than senior executives and provide notice to such workers,” he said.
Dr. Marcus hopes the ban improves doctors’ lives. “Your employer is going to have to treat you better because they know that you can easily go across town to a place that has a higher salary, and your patient can go with you.”
A version of this article appeared on Medscape.com.
But business groups have vowed to challenge the decision in court.
The proposed final rule passed on a 3-2 vote, with the dissenting commissioners disputing the FTC’s authority to broadly ban noncompetes.
Tensions around noncompetes have been building for years. In 2021, President Biden issued an executive order supporting measures to improve economic competition, in which he urged the FTC to consider its rulemaking authority to address noncompete clauses that unfairly limit workers’ mobility. In January 2023, per that directive, the agency proposed ending the restrictive covenants.
While the FTC estimates that the final rule will reduce healthcare costs by up to $194 billion over the next decade and increase worker earnings by $300 million annually, the ruling faces legal hurdles.
US Chamber of Commerce president and CEO Suzanne P. Clark said in a statement that the move is a “blatant power grab” that will undermine competitive business practices, adding that the Chamber will sue to block the measure.
The FTC received more than 26,000 comments on noncompetes during the public feedback period, with about 25,000 supporting the measure, said Benjamin Cady, JD, an FTC attorney.
Mr. Cady called the feedback “compelling,” citing instances of workers who were forced to commute long distances, uproot their families, or risk expensive litigation for wanting to pursue job opportunities.
For example, a comment from a physician working in Appalachia highlights the potential real-life implications of the agreements. “With hospital systems merging, providers with aggressive noncompetes must abandon the community that they serve if they [choose] to leave their employer. Healthcare providers feel trapped in their current employment situation, leading to significant burnout that can shorten their [career] longevity.”
Commissioner Alvaro Bedoya said physicians have had their lives upended by cumbersome noncompetes, often having to move out of state to practice. “A pandemic killed a million people in this country, and there are doctors who cannot work because of a noncompete,” he said.
It’s unclear whether physicians and others who work for nonprofit healthcare groups or hospitals will be covered by the new ban. FTC Commissioner Rebecca Slaughter acknowledged that the agency’s jurisdictional limitations mean that employees of “certain nonprofit organizations” may not benefit from the rule.
“We want to be transparent about the limitation and recognize there are workers, especially healthcare workers, who are bound by anticompetitive and unfair noncompete clauses, that our rule will struggle to reach,” she said. To cover nonprofit healthcare employees, Ms. Slaughter urged Congress to pass legislation banning noncompetes, such as the Workforce Mobility Act of 2021 and the Freedom to Compete Act of 2023.
The FTC final rule will take effect 120 days after it is published in the federal register, and new noncompete agreements will be banned as of this date. However, existing contracts for senior executives will remain in effect because these individuals are less likely to experience “acute harm” due to their ability to negotiate accordingly, said Mr. Cady.
States, AMA Take Aim at Noncompetes
Before the federal ban, several states had already passed legislation limiting the reach of noncompetes. According to a recent article in the Journal of the American College of Cardiology, 12 states prohibit noncompete clauses for physicians: Alabama, California, Colorado, Delaware, Massachusetts, Montana, New Hampshire, New Mexico, North Dakota, Oklahoma, Rhode Island, and South Dakota.
The remaining states allow noncompetes in some form, often excluding them for employees earning below a certain threshold. For example, in Oregon, noncompete agreements may apply to employees earning more than $113,241. Most states have provisions to adjust the threshold annually. The District of Columbia permits 2-year noncompetes for “medical specialists” earning over $250,000 annually.
Indiana employers can no longer enter into noncompete agreements with primary care providers. Other specialties may be subject to the clauses, except when the physician terminates the contract for cause or when an employer terminates the contract without cause.
Rachel Marcus, MD, a cardiologist in Washington, DC, found out how limiting her employment contract’s noncompete clause was when she wanted to leave a former position. Due to the restrictions, she told this news organization that she couldn’t work locally for a competitor for 2 years. The closest location she could seek employment without violating the agreement was Baltimore, approximately 40 miles away.
Dr. Marcus ultimately moved to another position within the same organization because of the company’s reputation for being “aggressive” in their enforcement actions.
Although the American Medical Association (AMA) does not support a total ban, its House of Delegates adopted policies last year to support the prohibition of noncompete contracts for physicians employed by for-profit or nonprofit hospitals, hospital systems, or staffing companies.
Challenges Await
The American Hospital Association, which opposed the proposed rule, called it “bad policy.” The decision “will likely be short-lived, with courts almost certain to stop it before it can do damage to hospitals’ ability to care for their patients and communities,” the association said in a statement.
To ease the transition to the new rule, the FTC also released a model language for employers to use when discussing the changes with their employees. “All employers need to do to comply with the rule is to stop enforcing existing noncompetes with workers other than senior executives and provide notice to such workers,” he said.
Dr. Marcus hopes the ban improves doctors’ lives. “Your employer is going to have to treat you better because they know that you can easily go across town to a place that has a higher salary, and your patient can go with you.”
A version of this article appeared on Medscape.com.
Are Women Better Doctors Than Men?
This transcript has been edited for clarity.
It’s a battle of the sexes today as we dive into a paper that makes you say, “Wow, what an interesting study” and also “Boy, am I glad I didn’t do that study.” That’s because studies like this are always somewhat fraught; they say something about medicine but also something about society — and that makes this a bit precarious. But that’s never stopped us before. So, let’s go ahead and try to answer the question: Do women make better doctors than men?
On the surface, this question seems nearly impossible to answer. It’s too broad; what does it mean to be a “better” doctor? At first blush it seems that there are just too many variables to control for here: the type of doctor, the type of patient, the clinical scenario, and so on.
But this study, “Comparison of hospital mortality and readmission rates by physician and patient sex,” which appears in Annals of Internal Medicine, uses a fairly ingenious method to cut through all the bias by leveraging two simple facts: First, hospital medicine is largely conducted by hospitalists these days; second, due to the shift-based nature of hospitalist work, the hospitalist you get when you are admitted to the hospital is pretty much random.
In other words, if you are admitted to the hospital for an acute illness and get a hospitalist as your attending, you have no control over whether it is a man or a woman. Is this a randomized trial? No, but it’s not bad.
Researchers used Medicare claims data to identify adults over age 65 who had nonelective hospital admissions throughout the United States. The claims revealed the sex of the patient and the name of the attending physician. By linking to a medical provider database, they could determine the sex of the provider.
The goal was to look at outcomes across four dyads:
- Male patient – male doctor
- Male patient – female doctor
- Female patient – male doctor
- Female patient – female doctor
The primary outcome was 30-day mortality.
I told you that focusing on hospitalists produces some pseudorandomization, but let’s look at the data to be sure. Just under a million patients were treated by approximately 50,000 physicians, 30% of whom were female. And, though female patients and male patients differed, they did not differ with respect to the sex of their hospitalist. So, by physician sex, patients were similar in mean age, race, ethnicity, household income, eligibility for Medicaid, and comorbid conditions. The authors even created a “predicted mortality” score which was similar across the groups as well.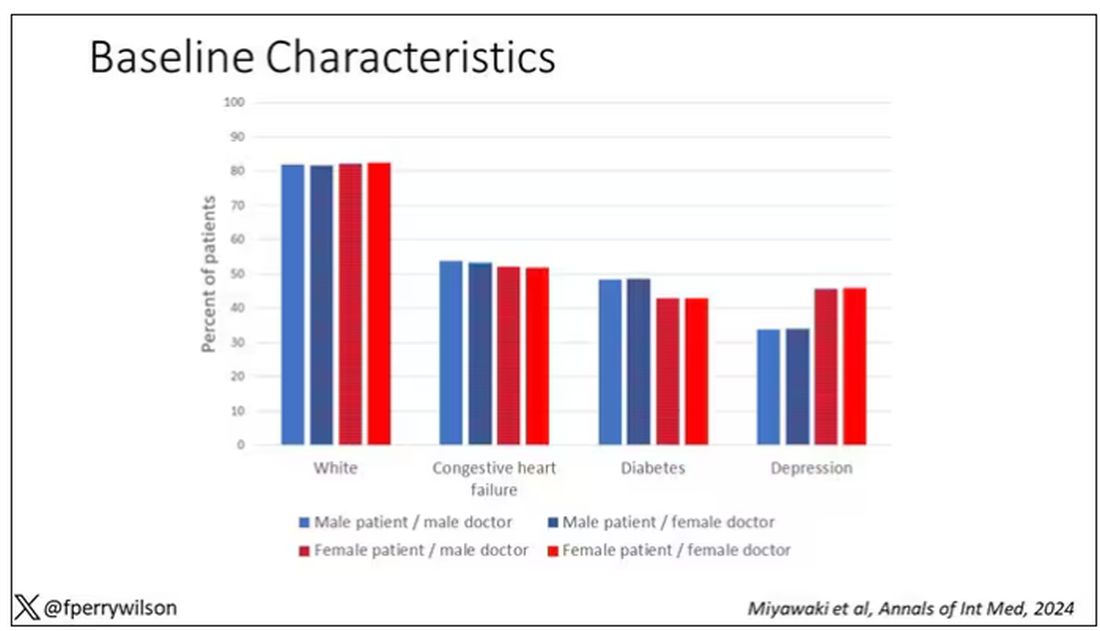
Now, the female physicians were a bit different from the male physicians. The female hospitalists were slightly more likely to have an osteopathic degree, had slightly fewer admissions per year, and were a bit younger.
So, we have broadly similar patients regardless of who their hospitalist was, but hospitalists differ by factors other than their sex. Fine.
I’ve graphed the results here. 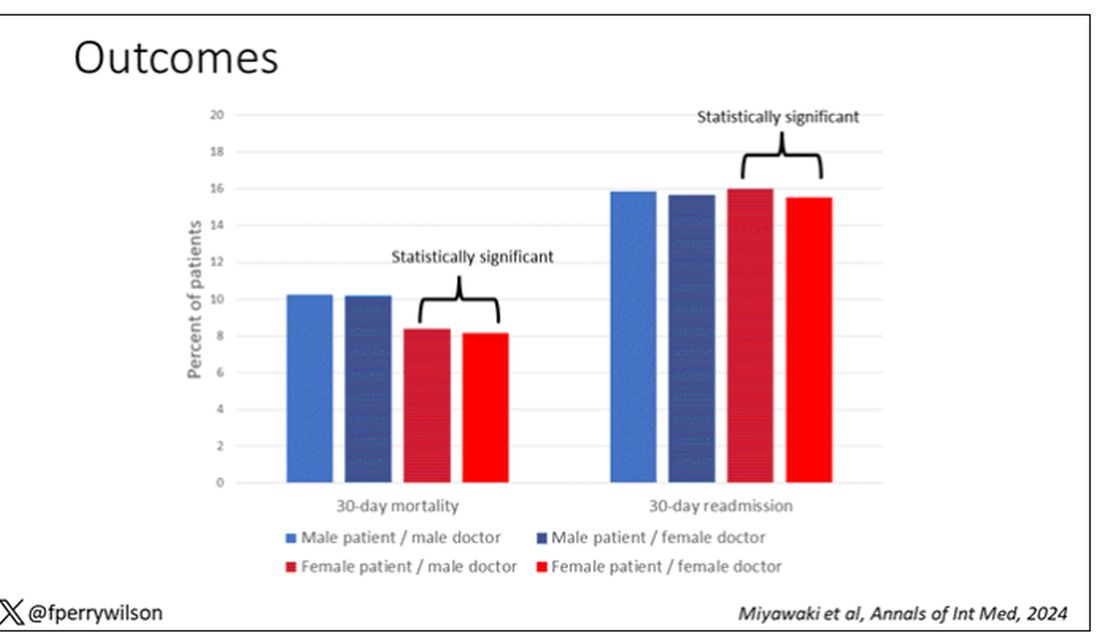
This is a relatively small effect, to be sure, but if you multiply it across the millions of hospitalist admissions per year, you can start to put up some real numbers.
So, what is going on here? I see four broad buckets of possibilities.
Let’s start with the obvious explanation: Women, on average, are better doctors than men. I am married to a woman doctor, and based on my personal experience, this explanation is undoubtedly true. But why would that be?
The authors cite data that suggest that female physicians are less likely than male physicians to dismiss patient concerns — and in particular, the concerns of female patients — perhaps leading to fewer missed diagnoses. But this is impossible to measure with administrative data, so this study can no more tell us whether these female hospitalists are more attentive than their male counterparts than it can suggest that the benefit is mediated by the shorter average height of female physicians. Perhaps the key is being closer to the patient?
The second possibility here is that this has nothing to do with the sex of the physician at all; it has to do with those other things that associate with the sex of the physician. We know, for example, that the female physicians saw fewer patients per year than the male physicians, but the study authors adjusted for this in the statistical models. Still, other unmeasured factors (confounders) could be present. By the way, confounders wouldn’t necessarily change the primary finding — you are better off being cared for by female physicians. It’s just not because they are female; it’s a convenient marker for some other quality, such as age.
The third possibility is that the study represents a phenomenon called collider bias. The idea here is that physicians only get into the study if they are hospitalists, and the quality of physicians who choose to become a hospitalist may differ by sex. When deciding on a specialty, a talented resident considering certain lifestyle issues may find hospital medicine particularly attractive — and that draw toward a more lifestyle-friendly specialty may differ by sex, as some prior studies have shown. If true, the pool of women hospitalists may be better than their male counterparts because male physicians of that caliber don’t become hospitalists.
Okay, don’t write in. I’m just trying to cite examples of how to think about collider bias. I can’t prove that this is the case, and in fact the authors do a sensitivity analysis of all physicians, not just hospitalists, and show the same thing. So this is probably not true, but epidemiology is fun, right?
And the fourth possibility: This is nothing but statistical noise. The effect size is incredibly small and just on the border of statistical significance. Especially when you’re working with very large datasets like this, you’ve got to be really careful about overinterpreting statistically significant findings that are nevertheless of small magnitude.
Regardless, it’s an interesting study, one that made me think and, of course, worry a bit about how I would present it. Forgive me if I’ve been indelicate in handling the complex issues of sex, gender, and society here. But I’m not sure what you expect; after all, I’m only a male doctor.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
It’s a battle of the sexes today as we dive into a paper that makes you say, “Wow, what an interesting study” and also “Boy, am I glad I didn’t do that study.” That’s because studies like this are always somewhat fraught; they say something about medicine but also something about society — and that makes this a bit precarious. But that’s never stopped us before. So, let’s go ahead and try to answer the question: Do women make better doctors than men?
On the surface, this question seems nearly impossible to answer. It’s too broad; what does it mean to be a “better” doctor? At first blush it seems that there are just too many variables to control for here: the type of doctor, the type of patient, the clinical scenario, and so on.
But this study, “Comparison of hospital mortality and readmission rates by physician and patient sex,” which appears in Annals of Internal Medicine, uses a fairly ingenious method to cut through all the bias by leveraging two simple facts: First, hospital medicine is largely conducted by hospitalists these days; second, due to the shift-based nature of hospitalist work, the hospitalist you get when you are admitted to the hospital is pretty much random.
In other words, if you are admitted to the hospital for an acute illness and get a hospitalist as your attending, you have no control over whether it is a man or a woman. Is this a randomized trial? No, but it’s not bad.
Researchers used Medicare claims data to identify adults over age 65 who had nonelective hospital admissions throughout the United States. The claims revealed the sex of the patient and the name of the attending physician. By linking to a medical provider database, they could determine the sex of the provider.
The goal was to look at outcomes across four dyads:
- Male patient – male doctor
- Male patient – female doctor
- Female patient – male doctor
- Female patient – female doctor
The primary outcome was 30-day mortality.
I told you that focusing on hospitalists produces some pseudorandomization, but let’s look at the data to be sure. Just under a million patients were treated by approximately 50,000 physicians, 30% of whom were female. And, though female patients and male patients differed, they did not differ with respect to the sex of their hospitalist. So, by physician sex, patients were similar in mean age, race, ethnicity, household income, eligibility for Medicaid, and comorbid conditions. The authors even created a “predicted mortality” score which was similar across the groups as well.
Now, the female physicians were a bit different from the male physicians. The female hospitalists were slightly more likely to have an osteopathic degree, had slightly fewer admissions per year, and were a bit younger.
So, we have broadly similar patients regardless of who their hospitalist was, but hospitalists differ by factors other than their sex. Fine.
I’ve graphed the results here.
This is a relatively small effect, to be sure, but if you multiply it across the millions of hospitalist admissions per year, you can start to put up some real numbers.
So, what is going on here? I see four broad buckets of possibilities.
Let’s start with the obvious explanation: Women, on average, are better doctors than men. I am married to a woman doctor, and based on my personal experience, this explanation is undoubtedly true. But why would that be?
The authors cite data that suggest that female physicians are less likely than male physicians to dismiss patient concerns — and in particular, the concerns of female patients — perhaps leading to fewer missed diagnoses. But this is impossible to measure with administrative data, so this study can no more tell us whether these female hospitalists are more attentive than their male counterparts than it can suggest that the benefit is mediated by the shorter average height of female physicians. Perhaps the key is being closer to the patient?
The second possibility here is that this has nothing to do with the sex of the physician at all; it has to do with those other things that associate with the sex of the physician. We know, for example, that the female physicians saw fewer patients per year than the male physicians, but the study authors adjusted for this in the statistical models. Still, other unmeasured factors (confounders) could be present. By the way, confounders wouldn’t necessarily change the primary finding — you are better off being cared for by female physicians. It’s just not because they are female; it’s a convenient marker for some other quality, such as age.
The third possibility is that the study represents a phenomenon called collider bias. The idea here is that physicians only get into the study if they are hospitalists, and the quality of physicians who choose to become a hospitalist may differ by sex. When deciding on a specialty, a talented resident considering certain lifestyle issues may find hospital medicine particularly attractive — and that draw toward a more lifestyle-friendly specialty may differ by sex, as some prior studies have shown. If true, the pool of women hospitalists may be better than their male counterparts because male physicians of that caliber don’t become hospitalists.
Okay, don’t write in. I’m just trying to cite examples of how to think about collider bias. I can’t prove that this is the case, and in fact the authors do a sensitivity analysis of all physicians, not just hospitalists, and show the same thing. So this is probably not true, but epidemiology is fun, right?
And the fourth possibility: This is nothing but statistical noise. The effect size is incredibly small and just on the border of statistical significance. Especially when you’re working with very large datasets like this, you’ve got to be really careful about overinterpreting statistically significant findings that are nevertheless of small magnitude.
Regardless, it’s an interesting study, one that made me think and, of course, worry a bit about how I would present it. Forgive me if I’ve been indelicate in handling the complex issues of sex, gender, and society here. But I’m not sure what you expect; after all, I’m only a male doctor.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
It’s a battle of the sexes today as we dive into a paper that makes you say, “Wow, what an interesting study” and also “Boy, am I glad I didn’t do that study.” That’s because studies like this are always somewhat fraught; they say something about medicine but also something about society — and that makes this a bit precarious. But that’s never stopped us before. So, let’s go ahead and try to answer the question: Do women make better doctors than men?
On the surface, this question seems nearly impossible to answer. It’s too broad; what does it mean to be a “better” doctor? At first blush it seems that there are just too many variables to control for here: the type of doctor, the type of patient, the clinical scenario, and so on.
But this study, “Comparison of hospital mortality and readmission rates by physician and patient sex,” which appears in Annals of Internal Medicine, uses a fairly ingenious method to cut through all the bias by leveraging two simple facts: First, hospital medicine is largely conducted by hospitalists these days; second, due to the shift-based nature of hospitalist work, the hospitalist you get when you are admitted to the hospital is pretty much random.
In other words, if you are admitted to the hospital for an acute illness and get a hospitalist as your attending, you have no control over whether it is a man or a woman. Is this a randomized trial? No, but it’s not bad.
Researchers used Medicare claims data to identify adults over age 65 who had nonelective hospital admissions throughout the United States. The claims revealed the sex of the patient and the name of the attending physician. By linking to a medical provider database, they could determine the sex of the provider.
The goal was to look at outcomes across four dyads:
- Male patient – male doctor
- Male patient – female doctor
- Female patient – male doctor
- Female patient – female doctor
The primary outcome was 30-day mortality.
I told you that focusing on hospitalists produces some pseudorandomization, but let’s look at the data to be sure. Just under a million patients were treated by approximately 50,000 physicians, 30% of whom were female. And, though female patients and male patients differed, they did not differ with respect to the sex of their hospitalist. So, by physician sex, patients were similar in mean age, race, ethnicity, household income, eligibility for Medicaid, and comorbid conditions. The authors even created a “predicted mortality” score which was similar across the groups as well.
Now, the female physicians were a bit different from the male physicians. The female hospitalists were slightly more likely to have an osteopathic degree, had slightly fewer admissions per year, and were a bit younger.
So, we have broadly similar patients regardless of who their hospitalist was, but hospitalists differ by factors other than their sex. Fine.
I’ve graphed the results here.
This is a relatively small effect, to be sure, but if you multiply it across the millions of hospitalist admissions per year, you can start to put up some real numbers.
So, what is going on here? I see four broad buckets of possibilities.
Let’s start with the obvious explanation: Women, on average, are better doctors than men. I am married to a woman doctor, and based on my personal experience, this explanation is undoubtedly true. But why would that be?
The authors cite data that suggest that female physicians are less likely than male physicians to dismiss patient concerns — and in particular, the concerns of female patients — perhaps leading to fewer missed diagnoses. But this is impossible to measure with administrative data, so this study can no more tell us whether these female hospitalists are more attentive than their male counterparts than it can suggest that the benefit is mediated by the shorter average height of female physicians. Perhaps the key is being closer to the patient?
The second possibility here is that this has nothing to do with the sex of the physician at all; it has to do with those other things that associate with the sex of the physician. We know, for example, that the female physicians saw fewer patients per year than the male physicians, but the study authors adjusted for this in the statistical models. Still, other unmeasured factors (confounders) could be present. By the way, confounders wouldn’t necessarily change the primary finding — you are better off being cared for by female physicians. It’s just not because they are female; it’s a convenient marker for some other quality, such as age.
The third possibility is that the study represents a phenomenon called collider bias. The idea here is that physicians only get into the study if they are hospitalists, and the quality of physicians who choose to become a hospitalist may differ by sex. When deciding on a specialty, a talented resident considering certain lifestyle issues may find hospital medicine particularly attractive — and that draw toward a more lifestyle-friendly specialty may differ by sex, as some prior studies have shown. If true, the pool of women hospitalists may be better than their male counterparts because male physicians of that caliber don’t become hospitalists.
Okay, don’t write in. I’m just trying to cite examples of how to think about collider bias. I can’t prove that this is the case, and in fact the authors do a sensitivity analysis of all physicians, not just hospitalists, and show the same thing. So this is probably not true, but epidemiology is fun, right?
And the fourth possibility: This is nothing but statistical noise. The effect size is incredibly small and just on the border of statistical significance. Especially when you’re working with very large datasets like this, you’ve got to be really careful about overinterpreting statistically significant findings that are nevertheless of small magnitude.
Regardless, it’s an interesting study, one that made me think and, of course, worry a bit about how I would present it. Forgive me if I’ve been indelicate in handling the complex issues of sex, gender, and society here. But I’m not sure what you expect; after all, I’m only a male doctor.
Dr. Wilson is associate professor of medicine and public health and director of the Clinical and Translational Research Accelerator at Yale University, New Haven, Conn. He has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Tiny Doses of Metabolically Armed CAR T Show Benefits
“Our study showed a manageable safety profile in r/r DLBCL/B-ALL, with promising breakthrough efficacy of a 100% complete remission in all dose groups,” said first author Jingjing Ren, MD, PhD, associate director of research and development with Leman Biotech in Shenzhen, China. Dr. Ren presented these findings at the American Association for Cancer Research annual meeting held in San Diego.
While CD19 CAR T-cell therapy has been transformative in the treatment of relapsed B -cell hematological malignancies in recent years, more than half of patients relapse within a year because of inadequate CAR T persistence.
To address the problem, Dr. Ren and her colleagues developed a metabolically armed, interleukin (IL)-10-expressing CAR T-cell product called Meta10-19 for the treatment patients with r/r DLBCL or r/r B-ALL.
According to the authors, the IL-10-expressing CAR T-cells trigger “stem-like memory responses” in various lymphoid organs, which prompt a “robust tumor eradication and durable protection,” and hence, better persistence.
Preclinical studies in mice showed the Meta10-19 CAR T-cells exhibited substantially higher expansion of approximately 100-fold compared with a control CD19 CAR-T product.
Therefore, “we dramatically reduced the dose to approximately 1% to 5% of commercial products for the IL-10-expressing CD19 CAR-T for patients,” coauthor Yugang Guo, PhD, cofounder and president of Leman Biotech said in an interview.
For the ongoing, open-label clinical trial, 12 adult patients with r/r DLBCL or r/r B-ALL and confirmed CD19 expression at a hospital center in China were enrolled between December 2022 and November 2023 and treated in three cohorts, receiving doses that corresponded to 1%, 2.5%, or 5% of the doses of other commercialized CAR-T infusion products.
All patients also underwent lympho-depleting chemotherapy with cyclophosphamide and fludarabine prior to the CAR T-cell infusion.
Six patients had r/r DLBCL and the other six had r/r B-ALL; their median age was 47 and their median time since diagnosis was 1 year.
In the single-arm, intent-to-treat analysis, the treatment induced a complete remission in all 12 patients, as evaluated by PET-CT scan, nuclear magnetic resonance (NMR) spectroscopy, or minimal residual disease assessment of bone marrow.
The median time to best response was 1 month (range 0.5 to 2.2 months).
There were no cases of severe cytokine storm syndrome or neurotoxicity, which are among key limitations with current commercial CAR-T products.
All of the patients continued to have a complete remission at 3 months. Two of the 12 patients, both with B-ALL, experienced relapses, one after 4.7 months and the other at 8 months.
The authors reported that the first treated patient had maintained continuous remission as of 9 months.
In comparison with the much higher full doses of commercial CD19 CAR-T products, only about 50% of patients with DLBCL and 70% of B-ALL patients have been shown to achieve CR at 3 months, the authors reported.
“Our IL-10 expressing CAR-T sustains CR at 3 months post infusion in the context of not following allogeneic hematopoietic stem cell transplant, which suggests IL-10 expressing CAR-T is more resistant to relapse,” Dr. Guo said.
In terms of safety, six patients with DLBCL and four with B-ALL experienced grade 1 cytokine release syndrome (CRS), and two patients with B-ALL developed grade 2 CRS. There were no grade 3 or 4 CRS cases.
One patient with B-ALL developed grade 3 ICANS.
Grade 3-4 cytopenias occurred in most patients, but all were limited to no later than 90 days.
“We observed reduced CRS, with no level 3 or 4, or ICANS,” Dr. Guo said. “There was increased cytopenia, but still manageable, compared with commercial products.”
Of note, the Meta10-19 cells showed efficacy in the extremely low infusion doses even among patients with bulky mass (≥ 7.5 cm) of DLBCL, which is associated with an increased risk of relapse.
One patient had primary central nervous system lymphoma (PCNSL), a rare form of DLBCL that is known to have the worst prognosis of all non-Hodgkin lymphomas.
Due to the unique nature of CNS primary tumors, the CAR T-cell infusion dose was further reduced to 1% of the standard dose for the patient.
The patient maintained complete remission for more than 8 months before relapsing in periphery blood, but not in the CNS, Dr. Guo noted.
“Luckily, this relapse has been easily controlled by chemotherapy, and the patient is maintaining complete remission again now,” Dr. Guo said.
Mechanisms?
Dr. Guo noted that the mechanism believed to explain the improvements despite such low doses is that “IL-10-expressing CAR-T exhibits enhanced proliferation, cytotoxicity, and stem-like antitumor memory due to enhanced metabolic activities of oxidative phosphorylation.”
The authors noted that a key major factor limiting accessibility to CAR-T therapies is the lengthy production cycle and high costs; however, the “extremely low doses of 1% to 5% can significantly reduce the production cycle and cost of CAR T-cell therapies, increasing accessibility,” they wrote in a press statement.
Currently, more than 20 patients have achieved a CR overall, and studies with a larger cohort and longer follow-up are ongoing, Dr. Guo reported.
The research team plans to launch further clinical investigation this year into patients with solid tumors.
Commenting on the study, Hongbo Chi, PhD, the Robert G. Webster Endowed Chair in Immunology at St. Jude Children’s Research Hospital in Memphis, Tennessee, noted that, based on the abstract, “the effects are quite remarkable, considering the therapeutic efficacy observed even at the low dose.
“Results from more patients are needed to fully validate these findings, but the results to date are very encouraging,” he said.
The study was sponsored by Leman Biotech. Dr. Chi had no disclosures to report.
“Our study showed a manageable safety profile in r/r DLBCL/B-ALL, with promising breakthrough efficacy of a 100% complete remission in all dose groups,” said first author Jingjing Ren, MD, PhD, associate director of research and development with Leman Biotech in Shenzhen, China. Dr. Ren presented these findings at the American Association for Cancer Research annual meeting held in San Diego.
While CD19 CAR T-cell therapy has been transformative in the treatment of relapsed B -cell hematological malignancies in recent years, more than half of patients relapse within a year because of inadequate CAR T persistence.
To address the problem, Dr. Ren and her colleagues developed a metabolically armed, interleukin (IL)-10-expressing CAR T-cell product called Meta10-19 for the treatment patients with r/r DLBCL or r/r B-ALL.
According to the authors, the IL-10-expressing CAR T-cells trigger “stem-like memory responses” in various lymphoid organs, which prompt a “robust tumor eradication and durable protection,” and hence, better persistence.
Preclinical studies in mice showed the Meta10-19 CAR T-cells exhibited substantially higher expansion of approximately 100-fold compared with a control CD19 CAR-T product.
Therefore, “we dramatically reduced the dose to approximately 1% to 5% of commercial products for the IL-10-expressing CD19 CAR-T for patients,” coauthor Yugang Guo, PhD, cofounder and president of Leman Biotech said in an interview.
For the ongoing, open-label clinical trial, 12 adult patients with r/r DLBCL or r/r B-ALL and confirmed CD19 expression at a hospital center in China were enrolled between December 2022 and November 2023 and treated in three cohorts, receiving doses that corresponded to 1%, 2.5%, or 5% of the doses of other commercialized CAR-T infusion products.
All patients also underwent lympho-depleting chemotherapy with cyclophosphamide and fludarabine prior to the CAR T-cell infusion.
Six patients had r/r DLBCL and the other six had r/r B-ALL; their median age was 47 and their median time since diagnosis was 1 year.
In the single-arm, intent-to-treat analysis, the treatment induced a complete remission in all 12 patients, as evaluated by PET-CT scan, nuclear magnetic resonance (NMR) spectroscopy, or minimal residual disease assessment of bone marrow.
The median time to best response was 1 month (range 0.5 to 2.2 months).
There were no cases of severe cytokine storm syndrome or neurotoxicity, which are among key limitations with current commercial CAR-T products.
All of the patients continued to have a complete remission at 3 months. Two of the 12 patients, both with B-ALL, experienced relapses, one after 4.7 months and the other at 8 months.
The authors reported that the first treated patient had maintained continuous remission as of 9 months.
In comparison with the much higher full doses of commercial CD19 CAR-T products, only about 50% of patients with DLBCL and 70% of B-ALL patients have been shown to achieve CR at 3 months, the authors reported.
“Our IL-10 expressing CAR-T sustains CR at 3 months post infusion in the context of not following allogeneic hematopoietic stem cell transplant, which suggests IL-10 expressing CAR-T is more resistant to relapse,” Dr. Guo said.
In terms of safety, six patients with DLBCL and four with B-ALL experienced grade 1 cytokine release syndrome (CRS), and two patients with B-ALL developed grade 2 CRS. There were no grade 3 or 4 CRS cases.
One patient with B-ALL developed grade 3 ICANS.
Grade 3-4 cytopenias occurred in most patients, but all were limited to no later than 90 days.
“We observed reduced CRS, with no level 3 or 4, or ICANS,” Dr. Guo said. “There was increased cytopenia, but still manageable, compared with commercial products.”
Of note, the Meta10-19 cells showed efficacy in the extremely low infusion doses even among patients with bulky mass (≥ 7.5 cm) of DLBCL, which is associated with an increased risk of relapse.
One patient had primary central nervous system lymphoma (PCNSL), a rare form of DLBCL that is known to have the worst prognosis of all non-Hodgkin lymphomas.
Due to the unique nature of CNS primary tumors, the CAR T-cell infusion dose was further reduced to 1% of the standard dose for the patient.
The patient maintained complete remission for more than 8 months before relapsing in periphery blood, but not in the CNS, Dr. Guo noted.
“Luckily, this relapse has been easily controlled by chemotherapy, and the patient is maintaining complete remission again now,” Dr. Guo said.
Mechanisms?
Dr. Guo noted that the mechanism believed to explain the improvements despite such low doses is that “IL-10-expressing CAR-T exhibits enhanced proliferation, cytotoxicity, and stem-like antitumor memory due to enhanced metabolic activities of oxidative phosphorylation.”
The authors noted that a key major factor limiting accessibility to CAR-T therapies is the lengthy production cycle and high costs; however, the “extremely low doses of 1% to 5% can significantly reduce the production cycle and cost of CAR T-cell therapies, increasing accessibility,” they wrote in a press statement.
Currently, more than 20 patients have achieved a CR overall, and studies with a larger cohort and longer follow-up are ongoing, Dr. Guo reported.
The research team plans to launch further clinical investigation this year into patients with solid tumors.
Commenting on the study, Hongbo Chi, PhD, the Robert G. Webster Endowed Chair in Immunology at St. Jude Children’s Research Hospital in Memphis, Tennessee, noted that, based on the abstract, “the effects are quite remarkable, considering the therapeutic efficacy observed even at the low dose.
“Results from more patients are needed to fully validate these findings, but the results to date are very encouraging,” he said.
The study was sponsored by Leman Biotech. Dr. Chi had no disclosures to report.
“Our study showed a manageable safety profile in r/r DLBCL/B-ALL, with promising breakthrough efficacy of a 100% complete remission in all dose groups,” said first author Jingjing Ren, MD, PhD, associate director of research and development with Leman Biotech in Shenzhen, China. Dr. Ren presented these findings at the American Association for Cancer Research annual meeting held in San Diego.
While CD19 CAR T-cell therapy has been transformative in the treatment of relapsed B -cell hematological malignancies in recent years, more than half of patients relapse within a year because of inadequate CAR T persistence.
To address the problem, Dr. Ren and her colleagues developed a metabolically armed, interleukin (IL)-10-expressing CAR T-cell product called Meta10-19 for the treatment patients with r/r DLBCL or r/r B-ALL.
According to the authors, the IL-10-expressing CAR T-cells trigger “stem-like memory responses” in various lymphoid organs, which prompt a “robust tumor eradication and durable protection,” and hence, better persistence.
Preclinical studies in mice showed the Meta10-19 CAR T-cells exhibited substantially higher expansion of approximately 100-fold compared with a control CD19 CAR-T product.
Therefore, “we dramatically reduced the dose to approximately 1% to 5% of commercial products for the IL-10-expressing CD19 CAR-T for patients,” coauthor Yugang Guo, PhD, cofounder and president of Leman Biotech said in an interview.
For the ongoing, open-label clinical trial, 12 adult patients with r/r DLBCL or r/r B-ALL and confirmed CD19 expression at a hospital center in China were enrolled between December 2022 and November 2023 and treated in three cohorts, receiving doses that corresponded to 1%, 2.5%, or 5% of the doses of other commercialized CAR-T infusion products.
All patients also underwent lympho-depleting chemotherapy with cyclophosphamide and fludarabine prior to the CAR T-cell infusion.
Six patients had r/r DLBCL and the other six had r/r B-ALL; their median age was 47 and their median time since diagnosis was 1 year.
In the single-arm, intent-to-treat analysis, the treatment induced a complete remission in all 12 patients, as evaluated by PET-CT scan, nuclear magnetic resonance (NMR) spectroscopy, or minimal residual disease assessment of bone marrow.
The median time to best response was 1 month (range 0.5 to 2.2 months).
There were no cases of severe cytokine storm syndrome or neurotoxicity, which are among key limitations with current commercial CAR-T products.
All of the patients continued to have a complete remission at 3 months. Two of the 12 patients, both with B-ALL, experienced relapses, one after 4.7 months and the other at 8 months.
The authors reported that the first treated patient had maintained continuous remission as of 9 months.
In comparison with the much higher full doses of commercial CD19 CAR-T products, only about 50% of patients with DLBCL and 70% of B-ALL patients have been shown to achieve CR at 3 months, the authors reported.
“Our IL-10 expressing CAR-T sustains CR at 3 months post infusion in the context of not following allogeneic hematopoietic stem cell transplant, which suggests IL-10 expressing CAR-T is more resistant to relapse,” Dr. Guo said.
In terms of safety, six patients with DLBCL and four with B-ALL experienced grade 1 cytokine release syndrome (CRS), and two patients with B-ALL developed grade 2 CRS. There were no grade 3 or 4 CRS cases.
One patient with B-ALL developed grade 3 ICANS.
Grade 3-4 cytopenias occurred in most patients, but all were limited to no later than 90 days.
“We observed reduced CRS, with no level 3 or 4, or ICANS,” Dr. Guo said. “There was increased cytopenia, but still manageable, compared with commercial products.”
Of note, the Meta10-19 cells showed efficacy in the extremely low infusion doses even among patients with bulky mass (≥ 7.5 cm) of DLBCL, which is associated with an increased risk of relapse.
One patient had primary central nervous system lymphoma (PCNSL), a rare form of DLBCL that is known to have the worst prognosis of all non-Hodgkin lymphomas.
Due to the unique nature of CNS primary tumors, the CAR T-cell infusion dose was further reduced to 1% of the standard dose for the patient.
The patient maintained complete remission for more than 8 months before relapsing in periphery blood, but not in the CNS, Dr. Guo noted.
“Luckily, this relapse has been easily controlled by chemotherapy, and the patient is maintaining complete remission again now,” Dr. Guo said.
Mechanisms?
Dr. Guo noted that the mechanism believed to explain the improvements despite such low doses is that “IL-10-expressing CAR-T exhibits enhanced proliferation, cytotoxicity, and stem-like antitumor memory due to enhanced metabolic activities of oxidative phosphorylation.”
The authors noted that a key major factor limiting accessibility to CAR-T therapies is the lengthy production cycle and high costs; however, the “extremely low doses of 1% to 5% can significantly reduce the production cycle and cost of CAR T-cell therapies, increasing accessibility,” they wrote in a press statement.
Currently, more than 20 patients have achieved a CR overall, and studies with a larger cohort and longer follow-up are ongoing, Dr. Guo reported.
The research team plans to launch further clinical investigation this year into patients with solid tumors.
Commenting on the study, Hongbo Chi, PhD, the Robert G. Webster Endowed Chair in Immunology at St. Jude Children’s Research Hospital in Memphis, Tennessee, noted that, based on the abstract, “the effects are quite remarkable, considering the therapeutic efficacy observed even at the low dose.
“Results from more patients are needed to fully validate these findings, but the results to date are very encouraging,” he said.
The study was sponsored by Leman Biotech. Dr. Chi had no disclosures to report.
FROM AACR 2024
Weighing the Benefits of Integrating AI-based Clinical Notes Into Your Practice
Picture a healthcare system where physicians aren’t bogged down by excessive charting but are instead fully present with their patients, offering undivided attention and personalized care. In a recent X post, Stuart Blitz, COO and co-founder of Hone Health, sparked a thought-provoking conversation. “The problem with US healthcare is physicians are burned out since they spend way too much time charting, not enough with patients,” he wrote. “If you created a health system that did zero charting, you’d attract the best physicians and all patients would go there. Who is working on this?”
This resonates with many in the medical community, myself included, because the strain of extensive documentation detracts from patient care. Having worked in both large and small healthcare systems, I know the burden of extensive charting is a palpable challenge, often detracting from the time we can devote to our patients.
The first part of this two-part series examines the overarching benefits of artificial intelligence (AI)–based clinical documentation in modern healthcare, a field witnessing a paradigm shift thanks to advancements in AI.
Transformative Evolution of Clinical Documentation
The transition from manual documentation to AI-driven solutions marks a significant shift in the field, with a number of products in development including Nuance, Abridge, Ambience, ScribeAmerica, 3M, and DeepScribe. These tools use ambient clinical intelligence (ACI) to automate documentation, capturing patient conversations and translating them into structured clinical summaries. This innovation aligns with the vision of reducing charting burdens and enhancing patient-physician interactions.
How does it work? ACI refers to a sophisticated form of AI applied in healthcare settings, particularly focusing on enhancing the clinical documentation process without disrupting the natural flow of the consultation. Here’s a technical yet practical breakdown of ACI and the algorithms it typically employs:
Data capture and processing: ACI systems employ various sensors and processing units, typically integrated into clinical settings. These sensors, like microphones and cameras, gather diverse data such as audio from patient-doctor dialogues and visual cues. This information is then processed in real-time or near–real-time.
Natural language processing (NLP): A core component of ACI is advanced NLP algorithms. These algorithms analyze the captured audio data, transcribing spoken words into text. NLP goes beyond mere transcription; it involves understanding context, extracting relevant medical information (like symptoms, diagnoses, and treatment plans), and interpreting the nuances of human language.
Deep learning: Machine learning, particularly deep-learning techniques, are employed to improve the accuracy of ACI systems continually. These algorithms can learn from vast datasets of clinical interactions, enhancing their ability to transcribe and interpret future conversations accurately. As they learn, they become better at understanding different accents, complex medical terms, and variations in speech patterns.
Integration with electronic health records (EHRs): ACI systems are often designed to integrate seamlessly with existing EHR systems. They can automatically populate patient records with information from patient-clinician interactions, reducing manual entry and potential errors.
Customization and personalization: Many ACI systems offer customizable templates or allow clinicians to tailor documentation workflows. This flexibility ensures that the output aligns with the specific needs and preferences of healthcare providers.
Ethical and privacy considerations: ACI systems must navigate significant ethical and privacy concerns, especially related to patient consent and data security. These systems need to comply with healthcare privacy regulations such as HIPAA. They need to securely manage sensitive patient data and restrict access to authorized personnel only.
Broad-Spectrum Benefits of AI in Documentation
- Reducing clinician burnout: By automating the documentation process, AI tools like DAX Copilot alleviate a significant contributor to physician burnout, enabling clinicians to focus more on patient care.
- Enhanced patient care: With AI handling documentation, clinicians can engage more with their patients, leading to improved care quality and patient satisfaction.
- Data accuracy and quality: AI-driven documentation captures detailed patient encounters accurately, ensuring high-quality and comprehensive medical records.
- Response to the growing need for efficient healthcare: AI-based documentation is a direct response to the growing call for more efficient healthcare practices, where clinicians spend less time on paperwork and more with patients.
The shift toward AI-based clinical documentation represents a critical step in addressing the inefficiencies in healthcare systems. It’s a move towards a more patient-centered approach, where clinicians can focus more on patient care by reducing the time spent on excessive charting. Hopefully, we can integrate these solutions into our clinics at a large enough scale to make such an impact.
In the next column, we will explore in-depth insights from Kenneth Harper at Nuance on the technical implementation of these tools, with DAX as an example.
I would love to read your comments on AI in clinical trials as well as other AI-related topics. Write me at Arturo.ai.medtech@gmail.com or find me on X @DrBonillaOnc.
Dr. Loaiza-Bonilla is the co-founder and chief medical officer at Massive Bio, a company connecting patients to clinical trials using artificial intelligence. His research and professional interests focus on precision medicine, clinical trial design, digital health, entrepreneurship, and patient advocacy. Dr Loaiza-Bonilla serves as medical director of oncology research at Capital Health in New Jersey, where he maintains a connection to patient care by attending to patients 2 days a week. He has served as a consultant for Verify, PSI CRO, Bayer, AstraZeneca, Cardinal Health, BrightInsight, The Lynx Group, Fresenius, Pfizer, Ipsen, and Guardant; served as a speaker or a member of a speakers bureau for Amgen, Guardant, Eisai, Ipsen, Natera, Merck, Bristol-Myers Squibb, and AstraZeneca. He holds a 5% or greater equity interest in Massive Bio.
A version of this article appeared on Medscape.com.
Picture a healthcare system where physicians aren’t bogged down by excessive charting but are instead fully present with their patients, offering undivided attention and personalized care. In a recent X post, Stuart Blitz, COO and co-founder of Hone Health, sparked a thought-provoking conversation. “The problem with US healthcare is physicians are burned out since they spend way too much time charting, not enough with patients,” he wrote. “If you created a health system that did zero charting, you’d attract the best physicians and all patients would go there. Who is working on this?”
This resonates with many in the medical community, myself included, because the strain of extensive documentation detracts from patient care. Having worked in both large and small healthcare systems, I know the burden of extensive charting is a palpable challenge, often detracting from the time we can devote to our patients.
The first part of this two-part series examines the overarching benefits of artificial intelligence (AI)–based clinical documentation in modern healthcare, a field witnessing a paradigm shift thanks to advancements in AI.
Transformative Evolution of Clinical Documentation
The transition from manual documentation to AI-driven solutions marks a significant shift in the field, with a number of products in development including Nuance, Abridge, Ambience, ScribeAmerica, 3M, and DeepScribe. These tools use ambient clinical intelligence (ACI) to automate documentation, capturing patient conversations and translating them into structured clinical summaries. This innovation aligns with the vision of reducing charting burdens and enhancing patient-physician interactions.
How does it work? ACI refers to a sophisticated form of AI applied in healthcare settings, particularly focusing on enhancing the clinical documentation process without disrupting the natural flow of the consultation. Here’s a technical yet practical breakdown of ACI and the algorithms it typically employs:
Data capture and processing: ACI systems employ various sensors and processing units, typically integrated into clinical settings. These sensors, like microphones and cameras, gather diverse data such as audio from patient-doctor dialogues and visual cues. This information is then processed in real-time or near–real-time.
Natural language processing (NLP): A core component of ACI is advanced NLP algorithms. These algorithms analyze the captured audio data, transcribing spoken words into text. NLP goes beyond mere transcription; it involves understanding context, extracting relevant medical information (like symptoms, diagnoses, and treatment plans), and interpreting the nuances of human language.
Deep learning: Machine learning, particularly deep-learning techniques, are employed to improve the accuracy of ACI systems continually. These algorithms can learn from vast datasets of clinical interactions, enhancing their ability to transcribe and interpret future conversations accurately. As they learn, they become better at understanding different accents, complex medical terms, and variations in speech patterns.
Integration with electronic health records (EHRs): ACI systems are often designed to integrate seamlessly with existing EHR systems. They can automatically populate patient records with information from patient-clinician interactions, reducing manual entry and potential errors.
Customization and personalization: Many ACI systems offer customizable templates or allow clinicians to tailor documentation workflows. This flexibility ensures that the output aligns with the specific needs and preferences of healthcare providers.
Ethical and privacy considerations: ACI systems must navigate significant ethical and privacy concerns, especially related to patient consent and data security. These systems need to comply with healthcare privacy regulations such as HIPAA. They need to securely manage sensitive patient data and restrict access to authorized personnel only.
Broad-Spectrum Benefits of AI in Documentation
- Reducing clinician burnout: By automating the documentation process, AI tools like DAX Copilot alleviate a significant contributor to physician burnout, enabling clinicians to focus more on patient care.
- Enhanced patient care: With AI handling documentation, clinicians can engage more with their patients, leading to improved care quality and patient satisfaction.
- Data accuracy and quality: AI-driven documentation captures detailed patient encounters accurately, ensuring high-quality and comprehensive medical records.
- Response to the growing need for efficient healthcare: AI-based documentation is a direct response to the growing call for more efficient healthcare practices, where clinicians spend less time on paperwork and more with patients.
The shift toward AI-based clinical documentation represents a critical step in addressing the inefficiencies in healthcare systems. It’s a move towards a more patient-centered approach, where clinicians can focus more on patient care by reducing the time spent on excessive charting. Hopefully, we can integrate these solutions into our clinics at a large enough scale to make such an impact.
In the next column, we will explore in-depth insights from Kenneth Harper at Nuance on the technical implementation of these tools, with DAX as an example.
I would love to read your comments on AI in clinical trials as well as other AI-related topics. Write me at Arturo.ai.medtech@gmail.com or find me on X @DrBonillaOnc.
Dr. Loaiza-Bonilla is the co-founder and chief medical officer at Massive Bio, a company connecting patients to clinical trials using artificial intelligence. His research and professional interests focus on precision medicine, clinical trial design, digital health, entrepreneurship, and patient advocacy. Dr Loaiza-Bonilla serves as medical director of oncology research at Capital Health in New Jersey, where he maintains a connection to patient care by attending to patients 2 days a week. He has served as a consultant for Verify, PSI CRO, Bayer, AstraZeneca, Cardinal Health, BrightInsight, The Lynx Group, Fresenius, Pfizer, Ipsen, and Guardant; served as a speaker or a member of a speakers bureau for Amgen, Guardant, Eisai, Ipsen, Natera, Merck, Bristol-Myers Squibb, and AstraZeneca. He holds a 5% or greater equity interest in Massive Bio.
A version of this article appeared on Medscape.com.
Picture a healthcare system where physicians aren’t bogged down by excessive charting but are instead fully present with their patients, offering undivided attention and personalized care. In a recent X post, Stuart Blitz, COO and co-founder of Hone Health, sparked a thought-provoking conversation. “The problem with US healthcare is physicians are burned out since they spend way too much time charting, not enough with patients,” he wrote. “If you created a health system that did zero charting, you’d attract the best physicians and all patients would go there. Who is working on this?”
This resonates with many in the medical community, myself included, because the strain of extensive documentation detracts from patient care. Having worked in both large and small healthcare systems, I know the burden of extensive charting is a palpable challenge, often detracting from the time we can devote to our patients.
The first part of this two-part series examines the overarching benefits of artificial intelligence (AI)–based clinical documentation in modern healthcare, a field witnessing a paradigm shift thanks to advancements in AI.
Transformative Evolution of Clinical Documentation
The transition from manual documentation to AI-driven solutions marks a significant shift in the field, with a number of products in development including Nuance, Abridge, Ambience, ScribeAmerica, 3M, and DeepScribe. These tools use ambient clinical intelligence (ACI) to automate documentation, capturing patient conversations and translating them into structured clinical summaries. This innovation aligns with the vision of reducing charting burdens and enhancing patient-physician interactions.
How does it work? ACI refers to a sophisticated form of AI applied in healthcare settings, particularly focusing on enhancing the clinical documentation process without disrupting the natural flow of the consultation. Here’s a technical yet practical breakdown of ACI and the algorithms it typically employs:
Data capture and processing: ACI systems employ various sensors and processing units, typically integrated into clinical settings. These sensors, like microphones and cameras, gather diverse data such as audio from patient-doctor dialogues and visual cues. This information is then processed in real-time or near–real-time.
Natural language processing (NLP): A core component of ACI is advanced NLP algorithms. These algorithms analyze the captured audio data, transcribing spoken words into text. NLP goes beyond mere transcription; it involves understanding context, extracting relevant medical information (like symptoms, diagnoses, and treatment plans), and interpreting the nuances of human language.
Deep learning: Machine learning, particularly deep-learning techniques, are employed to improve the accuracy of ACI systems continually. These algorithms can learn from vast datasets of clinical interactions, enhancing their ability to transcribe and interpret future conversations accurately. As they learn, they become better at understanding different accents, complex medical terms, and variations in speech patterns.
Integration with electronic health records (EHRs): ACI systems are often designed to integrate seamlessly with existing EHR systems. They can automatically populate patient records with information from patient-clinician interactions, reducing manual entry and potential errors.
Customization and personalization: Many ACI systems offer customizable templates or allow clinicians to tailor documentation workflows. This flexibility ensures that the output aligns with the specific needs and preferences of healthcare providers.
Ethical and privacy considerations: ACI systems must navigate significant ethical and privacy concerns, especially related to patient consent and data security. These systems need to comply with healthcare privacy regulations such as HIPAA. They need to securely manage sensitive patient data and restrict access to authorized personnel only.
Broad-Spectrum Benefits of AI in Documentation
- Reducing clinician burnout: By automating the documentation process, AI tools like DAX Copilot alleviate a significant contributor to physician burnout, enabling clinicians to focus more on patient care.
- Enhanced patient care: With AI handling documentation, clinicians can engage more with their patients, leading to improved care quality and patient satisfaction.
- Data accuracy and quality: AI-driven documentation captures detailed patient encounters accurately, ensuring high-quality and comprehensive medical records.
- Response to the growing need for efficient healthcare: AI-based documentation is a direct response to the growing call for more efficient healthcare practices, where clinicians spend less time on paperwork and more with patients.
The shift toward AI-based clinical documentation represents a critical step in addressing the inefficiencies in healthcare systems. It’s a move towards a more patient-centered approach, where clinicians can focus more on patient care by reducing the time spent on excessive charting. Hopefully, we can integrate these solutions into our clinics at a large enough scale to make such an impact.
In the next column, we will explore in-depth insights from Kenneth Harper at Nuance on the technical implementation of these tools, with DAX as an example.
I would love to read your comments on AI in clinical trials as well as other AI-related topics. Write me at Arturo.ai.medtech@gmail.com or find me on X @DrBonillaOnc.
Dr. Loaiza-Bonilla is the co-founder and chief medical officer at Massive Bio, a company connecting patients to clinical trials using artificial intelligence. His research and professional interests focus on precision medicine, clinical trial design, digital health, entrepreneurship, and patient advocacy. Dr Loaiza-Bonilla serves as medical director of oncology research at Capital Health in New Jersey, where he maintains a connection to patient care by attending to patients 2 days a week. He has served as a consultant for Verify, PSI CRO, Bayer, AstraZeneca, Cardinal Health, BrightInsight, The Lynx Group, Fresenius, Pfizer, Ipsen, and Guardant; served as a speaker or a member of a speakers bureau for Amgen, Guardant, Eisai, Ipsen, Natera, Merck, Bristol-Myers Squibb, and AstraZeneca. He holds a 5% or greater equity interest in Massive Bio.
A version of this article appeared on Medscape.com.
New Federal Rule Delivers Workplace Support, Time Off for Pregnant Docs
Pregnant physicians may receive more workplace accommodations and protection against discrimination thanks to an updated rule from the US Equal Employment Opportunity Commission (EEOC). The guidelines could prevent women from losing critical career momentum.
The Pregnant Workers Fairness Act (PWFA) aims to help workers balance professional demands with healthy pregnancies. It requires employers to provide reasonable accommodations for a “worker’s known limitations,” including physical or mental conditions associated with “pregnancy, childbirth, or related medical conditions.”
Reasonable accommodations vary but may involve time off to attend healthcare appointments or recover from childbirth, extra breaks during a shift, shorter work hours, or the ability to sit instead of stand. Private and public sector employers, including state and local governments, federal agencies, and employment agencies, must abide by the new guidelines unless they can provide evidence that doing so will cause undue hardship.
Female doctors have historically encountered significant barriers to family planning. Years of training cause them to delay having children, often leading to higher rates of infertility, miscarriage, and pregnancy complications than in the general population.
Some specialties, like surgeons, are particularly at risk, with 42% reporting at least one pregnancy loss. Most surgeons work their regular schedules until delivery despite desiring workload reductions, commonly citing unsupportive workplaces as a reason for not seeking accommodations.
Trauma surgeon Qaali Hussein, MD, became pregnant with her first child during her intern year in 2008. She told this news organization that her residency program didn’t even have a maternity policy at the time, and her male supervisor was certain that motherhood would end her surgical career.
She shared how “women usually waited until the end of their training to get pregnant. No one had ever gotten pregnant during the program and returned from maternity leave. I was the first to do so, so there wasn’t a policy or any program support to say, ‘What can we do to help?’ ”
Dr. Hussein used her vacation and sick time, returning to work 4 weeks after delivery. She had five more children, including twins her chief year and another baby during fellowship training in 2014.
Each subsequent pregnancy was met with the same response from program leadership, she recalled. “They’d say, ‘This is it. You may have been able to do the first and second child, but this one will be impossible.’ ”
After the PWFA regulations first became enforceable in June, the EEOC accepted public feedback. The guidelines received nearly 100,000 comments, spurred mainly by the inclusion of abortion care as a qualifying condition for which an employee could receive accommodations. About 54,000 comments called for abortion to be excluded from the final rule, and 40,000 supported keeping the clause.
The EEOC issued the final rule on April 15. It includes abortion care. However, the updated rule “does not require any employee to have — or not to have — an abortion, does not require taxpayers to pay for any abortions, and does not compel health care providers to provide any abortions,” the unpublished version of the final rule said. It is scheduled to take effect 60 days after its publication in the Federal Register on April 19.
Increasing Support for Doctor-Moms
The PWFA supplements other EEOC protections, such as pregnancy discrimination under Title VII of the Civil Rights Act of 1964 and access to reasonable accommodations under the Americans with Disabilities Act. In addition, it builds upon Department of Labor regulations, like the PUMP Act for breastfeeding employees and the Family and Medical Leave Act, which provides 12 weeks of unpaid, job-protected leave for the arrival of a child or certain medical conditions.
FMLA applies only to employees who have worked full-time for at least 12 months for an employer with 50 or more employees. Meanwhile, the unpaid, job-protected leave under the PWFA has no waiting period, lowers the required number of employees to 15, and permits accommodations for up to 40 weeks.
Employers are encouraged to honor “common and simple” requests, like using a closer parking space or pumping or nursing at work, without requiring a doctor’s note, the rule said.
Efforts to improve family leave policies for physicians and residents have been gaining traction. In 2021, the American Board of Medical Specialties began requiring its member boards with training programs lasting 2 or more years to allow at least 6 weeks off for parental, caregiver, and medical leave. This time can be taken without exhausting vacation or sick leave or requiring an extension in training. Over half of the 24 member boards permit leave beyond 6 weeks, including the American Boards of Allergy and Immunology, Emergency Medicine, Family Medicine, Radiology, and Surgery.
Estefania Oliveros, MD, MSc, cardiologist and assistant professor at the Lewis Katz School of Medicine at Temple University, Philadelphia, told this news organization that the Accreditation Council for Graduate Medical Education also requires that residents and fellows receive 6 weeks of paid leave.
“We add to that vacation time, so it gives them at least 8 weeks,” she said. The school has created spaces for nursing mothers — something neither she nor Dr. Hussein had access to when breastfeeding — and encourages the attendings to be proactive in excusing pregnant fellows for appointments.
This differs significantly from her fellowship training experience 6 years ago at another institution, where she worked without accommodations until the day before her cesarean delivery. Dr. Oliveros had to use all her vacation time for recovery, returning to the program after 4 weeks instead of the recommended 6.
“And that’s the story you hear all the time. Not because people are ill-intended; I just don’t think the system is designed to accommodate women, so we lose a lot of talent that way,” said Dr. Oliveros, whose 2019 survey in the Journal of the American College of Cardiology called for more support and protections for pregnant doctors.
Both doctors believe the PWFA will be beneficial but only if leadership in the field takes up the cause.
“The cultures of these institutions determine whether women feel safe or even confident enough to have children in medical school or residency,” said Dr. Hussein.
A version of this article appeared on Medscape.com.
Pregnant physicians may receive more workplace accommodations and protection against discrimination thanks to an updated rule from the US Equal Employment Opportunity Commission (EEOC). The guidelines could prevent women from losing critical career momentum.
The Pregnant Workers Fairness Act (PWFA) aims to help workers balance professional demands with healthy pregnancies. It requires employers to provide reasonable accommodations for a “worker’s known limitations,” including physical or mental conditions associated with “pregnancy, childbirth, or related medical conditions.”
Reasonable accommodations vary but may involve time off to attend healthcare appointments or recover from childbirth, extra breaks during a shift, shorter work hours, or the ability to sit instead of stand. Private and public sector employers, including state and local governments, federal agencies, and employment agencies, must abide by the new guidelines unless they can provide evidence that doing so will cause undue hardship.
Female doctors have historically encountered significant barriers to family planning. Years of training cause them to delay having children, often leading to higher rates of infertility, miscarriage, and pregnancy complications than in the general population.
Some specialties, like surgeons, are particularly at risk, with 42% reporting at least one pregnancy loss. Most surgeons work their regular schedules until delivery despite desiring workload reductions, commonly citing unsupportive workplaces as a reason for not seeking accommodations.
Trauma surgeon Qaali Hussein, MD, became pregnant with her first child during her intern year in 2008. She told this news organization that her residency program didn’t even have a maternity policy at the time, and her male supervisor was certain that motherhood would end her surgical career.
She shared how “women usually waited until the end of their training to get pregnant. No one had ever gotten pregnant during the program and returned from maternity leave. I was the first to do so, so there wasn’t a policy or any program support to say, ‘What can we do to help?’ ”
Dr. Hussein used her vacation and sick time, returning to work 4 weeks after delivery. She had five more children, including twins her chief year and another baby during fellowship training in 2014.
Each subsequent pregnancy was met with the same response from program leadership, she recalled. “They’d say, ‘This is it. You may have been able to do the first and second child, but this one will be impossible.’ ”
After the PWFA regulations first became enforceable in June, the EEOC accepted public feedback. The guidelines received nearly 100,000 comments, spurred mainly by the inclusion of abortion care as a qualifying condition for which an employee could receive accommodations. About 54,000 comments called for abortion to be excluded from the final rule, and 40,000 supported keeping the clause.
The EEOC issued the final rule on April 15. It includes abortion care. However, the updated rule “does not require any employee to have — or not to have — an abortion, does not require taxpayers to pay for any abortions, and does not compel health care providers to provide any abortions,” the unpublished version of the final rule said. It is scheduled to take effect 60 days after its publication in the Federal Register on April 19.
Increasing Support for Doctor-Moms
The PWFA supplements other EEOC protections, such as pregnancy discrimination under Title VII of the Civil Rights Act of 1964 and access to reasonable accommodations under the Americans with Disabilities Act. In addition, it builds upon Department of Labor regulations, like the PUMP Act for breastfeeding employees and the Family and Medical Leave Act, which provides 12 weeks of unpaid, job-protected leave for the arrival of a child or certain medical conditions.
FMLA applies only to employees who have worked full-time for at least 12 months for an employer with 50 or more employees. Meanwhile, the unpaid, job-protected leave under the PWFA has no waiting period, lowers the required number of employees to 15, and permits accommodations for up to 40 weeks.
Employers are encouraged to honor “common and simple” requests, like using a closer parking space or pumping or nursing at work, without requiring a doctor’s note, the rule said.
Efforts to improve family leave policies for physicians and residents have been gaining traction. In 2021, the American Board of Medical Specialties began requiring its member boards with training programs lasting 2 or more years to allow at least 6 weeks off for parental, caregiver, and medical leave. This time can be taken without exhausting vacation or sick leave or requiring an extension in training. Over half of the 24 member boards permit leave beyond 6 weeks, including the American Boards of Allergy and Immunology, Emergency Medicine, Family Medicine, Radiology, and Surgery.
Estefania Oliveros, MD, MSc, cardiologist and assistant professor at the Lewis Katz School of Medicine at Temple University, Philadelphia, told this news organization that the Accreditation Council for Graduate Medical Education also requires that residents and fellows receive 6 weeks of paid leave.
“We add to that vacation time, so it gives them at least 8 weeks,” she said. The school has created spaces for nursing mothers — something neither she nor Dr. Hussein had access to when breastfeeding — and encourages the attendings to be proactive in excusing pregnant fellows for appointments.
This differs significantly from her fellowship training experience 6 years ago at another institution, where she worked without accommodations until the day before her cesarean delivery. Dr. Oliveros had to use all her vacation time for recovery, returning to the program after 4 weeks instead of the recommended 6.
“And that’s the story you hear all the time. Not because people are ill-intended; I just don’t think the system is designed to accommodate women, so we lose a lot of talent that way,” said Dr. Oliveros, whose 2019 survey in the Journal of the American College of Cardiology called for more support and protections for pregnant doctors.
Both doctors believe the PWFA will be beneficial but only if leadership in the field takes up the cause.
“The cultures of these institutions determine whether women feel safe or even confident enough to have children in medical school or residency,” said Dr. Hussein.
A version of this article appeared on Medscape.com.
Pregnant physicians may receive more workplace accommodations and protection against discrimination thanks to an updated rule from the US Equal Employment Opportunity Commission (EEOC). The guidelines could prevent women from losing critical career momentum.
The Pregnant Workers Fairness Act (PWFA) aims to help workers balance professional demands with healthy pregnancies. It requires employers to provide reasonable accommodations for a “worker’s known limitations,” including physical or mental conditions associated with “pregnancy, childbirth, or related medical conditions.”
Reasonable accommodations vary but may involve time off to attend healthcare appointments or recover from childbirth, extra breaks during a shift, shorter work hours, or the ability to sit instead of stand. Private and public sector employers, including state and local governments, federal agencies, and employment agencies, must abide by the new guidelines unless they can provide evidence that doing so will cause undue hardship.
Female doctors have historically encountered significant barriers to family planning. Years of training cause them to delay having children, often leading to higher rates of infertility, miscarriage, and pregnancy complications than in the general population.
Some specialties, like surgeons, are particularly at risk, with 42% reporting at least one pregnancy loss. Most surgeons work their regular schedules until delivery despite desiring workload reductions, commonly citing unsupportive workplaces as a reason for not seeking accommodations.
Trauma surgeon Qaali Hussein, MD, became pregnant with her first child during her intern year in 2008. She told this news organization that her residency program didn’t even have a maternity policy at the time, and her male supervisor was certain that motherhood would end her surgical career.
She shared how “women usually waited until the end of their training to get pregnant. No one had ever gotten pregnant during the program and returned from maternity leave. I was the first to do so, so there wasn’t a policy or any program support to say, ‘What can we do to help?’ ”
Dr. Hussein used her vacation and sick time, returning to work 4 weeks after delivery. She had five more children, including twins her chief year and another baby during fellowship training in 2014.
Each subsequent pregnancy was met with the same response from program leadership, she recalled. “They’d say, ‘This is it. You may have been able to do the first and second child, but this one will be impossible.’ ”
After the PWFA regulations first became enforceable in June, the EEOC accepted public feedback. The guidelines received nearly 100,000 comments, spurred mainly by the inclusion of abortion care as a qualifying condition for which an employee could receive accommodations. About 54,000 comments called for abortion to be excluded from the final rule, and 40,000 supported keeping the clause.
The EEOC issued the final rule on April 15. It includes abortion care. However, the updated rule “does not require any employee to have — or not to have — an abortion, does not require taxpayers to pay for any abortions, and does not compel health care providers to provide any abortions,” the unpublished version of the final rule said. It is scheduled to take effect 60 days after its publication in the Federal Register on April 19.
Increasing Support for Doctor-Moms
The PWFA supplements other EEOC protections, such as pregnancy discrimination under Title VII of the Civil Rights Act of 1964 and access to reasonable accommodations under the Americans with Disabilities Act. In addition, it builds upon Department of Labor regulations, like the PUMP Act for breastfeeding employees and the Family and Medical Leave Act, which provides 12 weeks of unpaid, job-protected leave for the arrival of a child or certain medical conditions.
FMLA applies only to employees who have worked full-time for at least 12 months for an employer with 50 or more employees. Meanwhile, the unpaid, job-protected leave under the PWFA has no waiting period, lowers the required number of employees to 15, and permits accommodations for up to 40 weeks.
Employers are encouraged to honor “common and simple” requests, like using a closer parking space or pumping or nursing at work, without requiring a doctor’s note, the rule said.
Efforts to improve family leave policies for physicians and residents have been gaining traction. In 2021, the American Board of Medical Specialties began requiring its member boards with training programs lasting 2 or more years to allow at least 6 weeks off for parental, caregiver, and medical leave. This time can be taken without exhausting vacation or sick leave or requiring an extension in training. Over half of the 24 member boards permit leave beyond 6 weeks, including the American Boards of Allergy and Immunology, Emergency Medicine, Family Medicine, Radiology, and Surgery.
Estefania Oliveros, MD, MSc, cardiologist and assistant professor at the Lewis Katz School of Medicine at Temple University, Philadelphia, told this news organization that the Accreditation Council for Graduate Medical Education also requires that residents and fellows receive 6 weeks of paid leave.
“We add to that vacation time, so it gives them at least 8 weeks,” she said. The school has created spaces for nursing mothers — something neither she nor Dr. Hussein had access to when breastfeeding — and encourages the attendings to be proactive in excusing pregnant fellows for appointments.
This differs significantly from her fellowship training experience 6 years ago at another institution, where she worked without accommodations until the day before her cesarean delivery. Dr. Oliveros had to use all her vacation time for recovery, returning to the program after 4 weeks instead of the recommended 6.
“And that’s the story you hear all the time. Not because people are ill-intended; I just don’t think the system is designed to accommodate women, so we lose a lot of talent that way,” said Dr. Oliveros, whose 2019 survey in the Journal of the American College of Cardiology called for more support and protections for pregnant doctors.
Both doctors believe the PWFA will be beneficial but only if leadership in the field takes up the cause.
“The cultures of these institutions determine whether women feel safe or even confident enough to have children in medical school or residency,” said Dr. Hussein.
A version of this article appeared on Medscape.com.
Keratoacanthoma, SCC Relatively Rare With PD-1/PD-L1 Inhibitors, Study Suggests
TOPLINE:
(AEs) reported to the US Food and Drug Administration (FDA).
METHODOLOGY:
- The risk for dermatologic immune-related side effects may be increased with immunologic-modifying drugs.
- To determine if there are significant signals between keratoacanthomas and cSCCs and PD-1/PD-L1 inhibitors, researchers analyzed AEs associated with these agents reported to the FDA’s Adverse Event Reporting System (FAERS) between January 2004 and May 2023.
- Pharmacovigilance signals were identified, and a significant signal was defined as the lower 95% CI of a reporting odds ratio (ROR) greater than one or the lower 95% CI of an information component (IC) greater than 0.
TAKEAWAY:
- Of the 158,000 reports of PD-1/PD-L1 inhibitor use, 43 were in patients who developed a keratoacanthoma (mean age, 77 years; 39% women) and 83 were in patients who developed cSCC (mean age, 71 years; 41% women). Patients aged 60-79 years were most likely to develop keratoacanthomas and cSCC on these treatments.
- A PD-1/PD-L1 inhibitor was listed as the suspect drug in all 43 keratoacanthoma reports and in 70 of 83 cSCC reports (the remaining 13 listed them as the concomitant drug).
- Significant signals were reported for both keratoacanthoma (ROR, 9.7; IC, 1.9) and cSCC (ROR, 3.0; IC, 0.9) with PD-1/PD-L1 inhibitor use.
- Of the reports where this information was available, all 10 cases of PD-1/PD-L1 inhibitor–linked keratoacanthoma and 10 of 17 cases (59%) of PD-1/PD-L1 inhibitor–linked cSCC, resolution was noted following discontinuation or dose reduction of the inhibitor.
IN PRACTICE:
“Given the large number of patients receiving immunotherapy, FAERS recording only 43 patients developing keratoacanthoma and 83 patients developing cSCC highlights that these conditions are relatively rare adverse events,” the authors wrote but added that more studies are needed to confirm these results.
SOURCE:
The study, led by Pushkar Aggarwal, MD, MBA, of the Department of Dermatology, University of Cincinnati, Cincinnati, Ohio, was published online in JAMA Dermatology.
LIMITATIONS:
The data obtained from FAERS did not contain information on all AEs from drugs. In addition, a causal association could not be determined.
DISCLOSURES:
The funding source was not reported. The authors did not report any conflicts of interest.
A version of this article appeared on Medscape.com.
TOPLINE:
(AEs) reported to the US Food and Drug Administration (FDA).
METHODOLOGY:
- The risk for dermatologic immune-related side effects may be increased with immunologic-modifying drugs.
- To determine if there are significant signals between keratoacanthomas and cSCCs and PD-1/PD-L1 inhibitors, researchers analyzed AEs associated with these agents reported to the FDA’s Adverse Event Reporting System (FAERS) between January 2004 and May 2023.
- Pharmacovigilance signals were identified, and a significant signal was defined as the lower 95% CI of a reporting odds ratio (ROR) greater than one or the lower 95% CI of an information component (IC) greater than 0.
TAKEAWAY:
- Of the 158,000 reports of PD-1/PD-L1 inhibitor use, 43 were in patients who developed a keratoacanthoma (mean age, 77 years; 39% women) and 83 were in patients who developed cSCC (mean age, 71 years; 41% women). Patients aged 60-79 years were most likely to develop keratoacanthomas and cSCC on these treatments.
- A PD-1/PD-L1 inhibitor was listed as the suspect drug in all 43 keratoacanthoma reports and in 70 of 83 cSCC reports (the remaining 13 listed them as the concomitant drug).
- Significant signals were reported for both keratoacanthoma (ROR, 9.7; IC, 1.9) and cSCC (ROR, 3.0; IC, 0.9) with PD-1/PD-L1 inhibitor use.
- Of the reports where this information was available, all 10 cases of PD-1/PD-L1 inhibitor–linked keratoacanthoma and 10 of 17 cases (59%) of PD-1/PD-L1 inhibitor–linked cSCC, resolution was noted following discontinuation or dose reduction of the inhibitor.
IN PRACTICE:
“Given the large number of patients receiving immunotherapy, FAERS recording only 43 patients developing keratoacanthoma and 83 patients developing cSCC highlights that these conditions are relatively rare adverse events,” the authors wrote but added that more studies are needed to confirm these results.
SOURCE:
The study, led by Pushkar Aggarwal, MD, MBA, of the Department of Dermatology, University of Cincinnati, Cincinnati, Ohio, was published online in JAMA Dermatology.
LIMITATIONS:
The data obtained from FAERS did not contain information on all AEs from drugs. In addition, a causal association could not be determined.
DISCLOSURES:
The funding source was not reported. The authors did not report any conflicts of interest.
A version of this article appeared on Medscape.com.
TOPLINE:
(AEs) reported to the US Food and Drug Administration (FDA).
METHODOLOGY:
- The risk for dermatologic immune-related side effects may be increased with immunologic-modifying drugs.
- To determine if there are significant signals between keratoacanthomas and cSCCs and PD-1/PD-L1 inhibitors, researchers analyzed AEs associated with these agents reported to the FDA’s Adverse Event Reporting System (FAERS) between January 2004 and May 2023.
- Pharmacovigilance signals were identified, and a significant signal was defined as the lower 95% CI of a reporting odds ratio (ROR) greater than one or the lower 95% CI of an information component (IC) greater than 0.
TAKEAWAY:
- Of the 158,000 reports of PD-1/PD-L1 inhibitor use, 43 were in patients who developed a keratoacanthoma (mean age, 77 years; 39% women) and 83 were in patients who developed cSCC (mean age, 71 years; 41% women). Patients aged 60-79 years were most likely to develop keratoacanthomas and cSCC on these treatments.
- A PD-1/PD-L1 inhibitor was listed as the suspect drug in all 43 keratoacanthoma reports and in 70 of 83 cSCC reports (the remaining 13 listed them as the concomitant drug).
- Significant signals were reported for both keratoacanthoma (ROR, 9.7; IC, 1.9) and cSCC (ROR, 3.0; IC, 0.9) with PD-1/PD-L1 inhibitor use.
- Of the reports where this information was available, all 10 cases of PD-1/PD-L1 inhibitor–linked keratoacanthoma and 10 of 17 cases (59%) of PD-1/PD-L1 inhibitor–linked cSCC, resolution was noted following discontinuation or dose reduction of the inhibitor.
IN PRACTICE:
“Given the large number of patients receiving immunotherapy, FAERS recording only 43 patients developing keratoacanthoma and 83 patients developing cSCC highlights that these conditions are relatively rare adverse events,” the authors wrote but added that more studies are needed to confirm these results.
SOURCE:
The study, led by Pushkar Aggarwal, MD, MBA, of the Department of Dermatology, University of Cincinnati, Cincinnati, Ohio, was published online in JAMA Dermatology.
LIMITATIONS:
The data obtained from FAERS did not contain information on all AEs from drugs. In addition, a causal association could not be determined.
DISCLOSURES:
The funding source was not reported. The authors did not report any conflicts of interest.
A version of this article appeared on Medscape.com.
Adding ACEI to Chemotherapy Does Not Prevent Cardiotoxicity
a new randomized trial showed.
The results suggested adding an ACE inhibitor doesn’t affect cardiac injury or cardiac function outcomes “and should not be used as a preventative strategy” in these patients, David Austin, MD, consultant cardiologist, Academic Cardiovascular Unit, The James Cook University Hospital, Middlesbrough, England, and chief investigator for the PROACT study, told this news organization.
But while these negative results are disappointing, he said, “we now have a definitive result in a robustly conducted trial that will take the field forward.”
The findings were presented on April 8, 2024, at the American College of Cardiology (ACC) Scientific Session 2024.
Anthracyclines, which are extracted from Streptomyces bacterium, are chemotherapy drugs widely used to treat several types of cancer. Doxorubicin is among the most clinically important anthracyclines.
While extremely effective, anthracyclines can cause irreversible damage to cardiac cells and ultimately impair cardiac function and even cause heart failure, which may only be evident years after exposure. “Cardiac injury is very common in patients treated with high dose anthracyclines,” noted Dr. Austin.
The open-label PROACT study included 111 adult patients, mean age 58 years and predominantly White and women, being treated for breast cancer (62%) or NHL (38%) at National Health Service hospitals in England with high-dose anthracycline-based chemotherapy.
Patients were randomized to standard care (six cycles of high-dose doxorubicin-equivalent anthracycline-based chemotherapy) plus the ACE inhibitor enalapril maleate or standard care alone. The mean chemotherapy dose was 328 mg/m2; any dose greater than 300 is considered high.
The starting dose of enalapril was 2.5 mg twice a day, which was titrated up to a maximum of 10 mg twice a day. The ACE inhibitor was started at least 2 days before chemotherapy began and finished 3 weeks after the last anthracycline dose.
During the study, enalapril was titrated to 20 mg in more than 75% of patients, with the mean dose being 17.7 mg.
Myocardial Injury Outcome
The primary outcome was myocardial injury measured by the presence (≥ 14 ng/L) of high sensitivity cardiac troponin T (cTnT) during anthracycline treatment and 1 month after the last dose of anthracycline.
cTnT is highly expressed in cardiomyocytes and has become a preferred biomarker for detecting acute myocardial infarction and other causes of myocardial injury.
Blood sampling for cTnT and cardiac troponin I (cTnI) was performed at baseline, within 72 hours prior to chemotherapy and at trial completion. All patients had negative troponin results at baseline, indicating no heart damage.
A majority of patients experienced elevations in troponin (78% in the enalapril group and 83% in the standard of care group), but there was no statistically significant difference between groups (adjusted odds ratio [OR], 0.65; 95% CI, 0.23-1.78; P = .405).
There was also no significant difference between groups in terms of cTnI, a secondary endpoint. However, the proportion of patients testing positive for cTnI (47% in the enalapril group and 45% in controls) was substantially lower than that for cTnT.
Large Discrepancy
The “large discrepancy in the rate of injury” with cTnT “has implications for the clinical interpretation of cardiac biomarkers in routine practice, and we should proceed with caution,” Dr. Austin told this news organization.
The finding has implications because guidelines don’t currently differentiate based on the type of troponin, Dr. Austin said in a press release. “I was surprised by the difference, and I think this raises the question of what troponin we should be using.”
Secondary outcomes focused on cardiac function, measured using echocardiography and included left ventricular global longitudinal strain (LVGLS) and left ventricular ejection fraction (LVEF). These were measured at baseline, 4 weeks after the last anthracycline dose and 1 year after the final chemotherapy.
There was no between-group difference in LVGLS cardiac function (21% for enalapril vs 22% for standard of care; adjusted OR, 0.95; 95% CI, 0.33-2.74; P = .921). This was also true for LVEF (4% for enalapril vs 0% for standard of care group; adjusted OR, 4.89; 95% CI, 0.40-674.62; P = .236).
Asked what the research team plans to do next, Dr. Austin said “the immediate first step” is to continue following PROACT patients. “We know heart failure events and cardiac dysfunction can occur later down the line.”
Due to the challenge of enrolling patients into trials like PROACT, “we should come together as a sort of a broader cardiovascular/oncology academic community to try to understand how we can better recruit patients into these studies,” said Dr. Austin.
“We need to solve that problem before we then go on to maybe examine other potential preventative therapies.”
He doesn’t think an alternative ACE inhibitor would prove beneficial. “We need to look elsewhere for effective therapies in this area.”
He noted these new findings are “broadly consistent” with other trials that investigated angiotensin receptor blockers.
Tough Population
Commenting on the study during a media briefing, Anita Deswal, chair, medicine, Department of Cardiology, Division of Internal Medicine, The University of Texas, commended the researchers for managing to enroll patients with cancer as this is “a tough” population to get to agree to being in a clinical trial.
“These patients are often overwhelmed financially, physically, and emotionally with the cancer diagnosis, as well as the cancer therapy and, therefore, to enroll them in something to prevent, maybe, some potential cardiac toxicity down the line, is really hard.”
Past trials investigating neuro-hormonal blockers to prevent cardiotoxicity have been criticized for enrolling patients at “too low risk,” said Dr. Deswal. “But investigators here went that step beyond and enrolled patients who were going to receive higher doses of anthracyclines, so kudos to that.”
And she noted investigators managed to get patients on almost the maximum dose of enalapril. “So, the drug was poised to have an effect — if it was there.”
The negative results may have something to do with endpoints. “Maybe we haven’t quite figured out what are the cutoffs for high sensitivity troponin I that identify patients truly at risk” of developing heart failure in the future.
Commenting on the study for this news organization, Anu Lala, MD, assistant professor of medicine at the Icahn School of Medicine at Mount Sinai, New York City, said the results may come as a surprise to some.
“ACE inhibitors are considered cardioprotective and for this reason are often used prophylactically in patients receiving chemotherapy.”
Dr. Lala agrees troponin may not be the right endpoint. “Another question is whether clinical outcomes should be followed in addition to symptoms or onset of any heart failure symptoms, which may hold greater prognostic significance.”
The study was funded by the National Institute for Health and Care Research.
A version of this article appeared on Medscape.com.
a new randomized trial showed.
The results suggested adding an ACE inhibitor doesn’t affect cardiac injury or cardiac function outcomes “and should not be used as a preventative strategy” in these patients, David Austin, MD, consultant cardiologist, Academic Cardiovascular Unit, The James Cook University Hospital, Middlesbrough, England, and chief investigator for the PROACT study, told this news organization.
But while these negative results are disappointing, he said, “we now have a definitive result in a robustly conducted trial that will take the field forward.”
The findings were presented on April 8, 2024, at the American College of Cardiology (ACC) Scientific Session 2024.
Anthracyclines, which are extracted from Streptomyces bacterium, are chemotherapy drugs widely used to treat several types of cancer. Doxorubicin is among the most clinically important anthracyclines.
While extremely effective, anthracyclines can cause irreversible damage to cardiac cells and ultimately impair cardiac function and even cause heart failure, which may only be evident years after exposure. “Cardiac injury is very common in patients treated with high dose anthracyclines,” noted Dr. Austin.
The open-label PROACT study included 111 adult patients, mean age 58 years and predominantly White and women, being treated for breast cancer (62%) or NHL (38%) at National Health Service hospitals in England with high-dose anthracycline-based chemotherapy.
Patients were randomized to standard care (six cycles of high-dose doxorubicin-equivalent anthracycline-based chemotherapy) plus the ACE inhibitor enalapril maleate or standard care alone. The mean chemotherapy dose was 328 mg/m2; any dose greater than 300 is considered high.
The starting dose of enalapril was 2.5 mg twice a day, which was titrated up to a maximum of 10 mg twice a day. The ACE inhibitor was started at least 2 days before chemotherapy began and finished 3 weeks after the last anthracycline dose.
During the study, enalapril was titrated to 20 mg in more than 75% of patients, with the mean dose being 17.7 mg.
Myocardial Injury Outcome
The primary outcome was myocardial injury measured by the presence (≥ 14 ng/L) of high sensitivity cardiac troponin T (cTnT) during anthracycline treatment and 1 month after the last dose of anthracycline.
cTnT is highly expressed in cardiomyocytes and has become a preferred biomarker for detecting acute myocardial infarction and other causes of myocardial injury.
Blood sampling for cTnT and cardiac troponin I (cTnI) was performed at baseline, within 72 hours prior to chemotherapy and at trial completion. All patients had negative troponin results at baseline, indicating no heart damage.
A majority of patients experienced elevations in troponin (78% in the enalapril group and 83% in the standard of care group), but there was no statistically significant difference between groups (adjusted odds ratio [OR], 0.65; 95% CI, 0.23-1.78; P = .405).
There was also no significant difference between groups in terms of cTnI, a secondary endpoint. However, the proportion of patients testing positive for cTnI (47% in the enalapril group and 45% in controls) was substantially lower than that for cTnT.
Large Discrepancy
The “large discrepancy in the rate of injury” with cTnT “has implications for the clinical interpretation of cardiac biomarkers in routine practice, and we should proceed with caution,” Dr. Austin told this news organization.
The finding has implications because guidelines don’t currently differentiate based on the type of troponin, Dr. Austin said in a press release. “I was surprised by the difference, and I think this raises the question of what troponin we should be using.”
Secondary outcomes focused on cardiac function, measured using echocardiography and included left ventricular global longitudinal strain (LVGLS) and left ventricular ejection fraction (LVEF). These were measured at baseline, 4 weeks after the last anthracycline dose and 1 year after the final chemotherapy.
There was no between-group difference in LVGLS cardiac function (21% for enalapril vs 22% for standard of care; adjusted OR, 0.95; 95% CI, 0.33-2.74; P = .921). This was also true for LVEF (4% for enalapril vs 0% for standard of care group; adjusted OR, 4.89; 95% CI, 0.40-674.62; P = .236).
Asked what the research team plans to do next, Dr. Austin said “the immediate first step” is to continue following PROACT patients. “We know heart failure events and cardiac dysfunction can occur later down the line.”
Due to the challenge of enrolling patients into trials like PROACT, “we should come together as a sort of a broader cardiovascular/oncology academic community to try to understand how we can better recruit patients into these studies,” said Dr. Austin.
“We need to solve that problem before we then go on to maybe examine other potential preventative therapies.”
He doesn’t think an alternative ACE inhibitor would prove beneficial. “We need to look elsewhere for effective therapies in this area.”
He noted these new findings are “broadly consistent” with other trials that investigated angiotensin receptor blockers.
Tough Population
Commenting on the study during a media briefing, Anita Deswal, chair, medicine, Department of Cardiology, Division of Internal Medicine, The University of Texas, commended the researchers for managing to enroll patients with cancer as this is “a tough” population to get to agree to being in a clinical trial.
“These patients are often overwhelmed financially, physically, and emotionally with the cancer diagnosis, as well as the cancer therapy and, therefore, to enroll them in something to prevent, maybe, some potential cardiac toxicity down the line, is really hard.”
Past trials investigating neuro-hormonal blockers to prevent cardiotoxicity have been criticized for enrolling patients at “too low risk,” said Dr. Deswal. “But investigators here went that step beyond and enrolled patients who were going to receive higher doses of anthracyclines, so kudos to that.”
And she noted investigators managed to get patients on almost the maximum dose of enalapril. “So, the drug was poised to have an effect — if it was there.”
The negative results may have something to do with endpoints. “Maybe we haven’t quite figured out what are the cutoffs for high sensitivity troponin I that identify patients truly at risk” of developing heart failure in the future.
Commenting on the study for this news organization, Anu Lala, MD, assistant professor of medicine at the Icahn School of Medicine at Mount Sinai, New York City, said the results may come as a surprise to some.
“ACE inhibitors are considered cardioprotective and for this reason are often used prophylactically in patients receiving chemotherapy.”
Dr. Lala agrees troponin may not be the right endpoint. “Another question is whether clinical outcomes should be followed in addition to symptoms or onset of any heart failure symptoms, which may hold greater prognostic significance.”
The study was funded by the National Institute for Health and Care Research.
A version of this article appeared on Medscape.com.
a new randomized trial showed.
The results suggested adding an ACE inhibitor doesn’t affect cardiac injury or cardiac function outcomes “and should not be used as a preventative strategy” in these patients, David Austin, MD, consultant cardiologist, Academic Cardiovascular Unit, The James Cook University Hospital, Middlesbrough, England, and chief investigator for the PROACT study, told this news organization.
But while these negative results are disappointing, he said, “we now have a definitive result in a robustly conducted trial that will take the field forward.”
The findings were presented on April 8, 2024, at the American College of Cardiology (ACC) Scientific Session 2024.
Anthracyclines, which are extracted from Streptomyces bacterium, are chemotherapy drugs widely used to treat several types of cancer. Doxorubicin is among the most clinically important anthracyclines.
While extremely effective, anthracyclines can cause irreversible damage to cardiac cells and ultimately impair cardiac function and even cause heart failure, which may only be evident years after exposure. “Cardiac injury is very common in patients treated with high dose anthracyclines,” noted Dr. Austin.
The open-label PROACT study included 111 adult patients, mean age 58 years and predominantly White and women, being treated for breast cancer (62%) or NHL (38%) at National Health Service hospitals in England with high-dose anthracycline-based chemotherapy.
Patients were randomized to standard care (six cycles of high-dose doxorubicin-equivalent anthracycline-based chemotherapy) plus the ACE inhibitor enalapril maleate or standard care alone. The mean chemotherapy dose was 328 mg/m2; any dose greater than 300 is considered high.
The starting dose of enalapril was 2.5 mg twice a day, which was titrated up to a maximum of 10 mg twice a day. The ACE inhibitor was started at least 2 days before chemotherapy began and finished 3 weeks after the last anthracycline dose.
During the study, enalapril was titrated to 20 mg in more than 75% of patients, with the mean dose being 17.7 mg.
Myocardial Injury Outcome
The primary outcome was myocardial injury measured by the presence (≥ 14 ng/L) of high sensitivity cardiac troponin T (cTnT) during anthracycline treatment and 1 month after the last dose of anthracycline.
cTnT is highly expressed in cardiomyocytes and has become a preferred biomarker for detecting acute myocardial infarction and other causes of myocardial injury.
Blood sampling for cTnT and cardiac troponin I (cTnI) was performed at baseline, within 72 hours prior to chemotherapy and at trial completion. All patients had negative troponin results at baseline, indicating no heart damage.
A majority of patients experienced elevations in troponin (78% in the enalapril group and 83% in the standard of care group), but there was no statistically significant difference between groups (adjusted odds ratio [OR], 0.65; 95% CI, 0.23-1.78; P = .405).
There was also no significant difference between groups in terms of cTnI, a secondary endpoint. However, the proportion of patients testing positive for cTnI (47% in the enalapril group and 45% in controls) was substantially lower than that for cTnT.
Large Discrepancy
The “large discrepancy in the rate of injury” with cTnT “has implications for the clinical interpretation of cardiac biomarkers in routine practice, and we should proceed with caution,” Dr. Austin told this news organization.
The finding has implications because guidelines don’t currently differentiate based on the type of troponin, Dr. Austin said in a press release. “I was surprised by the difference, and I think this raises the question of what troponin we should be using.”
Secondary outcomes focused on cardiac function, measured using echocardiography and included left ventricular global longitudinal strain (LVGLS) and left ventricular ejection fraction (LVEF). These were measured at baseline, 4 weeks after the last anthracycline dose and 1 year after the final chemotherapy.
There was no between-group difference in LVGLS cardiac function (21% for enalapril vs 22% for standard of care; adjusted OR, 0.95; 95% CI, 0.33-2.74; P = .921). This was also true for LVEF (4% for enalapril vs 0% for standard of care group; adjusted OR, 4.89; 95% CI, 0.40-674.62; P = .236).
Asked what the research team plans to do next, Dr. Austin said “the immediate first step” is to continue following PROACT patients. “We know heart failure events and cardiac dysfunction can occur later down the line.”
Due to the challenge of enrolling patients into trials like PROACT, “we should come together as a sort of a broader cardiovascular/oncology academic community to try to understand how we can better recruit patients into these studies,” said Dr. Austin.
“We need to solve that problem before we then go on to maybe examine other potential preventative therapies.”
He doesn’t think an alternative ACE inhibitor would prove beneficial. “We need to look elsewhere for effective therapies in this area.”
He noted these new findings are “broadly consistent” with other trials that investigated angiotensin receptor blockers.
Tough Population
Commenting on the study during a media briefing, Anita Deswal, chair, medicine, Department of Cardiology, Division of Internal Medicine, The University of Texas, commended the researchers for managing to enroll patients with cancer as this is “a tough” population to get to agree to being in a clinical trial.
“These patients are often overwhelmed financially, physically, and emotionally with the cancer diagnosis, as well as the cancer therapy and, therefore, to enroll them in something to prevent, maybe, some potential cardiac toxicity down the line, is really hard.”
Past trials investigating neuro-hormonal blockers to prevent cardiotoxicity have been criticized for enrolling patients at “too low risk,” said Dr. Deswal. “But investigators here went that step beyond and enrolled patients who were going to receive higher doses of anthracyclines, so kudos to that.”
And she noted investigators managed to get patients on almost the maximum dose of enalapril. “So, the drug was poised to have an effect — if it was there.”
The negative results may have something to do with endpoints. “Maybe we haven’t quite figured out what are the cutoffs for high sensitivity troponin I that identify patients truly at risk” of developing heart failure in the future.
Commenting on the study for this news organization, Anu Lala, MD, assistant professor of medicine at the Icahn School of Medicine at Mount Sinai, New York City, said the results may come as a surprise to some.
“ACE inhibitors are considered cardioprotective and for this reason are often used prophylactically in patients receiving chemotherapy.”
Dr. Lala agrees troponin may not be the right endpoint. “Another question is whether clinical outcomes should be followed in addition to symptoms or onset of any heart failure symptoms, which may hold greater prognostic significance.”
The study was funded by the National Institute for Health and Care Research.
A version of this article appeared on Medscape.com.
FROM THE ACC 2024
Timing Is Everything: CAR T for Follicular Lymphoma
“CAR T-cells offer patients with relapsed or refractory follicular lymphoma the most durable responses and improved chance of survival beyond all other available therapies. This holds true for a broad range of high-risk disease features in patients with relapsed or refractory FL. Furthermore, it accomplishes this with a single infusion, and a discrete toxicity that is predictable, reversible and manageable,” said Caron Jacobson, MD, MMSc, of the Dana-Farber Cancer Institute in Boston.
Presenting at the Great Debates & Updates Hematologic Malignancies conference, held April 5-6 in New York City, Dr. Jacobson argued that more patients with R/R FL should be treated with CAR T.

She cited follow-up results from the ZUMA-5 study indicating that patients with R/R FL treated with the CAR T axicabtagene ciloleucel (YESCARTA; Kite Pharma) have a median progression free survival (PFS) of 57.3 months and a complete response rate (CR) of 80%. Furthermore, the lymphoma-specific four-year PFS appears to be reaching a plateau, suggesting that some patients treated with the agent may be cured.
The most significant drawback of treatment with axicabtagene ciloleucel is cytokine release syndrome (CRS) and neurotoxicity, which occurred at grade three and higher in 6% and 15%, of ZUMA-5 participants, respectively.
Two newer studies of anti-CD-19 CAR T-cell therapy in R/R FL, tisagenlecleucel in ELARA and lisocabtagene maraleucel in TRANSCEND FL, suggest that other CAR T-cell treatments can be as effective as axicabtagene ciloleucel, but with fewer side effects.
At a median follow up of 29 months, CR among patients in the ELARA study was 68.1%, and the overall response rate (ORR) was 86.2%. Fewer than half of patients had any CRS, and none had grade three or higher. Only 10% of patients had serious neurologic events, with only 1% of these events rated as grade three or higher.
At a median of 18.1 months, patients in the TRANSCEND FL study had a CR of 94% and an ORR of 97%. Over 58% of patients had CRS but it was grade three or higher only 1% of the time (one patient); 15% of patients had neurologic toxicity, but it was grade three or higher only 2% of the time (three patients).
Dr. Jacobson’s opponent in the debate, Peter Martin, MD, of NewYork–Presbyterian Hospital, Weill Cornell Medicine in New York City, acknowledged the strong performance of CAR T in R/R FL patients but argued that they should be used only in a small subset of patients.
“About 20% of patients will experience an early recurrence or progression of diseases within 24 months (PoD-24) which is associated with worse outcomes. About half of those patients experienced transformation, so they have diffuse large B-cell lymphoma, and they’re getting CAR T-cells. In the end, only 10% of patients with follicular lymphoma are relapsed or refractory and should consider getting Car T-cell therapy,” said Dr. Martin, who focused the rest of his presentation on the best options for treating patients with indolent R/R FL who did not have PoD-24.

He said these patients may be able to avoid the side effects of CAR T and perform well when treated with lenalidomide rituximab (R2) or bispecific antibodies. Data from the MAGNIFY trial of patients with R/R FL indicate that patients treated with R2 who did not experience relapse less than 24 months after starting treatment and were not heavily refractory to rituximab achieved a median PFS of over 4 years, with grade 3 or higher adverse events occurring in 5% of patients or less.
Treatment with bispecific antibodies, although inferior in performance to CAR T-cell therapy, may offer durable responses in some R/R CL patients without the risk of side effects associated with CAR T.
Mosunetuzumab, a bispecific antibody that is currently approved for follicular lymphoma, is designed with step-up dosing to reduce cytokine release syndrome and “achieved a complete response rate of 60% and a median PFS that looks like it’s probably about two years,” said Dr. Martin, noting that some patients continue to do well after the 3-year mark and speculated that “there will be some really long-term responders.”
In addition to the possibly durable nature of bispecific antibodies, they induce cytokine release syndrome at a much lower rate than CAR T, and most side effects are manageable in an outpatient setting, “usually just with Tylenol occasionally with a dose of steroids,” said Dr. Martin.
He contrasted this response with CAR T-cell therapy, which requires referral and travel to a specialized center for at least 1 month around the time of therapy.
Despite the differences of opinion between the presenters about whether CAR T should be used more or less in R/R FL, essentially the two specialists were making recommendations for different patient groups.
Dr. Jacobson observed that “Dr. Martin is looking at the 80% of people who do really well with follicular lymphoma." Those are the people who don’t require a third line of therapy. They are the people who don’t have PoD-24. I’m looking at the 20% of people who either do require a third line of therapy or who do have PoD-24, and we’re not treating nearly enough of those patients with follicular lymphoma.
“We’re actually arguing about treatment strategies for different populations of patients. And I think ultimately, we agree more than we disagree in the end,” she concluded.
The notion that CAR T, chemotherapy, and bispecific antibodies all have a place in treating R/R FL patients is supported by Charalambos (Babis) Andreadis, MD, hematologist at the University of California San Francisco’s Helen Diller Family Comprehensive Care Center. “If I had a patient with follicular who relapsed 24 months or later after primary therapy and had active disease that needed treatment, most providers would do a lenalidomide-based or chemo-based regimen. Down the line either bispecific or CAR T would be appropriate in third line,” said Dr. Andreadis.
However, he noted,“for someone who is an early progressor, I would similarly not be able to use either [chemotherapy or bispecific antibodies] in second line [therapy] but would definitely think that early CART would be a good option to consider given the longevity of the observed responses so far.”
Dr. Martin disclosed ties with AbbVie, AstraZeneca, BeiGene, Daiichi Sankyo, Epizyme, Genentech, Janssen, Merck, and PeproMene. Dr. Jacobson reported relationships with AbbVie, Abintus Bio, ADC Therapeutics, Appia Bio, AstraZeneca, BMS/Celgene, Caribou Bio, Daiichi Sankyo, ImmPACT Bio, Ipsen, Janssen, Kite/Gilead, MorphoSys, Novartis, Sana, Synthekine, Kite/Gilead, and Pfizer. Dr. Andreadis had no disclosures.
“CAR T-cells offer patients with relapsed or refractory follicular lymphoma the most durable responses and improved chance of survival beyond all other available therapies. This holds true for a broad range of high-risk disease features in patients with relapsed or refractory FL. Furthermore, it accomplishes this with a single infusion, and a discrete toxicity that is predictable, reversible and manageable,” said Caron Jacobson, MD, MMSc, of the Dana-Farber Cancer Institute in Boston.
Presenting at the Great Debates & Updates Hematologic Malignancies conference, held April 5-6 in New York City, Dr. Jacobson argued that more patients with R/R FL should be treated with CAR T.
She cited follow-up results from the ZUMA-5 study indicating that patients with R/R FL treated with the CAR T axicabtagene ciloleucel (YESCARTA; Kite Pharma) have a median progression free survival (PFS) of 57.3 months and a complete response rate (CR) of 80%. Furthermore, the lymphoma-specific four-year PFS appears to be reaching a plateau, suggesting that some patients treated with the agent may be cured.
The most significant drawback of treatment with axicabtagene ciloleucel is cytokine release syndrome (CRS) and neurotoxicity, which occurred at grade three and higher in 6% and 15%, of ZUMA-5 participants, respectively.
Two newer studies of anti-CD-19 CAR T-cell therapy in R/R FL, tisagenlecleucel in ELARA and lisocabtagene maraleucel in TRANSCEND FL, suggest that other CAR T-cell treatments can be as effective as axicabtagene ciloleucel, but with fewer side effects.
At a median follow up of 29 months, CR among patients in the ELARA study was 68.1%, and the overall response rate (ORR) was 86.2%. Fewer than half of patients had any CRS, and none had grade three or higher. Only 10% of patients had serious neurologic events, with only 1% of these events rated as grade three or higher.
At a median of 18.1 months, patients in the TRANSCEND FL study had a CR of 94% and an ORR of 97%. Over 58% of patients had CRS but it was grade three or higher only 1% of the time (one patient); 15% of patients had neurologic toxicity, but it was grade three or higher only 2% of the time (three patients).
Dr. Jacobson’s opponent in the debate, Peter Martin, MD, of NewYork–Presbyterian Hospital, Weill Cornell Medicine in New York City, acknowledged the strong performance of CAR T in R/R FL patients but argued that they should be used only in a small subset of patients.
“About 20% of patients will experience an early recurrence or progression of diseases within 24 months (PoD-24) which is associated with worse outcomes. About half of those patients experienced transformation, so they have diffuse large B-cell lymphoma, and they’re getting CAR T-cells. In the end, only 10% of patients with follicular lymphoma are relapsed or refractory and should consider getting Car T-cell therapy,” said Dr. Martin, who focused the rest of his presentation on the best options for treating patients with indolent R/R FL who did not have PoD-24.
He said these patients may be able to avoid the side effects of CAR T and perform well when treated with lenalidomide rituximab (R2) or bispecific antibodies. Data from the MAGNIFY trial of patients with R/R FL indicate that patients treated with R2 who did not experience relapse less than 24 months after starting treatment and were not heavily refractory to rituximab achieved a median PFS of over 4 years, with grade 3 or higher adverse events occurring in 5% of patients or less.
Treatment with bispecific antibodies, although inferior in performance to CAR T-cell therapy, may offer durable responses in some R/R CL patients without the risk of side effects associated with CAR T.
Mosunetuzumab, a bispecific antibody that is currently approved for follicular lymphoma, is designed with step-up dosing to reduce cytokine release syndrome and “achieved a complete response rate of 60% and a median PFS that looks like it’s probably about two years,” said Dr. Martin, noting that some patients continue to do well after the 3-year mark and speculated that “there will be some really long-term responders.”
In addition to the possibly durable nature of bispecific antibodies, they induce cytokine release syndrome at a much lower rate than CAR T, and most side effects are manageable in an outpatient setting, “usually just with Tylenol occasionally with a dose of steroids,” said Dr. Martin.
He contrasted this response with CAR T-cell therapy, which requires referral and travel to a specialized center for at least 1 month around the time of therapy.
Despite the differences of opinion between the presenters about whether CAR T should be used more or less in R/R FL, essentially the two specialists were making recommendations for different patient groups.
Dr. Jacobson observed that “Dr. Martin is looking at the 80% of people who do really well with follicular lymphoma." Those are the people who don’t require a third line of therapy. They are the people who don’t have PoD-24. I’m looking at the 20% of people who either do require a third line of therapy or who do have PoD-24, and we’re not treating nearly enough of those patients with follicular lymphoma.
“We’re actually arguing about treatment strategies for different populations of patients. And I think ultimately, we agree more than we disagree in the end,” she concluded.
The notion that CAR T, chemotherapy, and bispecific antibodies all have a place in treating R/R FL patients is supported by Charalambos (Babis) Andreadis, MD, hematologist at the University of California San Francisco’s Helen Diller Family Comprehensive Care Center. “If I had a patient with follicular who relapsed 24 months or later after primary therapy and had active disease that needed treatment, most providers would do a lenalidomide-based or chemo-based regimen. Down the line either bispecific or CAR T would be appropriate in third line,” said Dr. Andreadis.
However, he noted,“for someone who is an early progressor, I would similarly not be able to use either [chemotherapy or bispecific antibodies] in second line [therapy] but would definitely think that early CART would be a good option to consider given the longevity of the observed responses so far.”
Dr. Martin disclosed ties with AbbVie, AstraZeneca, BeiGene, Daiichi Sankyo, Epizyme, Genentech, Janssen, Merck, and PeproMene. Dr. Jacobson reported relationships with AbbVie, Abintus Bio, ADC Therapeutics, Appia Bio, AstraZeneca, BMS/Celgene, Caribou Bio, Daiichi Sankyo, ImmPACT Bio, Ipsen, Janssen, Kite/Gilead, MorphoSys, Novartis, Sana, Synthekine, Kite/Gilead, and Pfizer. Dr. Andreadis had no disclosures.
“CAR T-cells offer patients with relapsed or refractory follicular lymphoma the most durable responses and improved chance of survival beyond all other available therapies. This holds true for a broad range of high-risk disease features in patients with relapsed or refractory FL. Furthermore, it accomplishes this with a single infusion, and a discrete toxicity that is predictable, reversible and manageable,” said Caron Jacobson, MD, MMSc, of the Dana-Farber Cancer Institute in Boston.
Presenting at the Great Debates & Updates Hematologic Malignancies conference, held April 5-6 in New York City, Dr. Jacobson argued that more patients with R/R FL should be treated with CAR T.
She cited follow-up results from the ZUMA-5 study indicating that patients with R/R FL treated with the CAR T axicabtagene ciloleucel (YESCARTA; Kite Pharma) have a median progression free survival (PFS) of 57.3 months and a complete response rate (CR) of 80%. Furthermore, the lymphoma-specific four-year PFS appears to be reaching a plateau, suggesting that some patients treated with the agent may be cured.
The most significant drawback of treatment with axicabtagene ciloleucel is cytokine release syndrome (CRS) and neurotoxicity, which occurred at grade three and higher in 6% and 15%, of ZUMA-5 participants, respectively.
Two newer studies of anti-CD-19 CAR T-cell therapy in R/R FL, tisagenlecleucel in ELARA and lisocabtagene maraleucel in TRANSCEND FL, suggest that other CAR T-cell treatments can be as effective as axicabtagene ciloleucel, but with fewer side effects.
At a median follow up of 29 months, CR among patients in the ELARA study was 68.1%, and the overall response rate (ORR) was 86.2%. Fewer than half of patients had any CRS, and none had grade three or higher. Only 10% of patients had serious neurologic events, with only 1% of these events rated as grade three or higher.
At a median of 18.1 months, patients in the TRANSCEND FL study had a CR of 94% and an ORR of 97%. Over 58% of patients had CRS but it was grade three or higher only 1% of the time (one patient); 15% of patients had neurologic toxicity, but it was grade three or higher only 2% of the time (three patients).
Dr. Jacobson’s opponent in the debate, Peter Martin, MD, of NewYork–Presbyterian Hospital, Weill Cornell Medicine in New York City, acknowledged the strong performance of CAR T in R/R FL patients but argued that they should be used only in a small subset of patients.
“About 20% of patients will experience an early recurrence or progression of diseases within 24 months (PoD-24) which is associated with worse outcomes. About half of those patients experienced transformation, so they have diffuse large B-cell lymphoma, and they’re getting CAR T-cells. In the end, only 10% of patients with follicular lymphoma are relapsed or refractory and should consider getting Car T-cell therapy,” said Dr. Martin, who focused the rest of his presentation on the best options for treating patients with indolent R/R FL who did not have PoD-24.
He said these patients may be able to avoid the side effects of CAR T and perform well when treated with lenalidomide rituximab (R2) or bispecific antibodies. Data from the MAGNIFY trial of patients with R/R FL indicate that patients treated with R2 who did not experience relapse less than 24 months after starting treatment and were not heavily refractory to rituximab achieved a median PFS of over 4 years, with grade 3 or higher adverse events occurring in 5% of patients or less.
Treatment with bispecific antibodies, although inferior in performance to CAR T-cell therapy, may offer durable responses in some R/R CL patients without the risk of side effects associated with CAR T.
Mosunetuzumab, a bispecific antibody that is currently approved for follicular lymphoma, is designed with step-up dosing to reduce cytokine release syndrome and “achieved a complete response rate of 60% and a median PFS that looks like it’s probably about two years,” said Dr. Martin, noting that some patients continue to do well after the 3-year mark and speculated that “there will be some really long-term responders.”
In addition to the possibly durable nature of bispecific antibodies, they induce cytokine release syndrome at a much lower rate than CAR T, and most side effects are manageable in an outpatient setting, “usually just with Tylenol occasionally with a dose of steroids,” said Dr. Martin.
He contrasted this response with CAR T-cell therapy, which requires referral and travel to a specialized center for at least 1 month around the time of therapy.
Despite the differences of opinion between the presenters about whether CAR T should be used more or less in R/R FL, essentially the two specialists were making recommendations for different patient groups.
Dr. Jacobson observed that “Dr. Martin is looking at the 80% of people who do really well with follicular lymphoma." Those are the people who don’t require a third line of therapy. They are the people who don’t have PoD-24. I’m looking at the 20% of people who either do require a third line of therapy or who do have PoD-24, and we’re not treating nearly enough of those patients with follicular lymphoma.
“We’re actually arguing about treatment strategies for different populations of patients. And I think ultimately, we agree more than we disagree in the end,” she concluded.
The notion that CAR T, chemotherapy, and bispecific antibodies all have a place in treating R/R FL patients is supported by Charalambos (Babis) Andreadis, MD, hematologist at the University of California San Francisco’s Helen Diller Family Comprehensive Care Center. “If I had a patient with follicular who relapsed 24 months or later after primary therapy and had active disease that needed treatment, most providers would do a lenalidomide-based or chemo-based regimen. Down the line either bispecific or CAR T would be appropriate in third line,” said Dr. Andreadis.
However, he noted,“for someone who is an early progressor, I would similarly not be able to use either [chemotherapy or bispecific antibodies] in second line [therapy] but would definitely think that early CART would be a good option to consider given the longevity of the observed responses so far.”
Dr. Martin disclosed ties with AbbVie, AstraZeneca, BeiGene, Daiichi Sankyo, Epizyme, Genentech, Janssen, Merck, and PeproMene. Dr. Jacobson reported relationships with AbbVie, Abintus Bio, ADC Therapeutics, Appia Bio, AstraZeneca, BMS/Celgene, Caribou Bio, Daiichi Sankyo, ImmPACT Bio, Ipsen, Janssen, Kite/Gilead, MorphoSys, Novartis, Sana, Synthekine, Kite/Gilead, and Pfizer. Dr. Andreadis had no disclosures.
Most Targeted Cancer Drugs Lack Substantial Clinical Benefit
TOPLINE:
METHODOLOGY:
- The strength and quality of evidence supporting genome-targeted cancer drug approvals vary. A big reason is the growing number of cancer drug approvals based on surrogate endpoints, such as disease-free and progression-free survival, instead of clinical endpoints, such as overall survival or quality of life. The US Food and Drug Administration (FDA) has also approved genome-targeted cancer drugs based on phase 1 or single-arm trials.
- Given these less rigorous considerations for approval, “the validity and value of the targets and surrogate measures underlying FDA genome-targeted cancer drug approvals are uncertain,” the researchers explained.
- In the current analysis, researchers assessed the validity of the molecular targets as well as the clinical benefits of genome-targeted cancer drugs approved in the United States from 2015 to 2022 based on results from pivotal trials.
- The researchers evaluated the strength of evidence supporting molecular targetability using the European Society for Medical Oncology (ESMO) Scale for Clinical Actionability of Molecular Targets (ESCAT) and the clinical benefit using the ESMO–Magnitude of Clinical Benefit Scale (ESMO-MCBS).
- The authors defined a substantial clinical benefit as an A or B grade for curative intent and a 4 or 5 for noncurative intent. High-benefit genomic-based cancer treatments were defined as those associated with a substantial clinical benefit (ESMO-MCBS) and that qualified as ESCAT category level I-A (a clinical benefit based on prospective randomized data) or I-B (prospective nonrandomized data).
TAKEAWAY:
- The analyses focused on 50 molecular-targeted cancer drugs covering 84 indications. Of which, 45 indications (54%) were approved based on phase 1 or 2 pivotal trials, 45 (54%) were supported by single-arm pivotal trials and the remaining 39 (46%) by randomized trial, and 48 (57%) were approved based on subgroup analyses.
- Among the 84 indications, more than half (55%) of the pivotal trials supporting approval used overall response rate as a primary endpoint, 31% used progression-free survival, and 6% used disease-free survival. Only seven indications (8%) were supported by pivotal trials demonstrating an improvement in overall survival.
- Among the 84 trials, 24 (29%) met the ESMO-MCBS threshold for substantial clinical benefit.
- Overall, when combining all ratings, only 24 of the 84 indications (29%) were considered high-benefit genomic-based cancer treatments.
IN PRACTICE:
“We applied the ESMO-MCBS and ESCAT value frameworks to identify therapies and molecular targets providing high clinical value that should be widely available to patients” and “found that drug indications supported by these characteristics represent a minority of cancer drug approvals in recent years,” the authors said. Using these value frameworks could help payers, governments, and individual patients “prioritize the availability of high-value molecular-targeted therapies.”
SOURCE:
The study, with first author Ariadna Tibau, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School, Boston, was published online in JAMA Oncology.
LIMITATIONS:
The study evaluated only trials that supported regulatory approval and did not include outcomes of postapproval clinical studies, which could lead to changes in ESMO-MCBS grades and ESCAT levels of evidence over time.
DISCLOSURES:
The study was funded by the Kaiser Permanente Institute for Health Policy, Arnold Ventures, and the Commonwealth Fund. The authors had no relevant disclosures.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The strength and quality of evidence supporting genome-targeted cancer drug approvals vary. A big reason is the growing number of cancer drug approvals based on surrogate endpoints, such as disease-free and progression-free survival, instead of clinical endpoints, such as overall survival or quality of life. The US Food and Drug Administration (FDA) has also approved genome-targeted cancer drugs based on phase 1 or single-arm trials.
- Given these less rigorous considerations for approval, “the validity and value of the targets and surrogate measures underlying FDA genome-targeted cancer drug approvals are uncertain,” the researchers explained.
- In the current analysis, researchers assessed the validity of the molecular targets as well as the clinical benefits of genome-targeted cancer drugs approved in the United States from 2015 to 2022 based on results from pivotal trials.
- The researchers evaluated the strength of evidence supporting molecular targetability using the European Society for Medical Oncology (ESMO) Scale for Clinical Actionability of Molecular Targets (ESCAT) and the clinical benefit using the ESMO–Magnitude of Clinical Benefit Scale (ESMO-MCBS).
- The authors defined a substantial clinical benefit as an A or B grade for curative intent and a 4 or 5 for noncurative intent. High-benefit genomic-based cancer treatments were defined as those associated with a substantial clinical benefit (ESMO-MCBS) and that qualified as ESCAT category level I-A (a clinical benefit based on prospective randomized data) or I-B (prospective nonrandomized data).
TAKEAWAY:
- The analyses focused on 50 molecular-targeted cancer drugs covering 84 indications. Of which, 45 indications (54%) were approved based on phase 1 or 2 pivotal trials, 45 (54%) were supported by single-arm pivotal trials and the remaining 39 (46%) by randomized trial, and 48 (57%) were approved based on subgroup analyses.
- Among the 84 indications, more than half (55%) of the pivotal trials supporting approval used overall response rate as a primary endpoint, 31% used progression-free survival, and 6% used disease-free survival. Only seven indications (8%) were supported by pivotal trials demonstrating an improvement in overall survival.
- Among the 84 trials, 24 (29%) met the ESMO-MCBS threshold for substantial clinical benefit.
- Overall, when combining all ratings, only 24 of the 84 indications (29%) were considered high-benefit genomic-based cancer treatments.
IN PRACTICE:
“We applied the ESMO-MCBS and ESCAT value frameworks to identify therapies and molecular targets providing high clinical value that should be widely available to patients” and “found that drug indications supported by these characteristics represent a minority of cancer drug approvals in recent years,” the authors said. Using these value frameworks could help payers, governments, and individual patients “prioritize the availability of high-value molecular-targeted therapies.”
SOURCE:
The study, with first author Ariadna Tibau, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School, Boston, was published online in JAMA Oncology.
LIMITATIONS:
The study evaluated only trials that supported regulatory approval and did not include outcomes of postapproval clinical studies, which could lead to changes in ESMO-MCBS grades and ESCAT levels of evidence over time.
DISCLOSURES:
The study was funded by the Kaiser Permanente Institute for Health Policy, Arnold Ventures, and the Commonwealth Fund. The authors had no relevant disclosures.
A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- The strength and quality of evidence supporting genome-targeted cancer drug approvals vary. A big reason is the growing number of cancer drug approvals based on surrogate endpoints, such as disease-free and progression-free survival, instead of clinical endpoints, such as overall survival or quality of life. The US Food and Drug Administration (FDA) has also approved genome-targeted cancer drugs based on phase 1 or single-arm trials.
- Given these less rigorous considerations for approval, “the validity and value of the targets and surrogate measures underlying FDA genome-targeted cancer drug approvals are uncertain,” the researchers explained.
- In the current analysis, researchers assessed the validity of the molecular targets as well as the clinical benefits of genome-targeted cancer drugs approved in the United States from 2015 to 2022 based on results from pivotal trials.
- The researchers evaluated the strength of evidence supporting molecular targetability using the European Society for Medical Oncology (ESMO) Scale for Clinical Actionability of Molecular Targets (ESCAT) and the clinical benefit using the ESMO–Magnitude of Clinical Benefit Scale (ESMO-MCBS).
- The authors defined a substantial clinical benefit as an A or B grade for curative intent and a 4 or 5 for noncurative intent. High-benefit genomic-based cancer treatments were defined as those associated with a substantial clinical benefit (ESMO-MCBS) and that qualified as ESCAT category level I-A (a clinical benefit based on prospective randomized data) or I-B (prospective nonrandomized data).
TAKEAWAY:
- The analyses focused on 50 molecular-targeted cancer drugs covering 84 indications. Of which, 45 indications (54%) were approved based on phase 1 or 2 pivotal trials, 45 (54%) were supported by single-arm pivotal trials and the remaining 39 (46%) by randomized trial, and 48 (57%) were approved based on subgroup analyses.
- Among the 84 indications, more than half (55%) of the pivotal trials supporting approval used overall response rate as a primary endpoint, 31% used progression-free survival, and 6% used disease-free survival. Only seven indications (8%) were supported by pivotal trials demonstrating an improvement in overall survival.
- Among the 84 trials, 24 (29%) met the ESMO-MCBS threshold for substantial clinical benefit.
- Overall, when combining all ratings, only 24 of the 84 indications (29%) were considered high-benefit genomic-based cancer treatments.
IN PRACTICE:
“We applied the ESMO-MCBS and ESCAT value frameworks to identify therapies and molecular targets providing high clinical value that should be widely available to patients” and “found that drug indications supported by these characteristics represent a minority of cancer drug approvals in recent years,” the authors said. Using these value frameworks could help payers, governments, and individual patients “prioritize the availability of high-value molecular-targeted therapies.”
SOURCE:
The study, with first author Ariadna Tibau, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School, Boston, was published online in JAMA Oncology.
LIMITATIONS:
The study evaluated only trials that supported regulatory approval and did not include outcomes of postapproval clinical studies, which could lead to changes in ESMO-MCBS grades and ESCAT levels of evidence over time.
DISCLOSURES:
The study was funded by the Kaiser Permanente Institute for Health Policy, Arnold Ventures, and the Commonwealth Fund. The authors had no relevant disclosures.
A version of this article appeared on Medscape.com.