User login
Cannabis: Doctors tell FDA to get out of the weeds
The Food and Drug Administration held its first-ever public hearing about products containing cannabis or cannabis-derived compounds – and it got an earful.
Hearing from over 100 individuals, the all-staff FDA panel was asked repeatedly to take the lead in bringing order to a confused morass of state and local cannabis regulations. The regulatory landscape currently contains many voids that slow research and put consumers at risk, many witnesses testified.
The federal Farm Bill of 2018 legalized the cultivation of hemp – cannabis with very low 9-tetrahydrocannabinol (THC) content – with regulatory restrictions.
However, the Farm Bill did not legalize low-THC cannabis products, said FDA Acting Commissioner Norman Sharpless, MD. The agency has concluded that both THC and cannabidiol (CBD) are drugs – not dietary supplements – and any exception to these provisions “would be new terrain for the FDA,” he said.

And although restrictions on CBD sales have generally not been enforced, “under current law, CBD and THC cannot lawfully be added to a food or marketed as a dietary supplement,” said Dr. Sharpless.
Though the FDA could choose to carve out regulatory exceptions, it has not yet done so.
Stakeholders who gave testimony included not just physicians, scientists, consumers, and advocates, but also growers, manufacturers, distributors, and retailers – as well as the legal firms that represent these interests.
Broadly, physicians and scientists encouraged the FDA to move forward with classifying CBD and most CBD-containing products as drugs, rather than dietary supplements. In general, the opposite approach was promoted by agriculture and manufacturing representatives who testified.
However, all were united in asking the FDA for clarity – and alacrity.
Again and again, speakers asked the FDA to move posthaste in tidying up the current clutter of regulations. Ryan Vandrey, PhD, of Johns Hopkins University, Baltimore, explained that today, “Hemp-derived CBD is unscheduled, Epidiolex is Schedule V, and synthetic CBD is Schedule I in the DEA’s current framework.”
Kevin Chapman, MD, of Children’s Hospital Colorado, representing the American Epilepsy Society, called for regulation of CBD as a drug, and an accelerated clinical trial path. He noted that many families of children with epilepsy other than Lennox-Gastaut or Dravet syndrome (the only approved Epidiolex indications) are dosing other CBD products, “making it up as they go along."

Jacqueline French MD, chief scientific officer of the Epilepsy Foundation, agreed that many families of children with epilepsy are doing their best to find consistent and unadulterated CBD products. She said her worry is that “abrupt withdrawal of CBD from the market could lead to seizure worsening, injury, or even death for patients who now rely on non-pharmaceutical CBD for seizure control.”
Dr. French and Dr. Chapman each made the point that without insurance reimbursement, Epidiolex costs families over $30,000 annually, while CBD is a small fraction of that – as little as $1,000, said Dr. Chapman.
The lack of standards for CBD preparations means the content of oils, tinctures, and e-cigarette products can vary widely. Michelle Peace, PhD, a forensic scientist at Virginia Commonwealth University, Richmond, receives funding from the U.S. Department of Justice to study licit and illicit drug use with e-cigarettes. She and her colleagues have found dextromethorphan and the potent synthetic cannabinoid 5F-ADB in vaping supplies advertised as containing only CBD.
In a recent investigation prompted by multiple consumer reports of hallucinations after vaping CBD-labeled products, “17 of 18 samples contained a synthetic cannabinoid. Clinics will not find these kinds of drugs when they just do drug testing,” Dr. Peace said.
Another sampling of CBD products available from retail outlets showed that claims often bore little relation to cannabinoid content, said Bill Gurley, PhD, co-director of the Center for Dietary Supplement Research at the University of Arkansas, Little Rock. While several products that claimed to have CBD actually contained none at all, some had many more CBD than the labeled amount – 228 times more, in one instance. One tested product was actually 45% THC, with little CBD content.
The potential for CBD to interact with many other drugs is cause for concern, noted several presenters. Barry Gidal, PharmD, director of pharmacy practice at the University of Wisconsin, Madison, said that “CBD is a complicated molecule. It has a complicated biotransformation pathway” through at least 9 enzymes within the hepatic cytochrome P450 system.
“It wasn’t until the Epidiolex development program that we began to understand the clinical ramifications of this…. The effect of CBD on other drugs may go beyond the anti-seizure drugs that have been studied so far,” said Dr. Gidal. He pointed to recent published case reports of potential CBD-drug interactions reporting elevated international normalized ratios for a patient on warfarin using CBD, and another report of elevated tacrolimus levels in a patient using CBD.
The way forward
A variety of regulatory pathways were proposed at the hearing. To prevent adulteration and contamination issues, many advocated standardized good manufacturing practices (GMPs), product reporting, and identification or registration, and a centralized reporting registry for adverse events.
Patient advocates, physicians, and scientists called for an easing of access to cannabis for medical research. Currently, cannabis, still classified as a Schedule I substance by the Drug Enforcement Administration, is only legally available for this purpose through a supply maintained by the National Institute on Drug Abuse. A limited number of strains are available, and access requires a lengthy approval process.
Most discussion centered around CBD, though some presenters asked for smoother sailing for THC research as well, particularly as a potential adjunct or alternative to opioids for chronic pain. Cannabidiol has generally been recognized as non-psychoactive, and the FDA assigned it a very low probability of causing dependence or addiction problems in its own review of human data.
However, in his opening remarks, Dr. Sharpless warned that this fact does not make CBD a benign substance, and many questions remain unanswered.
“How much CBD is safe to consume in a day? What if someone applies a topical CBD lotion, consumes a CBD beverage or candy, and also consumes some CBD oil? How much is too much? How will it interact with other drugs the person might be taking? What if she’s pregnant? What if children access CBD products like gummy edibles? What happens when someone chronically uses CBD for prolonged periods? These and many other questions represent important and significant gaps in our knowledge,” said Dr. Sharpless.
The FDA has established a public docket where the public may submit comments and documents until July 2, 2019.
The Food and Drug Administration held its first-ever public hearing about products containing cannabis or cannabis-derived compounds – and it got an earful.
Hearing from over 100 individuals, the all-staff FDA panel was asked repeatedly to take the lead in bringing order to a confused morass of state and local cannabis regulations. The regulatory landscape currently contains many voids that slow research and put consumers at risk, many witnesses testified.
The federal Farm Bill of 2018 legalized the cultivation of hemp – cannabis with very low 9-tetrahydrocannabinol (THC) content – with regulatory restrictions.
However, the Farm Bill did not legalize low-THC cannabis products, said FDA Acting Commissioner Norman Sharpless, MD. The agency has concluded that both THC and cannabidiol (CBD) are drugs – not dietary supplements – and any exception to these provisions “would be new terrain for the FDA,” he said.
And although restrictions on CBD sales have generally not been enforced, “under current law, CBD and THC cannot lawfully be added to a food or marketed as a dietary supplement,” said Dr. Sharpless.
Though the FDA could choose to carve out regulatory exceptions, it has not yet done so.
Stakeholders who gave testimony included not just physicians, scientists, consumers, and advocates, but also growers, manufacturers, distributors, and retailers – as well as the legal firms that represent these interests.
Broadly, physicians and scientists encouraged the FDA to move forward with classifying CBD and most CBD-containing products as drugs, rather than dietary supplements. In general, the opposite approach was promoted by agriculture and manufacturing representatives who testified.
However, all were united in asking the FDA for clarity – and alacrity.
Again and again, speakers asked the FDA to move posthaste in tidying up the current clutter of regulations. Ryan Vandrey, PhD, of Johns Hopkins University, Baltimore, explained that today, “Hemp-derived CBD is unscheduled, Epidiolex is Schedule V, and synthetic CBD is Schedule I in the DEA’s current framework.”
Kevin Chapman, MD, of Children’s Hospital Colorado, representing the American Epilepsy Society, called for regulation of CBD as a drug, and an accelerated clinical trial path. He noted that many families of children with epilepsy other than Lennox-Gastaut or Dravet syndrome (the only approved Epidiolex indications) are dosing other CBD products, “making it up as they go along."
Jacqueline French MD, chief scientific officer of the Epilepsy Foundation, agreed that many families of children with epilepsy are doing their best to find consistent and unadulterated CBD products. She said her worry is that “abrupt withdrawal of CBD from the market could lead to seizure worsening, injury, or even death for patients who now rely on non-pharmaceutical CBD for seizure control.”
Dr. French and Dr. Chapman each made the point that without insurance reimbursement, Epidiolex costs families over $30,000 annually, while CBD is a small fraction of that – as little as $1,000, said Dr. Chapman.
The lack of standards for CBD preparations means the content of oils, tinctures, and e-cigarette products can vary widely. Michelle Peace, PhD, a forensic scientist at Virginia Commonwealth University, Richmond, receives funding from the U.S. Department of Justice to study licit and illicit drug use with e-cigarettes. She and her colleagues have found dextromethorphan and the potent synthetic cannabinoid 5F-ADB in vaping supplies advertised as containing only CBD.
In a recent investigation prompted by multiple consumer reports of hallucinations after vaping CBD-labeled products, “17 of 18 samples contained a synthetic cannabinoid. Clinics will not find these kinds of drugs when they just do drug testing,” Dr. Peace said.
Another sampling of CBD products available from retail outlets showed that claims often bore little relation to cannabinoid content, said Bill Gurley, PhD, co-director of the Center for Dietary Supplement Research at the University of Arkansas, Little Rock. While several products that claimed to have CBD actually contained none at all, some had many more CBD than the labeled amount – 228 times more, in one instance. One tested product was actually 45% THC, with little CBD content.
The potential for CBD to interact with many other drugs is cause for concern, noted several presenters. Barry Gidal, PharmD, director of pharmacy practice at the University of Wisconsin, Madison, said that “CBD is a complicated molecule. It has a complicated biotransformation pathway” through at least 9 enzymes within the hepatic cytochrome P450 system.
“It wasn’t until the Epidiolex development program that we began to understand the clinical ramifications of this…. The effect of CBD on other drugs may go beyond the anti-seizure drugs that have been studied so far,” said Dr. Gidal. He pointed to recent published case reports of potential CBD-drug interactions reporting elevated international normalized ratios for a patient on warfarin using CBD, and another report of elevated tacrolimus levels in a patient using CBD.
The way forward
A variety of regulatory pathways were proposed at the hearing. To prevent adulteration and contamination issues, many advocated standardized good manufacturing practices (GMPs), product reporting, and identification or registration, and a centralized reporting registry for adverse events.
Patient advocates, physicians, and scientists called for an easing of access to cannabis for medical research. Currently, cannabis, still classified as a Schedule I substance by the Drug Enforcement Administration, is only legally available for this purpose through a supply maintained by the National Institute on Drug Abuse. A limited number of strains are available, and access requires a lengthy approval process.
Most discussion centered around CBD, though some presenters asked for smoother sailing for THC research as well, particularly as a potential adjunct or alternative to opioids for chronic pain. Cannabidiol has generally been recognized as non-psychoactive, and the FDA assigned it a very low probability of causing dependence or addiction problems in its own review of human data.
However, in his opening remarks, Dr. Sharpless warned that this fact does not make CBD a benign substance, and many questions remain unanswered.
“How much CBD is safe to consume in a day? What if someone applies a topical CBD lotion, consumes a CBD beverage or candy, and also consumes some CBD oil? How much is too much? How will it interact with other drugs the person might be taking? What if she’s pregnant? What if children access CBD products like gummy edibles? What happens when someone chronically uses CBD for prolonged periods? These and many other questions represent important and significant gaps in our knowledge,” said Dr. Sharpless.
The FDA has established a public docket where the public may submit comments and documents until July 2, 2019.
The Food and Drug Administration held its first-ever public hearing about products containing cannabis or cannabis-derived compounds – and it got an earful.
Hearing from over 100 individuals, the all-staff FDA panel was asked repeatedly to take the lead in bringing order to a confused morass of state and local cannabis regulations. The regulatory landscape currently contains many voids that slow research and put consumers at risk, many witnesses testified.
The federal Farm Bill of 2018 legalized the cultivation of hemp – cannabis with very low 9-tetrahydrocannabinol (THC) content – with regulatory restrictions.
However, the Farm Bill did not legalize low-THC cannabis products, said FDA Acting Commissioner Norman Sharpless, MD. The agency has concluded that both THC and cannabidiol (CBD) are drugs – not dietary supplements – and any exception to these provisions “would be new terrain for the FDA,” he said.
And although restrictions on CBD sales have generally not been enforced, “under current law, CBD and THC cannot lawfully be added to a food or marketed as a dietary supplement,” said Dr. Sharpless.
Though the FDA could choose to carve out regulatory exceptions, it has not yet done so.
Stakeholders who gave testimony included not just physicians, scientists, consumers, and advocates, but also growers, manufacturers, distributors, and retailers – as well as the legal firms that represent these interests.
Broadly, physicians and scientists encouraged the FDA to move forward with classifying CBD and most CBD-containing products as drugs, rather than dietary supplements. In general, the opposite approach was promoted by agriculture and manufacturing representatives who testified.
However, all were united in asking the FDA for clarity – and alacrity.
Again and again, speakers asked the FDA to move posthaste in tidying up the current clutter of regulations. Ryan Vandrey, PhD, of Johns Hopkins University, Baltimore, explained that today, “Hemp-derived CBD is unscheduled, Epidiolex is Schedule V, and synthetic CBD is Schedule I in the DEA’s current framework.”
Kevin Chapman, MD, of Children’s Hospital Colorado, representing the American Epilepsy Society, called for regulation of CBD as a drug, and an accelerated clinical trial path. He noted that many families of children with epilepsy other than Lennox-Gastaut or Dravet syndrome (the only approved Epidiolex indications) are dosing other CBD products, “making it up as they go along."
Jacqueline French MD, chief scientific officer of the Epilepsy Foundation, agreed that many families of children with epilepsy are doing their best to find consistent and unadulterated CBD products. She said her worry is that “abrupt withdrawal of CBD from the market could lead to seizure worsening, injury, or even death for patients who now rely on non-pharmaceutical CBD for seizure control.”
Dr. French and Dr. Chapman each made the point that without insurance reimbursement, Epidiolex costs families over $30,000 annually, while CBD is a small fraction of that – as little as $1,000, said Dr. Chapman.
The lack of standards for CBD preparations means the content of oils, tinctures, and e-cigarette products can vary widely. Michelle Peace, PhD, a forensic scientist at Virginia Commonwealth University, Richmond, receives funding from the U.S. Department of Justice to study licit and illicit drug use with e-cigarettes. She and her colleagues have found dextromethorphan and the potent synthetic cannabinoid 5F-ADB in vaping supplies advertised as containing only CBD.
In a recent investigation prompted by multiple consumer reports of hallucinations after vaping CBD-labeled products, “17 of 18 samples contained a synthetic cannabinoid. Clinics will not find these kinds of drugs when they just do drug testing,” Dr. Peace said.
Another sampling of CBD products available from retail outlets showed that claims often bore little relation to cannabinoid content, said Bill Gurley, PhD, co-director of the Center for Dietary Supplement Research at the University of Arkansas, Little Rock. While several products that claimed to have CBD actually contained none at all, some had many more CBD than the labeled amount – 228 times more, in one instance. One tested product was actually 45% THC, with little CBD content.
The potential for CBD to interact with many other drugs is cause for concern, noted several presenters. Barry Gidal, PharmD, director of pharmacy practice at the University of Wisconsin, Madison, said that “CBD is a complicated molecule. It has a complicated biotransformation pathway” through at least 9 enzymes within the hepatic cytochrome P450 system.
“It wasn’t until the Epidiolex development program that we began to understand the clinical ramifications of this…. The effect of CBD on other drugs may go beyond the anti-seizure drugs that have been studied so far,” said Dr. Gidal. He pointed to recent published case reports of potential CBD-drug interactions reporting elevated international normalized ratios for a patient on warfarin using CBD, and another report of elevated tacrolimus levels in a patient using CBD.
The way forward
A variety of regulatory pathways were proposed at the hearing. To prevent adulteration and contamination issues, many advocated standardized good manufacturing practices (GMPs), product reporting, and identification or registration, and a centralized reporting registry for adverse events.
Patient advocates, physicians, and scientists called for an easing of access to cannabis for medical research. Currently, cannabis, still classified as a Schedule I substance by the Drug Enforcement Administration, is only legally available for this purpose through a supply maintained by the National Institute on Drug Abuse. A limited number of strains are available, and access requires a lengthy approval process.
Most discussion centered around CBD, though some presenters asked for smoother sailing for THC research as well, particularly as a potential adjunct or alternative to opioids for chronic pain. Cannabidiol has generally been recognized as non-psychoactive, and the FDA assigned it a very low probability of causing dependence or addiction problems in its own review of human data.
However, in his opening remarks, Dr. Sharpless warned that this fact does not make CBD a benign substance, and many questions remain unanswered.
“How much CBD is safe to consume in a day? What if someone applies a topical CBD lotion, consumes a CBD beverage or candy, and also consumes some CBD oil? How much is too much? How will it interact with other drugs the person might be taking? What if she’s pregnant? What if children access CBD products like gummy edibles? What happens when someone chronically uses CBD for prolonged periods? These and many other questions represent important and significant gaps in our knowledge,” said Dr. Sharpless.
The FDA has established a public docket where the public may submit comments and documents until July 2, 2019.
FROM AN FDA PUBLIC HEARING
CDC creates interactive education module to improve RMSF recognition
The Centers for Disease Control and Prevention has created a first-of-its-kind interactive training module to help physicians both recognize and diagnose Rocky Mountain spotted fever (RMSF).
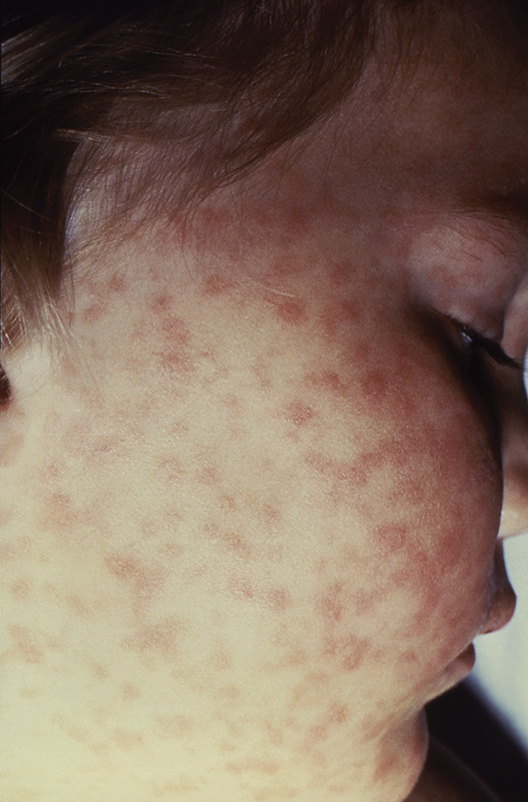
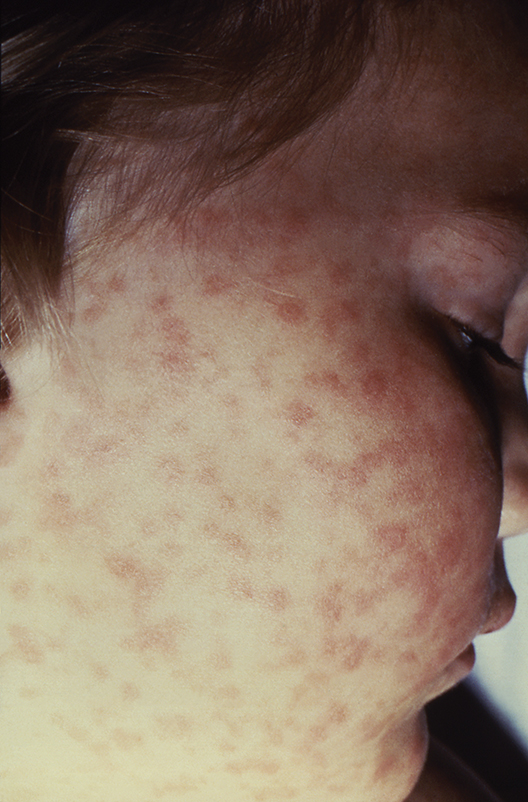
A record number of cases of RMSF were reported to the CDC in 2017 (6,248, up from 4,269 in 2016), but less than 1% of those cases had sufficient laboratory evidence to be confirmed. The CDC education module includes scenarios based on real cases to aid providers in recognizing RMSF and differentiating it from similar diseases. CME is available for physicians, nurse practitioners, physician assistants, veterinarians, nurses, epidemiologists, public health professionals, educators, and health communicators.
The disease initially presents with nonspecific symptoms such as fever, headache, or rash, but if left untreated, patients may require the amputation of fingers, toes, or limbs because of low blood flow; heart and lung specialty care; and ICU management. About 20% of untreated cases are fatal; half of these deaths occur within 8 days of initial presentation.
“Rocky Mountain spotted fever can be deadly if not treated early – yet cases often go unrecognized because the signs and symptoms are similar to those of many other diseases. With tickborne diseases on the rise in the U.S., this training will better equip health care providers to identify, diagnose, and treat this potentially fatal disease,” said CDC director Robert R. Redfield, MD.
Find the full press release on the CDC website.
The Centers for Disease Control and Prevention has created a first-of-its-kind interactive training module to help physicians both recognize and diagnose Rocky Mountain spotted fever (RMSF).
A record number of cases of RMSF were reported to the CDC in 2017 (6,248, up from 4,269 in 2016), but less than 1% of those cases had sufficient laboratory evidence to be confirmed. The CDC education module includes scenarios based on real cases to aid providers in recognizing RMSF and differentiating it from similar diseases. CME is available for physicians, nurse practitioners, physician assistants, veterinarians, nurses, epidemiologists, public health professionals, educators, and health communicators.
The disease initially presents with nonspecific symptoms such as fever, headache, or rash, but if left untreated, patients may require the amputation of fingers, toes, or limbs because of low blood flow; heart and lung specialty care; and ICU management. About 20% of untreated cases are fatal; half of these deaths occur within 8 days of initial presentation.
“Rocky Mountain spotted fever can be deadly if not treated early – yet cases often go unrecognized because the signs and symptoms are similar to those of many other diseases. With tickborne diseases on the rise in the U.S., this training will better equip health care providers to identify, diagnose, and treat this potentially fatal disease,” said CDC director Robert R. Redfield, MD.
Find the full press release on the CDC website.
The Centers for Disease Control and Prevention has created a first-of-its-kind interactive training module to help physicians both recognize and diagnose Rocky Mountain spotted fever (RMSF).
A record number of cases of RMSF were reported to the CDC in 2017 (6,248, up from 4,269 in 2016), but less than 1% of those cases had sufficient laboratory evidence to be confirmed. The CDC education module includes scenarios based on real cases to aid providers in recognizing RMSF and differentiating it from similar diseases. CME is available for physicians, nurse practitioners, physician assistants, veterinarians, nurses, epidemiologists, public health professionals, educators, and health communicators.
The disease initially presents with nonspecific symptoms such as fever, headache, or rash, but if left untreated, patients may require the amputation of fingers, toes, or limbs because of low blood flow; heart and lung specialty care; and ICU management. About 20% of untreated cases are fatal; half of these deaths occur within 8 days of initial presentation.
“Rocky Mountain spotted fever can be deadly if not treated early – yet cases often go unrecognized because the signs and symptoms are similar to those of many other diseases. With tickborne diseases on the rise in the U.S., this training will better equip health care providers to identify, diagnose, and treat this potentially fatal disease,” said CDC director Robert R. Redfield, MD.
Find the full press release on the CDC website.
FDA grants Priority Review to Vascepa for cardiovascular risk reduction
The Food and Drug Administration has granted a Priority Review to the supplemental new drug application for icosapent ethyl (Vascepa).

If approved, Vascepa – which is produced by Amarin – would be the first drug indicated to reduce residual cardiovascular risk in patients with LDL cholesterol managed by statins who still have persistent elevated triglycerides. The drug is now approved for reducing triglyceride levels in patients with baseline values of 500 mg/dL or greater.
The Priority Review is based on results of REDUCE-IT, a landmark cardiovascular outcomes trial whose primary results were presented at the American Heart Association scientific sessions last November and published in the New England Journal of Medicine. Vascepa achieved the primary study endpoint, reducing the relative risk for the first occurrence of a major adverse cardiovascular event significantly, by 25%.
The drug also met the study’s key secondary endpoint, reducing the incidence of a composite of cardiovascular death, nonfatal heart attack, and nonfatal stroke by 26%. Significant adverse events associated with Vascepa in the trial were peripheral edema, constipation, and atrial fibrillation.
Vascepa is currently indicated as an adjunct to diet to reduce triglyceride in adults with severe hypertriglyceridemia, a significantly smaller population than that represented in REDUCE-IT.
“We expect earlier approval of an expanded indication for Vascepa to lead to faster improvements in care for millions of patients with residual cardiovascular risk after statin therapy,” John F. Thero, president and CEO of Amarin, said in the statement.
The FDA is expected to issue a complete response by the end of September. Find the full press release on the Amarin website.
The Food and Drug Administration has granted a Priority Review to the supplemental new drug application for icosapent ethyl (Vascepa).
If approved, Vascepa – which is produced by Amarin – would be the first drug indicated to reduce residual cardiovascular risk in patients with LDL cholesterol managed by statins who still have persistent elevated triglycerides. The drug is now approved for reducing triglyceride levels in patients with baseline values of 500 mg/dL or greater.
The Priority Review is based on results of REDUCE-IT, a landmark cardiovascular outcomes trial whose primary results were presented at the American Heart Association scientific sessions last November and published in the New England Journal of Medicine. Vascepa achieved the primary study endpoint, reducing the relative risk for the first occurrence of a major adverse cardiovascular event significantly, by 25%.
The drug also met the study’s key secondary endpoint, reducing the incidence of a composite of cardiovascular death, nonfatal heart attack, and nonfatal stroke by 26%. Significant adverse events associated with Vascepa in the trial were peripheral edema, constipation, and atrial fibrillation.
Vascepa is currently indicated as an adjunct to diet to reduce triglyceride in adults with severe hypertriglyceridemia, a significantly smaller population than that represented in REDUCE-IT.
“We expect earlier approval of an expanded indication for Vascepa to lead to faster improvements in care for millions of patients with residual cardiovascular risk after statin therapy,” John F. Thero, president and CEO of Amarin, said in the statement.
The FDA is expected to issue a complete response by the end of September. Find the full press release on the Amarin website.
The Food and Drug Administration has granted a Priority Review to the supplemental new drug application for icosapent ethyl (Vascepa).
If approved, Vascepa – which is produced by Amarin – would be the first drug indicated to reduce residual cardiovascular risk in patients with LDL cholesterol managed by statins who still have persistent elevated triglycerides. The drug is now approved for reducing triglyceride levels in patients with baseline values of 500 mg/dL or greater.
The Priority Review is based on results of REDUCE-IT, a landmark cardiovascular outcomes trial whose primary results were presented at the American Heart Association scientific sessions last November and published in the New England Journal of Medicine. Vascepa achieved the primary study endpoint, reducing the relative risk for the first occurrence of a major adverse cardiovascular event significantly, by 25%.
The drug also met the study’s key secondary endpoint, reducing the incidence of a composite of cardiovascular death, nonfatal heart attack, and nonfatal stroke by 26%. Significant adverse events associated with Vascepa in the trial were peripheral edema, constipation, and atrial fibrillation.
Vascepa is currently indicated as an adjunct to diet to reduce triglyceride in adults with severe hypertriglyceridemia, a significantly smaller population than that represented in REDUCE-IT.
“We expect earlier approval of an expanded indication for Vascepa to lead to faster improvements in care for millions of patients with residual cardiovascular risk after statin therapy,” John F. Thero, president and CEO of Amarin, said in the statement.
The FDA is expected to issue a complete response by the end of September. Find the full press release on the Amarin website.
FDA approves first drug for steroid-refractory acute GVHD
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
FDA approves lenalidomide/rituximab for previously treated FL, MZL
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
The Food and Drug Administration has approved lenalidomide (Revlimid), in combination with rituximab, for the treatment of adult patients with previously treated follicular or marginal zone lymphoma.
FDA approval is based on results from the randomized, double-blind, phase 3 AUGMENT trial, which evaluated lenalidomide/rituximab versus rituximab and placebo in patients with previously treated follicular or marginal zone lymphoma. The median progression-free survival in those receiving lenalidomide/rituximab was 39.4 months, compared with 14.1 months for those receiving rituximab/placebo (odds ratio, 0.46; 95% confidence interval, 0.34-0.62; P less than .0001).
A numeric trend was seen in overall survival over the follow-up period of 28.3 months (16 vs. 26 deaths; hazard ratio, 0.61; 95% CI, 0.33-1.13).
The most common adverse events associated with lenalidomide/rituximab are neutropenia, diarrhea, constipation, cough, fatigue, rash, pyrexia, leukopenia, pruritus, upper respiratory tract infections, abdominal pain, anemia, headache, and thrombocytopenia. Lenalidomide also contains a boxed warning for embryo-fetal toxicity, hematologic toxicity, and venous and arterial thromboembolism.
“Chemotherapy continues to be a standard of care for indolent forms of NHL, but most patients will relapse or become refractory to their current treatment. This approval represents a new therapeutic option for previously treated patients with follicular and marginal zone lymphomas, including those who relapse or no longer respond to initial treatment,” Meghan Gutierrez, CEO of the Lymphoma Research Foundation, said in a statement.
FDA expands use of Vraylar to treatment of bipolar-associated depressive episodes
The Food and Drug Administration on May 28 approved a supplemental New Drug Application for cariprazine (Vraylar) for the treatment of depressive episodes associated with bipolar I disorder.
Approval for the expanded label was based on results of the RGH-MD-53, RGH-MD-54, and RGH-MD-56 clinical trials, in which cariprazine was compared with placebo over a 6-week period in patients with bipolar I disorder. In all three trials, patients receiving 1.5 mg cariprazine had significantly greater improvement in their Montgomery-Åsberg Depression Rating scale after 6 weeks, compared with patients receiving placebo.
Cariprazine previously was indicated for the treatment of manic or mixed episodes associated with bipolar I disorder in adults. The most common adverse reactions reported in the clinical trials were nausea, akathisia, restlessness, and extrapyramidal symptoms; these symptoms are similar to those on the Vraylar label.
“Treating bipolar disorder can be very difficult, because people living with the illness experience a range of depressive and manic symptoms, sometimes both at the same time, and , specifically manic, mixed, and depressive episodes, with just one medication,” Stephen M. Stahl, MD, PhD, professor of psychiatry at the University of California, San Diego, said in the press release.
Find the full press release on the Allergan website.
The Food and Drug Administration on May 28 approved a supplemental New Drug Application for cariprazine (Vraylar) for the treatment of depressive episodes associated with bipolar I disorder.
Approval for the expanded label was based on results of the RGH-MD-53, RGH-MD-54, and RGH-MD-56 clinical trials, in which cariprazine was compared with placebo over a 6-week period in patients with bipolar I disorder. In all three trials, patients receiving 1.5 mg cariprazine had significantly greater improvement in their Montgomery-Åsberg Depression Rating scale after 6 weeks, compared with patients receiving placebo.
Cariprazine previously was indicated for the treatment of manic or mixed episodes associated with bipolar I disorder in adults. The most common adverse reactions reported in the clinical trials were nausea, akathisia, restlessness, and extrapyramidal symptoms; these symptoms are similar to those on the Vraylar label.
“Treating bipolar disorder can be very difficult, because people living with the illness experience a range of depressive and manic symptoms, sometimes both at the same time, and , specifically manic, mixed, and depressive episodes, with just one medication,” Stephen M. Stahl, MD, PhD, professor of psychiatry at the University of California, San Diego, said in the press release.
Find the full press release on the Allergan website.
The Food and Drug Administration on May 28 approved a supplemental New Drug Application for cariprazine (Vraylar) for the treatment of depressive episodes associated with bipolar I disorder.
Approval for the expanded label was based on results of the RGH-MD-53, RGH-MD-54, and RGH-MD-56 clinical trials, in which cariprazine was compared with placebo over a 6-week period in patients with bipolar I disorder. In all three trials, patients receiving 1.5 mg cariprazine had significantly greater improvement in their Montgomery-Åsberg Depression Rating scale after 6 weeks, compared with patients receiving placebo.
Cariprazine previously was indicated for the treatment of manic or mixed episodes associated with bipolar I disorder in adults. The most common adverse reactions reported in the clinical trials were nausea, akathisia, restlessness, and extrapyramidal symptoms; these symptoms are similar to those on the Vraylar label.
“Treating bipolar disorder can be very difficult, because people living with the illness experience a range of depressive and manic symptoms, sometimes both at the same time, and , specifically manic, mixed, and depressive episodes, with just one medication,” Stephen M. Stahl, MD, PhD, professor of psychiatry at the University of California, San Diego, said in the press release.
Find the full press release on the Allergan website.
FDA approves PI3K inhibitor alpelisib for breast cancer
The Food and Drug Administration has approved the first PI3K inhibitor for the treatment of breast cancer.
The drug, alpelisib (Piqray), was approved for use in combination with fulvestrant for men and postmenopausal women who have hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative, PIK3CA-mutated, advanced or metastatic breast cancer after progression on, or after, an endocrine-based regimen. The approval was announced by the FDA in a statement.
The agency also approved a diagnostic test – the therascreen PIK3CA RGQ PCR Kit – for detecting the PIK3CA mutation. The approval is based on results from the SOLAR-1 trial. In 572 HR-positive, HER2-negative patients with advanced or metastatic breast cancer who progressed after, or during, treatment with an aromatase inhibitor, the addition of alpelisib to fulvestrant significantly improved progression-free survival in patients with PIK3CA mutated tumors.
Median progression-free survival was 5.7 months on fulvestrant plus placebo, versus 11 months with fulvestrant plus alpelisib (hazard ratio for progression or death, 0.65; 95% confidence interval, 0.50-0.85; P less than .001). The results of the trial were recently published in the New England Journal of Medicine (2019 May 16;380[20]:1929-40).
In the FDA statement, Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, noted that alpelisib is the “first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer.”
The drug’s application was approved under the Real-Time Oncology Review pilot program, which allows the FDA to begin analyzing efficacy and safety databases before a new drug application is even submitted.
The FDA noted that patients should not start treatment on alpelisib if they have a history of severe skin reactions, such as Stevens-Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis.
The Food and Drug Administration has approved the first PI3K inhibitor for the treatment of breast cancer.
The drug, alpelisib (Piqray), was approved for use in combination with fulvestrant for men and postmenopausal women who have hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative, PIK3CA-mutated, advanced or metastatic breast cancer after progression on, or after, an endocrine-based regimen. The approval was announced by the FDA in a statement.
The agency also approved a diagnostic test – the therascreen PIK3CA RGQ PCR Kit – for detecting the PIK3CA mutation. The approval is based on results from the SOLAR-1 trial. In 572 HR-positive, HER2-negative patients with advanced or metastatic breast cancer who progressed after, or during, treatment with an aromatase inhibitor, the addition of alpelisib to fulvestrant significantly improved progression-free survival in patients with PIK3CA mutated tumors.
Median progression-free survival was 5.7 months on fulvestrant plus placebo, versus 11 months with fulvestrant plus alpelisib (hazard ratio for progression or death, 0.65; 95% confidence interval, 0.50-0.85; P less than .001). The results of the trial were recently published in the New England Journal of Medicine (2019 May 16;380[20]:1929-40).
In the FDA statement, Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, noted that alpelisib is the “first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer.”
The drug’s application was approved under the Real-Time Oncology Review pilot program, which allows the FDA to begin analyzing efficacy and safety databases before a new drug application is even submitted.
The FDA noted that patients should not start treatment on alpelisib if they have a history of severe skin reactions, such as Stevens-Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis.
The Food and Drug Administration has approved the first PI3K inhibitor for the treatment of breast cancer.
The drug, alpelisib (Piqray), was approved for use in combination with fulvestrant for men and postmenopausal women who have hormone receptor (HR)–positive, human epidermal growth factor receptor 2 (HER2)–negative, PIK3CA-mutated, advanced or metastatic breast cancer after progression on, or after, an endocrine-based regimen. The approval was announced by the FDA in a statement.
The agency also approved a diagnostic test – the therascreen PIK3CA RGQ PCR Kit – for detecting the PIK3CA mutation. The approval is based on results from the SOLAR-1 trial. In 572 HR-positive, HER2-negative patients with advanced or metastatic breast cancer who progressed after, or during, treatment with an aromatase inhibitor, the addition of alpelisib to fulvestrant significantly improved progression-free survival in patients with PIK3CA mutated tumors.
Median progression-free survival was 5.7 months on fulvestrant plus placebo, versus 11 months with fulvestrant plus alpelisib (hazard ratio for progression or death, 0.65; 95% confidence interval, 0.50-0.85; P less than .001). The results of the trial were recently published in the New England Journal of Medicine (2019 May 16;380[20]:1929-40).
In the FDA statement, Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, noted that alpelisib is the “first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer.”
The drug’s application was approved under the Real-Time Oncology Review pilot program, which allows the FDA to begin analyzing efficacy and safety databases before a new drug application is even submitted.
The FDA noted that patients should not start treatment on alpelisib if they have a history of severe skin reactions, such as Stevens-Johnson syndrome, erythema multiforme, or toxic epidermal necrolysis.
FDA announces clearance of modified endoscope connector
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
In a letter published April 18, the FDA had written that the original version of the product, the Erbe USA ERBEFLO port connector, was the only one of its type on the market that did not feature a method of backflow prevention, as recommended by new FDA guidelines. As such, the original ERBEFLO device did not adequately reduce the risk of cross-contamination; blood, stool, or other fluids from previous patients could travel through the endoscopy channels, contaminating the connector, tubing, and water bottle.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
In a letter published April 18, the FDA had written that the original version of the product, the Erbe USA ERBEFLO port connector, was the only one of its type on the market that did not feature a method of backflow prevention, as recommended by new FDA guidelines. As such, the original ERBEFLO device did not adequately reduce the risk of cross-contamination; blood, stool, or other fluids from previous patients could travel through the endoscopy channels, contaminating the connector, tubing, and water bottle.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
which was designed to reduce the risk of cross-contamination previously identified by the FDA.
In a letter published April 18, the FDA had written that the original version of the product, the Erbe USA ERBEFLO port connector, was the only one of its type on the market that did not feature a method of backflow prevention, as recommended by new FDA guidelines. As such, the original ERBEFLO device did not adequately reduce the risk of cross-contamination; blood, stool, or other fluids from previous patients could travel through the endoscopy channels, contaminating the connector, tubing, and water bottle.
The FDA approval of the modified ERBEFLO port connector is based on a review of the functional and simulated use testing of the modified device design. The effectiveness of the device at reducing the risk of backflow and contamination is also supported by simulated testing.
Revised labeling included with the product identifies compatible endoscopes and accessories and provides warnings to ensure proper usage.
“The clearance of the modified ERBEFLO 24-hour use port connector provides another option for health care facilities whose staff understand and can fully implement the instructions for use to reduce the risk of cross-contamination and infection,” the FDA said in the May 23 update letter.
FDA approves NovoTTF-100L System for advanced mesothelioma
The Food and Drug Administration has approved the NovoTTF-100L System in combination with pemetrexed plus platinum-based chemotherapy for the first-line treatment of unresectable, locally advanced or metastatic, malignant pleural mesothelioma (MPM).
The NovoTTF-100L System uses electric fields tuned to specific frequencies to disrupt solid tumor cancer cell division, Novocure, makers of NovoTTF-100L, said in a press release.
FDA approval was based on the single-arm STELLAR registration trial, which included 80 patients with unresectable and previously untreated MPM who were candidates for treatment with pemetrexed and cisplatin or carboplatin.
Median overall survival among all patients treated with NovoTTF-100L plus chemotherapy was 18.2 months (95% confidence interval, 12.1-25.8). The disease control rate in the 72 patients with at least one follow-up CT scan performed was 97%; 40% of patients had a partial response, 57% had stable disease, and 3% had progressive disease. The median progression free survival was 7.6 months.
The most common adverse events observed with the NovoTTF-100L System in combination with chemotherapy in patients with MPM were anemia, constipation, nausea, asthenia, chest pain, fatigue, device skin reaction, pruritus, and cough.
Other potential adverse effects associated with the use of the NovoTTF-100L System include: treatment related skin toxicity, allergic reaction to the plaster or to the gel, electrode overheating leading to pain and/or local skin burns, infections at sites of electrode contact with the skin, local warmth and tingling sensation beneath the electrodes, muscle twitching, medical site reaction, and skin breakdown/skin ulcer.
The NovoTTF-100L System can be prescribed only by a health care provider who has completed the required certification training provided by Novocure, the company said in the press release.
The Food and Drug Administration has approved the NovoTTF-100L System in combination with pemetrexed plus platinum-based chemotherapy for the first-line treatment of unresectable, locally advanced or metastatic, malignant pleural mesothelioma (MPM).
The NovoTTF-100L System uses electric fields tuned to specific frequencies to disrupt solid tumor cancer cell division, Novocure, makers of NovoTTF-100L, said in a press release.
FDA approval was based on the single-arm STELLAR registration trial, which included 80 patients with unresectable and previously untreated MPM who were candidates for treatment with pemetrexed and cisplatin or carboplatin.
Median overall survival among all patients treated with NovoTTF-100L plus chemotherapy was 18.2 months (95% confidence interval, 12.1-25.8). The disease control rate in the 72 patients with at least one follow-up CT scan performed was 97%; 40% of patients had a partial response, 57% had stable disease, and 3% had progressive disease. The median progression free survival was 7.6 months.
The most common adverse events observed with the NovoTTF-100L System in combination with chemotherapy in patients with MPM were anemia, constipation, nausea, asthenia, chest pain, fatigue, device skin reaction, pruritus, and cough.
Other potential adverse effects associated with the use of the NovoTTF-100L System include: treatment related skin toxicity, allergic reaction to the plaster or to the gel, electrode overheating leading to pain and/or local skin burns, infections at sites of electrode contact with the skin, local warmth and tingling sensation beneath the electrodes, muscle twitching, medical site reaction, and skin breakdown/skin ulcer.
The NovoTTF-100L System can be prescribed only by a health care provider who has completed the required certification training provided by Novocure, the company said in the press release.
The Food and Drug Administration has approved the NovoTTF-100L System in combination with pemetrexed plus platinum-based chemotherapy for the first-line treatment of unresectable, locally advanced or metastatic, malignant pleural mesothelioma (MPM).
The NovoTTF-100L System uses electric fields tuned to specific frequencies to disrupt solid tumor cancer cell division, Novocure, makers of NovoTTF-100L, said in a press release.
FDA approval was based on the single-arm STELLAR registration trial, which included 80 patients with unresectable and previously untreated MPM who were candidates for treatment with pemetrexed and cisplatin or carboplatin.
Median overall survival among all patients treated with NovoTTF-100L plus chemotherapy was 18.2 months (95% confidence interval, 12.1-25.8). The disease control rate in the 72 patients with at least one follow-up CT scan performed was 97%; 40% of patients had a partial response, 57% had stable disease, and 3% had progressive disease. The median progression free survival was 7.6 months.
The most common adverse events observed with the NovoTTF-100L System in combination with chemotherapy in patients with MPM were anemia, constipation, nausea, asthenia, chest pain, fatigue, device skin reaction, pruritus, and cough.
Other potential adverse effects associated with the use of the NovoTTF-100L System include: treatment related skin toxicity, allergic reaction to the plaster or to the gel, electrode overheating leading to pain and/or local skin burns, infections at sites of electrode contact with the skin, local warmth and tingling sensation beneath the electrodes, muscle twitching, medical site reaction, and skin breakdown/skin ulcer.
The NovoTTF-100L System can be prescribed only by a health care provider who has completed the required certification training provided by Novocure, the company said in the press release.
FDA approves Zolgensma for infantile-onset SMA treatment
The Food and Drug Administration has approved Zolgensma (onasemnogene abeparvovec-xioi), the first gene therapy for the treatment of infantile-onset spinal muscular atrophy in children aged less than 2 years.
The FDA granted the approval of Zolgensma to AveXis Inc.
Spinal muscular atrophy (SMA) is a genetic disorder caused by a mutation in the SMN1 gene, which encodes the survival motor neuron protein. This protein is necessary for motor function throughout the body; without it, motor neurons die, causing severe, often fatal muscle weakness. Infantile-onset SMA is the most severe and most common form of the disease; children will have difficulty holding their head up, swallowing, or breathing. Symptoms can be present at birth or appear by 6 months.
FDA approval of Zolgensma is based on results of a pair of clinical trials – one ongoing, one completed – comprising 36 patients with infantile-onset SMA aged between 2 weeks and 8 months at study entry. Of the 21 patients initially enrolled in the ongoing trial, 19 remain, aged between 9.4 and 18.5 months; most of these patients are at least 14 months. Compared with natural disease course, patients treated with Zolgensma are more likely to reach developmental motor milestones such as head control and the ability to sit without support.
The most common adverse events associated with Zolgensma include elevated liver enzymes and vomiting. The labeling includes a warning that acute serious liver injury can occur, and patients with preexisting liver conditions are at a higher risk for serious liver injury. Liver function should be monitored for at least 3 months following initiation of Zolgensma treatment.
“Children with SMA experience difficulty performing essential functions of life. Most children with this disease do not survive past early childhood due to respiratory failure. Patients with SMA now have another treatment option to minimize the progression of SMA and improve survival,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved Zolgensma (onasemnogene abeparvovec-xioi), the first gene therapy for the treatment of infantile-onset spinal muscular atrophy in children aged less than 2 years.
The FDA granted the approval of Zolgensma to AveXis Inc.
Spinal muscular atrophy (SMA) is a genetic disorder caused by a mutation in the SMN1 gene, which encodes the survival motor neuron protein. This protein is necessary for motor function throughout the body; without it, motor neurons die, causing severe, often fatal muscle weakness. Infantile-onset SMA is the most severe and most common form of the disease; children will have difficulty holding their head up, swallowing, or breathing. Symptoms can be present at birth or appear by 6 months.
FDA approval of Zolgensma is based on results of a pair of clinical trials – one ongoing, one completed – comprising 36 patients with infantile-onset SMA aged between 2 weeks and 8 months at study entry. Of the 21 patients initially enrolled in the ongoing trial, 19 remain, aged between 9.4 and 18.5 months; most of these patients are at least 14 months. Compared with natural disease course, patients treated with Zolgensma are more likely to reach developmental motor milestones such as head control and the ability to sit without support.
The most common adverse events associated with Zolgensma include elevated liver enzymes and vomiting. The labeling includes a warning that acute serious liver injury can occur, and patients with preexisting liver conditions are at a higher risk for serious liver injury. Liver function should be monitored for at least 3 months following initiation of Zolgensma treatment.
“Children with SMA experience difficulty performing essential functions of life. Most children with this disease do not survive past early childhood due to respiratory failure. Patients with SMA now have another treatment option to minimize the progression of SMA and improve survival,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved Zolgensma (onasemnogene abeparvovec-xioi), the first gene therapy for the treatment of infantile-onset spinal muscular atrophy in children aged less than 2 years.
The FDA granted the approval of Zolgensma to AveXis Inc.
Spinal muscular atrophy (SMA) is a genetic disorder caused by a mutation in the SMN1 gene, which encodes the survival motor neuron protein. This protein is necessary for motor function throughout the body; without it, motor neurons die, causing severe, often fatal muscle weakness. Infantile-onset SMA is the most severe and most common form of the disease; children will have difficulty holding their head up, swallowing, or breathing. Symptoms can be present at birth or appear by 6 months.
FDA approval of Zolgensma is based on results of a pair of clinical trials – one ongoing, one completed – comprising 36 patients with infantile-onset SMA aged between 2 weeks and 8 months at study entry. Of the 21 patients initially enrolled in the ongoing trial, 19 remain, aged between 9.4 and 18.5 months; most of these patients are at least 14 months. Compared with natural disease course, patients treated with Zolgensma are more likely to reach developmental motor milestones such as head control and the ability to sit without support.
The most common adverse events associated with Zolgensma include elevated liver enzymes and vomiting. The labeling includes a warning that acute serious liver injury can occur, and patients with preexisting liver conditions are at a higher risk for serious liver injury. Liver function should be monitored for at least 3 months following initiation of Zolgensma treatment.
“Children with SMA experience difficulty performing essential functions of life. Most children with this disease do not survive past early childhood due to respiratory failure. Patients with SMA now have another treatment option to minimize the progression of SMA and improve survival,” Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, said in the press release.
Find the full press release on the FDA website.