User login
Many Hurdles Exist to Treating Lung Cancer With CAR T Cells
These hurdles include finding the right targets, minimizing the risks of the treatment, and reducing the enormous burdens getting these therapies places on patients.
“Precision immunotherapy,” or unleashing the immune system in a highly specific manner, “is obviously, in a way, a holy grail” in lung cancer, said Martin Forster, MD, PhD, who cochaired a session on the topic at the World Conference on Lung Cancer (WCLC) 2024.
He underlined, however, that “immunology is very complex, as is cancer biology,” and consequently, there are different avenues being explored, including CAR T-cell therapies, T-cell receptor therapies, and tumor-infiltrating lymphocytes, among others.
Antibody technology is also being harnessed to target chemotherapy, via antibody-drug conjugates, noted Forster, who is clinical lead of the early phase clinical trials programme at University College London in England.
Moreover, investigators are looking at combining various therapies, such as immune checkpoint inhibitors with T cell–engaging approaches.
He highlighted, however, that the ideal target for these approaches is something that is recognized by the immune system as being foreign, but is found within the cancer, “and you also want it ideally to be in all of the cancer cells.”
A good example is a clonal change, meaning an early evolutionary genetic alteration in the tumor that is present in all the cells, Forster said.
Identifying the Right Target
“One of the big challenges in all forms of targeted immunotherapy is around selecting the target and developing the right product for the right target,” Forster emphasized.
“This concept works really well in hematological malignancies” but is “proving to be more challenging to deliver within solid malignancies,” he added.
“The reason why so many lung tumors are resistant to immunotherapy is because they ‘re immunologically cold,” Roy Herbst, MD, PhD, Department of Medical Oncology, Yale Comprehensive Cancer Center, New Haven, Connecticut, said in an interview.
“There are no T cells in the tumor,” he explained, so it “doesn’t really matter how much you block checkpoint inhibitors, you still have to have a T cell in there in order to have effect.”
To overcome this problem CAR T-cell therapies are engineered to target a tumor, Herbst continued, but that “is a little hard in lung cancer because you need to have a unique antigen that’s on a lung tumor that’s not present on normal cells.”
Charu Aggarwal, MD, MPH, Leslye M. Heisler Associate Professor for Lung Cancer Excellence, Penn Medicine, Philadelphia, Pennsylvania, agreed, saying that there is “a lot of excitement with CAR T-cell therapies, and the promise of cure,” but “the biology is not as simple as we think.”
“For example, it’s not as simple as CD20 or CD19 targeting,” she said in an interview. “Most of the antigens that are being targeted in the solid tumor world, unfortunately, are also expressed on normal tissue. So there is always this potential for toxicity.”
A Question of Time
Another aspect of CAR T-cell therapy that is proving difficult is its delivery.
Forster outlined that the process involves first leukapheresis, in which T cells are obtained from a blood draw. These are then genetically modified to express chimeric antigen receptors before being multiplied in the laboratory and introduced to the patient.
This process can take several weeks, during which patients may require bridging treatment, such as chemotherapy or radiotherapy, to keep their cancer under control. “Sometimes, patients with solid tumors who are in later lines of therapy may not have the luxury of time to be able to wait for all of these steps,” Aggarwal said.
There is also the question of whether a bespoke treatment can be scaled up so that it can be delivered to more patients in a more timely manner.
“There are certainly lessons to be learned from use of off-the-shelf CAR T-cell products” in hematologic malignancies, she noted, “but we’re just not there yet in lung cancer.”
Life-Threatening Toxicities
To improve the chances of engraftment when the CAR T cells are introduced, patients will require prior lymphodepletion with chemotherapy.
This, Forster said, is a “relatively intensive part of treatment.” However, “if you just give immune cells to somebody, when the host body is already full of immune cells,” the CAR T cells are unlikely to engraft, and “so you need to create space for those cells to develop.”
“What you want is not an immediate effect” but rather an immune “memory” that will give an ongoing benefit, he underscored.
Many patients will need to stay in the hospital one or more nights “because when you bring T cells to a tumor, you get cytokine release syndrome [CRS],” Herbst said. This can cause hypotension, fever, and chills, similar to a viral response.
“So patients can get sick,” which in turn requires treatment and follow-up. That puts a “big burden on the health system” and is a major issue, Herbst said.
Patients are also at a risk for “significant neurotoxicity,” said session cochair Amy Moore, PhD, vice president of Global Engagement and Patient Partnerships, LUNGevity Foundation, Chicago. This, alongside CRS, “can be life threatening for our patients.”
Lengthy hospital stays also have a psychosocial impact on the patient and their quality of life, she emphasized, especially when they are treated in a center far away from family and loved ones.
“We’ve also heard anecdotally some reports recently of secondary malignancies” with CAR T cell and other therapies, and that’s something that needs to be monitored as more patients go on these treatments, she said.
‘At What Cost’ to Patients?
The difficulties faced by patients in receiving CAR T-cell therapy go far beyond the practicalities of generating the cells or the risks associated with lymphodepletion, however.
“These therapies are extraordinarily expensive,” although that has to be weighed against the cost of years of ongoing treatment with immunotherapy, Moore said.
Moreover, as CAR T-cell therapies are a “last resort” option, patients have to “exhaust all other treatments” before being eligible, she continued. There’s significant prior authorization challenges, which means patients “have to go through many hurdles before they can qualify for treatment with these therapies.”
This typically involves having numerous laboratory tests, which can add up to out-of-pocket expenses for patients often reaching tens of thousands of dollars, Moore said.
Another issue is that they must be administered in certified treatment centers, and there are a limited number of those in the United States, she added.
This increases the risk of heightening disparities, as patients are “forced to travel, seek lodging, and have meal expenses,” and the costs “are not trivial,” Moore underlined. “It can rack up quickly and mount to $10,000 or more.”
For physicians, there are difficulties in terms of the logistics of following up with those patients who need to be treated at centers on the other side of the country, uncertainties around reimbursement, and restrictions in terms of staff time and resources, among others.
“I’m as excited as you are at the science,” but it is the implementation that is at issue, Moore said. In other words, there is the offer of a cure with CAR T-cell therapy, but “at what cost?”
“For patients, these considerations are real and they’re significant” and “we have to ensure that what we’re doing is in service of people with cancer,” she emphasized.
No funding was declared. Aggarwal declared relationships with Genentech, Celgene, AstraZeneca, Daiichi Sankyo, Turning Point, Janssen, Pfizer, Lilly, Merck, Regeneron/Sanofi, Eisai, BeiGene, Boehringer Ingelheim, Blueprint Genetics, and Shionogi. Forster declared relationships with AstraZeneca, Boehringer Ingelheim, Merck, MSD, Achilles, Amgen, Bayer, Bristol-Myers Squibb, Celgene, EQRx, GSK, Immutep, Janssen, Merck, Oxford Vacmedix, PharmaMar, Roche, Takeda, Syncorp, Transgene, and Ultrahuman. Moore declared no relevant financial relationships.
These hurdles include finding the right targets, minimizing the risks of the treatment, and reducing the enormous burdens getting these therapies places on patients.
“Precision immunotherapy,” or unleashing the immune system in a highly specific manner, “is obviously, in a way, a holy grail” in lung cancer, said Martin Forster, MD, PhD, who cochaired a session on the topic at the World Conference on Lung Cancer (WCLC) 2024.
He underlined, however, that “immunology is very complex, as is cancer biology,” and consequently, there are different avenues being explored, including CAR T-cell therapies, T-cell receptor therapies, and tumor-infiltrating lymphocytes, among others.
Antibody technology is also being harnessed to target chemotherapy, via antibody-drug conjugates, noted Forster, who is clinical lead of the early phase clinical trials programme at University College London in England.
Moreover, investigators are looking at combining various therapies, such as immune checkpoint inhibitors with T cell–engaging approaches.
He highlighted, however, that the ideal target for these approaches is something that is recognized by the immune system as being foreign, but is found within the cancer, “and you also want it ideally to be in all of the cancer cells.”
A good example is a clonal change, meaning an early evolutionary genetic alteration in the tumor that is present in all the cells, Forster said.
Identifying the Right Target
“One of the big challenges in all forms of targeted immunotherapy is around selecting the target and developing the right product for the right target,” Forster emphasized.
“This concept works really well in hematological malignancies” but is “proving to be more challenging to deliver within solid malignancies,” he added.
“The reason why so many lung tumors are resistant to immunotherapy is because they ‘re immunologically cold,” Roy Herbst, MD, PhD, Department of Medical Oncology, Yale Comprehensive Cancer Center, New Haven, Connecticut, said in an interview.
“There are no T cells in the tumor,” he explained, so it “doesn’t really matter how much you block checkpoint inhibitors, you still have to have a T cell in there in order to have effect.”
To overcome this problem CAR T-cell therapies are engineered to target a tumor, Herbst continued, but that “is a little hard in lung cancer because you need to have a unique antigen that’s on a lung tumor that’s not present on normal cells.”
Charu Aggarwal, MD, MPH, Leslye M. Heisler Associate Professor for Lung Cancer Excellence, Penn Medicine, Philadelphia, Pennsylvania, agreed, saying that there is “a lot of excitement with CAR T-cell therapies, and the promise of cure,” but “the biology is not as simple as we think.”
“For example, it’s not as simple as CD20 or CD19 targeting,” she said in an interview. “Most of the antigens that are being targeted in the solid tumor world, unfortunately, are also expressed on normal tissue. So there is always this potential for toxicity.”
A Question of Time
Another aspect of CAR T-cell therapy that is proving difficult is its delivery.
Forster outlined that the process involves first leukapheresis, in which T cells are obtained from a blood draw. These are then genetically modified to express chimeric antigen receptors before being multiplied in the laboratory and introduced to the patient.
This process can take several weeks, during which patients may require bridging treatment, such as chemotherapy or radiotherapy, to keep their cancer under control. “Sometimes, patients with solid tumors who are in later lines of therapy may not have the luxury of time to be able to wait for all of these steps,” Aggarwal said.
There is also the question of whether a bespoke treatment can be scaled up so that it can be delivered to more patients in a more timely manner.
“There are certainly lessons to be learned from use of off-the-shelf CAR T-cell products” in hematologic malignancies, she noted, “but we’re just not there yet in lung cancer.”
Life-Threatening Toxicities
To improve the chances of engraftment when the CAR T cells are introduced, patients will require prior lymphodepletion with chemotherapy.
This, Forster said, is a “relatively intensive part of treatment.” However, “if you just give immune cells to somebody, when the host body is already full of immune cells,” the CAR T cells are unlikely to engraft, and “so you need to create space for those cells to develop.”
“What you want is not an immediate effect” but rather an immune “memory” that will give an ongoing benefit, he underscored.
Many patients will need to stay in the hospital one or more nights “because when you bring T cells to a tumor, you get cytokine release syndrome [CRS],” Herbst said. This can cause hypotension, fever, and chills, similar to a viral response.
“So patients can get sick,” which in turn requires treatment and follow-up. That puts a “big burden on the health system” and is a major issue, Herbst said.
Patients are also at a risk for “significant neurotoxicity,” said session cochair Amy Moore, PhD, vice president of Global Engagement and Patient Partnerships, LUNGevity Foundation, Chicago. This, alongside CRS, “can be life threatening for our patients.”
Lengthy hospital stays also have a psychosocial impact on the patient and their quality of life, she emphasized, especially when they are treated in a center far away from family and loved ones.
“We’ve also heard anecdotally some reports recently of secondary malignancies” with CAR T cell and other therapies, and that’s something that needs to be monitored as more patients go on these treatments, she said.
‘At What Cost’ to Patients?
The difficulties faced by patients in receiving CAR T-cell therapy go far beyond the practicalities of generating the cells or the risks associated with lymphodepletion, however.
“These therapies are extraordinarily expensive,” although that has to be weighed against the cost of years of ongoing treatment with immunotherapy, Moore said.
Moreover, as CAR T-cell therapies are a “last resort” option, patients have to “exhaust all other treatments” before being eligible, she continued. There’s significant prior authorization challenges, which means patients “have to go through many hurdles before they can qualify for treatment with these therapies.”
This typically involves having numerous laboratory tests, which can add up to out-of-pocket expenses for patients often reaching tens of thousands of dollars, Moore said.
Another issue is that they must be administered in certified treatment centers, and there are a limited number of those in the United States, she added.
This increases the risk of heightening disparities, as patients are “forced to travel, seek lodging, and have meal expenses,” and the costs “are not trivial,” Moore underlined. “It can rack up quickly and mount to $10,000 or more.”
For physicians, there are difficulties in terms of the logistics of following up with those patients who need to be treated at centers on the other side of the country, uncertainties around reimbursement, and restrictions in terms of staff time and resources, among others.
“I’m as excited as you are at the science,” but it is the implementation that is at issue, Moore said. In other words, there is the offer of a cure with CAR T-cell therapy, but “at what cost?”
“For patients, these considerations are real and they’re significant” and “we have to ensure that what we’re doing is in service of people with cancer,” she emphasized.
No funding was declared. Aggarwal declared relationships with Genentech, Celgene, AstraZeneca, Daiichi Sankyo, Turning Point, Janssen, Pfizer, Lilly, Merck, Regeneron/Sanofi, Eisai, BeiGene, Boehringer Ingelheim, Blueprint Genetics, and Shionogi. Forster declared relationships with AstraZeneca, Boehringer Ingelheim, Merck, MSD, Achilles, Amgen, Bayer, Bristol-Myers Squibb, Celgene, EQRx, GSK, Immutep, Janssen, Merck, Oxford Vacmedix, PharmaMar, Roche, Takeda, Syncorp, Transgene, and Ultrahuman. Moore declared no relevant financial relationships.
These hurdles include finding the right targets, minimizing the risks of the treatment, and reducing the enormous burdens getting these therapies places on patients.
“Precision immunotherapy,” or unleashing the immune system in a highly specific manner, “is obviously, in a way, a holy grail” in lung cancer, said Martin Forster, MD, PhD, who cochaired a session on the topic at the World Conference on Lung Cancer (WCLC) 2024.
He underlined, however, that “immunology is very complex, as is cancer biology,” and consequently, there are different avenues being explored, including CAR T-cell therapies, T-cell receptor therapies, and tumor-infiltrating lymphocytes, among others.
Antibody technology is also being harnessed to target chemotherapy, via antibody-drug conjugates, noted Forster, who is clinical lead of the early phase clinical trials programme at University College London in England.
Moreover, investigators are looking at combining various therapies, such as immune checkpoint inhibitors with T cell–engaging approaches.
He highlighted, however, that the ideal target for these approaches is something that is recognized by the immune system as being foreign, but is found within the cancer, “and you also want it ideally to be in all of the cancer cells.”
A good example is a clonal change, meaning an early evolutionary genetic alteration in the tumor that is present in all the cells, Forster said.
Identifying the Right Target
“One of the big challenges in all forms of targeted immunotherapy is around selecting the target and developing the right product for the right target,” Forster emphasized.
“This concept works really well in hematological malignancies” but is “proving to be more challenging to deliver within solid malignancies,” he added.
“The reason why so many lung tumors are resistant to immunotherapy is because they ‘re immunologically cold,” Roy Herbst, MD, PhD, Department of Medical Oncology, Yale Comprehensive Cancer Center, New Haven, Connecticut, said in an interview.
“There are no T cells in the tumor,” he explained, so it “doesn’t really matter how much you block checkpoint inhibitors, you still have to have a T cell in there in order to have effect.”
To overcome this problem CAR T-cell therapies are engineered to target a tumor, Herbst continued, but that “is a little hard in lung cancer because you need to have a unique antigen that’s on a lung tumor that’s not present on normal cells.”
Charu Aggarwal, MD, MPH, Leslye M. Heisler Associate Professor for Lung Cancer Excellence, Penn Medicine, Philadelphia, Pennsylvania, agreed, saying that there is “a lot of excitement with CAR T-cell therapies, and the promise of cure,” but “the biology is not as simple as we think.”
“For example, it’s not as simple as CD20 or CD19 targeting,” she said in an interview. “Most of the antigens that are being targeted in the solid tumor world, unfortunately, are also expressed on normal tissue. So there is always this potential for toxicity.”
A Question of Time
Another aspect of CAR T-cell therapy that is proving difficult is its delivery.
Forster outlined that the process involves first leukapheresis, in which T cells are obtained from a blood draw. These are then genetically modified to express chimeric antigen receptors before being multiplied in the laboratory and introduced to the patient.
This process can take several weeks, during which patients may require bridging treatment, such as chemotherapy or radiotherapy, to keep their cancer under control. “Sometimes, patients with solid tumors who are in later lines of therapy may not have the luxury of time to be able to wait for all of these steps,” Aggarwal said.
There is also the question of whether a bespoke treatment can be scaled up so that it can be delivered to more patients in a more timely manner.
“There are certainly lessons to be learned from use of off-the-shelf CAR T-cell products” in hematologic malignancies, she noted, “but we’re just not there yet in lung cancer.”
Life-Threatening Toxicities
To improve the chances of engraftment when the CAR T cells are introduced, patients will require prior lymphodepletion with chemotherapy.
This, Forster said, is a “relatively intensive part of treatment.” However, “if you just give immune cells to somebody, when the host body is already full of immune cells,” the CAR T cells are unlikely to engraft, and “so you need to create space for those cells to develop.”
“What you want is not an immediate effect” but rather an immune “memory” that will give an ongoing benefit, he underscored.
Many patients will need to stay in the hospital one or more nights “because when you bring T cells to a tumor, you get cytokine release syndrome [CRS],” Herbst said. This can cause hypotension, fever, and chills, similar to a viral response.
“So patients can get sick,” which in turn requires treatment and follow-up. That puts a “big burden on the health system” and is a major issue, Herbst said.
Patients are also at a risk for “significant neurotoxicity,” said session cochair Amy Moore, PhD, vice president of Global Engagement and Patient Partnerships, LUNGevity Foundation, Chicago. This, alongside CRS, “can be life threatening for our patients.”
Lengthy hospital stays also have a psychosocial impact on the patient and their quality of life, she emphasized, especially when they are treated in a center far away from family and loved ones.
“We’ve also heard anecdotally some reports recently of secondary malignancies” with CAR T cell and other therapies, and that’s something that needs to be monitored as more patients go on these treatments, she said.
‘At What Cost’ to Patients?
The difficulties faced by patients in receiving CAR T-cell therapy go far beyond the practicalities of generating the cells or the risks associated with lymphodepletion, however.
“These therapies are extraordinarily expensive,” although that has to be weighed against the cost of years of ongoing treatment with immunotherapy, Moore said.
Moreover, as CAR T-cell therapies are a “last resort” option, patients have to “exhaust all other treatments” before being eligible, she continued. There’s significant prior authorization challenges, which means patients “have to go through many hurdles before they can qualify for treatment with these therapies.”
This typically involves having numerous laboratory tests, which can add up to out-of-pocket expenses for patients often reaching tens of thousands of dollars, Moore said.
Another issue is that they must be administered in certified treatment centers, and there are a limited number of those in the United States, she added.
This increases the risk of heightening disparities, as patients are “forced to travel, seek lodging, and have meal expenses,” and the costs “are not trivial,” Moore underlined. “It can rack up quickly and mount to $10,000 or more.”
For physicians, there are difficulties in terms of the logistics of following up with those patients who need to be treated at centers on the other side of the country, uncertainties around reimbursement, and restrictions in terms of staff time and resources, among others.
“I’m as excited as you are at the science,” but it is the implementation that is at issue, Moore said. In other words, there is the offer of a cure with CAR T-cell therapy, but “at what cost?”
“For patients, these considerations are real and they’re significant” and “we have to ensure that what we’re doing is in service of people with cancer,” she emphasized.
No funding was declared. Aggarwal declared relationships with Genentech, Celgene, AstraZeneca, Daiichi Sankyo, Turning Point, Janssen, Pfizer, Lilly, Merck, Regeneron/Sanofi, Eisai, BeiGene, Boehringer Ingelheim, Blueprint Genetics, and Shionogi. Forster declared relationships with AstraZeneca, Boehringer Ingelheim, Merck, MSD, Achilles, Amgen, Bayer, Bristol-Myers Squibb, Celgene, EQRx, GSK, Immutep, Janssen, Merck, Oxford Vacmedix, PharmaMar, Roche, Takeda, Syncorp, Transgene, and Ultrahuman. Moore declared no relevant financial relationships.
FROM WCLC 2024
A Simple Blood Test May Predict Cancer Risk in T2D
TOPLINE:
potentially enabling the identification of higher-risk individuals through a simple blood test.
METHODOLOGY:
- T2D is associated with an increased risk for obesity-related cancers, including breast, renal, uterine, thyroid, ovarian, and gastrointestinal cancers, as well as multiple myeloma, possibly because of chronic low-grade inflammation.
- Researchers explored whether the markers of inflammation IL-6, tumor necrosis factor alpha (TNF-alpha), and high-sensitivity C-reactive protein (hsCRP) can serve as predictive biomarkers for obesity-related cancers in patients recently diagnosed with T2D.
- They identified patients with recent-onset T2D and no prior history of cancer participating in the ongoing Danish Centre for Strategic Research in Type 2 Diabetes cohort study.
- At study initiation, plasma levels of IL-6 and TNF-alpha were measured using Meso Scale Discovery assays, and serum levels of hsCRP were measured using immunofluorometric assays.
TAKEAWAY:
- Among 6,466 eligible patients (40.5% women; median age, 60.9 years), 327 developed obesity-related cancers over a median follow-up of 8.8 years.
- Each SD increase in log-transformed IL-6 levels increased the risk for obesity-related cancers by 19%.
- The researchers did not find a strong association between TNF-alpha or hsCRP and obesity-related cancers.
- The addition of baseline IL-6 levels to other well-known risk factors for obesity-related cancers improved the performance of a cancer prediction model from 0.685 to 0.693, translating to a small but important increase in the ability to predict whether an individual would develop one of these cancers.
IN PRACTICE:
“In future, a simple blood test could identify those at higher risk of the cancers,” said the study’s lead author in an accompanying press release.
SOURCE:
The study was led by Mathilde D. Bennetsen, Steno Diabetes Center Odense, Odense University Hospital, Odense, Denmark, and published online on August 27 as an early release from the European Association for the Study of Diabetes (EASD) 2024 Annual Meeting.
LIMITATIONS:
No limitations were discussed in this abstract. However, the reliance on registry data may have introduced potential biases related to data accuracy and completeness.
DISCLOSURES:
The Danish Centre for Strategic Research in Type 2 Diabetes was supported by grants from the Danish Agency for Science and the Novo Nordisk Foundation. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
potentially enabling the identification of higher-risk individuals through a simple blood test.
METHODOLOGY:
- T2D is associated with an increased risk for obesity-related cancers, including breast, renal, uterine, thyroid, ovarian, and gastrointestinal cancers, as well as multiple myeloma, possibly because of chronic low-grade inflammation.
- Researchers explored whether the markers of inflammation IL-6, tumor necrosis factor alpha (TNF-alpha), and high-sensitivity C-reactive protein (hsCRP) can serve as predictive biomarkers for obesity-related cancers in patients recently diagnosed with T2D.
- They identified patients with recent-onset T2D and no prior history of cancer participating in the ongoing Danish Centre for Strategic Research in Type 2 Diabetes cohort study.
- At study initiation, plasma levels of IL-6 and TNF-alpha were measured using Meso Scale Discovery assays, and serum levels of hsCRP were measured using immunofluorometric assays.
TAKEAWAY:
- Among 6,466 eligible patients (40.5% women; median age, 60.9 years), 327 developed obesity-related cancers over a median follow-up of 8.8 years.
- Each SD increase in log-transformed IL-6 levels increased the risk for obesity-related cancers by 19%.
- The researchers did not find a strong association between TNF-alpha or hsCRP and obesity-related cancers.
- The addition of baseline IL-6 levels to other well-known risk factors for obesity-related cancers improved the performance of a cancer prediction model from 0.685 to 0.693, translating to a small but important increase in the ability to predict whether an individual would develop one of these cancers.
IN PRACTICE:
“In future, a simple blood test could identify those at higher risk of the cancers,” said the study’s lead author in an accompanying press release.
SOURCE:
The study was led by Mathilde D. Bennetsen, Steno Diabetes Center Odense, Odense University Hospital, Odense, Denmark, and published online on August 27 as an early release from the European Association for the Study of Diabetes (EASD) 2024 Annual Meeting.
LIMITATIONS:
No limitations were discussed in this abstract. However, the reliance on registry data may have introduced potential biases related to data accuracy and completeness.
DISCLOSURES:
The Danish Centre for Strategic Research in Type 2 Diabetes was supported by grants from the Danish Agency for Science and the Novo Nordisk Foundation. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
potentially enabling the identification of higher-risk individuals through a simple blood test.
METHODOLOGY:
- T2D is associated with an increased risk for obesity-related cancers, including breast, renal, uterine, thyroid, ovarian, and gastrointestinal cancers, as well as multiple myeloma, possibly because of chronic low-grade inflammation.
- Researchers explored whether the markers of inflammation IL-6, tumor necrosis factor alpha (TNF-alpha), and high-sensitivity C-reactive protein (hsCRP) can serve as predictive biomarkers for obesity-related cancers in patients recently diagnosed with T2D.
- They identified patients with recent-onset T2D and no prior history of cancer participating in the ongoing Danish Centre for Strategic Research in Type 2 Diabetes cohort study.
- At study initiation, plasma levels of IL-6 and TNF-alpha were measured using Meso Scale Discovery assays, and serum levels of hsCRP were measured using immunofluorometric assays.
TAKEAWAY:
- Among 6,466 eligible patients (40.5% women; median age, 60.9 years), 327 developed obesity-related cancers over a median follow-up of 8.8 years.
- Each SD increase in log-transformed IL-6 levels increased the risk for obesity-related cancers by 19%.
- The researchers did not find a strong association between TNF-alpha or hsCRP and obesity-related cancers.
- The addition of baseline IL-6 levels to other well-known risk factors for obesity-related cancers improved the performance of a cancer prediction model from 0.685 to 0.693, translating to a small but important increase in the ability to predict whether an individual would develop one of these cancers.
IN PRACTICE:
“In future, a simple blood test could identify those at higher risk of the cancers,” said the study’s lead author in an accompanying press release.
SOURCE:
The study was led by Mathilde D. Bennetsen, Steno Diabetes Center Odense, Odense University Hospital, Odense, Denmark, and published online on August 27 as an early release from the European Association for the Study of Diabetes (EASD) 2024 Annual Meeting.
LIMITATIONS:
No limitations were discussed in this abstract. However, the reliance on registry data may have introduced potential biases related to data accuracy and completeness.
DISCLOSURES:
The Danish Centre for Strategic Research in Type 2 Diabetes was supported by grants from the Danish Agency for Science and the Novo Nordisk Foundation. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Cancer Treatment 101: A Primer for Non-Oncologists
The remaining 700,000 or so often proceed to chemotherapy either immediately or upon cancer recurrence, spread, or newly recognized metastases. “Cures” after that point are rare.
I’m speaking in generalities, understanding that each cancer and each patient is unique.
Chemotherapy
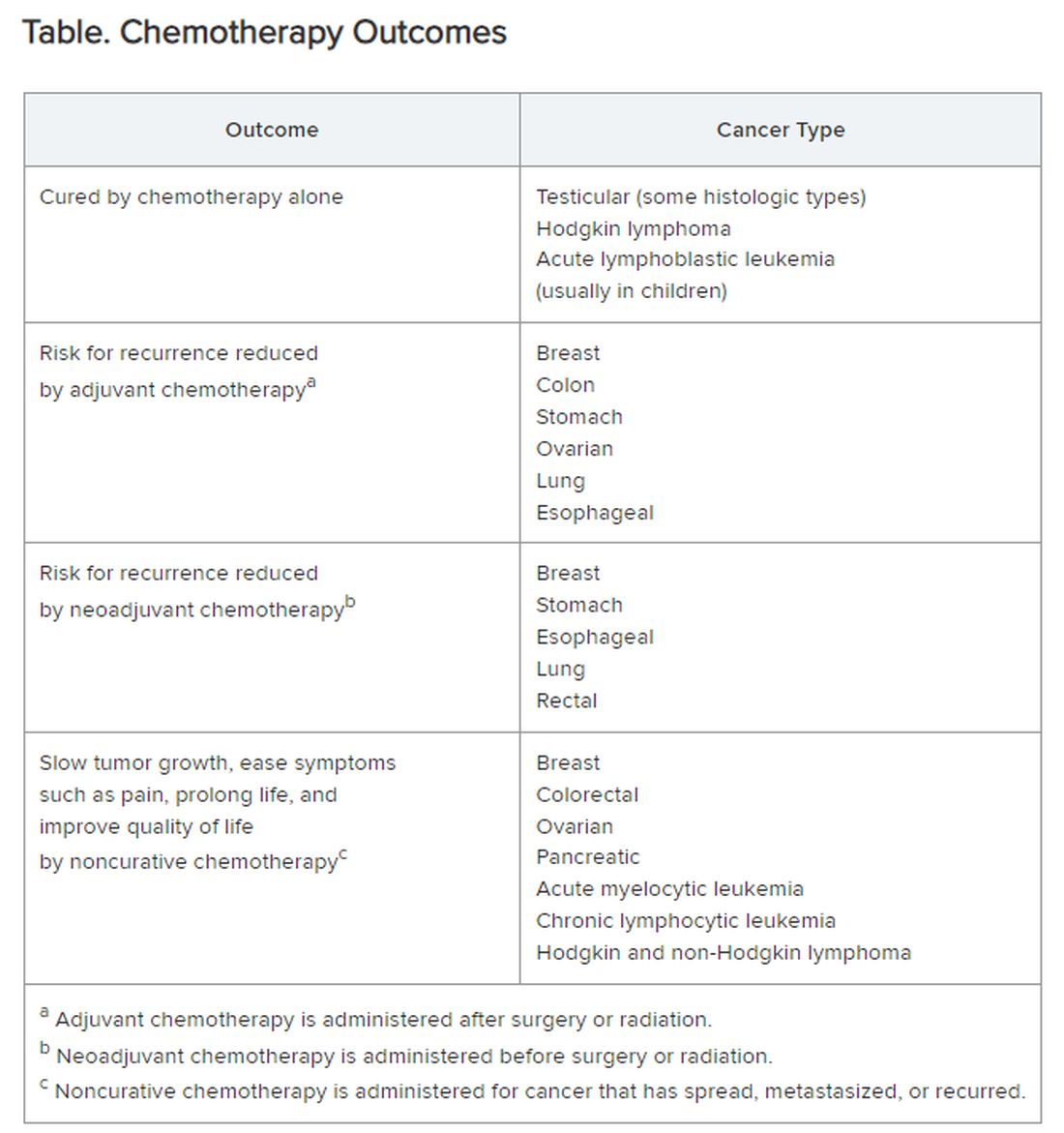
Chemotherapy alone can cure a small number of cancer types. When added to radiation or surgery, chemotherapy can help to cure a wider range of cancer types. As an add-on, chemotherapy can extend the length and quality of life for many patients with cancer. Since chemotherapy is by definition “toxic,” it can also shorten the duration or harm the quality of life and provide false hope. The Table summarizes what chemotherapy can and cannot achieve in selected cancer types.
Careful, compassionate communication between patient and physician is key. Goals and expectations must be clearly understood.
Organized chemotherapeutic efforts are further categorized as first line, second line, and third line.
First-line treatment. The initial round of recommended chemotherapy for a specific cancer. It is typically considered the most effective treatment for that type and stage of cancer on the basis of current research and clinical trials.
Second-line treatment. This is the treatment used if the first-line chemotherapy doesn’t work as desired. Reasons to switch to second-line chemo include:
- Lack of response (the tumor failed to shrink).
- Progression (the cancer may have grown or spread further).
- Adverse side effects were too severe to continue.
The drugs used in second-line chemo will typically be different from those used in first line, sometimes because cancer cells can develop resistance to chemotherapy drugs over time. Moreover, the goal of second-line chemo may differ from that of first-line therapy. Rather than chiefly aiming for a cure, second-line treatment might focus on slowing cancer growth, managing symptoms, or improving quality of life. Unfortunately, not every type of cancer has a readily available second-line option.
Third-line treatment. Third-line options come into play when both the initial course of chemo (first line) and the subsequent treatment (second line) have failed to achieve remission or control the cancer’s spread. Owing to the progressive nature of advanced cancers, patients might not be eligible or healthy enough for third-line therapy. Depending on cancer type, the patient’s general health, and response to previous treatments, third-line options could include:
- New or different chemotherapy drugs compared with prior lines.
- Surgery to debulk the tumor.
- Radiation for symptom control.
- Targeted therapy: drugs designed to target specific vulnerabilities in cancer cells.
- Immunotherapy: agents that help the body’s immune system fight cancer cells.
- Clinical trials testing new or investigational treatments, which may be applicable at any time, depending on the questions being addressed.
The goals of third-line therapy may shift from aiming for a cure to managing symptoms, improving quality of life, and potentially slowing cancer growth. The decision to pursue third-line therapy involves careful consideration by the doctor and patient, weighing the potential benefits and risks of treatment considering the individual’s overall health and specific situation.
It’s important to have realistic expectations about the potential outcomes of third-line therapy. Although remission may be unlikely, third-line therapy can still play a role in managing the disease.
Navigating advanced cancer treatment is very complex. The patient and physician must together consider detailed explanations and clarifications to set expectations and make informed decisions about care.
Interventions to Consider Earlier
In traditional clinical oncology practice, other interventions are possible, but these may not be offered until treatment has reached the third line:
- Molecular testing.
- Palliation.
- Clinical trials.
- Innovative testing to guide targeted therapy by ascertaining which agents are most likely (or not likely at all) to be effective.
I would argue that the patient’s interests are better served by considering and offering these other interventions much earlier, even before starting first-line chemotherapy.
Molecular testing. The best time for molecular testing of a new malignant tumor is typically at the time of diagnosis. Here’s why:
- Molecular testing helps identify specific genetic mutations in the cancer cells. This information can be crucial for selecting targeted therapies that are most effective against those specific mutations. Early detection allows for the most treatment options. For example, for non–small cell lung cancer, early is best because treatment and outcomes may well be changed by test results.
- Knowing the tumor’s molecular makeup can help determine whether a patient qualifies for clinical trials of new drugs designed for specific mutations.
- Some molecular markers can offer information about the tumor’s aggressiveness and potential for metastasis so that prognosis can be informed.
Molecular testing can be a valuable tool throughout a cancer patient’s journey. With genetically diverse tumors, the initial biopsy might not capture the full picture. Molecular testing of circulating tumor DNA can be used to monitor a patient’s response to treatment and detect potential mutations that might arise during treatment resistance. Retesting after metastasis can provide additional information that can aid in treatment decisions.
Palliative care. The ideal time to discuss palliative care with a patient with cancer is early in the diagnosis and treatment process. Palliative care is not the same as hospice care; it isn’t just about end-of-life. Palliative care focuses on improving a patient’s quality of life throughout cancer treatment. Palliative care specialists can address a wide range of symptoms a patient might experience from cancer or its treatment, including pain, fatigue, nausea, and anxiety.
Early discussions allow for a more comprehensive care plan. Open communication about all treatment options, including palliative care, empowers patients to make informed decisions about their care goals and preferences.
Specific situations where discussing palliative care might be appropriate are:
- Soon after a cancer diagnosis.
- If the patient experiences significant side effects from cancer treatment.
- When considering different treatment options, palliative care can complement those treatments.
- In advanced stages of cancer, to focus on comfort and quality of life.
Clinical trials. Participation in a clinical trial to explore new or investigational treatments should always be considered.
In theory, clinical trials should be an option at any time in the patient’s course. But the organized clinical trial experience may not be available or appropriate. Then, the individual becomes a de facto “clinical trial with an n of 1.” Read this brief open-access blog post at Cancer Commons to learn more about that circumstance.
Innovative testing. The best choice of chemotherapeutic or targeted therapies is often unclear. The clinician is likely to follow published guidelines, often from the National Comprehensive Cancer Network.
These are evidence based and driven by consensus of experts. But guideline-recommended therapy is not always effective, and weeks or months can pass before this ineffectiveness becomes apparent. Thus, many researchers and companies are seeking methods of testing each patient’s specific cancer to determine in advance, or very quickly, whether a particular drug is likely to be effective.
Read more about these leading innovations:
SAGE Oncotest: Entering the Next Generation of Tailored Cancer Treatment
Alibrex: A New Blood Test to Reveal Whether a Cancer Treatment is Working
PARIS Test Uses Lab-Grown Mini-Tumors to Find a Patient’s Best Treatment
Using Live Cells from Patients to Find the Right Cancer Drug
Other innovative therapies under investigation could even be agnostic to cancer type:
Treating Pancreatic Cancer: Could Metabolism — Not Genomics — Be the Key?
High-Energy Blue Light Powers a Promising New Treatment to Destroy Cancer Cells
All-Clear Follow-Up: Hydrogen Peroxide Appears to Treat Oral and Skin Lesions
Cancer is a tough nut to crack. Many people and organizations are trying very hard. So much is being learned. Some approaches will be effective. We can all hope.
Dr. Lundberg, editor in chief, Cancer Commons, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
The remaining 700,000 or so often proceed to chemotherapy either immediately or upon cancer recurrence, spread, or newly recognized metastases. “Cures” after that point are rare.
I’m speaking in generalities, understanding that each cancer and each patient is unique.
Chemotherapy
Chemotherapy alone can cure a small number of cancer types. When added to radiation or surgery, chemotherapy can help to cure a wider range of cancer types. As an add-on, chemotherapy can extend the length and quality of life for many patients with cancer. Since chemotherapy is by definition “toxic,” it can also shorten the duration or harm the quality of life and provide false hope. The Table summarizes what chemotherapy can and cannot achieve in selected cancer types.
Careful, compassionate communication between patient and physician is key. Goals and expectations must be clearly understood.
Organized chemotherapeutic efforts are further categorized as first line, second line, and third line.
First-line treatment. The initial round of recommended chemotherapy for a specific cancer. It is typically considered the most effective treatment for that type and stage of cancer on the basis of current research and clinical trials.
Second-line treatment. This is the treatment used if the first-line chemotherapy doesn’t work as desired. Reasons to switch to second-line chemo include:
- Lack of response (the tumor failed to shrink).
- Progression (the cancer may have grown or spread further).
- Adverse side effects were too severe to continue.
The drugs used in second-line chemo will typically be different from those used in first line, sometimes because cancer cells can develop resistance to chemotherapy drugs over time. Moreover, the goal of second-line chemo may differ from that of first-line therapy. Rather than chiefly aiming for a cure, second-line treatment might focus on slowing cancer growth, managing symptoms, or improving quality of life. Unfortunately, not every type of cancer has a readily available second-line option.
Third-line treatment. Third-line options come into play when both the initial course of chemo (first line) and the subsequent treatment (second line) have failed to achieve remission or control the cancer’s spread. Owing to the progressive nature of advanced cancers, patients might not be eligible or healthy enough for third-line therapy. Depending on cancer type, the patient’s general health, and response to previous treatments, third-line options could include:
- New or different chemotherapy drugs compared with prior lines.
- Surgery to debulk the tumor.
- Radiation for symptom control.
- Targeted therapy: drugs designed to target specific vulnerabilities in cancer cells.
- Immunotherapy: agents that help the body’s immune system fight cancer cells.
- Clinical trials testing new or investigational treatments, which may be applicable at any time, depending on the questions being addressed.
The goals of third-line therapy may shift from aiming for a cure to managing symptoms, improving quality of life, and potentially slowing cancer growth. The decision to pursue third-line therapy involves careful consideration by the doctor and patient, weighing the potential benefits and risks of treatment considering the individual’s overall health and specific situation.
It’s important to have realistic expectations about the potential outcomes of third-line therapy. Although remission may be unlikely, third-line therapy can still play a role in managing the disease.
Navigating advanced cancer treatment is very complex. The patient and physician must together consider detailed explanations and clarifications to set expectations and make informed decisions about care.
Interventions to Consider Earlier
In traditional clinical oncology practice, other interventions are possible, but these may not be offered until treatment has reached the third line:
- Molecular testing.
- Palliation.
- Clinical trials.
- Innovative testing to guide targeted therapy by ascertaining which agents are most likely (or not likely at all) to be effective.
I would argue that the patient’s interests are better served by considering and offering these other interventions much earlier, even before starting first-line chemotherapy.
Molecular testing. The best time for molecular testing of a new malignant tumor is typically at the time of diagnosis. Here’s why:
- Molecular testing helps identify specific genetic mutations in the cancer cells. This information can be crucial for selecting targeted therapies that are most effective against those specific mutations. Early detection allows for the most treatment options. For example, for non–small cell lung cancer, early is best because treatment and outcomes may well be changed by test results.
- Knowing the tumor’s molecular makeup can help determine whether a patient qualifies for clinical trials of new drugs designed for specific mutations.
- Some molecular markers can offer information about the tumor’s aggressiveness and potential for metastasis so that prognosis can be informed.
Molecular testing can be a valuable tool throughout a cancer patient’s journey. With genetically diverse tumors, the initial biopsy might not capture the full picture. Molecular testing of circulating tumor DNA can be used to monitor a patient’s response to treatment and detect potential mutations that might arise during treatment resistance. Retesting after metastasis can provide additional information that can aid in treatment decisions.
Palliative care. The ideal time to discuss palliative care with a patient with cancer is early in the diagnosis and treatment process. Palliative care is not the same as hospice care; it isn’t just about end-of-life. Palliative care focuses on improving a patient’s quality of life throughout cancer treatment. Palliative care specialists can address a wide range of symptoms a patient might experience from cancer or its treatment, including pain, fatigue, nausea, and anxiety.
Early discussions allow for a more comprehensive care plan. Open communication about all treatment options, including palliative care, empowers patients to make informed decisions about their care goals and preferences.
Specific situations where discussing palliative care might be appropriate are:
- Soon after a cancer diagnosis.
- If the patient experiences significant side effects from cancer treatment.
- When considering different treatment options, palliative care can complement those treatments.
- In advanced stages of cancer, to focus on comfort and quality of life.
Clinical trials. Participation in a clinical trial to explore new or investigational treatments should always be considered.
In theory, clinical trials should be an option at any time in the patient’s course. But the organized clinical trial experience may not be available or appropriate. Then, the individual becomes a de facto “clinical trial with an n of 1.” Read this brief open-access blog post at Cancer Commons to learn more about that circumstance.
Innovative testing. The best choice of chemotherapeutic or targeted therapies is often unclear. The clinician is likely to follow published guidelines, often from the National Comprehensive Cancer Network.
These are evidence based and driven by consensus of experts. But guideline-recommended therapy is not always effective, and weeks or months can pass before this ineffectiveness becomes apparent. Thus, many researchers and companies are seeking methods of testing each patient’s specific cancer to determine in advance, or very quickly, whether a particular drug is likely to be effective.
Read more about these leading innovations:
SAGE Oncotest: Entering the Next Generation of Tailored Cancer Treatment
Alibrex: A New Blood Test to Reveal Whether a Cancer Treatment is Working
PARIS Test Uses Lab-Grown Mini-Tumors to Find a Patient’s Best Treatment
Using Live Cells from Patients to Find the Right Cancer Drug
Other innovative therapies under investigation could even be agnostic to cancer type:
Treating Pancreatic Cancer: Could Metabolism — Not Genomics — Be the Key?
High-Energy Blue Light Powers a Promising New Treatment to Destroy Cancer Cells
All-Clear Follow-Up: Hydrogen Peroxide Appears to Treat Oral and Skin Lesions
Cancer is a tough nut to crack. Many people and organizations are trying very hard. So much is being learned. Some approaches will be effective. We can all hope.
Dr. Lundberg, editor in chief, Cancer Commons, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
The remaining 700,000 or so often proceed to chemotherapy either immediately or upon cancer recurrence, spread, or newly recognized metastases. “Cures” after that point are rare.
I’m speaking in generalities, understanding that each cancer and each patient is unique.
Chemotherapy
Chemotherapy alone can cure a small number of cancer types. When added to radiation or surgery, chemotherapy can help to cure a wider range of cancer types. As an add-on, chemotherapy can extend the length and quality of life for many patients with cancer. Since chemotherapy is by definition “toxic,” it can also shorten the duration or harm the quality of life and provide false hope. The Table summarizes what chemotherapy can and cannot achieve in selected cancer types.
Careful, compassionate communication between patient and physician is key. Goals and expectations must be clearly understood.
Organized chemotherapeutic efforts are further categorized as first line, second line, and third line.
First-line treatment. The initial round of recommended chemotherapy for a specific cancer. It is typically considered the most effective treatment for that type and stage of cancer on the basis of current research and clinical trials.
Second-line treatment. This is the treatment used if the first-line chemotherapy doesn’t work as desired. Reasons to switch to second-line chemo include:
- Lack of response (the tumor failed to shrink).
- Progression (the cancer may have grown or spread further).
- Adverse side effects were too severe to continue.
The drugs used in second-line chemo will typically be different from those used in first line, sometimes because cancer cells can develop resistance to chemotherapy drugs over time. Moreover, the goal of second-line chemo may differ from that of first-line therapy. Rather than chiefly aiming for a cure, second-line treatment might focus on slowing cancer growth, managing symptoms, or improving quality of life. Unfortunately, not every type of cancer has a readily available second-line option.
Third-line treatment. Third-line options come into play when both the initial course of chemo (first line) and the subsequent treatment (second line) have failed to achieve remission or control the cancer’s spread. Owing to the progressive nature of advanced cancers, patients might not be eligible or healthy enough for third-line therapy. Depending on cancer type, the patient’s general health, and response to previous treatments, third-line options could include:
- New or different chemotherapy drugs compared with prior lines.
- Surgery to debulk the tumor.
- Radiation for symptom control.
- Targeted therapy: drugs designed to target specific vulnerabilities in cancer cells.
- Immunotherapy: agents that help the body’s immune system fight cancer cells.
- Clinical trials testing new or investigational treatments, which may be applicable at any time, depending on the questions being addressed.
The goals of third-line therapy may shift from aiming for a cure to managing symptoms, improving quality of life, and potentially slowing cancer growth. The decision to pursue third-line therapy involves careful consideration by the doctor and patient, weighing the potential benefits and risks of treatment considering the individual’s overall health and specific situation.
It’s important to have realistic expectations about the potential outcomes of third-line therapy. Although remission may be unlikely, third-line therapy can still play a role in managing the disease.
Navigating advanced cancer treatment is very complex. The patient and physician must together consider detailed explanations and clarifications to set expectations and make informed decisions about care.
Interventions to Consider Earlier
In traditional clinical oncology practice, other interventions are possible, but these may not be offered until treatment has reached the third line:
- Molecular testing.
- Palliation.
- Clinical trials.
- Innovative testing to guide targeted therapy by ascertaining which agents are most likely (or not likely at all) to be effective.
I would argue that the patient’s interests are better served by considering and offering these other interventions much earlier, even before starting first-line chemotherapy.
Molecular testing. The best time for molecular testing of a new malignant tumor is typically at the time of diagnosis. Here’s why:
- Molecular testing helps identify specific genetic mutations in the cancer cells. This information can be crucial for selecting targeted therapies that are most effective against those specific mutations. Early detection allows for the most treatment options. For example, for non–small cell lung cancer, early is best because treatment and outcomes may well be changed by test results.
- Knowing the tumor’s molecular makeup can help determine whether a patient qualifies for clinical trials of new drugs designed for specific mutations.
- Some molecular markers can offer information about the tumor’s aggressiveness and potential for metastasis so that prognosis can be informed.
Molecular testing can be a valuable tool throughout a cancer patient’s journey. With genetically diverse tumors, the initial biopsy might not capture the full picture. Molecular testing of circulating tumor DNA can be used to monitor a patient’s response to treatment and detect potential mutations that might arise during treatment resistance. Retesting after metastasis can provide additional information that can aid in treatment decisions.
Palliative care. The ideal time to discuss palliative care with a patient with cancer is early in the diagnosis and treatment process. Palliative care is not the same as hospice care; it isn’t just about end-of-life. Palliative care focuses on improving a patient’s quality of life throughout cancer treatment. Palliative care specialists can address a wide range of symptoms a patient might experience from cancer or its treatment, including pain, fatigue, nausea, and anxiety.
Early discussions allow for a more comprehensive care plan. Open communication about all treatment options, including palliative care, empowers patients to make informed decisions about their care goals and preferences.
Specific situations where discussing palliative care might be appropriate are:
- Soon after a cancer diagnosis.
- If the patient experiences significant side effects from cancer treatment.
- When considering different treatment options, palliative care can complement those treatments.
- In advanced stages of cancer, to focus on comfort and quality of life.
Clinical trials. Participation in a clinical trial to explore new or investigational treatments should always be considered.
In theory, clinical trials should be an option at any time in the patient’s course. But the organized clinical trial experience may not be available or appropriate. Then, the individual becomes a de facto “clinical trial with an n of 1.” Read this brief open-access blog post at Cancer Commons to learn more about that circumstance.
Innovative testing. The best choice of chemotherapeutic or targeted therapies is often unclear. The clinician is likely to follow published guidelines, often from the National Comprehensive Cancer Network.
These are evidence based and driven by consensus of experts. But guideline-recommended therapy is not always effective, and weeks or months can pass before this ineffectiveness becomes apparent. Thus, many researchers and companies are seeking methods of testing each patient’s specific cancer to determine in advance, or very quickly, whether a particular drug is likely to be effective.
Read more about these leading innovations:
SAGE Oncotest: Entering the Next Generation of Tailored Cancer Treatment
Alibrex: A New Blood Test to Reveal Whether a Cancer Treatment is Working
PARIS Test Uses Lab-Grown Mini-Tumors to Find a Patient’s Best Treatment
Using Live Cells from Patients to Find the Right Cancer Drug
Other innovative therapies under investigation could even be agnostic to cancer type:
Treating Pancreatic Cancer: Could Metabolism — Not Genomics — Be the Key?
High-Energy Blue Light Powers a Promising New Treatment to Destroy Cancer Cells
All-Clear Follow-Up: Hydrogen Peroxide Appears to Treat Oral and Skin Lesions
Cancer is a tough nut to crack. Many people and organizations are trying very hard. So much is being learned. Some approaches will be effective. We can all hope.
Dr. Lundberg, editor in chief, Cancer Commons, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Diagnosing, Treating Rashes In Patients on Immune Checkpoint Inhibitors
WASHINGTON, DC — and with judicious usage and dosing of prednisone when deemed necessary, Blair Allais, MD, said during a session on supportive oncodermatology at the ElderDerm conference on dermatology in the older patient hosted by the George Washington University School of Medicine and Health Sciences, Washington, DC.
“It’s important when you see these patients to be as specific as possible” based on morphology and histopathology, and to treat the rashes in a similar way as in the non-ICI setting,” said Dr. Allais, a dermato-oncologist at the Inova Schar Cancer Institute, Fairfax, Virginia.
cirAEs are the most frequently reported and most visible adverse effects of checkpoint inhibition — a treatment that has emerged as a standard therapy for many malignancies since the first ICI was approved in 2011 for metastatic melanoma.
And contrary to what the phenomenon of immunosenescence might suggest, older patients are no less prone to cirAEs than younger patients. “You’d think you’d have fewer rashes and side effects as you age, but that’s not true,” said Dr. Allais, who completed a fellowship in cutaneous oncology after her dermatology residency.
A 2021 multicenter international cohort study of over 900 patients aged ≥ 80 years treated with single-agent ICIs for cancer did not find any significant differences in the development of immune-related adverse events among those younger than 85, those aged 85-89 years, and those 90 and older. Neither did the ELDERS study in the United Kingdom; this prospective observational study found similar rates of high-grade and low-grade immune toxicity in its two cohorts of patients ≥ 70 and < 70 years of age.
At the meeting, Dr. Allais, who coauthored a 2023 review of cirAEs from ICIs, reviewed recent developments and provided the following advice:
New diagnostic criteria: “Really exciting” news for more precise diagnosis and optimal therapy of cirAEs, Dr. Allais said, is a position paper published in the Journal for ImmunoTherapy of Cancer that offers consensus-based diagnostic criteria for the 10 most common types of dermatologic immune-related adverse events and an overall diagnostic framework. “Luckily, through the work of a Delphi consensus group, we can now have [more diagnostic specificity],” which is important for both clinical care and research, she said.
Most cirAEs have typically been reported nonspecifically as “rash,” but diagnosing a rash subtype is “critical in tailoring appropriate therapy that it is both effective and the least detrimental to the oncology treatment plan for patients with cancer,” the group’s coauthors wrote.
The 10 core diagnoses include psoriasis, eczematous dermatitis, vitiligo, Grover disease, eruptive atypical squamous proliferation, and bullous pemphigoid. Outside of the core diagnoses are other nonspecific presentations that require evaluation to arrive at a diagnosis, if possible, or to reveal data that can allow for targeted therapy and severity grading, the group explains in its paper.
“To prednisone or not to prednisone”: The development of cirAEs is associated with reduced mortality and improved cancer outcomes, making the use of immunosuppressants such as corticosteroids a therapeutic dilemma. “Patients who get these rashes usually do better with respect to their cancer, so the concern has been, if we affect how they respond to their immunotherapy, we may minimize that improvement in mortality,” said Dr. Allais, also assistant professor at the University of Virginia, Charlottesville, and clinical assistant professor of dermatology at George Washington University.
A widely discussed study published in 2015 reported on 254 patients with melanoma who developed an immune-related adverse event during treatment with ipilimumab — approximately one third of whom required systemic corticosteroids — and concluded that systemic corticosteroids did not affect overall survival or time to (cancer) treatment failure. This study from Memorial Sloan Kettering Cancer Center, New York City, “was the first large study looking at this question,” she said, and the subsequent message for several years in conferences and the literature was that steroids do not affect the efficacy of checkpoint inhibitors.
“But the study was not without limitations,” Dr. Allais said, “because the patients who got prednisone were mainly those with higher-grade toxicities,” while those not treated with corticosteroids had either no toxicities or low-grade toxicities. “If higher-grade toxicities were associated with better (antitumor) response, the steroids may have just [blunted] that benefit.”
The current totality of data available in the literature suggests that corticosteroids may indeed have an impact on the efficacy of ICI therapy. “Subsequent studies have come out in the community that have shown that we should probably think twice about giving prednisone to some patients, particularly within the first 50 days of ICI treatment, and that we should be mindful of the dose,” Dr. Allais said.
The takeaways from these studies — all published in the past few years — are to use prednisone early and liberally for life-threatening toxicity, to use it at the lowest dose and for the shortest course when there is not an appropriate alternative, to avoid it for diagnoses that are not treated with prednisone outside the ICI setting, and to “have a plan” for a steroid-sparing agent to use after prednisone, she said.
Dr. Allais recommends heightened consideration during the first 50 days of ICI treatment based on a multicenter retrospective study that found a significant association between use of high-dose glucocorticoids (≥ 60 mg prednisone equivalent once a day) within 8 weeks of anti–programmed cell death protein 1 (PD-1) monotherapy initiation and poorer progression-free and overall survival. The study covered a cohort of 947 patients with advanced melanoma treated with anti–PD-1 monotherapy between 2009 and 2019, 54% of whom developed immune-related adverse events.
This study and other recent studies addressing the association between steroids and survival outcomes in patients with immune-related adverse events during ICI therapy are described in Dr. Allais’ 2023 review of cirAEs from ICIs.
Approach to morbilliform eruptions: This rash is “super common” in patients on ICIs, occurring generally within 2-3 weeks of starting treatment. “It tends to be self-limited and can recur with future infusions,” Dr. Allais said.
Systemic steroids should be reserved for severe or refractory eruptions. “Usually, I treat the patients with topical steroids, and I manage their expectations (that the rash may recur with subsequent infusions), but I closely follow them up” within 2-3 weeks, she said. It’s important to rule out a severe cutaneous adverse drug eruption, of course, and to start high-dose systemic steroids immediately if necessary. “Antibiotics are a big culprit” and often can be discontinued.
Soak and smear: “I’m obsessed” with this technique of a 20-minute soak in plain water followed by application of steroid ointment, said Dr. Allais, referring to a small study published in 2005 that reported a complete response after 2 weeks in 60% of patients with psoriasis, atopic dermatitis, and other inflammatory skin conditions (none had cancer), who had failed prior systemic therapy. All patients had at least a 75% response.
The method offers a way to “avoid the systemic immunosuppression we’d get with prednisone,” she said. One just needs to make sure the older patient can get in and out of their tub safely.
ICI-induced bullous pemphigoid (BP): BP occurs more frequently in the ICI setting, compared with the general population, with a median time to development of 8.5 months after ICI initiation. It is associated in this setting with improved tumor response, but “many oncologists stop anticancer treatment because of this diagnosis,” she said.
In the supportive oncodermatology space, however, ICI-induced BP exemplifies the value of tailored treatment regimens, she said. A small multi-institutional retrospective cohort study published in 2023 identified 35 cases of ICI-BP among 5636 ICI-treated patients and found that 8 out of 11 patients who received biologic therapy (rituximab, omalizumab, or dupilumab) had a complete response to ICI-BP without flares following subsequent ICI cycles. And while statistical significance was not reached, the study showed that no cancer-related outcomes were worsened.
“If you see someone with ICI-induced BP and they have a lot of involvement, you could start them on steroids and get that steroid-sparing agent initiated for approval. ... And if IgE is elevated, you might reach for omalizumab,” said Dr. Allais, noting that her favored treatment overall is dupilumab.
Risk factors for the development of ICI-induced BP include age > 70, skin cancer, and having an initial response to ICI on first imaging, the latter of which “I find fascinating ... because imaging occurs within the first 12 weeks of treatment, but we don’t see BP popping up until 8.5 months into treatment,” she noted. “So maybe there’s a baseline risk factor that could predispose them.”
Caution with antibiotics: “I try to avoid antibiotics in the ICI setting,” Dr. Allais said, in deference to the “ever-important microbiome.” Studies have demonstrated that the microbiomes of responders to ICI treatment are different from those of nonresponders, she said.
And a “fascinating” study of patients with melanoma undergoing ICI therapy showed not only a higher abundance of Ruminococcaceae bacteria in responders vs nonresponders but a significant impact of dietary fiber. High dietary fiber was associated with significantly improved overall survival in the patients on ICI, with the most pronounced benefit in patients with good fiber intake and no probiotic use. “Even wilder, their T cells changed,” she said. “They had a high expression of genes related to T-cell activation ... so more tumor-infiltrating lymphocytes.”
A retrospective study of 568 patients with stages III and IV melanoma treated with ICI showed that those exposed to antibiotics prior to ICI had significantly worse overall survival than those not exposed to antibiotics. “Think before you give them,” Dr. Allais said. “And try to tell your older patients to eat beans and greens.”
Dr. Allais reported having no relevant disclosures.
A version of this article first appeared on Medscape.com.
WASHINGTON, DC — and with judicious usage and dosing of prednisone when deemed necessary, Blair Allais, MD, said during a session on supportive oncodermatology at the ElderDerm conference on dermatology in the older patient hosted by the George Washington University School of Medicine and Health Sciences, Washington, DC.
“It’s important when you see these patients to be as specific as possible” based on morphology and histopathology, and to treat the rashes in a similar way as in the non-ICI setting,” said Dr. Allais, a dermato-oncologist at the Inova Schar Cancer Institute, Fairfax, Virginia.
cirAEs are the most frequently reported and most visible adverse effects of checkpoint inhibition — a treatment that has emerged as a standard therapy for many malignancies since the first ICI was approved in 2011 for metastatic melanoma.
And contrary to what the phenomenon of immunosenescence might suggest, older patients are no less prone to cirAEs than younger patients. “You’d think you’d have fewer rashes and side effects as you age, but that’s not true,” said Dr. Allais, who completed a fellowship in cutaneous oncology after her dermatology residency.
A 2021 multicenter international cohort study of over 900 patients aged ≥ 80 years treated with single-agent ICIs for cancer did not find any significant differences in the development of immune-related adverse events among those younger than 85, those aged 85-89 years, and those 90 and older. Neither did the ELDERS study in the United Kingdom; this prospective observational study found similar rates of high-grade and low-grade immune toxicity in its two cohorts of patients ≥ 70 and < 70 years of age.
At the meeting, Dr. Allais, who coauthored a 2023 review of cirAEs from ICIs, reviewed recent developments and provided the following advice:
New diagnostic criteria: “Really exciting” news for more precise diagnosis and optimal therapy of cirAEs, Dr. Allais said, is a position paper published in the Journal for ImmunoTherapy of Cancer that offers consensus-based diagnostic criteria for the 10 most common types of dermatologic immune-related adverse events and an overall diagnostic framework. “Luckily, through the work of a Delphi consensus group, we can now have [more diagnostic specificity],” which is important for both clinical care and research, she said.
Most cirAEs have typically been reported nonspecifically as “rash,” but diagnosing a rash subtype is “critical in tailoring appropriate therapy that it is both effective and the least detrimental to the oncology treatment plan for patients with cancer,” the group’s coauthors wrote.
The 10 core diagnoses include psoriasis, eczematous dermatitis, vitiligo, Grover disease, eruptive atypical squamous proliferation, and bullous pemphigoid. Outside of the core diagnoses are other nonspecific presentations that require evaluation to arrive at a diagnosis, if possible, or to reveal data that can allow for targeted therapy and severity grading, the group explains in its paper.
“To prednisone or not to prednisone”: The development of cirAEs is associated with reduced mortality and improved cancer outcomes, making the use of immunosuppressants such as corticosteroids a therapeutic dilemma. “Patients who get these rashes usually do better with respect to their cancer, so the concern has been, if we affect how they respond to their immunotherapy, we may minimize that improvement in mortality,” said Dr. Allais, also assistant professor at the University of Virginia, Charlottesville, and clinical assistant professor of dermatology at George Washington University.
A widely discussed study published in 2015 reported on 254 patients with melanoma who developed an immune-related adverse event during treatment with ipilimumab — approximately one third of whom required systemic corticosteroids — and concluded that systemic corticosteroids did not affect overall survival or time to (cancer) treatment failure. This study from Memorial Sloan Kettering Cancer Center, New York City, “was the first large study looking at this question,” she said, and the subsequent message for several years in conferences and the literature was that steroids do not affect the efficacy of checkpoint inhibitors.
“But the study was not without limitations,” Dr. Allais said, “because the patients who got prednisone were mainly those with higher-grade toxicities,” while those not treated with corticosteroids had either no toxicities or low-grade toxicities. “If higher-grade toxicities were associated with better (antitumor) response, the steroids may have just [blunted] that benefit.”
The current totality of data available in the literature suggests that corticosteroids may indeed have an impact on the efficacy of ICI therapy. “Subsequent studies have come out in the community that have shown that we should probably think twice about giving prednisone to some patients, particularly within the first 50 days of ICI treatment, and that we should be mindful of the dose,” Dr. Allais said.
The takeaways from these studies — all published in the past few years — are to use prednisone early and liberally for life-threatening toxicity, to use it at the lowest dose and for the shortest course when there is not an appropriate alternative, to avoid it for diagnoses that are not treated with prednisone outside the ICI setting, and to “have a plan” for a steroid-sparing agent to use after prednisone, she said.
Dr. Allais recommends heightened consideration during the first 50 days of ICI treatment based on a multicenter retrospective study that found a significant association between use of high-dose glucocorticoids (≥ 60 mg prednisone equivalent once a day) within 8 weeks of anti–programmed cell death protein 1 (PD-1) monotherapy initiation and poorer progression-free and overall survival. The study covered a cohort of 947 patients with advanced melanoma treated with anti–PD-1 monotherapy between 2009 and 2019, 54% of whom developed immune-related adverse events.
This study and other recent studies addressing the association between steroids and survival outcomes in patients with immune-related adverse events during ICI therapy are described in Dr. Allais’ 2023 review of cirAEs from ICIs.
Approach to morbilliform eruptions: This rash is “super common” in patients on ICIs, occurring generally within 2-3 weeks of starting treatment. “It tends to be self-limited and can recur with future infusions,” Dr. Allais said.
Systemic steroids should be reserved for severe or refractory eruptions. “Usually, I treat the patients with topical steroids, and I manage their expectations (that the rash may recur with subsequent infusions), but I closely follow them up” within 2-3 weeks, she said. It’s important to rule out a severe cutaneous adverse drug eruption, of course, and to start high-dose systemic steroids immediately if necessary. “Antibiotics are a big culprit” and often can be discontinued.
Soak and smear: “I’m obsessed” with this technique of a 20-minute soak in plain water followed by application of steroid ointment, said Dr. Allais, referring to a small study published in 2005 that reported a complete response after 2 weeks in 60% of patients with psoriasis, atopic dermatitis, and other inflammatory skin conditions (none had cancer), who had failed prior systemic therapy. All patients had at least a 75% response.
The method offers a way to “avoid the systemic immunosuppression we’d get with prednisone,” she said. One just needs to make sure the older patient can get in and out of their tub safely.
ICI-induced bullous pemphigoid (BP): BP occurs more frequently in the ICI setting, compared with the general population, with a median time to development of 8.5 months after ICI initiation. It is associated in this setting with improved tumor response, but “many oncologists stop anticancer treatment because of this diagnosis,” she said.
In the supportive oncodermatology space, however, ICI-induced BP exemplifies the value of tailored treatment regimens, she said. A small multi-institutional retrospective cohort study published in 2023 identified 35 cases of ICI-BP among 5636 ICI-treated patients and found that 8 out of 11 patients who received biologic therapy (rituximab, omalizumab, or dupilumab) had a complete response to ICI-BP without flares following subsequent ICI cycles. And while statistical significance was not reached, the study showed that no cancer-related outcomes were worsened.
“If you see someone with ICI-induced BP and they have a lot of involvement, you could start them on steroids and get that steroid-sparing agent initiated for approval. ... And if IgE is elevated, you might reach for omalizumab,” said Dr. Allais, noting that her favored treatment overall is dupilumab.
Risk factors for the development of ICI-induced BP include age > 70, skin cancer, and having an initial response to ICI on first imaging, the latter of which “I find fascinating ... because imaging occurs within the first 12 weeks of treatment, but we don’t see BP popping up until 8.5 months into treatment,” she noted. “So maybe there’s a baseline risk factor that could predispose them.”
Caution with antibiotics: “I try to avoid antibiotics in the ICI setting,” Dr. Allais said, in deference to the “ever-important microbiome.” Studies have demonstrated that the microbiomes of responders to ICI treatment are different from those of nonresponders, she said.
And a “fascinating” study of patients with melanoma undergoing ICI therapy showed not only a higher abundance of Ruminococcaceae bacteria in responders vs nonresponders but a significant impact of dietary fiber. High dietary fiber was associated with significantly improved overall survival in the patients on ICI, with the most pronounced benefit in patients with good fiber intake and no probiotic use. “Even wilder, their T cells changed,” she said. “They had a high expression of genes related to T-cell activation ... so more tumor-infiltrating lymphocytes.”
A retrospective study of 568 patients with stages III and IV melanoma treated with ICI showed that those exposed to antibiotics prior to ICI had significantly worse overall survival than those not exposed to antibiotics. “Think before you give them,” Dr. Allais said. “And try to tell your older patients to eat beans and greens.”
Dr. Allais reported having no relevant disclosures.
A version of this article first appeared on Medscape.com.
WASHINGTON, DC — and with judicious usage and dosing of prednisone when deemed necessary, Blair Allais, MD, said during a session on supportive oncodermatology at the ElderDerm conference on dermatology in the older patient hosted by the George Washington University School of Medicine and Health Sciences, Washington, DC.
“It’s important when you see these patients to be as specific as possible” based on morphology and histopathology, and to treat the rashes in a similar way as in the non-ICI setting,” said Dr. Allais, a dermato-oncologist at the Inova Schar Cancer Institute, Fairfax, Virginia.
cirAEs are the most frequently reported and most visible adverse effects of checkpoint inhibition — a treatment that has emerged as a standard therapy for many malignancies since the first ICI was approved in 2011 for metastatic melanoma.
And contrary to what the phenomenon of immunosenescence might suggest, older patients are no less prone to cirAEs than younger patients. “You’d think you’d have fewer rashes and side effects as you age, but that’s not true,” said Dr. Allais, who completed a fellowship in cutaneous oncology after her dermatology residency.
A 2021 multicenter international cohort study of over 900 patients aged ≥ 80 years treated with single-agent ICIs for cancer did not find any significant differences in the development of immune-related adverse events among those younger than 85, those aged 85-89 years, and those 90 and older. Neither did the ELDERS study in the United Kingdom; this prospective observational study found similar rates of high-grade and low-grade immune toxicity in its two cohorts of patients ≥ 70 and < 70 years of age.
At the meeting, Dr. Allais, who coauthored a 2023 review of cirAEs from ICIs, reviewed recent developments and provided the following advice:
New diagnostic criteria: “Really exciting” news for more precise diagnosis and optimal therapy of cirAEs, Dr. Allais said, is a position paper published in the Journal for ImmunoTherapy of Cancer that offers consensus-based diagnostic criteria for the 10 most common types of dermatologic immune-related adverse events and an overall diagnostic framework. “Luckily, through the work of a Delphi consensus group, we can now have [more diagnostic specificity],” which is important for both clinical care and research, she said.
Most cirAEs have typically been reported nonspecifically as “rash,” but diagnosing a rash subtype is “critical in tailoring appropriate therapy that it is both effective and the least detrimental to the oncology treatment plan for patients with cancer,” the group’s coauthors wrote.
The 10 core diagnoses include psoriasis, eczematous dermatitis, vitiligo, Grover disease, eruptive atypical squamous proliferation, and bullous pemphigoid. Outside of the core diagnoses are other nonspecific presentations that require evaluation to arrive at a diagnosis, if possible, or to reveal data that can allow for targeted therapy and severity grading, the group explains in its paper.
“To prednisone or not to prednisone”: The development of cirAEs is associated with reduced mortality and improved cancer outcomes, making the use of immunosuppressants such as corticosteroids a therapeutic dilemma. “Patients who get these rashes usually do better with respect to their cancer, so the concern has been, if we affect how they respond to their immunotherapy, we may minimize that improvement in mortality,” said Dr. Allais, also assistant professor at the University of Virginia, Charlottesville, and clinical assistant professor of dermatology at George Washington University.
A widely discussed study published in 2015 reported on 254 patients with melanoma who developed an immune-related adverse event during treatment with ipilimumab — approximately one third of whom required systemic corticosteroids — and concluded that systemic corticosteroids did not affect overall survival or time to (cancer) treatment failure. This study from Memorial Sloan Kettering Cancer Center, New York City, “was the first large study looking at this question,” she said, and the subsequent message for several years in conferences and the literature was that steroids do not affect the efficacy of checkpoint inhibitors.
“But the study was not without limitations,” Dr. Allais said, “because the patients who got prednisone were mainly those with higher-grade toxicities,” while those not treated with corticosteroids had either no toxicities or low-grade toxicities. “If higher-grade toxicities were associated with better (antitumor) response, the steroids may have just [blunted] that benefit.”
The current totality of data available in the literature suggests that corticosteroids may indeed have an impact on the efficacy of ICI therapy. “Subsequent studies have come out in the community that have shown that we should probably think twice about giving prednisone to some patients, particularly within the first 50 days of ICI treatment, and that we should be mindful of the dose,” Dr. Allais said.
The takeaways from these studies — all published in the past few years — are to use prednisone early and liberally for life-threatening toxicity, to use it at the lowest dose and for the shortest course when there is not an appropriate alternative, to avoid it for diagnoses that are not treated with prednisone outside the ICI setting, and to “have a plan” for a steroid-sparing agent to use after prednisone, she said.
Dr. Allais recommends heightened consideration during the first 50 days of ICI treatment based on a multicenter retrospective study that found a significant association between use of high-dose glucocorticoids (≥ 60 mg prednisone equivalent once a day) within 8 weeks of anti–programmed cell death protein 1 (PD-1) monotherapy initiation and poorer progression-free and overall survival. The study covered a cohort of 947 patients with advanced melanoma treated with anti–PD-1 monotherapy between 2009 and 2019, 54% of whom developed immune-related adverse events.
This study and other recent studies addressing the association between steroids and survival outcomes in patients with immune-related adverse events during ICI therapy are described in Dr. Allais’ 2023 review of cirAEs from ICIs.
Approach to morbilliform eruptions: This rash is “super common” in patients on ICIs, occurring generally within 2-3 weeks of starting treatment. “It tends to be self-limited and can recur with future infusions,” Dr. Allais said.
Systemic steroids should be reserved for severe or refractory eruptions. “Usually, I treat the patients with topical steroids, and I manage their expectations (that the rash may recur with subsequent infusions), but I closely follow them up” within 2-3 weeks, she said. It’s important to rule out a severe cutaneous adverse drug eruption, of course, and to start high-dose systemic steroids immediately if necessary. “Antibiotics are a big culprit” and often can be discontinued.
Soak and smear: “I’m obsessed” with this technique of a 20-minute soak in plain water followed by application of steroid ointment, said Dr. Allais, referring to a small study published in 2005 that reported a complete response after 2 weeks in 60% of patients with psoriasis, atopic dermatitis, and other inflammatory skin conditions (none had cancer), who had failed prior systemic therapy. All patients had at least a 75% response.
The method offers a way to “avoid the systemic immunosuppression we’d get with prednisone,” she said. One just needs to make sure the older patient can get in and out of their tub safely.
ICI-induced bullous pemphigoid (BP): BP occurs more frequently in the ICI setting, compared with the general population, with a median time to development of 8.5 months after ICI initiation. It is associated in this setting with improved tumor response, but “many oncologists stop anticancer treatment because of this diagnosis,” she said.
In the supportive oncodermatology space, however, ICI-induced BP exemplifies the value of tailored treatment regimens, she said. A small multi-institutional retrospective cohort study published in 2023 identified 35 cases of ICI-BP among 5636 ICI-treated patients and found that 8 out of 11 patients who received biologic therapy (rituximab, omalizumab, or dupilumab) had a complete response to ICI-BP without flares following subsequent ICI cycles. And while statistical significance was not reached, the study showed that no cancer-related outcomes were worsened.
“If you see someone with ICI-induced BP and they have a lot of involvement, you could start them on steroids and get that steroid-sparing agent initiated for approval. ... And if IgE is elevated, you might reach for omalizumab,” said Dr. Allais, noting that her favored treatment overall is dupilumab.
Risk factors for the development of ICI-induced BP include age > 70, skin cancer, and having an initial response to ICI on first imaging, the latter of which “I find fascinating ... because imaging occurs within the first 12 weeks of treatment, but we don’t see BP popping up until 8.5 months into treatment,” she noted. “So maybe there’s a baseline risk factor that could predispose them.”
Caution with antibiotics: “I try to avoid antibiotics in the ICI setting,” Dr. Allais said, in deference to the “ever-important microbiome.” Studies have demonstrated that the microbiomes of responders to ICI treatment are different from those of nonresponders, she said.
And a “fascinating” study of patients with melanoma undergoing ICI therapy showed not only a higher abundance of Ruminococcaceae bacteria in responders vs nonresponders but a significant impact of dietary fiber. High dietary fiber was associated with significantly improved overall survival in the patients on ICI, with the most pronounced benefit in patients with good fiber intake and no probiotic use. “Even wilder, their T cells changed,” she said. “They had a high expression of genes related to T-cell activation ... so more tumor-infiltrating lymphocytes.”
A retrospective study of 568 patients with stages III and IV melanoma treated with ICI showed that those exposed to antibiotics prior to ICI had significantly worse overall survival than those not exposed to antibiotics. “Think before you give them,” Dr. Allais said. “And try to tell your older patients to eat beans and greens.”
Dr. Allais reported having no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM ELDERDERM 2024
Jeffrey Weber, MD, PhD, Giant of Cancer Care, Dies
Dr. Weber, a melanoma and cancer immunotherapy specialist, served as deputy director of the Laura and Isaac Perlmutter Cancer Center at New York University (NYU) Langone Medical Center in New York City. He also held positions as the Laura and Isaac Perlmutter Professor of Oncology in the Department of Medicine at the NYU Grossman School of Medicine, director of the Experimental Therapeutics Program, and co-leader of the Clinical Melanoma Program Board at NYU Langone Health.
Dr. Weber was a principal investigator on many studies, including pivotal clinical drug trials in melanoma and trials focused on managing autoimmune side effects from immunotherapy. He published more than 150 articles in top peer-reviewed journals.
For many years, Dr. Weber hosted the popular “Weber on Oncology” series of video contributions for Medscape Oncology, sharing updates and insights on noteworthy research and breakthroughs in melanoma.
“The Melanoma Research Alliance mourns the passing of Dr. Jeffrey S. Weber, a true pioneer in the field of cancer immunotherapy and an extraordinary leader in melanoma research. His contributions have forever changed the landscape of melanoma treatment, bringing groundbreaking advances from the lab into clinical practice and offering hope to countless patients,” the Melanoma Research Alliance posted on LinkedIn.
Many X users also shared condolences and memories of Dr. Weber, praising his numerous contributions and accomplishments.
“[Cancer Research Institute] mourns the loss of Dr. Jeffrey S. Weber ... [a]s an accomplished physician scientist, Dr. Weber drove advances in melanoma research, and played an active role in educating patients about the lifesaving power of immunotherapy,” the Cancer Research Institute posted.
A colleague noted that “[h]e was involved in the early days of cytokine and cell therapy and most recently led studies of personalized vaccines for melanoma patients. ... He was a great friend and colleague to many of us in the melanoma and immunotherapy field and we will remember him as a pioneer, thought leader and compassionate physician.”
A version of this article first appeared on Medscape.com.
Dr. Weber, a melanoma and cancer immunotherapy specialist, served as deputy director of the Laura and Isaac Perlmutter Cancer Center at New York University (NYU) Langone Medical Center in New York City. He also held positions as the Laura and Isaac Perlmutter Professor of Oncology in the Department of Medicine at the NYU Grossman School of Medicine, director of the Experimental Therapeutics Program, and co-leader of the Clinical Melanoma Program Board at NYU Langone Health.
Dr. Weber was a principal investigator on many studies, including pivotal clinical drug trials in melanoma and trials focused on managing autoimmune side effects from immunotherapy. He published more than 150 articles in top peer-reviewed journals.
For many years, Dr. Weber hosted the popular “Weber on Oncology” series of video contributions for Medscape Oncology, sharing updates and insights on noteworthy research and breakthroughs in melanoma.
“The Melanoma Research Alliance mourns the passing of Dr. Jeffrey S. Weber, a true pioneer in the field of cancer immunotherapy and an extraordinary leader in melanoma research. His contributions have forever changed the landscape of melanoma treatment, bringing groundbreaking advances from the lab into clinical practice and offering hope to countless patients,” the Melanoma Research Alliance posted on LinkedIn.
Many X users also shared condolences and memories of Dr. Weber, praising his numerous contributions and accomplishments.
“[Cancer Research Institute] mourns the loss of Dr. Jeffrey S. Weber ... [a]s an accomplished physician scientist, Dr. Weber drove advances in melanoma research, and played an active role in educating patients about the lifesaving power of immunotherapy,” the Cancer Research Institute posted.
A colleague noted that “[h]e was involved in the early days of cytokine and cell therapy and most recently led studies of personalized vaccines for melanoma patients. ... He was a great friend and colleague to many of us in the melanoma and immunotherapy field and we will remember him as a pioneer, thought leader and compassionate physician.”
A version of this article first appeared on Medscape.com.
Dr. Weber, a melanoma and cancer immunotherapy specialist, served as deputy director of the Laura and Isaac Perlmutter Cancer Center at New York University (NYU) Langone Medical Center in New York City. He also held positions as the Laura and Isaac Perlmutter Professor of Oncology in the Department of Medicine at the NYU Grossman School of Medicine, director of the Experimental Therapeutics Program, and co-leader of the Clinical Melanoma Program Board at NYU Langone Health.
Dr. Weber was a principal investigator on many studies, including pivotal clinical drug trials in melanoma and trials focused on managing autoimmune side effects from immunotherapy. He published more than 150 articles in top peer-reviewed journals.
For many years, Dr. Weber hosted the popular “Weber on Oncology” series of video contributions for Medscape Oncology, sharing updates and insights on noteworthy research and breakthroughs in melanoma.
“The Melanoma Research Alliance mourns the passing of Dr. Jeffrey S. Weber, a true pioneer in the field of cancer immunotherapy and an extraordinary leader in melanoma research. His contributions have forever changed the landscape of melanoma treatment, bringing groundbreaking advances from the lab into clinical practice and offering hope to countless patients,” the Melanoma Research Alliance posted on LinkedIn.
Many X users also shared condolences and memories of Dr. Weber, praising his numerous contributions and accomplishments.
“[Cancer Research Institute] mourns the loss of Dr. Jeffrey S. Weber ... [a]s an accomplished physician scientist, Dr. Weber drove advances in melanoma research, and played an active role in educating patients about the lifesaving power of immunotherapy,” the Cancer Research Institute posted.
A colleague noted that “[h]e was involved in the early days of cytokine and cell therapy and most recently led studies of personalized vaccines for melanoma patients. ... He was a great friend and colleague to many of us in the melanoma and immunotherapy field and we will remember him as a pioneer, thought leader and compassionate physician.”
A version of this article first appeared on Medscape.com.
Immunotherapy and Survival in Advanced NSCLC: Does Obesity Matter?
TOPLINE:
Overall, however, compared with low body mass index (BMI), overweight or obesity was associated with a lower risk for mortality among patients receiving either therapy.
METHODOLOGY:
- The association between BMI and overall survival in patients with cancer who receive immunotherapy or conventional chemotherapy in the frontline remains unclear. Patients with cancer and obesity are generally considered to have a worse prognosis, but some data suggest an obesity paradox, where patients with cancer and a higher BMI demonstrate better overall survival following immunotherapy or chemotherapy.
- To clarify whether (or how) BMI affects overall survival outcomes and the optimal frontline treatment choice, researchers evaluated 31,257 patients with advanced NSCLC from Japan who received immune checkpoint inhibitors (n = 12,816) or conventional chemotherapy (n = 18,441).
- Patient outcomes were assessed according to weight categories and frontline therapy type (immune checkpoint inhibitors or conventional chemotherapy), with overall survival as the primary outcome.
- A BMI < 18.5 was considered underweight, 18.5-24.9 was considered normal weight, 25.0-29.9 was considered overweight, and ≥ 30.0 was considered obese.
TAKEAWAY:
- In the overall population, regardless of weight, patients who received chemotherapy had a higher mortality rate than those who received immunotherapy — 35.9% vs 28.0%, respectively — over a follow-up of 3 years.
- However, overweight or obesity was associated with a lower risk for mortality compared with a lower BMI among patients with advanced NSCLC, regardless of whether they received immune checkpoint inhibitor therapy or conventional chemotherapy.
- Among patients who received immunotherapy, the risk for mortality decreased steadily as BMI increased from 15 to 24 and then increased at higher BMIs, indicating a U-shaped association.
- Immunotherapy was associated with a significant improvement in overall survival compared with conventional chemotherapy among patients with a BMI < 28; however, researchers observed no difference in overall survival between the two therapies in those with a BMI ≥ 28.
IN PRACTICE:
Overall, “these results support the presence of the obesity paradox in patients with [advanced] NSCLC who underwent either therapy,” the authors concluded.
But when focused on patients in the higher BMI group, there was no overall survival benefit with the frontline immunotherapy vs the conventional chemotherapy. “Immunotherapy therapy may not necessarily be the optimal first-line therapy for patients with overweight or obesity,” the authors wrote, adding that “the use of conventional chemotherapy should also be considered.”
SOURCE:
The study, led by Yasutaka Ihara, PharmD, Osaka Metropolitan University, Osaka, Japan, was published online in JAMA Network Open.
LIMITATIONS:
Retrospective design has inherent bias. PD-L1 status was not known, and the inclusion of Japanese population may have limited the generalizability of the findings.
DISCLOSURES:
This study received funding from the Graduate School of Medicine, Osaka Metropolitan University. Several authors reported receiving personal fees from various pharmaceutical sources.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Overall, however, compared with low body mass index (BMI), overweight or obesity was associated with a lower risk for mortality among patients receiving either therapy.
METHODOLOGY:
- The association between BMI and overall survival in patients with cancer who receive immunotherapy or conventional chemotherapy in the frontline remains unclear. Patients with cancer and obesity are generally considered to have a worse prognosis, but some data suggest an obesity paradox, where patients with cancer and a higher BMI demonstrate better overall survival following immunotherapy or chemotherapy.
- To clarify whether (or how) BMI affects overall survival outcomes and the optimal frontline treatment choice, researchers evaluated 31,257 patients with advanced NSCLC from Japan who received immune checkpoint inhibitors (n = 12,816) or conventional chemotherapy (n = 18,441).
- Patient outcomes were assessed according to weight categories and frontline therapy type (immune checkpoint inhibitors or conventional chemotherapy), with overall survival as the primary outcome.
- A BMI < 18.5 was considered underweight, 18.5-24.9 was considered normal weight, 25.0-29.9 was considered overweight, and ≥ 30.0 was considered obese.
TAKEAWAY:
- In the overall population, regardless of weight, patients who received chemotherapy had a higher mortality rate than those who received immunotherapy — 35.9% vs 28.0%, respectively — over a follow-up of 3 years.
- However, overweight or obesity was associated with a lower risk for mortality compared with a lower BMI among patients with advanced NSCLC, regardless of whether they received immune checkpoint inhibitor therapy or conventional chemotherapy.
- Among patients who received immunotherapy, the risk for mortality decreased steadily as BMI increased from 15 to 24 and then increased at higher BMIs, indicating a U-shaped association.
- Immunotherapy was associated with a significant improvement in overall survival compared with conventional chemotherapy among patients with a BMI < 28; however, researchers observed no difference in overall survival between the two therapies in those with a BMI ≥ 28.
IN PRACTICE:
Overall, “these results support the presence of the obesity paradox in patients with [advanced] NSCLC who underwent either therapy,” the authors concluded.
But when focused on patients in the higher BMI group, there was no overall survival benefit with the frontline immunotherapy vs the conventional chemotherapy. “Immunotherapy therapy may not necessarily be the optimal first-line therapy for patients with overweight or obesity,” the authors wrote, adding that “the use of conventional chemotherapy should also be considered.”
SOURCE:
The study, led by Yasutaka Ihara, PharmD, Osaka Metropolitan University, Osaka, Japan, was published online in JAMA Network Open.
LIMITATIONS:
Retrospective design has inherent bias. PD-L1 status was not known, and the inclusion of Japanese population may have limited the generalizability of the findings.
DISCLOSURES:
This study received funding from the Graduate School of Medicine, Osaka Metropolitan University. Several authors reported receiving personal fees from various pharmaceutical sources.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
TOPLINE:
Overall, however, compared with low body mass index (BMI), overweight or obesity was associated with a lower risk for mortality among patients receiving either therapy.
METHODOLOGY:
- The association between BMI and overall survival in patients with cancer who receive immunotherapy or conventional chemotherapy in the frontline remains unclear. Patients with cancer and obesity are generally considered to have a worse prognosis, but some data suggest an obesity paradox, where patients with cancer and a higher BMI demonstrate better overall survival following immunotherapy or chemotherapy.
- To clarify whether (or how) BMI affects overall survival outcomes and the optimal frontline treatment choice, researchers evaluated 31,257 patients with advanced NSCLC from Japan who received immune checkpoint inhibitors (n = 12,816) or conventional chemotherapy (n = 18,441).
- Patient outcomes were assessed according to weight categories and frontline therapy type (immune checkpoint inhibitors or conventional chemotherapy), with overall survival as the primary outcome.
- A BMI < 18.5 was considered underweight, 18.5-24.9 was considered normal weight, 25.0-29.9 was considered overweight, and ≥ 30.0 was considered obese.
TAKEAWAY:
- In the overall population, regardless of weight, patients who received chemotherapy had a higher mortality rate than those who received immunotherapy — 35.9% vs 28.0%, respectively — over a follow-up of 3 years.
- However, overweight or obesity was associated with a lower risk for mortality compared with a lower BMI among patients with advanced NSCLC, regardless of whether they received immune checkpoint inhibitor therapy or conventional chemotherapy.
- Among patients who received immunotherapy, the risk for mortality decreased steadily as BMI increased from 15 to 24 and then increased at higher BMIs, indicating a U-shaped association.
- Immunotherapy was associated with a significant improvement in overall survival compared with conventional chemotherapy among patients with a BMI < 28; however, researchers observed no difference in overall survival between the two therapies in those with a BMI ≥ 28.
IN PRACTICE:
Overall, “these results support the presence of the obesity paradox in patients with [advanced] NSCLC who underwent either therapy,” the authors concluded.
But when focused on patients in the higher BMI group, there was no overall survival benefit with the frontline immunotherapy vs the conventional chemotherapy. “Immunotherapy therapy may not necessarily be the optimal first-line therapy for patients with overweight or obesity,” the authors wrote, adding that “the use of conventional chemotherapy should also be considered.”
SOURCE:
The study, led by Yasutaka Ihara, PharmD, Osaka Metropolitan University, Osaka, Japan, was published online in JAMA Network Open.
LIMITATIONS:
Retrospective design has inherent bias. PD-L1 status was not known, and the inclusion of Japanese population may have limited the generalizability of the findings.
DISCLOSURES:
This study received funding from the Graduate School of Medicine, Osaka Metropolitan University. Several authors reported receiving personal fees from various pharmaceutical sources.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article first appeared on Medscape.com.
FDA Approves Lymphir for R/R Cutaneous T-Cell Lymphoma
The immunotherapy is a reformulation of denileukin diftitox (Ontak), initially approved in 1999 for certain patients with persistent or recurrent cutaneous T-cell lymphoma. In 2014, the original formulation was voluntarily withdrawn from the US market. Citius acquired rights to market a reformulated product outside of Asia in 2021.
This is the first indication for Lymphir, which targets interleukin-2 receptors on malignant T cells.
This approval marks “a significant milestone” for patients with cutaneous T-cell lymphoma, a rare cancer, company CEO Leonard Mazur said in a press release announcing the approval. “The introduction of Lymphir, with its potential to rapidly reduce skin disease and control symptomatic itching without cumulative toxicity, is expected to expand the [cutaneous T-cell lymphoma] treatment landscape and grow the overall market, currently estimated to be $300-$400 million.”
Approval was based on the single-arm, open-label 302 study in 69 patients who had a median of four prior anticancer therapies. Patients received 9 mcg/kg daily from day 1 to day 5 of 21-day cycles until disease progression or unacceptable toxicity.
The objective response rate was 36.2%, including complete responses in 8.7% of patients. Responses lasted 6 months or longer in 52% of patients. Over 80% of subjects had a decrease in skin tumor burden, and almost a third had clinically significant improvements in pruritus.
Adverse events occurring in 20% or more of patients include increased transaminases, decreased albumin, decreased hemoglobin, nausea, edema, fatigue, musculoskeletal pain, rash, chills, constipation, pyrexia, and capillary leak syndrome.
Labeling carries a boxed warning of capillary leak syndrome. Other warnings include visual impairment, infusion reactions, hepatotoxicity, and embryo-fetal toxicity. Citius is under a postmarketing requirement to characterize the risk for visual impairment.
The company expects to launch the agent within 5 months.
A version of this article first appeared on Medscape.com.
The immunotherapy is a reformulation of denileukin diftitox (Ontak), initially approved in 1999 for certain patients with persistent or recurrent cutaneous T-cell lymphoma. In 2014, the original formulation was voluntarily withdrawn from the US market. Citius acquired rights to market a reformulated product outside of Asia in 2021.
This is the first indication for Lymphir, which targets interleukin-2 receptors on malignant T cells.
This approval marks “a significant milestone” for patients with cutaneous T-cell lymphoma, a rare cancer, company CEO Leonard Mazur said in a press release announcing the approval. “The introduction of Lymphir, with its potential to rapidly reduce skin disease and control symptomatic itching without cumulative toxicity, is expected to expand the [cutaneous T-cell lymphoma] treatment landscape and grow the overall market, currently estimated to be $300-$400 million.”
Approval was based on the single-arm, open-label 302 study in 69 patients who had a median of four prior anticancer therapies. Patients received 9 mcg/kg daily from day 1 to day 5 of 21-day cycles until disease progression or unacceptable toxicity.
The objective response rate was 36.2%, including complete responses in 8.7% of patients. Responses lasted 6 months or longer in 52% of patients. Over 80% of subjects had a decrease in skin tumor burden, and almost a third had clinically significant improvements in pruritus.
Adverse events occurring in 20% or more of patients include increased transaminases, decreased albumin, decreased hemoglobin, nausea, edema, fatigue, musculoskeletal pain, rash, chills, constipation, pyrexia, and capillary leak syndrome.
Labeling carries a boxed warning of capillary leak syndrome. Other warnings include visual impairment, infusion reactions, hepatotoxicity, and embryo-fetal toxicity. Citius is under a postmarketing requirement to characterize the risk for visual impairment.
The company expects to launch the agent within 5 months.
A version of this article first appeared on Medscape.com.
The immunotherapy is a reformulation of denileukin diftitox (Ontak), initially approved in 1999 for certain patients with persistent or recurrent cutaneous T-cell lymphoma. In 2014, the original formulation was voluntarily withdrawn from the US market. Citius acquired rights to market a reformulated product outside of Asia in 2021.
This is the first indication for Lymphir, which targets interleukin-2 receptors on malignant T cells.
This approval marks “a significant milestone” for patients with cutaneous T-cell lymphoma, a rare cancer, company CEO Leonard Mazur said in a press release announcing the approval. “The introduction of Lymphir, with its potential to rapidly reduce skin disease and control symptomatic itching without cumulative toxicity, is expected to expand the [cutaneous T-cell lymphoma] treatment landscape and grow the overall market, currently estimated to be $300-$400 million.”
Approval was based on the single-arm, open-label 302 study in 69 patients who had a median of four prior anticancer therapies. Patients received 9 mcg/kg daily from day 1 to day 5 of 21-day cycles until disease progression or unacceptable toxicity.
The objective response rate was 36.2%, including complete responses in 8.7% of patients. Responses lasted 6 months or longer in 52% of patients. Over 80% of subjects had a decrease in skin tumor burden, and almost a third had clinically significant improvements in pruritus.
Adverse events occurring in 20% or more of patients include increased transaminases, decreased albumin, decreased hemoglobin, nausea, edema, fatigue, musculoskeletal pain, rash, chills, constipation, pyrexia, and capillary leak syndrome.
Labeling carries a boxed warning of capillary leak syndrome. Other warnings include visual impairment, infusion reactions, hepatotoxicity, and embryo-fetal toxicity. Citius is under a postmarketing requirement to characterize the risk for visual impairment.
The company expects to launch the agent within 5 months.
A version of this article first appeared on Medscape.com.
Immunotherapy May Be Overused in Dying Patients With Cancer
Chemotherapy has fallen out of favor for treating cancer toward the end of life. The toxicity is too high, and the benefit, if any, is often too low.
Immunotherapy, however, has been taking its place.
This means “there are patients who are getting immunotherapy who shouldn’t,” said Yale University, New Haven, Connecticut, surgical oncologist Sajid Khan, MD, senior investigator on a recent study that highlighted the growing use of these agents in patients’ last month of life.
What’s driving this trend, and how can oncologists avoid overtreatment with immunotherapy at the end of life?
The N-of-1 Patient
With immunotherapy at the end of life, “each of us has had our N-of-1” where a patient bounces back with a remarkable and durable response, said Don Dizon, MD, a gynecologic oncologist at Brown University, Providence, Rhode Island.
He recalled a patient with sarcoma who did not respond to chemotherapy. But after Dr. Dizon started her on immunotherapy, everything turned around. She has now been in remission for 8 years and counting.
The possibility of an unexpected or remarkable responder is seductive. And the improved safety of immunotherapy over chemotherapy adds to the allure.
Meanwhile, patients are often desperate. It’s rare for someone to be ready to stop treatment, Dr. Dizon said. Everybody “hopes that they’re going to be the exceptional responder.”
At the end of the day, the question often becomes: “Why not try immunotherapy? What’s there to lose?”
This thinking may be prompting broader use of immunotherapy in late-stage disease, even in instances with no Food and Drug Administration indication and virtually no supportive data, such as for metastatic ovarian cancer, Dr. Dizon said.
Back to Earth
The problem with the hopeful approach is that end-of-life turnarounds with immunotherapy are rare, and there’s no way at the moment to predict who will have one, said Laura Petrillo, MD, a palliative care physician at Massachusetts General Hospital, Boston.
Even though immunotherapy generally comes with fewer adverse events than chemotherapy, catastrophic side effects are still possible.
Dr. Petrillo recalled a 95-year-old woman with metastatic cancer who was largely asymptomatic.
She had a qualifying mutation for a checkpoint inhibitor, so her oncologist started her on one. The patient never bounced back from the severe colitis the agent caused, and she died of complications in the hospital.
Although such reactions with immunotherapy are uncommon, less serious problems caused by the agents can still have a major impact on a person’s quality of life. Low-grade diarrhea, for instance, may not sound too bad, but in a patient’s daily life, it can translate to six or more episodes a day.
Even with no side effects, prescribing immunotherapy can mean that patients with limited time left spend a good portion of it at an infusion clinic instead of at home. These patients are also less likely to be referred to hospice and more likely to be admitted to and die in the hospital.
And with treatments that can cost $20,000 per dose, financial toxicity becomes a big concern.
In short, some of the reasons why chemotherapy is not recommended at the end of life also apply to immunotherapy, Dr. Petrillo said.
Prescribing Decisions
Recent research highlights the growing use of immunotherapy at the end of life.
Dr. Khan’s retrospective study found, for instance, that the percentage of patients starting immunotherapy in the last 30 days of life increased by about fourfold to fivefold over the study period for the three cancers analyzed — stage IV melanoma, lung, and kidney cancers.
Among the population that died within 30 days, the percentage receiving immunotherapy increased over the study periods — 0.8%-4.3% for melanoma, 0.9%-3.2% for NSCLC, and 0.5%-2.6% for kidney cell carcinoma — prompting the conclusion that immunotherapy prescriptions in the last month of life are on the rise.
Prescribing immunotherapy in patients who ultimately died within 1 month occurred more frequently at low-volume, nonacademic centers than at academic or high-volume centers, and outcomes varied by practice setting.
Patients had better survival outcomes overall when receiving immunotherapy at academic or high-volume centers — a finding Dr. Khan said is worth investigating further. Possible explanations include better management of severe immune-related side effects at larger centers and more caution when prescribing immunotherapy to “borderline” candidates, such as those with several comorbidities.
Importantly, given the retrospective design, Dr. Khan and colleagues already knew which patients prescribed immunotherapy died within 30 days of initiating treatment.
More specifically, 5192 of 71,204 patients who received immunotherapy (7.3%) died within a month of initiating therapy, while 66,012 (92.7%) lived beyond that point.
The study, however, did not assess how the remaining 92.7% who lived beyond 30 days fared on immunotherapy and the differences between those who lived less than 30 days and those who survived longer.
Knowing the outcome of patients at the outset of the analysis still leaves open the question of when immunotherapy can extend life and when it can’t for the patient in front of you.
To avoid overtreating at the end of life, it’s important to have “the same standard that you have for giving chemotherapy. You have to treat it with the same respect,” said Moshe Chasky, MD, a community medical oncologist with Alliance Cancer Specialists in Philadelphia, Pennsylvania. “You can’t just be throwing” immunotherapy around “at the end of life.”
While there are no clear predictors of risk and benefit, there are some factors to help guide decisions.
As with chemotherapy, Dr. Petrillo said performance status is key. Dr. Petrillo and colleagues found that median overall survival with immune checkpoint inhibitors for advanced non–small cell lung cancer was 14.3 months in patients with an Eastern Cooperative Oncology Group performance score of 0-1 but only 4.5 months with scores of ≥ 2.
Dr. Khan also found that immunotherapy survival is, unsurprisingly, worse in patients with high metastatic burdens and more comorbidities.
“You should still consider immunotherapy for metastatic melanoma, non–small cell lung cancer, and renal cell carcinoma,” Dr. Khan said. The message here is to “think twice before using” it, especially in comorbid patients with widespread metastases.
“Just because something can be done doesn’t always mean it should be done,” he said.
At Yale, when Dr. Khan works, immunotherapy decisions are considered by a multidisciplinary tumor board. At Mass General, immunotherapy has generally moved to the frontline setting, and the hospital no longer prescribes checkpoint inhibitors to hospitalized patients because the cost is too high relative to the potential benefit, Dr. Petrillo explained.
Still, with all the uncertainties about risk and benefit, counseling patients is a challenge. Dr. Dizon called it “the epitome of shared decision-making.”
Dr. Petrillo noted that it’s critical not to counsel patients based solely on the anecdotal patients who do surprisingly well.
“It’s hard to mention that and not have that be what somebody anchors on,” she said. But that speaks to “how desperate people can feel, how hopeful they can be.”
Dr. Khan, Dr. Petrillo, and Dr. Chasky all reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Chemotherapy has fallen out of favor for treating cancer toward the end of life. The toxicity is too high, and the benefit, if any, is often too low.
Immunotherapy, however, has been taking its place.
This means “there are patients who are getting immunotherapy who shouldn’t,” said Yale University, New Haven, Connecticut, surgical oncologist Sajid Khan, MD, senior investigator on a recent study that highlighted the growing use of these agents in patients’ last month of life.
What’s driving this trend, and how can oncologists avoid overtreatment with immunotherapy at the end of life?
The N-of-1 Patient
With immunotherapy at the end of life, “each of us has had our N-of-1” where a patient bounces back with a remarkable and durable response, said Don Dizon, MD, a gynecologic oncologist at Brown University, Providence, Rhode Island.
He recalled a patient with sarcoma who did not respond to chemotherapy. But after Dr. Dizon started her on immunotherapy, everything turned around. She has now been in remission for 8 years and counting.
The possibility of an unexpected or remarkable responder is seductive. And the improved safety of immunotherapy over chemotherapy adds to the allure.
Meanwhile, patients are often desperate. It’s rare for someone to be ready to stop treatment, Dr. Dizon said. Everybody “hopes that they’re going to be the exceptional responder.”
At the end of the day, the question often becomes: “Why not try immunotherapy? What’s there to lose?”
This thinking may be prompting broader use of immunotherapy in late-stage disease, even in instances with no Food and Drug Administration indication and virtually no supportive data, such as for metastatic ovarian cancer, Dr. Dizon said.
Back to Earth
The problem with the hopeful approach is that end-of-life turnarounds with immunotherapy are rare, and there’s no way at the moment to predict who will have one, said Laura Petrillo, MD, a palliative care physician at Massachusetts General Hospital, Boston.
Even though immunotherapy generally comes with fewer adverse events than chemotherapy, catastrophic side effects are still possible.
Dr. Petrillo recalled a 95-year-old woman with metastatic cancer who was largely asymptomatic.
She had a qualifying mutation for a checkpoint inhibitor, so her oncologist started her on one. The patient never bounced back from the severe colitis the agent caused, and she died of complications in the hospital.
Although such reactions with immunotherapy are uncommon, less serious problems caused by the agents can still have a major impact on a person’s quality of life. Low-grade diarrhea, for instance, may not sound too bad, but in a patient’s daily life, it can translate to six or more episodes a day.
Even with no side effects, prescribing immunotherapy can mean that patients with limited time left spend a good portion of it at an infusion clinic instead of at home. These patients are also less likely to be referred to hospice and more likely to be admitted to and die in the hospital.
And with treatments that can cost $20,000 per dose, financial toxicity becomes a big concern.
In short, some of the reasons why chemotherapy is not recommended at the end of life also apply to immunotherapy, Dr. Petrillo said.
Prescribing Decisions
Recent research highlights the growing use of immunotherapy at the end of life.
Dr. Khan’s retrospective study found, for instance, that the percentage of patients starting immunotherapy in the last 30 days of life increased by about fourfold to fivefold over the study period for the three cancers analyzed — stage IV melanoma, lung, and kidney cancers.
Among the population that died within 30 days, the percentage receiving immunotherapy increased over the study periods — 0.8%-4.3% for melanoma, 0.9%-3.2% for NSCLC, and 0.5%-2.6% for kidney cell carcinoma — prompting the conclusion that immunotherapy prescriptions in the last month of life are on the rise.
Prescribing immunotherapy in patients who ultimately died within 1 month occurred more frequently at low-volume, nonacademic centers than at academic or high-volume centers, and outcomes varied by practice setting.
Patients had better survival outcomes overall when receiving immunotherapy at academic or high-volume centers — a finding Dr. Khan said is worth investigating further. Possible explanations include better management of severe immune-related side effects at larger centers and more caution when prescribing immunotherapy to “borderline” candidates, such as those with several comorbidities.
Importantly, given the retrospective design, Dr. Khan and colleagues already knew which patients prescribed immunotherapy died within 30 days of initiating treatment.
More specifically, 5192 of 71,204 patients who received immunotherapy (7.3%) died within a month of initiating therapy, while 66,012 (92.7%) lived beyond that point.
The study, however, did not assess how the remaining 92.7% who lived beyond 30 days fared on immunotherapy and the differences between those who lived less than 30 days and those who survived longer.
Knowing the outcome of patients at the outset of the analysis still leaves open the question of when immunotherapy can extend life and when it can’t for the patient in front of you.
To avoid overtreating at the end of life, it’s important to have “the same standard that you have for giving chemotherapy. You have to treat it with the same respect,” said Moshe Chasky, MD, a community medical oncologist with Alliance Cancer Specialists in Philadelphia, Pennsylvania. “You can’t just be throwing” immunotherapy around “at the end of life.”
While there are no clear predictors of risk and benefit, there are some factors to help guide decisions.
As with chemotherapy, Dr. Petrillo said performance status is key. Dr. Petrillo and colleagues found that median overall survival with immune checkpoint inhibitors for advanced non–small cell lung cancer was 14.3 months in patients with an Eastern Cooperative Oncology Group performance score of 0-1 but only 4.5 months with scores of ≥ 2.
Dr. Khan also found that immunotherapy survival is, unsurprisingly, worse in patients with high metastatic burdens and more comorbidities.
“You should still consider immunotherapy for metastatic melanoma, non–small cell lung cancer, and renal cell carcinoma,” Dr. Khan said. The message here is to “think twice before using” it, especially in comorbid patients with widespread metastases.
“Just because something can be done doesn’t always mean it should be done,” he said.
At Yale, when Dr. Khan works, immunotherapy decisions are considered by a multidisciplinary tumor board. At Mass General, immunotherapy has generally moved to the frontline setting, and the hospital no longer prescribes checkpoint inhibitors to hospitalized patients because the cost is too high relative to the potential benefit, Dr. Petrillo explained.
Still, with all the uncertainties about risk and benefit, counseling patients is a challenge. Dr. Dizon called it “the epitome of shared decision-making.”
Dr. Petrillo noted that it’s critical not to counsel patients based solely on the anecdotal patients who do surprisingly well.
“It’s hard to mention that and not have that be what somebody anchors on,” she said. But that speaks to “how desperate people can feel, how hopeful they can be.”
Dr. Khan, Dr. Petrillo, and Dr. Chasky all reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Chemotherapy has fallen out of favor for treating cancer toward the end of life. The toxicity is too high, and the benefit, if any, is often too low.
Immunotherapy, however, has been taking its place.
This means “there are patients who are getting immunotherapy who shouldn’t,” said Yale University, New Haven, Connecticut, surgical oncologist Sajid Khan, MD, senior investigator on a recent study that highlighted the growing use of these agents in patients’ last month of life.
What’s driving this trend, and how can oncologists avoid overtreatment with immunotherapy at the end of life?
The N-of-1 Patient
With immunotherapy at the end of life, “each of us has had our N-of-1” where a patient bounces back with a remarkable and durable response, said Don Dizon, MD, a gynecologic oncologist at Brown University, Providence, Rhode Island.
He recalled a patient with sarcoma who did not respond to chemotherapy. But after Dr. Dizon started her on immunotherapy, everything turned around. She has now been in remission for 8 years and counting.
The possibility of an unexpected or remarkable responder is seductive. And the improved safety of immunotherapy over chemotherapy adds to the allure.
Meanwhile, patients are often desperate. It’s rare for someone to be ready to stop treatment, Dr. Dizon said. Everybody “hopes that they’re going to be the exceptional responder.”
At the end of the day, the question often becomes: “Why not try immunotherapy? What’s there to lose?”
This thinking may be prompting broader use of immunotherapy in late-stage disease, even in instances with no Food and Drug Administration indication and virtually no supportive data, such as for metastatic ovarian cancer, Dr. Dizon said.
Back to Earth
The problem with the hopeful approach is that end-of-life turnarounds with immunotherapy are rare, and there’s no way at the moment to predict who will have one, said Laura Petrillo, MD, a palliative care physician at Massachusetts General Hospital, Boston.
Even though immunotherapy generally comes with fewer adverse events than chemotherapy, catastrophic side effects are still possible.
Dr. Petrillo recalled a 95-year-old woman with metastatic cancer who was largely asymptomatic.
She had a qualifying mutation for a checkpoint inhibitor, so her oncologist started her on one. The patient never bounced back from the severe colitis the agent caused, and she died of complications in the hospital.
Although such reactions with immunotherapy are uncommon, less serious problems caused by the agents can still have a major impact on a person’s quality of life. Low-grade diarrhea, for instance, may not sound too bad, but in a patient’s daily life, it can translate to six or more episodes a day.
Even with no side effects, prescribing immunotherapy can mean that patients with limited time left spend a good portion of it at an infusion clinic instead of at home. These patients are also less likely to be referred to hospice and more likely to be admitted to and die in the hospital.
And with treatments that can cost $20,000 per dose, financial toxicity becomes a big concern.
In short, some of the reasons why chemotherapy is not recommended at the end of life also apply to immunotherapy, Dr. Petrillo said.
Prescribing Decisions
Recent research highlights the growing use of immunotherapy at the end of life.
Dr. Khan’s retrospective study found, for instance, that the percentage of patients starting immunotherapy in the last 30 days of life increased by about fourfold to fivefold over the study period for the three cancers analyzed — stage IV melanoma, lung, and kidney cancers.
Among the population that died within 30 days, the percentage receiving immunotherapy increased over the study periods — 0.8%-4.3% for melanoma, 0.9%-3.2% for NSCLC, and 0.5%-2.6% for kidney cell carcinoma — prompting the conclusion that immunotherapy prescriptions in the last month of life are on the rise.
Prescribing immunotherapy in patients who ultimately died within 1 month occurred more frequently at low-volume, nonacademic centers than at academic or high-volume centers, and outcomes varied by practice setting.
Patients had better survival outcomes overall when receiving immunotherapy at academic or high-volume centers — a finding Dr. Khan said is worth investigating further. Possible explanations include better management of severe immune-related side effects at larger centers and more caution when prescribing immunotherapy to “borderline” candidates, such as those with several comorbidities.
Importantly, given the retrospective design, Dr. Khan and colleagues already knew which patients prescribed immunotherapy died within 30 days of initiating treatment.
More specifically, 5192 of 71,204 patients who received immunotherapy (7.3%) died within a month of initiating therapy, while 66,012 (92.7%) lived beyond that point.
The study, however, did not assess how the remaining 92.7% who lived beyond 30 days fared on immunotherapy and the differences between those who lived less than 30 days and those who survived longer.
Knowing the outcome of patients at the outset of the analysis still leaves open the question of when immunotherapy can extend life and when it can’t for the patient in front of you.
To avoid overtreating at the end of life, it’s important to have “the same standard that you have for giving chemotherapy. You have to treat it with the same respect,” said Moshe Chasky, MD, a community medical oncologist with Alliance Cancer Specialists in Philadelphia, Pennsylvania. “You can’t just be throwing” immunotherapy around “at the end of life.”
While there are no clear predictors of risk and benefit, there are some factors to help guide decisions.
As with chemotherapy, Dr. Petrillo said performance status is key. Dr. Petrillo and colleagues found that median overall survival with immune checkpoint inhibitors for advanced non–small cell lung cancer was 14.3 months in patients with an Eastern Cooperative Oncology Group performance score of 0-1 but only 4.5 months with scores of ≥ 2.
Dr. Khan also found that immunotherapy survival is, unsurprisingly, worse in patients with high metastatic burdens and more comorbidities.
“You should still consider immunotherapy for metastatic melanoma, non–small cell lung cancer, and renal cell carcinoma,” Dr. Khan said. The message here is to “think twice before using” it, especially in comorbid patients with widespread metastases.
“Just because something can be done doesn’t always mean it should be done,” he said.
At Yale, when Dr. Khan works, immunotherapy decisions are considered by a multidisciplinary tumor board. At Mass General, immunotherapy has generally moved to the frontline setting, and the hospital no longer prescribes checkpoint inhibitors to hospitalized patients because the cost is too high relative to the potential benefit, Dr. Petrillo explained.
Still, with all the uncertainties about risk and benefit, counseling patients is a challenge. Dr. Dizon called it “the epitome of shared decision-making.”
Dr. Petrillo noted that it’s critical not to counsel patients based solely on the anecdotal patients who do surprisingly well.
“It’s hard to mention that and not have that be what somebody anchors on,” she said. But that speaks to “how desperate people can feel, how hopeful they can be.”
Dr. Khan, Dr. Petrillo, and Dr. Chasky all reported no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Modest Gains Shown in Breast Cancer Immunotherapy Trials
TOPLINE:
particularly among single-center studies which are more likely to go unreported, and many phase 2 studies failing to translate into successful phase 3 trials.
METHODOLOGY:
- Few immunotherapy agents — only pembrolizumab in the United States, as of December 2023, and atezolizumab in Europe — have received approvals for use in patients with breast cancer, indicating low returns on the large number of breast cancer immunotherapy trials launched in the early 2010s.
- In this cross-sectional study, researchers evaluated 331 immunotherapy trials, initiated between January 2004 and April 2023, that enrolled 48,844 patients with breast cancer.
- Of these, 47 were phase 1 trials, 242 were phase 2 trials, and 42 were phase 3 trials.
- A trial was considered reported if the results were posted on ClinicalTrial.gov or reported as an abstract or a manuscript.
- Overall, 120 trials met their completion date up to November 2022; of these, 30 (25%) failed to report outcomes, which included two phase 3 trials.
TAKEAWAY:
- Phase 1 trials had the highest rate of nonreporting (31.8%), followed by phase 2 (23.6%) and phase 3 (22.2%) trials.
- Single-center studies were more likely to be unreported than multicenter studies (35.2% vs 15.0%; P = .02).
- Of 90 reported trials, 47 (52.2%) met their primary endpoints and 43 (47.8%) did not.
- The majority, 17 out of 19 (89.5%), of the reported randomized trials had negative results.
IN PRACTICE:
“The findings of this study suggest that the large number of immunotherapy trials being run have yielded modest clinical impact,” the authors wrote. “More selective initiation of phase 2 trials, grounded in preclinical and biomarker observations and with optimal statistical designs for early efficacy assessment, is needed to increase trial efficiency.”
SOURCE:
The study, led by Marco Mariani, MD, Università Vita-Salute San Raffaele, Milan, Italy, was published online in JAMA Network Open.
LIMITATIONS:
The study’s reliance on ClinicalTrials.gov as the primary source of trial data might have resulted in some trials being overlooked. In addition, manual data extraction could cause inaccuracies and potentially introduced biases in the interpretation of trial results. Primary study completion date cutoff of December 2022 could have excluded significant data from more recent trials.
DISCLOSURES:
This study received support via Susan Komen Leadership Grant and the Fondazione AIRC per la Ricerca sul Cancro. Several authors reported receiving grants and personal fees and having other ties with various sources.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
particularly among single-center studies which are more likely to go unreported, and many phase 2 studies failing to translate into successful phase 3 trials.
METHODOLOGY:
- Few immunotherapy agents — only pembrolizumab in the United States, as of December 2023, and atezolizumab in Europe — have received approvals for use in patients with breast cancer, indicating low returns on the large number of breast cancer immunotherapy trials launched in the early 2010s.
- In this cross-sectional study, researchers evaluated 331 immunotherapy trials, initiated between January 2004 and April 2023, that enrolled 48,844 patients with breast cancer.
- Of these, 47 were phase 1 trials, 242 were phase 2 trials, and 42 were phase 3 trials.
- A trial was considered reported if the results were posted on ClinicalTrial.gov or reported as an abstract or a manuscript.
- Overall, 120 trials met their completion date up to November 2022; of these, 30 (25%) failed to report outcomes, which included two phase 3 trials.
TAKEAWAY:
- Phase 1 trials had the highest rate of nonreporting (31.8%), followed by phase 2 (23.6%) and phase 3 (22.2%) trials.
- Single-center studies were more likely to be unreported than multicenter studies (35.2% vs 15.0%; P = .02).
- Of 90 reported trials, 47 (52.2%) met their primary endpoints and 43 (47.8%) did not.
- The majority, 17 out of 19 (89.5%), of the reported randomized trials had negative results.
IN PRACTICE:
“The findings of this study suggest that the large number of immunotherapy trials being run have yielded modest clinical impact,” the authors wrote. “More selective initiation of phase 2 trials, grounded in preclinical and biomarker observations and with optimal statistical designs for early efficacy assessment, is needed to increase trial efficiency.”
SOURCE:
The study, led by Marco Mariani, MD, Università Vita-Salute San Raffaele, Milan, Italy, was published online in JAMA Network Open.
LIMITATIONS:
The study’s reliance on ClinicalTrials.gov as the primary source of trial data might have resulted in some trials being overlooked. In addition, manual data extraction could cause inaccuracies and potentially introduced biases in the interpretation of trial results. Primary study completion date cutoff of December 2022 could have excluded significant data from more recent trials.
DISCLOSURES:
This study received support via Susan Komen Leadership Grant and the Fondazione AIRC per la Ricerca sul Cancro. Several authors reported receiving grants and personal fees and having other ties with various sources.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
particularly among single-center studies which are more likely to go unreported, and many phase 2 studies failing to translate into successful phase 3 trials.
METHODOLOGY:
- Few immunotherapy agents — only pembrolizumab in the United States, as of December 2023, and atezolizumab in Europe — have received approvals for use in patients with breast cancer, indicating low returns on the large number of breast cancer immunotherapy trials launched in the early 2010s.
- In this cross-sectional study, researchers evaluated 331 immunotherapy trials, initiated between January 2004 and April 2023, that enrolled 48,844 patients with breast cancer.
- Of these, 47 were phase 1 trials, 242 were phase 2 trials, and 42 were phase 3 trials.
- A trial was considered reported if the results were posted on ClinicalTrial.gov or reported as an abstract or a manuscript.
- Overall, 120 trials met their completion date up to November 2022; of these, 30 (25%) failed to report outcomes, which included two phase 3 trials.
TAKEAWAY:
- Phase 1 trials had the highest rate of nonreporting (31.8%), followed by phase 2 (23.6%) and phase 3 (22.2%) trials.
- Single-center studies were more likely to be unreported than multicenter studies (35.2% vs 15.0%; P = .02).
- Of 90 reported trials, 47 (52.2%) met their primary endpoints and 43 (47.8%) did not.
- The majority, 17 out of 19 (89.5%), of the reported randomized trials had negative results.
IN PRACTICE:
“The findings of this study suggest that the large number of immunotherapy trials being run have yielded modest clinical impact,” the authors wrote. “More selective initiation of phase 2 trials, grounded in preclinical and biomarker observations and with optimal statistical designs for early efficacy assessment, is needed to increase trial efficiency.”
SOURCE:
The study, led by Marco Mariani, MD, Università Vita-Salute San Raffaele, Milan, Italy, was published online in JAMA Network Open.
LIMITATIONS:
The study’s reliance on ClinicalTrials.gov as the primary source of trial data might have resulted in some trials being overlooked. In addition, manual data extraction could cause inaccuracies and potentially introduced biases in the interpretation of trial results. Primary study completion date cutoff of December 2022 could have excluded significant data from more recent trials.
DISCLOSURES:
This study received support via Susan Komen Leadership Grant and the Fondazione AIRC per la Ricerca sul Cancro. Several authors reported receiving grants and personal fees and having other ties with various sources.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
FDA Approves First Engineered Cell Therapy for a Solid Tumor
Afami-cel — the first engineered cell therapy for a solid tumor — is indicated specifically for adults with unresectable or metastatic synovial sarcoma who have received prior chemotherapy, are positive for several human leukocyte antigens (HLAs), and whose tumors express melanoma-associated antigen A4, as determined by FDA-authorized companion diagnostic devices.
The single-dose treatment targets solid tumors expressing melanoma-associated antigen A4, a protein highly expressed in synovial sarcoma.
Synovial sarcoma is a rare form of cancer, which affects about 1000 people in the US each year. Malignant cells develop and form a tumor in soft tissues, often in the extremities.
“Adults with metastatic synovial sarcoma, a life-threatening form of cancer, often face limited treatment options in addition to the risk of cancer spread or recurrence,” Nicole Verdun, MD, director of the Office of Therapeutic Products in the FDA’s Center for Biologics Evaluation and Research, said in the agency press release announcing the approval. “Today’s approval represents a significant milestone in the development of an innovative, safe and effective therapy for patients with this rare but potentially fatal disease.”
T-cell receptor therapy, like chimeric antigen receptor (CAR) T-cell (CAR-T) therapy, involves altering patient T cells to fight cancer. While CAR-T therapy inserts an artificial receptor to target a specific surface protein on cancer cells, the T-cell receptor therapy modifies existing receptors to recognize an array of antigens on the surface of cancer cells — a promising strategy for targeting solid tumors.
The accelerated approval of afami-cel was based on the phase 2 SPEARHEAD-1 trial in 44 patients with synovial sarcoma who received a single infusion of the therapy. The trial had enrolled 52 patients, but 8 did not receive afami-cel, including 3 who died and 1 who withdrew.
According to the FDA announcement, the overall response rate was 43.2%, with a median time to response of 4.9 weeks. The median duration of response was 6 months (95% CI, 4.6 months to not reached). Among patients who responded, 39% had a duration of response of 12 months or longer.
“These results suggest that a one-time treatment with afami-cel has the potential to extend life while allowing responders to go off chemotherapy,” said lead investigator Sandra D’Angelo, MD, a sarcoma specialist at Memorial Sloan Kettering Cancer Center in New York City, in a company press release.
The prescribing information includes a boxed warning for serious or fatal cytokine release syndrome.
The most common nonlaboratory adverse reactions, occurring in at least 20% of patients, included cytokine release syndrome, nausea, vomiting, fatigue, infections, pyrexia, constipation, dyspnea, tachycardia, hypotension, diarrhea, and edema. The most common grade 3 or 4 laboratory abnormalities, occurring in at least 20% of patients, included decreased lymphocyte count, neutrophil count, white cell blood count, red blood cell, and platelet count.
The recommended dose is between 2.68x109 to 10x109 MAGE-A4 T-cell receptor–positive T-cells. The FDA notice specifies not using a leukodepleting filter or prophylactic systemic corticosteroids.
The list price for the one-time therapy is $727,000, according to Fierce Pharma.
A version of this article first appeared on Medscape.com.
Afami-cel — the first engineered cell therapy for a solid tumor — is indicated specifically for adults with unresectable or metastatic synovial sarcoma who have received prior chemotherapy, are positive for several human leukocyte antigens (HLAs), and whose tumors express melanoma-associated antigen A4, as determined by FDA-authorized companion diagnostic devices.
The single-dose treatment targets solid tumors expressing melanoma-associated antigen A4, a protein highly expressed in synovial sarcoma.
Synovial sarcoma is a rare form of cancer, which affects about 1000 people in the US each year. Malignant cells develop and form a tumor in soft tissues, often in the extremities.
“Adults with metastatic synovial sarcoma, a life-threatening form of cancer, often face limited treatment options in addition to the risk of cancer spread or recurrence,” Nicole Verdun, MD, director of the Office of Therapeutic Products in the FDA’s Center for Biologics Evaluation and Research, said in the agency press release announcing the approval. “Today’s approval represents a significant milestone in the development of an innovative, safe and effective therapy for patients with this rare but potentially fatal disease.”
T-cell receptor therapy, like chimeric antigen receptor (CAR) T-cell (CAR-T) therapy, involves altering patient T cells to fight cancer. While CAR-T therapy inserts an artificial receptor to target a specific surface protein on cancer cells, the T-cell receptor therapy modifies existing receptors to recognize an array of antigens on the surface of cancer cells — a promising strategy for targeting solid tumors.
The accelerated approval of afami-cel was based on the phase 2 SPEARHEAD-1 trial in 44 patients with synovial sarcoma who received a single infusion of the therapy. The trial had enrolled 52 patients, but 8 did not receive afami-cel, including 3 who died and 1 who withdrew.
According to the FDA announcement, the overall response rate was 43.2%, with a median time to response of 4.9 weeks. The median duration of response was 6 months (95% CI, 4.6 months to not reached). Among patients who responded, 39% had a duration of response of 12 months or longer.
“These results suggest that a one-time treatment with afami-cel has the potential to extend life while allowing responders to go off chemotherapy,” said lead investigator Sandra D’Angelo, MD, a sarcoma specialist at Memorial Sloan Kettering Cancer Center in New York City, in a company press release.
The prescribing information includes a boxed warning for serious or fatal cytokine release syndrome.
The most common nonlaboratory adverse reactions, occurring in at least 20% of patients, included cytokine release syndrome, nausea, vomiting, fatigue, infections, pyrexia, constipation, dyspnea, tachycardia, hypotension, diarrhea, and edema. The most common grade 3 or 4 laboratory abnormalities, occurring in at least 20% of patients, included decreased lymphocyte count, neutrophil count, white cell blood count, red blood cell, and platelet count.
The recommended dose is between 2.68x109 to 10x109 MAGE-A4 T-cell receptor–positive T-cells. The FDA notice specifies not using a leukodepleting filter or prophylactic systemic corticosteroids.
The list price for the one-time therapy is $727,000, according to Fierce Pharma.
A version of this article first appeared on Medscape.com.
Afami-cel — the first engineered cell therapy for a solid tumor — is indicated specifically for adults with unresectable or metastatic synovial sarcoma who have received prior chemotherapy, are positive for several human leukocyte antigens (HLAs), and whose tumors express melanoma-associated antigen A4, as determined by FDA-authorized companion diagnostic devices.
The single-dose treatment targets solid tumors expressing melanoma-associated antigen A4, a protein highly expressed in synovial sarcoma.
Synovial sarcoma is a rare form of cancer, which affects about 1000 people in the US each year. Malignant cells develop and form a tumor in soft tissues, often in the extremities.
“Adults with metastatic synovial sarcoma, a life-threatening form of cancer, often face limited treatment options in addition to the risk of cancer spread or recurrence,” Nicole Verdun, MD, director of the Office of Therapeutic Products in the FDA’s Center for Biologics Evaluation and Research, said in the agency press release announcing the approval. “Today’s approval represents a significant milestone in the development of an innovative, safe and effective therapy for patients with this rare but potentially fatal disease.”
T-cell receptor therapy, like chimeric antigen receptor (CAR) T-cell (CAR-T) therapy, involves altering patient T cells to fight cancer. While CAR-T therapy inserts an artificial receptor to target a specific surface protein on cancer cells, the T-cell receptor therapy modifies existing receptors to recognize an array of antigens on the surface of cancer cells — a promising strategy for targeting solid tumors.
The accelerated approval of afami-cel was based on the phase 2 SPEARHEAD-1 trial in 44 patients with synovial sarcoma who received a single infusion of the therapy. The trial had enrolled 52 patients, but 8 did not receive afami-cel, including 3 who died and 1 who withdrew.
According to the FDA announcement, the overall response rate was 43.2%, with a median time to response of 4.9 weeks. The median duration of response was 6 months (95% CI, 4.6 months to not reached). Among patients who responded, 39% had a duration of response of 12 months or longer.
“These results suggest that a one-time treatment with afami-cel has the potential to extend life while allowing responders to go off chemotherapy,” said lead investigator Sandra D’Angelo, MD, a sarcoma specialist at Memorial Sloan Kettering Cancer Center in New York City, in a company press release.
The prescribing information includes a boxed warning for serious or fatal cytokine release syndrome.
The most common nonlaboratory adverse reactions, occurring in at least 20% of patients, included cytokine release syndrome, nausea, vomiting, fatigue, infections, pyrexia, constipation, dyspnea, tachycardia, hypotension, diarrhea, and edema. The most common grade 3 or 4 laboratory abnormalities, occurring in at least 20% of patients, included decreased lymphocyte count, neutrophil count, white cell blood count, red blood cell, and platelet count.
The recommended dose is between 2.68x109 to 10x109 MAGE-A4 T-cell receptor–positive T-cells. The FDA notice specifies not using a leukodepleting filter or prophylactic systemic corticosteroids.
The list price for the one-time therapy is $727,000, according to Fierce Pharma.
A version of this article first appeared on Medscape.com.