User login
For MD-IQ use only
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
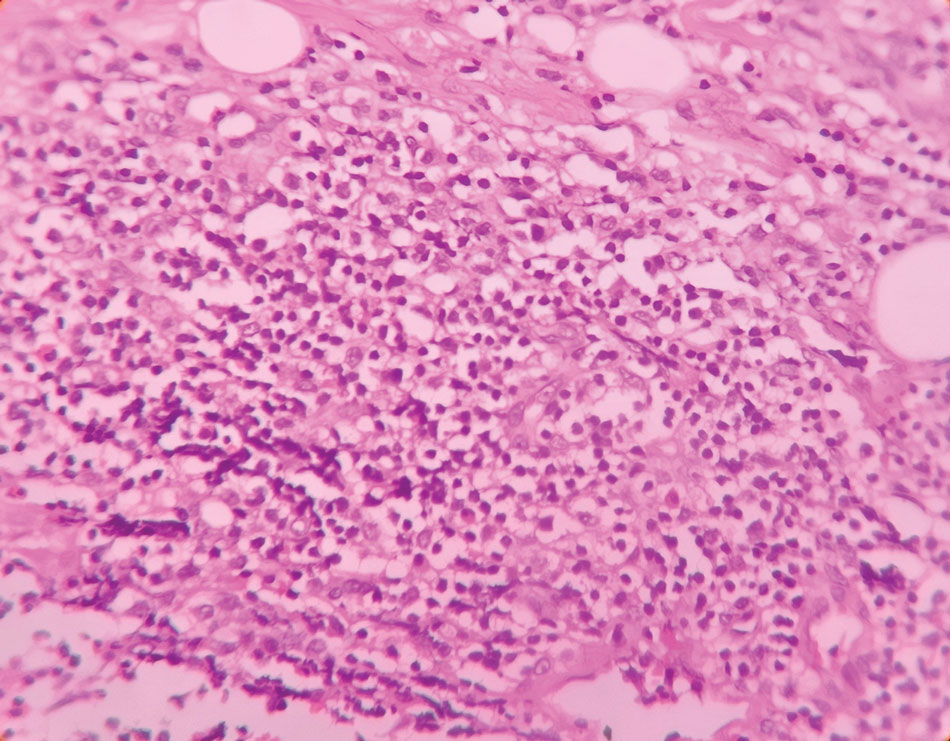
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
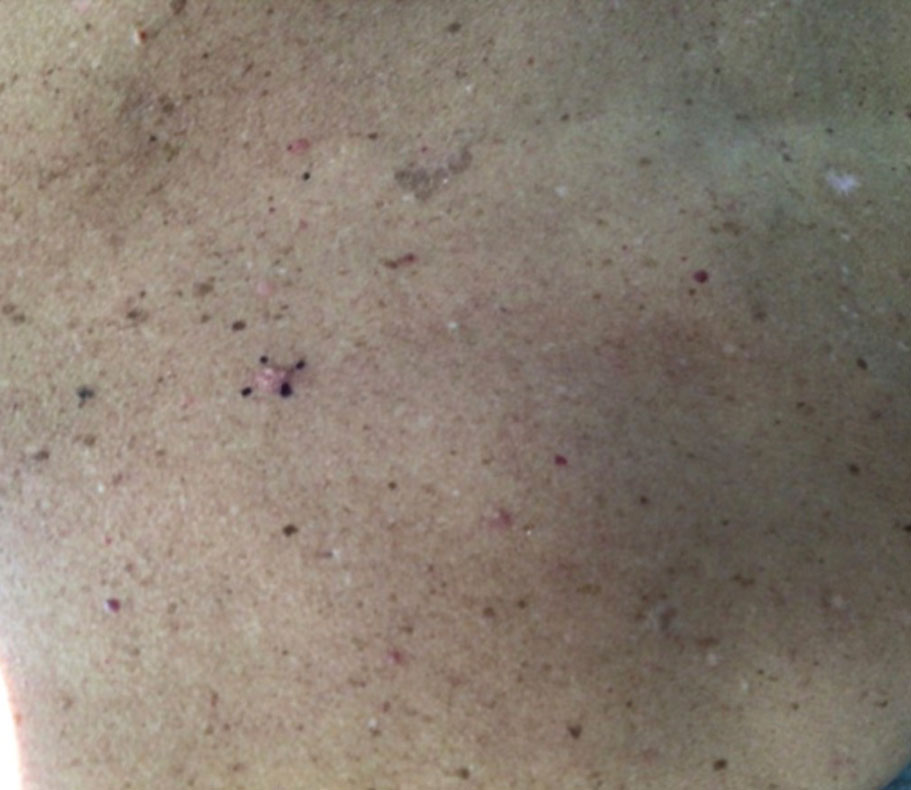
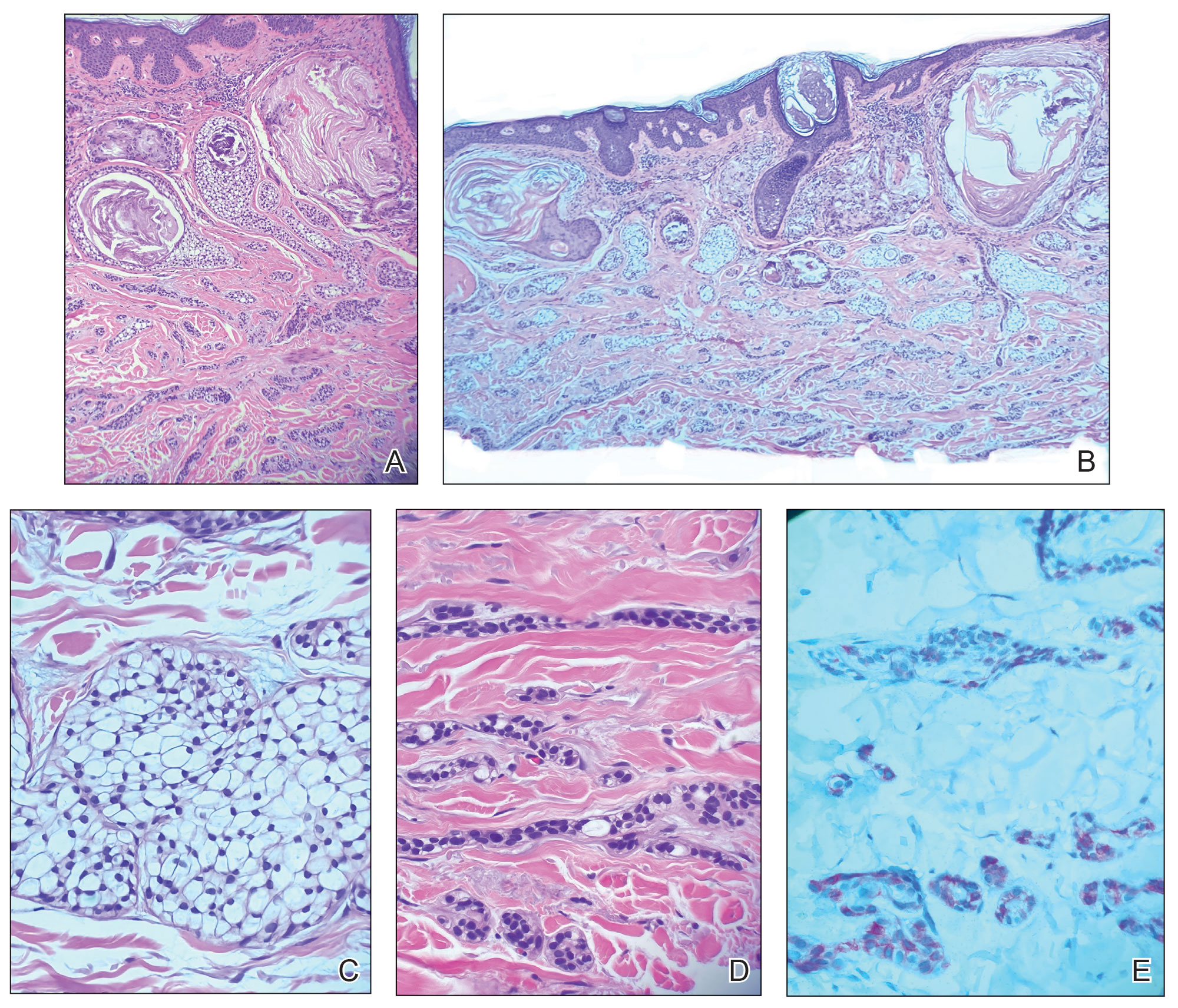
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
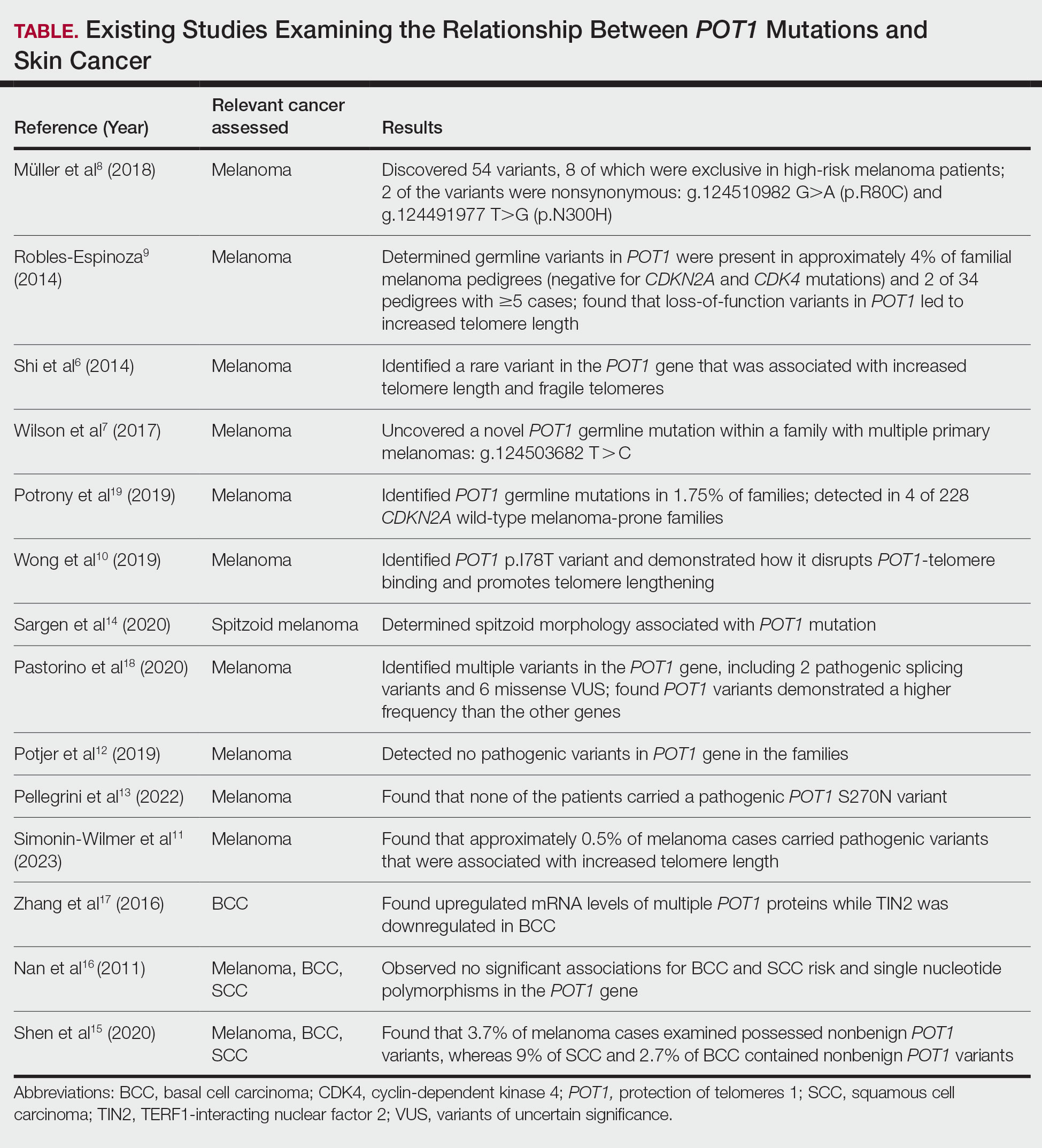
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
PRACTICE POINTS
- Dermatologists should consider referring patients with both a history of skin cancer and a strong family history of internal malignancy for genetic testing for POT1 (protection of telomeres 1) mutations.
- Although melanoma, chronic lymphocytic leukemia, angiosarcoma, and gliomas are most commonly associated with POT1 mutations, this case suggests a broader and more heterogeneous malignancy spectrum than previously recognized.

Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
THE DIAGNOSIS: Mycobacteria infection
Despite the initial biopsy for tissue culture showing no growth, a subsequent biopsy performed 1 month later yielded a positive result. Mycobacterium marinum was identified through organism genome sequencing. The patient was further treated by infectious disease with clarithromycin and ethambutol, with complete resolution of the lesions.
Although initial staining with acid-fast bacilli and tissue culture were negative, we suspected a diagnosis of mycobacterial infection with sporotrichoid spread of multiple nodular and ulcerated lesions that was unresponsive to antibiotics. Performing a tissue culture is crucial for diagnosing mycobacterial skin and soft-tissue infections, as an acid-fast bacilli stain alone cannot distinguish between different mycobacterial species. Lowenstein-Jensen agar is a selective medium specifically used for the culture and isolation of Mycobacterium species. The strict temperature requirement of 30 °C to 32 °C (86-89.6 °F) for the growth of this organism suggests that the infection predominantly affects the limbs, which tend to have a slightly lower temperature compared to the core of the body.1 In our case, the histologic findings and clinical history suggested granulomatous involvement due to fungi or mycobacteria.
Cutaneous leishmaniasis is characterized by ulcers with possible accompanying nodular lymphangitis; however, the patient did not have relevant travel history. Leishmaniasis results from a parasite transmitted by a sandfly, with most cases occurring in Afghanistan, Algeria, Brazil, Iran, Pakistan, Peru, Saudi Arabia, and Syria.2
Ecthyma gangrenosum is characterized by tender necrotic plaques seen predominantly in immunocompromised patients and is associated with Pseudomonas aeruginosa bacteremia.3 Our patient had lesions present for a duration of 5 months, which is inconsistent with the more rapidly progressing course of ecthyma gangrenosum.
Leukocytoclastic vasculitis may manifest with palpable purpura of the lower extremities. An infectious trigger, such as Mycobacterium, may lead to a leukocytoclastic vasculitis. The histopathologic findings classically demonstrate neutrophil deposition in vessel walls, deposition of fibrin in the vessel lumen, and nuclear debris.4
Despite the presence of granulomatous changes in our patient, the presentation of ulcerated nodules in a sporotrichoid pattern on one extremity suggests a diagnosis of infectious etiology rather than sarcoidosis.
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
- Gonçalves IC, Furtado I, Gonçalves MJ, et al. Mycobacterium marinum cutaneous infection: a series of three cases and literature review. Cureus. 2022;14:E31787. doi:10.7759/cureus.31787
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007 /s40257-022-00726-8
- Vaiman M, Lazarovitch T, Heller L, et al. Ecthyma gangrenosum and ecthyma-like lesions: review article. Eur J Clin Microbiol Infect Dis. 2015;34:633-639.
- Baigrie D, Goyal A, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. Updated August 8, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK482159/
Ulcerated Lesions on the Right Leg
Ulcerated Lesions on the Right Leg
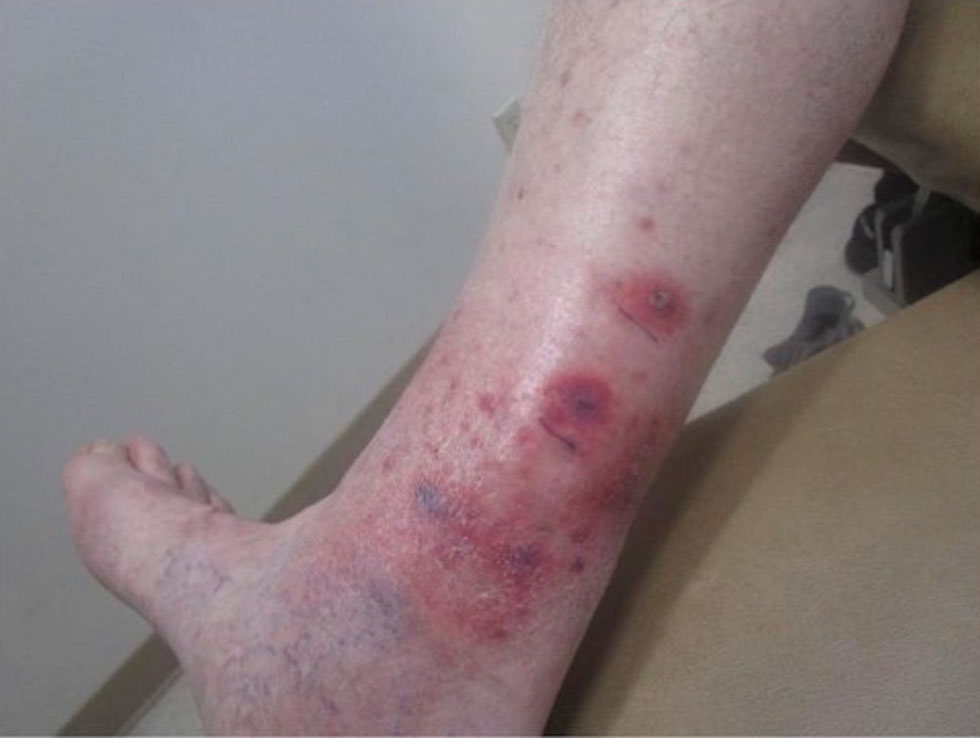
A 78-year-old man was referred to our dermatology clinic for evaluation of nontender erythematous plaques and nodules with central ulceration on the right leg of 5 months’ duration. The patient’s medical history was remarkable for hyperlipidemia, gastroesophageal reflux disease, prostate cancer, and colon cancer status post resection. He denied any relevant travel history but noted that he was an avid hiker and suspected he may have obtained a puncture wound from a bush or a mosquito bite prior to the appearance of the lesions. Previous therapies prescribed by outside physicians and our practice included trimethoprim/sulfamethoxazole, ceftriaxone, levofloxacin, mupirocin, and topical corticosteroids, all with minimal benefit. Clinical examination on initial presentation revealed multiple ulcerations of the lower extremities present for more than 2 months. Punch biopsy of a sample lesion at the current presentation revealed granulomatous change, focal necrosis, and a mixed inflammatory cell infiltrate. Grocott-Gomori methenamine silver and periodic acid–Schiff stains were negative for fungal organisms. The initial acid-fast bacilli stain was negative for mycobacteria, and tissue culture showed no growth.

Multiple Grouped Erythematous to Violaceous Preauricular Papules
Multiple Grouped Erythematous to Violaceous Preauricular Papules
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
THE DIAGNOSIS: Angiolymphoid Hyperplasia With Eosinophilia
Angiolymphoid hyperplasia with eosinophilia (ALHE) is a rare, benign, inflammatory vascular proliferation with lymphocytic and eosinophilic infiltration. Bleeding and pruritus associated with ALHE can substantially affect a patient’s quality of life, necessitating correct diagnosis and effective treatment.1 The etiopathogenesis of ALHE is poorly understood, and it often is attributed to an underlying vascular malformation or local trauma. Vascular proliferation due to hyperestrogenemia could explain why pregnancy is considered a predisposing factor for ALHE.1,2
Angiolymphoid hyperplasia with eosinophilia typically manifests with solitary or multiple pink to red-brown, dome-shaped papules or nodules occurring most frequently on the head and neck. Lesions may be either asymptomatic or associated with pruritus, pain, and spontaneous bleeding.1 Dermoscopy is crucial to diagnosis. The most frequent dermoscopic findings include a polymorphic vascular pattern such as dotted and linear irregular vessels over a pink background, white lines, white dots, white structureless areas, and red-purple lacunae.2,3 Histopathology will demonstrate a vascular proliferation with plump epithelioid endothelial cells showing abundant eosinophilic cytoplasm, accompanied by a variable lymphocytic and eosinophilic inflammatory infiltrate (Figure 1).1
In our case, dermoscopic-histopathologic correlation suggested that the polymorphic vascular pattern and clods on a pink background corresponded to thin- and thick-walled vessels containing plump endothelial cells and intraluminal erythrocytes within the superficial and deep dermis. White structures could represent underlying fibrosis and altered dermal collagen due to vascular proliferation. The brown pigment network and peripheral brownish pigmentation were most likely secondary to increased melanin and accentuation of the pigment network in the setting of Fitzpatrick skin types IV to V, although pruritic trauma with postinflammatory hyperpigmentation may also have contributed, making dermoscopic-histopathologic correlation challenging.
Surgical excision is considered the primary treatment modality for ALHE, with the lowest recurrence rates.1 Alternative therapeutic options include intralesional steroids, cryotherapy, sclerotherapy, radiofrequency, pulsed dye laser, and carbon dioxide laser, with varying efficacy reported.1 Our patient was treated with a combination of a long-pulse Nd:YAG laser (pulse width of 30 ms) to target the vascular component, followed by a single session with an ablative Er:YAG laser. After 4 weeks, healing with good cosmetic results was observed (Figure 2). At 6-month follow-up, there was no recurrence of the lesions.
Kimura disease, often considered the closest differential diagnosis for ALHE, is a rare lymphoproliferative fibroinflammatory condition. Patients present with subcutaneous nodules on the head and neck, often associated with lymphadenopathy. Elevated serum IgE levels and peripheral blood eosinophilia are common.1 Another consideration in the differential diagnosis is cutaneous bacillary angiomatosis caused by Bartonella species, a vascular proliferative condition that mostly affects individuals with HIV, transplant recipients, and those taking immunosuppressive medications.4 Pyogenic granuloma, also known as lobular capillary haemangioma, is another benign vascular proliferation that resembles ALHE. Clinically, it manifests as a solitary, painless, flesh-colored to erythematous papulonodule; however, multiple grouped lesions also can occur. The lesions often are associated with bleeding and erosions.5 Epithelioid hemangioendothelioma is a rare vascular tumor most frequently manifesting in the liver, lungs, or bones, and very rarely is limited to skin. Cutaneous epithelioid hemangioendothelioma mimics ALHE and may manifest as a solitary erythematous mass, multiple dome-shaped masses, or dermal nodules.6
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
- Brahs A, Sledge B, Mullen H, et al. Angiolymphoid hyperplasia with eosinophilia: many syllables, many unanswered questions. J Clin Aesthet Dermatol. 2021;14:49-54.
- Kalantri M, Khopkar U. Spectrum of dermoscopic pattern in a patient with angiolymphoid hyperplasia with tissue eosinophilia. Indian J Dermatol. 2020;65:556-558.
- Chauhan P, Vinay K, Jindal R, et al. Dermoscopic characterisation of angiolymphoid hyperplasia in skin of colour: a case series of six patients with review of literature. Indian J Dermatol Venereol Leprol. 2024;90:848.
- Ramírez Ramírez CR, Saavedra S, Ramírez Ronda CH. Bacillary angiomatosis: microbiology, histopathology, clinical presentation, diagnosis and management. Bol Asoc Med PR. 1996;88:46-51.
- Leung AKC, Barankin B, Hon KL. Pyogenic granuloma. Clinics Mother Child Health. 2014;11:E106. doi:10.4172/2090-7214.1000e106
- Kumar V, Kachhawa D, Rekha S, et al. Cutaneous epithelioid hemangioendothelioma: a rare presentation. Indian J Dermatol Venereol Leprol. 2018;84:739-742.
Multiple Grouped Erythematous to Violaceous Preauricular Papules
Multiple Grouped Erythematous to Violaceous Preauricular Papules
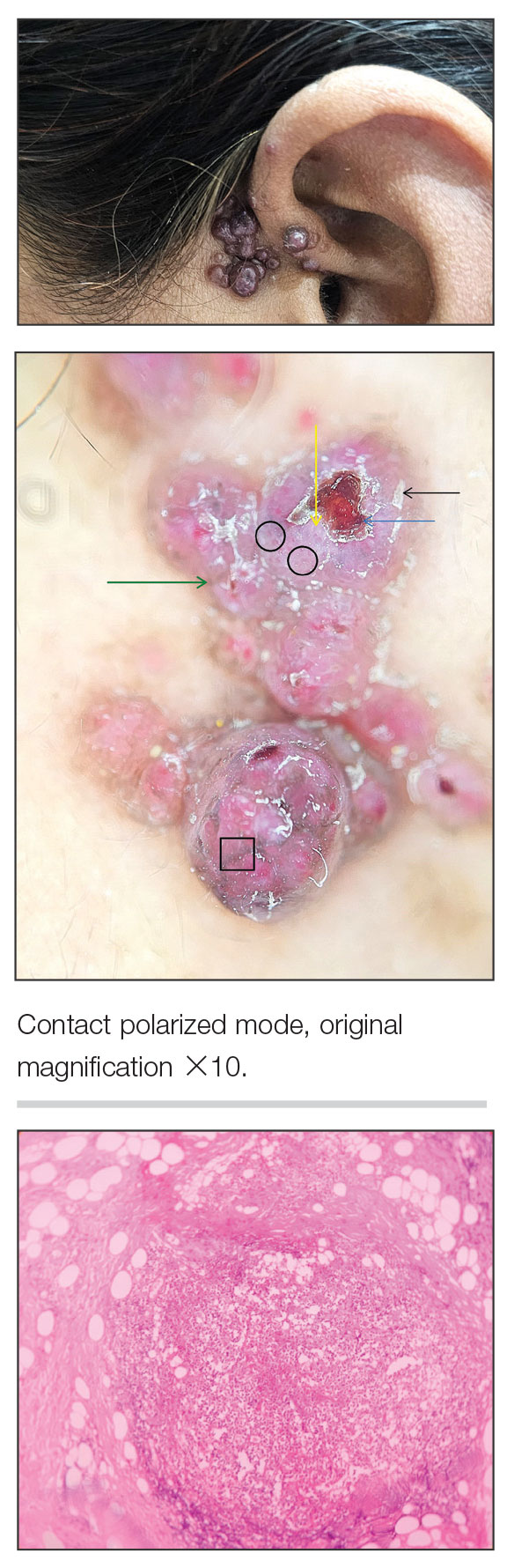
A 35-year-old woman presented with an insidious onset of multiple grouped erythematous to violaceous papules over the left preauricular area of 3 months’ duration (top quiz image). The lesions were soft, itchy, nontender, and friable and were associated with bleeding on excoriation and preauricular lymphadenopathy. Serology for HIV was nonreactive, and Gram staining revealed no bacilli. Laboratory assessment including a complete blood count, urinalysis, and liver and renal function tests was normal.
On dermoscopy (middle quiz image), multiple linear and dotted vessels (circle), reddish lacunae (clods), hemorrhagic crusting (blue arrow), white scaling (black arrow), a brown pigment network (square), white structureless areas (yellow arrow), and white lines were seen over a pale-pink background (green arrow). Scaling and crusting over some lesions, along with a peripheral rim of scaling and brownish pigmentation, also was appreciated. Histopathology revealed a proliferation of vascular channels admixed with lymphocytes, plasma cells, and eosinophils along with a proliferation of thin- and thick-walled blood vessels in the superficial as well as deep dermis (bottom quiz image).

Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
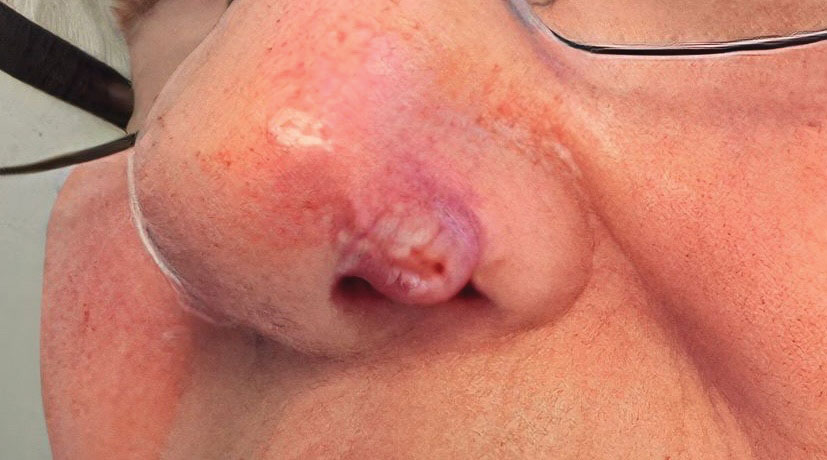
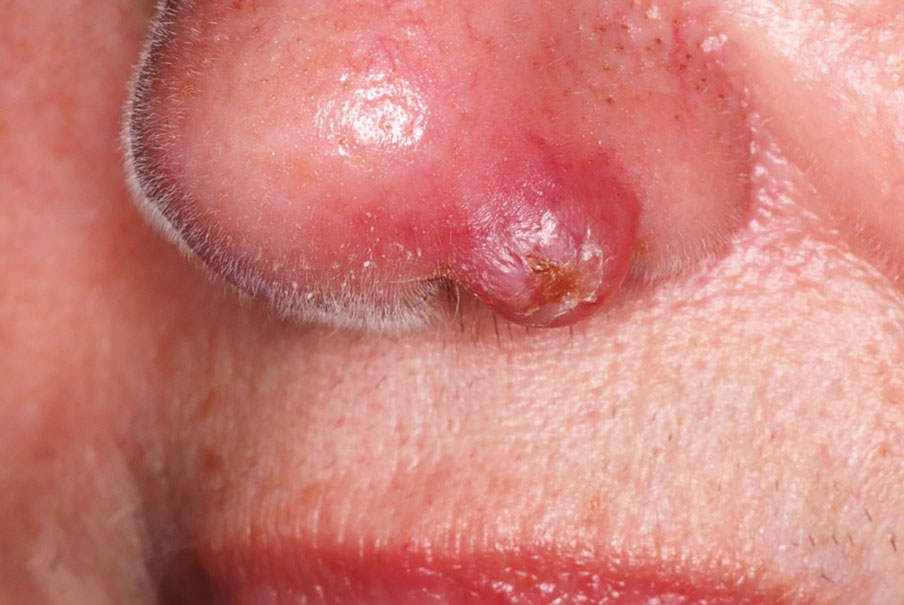
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
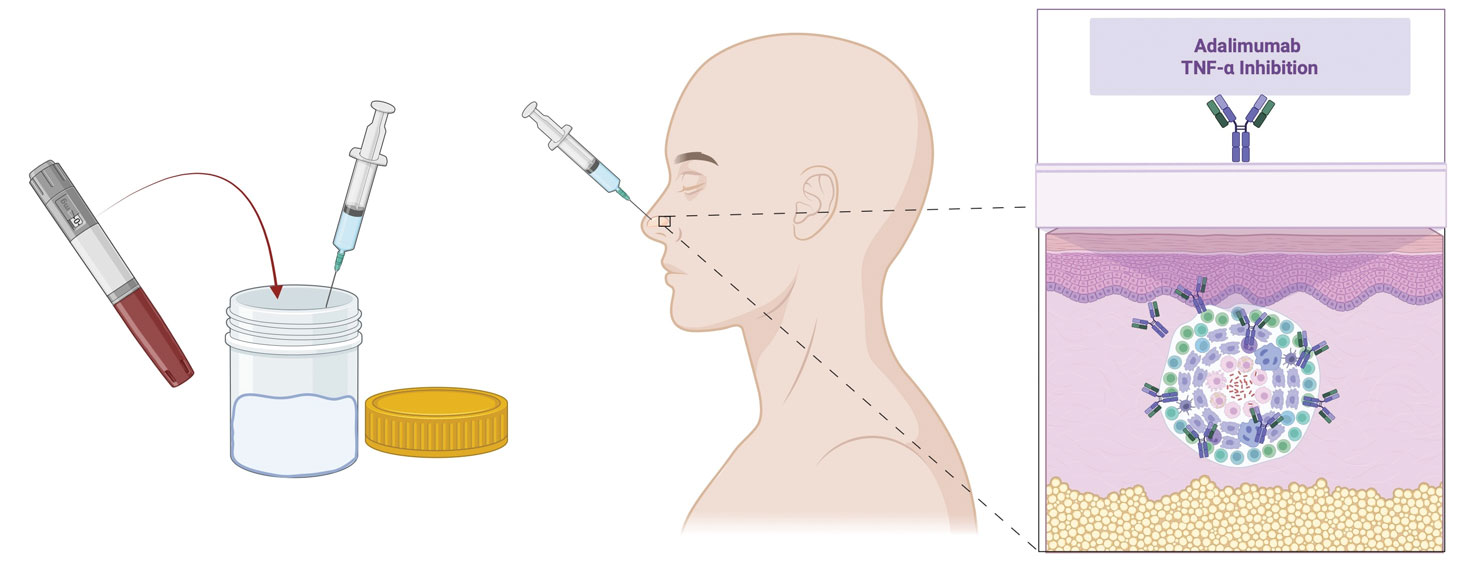
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
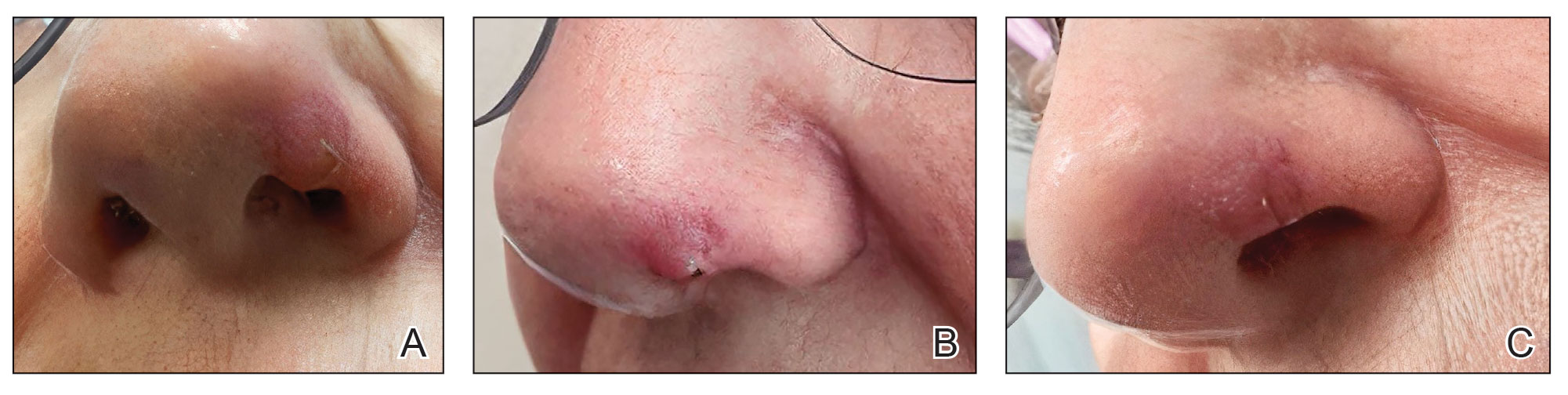
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
Practice Gap
Chronic localized granulomatous inflammation can be difficult to manage, particularly when manifesting on the face. Intralesional corticosteroids may lead to atrophy and dyspigmentation and therefore must be used cautiously in cosmetically sensitive areas.1 Surgical removal can lead to recurrence, and systemic agents may carry risks disproportionate to disease burden. Although tumor necrosis factor (TNF) α inhibitors are effective systemically, their localized use in cutaneous granulomatous dermatoses remains underreported.1-3 We describe a technique using intralesional injection of adalimumab to treat chronic refractory cutaneous granulomatous inflammation.
The Technique
A 69-year-old woman presented with a crusted erythematous papule with surrounding inflammation on the left nasal ala of 5 years’ duration (Figure 1). Histopathology demonstrated a localized cutaneous granulomatous process. There was no clinical, radiographic, or laboratory evidence of systemic sarcoidosis. Infectious causes were excluded through negative tissue cultures and special stains, including auramine-rhodamine. Over a 3-month period following initial presentation, the lesion proved refractory to intralesional 5-fluorouracil, intralesional triamcinolone acetonide, pentoxifylline, N-acetylcysteine, and shave excision (Figure 2).
At 3-month follow-up, given the lesion’s persistence despite local and systemic anti-inflammatory approaches and our intent to avoid repeated corticosteroid exposure or more aggressive surgery in a cosmetically sensitive facial site, we attempted treatment with intralesional adalimumab. A 40-mg/0.4-mL dose of adalimumab was withdrawn directly from a prefilled autoinjector and placed into a sterile container, then transferred to a syringe fitted with a 30-gauge needle. Finally, the full 0.4 mL was injected intralesionally (Figure 3) until complete blanching of the lesion was achieved.
At 1-month follow-up, the lesion demonstrated decreased erythema and crusting (Figure 4A). The patient subsequently underwent 12 adalimumab injections over an 18-month period with marked reduction in size and erythema of the lesion without complications (Figure 4B). In addition, doxycycline 100 mg/d was started 11 months after the first adalimumab injection to address mild residual inflammation (Figure 4C); after 4 months, the dose was reduced to 50 mg/d due to gastrointestinal adverse effects. Doxycycline was maintained for 3 additional months with persistent improvement of the lesion.
Practice Implication
Intralesional administration of adalimumab may represent a useful therapeutic option for localized refractory granulomatous inflammation, particularly in sensitive areas such as the face, where conventional therapies may be limited by adverse effects or suboptimal response. Localized delivery of TNF-α inhibition directly to the site of inflammation may allow for clinical improvement while minimizing systemic exposure associated with biologic therapy.2 This approach may be particularly advantageous in cases in which repeated intralesional corticosteroid injections raise concern for atrophy or dyspigmentation, or when surgical intervention carries a risk for recurrence or cosmetic morbidity.1,2 Given the established role of TNF-α in granuloma formation and maintenance, intralesional adalimumab provides a biologically plausible targeted therapeutic strategy. Further studies are needed to evaluate the potential applications in other cutaneous granulomatous dermatoses.2,3
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
- Philips MA, Lynch J, Azmi FH. Ulcerative cutaneous sarcoidosis responding to adalimumab. J Am Acad Dermatol. 2005;53:917. doi:10.1016/j.jaad.2005.02.023
- Balan K, Sagut P, Ederle AC, et al. Cutaneous sarcoidosis treated with intralesional adalimumab. Int J Dermatol. 2025;64:1120-1121. doi:10.1111/ijd.17549
- Dunn C, Whitney Z, Foss M, et al. Intralesional certolizumab for refractory lupus pernio. JAMA Dermatol. 2023;159:890-891. doi:10.1001 /jamadermatol.2023.0987
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation
Using Intralesional Adalimumab for Chronic Refractory Cutaneous Granulomatous Inflammation

Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful nodules, abscesses, scarring, and sinus tracts that commonly manifest in the axillary, inguinal, perianal, and inframammary regions.1 Hidradenitis suppurativa has been associated with several metabolic and cardiovascular comorbidities as well as polycystic ovary syndrome (PCOS)(recently renamed polyendocrine metabolic ovarian syndrome),2,3 a condition characterized by hyperandrogenism, chronic anovulation, and polycystic ovaries.2 Multiple comorbidities of PCOS overlap with those of HS, including type 2 diabetes, cardiovascular disease, and metabolic syndrome.1,3-5 While HS may be associated with PCOS, there is limited literature analyzing the association between these conditions. This study aimed to analyze the association between HS and PCOS using data from the National Institute of Health’s All of Us Research Program database (https://allofus.nih.gov/). While other studies have looked at the association between HS and PCOS, ours is among the first to analyze the relationship between multiple race/ ethnicity groups, which is especially important given racial disparities in HS and comorbid diseases.
Methods
A cross-sectional, population-based study of females included in the All of Us Research Program database was conducted. Patients with HS were identified using the Systematized Nomenclature of Medicine–Clinical Terms (SNOMED CT) code 59393003, while PCOS was identified with the code 237055002. Type 2 diabetes was identified with the following SNOMED CT codes: 44054006, 313436004, 237599002, 199230006, 359642000, and 81531005. Obesity was identified with the following codes: 414916001, 238136002, 190966007, 296526005, 294493008, 238134004, 83911000119104, and 415530009. Male patients and those who did not answer questions regarding sociodemographic variables were excluded from the final analysis. P values were calculated using Pearson χ2 tests. Multivariate logistic regression was used to calculate adjusted odds ratios and unadjusted odds ratios to analyze the association between HS and PCOS while controlling for age, race/ethnicity, smoking status, type 2 diabetes, and obesity. Statistical analyses were conducted using a 95% CI.
Results
The final analysis included 78,742 patients. The prevalence of PCOS was 5.64% in the HS group vs 0.93% in the non-HS group (eTable 1). Individuals with HS had higher rates of smoking cigarettes (57.71% vs 37.67%), obesity (51.08% vs 17.22%), and type 2 diabetes (20.73% vs 9.11%) than individuals without HS, respectively.
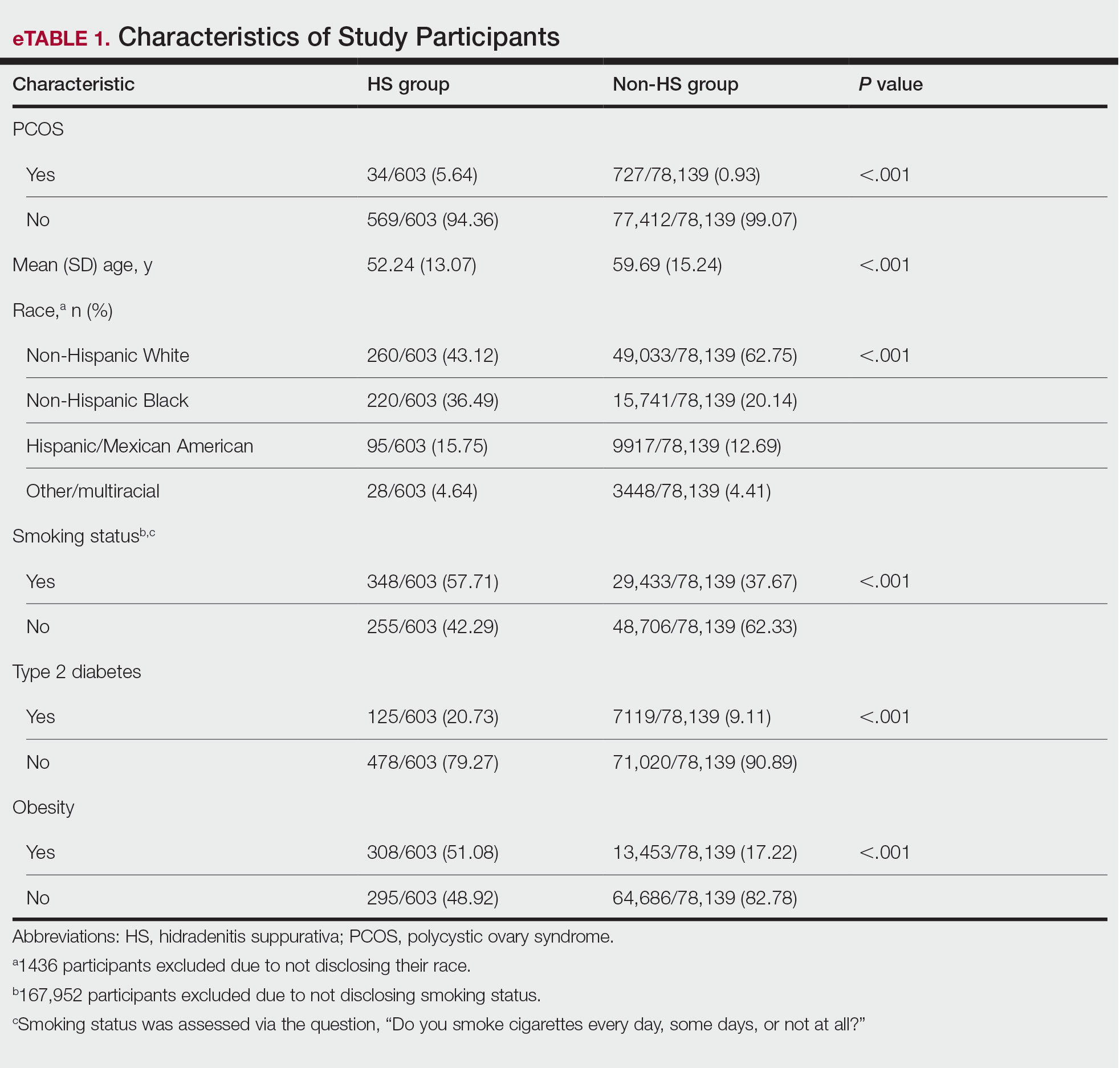
Multivariate logistic regression analyses revealed that individuals with HS were 2.06 times more likely to have PCOS after adjusting for sociodemographic variables and comorbidities (95% CI, 1.41-3.02; P<.001). Adjusted subgroup analyses by race/ethnicity did not yield statistically significant results; however, unadjusted analyses revealed that individuals with HS had significantly increased odds of PCOS across all race/ethnicity groups (eTable 2). Interaction terms analysis to determine if the relationship between HS and PCOS differs by race/ ethnicity did not yield statistically significant results. However, independent of HS status, non-Hispanic Black and Hispanic patients were less likely to have PCOS compared to White individuals (adjusted odds ratio, 0.37 and 0.56, respectively; P<.001). Disparities in access to care could have led to underdiagnosis of PCOS among non-Hispanic Black and Hispanic patients. Lastly, individuals with type 2 diabetes were 10.43 times more likely to have PCOS than those without, while patients with obesity were 11.14 times more likely to have PCOS than those without.

Comment
This study demonstrated that females with HS are 2.06 times more likely to have PCOS than those without HS, even after controlling for important sociodemographic variables and comorbidities. While adjusted subgroup analyses did not yield statistically significant results, unadjusted analyses demonstrated increased odds of PCOS in patients with HS across all race/ethnicity groups, suggesting that sociodemographic variables and comorbidities substantially influence the relationship between HS and PCOS; for instance, patients with type 2 diabetes and obesity are approximately 10- to 11-fold more likely to have PCOS than patients without these conditions. Non-Hispanic Black and Hispanic patients were less likely to have PCOS compared with White patients, indicating possible underdiagnosis of PCOS in these populations and highlighting the need for increased PCOS screening. Limitations of this study include the reliance on SNOMED CT codes, which may have led to underdiagnosis of HS or PCOS, as well as the inability to differentiate between mild and severe HS in the database.
Hyperandrogenism is believed to contribute to the pathogenesis of both HS and PCOS, supporting the potential use of antiandrogen therapies, such as spironolactone, in managing both conditions.2,3 Furthermore, oral contraceptives may have a role in managing both conditions. In HS, oral contraceptives help to mitigate flares associated with hormonal changes during menstruation, while in PCOS, they are used to regulate the hormonal cycle and reduce hirsutism.2-4 However, not all women experience menstrual flares of HS, suggesting that variations in HS phenotypes may influence individual responses to hormonal changes.1 Additionally, the considerable overlap in metabolic and cardiovascular comorbidities between HS and PCOS indicates that shared pathomechanisms may contribute to the association between these conditions.1,2 For example, proinflammatory adipokines released in both HS and PCOS may contribute to inflammation, cardiovascular disease, and insulin resistance.3,5
Conclusion
Further research is needed to better understand the shared pathophysiology that links these 2 diseases and to identify targeted approaches for optimizing management and improving patient outcomes. The association between HS and PCOS highlights the importance of screening for metabolic and reproductive comorbidities in patients with HS. Early recognition and management of both HS and PCOS can improve long-term outcomes.
- van Straalen KR, Prens EP, Gudjonsson JE. Insights into hidradenitis suppurativa. J Allergy Clin Immunol. 2022;149:1150-1161. doi:10.1016 /j.jaci.2022.02.003
- Choudhari R, Tayade S, Tiwari A, et al. Diagnosis, management, and associated comorbidities of polycystic ovary syndrome: a narrative review. Cureus. 2024;16:e58733. doi:10.7759/cureus.58733
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Montero-Vilchez T, Valenzuela-Amigo A, Cuenca-Barrales C, et al. The role of oral contraceptive pills in hidradenitis suppurativa: a cohort study. Life (Basel). 2021;11:697. doi:10.3390/life11070697
- Randeva HS, Tan BK, Weickert MO, et al. Cardiometabolic aspects of the polycystic ovary syndrome. Endocr Rev. 2012;33:812-841. doi:10.1210/er.2012-1003
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful nodules, abscesses, scarring, and sinus tracts that commonly manifest in the axillary, inguinal, perianal, and inframammary regions.1 Hidradenitis suppurativa has been associated with several metabolic and cardiovascular comorbidities as well as polycystic ovary syndrome (PCOS)(recently renamed polyendocrine metabolic ovarian syndrome),2,3 a condition characterized by hyperandrogenism, chronic anovulation, and polycystic ovaries.2 Multiple comorbidities of PCOS overlap with those of HS, including type 2 diabetes, cardiovascular disease, and metabolic syndrome.1,3-5 While HS may be associated with PCOS, there is limited literature analyzing the association between these conditions. This study aimed to analyze the association between HS and PCOS using data from the National Institute of Health’s All of Us Research Program database (https://allofus.nih.gov/). While other studies have looked at the association between HS and PCOS, ours is among the first to analyze the relationship between multiple race/ ethnicity groups, which is especially important given racial disparities in HS and comorbid diseases.
Methods
A cross-sectional, population-based study of females included in the All of Us Research Program database was conducted. Patients with HS were identified using the Systematized Nomenclature of Medicine–Clinical Terms (SNOMED CT) code 59393003, while PCOS was identified with the code 237055002. Type 2 diabetes was identified with the following SNOMED CT codes: 44054006, 313436004, 237599002, 199230006, 359642000, and 81531005. Obesity was identified with the following codes: 414916001, 238136002, 190966007, 296526005, 294493008, 238134004, 83911000119104, and 415530009. Male patients and those who did not answer questions regarding sociodemographic variables were excluded from the final analysis. P values were calculated using Pearson χ2 tests. Multivariate logistic regression was used to calculate adjusted odds ratios and unadjusted odds ratios to analyze the association between HS and PCOS while controlling for age, race/ethnicity, smoking status, type 2 diabetes, and obesity. Statistical analyses were conducted using a 95% CI.
Results
The final analysis included 78,742 patients. The prevalence of PCOS was 5.64% in the HS group vs 0.93% in the non-HS group (eTable 1). Individuals with HS had higher rates of smoking cigarettes (57.71% vs 37.67%), obesity (51.08% vs 17.22%), and type 2 diabetes (20.73% vs 9.11%) than individuals without HS, respectively.
Multivariate logistic regression analyses revealed that individuals with HS were 2.06 times more likely to have PCOS after adjusting for sociodemographic variables and comorbidities (95% CI, 1.41-3.02; P<.001). Adjusted subgroup analyses by race/ethnicity did not yield statistically significant results; however, unadjusted analyses revealed that individuals with HS had significantly increased odds of PCOS across all race/ethnicity groups (eTable 2). Interaction terms analysis to determine if the relationship between HS and PCOS differs by race/ ethnicity did not yield statistically significant results. However, independent of HS status, non-Hispanic Black and Hispanic patients were less likely to have PCOS compared to White individuals (adjusted odds ratio, 0.37 and 0.56, respectively; P<.001). Disparities in access to care could have led to underdiagnosis of PCOS among non-Hispanic Black and Hispanic patients. Lastly, individuals with type 2 diabetes were 10.43 times more likely to have PCOS than those without, while patients with obesity were 11.14 times more likely to have PCOS than those without.
Comment
This study demonstrated that females with HS are 2.06 times more likely to have PCOS than those without HS, even after controlling for important sociodemographic variables and comorbidities. While adjusted subgroup analyses did not yield statistically significant results, unadjusted analyses demonstrated increased odds of PCOS in patients with HS across all race/ethnicity groups, suggesting that sociodemographic variables and comorbidities substantially influence the relationship between HS and PCOS; for instance, patients with type 2 diabetes and obesity are approximately 10- to 11-fold more likely to have PCOS than patients without these conditions. Non-Hispanic Black and Hispanic patients were less likely to have PCOS compared with White patients, indicating possible underdiagnosis of PCOS in these populations and highlighting the need for increased PCOS screening. Limitations of this study include the reliance on SNOMED CT codes, which may have led to underdiagnosis of HS or PCOS, as well as the inability to differentiate between mild and severe HS in the database.
Hyperandrogenism is believed to contribute to the pathogenesis of both HS and PCOS, supporting the potential use of antiandrogen therapies, such as spironolactone, in managing both conditions.2,3 Furthermore, oral contraceptives may have a role in managing both conditions. In HS, oral contraceptives help to mitigate flares associated with hormonal changes during menstruation, while in PCOS, they are used to regulate the hormonal cycle and reduce hirsutism.2-4 However, not all women experience menstrual flares of HS, suggesting that variations in HS phenotypes may influence individual responses to hormonal changes.1 Additionally, the considerable overlap in metabolic and cardiovascular comorbidities between HS and PCOS indicates that shared pathomechanisms may contribute to the association between these conditions.1,2 For example, proinflammatory adipokines released in both HS and PCOS may contribute to inflammation, cardiovascular disease, and insulin resistance.3,5
Conclusion
Further research is needed to better understand the shared pathophysiology that links these 2 diseases and to identify targeted approaches for optimizing management and improving patient outcomes. The association between HS and PCOS highlights the importance of screening for metabolic and reproductive comorbidities in patients with HS. Early recognition and management of both HS and PCOS can improve long-term outcomes.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by painful nodules, abscesses, scarring, and sinus tracts that commonly manifest in the axillary, inguinal, perianal, and inframammary regions.1 Hidradenitis suppurativa has been associated with several metabolic and cardiovascular comorbidities as well as polycystic ovary syndrome (PCOS)(recently renamed polyendocrine metabolic ovarian syndrome),2,3 a condition characterized by hyperandrogenism, chronic anovulation, and polycystic ovaries.2 Multiple comorbidities of PCOS overlap with those of HS, including type 2 diabetes, cardiovascular disease, and metabolic syndrome.1,3-5 While HS may be associated with PCOS, there is limited literature analyzing the association between these conditions. This study aimed to analyze the association between HS and PCOS using data from the National Institute of Health’s All of Us Research Program database (https://allofus.nih.gov/). While other studies have looked at the association between HS and PCOS, ours is among the first to analyze the relationship between multiple race/ ethnicity groups, which is especially important given racial disparities in HS and comorbid diseases.
Methods
A cross-sectional, population-based study of females included in the All of Us Research Program database was conducted. Patients with HS were identified using the Systematized Nomenclature of Medicine–Clinical Terms (SNOMED CT) code 59393003, while PCOS was identified with the code 237055002. Type 2 diabetes was identified with the following SNOMED CT codes: 44054006, 313436004, 237599002, 199230006, 359642000, and 81531005. Obesity was identified with the following codes: 414916001, 238136002, 190966007, 296526005, 294493008, 238134004, 83911000119104, and 415530009. Male patients and those who did not answer questions regarding sociodemographic variables were excluded from the final analysis. P values were calculated using Pearson χ2 tests. Multivariate logistic regression was used to calculate adjusted odds ratios and unadjusted odds ratios to analyze the association between HS and PCOS while controlling for age, race/ethnicity, smoking status, type 2 diabetes, and obesity. Statistical analyses were conducted using a 95% CI.
Results
The final analysis included 78,742 patients. The prevalence of PCOS was 5.64% in the HS group vs 0.93% in the non-HS group (eTable 1). Individuals with HS had higher rates of smoking cigarettes (57.71% vs 37.67%), obesity (51.08% vs 17.22%), and type 2 diabetes (20.73% vs 9.11%) than individuals without HS, respectively.
Multivariate logistic regression analyses revealed that individuals with HS were 2.06 times more likely to have PCOS after adjusting for sociodemographic variables and comorbidities (95% CI, 1.41-3.02; P<.001). Adjusted subgroup analyses by race/ethnicity did not yield statistically significant results; however, unadjusted analyses revealed that individuals with HS had significantly increased odds of PCOS across all race/ethnicity groups (eTable 2). Interaction terms analysis to determine if the relationship between HS and PCOS differs by race/ ethnicity did not yield statistically significant results. However, independent of HS status, non-Hispanic Black and Hispanic patients were less likely to have PCOS compared to White individuals (adjusted odds ratio, 0.37 and 0.56, respectively; P<.001). Disparities in access to care could have led to underdiagnosis of PCOS among non-Hispanic Black and Hispanic patients. Lastly, individuals with type 2 diabetes were 10.43 times more likely to have PCOS than those without, while patients with obesity were 11.14 times more likely to have PCOS than those without.
Comment
This study demonstrated that females with HS are 2.06 times more likely to have PCOS than those without HS, even after controlling for important sociodemographic variables and comorbidities. While adjusted subgroup analyses did not yield statistically significant results, unadjusted analyses demonstrated increased odds of PCOS in patients with HS across all race/ethnicity groups, suggesting that sociodemographic variables and comorbidities substantially influence the relationship between HS and PCOS; for instance, patients with type 2 diabetes and obesity are approximately 10- to 11-fold more likely to have PCOS than patients without these conditions. Non-Hispanic Black and Hispanic patients were less likely to have PCOS compared with White patients, indicating possible underdiagnosis of PCOS in these populations and highlighting the need for increased PCOS screening. Limitations of this study include the reliance on SNOMED CT codes, which may have led to underdiagnosis of HS or PCOS, as well as the inability to differentiate between mild and severe HS in the database.
Hyperandrogenism is believed to contribute to the pathogenesis of both HS and PCOS, supporting the potential use of antiandrogen therapies, such as spironolactone, in managing both conditions.2,3 Furthermore, oral contraceptives may have a role in managing both conditions. In HS, oral contraceptives help to mitigate flares associated with hormonal changes during menstruation, while in PCOS, they are used to regulate the hormonal cycle and reduce hirsutism.2-4 However, not all women experience menstrual flares of HS, suggesting that variations in HS phenotypes may influence individual responses to hormonal changes.1 Additionally, the considerable overlap in metabolic and cardiovascular comorbidities between HS and PCOS indicates that shared pathomechanisms may contribute to the association between these conditions.1,2 For example, proinflammatory adipokines released in both HS and PCOS may contribute to inflammation, cardiovascular disease, and insulin resistance.3,5
Conclusion
Further research is needed to better understand the shared pathophysiology that links these 2 diseases and to identify targeted approaches for optimizing management and improving patient outcomes. The association between HS and PCOS highlights the importance of screening for metabolic and reproductive comorbidities in patients with HS. Early recognition and management of both HS and PCOS can improve long-term outcomes.
- van Straalen KR, Prens EP, Gudjonsson JE. Insights into hidradenitis suppurativa. J Allergy Clin Immunol. 2022;149:1150-1161. doi:10.1016 /j.jaci.2022.02.003
- Choudhari R, Tayade S, Tiwari A, et al. Diagnosis, management, and associated comorbidities of polycystic ovary syndrome: a narrative review. Cureus. 2024;16:e58733. doi:10.7759/cureus.58733
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Montero-Vilchez T, Valenzuela-Amigo A, Cuenca-Barrales C, et al. The role of oral contraceptive pills in hidradenitis suppurativa: a cohort study. Life (Basel). 2021;11:697. doi:10.3390/life11070697
- Randeva HS, Tan BK, Weickert MO, et al. Cardiometabolic aspects of the polycystic ovary syndrome. Endocr Rev. 2012;33:812-841. doi:10.1210/er.2012-1003
- van Straalen KR, Prens EP, Gudjonsson JE. Insights into hidradenitis suppurativa. J Allergy Clin Immunol. 2022;149:1150-1161. doi:10.1016 /j.jaci.2022.02.003
- Choudhari R, Tayade S, Tiwari A, et al. Diagnosis, management, and associated comorbidities of polycystic ovary syndrome: a narrative review. Cureus. 2024;16:e58733. doi:10.7759/cureus.58733
- Abu Rached N, Gambichler T, Dietrich JW, et al. The role of hormones in hidradenitis suppurativa: a systematic review. Int J Mol Sci. 2022;23:15250. doi:10.3390/ijms232315250
- Montero-Vilchez T, Valenzuela-Amigo A, Cuenca-Barrales C, et al. The role of oral contraceptive pills in hidradenitis suppurativa: a cohort study. Life (Basel). 2021;11:697. doi:10.3390/life11070697
- Randeva HS, Tan BK, Weickert MO, et al. Cardiometabolic aspects of the polycystic ovary syndrome. Endocr Rev. 2012;33:812-841. doi:10.1210/er.2012-1003
Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
Association Between Hidradenitis Suppurativa and Polycystic Ovary Syndrome
PRACTICE POINTS
- Patients with hidradenitis suppurativa were 2.06 times more likely to have polycystic ovary syndrome (PCOS) than patients without HS after controlling for age, race/ ethnicity, tobacco use, type 2 diabetes, and obesity.
- Non-Hispanic Black and Hispanic patients were less likely than White patients to have a diagnosis of PCOS, potentially reflecting underdiagnosis in these populations.
- Individuals with type 2 diabetes and obesity were 10.43 and 11.14 times more likely, respectively, to have PCOS.

AI Scribes or VHA Docs: Which Created Better Clinical Notes?
Artificial intelligence (AI) scribes produced lower-quality documentation of clinical notes than human clinicians, and especially struggled in settings with background noise or clinicians wearing masks, a new Veterans Health Administration (VHA) study finds.
In 5 simulated clinical cases, notes written by various AI programs scored lower than reports produced by humans on the modified Physician Documentation Quality Instrument (PDQI-9), a measurement of note quality scale, reported Ashok Reddy, MD, MSc, of the University of Washington and Veterans Affairs Puget Sound Health Care System, Seattle, et al in the April issue of Annals of Internal Medicine.
AI scribes scored lower compared with humans across all domains, including accuracy, thoroughness, and usefulness. There was an especially large gap in scores on the 50-point PDQI-9 in an acute low back pain case (human, 43.8 points; AI, 20.3 points; difference, 23.5 points).
“For clinicians, AI scribes should be regarded as tools for generating draft documentation that requires review and editing, rather than as a substitute for clinician-authored notes,” the authors wrote. “Although ambient AI scribes hold promise for reducing clinician burden, rigorous and ongoing evaluation of their quality is essential to ensure that these tools enhance rather than compromise the quality of clinical care.”
AI Scribe Use is Widespread
Taylor N. Anderson, MD, a clinical informatics fellow at Oregon Health & Science University, Portland, is familiar with the study findings and noted that the use of AI scribes in medicine has grown rapidly. All major health organizations are either using it or facing “enormous pressure” from clinicians to do so, she told Federal Practitioner.
Previous research has linked the use of AI scribes for clinical notes to less electronic health record usage and documentation time for clinicians, leading to more time for patient visits. Still, the quality of clinical notes written by AI is “quite variable across vendors,” Anderson said.
Anderson led a 2025 study that examined 5 AI scribe platforms and found an average of 3.0 errors per case with “potential for moderate-to-severe harm.”
For the new study on the simulated cases, part of a VHA-sponsored “technology sprint” via Challenge.gov, researchers developed audio descriptions of 5 clinical cases reflecting common patient encounters in primary care: acute low back pain, chest pain, a new diagnosis of diabetes, a pharmacy consultation, and a follow-up with a nurse case manager for heart failure.
Two cases included non-English accents, 1 included background noise, and 1 featured speech through a medical mask. All the “patients” were played by what the authors described as “trained standardized patient actors.”
For each case, 3 humans and 11 AI scribe programs produced clinical notes. The clinical notes were then evaluated by 6 raters.
Researchers found that AI scribe-generated notes scored worse than human-generated notes across all 10 domains of the modified PDQI-9 (accuracy, thoroughness, usefulness, organization, comprehensiveness, succinctness, synthesization, internal consistency, and freedom from hallucination and bias).
There were especially large gaps between the AI and human notes in the domains of thoroughness, organization, and usefulness. Even wider gaps were observed for the encounters with noise and mask usage.
“These findings highlight that although ambient AI scribes can generate complete notes, the overall quality remains broadly below that of human-authored documentation,” the authors wrote.
No Comparison Between AI Scribes
The researchers noted that “given contractual limitations, we cannot interpret the results for specific vendors.” They also noted that the study did not use professional scribes, who may produce even higher-quality results, and the humans were not producing notes in a real-world clinical environment.
Anderson, the clinical informatics fellow, pointed out that the study does not examine the common scenario in which a clinician edits notes produced by an AI scribe. In fact, she said, there is no current research on this, failing to examine “the postediting note that would actually go into the chart.”
In an accompanying commentary, collaborative scientist Aaron Tierney, PhD, and Kristine Lee, MD, an associate executive director, both with the Permanente Medical Group, California, called for future research to focus on “real-world performance, promote the development of documentation policies that prioritize patient care over billing requirements, and systematically incorporate patient perspectives into assessments of quality.”
Why AI Misses the Mark
In an interview with Federal Practitioner, AI researcher Maxim Topaz, PhD, RN, MA, an associate professor of Nursing and Data Science at Columbia University School of Nursing, New York City, who is familiar with the study but did not participate in it, praised the research.
He pointed out that AI has trouble accurately representing clinical encounters because they “tend to fill gaps with plausible-sounding language, which can mask omissions and make errors harder to catch.” Also, “ambient scribes can only document what is verbalized aloud. Physical exam findings the clinician notices but does not narrate, nonverbal cues, and patient-initiated concerns that drift past in conversation are systematically underrepresented.”
Moving forward, Topaz advised clinicians to “treat AI-generated notes as a first draft, not a finished product. Read them carefully, especially for omissions, which the current evidence suggests are by far the most common error type and which are harder to spot than fabrications because the surrounding note still reads coherently.”
Two study authors disclosed employment by the US Department of Veterans Affairs. Other authors had no disclosures. The commentary authors have no disclosures. Anderson has no disclosures. Topaz discloses relationships with the National Institutes of Health and other federal sources.
Artificial intelligence (AI) scribes produced lower-quality documentation of clinical notes than human clinicians, and especially struggled in settings with background noise or clinicians wearing masks, a new Veterans Health Administration (VHA) study finds.
In 5 simulated clinical cases, notes written by various AI programs scored lower than reports produced by humans on the modified Physician Documentation Quality Instrument (PDQI-9), a measurement of note quality scale, reported Ashok Reddy, MD, MSc, of the University of Washington and Veterans Affairs Puget Sound Health Care System, Seattle, et al in the April issue of Annals of Internal Medicine.
AI scribes scored lower compared with humans across all domains, including accuracy, thoroughness, and usefulness. There was an especially large gap in scores on the 50-point PDQI-9 in an acute low back pain case (human, 43.8 points; AI, 20.3 points; difference, 23.5 points).
“For clinicians, AI scribes should be regarded as tools for generating draft documentation that requires review and editing, rather than as a substitute for clinician-authored notes,” the authors wrote. “Although ambient AI scribes hold promise for reducing clinician burden, rigorous and ongoing evaluation of their quality is essential to ensure that these tools enhance rather than compromise the quality of clinical care.”
AI Scribe Use is Widespread
Taylor N. Anderson, MD, a clinical informatics fellow at Oregon Health & Science University, Portland, is familiar with the study findings and noted that the use of AI scribes in medicine has grown rapidly. All major health organizations are either using it or facing “enormous pressure” from clinicians to do so, she told Federal Practitioner.
Previous research has linked the use of AI scribes for clinical notes to less electronic health record usage and documentation time for clinicians, leading to more time for patient visits. Still, the quality of clinical notes written by AI is “quite variable across vendors,” Anderson said.
Anderson led a 2025 study that examined 5 AI scribe platforms and found an average of 3.0 errors per case with “potential for moderate-to-severe harm.”
For the new study on the simulated cases, part of a VHA-sponsored “technology sprint” via Challenge.gov, researchers developed audio descriptions of 5 clinical cases reflecting common patient encounters in primary care: acute low back pain, chest pain, a new diagnosis of diabetes, a pharmacy consultation, and a follow-up with a nurse case manager for heart failure.
Two cases included non-English accents, 1 included background noise, and 1 featured speech through a medical mask. All the “patients” were played by what the authors described as “trained standardized patient actors.”
For each case, 3 humans and 11 AI scribe programs produced clinical notes. The clinical notes were then evaluated by 6 raters.
Researchers found that AI scribe-generated notes scored worse than human-generated notes across all 10 domains of the modified PDQI-9 (accuracy, thoroughness, usefulness, organization, comprehensiveness, succinctness, synthesization, internal consistency, and freedom from hallucination and bias).
There were especially large gaps between the AI and human notes in the domains of thoroughness, organization, and usefulness. Even wider gaps were observed for the encounters with noise and mask usage.
“These findings highlight that although ambient AI scribes can generate complete notes, the overall quality remains broadly below that of human-authored documentation,” the authors wrote.
No Comparison Between AI Scribes
The researchers noted that “given contractual limitations, we cannot interpret the results for specific vendors.” They also noted that the study did not use professional scribes, who may produce even higher-quality results, and the humans were not producing notes in a real-world clinical environment.
Anderson, the clinical informatics fellow, pointed out that the study does not examine the common scenario in which a clinician edits notes produced by an AI scribe. In fact, she said, there is no current research on this, failing to examine “the postediting note that would actually go into the chart.”
In an accompanying commentary, collaborative scientist Aaron Tierney, PhD, and Kristine Lee, MD, an associate executive director, both with the Permanente Medical Group, California, called for future research to focus on “real-world performance, promote the development of documentation policies that prioritize patient care over billing requirements, and systematically incorporate patient perspectives into assessments of quality.”
Why AI Misses the Mark
In an interview with Federal Practitioner, AI researcher Maxim Topaz, PhD, RN, MA, an associate professor of Nursing and Data Science at Columbia University School of Nursing, New York City, who is familiar with the study but did not participate in it, praised the research.
He pointed out that AI has trouble accurately representing clinical encounters because they “tend to fill gaps with plausible-sounding language, which can mask omissions and make errors harder to catch.” Also, “ambient scribes can only document what is verbalized aloud. Physical exam findings the clinician notices but does not narrate, nonverbal cues, and patient-initiated concerns that drift past in conversation are systematically underrepresented.”
Moving forward, Topaz advised clinicians to “treat AI-generated notes as a first draft, not a finished product. Read them carefully, especially for omissions, which the current evidence suggests are by far the most common error type and which are harder to spot than fabrications because the surrounding note still reads coherently.”
Two study authors disclosed employment by the US Department of Veterans Affairs. Other authors had no disclosures. The commentary authors have no disclosures. Anderson has no disclosures. Topaz discloses relationships with the National Institutes of Health and other federal sources.
Artificial intelligence (AI) scribes produced lower-quality documentation of clinical notes than human clinicians, and especially struggled in settings with background noise or clinicians wearing masks, a new Veterans Health Administration (VHA) study finds.
In 5 simulated clinical cases, notes written by various AI programs scored lower than reports produced by humans on the modified Physician Documentation Quality Instrument (PDQI-9), a measurement of note quality scale, reported Ashok Reddy, MD, MSc, of the University of Washington and Veterans Affairs Puget Sound Health Care System, Seattle, et al in the April issue of Annals of Internal Medicine.
AI scribes scored lower compared with humans across all domains, including accuracy, thoroughness, and usefulness. There was an especially large gap in scores on the 50-point PDQI-9 in an acute low back pain case (human, 43.8 points; AI, 20.3 points; difference, 23.5 points).
“For clinicians, AI scribes should be regarded as tools for generating draft documentation that requires review and editing, rather than as a substitute for clinician-authored notes,” the authors wrote. “Although ambient AI scribes hold promise for reducing clinician burden, rigorous and ongoing evaluation of their quality is essential to ensure that these tools enhance rather than compromise the quality of clinical care.”
AI Scribe Use is Widespread
Taylor N. Anderson, MD, a clinical informatics fellow at Oregon Health & Science University, Portland, is familiar with the study findings and noted that the use of AI scribes in medicine has grown rapidly. All major health organizations are either using it or facing “enormous pressure” from clinicians to do so, she told Federal Practitioner.
Previous research has linked the use of AI scribes for clinical notes to less electronic health record usage and documentation time for clinicians, leading to more time for patient visits. Still, the quality of clinical notes written by AI is “quite variable across vendors,” Anderson said.
Anderson led a 2025 study that examined 5 AI scribe platforms and found an average of 3.0 errors per case with “potential for moderate-to-severe harm.”
For the new study on the simulated cases, part of a VHA-sponsored “technology sprint” via Challenge.gov, researchers developed audio descriptions of 5 clinical cases reflecting common patient encounters in primary care: acute low back pain, chest pain, a new diagnosis of diabetes, a pharmacy consultation, and a follow-up with a nurse case manager for heart failure.
Two cases included non-English accents, 1 included background noise, and 1 featured speech through a medical mask. All the “patients” were played by what the authors described as “trained standardized patient actors.”
For each case, 3 humans and 11 AI scribe programs produced clinical notes. The clinical notes were then evaluated by 6 raters.
Researchers found that AI scribe-generated notes scored worse than human-generated notes across all 10 domains of the modified PDQI-9 (accuracy, thoroughness, usefulness, organization, comprehensiveness, succinctness, synthesization, internal consistency, and freedom from hallucination and bias).
There were especially large gaps between the AI and human notes in the domains of thoroughness, organization, and usefulness. Even wider gaps were observed for the encounters with noise and mask usage.
“These findings highlight that although ambient AI scribes can generate complete notes, the overall quality remains broadly below that of human-authored documentation,” the authors wrote.
No Comparison Between AI Scribes
The researchers noted that “given contractual limitations, we cannot interpret the results for specific vendors.” They also noted that the study did not use professional scribes, who may produce even higher-quality results, and the humans were not producing notes in a real-world clinical environment.
Anderson, the clinical informatics fellow, pointed out that the study does not examine the common scenario in which a clinician edits notes produced by an AI scribe. In fact, she said, there is no current research on this, failing to examine “the postediting note that would actually go into the chart.”
In an accompanying commentary, collaborative scientist Aaron Tierney, PhD, and Kristine Lee, MD, an associate executive director, both with the Permanente Medical Group, California, called for future research to focus on “real-world performance, promote the development of documentation policies that prioritize patient care over billing requirements, and systematically incorporate patient perspectives into assessments of quality.”
Why AI Misses the Mark
In an interview with Federal Practitioner, AI researcher Maxim Topaz, PhD, RN, MA, an associate professor of Nursing and Data Science at Columbia University School of Nursing, New York City, who is familiar with the study but did not participate in it, praised the research.
He pointed out that AI has trouble accurately representing clinical encounters because they “tend to fill gaps with plausible-sounding language, which can mask omissions and make errors harder to catch.” Also, “ambient scribes can only document what is verbalized aloud. Physical exam findings the clinician notices but does not narrate, nonverbal cues, and patient-initiated concerns that drift past in conversation are systematically underrepresented.”
Moving forward, Topaz advised clinicians to “treat AI-generated notes as a first draft, not a finished product. Read them carefully, especially for omissions, which the current evidence suggests are by far the most common error type and which are harder to spot than fabrications because the surrounding note still reads coherently.”
Two study authors disclosed employment by the US Department of Veterans Affairs. Other authors had no disclosures. The commentary authors have no disclosures. Anderson has no disclosures. Topaz discloses relationships with the National Institutes of Health and other federal sources.
Underground Hospitals: Is Combat Medicine Entering a New Era?
Drone warfare and repeated attacks on medical infrastructure are reshaping battlefield medicine in Ukraine, driving the development of underground military hospitals designed to stabilize and treat wounded soldiers close to active combat zones, rather than relying on rapid evacuation.
Since the start of Russia’s full-scale invasion of Ukraine, the World Health Organization has documented nearly 3000 attacks on healthcare facilities and violations of the Geneva Conventions that protect medical personnel and healthcare infrastructure during armed conflict.
In response, Ukraine has developed underground military hospitals designed to withstand bombardment and maintain the continuity of medical care. By combining infrastructure inherited from the Cold War with rapidly constructed new facilities, the country has managed to preserve healthcare capacity and support military operations close to the frontlines.
Underground Hospital
In September 2024, the Ukrainian Ministry of Defense, in partnership with the Metinvest Group, opened Ukraine’s first underground military stabilization hospital near the front lines. The project was developed under Metinvest’s military support initiative, known as the Steel Front, which supplies protective infrastructure and equipment for frontline operations.
In addition to producing steel bunkers for these facilities, the company manufactures military support equipment, including mine clearing plows, drone protection screens, systems designed to intercept loitering munitions, armor plates, and vehicle reinforcements for frontline operations.
The underground hospital consists of six steel bunkers, each measuring 7.6 m in length and 2.5 m in diameter, with a total area of 500 m2. The structures function as multifunctional units designed to maintain operational capability in high-threat environments. The facility includes ventilation, water supply, drainage, and electrical systems. During construction and installation, security measures aimed to reduce detectability and lower the risk for attack. The hospital also incorporates electronic warfare systems intended to strengthen operational protection.
The total investment reached 20 million Ukrainian hryvnias, approximately 385,000 euros. Of these, 7 million hryvnias funded medical equipment, while 13 million supported metal structures, construction materials, and infrastructure.
The hospital is equipped with oxygen concentrators, ventilators, cardiac monitors, defibrillators, surgical equipment, lighting systems, sterilizers, patient warming systems, and medical furniture. The complex includes two operating rooms, two resuscitation stations, a work area, and a staff rest area. Depending on the staffing and operational configuration, the hospital can stabilize wounded individuals and perform up to four simultaneous procedures. The design follows North Atlantic Treaty Organization standards for second-level field hospitals, designated Role/Echelon 2.
In a statement released by the Metinvest Group after the facility opened in 2024, Roman Kuzev, acting commander of the “East” medical task force, said: “This underground hospital is the best stabilization center available. This will allow us to provide medical care to over 100 patients a day, saving hundreds of lives for our heroes. I hope the number of such facilities will grow.”
Kuzev’s expectations materialized in 2025, when the Metinvest Group completed the construction of a second underground military hospital in one of the most active frontline sectors. The new facility provides greater protection and camouflage, and incorporates structural modifications based on lessons learned from the first hospital. It is buried more than 6 m underground and reinforced with additional protective layers.
The hospital includes four functional units housing surgical and stabilization areas, a delivery room, and a break area for healthcare personnel. The facility covers 350 m2 and required an investment exceeding 21 million Ukrainian hryvnias.
The center can simultaneously support up to three surgical procedures of varying complexities. Military authorities supplied equipment, including high-flow infusion pumps, x-ray systems, oxygen concentrators, defibrillators, and additional devices. Medical services are provided by teams of up to 20 professionals, including orthopedic surgeons, general surgeons, anesthesiologists, surgical nurses, and nursing assistants.
Historic Origin
World War I marked a turning point in modern warfare by introducing technologies that increased battlefield violence to unprecedented levels. The widespread use of machine guns, poisonous gas, tanks, and trench warfare has turned the battlefield into an extremely deadly environment.
At the same time, the conflict drove advances in military medicine that continue to influence practice today, including blood transfusions, psychological support for soldiers experiencing so called “shell shock,” and the development of field hospitals and mobile medical units.
One of the earliest documented underground hospitals was established in Arras, France, where a network of preexisting tunnels known as boves was expanded by New Zealand engineers to provide Allied forces with a tactical advantage. The tunnels were designed to shelter troops in preparation for the 1917 Arras Offensive, allowing them to assemble safely without being detected by German forces.
The underground hospital in Arras, which opened in 1916, includes waiting rooms, operating rooms, rest areas, spaces accommodating up to 700 stretchers, and a morgue. It also features internal electrical and plumbing systems, making it one of the most advanced medical facilities of its time.
Shift in Care
The expanding use of drones on the battlefield has increased the risks linked to casualty evacuation, particularly aeromedical evacuation, reducing the effectiveness of traditional military care models. In response, Ukraine has adopted an approach centered on extended field care and the development of a decentralized medical system, supported by close collaboration with the private sector to rapidly secure resources and infrastructure.
These strategies represent a shift in military medicine toward prolonged onsite stabilization rather than rapid evacuation. The combined use of underground facilities and repurposed infrastructure has helped maintain medical capacity under high threat conditions, improving survival among wounded individuals, and strengthening healthcare system resilience during conflict, according to US Army reports.
In addition to serving as a model for this shift in military medicine, the underground hospital project received the Partnership for Sustainability Award 2025 in Ukraine from the United Nations Global Compact in the “Rebuilding Ukraine” category. The award, presented by the United Nations network that promotes corporate sustainability and Sustainable Development Goals, recognizes private sector initiatives that support postwar reconstruction and strengthen social and institutional resilience.
The project was recognized for its contribution to saving lives and strengthening medical capacity in areas affected by active hostility.
This article was translated from El Médico Interactivo on Univadis, part of the Medscape Professional Network.
A version of this article appeared on Medscape.com.
Drone warfare and repeated attacks on medical infrastructure are reshaping battlefield medicine in Ukraine, driving the development of underground military hospitals designed to stabilize and treat wounded soldiers close to active combat zones, rather than relying on rapid evacuation.
Since the start of Russia’s full-scale invasion of Ukraine, the World Health Organization has documented nearly 3000 attacks on healthcare facilities and violations of the Geneva Conventions that protect medical personnel and healthcare infrastructure during armed conflict.
In response, Ukraine has developed underground military hospitals designed to withstand bombardment and maintain the continuity of medical care. By combining infrastructure inherited from the Cold War with rapidly constructed new facilities, the country has managed to preserve healthcare capacity and support military operations close to the frontlines.
Underground Hospital
In September 2024, the Ukrainian Ministry of Defense, in partnership with the Metinvest Group, opened Ukraine’s first underground military stabilization hospital near the front lines. The project was developed under Metinvest’s military support initiative, known as the Steel Front, which supplies protective infrastructure and equipment for frontline operations.
In addition to producing steel bunkers for these facilities, the company manufactures military support equipment, including mine clearing plows, drone protection screens, systems designed to intercept loitering munitions, armor plates, and vehicle reinforcements for frontline operations.
The underground hospital consists of six steel bunkers, each measuring 7.6 m in length and 2.5 m in diameter, with a total area of 500 m2. The structures function as multifunctional units designed to maintain operational capability in high-threat environments. The facility includes ventilation, water supply, drainage, and electrical systems. During construction and installation, security measures aimed to reduce detectability and lower the risk for attack. The hospital also incorporates electronic warfare systems intended to strengthen operational protection.
The total investment reached 20 million Ukrainian hryvnias, approximately 385,000 euros. Of these, 7 million hryvnias funded medical equipment, while 13 million supported metal structures, construction materials, and infrastructure.
The hospital is equipped with oxygen concentrators, ventilators, cardiac monitors, defibrillators, surgical equipment, lighting systems, sterilizers, patient warming systems, and medical furniture. The complex includes two operating rooms, two resuscitation stations, a work area, and a staff rest area. Depending on the staffing and operational configuration, the hospital can stabilize wounded individuals and perform up to four simultaneous procedures. The design follows North Atlantic Treaty Organization standards for second-level field hospitals, designated Role/Echelon 2.
In a statement released by the Metinvest Group after the facility opened in 2024, Roman Kuzev, acting commander of the “East” medical task force, said: “This underground hospital is the best stabilization center available. This will allow us to provide medical care to over 100 patients a day, saving hundreds of lives for our heroes. I hope the number of such facilities will grow.”
Kuzev’s expectations materialized in 2025, when the Metinvest Group completed the construction of a second underground military hospital in one of the most active frontline sectors. The new facility provides greater protection and camouflage, and incorporates structural modifications based on lessons learned from the first hospital. It is buried more than 6 m underground and reinforced with additional protective layers.
The hospital includes four functional units housing surgical and stabilization areas, a delivery room, and a break area for healthcare personnel. The facility covers 350 m2 and required an investment exceeding 21 million Ukrainian hryvnias.
The center can simultaneously support up to three surgical procedures of varying complexities. Military authorities supplied equipment, including high-flow infusion pumps, x-ray systems, oxygen concentrators, defibrillators, and additional devices. Medical services are provided by teams of up to 20 professionals, including orthopedic surgeons, general surgeons, anesthesiologists, surgical nurses, and nursing assistants.
Historic Origin
World War I marked a turning point in modern warfare by introducing technologies that increased battlefield violence to unprecedented levels. The widespread use of machine guns, poisonous gas, tanks, and trench warfare has turned the battlefield into an extremely deadly environment.
At the same time, the conflict drove advances in military medicine that continue to influence practice today, including blood transfusions, psychological support for soldiers experiencing so called “shell shock,” and the development of field hospitals and mobile medical units.
One of the earliest documented underground hospitals was established in Arras, France, where a network of preexisting tunnels known as boves was expanded by New Zealand engineers to provide Allied forces with a tactical advantage. The tunnels were designed to shelter troops in preparation for the 1917 Arras Offensive, allowing them to assemble safely without being detected by German forces.
The underground hospital in Arras, which opened in 1916, includes waiting rooms, operating rooms, rest areas, spaces accommodating up to 700 stretchers, and a morgue. It also features internal electrical and plumbing systems, making it one of the most advanced medical facilities of its time.
Shift in Care
The expanding use of drones on the battlefield has increased the risks linked to casualty evacuation, particularly aeromedical evacuation, reducing the effectiveness of traditional military care models. In response, Ukraine has adopted an approach centered on extended field care and the development of a decentralized medical system, supported by close collaboration with the private sector to rapidly secure resources and infrastructure.
These strategies represent a shift in military medicine toward prolonged onsite stabilization rather than rapid evacuation. The combined use of underground facilities and repurposed infrastructure has helped maintain medical capacity under high threat conditions, improving survival among wounded individuals, and strengthening healthcare system resilience during conflict, according to US Army reports.
In addition to serving as a model for this shift in military medicine, the underground hospital project received the Partnership for Sustainability Award 2025 in Ukraine from the United Nations Global Compact in the “Rebuilding Ukraine” category. The award, presented by the United Nations network that promotes corporate sustainability and Sustainable Development Goals, recognizes private sector initiatives that support postwar reconstruction and strengthen social and institutional resilience.
The project was recognized for its contribution to saving lives and strengthening medical capacity in areas affected by active hostility.
This article was translated from El Médico Interactivo on Univadis, part of the Medscape Professional Network.
A version of this article appeared on Medscape.com.
Drone warfare and repeated attacks on medical infrastructure are reshaping battlefield medicine in Ukraine, driving the development of underground military hospitals designed to stabilize and treat wounded soldiers close to active combat zones, rather than relying on rapid evacuation.
Since the start of Russia’s full-scale invasion of Ukraine, the World Health Organization has documented nearly 3000 attacks on healthcare facilities and violations of the Geneva Conventions that protect medical personnel and healthcare infrastructure during armed conflict.
In response, Ukraine has developed underground military hospitals designed to withstand bombardment and maintain the continuity of medical care. By combining infrastructure inherited from the Cold War with rapidly constructed new facilities, the country has managed to preserve healthcare capacity and support military operations close to the frontlines.
Underground Hospital
In September 2024, the Ukrainian Ministry of Defense, in partnership with the Metinvest Group, opened Ukraine’s first underground military stabilization hospital near the front lines. The project was developed under Metinvest’s military support initiative, known as the Steel Front, which supplies protective infrastructure and equipment for frontline operations.
In addition to producing steel bunkers for these facilities, the company manufactures military support equipment, including mine clearing plows, drone protection screens, systems designed to intercept loitering munitions, armor plates, and vehicle reinforcements for frontline operations.
The underground hospital consists of six steel bunkers, each measuring 7.6 m in length and 2.5 m in diameter, with a total area of 500 m2. The structures function as multifunctional units designed to maintain operational capability in high-threat environments. The facility includes ventilation, water supply, drainage, and electrical systems. During construction and installation, security measures aimed to reduce detectability and lower the risk for attack. The hospital also incorporates electronic warfare systems intended to strengthen operational protection.
The total investment reached 20 million Ukrainian hryvnias, approximately 385,000 euros. Of these, 7 million hryvnias funded medical equipment, while 13 million supported metal structures, construction materials, and infrastructure.
The hospital is equipped with oxygen concentrators, ventilators, cardiac monitors, defibrillators, surgical equipment, lighting systems, sterilizers, patient warming systems, and medical furniture. The complex includes two operating rooms, two resuscitation stations, a work area, and a staff rest area. Depending on the staffing and operational configuration, the hospital can stabilize wounded individuals and perform up to four simultaneous procedures. The design follows North Atlantic Treaty Organization standards for second-level field hospitals, designated Role/Echelon 2.
In a statement released by the Metinvest Group after the facility opened in 2024, Roman Kuzev, acting commander of the “East” medical task force, said: “This underground hospital is the best stabilization center available. This will allow us to provide medical care to over 100 patients a day, saving hundreds of lives for our heroes. I hope the number of such facilities will grow.”
Kuzev’s expectations materialized in 2025, when the Metinvest Group completed the construction of a second underground military hospital in one of the most active frontline sectors. The new facility provides greater protection and camouflage, and incorporates structural modifications based on lessons learned from the first hospital. It is buried more than 6 m underground and reinforced with additional protective layers.
The hospital includes four functional units housing surgical and stabilization areas, a delivery room, and a break area for healthcare personnel. The facility covers 350 m2 and required an investment exceeding 21 million Ukrainian hryvnias.
The center can simultaneously support up to three surgical procedures of varying complexities. Military authorities supplied equipment, including high-flow infusion pumps, x-ray systems, oxygen concentrators, defibrillators, and additional devices. Medical services are provided by teams of up to 20 professionals, including orthopedic surgeons, general surgeons, anesthesiologists, surgical nurses, and nursing assistants.
Historic Origin
World War I marked a turning point in modern warfare by introducing technologies that increased battlefield violence to unprecedented levels. The widespread use of machine guns, poisonous gas, tanks, and trench warfare has turned the battlefield into an extremely deadly environment.
At the same time, the conflict drove advances in military medicine that continue to influence practice today, including blood transfusions, psychological support for soldiers experiencing so called “shell shock,” and the development of field hospitals and mobile medical units.
One of the earliest documented underground hospitals was established in Arras, France, where a network of preexisting tunnels known as boves was expanded by New Zealand engineers to provide Allied forces with a tactical advantage. The tunnels were designed to shelter troops in preparation for the 1917 Arras Offensive, allowing them to assemble safely without being detected by German forces.
The underground hospital in Arras, which opened in 1916, includes waiting rooms, operating rooms, rest areas, spaces accommodating up to 700 stretchers, and a morgue. It also features internal electrical and plumbing systems, making it one of the most advanced medical facilities of its time.
Shift in Care
The expanding use of drones on the battlefield has increased the risks linked to casualty evacuation, particularly aeromedical evacuation, reducing the effectiveness of traditional military care models. In response, Ukraine has adopted an approach centered on extended field care and the development of a decentralized medical system, supported by close collaboration with the private sector to rapidly secure resources and infrastructure.
These strategies represent a shift in military medicine toward prolonged onsite stabilization rather than rapid evacuation. The combined use of underground facilities and repurposed infrastructure has helped maintain medical capacity under high threat conditions, improving survival among wounded individuals, and strengthening healthcare system resilience during conflict, according to US Army reports.
In addition to serving as a model for this shift in military medicine, the underground hospital project received the Partnership for Sustainability Award 2025 in Ukraine from the United Nations Global Compact in the “Rebuilding Ukraine” category. The award, presented by the United Nations network that promotes corporate sustainability and Sustainable Development Goals, recognizes private sector initiatives that support postwar reconstruction and strengthen social and institutional resilience.
The project was recognized for its contribution to saving lives and strengthening medical capacity in areas affected by active hostility.
This article was translated from El Médico Interactivo on Univadis, part of the Medscape Professional Network.
A version of this article appeared on Medscape.com.
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
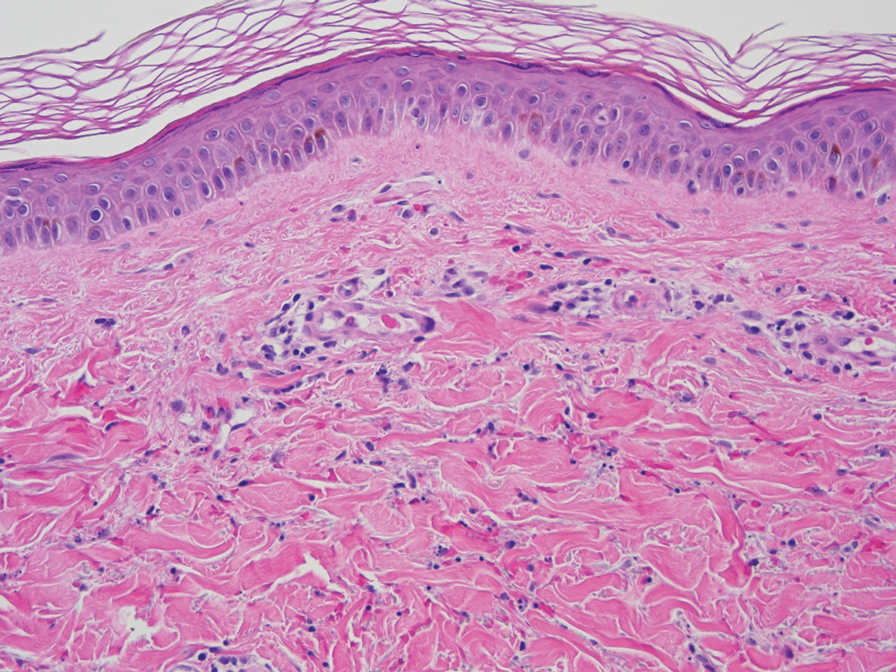
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
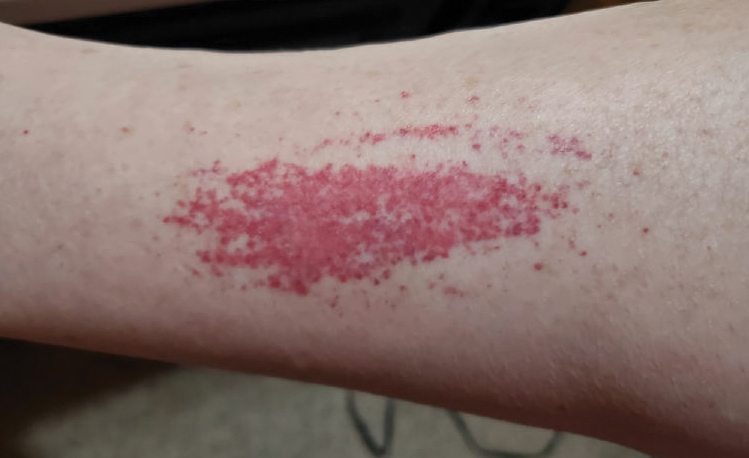
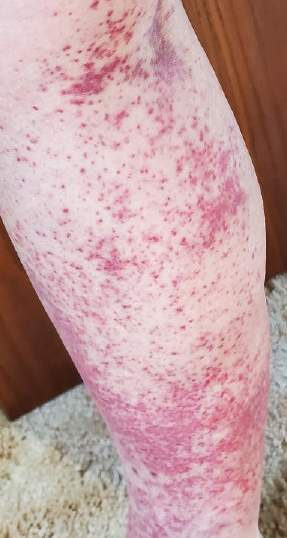
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Practice Points
- Elevation of the erythrocyte sedimentation rate (ESR) is nonspecific for inflammation and may be observed in the setting of increased immunoglobulin levels.
- Patients with elevated ESR and clinical evidence of recurrent petechiae and purpura should be screened for monoclonal and polyclonal gammopathies.

A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
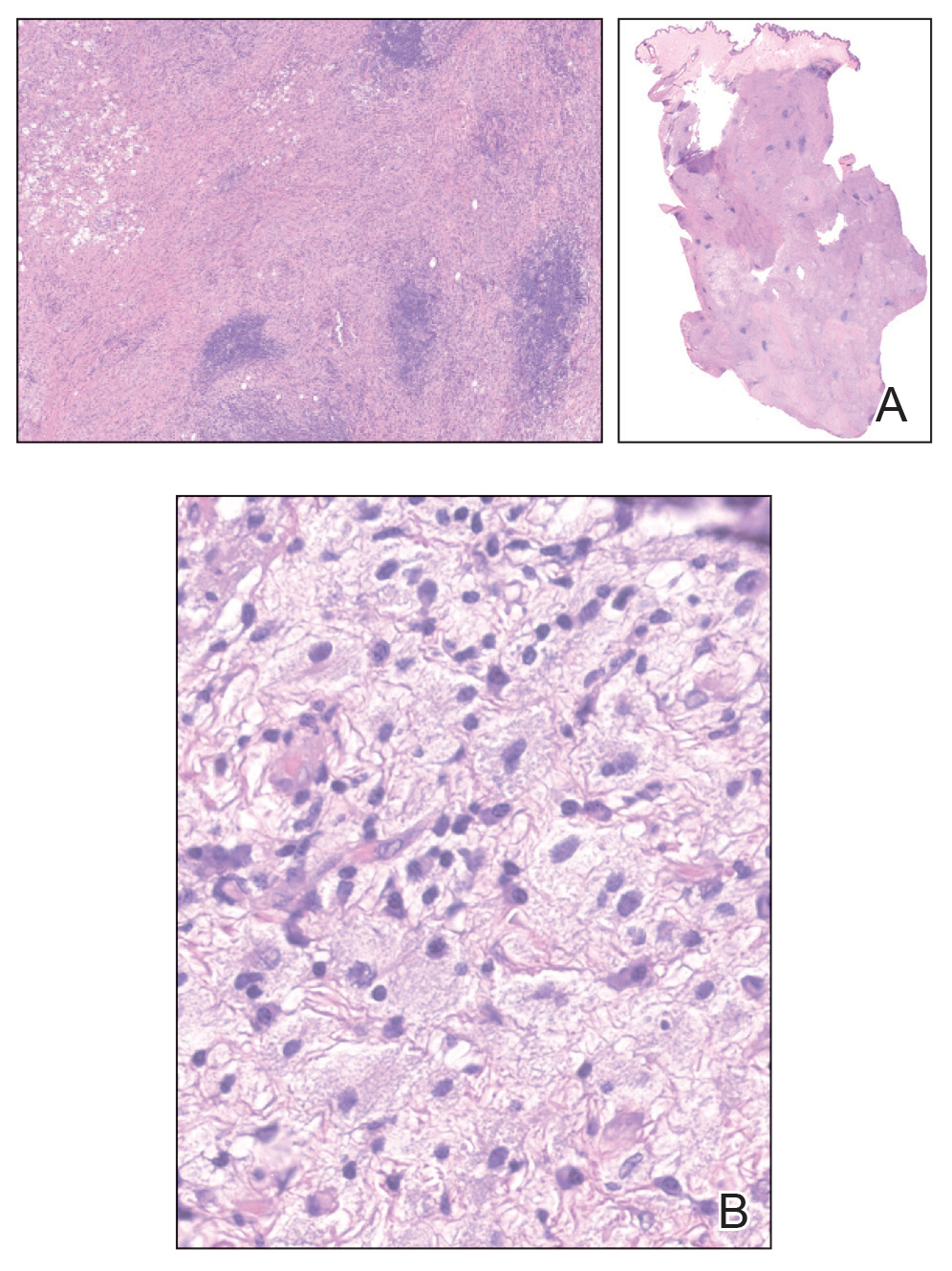
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
A 34-year-old man presented to our dermatology clinic for evaluation of a lesion in the right axilla of 1 year’s duration that had recently increased in size. The lesion was nontender and intermittently pruritic and was associated with focal hypohidrosis. The patient denied any fevers, chills, or recent weight change. His medical history was otherwise unremarkable. His only medications were daily ashwagandha and vitamin B and C supplements. On physical examination, a firm, 6-cm, subcutaneous nodule was noted in the right axilla with central alopecia and without a clear punctum. He had no palpable cervical, postauricular, or inguinal lymphadenopathy. The left axilla was clear, and there were no other relevant skin findings. Laboratory testing including a complete blood count, comprehensive metabolic panel, and sexually transmitted infections panel was unremarkable. Ultrasonography and subsequent magnetic resonance imaging of the right axilla showed a 4.9-cm nodule located in the subcutaneous fat with minimal deep infiltration and relatively smooth margins. An incisional biopsy of the lesion was performed.
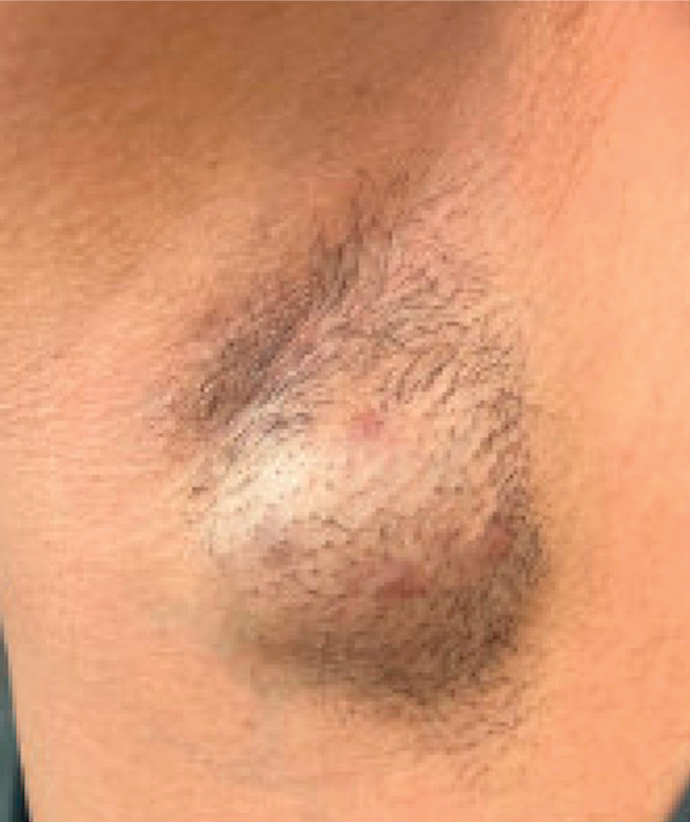

Xylazine-Induced Skin Necrosis
Xylazine-Induced Skin Necrosis
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
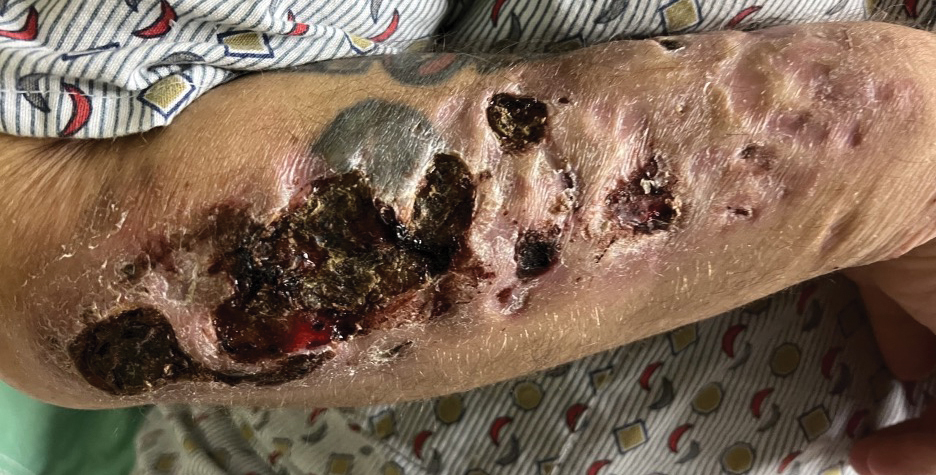
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
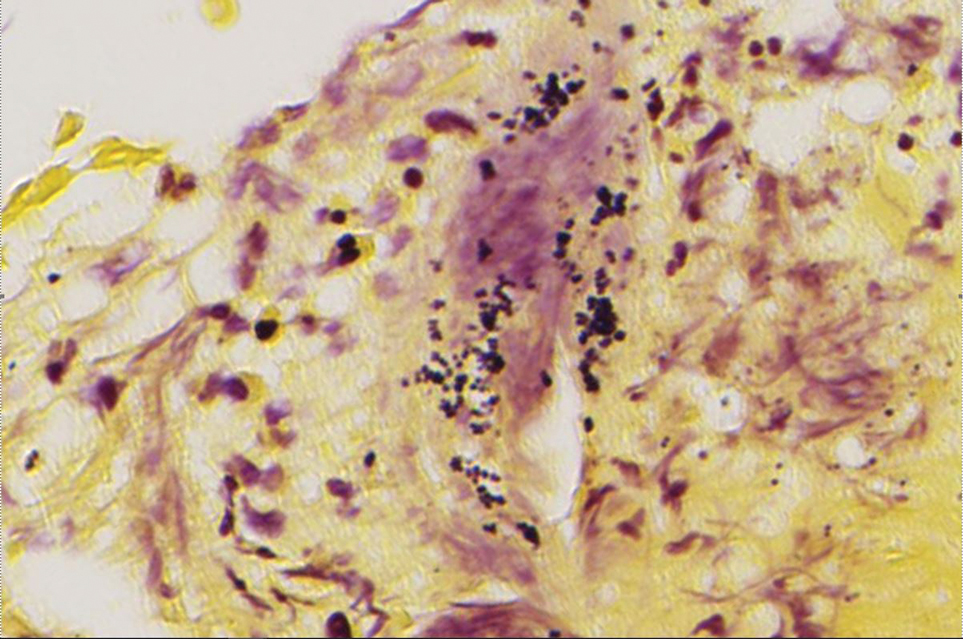
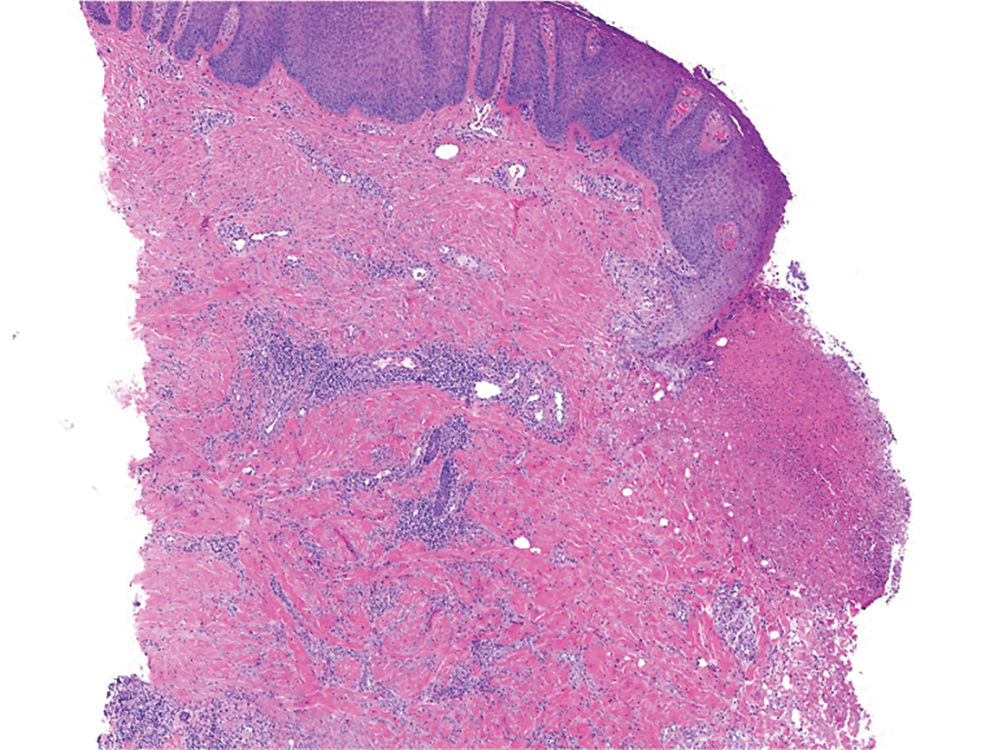
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
Xylazine-Induced Skin Necrosis
Xylazine-Induced Skin Necrosis
Practice Points
- Dermatologists should be aware of the potential for xylazine to cause ulcers in patients with a history of intravenous drug use.
- Early recognition of xylazine skin ulcers is imperative, as delayed diagnosis increases morbidity such as soft-tissue and bone infection, sepsis, and death.
