User login
Teva expands its recall of losartan lots
The Food and Drug Administration has announced that , according to a release.

The recall for this and other angiotensin II receptor blockers was initiated by Teva on April 25, 2019, because of detection of unacceptable levels of the possibly cancer-causing impurity N-Nitroso-N-methyl-4-aminobutyric acid (NMBA). Teva expanded this recall on June 10, with another update issued on June 12.
Losartan is not the only ARB found to contain NMBA; a full list of all ARBs affected can be found on the FDA website and currently includes more than 1,100 lots being recalled. The list can be searched and sorted by such considerations as medicine in question, company involved, and lot number.
The Food and Drug Administration has announced that , according to a release.
The recall for this and other angiotensin II receptor blockers was initiated by Teva on April 25, 2019, because of detection of unacceptable levels of the possibly cancer-causing impurity N-Nitroso-N-methyl-4-aminobutyric acid (NMBA). Teva expanded this recall on June 10, with another update issued on June 12.
Losartan is not the only ARB found to contain NMBA; a full list of all ARBs affected can be found on the FDA website and currently includes more than 1,100 lots being recalled. The list can be searched and sorted by such considerations as medicine in question, company involved, and lot number.
The Food and Drug Administration has announced that , according to a release.
The recall for this and other angiotensin II receptor blockers was initiated by Teva on April 25, 2019, because of detection of unacceptable levels of the possibly cancer-causing impurity N-Nitroso-N-methyl-4-aminobutyric acid (NMBA). Teva expanded this recall on June 10, with another update issued on June 12.
Losartan is not the only ARB found to contain NMBA; a full list of all ARBs affected can be found on the FDA website and currently includes more than 1,100 lots being recalled. The list can be searched and sorted by such considerations as medicine in question, company involved, and lot number.
FDA approves trastuzumab-anns for HER2-positive breast, gastric cancer
The Food and Drug Administration has approved Amgen’s trastuzumab-anns as a trastuzumab biosimilar for the treatment of HER2-positive breast cancer and gastric cancer.

This biosimilar, to be marketed as Kanjinti, is the fifth trastuzumab biosimilar to be approved by the agency, according to the FDA.
Approval was based in part on the LILAC study, which demonstrated that the biosimilar, previously called ABP-980, had similar efficacy and comparable cardiac safety with trastuzumab.
In the phase 3 study, 725 patients with HER2-positive early breast cancer were randomized to neoadjuvant treatment with trastuzumab-anns or trastuzumab, plus paclitaxel, for four cycles following four cycles of chemotherapy. The primary pathological complete response endpoint was achieved in 48% of those in the biosimilar arm, compared with 40.5% in the trastuzumab arm. Patients then went on to receive adjuvant treatment with ABP 980 or trastuzumab every 3 weeks for up to 1 year following surgery.
Grade 3 or worse adverse events during the neoadjuvant phase occurred in 15% of patients in the ABP 980 group and 14% in the trastuzumab group. The most frequent grade 3 event in both study arms was neutropenia. In the adjuvant phase, grade 3 or worse adverse events occurred in 9% of those continuing ABP 980 and in 6% of those continuing trastuzumab. The most frequent events in both arms were infections, infestations, and neutropenia.
Trastuzumab-anns is indicated for adjuvant treatment of HER2-overexpressing node positive or node negative breast cancer, first-line treatment of HER2-overexpressing metastatic breast cancer, and first-line treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma. The FDA indicates patients should be selected based on an FDA-approved companion diagnostic for a trastuzumab product.
The biosimilar includes a boxed warning for cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity.
The Food and Drug Administration has approved Amgen’s trastuzumab-anns as a trastuzumab biosimilar for the treatment of HER2-positive breast cancer and gastric cancer.
This biosimilar, to be marketed as Kanjinti, is the fifth trastuzumab biosimilar to be approved by the agency, according to the FDA.
Approval was based in part on the LILAC study, which demonstrated that the biosimilar, previously called ABP-980, had similar efficacy and comparable cardiac safety with trastuzumab.
In the phase 3 study, 725 patients with HER2-positive early breast cancer were randomized to neoadjuvant treatment with trastuzumab-anns or trastuzumab, plus paclitaxel, for four cycles following four cycles of chemotherapy. The primary pathological complete response endpoint was achieved in 48% of those in the biosimilar arm, compared with 40.5% in the trastuzumab arm. Patients then went on to receive adjuvant treatment with ABP 980 or trastuzumab every 3 weeks for up to 1 year following surgery.
Grade 3 or worse adverse events during the neoadjuvant phase occurred in 15% of patients in the ABP 980 group and 14% in the trastuzumab group. The most frequent grade 3 event in both study arms was neutropenia. In the adjuvant phase, grade 3 or worse adverse events occurred in 9% of those continuing ABP 980 and in 6% of those continuing trastuzumab. The most frequent events in both arms were infections, infestations, and neutropenia.
Trastuzumab-anns is indicated for adjuvant treatment of HER2-overexpressing node positive or node negative breast cancer, first-line treatment of HER2-overexpressing metastatic breast cancer, and first-line treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma. The FDA indicates patients should be selected based on an FDA-approved companion diagnostic for a trastuzumab product.
The biosimilar includes a boxed warning for cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity.
The Food and Drug Administration has approved Amgen’s trastuzumab-anns as a trastuzumab biosimilar for the treatment of HER2-positive breast cancer and gastric cancer.
This biosimilar, to be marketed as Kanjinti, is the fifth trastuzumab biosimilar to be approved by the agency, according to the FDA.
Approval was based in part on the LILAC study, which demonstrated that the biosimilar, previously called ABP-980, had similar efficacy and comparable cardiac safety with trastuzumab.
In the phase 3 study, 725 patients with HER2-positive early breast cancer were randomized to neoadjuvant treatment with trastuzumab-anns or trastuzumab, plus paclitaxel, for four cycles following four cycles of chemotherapy. The primary pathological complete response endpoint was achieved in 48% of those in the biosimilar arm, compared with 40.5% in the trastuzumab arm. Patients then went on to receive adjuvant treatment with ABP 980 or trastuzumab every 3 weeks for up to 1 year following surgery.
Grade 3 or worse adverse events during the neoadjuvant phase occurred in 15% of patients in the ABP 980 group and 14% in the trastuzumab group. The most frequent grade 3 event in both study arms was neutropenia. In the adjuvant phase, grade 3 or worse adverse events occurred in 9% of those continuing ABP 980 and in 6% of those continuing trastuzumab. The most frequent events in both arms were infections, infestations, and neutropenia.
Trastuzumab-anns is indicated for adjuvant treatment of HER2-overexpressing node positive or node negative breast cancer, first-line treatment of HER2-overexpressing metastatic breast cancer, and first-line treatment of patients with HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma. The FDA indicates patients should be selected based on an FDA-approved companion diagnostic for a trastuzumab product.
The biosimilar includes a boxed warning for cardiomyopathy, infusion reactions, embryo-fetal toxicity, and pulmonary toxicity.
CDC activates Emergency Operations Center for Congo Ebola outbreak
in the Democratic Republic of the Congo (DRC). With over 2,000 confirmed cases, the outbreak is the second largest ever recorded.
It recently spread to neighboring Uganda, by a family who crossed the border from the DRC.
As of June 11, 187 CDC staff have completed 278 deployments to the DRC, Uganda, and other neighboring countries, as well as to the World Health Organization in Geneva.

“We are activating the Emergency Operations Center at CDC headquarters to provide enhanced operational support to our” Ebola response team in the Congo. The level 3 activation – the lowest level – “allows the agency to provide increased operational support” and “logistics planning for a longer term, sustained effort,” CDC said in a press release.
Activation “does not mean that the threat of Ebola to the United States has increased.” The risk of global spread remains low, CDC said.
The outbreak is occurring in an area of armed conflict and other problems that complicate public health efforts and increase the risk of disease spread.
in the Democratic Republic of the Congo (DRC). With over 2,000 confirmed cases, the outbreak is the second largest ever recorded.
It recently spread to neighboring Uganda, by a family who crossed the border from the DRC.
As of June 11, 187 CDC staff have completed 278 deployments to the DRC, Uganda, and other neighboring countries, as well as to the World Health Organization in Geneva.
“We are activating the Emergency Operations Center at CDC headquarters to provide enhanced operational support to our” Ebola response team in the Congo. The level 3 activation – the lowest level – “allows the agency to provide increased operational support” and “logistics planning for a longer term, sustained effort,” CDC said in a press release.
Activation “does not mean that the threat of Ebola to the United States has increased.” The risk of global spread remains low, CDC said.
The outbreak is occurring in an area of armed conflict and other problems that complicate public health efforts and increase the risk of disease spread.
in the Democratic Republic of the Congo (DRC). With over 2,000 confirmed cases, the outbreak is the second largest ever recorded.
It recently spread to neighboring Uganda, by a family who crossed the border from the DRC.
As of June 11, 187 CDC staff have completed 278 deployments to the DRC, Uganda, and other neighboring countries, as well as to the World Health Organization in Geneva.
“We are activating the Emergency Operations Center at CDC headquarters to provide enhanced operational support to our” Ebola response team in the Congo. The level 3 activation – the lowest level – “allows the agency to provide increased operational support” and “logistics planning for a longer term, sustained effort,” CDC said in a press release.
Activation “does not mean that the threat of Ebola to the United States has increased.” The risk of global spread remains low, CDC said.
The outbreak is occurring in an area of armed conflict and other problems that complicate public health efforts and increase the risk of disease spread.
FDA approves Keytruda for metastatic HNSCC
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the first-line treatment of patients with metastatic or unresectable recurrent head and neck squamous cell carcinoma (HNSCC).
FDA approval was based on results of the randomized, multicenter, three-arm, open‑label, active‑controlled KEYNOTE-048 trial. The 882 patients in the trial had metastatic HNSCC, and they received either single-agent pembrolizumab; pembrolizumab, carboplatin or cisplatin, and platinum and fluorouracil; or cetuximab, carboplatin or cisplatin, and platinum and fluorouracil.
Patients who received pembrolizumab plus chemotherapy had a mean overall survival of 13.0 months, compared with 10.7 months in the cetuximab plus chemotherapy group (hazard ratio, 0.77; 95% CI, 0.63-0.93; P = .0067).
In the patient subgroups that received single-agent pembrolizumab, overall survival was 12.3 months in patients with a Combined Positive Score of at least 1, compared with 10.3 months for the cetuximab plus chemotherapy group (HR, 0.78; 95% CI, 0.64-0.96; P = .0171). In patients with a Combined Positive Score of at least 20, overall survival was 14.9 months in the pembrolizumab-only group and 10.7 months in the cetuximab plus chemotherapy group (HR, 0.61; 95% CI, 0.45-0.83; P = .0015).
The most common adverse events in patients who received single-agent pembrolizumab were fatigue, constipation, and rash. In patients who received pembrolizumab plus chemotherapy, the most common adverse events were nausea, fatigue, constipation, vomiting, mucosal inflammation, diarrhea, decreased appetite, stomatitis, and cough.
Pembrolizumab is approved for use in combination with platinum and fluorouracil for all patients and as a single agent for patients whose tumors express programmed death–ligand 1, the FDA said.
The FDA also expanded the intended use for the PD-L1 IHC 22C3 pharmDx kit to include use as a companion diagnostic device for selecting patients with HNSCC for treatment with pembrolizumab as a single agent.
Find the full press release on the FDA website.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the first-line treatment of patients with metastatic or unresectable recurrent head and neck squamous cell carcinoma (HNSCC).
FDA approval was based on results of the randomized, multicenter, three-arm, open‑label, active‑controlled KEYNOTE-048 trial. The 882 patients in the trial had metastatic HNSCC, and they received either single-agent pembrolizumab; pembrolizumab, carboplatin or cisplatin, and platinum and fluorouracil; or cetuximab, carboplatin or cisplatin, and platinum and fluorouracil.
Patients who received pembrolizumab plus chemotherapy had a mean overall survival of 13.0 months, compared with 10.7 months in the cetuximab plus chemotherapy group (hazard ratio, 0.77; 95% CI, 0.63-0.93; P = .0067).
In the patient subgroups that received single-agent pembrolizumab, overall survival was 12.3 months in patients with a Combined Positive Score of at least 1, compared with 10.3 months for the cetuximab plus chemotherapy group (HR, 0.78; 95% CI, 0.64-0.96; P = .0171). In patients with a Combined Positive Score of at least 20, overall survival was 14.9 months in the pembrolizumab-only group and 10.7 months in the cetuximab plus chemotherapy group (HR, 0.61; 95% CI, 0.45-0.83; P = .0015).
The most common adverse events in patients who received single-agent pembrolizumab were fatigue, constipation, and rash. In patients who received pembrolizumab plus chemotherapy, the most common adverse events were nausea, fatigue, constipation, vomiting, mucosal inflammation, diarrhea, decreased appetite, stomatitis, and cough.
Pembrolizumab is approved for use in combination with platinum and fluorouracil for all patients and as a single agent for patients whose tumors express programmed death–ligand 1, the FDA said.
The FDA also expanded the intended use for the PD-L1 IHC 22C3 pharmDx kit to include use as a companion diagnostic device for selecting patients with HNSCC for treatment with pembrolizumab as a single agent.
Find the full press release on the FDA website.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the first-line treatment of patients with metastatic or unresectable recurrent head and neck squamous cell carcinoma (HNSCC).
FDA approval was based on results of the randomized, multicenter, three-arm, open‑label, active‑controlled KEYNOTE-048 trial. The 882 patients in the trial had metastatic HNSCC, and they received either single-agent pembrolizumab; pembrolizumab, carboplatin or cisplatin, and platinum and fluorouracil; or cetuximab, carboplatin or cisplatin, and platinum and fluorouracil.
Patients who received pembrolizumab plus chemotherapy had a mean overall survival of 13.0 months, compared with 10.7 months in the cetuximab plus chemotherapy group (hazard ratio, 0.77; 95% CI, 0.63-0.93; P = .0067).
In the patient subgroups that received single-agent pembrolizumab, overall survival was 12.3 months in patients with a Combined Positive Score of at least 1, compared with 10.3 months for the cetuximab plus chemotherapy group (HR, 0.78; 95% CI, 0.64-0.96; P = .0171). In patients with a Combined Positive Score of at least 20, overall survival was 14.9 months in the pembrolizumab-only group and 10.7 months in the cetuximab plus chemotherapy group (HR, 0.61; 95% CI, 0.45-0.83; P = .0015).
The most common adverse events in patients who received single-agent pembrolizumab were fatigue, constipation, and rash. In patients who received pembrolizumab plus chemotherapy, the most common adverse events were nausea, fatigue, constipation, vomiting, mucosal inflammation, diarrhea, decreased appetite, stomatitis, and cough.
Pembrolizumab is approved for use in combination with platinum and fluorouracil for all patients and as a single agent for patients whose tumors express programmed death–ligand 1, the FDA said.
The FDA also expanded the intended use for the PD-L1 IHC 22C3 pharmDx kit to include use as a companion diagnostic device for selecting patients with HNSCC for treatment with pembrolizumab as a single agent.
Find the full press release on the FDA website.
FDA approves Polivy for DLBCL
The Food and Drug Administration has granted accelerated approval to Polivy (polatuzumab vedotin-piiq), in conjunction with bendamustine and a rituximab product, for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have undergone at least two prior therapies.
The FDA approval is based on results of an open-label, multicenter clinical trial of 80 patients with DLBCL who had undergone at least one prior regimen. Patients received either Polivy plus bendamustine and rituximab or only bendamustine and rituximab for six 21-day cycles. At the completion of therapy, 40% of patients who received Polivy in conjunction with bendamustine and rituximab achieved a complete response, compared with 18% of patients who received only bendamustine and rituximab; total response was 63% in those who received Polivy and 25% in those who did not.
The most common adverse events included neutropenia, thrombocytopenia, anemia, peripheral neuropathy, fatigue, diarrhea, pyrexia, decreased appetite, and pneumonia. Serious adverse events occurred in 64% of patients, most commonly from infection; the most common cause for treatment discontinuation was cytopenia.
The recommended dose of Polivy is 1.8 mg/kg as an intravenous infusion over 90 minutes every 21 days for six cycles in combination with bendamustine and a rituximab product, the FDA said. Subsequent infusions may be administered over 30 minutes if the previous infusion is tolerated.
Find the full press release on the FDA website.
The Food and Drug Administration has granted accelerated approval to Polivy (polatuzumab vedotin-piiq), in conjunction with bendamustine and a rituximab product, for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have undergone at least two prior therapies.
The FDA approval is based on results of an open-label, multicenter clinical trial of 80 patients with DLBCL who had undergone at least one prior regimen. Patients received either Polivy plus bendamustine and rituximab or only bendamustine and rituximab for six 21-day cycles. At the completion of therapy, 40% of patients who received Polivy in conjunction with bendamustine and rituximab achieved a complete response, compared with 18% of patients who received only bendamustine and rituximab; total response was 63% in those who received Polivy and 25% in those who did not.
The most common adverse events included neutropenia, thrombocytopenia, anemia, peripheral neuropathy, fatigue, diarrhea, pyrexia, decreased appetite, and pneumonia. Serious adverse events occurred in 64% of patients, most commonly from infection; the most common cause for treatment discontinuation was cytopenia.
The recommended dose of Polivy is 1.8 mg/kg as an intravenous infusion over 90 minutes every 21 days for six cycles in combination with bendamustine and a rituximab product, the FDA said. Subsequent infusions may be administered over 30 minutes if the previous infusion is tolerated.
Find the full press release on the FDA website.
The Food and Drug Administration has granted accelerated approval to Polivy (polatuzumab vedotin-piiq), in conjunction with bendamustine and a rituximab product, for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have undergone at least two prior therapies.
The FDA approval is based on results of an open-label, multicenter clinical trial of 80 patients with DLBCL who had undergone at least one prior regimen. Patients received either Polivy plus bendamustine and rituximab or only bendamustine and rituximab for six 21-day cycles. At the completion of therapy, 40% of patients who received Polivy in conjunction with bendamustine and rituximab achieved a complete response, compared with 18% of patients who received only bendamustine and rituximab; total response was 63% in those who received Polivy and 25% in those who did not.
The most common adverse events included neutropenia, thrombocytopenia, anemia, peripheral neuropathy, fatigue, diarrhea, pyrexia, decreased appetite, and pneumonia. Serious adverse events occurred in 64% of patients, most commonly from infection; the most common cause for treatment discontinuation was cytopenia.
The recommended dose of Polivy is 1.8 mg/kg as an intravenous infusion over 90 minutes every 21 days for six cycles in combination with bendamustine and a rituximab product, the FDA said. Subsequent infusions may be administered over 30 minutes if the previous infusion is tolerated.
Find the full press release on the FDA website.
FDA approves IB-Stim device for abdominal pain in adolescents with IBS
The IB-Stim device has been approved to aid in the reduction of functional abdominal pain in patients 11-18 years of age with irritable bowel syndrome (IBS), according to the U.S. Food and Drug Administration.
“This device offers a safe option for treatment of adolescents experiencing pain from IBS through the use of mild nerve stimulation,” Carlos Peña, PhD, director of the Office of Neurological and Physical Medicine Devices in the FDA’s Center for Devices and Radiological Health, said in a press release.
The prescription-only device has a single-use electrical nerve stimulator that is placed behind the patient’s ear. Stimulating nerve bundles in and around the ear is thought to provide pain relief. The battery-powered chip of the device emits low-frequency electrical pulses continuously for 5 days, at which time it is replaced. Patients can use the device for up to 3 consecutive weeks to reduce functional abdominal pain associated with IBS.
The FDA reviewed data from 50 patients, aged 11-18 years, with IBS; 27 patients were treated with the device and 23 patients received a placebo device. The study measured change from baseline to the end of the third week in worst abdominal pain, usual pain, and Pain Frequency Severity Duration (PFSD) scores. Patients were allowed to continue stable doses of medication to treat chronic abdominal pain.
IB-Stim treatment resulted in at least a 30% decrease in usual pain at the end of 3 weeks in 52% of treated patients, compared with 30% of patients who received the placebo, and at least a 30% decrease in worst pain in 59% of treated patients, compared with 26% of patients who received the placebo.
The treatment group also had greater changes in composite PFSD scores at the end of three weeks. During the study, six patients reported mild ear discomfort, and three patients reported adhesive allergy at the site of application, according to the press release.
The device is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris.
The FDA granted marketing authorization of the IB-Stim to Innovative Health Solutions.
The IB-Stim device has been approved to aid in the reduction of functional abdominal pain in patients 11-18 years of age with irritable bowel syndrome (IBS), according to the U.S. Food and Drug Administration.
“This device offers a safe option for treatment of adolescents experiencing pain from IBS through the use of mild nerve stimulation,” Carlos Peña, PhD, director of the Office of Neurological and Physical Medicine Devices in the FDA’s Center for Devices and Radiological Health, said in a press release.
The prescription-only device has a single-use electrical nerve stimulator that is placed behind the patient’s ear. Stimulating nerve bundles in and around the ear is thought to provide pain relief. The battery-powered chip of the device emits low-frequency electrical pulses continuously for 5 days, at which time it is replaced. Patients can use the device for up to 3 consecutive weeks to reduce functional abdominal pain associated with IBS.
The FDA reviewed data from 50 patients, aged 11-18 years, with IBS; 27 patients were treated with the device and 23 patients received a placebo device. The study measured change from baseline to the end of the third week in worst abdominal pain, usual pain, and Pain Frequency Severity Duration (PFSD) scores. Patients were allowed to continue stable doses of medication to treat chronic abdominal pain.
IB-Stim treatment resulted in at least a 30% decrease in usual pain at the end of 3 weeks in 52% of treated patients, compared with 30% of patients who received the placebo, and at least a 30% decrease in worst pain in 59% of treated patients, compared with 26% of patients who received the placebo.
The treatment group also had greater changes in composite PFSD scores at the end of three weeks. During the study, six patients reported mild ear discomfort, and three patients reported adhesive allergy at the site of application, according to the press release.
The device is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris.
The FDA granted marketing authorization of the IB-Stim to Innovative Health Solutions.
The IB-Stim device has been approved to aid in the reduction of functional abdominal pain in patients 11-18 years of age with irritable bowel syndrome (IBS), according to the U.S. Food and Drug Administration.
“This device offers a safe option for treatment of adolescents experiencing pain from IBS through the use of mild nerve stimulation,” Carlos Peña, PhD, director of the Office of Neurological and Physical Medicine Devices in the FDA’s Center for Devices and Radiological Health, said in a press release.
The prescription-only device has a single-use electrical nerve stimulator that is placed behind the patient’s ear. Stimulating nerve bundles in and around the ear is thought to provide pain relief. The battery-powered chip of the device emits low-frequency electrical pulses continuously for 5 days, at which time it is replaced. Patients can use the device for up to 3 consecutive weeks to reduce functional abdominal pain associated with IBS.
The FDA reviewed data from 50 patients, aged 11-18 years, with IBS; 27 patients were treated with the device and 23 patients received a placebo device. The study measured change from baseline to the end of the third week in worst abdominal pain, usual pain, and Pain Frequency Severity Duration (PFSD) scores. Patients were allowed to continue stable doses of medication to treat chronic abdominal pain.
IB-Stim treatment resulted in at least a 30% decrease in usual pain at the end of 3 weeks in 52% of treated patients, compared with 30% of patients who received the placebo, and at least a 30% decrease in worst pain in 59% of treated patients, compared with 26% of patients who received the placebo.
The treatment group also had greater changes in composite PFSD scores at the end of three weeks. During the study, six patients reported mild ear discomfort, and three patients reported adhesive allergy at the site of application, according to the press release.
The device is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris.
The FDA granted marketing authorization of the IB-Stim to Innovative Health Solutions.
The costs of surviving cancer
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
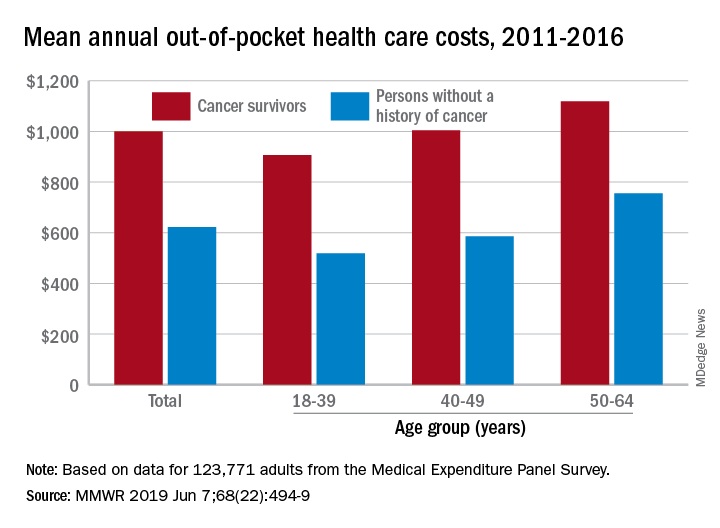
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
Cancer survivors have significantly higher out-of-pocket medical costs than those with no history of cancer, and a quarter of those survivors have some type of material hardship related to their diagnosis, according to the Centers for Disease Control and Prevention.
Along with those material financial hardships – the need to borrow money, go into debt, or declare bankruptcy – more than 34% of cancer survivors aged 18-64 years experienced psychological financial hardship, defined as worry about large medical bills, in 2011 and 2016, Donatus U. Ekwueme, PhD, and his associates reported in the Morbidity and Mortality Weekly Report.
Cancer survivors spend 60% more out of pocket than those with no cancer history: $1,000 a year from 2011 to 2016, compared with $622 for adults without a history of cancer. Spending was lowest among younger people (18-39 years) and increased with age, but the prevalence of both material and psychological hardships was highest in the middle age group (40-49 years) and lowest in the oldest group (50-64 years), they said.
Women had higher out-of-pocket costs than men, although the difference was smaller for those with cancer ($1,023 vs. $976) than for those without ($721 vs. $519). Material and psychological hardships were both more common among women, said Dr. Ekwueme of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates.
Mean out-of-pocket spending was much higher for cancer survivors with private health insurance ($1,114) than for survivors with public insurance ($471), but material hardship was much more prevalent among those with public insurance (33.1% vs. 21.9%). Rates of psychological hardship, however, were much closer: 35.9% for those with public insurance and 32.5% for those with private insurance, the investigators said.
“The number of Americans with a history of cancer is projected to increase in the next decade, and the economic burden associated with living with a cancer diagnosis will likely increase as well,” they wrote, and interventions such as “systematic screening for financial hardship at cancer diagnosis and throughout the cancer care trajectory [are needed] to minimize financial hardship for cancer survivors.”
The analysis was based on data for 123,771 adults aged 18-64 years from the Medical Expenditure Panel Survey. Out-of-pocket costs were calculated using data from 2011 to 2016, with all costs adjusted to 2016 dollars, but the hardship calculations involved data from only 2011 and 2016.
SOURCE: Ekwueme DU et al. MMWR 2019 Jun 7;68(22):494-9.
FROM MMWR
Stewart Tepper: Emgality approval ‘very exciting’
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.

It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
The drug, a humanized monoclonal antibody that binds to calcitonin gene-related peptide (CGRP), is administered by self-injection in 300-mg doses.
Galcanezumab is the first medication for episodic cluster headache that reduces the frequency of attacks, the agency said in an announcement.
Cluster headache can be more intense than migraine. The pain is unilateral and occurs in the orbital, supraorbital, or temporal regions. It reaches its peak intensity within 5-10 minutes and generally lasts for 30-90 minutes. Symptoms include a burning sensation, conjunctival injection, rhinorrhea, and photosensitivity. Patients often have one to three of these headaches per day, and the headaches appear to be linked to the circadian rhythm. An episodic cluster cycle can last for weeks to months of daily or near daily attacks.
A study presented at the recent meeting of the American Academy of Neurology provided evidence of the drug’s efficacy in cluster headache. In this trial, researchers randomized 106 patients with episodic cluster headache to galcanezumab or placebo. The baseline cluster headache frequency was 17.3 attacks per week, and galcanezumab reduced this frequency to 9.1 attacks per week, compared with 12.1 attacks per week with placebo. The most common side effect reported in this and other clinical trials was injection-site reactions.
Galcanezumab entails a risk of hypersensitivity reactions, according to the FDA. These reactions may occur several days after administration and may be prolonged. “If a serious hypersensitivity reaction occurs, treatment should be discontinued,” the agency said.
“It’s a very exciting day. There had never been a drug approved for prevention of cluster headache,” said Stewart J. Tepper, MD, professor of neurology at the Geisel School of Medicine at Dartmouth and director of the Dartmouth Headache Center, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
It is difficult to achieve therapeutic concentrations of current preventive medications that do not have FDA approval for this indication, such as verapamil, lithium, or antiepileptic drugs. Galcanezumab, in contrast, works quickly. It is important to note that the approval was for preventive treatment of episodic cluster headache, not for prevention of chronic cluster headache, and not for acute treatment, Dr. Tepper said.
“It’s important to get optimal therapy for cluster headache. It is one of the most disabling, terrible disorders on Earth,” Dr. Tepper said. “The importance [of this approval] cannot be overestimated.”
When asked for comment, Alan M. Rapoport, MD, clinical professor of neurology at the University of California, Los Angeles, said “If this monoclonal antibody to the CGRP ligand works as well in real life as in the trial, it will be an important advance in the treatment of cluster headache.”
Prior to the approval of galcanezumab, noninvasive vagal nerve stimulation was approved in November 2018 for adjunctive use in the preventive treatment of cluster headache in adults.
The FDA granted the application for galcanezumab using a Priority Review and Breakthrough Therapy designation. The agency approved galcanezumab for the preventive treatment of migraine in adults in September 2018. The drug appears to have a similar safety profile in both patient populations. Eli Lilly, which is based in Indianapolis, Indiana, manufactures the drug.
This article was updated June 5, 2019.
FDA: Vinpocetine associated with fetal harms, miscarriage
according to a statement from the agency.
This warning is based on data reviewed by the FDA, including a report from the National Institutes of Health’s National Toxicology Program, that show associations between vinpocetine use and decreased fetal weight and increased risk of miscarriage in animals. The agency is particularly concerned because products containing this ingredient, including those marketed as improving energy and memory, are widely available to women of childbearing age. As a result, the agency has recommended these women not take vinpocetine.
Vinpocetine is a synthetically produced compound used in dietary supplements either on its own or in combination and may be referred to as Vinca minor extract, lesser periwinkle extract, or common periwinkle extract on product labels. Although vinpocetine is regulated in some countries as a prescription drug, when it’s sold in dietary supplements in the United States, the FDA does not usually review those products or their labeling before they become available to consumers under the same safety and effectiveness standards used to evaluate drug products.
“Today’s safety warning is just one of many steps the FDA is taking to adapt to the realities of the evolving dietary supplement industry,” according to the agency’s statement. “Protecting the public from unsafe dietary supplements remains a top priority for the FDA.”
The full statement regarding vinpocetine and its risks can be found on the FDA website.
according to a statement from the agency.
This warning is based on data reviewed by the FDA, including a report from the National Institutes of Health’s National Toxicology Program, that show associations between vinpocetine use and decreased fetal weight and increased risk of miscarriage in animals. The agency is particularly concerned because products containing this ingredient, including those marketed as improving energy and memory, are widely available to women of childbearing age. As a result, the agency has recommended these women not take vinpocetine.
Vinpocetine is a synthetically produced compound used in dietary supplements either on its own or in combination and may be referred to as Vinca minor extract, lesser periwinkle extract, or common periwinkle extract on product labels. Although vinpocetine is regulated in some countries as a prescription drug, when it’s sold in dietary supplements in the United States, the FDA does not usually review those products or their labeling before they become available to consumers under the same safety and effectiveness standards used to evaluate drug products.
“Today’s safety warning is just one of many steps the FDA is taking to adapt to the realities of the evolving dietary supplement industry,” according to the agency’s statement. “Protecting the public from unsafe dietary supplements remains a top priority for the FDA.”
The full statement regarding vinpocetine and its risks can be found on the FDA website.
according to a statement from the agency.
This warning is based on data reviewed by the FDA, including a report from the National Institutes of Health’s National Toxicology Program, that show associations between vinpocetine use and decreased fetal weight and increased risk of miscarriage in animals. The agency is particularly concerned because products containing this ingredient, including those marketed as improving energy and memory, are widely available to women of childbearing age. As a result, the agency has recommended these women not take vinpocetine.
Vinpocetine is a synthetically produced compound used in dietary supplements either on its own or in combination and may be referred to as Vinca minor extract, lesser periwinkle extract, or common periwinkle extract on product labels. Although vinpocetine is regulated in some countries as a prescription drug, when it’s sold in dietary supplements in the United States, the FDA does not usually review those products or their labeling before they become available to consumers under the same safety and effectiveness standards used to evaluate drug products.
“Today’s safety warning is just one of many steps the FDA is taking to adapt to the realities of the evolving dietary supplement industry,” according to the agency’s statement. “Protecting the public from unsafe dietary supplements remains a top priority for the FDA.”
The full statement regarding vinpocetine and its risks can be found on the FDA website.
FDA launches call center project to streamline Expanded Access request process
CHICAGO – The Food and Drug Administration launched a new call center project to assist physicians seeking to help cancer patients access unapproved therapies.

Entitled “Project Facilitate,” the program aims to create a single point of contact with FDA oncology staff who can guide physicians through the process of submitting Expanded Access (EA) requests on behalf of individual patients.
“This is a pilot program to provide continuous support to health care professionals throughout the entire Expanded Access process,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products said during the unveiling of the project during a press briefing at the annual meeting of the American Society of Clinical Oncology.
Physicians utilizing Project Facilitate can expect a “concierge service” experience including advice on the information needed to complete requests, assistance completing forms, pharma/biotech contact information, independent review board resource options, and follow-up on patient outcomes.
The project will work in synergy with the Reagan-Udall EA Navigator website, an “online road map” for physicians and patients that was launched 2 years ago “to facilitate and coordinate and collaborate with the FDA to advance the science mission of FDA,” and which has been expanded in conjunction with Project Facilitate, Ellen V. Sigal, PhD, chair of the board of the Reagan-Udall Foundation for the FDA, said at the press briefing.
“EA Navigator delivers transparent, concise, and searchable information provided by companies about their Expanded Access policies,” Dr. Sigal said. “Today I’m pleased to announce that the Navigator now features Expanded Access opportunities listed in ClinicalTrials.gov for companies in the directory.
“For the first time, those who need quick access to drug availability and Expanded Access options will find it in one place without having to visit site by site by site, or sift through thousands of studies that don’t merit their needs,” she added, noting that EA Navigator will often be the first step for physicians before they engage with Project Facilitate.
Project Facilitate can be reached Monday-Friday, 9 a.m.-5 p.m. ET at 240-402-0004, or by email at OncProjectFacilitate@fda.hhs.gov.
CHICAGO – The Food and Drug Administration launched a new call center project to assist physicians seeking to help cancer patients access unapproved therapies.
Entitled “Project Facilitate,” the program aims to create a single point of contact with FDA oncology staff who can guide physicians through the process of submitting Expanded Access (EA) requests on behalf of individual patients.
“This is a pilot program to provide continuous support to health care professionals throughout the entire Expanded Access process,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products said during the unveiling of the project during a press briefing at the annual meeting of the American Society of Clinical Oncology.
Physicians utilizing Project Facilitate can expect a “concierge service” experience including advice on the information needed to complete requests, assistance completing forms, pharma/biotech contact information, independent review board resource options, and follow-up on patient outcomes.
The project will work in synergy with the Reagan-Udall EA Navigator website, an “online road map” for physicians and patients that was launched 2 years ago “to facilitate and coordinate and collaborate with the FDA to advance the science mission of FDA,” and which has been expanded in conjunction with Project Facilitate, Ellen V. Sigal, PhD, chair of the board of the Reagan-Udall Foundation for the FDA, said at the press briefing.
“EA Navigator delivers transparent, concise, and searchable information provided by companies about their Expanded Access policies,” Dr. Sigal said. “Today I’m pleased to announce that the Navigator now features Expanded Access opportunities listed in ClinicalTrials.gov for companies in the directory.
“For the first time, those who need quick access to drug availability and Expanded Access options will find it in one place without having to visit site by site by site, or sift through thousands of studies that don’t merit their needs,” she added, noting that EA Navigator will often be the first step for physicians before they engage with Project Facilitate.
Project Facilitate can be reached Monday-Friday, 9 a.m.-5 p.m. ET at 240-402-0004, or by email at OncProjectFacilitate@fda.hhs.gov.
CHICAGO – The Food and Drug Administration launched a new call center project to assist physicians seeking to help cancer patients access unapproved therapies.
Entitled “Project Facilitate,” the program aims to create a single point of contact with FDA oncology staff who can guide physicians through the process of submitting Expanded Access (EA) requests on behalf of individual patients.
“This is a pilot program to provide continuous support to health care professionals throughout the entire Expanded Access process,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products said during the unveiling of the project during a press briefing at the annual meeting of the American Society of Clinical Oncology.
Physicians utilizing Project Facilitate can expect a “concierge service” experience including advice on the information needed to complete requests, assistance completing forms, pharma/biotech contact information, independent review board resource options, and follow-up on patient outcomes.
The project will work in synergy with the Reagan-Udall EA Navigator website, an “online road map” for physicians and patients that was launched 2 years ago “to facilitate and coordinate and collaborate with the FDA to advance the science mission of FDA,” and which has been expanded in conjunction with Project Facilitate, Ellen V. Sigal, PhD, chair of the board of the Reagan-Udall Foundation for the FDA, said at the press briefing.
“EA Navigator delivers transparent, concise, and searchable information provided by companies about their Expanded Access policies,” Dr. Sigal said. “Today I’m pleased to announce that the Navigator now features Expanded Access opportunities listed in ClinicalTrials.gov for companies in the directory.
“For the first time, those who need quick access to drug availability and Expanded Access options will find it in one place without having to visit site by site by site, or sift through thousands of studies that don’t merit their needs,” she added, noting that EA Navigator will often be the first step for physicians before they engage with Project Facilitate.
Project Facilitate can be reached Monday-Friday, 9 a.m.-5 p.m. ET at 240-402-0004, or by email at OncProjectFacilitate@fda.hhs.gov.
REPORTING FROM ASCO 2019