User login
Mobile Medical Apps for Patient Education: A Graded Review of Available Dermatology Apps
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
Practice Points
- Mobile dermatology apps for educational purposes should be objectively reviewed before being used by patients.
- In our study, only 9 (20.5%) of the 44 dermatology apps evaluated were considered adequate for patient information based on our grading criteria.
VIDEO: Rivaroxaban plus aspirin halves ischemic strokes
LOS ANGELES – Combined treatment with a low dosage of the anticoagulant rivaroxaban plus aspirin cut the incidence of ischemic strokes nearly in half, compared with aspirin alone, in a multicenter, randomized trial of more than 27,000 patients with stable atherosclerotic vascular disease.
This dramatic reduction in ischemic strokes as well as in all-cause strokes by adding low-dose rivaroxaban(Xarelto) occurred without any significant increase in hemorrhagic strokes but with a small increase in total major bleeding events, such as gastrointestinal bleeds, Mike Sharma, MD, said at the International Stroke Conference, sponsored by the American Heart Association.
“There was a consistent effect across all strata of stroke risk. For patients who had a prior stroke, it’s pretty clear to use rivaroxaban plus aspirin because it had a big benefit” with no increase in intracranial hemorrhages, Dr. Sharma said in a video interview.
“We think these results will fundamentally change how we approach stroke prevention,” added Dr. Sharma, a stroke neurologist in the Population Health Research Institute of McMaster University in Hamilton, Ont.
The results he reported came from a secondary analysis of data collected in the COMPASS (Rivaroxaban for the Prevention of Major Cardiovascular Events in Coronary or Peripheral Artery Disease) trial, which enrolled 27,395 patients with stable coronary or peripheral artery disease at 602 centers in 33 countries.
The primary outcome of the trial, reported in 2017, was the combined rate of cardiovascular death, MI, or stroke during an average 23 months of follow-up, which occurred in 4.1% of patients treated with 2.5 mg rivaroxaban twice daily plus 100 mg aspirin once daily, 4.9% of patients who received 5.0 mg rivaroxaban twice daily, and 5.4% in patients who received 100 mg aspirin daily, a statistically significant 24% relative risk reduction in the combined treatment group, compared with aspirin only. The rivaroxaban only–treated patients did not significantly differ from the control patients who received only aspirin (N Engl J Med. 2017 Oct 5;377[14]:1319-30). The main results showed a 1.2% increase in the rate of major bleeds in patients treated with rivaroxaban plus aspirin, compared with aspirin only, but the rate of nonfatal symptomatic intracranial hemorrhages was identical in the two treatment groups.
The new results Dr. Sharma reported at the conference focused on various measures of stroke. The rate of all strokes was 42% lower among the patients treated with rivaroxaban plus aspirin, compared with the aspirin alone patients, and ischemic strokes were 49% lower with the dual therapy, compared with aspirin only. Both differences were statistically significant. In contrast, the rivaroxaban alone regimen did not significantly reduce all-cause strokes. It did significantly reduce ischemic strokes, compared with aspirin only, but it also significantly increased hemorrhagic strokes, compared with aspirin only, an adverse effect not caused by the combination of low-dose rivaroxaban plus aspirin.
Rivaroxaban plus aspirin surpassed aspirin alone for preventing both mild and severe strokes and for preventing strokes both in patients with a history of a prior stroke and in those who never had a prior stroke. The stroke reduction produced by rivaroxaban plus aspirin was greatest in the highest risk patients – those with a prior stroke. On the combined regimen, these patients had an average stroke incidence of 0.7% per year, compared with an annual 3.4% rate among the patients on aspirin only, a 2.7% absolute reduction by using rivaroxaban plus aspirin that translated into a number needed to treat of 37 patients with a history of stroke to prevent one new stroke per year.
The 2017 report of the main COMPASS results included a net clinical benefit analysis that factored together the primary endpoint events and major bleeding events. The net rate of all these events was 4.7% with rivaroxaban plus aspirin and 5.9% with aspirin only, a statistically significant 20% relative risk reduction for all adverse outcomes with dual therapy. This net clinical benefit suggests that adding rivaroxaban has a cost-effective benefit. Assessment of rivaroxaban’s cost benefit in COMPASS is in process, Dr. Sharma said.
Rivaroxaban received Food and Drug Administration marketing approval in 2011 for preventing deep vein thrombosis and preventing stroke in patients with atrial fibrillation at dosages higher than what was used in COMPASS. The approved rivaroxaban dosage for preventing deep vein thrombosis is 10 mg/day, and 20 mg/day for preventing stroke in atrial fibrillation patients. The 2.5-mg formulation of rivaroxaban that was given twice daily had the best safety and efficacy in COMPASS, but it is not available now on the U.S. market, although it is available in Europe. Johnson & Johnson, which markets rivaroxaban globally with Bayer, submitted an application to the FDA in December for marketing approval of the 2.5-mg formulation in twice-daily dosing for use as in the COMPASS trial.
COMPASS was sponsored by Bayer, the company that markets rivaroxaban in collaboration with Johnson & Johnson. Dr. Sharma has been a consultant or adviser to Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, and Daiichi-Sankyo.
SOURCE: Sharma M et al. ISC 2018, Abstract LB7.
LOS ANGELES – Combined treatment with a low dosage of the anticoagulant rivaroxaban plus aspirin cut the incidence of ischemic strokes nearly in half, compared with aspirin alone, in a multicenter, randomized trial of more than 27,000 patients with stable atherosclerotic vascular disease.
This dramatic reduction in ischemic strokes as well as in all-cause strokes by adding low-dose rivaroxaban(Xarelto) occurred without any significant increase in hemorrhagic strokes but with a small increase in total major bleeding events, such as gastrointestinal bleeds, Mike Sharma, MD, said at the International Stroke Conference, sponsored by the American Heart Association.
“There was a consistent effect across all strata of stroke risk. For patients who had a prior stroke, it’s pretty clear to use rivaroxaban plus aspirin because it had a big benefit” with no increase in intracranial hemorrhages, Dr. Sharma said in a video interview.
“We think these results will fundamentally change how we approach stroke prevention,” added Dr. Sharma, a stroke neurologist in the Population Health Research Institute of McMaster University in Hamilton, Ont.
The results he reported came from a secondary analysis of data collected in the COMPASS (Rivaroxaban for the Prevention of Major Cardiovascular Events in Coronary or Peripheral Artery Disease) trial, which enrolled 27,395 patients with stable coronary or peripheral artery disease at 602 centers in 33 countries.
The primary outcome of the trial, reported in 2017, was the combined rate of cardiovascular death, MI, or stroke during an average 23 months of follow-up, which occurred in 4.1% of patients treated with 2.5 mg rivaroxaban twice daily plus 100 mg aspirin once daily, 4.9% of patients who received 5.0 mg rivaroxaban twice daily, and 5.4% in patients who received 100 mg aspirin daily, a statistically significant 24% relative risk reduction in the combined treatment group, compared with aspirin only. The rivaroxaban only–treated patients did not significantly differ from the control patients who received only aspirin (N Engl J Med. 2017 Oct 5;377[14]:1319-30). The main results showed a 1.2% increase in the rate of major bleeds in patients treated with rivaroxaban plus aspirin, compared with aspirin only, but the rate of nonfatal symptomatic intracranial hemorrhages was identical in the two treatment groups.
The new results Dr. Sharma reported at the conference focused on various measures of stroke. The rate of all strokes was 42% lower among the patients treated with rivaroxaban plus aspirin, compared with the aspirin alone patients, and ischemic strokes were 49% lower with the dual therapy, compared with aspirin only. Both differences were statistically significant. In contrast, the rivaroxaban alone regimen did not significantly reduce all-cause strokes. It did significantly reduce ischemic strokes, compared with aspirin only, but it also significantly increased hemorrhagic strokes, compared with aspirin only, an adverse effect not caused by the combination of low-dose rivaroxaban plus aspirin.
Rivaroxaban plus aspirin surpassed aspirin alone for preventing both mild and severe strokes and for preventing strokes both in patients with a history of a prior stroke and in those who never had a prior stroke. The stroke reduction produced by rivaroxaban plus aspirin was greatest in the highest risk patients – those with a prior stroke. On the combined regimen, these patients had an average stroke incidence of 0.7% per year, compared with an annual 3.4% rate among the patients on aspirin only, a 2.7% absolute reduction by using rivaroxaban plus aspirin that translated into a number needed to treat of 37 patients with a history of stroke to prevent one new stroke per year.
The 2017 report of the main COMPASS results included a net clinical benefit analysis that factored together the primary endpoint events and major bleeding events. The net rate of all these events was 4.7% with rivaroxaban plus aspirin and 5.9% with aspirin only, a statistically significant 20% relative risk reduction for all adverse outcomes with dual therapy. This net clinical benefit suggests that adding rivaroxaban has a cost-effective benefit. Assessment of rivaroxaban’s cost benefit in COMPASS is in process, Dr. Sharma said.
Rivaroxaban received Food and Drug Administration marketing approval in 2011 for preventing deep vein thrombosis and preventing stroke in patients with atrial fibrillation at dosages higher than what was used in COMPASS. The approved rivaroxaban dosage for preventing deep vein thrombosis is 10 mg/day, and 20 mg/day for preventing stroke in atrial fibrillation patients. The 2.5-mg formulation of rivaroxaban that was given twice daily had the best safety and efficacy in COMPASS, but it is not available now on the U.S. market, although it is available in Europe. Johnson & Johnson, which markets rivaroxaban globally with Bayer, submitted an application to the FDA in December for marketing approval of the 2.5-mg formulation in twice-daily dosing for use as in the COMPASS trial.
COMPASS was sponsored by Bayer, the company that markets rivaroxaban in collaboration with Johnson & Johnson. Dr. Sharma has been a consultant or adviser to Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, and Daiichi-Sankyo.
SOURCE: Sharma M et al. ISC 2018, Abstract LB7.
LOS ANGELES – Combined treatment with a low dosage of the anticoagulant rivaroxaban plus aspirin cut the incidence of ischemic strokes nearly in half, compared with aspirin alone, in a multicenter, randomized trial of more than 27,000 patients with stable atherosclerotic vascular disease.
This dramatic reduction in ischemic strokes as well as in all-cause strokes by adding low-dose rivaroxaban(Xarelto) occurred without any significant increase in hemorrhagic strokes but with a small increase in total major bleeding events, such as gastrointestinal bleeds, Mike Sharma, MD, said at the International Stroke Conference, sponsored by the American Heart Association.
“There was a consistent effect across all strata of stroke risk. For patients who had a prior stroke, it’s pretty clear to use rivaroxaban plus aspirin because it had a big benefit” with no increase in intracranial hemorrhages, Dr. Sharma said in a video interview.
“We think these results will fundamentally change how we approach stroke prevention,” added Dr. Sharma, a stroke neurologist in the Population Health Research Institute of McMaster University in Hamilton, Ont.
The results he reported came from a secondary analysis of data collected in the COMPASS (Rivaroxaban for the Prevention of Major Cardiovascular Events in Coronary or Peripheral Artery Disease) trial, which enrolled 27,395 patients with stable coronary or peripheral artery disease at 602 centers in 33 countries.
The primary outcome of the trial, reported in 2017, was the combined rate of cardiovascular death, MI, or stroke during an average 23 months of follow-up, which occurred in 4.1% of patients treated with 2.5 mg rivaroxaban twice daily plus 100 mg aspirin once daily, 4.9% of patients who received 5.0 mg rivaroxaban twice daily, and 5.4% in patients who received 100 mg aspirin daily, a statistically significant 24% relative risk reduction in the combined treatment group, compared with aspirin only. The rivaroxaban only–treated patients did not significantly differ from the control patients who received only aspirin (N Engl J Med. 2017 Oct 5;377[14]:1319-30). The main results showed a 1.2% increase in the rate of major bleeds in patients treated with rivaroxaban plus aspirin, compared with aspirin only, but the rate of nonfatal symptomatic intracranial hemorrhages was identical in the two treatment groups.
The new results Dr. Sharma reported at the conference focused on various measures of stroke. The rate of all strokes was 42% lower among the patients treated with rivaroxaban plus aspirin, compared with the aspirin alone patients, and ischemic strokes were 49% lower with the dual therapy, compared with aspirin only. Both differences were statistically significant. In contrast, the rivaroxaban alone regimen did not significantly reduce all-cause strokes. It did significantly reduce ischemic strokes, compared with aspirin only, but it also significantly increased hemorrhagic strokes, compared with aspirin only, an adverse effect not caused by the combination of low-dose rivaroxaban plus aspirin.
Rivaroxaban plus aspirin surpassed aspirin alone for preventing both mild and severe strokes and for preventing strokes both in patients with a history of a prior stroke and in those who never had a prior stroke. The stroke reduction produced by rivaroxaban plus aspirin was greatest in the highest risk patients – those with a prior stroke. On the combined regimen, these patients had an average stroke incidence of 0.7% per year, compared with an annual 3.4% rate among the patients on aspirin only, a 2.7% absolute reduction by using rivaroxaban plus aspirin that translated into a number needed to treat of 37 patients with a history of stroke to prevent one new stroke per year.
The 2017 report of the main COMPASS results included a net clinical benefit analysis that factored together the primary endpoint events and major bleeding events. The net rate of all these events was 4.7% with rivaroxaban plus aspirin and 5.9% with aspirin only, a statistically significant 20% relative risk reduction for all adverse outcomes with dual therapy. This net clinical benefit suggests that adding rivaroxaban has a cost-effective benefit. Assessment of rivaroxaban’s cost benefit in COMPASS is in process, Dr. Sharma said.
Rivaroxaban received Food and Drug Administration marketing approval in 2011 for preventing deep vein thrombosis and preventing stroke in patients with atrial fibrillation at dosages higher than what was used in COMPASS. The approved rivaroxaban dosage for preventing deep vein thrombosis is 10 mg/day, and 20 mg/day for preventing stroke in atrial fibrillation patients. The 2.5-mg formulation of rivaroxaban that was given twice daily had the best safety and efficacy in COMPASS, but it is not available now on the U.S. market, although it is available in Europe. Johnson & Johnson, which markets rivaroxaban globally with Bayer, submitted an application to the FDA in December for marketing approval of the 2.5-mg formulation in twice-daily dosing for use as in the COMPASS trial.
COMPASS was sponsored by Bayer, the company that markets rivaroxaban in collaboration with Johnson & Johnson. Dr. Sharma has been a consultant or adviser to Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, and Daiichi-Sankyo.
SOURCE: Sharma M et al. ISC 2018, Abstract LB7.
REPORTING FROM ISC 2018
Key clinical point: Rivaroxaban plus aspirin cuts strokes in patients with stable atherosclerotic vascular disease.
Major finding: Rivaroxaban plus aspirin cut the rate of ischemic strokes by 49%, compared with aspirin only.
Study details: Secondary analysis from the COMPASS trial, a multicenter, randomized trial with 27,395 patients.
Disclosures: COMPASS was sponsored by Bayer, the company that markets rivaroxaban in collaboration with Johnson & Johnson. Dr. Sharma has been a consultant or adviser to Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, and Daiichi-Sankyo.
Source: Sharma M et al. ISC 2018, Abstract LB7.

US Dermatology Residency Program Rankings Based on Academic Achievement
Rankings of US residency programs based on academic achievement are a resource for fourth-year medical students applying for residency through the National Resident Matching Program. They also highlight the leading academic training programs in each medical specialty. Currently, the Doximity Residency Navigator (https://residency.doximity.com) provides rankings of US residency programs based on either subjective or objective criteria. The subjective rankings utilize current resident and recent alumni satisfaction surveys as well as nominations from board-certified Doximity members who were asked to nominate up to 5 residency programs in their specialty that offer the best clinical training. The objective rankings are based on measurement of research output, which is calculated from the collective h-index of publications authored by graduating alumni within the last 15 years as well as the amount of research funding awarded.1
Aquino et al2 provided a ranking of US dermatology residency programs using alternative objective data measures (as of December 31, 2008) from the Doximity algorithm, including National Institutes of Health (NIH) and Dermatology Foundation (DF) funding, number of publications by full-time faculty members, number of faculty lectures given at annual meetings of 5 societies, and number of full-time faculty members serving on the editorial boards of 6 dermatology journals. The current study is an update to those rankings utilizing data from 2014.
Methods
The following data for each dermatology residency program were obtained to formulate the rankings: number of full-time faculty members, amount of NIH funding received in 2014 (https://report.nih.gov/), number of publications by full-time faculty members in 2014 (http://www.ncbi.nlm.nih.gov/pubmed/), and the number of faculty lectures given at annual meetings of 5 societies in 2014 (American Academy of Dermatology, the Society for Investigative Dermatology, the American Society of Dermatopathology, the Society for Pediatric Dermatology, and the American Society for Dermatologic Surgery). This study was approved by the institutional review board at Kaiser Permanente Southern California.
The names of all US dermatology residency programs were obtained as of December 31, 2014, from FREIDA Online using the search term dermatology. An email was sent to a representative from each residency program (eg, residency program coordinator, program director, full-time faculty member) requesting confirmation of a list of full-time faculty members in the program, excluding part-time and volunteer faculty. If a response was not obtained or the representative declined to participate, a list was compiled using available information from that residency program’s website.
National Institutes of Health funding for 2014 was obtained for individual faculty members from the NIH Research Portfolio Online Reporting Tools expenditures and reports (https://projectreporter.nih.gov/reporter.cfm) by searching the first and last name of each full-time faculty member along with their affiliated institution. The search results were filtered to only include NIH funding for full-time faculty members listed as principal investigators rather than as coinvestigators. The fiscal year total cost by institute/center for each full-time faculty member’s projects was summated to obtain the total NIH funding for the program.
The total number of publications by full-time faculty members in 2014 was obtained utilizing a PubMed search of articles indexed for MEDLINE using each faculty member’s first and last name. The authors’ affiliations were verified for each publication, and the number of publications was summed for all full-time faculty members at each residency program. If multiple authors from the same program coauthored an article, it was only counted once toward the total number of faculty publications from that program.
Program brochures for the 2014 meetings of the 5 societies were reviewed to quantify the number of lectures given by full-time faculty members in each program.
Each residency program was assigned a score from 0 to 1.0 for each of the 4 factors of academic achievement analyzed. The program with the highest number of faculty publications was assigned a score of 1.0 and the program with the lowest number of publications was assigned a score of 0. The programs in between were subsequently assigned scores from 0 to 1.0 based on the number of publications as a percentage of the number of publications from the program with the most publications.
A weighted ranking scheme was used to rank residency programs based on the relative importance of each factor. There were 3 factors that were deemed to be the most reflective of academic achievement among dermatology residency programs: amount of NIH funding received in 2014, number of publications by full-time faculty members in 2014, and number of faculty lectures given at society meetings in 2014; thus, these factors were given a weight of 1.0. The remaining factor— total number of full-time faculty members—was given a weight of 0.5. Values were totaled and programs were ranked based on the sum of these values. All quantitative analyses were performed using an electronic spreadsheet program.
Results
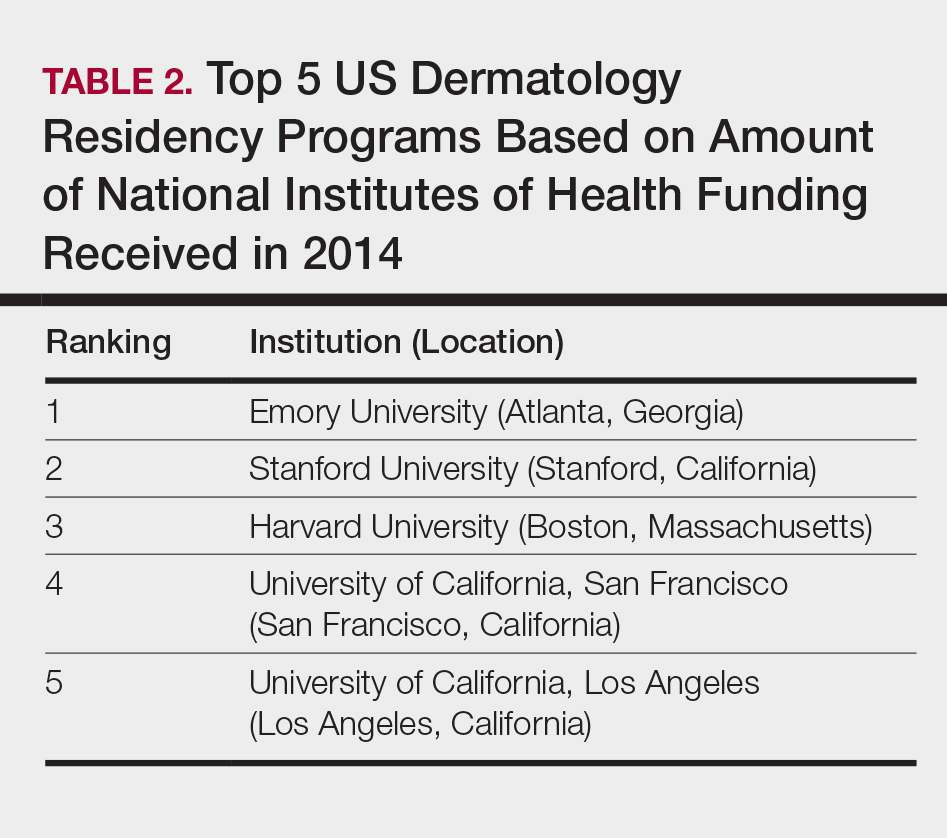
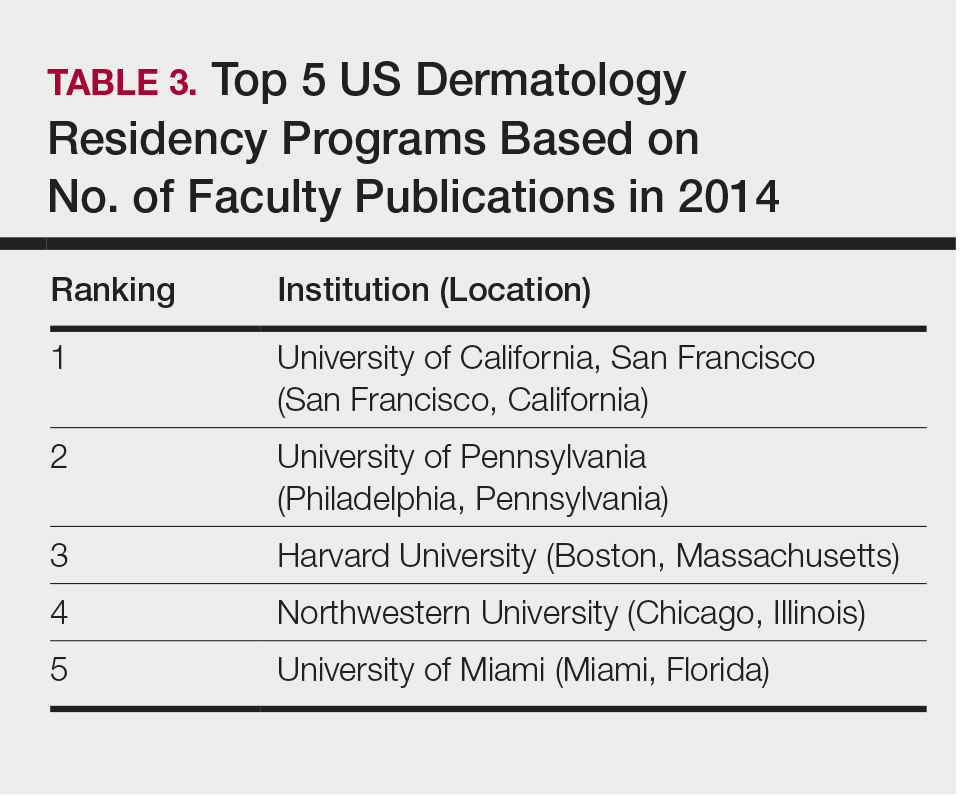
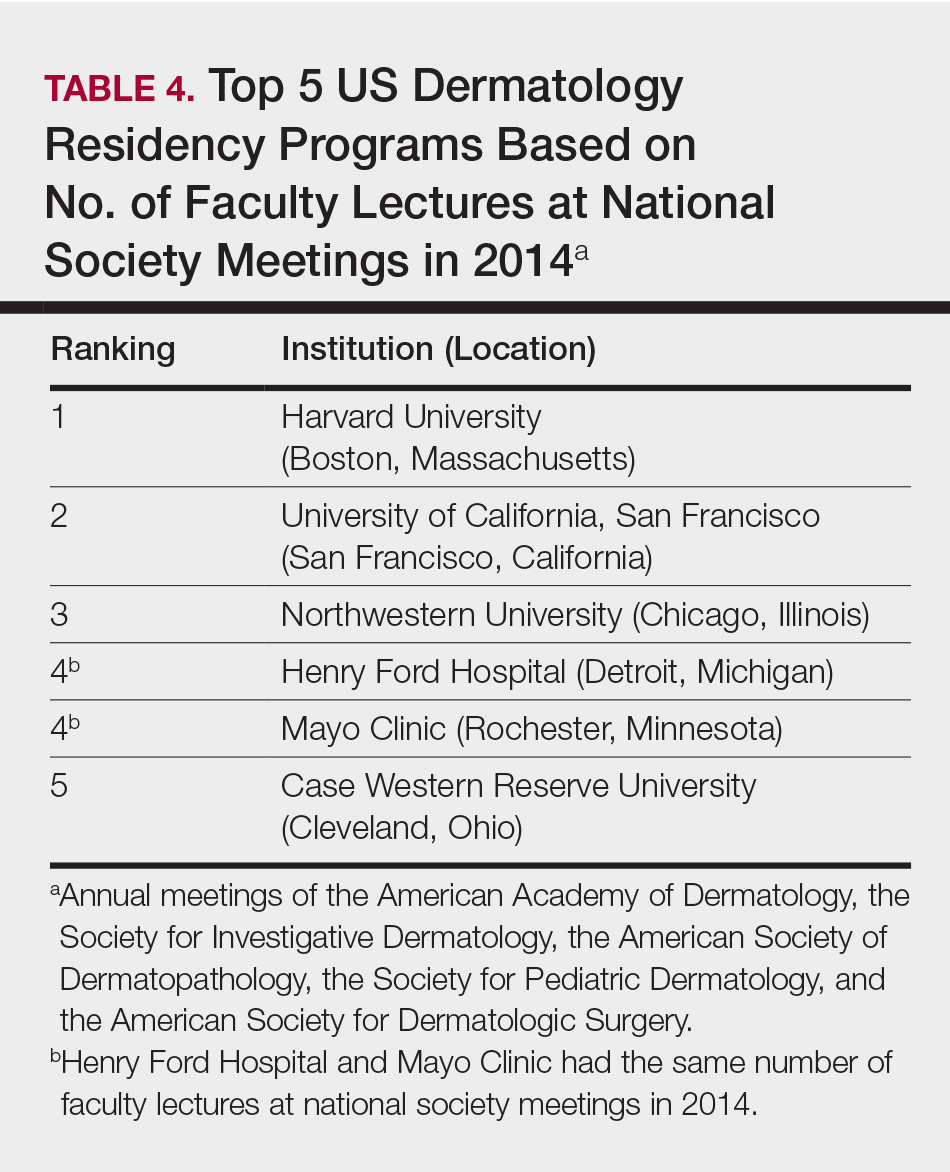
The overall ranking of the top 20 US dermatology residency programs in 2014 is presented in Table 1. The top 5 programs based on each of the 3 factors most reflective of academic achievement used in the weighted ranking algorithm are presented in Tables 2 through 4.

Comment
The ranking of US residency programs involves using data in an unbiased manner while also accounting for important subjective measures. In a 2015 survey of residency applicants (n=6285), the 5 most important factors for applicants in selecting a program were the program’s ability to prepare residents for future training or position, resident esprit de corps, faculty availability and involvement in teaching, depth and breadth of faculty, and variety of patients and clinical resources.3 However, these subjective measures are difficult to quantify in a standardized fashion. In its ranking of residency programs, the Doximity Residency Navigator utilizes surveys of current residents and recent alumni as well as nominations from board-certified Doximity members.1
One of the main issues in utilizing survey data to rank residency programs is the inherent bias that most residents and alumni possess toward their own program. Moreover, the question arises whether most residents, faculty members, or recent alumni of residency programs have sufficient knowledge of other programs to rank them in a well-informed manner.
Wu et al4 used data from 2004 to perform the first algorithmic ranking of US dermatology programs, which was based on publications in 2001 to 2004, the amount of NIH funding in 2004, DF grants in 2001 to 2004, faculty lectures delivered at national conferences in 2004, and number of full-time faculty members on the editorial boards of the top 3 US dermatology journals and the top 4 subspecialty journals. Aquino et al2 provided updated rankings that utilized a weighted algorithm to collect data from 2008 related to a number of factors, including annual amount of NIH and DF funding received, number of publications by full-time faculty members, number of faculty lectures given at 5 annual society meetings, and number of full-time faculty members who were on the editorial boards of 6 dermatology journals with the highest impact factors. The top 5 ranked programs based on the 2008 data were the University of California, San Francisco (San Francisco, California); Northwestern University (Chicago, Illinois); University of Pennsylvania (Philadelphia, Pennsylvania); Yale University (New Haven, Connecticut); and Stanford University (Stanford, California).2
The current ranking algorithm is more indicative of a residency program’s commitment to research and scholarship, with an assumption that successful clinical training is offered. Leading researchers in the field also are usually known to be clinical experts, but the current data does not take into account the frequency, quality, or methodology of teaching provided to residents. Perhaps the most objective measure reflecting the quality of resident education would be American Board of Dermatology examination scores, but these data are not publically available. Additional factors such as the percentage of residents who received fellowship positions; diversity of the patient population; and number and extent of surgical, cosmetic, or laser procedures performed also are not readily available. Doximity provides board pass rates for each residency program, but these data are self-reported and are not taken into account in their rankings.1
The current study aimed to utilize publicly available data to rank US dermatology residency programs based on objective measures of academic achievement. A recent study showed that 531 of 793 applicants (67%) to emergency medicine residency programs were aware of the Doximity residency rankings.One-quarter of these applicants made changes to their rank list based on this data, demonstrating that residency rankings may impact applicant decision-making.5 In the future, the most accurate and unbiased rankings may be performed if each residency program joins a cooperative effort to provide more objective data about the training they provide and utilizes a standardized survey system for current residents and recent graduates to evaluate important subjective measures.
Conclusion
Based on our weighted ranking algorithm, the top 5 dermatology residency programs in 2014 were Harvard University (Boston, Massachusetts); University of California, San Francisco (San Francisco, California); Stanford University (Stanford, California); University of Pennsylvania (Philadelphia, Pennsylvania); and Emory University (Atlanta, Georgia).
Acknowledgments
We thank all of the program coordinators, full-time faculty members, program directors, and chairs who provided responses to our inquiries for additional information about their residency programs.
- Residency navigator 2017-2018. Doximity website. https://residency.doximity.com. Accessed January 19, 2018.
- Aquino LL, Wen G, Wu JJ. US dermatology residency program rankings. Cutis. 2014;94:189-194.
- Phitayakorn R, Macklin EA, Goldsmith J, et al. Applicants’ self-reported priorities in selecting a residency program. J Grad Med Educ. 2015;7:21-26.
- Wu JJ, Ramirez CC, Alonso CA, et al. Ranking the dermatology programs based on measurements of academic achievement. Dermatol Online J. 2007;13:3.
- Peterson WJ, Hopson LR, Khandelwal S. Impact of Doximity residency rankings on emergency medicine applicant rank lists [published online May 5, 2016]. West J Emerg Med. 2016;17:350-354.
Rankings of US residency programs based on academic achievement are a resource for fourth-year medical students applying for residency through the National Resident Matching Program. They also highlight the leading academic training programs in each medical specialty. Currently, the Doximity Residency Navigator (https://residency.doximity.com) provides rankings of US residency programs based on either subjective or objective criteria. The subjective rankings utilize current resident and recent alumni satisfaction surveys as well as nominations from board-certified Doximity members who were asked to nominate up to 5 residency programs in their specialty that offer the best clinical training. The objective rankings are based on measurement of research output, which is calculated from the collective h-index of publications authored by graduating alumni within the last 15 years as well as the amount of research funding awarded.1
Aquino et al2 provided a ranking of US dermatology residency programs using alternative objective data measures (as of December 31, 2008) from the Doximity algorithm, including National Institutes of Health (NIH) and Dermatology Foundation (DF) funding, number of publications by full-time faculty members, number of faculty lectures given at annual meetings of 5 societies, and number of full-time faculty members serving on the editorial boards of 6 dermatology journals. The current study is an update to those rankings utilizing data from 2014.
Methods
The following data for each dermatology residency program were obtained to formulate the rankings: number of full-time faculty members, amount of NIH funding received in 2014 (https://report.nih.gov/), number of publications by full-time faculty members in 2014 (http://www.ncbi.nlm.nih.gov/pubmed/), and the number of faculty lectures given at annual meetings of 5 societies in 2014 (American Academy of Dermatology, the Society for Investigative Dermatology, the American Society of Dermatopathology, the Society for Pediatric Dermatology, and the American Society for Dermatologic Surgery). This study was approved by the institutional review board at Kaiser Permanente Southern California.
The names of all US dermatology residency programs were obtained as of December 31, 2014, from FREIDA Online using the search term dermatology. An email was sent to a representative from each residency program (eg, residency program coordinator, program director, full-time faculty member) requesting confirmation of a list of full-time faculty members in the program, excluding part-time and volunteer faculty. If a response was not obtained or the representative declined to participate, a list was compiled using available information from that residency program’s website.
National Institutes of Health funding for 2014 was obtained for individual faculty members from the NIH Research Portfolio Online Reporting Tools expenditures and reports (https://projectreporter.nih.gov/reporter.cfm) by searching the first and last name of each full-time faculty member along with their affiliated institution. The search results were filtered to only include NIH funding for full-time faculty members listed as principal investigators rather than as coinvestigators. The fiscal year total cost by institute/center for each full-time faculty member’s projects was summated to obtain the total NIH funding for the program.
The total number of publications by full-time faculty members in 2014 was obtained utilizing a PubMed search of articles indexed for MEDLINE using each faculty member’s first and last name. The authors’ affiliations were verified for each publication, and the number of publications was summed for all full-time faculty members at each residency program. If multiple authors from the same program coauthored an article, it was only counted once toward the total number of faculty publications from that program.
Program brochures for the 2014 meetings of the 5 societies were reviewed to quantify the number of lectures given by full-time faculty members in each program.
Each residency program was assigned a score from 0 to 1.0 for each of the 4 factors of academic achievement analyzed. The program with the highest number of faculty publications was assigned a score of 1.0 and the program with the lowest number of publications was assigned a score of 0. The programs in between were subsequently assigned scores from 0 to 1.0 based on the number of publications as a percentage of the number of publications from the program with the most publications.
A weighted ranking scheme was used to rank residency programs based on the relative importance of each factor. There were 3 factors that were deemed to be the most reflective of academic achievement among dermatology residency programs: amount of NIH funding received in 2014, number of publications by full-time faculty members in 2014, and number of faculty lectures given at society meetings in 2014; thus, these factors were given a weight of 1.0. The remaining factor— total number of full-time faculty members—was given a weight of 0.5. Values were totaled and programs were ranked based on the sum of these values. All quantitative analyses were performed using an electronic spreadsheet program.
Results
The overall ranking of the top 20 US dermatology residency programs in 2014 is presented in Table 1. The top 5 programs based on each of the 3 factors most reflective of academic achievement used in the weighted ranking algorithm are presented in Tables 2 through 4.
Comment
The ranking of US residency programs involves using data in an unbiased manner while also accounting for important subjective measures. In a 2015 survey of residency applicants (n=6285), the 5 most important factors for applicants in selecting a program were the program’s ability to prepare residents for future training or position, resident esprit de corps, faculty availability and involvement in teaching, depth and breadth of faculty, and variety of patients and clinical resources.3 However, these subjective measures are difficult to quantify in a standardized fashion. In its ranking of residency programs, the Doximity Residency Navigator utilizes surveys of current residents and recent alumni as well as nominations from board-certified Doximity members.1
One of the main issues in utilizing survey data to rank residency programs is the inherent bias that most residents and alumni possess toward their own program. Moreover, the question arises whether most residents, faculty members, or recent alumni of residency programs have sufficient knowledge of other programs to rank them in a well-informed manner.
Wu et al4 used data from 2004 to perform the first algorithmic ranking of US dermatology programs, which was based on publications in 2001 to 2004, the amount of NIH funding in 2004, DF grants in 2001 to 2004, faculty lectures delivered at national conferences in 2004, and number of full-time faculty members on the editorial boards of the top 3 US dermatology journals and the top 4 subspecialty journals. Aquino et al2 provided updated rankings that utilized a weighted algorithm to collect data from 2008 related to a number of factors, including annual amount of NIH and DF funding received, number of publications by full-time faculty members, number of faculty lectures given at 5 annual society meetings, and number of full-time faculty members who were on the editorial boards of 6 dermatology journals with the highest impact factors. The top 5 ranked programs based on the 2008 data were the University of California, San Francisco (San Francisco, California); Northwestern University (Chicago, Illinois); University of Pennsylvania (Philadelphia, Pennsylvania); Yale University (New Haven, Connecticut); and Stanford University (Stanford, California).2
The current ranking algorithm is more indicative of a residency program’s commitment to research and scholarship, with an assumption that successful clinical training is offered. Leading researchers in the field also are usually known to be clinical experts, but the current data does not take into account the frequency, quality, or methodology of teaching provided to residents. Perhaps the most objective measure reflecting the quality of resident education would be American Board of Dermatology examination scores, but these data are not publically available. Additional factors such as the percentage of residents who received fellowship positions; diversity of the patient population; and number and extent of surgical, cosmetic, or laser procedures performed also are not readily available. Doximity provides board pass rates for each residency program, but these data are self-reported and are not taken into account in their rankings.1
The current study aimed to utilize publicly available data to rank US dermatology residency programs based on objective measures of academic achievement. A recent study showed that 531 of 793 applicants (67%) to emergency medicine residency programs were aware of the Doximity residency rankings.One-quarter of these applicants made changes to their rank list based on this data, demonstrating that residency rankings may impact applicant decision-making.5 In the future, the most accurate and unbiased rankings may be performed if each residency program joins a cooperative effort to provide more objective data about the training they provide and utilizes a standardized survey system for current residents and recent graduates to evaluate important subjective measures.
Conclusion
Based on our weighted ranking algorithm, the top 5 dermatology residency programs in 2014 were Harvard University (Boston, Massachusetts); University of California, San Francisco (San Francisco, California); Stanford University (Stanford, California); University of Pennsylvania (Philadelphia, Pennsylvania); and Emory University (Atlanta, Georgia).
Acknowledgments
We thank all of the program coordinators, full-time faculty members, program directors, and chairs who provided responses to our inquiries for additional information about their residency programs.
Rankings of US residency programs based on academic achievement are a resource for fourth-year medical students applying for residency through the National Resident Matching Program. They also highlight the leading academic training programs in each medical specialty. Currently, the Doximity Residency Navigator (https://residency.doximity.com) provides rankings of US residency programs based on either subjective or objective criteria. The subjective rankings utilize current resident and recent alumni satisfaction surveys as well as nominations from board-certified Doximity members who were asked to nominate up to 5 residency programs in their specialty that offer the best clinical training. The objective rankings are based on measurement of research output, which is calculated from the collective h-index of publications authored by graduating alumni within the last 15 years as well as the amount of research funding awarded.1
Aquino et al2 provided a ranking of US dermatology residency programs using alternative objective data measures (as of December 31, 2008) from the Doximity algorithm, including National Institutes of Health (NIH) and Dermatology Foundation (DF) funding, number of publications by full-time faculty members, number of faculty lectures given at annual meetings of 5 societies, and number of full-time faculty members serving on the editorial boards of 6 dermatology journals. The current study is an update to those rankings utilizing data from 2014.
Methods
The following data for each dermatology residency program were obtained to formulate the rankings: number of full-time faculty members, amount of NIH funding received in 2014 (https://report.nih.gov/), number of publications by full-time faculty members in 2014 (http://www.ncbi.nlm.nih.gov/pubmed/), and the number of faculty lectures given at annual meetings of 5 societies in 2014 (American Academy of Dermatology, the Society for Investigative Dermatology, the American Society of Dermatopathology, the Society for Pediatric Dermatology, and the American Society for Dermatologic Surgery). This study was approved by the institutional review board at Kaiser Permanente Southern California.
The names of all US dermatology residency programs were obtained as of December 31, 2014, from FREIDA Online using the search term dermatology. An email was sent to a representative from each residency program (eg, residency program coordinator, program director, full-time faculty member) requesting confirmation of a list of full-time faculty members in the program, excluding part-time and volunteer faculty. If a response was not obtained or the representative declined to participate, a list was compiled using available information from that residency program’s website.
National Institutes of Health funding for 2014 was obtained for individual faculty members from the NIH Research Portfolio Online Reporting Tools expenditures and reports (https://projectreporter.nih.gov/reporter.cfm) by searching the first and last name of each full-time faculty member along with their affiliated institution. The search results were filtered to only include NIH funding for full-time faculty members listed as principal investigators rather than as coinvestigators. The fiscal year total cost by institute/center for each full-time faculty member’s projects was summated to obtain the total NIH funding for the program.
The total number of publications by full-time faculty members in 2014 was obtained utilizing a PubMed search of articles indexed for MEDLINE using each faculty member’s first and last name. The authors’ affiliations were verified for each publication, and the number of publications was summed for all full-time faculty members at each residency program. If multiple authors from the same program coauthored an article, it was only counted once toward the total number of faculty publications from that program.
Program brochures for the 2014 meetings of the 5 societies were reviewed to quantify the number of lectures given by full-time faculty members in each program.
Each residency program was assigned a score from 0 to 1.0 for each of the 4 factors of academic achievement analyzed. The program with the highest number of faculty publications was assigned a score of 1.0 and the program with the lowest number of publications was assigned a score of 0. The programs in between were subsequently assigned scores from 0 to 1.0 based on the number of publications as a percentage of the number of publications from the program with the most publications.
A weighted ranking scheme was used to rank residency programs based on the relative importance of each factor. There were 3 factors that were deemed to be the most reflective of academic achievement among dermatology residency programs: amount of NIH funding received in 2014, number of publications by full-time faculty members in 2014, and number of faculty lectures given at society meetings in 2014; thus, these factors were given a weight of 1.0. The remaining factor— total number of full-time faculty members—was given a weight of 0.5. Values were totaled and programs were ranked based on the sum of these values. All quantitative analyses were performed using an electronic spreadsheet program.
Results
The overall ranking of the top 20 US dermatology residency programs in 2014 is presented in Table 1. The top 5 programs based on each of the 3 factors most reflective of academic achievement used in the weighted ranking algorithm are presented in Tables 2 through 4.
Comment
The ranking of US residency programs involves using data in an unbiased manner while also accounting for important subjective measures. In a 2015 survey of residency applicants (n=6285), the 5 most important factors for applicants in selecting a program were the program’s ability to prepare residents for future training or position, resident esprit de corps, faculty availability and involvement in teaching, depth and breadth of faculty, and variety of patients and clinical resources.3 However, these subjective measures are difficult to quantify in a standardized fashion. In its ranking of residency programs, the Doximity Residency Navigator utilizes surveys of current residents and recent alumni as well as nominations from board-certified Doximity members.1
One of the main issues in utilizing survey data to rank residency programs is the inherent bias that most residents and alumni possess toward their own program. Moreover, the question arises whether most residents, faculty members, or recent alumni of residency programs have sufficient knowledge of other programs to rank them in a well-informed manner.
Wu et al4 used data from 2004 to perform the first algorithmic ranking of US dermatology programs, which was based on publications in 2001 to 2004, the amount of NIH funding in 2004, DF grants in 2001 to 2004, faculty lectures delivered at national conferences in 2004, and number of full-time faculty members on the editorial boards of the top 3 US dermatology journals and the top 4 subspecialty journals. Aquino et al2 provided updated rankings that utilized a weighted algorithm to collect data from 2008 related to a number of factors, including annual amount of NIH and DF funding received, number of publications by full-time faculty members, number of faculty lectures given at 5 annual society meetings, and number of full-time faculty members who were on the editorial boards of 6 dermatology journals with the highest impact factors. The top 5 ranked programs based on the 2008 data were the University of California, San Francisco (San Francisco, California); Northwestern University (Chicago, Illinois); University of Pennsylvania (Philadelphia, Pennsylvania); Yale University (New Haven, Connecticut); and Stanford University (Stanford, California).2
The current ranking algorithm is more indicative of a residency program’s commitment to research and scholarship, with an assumption that successful clinical training is offered. Leading researchers in the field also are usually known to be clinical experts, but the current data does not take into account the frequency, quality, or methodology of teaching provided to residents. Perhaps the most objective measure reflecting the quality of resident education would be American Board of Dermatology examination scores, but these data are not publically available. Additional factors such as the percentage of residents who received fellowship positions; diversity of the patient population; and number and extent of surgical, cosmetic, or laser procedures performed also are not readily available. Doximity provides board pass rates for each residency program, but these data are self-reported and are not taken into account in their rankings.1
The current study aimed to utilize publicly available data to rank US dermatology residency programs based on objective measures of academic achievement. A recent study showed that 531 of 793 applicants (67%) to emergency medicine residency programs were aware of the Doximity residency rankings.One-quarter of these applicants made changes to their rank list based on this data, demonstrating that residency rankings may impact applicant decision-making.5 In the future, the most accurate and unbiased rankings may be performed if each residency program joins a cooperative effort to provide more objective data about the training they provide and utilizes a standardized survey system for current residents and recent graduates to evaluate important subjective measures.
Conclusion
Based on our weighted ranking algorithm, the top 5 dermatology residency programs in 2014 were Harvard University (Boston, Massachusetts); University of California, San Francisco (San Francisco, California); Stanford University (Stanford, California); University of Pennsylvania (Philadelphia, Pennsylvania); and Emory University (Atlanta, Georgia).
Acknowledgments
We thank all of the program coordinators, full-time faculty members, program directors, and chairs who provided responses to our inquiries for additional information about their residency programs.
- Residency navigator 2017-2018. Doximity website. https://residency.doximity.com. Accessed January 19, 2018.
- Aquino LL, Wen G, Wu JJ. US dermatology residency program rankings. Cutis. 2014;94:189-194.
- Phitayakorn R, Macklin EA, Goldsmith J, et al. Applicants’ self-reported priorities in selecting a residency program. J Grad Med Educ. 2015;7:21-26.
- Wu JJ, Ramirez CC, Alonso CA, et al. Ranking the dermatology programs based on measurements of academic achievement. Dermatol Online J. 2007;13:3.
- Peterson WJ, Hopson LR, Khandelwal S. Impact of Doximity residency rankings on emergency medicine applicant rank lists [published online May 5, 2016]. West J Emerg Med. 2016;17:350-354.
- Residency navigator 2017-2018. Doximity website. https://residency.doximity.com. Accessed January 19, 2018.
- Aquino LL, Wen G, Wu JJ. US dermatology residency program rankings. Cutis. 2014;94:189-194.
- Phitayakorn R, Macklin EA, Goldsmith J, et al. Applicants’ self-reported priorities in selecting a residency program. J Grad Med Educ. 2015;7:21-26.
- Wu JJ, Ramirez CC, Alonso CA, et al. Ranking the dermatology programs based on measurements of academic achievement. Dermatol Online J. 2007;13:3.
- Peterson WJ, Hopson LR, Khandelwal S. Impact of Doximity residency rankings on emergency medicine applicant rank lists [published online May 5, 2016]. West J Emerg Med. 2016;17:350-354.
Practice Points
- Dermatology is not among the many hospital-based adult specialties that are routinely ranked annually by US News & World Report.
- In the current study, US dermatology residency programs were ranked based on various academic factors, including the number of full-time faculty members, amount of National Institutes of Health funding received in 2014, number of publications by full-time faculty members in 2014, and the number of faculty lectures given at annual meetings of 5 societies in 2014.

VIDEO: Dupilumab or cyclosporine for treating atopic dermatitis?
KAUAI, HAWAII – Sometimes, older is better, according to Eric Simpson, MD, professor of dermatology at Oregon Health & Science University, Portland.
Dr. Simpson was a key investigator in trials that were the basis of dupilumab’s approval in 2017 for adults with moderate to severe atopic dermatitis (AD), but there’s still a role for cyclosporine and other old standbys, he said in a video interview at the Hawaii Dermatology Seminar, provided by Global Academy for Medical Education/Skin Disease Education Foundation.
He said he’s asked all the time how to pick a systemic treatment for AD when topicals aren’t doing the trick. In the interview, he explained how dupilumab (Dupixent) fits into the picture, and how to select the right systemic therapy for the right patient. There are not a lot of data yet pointing to one option over the others for first-line treatment; a lot of it comes down to clinical smarts and patient preference.
Dr. Simpson is a consultant and/or investigator for a number of companies, including Eli Lilly, Pfizer, Novartis, and dupilumab manufacturer, Regeneron.
SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
KAUAI, HAWAII – Sometimes, older is better, according to Eric Simpson, MD, professor of dermatology at Oregon Health & Science University, Portland.
Dr. Simpson was a key investigator in trials that were the basis of dupilumab’s approval in 2017 for adults with moderate to severe atopic dermatitis (AD), but there’s still a role for cyclosporine and other old standbys, he said in a video interview at the Hawaii Dermatology Seminar, provided by Global Academy for Medical Education/Skin Disease Education Foundation.
He said he’s asked all the time how to pick a systemic treatment for AD when topicals aren’t doing the trick. In the interview, he explained how dupilumab (Dupixent) fits into the picture, and how to select the right systemic therapy for the right patient. There are not a lot of data yet pointing to one option over the others for first-line treatment; a lot of it comes down to clinical smarts and patient preference.
Dr. Simpson is a consultant and/or investigator for a number of companies, including Eli Lilly, Pfizer, Novartis, and dupilumab manufacturer, Regeneron.
SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
KAUAI, HAWAII – Sometimes, older is better, according to Eric Simpson, MD, professor of dermatology at Oregon Health & Science University, Portland.
Dr. Simpson was a key investigator in trials that were the basis of dupilumab’s approval in 2017 for adults with moderate to severe atopic dermatitis (AD), but there’s still a role for cyclosporine and other old standbys, he said in a video interview at the Hawaii Dermatology Seminar, provided by Global Academy for Medical Education/Skin Disease Education Foundation.
He said he’s asked all the time how to pick a systemic treatment for AD when topicals aren’t doing the trick. In the interview, he explained how dupilumab (Dupixent) fits into the picture, and how to select the right systemic therapy for the right patient. There are not a lot of data yet pointing to one option over the others for first-line treatment; a lot of it comes down to clinical smarts and patient preference.
Dr. Simpson is a consultant and/or investigator for a number of companies, including Eli Lilly, Pfizer, Novartis, and dupilumab manufacturer, Regeneron.
SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
REPORTING FROM SDEF HAWAII DERMATOLOGY SEMINAR
Pain-Minimizing Strategies for Nail Surgery
Nail surgery is an important part of dermatologic training and clinical practice, both for diagnosis and treatment of nail disorders as well as benign and malignant nail tumors. Patient comfort is essential prior to the procedure and while administering local anesthetics. Effective anesthesia facilitates nail unit biopsies, excisions, and other surgical nail procedures. Pain management immediately following the procedure and during the postoperative period are equally important.
Patients who undergo nail surgery may experience anxiety due to fear of a cancer diagnosis, pain during the surgery, or disfigurement from the procedure. This anxiety may lead to increased blood pressure, a decreased pain threshold, and mental and physical discomfort.1 A detailed explanation of the procedure itself as well as expectations following the surgery are helpful in diminishing these fears. Administration of a fast-acting benzodiazepine also may be helpful in these patients to decrease anxiety prior to the procedure.2
Attaining adequate anesthesia requires an understanding of digital anatomy, particularly innervation. Innervation of the digits is supplied by the volar and dorsal nerves, which divide into 3 branches at the distal interphalangeal joint, innervating the nail bed, the digital tip, and the pulp.3 Pacinian and Ruffini corpuscles and free-ended nociceptors activate nerve fibers that transmit pain impulses.4,5 Local anesthetics block pain transmission by impeding voltage-gated sodium channels located at free nerve endings. Pain from anesthesia may be due to both needle insertion and fluid infiltration.
Simple measures can maximize patient comfort during digital anesthesia. Both audiovisual distraction and interpersonal interaction can help to put the patient at ease.6,7 Application of topical anesthetic cream (1–2 hours prior to the procedure under occlusion),8 ice (at least 6 minutes),9 or an ethyl chloride spray can be applied to the nail folds prior to needle insertion to alleviate injection pain, but these methods do little for infiltration pain. Use of an ethyl chloride spray may be the preferred technique due to the rapidity of the analgesic effects (Figure).10 A vibrating massager also can be applied in close proximity to the site of needle insertion.11
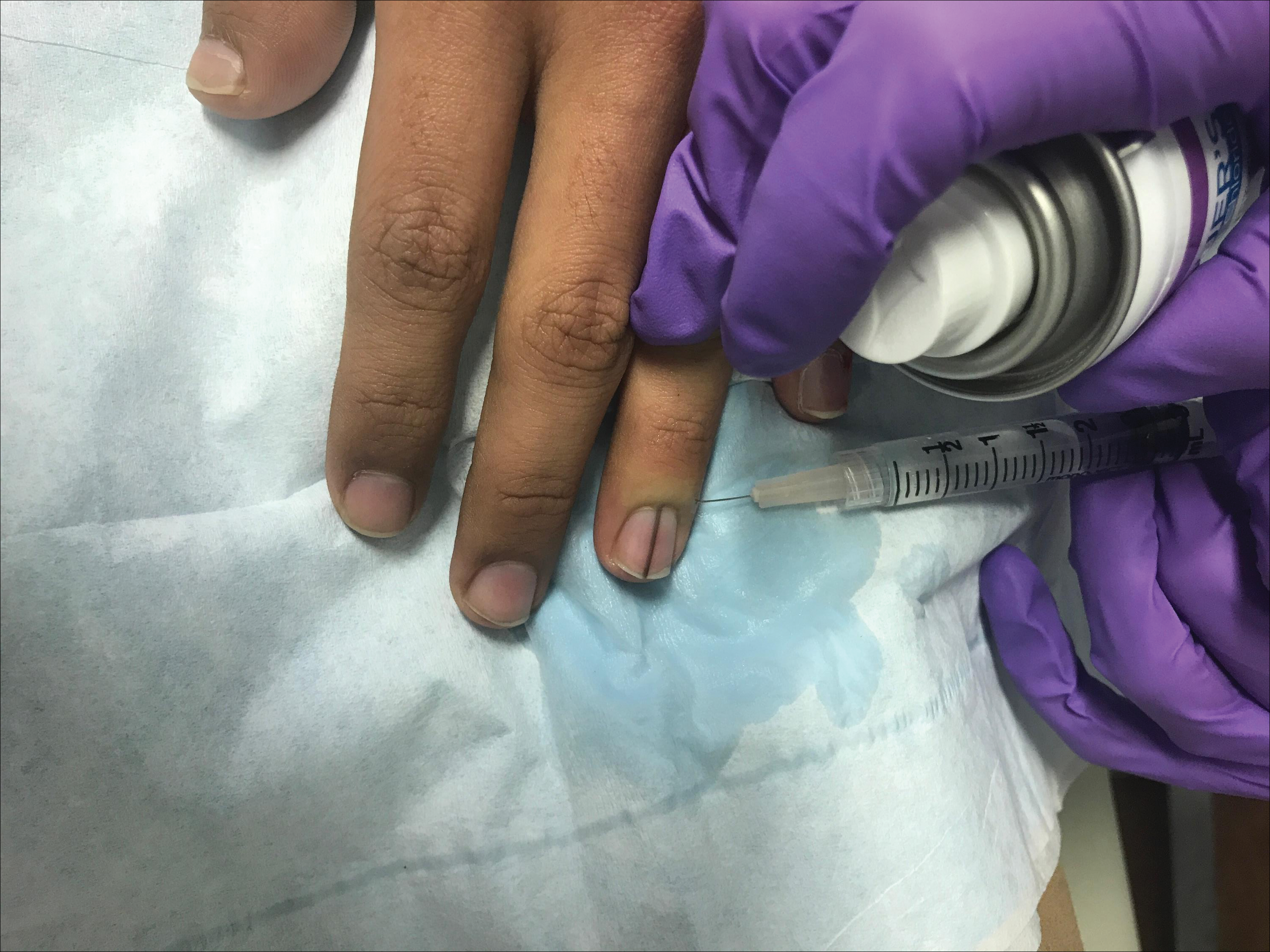
Proper anesthetic preparation and technique also can minimize pain during injection. Because lidocaine 1% is acidic (pH, 6.09), buffering with sodium bicarbonate 8.4% can result in decreased injection pain and faster onset of action.6,12 Warming the anesthetic using a water bath, incubator, or autoclave can decrease pain without degradation of lidocaine or epinephrine.13 At a minimum, 30-gauge needles are preferred to minimize pain from needle insertion. Use of 33-gauge needles has shown benefit for injecting the face and scalp and may prove to be helpful injecting sensitive areas such as the digits.14 A slow injection technique is more comfortable for the patient, as rapid injection causes tissue distention.11
The ideal anesthetic for nail surgery would have a fast onset and a long duration of action, which would allow for shorter operation time as well as alleviation of pain postprocedure and some degree of vasoconstriction to help maintain a bloodless field. Lidocaine has the fastest time of onset (<1–3 minutes) but a short duration of action (30–120 minutes) and a vasodilatory effect. Bupivacaine takes 2 to 5 minutes to take effect and has a long duration of action (120–240 minutes) but a risk for cardiotoxicity. Ropivacaine is the preferred anesthetic by some nail surgeons because of its intermediate time of onset (1–15 minutes), long duration of action (120–360 minutes), and the benefit of some vasoconstriction.5,15 The addition of epinephrine has 2 main advantages: vasoconstriction and prolongation of anesthetic effects; the latter may help to alleviate postoperative pain. If there are no contraindications to its use (ie, severe hypertension, Raynaud phenomenon), it can be used safely in digital anesthesia without risk for ischemia or infarction.11
Digital anesthesia can be achieved by infiltration or using nerve blocks. One major difference between these 2 approaches is the time of onset of anesthesia, with the former being nearly instantaneous and the latter taking up to 15 minutes.16 There also usually is more prolonged pain at the site of needle insertion with nerve blocks compared to infiltration. The type of nail surgery being performed, the digit involved, and surgeon preference will determine the anesthetic method of choice.17
Pain management immediately following the procedure and for several days after is essential. Use of a longer-acting anesthetic, such as bupivacaine or ropivacaine, will provide anesthesia for several hours. A well-padded dressing serves to absorb blood and protect the nail and distal digit from trauma, as even minor trauma can exacerbate pain and bleeding. The patient should be instructed to apply ice to the surgical site and keep the ipsilateral extremity elevated for the next 2 days to reduce edema and pain.15 Written instructions are helpful, as anxiety during and after the procedure may limit the patient’s understanding and recollection of the verbal postoperative instructions. To maximize readability of the information, the National Institutes of Health and American Medical Association recommend that the instructions be written at a fourth- to sixth-grade reading level.18,19
A single dose of ibuprofen (400 mg) or acetaminophen (500 mg to 1 g) immediately before or after the procedure can reduce opioid use and postoperative pain.20 Gabapentin (300–1200 mg) given 1 to 2 hours before surgery may be considered in patients who are at high risk for postsurgical pain.21 Acetaminophen or nonsteroidal anti-inflammatory drugs (eg, ibuprofen [200–400 mg]) administered every 4 to 6 hours provides considerable pain reduction postprocedure. Nonsteroidal anti-inflammatory drugs may be superior to acetaminophen for pain control22 and carry a low risk for postoperative bleeding.23 Additionally, a combination of acetaminophen with a nonsteroidal anti-inflammatory drug for 3 doses may be more effective than either drug alone.24 Some patients may require an opioid combination, such as codeine plus acetaminophen, for a short time (up to 3 days) for pain relief following surgery. Excessive pain or pain lasting than more than 3 days is not normal or expected; in these cases, patients should return to the office to rule out ischemia or infection.
It is important to implement pain-minimizing strategies for nail surgeries. Because many of these approaches are derived from other surgical specialties, well-controlled clinical trials in patients undergoing nail surgery will be necessary to improve outcomes.
- Goktay F, Altan ZM, Talas A, et al. Anxiety among patients undergoing nail surgery and skin punch biopsy: effects of age, gender, educational status, and previous experience. J Cutan Med Surg. 2016;20:35-39.
- Ravitskiy L, Phillips PK, Roenigk RK, et al. The use of oral midazolam for perioperative anxiolysis of healthy patients undergoing Mohs surgery: conclusions from randomized controlled and prospective studies. J Am Acad Dermatol. 2011;64:310-322.
- Richert B. Anesthesia of the nail apparatus. In: Richert B, Di Chiacchio N, Haneke E, eds. Nail Surgery. New York, NY: Informa Healthcare; 2010:24-30.
- Egekvist H, Bjerring P, Arendt-Nielsen L. Pain and mechanical injury of human skin following needle insertions. Eur J Pain. 1999;3:41-49.
- Soriano TT, Beynet DP. Anesthesia and analgesia. In: Robinson J, Hanke CW, Siegel D, et al, eds. Surgery of the Skin. 2nd ed. New York, NY: Elsevier; 2010:43-63.
- Strazar AR, Leynes PG, Lalonde DH. Minimizing the pain of local anesthesia injection. Plast Reconstr Surg. 2013;132:675-684.
- Drahota A, Galloway E, Stores R, et al. Audiovisual distraction as an adjunct to pain and anxiety relief during minor surgery. Foot (Edinb). 2008;18:211-219.
- Browne J, Fung M, Donnelly M, et al. The use of EMLA reduces the pain associated with digital ring block for ingrowing toenail correction. Eur J Anaesthesiol. 2000;17:182-184.
- Hayward SC, Landorf KB, Redmond AC. Ice reduces needle-stick pain associated with a digital nerve block of the hallux. Foot. 2006;16:145-148.
- Kose O, Saylan S, Ediz N, et al. Effects of topical alkane vapocoolant spray on pain intensity prior to digital nerve block for ingrown nail surgery. Foot Ankle Spec. 2010;3:73-75.
- Jellinek NJ, Velez NF. Nail surgery: best way to obtain effective anesthesia. Dermatol Clin. 2015;33:265-271.
- Strazar R, Lalonde D. Minimizing injection pain in local anesthesia. CMAJ. 2012;184:2016.
- Hogan ME, vanderVaart S, Perampaladas K, et al. Systematic review and meta-analysis of the effect of warming local anesthetics on injection pain. Ann Emerg Med. 2011;58:86-98.e1.
- Zelickson BR, Goldberg LH, Rubenzik MK, et al. Finer needles reduce pain associated with injection of local anesthetic using a minimal insertion injection technique [published online October 6, 2017]. Dermatol Surg. doi:10.1097/DSS.0000000000001279.
- Haneke E. Nail surgery. Clin Dermatol. 2013;31:516-525.
- Vinycomb TI, Sahhar LJ. Comparison of local anesthetics for digital nerve blocks: a systematic review. J Hand Surg Am. 2014;39:744-51.e5.
- Jellinek NJ. Nail surgery: practical tips and treatment options. Dermatol Ther. 2007;20:68-74.
- How to write easy-to-read health materials. Medline Plus website. https://medlineplus.gov/etr.html. Updated June 28, 2017.
Accessed January 29, 2018. - Weis BD. Health Literacy: A Manual for Clinicians. Chicago, IL: American Medical Foundation, American Medical Association; 2003.
- Rosero EB, Joshi GP. Preemptive, preventive, multimodal analgesia: what do they really mean? Plast Reconstr Surg. 2014;134(4 suppl 2):85S-93S.
- Straube S, Derry S, Moore RA, et al. Single dose oral gabapentin for established acute postoperative pain in adults [published online May 12 2010]. Cochrane Database Syst Rev. doi:10.1002/14651858.CD008183.pub2.
- Bailey E, Worthington H, Coulthard P. Ibuprofen and/or paracetamol (acetaminophen) for pain relief after surgical removal of lower wisdom teeth, a Cochrane systematic review. Br Dent J. 2014;216:451-455.
- Glass JS, Hardy CL, Meeks NM, et al. Acute pain management in dermatology: risk assessment and treatment. J Am Acad Dermatol. 2015;73:543-560; quiz 561-562.
- Sniezek PJ, Brodland DG, Zitelli JA. A randomized controlled trial comparing acetaminophen, acetaminophen and ibuprofen, and acetaminophen and codeine for postoperative pain relief after Mohs surgery and cutaneous reconstruction. Dermatol Surg. 2011;37:1007-1013.
Nail surgery is an important part of dermatologic training and clinical practice, both for diagnosis and treatment of nail disorders as well as benign and malignant nail tumors. Patient comfort is essential prior to the procedure and while administering local anesthetics. Effective anesthesia facilitates nail unit biopsies, excisions, and other surgical nail procedures. Pain management immediately following the procedure and during the postoperative period are equally important.
Patients who undergo nail surgery may experience anxiety due to fear of a cancer diagnosis, pain during the surgery, or disfigurement from the procedure. This anxiety may lead to increased blood pressure, a decreased pain threshold, and mental and physical discomfort.1 A detailed explanation of the procedure itself as well as expectations following the surgery are helpful in diminishing these fears. Administration of a fast-acting benzodiazepine also may be helpful in these patients to decrease anxiety prior to the procedure.2
Attaining adequate anesthesia requires an understanding of digital anatomy, particularly innervation. Innervation of the digits is supplied by the volar and dorsal nerves, which divide into 3 branches at the distal interphalangeal joint, innervating the nail bed, the digital tip, and the pulp.3 Pacinian and Ruffini corpuscles and free-ended nociceptors activate nerve fibers that transmit pain impulses.4,5 Local anesthetics block pain transmission by impeding voltage-gated sodium channels located at free nerve endings. Pain from anesthesia may be due to both needle insertion and fluid infiltration.
Simple measures can maximize patient comfort during digital anesthesia. Both audiovisual distraction and interpersonal interaction can help to put the patient at ease.6,7 Application of topical anesthetic cream (1–2 hours prior to the procedure under occlusion),8 ice (at least 6 minutes),9 or an ethyl chloride spray can be applied to the nail folds prior to needle insertion to alleviate injection pain, but these methods do little for infiltration pain. Use of an ethyl chloride spray may be the preferred technique due to the rapidity of the analgesic effects (Figure).10 A vibrating massager also can be applied in close proximity to the site of needle insertion.11
Proper anesthetic preparation and technique also can minimize pain during injection. Because lidocaine 1% is acidic (pH, 6.09), buffering with sodium bicarbonate 8.4% can result in decreased injection pain and faster onset of action.6,12 Warming the anesthetic using a water bath, incubator, or autoclave can decrease pain without degradation of lidocaine or epinephrine.13 At a minimum, 30-gauge needles are preferred to minimize pain from needle insertion. Use of 33-gauge needles has shown benefit for injecting the face and scalp and may prove to be helpful injecting sensitive areas such as the digits.14 A slow injection technique is more comfortable for the patient, as rapid injection causes tissue distention.11
The ideal anesthetic for nail surgery would have a fast onset and a long duration of action, which would allow for shorter operation time as well as alleviation of pain postprocedure and some degree of vasoconstriction to help maintain a bloodless field. Lidocaine has the fastest time of onset (<1–3 minutes) but a short duration of action (30–120 minutes) and a vasodilatory effect. Bupivacaine takes 2 to 5 minutes to take effect and has a long duration of action (120–240 minutes) but a risk for cardiotoxicity. Ropivacaine is the preferred anesthetic by some nail surgeons because of its intermediate time of onset (1–15 minutes), long duration of action (120–360 minutes), and the benefit of some vasoconstriction.5,15 The addition of epinephrine has 2 main advantages: vasoconstriction and prolongation of anesthetic effects; the latter may help to alleviate postoperative pain. If there are no contraindications to its use (ie, severe hypertension, Raynaud phenomenon), it can be used safely in digital anesthesia without risk for ischemia or infarction.11
Digital anesthesia can be achieved by infiltration or using nerve blocks. One major difference between these 2 approaches is the time of onset of anesthesia, with the former being nearly instantaneous and the latter taking up to 15 minutes.16 There also usually is more prolonged pain at the site of needle insertion with nerve blocks compared to infiltration. The type of nail surgery being performed, the digit involved, and surgeon preference will determine the anesthetic method of choice.17
Pain management immediately following the procedure and for several days after is essential. Use of a longer-acting anesthetic, such as bupivacaine or ropivacaine, will provide anesthesia for several hours. A well-padded dressing serves to absorb blood and protect the nail and distal digit from trauma, as even minor trauma can exacerbate pain and bleeding. The patient should be instructed to apply ice to the surgical site and keep the ipsilateral extremity elevated for the next 2 days to reduce edema and pain.15 Written instructions are helpful, as anxiety during and after the procedure may limit the patient’s understanding and recollection of the verbal postoperative instructions. To maximize readability of the information, the National Institutes of Health and American Medical Association recommend that the instructions be written at a fourth- to sixth-grade reading level.18,19
A single dose of ibuprofen (400 mg) or acetaminophen (500 mg to 1 g) immediately before or after the procedure can reduce opioid use and postoperative pain.20 Gabapentin (300–1200 mg) given 1 to 2 hours before surgery may be considered in patients who are at high risk for postsurgical pain.21 Acetaminophen or nonsteroidal anti-inflammatory drugs (eg, ibuprofen [200–400 mg]) administered every 4 to 6 hours provides considerable pain reduction postprocedure. Nonsteroidal anti-inflammatory drugs may be superior to acetaminophen for pain control22 and carry a low risk for postoperative bleeding.23 Additionally, a combination of acetaminophen with a nonsteroidal anti-inflammatory drug for 3 doses may be more effective than either drug alone.24 Some patients may require an opioid combination, such as codeine plus acetaminophen, for a short time (up to 3 days) for pain relief following surgery. Excessive pain or pain lasting than more than 3 days is not normal or expected; in these cases, patients should return to the office to rule out ischemia or infection.
It is important to implement pain-minimizing strategies for nail surgeries. Because many of these approaches are derived from other surgical specialties, well-controlled clinical trials in patients undergoing nail surgery will be necessary to improve outcomes.
Nail surgery is an important part of dermatologic training and clinical practice, both for diagnosis and treatment of nail disorders as well as benign and malignant nail tumors. Patient comfort is essential prior to the procedure and while administering local anesthetics. Effective anesthesia facilitates nail unit biopsies, excisions, and other surgical nail procedures. Pain management immediately following the procedure and during the postoperative period are equally important.
Patients who undergo nail surgery may experience anxiety due to fear of a cancer diagnosis, pain during the surgery, or disfigurement from the procedure. This anxiety may lead to increased blood pressure, a decreased pain threshold, and mental and physical discomfort.1 A detailed explanation of the procedure itself as well as expectations following the surgery are helpful in diminishing these fears. Administration of a fast-acting benzodiazepine also may be helpful in these patients to decrease anxiety prior to the procedure.2
Attaining adequate anesthesia requires an understanding of digital anatomy, particularly innervation. Innervation of the digits is supplied by the volar and dorsal nerves, which divide into 3 branches at the distal interphalangeal joint, innervating the nail bed, the digital tip, and the pulp.3 Pacinian and Ruffini corpuscles and free-ended nociceptors activate nerve fibers that transmit pain impulses.4,5 Local anesthetics block pain transmission by impeding voltage-gated sodium channels located at free nerve endings. Pain from anesthesia may be due to both needle insertion and fluid infiltration.
Simple measures can maximize patient comfort during digital anesthesia. Both audiovisual distraction and interpersonal interaction can help to put the patient at ease.6,7 Application of topical anesthetic cream (1–2 hours prior to the procedure under occlusion),8 ice (at least 6 minutes),9 or an ethyl chloride spray can be applied to the nail folds prior to needle insertion to alleviate injection pain, but these methods do little for infiltration pain. Use of an ethyl chloride spray may be the preferred technique due to the rapidity of the analgesic effects (Figure).10 A vibrating massager also can be applied in close proximity to the site of needle insertion.11
Proper anesthetic preparation and technique also can minimize pain during injection. Because lidocaine 1% is acidic (pH, 6.09), buffering with sodium bicarbonate 8.4% can result in decreased injection pain and faster onset of action.6,12 Warming the anesthetic using a water bath, incubator, or autoclave can decrease pain without degradation of lidocaine or epinephrine.13 At a minimum, 30-gauge needles are preferred to minimize pain from needle insertion. Use of 33-gauge needles has shown benefit for injecting the face and scalp and may prove to be helpful injecting sensitive areas such as the digits.14 A slow injection technique is more comfortable for the patient, as rapid injection causes tissue distention.11
The ideal anesthetic for nail surgery would have a fast onset and a long duration of action, which would allow for shorter operation time as well as alleviation of pain postprocedure and some degree of vasoconstriction to help maintain a bloodless field. Lidocaine has the fastest time of onset (<1–3 minutes) but a short duration of action (30–120 minutes) and a vasodilatory effect. Bupivacaine takes 2 to 5 minutes to take effect and has a long duration of action (120–240 minutes) but a risk for cardiotoxicity. Ropivacaine is the preferred anesthetic by some nail surgeons because of its intermediate time of onset (1–15 minutes), long duration of action (120–360 minutes), and the benefit of some vasoconstriction.5,15 The addition of epinephrine has 2 main advantages: vasoconstriction and prolongation of anesthetic effects; the latter may help to alleviate postoperative pain. If there are no contraindications to its use (ie, severe hypertension, Raynaud phenomenon), it can be used safely in digital anesthesia without risk for ischemia or infarction.11
Digital anesthesia can be achieved by infiltration or using nerve blocks. One major difference between these 2 approaches is the time of onset of anesthesia, with the former being nearly instantaneous and the latter taking up to 15 minutes.16 There also usually is more prolonged pain at the site of needle insertion with nerve blocks compared to infiltration. The type of nail surgery being performed, the digit involved, and surgeon preference will determine the anesthetic method of choice.17
Pain management immediately following the procedure and for several days after is essential. Use of a longer-acting anesthetic, such as bupivacaine or ropivacaine, will provide anesthesia for several hours. A well-padded dressing serves to absorb blood and protect the nail and distal digit from trauma, as even minor trauma can exacerbate pain and bleeding. The patient should be instructed to apply ice to the surgical site and keep the ipsilateral extremity elevated for the next 2 days to reduce edema and pain.15 Written instructions are helpful, as anxiety during and after the procedure may limit the patient’s understanding and recollection of the verbal postoperative instructions. To maximize readability of the information, the National Institutes of Health and American Medical Association recommend that the instructions be written at a fourth- to sixth-grade reading level.18,19
A single dose of ibuprofen (400 mg) or acetaminophen (500 mg to 1 g) immediately before or after the procedure can reduce opioid use and postoperative pain.20 Gabapentin (300–1200 mg) given 1 to 2 hours before surgery may be considered in patients who are at high risk for postsurgical pain.21 Acetaminophen or nonsteroidal anti-inflammatory drugs (eg, ibuprofen [200–400 mg]) administered every 4 to 6 hours provides considerable pain reduction postprocedure. Nonsteroidal anti-inflammatory drugs may be superior to acetaminophen for pain control22 and carry a low risk for postoperative bleeding.23 Additionally, a combination of acetaminophen with a nonsteroidal anti-inflammatory drug for 3 doses may be more effective than either drug alone.24 Some patients may require an opioid combination, such as codeine plus acetaminophen, for a short time (up to 3 days) for pain relief following surgery. Excessive pain or pain lasting than more than 3 days is not normal or expected; in these cases, patients should return to the office to rule out ischemia or infection.
It is important to implement pain-minimizing strategies for nail surgeries. Because many of these approaches are derived from other surgical specialties, well-controlled clinical trials in patients undergoing nail surgery will be necessary to improve outcomes.
- Goktay F, Altan ZM, Talas A, et al. Anxiety among patients undergoing nail surgery and skin punch biopsy: effects of age, gender, educational status, and previous experience. J Cutan Med Surg. 2016;20:35-39.
- Ravitskiy L, Phillips PK, Roenigk RK, et al. The use of oral midazolam for perioperative anxiolysis of healthy patients undergoing Mohs surgery: conclusions from randomized controlled and prospective studies. J Am Acad Dermatol. 2011;64:310-322.
- Richert B. Anesthesia of the nail apparatus. In: Richert B, Di Chiacchio N, Haneke E, eds. Nail Surgery. New York, NY: Informa Healthcare; 2010:24-30.
- Egekvist H, Bjerring P, Arendt-Nielsen L. Pain and mechanical injury of human skin following needle insertions. Eur J Pain. 1999;3:41-49.
- Soriano TT, Beynet DP. Anesthesia and analgesia. In: Robinson J, Hanke CW, Siegel D, et al, eds. Surgery of the Skin. 2nd ed. New York, NY: Elsevier; 2010:43-63.
- Strazar AR, Leynes PG, Lalonde DH. Minimizing the pain of local anesthesia injection. Plast Reconstr Surg. 2013;132:675-684.
- Drahota A, Galloway E, Stores R, et al. Audiovisual distraction as an adjunct to pain and anxiety relief during minor surgery. Foot (Edinb). 2008;18:211-219.
- Browne J, Fung M, Donnelly M, et al. The use of EMLA reduces the pain associated with digital ring block for ingrowing toenail correction. Eur J Anaesthesiol. 2000;17:182-184.
- Hayward SC, Landorf KB, Redmond AC. Ice reduces needle-stick pain associated with a digital nerve block of the hallux. Foot. 2006;16:145-148.
- Kose O, Saylan S, Ediz N, et al. Effects of topical alkane vapocoolant spray on pain intensity prior to digital nerve block for ingrown nail surgery. Foot Ankle Spec. 2010;3:73-75.
- Jellinek NJ, Velez NF. Nail surgery: best way to obtain effective anesthesia. Dermatol Clin. 2015;33:265-271.
- Strazar R, Lalonde D. Minimizing injection pain in local anesthesia. CMAJ. 2012;184:2016.
- Hogan ME, vanderVaart S, Perampaladas K, et al. Systematic review and meta-analysis of the effect of warming local anesthetics on injection pain. Ann Emerg Med. 2011;58:86-98.e1.
- Zelickson BR, Goldberg LH, Rubenzik MK, et al. Finer needles reduce pain associated with injection of local anesthetic using a minimal insertion injection technique [published online October 6, 2017]. Dermatol Surg. doi:10.1097/DSS.0000000000001279.
- Haneke E. Nail surgery. Clin Dermatol. 2013;31:516-525.
- Vinycomb TI, Sahhar LJ. Comparison of local anesthetics for digital nerve blocks: a systematic review. J Hand Surg Am. 2014;39:744-51.e5.
- Jellinek NJ. Nail surgery: practical tips and treatment options. Dermatol Ther. 2007;20:68-74.
- How to write easy-to-read health materials. Medline Plus website. https://medlineplus.gov/etr.html. Updated June 28, 2017.
Accessed January 29, 2018. - Weis BD. Health Literacy: A Manual for Clinicians. Chicago, IL: American Medical Foundation, American Medical Association; 2003.
- Rosero EB, Joshi GP. Preemptive, preventive, multimodal analgesia: what do they really mean? Plast Reconstr Surg. 2014;134(4 suppl 2):85S-93S.
- Straube S, Derry S, Moore RA, et al. Single dose oral gabapentin for established acute postoperative pain in adults [published online May 12 2010]. Cochrane Database Syst Rev. doi:10.1002/14651858.CD008183.pub2.
- Bailey E, Worthington H, Coulthard P. Ibuprofen and/or paracetamol (acetaminophen) for pain relief after surgical removal of lower wisdom teeth, a Cochrane systematic review. Br Dent J. 2014;216:451-455.
- Glass JS, Hardy CL, Meeks NM, et al. Acute pain management in dermatology: risk assessment and treatment. J Am Acad Dermatol. 2015;73:543-560; quiz 561-562.
- Sniezek PJ, Brodland DG, Zitelli JA. A randomized controlled trial comparing acetaminophen, acetaminophen and ibuprofen, and acetaminophen and codeine for postoperative pain relief after Mohs surgery and cutaneous reconstruction. Dermatol Surg. 2011;37:1007-1013.
- Goktay F, Altan ZM, Talas A, et al. Anxiety among patients undergoing nail surgery and skin punch biopsy: effects of age, gender, educational status, and previous experience. J Cutan Med Surg. 2016;20:35-39.
- Ravitskiy L, Phillips PK, Roenigk RK, et al. The use of oral midazolam for perioperative anxiolysis of healthy patients undergoing Mohs surgery: conclusions from randomized controlled and prospective studies. J Am Acad Dermatol. 2011;64:310-322.
- Richert B. Anesthesia of the nail apparatus. In: Richert B, Di Chiacchio N, Haneke E, eds. Nail Surgery. New York, NY: Informa Healthcare; 2010:24-30.
- Egekvist H, Bjerring P, Arendt-Nielsen L. Pain and mechanical injury of human skin following needle insertions. Eur J Pain. 1999;3:41-49.
- Soriano TT, Beynet DP. Anesthesia and analgesia. In: Robinson J, Hanke CW, Siegel D, et al, eds. Surgery of the Skin. 2nd ed. New York, NY: Elsevier; 2010:43-63.
- Strazar AR, Leynes PG, Lalonde DH. Minimizing the pain of local anesthesia injection. Plast Reconstr Surg. 2013;132:675-684.
- Drahota A, Galloway E, Stores R, et al. Audiovisual distraction as an adjunct to pain and anxiety relief during minor surgery. Foot (Edinb). 2008;18:211-219.
- Browne J, Fung M, Donnelly M, et al. The use of EMLA reduces the pain associated with digital ring block for ingrowing toenail correction. Eur J Anaesthesiol. 2000;17:182-184.
- Hayward SC, Landorf KB, Redmond AC. Ice reduces needle-stick pain associated with a digital nerve block of the hallux. Foot. 2006;16:145-148.
- Kose O, Saylan S, Ediz N, et al. Effects of topical alkane vapocoolant spray on pain intensity prior to digital nerve block for ingrown nail surgery. Foot Ankle Spec. 2010;3:73-75.
- Jellinek NJ, Velez NF. Nail surgery: best way to obtain effective anesthesia. Dermatol Clin. 2015;33:265-271.
- Strazar R, Lalonde D. Minimizing injection pain in local anesthesia. CMAJ. 2012;184:2016.
- Hogan ME, vanderVaart S, Perampaladas K, et al. Systematic review and meta-analysis of the effect of warming local anesthetics on injection pain. Ann Emerg Med. 2011;58:86-98.e1.
- Zelickson BR, Goldberg LH, Rubenzik MK, et al. Finer needles reduce pain associated with injection of local anesthetic using a minimal insertion injection technique [published online October 6, 2017]. Dermatol Surg. doi:10.1097/DSS.0000000000001279.
- Haneke E. Nail surgery. Clin Dermatol. 2013;31:516-525.
- Vinycomb TI, Sahhar LJ. Comparison of local anesthetics for digital nerve blocks: a systematic review. J Hand Surg Am. 2014;39:744-51.e5.
- Jellinek NJ. Nail surgery: practical tips and treatment options. Dermatol Ther. 2007;20:68-74.
- How to write easy-to-read health materials. Medline Plus website. https://medlineplus.gov/etr.html. Updated June 28, 2017.
Accessed January 29, 2018. - Weis BD. Health Literacy: A Manual for Clinicians. Chicago, IL: American Medical Foundation, American Medical Association; 2003.
- Rosero EB, Joshi GP. Preemptive, preventive, multimodal analgesia: what do they really mean? Plast Reconstr Surg. 2014;134(4 suppl 2):85S-93S.
- Straube S, Derry S, Moore RA, et al. Single dose oral gabapentin for established acute postoperative pain in adults [published online May 12 2010]. Cochrane Database Syst Rev. doi:10.1002/14651858.CD008183.pub2.
- Bailey E, Worthington H, Coulthard P. Ibuprofen and/or paracetamol (acetaminophen) for pain relief after surgical removal of lower wisdom teeth, a Cochrane systematic review. Br Dent J. 2014;216:451-455.
- Glass JS, Hardy CL, Meeks NM, et al. Acute pain management in dermatology: risk assessment and treatment. J Am Acad Dermatol. 2015;73:543-560; quiz 561-562.
- Sniezek PJ, Brodland DG, Zitelli JA. A randomized controlled trial comparing acetaminophen, acetaminophen and ibuprofen, and acetaminophen and codeine for postoperative pain relief after Mohs surgery and cutaneous reconstruction. Dermatol Surg. 2011;37:1007-1013.
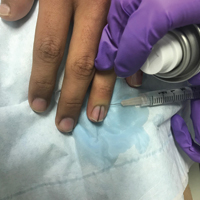
MDedge Daily News: Flu set to break hospital records
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Hospitals race toward a new flu record, the drug pipeline is filling up for inflammatory bowel disease, there’s bad news for cardiovascular prevention in type 2 diabetes, and research points to elective induction at 39 weeks.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Hospitals race toward a new flu record, the drug pipeline is filling up for inflammatory bowel disease, there’s bad news for cardiovascular prevention in type 2 diabetes, and research points to elective induction at 39 weeks.
Listen to the MDedge Daily News podcast for all the details on today’s top news.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Hospitals race toward a new flu record, the drug pipeline is filling up for inflammatory bowel disease, there’s bad news for cardiovascular prevention in type 2 diabetes, and research points to elective induction at 39 weeks.
Listen to the MDedge Daily News podcast for all the details on today’s top news.

ASCO expands recommendations on bone-modifying agents in myeloma
Bisphosphonates should be prescribed for any patient receiving treatment for active multiple myeloma, regardless of whether or not there is evidence of lytic bone destruction or spinal compression fracture, according to updated guidelines from the American Society of Clinical Oncology.
Previous guidelines from the society, last updated in 2007, recommended the use of intravenous bisphosphonates for patients with myeloma with evidence of bone disease, according to the expert panel that drafted the update.

“Fewer adverse events related to renal toxicity have been noted with denosumab, compared with zoledronic acid,” and “this may be preferred in patients with compromised renal function,” wrote the expert panel, led by cochairs Kenneth C. Anderson, MD, of Dana-Farber Cancer Institute, Boston, and Robert A. Kyle, MD, of Mayo Clinic, Rochester, Minn.
ASCO guidelines on bisphosphonates in myeloma were first drafted in 2002 and then updated in 2007. The new recommendations on bone-modifying therapy in myeloma are based on review of an additional 35 publications. The new guidelines are “consistent with the previous recommendations” while updating indications for therapy and information on denosumab, according to the expert panel.
Evidence that myeloma patients without lytic bone disease will benefit from intravenous bisphosphonates comes from the randomized MRC IX trial, in which patients who received zoledronic acid had reduced skeletal-related events at relapse and improved progression-free survival.
Denosumab, a receptor activator of nuclear factor kappa-B ligand (RANKL) inhibitor, was noninferior to zoledronic acid for prevention of skeletal-related events in a randomized phase 3 clinical trial; however, it is “more expensive than zoledronic acid or pamidronate and must be considered in treatment decisions,” the guidelines authors wrote.
The total price in the United States for a 1-year treatment cycle of denosumab is just under $26,000, according to data included in the ASCO guideline. By comparison, the 1-year treatment cycle price for the bisphosphonates ranges from $214 to $697, depending on the regimen.
When intravenous bisphosphonate therapy is warranted, the guideline-recommended schedule is infusion of zoledronic acid 4 mg over at least 15 minutes, or pamidronate 90 mg over 2 hours, every 3-4 weeks.
The guidelines also address osteonecrosis of the jaw (ONJ), a major complication observed not only with the potent bisphosphonates pamidronate and zoledronic acid, but also with denosumab.
The panel said they were in agreement with revised labels from the Food and Drug Administration for zoledronic acid and pamidronate, among other papers or statements addressing ONJ and noted that patients need a comprehensive dental exam and preventive dentistry as appropriate before starting bone-modifying therapy.
“The risk of ONJ has prompted the use of less-frequent dosing of zoledronic acid, which may be an option for patients,” they said in their report.
Guideline authors reported ties to Amgen, Celgene, Millennium Pharmaceuticals, Gilead Sciences, Bristol-Myers Squibb, Novartis, Pfizer, and others.
hematologynews@frontlinemedcom.com
SOURCE: Anderson K et al. J Clin Oncol. 2018 Jan 17:JCO2017766402. doi: 10.1200/JCO.2017.76.6402.
Bisphosphonates should be prescribed for any patient receiving treatment for active multiple myeloma, regardless of whether or not there is evidence of lytic bone destruction or spinal compression fracture, according to updated guidelines from the American Society of Clinical Oncology.
Previous guidelines from the society, last updated in 2007, recommended the use of intravenous bisphosphonates for patients with myeloma with evidence of bone disease, according to the expert panel that drafted the update.
“Fewer adverse events related to renal toxicity have been noted with denosumab, compared with zoledronic acid,” and “this may be preferred in patients with compromised renal function,” wrote the expert panel, led by cochairs Kenneth C. Anderson, MD, of Dana-Farber Cancer Institute, Boston, and Robert A. Kyle, MD, of Mayo Clinic, Rochester, Minn.
ASCO guidelines on bisphosphonates in myeloma were first drafted in 2002 and then updated in 2007. The new recommendations on bone-modifying therapy in myeloma are based on review of an additional 35 publications. The new guidelines are “consistent with the previous recommendations” while updating indications for therapy and information on denosumab, according to the expert panel.
Evidence that myeloma patients without lytic bone disease will benefit from intravenous bisphosphonates comes from the randomized MRC IX trial, in which patients who received zoledronic acid had reduced skeletal-related events at relapse and improved progression-free survival.
Denosumab, a receptor activator of nuclear factor kappa-B ligand (RANKL) inhibitor, was noninferior to zoledronic acid for prevention of skeletal-related events in a randomized phase 3 clinical trial; however, it is “more expensive than zoledronic acid or pamidronate and must be considered in treatment decisions,” the guidelines authors wrote.
The total price in the United States for a 1-year treatment cycle of denosumab is just under $26,000, according to data included in the ASCO guideline. By comparison, the 1-year treatment cycle price for the bisphosphonates ranges from $214 to $697, depending on the regimen.
When intravenous bisphosphonate therapy is warranted, the guideline-recommended schedule is infusion of zoledronic acid 4 mg over at least 15 minutes, or pamidronate 90 mg over 2 hours, every 3-4 weeks.
The guidelines also address osteonecrosis of the jaw (ONJ), a major complication observed not only with the potent bisphosphonates pamidronate and zoledronic acid, but also with denosumab.
The panel said they were in agreement with revised labels from the Food and Drug Administration for zoledronic acid and pamidronate, among other papers or statements addressing ONJ and noted that patients need a comprehensive dental exam and preventive dentistry as appropriate before starting bone-modifying therapy.
“The risk of ONJ has prompted the use of less-frequent dosing of zoledronic acid, which may be an option for patients,” they said in their report.
Guideline authors reported ties to Amgen, Celgene, Millennium Pharmaceuticals, Gilead Sciences, Bristol-Myers Squibb, Novartis, Pfizer, and others.
hematologynews@frontlinemedcom.com
SOURCE: Anderson K et al. J Clin Oncol. 2018 Jan 17:JCO2017766402. doi: 10.1200/JCO.2017.76.6402.
Bisphosphonates should be prescribed for any patient receiving treatment for active multiple myeloma, regardless of whether or not there is evidence of lytic bone destruction or spinal compression fracture, according to updated guidelines from the American Society of Clinical Oncology.
Previous guidelines from the society, last updated in 2007, recommended the use of intravenous bisphosphonates for patients with myeloma with evidence of bone disease, according to the expert panel that drafted the update.
“Fewer adverse events related to renal toxicity have been noted with denosumab, compared with zoledronic acid,” and “this may be preferred in patients with compromised renal function,” wrote the expert panel, led by cochairs Kenneth C. Anderson, MD, of Dana-Farber Cancer Institute, Boston, and Robert A. Kyle, MD, of Mayo Clinic, Rochester, Minn.
ASCO guidelines on bisphosphonates in myeloma were first drafted in 2002 and then updated in 2007. The new recommendations on bone-modifying therapy in myeloma are based on review of an additional 35 publications. The new guidelines are “consistent with the previous recommendations” while updating indications for therapy and information on denosumab, according to the expert panel.
Evidence that myeloma patients without lytic bone disease will benefit from intravenous bisphosphonates comes from the randomized MRC IX trial, in which patients who received zoledronic acid had reduced skeletal-related events at relapse and improved progression-free survival.
Denosumab, a receptor activator of nuclear factor kappa-B ligand (RANKL) inhibitor, was noninferior to zoledronic acid for prevention of skeletal-related events in a randomized phase 3 clinical trial; however, it is “more expensive than zoledronic acid or pamidronate and must be considered in treatment decisions,” the guidelines authors wrote.
The total price in the United States for a 1-year treatment cycle of denosumab is just under $26,000, according to data included in the ASCO guideline. By comparison, the 1-year treatment cycle price for the bisphosphonates ranges from $214 to $697, depending on the regimen.
When intravenous bisphosphonate therapy is warranted, the guideline-recommended schedule is infusion of zoledronic acid 4 mg over at least 15 minutes, or pamidronate 90 mg over 2 hours, every 3-4 weeks.
The guidelines also address osteonecrosis of the jaw (ONJ), a major complication observed not only with the potent bisphosphonates pamidronate and zoledronic acid, but also with denosumab.
The panel said they were in agreement with revised labels from the Food and Drug Administration for zoledronic acid and pamidronate, among other papers or statements addressing ONJ and noted that patients need a comprehensive dental exam and preventive dentistry as appropriate before starting bone-modifying therapy.
“The risk of ONJ has prompted the use of less-frequent dosing of zoledronic acid, which may be an option for patients,” they said in their report.
Guideline authors reported ties to Amgen, Celgene, Millennium Pharmaceuticals, Gilead Sciences, Bristol-Myers Squibb, Novartis, Pfizer, and others.
hematologynews@frontlinemedcom.com
SOURCE: Anderson K et al. J Clin Oncol. 2018 Jan 17:JCO2017766402. doi: 10.1200/JCO.2017.76.6402.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
nPEP for HIV: Updated CDC guidelines available for primary care physicians
In 2016, the Centers for Disease Control and Prevention provided health care providers with updated recommendations for nonoccupational postexposure prophylaxis (nPEP) with antiretroviral drugs to prevent transmission of HIV following sexual interaction, injection-drug use, or other nonoccupational exposures.1 The new recommendations include the use of more effective and more tolerable drug regimens that employ antiretroviral medications that were approved since the previous guidelines came out in 2005; they also provide updated guidance on exposure assessment, baseline and follow-up HIV testing, and longer-term prevention measures, such as pre-exposure prophylaxis (PrEP).
Screening for HIV infection has been expanding broadly in all health care settings over the past decade, so primary care physicians play an increasingly vital role in preventing HIV infection. Today, primary care physicians are also often the most likely “go-to” health care provider when patients think they may have been exposed to HIV. Clinically, this is an emergency situation, so time is of the essence: Treatment with three powerful antiretrovirals must be initiated within a few hours of – but no later than 72 hours after – an isolated exposure to blood, genital secretions, or other potentially infectious body fluids that may contain HIV.
The key issue for primary care physicians, especially those who have never prescribed PEP before, is advance planning. What you do up front, in terms of organizing materials and training staff, is worth the effort because there is so much at stake – for your patients and for society. The good news is that once you have an established nPEP protocol in place, it stays in place. When a patient asks for help, the protocol kicks in automatically.
Getting ready for nPEP
Prepare your staff:
- Educate your whole staff about the urgency of seeing potential nPEP patients immediately.
- Choose the staff person in your office who will submit requests for PEP medications to the pharmacy and/or pharmaceutical companies; your financial reimbursement staff person is likely a good candidate for this job.
- Learn about patient assistance programs (for uninsured or underinsured patients) and crime victims compensation programs (reimbursement or emergency awards for victims of violent crimes, including rape, for various out-of-pocket expenses including medical expenses).
Keep paperwork and materials on hand:
- Have information and forms for patient assistance programs for pharmaceutical companies supplying the drugs. Pharmaceutical companies are aware of the urgency for nPEP medications and are ready to respond immediately. They may mail the medication so it arrives the next day or, more likely, fax a voucher or other information for the patient to present to a local pharmacist who will fill the prescription.
- Have information on your state’s crime victims compensation program available.
- Consider keeping nPEP Starter Packs (with an initial 3-7 days’ worth of medication) readily available in your office.
Rapid evaluation of patients seeking care after potential exposure to HIV
Effective delivery of nPEP requires prompt initial evaluation of patients and assessment of HIV transmission risk. Take a methodical, step-by-step history of the exposure to address the following basic questions:
- Date and time of exposure? nPEP should be initiated as soon as possible after HIV exposure; it is unlikely to be effective if not initiated within 72 hours or less.
- Frequency of exposure? Type/route of exposure? nPEP is generally reserved for isolated or infrequent exposures that present a substantial risk for HIV acquisition (see Table 1 on HIV acquisition risk below).
- HIV status of exposure source? If the source is positive, is the source person on HIV treatment with antiretroviral therapy? If unknown, is the source person an injecting drug user or a man who has sex with men (MSM)?
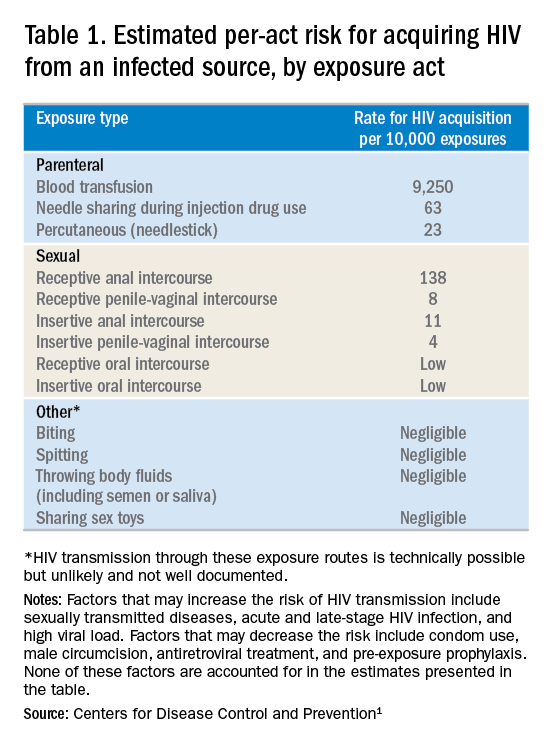
Based on the initial evaluation, is nPEP recommended?
Answers to the questions asked during the initial evaluation of the patient will determine whether nPEP is indicated. Along with its updated recommendations, the CDC provided an algorithm to help guide evaluation and treatment.
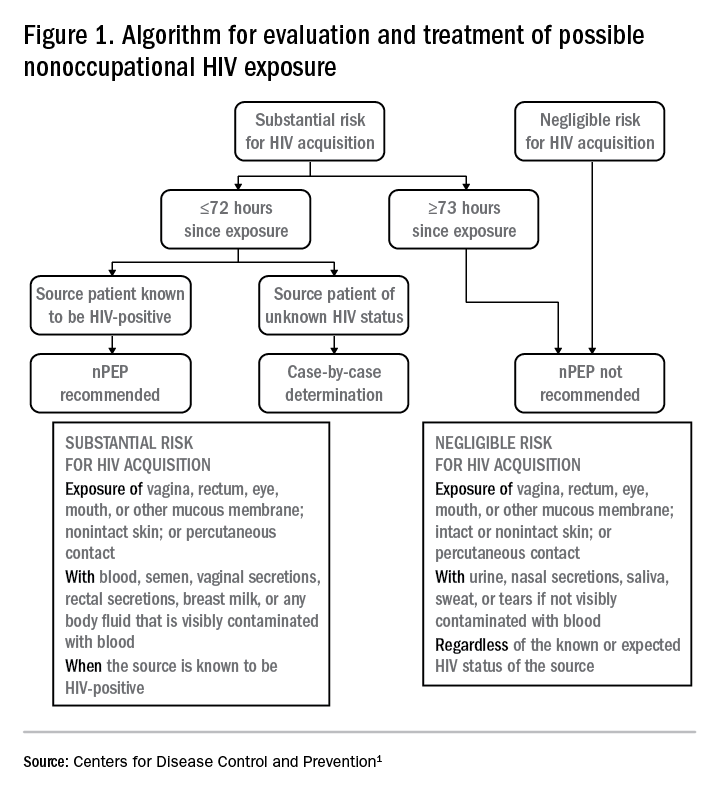
Preferred HIV test
Administer an HIV test to all patients considered for nPEP, preferably the rapid combined antigen and antibody test (Ag/Ab), or just the antibody test if the Ag/Ab test is not available. nPEP is indicated only for persons without HIV infections. However, if results are not available during the initial evaluation, assume the patient is not infected. If indicated and started, nPEP can be discontinued if tests later shown the patient already has an HIV infection.
Laboratory testing
If nPEP is indicated, conduct laboratory testing. Lab testing is required to document the patient’s HIV status (and that of the source person, when available), identify and manage other conditions potentially resulting from exposure, identify conditions that may affect the nPEP medication regimen, and monitor safety or toxicities to the prescribed regimen.
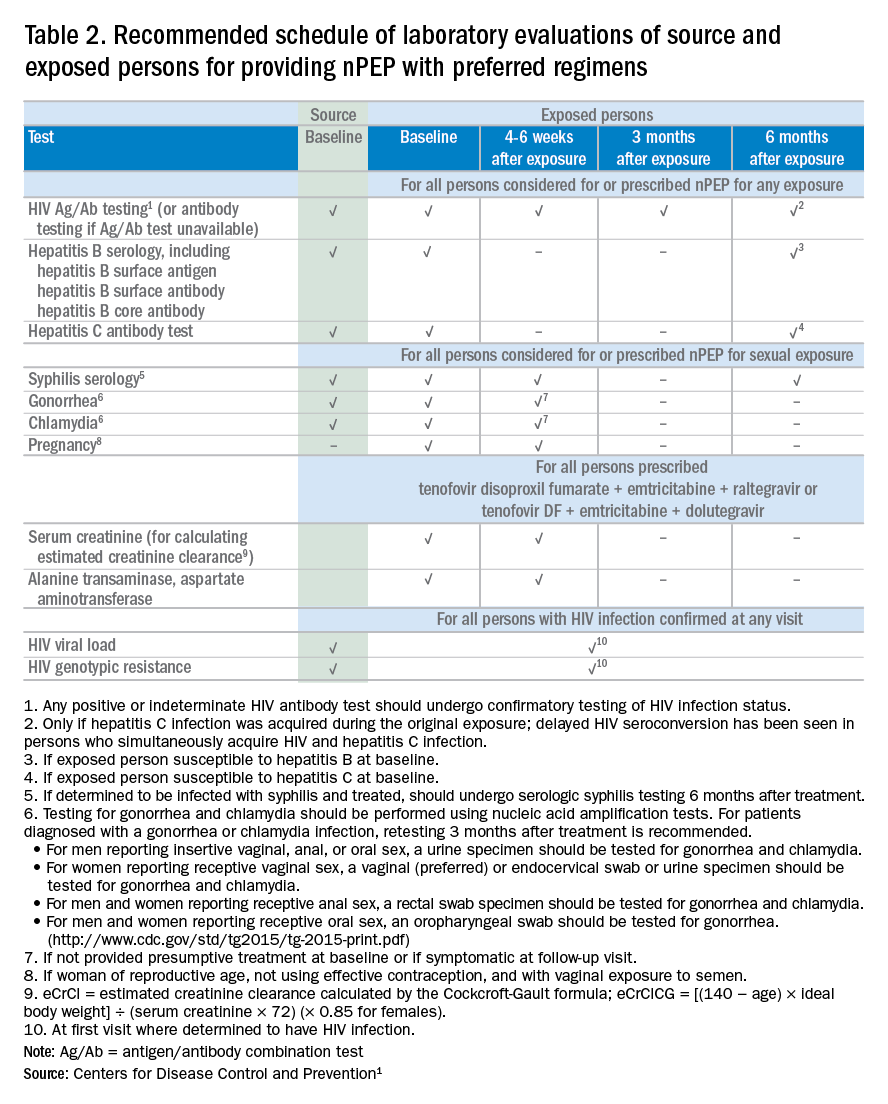
nPEP treatment regimen for otherwise healthy adults and adolescents
In the absence of randomized clinical trials, data from a case/control study demonstrating an 81% reduction of HIV transmission after use of occupational PEP among hospital workers remains the strongest evidence for the benefit of nPEP.1,2 For patients offered nPEP, recommended treatment includes prescribing either of the following regimens for 28 days:
- Preferred regimen: tenofovir disoproxil fumarate (TDF) (300 mg) with emtricitabine (FTC) (200 mg) once daily plus either raltegravir (RAL) 400 mg twice daily or dolutegravir (DTG) 50 mg daily.
- Alternative regimen: TDF (300 mg) with FTC (200 mg) once daily plus darunavir (DRV) (800 mg) and ritonavir (RTV) (100 mg) once daily.
Additional considerations and nPEP treatment regimens for children, patients with decreased renal function, and pregnant women are included in the CDC guidelines.
Crucial Information for Patients on nPEP
Emphasize the importance of proper dosing and adherence.
Review the patient information for each drug in the regimen, specifically the black boxes, warnings, and side effects, and counsel your patients accordingly.
Transitioning from nPEP to PrEP or from PrEP to nPEP
If you have a patient who engages in behavior that places them at risk for frequent, recurrent exposures to HIV, consider transitioning them to PrEP (pre-exposure prophylaxis) following their 28-day course of nPEP.3 PrEP is a two-drug regimen taken daily on an ongoing basis.
Additionally, for patients who are already on PrEP but who have not taken their medications within a week before the possible exposure, consider initiating nPEP for 28 days and then reintroducing PrEP if their HIV status is negative and the problems with adherence can be addressed moving forward.
Raising Awareness About nPEP
Many people never expect to be exposed to HIV and may not know about the availability of PEP in an emergency situation. You can help raise awareness by making educational materials available in your waiting rooms and exam rooms. Brochures and other HIV/AIDS educational materials for patients are available from the CDC Act Against AIDS campaign.
Summary

Dr. Dominguez is a Captain, U.S. Public Health Service, epidemiology branch, division of HIV/AIDS prevention, CDC.
Additional resources
- The CDC recommends that everyone between the ages of 13 and 64 get tested for HIV at least once as part of routine health care. As part of its Act Against AIDS initiative, the CDC developed the HIV Screening Standard Care program, which provides free tools and resources to help clinicians and nurses incorporate routine HIV screening into primary care settings.
- HIV guidelines and recommendations .
- Postexposure prophylaxis (PEP)
- Pre-Exposure prophylaxis (PrEP)
References
1. Centers for Disease Control and Prevention. Updated guidelines for antiretroviral postexposure prophylaxis after sexual, injection drug use, or other nonoccupational exposure to HIV. United States, 2016. Accessed March 6, 2017.
2. Cardo DM et al. New Engl J Med. 1997;337(21):1485-90.
3. Centers for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States–2014: a clinical practice guideline. Accessed March 6, 2017.
In 2016, the Centers for Disease Control and Prevention provided health care providers with updated recommendations for nonoccupational postexposure prophylaxis (nPEP) with antiretroviral drugs to prevent transmission of HIV following sexual interaction, injection-drug use, or other nonoccupational exposures.1 The new recommendations include the use of more effective and more tolerable drug regimens that employ antiretroviral medications that were approved since the previous guidelines came out in 2005; they also provide updated guidance on exposure assessment, baseline and follow-up HIV testing, and longer-term prevention measures, such as pre-exposure prophylaxis (PrEP).
Screening for HIV infection has been expanding broadly in all health care settings over the past decade, so primary care physicians play an increasingly vital role in preventing HIV infection. Today, primary care physicians are also often the most likely “go-to” health care provider when patients think they may have been exposed to HIV. Clinically, this is an emergency situation, so time is of the essence: Treatment with three powerful antiretrovirals must be initiated within a few hours of – but no later than 72 hours after – an isolated exposure to blood, genital secretions, or other potentially infectious body fluids that may contain HIV.
The key issue for primary care physicians, especially those who have never prescribed PEP before, is advance planning. What you do up front, in terms of organizing materials and training staff, is worth the effort because there is so much at stake – for your patients and for society. The good news is that once you have an established nPEP protocol in place, it stays in place. When a patient asks for help, the protocol kicks in automatically.
Getting ready for nPEP
Prepare your staff:
- Educate your whole staff about the urgency of seeing potential nPEP patients immediately.
- Choose the staff person in your office who will submit requests for PEP medications to the pharmacy and/or pharmaceutical companies; your financial reimbursement staff person is likely a good candidate for this job.
- Learn about patient assistance programs (for uninsured or underinsured patients) and crime victims compensation programs (reimbursement or emergency awards for victims of violent crimes, including rape, for various out-of-pocket expenses including medical expenses).
Keep paperwork and materials on hand:
- Have information and forms for patient assistance programs for pharmaceutical companies supplying the drugs. Pharmaceutical companies are aware of the urgency for nPEP medications and are ready to respond immediately. They may mail the medication so it arrives the next day or, more likely, fax a voucher or other information for the patient to present to a local pharmacist who will fill the prescription.
- Have information on your state’s crime victims compensation program available.
- Consider keeping nPEP Starter Packs (with an initial 3-7 days’ worth of medication) readily available in your office.
Rapid evaluation of patients seeking care after potential exposure to HIV
Effective delivery of nPEP requires prompt initial evaluation of patients and assessment of HIV transmission risk. Take a methodical, step-by-step history of the exposure to address the following basic questions:
- Date and time of exposure? nPEP should be initiated as soon as possible after HIV exposure; it is unlikely to be effective if not initiated within 72 hours or less.
- Frequency of exposure? Type/route of exposure? nPEP is generally reserved for isolated or infrequent exposures that present a substantial risk for HIV acquisition (see Table 1 on HIV acquisition risk below).
- HIV status of exposure source? If the source is positive, is the source person on HIV treatment with antiretroviral therapy? If unknown, is the source person an injecting drug user or a man who has sex with men (MSM)?
Based on the initial evaluation, is nPEP recommended?
Answers to the questions asked during the initial evaluation of the patient will determine whether nPEP is indicated. Along with its updated recommendations, the CDC provided an algorithm to help guide evaluation and treatment.
Preferred HIV test
Administer an HIV test to all patients considered for nPEP, preferably the rapid combined antigen and antibody test (Ag/Ab), or just the antibody test if the Ag/Ab test is not available. nPEP is indicated only for persons without HIV infections. However, if results are not available during the initial evaluation, assume the patient is not infected. If indicated and started, nPEP can be discontinued if tests later shown the patient already has an HIV infection.
Laboratory testing
If nPEP is indicated, conduct laboratory testing. Lab testing is required to document the patient’s HIV status (and that of the source person, when available), identify and manage other conditions potentially resulting from exposure, identify conditions that may affect the nPEP medication regimen, and monitor safety or toxicities to the prescribed regimen.
nPEP treatment regimen for otherwise healthy adults and adolescents
In the absence of randomized clinical trials, data from a case/control study demonstrating an 81% reduction of HIV transmission after use of occupational PEP among hospital workers remains the strongest evidence for the benefit of nPEP.1,2 For patients offered nPEP, recommended treatment includes prescribing either of the following regimens for 28 days:
- Preferred regimen: tenofovir disoproxil fumarate (TDF) (300 mg) with emtricitabine (FTC) (200 mg) once daily plus either raltegravir (RAL) 400 mg twice daily or dolutegravir (DTG) 50 mg daily.
- Alternative regimen: TDF (300 mg) with FTC (200 mg) once daily plus darunavir (DRV) (800 mg) and ritonavir (RTV) (100 mg) once daily.
Additional considerations and nPEP treatment regimens for children, patients with decreased renal function, and pregnant women are included in the CDC guidelines.
Crucial Information for Patients on nPEP
Emphasize the importance of proper dosing and adherence.
Review the patient information for each drug in the regimen, specifically the black boxes, warnings, and side effects, and counsel your patients accordingly.
Transitioning from nPEP to PrEP or from PrEP to nPEP
If you have a patient who engages in behavior that places them at risk for frequent, recurrent exposures to HIV, consider transitioning them to PrEP (pre-exposure prophylaxis) following their 28-day course of nPEP.3 PrEP is a two-drug regimen taken daily on an ongoing basis.
Additionally, for patients who are already on PrEP but who have not taken their medications within a week before the possible exposure, consider initiating nPEP for 28 days and then reintroducing PrEP if their HIV status is negative and the problems with adherence can be addressed moving forward.
Raising Awareness About nPEP
Many people never expect to be exposed to HIV and may not know about the availability of PEP in an emergency situation. You can help raise awareness by making educational materials available in your waiting rooms and exam rooms. Brochures and other HIV/AIDS educational materials for patients are available from the CDC Act Against AIDS campaign.
Summary
Dr. Dominguez is a Captain, U.S. Public Health Service, epidemiology branch, division of HIV/AIDS prevention, CDC.
Additional resources
- The CDC recommends that everyone between the ages of 13 and 64 get tested for HIV at least once as part of routine health care. As part of its Act Against AIDS initiative, the CDC developed the HIV Screening Standard Care program, which provides free tools and resources to help clinicians and nurses incorporate routine HIV screening into primary care settings.
- HIV guidelines and recommendations .
- Postexposure prophylaxis (PEP)
- Pre-Exposure prophylaxis (PrEP)
References
1. Centers for Disease Control and Prevention. Updated guidelines for antiretroviral postexposure prophylaxis after sexual, injection drug use, or other nonoccupational exposure to HIV. United States, 2016. Accessed March 6, 2017.
2. Cardo DM et al. New Engl J Med. 1997;337(21):1485-90.
3. Centers for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States–2014: a clinical practice guideline. Accessed March 6, 2017.
In 2016, the Centers for Disease Control and Prevention provided health care providers with updated recommendations for nonoccupational postexposure prophylaxis (nPEP) with antiretroviral drugs to prevent transmission of HIV following sexual interaction, injection-drug use, or other nonoccupational exposures.1 The new recommendations include the use of more effective and more tolerable drug regimens that employ antiretroviral medications that were approved since the previous guidelines came out in 2005; they also provide updated guidance on exposure assessment, baseline and follow-up HIV testing, and longer-term prevention measures, such as pre-exposure prophylaxis (PrEP).
Screening for HIV infection has been expanding broadly in all health care settings over the past decade, so primary care physicians play an increasingly vital role in preventing HIV infection. Today, primary care physicians are also often the most likely “go-to” health care provider when patients think they may have been exposed to HIV. Clinically, this is an emergency situation, so time is of the essence: Treatment with three powerful antiretrovirals must be initiated within a few hours of – but no later than 72 hours after – an isolated exposure to blood, genital secretions, or other potentially infectious body fluids that may contain HIV.
The key issue for primary care physicians, especially those who have never prescribed PEP before, is advance planning. What you do up front, in terms of organizing materials and training staff, is worth the effort because there is so much at stake – for your patients and for society. The good news is that once you have an established nPEP protocol in place, it stays in place. When a patient asks for help, the protocol kicks in automatically.
Getting ready for nPEP
Prepare your staff:
- Educate your whole staff about the urgency of seeing potential nPEP patients immediately.
- Choose the staff person in your office who will submit requests for PEP medications to the pharmacy and/or pharmaceutical companies; your financial reimbursement staff person is likely a good candidate for this job.
- Learn about patient assistance programs (for uninsured or underinsured patients) and crime victims compensation programs (reimbursement or emergency awards for victims of violent crimes, including rape, for various out-of-pocket expenses including medical expenses).
Keep paperwork and materials on hand:
- Have information and forms for patient assistance programs for pharmaceutical companies supplying the drugs. Pharmaceutical companies are aware of the urgency for nPEP medications and are ready to respond immediately. They may mail the medication so it arrives the next day or, more likely, fax a voucher or other information for the patient to present to a local pharmacist who will fill the prescription.
- Have information on your state’s crime victims compensation program available.
- Consider keeping nPEP Starter Packs (with an initial 3-7 days’ worth of medication) readily available in your office.
Rapid evaluation of patients seeking care after potential exposure to HIV
Effective delivery of nPEP requires prompt initial evaluation of patients and assessment of HIV transmission risk. Take a methodical, step-by-step history of the exposure to address the following basic questions:
- Date and time of exposure? nPEP should be initiated as soon as possible after HIV exposure; it is unlikely to be effective if not initiated within 72 hours or less.
- Frequency of exposure? Type/route of exposure? nPEP is generally reserved for isolated or infrequent exposures that present a substantial risk for HIV acquisition (see Table 1 on HIV acquisition risk below).
- HIV status of exposure source? If the source is positive, is the source person on HIV treatment with antiretroviral therapy? If unknown, is the source person an injecting drug user or a man who has sex with men (MSM)?
Based on the initial evaluation, is nPEP recommended?
Answers to the questions asked during the initial evaluation of the patient will determine whether nPEP is indicated. Along with its updated recommendations, the CDC provided an algorithm to help guide evaluation and treatment.
Preferred HIV test
Administer an HIV test to all patients considered for nPEP, preferably the rapid combined antigen and antibody test (Ag/Ab), or just the antibody test if the Ag/Ab test is not available. nPEP is indicated only for persons without HIV infections. However, if results are not available during the initial evaluation, assume the patient is not infected. If indicated and started, nPEP can be discontinued if tests later shown the patient already has an HIV infection.
Laboratory testing
If nPEP is indicated, conduct laboratory testing. Lab testing is required to document the patient’s HIV status (and that of the source person, when available), identify and manage other conditions potentially resulting from exposure, identify conditions that may affect the nPEP medication regimen, and monitor safety or toxicities to the prescribed regimen.
nPEP treatment regimen for otherwise healthy adults and adolescents
In the absence of randomized clinical trials, data from a case/control study demonstrating an 81% reduction of HIV transmission after use of occupational PEP among hospital workers remains the strongest evidence for the benefit of nPEP.1,2 For patients offered nPEP, recommended treatment includes prescribing either of the following regimens for 28 days:
- Preferred regimen: tenofovir disoproxil fumarate (TDF) (300 mg) with emtricitabine (FTC) (200 mg) once daily plus either raltegravir (RAL) 400 mg twice daily or dolutegravir (DTG) 50 mg daily.
- Alternative regimen: TDF (300 mg) with FTC (200 mg) once daily plus darunavir (DRV) (800 mg) and ritonavir (RTV) (100 mg) once daily.
Additional considerations and nPEP treatment regimens for children, patients with decreased renal function, and pregnant women are included in the CDC guidelines.
Crucial Information for Patients on nPEP
Emphasize the importance of proper dosing and adherence.
Review the patient information for each drug in the regimen, specifically the black boxes, warnings, and side effects, and counsel your patients accordingly.
Transitioning from nPEP to PrEP or from PrEP to nPEP
If you have a patient who engages in behavior that places them at risk for frequent, recurrent exposures to HIV, consider transitioning them to PrEP (pre-exposure prophylaxis) following their 28-day course of nPEP.3 PrEP is a two-drug regimen taken daily on an ongoing basis.
Additionally, for patients who are already on PrEP but who have not taken their medications within a week before the possible exposure, consider initiating nPEP for 28 days and then reintroducing PrEP if their HIV status is negative and the problems with adherence can be addressed moving forward.
Raising Awareness About nPEP
Many people never expect to be exposed to HIV and may not know about the availability of PEP in an emergency situation. You can help raise awareness by making educational materials available in your waiting rooms and exam rooms. Brochures and other HIV/AIDS educational materials for patients are available from the CDC Act Against AIDS campaign.
Summary
Dr. Dominguez is a Captain, U.S. Public Health Service, epidemiology branch, division of HIV/AIDS prevention, CDC.
Additional resources
- The CDC recommends that everyone between the ages of 13 and 64 get tested for HIV at least once as part of routine health care. As part of its Act Against AIDS initiative, the CDC developed the HIV Screening Standard Care program, which provides free tools and resources to help clinicians and nurses incorporate routine HIV screening into primary care settings.
- HIV guidelines and recommendations .
- Postexposure prophylaxis (PEP)
- Pre-Exposure prophylaxis (PrEP)
References
1. Centers for Disease Control and Prevention. Updated guidelines for antiretroviral postexposure prophylaxis after sexual, injection drug use, or other nonoccupational exposure to HIV. United States, 2016. Accessed March 6, 2017.
2. Cardo DM et al. New Engl J Med. 1997;337(21):1485-90.
3. Centers for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States–2014: a clinical practice guideline. Accessed March 6, 2017.
VIDEO: Retinal infarctions get missed as stroke harbingers
LOS ANGELES – Retinal infarctions are often going missed as important red flags for future ischemic strokes.
Among U.S. Medicare beneficiaries older than 65 years who had a retinal infarction (RI), “only one-third underwent adequate stroke risk factor evaluation,” Alexander E. Merkler, MD, reported in a poster presented at the International Stroke Conference sponsored by the American Heart Association. And fewer than 10% underwent assessment by a neurologist, based on a review of 5,688 of these older Medicare beneficiaries who had a RI sometime during 2009-2015.
The high-risk profile of these patients was affirmed by a 1% ischemic stroke incidence during the 90 days following their RI diagnosis, a rate roughly fourfold higher than in similar patients without a recent RI.
“A lot of people don’t recognize that a retinal infarction is a type of stroke,” Dr. Merkler said in a video interview. To test this hypothesis, Dr. Merkler and his associates examined the follow-up run on elderly Medicare beneficiaries following a RI diagnosis.”The guidelines recommend evaluating why these patients had a stroke [a retinal infarction] and treating risk factors to reduce the risk of a future stroke,” said Dr. Merkler, a neurologist at Weill Cornell Medicine in New York.
The review showed that 34% of the RI patients underwent cervical carotid imaging, 29% had heart rhythm monitoring, 23% underwent echocardiography, and 8% had assessment by a neurologist.
Dr. Merkler had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SOURCE: Merkler A et al. ISC 2018 Abstract TMP76 (Stroke. 2018 Jan;49[Suppl 1]:ATMP76).
LOS ANGELES – Retinal infarctions are often going missed as important red flags for future ischemic strokes.
Among U.S. Medicare beneficiaries older than 65 years who had a retinal infarction (RI), “only one-third underwent adequate stroke risk factor evaluation,” Alexander E. Merkler, MD, reported in a poster presented at the International Stroke Conference sponsored by the American Heart Association. And fewer than 10% underwent assessment by a neurologist, based on a review of 5,688 of these older Medicare beneficiaries who had a RI sometime during 2009-2015.
The high-risk profile of these patients was affirmed by a 1% ischemic stroke incidence during the 90 days following their RI diagnosis, a rate roughly fourfold higher than in similar patients without a recent RI.
“A lot of people don’t recognize that a retinal infarction is a type of stroke,” Dr. Merkler said in a video interview. To test this hypothesis, Dr. Merkler and his associates examined the follow-up run on elderly Medicare beneficiaries following a RI diagnosis.”The guidelines recommend evaluating why these patients had a stroke [a retinal infarction] and treating risk factors to reduce the risk of a future stroke,” said Dr. Merkler, a neurologist at Weill Cornell Medicine in New York.
The review showed that 34% of the RI patients underwent cervical carotid imaging, 29% had heart rhythm monitoring, 23% underwent echocardiography, and 8% had assessment by a neurologist.
Dr. Merkler had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SOURCE: Merkler A et al. ISC 2018 Abstract TMP76 (Stroke. 2018 Jan;49[Suppl 1]:ATMP76).
LOS ANGELES – Retinal infarctions are often going missed as important red flags for future ischemic strokes.
Among U.S. Medicare beneficiaries older than 65 years who had a retinal infarction (RI), “only one-third underwent adequate stroke risk factor evaluation,” Alexander E. Merkler, MD, reported in a poster presented at the International Stroke Conference sponsored by the American Heart Association. And fewer than 10% underwent assessment by a neurologist, based on a review of 5,688 of these older Medicare beneficiaries who had a RI sometime during 2009-2015.
The high-risk profile of these patients was affirmed by a 1% ischemic stroke incidence during the 90 days following their RI diagnosis, a rate roughly fourfold higher than in similar patients without a recent RI.
“A lot of people don’t recognize that a retinal infarction is a type of stroke,” Dr. Merkler said in a video interview. To test this hypothesis, Dr. Merkler and his associates examined the follow-up run on elderly Medicare beneficiaries following a RI diagnosis.”The guidelines recommend evaluating why these patients had a stroke [a retinal infarction] and treating risk factors to reduce the risk of a future stroke,” said Dr. Merkler, a neurologist at Weill Cornell Medicine in New York.
The review showed that 34% of the RI patients underwent cervical carotid imaging, 29% had heart rhythm monitoring, 23% underwent echocardiography, and 8% had assessment by a neurologist.
Dr. Merkler had no disclosures.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SOURCE: Merkler A et al. ISC 2018 Abstract TMP76 (Stroke. 2018 Jan;49[Suppl 1]:ATMP76).
REPORTING FROM ISC 2018
Key clinical point: Retinal infarction patients often fail to undergo stroke assessment.
Major finding: One-third of Medicare beneficiaries with retinal infarction received adequate evaluation for stroke risk factors.
Study details: Review of 5,688 Medicare patients with a retinal infarction during 2009-2015.
Disclosures: Dr. Merkler had no disclosures.
Source: Merkler A et al. ISC 2018 Abstract TMP76 (Stroke. 2018 Jan;49[Suppl 1]:ATMP76).
Smallpox Vaccine Complications: The Dermatologist’s Role in Diagnosis and Management
The practice of variolation, or inoculation of the smallpox virus from a pustule into a healthy person, was described as early as 1500
Immunization
Vaccinia is an orthopoxvirus, distinct from the smallpox virus variola, with cross-protective immunity after infection. The smallpox vaccine that is available today is a second-generation vaccinia virus derived from plaque purification cloning from the first-generation version originally licensed in 1932, which was central to eradication.5 Today’s vaccine is administered using a bifurcated needle to puncture the epidermis 15 times. Ideally, a papule forms at the inoculation site 3 to 5 days later, progresses to a vesicle and then a pustule, and finally crusts and reaches maximum size by day 10. The crust separates from the skin at 14 to 21 days, at which time the virus can no longer be isolated from the wound. United States Department of Defense surveillance of the first 450,000 vaccinated personnel noted 1% of recipients developed cutaneous eruptions beyond the vaccination site, 5% developed a localized rash, and 1% experienced a generalized eruption.2 Adverse reactions included generalized vaccinia, erythema multiforme (EM), autoinoculation (including ocular vaccinia), and contact vaccinia. There were no cases of eczema vaccinatum (EV) or progressive vaccinia (PV) reported, and no deaths were attributed to these initial vaccines.2
Immunologic Response
Vaccinia replicates in keratinocytes, spreading from cell to cell, resulting in necrosis and vesicle formation. Components of both cellular and humoral immune responses are in place by 10 days after immunization. Deficiencies in these responses result in vaccine complications secondary to vaccine escape and replication beyond the inoculation site.6 A helper T cell TH2-predominant cytokine response in atopic individuals is the likely pathogenesis required for the rapid viral spread for EV.7 Similarly, patients with cell-mediated immunity deficiencies cannot sufficiently produce enough cytotoxic T cells to eliminate an established infection, which can result in PV. Despite the effectiveness of intravenous vaccinia immunoglobulins (VIGIVs) when administered to patients with certain vaccine complications, observations that children with severe X-linked agammaglobulinemia (Bruton disease) have normal responses to vaccination suggest that antibody production is least important in viral control.8 Simian models also suggest that B-cell depletion has no impact on lesion dissemination, as lesion size is inversely correlated with T-cell count.9
Eczema Vaccinatum
A national survey estimated the prevalence of eczema in the United States at 31.6 million individuals,10 with 2- to 3-fold increases in incidence since the 1970s.11 Due to the risk for developing EV, the Advisory Committee on Immunization Practices considers personal history of eczema or contact with a family member who has eczema (either currently or in the past) contraindications to nonemergency administration of the vaccine.12,13 However, atopic conditions in general are underrecognized, with only approximately one-third of patients carrying an official diagnosis from a physician.10 Despite a large atopic and vaccinated population, EV remains relatively uncommon at 10 to 39 cases per million vaccines.6
The EV rash classically involves the midface, neck, and antecubital and popliteal fossae but can present in any location. The lesions start as papules that quickly progress to vesicles and pustules with crusting on an erythematous base. Given the extent of denudation of the epidermis, impetiginization can occur. Death rates as high as 30% have been reported14 but have only occurred in instances of secondary contact transmission with no deaths occurring in the primary vaccinees.15 In a case published in 2008, a 2-year-old boy developed the first documented EV case under the new program after exposure to his father’s predeployment vaccine.16 A similar rash is shown in Figure 1 with notable vesicles and pustules. The child required burn patient–type management, VIGIV, and treatment with cidofovir and an investigational antiorthopox agent. He was discharged from the hospital after 48 days without sequelae or considerable scarring.16 If a family member has a contraindication barring secondary contact with the vaccine, the US Department of Defense’s policy defers vaccination in active-duty members until they reach their deployment destination, at which point the inoculation is administered.
Progressive Vaccinia
Progressive vaccinia is also known as vaccinia necrosum or vaccinia gangrenosum. It is a dreaded but uncommon complication, occurring once in every 1 million vaccinations. It carries an overall case fatality rate of 15%,17 but it nearly always is fatal in patients with severe T-cell defects.18 Progressive vaccinia occurs exclusively in patients with cell-mediated immunodeficiency, with the severity of the acute illness correlating with the severity of immunodeficiency. In patients with cell-mediated immunodeficiency but intact humoral immunity, progression can be limited to expansion of the lesion, as it is thought that antibody production restricts viremia.18 Progressive vaccinia should be suspected in a patient if the vaccine site shows no signs of improvement by 14 days.19 The PV lesions do not heal and may progress or recur in patients with signs of prior healing. The leading edge has confluent vesicles, and the center of the lesion develops necrosis with thick black eschar formation. Most specifically, there is no surrounding inflammation; however, inflammation can develop later as a response to treatment or secondary infection. Figure 2 shows a PV lesion with black eschar and a transition to intact dermis without inflammation.
The first known case of PV since the 1960s vaccination campaign occurred in an active-duty Marine vaccinated with vaccinia before a diagnosis of acute myelogenous leukemia was recognized 2 weeks later.19 The vaccine site was stable in size and crusted when he received neutropenia-inducing chemotherapy 6.5 weeks after vaccination. The site then progressed in a manner typical for PV with central necrosis and a lack of inflammation at the expanding painless wound edge.19 This classic appearance with progression of satellite lesions prompted the treatment team to obtain wound and serum samples, which yielded the orthopox virus from polymerase chain reaction and viral culture. He required 2 months of care in an intensive care unit and received treatment with topical imiquimod, VIGIV, a topical and intravenous antiorthopox agent, and a second investigational antiorthopox agent; the patient ultimately survived.17,20
Generalized Vaccinia
Generalized vaccinia (GV) typically is a benign vaccine complication resulting from viremic spread from the initial inoculation site and is most commonly seen in healthy patients. Generalized vaccinia is only life threatening in immunocompromised patients. The incidence of GV is 23.4 to 241.5 patients per million vaccines.6 The majority of GV cases occur 5 to 12 days after vaccination when small distant pustules or vesicles appear on any part of the body, including the palms and soles. The lesions usually are smaller than the primary vaccination site and resolve more quickly. Generalized vaccinia can have a few to several hundred pocks, though the rash is rarely as diffuse as EV presentations.3 Given that EV can present diffusely on skin unaffected by atopic dermatitis, GV can be difficult to distinguish from EV. Features more common to EV include more systemically ill patients, increased numbers of lesions, and lesions that become confluent in an atopic distribution. It has been suggested that GV can be differentiated from vesicular or vesiculopapular EM because GV does not develop flaccid bullae and EM typically has targetoid lesions.18 Mild GV disease requires no treatment, but VIGIV can be used in more extensive cases.
Localized Reactions Due to Viral Replication
Accidental autoinoculation can occur when patients touch the vaccination site and then themselves, transferring virus particles to areas of compromised skin integrity, most commonly on the face, eyes, hands, genitalia, anus, or any other broken skin. Autoinoculation happens with some frequency and is of limited clinical concern unless there is ocular involvement. Keratitis develops in 6% of ocular vaccinia cases, and VIGIV is contraindicated, as rabbit models suggest that antigen-antibody precipitates in the cornea can cause scarring.21 Instead, trifluorothymidine is an effective topical treatment available for ocular vaccinia.
A robust response or “take” is defined as a reaction having redness, swelling, and warmth more than 3 inches in diameter at the inoculation site, peaking 6 to 12 days after inoculation with spontaneous regression occurring 1 to 3 days after.22,23 A robust take frequently is of concern to the clinician, as it can be difficult to discern from secondary infection. Secondary infections are uncommon, and a robust take is secondary to viral, not bacterial, cellulitis. Unfortunately, there are no diagnostics that have utility in distinguishing between the two, and the decision to administer empiric antibiotics might be unavoidable in light of the consequences of an untreated, rapidly progressive bacterial cellulitis. Milder cases in the setting of no constitutional symptoms could be safely monitored if close follow-up is assured.
Generalized Skin Reactions Without Viral Replication
Development of erythematous, pruritic, urticarial, and diffuse targetlike lesions of EM is common in first-time vaccinees. Often misdiagnosed as GV, EM is an immunologically mediated, not virally mediated, process. The most common infectious cause prompting EM is herpes simplex virus type 1. In the setting of a live-virus vaccine, it is difficult to determine if the vaccine prompted herpes simplex virus type 1 viral shedding and associated EM or if the vaccinia vaccine is more directly the cause of EM.24 Symptoms typically are mild, but more severe reactions may require treatment with corticosteroids. Stevens-Johnson syndrome with a severe bullous eruption has been linked to vaccinia24 but fortunately is rare. Morbilliform eruptions, urticaria, and angioedema also can occur.
Final Thoughts
Given current world events and ongoing bioterrorism threats, the smallpox vaccine program continues indefinitely. With a brisk military deployment tempo, a larger population of new vaccinees naturally will yield more cutaneous reactions. Military members, civilian health care workers, and members of the National Guard and National Reserves will develop complications and present to dermatologists for care. The historical pool of providers accustomed to seeing these complications from the 1960s eradication campaign is scant. Military and civilian dermatologists alike are uniquely poised to be the experts on protean manifestations of vaccinia reactions.
- Voigt EA, Kennedy RB, Poland GA. Defending against smallpox: a focus on vaccines. Expert Rev Vaccines. 2016;15:1197-1211.
- Grabenstein J, Wikenwerder W Jr. US military smallpox vaccination program experience. JAMA. 2003;289:3278-3282.
- Kelly CD, Egan C, Davis SW, et al. Laboratory confirmation of generalized vaccinia following smallpox vaccination. J Clin Microbiol. 2004;42:1373-1375.
- Slike BM, Creegan M, Marovich M, et al. Humoral immunity to primary smallpox vaccination: impact of childhood versus adult immunization on vaccinia vector vaccine development in military populations. PLoS One. 2017;12:E0169247.
- Notice to readers: newly licensed vaccine to replace old smallpox vaccine. MMWR. 2008;57:207-208.
- Bray M. Pathogenesis and potential antiviral therapy of complications of smallpox vaccination. Antiviral Res. 2003;58:101-114.
- Engler R, Kenner J, Leung D. Smallpox vaccination: risk considerations for patients with atopic dermatitis. J Allergy Clin Immunol. 2002;110:357-365.
- Bray M, Wright ME. Progressive vaccinia. Clin Infect Dis. 2003;36:766-774.
- Gordon S, Cecchinato V, Andresen V, et al. Smallpox vaccine safety is dependent on T cells and not B cells. J Infect Dis. 2011;203:1043-1053.
- Hanifin J, Reed M. A population-based survey of eczema prevalence in the United States. Dermatitis. 2007;82:82-91.
- Avena-Woods C. Overview of atopic dermatitis. Am J Manag Care. 2017;23(8 suppl):S115-S123.
- Wharton M, Strikas RA, Harpaz R, et al; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Recommendations for using smallpox vaccine in a pre-event vaccination program. Supplemental recommendations of the Advisory Committee on Immunization Practices (ACIP) and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep. 2003;52:1-16.
- Petersen BW, Harms TJ, Reynolds MG, et al. Use of vaccinia virus smallpox vaccine in laboratory and health care personnel at risk for occupation exposure to orthopoxviruses—recommendations of the Advisory Committee on Immunizations Practices (ACIP), 2015. MMWR Morb Mortal Wkly Rep. 2016;65:257-262.
- Nell P, Kohl KS, Graham PL, et al; Brighton Collaboration Vaccinia Virus Vaccine Adverse Event Working Group for Eczema Vaccinatum. Eczema vaccinatum as an adverse event following exposure to vaccinia virus: case definition and guidelines of data collection analysis, and presentation of immunization safety data. Vaccine. 2007:25;5725-5734.
- Aragón TJ, Ulrich S, Fernyak S, et al. Risks of serious complications and death from smallpox vaccination: a systematic review of the United States experience, 1963-1968. BMC Public Health. 2003;3:26.
- Vora S, Damon I, Fulginiti V, et al. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin Infect Dis. 2008;46:1555-1561.
- Centers for Disease Control and Prevention (CDC). Progressive vaccinia in a military smallpox vaccinee—United States 2009. MMWR Morb Mortal Wkly Rep. 2009;58:532-536.
- Fulginiti VA, Papier A, Lane M, et al. Smallpox vaccination: a review, part II. adverse events. Clin Infect Dis. 2003;37:251-271.
- Nell P, Kohl KS, Graham PL, et al; Brighton Collaboration Vaccinia Virus Vaccine Adverse Event Working Group for Progressive Vaccinia. Progressive vaccinia as an adverse event following exposure to vaccinia virus: case definition and guidelines of data collection, analysis, and presentation of immunization safety data. Vaccine. 2007;25:5735-5744.
- Lederman ER, Davidson W, Groff HL, et al. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J Infect Dis. 2012;206:E1372-E1385.
- Lane ML, Goldstein J. Adverse events occurring after smallpox vaccination. Semin Ped Infect Dis. 2003;14:189-195.
- Vaccine adverse events. CDC website. http://www.cdc.gov/smallpox/clinicians/vaccine-adverse-events5.html. Accessed January 3, 2018.
- Cono J, Casey CG, Bell DM. Smallpox vaccination and adversereactions, guidance for clinicians. CDC website. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5204a1.htm. Accessed January 3, 2018.
- Rosenblatt AE, Stein SL. Cutaneous reactions to vaccinations. Clin Dermatol. 2015;33:327-332.
The practice of variolation, or inoculation of the smallpox virus from a pustule into a healthy person, was described as early as 1500
Immunization
Vaccinia is an orthopoxvirus, distinct from the smallpox virus variola, with cross-protective immunity after infection. The smallpox vaccine that is available today is a second-generation vaccinia virus derived from plaque purification cloning from the first-generation version originally licensed in 1932, which was central to eradication.5 Today’s vaccine is administered using a bifurcated needle to puncture the epidermis 15 times. Ideally, a papule forms at the inoculation site 3 to 5 days later, progresses to a vesicle and then a pustule, and finally crusts and reaches maximum size by day 10. The crust separates from the skin at 14 to 21 days, at which time the virus can no longer be isolated from the wound. United States Department of Defense surveillance of the first 450,000 vaccinated personnel noted 1% of recipients developed cutaneous eruptions beyond the vaccination site, 5% developed a localized rash, and 1% experienced a generalized eruption.2 Adverse reactions included generalized vaccinia, erythema multiforme (EM), autoinoculation (including ocular vaccinia), and contact vaccinia. There were no cases of eczema vaccinatum (EV) or progressive vaccinia (PV) reported, and no deaths were attributed to these initial vaccines.2
Immunologic Response
Vaccinia replicates in keratinocytes, spreading from cell to cell, resulting in necrosis and vesicle formation. Components of both cellular and humoral immune responses are in place by 10 days after immunization. Deficiencies in these responses result in vaccine complications secondary to vaccine escape and replication beyond the inoculation site.6 A helper T cell TH2-predominant cytokine response in atopic individuals is the likely pathogenesis required for the rapid viral spread for EV.7 Similarly, patients with cell-mediated immunity deficiencies cannot sufficiently produce enough cytotoxic T cells to eliminate an established infection, which can result in PV. Despite the effectiveness of intravenous vaccinia immunoglobulins (VIGIVs) when administered to patients with certain vaccine complications, observations that children with severe X-linked agammaglobulinemia (Bruton disease) have normal responses to vaccination suggest that antibody production is least important in viral control.8 Simian models also suggest that B-cell depletion has no impact on lesion dissemination, as lesion size is inversely correlated with T-cell count.9
Eczema Vaccinatum
A national survey estimated the prevalence of eczema in the United States at 31.6 million individuals,10 with 2- to 3-fold increases in incidence since the 1970s.11 Due to the risk for developing EV, the Advisory Committee on Immunization Practices considers personal history of eczema or contact with a family member who has eczema (either currently or in the past) contraindications to nonemergency administration of the vaccine.12,13 However, atopic conditions in general are underrecognized, with only approximately one-third of patients carrying an official diagnosis from a physician.10 Despite a large atopic and vaccinated population, EV remains relatively uncommon at 10 to 39 cases per million vaccines.6
The EV rash classically involves the midface, neck, and antecubital and popliteal fossae but can present in any location. The lesions start as papules that quickly progress to vesicles and pustules with crusting on an erythematous base. Given the extent of denudation of the epidermis, impetiginization can occur. Death rates as high as 30% have been reported14 but have only occurred in instances of secondary contact transmission with no deaths occurring in the primary vaccinees.15 In a case published in 2008, a 2-year-old boy developed the first documented EV case under the new program after exposure to his father’s predeployment vaccine.16 A similar rash is shown in Figure 1 with notable vesicles and pustules. The child required burn patient–type management, VIGIV, and treatment with cidofovir and an investigational antiorthopox agent. He was discharged from the hospital after 48 days without sequelae or considerable scarring.16 If a family member has a contraindication barring secondary contact with the vaccine, the US Department of Defense’s policy defers vaccination in active-duty members until they reach their deployment destination, at which point the inoculation is administered.
Progressive Vaccinia
Progressive vaccinia is also known as vaccinia necrosum or vaccinia gangrenosum. It is a dreaded but uncommon complication, occurring once in every 1 million vaccinations. It carries an overall case fatality rate of 15%,17 but it nearly always is fatal in patients with severe T-cell defects.18 Progressive vaccinia occurs exclusively in patients with cell-mediated immunodeficiency, with the severity of the acute illness correlating with the severity of immunodeficiency. In patients with cell-mediated immunodeficiency but intact humoral immunity, progression can be limited to expansion of the lesion, as it is thought that antibody production restricts viremia.18 Progressive vaccinia should be suspected in a patient if the vaccine site shows no signs of improvement by 14 days.19 The PV lesions do not heal and may progress or recur in patients with signs of prior healing. The leading edge has confluent vesicles, and the center of the lesion develops necrosis with thick black eschar formation. Most specifically, there is no surrounding inflammation; however, inflammation can develop later as a response to treatment or secondary infection. Figure 2 shows a PV lesion with black eschar and a transition to intact dermis without inflammation.
The first known case of PV since the 1960s vaccination campaign occurred in an active-duty Marine vaccinated with vaccinia before a diagnosis of acute myelogenous leukemia was recognized 2 weeks later.19 The vaccine site was stable in size and crusted when he received neutropenia-inducing chemotherapy 6.5 weeks after vaccination. The site then progressed in a manner typical for PV with central necrosis and a lack of inflammation at the expanding painless wound edge.19 This classic appearance with progression of satellite lesions prompted the treatment team to obtain wound and serum samples, which yielded the orthopox virus from polymerase chain reaction and viral culture. He required 2 months of care in an intensive care unit and received treatment with topical imiquimod, VIGIV, a topical and intravenous antiorthopox agent, and a second investigational antiorthopox agent; the patient ultimately survived.17,20
Generalized Vaccinia
Generalized vaccinia (GV) typically is a benign vaccine complication resulting from viremic spread from the initial inoculation site and is most commonly seen in healthy patients. Generalized vaccinia is only life threatening in immunocompromised patients. The incidence of GV is 23.4 to 241.5 patients per million vaccines.6 The majority of GV cases occur 5 to 12 days after vaccination when small distant pustules or vesicles appear on any part of the body, including the palms and soles. The lesions usually are smaller than the primary vaccination site and resolve more quickly. Generalized vaccinia can have a few to several hundred pocks, though the rash is rarely as diffuse as EV presentations.3 Given that EV can present diffusely on skin unaffected by atopic dermatitis, GV can be difficult to distinguish from EV. Features more common to EV include more systemically ill patients, increased numbers of lesions, and lesions that become confluent in an atopic distribution. It has been suggested that GV can be differentiated from vesicular or vesiculopapular EM because GV does not develop flaccid bullae and EM typically has targetoid lesions.18 Mild GV disease requires no treatment, but VIGIV can be used in more extensive cases.
Localized Reactions Due to Viral Replication
Accidental autoinoculation can occur when patients touch the vaccination site and then themselves, transferring virus particles to areas of compromised skin integrity, most commonly on the face, eyes, hands, genitalia, anus, or any other broken skin. Autoinoculation happens with some frequency and is of limited clinical concern unless there is ocular involvement. Keratitis develops in 6% of ocular vaccinia cases, and VIGIV is contraindicated, as rabbit models suggest that antigen-antibody precipitates in the cornea can cause scarring.21 Instead, trifluorothymidine is an effective topical treatment available for ocular vaccinia.
A robust response or “take” is defined as a reaction having redness, swelling, and warmth more than 3 inches in diameter at the inoculation site, peaking 6 to 12 days after inoculation with spontaneous regression occurring 1 to 3 days after.22,23 A robust take frequently is of concern to the clinician, as it can be difficult to discern from secondary infection. Secondary infections are uncommon, and a robust take is secondary to viral, not bacterial, cellulitis. Unfortunately, there are no diagnostics that have utility in distinguishing between the two, and the decision to administer empiric antibiotics might be unavoidable in light of the consequences of an untreated, rapidly progressive bacterial cellulitis. Milder cases in the setting of no constitutional symptoms could be safely monitored if close follow-up is assured.
Generalized Skin Reactions Without Viral Replication
Development of erythematous, pruritic, urticarial, and diffuse targetlike lesions of EM is common in first-time vaccinees. Often misdiagnosed as GV, EM is an immunologically mediated, not virally mediated, process. The most common infectious cause prompting EM is herpes simplex virus type 1. In the setting of a live-virus vaccine, it is difficult to determine if the vaccine prompted herpes simplex virus type 1 viral shedding and associated EM or if the vaccinia vaccine is more directly the cause of EM.24 Symptoms typically are mild, but more severe reactions may require treatment with corticosteroids. Stevens-Johnson syndrome with a severe bullous eruption has been linked to vaccinia24 but fortunately is rare. Morbilliform eruptions, urticaria, and angioedema also can occur.
Final Thoughts
Given current world events and ongoing bioterrorism threats, the smallpox vaccine program continues indefinitely. With a brisk military deployment tempo, a larger population of new vaccinees naturally will yield more cutaneous reactions. Military members, civilian health care workers, and members of the National Guard and National Reserves will develop complications and present to dermatologists for care. The historical pool of providers accustomed to seeing these complications from the 1960s eradication campaign is scant. Military and civilian dermatologists alike are uniquely poised to be the experts on protean manifestations of vaccinia reactions.
The practice of variolation, or inoculation of the smallpox virus from a pustule into a healthy person, was described as early as 1500
Immunization
Vaccinia is an orthopoxvirus, distinct from the smallpox virus variola, with cross-protective immunity after infection. The smallpox vaccine that is available today is a second-generation vaccinia virus derived from plaque purification cloning from the first-generation version originally licensed in 1932, which was central to eradication.5 Today’s vaccine is administered using a bifurcated needle to puncture the epidermis 15 times. Ideally, a papule forms at the inoculation site 3 to 5 days later, progresses to a vesicle and then a pustule, and finally crusts and reaches maximum size by day 10. The crust separates from the skin at 14 to 21 days, at which time the virus can no longer be isolated from the wound. United States Department of Defense surveillance of the first 450,000 vaccinated personnel noted 1% of recipients developed cutaneous eruptions beyond the vaccination site, 5% developed a localized rash, and 1% experienced a generalized eruption.2 Adverse reactions included generalized vaccinia, erythema multiforme (EM), autoinoculation (including ocular vaccinia), and contact vaccinia. There were no cases of eczema vaccinatum (EV) or progressive vaccinia (PV) reported, and no deaths were attributed to these initial vaccines.2
Immunologic Response
Vaccinia replicates in keratinocytes, spreading from cell to cell, resulting in necrosis and vesicle formation. Components of both cellular and humoral immune responses are in place by 10 days after immunization. Deficiencies in these responses result in vaccine complications secondary to vaccine escape and replication beyond the inoculation site.6 A helper T cell TH2-predominant cytokine response in atopic individuals is the likely pathogenesis required for the rapid viral spread for EV.7 Similarly, patients with cell-mediated immunity deficiencies cannot sufficiently produce enough cytotoxic T cells to eliminate an established infection, which can result in PV. Despite the effectiveness of intravenous vaccinia immunoglobulins (VIGIVs) when administered to patients with certain vaccine complications, observations that children with severe X-linked agammaglobulinemia (Bruton disease) have normal responses to vaccination suggest that antibody production is least important in viral control.8 Simian models also suggest that B-cell depletion has no impact on lesion dissemination, as lesion size is inversely correlated with T-cell count.9
Eczema Vaccinatum
A national survey estimated the prevalence of eczema in the United States at 31.6 million individuals,10 with 2- to 3-fold increases in incidence since the 1970s.11 Due to the risk for developing EV, the Advisory Committee on Immunization Practices considers personal history of eczema or contact with a family member who has eczema (either currently or in the past) contraindications to nonemergency administration of the vaccine.12,13 However, atopic conditions in general are underrecognized, with only approximately one-third of patients carrying an official diagnosis from a physician.10 Despite a large atopic and vaccinated population, EV remains relatively uncommon at 10 to 39 cases per million vaccines.6
The EV rash classically involves the midface, neck, and antecubital and popliteal fossae but can present in any location. The lesions start as papules that quickly progress to vesicles and pustules with crusting on an erythematous base. Given the extent of denudation of the epidermis, impetiginization can occur. Death rates as high as 30% have been reported14 but have only occurred in instances of secondary contact transmission with no deaths occurring in the primary vaccinees.15 In a case published in 2008, a 2-year-old boy developed the first documented EV case under the new program after exposure to his father’s predeployment vaccine.16 A similar rash is shown in Figure 1 with notable vesicles and pustules. The child required burn patient–type management, VIGIV, and treatment with cidofovir and an investigational antiorthopox agent. He was discharged from the hospital after 48 days without sequelae or considerable scarring.16 If a family member has a contraindication barring secondary contact with the vaccine, the US Department of Defense’s policy defers vaccination in active-duty members until they reach their deployment destination, at which point the inoculation is administered.
Progressive Vaccinia
Progressive vaccinia is also known as vaccinia necrosum or vaccinia gangrenosum. It is a dreaded but uncommon complication, occurring once in every 1 million vaccinations. It carries an overall case fatality rate of 15%,17 but it nearly always is fatal in patients with severe T-cell defects.18 Progressive vaccinia occurs exclusively in patients with cell-mediated immunodeficiency, with the severity of the acute illness correlating with the severity of immunodeficiency. In patients with cell-mediated immunodeficiency but intact humoral immunity, progression can be limited to expansion of the lesion, as it is thought that antibody production restricts viremia.18 Progressive vaccinia should be suspected in a patient if the vaccine site shows no signs of improvement by 14 days.19 The PV lesions do not heal and may progress or recur in patients with signs of prior healing. The leading edge has confluent vesicles, and the center of the lesion develops necrosis with thick black eschar formation. Most specifically, there is no surrounding inflammation; however, inflammation can develop later as a response to treatment or secondary infection. Figure 2 shows a PV lesion with black eschar and a transition to intact dermis without inflammation.
The first known case of PV since the 1960s vaccination campaign occurred in an active-duty Marine vaccinated with vaccinia before a diagnosis of acute myelogenous leukemia was recognized 2 weeks later.19 The vaccine site was stable in size and crusted when he received neutropenia-inducing chemotherapy 6.5 weeks after vaccination. The site then progressed in a manner typical for PV with central necrosis and a lack of inflammation at the expanding painless wound edge.19 This classic appearance with progression of satellite lesions prompted the treatment team to obtain wound and serum samples, which yielded the orthopox virus from polymerase chain reaction and viral culture. He required 2 months of care in an intensive care unit and received treatment with topical imiquimod, VIGIV, a topical and intravenous antiorthopox agent, and a second investigational antiorthopox agent; the patient ultimately survived.17,20
Generalized Vaccinia
Generalized vaccinia (GV) typically is a benign vaccine complication resulting from viremic spread from the initial inoculation site and is most commonly seen in healthy patients. Generalized vaccinia is only life threatening in immunocompromised patients. The incidence of GV is 23.4 to 241.5 patients per million vaccines.6 The majority of GV cases occur 5 to 12 days after vaccination when small distant pustules or vesicles appear on any part of the body, including the palms and soles. The lesions usually are smaller than the primary vaccination site and resolve more quickly. Generalized vaccinia can have a few to several hundred pocks, though the rash is rarely as diffuse as EV presentations.3 Given that EV can present diffusely on skin unaffected by atopic dermatitis, GV can be difficult to distinguish from EV. Features more common to EV include more systemically ill patients, increased numbers of lesions, and lesions that become confluent in an atopic distribution. It has been suggested that GV can be differentiated from vesicular or vesiculopapular EM because GV does not develop flaccid bullae and EM typically has targetoid lesions.18 Mild GV disease requires no treatment, but VIGIV can be used in more extensive cases.
Localized Reactions Due to Viral Replication
Accidental autoinoculation can occur when patients touch the vaccination site and then themselves, transferring virus particles to areas of compromised skin integrity, most commonly on the face, eyes, hands, genitalia, anus, or any other broken skin. Autoinoculation happens with some frequency and is of limited clinical concern unless there is ocular involvement. Keratitis develops in 6% of ocular vaccinia cases, and VIGIV is contraindicated, as rabbit models suggest that antigen-antibody precipitates in the cornea can cause scarring.21 Instead, trifluorothymidine is an effective topical treatment available for ocular vaccinia.
A robust response or “take” is defined as a reaction having redness, swelling, and warmth more than 3 inches in diameter at the inoculation site, peaking 6 to 12 days after inoculation with spontaneous regression occurring 1 to 3 days after.22,23 A robust take frequently is of concern to the clinician, as it can be difficult to discern from secondary infection. Secondary infections are uncommon, and a robust take is secondary to viral, not bacterial, cellulitis. Unfortunately, there are no diagnostics that have utility in distinguishing between the two, and the decision to administer empiric antibiotics might be unavoidable in light of the consequences of an untreated, rapidly progressive bacterial cellulitis. Milder cases in the setting of no constitutional symptoms could be safely monitored if close follow-up is assured.
Generalized Skin Reactions Without Viral Replication
Development of erythematous, pruritic, urticarial, and diffuse targetlike lesions of EM is common in first-time vaccinees. Often misdiagnosed as GV, EM is an immunologically mediated, not virally mediated, process. The most common infectious cause prompting EM is herpes simplex virus type 1. In the setting of a live-virus vaccine, it is difficult to determine if the vaccine prompted herpes simplex virus type 1 viral shedding and associated EM or if the vaccinia vaccine is more directly the cause of EM.24 Symptoms typically are mild, but more severe reactions may require treatment with corticosteroids. Stevens-Johnson syndrome with a severe bullous eruption has been linked to vaccinia24 but fortunately is rare. Morbilliform eruptions, urticaria, and angioedema also can occur.
Final Thoughts
Given current world events and ongoing bioterrorism threats, the smallpox vaccine program continues indefinitely. With a brisk military deployment tempo, a larger population of new vaccinees naturally will yield more cutaneous reactions. Military members, civilian health care workers, and members of the National Guard and National Reserves will develop complications and present to dermatologists for care. The historical pool of providers accustomed to seeing these complications from the 1960s eradication campaign is scant. Military and civilian dermatologists alike are uniquely poised to be the experts on protean manifestations of vaccinia reactions.
- Voigt EA, Kennedy RB, Poland GA. Defending against smallpox: a focus on vaccines. Expert Rev Vaccines. 2016;15:1197-1211.
- Grabenstein J, Wikenwerder W Jr. US military smallpox vaccination program experience. JAMA. 2003;289:3278-3282.
- Kelly CD, Egan C, Davis SW, et al. Laboratory confirmation of generalized vaccinia following smallpox vaccination. J Clin Microbiol. 2004;42:1373-1375.
- Slike BM, Creegan M, Marovich M, et al. Humoral immunity to primary smallpox vaccination: impact of childhood versus adult immunization on vaccinia vector vaccine development in military populations. PLoS One. 2017;12:E0169247.
- Notice to readers: newly licensed vaccine to replace old smallpox vaccine. MMWR. 2008;57:207-208.
- Bray M. Pathogenesis and potential antiviral therapy of complications of smallpox vaccination. Antiviral Res. 2003;58:101-114.
- Engler R, Kenner J, Leung D. Smallpox vaccination: risk considerations for patients with atopic dermatitis. J Allergy Clin Immunol. 2002;110:357-365.
- Bray M, Wright ME. Progressive vaccinia. Clin Infect Dis. 2003;36:766-774.
- Gordon S, Cecchinato V, Andresen V, et al. Smallpox vaccine safety is dependent on T cells and not B cells. J Infect Dis. 2011;203:1043-1053.
- Hanifin J, Reed M. A population-based survey of eczema prevalence in the United States. Dermatitis. 2007;82:82-91.
- Avena-Woods C. Overview of atopic dermatitis. Am J Manag Care. 2017;23(8 suppl):S115-S123.
- Wharton M, Strikas RA, Harpaz R, et al; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Recommendations for using smallpox vaccine in a pre-event vaccination program. Supplemental recommendations of the Advisory Committee on Immunization Practices (ACIP) and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep. 2003;52:1-16.
- Petersen BW, Harms TJ, Reynolds MG, et al. Use of vaccinia virus smallpox vaccine in laboratory and health care personnel at risk for occupation exposure to orthopoxviruses—recommendations of the Advisory Committee on Immunizations Practices (ACIP), 2015. MMWR Morb Mortal Wkly Rep. 2016;65:257-262.
- Nell P, Kohl KS, Graham PL, et al; Brighton Collaboration Vaccinia Virus Vaccine Adverse Event Working Group for Eczema Vaccinatum. Eczema vaccinatum as an adverse event following exposure to vaccinia virus: case definition and guidelines of data collection analysis, and presentation of immunization safety data. Vaccine. 2007:25;5725-5734.
- Aragón TJ, Ulrich S, Fernyak S, et al. Risks of serious complications and death from smallpox vaccination: a systematic review of the United States experience, 1963-1968. BMC Public Health. 2003;3:26.
- Vora S, Damon I, Fulginiti V, et al. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin Infect Dis. 2008;46:1555-1561.
- Centers for Disease Control and Prevention (CDC). Progressive vaccinia in a military smallpox vaccinee—United States 2009. MMWR Morb Mortal Wkly Rep. 2009;58:532-536.
- Fulginiti VA, Papier A, Lane M, et al. Smallpox vaccination: a review, part II. adverse events. Clin Infect Dis. 2003;37:251-271.
- Nell P, Kohl KS, Graham PL, et al; Brighton Collaboration Vaccinia Virus Vaccine Adverse Event Working Group for Progressive Vaccinia. Progressive vaccinia as an adverse event following exposure to vaccinia virus: case definition and guidelines of data collection, analysis, and presentation of immunization safety data. Vaccine. 2007;25:5735-5744.
- Lederman ER, Davidson W, Groff HL, et al. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J Infect Dis. 2012;206:E1372-E1385.
- Lane ML, Goldstein J. Adverse events occurring after smallpox vaccination. Semin Ped Infect Dis. 2003;14:189-195.
- Vaccine adverse events. CDC website. http://www.cdc.gov/smallpox/clinicians/vaccine-adverse-events5.html. Accessed January 3, 2018.
- Cono J, Casey CG, Bell DM. Smallpox vaccination and adversereactions, guidance for clinicians. CDC website. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5204a1.htm. Accessed January 3, 2018.
- Rosenblatt AE, Stein SL. Cutaneous reactions to vaccinations. Clin Dermatol. 2015;33:327-332.
- Voigt EA, Kennedy RB, Poland GA. Defending against smallpox: a focus on vaccines. Expert Rev Vaccines. 2016;15:1197-1211.
- Grabenstein J, Wikenwerder W Jr. US military smallpox vaccination program experience. JAMA. 2003;289:3278-3282.
- Kelly CD, Egan C, Davis SW, et al. Laboratory confirmation of generalized vaccinia following smallpox vaccination. J Clin Microbiol. 2004;42:1373-1375.
- Slike BM, Creegan M, Marovich M, et al. Humoral immunity to primary smallpox vaccination: impact of childhood versus adult immunization on vaccinia vector vaccine development in military populations. PLoS One. 2017;12:E0169247.
- Notice to readers: newly licensed vaccine to replace old smallpox vaccine. MMWR. 2008;57:207-208.
- Bray M. Pathogenesis and potential antiviral therapy of complications of smallpox vaccination. Antiviral Res. 2003;58:101-114.
- Engler R, Kenner J, Leung D. Smallpox vaccination: risk considerations for patients with atopic dermatitis. J Allergy Clin Immunol. 2002;110:357-365.
- Bray M, Wright ME. Progressive vaccinia. Clin Infect Dis. 2003;36:766-774.
- Gordon S, Cecchinato V, Andresen V, et al. Smallpox vaccine safety is dependent on T cells and not B cells. J Infect Dis. 2011;203:1043-1053.
- Hanifin J, Reed M. A population-based survey of eczema prevalence in the United States. Dermatitis. 2007;82:82-91.
- Avena-Woods C. Overview of atopic dermatitis. Am J Manag Care. 2017;23(8 suppl):S115-S123.
- Wharton M, Strikas RA, Harpaz R, et al; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Recommendations for using smallpox vaccine in a pre-event vaccination program. Supplemental recommendations of the Advisory Committee on Immunization Practices (ACIP) and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep. 2003;52:1-16.
- Petersen BW, Harms TJ, Reynolds MG, et al. Use of vaccinia virus smallpox vaccine in laboratory and health care personnel at risk for occupation exposure to orthopoxviruses—recommendations of the Advisory Committee on Immunizations Practices (ACIP), 2015. MMWR Morb Mortal Wkly Rep. 2016;65:257-262.
- Nell P, Kohl KS, Graham PL, et al; Brighton Collaboration Vaccinia Virus Vaccine Adverse Event Working Group for Eczema Vaccinatum. Eczema vaccinatum as an adverse event following exposure to vaccinia virus: case definition and guidelines of data collection analysis, and presentation of immunization safety data. Vaccine. 2007:25;5725-5734.
- Aragón TJ, Ulrich S, Fernyak S, et al. Risks of serious complications and death from smallpox vaccination: a systematic review of the United States experience, 1963-1968. BMC Public Health. 2003;3:26.
- Vora S, Damon I, Fulginiti V, et al. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin Infect Dis. 2008;46:1555-1561.
- Centers for Disease Control and Prevention (CDC). Progressive vaccinia in a military smallpox vaccinee—United States 2009. MMWR Morb Mortal Wkly Rep. 2009;58:532-536.
- Fulginiti VA, Papier A, Lane M, et al. Smallpox vaccination: a review, part II. adverse events. Clin Infect Dis. 2003;37:251-271.
- Nell P, Kohl KS, Graham PL, et al; Brighton Collaboration Vaccinia Virus Vaccine Adverse Event Working Group for Progressive Vaccinia. Progressive vaccinia as an adverse event following exposure to vaccinia virus: case definition and guidelines of data collection, analysis, and presentation of immunization safety data. Vaccine. 2007;25:5735-5744.
- Lederman ER, Davidson W, Groff HL, et al. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J Infect Dis. 2012;206:E1372-E1385.
- Lane ML, Goldstein J. Adverse events occurring after smallpox vaccination. Semin Ped Infect Dis. 2003;14:189-195.
- Vaccine adverse events. CDC website. http://www.cdc.gov/smallpox/clinicians/vaccine-adverse-events5.html. Accessed January 3, 2018.
- Cono J, Casey CG, Bell DM. Smallpox vaccination and adversereactions, guidance for clinicians. CDC website. http://www.cdc.gov/mmwr/preview/mmwrhtml/rr5204a1.htm. Accessed January 3, 2018.
- Rosenblatt AE, Stein SL. Cutaneous reactions to vaccinations. Clin Dermatol. 2015;33:327-332.
Practice Points
- Dermatologists should be aware that smallpox vaccinations are being administered to patients and may present with a myriad of cutaneous complications.
- Progressive vaccinia should be suspected if a smallpox inoculation has not healed after 14 days and, most specifically, if there is no inflammation surrounding the site.
- Generalized vaccinia generally is a benign condition seen in otherwise healthy patients and usually requires no treatment.
Atopic patients should be educated to avoid receiving routine smallpox vaccinations if they would be considered at risk for requiring the inoculation.