User login
FDA issues warning on insulin pump cybersecurity weakness
The Food and Drug Administration has issued a warning to patients and health care providers that a pair of Medtronic insulin pumps are being recalled because of potential cybersecurity risks, according to a press release.

The affected devices are the MiniMed 508 and MiniMed Paradigm series insulin pumps, which wirelessly connect to both the patient’s blood glucose meter and continuous glucose monitoring system. A remote controller and CareLink USB – a thumb-sized wireless device that plugs into a computer – are used to operate the devices; the remote controller sends insulin dosing commands to the pump and the CareLink USB can be used to download and share data with the patient’s health care provider.
The potential risk involves the wireless communication between the pumps and related devices such as the blood glucose meter and remote controller. The FDA has identified a cybersecurity vulnerability within the insulin pumps, and is concerned that a third party could connect to the device and change the pump’s settings. Insulin could be given in excess, causing hypoglycemia, or stopped, causing hyperglycemia or diabetic ketoacidosis.
Medtronic has identified 4,000 patients in the United States who are affected by the security weakness. Because the company is unable to adequately update or patch the device to remove the weakness, the FDA is working to ensure that Medtronic addresses the issue in any way possible, including helping patients with affected pumps switch to newer models.
“While we are not aware of patients who may have been harmed by this particular cybersecurity vulnerability, the risk of patient harm if such a vulnerability were left unaddressed is significant. The safety communication issued today contains recommendations for what actions patients and health care providers should take to avoid the risk this vulnerability could pose,” said Suzanne Schwartz, MD, MBA, deputy director of the Office of Strategic Partnerships and Technology Innovation.
Find the full press release on the FDA website.
The Food and Drug Administration has issued a warning to patients and health care providers that a pair of Medtronic insulin pumps are being recalled because of potential cybersecurity risks, according to a press release.
The affected devices are the MiniMed 508 and MiniMed Paradigm series insulin pumps, which wirelessly connect to both the patient’s blood glucose meter and continuous glucose monitoring system. A remote controller and CareLink USB – a thumb-sized wireless device that plugs into a computer – are used to operate the devices; the remote controller sends insulin dosing commands to the pump and the CareLink USB can be used to download and share data with the patient’s health care provider.
The potential risk involves the wireless communication between the pumps and related devices such as the blood glucose meter and remote controller. The FDA has identified a cybersecurity vulnerability within the insulin pumps, and is concerned that a third party could connect to the device and change the pump’s settings. Insulin could be given in excess, causing hypoglycemia, or stopped, causing hyperglycemia or diabetic ketoacidosis.
Medtronic has identified 4,000 patients in the United States who are affected by the security weakness. Because the company is unable to adequately update or patch the device to remove the weakness, the FDA is working to ensure that Medtronic addresses the issue in any way possible, including helping patients with affected pumps switch to newer models.
“While we are not aware of patients who may have been harmed by this particular cybersecurity vulnerability, the risk of patient harm if such a vulnerability were left unaddressed is significant. The safety communication issued today contains recommendations for what actions patients and health care providers should take to avoid the risk this vulnerability could pose,” said Suzanne Schwartz, MD, MBA, deputy director of the Office of Strategic Partnerships and Technology Innovation.
Find the full press release on the FDA website.
The Food and Drug Administration has issued a warning to patients and health care providers that a pair of Medtronic insulin pumps are being recalled because of potential cybersecurity risks, according to a press release.
The affected devices are the MiniMed 508 and MiniMed Paradigm series insulin pumps, which wirelessly connect to both the patient’s blood glucose meter and continuous glucose monitoring system. A remote controller and CareLink USB – a thumb-sized wireless device that plugs into a computer – are used to operate the devices; the remote controller sends insulin dosing commands to the pump and the CareLink USB can be used to download and share data with the patient’s health care provider.
The potential risk involves the wireless communication between the pumps and related devices such as the blood glucose meter and remote controller. The FDA has identified a cybersecurity vulnerability within the insulin pumps, and is concerned that a third party could connect to the device and change the pump’s settings. Insulin could be given in excess, causing hypoglycemia, or stopped, causing hyperglycemia or diabetic ketoacidosis.
Medtronic has identified 4,000 patients in the United States who are affected by the security weakness. Because the company is unable to adequately update or patch the device to remove the weakness, the FDA is working to ensure that Medtronic addresses the issue in any way possible, including helping patients with affected pumps switch to newer models.
“While we are not aware of patients who may have been harmed by this particular cybersecurity vulnerability, the risk of patient harm if such a vulnerability were left unaddressed is significant. The safety communication issued today contains recommendations for what actions patients and health care providers should take to avoid the risk this vulnerability could pose,” said Suzanne Schwartz, MD, MBA, deputy director of the Office of Strategic Partnerships and Technology Innovation.
Find the full press release on the FDA website.
FDA expands Doptelet approval to ITP patients with thrombocytopenia
The Food and Drug Administration has approved a supplemental New Drug Application expanding the indication of avatrombopag (Doptelet) to include treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) with insufficient response to previous therapy, according to Dova Pharmaceuticals.

FDA approval was based on results of a phase 3 trial in which a majority of patients who received avatrombopag achieved a platelet count of at least 50,000 per mcg after 8 days of therapy. In addition, efficacy was superior to patients in the placebo group in the maintenance of platelet counts during the 6-month treatment period.
Avatrombopag – an oral, thrombopoietin receptor agonist administered with food – was previously indicated for the treatment of chronic liver disease in adult patients who are scheduled to undergo a procedure. The most common adverse reactions in patients with ITP include headache, fatigue, contusion, epistaxis, upper respiratory tract infection, arthralgia, gingival bleeding, petechiae, and nasopharyngitis.
Find the full press release on the Dova Pharmaceuticals website.
The Food and Drug Administration has approved a supplemental New Drug Application expanding the indication of avatrombopag (Doptelet) to include treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) with insufficient response to previous therapy, according to Dova Pharmaceuticals.
FDA approval was based on results of a phase 3 trial in which a majority of patients who received avatrombopag achieved a platelet count of at least 50,000 per mcg after 8 days of therapy. In addition, efficacy was superior to patients in the placebo group in the maintenance of platelet counts during the 6-month treatment period.
Avatrombopag – an oral, thrombopoietin receptor agonist administered with food – was previously indicated for the treatment of chronic liver disease in adult patients who are scheduled to undergo a procedure. The most common adverse reactions in patients with ITP include headache, fatigue, contusion, epistaxis, upper respiratory tract infection, arthralgia, gingival bleeding, petechiae, and nasopharyngitis.
Find the full press release on the Dova Pharmaceuticals website.
The Food and Drug Administration has approved a supplemental New Drug Application expanding the indication of avatrombopag (Doptelet) to include treatment of thrombocytopenia in adults with chronic immune thrombocytopenia (ITP) with insufficient response to previous therapy, according to Dova Pharmaceuticals.
FDA approval was based on results of a phase 3 trial in which a majority of patients who received avatrombopag achieved a platelet count of at least 50,000 per mcg after 8 days of therapy. In addition, efficacy was superior to patients in the placebo group in the maintenance of platelet counts during the 6-month treatment period.
Avatrombopag – an oral, thrombopoietin receptor agonist administered with food – was previously indicated for the treatment of chronic liver disease in adult patients who are scheduled to undergo a procedure. The most common adverse reactions in patients with ITP include headache, fatigue, contusion, epistaxis, upper respiratory tract infection, arthralgia, gingival bleeding, petechiae, and nasopharyngitis.
Find the full press release on the Dova Pharmaceuticals website.
More than half of U.S. adults have never received HIV screening
Despite a 2006 recommendation by the Centers for Disease Control and Prevention recommending universal screening for HIV, less than half of U.S. adults have ever been tested for the disease, a new study found.
“HIV screening is a critical entry point to a range of HIV prevention and treatment options. For persons at ongoing risk for HIV infection exposure, annual screening also offers the opportunity to discuss options to reduce risk, including HIV preexposure prophylaxis,” Marc A. Pitasi, MPH, and investigators from the CDC wrote in the Morbidity and Mortality Weekly Report.
The CDC investigators analyzed data collected from the Behavioral Risk Factor Surveillance System in 2016-2017 to assess the percentage of adults screened for HIV ever and in the past year across the entirety of the United States, in 50 local jurisdictions where the majority of new U.S. HIV cases occur, and in seven states with a disproportionately high rate of HIV in rural populations.
The rate of ever testing was 38.9% across the entire country, 46.9% in the 50 local jurisdictions, and 35.5% in the seven states. The percentage of adults who had undergone HIV screening in the past year was significantly smaller at 10.1% across the entire country, 14.5% in the 50 local jurisdictions, and 9.3% in the seven states. This improved to 29.2%, 34.3%, and 26.2%, respectively, in adults who were tested in the past year who were also at risk for the disease.
The rate of ever testing varied widely among the 50 jurisdictions, ranging from 36.5% in Maricopa County, Ariz., to 70.7% in Washington, D.C. However, in addition to D.C., only Baltimore, Bronx County (N.Y.), and New York County reached or exceeded 60% coverage. Bronx County also had the highest rate of HIV screening in the past year at 31.3%, while Alameda County, Calif., had the lowest rate at 8.1%.
Of the seven states (Alabama, Arkansas, Kentucky, Mississippi, Missouri, Oklahoma, and South Carolina), Mississippi had the highest overall ever-testing rate at 40.2%, and Oklahoma had the lowest at 29.7%. People living in rural areas in those states were less likely to have ever been tested (32.1%) than those living in urban areas (37.2%). The difference between rural and urban areas increased for people at risk for HIV who had been screened in the past year, with 18.4% and 29.0%, respectively, reporting undergoing screening.
“These data provide a baseline from which to measure changes in screening in these jurisdictions and other parts of the United States over time. To achieve national goals and end the HIV epidemic in the United States, innovative and novel screening approaches might be needed to reach segments of the population that have never been tested for HIV,” the investigators concluded.
The investigators reported no conflicts of interest.
SOURCE: Pitasi MA et al. MMWR Morb Mortal Wkly Rep. 2019;68:561-7.
Despite a 2006 recommendation by the Centers for Disease Control and Prevention recommending universal screening for HIV, less than half of U.S. adults have ever been tested for the disease, a new study found.
“HIV screening is a critical entry point to a range of HIV prevention and treatment options. For persons at ongoing risk for HIV infection exposure, annual screening also offers the opportunity to discuss options to reduce risk, including HIV preexposure prophylaxis,” Marc A. Pitasi, MPH, and investigators from the CDC wrote in the Morbidity and Mortality Weekly Report.
The CDC investigators analyzed data collected from the Behavioral Risk Factor Surveillance System in 2016-2017 to assess the percentage of adults screened for HIV ever and in the past year across the entirety of the United States, in 50 local jurisdictions where the majority of new U.S. HIV cases occur, and in seven states with a disproportionately high rate of HIV in rural populations.
The rate of ever testing was 38.9% across the entire country, 46.9% in the 50 local jurisdictions, and 35.5% in the seven states. The percentage of adults who had undergone HIV screening in the past year was significantly smaller at 10.1% across the entire country, 14.5% in the 50 local jurisdictions, and 9.3% in the seven states. This improved to 29.2%, 34.3%, and 26.2%, respectively, in adults who were tested in the past year who were also at risk for the disease.
The rate of ever testing varied widely among the 50 jurisdictions, ranging from 36.5% in Maricopa County, Ariz., to 70.7% in Washington, D.C. However, in addition to D.C., only Baltimore, Bronx County (N.Y.), and New York County reached or exceeded 60% coverage. Bronx County also had the highest rate of HIV screening in the past year at 31.3%, while Alameda County, Calif., had the lowest rate at 8.1%.
Of the seven states (Alabama, Arkansas, Kentucky, Mississippi, Missouri, Oklahoma, and South Carolina), Mississippi had the highest overall ever-testing rate at 40.2%, and Oklahoma had the lowest at 29.7%. People living in rural areas in those states were less likely to have ever been tested (32.1%) than those living in urban areas (37.2%). The difference between rural and urban areas increased for people at risk for HIV who had been screened in the past year, with 18.4% and 29.0%, respectively, reporting undergoing screening.
“These data provide a baseline from which to measure changes in screening in these jurisdictions and other parts of the United States over time. To achieve national goals and end the HIV epidemic in the United States, innovative and novel screening approaches might be needed to reach segments of the population that have never been tested for HIV,” the investigators concluded.
The investigators reported no conflicts of interest.
SOURCE: Pitasi MA et al. MMWR Morb Mortal Wkly Rep. 2019;68:561-7.
Despite a 2006 recommendation by the Centers for Disease Control and Prevention recommending universal screening for HIV, less than half of U.S. adults have ever been tested for the disease, a new study found.
“HIV screening is a critical entry point to a range of HIV prevention and treatment options. For persons at ongoing risk for HIV infection exposure, annual screening also offers the opportunity to discuss options to reduce risk, including HIV preexposure prophylaxis,” Marc A. Pitasi, MPH, and investigators from the CDC wrote in the Morbidity and Mortality Weekly Report.
The CDC investigators analyzed data collected from the Behavioral Risk Factor Surveillance System in 2016-2017 to assess the percentage of adults screened for HIV ever and in the past year across the entirety of the United States, in 50 local jurisdictions where the majority of new U.S. HIV cases occur, and in seven states with a disproportionately high rate of HIV in rural populations.
The rate of ever testing was 38.9% across the entire country, 46.9% in the 50 local jurisdictions, and 35.5% in the seven states. The percentage of adults who had undergone HIV screening in the past year was significantly smaller at 10.1% across the entire country, 14.5% in the 50 local jurisdictions, and 9.3% in the seven states. This improved to 29.2%, 34.3%, and 26.2%, respectively, in adults who were tested in the past year who were also at risk for the disease.
The rate of ever testing varied widely among the 50 jurisdictions, ranging from 36.5% in Maricopa County, Ariz., to 70.7% in Washington, D.C. However, in addition to D.C., only Baltimore, Bronx County (N.Y.), and New York County reached or exceeded 60% coverage. Bronx County also had the highest rate of HIV screening in the past year at 31.3%, while Alameda County, Calif., had the lowest rate at 8.1%.
Of the seven states (Alabama, Arkansas, Kentucky, Mississippi, Missouri, Oklahoma, and South Carolina), Mississippi had the highest overall ever-testing rate at 40.2%, and Oklahoma had the lowest at 29.7%. People living in rural areas in those states were less likely to have ever been tested (32.1%) than those living in urban areas (37.2%). The difference between rural and urban areas increased for people at risk for HIV who had been screened in the past year, with 18.4% and 29.0%, respectively, reporting undergoing screening.
“These data provide a baseline from which to measure changes in screening in these jurisdictions and other parts of the United States over time. To achieve national goals and end the HIV epidemic in the United States, innovative and novel screening approaches might be needed to reach segments of the population that have never been tested for HIV,” the investigators concluded.
The investigators reported no conflicts of interest.
SOURCE: Pitasi MA et al. MMWR Morb Mortal Wkly Rep. 2019;68:561-7.
FROM MMWR
FDA approves drug to treat low sexual desire in women
The .
![]()
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment,” Hylton V. Joffe, MD, director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive, and Urologic Products, stated in a press release. “Today’s approval provides women with another treatment option for this condition.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not caused by a medical or psychiatric condition. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire, and generalized HSDD is a lack of desire that occurs regardless of the type of sexual activity, situation, or partner.
Vyleesi was studied in two 24-week, randomized, double-blind, placebo-controlled trials in 1,247 premenopausal women with acquired, generalized HSDD. The women used Vyleesi two or three times per month and no more than once a week. About one-quarter of patients treated with Vyleesi had an increase of 1.2 or more in their sexual desire score (scored on a range of 1.2 to 6.0, with higher scores indicating greater sexual desire), compared with about 17% of those who took placebo. About 35% of the patients treated with Vyleesi had a decrease of one or more in their distress score (scored on a range of 0-4, with higher scores indicating greater distress from low sexual desire) compared with about 31% of those who took placebo.
The drug is injected under the skin of the abdomen or thigh at least 45 minutes before anticipated sexual activity. Patients may decide the optimal time to use Vyleesi based on the duration of benefit and any side effects, such as nausea. Patients should not take more than one dose of Vyleesi within 24 hours, or more than eight doses per month. Patients should discontinue treatment after 8 weeks if they do not report an improvement in sexual desire and associated distress.
Vyleesi works by activating melanocortin receptors but the exact mechanism for improving sexual desire is unknown. Some side effects were reported. “The most common side effects of Vyleesi are nausea and vomiting, flushing, injection site reactions, and headache. About 40% of patients in the clinical trials experienced nausea, most commonly with the first Vyleesi injection, and 13% needed medications for the treatment of nausea. About 1% of patients treated with Vyleesi in the clinical trials reported darkening of the gums and parts of the skin, including the face and breasts, which did not go away in about half the patients after stopping treatment. Patients with dark skin were more likely to develop this side effect,” according to the press release.
A temporary increase in blood pressure in patients after dosing with Vyleesi was observed during the clinical trials and therefore the drug is not recommended in patients at high risk for cardiovascular disease. In addition, patients who take a naltrexone-containing medication by mouth to treat alcohol or opioid dependence should not use Vyleesi because it may significantly decrease the levels of naltrexone in the blood and could lead to naltrexone treatment failure.
The full press release can be found on the FDA website.
The .
![]()
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment,” Hylton V. Joffe, MD, director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive, and Urologic Products, stated in a press release. “Today’s approval provides women with another treatment option for this condition.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not caused by a medical or psychiatric condition. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire, and generalized HSDD is a lack of desire that occurs regardless of the type of sexual activity, situation, or partner.
Vyleesi was studied in two 24-week, randomized, double-blind, placebo-controlled trials in 1,247 premenopausal women with acquired, generalized HSDD. The women used Vyleesi two or three times per month and no more than once a week. About one-quarter of patients treated with Vyleesi had an increase of 1.2 or more in their sexual desire score (scored on a range of 1.2 to 6.0, with higher scores indicating greater sexual desire), compared with about 17% of those who took placebo. About 35% of the patients treated with Vyleesi had a decrease of one or more in their distress score (scored on a range of 0-4, with higher scores indicating greater distress from low sexual desire) compared with about 31% of those who took placebo.
The drug is injected under the skin of the abdomen or thigh at least 45 minutes before anticipated sexual activity. Patients may decide the optimal time to use Vyleesi based on the duration of benefit and any side effects, such as nausea. Patients should not take more than one dose of Vyleesi within 24 hours, or more than eight doses per month. Patients should discontinue treatment after 8 weeks if they do not report an improvement in sexual desire and associated distress.
Vyleesi works by activating melanocortin receptors but the exact mechanism for improving sexual desire is unknown. Some side effects were reported. “The most common side effects of Vyleesi are nausea and vomiting, flushing, injection site reactions, and headache. About 40% of patients in the clinical trials experienced nausea, most commonly with the first Vyleesi injection, and 13% needed medications for the treatment of nausea. About 1% of patients treated with Vyleesi in the clinical trials reported darkening of the gums and parts of the skin, including the face and breasts, which did not go away in about half the patients after stopping treatment. Patients with dark skin were more likely to develop this side effect,” according to the press release.
A temporary increase in blood pressure in patients after dosing with Vyleesi was observed during the clinical trials and therefore the drug is not recommended in patients at high risk for cardiovascular disease. In addition, patients who take a naltrexone-containing medication by mouth to treat alcohol or opioid dependence should not use Vyleesi because it may significantly decrease the levels of naltrexone in the blood and could lead to naltrexone treatment failure.
The full press release can be found on the FDA website.
The .
![]()
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment,” Hylton V. Joffe, MD, director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive, and Urologic Products, stated in a press release. “Today’s approval provides women with another treatment option for this condition.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not caused by a medical or psychiatric condition. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire, and generalized HSDD is a lack of desire that occurs regardless of the type of sexual activity, situation, or partner.
Vyleesi was studied in two 24-week, randomized, double-blind, placebo-controlled trials in 1,247 premenopausal women with acquired, generalized HSDD. The women used Vyleesi two or three times per month and no more than once a week. About one-quarter of patients treated with Vyleesi had an increase of 1.2 or more in their sexual desire score (scored on a range of 1.2 to 6.0, with higher scores indicating greater sexual desire), compared with about 17% of those who took placebo. About 35% of the patients treated with Vyleesi had a decrease of one or more in their distress score (scored on a range of 0-4, with higher scores indicating greater distress from low sexual desire) compared with about 31% of those who took placebo.
The drug is injected under the skin of the abdomen or thigh at least 45 minutes before anticipated sexual activity. Patients may decide the optimal time to use Vyleesi based on the duration of benefit and any side effects, such as nausea. Patients should not take more than one dose of Vyleesi within 24 hours, or more than eight doses per month. Patients should discontinue treatment after 8 weeks if they do not report an improvement in sexual desire and associated distress.
Vyleesi works by activating melanocortin receptors but the exact mechanism for improving sexual desire is unknown. Some side effects were reported. “The most common side effects of Vyleesi are nausea and vomiting, flushing, injection site reactions, and headache. About 40% of patients in the clinical trials experienced nausea, most commonly with the first Vyleesi injection, and 13% needed medications for the treatment of nausea. About 1% of patients treated with Vyleesi in the clinical trials reported darkening of the gums and parts of the skin, including the face and breasts, which did not go away in about half the patients after stopping treatment. Patients with dark skin were more likely to develop this side effect,” according to the press release.
A temporary increase in blood pressure in patients after dosing with Vyleesi was observed during the clinical trials and therefore the drug is not recommended in patients at high risk for cardiovascular disease. In addition, patients who take a naltrexone-containing medication by mouth to treat alcohol or opioid dependence should not use Vyleesi because it may significantly decrease the levels of naltrexone in the blood and could lead to naltrexone treatment failure.
The full press release can be found on the FDA website.
FDA panel: Continue using paclitaxel-eluting PAD devices, with caveats
GAITHERSBURG, MD. – There was sufficient evidence of a late mortality signal seen at 2-5 years post procedurally for paclitaxel-eluting stents and coated balloons to warrant a label change for the devices, the Food and Drug Administration’s Circulatory System Devices Panel unanimously agreed after 2 days of deliberation.

That signal was brought to light in a meta-analysis published last December by Konstantinos Katsanos, MD, of Patras University Hospital, Rion, Greece, and colleagues (J Am Heart Assoc. 2018;7:e011245). Although there were concerns about the quality of the industry data used in the study, the caliber of the analysis itself and the subsequent data presented by the FDA to the panel were deemed sufficient to recommend a warning of concern to patients and providers.
Much of the new data from industry and large database registries presented to the panel, which was chaired by Richard A. Lange, MD, indicated a lessening to no evidence of the mortality effect. But this evidence was deemed insufficient to counter the evidence of the randomized controlled trials individually and collectively as presented in the Katsanos meta-analysis and subsequent information presented by the FDA that examined various parameters in a variety of sensitivity analyses that confirmed the late mortality signal. There was also concern that the industry and the registry analyses presented were not peer reviewed.
However, the panel also determined that it would be inappropriate to pull the devices from the market and from general use for several reasons.
One key reason was that, according to the panel, there was no mechanistic cause apparent for the late mortality. In addition, no convincing dose-response data could be teased from the preclinical and clinical trials studied because of their variability of devices, application methods, and lack of appropriate tissue analysis across studies.
Finally, the industry data used to create the meta-analysis were considered to be fundamentally flawed: in blinding, in the relatively small numbers of patients, and in the large percentage of patients lost to follow-up. The latter could have dramatically influenced the perceived results, especially as the studies were not powered or designed to follow mortality over such a period of time, according to the panel.
These limitations to the signal were especially important to the panel because of the obvious benefits with regard to quality of life provided to patients from these devices, which were attested to during the 2-day meeting by numerous presenters from industry, medical organizations – including societies and nonprofits – and providers.
In responding to FDA requests on a variety of concerns, the panel reiterated that there was a credible mortality signal, but that they could not be confident about the magnitude and whether it was caused by the paclitaxel treatment or some factor in the design or conduct of the studies. In addition, the panel members felt that they could neither confirm nor eliminate a class effect, given the fact that the information was based on a meta-analysis and thus none of the included devices could safely be removed from consideration.
They suggested that further safety information should be obtained, potentially by assessing and perhaps altering data collection in 29 ongoing studies over the next 5 years or so in more than 10,000 patients.
In addition, several of the panel members felt that additional animal studies might be performed including the use of older rat models; and using animal models that mimicked the kind of comorbidities present in the treated population, such as diabetes and atherosclerosis. They suggested cross-company industry cooperation with the FDA on these models, including looking at drug interactions and mimicking the dose application of stents/balloons.
Both the FDA representative and the panel were especially concerned with the benefit/risk profile.
The recommendation to still market the devices with a label warning was warranted, according to many members of the panel. They pointed to the clear benefits in quality of life and the lowered need for revascularization despite the evidence of the mortality signal, which, while statistically significant, could not be pinned town with regard to mechanisms or specific causes of death.
Overall, there was a concern that there should be a dialogue between patients and their doctors to discuss clear short-term benefits with unknown long-term risk, and that the label should support this by clearly mentioning the mortality signal that was found, although there was no attempt to develop exact wording.
Panel member Joaquin E. Cigarroa, MD, head of cardiovascular medicine at Oregon Health & Science University, Portland, suggested with regard to labeling that a statement “that ‘there may be’ – not ‘there is’ – a late mortality signal, should be included.”
Panel member John C. Somberg, MD, program director of clinical research and bioinformatics at Rush University in Lake Bluff, Ill., stated: “The label should say something like, ‘when looking at a meta-analysis that combined all studies with stents and balloons that carried paclitaxel, there may be a late mortality, which must be balanced against an early and sustained benefit in terms of pain on walking and potential loss of circulation to your extremity.’ ”
Other panel members thought that the meta-analysis should not be privileged and that somehow the totality of the evidence should somehow be distilled down into the label, including the evidence against the signal.
“We’re meeting because of a signal, of a concern – an honest, well-meaning concern – of increased mortality. And my opinion is that the patients need to be informed of it,” said Dr. Lange, president, Texas Tech University, El Paso.
Some members of the panel felt that it may not be justifiable to use these devices in patients with low intrinsic risk and low recurrence risk, and the whole spectrum of patients may need to be considered in further studies to figure out the subgroups that have more benefits and more risks, and also to consider how to mitigate risks in patients who receive the device, whether through medical therapy or lifestyle modification.
In particular, Frank W. LoGerfo, MD, the William V. McDermott Distinguished Professor of Surgery at Harvard Medical School, Boston, stated: “Interventions for claudication should be extremely rare. It rarely progresses, and the pain should be worked through by exercise with low risk of limb loss.” He added that intervention with these devices “takes away options. Trading life for something that is not limb-threatening is something we should not be considering.”
There was no firm consensus on whether new randomized trials should be done, although they were of course the ideal solution. Kevin E. Kip, PhD, Distinguished USF Health Professor at the University of South Florida, Tampa, and others argued that, whether new trials were necessary or not, to deal with the safety question in a timely fashion, existing trials have to capture as much of the missing data as possible, and carry out follow-up out further.
FDA representative Bram Zuckerman, MD, director of the Office of Cardiovascular Devices at the Center for Devices and Radiological Health, indicated that those things might not be easily be accomplished due to regulatory constraints and the financial costs, and that to do so there would be need for community effort among stakeholders, including collaborative efforts with existing prospective registries such as that run by the Vascular Quality Initiative.
One overall conclusion by both the FDA and panel members was that the quality of these and other such studies going forward must improve, by standardizing definitions and data forms to make studies more uniform across the industry. They reemphasized the need to work with the registries to get common data included, and to incorporate of insurance provider and Social Security death data as much as possible to help alleviate the lost follow-up problem.
SOURCE: Webcasts of the complete 2 days of the FDA panel meeting are available online.
GAITHERSBURG, MD. – There was sufficient evidence of a late mortality signal seen at 2-5 years post procedurally for paclitaxel-eluting stents and coated balloons to warrant a label change for the devices, the Food and Drug Administration’s Circulatory System Devices Panel unanimously agreed after 2 days of deliberation.
That signal was brought to light in a meta-analysis published last December by Konstantinos Katsanos, MD, of Patras University Hospital, Rion, Greece, and colleagues (J Am Heart Assoc. 2018;7:e011245). Although there were concerns about the quality of the industry data used in the study, the caliber of the analysis itself and the subsequent data presented by the FDA to the panel were deemed sufficient to recommend a warning of concern to patients and providers.
Much of the new data from industry and large database registries presented to the panel, which was chaired by Richard A. Lange, MD, indicated a lessening to no evidence of the mortality effect. But this evidence was deemed insufficient to counter the evidence of the randomized controlled trials individually and collectively as presented in the Katsanos meta-analysis and subsequent information presented by the FDA that examined various parameters in a variety of sensitivity analyses that confirmed the late mortality signal. There was also concern that the industry and the registry analyses presented were not peer reviewed.
However, the panel also determined that it would be inappropriate to pull the devices from the market and from general use for several reasons.
One key reason was that, according to the panel, there was no mechanistic cause apparent for the late mortality. In addition, no convincing dose-response data could be teased from the preclinical and clinical trials studied because of their variability of devices, application methods, and lack of appropriate tissue analysis across studies.
Finally, the industry data used to create the meta-analysis were considered to be fundamentally flawed: in blinding, in the relatively small numbers of patients, and in the large percentage of patients lost to follow-up. The latter could have dramatically influenced the perceived results, especially as the studies were not powered or designed to follow mortality over such a period of time, according to the panel.
These limitations to the signal were especially important to the panel because of the obvious benefits with regard to quality of life provided to patients from these devices, which were attested to during the 2-day meeting by numerous presenters from industry, medical organizations – including societies and nonprofits – and providers.
In responding to FDA requests on a variety of concerns, the panel reiterated that there was a credible mortality signal, but that they could not be confident about the magnitude and whether it was caused by the paclitaxel treatment or some factor in the design or conduct of the studies. In addition, the panel members felt that they could neither confirm nor eliminate a class effect, given the fact that the information was based on a meta-analysis and thus none of the included devices could safely be removed from consideration.
They suggested that further safety information should be obtained, potentially by assessing and perhaps altering data collection in 29 ongoing studies over the next 5 years or so in more than 10,000 patients.
In addition, several of the panel members felt that additional animal studies might be performed including the use of older rat models; and using animal models that mimicked the kind of comorbidities present in the treated population, such as diabetes and atherosclerosis. They suggested cross-company industry cooperation with the FDA on these models, including looking at drug interactions and mimicking the dose application of stents/balloons.
Both the FDA representative and the panel were especially concerned with the benefit/risk profile.
The recommendation to still market the devices with a label warning was warranted, according to many members of the panel. They pointed to the clear benefits in quality of life and the lowered need for revascularization despite the evidence of the mortality signal, which, while statistically significant, could not be pinned town with regard to mechanisms or specific causes of death.
Overall, there was a concern that there should be a dialogue between patients and their doctors to discuss clear short-term benefits with unknown long-term risk, and that the label should support this by clearly mentioning the mortality signal that was found, although there was no attempt to develop exact wording.
Panel member Joaquin E. Cigarroa, MD, head of cardiovascular medicine at Oregon Health & Science University, Portland, suggested with regard to labeling that a statement “that ‘there may be’ – not ‘there is’ – a late mortality signal, should be included.”
Panel member John C. Somberg, MD, program director of clinical research and bioinformatics at Rush University in Lake Bluff, Ill., stated: “The label should say something like, ‘when looking at a meta-analysis that combined all studies with stents and balloons that carried paclitaxel, there may be a late mortality, which must be balanced against an early and sustained benefit in terms of pain on walking and potential loss of circulation to your extremity.’ ”
Other panel members thought that the meta-analysis should not be privileged and that somehow the totality of the evidence should somehow be distilled down into the label, including the evidence against the signal.
“We’re meeting because of a signal, of a concern – an honest, well-meaning concern – of increased mortality. And my opinion is that the patients need to be informed of it,” said Dr. Lange, president, Texas Tech University, El Paso.
Some members of the panel felt that it may not be justifiable to use these devices in patients with low intrinsic risk and low recurrence risk, and the whole spectrum of patients may need to be considered in further studies to figure out the subgroups that have more benefits and more risks, and also to consider how to mitigate risks in patients who receive the device, whether through medical therapy or lifestyle modification.
In particular, Frank W. LoGerfo, MD, the William V. McDermott Distinguished Professor of Surgery at Harvard Medical School, Boston, stated: “Interventions for claudication should be extremely rare. It rarely progresses, and the pain should be worked through by exercise with low risk of limb loss.” He added that intervention with these devices “takes away options. Trading life for something that is not limb-threatening is something we should not be considering.”
There was no firm consensus on whether new randomized trials should be done, although they were of course the ideal solution. Kevin E. Kip, PhD, Distinguished USF Health Professor at the University of South Florida, Tampa, and others argued that, whether new trials were necessary or not, to deal with the safety question in a timely fashion, existing trials have to capture as much of the missing data as possible, and carry out follow-up out further.
FDA representative Bram Zuckerman, MD, director of the Office of Cardiovascular Devices at the Center for Devices and Radiological Health, indicated that those things might not be easily be accomplished due to regulatory constraints and the financial costs, and that to do so there would be need for community effort among stakeholders, including collaborative efforts with existing prospective registries such as that run by the Vascular Quality Initiative.
One overall conclusion by both the FDA and panel members was that the quality of these and other such studies going forward must improve, by standardizing definitions and data forms to make studies more uniform across the industry. They reemphasized the need to work with the registries to get common data included, and to incorporate of insurance provider and Social Security death data as much as possible to help alleviate the lost follow-up problem.
SOURCE: Webcasts of the complete 2 days of the FDA panel meeting are available online.
GAITHERSBURG, MD. – There was sufficient evidence of a late mortality signal seen at 2-5 years post procedurally for paclitaxel-eluting stents and coated balloons to warrant a label change for the devices, the Food and Drug Administration’s Circulatory System Devices Panel unanimously agreed after 2 days of deliberation.
That signal was brought to light in a meta-analysis published last December by Konstantinos Katsanos, MD, of Patras University Hospital, Rion, Greece, and colleagues (J Am Heart Assoc. 2018;7:e011245). Although there were concerns about the quality of the industry data used in the study, the caliber of the analysis itself and the subsequent data presented by the FDA to the panel were deemed sufficient to recommend a warning of concern to patients and providers.
Much of the new data from industry and large database registries presented to the panel, which was chaired by Richard A. Lange, MD, indicated a lessening to no evidence of the mortality effect. But this evidence was deemed insufficient to counter the evidence of the randomized controlled trials individually and collectively as presented in the Katsanos meta-analysis and subsequent information presented by the FDA that examined various parameters in a variety of sensitivity analyses that confirmed the late mortality signal. There was also concern that the industry and the registry analyses presented were not peer reviewed.
However, the panel also determined that it would be inappropriate to pull the devices from the market and from general use for several reasons.
One key reason was that, according to the panel, there was no mechanistic cause apparent for the late mortality. In addition, no convincing dose-response data could be teased from the preclinical and clinical trials studied because of their variability of devices, application methods, and lack of appropriate tissue analysis across studies.
Finally, the industry data used to create the meta-analysis were considered to be fundamentally flawed: in blinding, in the relatively small numbers of patients, and in the large percentage of patients lost to follow-up. The latter could have dramatically influenced the perceived results, especially as the studies were not powered or designed to follow mortality over such a period of time, according to the panel.
These limitations to the signal were especially important to the panel because of the obvious benefits with regard to quality of life provided to patients from these devices, which were attested to during the 2-day meeting by numerous presenters from industry, medical organizations – including societies and nonprofits – and providers.
In responding to FDA requests on a variety of concerns, the panel reiterated that there was a credible mortality signal, but that they could not be confident about the magnitude and whether it was caused by the paclitaxel treatment or some factor in the design or conduct of the studies. In addition, the panel members felt that they could neither confirm nor eliminate a class effect, given the fact that the information was based on a meta-analysis and thus none of the included devices could safely be removed from consideration.
They suggested that further safety information should be obtained, potentially by assessing and perhaps altering data collection in 29 ongoing studies over the next 5 years or so in more than 10,000 patients.
In addition, several of the panel members felt that additional animal studies might be performed including the use of older rat models; and using animal models that mimicked the kind of comorbidities present in the treated population, such as diabetes and atherosclerosis. They suggested cross-company industry cooperation with the FDA on these models, including looking at drug interactions and mimicking the dose application of stents/balloons.
Both the FDA representative and the panel were especially concerned with the benefit/risk profile.
The recommendation to still market the devices with a label warning was warranted, according to many members of the panel. They pointed to the clear benefits in quality of life and the lowered need for revascularization despite the evidence of the mortality signal, which, while statistically significant, could not be pinned town with regard to mechanisms or specific causes of death.
Overall, there was a concern that there should be a dialogue between patients and their doctors to discuss clear short-term benefits with unknown long-term risk, and that the label should support this by clearly mentioning the mortality signal that was found, although there was no attempt to develop exact wording.
Panel member Joaquin E. Cigarroa, MD, head of cardiovascular medicine at Oregon Health & Science University, Portland, suggested with regard to labeling that a statement “that ‘there may be’ – not ‘there is’ – a late mortality signal, should be included.”
Panel member John C. Somberg, MD, program director of clinical research and bioinformatics at Rush University in Lake Bluff, Ill., stated: “The label should say something like, ‘when looking at a meta-analysis that combined all studies with stents and balloons that carried paclitaxel, there may be a late mortality, which must be balanced against an early and sustained benefit in terms of pain on walking and potential loss of circulation to your extremity.’ ”
Other panel members thought that the meta-analysis should not be privileged and that somehow the totality of the evidence should somehow be distilled down into the label, including the evidence against the signal.
“We’re meeting because of a signal, of a concern – an honest, well-meaning concern – of increased mortality. And my opinion is that the patients need to be informed of it,” said Dr. Lange, president, Texas Tech University, El Paso.
Some members of the panel felt that it may not be justifiable to use these devices in patients with low intrinsic risk and low recurrence risk, and the whole spectrum of patients may need to be considered in further studies to figure out the subgroups that have more benefits and more risks, and also to consider how to mitigate risks in patients who receive the device, whether through medical therapy or lifestyle modification.
In particular, Frank W. LoGerfo, MD, the William V. McDermott Distinguished Professor of Surgery at Harvard Medical School, Boston, stated: “Interventions for claudication should be extremely rare. It rarely progresses, and the pain should be worked through by exercise with low risk of limb loss.” He added that intervention with these devices “takes away options. Trading life for something that is not limb-threatening is something we should not be considering.”
There was no firm consensus on whether new randomized trials should be done, although they were of course the ideal solution. Kevin E. Kip, PhD, Distinguished USF Health Professor at the University of South Florida, Tampa, and others argued that, whether new trials were necessary or not, to deal with the safety question in a timely fashion, existing trials have to capture as much of the missing data as possible, and carry out follow-up out further.
FDA representative Bram Zuckerman, MD, director of the Office of Cardiovascular Devices at the Center for Devices and Radiological Health, indicated that those things might not be easily be accomplished due to regulatory constraints and the financial costs, and that to do so there would be need for community effort among stakeholders, including collaborative efforts with existing prospective registries such as that run by the Vascular Quality Initiative.
One overall conclusion by both the FDA and panel members was that the quality of these and other such studies going forward must improve, by standardizing definitions and data forms to make studies more uniform across the industry. They reemphasized the need to work with the registries to get common data included, and to incorporate of insurance provider and Social Security death data as much as possible to help alleviate the lost follow-up problem.
SOURCE: Webcasts of the complete 2 days of the FDA panel meeting are available online.
REPORTING FROM AN FDA PANEL MEETING
Liraglutide indication expanded to include children
according to a news release from the agency. The approval comes after granting the application Priority Review, which means the FDA aimed to take action on it within 6 months instead of the usual 10 months seen with a Standard Review.
Liraglutide injection has been approved for treatment of adults with type 2 diabetes since 2010, but this is the first noninsulin treatment for pediatric patients since metformin received approval for this population in 2000.
“The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with [type 2 diabetes],” said Lisa Yanoff, MD, acting director of the division of metabolism and endocrinology products in the agency’s Center for Drug Evaluation and Research.
The approval is based on efficacy and safety results from several placebo-controlled trials in adults, and one trial in 134 pediatric patients aged 10 years or older. In the latter trial, which ran for more than 26 weeks, hemoglobin A1c levels fell below 7% in approximately 64% of patients treated with liraglutide injection, compared with 37% of those who received placebo. Those results were seen regardless of whether patients concomitantly used insulin.
Although an increase in hypoglycemia risk has sometimes been seen in adult patients taking liraglutide injection with insulin or other drugs that raise insulin production, this heightened risk was seen in the pediatric patients regardless of whether they took other therapies for diabetes.
Liraglutide injection carries a Boxed Warning, the FDA’s strongest warning, for heightened risk of thyroid C-cell tumors. Therefore, the agency recommends against patients with history or family members with history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2 using this treatment. Among its other warnings are those pertaining to patients with renal impairment or kidney failure, pancreatitis, and/or acute gallbladder disease.
The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion, and constipation. The full prescribing information can be found on the FDA website.
according to a news release from the agency. The approval comes after granting the application Priority Review, which means the FDA aimed to take action on it within 6 months instead of the usual 10 months seen with a Standard Review.
Liraglutide injection has been approved for treatment of adults with type 2 diabetes since 2010, but this is the first noninsulin treatment for pediatric patients since metformin received approval for this population in 2000.
“The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with [type 2 diabetes],” said Lisa Yanoff, MD, acting director of the division of metabolism and endocrinology products in the agency’s Center for Drug Evaluation and Research.
The approval is based on efficacy and safety results from several placebo-controlled trials in adults, and one trial in 134 pediatric patients aged 10 years or older. In the latter trial, which ran for more than 26 weeks, hemoglobin A1c levels fell below 7% in approximately 64% of patients treated with liraglutide injection, compared with 37% of those who received placebo. Those results were seen regardless of whether patients concomitantly used insulin.
Although an increase in hypoglycemia risk has sometimes been seen in adult patients taking liraglutide injection with insulin or other drugs that raise insulin production, this heightened risk was seen in the pediatric patients regardless of whether they took other therapies for diabetes.
Liraglutide injection carries a Boxed Warning, the FDA’s strongest warning, for heightened risk of thyroid C-cell tumors. Therefore, the agency recommends against patients with history or family members with history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2 using this treatment. Among its other warnings are those pertaining to patients with renal impairment or kidney failure, pancreatitis, and/or acute gallbladder disease.
The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion, and constipation. The full prescribing information can be found on the FDA website.
according to a news release from the agency. The approval comes after granting the application Priority Review, which means the FDA aimed to take action on it within 6 months instead of the usual 10 months seen with a Standard Review.
Liraglutide injection has been approved for treatment of adults with type 2 diabetes since 2010, but this is the first noninsulin treatment for pediatric patients since metformin received approval for this population in 2000.
“The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with [type 2 diabetes],” said Lisa Yanoff, MD, acting director of the division of metabolism and endocrinology products in the agency’s Center for Drug Evaluation and Research.
The approval is based on efficacy and safety results from several placebo-controlled trials in adults, and one trial in 134 pediatric patients aged 10 years or older. In the latter trial, which ran for more than 26 weeks, hemoglobin A1c levels fell below 7% in approximately 64% of patients treated with liraglutide injection, compared with 37% of those who received placebo. Those results were seen regardless of whether patients concomitantly used insulin.
Although an increase in hypoglycemia risk has sometimes been seen in adult patients taking liraglutide injection with insulin or other drugs that raise insulin production, this heightened risk was seen in the pediatric patients regardless of whether they took other therapies for diabetes.
Liraglutide injection carries a Boxed Warning, the FDA’s strongest warning, for heightened risk of thyroid C-cell tumors. Therefore, the agency recommends against patients with history or family members with history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2 using this treatment. Among its other warnings are those pertaining to patients with renal impairment or kidney failure, pancreatitis, and/or acute gallbladder disease.
The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion, and constipation. The full prescribing information can be found on the FDA website.
FDA approves pembrolizumab for advanced SCLC
The Food and Drug Administration has granted accelerated approval to pembrolizumab for patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy.
Approval was based on an overall response rate of 19% among 83 patients with SCLC who had disease progression on or after two or more prior lines of therapy enrolled in two nonrandomized trials, according to the FDA.
SCLC cohorts in KEYNOTE-028 and KEYNOTE-158 received either pembrolizumab 200 mg intravenously every 3 weeks (n = 64) or 10 mg/kg intravenously every 2 weeks (n = 19). Treatment continued until documented disease progression, unacceptable toxicity, or for a maximum of 24 months.
The ORR was 19% (95% confidence interval, 11%-29%), while the complete response rate was 2%. Responses were durable for 6 months or longer in 94% of the 16 responding patients.
Common adverse reactions included fatigue, decreased appetite, cough, nausea, and constipation. The most frequent serious adverse reactions were pneumonia and pleural effusion.
The recommended dosage for SCLC treatment is 200 mg, administered as an intravenous infusion over 30 minutes every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression, the FDA said.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to pembrolizumab for patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy.
Approval was based on an overall response rate of 19% among 83 patients with SCLC who had disease progression on or after two or more prior lines of therapy enrolled in two nonrandomized trials, according to the FDA.
SCLC cohorts in KEYNOTE-028 and KEYNOTE-158 received either pembrolizumab 200 mg intravenously every 3 weeks (n = 64) or 10 mg/kg intravenously every 2 weeks (n = 19). Treatment continued until documented disease progression, unacceptable toxicity, or for a maximum of 24 months.
The ORR was 19% (95% confidence interval, 11%-29%), while the complete response rate was 2%. Responses were durable for 6 months or longer in 94% of the 16 responding patients.
Common adverse reactions included fatigue, decreased appetite, cough, nausea, and constipation. The most frequent serious adverse reactions were pneumonia and pleural effusion.
The recommended dosage for SCLC treatment is 200 mg, administered as an intravenous infusion over 30 minutes every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression, the FDA said.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to pembrolizumab for patients with metastatic small cell lung cancer (SCLC) with disease progression on or after platinum-based chemotherapy and at least one other prior line of therapy.
Approval was based on an overall response rate of 19% among 83 patients with SCLC who had disease progression on or after two or more prior lines of therapy enrolled in two nonrandomized trials, according to the FDA.
SCLC cohorts in KEYNOTE-028 and KEYNOTE-158 received either pembrolizumab 200 mg intravenously every 3 weeks (n = 64) or 10 mg/kg intravenously every 2 weeks (n = 19). Treatment continued until documented disease progression, unacceptable toxicity, or for a maximum of 24 months.
The ORR was 19% (95% confidence interval, 11%-29%), while the complete response rate was 2%. Responses were durable for 6 months or longer in 94% of the 16 responding patients.
Common adverse reactions included fatigue, decreased appetite, cough, nausea, and constipation. The most frequent serious adverse reactions were pneumonia and pleural effusion.
The recommended dosage for SCLC treatment is 200 mg, administered as an intravenous infusion over 30 minutes every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression, the FDA said.
Pembrolizumab is marketed as Keytruda by Merck.
Obesity and overweight declined among lower-income kids
The combined rate of , according to a study in JAMA.
Liping Pan, MD, MPH, of the Centers for Disease Control and Prevention and colleagues used data from the WIC Participant and Program Characteristics survey from 2010, 2012, 2014, and 2016 for 12,403,629 children aged 2-4 years from 50 states, Washington, D.C., and 5 territories. In addition to a –3.2% change (95% confidence interval, –3.3% to –3.2%) in adjusted prevalence difference for the combined rate of obesity and overweight seen between 2010 and 2016, the researchers found the crude prevalence decreased from 32.5% to 29.1%. A decrease was also seen for obesity alone (crude prevalence, 15.9% to 13.9%; adjusted prevalence difference, –1.9%; 95% CI, –1.9% to –1.8%).
One of the limitations of the study is that the characteristics of enrolled children might differ from those of children not enrolled in this WIC program; however, the researchers noted that they accounted for many demographic characteristics in the trend analyses.
“Reasons for the declines in obesity among young children in WIC remain undetermined but may include WIC food package revisions and local, state, and national initiatives,” they wrote.
SOURCE: Pan L et al. JAMA. 2019 Jun 18;321(23):2364-6.
The combined rate of , according to a study in JAMA.
Liping Pan, MD, MPH, of the Centers for Disease Control and Prevention and colleagues used data from the WIC Participant and Program Characteristics survey from 2010, 2012, 2014, and 2016 for 12,403,629 children aged 2-4 years from 50 states, Washington, D.C., and 5 territories. In addition to a –3.2% change (95% confidence interval, –3.3% to –3.2%) in adjusted prevalence difference for the combined rate of obesity and overweight seen between 2010 and 2016, the researchers found the crude prevalence decreased from 32.5% to 29.1%. A decrease was also seen for obesity alone (crude prevalence, 15.9% to 13.9%; adjusted prevalence difference, –1.9%; 95% CI, –1.9% to –1.8%).
One of the limitations of the study is that the characteristics of enrolled children might differ from those of children not enrolled in this WIC program; however, the researchers noted that they accounted for many demographic characteristics in the trend analyses.
“Reasons for the declines in obesity among young children in WIC remain undetermined but may include WIC food package revisions and local, state, and national initiatives,” they wrote.
SOURCE: Pan L et al. JAMA. 2019 Jun 18;321(23):2364-6.
The combined rate of , according to a study in JAMA.
Liping Pan, MD, MPH, of the Centers for Disease Control and Prevention and colleagues used data from the WIC Participant and Program Characteristics survey from 2010, 2012, 2014, and 2016 for 12,403,629 children aged 2-4 years from 50 states, Washington, D.C., and 5 territories. In addition to a –3.2% change (95% confidence interval, –3.3% to –3.2%) in adjusted prevalence difference for the combined rate of obesity and overweight seen between 2010 and 2016, the researchers found the crude prevalence decreased from 32.5% to 29.1%. A decrease was also seen for obesity alone (crude prevalence, 15.9% to 13.9%; adjusted prevalence difference, –1.9%; 95% CI, –1.9% to –1.8%).
One of the limitations of the study is that the characteristics of enrolled children might differ from those of children not enrolled in this WIC program; however, the researchers noted that they accounted for many demographic characteristics in the trend analyses.
“Reasons for the declines in obesity among young children in WIC remain undetermined but may include WIC food package revisions and local, state, and national initiatives,” they wrote.
SOURCE: Pan L et al. JAMA. 2019 Jun 18;321(23):2364-6.
FROM JAMA
FDA warns about fecal microbiota for transplantation
Officials at the Food and Drug Administration have issued a safety alert regarding the use of fecal microbiota for transplantation and the risk of serious adverse reactions because of transmission of multidrug-resistant organisms (MDROs).

According to the alert, which was issued on June 13, 2019, the agency became aware of two immunocompromised adult patients who received investigational fecal microbiota for transplantation (FMT) and developed extended-spectrum beta-lactamase (EBSL)–producing Escherichia coli. One of the patients died.
“This is certainly a theoretical risk that we’ve known about,” Lea Ann Chen, MD, a gastroenterologist at New York University, said in an interview. “This announcement is important, because we probably don’t counsel patients specifically about this risk. We say there is a risk for transmission of infectious agents in general, but I think that probably very few counsel patients about a risk for transmission of MDROs.”
The donor stool and FMT used in the two patients were not tested for ESBL-producing gram-negative organisms prior to use.
As a result of these serious adverse reactions, the FDA has determined that the following donor screening and stool testing protections are needed for any investigational use of FMT.
- Donor screening with questions that specifically address risk factors for colonization with MDROs, and exclusion of individuals at higher risk of colonization with MDROs.
- MDRO testing of donor stool and exclusion of stool that tests positive for MDRO. FDA scientists have determined the specific MDRO testing and frequency that should be implemented.
On June 14, the American Gastroenterological Association sent a communication about the FDA alert to its members, which stated that the AGA “is committed to advancing applications of the gut microbiome. Our top priority is ensuring patient safety from microbiome-based therapeutics, such as FMT. Through the AGA FMT National Registry, AGA is working with physicians and patients to track FMT usage, patient outcomes and adverse events. Associated with the registry is a biorepository of donor and patient stool samples, which will allow further investigation of unexpected events such as those described in FDA’s safety alert.”
Dr. Chen, who received the AGA Research Foundation’s 2016 Research Scholar Award for her work on the gut microbiome and inflammatory bowel disease, pointed out that FMT has also been studied as a way to prevent colonization and infection with certain drug resistant organisms, such as vancomycin-resistant Enterococcus.
“Therefore, it’s not that FMT is ‘bad;’ we just have to be more diligent about optimizing the safety of the procedure by screening for of multidrug-resistant organisms,” she said. “We also need to study the use of FMT more, so that we can fully understand the risks associated with the procedure. It’s an important and potentially lifesaving procedure for some, but it’s important that everyone go into the procedure understanding fully what the risks and benefits are.”
Suspected adverse events related to the administration of FMT products can be reported to the FDA at 1-800-332-1088 or via MedWatch.
Officials at the Food and Drug Administration have issued a safety alert regarding the use of fecal microbiota for transplantation and the risk of serious adverse reactions because of transmission of multidrug-resistant organisms (MDROs).
According to the alert, which was issued on June 13, 2019, the agency became aware of two immunocompromised adult patients who received investigational fecal microbiota for transplantation (FMT) and developed extended-spectrum beta-lactamase (EBSL)–producing Escherichia coli. One of the patients died.
“This is certainly a theoretical risk that we’ve known about,” Lea Ann Chen, MD, a gastroenterologist at New York University, said in an interview. “This announcement is important, because we probably don’t counsel patients specifically about this risk. We say there is a risk for transmission of infectious agents in general, but I think that probably very few counsel patients about a risk for transmission of MDROs.”
The donor stool and FMT used in the two patients were not tested for ESBL-producing gram-negative organisms prior to use.
As a result of these serious adverse reactions, the FDA has determined that the following donor screening and stool testing protections are needed for any investigational use of FMT.
- Donor screening with questions that specifically address risk factors for colonization with MDROs, and exclusion of individuals at higher risk of colonization with MDROs.
- MDRO testing of donor stool and exclusion of stool that tests positive for MDRO. FDA scientists have determined the specific MDRO testing and frequency that should be implemented.
On June 14, the American Gastroenterological Association sent a communication about the FDA alert to its members, which stated that the AGA “is committed to advancing applications of the gut microbiome. Our top priority is ensuring patient safety from microbiome-based therapeutics, such as FMT. Through the AGA FMT National Registry, AGA is working with physicians and patients to track FMT usage, patient outcomes and adverse events. Associated with the registry is a biorepository of donor and patient stool samples, which will allow further investigation of unexpected events such as those described in FDA’s safety alert.”
Dr. Chen, who received the AGA Research Foundation’s 2016 Research Scholar Award for her work on the gut microbiome and inflammatory bowel disease, pointed out that FMT has also been studied as a way to prevent colonization and infection with certain drug resistant organisms, such as vancomycin-resistant Enterococcus.
“Therefore, it’s not that FMT is ‘bad;’ we just have to be more diligent about optimizing the safety of the procedure by screening for of multidrug-resistant organisms,” she said. “We also need to study the use of FMT more, so that we can fully understand the risks associated with the procedure. It’s an important and potentially lifesaving procedure for some, but it’s important that everyone go into the procedure understanding fully what the risks and benefits are.”
Suspected adverse events related to the administration of FMT products can be reported to the FDA at 1-800-332-1088 or via MedWatch.
Officials at the Food and Drug Administration have issued a safety alert regarding the use of fecal microbiota for transplantation and the risk of serious adverse reactions because of transmission of multidrug-resistant organisms (MDROs).
According to the alert, which was issued on June 13, 2019, the agency became aware of two immunocompromised adult patients who received investigational fecal microbiota for transplantation (FMT) and developed extended-spectrum beta-lactamase (EBSL)–producing Escherichia coli. One of the patients died.
“This is certainly a theoretical risk that we’ve known about,” Lea Ann Chen, MD, a gastroenterologist at New York University, said in an interview. “This announcement is important, because we probably don’t counsel patients specifically about this risk. We say there is a risk for transmission of infectious agents in general, but I think that probably very few counsel patients about a risk for transmission of MDROs.”
The donor stool and FMT used in the two patients were not tested for ESBL-producing gram-negative organisms prior to use.
As a result of these serious adverse reactions, the FDA has determined that the following donor screening and stool testing protections are needed for any investigational use of FMT.
- Donor screening with questions that specifically address risk factors for colonization with MDROs, and exclusion of individuals at higher risk of colonization with MDROs.
- MDRO testing of donor stool and exclusion of stool that tests positive for MDRO. FDA scientists have determined the specific MDRO testing and frequency that should be implemented.
On June 14, the American Gastroenterological Association sent a communication about the FDA alert to its members, which stated that the AGA “is committed to advancing applications of the gut microbiome. Our top priority is ensuring patient safety from microbiome-based therapeutics, such as FMT. Through the AGA FMT National Registry, AGA is working with physicians and patients to track FMT usage, patient outcomes and adverse events. Associated with the registry is a biorepository of donor and patient stool samples, which will allow further investigation of unexpected events such as those described in FDA’s safety alert.”
Dr. Chen, who received the AGA Research Foundation’s 2016 Research Scholar Award for her work on the gut microbiome and inflammatory bowel disease, pointed out that FMT has also been studied as a way to prevent colonization and infection with certain drug resistant organisms, such as vancomycin-resistant Enterococcus.
“Therefore, it’s not that FMT is ‘bad;’ we just have to be more diligent about optimizing the safety of the procedure by screening for of multidrug-resistant organisms,” she said. “We also need to study the use of FMT more, so that we can fully understand the risks associated with the procedure. It’s an important and potentially lifesaving procedure for some, but it’s important that everyone go into the procedure understanding fully what the risks and benefits are.”
Suspected adverse events related to the administration of FMT products can be reported to the FDA at 1-800-332-1088 or via MedWatch.
U.S. travelers to Europe need up to date measles immunization
researchers at the Centers for Disease Control and Prevention recommend in a Pediatrics special report.
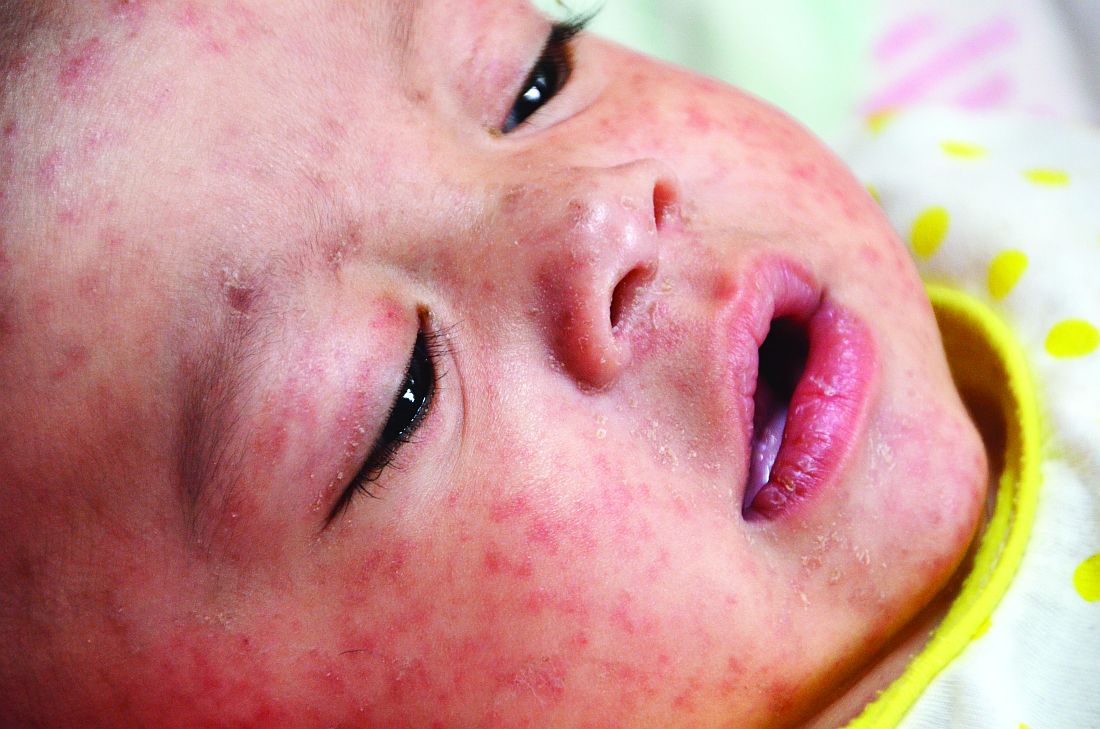
More than 41,000 measles cases and 37 deaths – primarily due to low immunization coverage – were reported in the World Health Organization European Region in the first 6 months of 2018, the highest incidence since the 1990s. Typical case counts since 2010 have ranged from 5,000 to 24,000 in this region, wrote Kristina M. Angelo, DO, MPH, of the Centers for Disease Control and Prevention Travelers’ Health Branch in Atlanta, and associates.
France, Italy and Greece – all particularly popular countries for U.S. vacationers to visit – have particularly high numbers of cases, as do Georgia, Russia, Serbia and, comprising the majority of cases, Ukraine. Italy, for example, is the 10th most popular destination worldwide for Americans, with an estimated 2.5 million American visitors in 2015.
“The large number of measles infections in the WHO European Region ... is a global concern because the European continent is the most common travel destination worldwide,” but is not perceived as a place with infectious disease risk. So travelers may not consider the need of a pretravel health consultation, including vaccination, they said.
But they need to, Dr. Angelo and associates state, and health care providers should be vigilant about checking for symptoms of measles among those who have recently returned from overseas. Given how highly contagious measles is, unvaccinated and under vaccinated travelers to Europe are susceptible to infection, as are any people they encounter back in the United States if the travelers come home sick.
Measles was eliminated in the United States in 2000, but that status is in jeopardy, CDC officials recently warned. The number of domestic measles cases has exceeded 1,000 just halfway through 2019, the highest count since 1992, nearly a decade before elimination.
“Avoiding international travel with nonimmune infants and performing early vaccination at 6 to 12 months of age per the ACIP [Advisory Committee on Immunization Practices] recommendations if travel is unavoidable are of utmost importance,” Dr. Angelo and colleagues advised. “Other at-risk populations (e.g., immunocompromised individuals and pregnant women), for whom vaccination against the measles virus is contraindicated, may consider alternative destinations or delay travel to measles-endemic destinations or areas with known, ongoing measles outbreaks.”
“Presumptive immunity to measles is defined as 1 or more of the following: birth before 1957, laboratory evidence of immunity or infection, 1 or more doses of a measles containing vaccine administered for preschool-aged children and low-risk adults, or 2 doses of measles vaccine among school-aged children and high-risk adults, including international travelers,” they explained.
In Europe, measles remains endemic in Belgium, Bosnia and Herzegovina, France, Georgia, Germany, Italy, Romania, the Russian Federation, Serbia and the Ukraine, the authors wrote.
“As long as measles remains endemic in other countries, the United States will be challenged by measles importations,” the authors wrote. Yet at least one past study in 2017 revealed a third of U.S. travelers to Europe left the country without being fully vaccinated against measles, most often due to vaccine refusal.
“The reason one-third of travelers to Europe missed an opportunity for measles vaccination remains unclear,” the authors wrote. “It may represent a lack of concern or awareness on the part of travelers and the health care providers about acquiring measles in Europe.”
Dr. Angelo and colleagues also emphasized the importance of returning U.S. travelers seeking health care if they have symptoms of measles, including fever and a rash.
Health care providers should ask all patients about recent international travel, they stated. “If measles is suspected, health care providers should isolate travelers immediately, placing them on airborne precautions until day 4 of the rash.” Providers may consider administering immunoglobulin for unvaccinated and undervaccinated travelers and monitor them for 21 days for development of measles symptoms.
The statement was funded by the CDC. The authors reported no relevant financial disclosures.
SOURCE: Angelo KM et al. Pediatrics. 2019 Jun 17. doi: /10.1542/peds.2019-0414.
researchers at the Centers for Disease Control and Prevention recommend in a Pediatrics special report.
More than 41,000 measles cases and 37 deaths – primarily due to low immunization coverage – were reported in the World Health Organization European Region in the first 6 months of 2018, the highest incidence since the 1990s. Typical case counts since 2010 have ranged from 5,000 to 24,000 in this region, wrote Kristina M. Angelo, DO, MPH, of the Centers for Disease Control and Prevention Travelers’ Health Branch in Atlanta, and associates.
France, Italy and Greece – all particularly popular countries for U.S. vacationers to visit – have particularly high numbers of cases, as do Georgia, Russia, Serbia and, comprising the majority of cases, Ukraine. Italy, for example, is the 10th most popular destination worldwide for Americans, with an estimated 2.5 million American visitors in 2015.
“The large number of measles infections in the WHO European Region ... is a global concern because the European continent is the most common travel destination worldwide,” but is not perceived as a place with infectious disease risk. So travelers may not consider the need of a pretravel health consultation, including vaccination, they said.
But they need to, Dr. Angelo and associates state, and health care providers should be vigilant about checking for symptoms of measles among those who have recently returned from overseas. Given how highly contagious measles is, unvaccinated and under vaccinated travelers to Europe are susceptible to infection, as are any people they encounter back in the United States if the travelers come home sick.
Measles was eliminated in the United States in 2000, but that status is in jeopardy, CDC officials recently warned. The number of domestic measles cases has exceeded 1,000 just halfway through 2019, the highest count since 1992, nearly a decade before elimination.
“Avoiding international travel with nonimmune infants and performing early vaccination at 6 to 12 months of age per the ACIP [Advisory Committee on Immunization Practices] recommendations if travel is unavoidable are of utmost importance,” Dr. Angelo and colleagues advised. “Other at-risk populations (e.g., immunocompromised individuals and pregnant women), for whom vaccination against the measles virus is contraindicated, may consider alternative destinations or delay travel to measles-endemic destinations or areas with known, ongoing measles outbreaks.”
“Presumptive immunity to measles is defined as 1 or more of the following: birth before 1957, laboratory evidence of immunity or infection, 1 or more doses of a measles containing vaccine administered for preschool-aged children and low-risk adults, or 2 doses of measles vaccine among school-aged children and high-risk adults, including international travelers,” they explained.
In Europe, measles remains endemic in Belgium, Bosnia and Herzegovina, France, Georgia, Germany, Italy, Romania, the Russian Federation, Serbia and the Ukraine, the authors wrote.
“As long as measles remains endemic in other countries, the United States will be challenged by measles importations,” the authors wrote. Yet at least one past study in 2017 revealed a third of U.S. travelers to Europe left the country without being fully vaccinated against measles, most often due to vaccine refusal.
“The reason one-third of travelers to Europe missed an opportunity for measles vaccination remains unclear,” the authors wrote. “It may represent a lack of concern or awareness on the part of travelers and the health care providers about acquiring measles in Europe.”
Dr. Angelo and colleagues also emphasized the importance of returning U.S. travelers seeking health care if they have symptoms of measles, including fever and a rash.
Health care providers should ask all patients about recent international travel, they stated. “If measles is suspected, health care providers should isolate travelers immediately, placing them on airborne precautions until day 4 of the rash.” Providers may consider administering immunoglobulin for unvaccinated and undervaccinated travelers and monitor them for 21 days for development of measles symptoms.
The statement was funded by the CDC. The authors reported no relevant financial disclosures.
SOURCE: Angelo KM et al. Pediatrics. 2019 Jun 17. doi: /10.1542/peds.2019-0414.
researchers at the Centers for Disease Control and Prevention recommend in a Pediatrics special report.
More than 41,000 measles cases and 37 deaths – primarily due to low immunization coverage – were reported in the World Health Organization European Region in the first 6 months of 2018, the highest incidence since the 1990s. Typical case counts since 2010 have ranged from 5,000 to 24,000 in this region, wrote Kristina M. Angelo, DO, MPH, of the Centers for Disease Control and Prevention Travelers’ Health Branch in Atlanta, and associates.
France, Italy and Greece – all particularly popular countries for U.S. vacationers to visit – have particularly high numbers of cases, as do Georgia, Russia, Serbia and, comprising the majority of cases, Ukraine. Italy, for example, is the 10th most popular destination worldwide for Americans, with an estimated 2.5 million American visitors in 2015.
“The large number of measles infections in the WHO European Region ... is a global concern because the European continent is the most common travel destination worldwide,” but is not perceived as a place with infectious disease risk. So travelers may not consider the need of a pretravel health consultation, including vaccination, they said.
But they need to, Dr. Angelo and associates state, and health care providers should be vigilant about checking for symptoms of measles among those who have recently returned from overseas. Given how highly contagious measles is, unvaccinated and under vaccinated travelers to Europe are susceptible to infection, as are any people they encounter back in the United States if the travelers come home sick.
Measles was eliminated in the United States in 2000, but that status is in jeopardy, CDC officials recently warned. The number of domestic measles cases has exceeded 1,000 just halfway through 2019, the highest count since 1992, nearly a decade before elimination.
“Avoiding international travel with nonimmune infants and performing early vaccination at 6 to 12 months of age per the ACIP [Advisory Committee on Immunization Practices] recommendations if travel is unavoidable are of utmost importance,” Dr. Angelo and colleagues advised. “Other at-risk populations (e.g., immunocompromised individuals and pregnant women), for whom vaccination against the measles virus is contraindicated, may consider alternative destinations or delay travel to measles-endemic destinations or areas with known, ongoing measles outbreaks.”
“Presumptive immunity to measles is defined as 1 or more of the following: birth before 1957, laboratory evidence of immunity or infection, 1 or more doses of a measles containing vaccine administered for preschool-aged children and low-risk adults, or 2 doses of measles vaccine among school-aged children and high-risk adults, including international travelers,” they explained.
In Europe, measles remains endemic in Belgium, Bosnia and Herzegovina, France, Georgia, Germany, Italy, Romania, the Russian Federation, Serbia and the Ukraine, the authors wrote.
“As long as measles remains endemic in other countries, the United States will be challenged by measles importations,” the authors wrote. Yet at least one past study in 2017 revealed a third of U.S. travelers to Europe left the country without being fully vaccinated against measles, most often due to vaccine refusal.
“The reason one-third of travelers to Europe missed an opportunity for measles vaccination remains unclear,” the authors wrote. “It may represent a lack of concern or awareness on the part of travelers and the health care providers about acquiring measles in Europe.”
Dr. Angelo and colleagues also emphasized the importance of returning U.S. travelers seeking health care if they have symptoms of measles, including fever and a rash.
Health care providers should ask all patients about recent international travel, they stated. “If measles is suspected, health care providers should isolate travelers immediately, placing them on airborne precautions until day 4 of the rash.” Providers may consider administering immunoglobulin for unvaccinated and undervaccinated travelers and monitor them for 21 days for development of measles symptoms.
The statement was funded by the CDC. The authors reported no relevant financial disclosures.
SOURCE: Angelo KM et al. Pediatrics. 2019 Jun 17. doi: /10.1542/peds.2019-0414.
FROM PEDIATRICS