User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Update on Management of Atopic Dermatitis in Young Children
Update on Management of Atopic Dermatitis in Young Children
Atopic dermatitis (AD) is a chronic inflammatory skin condition associated with skin barrier impairment and immune system dysregulation.1 Development of AD in young children can present challenges in determining appropriate treatment regimens. Natural remedies for AD often are promoted on social media over traditional treatments, including topical corticosteroids (TCSs), which can contribute to corticophobia.2 Dermatologists play a critical role not only in optimizing topical therapy but also addressing patient interest in natural approaches to AD, including diet-related questions. This article outlines the role of diet and probiotics in pediatric AD and reviews the topical treatments currently approved for this patient population.
Diet and Probiotics
With a growing focus on natural therapies for AD, dietary interventions have come to the forefront. A prevalent theme among patients and their families is addressing gut health and allergic triggers. Broad elimination diets have not shown clinical benefit in patients with AD regardless of age,3 and in children, they may result in nutritional deficiencies, poor growth, and increased risk for IgE-mediated food allergies.4 If a true food allergy is identified based on positive IgE and an acute clinical reaction, elimination of the allergen may provide some benefit.5
The link between gut microbiota and skin health has driven an interest in the role of probiotics in the treatment of pediatric AD. A meta-analysis of 20 articles concluded that, whether administered to infants or breastfeeding mothers, use of probiotics overall led to a significant reduction in AD risk in infants (P=.001). Lactobacillus and mixed strains were effective.6 While broad elimination diets are not used to treat AD, probiotic supplementation can be considered for prevention of AD.
Topical Corticosteroids
Topical corticosteroids are the cornerstone of AD treatment; however, corticophobia among patients is on the rise, leading to poor adherence and suboptimal control of AD.7 Mild cutaneous adverse effects (AEs) including skin atrophy, striae, and telangiectasias may occur. Rarely, systemic AEs occur due to absorption of TCSs into the bloodstream, mainly with application of potent steroids over large body surface areas or under occlusion.8 When the optimal potency of a TCS is chosen and used appropriately, incidence of AEs from TCS use is very low.9
Counseling parents about risk factors that can lead to AEs during treatment with TCSs and formulating regimens that minimize these risks while maintaining efficacy increases adherence and outcomes. Pulse maintenance dosing of TCSs typically involves application 1 to 2 times weekly to areas of the skin that are prone to frequent outbreaks. Pulse maintenance dosing can reduce the incidence of AD flares while also decreasing the total amount of topical medication needed as compared to the reactive approach alone, thereby reducing risk for AEs.8
Steroid-Sparing Topical Treatments
Although TCSs are considered first-line agents, recently there has been an advent of steroid-sparing topical agents approved by the US Food and Drug Administration (FDA) for pediatric patients with AD, including topical calcineurin inhibitors (TCIs), phosphodiesterase 4 inhibitors, a Janus kinase inhibitor, and aryl hydrocarbon receptor agonists. Offering steroid-sparing agents in these patients can help ease parental anxiety regarding TCS overuse.
Topical Calcineurin Inhibitors—Pimecrolimus cream 1% and tacrolimus ointment 0.03% are approved for patients aged 2 years and older and have anti-inflammatory and antipruritic effects equivalent to low-potency TCS. Tacrolimus ointment 0.1% is approved for patients aged 16 years and older with similar efficacy to a midpotency TCSs. Pimecrolimus cream 1% and tacrolimus ointment 0.03% often are used off-label in children younger than 2 years, as supported by clinical trials showing their safety and efficacy.10
Topical calcineurin inhibitors can replace or supplement TCSs, making TCIs a desirable option for avoidance of steroid-related AEs. The addition of a TCI to spot treatment or a pulse regimen in a young patient can reassure them and their caregivers that the provider is proactively reducing the risk of TCS overuse. The largest barrier to TCI use is the FDA’s black box warning based on the oral formulation of tacrolimus, citing a potential increased risk for lymphoma and skin cancer; however, there is no evidence for substantial systemic absorption of topical pimecrolimus or tacrolimus.11 Large task-force reviews have found no association between TCI use and development of malignancy.12,13 Based on the current data, counseling patients and their caregivers that this risk primarily is theoretical may help them more confidently integrate TCIs into their treatment regimen. Burning and tingling may occur in a minority of pediatric patients using TCIs for AD. Applying the medication to open wounds or inflamed skin increases the risk for stinging, but pretreatment with a short course of TCSs before transitioning to a TCI may boost tolerance.14
Phosphodiesterase 4 Inhibitors—Crisaborole ointment 2%, a phosphodiesterase 4 inhibitor, is approved for children aged 3 months and older with mild to moderate AD. Its use has been more limited than TCSs and TCIs, as local irritation including stinging and burning can occur in up to 50% of patients.15 One study comparing crisaborole 2% with tacrolimus 0.03% revealed greater improvement with tacrolimus.16 A second phosphodiesterase 4 inhibitor approved for once-daily use in children aged 6 years and older with mild to moderate AD is roflumilast cream 0.15%. Roflumilast reduces eczema severity and pruritus, with AEs also limited to application-site stinging and burning.17
Janus Kinase Inhibitor—Ruxolitinib cream 1.5%, a Janus kinase inhibitor, has been approved by the FDA since 2023 for twice-daily use in children aged 12 years and older with AD. Similar to TCIs, ruxolitinib cream carries a black box warning. Short-term safety data on ruxolitinib cream have revealed low levels of ruxolitinib concentration in plasma18; however, long-term studies on topical Janus kinase inhibitors for AD in pediatric and adult populations are lacking. To reduce the risk for systemic absorption, recommendations include limiting usage to 60 g per week and limiting treatment to less than 20% of the body surface area.19 Ruxolitinib has efficacy similar to or possibly superior to triamcinolone 0.1%.20 Ruxolitinib is emerging as a promising nonsteroidal option that potentially is highly efficacious and well tolerated without cutaneous AEs.
Aryl Hydrocarbon Receptor Agonist—Tapinarof cream 1% is an aryl hydrocarbon receptor agonist that has been approved by the FDA since 2024 for children aged 2 years and older as a once-daily treatment for moderate to severe AD. Adverse events include folliculitis, nasopharyngitis, and headache, which are mostly mild or moderate.21
Final Thoughts
Topical management of pediatric AD includes traditional therapy with TCSs and newer steroid-sparing agents, which can help address corticophobia. Anticipatory guidance regarding the safety and long-term effects of individual therapies is critical to ensuring patient adherence to treatment regimens. Probiotics may help prevent pediatric AD, but future studies are needed to determine their role in treatment.
- Weidinger S, Beck LA, Bieber T, et al. Atopic dermatitis. Nat Rev Dis Primers. 2018;4:1.
- Voillot P, Riche B, Portafax M, et al. Social media platforms listening study on atopic dermatitis: quantitative and qualitative findings. J Med Internet Res. 2022;24:E31140.
- Bath-Hextall F, Delamere FM, Williams HC. Dietary exclusions for improving established atopic eczema in adults and children: systematic review. Allergy. 2009;64:258-264.
- Rustad AM, Nickles MA, Bilimoria SN, et al. The role of diet modification in atopic dermatitis: navigating the complexity. Am J Clin Dermatol. 2022;23:27-36.
- Khan A, Adalsteinsson J, Whitaker-Worth DL. Atopic dermatitis and nutrition. Clin Dermatol. 2022;40:135-144.
- Chen L, Ni Y, Wu X, et al. Probiotics for the prevention of atopic dermatitis in infants from different geographic regions: a systematic review and meta-analysis. J Dermatolog Treat. 2022;33:2931-2939.
- Herzum A, Occella C, Gariazzo L, et al. Corticophobia among parents of children with atopic dermatitis: assessing major and minor risk factors for high TOPICOP scores. J Clin Med. 2023;12:6813.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Callen J, Chamlin S, Eichenfield LF, et al. A systematic review of the safety of topical therapies for atopic dermatitis. Br J Dermatol. 2007;156:203-221.
- Reitamo S, Rustin M, Ruzicka T, et al. Efficacy and safety of tacrolimus ointment compared with that of hydrocortisone butyrate ointment in adult patients with atopic dermatitis. J Allergy Clin Immunol. 2002;109:547-555.
- Thaçi D, Salgo R. Malignancy concerns of topical calcineurin inhibitors for atopic dermatitis: facts and controversies. Clin Dermatol. 2010;28:52-56.
- Berger TG, Duvic M, Van Voorhees AS, et al. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the AAD Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Fonacier L, Spergel J, Charlesworth EN, et al. Report of the Topical Calcineurin Inhibitor Task Force of the American College of Allergy, Asthma and Immunology and the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2005;115:1249-1253.
- Eichenfield LF, Lucky AW, Boguniewicz M, et al. Safety and efficacy of pimecrolimus (ASM 981) cream 1% in the treatment of mild and moderate atopic dermatitis in children and adolescents. J Am Acad Dermatol. 2002;46:495-504.
- Lin CPL, Gordon S, Her MJ, et al. A retrospective study: application site pain with the use of crisaborole, a topical phosphodiesterase 4 inhibitor. J Am Acad Dermatol. 2019;80:1451-1453.
- Ryan Wolf J, Chen A, Wieser J, et al. Improved patient- and caregiver-reported outcomes distinguish tacrolimus 0.03% from crisaborole in children with atopic dermatitis. J Eur Acad Dermatol Venereol. 2024;38:1364-1372.
- Simpson EL, Eichenfield LF, Alonso-Llamazares J, et al. Roflumilast cream, 0.15%, for atopic dermatitis in adults and children: INTEGUMENT-1 and INTEGUMENT-2 randomized clinical trials. JAMA Dermatol. 2024;160:1161-1170.
- Papp K, Szepietowski JC, Kircik L, et al. Long-term safety and disease control with ruxolitinib cream in atopic dermatitis: results from two phase 3 studies. J Am Acad Dermatol. 2023;88:1008-1016.
- Sidbury R, Alikhan A, Bercovitch L, et al. Guidelines of carefor the management of atopic dermatitis in adults with topical therapies. J Am Acad Dermatol. 2023;89:E1-E20.
- Sadeghi S, Mohandesi NA. Efficacy and safety of topical JAK inhibitors in the treatment of atopic dermatitis in paediatrics and adults: a systematic review. Exp Dermatol. 2023;32:599-610.
- Silverberg JI, Eichenfield LF, Hebert AA, et al. Tapinarof cream 1% once daily: significant efficacy in the treatment of moderate to severe atopic dermatitis in adults and children down to 2 years of age in the pivotal phase 3 ADORING trials. J Am Acad Dermatol. 2024;91:457-465.
Atopic dermatitis (AD) is a chronic inflammatory skin condition associated with skin barrier impairment and immune system dysregulation.1 Development of AD in young children can present challenges in determining appropriate treatment regimens. Natural remedies for AD often are promoted on social media over traditional treatments, including topical corticosteroids (TCSs), which can contribute to corticophobia.2 Dermatologists play a critical role not only in optimizing topical therapy but also addressing patient interest in natural approaches to AD, including diet-related questions. This article outlines the role of diet and probiotics in pediatric AD and reviews the topical treatments currently approved for this patient population.
Diet and Probiotics
With a growing focus on natural therapies for AD, dietary interventions have come to the forefront. A prevalent theme among patients and their families is addressing gut health and allergic triggers. Broad elimination diets have not shown clinical benefit in patients with AD regardless of age,3 and in children, they may result in nutritional deficiencies, poor growth, and increased risk for IgE-mediated food allergies.4 If a true food allergy is identified based on positive IgE and an acute clinical reaction, elimination of the allergen may provide some benefit.5
The link between gut microbiota and skin health has driven an interest in the role of probiotics in the treatment of pediatric AD. A meta-analysis of 20 articles concluded that, whether administered to infants or breastfeeding mothers, use of probiotics overall led to a significant reduction in AD risk in infants (P=.001). Lactobacillus and mixed strains were effective.6 While broad elimination diets are not used to treat AD, probiotic supplementation can be considered for prevention of AD.
Topical Corticosteroids
Topical corticosteroids are the cornerstone of AD treatment; however, corticophobia among patients is on the rise, leading to poor adherence and suboptimal control of AD.7 Mild cutaneous adverse effects (AEs) including skin atrophy, striae, and telangiectasias may occur. Rarely, systemic AEs occur due to absorption of TCSs into the bloodstream, mainly with application of potent steroids over large body surface areas or under occlusion.8 When the optimal potency of a TCS is chosen and used appropriately, incidence of AEs from TCS use is very low.9
Counseling parents about risk factors that can lead to AEs during treatment with TCSs and formulating regimens that minimize these risks while maintaining efficacy increases adherence and outcomes. Pulse maintenance dosing of TCSs typically involves application 1 to 2 times weekly to areas of the skin that are prone to frequent outbreaks. Pulse maintenance dosing can reduce the incidence of AD flares while also decreasing the total amount of topical medication needed as compared to the reactive approach alone, thereby reducing risk for AEs.8
Steroid-Sparing Topical Treatments
Although TCSs are considered first-line agents, recently there has been an advent of steroid-sparing topical agents approved by the US Food and Drug Administration (FDA) for pediatric patients with AD, including topical calcineurin inhibitors (TCIs), phosphodiesterase 4 inhibitors, a Janus kinase inhibitor, and aryl hydrocarbon receptor agonists. Offering steroid-sparing agents in these patients can help ease parental anxiety regarding TCS overuse.
Topical Calcineurin Inhibitors—Pimecrolimus cream 1% and tacrolimus ointment 0.03% are approved for patients aged 2 years and older and have anti-inflammatory and antipruritic effects equivalent to low-potency TCS. Tacrolimus ointment 0.1% is approved for patients aged 16 years and older with similar efficacy to a midpotency TCSs. Pimecrolimus cream 1% and tacrolimus ointment 0.03% often are used off-label in children younger than 2 years, as supported by clinical trials showing their safety and efficacy.10
Topical calcineurin inhibitors can replace or supplement TCSs, making TCIs a desirable option for avoidance of steroid-related AEs. The addition of a TCI to spot treatment or a pulse regimen in a young patient can reassure them and their caregivers that the provider is proactively reducing the risk of TCS overuse. The largest barrier to TCI use is the FDA’s black box warning based on the oral formulation of tacrolimus, citing a potential increased risk for lymphoma and skin cancer; however, there is no evidence for substantial systemic absorption of topical pimecrolimus or tacrolimus.11 Large task-force reviews have found no association between TCI use and development of malignancy.12,13 Based on the current data, counseling patients and their caregivers that this risk primarily is theoretical may help them more confidently integrate TCIs into their treatment regimen. Burning and tingling may occur in a minority of pediatric patients using TCIs for AD. Applying the medication to open wounds or inflamed skin increases the risk for stinging, but pretreatment with a short course of TCSs before transitioning to a TCI may boost tolerance.14
Phosphodiesterase 4 Inhibitors—Crisaborole ointment 2%, a phosphodiesterase 4 inhibitor, is approved for children aged 3 months and older with mild to moderate AD. Its use has been more limited than TCSs and TCIs, as local irritation including stinging and burning can occur in up to 50% of patients.15 One study comparing crisaborole 2% with tacrolimus 0.03% revealed greater improvement with tacrolimus.16 A second phosphodiesterase 4 inhibitor approved for once-daily use in children aged 6 years and older with mild to moderate AD is roflumilast cream 0.15%. Roflumilast reduces eczema severity and pruritus, with AEs also limited to application-site stinging and burning.17
Janus Kinase Inhibitor—Ruxolitinib cream 1.5%, a Janus kinase inhibitor, has been approved by the FDA since 2023 for twice-daily use in children aged 12 years and older with AD. Similar to TCIs, ruxolitinib cream carries a black box warning. Short-term safety data on ruxolitinib cream have revealed low levels of ruxolitinib concentration in plasma18; however, long-term studies on topical Janus kinase inhibitors for AD in pediatric and adult populations are lacking. To reduce the risk for systemic absorption, recommendations include limiting usage to 60 g per week and limiting treatment to less than 20% of the body surface area.19 Ruxolitinib has efficacy similar to or possibly superior to triamcinolone 0.1%.20 Ruxolitinib is emerging as a promising nonsteroidal option that potentially is highly efficacious and well tolerated without cutaneous AEs.
Aryl Hydrocarbon Receptor Agonist—Tapinarof cream 1% is an aryl hydrocarbon receptor agonist that has been approved by the FDA since 2024 for children aged 2 years and older as a once-daily treatment for moderate to severe AD. Adverse events include folliculitis, nasopharyngitis, and headache, which are mostly mild or moderate.21
Final Thoughts
Topical management of pediatric AD includes traditional therapy with TCSs and newer steroid-sparing agents, which can help address corticophobia. Anticipatory guidance regarding the safety and long-term effects of individual therapies is critical to ensuring patient adherence to treatment regimens. Probiotics may help prevent pediatric AD, but future studies are needed to determine their role in treatment.
Atopic dermatitis (AD) is a chronic inflammatory skin condition associated with skin barrier impairment and immune system dysregulation.1 Development of AD in young children can present challenges in determining appropriate treatment regimens. Natural remedies for AD often are promoted on social media over traditional treatments, including topical corticosteroids (TCSs), which can contribute to corticophobia.2 Dermatologists play a critical role not only in optimizing topical therapy but also addressing patient interest in natural approaches to AD, including diet-related questions. This article outlines the role of diet and probiotics in pediatric AD and reviews the topical treatments currently approved for this patient population.
Diet and Probiotics
With a growing focus on natural therapies for AD, dietary interventions have come to the forefront. A prevalent theme among patients and their families is addressing gut health and allergic triggers. Broad elimination diets have not shown clinical benefit in patients with AD regardless of age,3 and in children, they may result in nutritional deficiencies, poor growth, and increased risk for IgE-mediated food allergies.4 If a true food allergy is identified based on positive IgE and an acute clinical reaction, elimination of the allergen may provide some benefit.5
The link between gut microbiota and skin health has driven an interest in the role of probiotics in the treatment of pediatric AD. A meta-analysis of 20 articles concluded that, whether administered to infants or breastfeeding mothers, use of probiotics overall led to a significant reduction in AD risk in infants (P=.001). Lactobacillus and mixed strains were effective.6 While broad elimination diets are not used to treat AD, probiotic supplementation can be considered for prevention of AD.
Topical Corticosteroids
Topical corticosteroids are the cornerstone of AD treatment; however, corticophobia among patients is on the rise, leading to poor adherence and suboptimal control of AD.7 Mild cutaneous adverse effects (AEs) including skin atrophy, striae, and telangiectasias may occur. Rarely, systemic AEs occur due to absorption of TCSs into the bloodstream, mainly with application of potent steroids over large body surface areas or under occlusion.8 When the optimal potency of a TCS is chosen and used appropriately, incidence of AEs from TCS use is very low.9
Counseling parents about risk factors that can lead to AEs during treatment with TCSs and formulating regimens that minimize these risks while maintaining efficacy increases adherence and outcomes. Pulse maintenance dosing of TCSs typically involves application 1 to 2 times weekly to areas of the skin that are prone to frequent outbreaks. Pulse maintenance dosing can reduce the incidence of AD flares while also decreasing the total amount of topical medication needed as compared to the reactive approach alone, thereby reducing risk for AEs.8
Steroid-Sparing Topical Treatments
Although TCSs are considered first-line agents, recently there has been an advent of steroid-sparing topical agents approved by the US Food and Drug Administration (FDA) for pediatric patients with AD, including topical calcineurin inhibitors (TCIs), phosphodiesterase 4 inhibitors, a Janus kinase inhibitor, and aryl hydrocarbon receptor agonists. Offering steroid-sparing agents in these patients can help ease parental anxiety regarding TCS overuse.
Topical Calcineurin Inhibitors—Pimecrolimus cream 1% and tacrolimus ointment 0.03% are approved for patients aged 2 years and older and have anti-inflammatory and antipruritic effects equivalent to low-potency TCS. Tacrolimus ointment 0.1% is approved for patients aged 16 years and older with similar efficacy to a midpotency TCSs. Pimecrolimus cream 1% and tacrolimus ointment 0.03% often are used off-label in children younger than 2 years, as supported by clinical trials showing their safety and efficacy.10
Topical calcineurin inhibitors can replace or supplement TCSs, making TCIs a desirable option for avoidance of steroid-related AEs. The addition of a TCI to spot treatment or a pulse regimen in a young patient can reassure them and their caregivers that the provider is proactively reducing the risk of TCS overuse. The largest barrier to TCI use is the FDA’s black box warning based on the oral formulation of tacrolimus, citing a potential increased risk for lymphoma and skin cancer; however, there is no evidence for substantial systemic absorption of topical pimecrolimus or tacrolimus.11 Large task-force reviews have found no association between TCI use and development of malignancy.12,13 Based on the current data, counseling patients and their caregivers that this risk primarily is theoretical may help them more confidently integrate TCIs into their treatment regimen. Burning and tingling may occur in a minority of pediatric patients using TCIs for AD. Applying the medication to open wounds or inflamed skin increases the risk for stinging, but pretreatment with a short course of TCSs before transitioning to a TCI may boost tolerance.14
Phosphodiesterase 4 Inhibitors—Crisaborole ointment 2%, a phosphodiesterase 4 inhibitor, is approved for children aged 3 months and older with mild to moderate AD. Its use has been more limited than TCSs and TCIs, as local irritation including stinging and burning can occur in up to 50% of patients.15 One study comparing crisaborole 2% with tacrolimus 0.03% revealed greater improvement with tacrolimus.16 A second phosphodiesterase 4 inhibitor approved for once-daily use in children aged 6 years and older with mild to moderate AD is roflumilast cream 0.15%. Roflumilast reduces eczema severity and pruritus, with AEs also limited to application-site stinging and burning.17
Janus Kinase Inhibitor—Ruxolitinib cream 1.5%, a Janus kinase inhibitor, has been approved by the FDA since 2023 for twice-daily use in children aged 12 years and older with AD. Similar to TCIs, ruxolitinib cream carries a black box warning. Short-term safety data on ruxolitinib cream have revealed low levels of ruxolitinib concentration in plasma18; however, long-term studies on topical Janus kinase inhibitors for AD in pediatric and adult populations are lacking. To reduce the risk for systemic absorption, recommendations include limiting usage to 60 g per week and limiting treatment to less than 20% of the body surface area.19 Ruxolitinib has efficacy similar to or possibly superior to triamcinolone 0.1%.20 Ruxolitinib is emerging as a promising nonsteroidal option that potentially is highly efficacious and well tolerated without cutaneous AEs.
Aryl Hydrocarbon Receptor Agonist—Tapinarof cream 1% is an aryl hydrocarbon receptor agonist that has been approved by the FDA since 2024 for children aged 2 years and older as a once-daily treatment for moderate to severe AD. Adverse events include folliculitis, nasopharyngitis, and headache, which are mostly mild or moderate.21
Final Thoughts
Topical management of pediatric AD includes traditional therapy with TCSs and newer steroid-sparing agents, which can help address corticophobia. Anticipatory guidance regarding the safety and long-term effects of individual therapies is critical to ensuring patient adherence to treatment regimens. Probiotics may help prevent pediatric AD, but future studies are needed to determine their role in treatment.
- Weidinger S, Beck LA, Bieber T, et al. Atopic dermatitis. Nat Rev Dis Primers. 2018;4:1.
- Voillot P, Riche B, Portafax M, et al. Social media platforms listening study on atopic dermatitis: quantitative and qualitative findings. J Med Internet Res. 2022;24:E31140.
- Bath-Hextall F, Delamere FM, Williams HC. Dietary exclusions for improving established atopic eczema in adults and children: systematic review. Allergy. 2009;64:258-264.
- Rustad AM, Nickles MA, Bilimoria SN, et al. The role of diet modification in atopic dermatitis: navigating the complexity. Am J Clin Dermatol. 2022;23:27-36.
- Khan A, Adalsteinsson J, Whitaker-Worth DL. Atopic dermatitis and nutrition. Clin Dermatol. 2022;40:135-144.
- Chen L, Ni Y, Wu X, et al. Probiotics for the prevention of atopic dermatitis in infants from different geographic regions: a systematic review and meta-analysis. J Dermatolog Treat. 2022;33:2931-2939.
- Herzum A, Occella C, Gariazzo L, et al. Corticophobia among parents of children with atopic dermatitis: assessing major and minor risk factors for high TOPICOP scores. J Clin Med. 2023;12:6813.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Callen J, Chamlin S, Eichenfield LF, et al. A systematic review of the safety of topical therapies for atopic dermatitis. Br J Dermatol. 2007;156:203-221.
- Reitamo S, Rustin M, Ruzicka T, et al. Efficacy and safety of tacrolimus ointment compared with that of hydrocortisone butyrate ointment in adult patients with atopic dermatitis. J Allergy Clin Immunol. 2002;109:547-555.
- Thaçi D, Salgo R. Malignancy concerns of topical calcineurin inhibitors for atopic dermatitis: facts and controversies. Clin Dermatol. 2010;28:52-56.
- Berger TG, Duvic M, Van Voorhees AS, et al. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the AAD Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Fonacier L, Spergel J, Charlesworth EN, et al. Report of the Topical Calcineurin Inhibitor Task Force of the American College of Allergy, Asthma and Immunology and the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2005;115:1249-1253.
- Eichenfield LF, Lucky AW, Boguniewicz M, et al. Safety and efficacy of pimecrolimus (ASM 981) cream 1% in the treatment of mild and moderate atopic dermatitis in children and adolescents. J Am Acad Dermatol. 2002;46:495-504.
- Lin CPL, Gordon S, Her MJ, et al. A retrospective study: application site pain with the use of crisaborole, a topical phosphodiesterase 4 inhibitor. J Am Acad Dermatol. 2019;80:1451-1453.
- Ryan Wolf J, Chen A, Wieser J, et al. Improved patient- and caregiver-reported outcomes distinguish tacrolimus 0.03% from crisaborole in children with atopic dermatitis. J Eur Acad Dermatol Venereol. 2024;38:1364-1372.
- Simpson EL, Eichenfield LF, Alonso-Llamazares J, et al. Roflumilast cream, 0.15%, for atopic dermatitis in adults and children: INTEGUMENT-1 and INTEGUMENT-2 randomized clinical trials. JAMA Dermatol. 2024;160:1161-1170.
- Papp K, Szepietowski JC, Kircik L, et al. Long-term safety and disease control with ruxolitinib cream in atopic dermatitis: results from two phase 3 studies. J Am Acad Dermatol. 2023;88:1008-1016.
- Sidbury R, Alikhan A, Bercovitch L, et al. Guidelines of carefor the management of atopic dermatitis in adults with topical therapies. J Am Acad Dermatol. 2023;89:E1-E20.
- Sadeghi S, Mohandesi NA. Efficacy and safety of topical JAK inhibitors in the treatment of atopic dermatitis in paediatrics and adults: a systematic review. Exp Dermatol. 2023;32:599-610.
- Silverberg JI, Eichenfield LF, Hebert AA, et al. Tapinarof cream 1% once daily: significant efficacy in the treatment of moderate to severe atopic dermatitis in adults and children down to 2 years of age in the pivotal phase 3 ADORING trials. J Am Acad Dermatol. 2024;91:457-465.
- Weidinger S, Beck LA, Bieber T, et al. Atopic dermatitis. Nat Rev Dis Primers. 2018;4:1.
- Voillot P, Riche B, Portafax M, et al. Social media platforms listening study on atopic dermatitis: quantitative and qualitative findings. J Med Internet Res. 2022;24:E31140.
- Bath-Hextall F, Delamere FM, Williams HC. Dietary exclusions for improving established atopic eczema in adults and children: systematic review. Allergy. 2009;64:258-264.
- Rustad AM, Nickles MA, Bilimoria SN, et al. The role of diet modification in atopic dermatitis: navigating the complexity. Am J Clin Dermatol. 2022;23:27-36.
- Khan A, Adalsteinsson J, Whitaker-Worth DL. Atopic dermatitis and nutrition. Clin Dermatol. 2022;40:135-144.
- Chen L, Ni Y, Wu X, et al. Probiotics for the prevention of atopic dermatitis in infants from different geographic regions: a systematic review and meta-analysis. J Dermatolog Treat. 2022;33:2931-2939.
- Herzum A, Occella C, Gariazzo L, et al. Corticophobia among parents of children with atopic dermatitis: assessing major and minor risk factors for high TOPICOP scores. J Clin Med. 2023;12:6813.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Callen J, Chamlin S, Eichenfield LF, et al. A systematic review of the safety of topical therapies for atopic dermatitis. Br J Dermatol. 2007;156:203-221.
- Reitamo S, Rustin M, Ruzicka T, et al. Efficacy and safety of tacrolimus ointment compared with that of hydrocortisone butyrate ointment in adult patients with atopic dermatitis. J Allergy Clin Immunol. 2002;109:547-555.
- Thaçi D, Salgo R. Malignancy concerns of topical calcineurin inhibitors for atopic dermatitis: facts and controversies. Clin Dermatol. 2010;28:52-56.
- Berger TG, Duvic M, Van Voorhees AS, et al. The use of topical calcineurin inhibitors in dermatology: safety concerns. report of the AAD Association Task Force. J Am Acad Dermatol. 2006;54:818-823.
- Fonacier L, Spergel J, Charlesworth EN, et al. Report of the Topical Calcineurin Inhibitor Task Force of the American College of Allergy, Asthma and Immunology and the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2005;115:1249-1253.
- Eichenfield LF, Lucky AW, Boguniewicz M, et al. Safety and efficacy of pimecrolimus (ASM 981) cream 1% in the treatment of mild and moderate atopic dermatitis in children and adolescents. J Am Acad Dermatol. 2002;46:495-504.
- Lin CPL, Gordon S, Her MJ, et al. A retrospective study: application site pain with the use of crisaborole, a topical phosphodiesterase 4 inhibitor. J Am Acad Dermatol. 2019;80:1451-1453.
- Ryan Wolf J, Chen A, Wieser J, et al. Improved patient- and caregiver-reported outcomes distinguish tacrolimus 0.03% from crisaborole in children with atopic dermatitis. J Eur Acad Dermatol Venereol. 2024;38:1364-1372.
- Simpson EL, Eichenfield LF, Alonso-Llamazares J, et al. Roflumilast cream, 0.15%, for atopic dermatitis in adults and children: INTEGUMENT-1 and INTEGUMENT-2 randomized clinical trials. JAMA Dermatol. 2024;160:1161-1170.
- Papp K, Szepietowski JC, Kircik L, et al. Long-term safety and disease control with ruxolitinib cream in atopic dermatitis: results from two phase 3 studies. J Am Acad Dermatol. 2023;88:1008-1016.
- Sidbury R, Alikhan A, Bercovitch L, et al. Guidelines of carefor the management of atopic dermatitis in adults with topical therapies. J Am Acad Dermatol. 2023;89:E1-E20.
- Sadeghi S, Mohandesi NA. Efficacy and safety of topical JAK inhibitors in the treatment of atopic dermatitis in paediatrics and adults: a systematic review. Exp Dermatol. 2023;32:599-610.
- Silverberg JI, Eichenfield LF, Hebert AA, et al. Tapinarof cream 1% once daily: significant efficacy in the treatment of moderate to severe atopic dermatitis in adults and children down to 2 years of age in the pivotal phase 3 ADORING trials. J Am Acad Dermatol. 2024;91:457-465.
Update on Management of Atopic Dermatitis in Young Children
Update on Management of Atopic Dermatitis in Young Children

Flesh-Colored Lesion on the Ear
Flesh-Colored Lesion on the Ear
THE DIAGNOSIS: Gouty Tophus
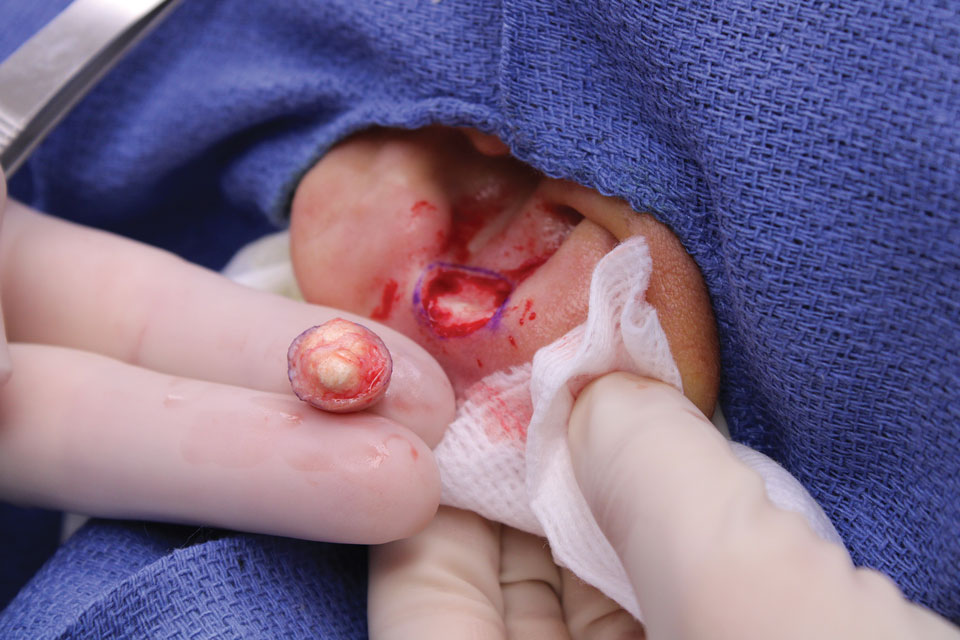
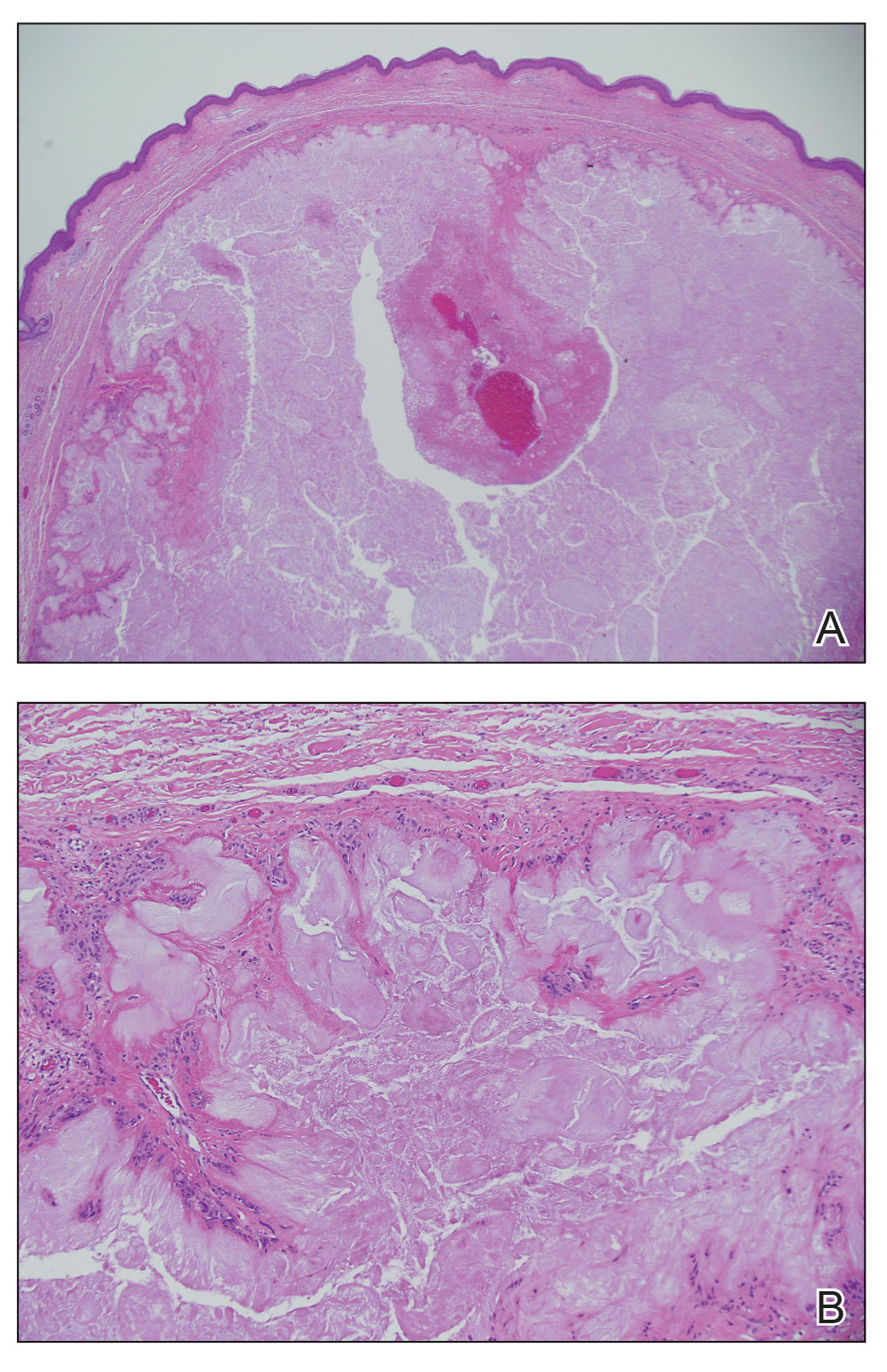
The lesion was excised and sent for histopathologic examination (eFigures 1 and 2), revealing aggregates of feathery, amorphous, pale-pink material, which confirmed the diagnosis of gouty tophus. The surgical site was left to heal by secondary intention. Upon further evaluation, the patient reported recurrent monoarticular joint pain in the ankles and feet, and laboratory workup revealed elevated serum uric acid. He was advised to follow up with his primary care physician to discuss systemic treatment options for gout.
Gout is an inflammatory arthritis characterized by the deposition of monosodium urate monohydrate crystals in the joints, soft tissue, and bone due to elevated serum uric acid. Uric acid is the final product of purine metabolism, and serum levels may be elevated due to excess production or underexcretion. Multiple genetic, environmental, and metabolic factors influence these processes.1 Collections of monosodium urate crystals may develop intra- or extra-articularly, the latter of which are known as gouty tophi. These nodules have a classic chalklike consistency and typically are seen in patients with untreated gout starting approximately 10 years after the first flare. The most common locations for subcutaneous gouty tophi are acral sites (eg, fingertips, ears) as well as the wrists, knees, and elbows (olecranon bursae). Rarely, gouty panniculitis also may develop.2
Histopathology of gouty tophi reveals nodular aggregates of acellular, amorphous, pale-pink material surrounded by palisading histiocytes and multinucleated giant cells. The presence of needlelike monosodium urate crystals, which display negative birefringence, is diagnostic. Unfortunately, these crystals are destroyed in routine formalin processing.3
There are limited data regarding treatment of gouty tophi. Urate-lowering systemic medications such as pegloticase may be beneficial, but more data are needed.4 We pursued surgical excision in our case for definitive diagnosis; however, it is not a common treatment for gouty tophi. Typically, urate-lowering therapy is utilized to resolve or shrink lesions over time.5
The differential diagnosis for gouty tophi includes epidermal inclusion cyst (EIC), the most common type of cutaneous cyst. Though EICs can manifest anywhere on the body, they are not as common on the ears as gouty tophi. Epidermal inclusion cysts clinically manifest as soft subcutaneous nodules, and a central punctum often is noted. These lesions are derived from the follicular infundibulum and histologically are characterized by a cystic cavity lined by a stratified squamous epithelium with a granular layer. The cavity contains loose laminated keratin material.6
Pseudocyst of the auricle is a benign cystic swelling of the pinna that can develop spontaneously but most often manifests following trauma to the area, which is believed to separate the tissue planes in the cartilage, allowing fluid to accumulate. This lesion typically is asymptomatic, though some patients report mild tenderness.7 Histology shows a cystic structure within the cartilage without an epithelial lining, and a perivascular inflammatory response often is observed.8
Pilomatricoma, also known as pilomatrixoma, is a benign tumor derived from the hair follicle matrix that manifests as a firm, slow-growing, painless subcutaneous nodule. It most often is found on the head and neck, commonly in the periauricular area.9 Though rare, it has been found on the auricle and external auditory canal.10 Histologically, pilomatricomas are well-defined tumors containing internal trabeculae. They contain populations of basaloid and ghost cells and often calcify, sometimes with resultant bone formation.9
Dermoid cysts are benign tumors that develop along lines of embryonic closure and often are diagnosed at birth or in early childhood. They most commonly manifest on the head and neck, typically in the supraorbital area. Rarely, they have been reported on the ear.6 Dermoid cysts may resemble EICs clinically and histopathologically, except that the cyst wall contains mature adnexal structures such as hair follicles and sebaceous glands.
- Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039-2052. doi:10.1016/S0140-6736(16)00346-9
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445. doi:10.1097/GOX.0000000000000420
- Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. doi:10.1186/s12891-019-2519-y
- Sriranganathan MK, Vinik O, Pardo Pardo J, et al. Interventions for tophi in gout. Cochrane Database Syst Rev. 2021;8:CD010069. doi:10.1002/14651858.CD010069.pub3
- Evidence review for surgical excision of tophi. Gout: diagnosis and management. National Institute for Health and Care Excellence (NICE). June 2022. Accessed October 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK583526/
- Cho Y, Lee DH. Clinical characteristics of idiopathic epidermoid and dermoid cysts of the ear. J Audiol Otol. 2017;21:77-80. doi:10.7874 /jao.2017.21.2.77
- Ballan A, Zogheib S, Hanna C, et al. Auricular pseudocysts: a systematic review of the literature. Int J Dermatol. 2022;61:109-117. doi:10.1111/ijd.15816
- Lim CM, Goh YH, Chao SS, et al. Pseudocyst of the auricle: a histologic perspective. Laryngoscope. 2004;114:1281-1284. doi:10.1097/00005537-200407000-00026
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018; 40:631-641. doi:10.1097/DAD.0000000000001118
- McInerney NJ, Nae A, Brennan S, et al. Pilomatricoma of the external auditory canal. Royal College of Surgeons in Ireland. 2023. doi:10.1016/j.xocr.2023.10053
THE DIAGNOSIS: Gouty Tophus
The lesion was excised and sent for histopathologic examination (eFigures 1 and 2), revealing aggregates of feathery, amorphous, pale-pink material, which confirmed the diagnosis of gouty tophus. The surgical site was left to heal by secondary intention. Upon further evaluation, the patient reported recurrent monoarticular joint pain in the ankles and feet, and laboratory workup revealed elevated serum uric acid. He was advised to follow up with his primary care physician to discuss systemic treatment options for gout.
Gout is an inflammatory arthritis characterized by the deposition of monosodium urate monohydrate crystals in the joints, soft tissue, and bone due to elevated serum uric acid. Uric acid is the final product of purine metabolism, and serum levels may be elevated due to excess production or underexcretion. Multiple genetic, environmental, and metabolic factors influence these processes.1 Collections of monosodium urate crystals may develop intra- or extra-articularly, the latter of which are known as gouty tophi. These nodules have a classic chalklike consistency and typically are seen in patients with untreated gout starting approximately 10 years after the first flare. The most common locations for subcutaneous gouty tophi are acral sites (eg, fingertips, ears) as well as the wrists, knees, and elbows (olecranon bursae). Rarely, gouty panniculitis also may develop.2
Histopathology of gouty tophi reveals nodular aggregates of acellular, amorphous, pale-pink material surrounded by palisading histiocytes and multinucleated giant cells. The presence of needlelike monosodium urate crystals, which display negative birefringence, is diagnostic. Unfortunately, these crystals are destroyed in routine formalin processing.3
There are limited data regarding treatment of gouty tophi. Urate-lowering systemic medications such as pegloticase may be beneficial, but more data are needed.4 We pursued surgical excision in our case for definitive diagnosis; however, it is not a common treatment for gouty tophi. Typically, urate-lowering therapy is utilized to resolve or shrink lesions over time.5
The differential diagnosis for gouty tophi includes epidermal inclusion cyst (EIC), the most common type of cutaneous cyst. Though EICs can manifest anywhere on the body, they are not as common on the ears as gouty tophi. Epidermal inclusion cysts clinically manifest as soft subcutaneous nodules, and a central punctum often is noted. These lesions are derived from the follicular infundibulum and histologically are characterized by a cystic cavity lined by a stratified squamous epithelium with a granular layer. The cavity contains loose laminated keratin material.6
Pseudocyst of the auricle is a benign cystic swelling of the pinna that can develop spontaneously but most often manifests following trauma to the area, which is believed to separate the tissue planes in the cartilage, allowing fluid to accumulate. This lesion typically is asymptomatic, though some patients report mild tenderness.7 Histology shows a cystic structure within the cartilage without an epithelial lining, and a perivascular inflammatory response often is observed.8
Pilomatricoma, also known as pilomatrixoma, is a benign tumor derived from the hair follicle matrix that manifests as a firm, slow-growing, painless subcutaneous nodule. It most often is found on the head and neck, commonly in the periauricular area.9 Though rare, it has been found on the auricle and external auditory canal.10 Histologically, pilomatricomas are well-defined tumors containing internal trabeculae. They contain populations of basaloid and ghost cells and often calcify, sometimes with resultant bone formation.9
Dermoid cysts are benign tumors that develop along lines of embryonic closure and often are diagnosed at birth or in early childhood. They most commonly manifest on the head and neck, typically in the supraorbital area. Rarely, they have been reported on the ear.6 Dermoid cysts may resemble EICs clinically and histopathologically, except that the cyst wall contains mature adnexal structures such as hair follicles and sebaceous glands.
THE DIAGNOSIS: Gouty Tophus
The lesion was excised and sent for histopathologic examination (eFigures 1 and 2), revealing aggregates of feathery, amorphous, pale-pink material, which confirmed the diagnosis of gouty tophus. The surgical site was left to heal by secondary intention. Upon further evaluation, the patient reported recurrent monoarticular joint pain in the ankles and feet, and laboratory workup revealed elevated serum uric acid. He was advised to follow up with his primary care physician to discuss systemic treatment options for gout.
Gout is an inflammatory arthritis characterized by the deposition of monosodium urate monohydrate crystals in the joints, soft tissue, and bone due to elevated serum uric acid. Uric acid is the final product of purine metabolism, and serum levels may be elevated due to excess production or underexcretion. Multiple genetic, environmental, and metabolic factors influence these processes.1 Collections of monosodium urate crystals may develop intra- or extra-articularly, the latter of which are known as gouty tophi. These nodules have a classic chalklike consistency and typically are seen in patients with untreated gout starting approximately 10 years after the first flare. The most common locations for subcutaneous gouty tophi are acral sites (eg, fingertips, ears) as well as the wrists, knees, and elbows (olecranon bursae). Rarely, gouty panniculitis also may develop.2
Histopathology of gouty tophi reveals nodular aggregates of acellular, amorphous, pale-pink material surrounded by palisading histiocytes and multinucleated giant cells. The presence of needlelike monosodium urate crystals, which display negative birefringence, is diagnostic. Unfortunately, these crystals are destroyed in routine formalin processing.3
There are limited data regarding treatment of gouty tophi. Urate-lowering systemic medications such as pegloticase may be beneficial, but more data are needed.4 We pursued surgical excision in our case for definitive diagnosis; however, it is not a common treatment for gouty tophi. Typically, urate-lowering therapy is utilized to resolve or shrink lesions over time.5
The differential diagnosis for gouty tophi includes epidermal inclusion cyst (EIC), the most common type of cutaneous cyst. Though EICs can manifest anywhere on the body, they are not as common on the ears as gouty tophi. Epidermal inclusion cysts clinically manifest as soft subcutaneous nodules, and a central punctum often is noted. These lesions are derived from the follicular infundibulum and histologically are characterized by a cystic cavity lined by a stratified squamous epithelium with a granular layer. The cavity contains loose laminated keratin material.6
Pseudocyst of the auricle is a benign cystic swelling of the pinna that can develop spontaneously but most often manifests following trauma to the area, which is believed to separate the tissue planes in the cartilage, allowing fluid to accumulate. This lesion typically is asymptomatic, though some patients report mild tenderness.7 Histology shows a cystic structure within the cartilage without an epithelial lining, and a perivascular inflammatory response often is observed.8
Pilomatricoma, also known as pilomatrixoma, is a benign tumor derived from the hair follicle matrix that manifests as a firm, slow-growing, painless subcutaneous nodule. It most often is found on the head and neck, commonly in the periauricular area.9 Though rare, it has been found on the auricle and external auditory canal.10 Histologically, pilomatricomas are well-defined tumors containing internal trabeculae. They contain populations of basaloid and ghost cells and often calcify, sometimes with resultant bone formation.9
Dermoid cysts are benign tumors that develop along lines of embryonic closure and often are diagnosed at birth or in early childhood. They most commonly manifest on the head and neck, typically in the supraorbital area. Rarely, they have been reported on the ear.6 Dermoid cysts may resemble EICs clinically and histopathologically, except that the cyst wall contains mature adnexal structures such as hair follicles and sebaceous glands.
- Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039-2052. doi:10.1016/S0140-6736(16)00346-9
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445. doi:10.1097/GOX.0000000000000420
- Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. doi:10.1186/s12891-019-2519-y
- Sriranganathan MK, Vinik O, Pardo Pardo J, et al. Interventions for tophi in gout. Cochrane Database Syst Rev. 2021;8:CD010069. doi:10.1002/14651858.CD010069.pub3
- Evidence review for surgical excision of tophi. Gout: diagnosis and management. National Institute for Health and Care Excellence (NICE). June 2022. Accessed October 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK583526/
- Cho Y, Lee DH. Clinical characteristics of idiopathic epidermoid and dermoid cysts of the ear. J Audiol Otol. 2017;21:77-80. doi:10.7874 /jao.2017.21.2.77
- Ballan A, Zogheib S, Hanna C, et al. Auricular pseudocysts: a systematic review of the literature. Int J Dermatol. 2022;61:109-117. doi:10.1111/ijd.15816
- Lim CM, Goh YH, Chao SS, et al. Pseudocyst of the auricle: a histologic perspective. Laryngoscope. 2004;114:1281-1284. doi:10.1097/00005537-200407000-00026
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018; 40:631-641. doi:10.1097/DAD.0000000000001118
- McInerney NJ, Nae A, Brennan S, et al. Pilomatricoma of the external auditory canal. Royal College of Surgeons in Ireland. 2023. doi:10.1016/j.xocr.2023.10053
- Dalbeth N, Merriman TR, Stamp LK. Gout. Lancet. 2016;388:2039-2052. doi:10.1016/S0140-6736(16)00346-9
- Gaviria JL, Ortega VG, Gaona J, et al. Unusual dermatological manifestations of gout: review of literature and a case report. Plast Reconstr Surg Glob Open. 2015;3:E445. doi:10.1097/GOX.0000000000000420
- Towiwat P, Chhana A, Dalbeth N. The anatomical pathology of gout: a systematic literature review. BMC Musculoskelet Disord. 2019;20:140. doi:10.1186/s12891-019-2519-y
- Sriranganathan MK, Vinik O, Pardo Pardo J, et al. Interventions for tophi in gout. Cochrane Database Syst Rev. 2021;8:CD010069. doi:10.1002/14651858.CD010069.pub3
- Evidence review for surgical excision of tophi. Gout: diagnosis and management. National Institute for Health and Care Excellence (NICE). June 2022. Accessed October 8, 2025. https://www.ncbi.nlm.nih.gov/books/NBK583526/
- Cho Y, Lee DH. Clinical characteristics of idiopathic epidermoid and dermoid cysts of the ear. J Audiol Otol. 2017;21:77-80. doi:10.7874 /jao.2017.21.2.77
- Ballan A, Zogheib S, Hanna C, et al. Auricular pseudocysts: a systematic review of the literature. Int J Dermatol. 2022;61:109-117. doi:10.1111/ijd.15816
- Lim CM, Goh YH, Chao SS, et al. Pseudocyst of the auricle: a histologic perspective. Laryngoscope. 2004;114:1281-1284. doi:10.1097/00005537-200407000-00026
- Jones CD, Ho W, Robertson BF, et al. Pilomatrixoma: a comprehensive review of the literature. Am J Dermatopathol. 2018; 40:631-641. doi:10.1097/DAD.0000000000001118
- McInerney NJ, Nae A, Brennan S, et al. Pilomatricoma of the external auditory canal. Royal College of Surgeons in Ireland. 2023. doi:10.1016/j.xocr.2023.10053
Flesh-Colored Lesion on the Ear
Flesh-Colored Lesion on the Ear
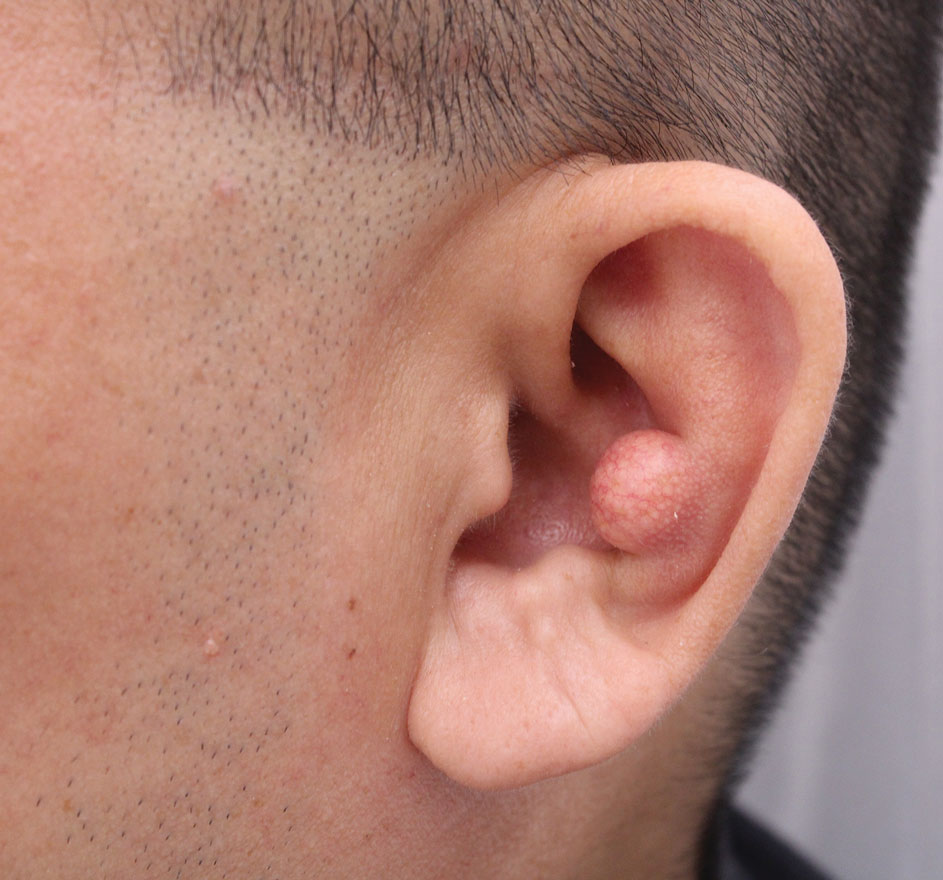
A 46-year-old man with a history of hypertension, hyperlipidemia, and type 2 diabetes presented to the dermatology clinic with a painless nodule on the left ear of 2 years’ duration. The patient denied any bleeding, drainage, or prior trauma to the area. He noted that the lesion had grown slowly over time. Physical examination revealed a 1.5×1.5-cm, flesh-colored, subcutaneous nodule with overlying telangiectasias on the left antihelix.

Smooth Symmetric Plaques on the Face, Trunk, and Extremities
Smooth Symmetric Plaques on the Face, Trunk, and Extremities
THE DIAGNOSIS: Lepromatous Leprosy
Histopathology showed collections of epithelioid to sarcoidal granulomas throughout the dermis and clustered around nerve bundles with a grenz zone at the dermoepidermal junction. Fite stain was positive for acid-fast bacteria, which were confirmed to be Mycobacterium leprae by by the National Hansen’s Disease program. Based on these findings, a diagnosis of lepromatous leprosy (LL) was made. The patient was treated by the infectious disease department with multidrug therapy that included monthly rifampin, moxifloxacin, and minocycline; weekly methotrexate with daily folic acid; and an extended prednisone taper with prophylactic cholecalciferol.
Lepromatous leprosy is characterized by high antibody titers to the acid-fast, gram-positive bacillus Mycobacterium leprae as well as a high bacillary load.1 Patients typically present with muscle weakness, anesthetic skin patches, and claw hands. Patients also may present with foot drop, ulcerations of the hands and feet, autonomic dysfunction with anhidrosis or impaired sweating, and localized alopecia.2 Over months to years, LL may progress to extensive sensory loss and indurated lesions that infiltrate the skin and cause thickening, especially on the face (known as leonine facies). Furthermore, LL is characterized by extensive bilaterally symmetric cutaneous lesions with poorly defined borders and raised indurated centers.3
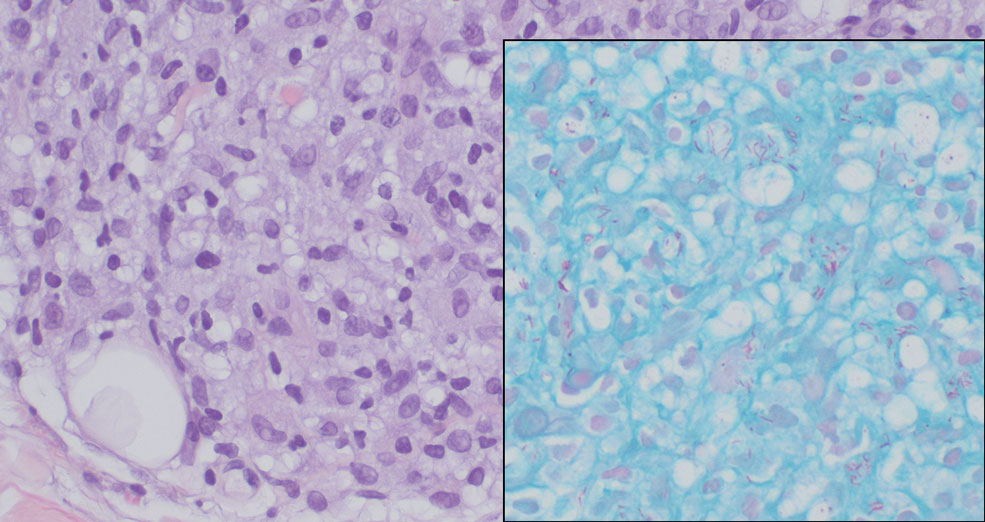
Lepromatous leprosy transmission is not fully understood but is thought to occur via airborne droplets from coughing/sneezing and nasal secretions.2 Histopathology generally shows a dense and diffuse granulomatous infiltrate that involves the dermis but is separated from the epidermis by a zone of collagen (grenz zone).3 Histology is characterized by the presence of lymphocytes and numerous foamy macrophages (lepra or Virchow cells) containing M leprae organisms. In persistent lesions, the high density of uncleared bacilli forms spherical cytoplasmic clumps known as globi within enlarged foamy histiocytes (Figure 1).4 The macrophages form granulomatous lesions in the skin and around nerve bundles, resulting in tissue damage and decreased sensation. The current standard of care for LL is a multidrug combination of dapsone, rifampin, and clofazimine. Early diagnosis and complete treatment of LL is crucial, as this approach typically leads to complete cure of the disease.
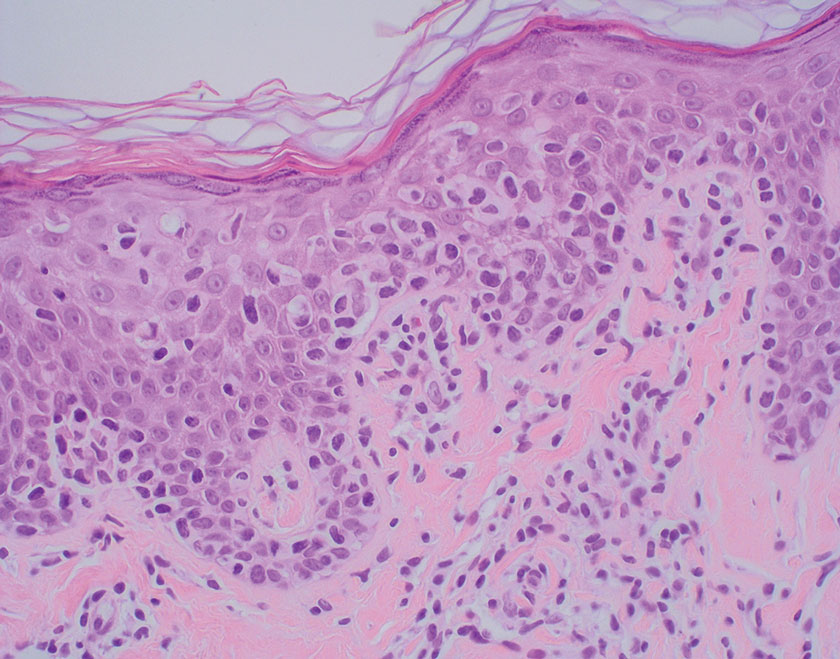
The differential diagnosis for LL includes granuloma annulare (GA), mycosis fungoides (MF), sarcoidosis, and subacute cutaneous lupus erythematosus (SCLE). Granuloma annulare is a noninfectious inflammatory granulomatous skin disease that manifests in a localized, generalized, or subcutaneous pattern. Localized GA is the most common form and manifests as self-resolving, flesh-colored or erythematous papules or plaques limited to the extremities.5,6 Generalized GA is defined by more than 10 widespread annular plaques involving the trunk and extremities and can persist for decades.6 This form can be associated with hyperlipidemia, diabetes, autoimmune disease and immunodeficiency (eg, HIV), and rarely with lymphoma or solid tumors. On histology, GA shows necrobiosis surrounded by palisading histiocytes and mucin (palisading GA) or patchy interstitial histiocytes and lymphocytes (interstitial GA)(Figure 2).6 This palisading pattern differs from the histiocytes in LL, which contain numerous acid-fast bacilli and bacterial clumps. Topical and intralesional corticosteroids are first-line therapies for GA.
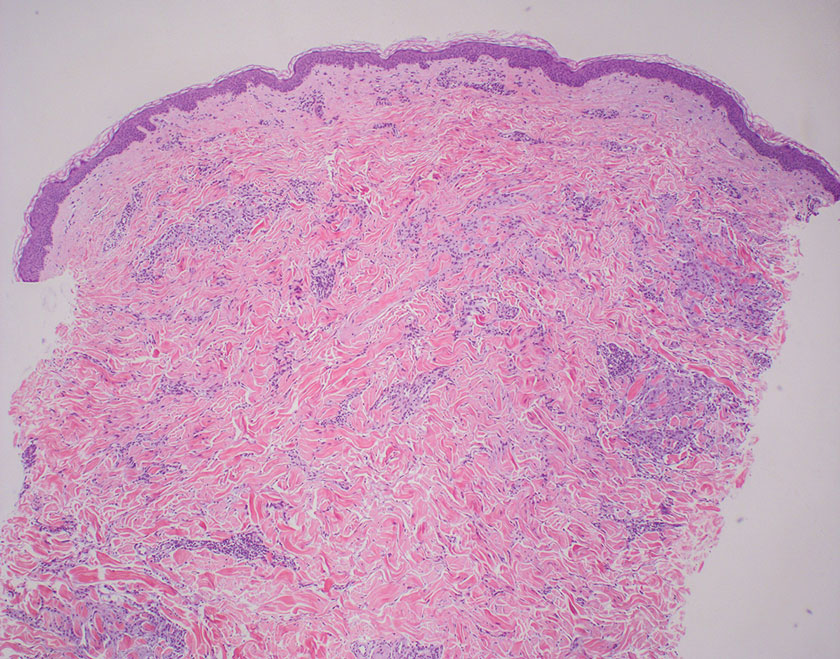
Mycosis fungoides is a cutaneous T-cell lymphoma characterized by proliferation of CD4+ T cells.7 In the early stages of MF, patients may present with multiple erythematous and scaly patches, plaques, or nodules that most commonly develop on unexposed areas of the skin, but specific variants frequently may cause lesions on the face or scalp.8 Tumors may be solitary, localized, or generalized and may be observed alongside patches and plaques or in the absence of cutaneous lesions.7 The pathologic features of MF include fibrosis of the papillary dermis, individual haloed atypical lymphocytes in the epidermis, and atypical lymphoid cells with cerebriform nuclei (Figure 3).9 Granulomatous MF is characterized by diffuse nodular and perivascular infiltrates of histiocytes with small lymphocytes without atypia, eosinophils, and plasma cells. Small lymphocytes with cerebriform nuclei and larger lymphocytes with hyperconvoluted nuclei also may be seen, in addition to multinucleated histiocytic giant cells. Although MF commonly manifests with epidermotropism, it typically is absent in granulomatous MF (GMF).10 Granulomatous MF may manifest similarly to LL. Noduloulcerative lesions and infiltration of atypical lymphocytes into the epidermis (epidermotropism) are much more common in GMF than in LL; however, although ulcerative nodules are not a common feature in patients with leprosy (except during reactional states [ie, Lucio phenomenon]) or secondary to neuropathies, they also can occur in LL.11 In GMF, the infiltrate does not follow a specific pattern, whereas LL infiltrates tend to follow a nerve distribution. Treatment for MF is determined by disease severity.12 First-line therapy includes local corticosteroids and phototherapy with UVB irradiation.
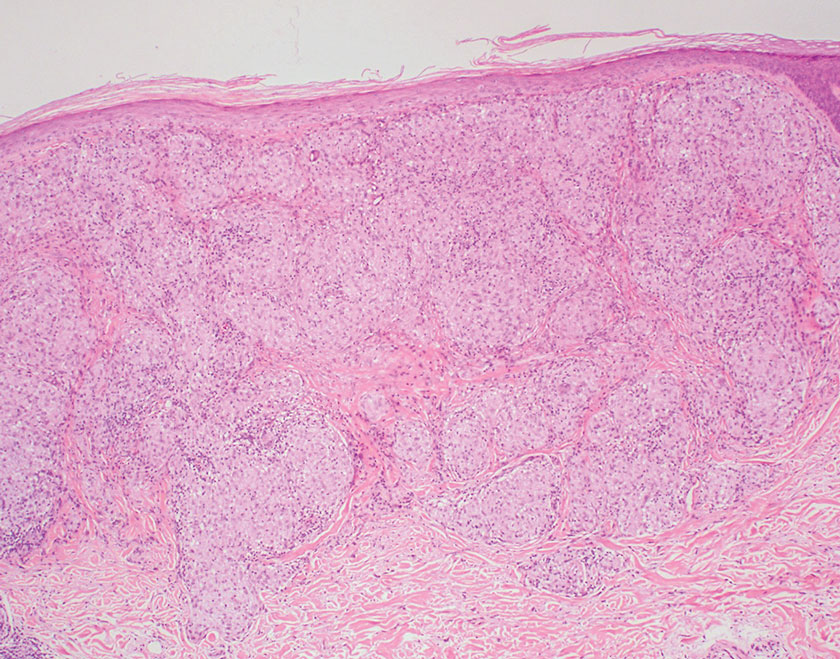
Sarcoidosis is a multisystem disease that demonstrates nonspecific clinical manifestations affecting the lungs, eyes, liver, and skin.13 Environmental exposures to silica and inorganic matter have been linked to an increased risk for sarcoidosis, with patients presenting with fatigue, fever, and arthralgia.13 Skin manifestations include subcutaneous nodules, polymorphous plaques, and erythema nodosum—nodosum—the most common cutaneous presentation of sarcoidosis. Erythema nodosum manifests as symmetrically distributed, nonulcerative, painful red nodules on the skin, especially the lower legs. The histopathology of sarcoidosis shows noncaseating granulomas with activated T-lymphocytes, epithelioid cells, and multinucleated giant cells (Figure 4). Although granulomas occur in both LL and sarcoidosis, those in sarcoidosis typically consist of epithelioid cells surrounded by a rim of lymphocytes, whereas LL granulomas contain foamy histiocytes and multinucleated giant cells. Treatment of sarcoidosis depends on disease progression and generally involves oral corticosteroids, followed by corticosteroid-sparing regimens.
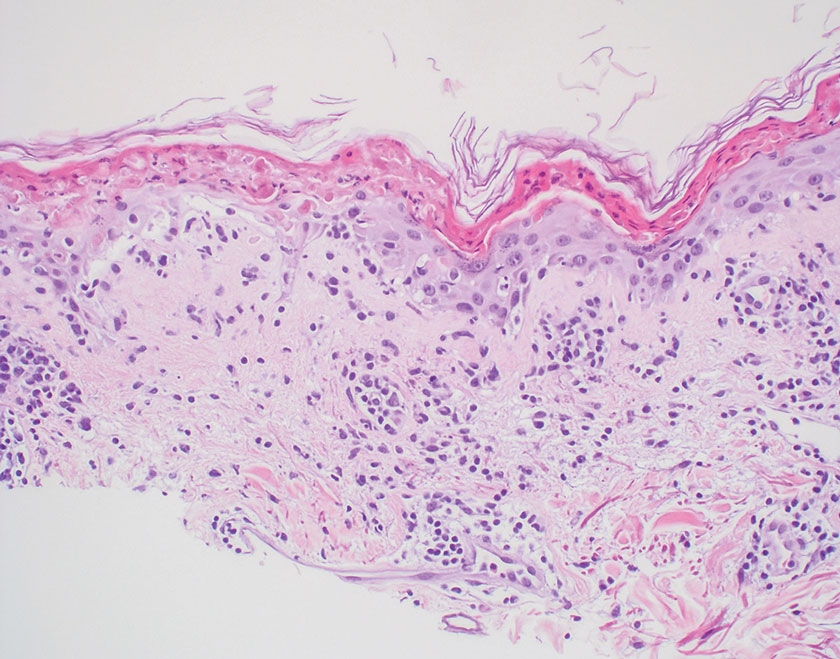
Subacute cutaneous lupus erythematosus is a chronic autoimmune disease that predominantly affects younger women. Common findings in SCLE include red scaly plaques and ring-shaped lesions on sun-exposed areas of the skin.14 Subacute cutaneous lupus erythematosus primarily is characterized by a photosensitive rash, often with arthralgia, myalgia, and/or oral ulcers; less commonly, a small percentage of patients can experience central nervous system involvement, vasculitis, or nephritis. The histologic findings of SCLE include hydropic degeneration of the basal cell layer and periadnexal infiltrates (Figure 5). The incidence of SCLE often is associated with anti-Ro (SSA) and anti-La (SSB) antibodies.15 Treatment of SCLE focuses on managing skin symptoms with corticosteroids, antimalarials, and sun protection.
- Bobosha K, Wilson L, van Meijgaarden KE, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8:E2773. doi:10.1371 /journal.pntd.0002773
- Fischer M. Leprosy–an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827. doi:10.1111/ddg.13301
- Jolly M, Pickard SA, Mikolaitis RA, et al. Lupus QoL-US benchmarks for US patients with systemic lupus erythematosus. J Rheumatol. 2010;37:1828-1833. doi:10.3899/jrheum.091443
- Chan MMF, Smoller BR. Overview of the histopathology and other laboratory investigations in leprosy. Curr Trop Med Rep. 2016;3:131-137. doi:10.1007/s40475-016-0086-y
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016; 75:457-465. doi:10.1016/j.jaad.2015.03.054
- Lukács J, Schliemann S, Elsner P. Treatment of generalized granuloma annulare–a systematic review. J Eur Acad Dermatol Venereol. 2015;29:1467-1480. doi:10.1111/jdv.12976
- Zinzani PL, Ferreri AJM, Cerroni L. Mycosis fungoides. Crit Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Ahn CS, ALSayyah A, Sangüeza OP. Mycosis fungoides: an updated review of clinicopathologic variants. Am J Dermatopathol. 2014;36:933- 951. doi:10.1097/DAD.0000000000000207
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohidrosis mimicking lepromatous leprosy. Indian J Dermatol Venereol Leprol. 2010;76:686. doi:10.4103/0378-6323.72470
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the cutaneous lymphoma histopathology task force group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001 /archdermatol.2008.46
- Miyashiro D, Cardona C, Valente N, et al. Ulcers in leprosy patients, an unrecognized clinical manifestation: a report of 8 cases. BMC Infect Dis. 2019;19:1013. doi:10.1186/s12879-019-4639-2
- Cerroni L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37:2-10. doi:10.12788/j.sder.2018.002
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390 /jcm9041081
- Zÿ ychowska M, Reich A. Dermoscopic features of acute, subacute, chronic and intermittent subtypes of cutaneous lupus erythematosus in Caucasians. J Clin Med. 2022;11:4088. doi:10.3390/jcm11144088
- Lazar AL. Subacute cutaneous lupus erythematosus: a facultative paraneoplastic dermatosis. Clin Dermatol. 2022;40:728-742. doi:10.1016 /j.clindermatol.2022.07.007
THE DIAGNOSIS: Lepromatous Leprosy
Histopathology showed collections of epithelioid to sarcoidal granulomas throughout the dermis and clustered around nerve bundles with a grenz zone at the dermoepidermal junction. Fite stain was positive for acid-fast bacteria, which were confirmed to be Mycobacterium leprae by by the National Hansen’s Disease program. Based on these findings, a diagnosis of lepromatous leprosy (LL) was made. The patient was treated by the infectious disease department with multidrug therapy that included monthly rifampin, moxifloxacin, and minocycline; weekly methotrexate with daily folic acid; and an extended prednisone taper with prophylactic cholecalciferol.
Lepromatous leprosy is characterized by high antibody titers to the acid-fast, gram-positive bacillus Mycobacterium leprae as well as a high bacillary load.1 Patients typically present with muscle weakness, anesthetic skin patches, and claw hands. Patients also may present with foot drop, ulcerations of the hands and feet, autonomic dysfunction with anhidrosis or impaired sweating, and localized alopecia.2 Over months to years, LL may progress to extensive sensory loss and indurated lesions that infiltrate the skin and cause thickening, especially on the face (known as leonine facies). Furthermore, LL is characterized by extensive bilaterally symmetric cutaneous lesions with poorly defined borders and raised indurated centers.3
Lepromatous leprosy transmission is not fully understood but is thought to occur via airborne droplets from coughing/sneezing and nasal secretions.2 Histopathology generally shows a dense and diffuse granulomatous infiltrate that involves the dermis but is separated from the epidermis by a zone of collagen (grenz zone).3 Histology is characterized by the presence of lymphocytes and numerous foamy macrophages (lepra or Virchow cells) containing M leprae organisms. In persistent lesions, the high density of uncleared bacilli forms spherical cytoplasmic clumps known as globi within enlarged foamy histiocytes (Figure 1).4 The macrophages form granulomatous lesions in the skin and around nerve bundles, resulting in tissue damage and decreased sensation. The current standard of care for LL is a multidrug combination of dapsone, rifampin, and clofazimine. Early diagnosis and complete treatment of LL is crucial, as this approach typically leads to complete cure of the disease.
The differential diagnosis for LL includes granuloma annulare (GA), mycosis fungoides (MF), sarcoidosis, and subacute cutaneous lupus erythematosus (SCLE). Granuloma annulare is a noninfectious inflammatory granulomatous skin disease that manifests in a localized, generalized, or subcutaneous pattern. Localized GA is the most common form and manifests as self-resolving, flesh-colored or erythematous papules or plaques limited to the extremities.5,6 Generalized GA is defined by more than 10 widespread annular plaques involving the trunk and extremities and can persist for decades.6 This form can be associated with hyperlipidemia, diabetes, autoimmune disease and immunodeficiency (eg, HIV), and rarely with lymphoma or solid tumors. On histology, GA shows necrobiosis surrounded by palisading histiocytes and mucin (palisading GA) or patchy interstitial histiocytes and lymphocytes (interstitial GA)(Figure 2).6 This palisading pattern differs from the histiocytes in LL, which contain numerous acid-fast bacilli and bacterial clumps. Topical and intralesional corticosteroids are first-line therapies for GA.
Mycosis fungoides is a cutaneous T-cell lymphoma characterized by proliferation of CD4+ T cells.7 In the early stages of MF, patients may present with multiple erythematous and scaly patches, plaques, or nodules that most commonly develop on unexposed areas of the skin, but specific variants frequently may cause lesions on the face or scalp.8 Tumors may be solitary, localized, or generalized and may be observed alongside patches and plaques or in the absence of cutaneous lesions.7 The pathologic features of MF include fibrosis of the papillary dermis, individual haloed atypical lymphocytes in the epidermis, and atypical lymphoid cells with cerebriform nuclei (Figure 3).9 Granulomatous MF is characterized by diffuse nodular and perivascular infiltrates of histiocytes with small lymphocytes without atypia, eosinophils, and plasma cells. Small lymphocytes with cerebriform nuclei and larger lymphocytes with hyperconvoluted nuclei also may be seen, in addition to multinucleated histiocytic giant cells. Although MF commonly manifests with epidermotropism, it typically is absent in granulomatous MF (GMF).10 Granulomatous MF may manifest similarly to LL. Noduloulcerative lesions and infiltration of atypical lymphocytes into the epidermis (epidermotropism) are much more common in GMF than in LL; however, although ulcerative nodules are not a common feature in patients with leprosy (except during reactional states [ie, Lucio phenomenon]) or secondary to neuropathies, they also can occur in LL.11 In GMF, the infiltrate does not follow a specific pattern, whereas LL infiltrates tend to follow a nerve distribution. Treatment for MF is determined by disease severity.12 First-line therapy includes local corticosteroids and phototherapy with UVB irradiation.
Sarcoidosis is a multisystem disease that demonstrates nonspecific clinical manifestations affecting the lungs, eyes, liver, and skin.13 Environmental exposures to silica and inorganic matter have been linked to an increased risk for sarcoidosis, with patients presenting with fatigue, fever, and arthralgia.13 Skin manifestations include subcutaneous nodules, polymorphous plaques, and erythema nodosum—nodosum—the most common cutaneous presentation of sarcoidosis. Erythema nodosum manifests as symmetrically distributed, nonulcerative, painful red nodules on the skin, especially the lower legs. The histopathology of sarcoidosis shows noncaseating granulomas with activated T-lymphocytes, epithelioid cells, and multinucleated giant cells (Figure 4). Although granulomas occur in both LL and sarcoidosis, those in sarcoidosis typically consist of epithelioid cells surrounded by a rim of lymphocytes, whereas LL granulomas contain foamy histiocytes and multinucleated giant cells. Treatment of sarcoidosis depends on disease progression and generally involves oral corticosteroids, followed by corticosteroid-sparing regimens.
Subacute cutaneous lupus erythematosus is a chronic autoimmune disease that predominantly affects younger women. Common findings in SCLE include red scaly plaques and ring-shaped lesions on sun-exposed areas of the skin.14 Subacute cutaneous lupus erythematosus primarily is characterized by a photosensitive rash, often with arthralgia, myalgia, and/or oral ulcers; less commonly, a small percentage of patients can experience central nervous system involvement, vasculitis, or nephritis. The histologic findings of SCLE include hydropic degeneration of the basal cell layer and periadnexal infiltrates (Figure 5). The incidence of SCLE often is associated with anti-Ro (SSA) and anti-La (SSB) antibodies.15 Treatment of SCLE focuses on managing skin symptoms with corticosteroids, antimalarials, and sun protection.
THE DIAGNOSIS: Lepromatous Leprosy
Histopathology showed collections of epithelioid to sarcoidal granulomas throughout the dermis and clustered around nerve bundles with a grenz zone at the dermoepidermal junction. Fite stain was positive for acid-fast bacteria, which were confirmed to be Mycobacterium leprae by by the National Hansen’s Disease program. Based on these findings, a diagnosis of lepromatous leprosy (LL) was made. The patient was treated by the infectious disease department with multidrug therapy that included monthly rifampin, moxifloxacin, and minocycline; weekly methotrexate with daily folic acid; and an extended prednisone taper with prophylactic cholecalciferol.
Lepromatous leprosy is characterized by high antibody titers to the acid-fast, gram-positive bacillus Mycobacterium leprae as well as a high bacillary load.1 Patients typically present with muscle weakness, anesthetic skin patches, and claw hands. Patients also may present with foot drop, ulcerations of the hands and feet, autonomic dysfunction with anhidrosis or impaired sweating, and localized alopecia.2 Over months to years, LL may progress to extensive sensory loss and indurated lesions that infiltrate the skin and cause thickening, especially on the face (known as leonine facies). Furthermore, LL is characterized by extensive bilaterally symmetric cutaneous lesions with poorly defined borders and raised indurated centers.3
Lepromatous leprosy transmission is not fully understood but is thought to occur via airborne droplets from coughing/sneezing and nasal secretions.2 Histopathology generally shows a dense and diffuse granulomatous infiltrate that involves the dermis but is separated from the epidermis by a zone of collagen (grenz zone).3 Histology is characterized by the presence of lymphocytes and numerous foamy macrophages (lepra or Virchow cells) containing M leprae organisms. In persistent lesions, the high density of uncleared bacilli forms spherical cytoplasmic clumps known as globi within enlarged foamy histiocytes (Figure 1).4 The macrophages form granulomatous lesions in the skin and around nerve bundles, resulting in tissue damage and decreased sensation. The current standard of care for LL is a multidrug combination of dapsone, rifampin, and clofazimine. Early diagnosis and complete treatment of LL is crucial, as this approach typically leads to complete cure of the disease.
The differential diagnosis for LL includes granuloma annulare (GA), mycosis fungoides (MF), sarcoidosis, and subacute cutaneous lupus erythematosus (SCLE). Granuloma annulare is a noninfectious inflammatory granulomatous skin disease that manifests in a localized, generalized, or subcutaneous pattern. Localized GA is the most common form and manifests as self-resolving, flesh-colored or erythematous papules or plaques limited to the extremities.5,6 Generalized GA is defined by more than 10 widespread annular plaques involving the trunk and extremities and can persist for decades.6 This form can be associated with hyperlipidemia, diabetes, autoimmune disease and immunodeficiency (eg, HIV), and rarely with lymphoma or solid tumors. On histology, GA shows necrobiosis surrounded by palisading histiocytes and mucin (palisading GA) or patchy interstitial histiocytes and lymphocytes (interstitial GA)(Figure 2).6 This palisading pattern differs from the histiocytes in LL, which contain numerous acid-fast bacilli and bacterial clumps. Topical and intralesional corticosteroids are first-line therapies for GA.
Mycosis fungoides is a cutaneous T-cell lymphoma characterized by proliferation of CD4+ T cells.7 In the early stages of MF, patients may present with multiple erythematous and scaly patches, plaques, or nodules that most commonly develop on unexposed areas of the skin, but specific variants frequently may cause lesions on the face or scalp.8 Tumors may be solitary, localized, or generalized and may be observed alongside patches and plaques or in the absence of cutaneous lesions.7 The pathologic features of MF include fibrosis of the papillary dermis, individual haloed atypical lymphocytes in the epidermis, and atypical lymphoid cells with cerebriform nuclei (Figure 3).9 Granulomatous MF is characterized by diffuse nodular and perivascular infiltrates of histiocytes with small lymphocytes without atypia, eosinophils, and plasma cells. Small lymphocytes with cerebriform nuclei and larger lymphocytes with hyperconvoluted nuclei also may be seen, in addition to multinucleated histiocytic giant cells. Although MF commonly manifests with epidermotropism, it typically is absent in granulomatous MF (GMF).10 Granulomatous MF may manifest similarly to LL. Noduloulcerative lesions and infiltration of atypical lymphocytes into the epidermis (epidermotropism) are much more common in GMF than in LL; however, although ulcerative nodules are not a common feature in patients with leprosy (except during reactional states [ie, Lucio phenomenon]) or secondary to neuropathies, they also can occur in LL.11 In GMF, the infiltrate does not follow a specific pattern, whereas LL infiltrates tend to follow a nerve distribution. Treatment for MF is determined by disease severity.12 First-line therapy includes local corticosteroids and phototherapy with UVB irradiation.
Sarcoidosis is a multisystem disease that demonstrates nonspecific clinical manifestations affecting the lungs, eyes, liver, and skin.13 Environmental exposures to silica and inorganic matter have been linked to an increased risk for sarcoidosis, with patients presenting with fatigue, fever, and arthralgia.13 Skin manifestations include subcutaneous nodules, polymorphous plaques, and erythema nodosum—nodosum—the most common cutaneous presentation of sarcoidosis. Erythema nodosum manifests as symmetrically distributed, nonulcerative, painful red nodules on the skin, especially the lower legs. The histopathology of sarcoidosis shows noncaseating granulomas with activated T-lymphocytes, epithelioid cells, and multinucleated giant cells (Figure 4). Although granulomas occur in both LL and sarcoidosis, those in sarcoidosis typically consist of epithelioid cells surrounded by a rim of lymphocytes, whereas LL granulomas contain foamy histiocytes and multinucleated giant cells. Treatment of sarcoidosis depends on disease progression and generally involves oral corticosteroids, followed by corticosteroid-sparing regimens.
Subacute cutaneous lupus erythematosus is a chronic autoimmune disease that predominantly affects younger women. Common findings in SCLE include red scaly plaques and ring-shaped lesions on sun-exposed areas of the skin.14 Subacute cutaneous lupus erythematosus primarily is characterized by a photosensitive rash, often with arthralgia, myalgia, and/or oral ulcers; less commonly, a small percentage of patients can experience central nervous system involvement, vasculitis, or nephritis. The histologic findings of SCLE include hydropic degeneration of the basal cell layer and periadnexal infiltrates (Figure 5). The incidence of SCLE often is associated with anti-Ro (SSA) and anti-La (SSB) antibodies.15 Treatment of SCLE focuses on managing skin symptoms with corticosteroids, antimalarials, and sun protection.
- Bobosha K, Wilson L, van Meijgaarden KE, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8:E2773. doi:10.1371 /journal.pntd.0002773
- Fischer M. Leprosy–an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827. doi:10.1111/ddg.13301
- Jolly M, Pickard SA, Mikolaitis RA, et al. Lupus QoL-US benchmarks for US patients with systemic lupus erythematosus. J Rheumatol. 2010;37:1828-1833. doi:10.3899/jrheum.091443
- Chan MMF, Smoller BR. Overview of the histopathology and other laboratory investigations in leprosy. Curr Trop Med Rep. 2016;3:131-137. doi:10.1007/s40475-016-0086-y
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016; 75:457-465. doi:10.1016/j.jaad.2015.03.054
- Lukács J, Schliemann S, Elsner P. Treatment of generalized granuloma annulare–a systematic review. J Eur Acad Dermatol Venereol. 2015;29:1467-1480. doi:10.1111/jdv.12976
- Zinzani PL, Ferreri AJM, Cerroni L. Mycosis fungoides. Crit Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Ahn CS, ALSayyah A, Sangüeza OP. Mycosis fungoides: an updated review of clinicopathologic variants. Am J Dermatopathol. 2014;36:933- 951. doi:10.1097/DAD.0000000000000207
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohidrosis mimicking lepromatous leprosy. Indian J Dermatol Venereol Leprol. 2010;76:686. doi:10.4103/0378-6323.72470
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the cutaneous lymphoma histopathology task force group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001 /archdermatol.2008.46
- Miyashiro D, Cardona C, Valente N, et al. Ulcers in leprosy patients, an unrecognized clinical manifestation: a report of 8 cases. BMC Infect Dis. 2019;19:1013. doi:10.1186/s12879-019-4639-2
- Cerroni L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37:2-10. doi:10.12788/j.sder.2018.002
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390 /jcm9041081
- Zÿ ychowska M, Reich A. Dermoscopic features of acute, subacute, chronic and intermittent subtypes of cutaneous lupus erythematosus in Caucasians. J Clin Med. 2022;11:4088. doi:10.3390/jcm11144088
- Lazar AL. Subacute cutaneous lupus erythematosus: a facultative paraneoplastic dermatosis. Clin Dermatol. 2022;40:728-742. doi:10.1016 /j.clindermatol.2022.07.007
- Bobosha K, Wilson L, van Meijgaarden KE, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8:E2773. doi:10.1371 /journal.pntd.0002773
- Fischer M. Leprosy–an overview of clinical features, diagnosis, and treatment. J Dtsch Dermatol Ges. 2017;15:801-827. doi:10.1111/ddg.13301
- Jolly M, Pickard SA, Mikolaitis RA, et al. Lupus QoL-US benchmarks for US patients with systemic lupus erythematosus. J Rheumatol. 2010;37:1828-1833. doi:10.3899/jrheum.091443
- Chan MMF, Smoller BR. Overview of the histopathology and other laboratory investigations in leprosy. Curr Trop Med Rep. 2016;3:131-137. doi:10.1007/s40475-016-0086-y
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016; 75:457-465. doi:10.1016/j.jaad.2015.03.054
- Lukács J, Schliemann S, Elsner P. Treatment of generalized granuloma annulare–a systematic review. J Eur Acad Dermatol Venereol. 2015;29:1467-1480. doi:10.1111/jdv.12976
- Zinzani PL, Ferreri AJM, Cerroni L. Mycosis fungoides. Crit Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Ahn CS, ALSayyah A, Sangüeza OP. Mycosis fungoides: an updated review of clinicopathologic variants. Am J Dermatopathol. 2014;36:933- 951. doi:10.1097/DAD.0000000000000207
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohidrosis mimicking lepromatous leprosy. Indian J Dermatol Venereol Leprol. 2010;76:686. doi:10.4103/0378-6323.72470
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the cutaneous lymphoma histopathology task force group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617. doi:10.1001 /archdermatol.2008.46
- Miyashiro D, Cardona C, Valente N, et al. Ulcers in leprosy patients, an unrecognized clinical manifestation: a report of 8 cases. BMC Infect Dis. 2019;19:1013. doi:10.1186/s12879-019-4639-2
- Cerroni L. Mycosis fungoides-clinical and histopathologic features, differential diagnosis, and treatment. Semin Cutan Med Surg. 2018;37:2-10. doi:10.12788/j.sder.2018.002
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: causes, diagnosis, clinical features, and treatments. J Clin Med. 2020;9:1081. doi:10.3390 /jcm9041081
- Zÿ ychowska M, Reich A. Dermoscopic features of acute, subacute, chronic and intermittent subtypes of cutaneous lupus erythematosus in Caucasians. J Clin Med. 2022;11:4088. doi:10.3390/jcm11144088
- Lazar AL. Subacute cutaneous lupus erythematosus: a facultative paraneoplastic dermatosis. Clin Dermatol. 2022;40:728-742. doi:10.1016 /j.clindermatol.2022.07.007
Smooth Symmetric Plaques on the Face, Trunk, and Extremities
Smooth Symmetric Plaques on the Face, Trunk, and Extremities
A 44-year-old woman presented to the dermatology clinic with a widespread red, itchy, bumpy rash of 1 year’s duration. Physical examination revealed smooth, coalescing, erythematous and edematous plaques on the face (notably the forehead, malar cheeks, and nose), back, arms, and legs. Several plaques on the back had central hypopigmentation. The patient also reported numbness and weakness in the fingers and toes, and hypoesthesia within the lesions was noted. A biopsy of one of the lesions on the left ventral forearm was performed.
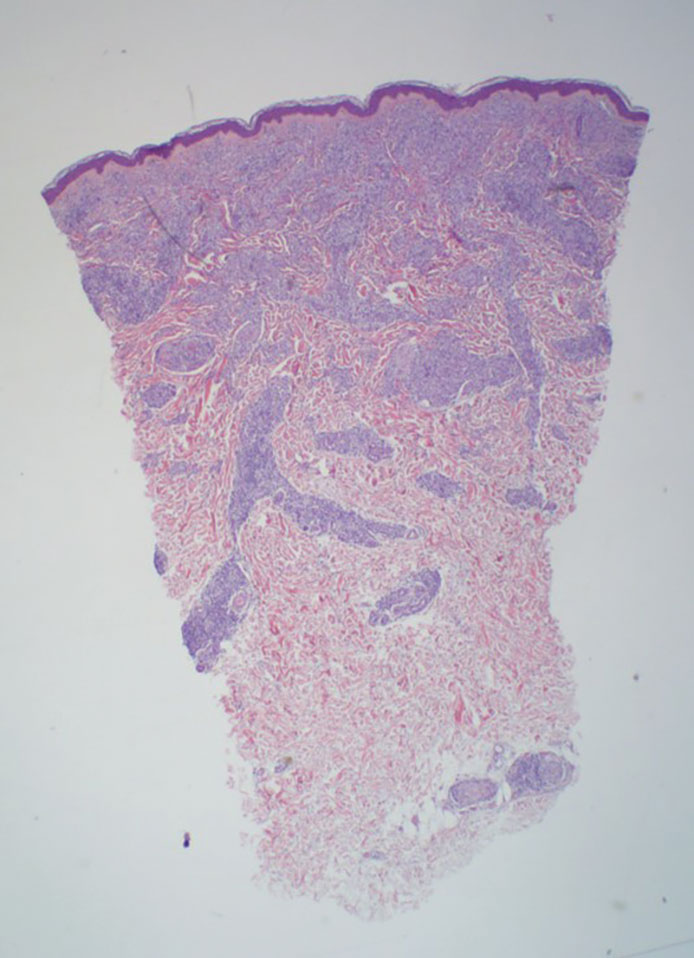

Acute Generalized Exanthematous Pustulosis Secondary to Application of Tapinarof Cream 1%
Acute Generalized Exanthematous Pustulosis Secondary to Application of Tapinarof Cream 1%
To the Editor:
For many years, topical treatment of plaque psoriasis was limited to steroids, calcineurin inhibitors, vitamin D analogs, retinoids, coal tar products, and anthralin. In recent years, 2 new nonsteroidal treatment options with alternative mechanisms of action, roflumilast 0.3% and tapinarof 1%, have been approved by the US Food and Drug Administration.1 Roflumilast 0.3%, a topical phosphodiesterase 4 inhibitor, was shown in phase 3 clinical trials to reach an Investigator Global Assessment response of 37.5% to 42.2% in 8 weeks using once-daily application with minimal cutaneous adverse effects.1 Furthermore, it has demonstrated efficacy in treating psoriasis in intertriginous areas in subset analyses.1 Tapinarof is an aryl hydrocarbon receptor agonist that suppresses Th17 cell differentiation by downregulating IL-17, IL-22, and IL-23.1 In phase 3 clinical trials, 35% to 40% of patients who used tapinarof cream 1% once daily demonstrated improvement in psoriasis compared with 6% who used the vehicle alone.2 In these studies, 18% to 24% of patients who used tapinarof cream 1% experienced folliculitis.2
Acute generalized exanthematous pustulosis (AGEP) is a nonfollicular pustular drug reaction with systemic symptoms that typically occurs within 2 weeks of exposure to an inciting medication. Systemic antibiotics are the most commonly reported cause of AGEP.3 There are few reports in the literature of AGEP induced by topical agents.4,5 We report a case of AGEP in a young man following the use of tapinarof cream 1%.
A 23-year-old man with a history of psoriasis presented to the emergency department with fever and a pustular rash. One week prior to presentation, he developed a pustular eruption around plaques of psoriasis on the arms and legs. The patient had been prescribed tapinarof cream 1% by an outside dermatologist and was applying the medication to the affected areas once daily for 1 month prior to onset of symptoms. He discontinued tapinarof a few days prior to the eruption starting, but the rash progressed centrifugally and was associated with fevers and fatigue despite treatment with a brief course of empiric cephalexin prescribed by his primary care provider.
At presentation to our institution, the patient had widespread erythematous patches studded with pustules located on the arms, legs, and flexural areas as well as plaques of psoriasis involving approximately 20% of the body surface area (Figure 1). Furthermore, the patient was noted to have large noninflammatory bullae along the legs. The new eruption occurred on areas that were both treated and spared from the tapinarof cream 1%. Laboratory evaluation showed neutrophil-predominant leukocytosis (white blood cell count, 15.9×103/µL [reference range, 4.0-11.0×103/µL]; absolute neutrophil count, 10.3×103/µL [reference range, 1.5-8.0×103/µL]), absolute eosinophilia (1930/µL [reference range, 0-0.5×103/µL]), hypocalcemia (8.4 mg/dL [reference range, 8.5-10.5 mg/dL]), and a mild transaminitis (aspartate aminotransferase, 37 IU/L [reference range, 10-40 IU/L]; alanine aminotransferase, 53 IU/L [reference range, 7-56 U/L]). Histopathology demonstrated spongiosis with subcorneal and intraepidermal pustules and mixed dermal inflammation containing eosinophils (Figure 2). Direct immunofluorescence revealed mild granular staining of C3 at the basement membrane zone.
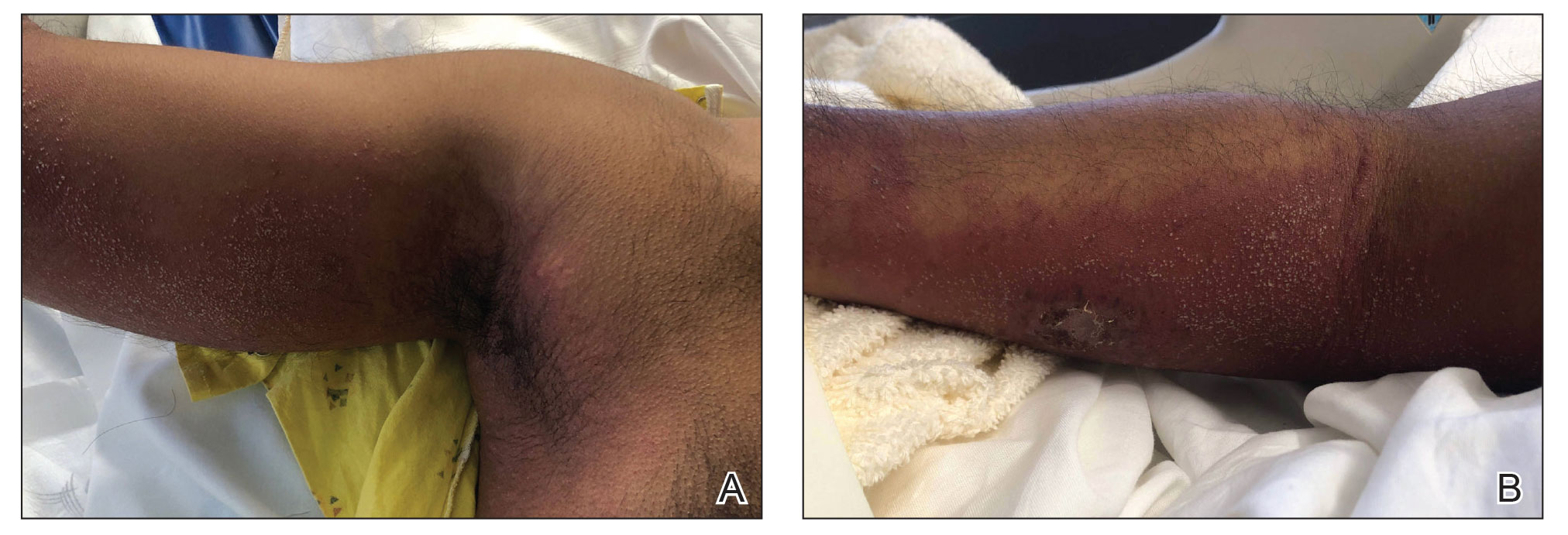
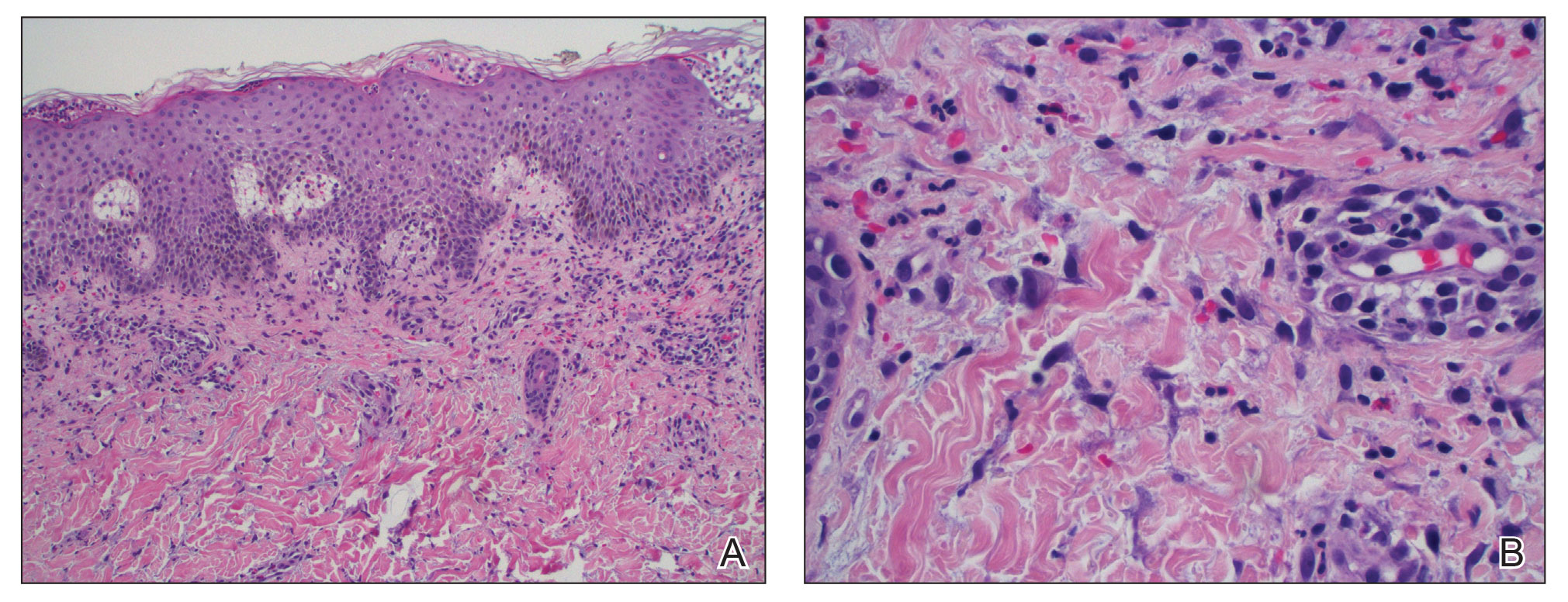
The patient was started on 1 mg/kg/d of prednisone tapered over 20 days, and he rapidly improved. Alanine aminotransferase levels peaked at 120 IU/L 2 weeks later. At that time, he had complete resolution of the original eruption and was transitioned to topical steroids for continued management of the psoriasis (Figure 3).
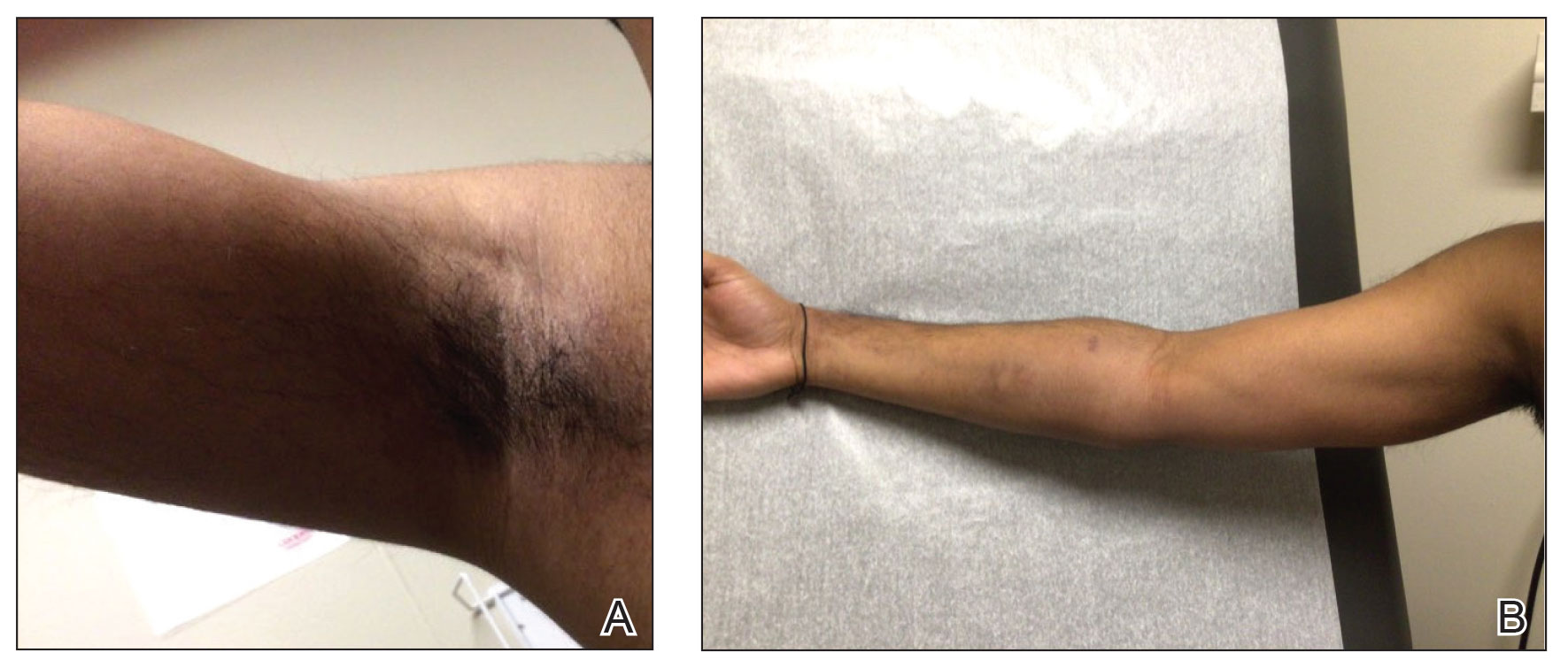
The differential diagnosis for our patient included AGEP, generalized pustular psoriasis (GPP), miliaria pustulosa, generalized cutaneous candidiasis, exuberant allergic contact dermatitis (ACD), and linear IgA bullous dermatosis (LABD). Based on the clinical manifestations, laboratory results, and histopathologic evaluation, we made the diagnosis of AGEP secondary to tapinarof with systemic absorption. Acute generalized exanthematous pustulosis has been reported with topical use of morphine and diphenhydramine, among other agents.4,5 To our knowledge, AGEP due to tapinarof cream 1% has not been reported. In the original clinical trials of tapinarof, folliculitis was contained to sites of application.2 Our patient developed pustules at sites distant to areas of application, as well as systemic symptoms and laboratory abnormalities, indicating a systemic reaction. It can be difficult to distinguish AGEP clinically and histologically from GPP. Both conditions can manifest with fever, hypocalcemia, and sterile pustules on a background of erythema that favors intertriginous areas.6 Infection, rapid oral steroid withdrawal, pregnancy, and rarely oral medications have been reported causes of GPP.6 Our patient did not have any of these exposures. There is overlap in the histology of AGEP and GPP. One retrospective series compared histologic samples to help distinguish these 2 entities. Reliable markers that favored AGEP over GPP included eosinophilic spongiosis, interface dermatitis, and dermal eosinophilia (>2/mm2).7 In contrast, the presence of CD161 positivity in the dermis with at least 10 cells favored a diagnosis of GPP.7 In our case, the presence of spongiosis with eosinophils in the dermis favored a diagnosis of AGEP over GPP.
Miliaria pustulosa is a benign condition caused by the occlusion of the epidermal portion of eccrine glands related to either high fever or hot and humid environmental conditions. While it can be present in intertriginous areas like AGEP, miliaria pustulosa can be seen extensively on the back, most commonly in immobile hospitalized patients.8 Generalized cutaneous candidiasis usually is caused by the yeast Candida albicans and can take on multiple morphologies, including folliculitis.9 The eruption may be disseminated but often is accentuated in intertriginous areas and the anogenital folds. Predisposing factors include immunosuppression, endocrinopathies, recent use of systemic antibiotics or steroids, chemotherapy, and indwelling catheters.9 Outside of recent antibiotic use, our patient did not have any risk factors for miliaria pustulosa, making this diagnosis unlikely.
Given the presence of overlapping bullae along the lower extremities, an exuberant ACD and LABD were considered. Bullae formation can occur in ACD secondary to robust inflammation and edema leading to acantholysis.10 While a delayed hypersensitivity reaction to topical tapinarof cream 1% was considered given that the patient used the medication for approximately 1 month prior to the onset of symptoms, it would be unlikely for ACD to present with a concomitant pustular eruption. Linear IgA bullous dermatosis is an autoimmune blistering disease in which antibodies target bullous pemphigoid antigen 2, and there is characteristically linear deposition of IgA at the dermal-epidermal junction that leads to subepidermal blistering.11 This often manifests clinically as widespread tense vesicles in an annular or string-of-pearls appearance. However, morphologies can vary, and large bullae may be seen. In adults, LABD typically is associated with inflammatory bowel disease, malignancy, or medications, notably vancomycin.11,12 Our patient did not have any of these predisposing factors, and his biopsy for direct immunofluorescence did not reveal the classic pattern described above.
Interestingly, there have been reports in the literature of bullous AGEP in the setting of oral anti-infectives. One report described a 62-year-old woman who developed widespread nonfollicular pustules with multiple tense serous blisters 24 hours after taking oral terbinafine.13 Another case described an 80-year-old woman with a similar presentation following a course of ciprofloxacin (although the timeline of medication administration was not described).14 In this case, patch testing to the culprit medication reproduced the response.14 In both cases, a biopsy revealed subcorneal and intraepidermal pustules with marked dermal edema.13,14 As previously described, spongiosis is a common feature of AGEP. We hypothesize that, similar to these reports, our patient had a robust inflammatory response leading to spongiosis, acantholysis, and blister formation secondary to AGEP.
Dermatologists should be aware of this case of AGEP secondary to tapinarof cream 1%, as reports in the literature are rare and it is a reminder that topical medications can cause serious systemic reactions.
- Lebwohl MG, Kircik LH, Moore AY, et al. Effect of roflumilast cream vs vehicle cream on chronic plaque psoriasis: the DERMIS-1 and DERMIS-2 randomized clinical trials. JAMA. 2022;328:1073-1084. doi:10.1001/jama.2022.15632
- Lebwohl MG, Stein Gold L, Strober B, et al. Phase 3 trials of tapinarof cream for plaque psoriasis. N Engl J Med. 2021;385:2219-2229. doi:10.1056/NEJMoa2103629
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848. doi:10.1016/j.jaad.2015.07.017
- Ghazawi FM, Colantonio S, Bradshaw S, et al. Acute generalized exanthematous pustulosis induced by topical morphine and confirmed by patch testing. Dermat Contact Atopic Occup Drug. 2020;31:E22-E23. doi:10.1097/DER.0000000000000573
- Hanafusa T, Igawa K, Azukizawa H, et al. Acute generalized exanthematous pustulosis induced by topical diphenhydramine. Eur J Dermatol. 2011;21:994-995. doi:10.1684/ejd.2011.1500
- Reynolds KA, Pithadia DJ, Lee EB, et al. Generalized pustular psoriasis: a review of the pathophysiology, clinical manifestations,diagnosis, and treatment. Cutis. 2022;110:19-25. doi:10.12788/cutis.0579
- Isom J, Braswell DS, Siroy A, et al. Clinical and histopathologic features differentiating acute generalized exanthematous pustulosis and pustular psoriasis: a retrospective series. J Am Acad Dermatol. 2020;83:265-267. doi:10.1016/j.jaad.2020.03.015
- Fealey RD, Hebert AA. Disorders of the eccrine sweat glands and sweating. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine.8th ed. McGraw-Hill; 2012:946.
- Elewski BE, Hughey LC, Marchiony Hunt K, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1329-1363.
- Elmas ÖF, Akdeniz N, Atasoy M, et al. Contact dermatitis: a great imitator. Clin Dermatol. 2020;38:176-192. doi:10.1016/j.clindermatol.2019.10.003
- Hull CM, Zone JZ. Dermatitis herpetiforms and linear IgA bullous dermatosis. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:527-537.
- Yamagami J, Nakamura Y, Nagao K, et al. Vancomycin mediates IgA autoreactivity in drug-induced linear IgA bullous dermatosis. J Invest Dermatol. 2018;138:1473-1480.
- Bullous acute generalized exanthematous pustulosis due to oral terbinafine. J Am Acad Dermatol. 2005;52:P115. doi:10.1016/j.jaad.2004.10.468
- Hausermann P, Scherer K, Weber M, et al. Ciprofloxacin-induced acute generalized exanthematous pustulosis mimicking bullous drug eruption confirmed by a positive patch test. Dermatology. 2005;211:277-280. doi:10.1159/000087024
To the Editor:
For many years, topical treatment of plaque psoriasis was limited to steroids, calcineurin inhibitors, vitamin D analogs, retinoids, coal tar products, and anthralin. In recent years, 2 new nonsteroidal treatment options with alternative mechanisms of action, roflumilast 0.3% and tapinarof 1%, have been approved by the US Food and Drug Administration.1 Roflumilast 0.3%, a topical phosphodiesterase 4 inhibitor, was shown in phase 3 clinical trials to reach an Investigator Global Assessment response of 37.5% to 42.2% in 8 weeks using once-daily application with minimal cutaneous adverse effects.1 Furthermore, it has demonstrated efficacy in treating psoriasis in intertriginous areas in subset analyses.1 Tapinarof is an aryl hydrocarbon receptor agonist that suppresses Th17 cell differentiation by downregulating IL-17, IL-22, and IL-23.1 In phase 3 clinical trials, 35% to 40% of patients who used tapinarof cream 1% once daily demonstrated improvement in psoriasis compared with 6% who used the vehicle alone.2 In these studies, 18% to 24% of patients who used tapinarof cream 1% experienced folliculitis.2
Acute generalized exanthematous pustulosis (AGEP) is a nonfollicular pustular drug reaction with systemic symptoms that typically occurs within 2 weeks of exposure to an inciting medication. Systemic antibiotics are the most commonly reported cause of AGEP.3 There are few reports in the literature of AGEP induced by topical agents.4,5 We report a case of AGEP in a young man following the use of tapinarof cream 1%.
A 23-year-old man with a history of psoriasis presented to the emergency department with fever and a pustular rash. One week prior to presentation, he developed a pustular eruption around plaques of psoriasis on the arms and legs. The patient had been prescribed tapinarof cream 1% by an outside dermatologist and was applying the medication to the affected areas once daily for 1 month prior to onset of symptoms. He discontinued tapinarof a few days prior to the eruption starting, but the rash progressed centrifugally and was associated with fevers and fatigue despite treatment with a brief course of empiric cephalexin prescribed by his primary care provider.
At presentation to our institution, the patient had widespread erythematous patches studded with pustules located on the arms, legs, and flexural areas as well as plaques of psoriasis involving approximately 20% of the body surface area (Figure 1). Furthermore, the patient was noted to have large noninflammatory bullae along the legs. The new eruption occurred on areas that were both treated and spared from the tapinarof cream 1%. Laboratory evaluation showed neutrophil-predominant leukocytosis (white blood cell count, 15.9×103/µL [reference range, 4.0-11.0×103/µL]; absolute neutrophil count, 10.3×103/µL [reference range, 1.5-8.0×103/µL]), absolute eosinophilia (1930/µL [reference range, 0-0.5×103/µL]), hypocalcemia (8.4 mg/dL [reference range, 8.5-10.5 mg/dL]), and a mild transaminitis (aspartate aminotransferase, 37 IU/L [reference range, 10-40 IU/L]; alanine aminotransferase, 53 IU/L [reference range, 7-56 U/L]). Histopathology demonstrated spongiosis with subcorneal and intraepidermal pustules and mixed dermal inflammation containing eosinophils (Figure 2). Direct immunofluorescence revealed mild granular staining of C3 at the basement membrane zone.
The patient was started on 1 mg/kg/d of prednisone tapered over 20 days, and he rapidly improved. Alanine aminotransferase levels peaked at 120 IU/L 2 weeks later. At that time, he had complete resolution of the original eruption and was transitioned to topical steroids for continued management of the psoriasis (Figure 3).
The differential diagnosis for our patient included AGEP, generalized pustular psoriasis (GPP), miliaria pustulosa, generalized cutaneous candidiasis, exuberant allergic contact dermatitis (ACD), and linear IgA bullous dermatosis (LABD). Based on the clinical manifestations, laboratory results, and histopathologic evaluation, we made the diagnosis of AGEP secondary to tapinarof with systemic absorption. Acute generalized exanthematous pustulosis has been reported with topical use of morphine and diphenhydramine, among other agents.4,5 To our knowledge, AGEP due to tapinarof cream 1% has not been reported. In the original clinical trials of tapinarof, folliculitis was contained to sites of application.2 Our patient developed pustules at sites distant to areas of application, as well as systemic symptoms and laboratory abnormalities, indicating a systemic reaction. It can be difficult to distinguish AGEP clinically and histologically from GPP. Both conditions can manifest with fever, hypocalcemia, and sterile pustules on a background of erythema that favors intertriginous areas.6 Infection, rapid oral steroid withdrawal, pregnancy, and rarely oral medications have been reported causes of GPP.6 Our patient did not have any of these exposures. There is overlap in the histology of AGEP and GPP. One retrospective series compared histologic samples to help distinguish these 2 entities. Reliable markers that favored AGEP over GPP included eosinophilic spongiosis, interface dermatitis, and dermal eosinophilia (>2/mm2).7 In contrast, the presence of CD161 positivity in the dermis with at least 10 cells favored a diagnosis of GPP.7 In our case, the presence of spongiosis with eosinophils in the dermis favored a diagnosis of AGEP over GPP.
Miliaria pustulosa is a benign condition caused by the occlusion of the epidermal portion of eccrine glands related to either high fever or hot and humid environmental conditions. While it can be present in intertriginous areas like AGEP, miliaria pustulosa can be seen extensively on the back, most commonly in immobile hospitalized patients.8 Generalized cutaneous candidiasis usually is caused by the yeast Candida albicans and can take on multiple morphologies, including folliculitis.9 The eruption may be disseminated but often is accentuated in intertriginous areas and the anogenital folds. Predisposing factors include immunosuppression, endocrinopathies, recent use of systemic antibiotics or steroids, chemotherapy, and indwelling catheters.9 Outside of recent antibiotic use, our patient did not have any risk factors for miliaria pustulosa, making this diagnosis unlikely.
Given the presence of overlapping bullae along the lower extremities, an exuberant ACD and LABD were considered. Bullae formation can occur in ACD secondary to robust inflammation and edema leading to acantholysis.10 While a delayed hypersensitivity reaction to topical tapinarof cream 1% was considered given that the patient used the medication for approximately 1 month prior to the onset of symptoms, it would be unlikely for ACD to present with a concomitant pustular eruption. Linear IgA bullous dermatosis is an autoimmune blistering disease in which antibodies target bullous pemphigoid antigen 2, and there is characteristically linear deposition of IgA at the dermal-epidermal junction that leads to subepidermal blistering.11 This often manifests clinically as widespread tense vesicles in an annular or string-of-pearls appearance. However, morphologies can vary, and large bullae may be seen. In adults, LABD typically is associated with inflammatory bowel disease, malignancy, or medications, notably vancomycin.11,12 Our patient did not have any of these predisposing factors, and his biopsy for direct immunofluorescence did not reveal the classic pattern described above.
Interestingly, there have been reports in the literature of bullous AGEP in the setting of oral anti-infectives. One report described a 62-year-old woman who developed widespread nonfollicular pustules with multiple tense serous blisters 24 hours after taking oral terbinafine.13 Another case described an 80-year-old woman with a similar presentation following a course of ciprofloxacin (although the timeline of medication administration was not described).14 In this case, patch testing to the culprit medication reproduced the response.14 In both cases, a biopsy revealed subcorneal and intraepidermal pustules with marked dermal edema.13,14 As previously described, spongiosis is a common feature of AGEP. We hypothesize that, similar to these reports, our patient had a robust inflammatory response leading to spongiosis, acantholysis, and blister formation secondary to AGEP.
Dermatologists should be aware of this case of AGEP secondary to tapinarof cream 1%, as reports in the literature are rare and it is a reminder that topical medications can cause serious systemic reactions.
To the Editor:
For many years, topical treatment of plaque psoriasis was limited to steroids, calcineurin inhibitors, vitamin D analogs, retinoids, coal tar products, and anthralin. In recent years, 2 new nonsteroidal treatment options with alternative mechanisms of action, roflumilast 0.3% and tapinarof 1%, have been approved by the US Food and Drug Administration.1 Roflumilast 0.3%, a topical phosphodiesterase 4 inhibitor, was shown in phase 3 clinical trials to reach an Investigator Global Assessment response of 37.5% to 42.2% in 8 weeks using once-daily application with minimal cutaneous adverse effects.1 Furthermore, it has demonstrated efficacy in treating psoriasis in intertriginous areas in subset analyses.1 Tapinarof is an aryl hydrocarbon receptor agonist that suppresses Th17 cell differentiation by downregulating IL-17, IL-22, and IL-23.1 In phase 3 clinical trials, 35% to 40% of patients who used tapinarof cream 1% once daily demonstrated improvement in psoriasis compared with 6% who used the vehicle alone.2 In these studies, 18% to 24% of patients who used tapinarof cream 1% experienced folliculitis.2
Acute generalized exanthematous pustulosis (AGEP) is a nonfollicular pustular drug reaction with systemic symptoms that typically occurs within 2 weeks of exposure to an inciting medication. Systemic antibiotics are the most commonly reported cause of AGEP.3 There are few reports in the literature of AGEP induced by topical agents.4,5 We report a case of AGEP in a young man following the use of tapinarof cream 1%.
A 23-year-old man with a history of psoriasis presented to the emergency department with fever and a pustular rash. One week prior to presentation, he developed a pustular eruption around plaques of psoriasis on the arms and legs. The patient had been prescribed tapinarof cream 1% by an outside dermatologist and was applying the medication to the affected areas once daily for 1 month prior to onset of symptoms. He discontinued tapinarof a few days prior to the eruption starting, but the rash progressed centrifugally and was associated with fevers and fatigue despite treatment with a brief course of empiric cephalexin prescribed by his primary care provider.
At presentation to our institution, the patient had widespread erythematous patches studded with pustules located on the arms, legs, and flexural areas as well as plaques of psoriasis involving approximately 20% of the body surface area (Figure 1). Furthermore, the patient was noted to have large noninflammatory bullae along the legs. The new eruption occurred on areas that were both treated and spared from the tapinarof cream 1%. Laboratory evaluation showed neutrophil-predominant leukocytosis (white blood cell count, 15.9×103/µL [reference range, 4.0-11.0×103/µL]; absolute neutrophil count, 10.3×103/µL [reference range, 1.5-8.0×103/µL]), absolute eosinophilia (1930/µL [reference range, 0-0.5×103/µL]), hypocalcemia (8.4 mg/dL [reference range, 8.5-10.5 mg/dL]), and a mild transaminitis (aspartate aminotransferase, 37 IU/L [reference range, 10-40 IU/L]; alanine aminotransferase, 53 IU/L [reference range, 7-56 U/L]). Histopathology demonstrated spongiosis with subcorneal and intraepidermal pustules and mixed dermal inflammation containing eosinophils (Figure 2). Direct immunofluorescence revealed mild granular staining of C3 at the basement membrane zone.
The patient was started on 1 mg/kg/d of prednisone tapered over 20 days, and he rapidly improved. Alanine aminotransferase levels peaked at 120 IU/L 2 weeks later. At that time, he had complete resolution of the original eruption and was transitioned to topical steroids for continued management of the psoriasis (Figure 3).
The differential diagnosis for our patient included AGEP, generalized pustular psoriasis (GPP), miliaria pustulosa, generalized cutaneous candidiasis, exuberant allergic contact dermatitis (ACD), and linear IgA bullous dermatosis (LABD). Based on the clinical manifestations, laboratory results, and histopathologic evaluation, we made the diagnosis of AGEP secondary to tapinarof with systemic absorption. Acute generalized exanthematous pustulosis has been reported with topical use of morphine and diphenhydramine, among other agents.4,5 To our knowledge, AGEP due to tapinarof cream 1% has not been reported. In the original clinical trials of tapinarof, folliculitis was contained to sites of application.2 Our patient developed pustules at sites distant to areas of application, as well as systemic symptoms and laboratory abnormalities, indicating a systemic reaction. It can be difficult to distinguish AGEP clinically and histologically from GPP. Both conditions can manifest with fever, hypocalcemia, and sterile pustules on a background of erythema that favors intertriginous areas.6 Infection, rapid oral steroid withdrawal, pregnancy, and rarely oral medications have been reported causes of GPP.6 Our patient did not have any of these exposures. There is overlap in the histology of AGEP and GPP. One retrospective series compared histologic samples to help distinguish these 2 entities. Reliable markers that favored AGEP over GPP included eosinophilic spongiosis, interface dermatitis, and dermal eosinophilia (>2/mm2).7 In contrast, the presence of CD161 positivity in the dermis with at least 10 cells favored a diagnosis of GPP.7 In our case, the presence of spongiosis with eosinophils in the dermis favored a diagnosis of AGEP over GPP.
Miliaria pustulosa is a benign condition caused by the occlusion of the epidermal portion of eccrine glands related to either high fever or hot and humid environmental conditions. While it can be present in intertriginous areas like AGEP, miliaria pustulosa can be seen extensively on the back, most commonly in immobile hospitalized patients.8 Generalized cutaneous candidiasis usually is caused by the yeast Candida albicans and can take on multiple morphologies, including folliculitis.9 The eruption may be disseminated but often is accentuated in intertriginous areas and the anogenital folds. Predisposing factors include immunosuppression, endocrinopathies, recent use of systemic antibiotics or steroids, chemotherapy, and indwelling catheters.9 Outside of recent antibiotic use, our patient did not have any risk factors for miliaria pustulosa, making this diagnosis unlikely.
Given the presence of overlapping bullae along the lower extremities, an exuberant ACD and LABD were considered. Bullae formation can occur in ACD secondary to robust inflammation and edema leading to acantholysis.10 While a delayed hypersensitivity reaction to topical tapinarof cream 1% was considered given that the patient used the medication for approximately 1 month prior to the onset of symptoms, it would be unlikely for ACD to present with a concomitant pustular eruption. Linear IgA bullous dermatosis is an autoimmune blistering disease in which antibodies target bullous pemphigoid antigen 2, and there is characteristically linear deposition of IgA at the dermal-epidermal junction that leads to subepidermal blistering.11 This often manifests clinically as widespread tense vesicles in an annular or string-of-pearls appearance. However, morphologies can vary, and large bullae may be seen. In adults, LABD typically is associated with inflammatory bowel disease, malignancy, or medications, notably vancomycin.11,12 Our patient did not have any of these predisposing factors, and his biopsy for direct immunofluorescence did not reveal the classic pattern described above.
Interestingly, there have been reports in the literature of bullous AGEP in the setting of oral anti-infectives. One report described a 62-year-old woman who developed widespread nonfollicular pustules with multiple tense serous blisters 24 hours after taking oral terbinafine.13 Another case described an 80-year-old woman with a similar presentation following a course of ciprofloxacin (although the timeline of medication administration was not described).14 In this case, patch testing to the culprit medication reproduced the response.14 In both cases, a biopsy revealed subcorneal and intraepidermal pustules with marked dermal edema.13,14 As previously described, spongiosis is a common feature of AGEP. We hypothesize that, similar to these reports, our patient had a robust inflammatory response leading to spongiosis, acantholysis, and blister formation secondary to AGEP.
Dermatologists should be aware of this case of AGEP secondary to tapinarof cream 1%, as reports in the literature are rare and it is a reminder that topical medications can cause serious systemic reactions.
- Lebwohl MG, Kircik LH, Moore AY, et al. Effect of roflumilast cream vs vehicle cream on chronic plaque psoriasis: the DERMIS-1 and DERMIS-2 randomized clinical trials. JAMA. 2022;328:1073-1084. doi:10.1001/jama.2022.15632
- Lebwohl MG, Stein Gold L, Strober B, et al. Phase 3 trials of tapinarof cream for plaque psoriasis. N Engl J Med. 2021;385:2219-2229. doi:10.1056/NEJMoa2103629
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848. doi:10.1016/j.jaad.2015.07.017
- Ghazawi FM, Colantonio S, Bradshaw S, et al. Acute generalized exanthematous pustulosis induced by topical morphine and confirmed by patch testing. Dermat Contact Atopic Occup Drug. 2020;31:E22-E23. doi:10.1097/DER.0000000000000573
- Hanafusa T, Igawa K, Azukizawa H, et al. Acute generalized exanthematous pustulosis induced by topical diphenhydramine. Eur J Dermatol. 2011;21:994-995. doi:10.1684/ejd.2011.1500
- Reynolds KA, Pithadia DJ, Lee EB, et al. Generalized pustular psoriasis: a review of the pathophysiology, clinical manifestations,diagnosis, and treatment. Cutis. 2022;110:19-25. doi:10.12788/cutis.0579
- Isom J, Braswell DS, Siroy A, et al. Clinical and histopathologic features differentiating acute generalized exanthematous pustulosis and pustular psoriasis: a retrospective series. J Am Acad Dermatol. 2020;83:265-267. doi:10.1016/j.jaad.2020.03.015
- Fealey RD, Hebert AA. Disorders of the eccrine sweat glands and sweating. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine.8th ed. McGraw-Hill; 2012:946.
- Elewski BE, Hughey LC, Marchiony Hunt K, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1329-1363.
- Elmas ÖF, Akdeniz N, Atasoy M, et al. Contact dermatitis: a great imitator. Clin Dermatol. 2020;38:176-192. doi:10.1016/j.clindermatol.2019.10.003
- Hull CM, Zone JZ. Dermatitis herpetiforms and linear IgA bullous dermatosis. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:527-537.
- Yamagami J, Nakamura Y, Nagao K, et al. Vancomycin mediates IgA autoreactivity in drug-induced linear IgA bullous dermatosis. J Invest Dermatol. 2018;138:1473-1480.
- Bullous acute generalized exanthematous pustulosis due to oral terbinafine. J Am Acad Dermatol. 2005;52:P115. doi:10.1016/j.jaad.2004.10.468
- Hausermann P, Scherer K, Weber M, et al. Ciprofloxacin-induced acute generalized exanthematous pustulosis mimicking bullous drug eruption confirmed by a positive patch test. Dermatology. 2005;211:277-280. doi:10.1159/000087024
- Lebwohl MG, Kircik LH, Moore AY, et al. Effect of roflumilast cream vs vehicle cream on chronic plaque psoriasis: the DERMIS-1 and DERMIS-2 randomized clinical trials. JAMA. 2022;328:1073-1084. doi:10.1001/jama.2022.15632
- Lebwohl MG, Stein Gold L, Strober B, et al. Phase 3 trials of tapinarof cream for plaque psoriasis. N Engl J Med. 2021;385:2219-2229. doi:10.1056/NEJMoa2103629
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848. doi:10.1016/j.jaad.2015.07.017
- Ghazawi FM, Colantonio S, Bradshaw S, et al. Acute generalized exanthematous pustulosis induced by topical morphine and confirmed by patch testing. Dermat Contact Atopic Occup Drug. 2020;31:E22-E23. doi:10.1097/DER.0000000000000573
- Hanafusa T, Igawa K, Azukizawa H, et al. Acute generalized exanthematous pustulosis induced by topical diphenhydramine. Eur J Dermatol. 2011;21:994-995. doi:10.1684/ejd.2011.1500
- Reynolds KA, Pithadia DJ, Lee EB, et al. Generalized pustular psoriasis: a review of the pathophysiology, clinical manifestations,diagnosis, and treatment. Cutis. 2022;110:19-25. doi:10.12788/cutis.0579
- Isom J, Braswell DS, Siroy A, et al. Clinical and histopathologic features differentiating acute generalized exanthematous pustulosis and pustular psoriasis: a retrospective series. J Am Acad Dermatol. 2020;83:265-267. doi:10.1016/j.jaad.2020.03.015
- Fealey RD, Hebert AA. Disorders of the eccrine sweat glands and sweating. In: Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine.8th ed. McGraw-Hill; 2012:946.
- Elewski BE, Hughey LC, Marchiony Hunt K, et al. Fungal diseases. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:1329-1363.
- Elmas ÖF, Akdeniz N, Atasoy M, et al. Contact dermatitis: a great imitator. Clin Dermatol. 2020;38:176-192. doi:10.1016/j.clindermatol.2019.10.003
- Hull CM, Zone JZ. Dermatitis herpetiforms and linear IgA bullous dermatosis. In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2017:527-537.
- Yamagami J, Nakamura Y, Nagao K, et al. Vancomycin mediates IgA autoreactivity in drug-induced linear IgA bullous dermatosis. J Invest Dermatol. 2018;138:1473-1480.
- Bullous acute generalized exanthematous pustulosis due to oral terbinafine. J Am Acad Dermatol. 2005;52:P115. doi:10.1016/j.jaad.2004.10.468
- Hausermann P, Scherer K, Weber M, et al. Ciprofloxacin-induced acute generalized exanthematous pustulosis mimicking bullous drug eruption confirmed by a positive patch test. Dermatology. 2005;211:277-280. doi:10.1159/000087024
Acute Generalized Exanthematous Pustulosis Secondary to Application of Tapinarof Cream 1%
Acute Generalized Exanthematous Pustulosis Secondary to Application of Tapinarof Cream 1%
PRACTICE POINTS
- Tapinarof cream 1% can be absorbed systemically and cause acute generalized exanthematous pustulosis (AGEP).
- Clinical configuration and histology can be useful to distinguish AGEP from mimickers.
- Topical application of drugs in general, particularly over large body surface areas, may lead to systemic drug eruptions.

Longitudinal Erythronychia Manifesting With Pain and Cold Sensitivity
The Diagnosis: Glomangiomyoma
The nail unit excision specimen showed collections of cuboidal cells and spindled cells within the corium that were consistent with a diagnosis of a glomangiomyoma, a rare glomus tumor variant (Figure). Glomus tumors are benign neoplasms comprising glomus bodies, which are arteriovenous anastomoses involved in thermoregulation.1 They develop in areas densely populated by glomus bodies, including the fingers, toes, and subungual areas. Glomus tumors most commonly develop in middle-aged women.2 Clinically, they manifest with a characteristic triad of intense pain, point tenderness, and cold sensitivity and may appear as reddish-pink or blue macules under the nail plate and/or longitudinal erythronychia.2-6 The presence of multiple glomus tumors is associated with neurofibromatosis type 1.7
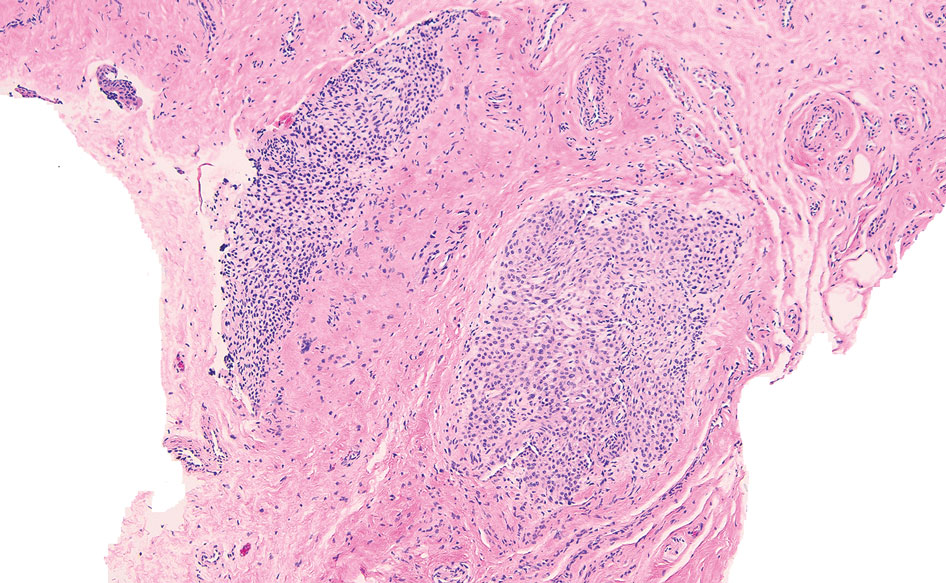
Advanced imaging including ultrasonography and magnetic resonance imaging (MRI) may help confirm the diagnosis but may not be cost effective, as excision with histopathology is needed to relieve symptoms and render a definitive diagnosis. Radiography is highly insensitive in identifying bone erosions associated with glomus tumors.8 With ultrasonography, glomus tumors appear hypoechoic; with Doppler ultrasonography, they appear hypervascular. With MRI, glomus tumors appear as well-defined nodular lesions with hypointense signal intensity on T1-weighted sequence and hyperintense signal intensity on T2-weighted sequence, with strong enhancement using gadolinium-based contrast.9,10 On histopathology, a glomus tumor appears as a nodular tumor with sheets of oval-nucleated cells arranged in multicellular layers surrounding blood vessels and are immunoreactive for α-smooth muscle actin, muscle-specific actin, and type IV collagen.11,12
There are several glomus tumor variants. The most common is a solid glomus tumor, which predominantly is composed of glomus cells, followed by glomangioma, which mainly is composed of blood vessels. Glomangiomyoma, which mostly is composed of smooth muscle cells, is the rarest variant.13
While glomus tumors are common in the subungual areas, it is an uncommon location for glomangiomyomas, which have been reported in the nail unit in only 7 prior case reports identified through searches of PubMed and Google Scholar using the terms glomangiomyoma, glomangiomyoma nail, and subungual glomangiomyoma (Table).13-19 Glomangiomyomas more commonly are described in solid organs, including the stomach, kidney, pancreas, and bladder.16 The mean age of patients with subungual glomangiomyomas, including our patient, was 40.4 years (range, 3-61 years), with the majority being female (75.0% [6/8]). Most patients presented with fingernail involvement (75.0% [6/8]), nail dystrophy (eg, nail plate thinning, longitudinal grooves, splinter hemorrhages, longitudinal erythronychia)(62.5% [5/8]), and intermittent pain and/or point tenderness in the affected nail (75.0% [6/8]).13-19 Notably, only our patient had longitudinal erythronychia as a clinical feature, and only one other case described MRI findings, which included a lobulated mass with intense contrast and distal phalanx destruction.18 One patient was a 3-year-old girl with a family history of generalized multiple glomangiomyomas. Although subungual glomangiomyoma was not confirmed on histopathology, the diagnosis in this patient was presumed based on her family history.13 On histopathology, glomangiomyomas are composed of oval-nucleated cells surrounding blood vessels. These oval-nucleated cells then gradually transition to smooth muscle cells.20
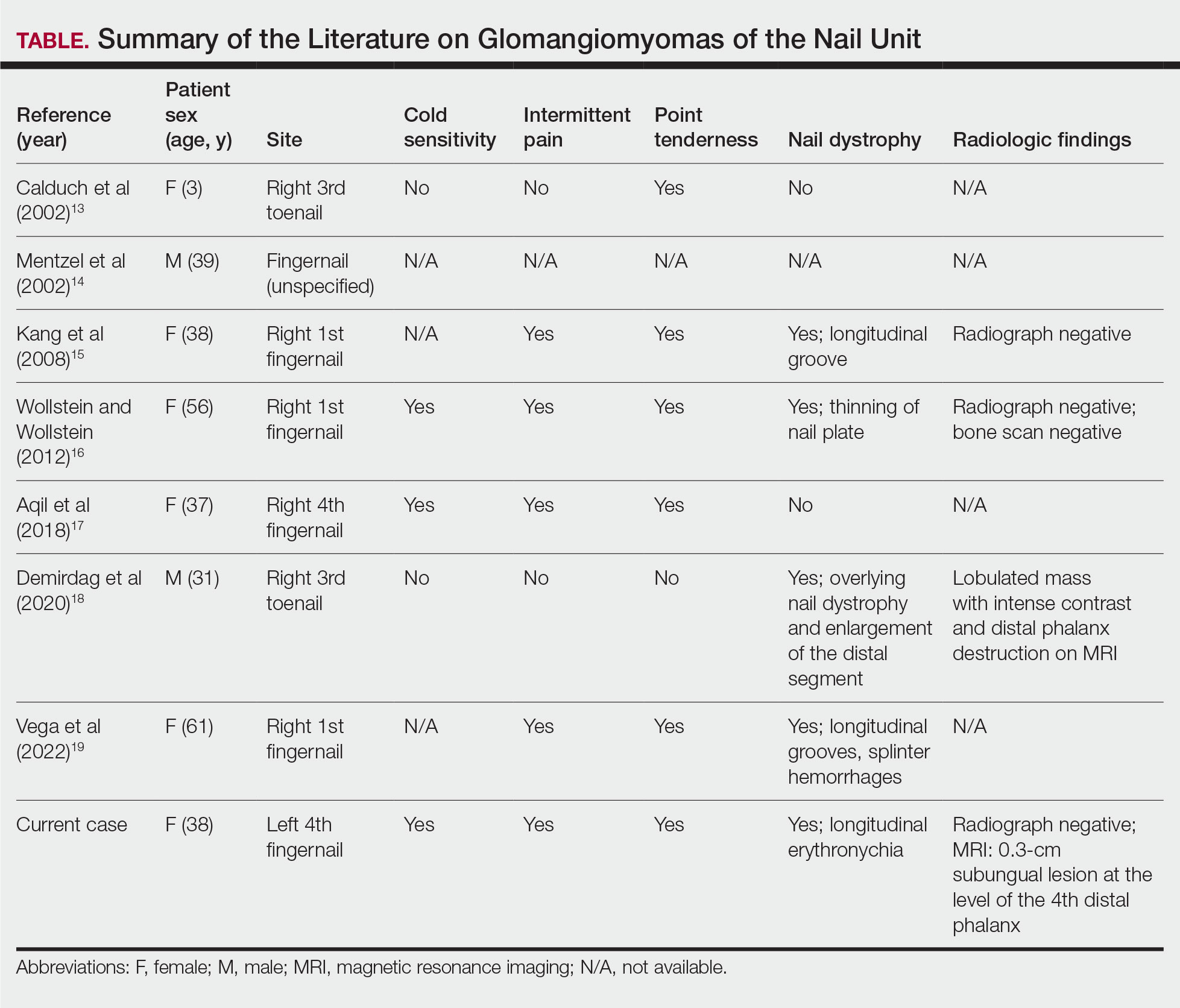
A myxoid cyst is composed of a pseudocyst, which lacks a cyst lining, and is a result of synovial fluid from the distal interphalangeal joint entering the pseudocyst space.2 It typically manifests with a longitudinal groove in the nail plate. A flesh-colored nodule may be appreciated between the cuticle and the distal interphalangeal joint.2 The depth of the longitudinal groove may vary depending on the volume of synovial fluid within the myxoid cyst.21 In a series of 35 cases of subungual myxoid cysts, none manifested with longitudinal erythronychia. Due to their composition, myxoid cysts can be distinguished easily from solid tumors of the nail unit via transillumination.22 Pain is a much less common with myxoid cysts vs glomus tumors, as the filling of the pseudocyst space with synovial fluid typically is gradual, allowing the surrounding tissue to accommodate and adapt over time.21 In equivocal cases, MRI or high-resolution ultrasonography may be used to distinguish myxoid cysts and glomus tumors.8 Histopathology shows accumulation of mucin in the dermis with surrounding fibrous stroma.23
Subungual neuromas are painful benign tumors that develop due to disorganized neural proliferation following disruption to peripheral nerves secondary to trauma or surgery. In 3 case reports, subungual neuromas manifested as painful subungual nodules, with proximal nail plate ridging, or onycholysis.24-26 Since neuromas have only rarely been described in the subungual region, reports of MRI and ultrasonography findings are unknown. Histopathology is needed to distinguish neuromas from glomus tumors. Histopathology shows an acapsular structure consisting of disorganized spindle-cell proliferation and nerve fibers arranged in a tangle of fascicles within fibrotic tissue.25 On immunochemistry, spindle cells typically are positive for cellular antigen protein S100.26
Leiomyomas are benign neoplasms derived from smooth muscle, typically localized to the uterus or gastrointestinal tract, and have been described rarely in the nail unit.27,28 It is hypothesized that subungual leiomyomas originate from the vascular smooth muscle in the subcutaneous layer of the nail unit.28 Like glomus tumors, leiomyomas of the subungual region often manifest with pain and longitudinal erythronychia.27-30 Subungual leiomyomas may be distinguished from glomus tumors via advanced imaging techniques, including ultrasonography and MRI. Cutaneous leiomyomas have been described with mild to moderate internal low flow vascularity on Doppler ultrasonography, while glomus tumors typically reveal high internal vascularity.28 Biopsy with histopathology is needed for definitive diagnosis. On histopathology, leiomyomas demonstrate bland-appearing spindle-shaped cells with elongated nuclei arranged in fascicles.27 They typically are positive for α-smooth muscle actin and caldesmon on immunostaining.
Eccrine spiradenomas are benign adnexal tumors likely of apocrine origin with limited case reports in the literature.31,32 Clinically, eccrine spiradenomas involving the nail unit may manifest with longitudinal nail splitting of the nail or as a papule on the proximal nail fold, with associated tenderness.31,32 In a report of a 50-year-old woman with a histopathologically confirmed eccrine spiradenoma manifesting with longitudinal splitting of the nail and pain in the proximal nail fold, the mass appeared hypoechoic on ultrasonography with increased intramass vascularity on Doppler, while MRI showed an intensely enhancing lesion.31 These imaging features, combined with a classically manifesting feature of pain, make eccrine spiradenomas difficult to distinguish from glomus tumors; therefore, histopathologic examination can provide a definitive diagnosis, and surgical excision is used for treatment.31 On histopathology, these tumors are well circumscribed and composed of both small dark basaloid cells with peripheral compact nuclei and larger cells with central pale nuclei, which may be arranged in tubules.31,32
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132: 1448-1452. doi:10.5858/2008-132-1448-gt
- Hare AQ, Rich P. Nail tumors. Dermatol Clin. 2021;39:281-292. doi:10.1016/j.det.2020.12.007
- Hazani R, Houle JM, Kasdan ML, et al. Glomus tumors of the hand. Eplasty. 2008;8:E48.
- Hwang JK, Lipner SR. Blue nail discoloration: literature review and diagnostic algorithms. Am J Clin Dermatol. 2023;24:419-441. doi:10.1007/s40257-023-00768-6
- Lipner SR, Scher RK. Longitudinal erythronychia of the fingernail. JAMA Dermatol. 2016;152:1271-1272. doi:10.1001/jamadermatol.2016.2747
- Jellinek NJ, Lipner SR. Longitudinal erythronychia: retrospective single-center study evaluating differential diagnosis and the likelihood of malignancy. Dermatol Surg. 2016;42:310-319. doi:10.1097 /DSS.0000000000000594
- Lipner SR, Scher RK. Subungual glomus tumors: underrecognized clinical findings in neurofibromatosis 1. J Am Acad Dermatol. 2021;84:E269. doi:10.1016/j.jaad.2020.08.129
- Dhami A, Vale SM, Richardson ML, et al. Comparing ultrasound with magnetic resonance imaging in the evaluation of subungual glomus tumors and subungual myxoid cysts. Skin Appendage Disord. 2023;9:262-267. doi:10.1159/000530397
- Baek HJ, Lee SJ, Cho KH, et al. Subungual tumors: clinicopathologic correlation with US and MR imaging findings. Radiographics. 2010;30:1621-1636. doi:10.1148/rg.306105514
- Patel T, Meena V, Meena P. Hand and foot glomus tumors: significance of MRI diagnosis followed by histopathological assessment. Cureus. 2022;14:E30038. doi:10.7759/cureus.30038
- Mravic M, LaChaud G, Nguyen A, et al. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23:181-188. doi:10.1177/1066896914567330
- Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12. doi:10.1097/00000478-200101000-00001
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mentzel T, Hügel H, Kutzner H. CD34-positive glomus tumor: clinicopathologic and immunohistochemical analysis of six cases with myxoid stromal changes. J Cutan Pathol. 2002;29:421-425. doi:10.1034 /j.1600-0560.2002.290706.x
- Kang TW, Lee KH, Park CJ. A case of subungual glomangiomyoma with myxoid stromal change. Korean J Dermatol. 2008;46:550-553.
- Wollstein A, Wollstein R. Subungual glomangiomyoma—a case report. Hand Surg. 2012;17:271-273. doi:10.1142/S021881041272032X
- Aqil N, Gallouj S, Moustaide K, et al. Painful tumors in a patient with neurofibromatosis type 1: a case report. J Med Case Rep. 2018;12:319. doi:10.1186/s13256-018-1847-0
- Demirdag HG, Akay BN, Kirmizi A, et al. Subungual glomangiomyoma. J Am Podiatr Med Assoc. 2020;110:Article_13. doi:10.7547/19-051
- Vega SML, Ruiz SJA, Ramírez CS, et al. Subungual glomangiomyoma: a case report. Dermatol Cosmet Med Quir. 2022;20:258-262.
- Chalise S, Jha A, Neupane PR. Glomangiomyoma of uncertain malignant potential in the urinary bladder: a case report. JNMA J Nepal Med Assoc. 2021;59:719-722. doi:10.31729/jnma.5388
- de Berker D, Goettman S, Baran R. Subungual myxoid cysts: clinical manifestations and response to therapy. J Am Acad Dermatol. 2002;46:394-398. doi:10.1067/mjd.2002.119652
- Gupta MK, Lipner SR. Transillumination for improved diagnosis of digital myxoid cysts. Cutis. 2020;105:82.
- Fernandez-Flores A, Saeb-Lima M. Mucin as a diagnostic clue in dermatopathology. J Cutan Pathol. 2016;43:1005-1016. doi:10.1111/cup.12782
- Choi R, Kim SR, Glusac EJ, et al. Subungual neuroma masquerading as green nail syndrome. JAAD Case Rep. 2022;20:17-19. doi:10.1016 /j.jdcr.2021.11.025
- Rashid RM, Rashid RM, Thomas V. Subungal traumatic neuroma. J Am Acad Dermatol. 2010;63:E7-E8. doi:10.1016/j.jaad.2010.01.028
- Whitehouse HJ, Urwin R, Stables G. Traumatic subungual neuroma. Clin Exp Dermatol. 2018;43:65-66. doi:10.1111/ced.13247
- Lipner SR, Ko D, Husain S. Subungual leiyomyoma presenting as erythronychia: case report and review of the literature. J Drugs Dermatol. 2019;18:465-467.
- Taleb E, Saldías C, Gonzalez S, et al. Sonographic characteristics of leiomyomatous tumors of skin and nail: a case series. Dermatol Pract Concept. 2022;12:e2022082. doi:10.5826/dpc.1203a82
- Baran R, Requena L, Drapé JL. Subungual angioleiomyoma masquerading as a glomus tumour. Br J Dermatol. 2000;142:1239-1241. doi:10.1046/ j.1365-2133.2000.03560.x
- Watabe D, Sakurai E, Mori S, et al. Subungual angioleiomyoma. Indian J Dermatol Venereol Leprol. 2017;83:74-75. doi:10.4103/0378-6323 .185045
- Jha AK, Sinha R, Kumar A, et al. Spiradenoma causing longitudinal splitting of the nail. Clin Exp Dermatol. 2016;41:754-756. doi:10.1111 /ced.12886
- Leach BC, Graham BS. Papular lesion of the proximal nail fold. eccrine spiradenoma. Arch Dermatol. 2004;140:1003-1008. doi:10.1001 /archderm.140.8.1003-a
The Diagnosis: Glomangiomyoma
The nail unit excision specimen showed collections of cuboidal cells and spindled cells within the corium that were consistent with a diagnosis of a glomangiomyoma, a rare glomus tumor variant (Figure). Glomus tumors are benign neoplasms comprising glomus bodies, which are arteriovenous anastomoses involved in thermoregulation.1 They develop in areas densely populated by glomus bodies, including the fingers, toes, and subungual areas. Glomus tumors most commonly develop in middle-aged women.2 Clinically, they manifest with a characteristic triad of intense pain, point tenderness, and cold sensitivity and may appear as reddish-pink or blue macules under the nail plate and/or longitudinal erythronychia.2-6 The presence of multiple glomus tumors is associated with neurofibromatosis type 1.7
Advanced imaging including ultrasonography and magnetic resonance imaging (MRI) may help confirm the diagnosis but may not be cost effective, as excision with histopathology is needed to relieve symptoms and render a definitive diagnosis. Radiography is highly insensitive in identifying bone erosions associated with glomus tumors.8 With ultrasonography, glomus tumors appear hypoechoic; with Doppler ultrasonography, they appear hypervascular. With MRI, glomus tumors appear as well-defined nodular lesions with hypointense signal intensity on T1-weighted sequence and hyperintense signal intensity on T2-weighted sequence, with strong enhancement using gadolinium-based contrast.9,10 On histopathology, a glomus tumor appears as a nodular tumor with sheets of oval-nucleated cells arranged in multicellular layers surrounding blood vessels and are immunoreactive for α-smooth muscle actin, muscle-specific actin, and type IV collagen.11,12
There are several glomus tumor variants. The most common is a solid glomus tumor, which predominantly is composed of glomus cells, followed by glomangioma, which mainly is composed of blood vessels. Glomangiomyoma, which mostly is composed of smooth muscle cells, is the rarest variant.13
While glomus tumors are common in the subungual areas, it is an uncommon location for glomangiomyomas, which have been reported in the nail unit in only 7 prior case reports identified through searches of PubMed and Google Scholar using the terms glomangiomyoma, glomangiomyoma nail, and subungual glomangiomyoma (Table).13-19 Glomangiomyomas more commonly are described in solid organs, including the stomach, kidney, pancreas, and bladder.16 The mean age of patients with subungual glomangiomyomas, including our patient, was 40.4 years (range, 3-61 years), with the majority being female (75.0% [6/8]). Most patients presented with fingernail involvement (75.0% [6/8]), nail dystrophy (eg, nail plate thinning, longitudinal grooves, splinter hemorrhages, longitudinal erythronychia)(62.5% [5/8]), and intermittent pain and/or point tenderness in the affected nail (75.0% [6/8]).13-19 Notably, only our patient had longitudinal erythronychia as a clinical feature, and only one other case described MRI findings, which included a lobulated mass with intense contrast and distal phalanx destruction.18 One patient was a 3-year-old girl with a family history of generalized multiple glomangiomyomas. Although subungual glomangiomyoma was not confirmed on histopathology, the diagnosis in this patient was presumed based on her family history.13 On histopathology, glomangiomyomas are composed of oval-nucleated cells surrounding blood vessels. These oval-nucleated cells then gradually transition to smooth muscle cells.20
A myxoid cyst is composed of a pseudocyst, which lacks a cyst lining, and is a result of synovial fluid from the distal interphalangeal joint entering the pseudocyst space.2 It typically manifests with a longitudinal groove in the nail plate. A flesh-colored nodule may be appreciated between the cuticle and the distal interphalangeal joint.2 The depth of the longitudinal groove may vary depending on the volume of synovial fluid within the myxoid cyst.21 In a series of 35 cases of subungual myxoid cysts, none manifested with longitudinal erythronychia. Due to their composition, myxoid cysts can be distinguished easily from solid tumors of the nail unit via transillumination.22 Pain is a much less common with myxoid cysts vs glomus tumors, as the filling of the pseudocyst space with synovial fluid typically is gradual, allowing the surrounding tissue to accommodate and adapt over time.21 In equivocal cases, MRI or high-resolution ultrasonography may be used to distinguish myxoid cysts and glomus tumors.8 Histopathology shows accumulation of mucin in the dermis with surrounding fibrous stroma.23
Subungual neuromas are painful benign tumors that develop due to disorganized neural proliferation following disruption to peripheral nerves secondary to trauma or surgery. In 3 case reports, subungual neuromas manifested as painful subungual nodules, with proximal nail plate ridging, or onycholysis.24-26 Since neuromas have only rarely been described in the subungual region, reports of MRI and ultrasonography findings are unknown. Histopathology is needed to distinguish neuromas from glomus tumors. Histopathology shows an acapsular structure consisting of disorganized spindle-cell proliferation and nerve fibers arranged in a tangle of fascicles within fibrotic tissue.25 On immunochemistry, spindle cells typically are positive for cellular antigen protein S100.26
Leiomyomas are benign neoplasms derived from smooth muscle, typically localized to the uterus or gastrointestinal tract, and have been described rarely in the nail unit.27,28 It is hypothesized that subungual leiomyomas originate from the vascular smooth muscle in the subcutaneous layer of the nail unit.28 Like glomus tumors, leiomyomas of the subungual region often manifest with pain and longitudinal erythronychia.27-30 Subungual leiomyomas may be distinguished from glomus tumors via advanced imaging techniques, including ultrasonography and MRI. Cutaneous leiomyomas have been described with mild to moderate internal low flow vascularity on Doppler ultrasonography, while glomus tumors typically reveal high internal vascularity.28 Biopsy with histopathology is needed for definitive diagnosis. On histopathology, leiomyomas demonstrate bland-appearing spindle-shaped cells with elongated nuclei arranged in fascicles.27 They typically are positive for α-smooth muscle actin and caldesmon on immunostaining.
Eccrine spiradenomas are benign adnexal tumors likely of apocrine origin with limited case reports in the literature.31,32 Clinically, eccrine spiradenomas involving the nail unit may manifest with longitudinal nail splitting of the nail or as a papule on the proximal nail fold, with associated tenderness.31,32 In a report of a 50-year-old woman with a histopathologically confirmed eccrine spiradenoma manifesting with longitudinal splitting of the nail and pain in the proximal nail fold, the mass appeared hypoechoic on ultrasonography with increased intramass vascularity on Doppler, while MRI showed an intensely enhancing lesion.31 These imaging features, combined with a classically manifesting feature of pain, make eccrine spiradenomas difficult to distinguish from glomus tumors; therefore, histopathologic examination can provide a definitive diagnosis, and surgical excision is used for treatment.31 On histopathology, these tumors are well circumscribed and composed of both small dark basaloid cells with peripheral compact nuclei and larger cells with central pale nuclei, which may be arranged in tubules.31,32
The Diagnosis: Glomangiomyoma
The nail unit excision specimen showed collections of cuboidal cells and spindled cells within the corium that were consistent with a diagnosis of a glomangiomyoma, a rare glomus tumor variant (Figure). Glomus tumors are benign neoplasms comprising glomus bodies, which are arteriovenous anastomoses involved in thermoregulation.1 They develop in areas densely populated by glomus bodies, including the fingers, toes, and subungual areas. Glomus tumors most commonly develop in middle-aged women.2 Clinically, they manifest with a characteristic triad of intense pain, point tenderness, and cold sensitivity and may appear as reddish-pink or blue macules under the nail plate and/or longitudinal erythronychia.2-6 The presence of multiple glomus tumors is associated with neurofibromatosis type 1.7
Advanced imaging including ultrasonography and magnetic resonance imaging (MRI) may help confirm the diagnosis but may not be cost effective, as excision with histopathology is needed to relieve symptoms and render a definitive diagnosis. Radiography is highly insensitive in identifying bone erosions associated with glomus tumors.8 With ultrasonography, glomus tumors appear hypoechoic; with Doppler ultrasonography, they appear hypervascular. With MRI, glomus tumors appear as well-defined nodular lesions with hypointense signal intensity on T1-weighted sequence and hyperintense signal intensity on T2-weighted sequence, with strong enhancement using gadolinium-based contrast.9,10 On histopathology, a glomus tumor appears as a nodular tumor with sheets of oval-nucleated cells arranged in multicellular layers surrounding blood vessels and are immunoreactive for α-smooth muscle actin, muscle-specific actin, and type IV collagen.11,12
There are several glomus tumor variants. The most common is a solid glomus tumor, which predominantly is composed of glomus cells, followed by glomangioma, which mainly is composed of blood vessels. Glomangiomyoma, which mostly is composed of smooth muscle cells, is the rarest variant.13
While glomus tumors are common in the subungual areas, it is an uncommon location for glomangiomyomas, which have been reported in the nail unit in only 7 prior case reports identified through searches of PubMed and Google Scholar using the terms glomangiomyoma, glomangiomyoma nail, and subungual glomangiomyoma (Table).13-19 Glomangiomyomas more commonly are described in solid organs, including the stomach, kidney, pancreas, and bladder.16 The mean age of patients with subungual glomangiomyomas, including our patient, was 40.4 years (range, 3-61 years), with the majority being female (75.0% [6/8]). Most patients presented with fingernail involvement (75.0% [6/8]), nail dystrophy (eg, nail plate thinning, longitudinal grooves, splinter hemorrhages, longitudinal erythronychia)(62.5% [5/8]), and intermittent pain and/or point tenderness in the affected nail (75.0% [6/8]).13-19 Notably, only our patient had longitudinal erythronychia as a clinical feature, and only one other case described MRI findings, which included a lobulated mass with intense contrast and distal phalanx destruction.18 One patient was a 3-year-old girl with a family history of generalized multiple glomangiomyomas. Although subungual glomangiomyoma was not confirmed on histopathology, the diagnosis in this patient was presumed based on her family history.13 On histopathology, glomangiomyomas are composed of oval-nucleated cells surrounding blood vessels. These oval-nucleated cells then gradually transition to smooth muscle cells.20
A myxoid cyst is composed of a pseudocyst, which lacks a cyst lining, and is a result of synovial fluid from the distal interphalangeal joint entering the pseudocyst space.2 It typically manifests with a longitudinal groove in the nail plate. A flesh-colored nodule may be appreciated between the cuticle and the distal interphalangeal joint.2 The depth of the longitudinal groove may vary depending on the volume of synovial fluid within the myxoid cyst.21 In a series of 35 cases of subungual myxoid cysts, none manifested with longitudinal erythronychia. Due to their composition, myxoid cysts can be distinguished easily from solid tumors of the nail unit via transillumination.22 Pain is a much less common with myxoid cysts vs glomus tumors, as the filling of the pseudocyst space with synovial fluid typically is gradual, allowing the surrounding tissue to accommodate and adapt over time.21 In equivocal cases, MRI or high-resolution ultrasonography may be used to distinguish myxoid cysts and glomus tumors.8 Histopathology shows accumulation of mucin in the dermis with surrounding fibrous stroma.23
Subungual neuromas are painful benign tumors that develop due to disorganized neural proliferation following disruption to peripheral nerves secondary to trauma or surgery. In 3 case reports, subungual neuromas manifested as painful subungual nodules, with proximal nail plate ridging, or onycholysis.24-26 Since neuromas have only rarely been described in the subungual region, reports of MRI and ultrasonography findings are unknown. Histopathology is needed to distinguish neuromas from glomus tumors. Histopathology shows an acapsular structure consisting of disorganized spindle-cell proliferation and nerve fibers arranged in a tangle of fascicles within fibrotic tissue.25 On immunochemistry, spindle cells typically are positive for cellular antigen protein S100.26
Leiomyomas are benign neoplasms derived from smooth muscle, typically localized to the uterus or gastrointestinal tract, and have been described rarely in the nail unit.27,28 It is hypothesized that subungual leiomyomas originate from the vascular smooth muscle in the subcutaneous layer of the nail unit.28 Like glomus tumors, leiomyomas of the subungual region often manifest with pain and longitudinal erythronychia.27-30 Subungual leiomyomas may be distinguished from glomus tumors via advanced imaging techniques, including ultrasonography and MRI. Cutaneous leiomyomas have been described with mild to moderate internal low flow vascularity on Doppler ultrasonography, while glomus tumors typically reveal high internal vascularity.28 Biopsy with histopathology is needed for definitive diagnosis. On histopathology, leiomyomas demonstrate bland-appearing spindle-shaped cells with elongated nuclei arranged in fascicles.27 They typically are positive for α-smooth muscle actin and caldesmon on immunostaining.
Eccrine spiradenomas are benign adnexal tumors likely of apocrine origin with limited case reports in the literature.31,32 Clinically, eccrine spiradenomas involving the nail unit may manifest with longitudinal nail splitting of the nail or as a papule on the proximal nail fold, with associated tenderness.31,32 In a report of a 50-year-old woman with a histopathologically confirmed eccrine spiradenoma manifesting with longitudinal splitting of the nail and pain in the proximal nail fold, the mass appeared hypoechoic on ultrasonography with increased intramass vascularity on Doppler, while MRI showed an intensely enhancing lesion.31 These imaging features, combined with a classically manifesting feature of pain, make eccrine spiradenomas difficult to distinguish from glomus tumors; therefore, histopathologic examination can provide a definitive diagnosis, and surgical excision is used for treatment.31 On histopathology, these tumors are well circumscribed and composed of both small dark basaloid cells with peripheral compact nuclei and larger cells with central pale nuclei, which may be arranged in tubules.31,32
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132: 1448-1452. doi:10.5858/2008-132-1448-gt
- Hare AQ, Rich P. Nail tumors. Dermatol Clin. 2021;39:281-292. doi:10.1016/j.det.2020.12.007
- Hazani R, Houle JM, Kasdan ML, et al. Glomus tumors of the hand. Eplasty. 2008;8:E48.
- Hwang JK, Lipner SR. Blue nail discoloration: literature review and diagnostic algorithms. Am J Clin Dermatol. 2023;24:419-441. doi:10.1007/s40257-023-00768-6
- Lipner SR, Scher RK. Longitudinal erythronychia of the fingernail. JAMA Dermatol. 2016;152:1271-1272. doi:10.1001/jamadermatol.2016.2747
- Jellinek NJ, Lipner SR. Longitudinal erythronychia: retrospective single-center study evaluating differential diagnosis and the likelihood of malignancy. Dermatol Surg. 2016;42:310-319. doi:10.1097 /DSS.0000000000000594
- Lipner SR, Scher RK. Subungual glomus tumors: underrecognized clinical findings in neurofibromatosis 1. J Am Acad Dermatol. 2021;84:E269. doi:10.1016/j.jaad.2020.08.129
- Dhami A, Vale SM, Richardson ML, et al. Comparing ultrasound with magnetic resonance imaging in the evaluation of subungual glomus tumors and subungual myxoid cysts. Skin Appendage Disord. 2023;9:262-267. doi:10.1159/000530397
- Baek HJ, Lee SJ, Cho KH, et al. Subungual tumors: clinicopathologic correlation with US and MR imaging findings. Radiographics. 2010;30:1621-1636. doi:10.1148/rg.306105514
- Patel T, Meena V, Meena P. Hand and foot glomus tumors: significance of MRI diagnosis followed by histopathological assessment. Cureus. 2022;14:E30038. doi:10.7759/cureus.30038
- Mravic M, LaChaud G, Nguyen A, et al. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23:181-188. doi:10.1177/1066896914567330
- Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12. doi:10.1097/00000478-200101000-00001
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mentzel T, Hügel H, Kutzner H. CD34-positive glomus tumor: clinicopathologic and immunohistochemical analysis of six cases with myxoid stromal changes. J Cutan Pathol. 2002;29:421-425. doi:10.1034 /j.1600-0560.2002.290706.x
- Kang TW, Lee KH, Park CJ. A case of subungual glomangiomyoma with myxoid stromal change. Korean J Dermatol. 2008;46:550-553.
- Wollstein A, Wollstein R. Subungual glomangiomyoma—a case report. Hand Surg. 2012;17:271-273. doi:10.1142/S021881041272032X
- Aqil N, Gallouj S, Moustaide K, et al. Painful tumors in a patient with neurofibromatosis type 1: a case report. J Med Case Rep. 2018;12:319. doi:10.1186/s13256-018-1847-0
- Demirdag HG, Akay BN, Kirmizi A, et al. Subungual glomangiomyoma. J Am Podiatr Med Assoc. 2020;110:Article_13. doi:10.7547/19-051
- Vega SML, Ruiz SJA, Ramírez CS, et al. Subungual glomangiomyoma: a case report. Dermatol Cosmet Med Quir. 2022;20:258-262.
- Chalise S, Jha A, Neupane PR. Glomangiomyoma of uncertain malignant potential in the urinary bladder: a case report. JNMA J Nepal Med Assoc. 2021;59:719-722. doi:10.31729/jnma.5388
- de Berker D, Goettman S, Baran R. Subungual myxoid cysts: clinical manifestations and response to therapy. J Am Acad Dermatol. 2002;46:394-398. doi:10.1067/mjd.2002.119652
- Gupta MK, Lipner SR. Transillumination for improved diagnosis of digital myxoid cysts. Cutis. 2020;105:82.
- Fernandez-Flores A, Saeb-Lima M. Mucin as a diagnostic clue in dermatopathology. J Cutan Pathol. 2016;43:1005-1016. doi:10.1111/cup.12782
- Choi R, Kim SR, Glusac EJ, et al. Subungual neuroma masquerading as green nail syndrome. JAAD Case Rep. 2022;20:17-19. doi:10.1016 /j.jdcr.2021.11.025
- Rashid RM, Rashid RM, Thomas V. Subungal traumatic neuroma. J Am Acad Dermatol. 2010;63:E7-E8. doi:10.1016/j.jaad.2010.01.028
- Whitehouse HJ, Urwin R, Stables G. Traumatic subungual neuroma. Clin Exp Dermatol. 2018;43:65-66. doi:10.1111/ced.13247
- Lipner SR, Ko D, Husain S. Subungual leiyomyoma presenting as erythronychia: case report and review of the literature. J Drugs Dermatol. 2019;18:465-467.
- Taleb E, Saldías C, Gonzalez S, et al. Sonographic characteristics of leiomyomatous tumors of skin and nail: a case series. Dermatol Pract Concept. 2022;12:e2022082. doi:10.5826/dpc.1203a82
- Baran R, Requena L, Drapé JL. Subungual angioleiomyoma masquerading as a glomus tumour. Br J Dermatol. 2000;142:1239-1241. doi:10.1046/ j.1365-2133.2000.03560.x
- Watabe D, Sakurai E, Mori S, et al. Subungual angioleiomyoma. Indian J Dermatol Venereol Leprol. 2017;83:74-75. doi:10.4103/0378-6323 .185045
- Jha AK, Sinha R, Kumar A, et al. Spiradenoma causing longitudinal splitting of the nail. Clin Exp Dermatol. 2016;41:754-756. doi:10.1111 /ced.12886
- Leach BC, Graham BS. Papular lesion of the proximal nail fold. eccrine spiradenoma. Arch Dermatol. 2004;140:1003-1008. doi:10.1001 /archderm.140.8.1003-a
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132: 1448-1452. doi:10.5858/2008-132-1448-gt
- Hare AQ, Rich P. Nail tumors. Dermatol Clin. 2021;39:281-292. doi:10.1016/j.det.2020.12.007
- Hazani R, Houle JM, Kasdan ML, et al. Glomus tumors of the hand. Eplasty. 2008;8:E48.
- Hwang JK, Lipner SR. Blue nail discoloration: literature review and diagnostic algorithms. Am J Clin Dermatol. 2023;24:419-441. doi:10.1007/s40257-023-00768-6
- Lipner SR, Scher RK. Longitudinal erythronychia of the fingernail. JAMA Dermatol. 2016;152:1271-1272. doi:10.1001/jamadermatol.2016.2747
- Jellinek NJ, Lipner SR. Longitudinal erythronychia: retrospective single-center study evaluating differential diagnosis and the likelihood of malignancy. Dermatol Surg. 2016;42:310-319. doi:10.1097 /DSS.0000000000000594
- Lipner SR, Scher RK. Subungual glomus tumors: underrecognized clinical findings in neurofibromatosis 1. J Am Acad Dermatol. 2021;84:E269. doi:10.1016/j.jaad.2020.08.129
- Dhami A, Vale SM, Richardson ML, et al. Comparing ultrasound with magnetic resonance imaging in the evaluation of subungual glomus tumors and subungual myxoid cysts. Skin Appendage Disord. 2023;9:262-267. doi:10.1159/000530397
- Baek HJ, Lee SJ, Cho KH, et al. Subungual tumors: clinicopathologic correlation with US and MR imaging findings. Radiographics. 2010;30:1621-1636. doi:10.1148/rg.306105514
- Patel T, Meena V, Meena P. Hand and foot glomus tumors: significance of MRI diagnosis followed by histopathological assessment. Cureus. 2022;14:E30038. doi:10.7759/cureus.30038
- Mravic M, LaChaud G, Nguyen A, et al. Clinical and histopathological diagnosis of glomus tumor: an institutional experience of 138 cases. Int J Surg Pathol. 2015;23:181-188. doi:10.1177/1066896914567330
- Folpe AL, Fanburg-Smith JC, Miettinen M, et al. Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol. 2001;25:1-12. doi:10.1097/00000478-200101000-00001
- Calduch L, Monteagudo C, Martínez-Ruiz E, et al. Familial generalized multiple glomangiomyoma: report of a new family, with immunohistochemical and ultrastructural studies and review of the literature. Pediatr Dermatol. 2002;19:402-408. doi:10.1046/j.1525-1470.2002.00114.x
- Mentzel T, Hügel H, Kutzner H. CD34-positive glomus tumor: clinicopathologic and immunohistochemical analysis of six cases with myxoid stromal changes. J Cutan Pathol. 2002;29:421-425. doi:10.1034 /j.1600-0560.2002.290706.x
- Kang TW, Lee KH, Park CJ. A case of subungual glomangiomyoma with myxoid stromal change. Korean J Dermatol. 2008;46:550-553.
- Wollstein A, Wollstein R. Subungual glomangiomyoma—a case report. Hand Surg. 2012;17:271-273. doi:10.1142/S021881041272032X
- Aqil N, Gallouj S, Moustaide K, et al. Painful tumors in a patient with neurofibromatosis type 1: a case report. J Med Case Rep. 2018;12:319. doi:10.1186/s13256-018-1847-0
- Demirdag HG, Akay BN, Kirmizi A, et al. Subungual glomangiomyoma. J Am Podiatr Med Assoc. 2020;110:Article_13. doi:10.7547/19-051
- Vega SML, Ruiz SJA, Ramírez CS, et al. Subungual glomangiomyoma: a case report. Dermatol Cosmet Med Quir. 2022;20:258-262.
- Chalise S, Jha A, Neupane PR. Glomangiomyoma of uncertain malignant potential in the urinary bladder: a case report. JNMA J Nepal Med Assoc. 2021;59:719-722. doi:10.31729/jnma.5388
- de Berker D, Goettman S, Baran R. Subungual myxoid cysts: clinical manifestations and response to therapy. J Am Acad Dermatol. 2002;46:394-398. doi:10.1067/mjd.2002.119652
- Gupta MK, Lipner SR. Transillumination for improved diagnosis of digital myxoid cysts. Cutis. 2020;105:82.
- Fernandez-Flores A, Saeb-Lima M. Mucin as a diagnostic clue in dermatopathology. J Cutan Pathol. 2016;43:1005-1016. doi:10.1111/cup.12782
- Choi R, Kim SR, Glusac EJ, et al. Subungual neuroma masquerading as green nail syndrome. JAAD Case Rep. 2022;20:17-19. doi:10.1016 /j.jdcr.2021.11.025
- Rashid RM, Rashid RM, Thomas V. Subungal traumatic neuroma. J Am Acad Dermatol. 2010;63:E7-E8. doi:10.1016/j.jaad.2010.01.028
- Whitehouse HJ, Urwin R, Stables G. Traumatic subungual neuroma. Clin Exp Dermatol. 2018;43:65-66. doi:10.1111/ced.13247
- Lipner SR, Ko D, Husain S. Subungual leiyomyoma presenting as erythronychia: case report and review of the literature. J Drugs Dermatol. 2019;18:465-467.
- Taleb E, Saldías C, Gonzalez S, et al. Sonographic characteristics of leiomyomatous tumors of skin and nail: a case series. Dermatol Pract Concept. 2022;12:e2022082. doi:10.5826/dpc.1203a82
- Baran R, Requena L, Drapé JL. Subungual angioleiomyoma masquerading as a glomus tumour. Br J Dermatol. 2000;142:1239-1241. doi:10.1046/ j.1365-2133.2000.03560.x
- Watabe D, Sakurai E, Mori S, et al. Subungual angioleiomyoma. Indian J Dermatol Venereol Leprol. 2017;83:74-75. doi:10.4103/0378-6323 .185045
- Jha AK, Sinha R, Kumar A, et al. Spiradenoma causing longitudinal splitting of the nail. Clin Exp Dermatol. 2016;41:754-756. doi:10.1111 /ced.12886
- Leach BC, Graham BS. Papular lesion of the proximal nail fold. eccrine spiradenoma. Arch Dermatol. 2004;140:1003-1008. doi:10.1001 /archderm.140.8.1003-a
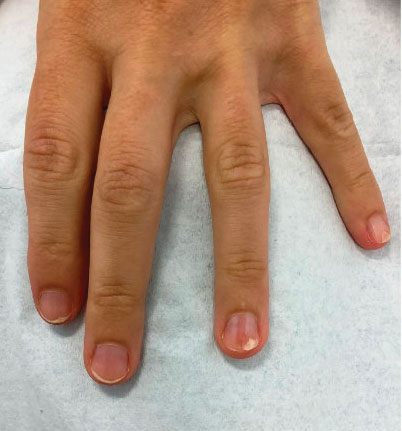
A 38-year-old woman presented to our nail specialty clinic with a red line and associated pain on the left fourth fingernail of 2 and 3 years’ duration, respectively. The patient described the pain as throbbing, with sensitivity to pressure and cold. She noted that the nail grew slowly and would sometimes split at the distal edge. She did not recall any discrete trauma to the digit or nail. The patient was right-handed, making the symptoms less likely to be due to overuse from daily activities. She had received no prior treatment for these symptoms.
The patient’s medical history included iron deficiency as well as acne and eczema. She had no personal or family history of skin cancer. Physical examination of the affected digit and nail revealed a longitudinal red line and distal onycholysis. With contact dermoscopy, the red line blanched. Pressure applied using a #11 scalpel blade elicited pinpoint tenderness (positive Love test), and application of an ice pack caused pain (positive cold test). A radiograph of the left hand was negative for bone erosions, and magnetic resonance imaging showed a 0.3-cm subungual lesion at the level of the fourth distal phalanx. An excision of the nail unit was performed.
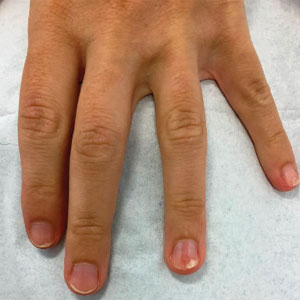
Rare Case of Necrobiotic Xanthogranuloma on the Scalp
Rare Case of Necrobiotic Xanthogranuloma on the Scalp
To the Editor:
Necrobiotic xanthogranuloma (NXG) is classified as a cutaneous non–Langerhans cell histiocytosis, often seen with monoclonal gammopathy of undetermined significance or multiple myeloma.1 Clinically, it appears as a red or yellow plaque with occasional ulceration and telangiectasias, most commonly seen periorbitally and on the trunk. On pathology, NXG appears as necrobiosis, giant cells, and various inflammatory cells extending into the subcutaneous tissue.2 In this article, we describe a rare presentation of NXG in location and skin type.
A 52-year-old woman with a history of systemic lupus erythematosus (SLE) presented with alopecia and a tender lesion on the scalp of 5 years’ duration (Figure 1). The patient had no history of a similar lesion, and no other lesions were present. A biopsy performed at an outside clinic a few weeks to months prior to the initial presentation to our clinic showed NXG (Figure 2). Evaluation at our clinic revealed a 4x4-cm orange-brown annular plaque on the left parietal scalp. Serum and urine protein electrophoresis studies were negative. The patient reported she was up to date with recommended screenings such as mammography and colonoscopy.
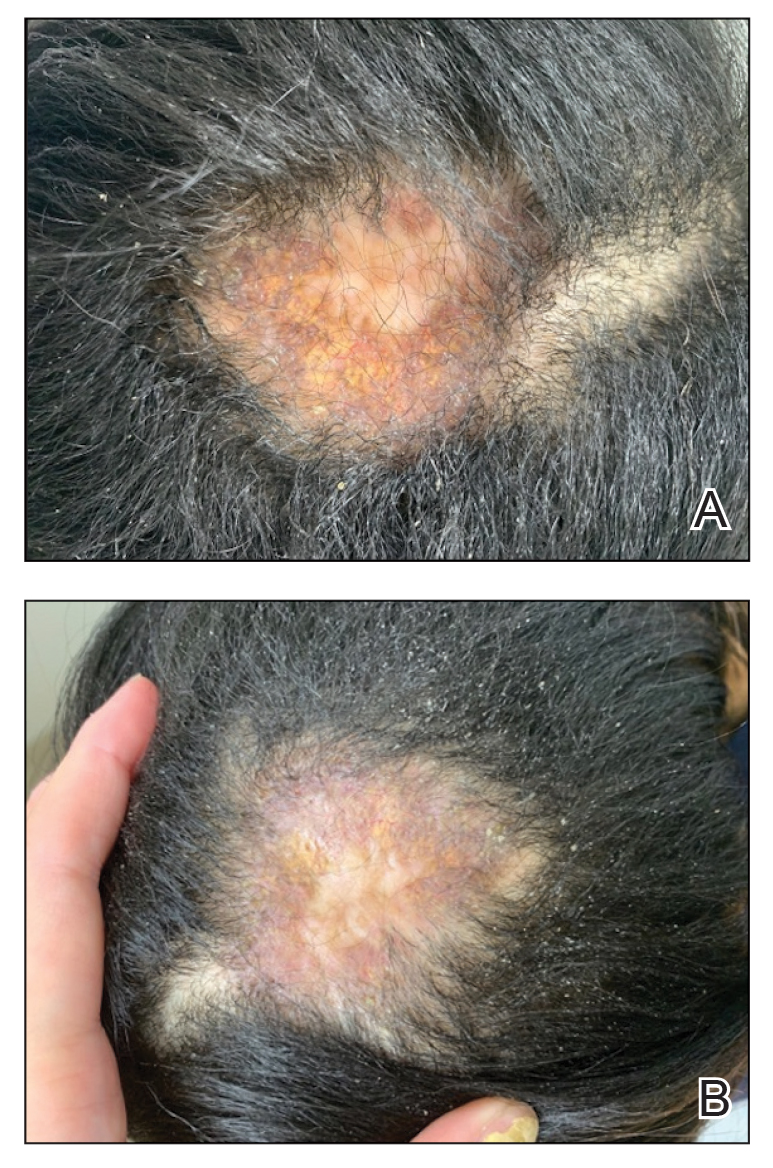
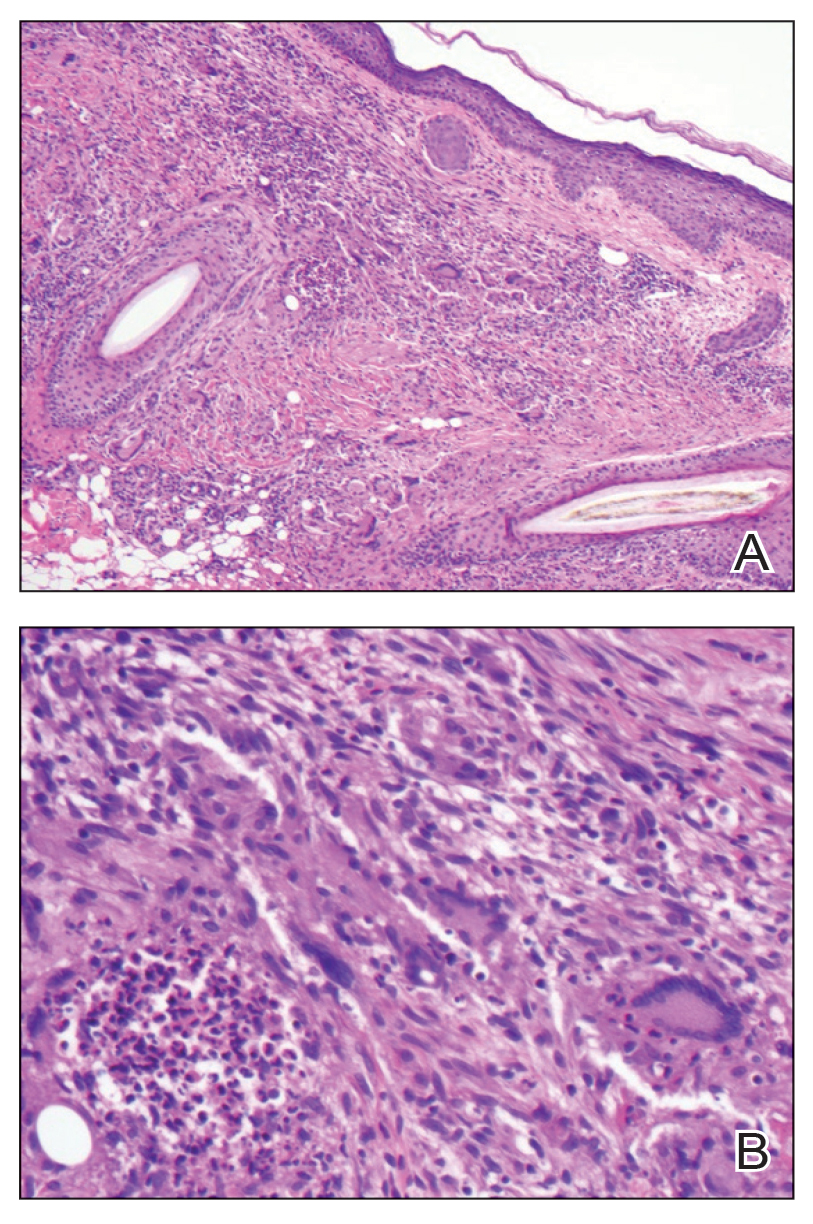
We started the patient on topical triamcinolone and topical ruxolitinib and administered intralesional triamcinolone. She was already taking hydroxychloroquine and leflunomide for SLE. Three weeks later, she returned with improved symptoms and appearance (Figure 1). She remained on intralesional triamcinolone and ruxolitinib and continues to experience improvement.
Necrobiotic xanthogranuloma is rare and typically is associated with monoclonal gammopathy.2 In one study, 83 of 100 of patients with NXG presented with or were found to have a monoclonal gammopathy.2 In another study, paraproteinemia was detected in 82.1% of patients.3 The majority of case reports and systematic reviews detail periorbital or thoracic lesions.4 The location on the scalp and lack of association with paraproteinemia make this a rare presentation of NXG. Studies may be warranted to explore any association of SLE with NXG if more cases present.
In a multicenter cross-sectional study and systematic review of 235 patients with NXG, 87% were White, 12% were Asian, and only 1% were Black or African American.3 The limited representation of skin of color raises concern for the possibility of missed diagnoses and delays in care.
Treatment of NXG often is multimodal with use of intravenous immunoglobulin, oral steroids, chlorambucil, melphalan, and other alkylating agents, and response is variable.3-6 Recent studies show treatment effectiveness with Janus kinase inhibitors in granulomatous dermatitides.7-9 As our patient was not responding to prior treatments, we decided to try ruxolitinib, and she has continued to improve with it.10,11 Interestingly, the patient experienced continued improvement with intralesional triamcinolone, which is not often reported in the literature.2-6 Overall, NXG is an extremely rare condition that requires special care in workup to rule out paraproteinemia and a thoughtful approach to treatment modalities.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Nelson CA, Zhong CS, Hashemi DA, et al. A multicenter cross-sectional study and systematic review of necrobiotic xanthogranuloma with proposed diagnostic criteria. JAMA Dermatol. 2020;156:270-279.
- Huynh KN, Nguyen BD. Histiocytosis and neoplasms of macrophagedendritic cell lineages: multimodality imaging with emphasis on PET/CT. Radiographics. 2021;41:576-594. doi: 10.1148/rg.2021200096
- Hilal T, DiCaudo DJ, Connolly SM, et al. Necrobiotic xanthogranuloma: a 30-year single-center experience. Ann Hematol. 2018;97:1471-1479.
- Oumeish OY, Oumeish I, Tarawneh M, et al. Necrobiotic xanthogranuloma associated with paraproteinemia and non- Hodgkin’s lymphoma developing into chronic lymphocytic leukemia: the first case reported in the literature and review of the literature. Int J Dermatol. 2006;45:306-310.
- Damsky W, Thakral D, McGeary MK, et al. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol. 2020;82:612-621. doi:10.1016 /j.jaad.2019.05.098
- Wang A, Rahman NT, McGeary MK, et al. Treatment of granuloma annulare and suppression of proinflammatory cytokine activity with tofacitinib. J Allergy Clin Immunol. 2021;147:1795-1809. doi:10.1016 /j.jaci.2020.10.012
- Stratman S, Amara S, Tan KJ, et al. Systemic Janus kinase inhibitors in the management of granuloma annulare. Arch Dermatol Res. 2025;317:743. doi:10.1007/s00403-025-04248-1
- McPhie ML, Swales WC, Gooderham MJ. Improvement of granulomatous skin conditions with tofacitinib in three patients: a case report. SAGE Open Med Case Rep. 2021;9:2050313X211039477. doi: 10.1177/2050313X211039477
- Sood S, Heung M, Georgakopoulos JR, et al. Use of Janus kinase inhibitors for granulomatous dermatoses: a systematic review. J Am Acad Dermatol. 2023;89:357-359. doi: 10.1016/j.jaad.2023.03.024
To the Editor:
Necrobiotic xanthogranuloma (NXG) is classified as a cutaneous non–Langerhans cell histiocytosis, often seen with monoclonal gammopathy of undetermined significance or multiple myeloma.1 Clinically, it appears as a red or yellow plaque with occasional ulceration and telangiectasias, most commonly seen periorbitally and on the trunk. On pathology, NXG appears as necrobiosis, giant cells, and various inflammatory cells extending into the subcutaneous tissue.2 In this article, we describe a rare presentation of NXG in location and skin type.
A 52-year-old woman with a history of systemic lupus erythematosus (SLE) presented with alopecia and a tender lesion on the scalp of 5 years’ duration (Figure 1). The patient had no history of a similar lesion, and no other lesions were present. A biopsy performed at an outside clinic a few weeks to months prior to the initial presentation to our clinic showed NXG (Figure 2). Evaluation at our clinic revealed a 4x4-cm orange-brown annular plaque on the left parietal scalp. Serum and urine protein electrophoresis studies were negative. The patient reported she was up to date with recommended screenings such as mammography and colonoscopy.
We started the patient on topical triamcinolone and topical ruxolitinib and administered intralesional triamcinolone. She was already taking hydroxychloroquine and leflunomide for SLE. Three weeks later, she returned with improved symptoms and appearance (Figure 1). She remained on intralesional triamcinolone and ruxolitinib and continues to experience improvement.
Necrobiotic xanthogranuloma is rare and typically is associated with monoclonal gammopathy.2 In one study, 83 of 100 of patients with NXG presented with or were found to have a monoclonal gammopathy.2 In another study, paraproteinemia was detected in 82.1% of patients.3 The majority of case reports and systematic reviews detail periorbital or thoracic lesions.4 The location on the scalp and lack of association with paraproteinemia make this a rare presentation of NXG. Studies may be warranted to explore any association of SLE with NXG if more cases present.
In a multicenter cross-sectional study and systematic review of 235 patients with NXG, 87% were White, 12% were Asian, and only 1% were Black or African American.3 The limited representation of skin of color raises concern for the possibility of missed diagnoses and delays in care.
Treatment of NXG often is multimodal with use of intravenous immunoglobulin, oral steroids, chlorambucil, melphalan, and other alkylating agents, and response is variable.3-6 Recent studies show treatment effectiveness with Janus kinase inhibitors in granulomatous dermatitides.7-9 As our patient was not responding to prior treatments, we decided to try ruxolitinib, and she has continued to improve with it.10,11 Interestingly, the patient experienced continued improvement with intralesional triamcinolone, which is not often reported in the literature.2-6 Overall, NXG is an extremely rare condition that requires special care in workup to rule out paraproteinemia and a thoughtful approach to treatment modalities.
To the Editor:
Necrobiotic xanthogranuloma (NXG) is classified as a cutaneous non–Langerhans cell histiocytosis, often seen with monoclonal gammopathy of undetermined significance or multiple myeloma.1 Clinically, it appears as a red or yellow plaque with occasional ulceration and telangiectasias, most commonly seen periorbitally and on the trunk. On pathology, NXG appears as necrobiosis, giant cells, and various inflammatory cells extending into the subcutaneous tissue.2 In this article, we describe a rare presentation of NXG in location and skin type.
A 52-year-old woman with a history of systemic lupus erythematosus (SLE) presented with alopecia and a tender lesion on the scalp of 5 years’ duration (Figure 1). The patient had no history of a similar lesion, and no other lesions were present. A biopsy performed at an outside clinic a few weeks to months prior to the initial presentation to our clinic showed NXG (Figure 2). Evaluation at our clinic revealed a 4x4-cm orange-brown annular plaque on the left parietal scalp. Serum and urine protein electrophoresis studies were negative. The patient reported she was up to date with recommended screenings such as mammography and colonoscopy.
We started the patient on topical triamcinolone and topical ruxolitinib and administered intralesional triamcinolone. She was already taking hydroxychloroquine and leflunomide for SLE. Three weeks later, she returned with improved symptoms and appearance (Figure 1). She remained on intralesional triamcinolone and ruxolitinib and continues to experience improvement.
Necrobiotic xanthogranuloma is rare and typically is associated with monoclonal gammopathy.2 In one study, 83 of 100 of patients with NXG presented with or were found to have a monoclonal gammopathy.2 In another study, paraproteinemia was detected in 82.1% of patients.3 The majority of case reports and systematic reviews detail periorbital or thoracic lesions.4 The location on the scalp and lack of association with paraproteinemia make this a rare presentation of NXG. Studies may be warranted to explore any association of SLE with NXG if more cases present.
In a multicenter cross-sectional study and systematic review of 235 patients with NXG, 87% were White, 12% were Asian, and only 1% were Black or African American.3 The limited representation of skin of color raises concern for the possibility of missed diagnoses and delays in care.
Treatment of NXG often is multimodal with use of intravenous immunoglobulin, oral steroids, chlorambucil, melphalan, and other alkylating agents, and response is variable.3-6 Recent studies show treatment effectiveness with Janus kinase inhibitors in granulomatous dermatitides.7-9 As our patient was not responding to prior treatments, we decided to try ruxolitinib, and she has continued to improve with it.10,11 Interestingly, the patient experienced continued improvement with intralesional triamcinolone, which is not often reported in the literature.2-6 Overall, NXG is an extremely rare condition that requires special care in workup to rule out paraproteinemia and a thoughtful approach to treatment modalities.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Nelson CA, Zhong CS, Hashemi DA, et al. A multicenter cross-sectional study and systematic review of necrobiotic xanthogranuloma with proposed diagnostic criteria. JAMA Dermatol. 2020;156:270-279.
- Huynh KN, Nguyen BD. Histiocytosis and neoplasms of macrophagedendritic cell lineages: multimodality imaging with emphasis on PET/CT. Radiographics. 2021;41:576-594. doi: 10.1148/rg.2021200096
- Hilal T, DiCaudo DJ, Connolly SM, et al. Necrobiotic xanthogranuloma: a 30-year single-center experience. Ann Hematol. 2018;97:1471-1479.
- Oumeish OY, Oumeish I, Tarawneh M, et al. Necrobiotic xanthogranuloma associated with paraproteinemia and non- Hodgkin’s lymphoma developing into chronic lymphocytic leukemia: the first case reported in the literature and review of the literature. Int J Dermatol. 2006;45:306-310.
- Damsky W, Thakral D, McGeary MK, et al. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol. 2020;82:612-621. doi:10.1016 /j.jaad.2019.05.098
- Wang A, Rahman NT, McGeary MK, et al. Treatment of granuloma annulare and suppression of proinflammatory cytokine activity with tofacitinib. J Allergy Clin Immunol. 2021;147:1795-1809. doi:10.1016 /j.jaci.2020.10.012
- Stratman S, Amara S, Tan KJ, et al. Systemic Janus kinase inhibitors in the management of granuloma annulare. Arch Dermatol Res. 2025;317:743. doi:10.1007/s00403-025-04248-1
- McPhie ML, Swales WC, Gooderham MJ. Improvement of granulomatous skin conditions with tofacitinib in three patients: a case report. SAGE Open Med Case Rep. 2021;9:2050313X211039477. doi: 10.1177/2050313X211039477
- Sood S, Heung M, Georgakopoulos JR, et al. Use of Janus kinase inhibitors for granulomatous dermatoses: a systematic review. J Am Acad Dermatol. 2023;89:357-359. doi: 10.1016/j.jaad.2023.03.024
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127:2672-2681.
- Spicknall KE, Mehregan DA. Necrobiotic xanthogranuloma. Int J Dermatol. 2009;48:1-10.
- Nelson CA, Zhong CS, Hashemi DA, et al. A multicenter cross-sectional study and systematic review of necrobiotic xanthogranuloma with proposed diagnostic criteria. JAMA Dermatol. 2020;156:270-279.
- Huynh KN, Nguyen BD. Histiocytosis and neoplasms of macrophagedendritic cell lineages: multimodality imaging with emphasis on PET/CT. Radiographics. 2021;41:576-594. doi: 10.1148/rg.2021200096
- Hilal T, DiCaudo DJ, Connolly SM, et al. Necrobiotic xanthogranuloma: a 30-year single-center experience. Ann Hematol. 2018;97:1471-1479.
- Oumeish OY, Oumeish I, Tarawneh M, et al. Necrobiotic xanthogranuloma associated with paraproteinemia and non- Hodgkin’s lymphoma developing into chronic lymphocytic leukemia: the first case reported in the literature and review of the literature. Int J Dermatol. 2006;45:306-310.
- Damsky W, Thakral D, McGeary MK, et al. Janus kinase inhibition induces disease remission in cutaneous sarcoidosis and granuloma annulare. J Am Acad Dermatol. 2020;82:612-621. doi:10.1016 /j.jaad.2019.05.098
- Wang A, Rahman NT, McGeary MK, et al. Treatment of granuloma annulare and suppression of proinflammatory cytokine activity with tofacitinib. J Allergy Clin Immunol. 2021;147:1795-1809. doi:10.1016 /j.jaci.2020.10.012
- Stratman S, Amara S, Tan KJ, et al. Systemic Janus kinase inhibitors in the management of granuloma annulare. Arch Dermatol Res. 2025;317:743. doi:10.1007/s00403-025-04248-1
- McPhie ML, Swales WC, Gooderham MJ. Improvement of granulomatous skin conditions with tofacitinib in three patients: a case report. SAGE Open Med Case Rep. 2021;9:2050313X211039477. doi: 10.1177/2050313X211039477
- Sood S, Heung M, Georgakopoulos JR, et al. Use of Janus kinase inhibitors for granulomatous dermatoses: a systematic review. J Am Acad Dermatol. 2023;89:357-359. doi: 10.1016/j.jaad.2023.03.024
Rare Case of Necrobiotic Xanthogranuloma on the Scalp
Rare Case of Necrobiotic Xanthogranuloma on the Scalp
PRACTICE POINTS
- In skin of color, necrobiotic xanthogranuloma can appear orange or brown compared to its yellow appearance in lighter skin types.
- When necrobiotic xanthogranuloma is suspected, a thorough malignancy workup should be conducted.

Direct Care Dermatology: Weighing the Pros and Cons for the Early-Career Physician
Direct Care Dermatology: Weighing the Pros and Cons for the Early-Career Physician
As the health care landscape continues to shift, direct care (also known as direct pay) models have emerged as attractive alternatives to traditional insurance-based practice. For dermatology residents poised to enter the workforce, the direct care model offers potential advantages in autonomy, patient relationships, and work-life balance, but not without considerable risks and operational challenges. This article explores the key benefits and drawbacks of starting a direct care dermatology practice, providing a framework to help early-career dermatologists determine whether this path aligns with their personal and professional goals.
The transition from dermatology residency to clinical practice allows for a variety of paths, from large academic institutions to private practice to corporate entities (private equity–owned groups). In recent years, the direct care model has gained traction, particularly among physicians seeking greater autonomy and a more sustainable pace of practice.
Direct care dermatology practices operate outside the constraints of third-party payers, offering patients transparent pricing and direct access to care in exchange for fees paid out of pocket. By eliminating insurance companies as the middleman, it allows for less overhead, longer visits with patients, and increased access to care; however, though this model may seem appealing, direct care practices are not without their own set of challenges, especially amid rising concerns over physician burnout and administrative burden.
This article explores the key benefits and drawbacks of starting a direct care dermatology practice, providing a framework to help early-career dermatologists determine whether this path aligns with their personal and professional goals.
Despite its appeal, starting a direct care practice is not without substantial risks and hurdles—particularly for residents just out of training. These challenges include financial risks and startup costs, market uncertainty, lack of mentorship or support, and limitations in treating complex dermatologic conditions.
Before committing to practicing via a direct care model, dermatology residents should reflect on the following:
- Risk tolerance: Are you comfortable navigating the business and financial risk?
- Location: Does your target community have patients willing and able to pay out of pocket?
- Scope of interest: Will a direct care practice align with your clinical passions?
- Support systems: Do you have access to mentors, legal and financial advisors, and operational support?
- Long-term goals: Are you building a lifestyle practice, a scalable business, or a stepping stone to a future opportunity?
Ultimately, the decision to pursue a direct care model requires careful reflection on personal values, financial preparedness, and the unique needs of the community one intends to serve.
The direct care dermatology model offers an appealing alternative to traditional practice, especially for those prioritizing autonomy, patient connection, and work-life balance; however, it demands an entrepreneurial spirit as well as careful planning and an acceptance of financial uncertainty—factors that may pose challenges for new graduates. For dermatology residents, the decision to pursue direct care should be grounded in personal values, practical considerations, and a clear understanding of both the opportunities and limitations of this evolving practice model.
- Sinsky CA, Colligan L, Li L, et al. Allocation of physician time in ambulatory practice: a time and motion study in 4 specialties. Ann Intern Med.
- Dorrell DN, Feldman S, Wei-ting Huang W. The most common causes of burnout among US academic dermatologists based on a survey study. J Am Acad of Dermatol. 2019;81:269-270.
- Carlasare LE. Defining the place of direct primary care in a value-based care system. WMJ. 2018;117:106-110.
As the health care landscape continues to shift, direct care (also known as direct pay) models have emerged as attractive alternatives to traditional insurance-based practice. For dermatology residents poised to enter the workforce, the direct care model offers potential advantages in autonomy, patient relationships, and work-life balance, but not without considerable risks and operational challenges. This article explores the key benefits and drawbacks of starting a direct care dermatology practice, providing a framework to help early-career dermatologists determine whether this path aligns with their personal and professional goals.
The transition from dermatology residency to clinical practice allows for a variety of paths, from large academic institutions to private practice to corporate entities (private equity–owned groups). In recent years, the direct care model has gained traction, particularly among physicians seeking greater autonomy and a more sustainable pace of practice.
Direct care dermatology practices operate outside the constraints of third-party payers, offering patients transparent pricing and direct access to care in exchange for fees paid out of pocket. By eliminating insurance companies as the middleman, it allows for less overhead, longer visits with patients, and increased access to care; however, though this model may seem appealing, direct care practices are not without their own set of challenges, especially amid rising concerns over physician burnout and administrative burden.
This article explores the key benefits and drawbacks of starting a direct care dermatology practice, providing a framework to help early-career dermatologists determine whether this path aligns with their personal and professional goals.
Despite its appeal, starting a direct care practice is not without substantial risks and hurdles—particularly for residents just out of training. These challenges include financial risks and startup costs, market uncertainty, lack of mentorship or support, and limitations in treating complex dermatologic conditions.
Before committing to practicing via a direct care model, dermatology residents should reflect on the following:
- Risk tolerance: Are you comfortable navigating the business and financial risk?
- Location: Does your target community have patients willing and able to pay out of pocket?
- Scope of interest: Will a direct care practice align with your clinical passions?
- Support systems: Do you have access to mentors, legal and financial advisors, and operational support?
- Long-term goals: Are you building a lifestyle practice, a scalable business, or a stepping stone to a future opportunity?
Ultimately, the decision to pursue a direct care model requires careful reflection on personal values, financial preparedness, and the unique needs of the community one intends to serve.
The direct care dermatology model offers an appealing alternative to traditional practice, especially for those prioritizing autonomy, patient connection, and work-life balance; however, it demands an entrepreneurial spirit as well as careful planning and an acceptance of financial uncertainty—factors that may pose challenges for new graduates. For dermatology residents, the decision to pursue direct care should be grounded in personal values, practical considerations, and a clear understanding of both the opportunities and limitations of this evolving practice model.
As the health care landscape continues to shift, direct care (also known as direct pay) models have emerged as attractive alternatives to traditional insurance-based practice. For dermatology residents poised to enter the workforce, the direct care model offers potential advantages in autonomy, patient relationships, and work-life balance, but not without considerable risks and operational challenges. This article explores the key benefits and drawbacks of starting a direct care dermatology practice, providing a framework to help early-career dermatologists determine whether this path aligns with their personal and professional goals.
The transition from dermatology residency to clinical practice allows for a variety of paths, from large academic institutions to private practice to corporate entities (private equity–owned groups). In recent years, the direct care model has gained traction, particularly among physicians seeking greater autonomy and a more sustainable pace of practice.
Direct care dermatology practices operate outside the constraints of third-party payers, offering patients transparent pricing and direct access to care in exchange for fees paid out of pocket. By eliminating insurance companies as the middleman, it allows for less overhead, longer visits with patients, and increased access to care; however, though this model may seem appealing, direct care practices are not without their own set of challenges, especially amid rising concerns over physician burnout and administrative burden.
This article explores the key benefits and drawbacks of starting a direct care dermatology practice, providing a framework to help early-career dermatologists determine whether this path aligns with their personal and professional goals.
Despite its appeal, starting a direct care practice is not without substantial risks and hurdles—particularly for residents just out of training. These challenges include financial risks and startup costs, market uncertainty, lack of mentorship or support, and limitations in treating complex dermatologic conditions.
Before committing to practicing via a direct care model, dermatology residents should reflect on the following:
- Risk tolerance: Are you comfortable navigating the business and financial risk?
- Location: Does your target community have patients willing and able to pay out of pocket?
- Scope of interest: Will a direct care practice align with your clinical passions?
- Support systems: Do you have access to mentors, legal and financial advisors, and operational support?
- Long-term goals: Are you building a lifestyle practice, a scalable business, or a stepping stone to a future opportunity?
Ultimately, the decision to pursue a direct care model requires careful reflection on personal values, financial preparedness, and the unique needs of the community one intends to serve.
The direct care dermatology model offers an appealing alternative to traditional practice, especially for those prioritizing autonomy, patient connection, and work-life balance; however, it demands an entrepreneurial spirit as well as careful planning and an acceptance of financial uncertainty—factors that may pose challenges for new graduates. For dermatology residents, the decision to pursue direct care should be grounded in personal values, practical considerations, and a clear understanding of both the opportunities and limitations of this evolving practice model.
- Sinsky CA, Colligan L, Li L, et al. Allocation of physician time in ambulatory practice: a time and motion study in 4 specialties. Ann Intern Med.
- Dorrell DN, Feldman S, Wei-ting Huang W. The most common causes of burnout among US academic dermatologists based on a survey study. J Am Acad of Dermatol. 2019;81:269-270.
- Carlasare LE. Defining the place of direct primary care in a value-based care system. WMJ. 2018;117:106-110.
- Sinsky CA, Colligan L, Li L, et al. Allocation of physician time in ambulatory practice: a time and motion study in 4 specialties. Ann Intern Med.
- Dorrell DN, Feldman S, Wei-ting Huang W. The most common causes of burnout among US academic dermatologists based on a survey study. J Am Acad of Dermatol. 2019;81:269-270.
- Carlasare LE. Defining the place of direct primary care in a value-based care system. WMJ. 2018;117:106-110.
Direct Care Dermatology: Weighing the Pros and Cons for the Early-Career Physician
Direct Care Dermatology: Weighing the Pros and Cons for the Early-Career Physician
PRACTICE POINTS
- Direct care practices may be the new horizon of health care.
- Starting a direct care practice offers autonomy but demands entrepreneurial readiness.
- New dermatologists can enjoy control over scheduling, pricing, and patient care, but success requires business acumen, financial planning, and comfort with risk.
Steatocystomas: Update on Clinical Manifestations, Diagnosis, and Management
Steatocystomas: Update on Clinical Manifestations, Diagnosis, and Management
Steatocystomas are small sebum-filled cysts that typically manifest in the dermis and originate from sebaceous follicles. Although commonly asymptomatic, these lesions can manifest with pruritus or become infected, predisposing patients to further complications.1 Steatocystomas can manifest as single (steatocystoma simplex [SS]) or numerous (steatocystoma multiplex [SM]) lesions; the lesions also can spontaneously rupture with characteristics that resemble hidradenitis suppurativa (HS)(steatocystoma multiplex suppurativa [SMS]).1,2
Steatocystomas are relatively rare, and there is limited consensus in the published literature on the etiology and management of this condition. In this article, we present a comprehensive review of steatocystomas in the current literature. We highlight important features to consider when making the diagnosis and also offer recommendations for best-practice treatment.
Historical Background
Although not explicitly identified by name, the first documentation of steatocystomas is a case report published in 1873. In this account, the author described a patient who presented with approximately 250 flesh-colored dermal cysts across the body that varied in size.3 In 1899, the term steatocystoma multiple—derived from Greek roots meaning “fatty bag”—was first used.4
In 1982, almost a century later, Brownstein5 reported some of the earliest cases of SS. This solitary subtype is identical to SM on a microscopic level; however, unlike SM, this variant occurs as a single lesion that typically forms in adulthood and in the absence of family history. Other benign adnexal tumors (eg, pilomatricomas, pilar cysts, and sebaceous hyperplasias) also can manifest as either solitary or multiple lesions.
In 1976, McDonald and Reed6 reported the first known cases of patients with both SM and HS. At the time, the co-occurrence of these conditions was viewed as coincidental, but there were postulations of a shared inflammatory process and hereditary link6; it was not until 1982 that the term steatocystoma multiplex suppurativum was coined to describe this variant.7 Although rare, there have been multiple documented instances of SMS since. It has been suggested that the convergence of these conditions may indicate a shared follicular proliferation defect.8 Ongoing investigation is warranted to explain the underlying pathogenesis of this unique variant.
Epidemiology
The available epidemiologic data primarily relate to SM, the most common steatocystoma variant. Nevertheless, SM is a relatively rare condition, and the exact incidence and prevalence remain unknown.8,9 Steatocystomas typically manifest in the first and second decades of life and have been observed in patients of both sexes, with studies demonstrating no notable sex bias.4,9
Etiology and Pathophysiology
Steatocystomas can occur sporadically or may be inherited as an autosomal-dominant condition.4 Typically, SS tends to manifest as an isolated occurrence without any inherent genetic predisposition.5 Alternatively, SM may develop sporadically or be associated with a mutation in the keratin 17 gene (KRT17).4 Steatocystoma multiplex also has been associated with at least 4 different missense mutations, including N92H, R94H, and R94C, located on the long (q) arm of chromosome 17.4,10-12
The keratin 17 gene is responsible for encoding the keratin 17 protein, a type I intermediate filament predominantly synthesized in the basal cells of epithelial tissue. This fibrous structural protein can regulate many processes, including inflammation and cell proliferation, and is found in regions such as the sebaceous glands, hair follicles, and eccrine sweat glands. Overexpression of KRT17 has been suggested in other cutaneous conditions, most notably psoriasis.12 Despite KRT17’s many roles, it remains unclear why SM typically manifests with a myriad of sebum-containing cysts as the primary symptom.12 Continued investigation into the genetic underpinnings of SM and the keratin 17 protein is necessary to further elucidate a more comprehensive understanding of this condition.
Hormonal influences have been suggested as a potential trigger for steatocystoma growth.4,13 This condition is associated with dysfunction of the sebaceous glands, and, correspondingly, the incidence of disease is highest in pubertal patients, in whom androgen levels and sebum production are elevated.4,13,14 Two cases of transgender men taking testosterone therapy presenting with steatocystomas provide additional clinical support for this association.15
Additionally, the use of immunomodulatory agents, such as ustekinumab (anti–interleukin 12/interleukin 23), has been shown to trigger SM. It is predicted that the reduced expression of certain interferons and interleukins may lead to downstream consequences in the keratin 17 pathway and lead to SM lesion formation in genetically susceptible individuals.16 Targeting these potential causes in the future may prove efficacious in the secondary prevention of familial SM manifestation or exacerbations.
Mutations in the KRT17 gene also have been implicated in pachyonychia congenita type 2 (PC-2).4 Marked by extensive systemic hyperkeratosis, PC-2 has been observed to coincide with SM in certain patients.4,5 Interestingly, the location of the KRT17 mutations are identical in both PC-2 and SM.4 Although most individuals with hereditary SM do not exhibit the characteristic features of PC-2, mild nail and dental abnormalities have been observed in some SM cases.4,10 This relationship suggests that SM may be a less severe variant of PC-2 or part of a complex polygenetic spectrum of disease.10 Further research is imperative to determine the exact nature and extent of the relationship between these conditions.
Clinical Manifestations
Steatocystomas are flesh-colored subcutaneous cysts that range in size from less than 3 mm to larger than 3 cm in diameter (Figure). They form within a single pilosebaceous unit and typically display firm attachment due to their origination in the dermis.2,7,17 Steatocystomas generally contain lipid material, and less frequently, keratin and hair shafts, distinguishing them as the only “true” sebaceous cysts.18 Their color can range from flesh-toned to yellow, with reports of occasional dark-blue shades and calcifications.19,20 Steatocystomas can persist indefinitely, and they usually are asymptomatic.
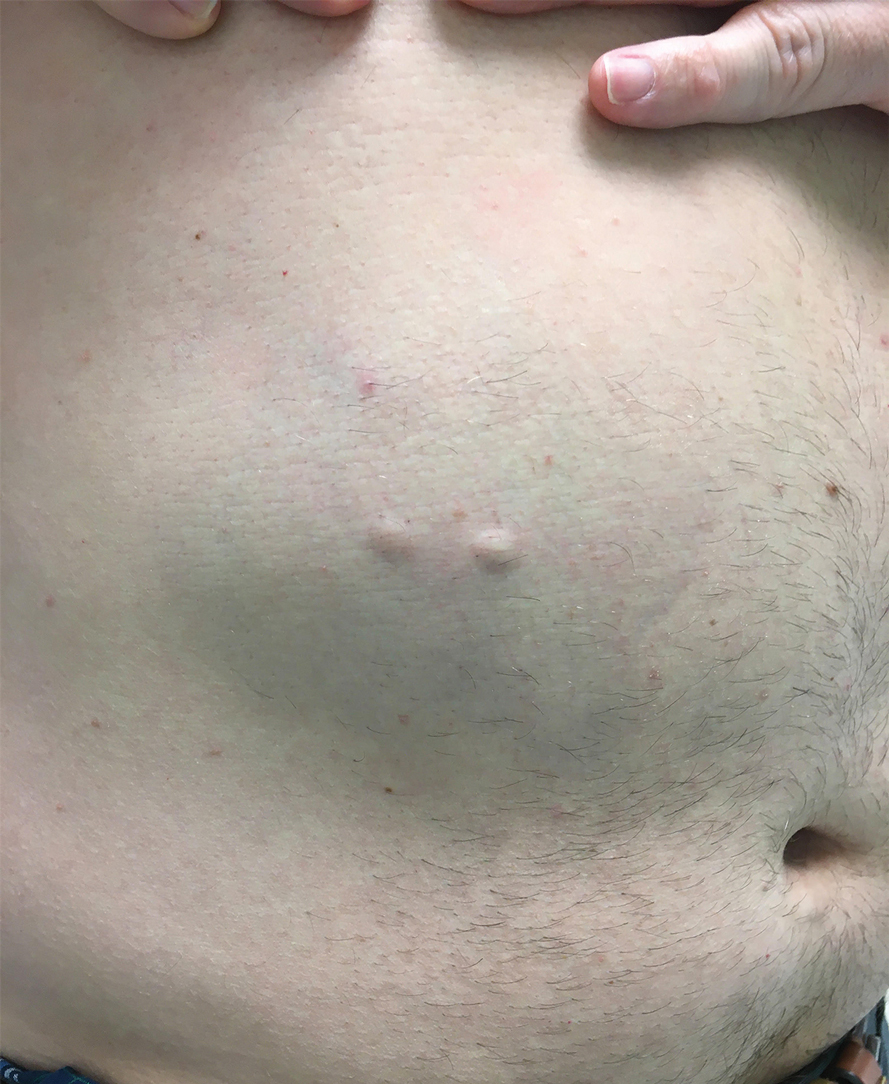
Diagnosis of steatocystoma is confirmed by biopsy.4 Steatocystomas are characterized by a dermal cyst lined by stratified squamous cell epithelium (eFigures 1 and 2).21 Classically they feature flattened sebaceous lobules, multinucleated giant cells, and abortive hair follicles. The lining of these cysts is marked by lymphocytic infiltrate and a dense, wrinkled, eosinophilic keratin cuticle that replaces the granular layer.22 The cyst maintains an epidermal connection through a follicular infundibulum characterized by clumps of keratinocytes, sebocytes, corneocytes, and/or hair follicles.7 Aspirated contents reveal crystalline structures and anucleate squamous cells upon microscopic analysis. That being said, variable histologic findings of steatocystomas have been described.23
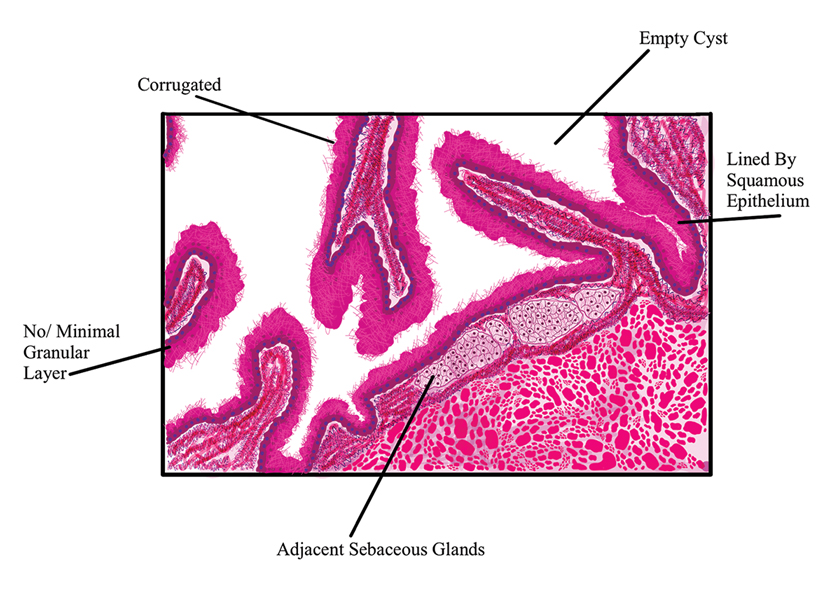
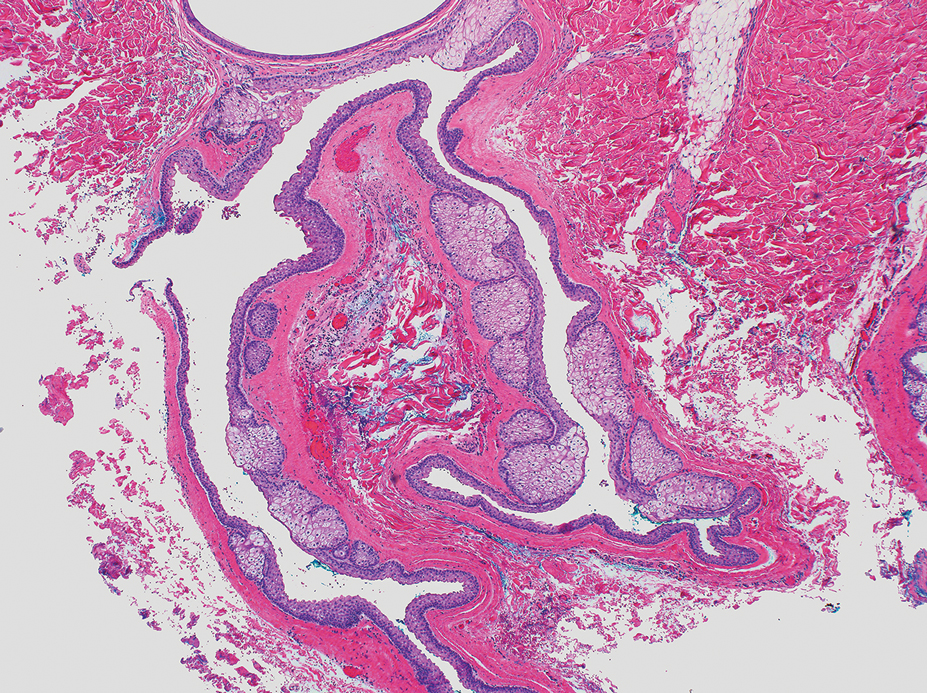
Steatocystoma simplex, as the name implies, classifies a single isolated steatocystoma. This subtype exhibits similar histopathologic and clinical features to the other subtypes of steatocystomas. Notably, SS is not associated with a genetic mutation and is not an inherited condition within families.5 Steatocystoma multiplex manifests with many steatocystomas, often distributed widely across the body.3,4 The chest, axillae, and groin are the most common locations; however, these cysts can manifest on the face, back, abdomen, and extremities.4,18-22 Rare occurrences of SM limited to the face, scalp, and distal extremities have been documented.18,21,24,25 Due to the possibility of an autosomal-dominant inheritance, it is advisable to take a comprehensive family history in patients for whom SM is in the differential.17
Steatocystoma multiplex—especially familial variants—has been shown to develop in conjunction with other dermatologic conditions, including eruptive vellus hair (EVH) cysts, persistent infantile milia, and epidermoid/dermoid cysts.26 While some investigators regard these as separate entities due to their varied genetic etiology, it has been suggested that these conditions may be related and that the diagnosis is determined by the location of cyst origin along the sebaceous ducts.26,27 Other dermatologic conditions and lesions that frequently manifest comorbidly with SM include hidrocystomas, syringomas, pilonidal cysts, lichen planus, nodulocystic acne, trichotillomania, trichoblastomas, trichoepithelioma, HS, keratoacanthomas, acrokeratosis verruciformis of Hopf, and embryonal hair formation. Steatocystoma multiplex, manifesting comorbidly with dental and orofacial malformations (eg, partial noneruption of secondary teeth, natal and defective teeth, and bilateral preauricular sinuses) has been classified as SM natal teeth syndrome.6
Steatocystoma multiplex suppurativa is a rare and serious variant of SM characterized by inflammation, cyst rupture, sinus tract formation, and scarring.24 Patients with SMS typically have multiple intact SM cysts, which can aid in differentiation from HS.2,24 Steatocystoma multiplex suppurativa is associated with more complications than SS and SM, including cyst perforation, development of purulent and/or foul-smelling discharge, infection, scarring, pain, and overall discomfort.2
Given its rarity and the potential manifestations that overlap with other conditions, steatocystomas easily can be misdiagnosed. In some clinical instances, EVHs may share similar characteristics with SM; however, certain distinguishing features exist, including a central tuft of protruding hairs and different expressed contents, such as the vellus hair shafts, from the cyst’s lumen.28 Furthermore, histologic examination of EVHs reveals epidermoid keratinization of the lining as well as a lack of sebaceous glands within the wall.28,29 Other similar conditions include epidermoid cysts, pilar cysts, lipomas, epidermal inclusion cysts, dermoid cysts, sebaceous hyperplasia, folliculitis, xanthomas, neurofibromatosis, and syringomas.30 Occasionally, SMS can be mistaken for HS or acne conglobata, and SM lesions with a facial distribution can mimic acne vulgaris.1,31 These conditions should be excluded before a diagnosis of SS, SM, or SMS is made.
Importantly, SM is visually indistinguishable from subcutaneous metastasis on physical examination, and there are reports of oncologic conditions (eg, pulmonary adenocarcinoma metastasized to the skin) being mistaken for SS or SM.32 Therefore, a thorough clinical examination, histopathologic analysis, and potential use of other imaging modalities such as ultrasonography (US) are needed to ensure an accurate diagnosis.
Ultrasonography has demonstrated utility in diagnosing steatocystomas.33-35 Steatocystomas have incidentally been found on routine mammograms and can demonstrate well-defined circular nodules with radiolucent characteristics and a thin radiodense outline.33,36 Homogeneous hypoechoic nodules within the dermis without posterior acoustic features generally are observed (eFigure 3).33,37 In patients declining biopsy, US may be useful in further characterization of an unknown lesion. Color Doppler US can be used to distinguish SMS from HS. Specifically, SM typically exhibits an absence of Doppler signaling due to a lack of vascularity, providing a helpful diagnostic clue for the SMS variant.33
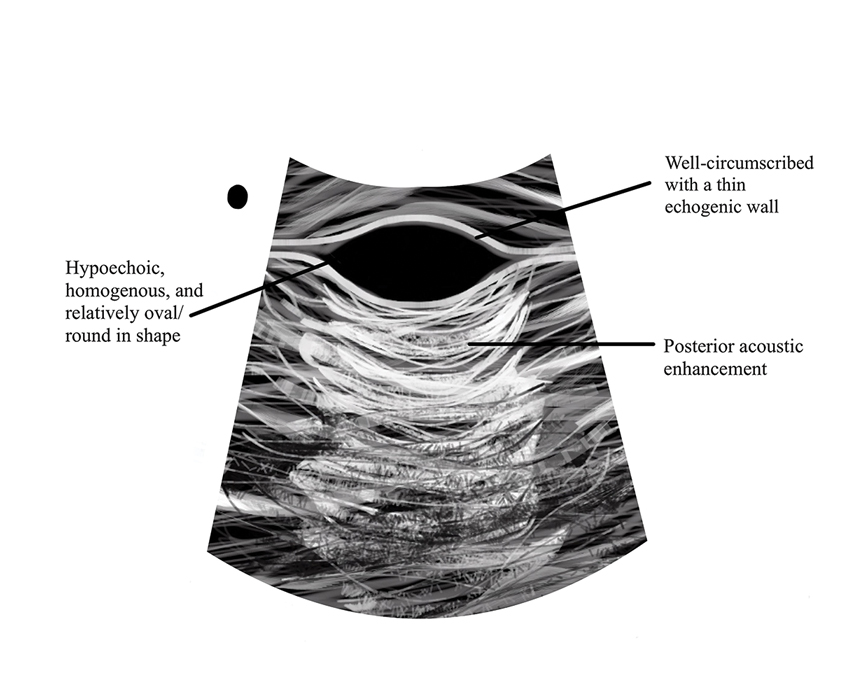
Management and Treatment Options
There is no established standard treatment for steatocystomas; therefore, the approach to management is contingent on clinical presentation and patient preferences. Various medical, surgical, and laser management options are available, each with its own advantages and limitations. Treatment of SM is difficult due to the large number of lesions.38 In many cases, continued observation is a viable treatment option, as most SS and SM lesions are asymptomatic; however, cosmetic concerns can be debilitating for patients with SM and may warrant intervention.39 More extensive medical and surgical management often are necessary in SMS due to associated morbidity. Discussing options and goals as well as setting realistic expectations with the patient are essential in determining the optimal approach.
Medical Management—In medical literature, oral isotretinoin (13-cis-retinoic acid) has been the mainstay of therapy for steatocystoma, as its effect on the size and activity of sebaceous glands is hypothesized to decrease disease activity.38,40 Interventional studies and case reports have exhibited varying degrees of effectiveness.1,38-41 Some reports depict a reduction in the formation of new lesions and a decrease in the size of pre-existing lesions, some show mild delayed therapeutic efficacy, and others suggest exacerbation of the condition.1,38-41 This outcome variability is attributed to isotretinoin’s preferential efficacy in treating inflammatory lesions.40,42
Tetracycline derivatives and intralesional steroid injections also have been employed with some efficacy in patients with focal inflammatory SM and SMS.43 There is limited evidence on the long-term outcomes of these interventions, and intralesional injections often are not recommended in conditions such as SM, in which there are many lesions present.
Surgical Management—Minimally invasive surgical procedures including drainage and resections have been used with varying efficacy in SS and SM. Typically, a 2- to 3-mm incision or sharp-tipped cautery is employed to puncture the cyst. Alternatively, radiofrequency probes with a 2.4-MHz frequency setting have been used to minimize incision size.44 The contents then are expressed with manual pressure or forceps, and the cyst sac is extracted using forceps and/or a vein hook (eFigure 4).44,45 The specific surgical techniques and their respective advantages and limitations are summarized in the eTable. Reported advantages and limitations of surgical techniques are derived from information provided by the authors of steatocystoma case reports, which are based on observations of a very limited sample size.
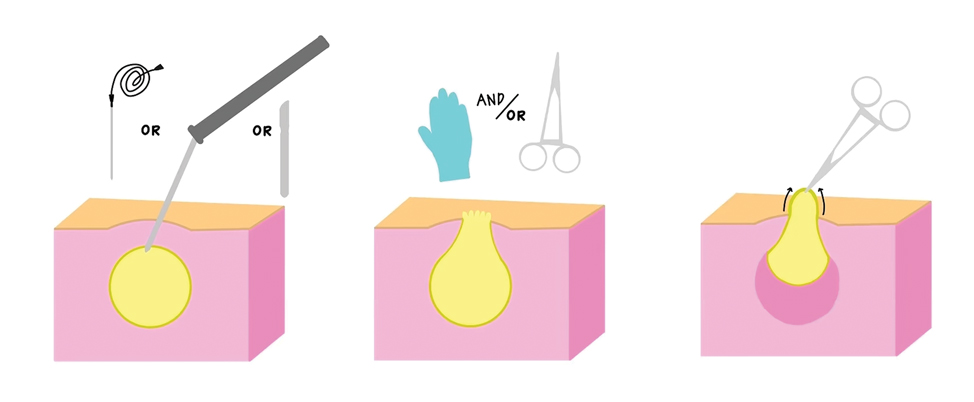
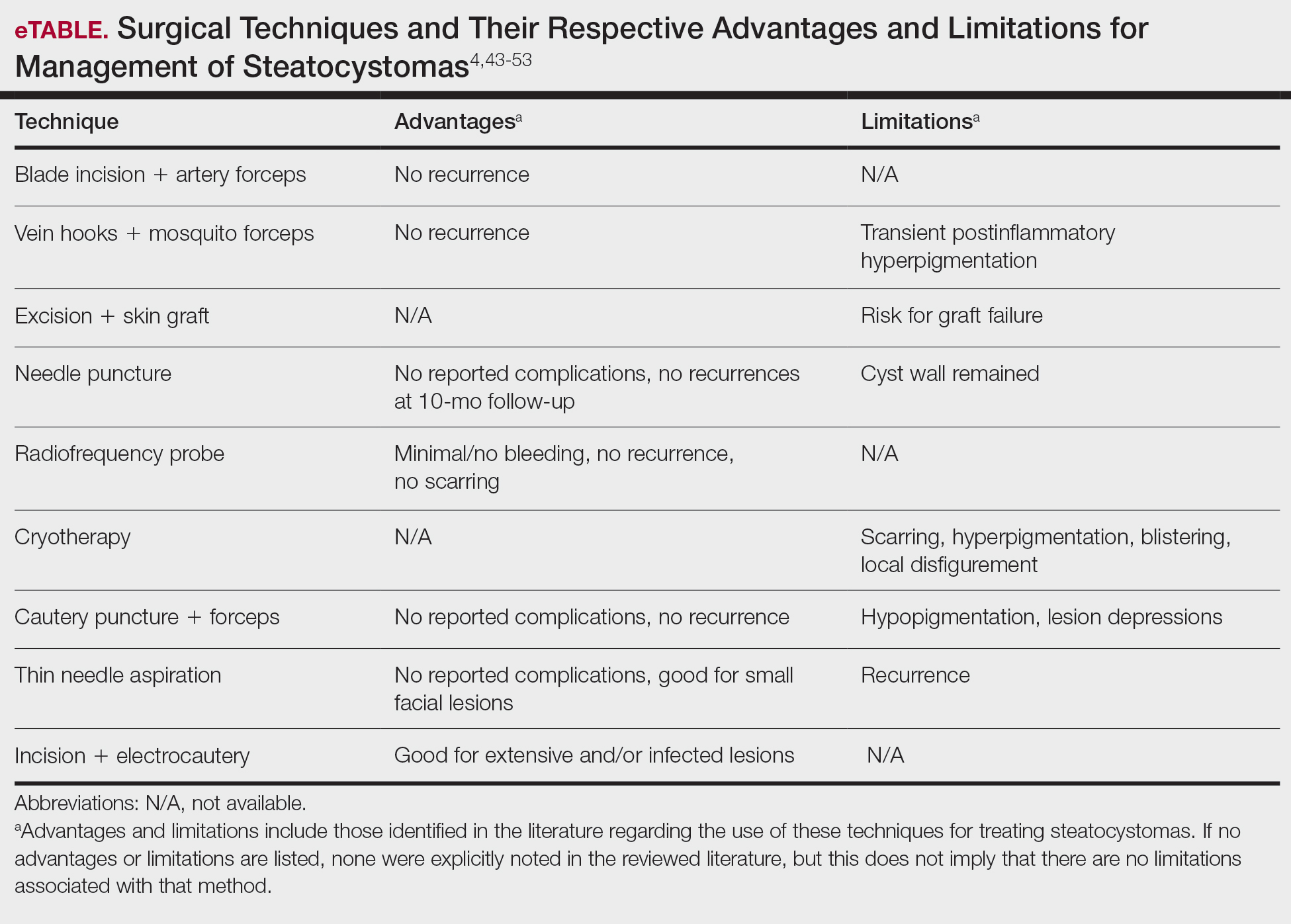
Laser Treatment—Various laser modalities have been used in the management of steatocystomas, including carbon dioxide lasers, erbium-doped yttrium aluminum garnet lasers, 1450-nm diode plus 1550-nm fractionated erbium-doped fiber lasers, and 1927-nm diode lasers.54,55-57 These lasers are used to perforate the cyst before extirpation and have displayed advantages in minimizing scar length.58 The super-pulse mode of carbon dioxide lasers demonstrates efficacy with minimal scarring and recurrence, and this mode is preferred to minimize thermal damage.54,59 Furthermore, this modality can be especially useful in patients whose condition is refractory to other noninvasive options.59 Similarly, the erbium-doped yttrium aluminum garnet laser was well tolerated with no complications noted.55 The 1927-nm diode laser also displayed good outcomes as well as no recurrence.57 With laser use, it is important to note that multiple treatments are needed to see optimal outcomes.54 Moreover, laser settings must be carefully considered, especially in patients with Fitzpatrick skin type III or higher, and topical anti-inflammatory agents should be considered posttreatment to minimize complications.54,59,60
Recommendations
For management of SS, we recommend conservative therapy of watchful observation, as scarring or postinflammatory pigment change may be brought on by medical or surgical therapy; however, if SS is cosmetically bothersome, laser or surgical excision can be done (eFigure 4).4,43-53 It is important to counsel the patient on risks/benefits. For SM, watchful observation also is indicated; however, systemic therapies aimed at prevention may be the most efficacious by limiting disease progression, and oral tetracycline or isotretinoin may be tried.4 Tetracyclines have the risk for photosensitivity and are teratogenic, while isotretinoin is extremely teratogenic, requires laboratory monitoring and regular pregnancy tests in women, and often causes substantial mucosal dryness. If lesions are bothersome or refractory to these therapies, intralesional steroids or surgical/laser procedures can be tried throughout multiple visits.43-53 For SMS, systemic therapies frequently are recommended. The risks of systemic tetracycline and isotretinoin therapies must be discussed. Patients with treatment-refractory SMS may require surgical excision or deroofing of sinus tracts.43-53 This management is similar to that of HS and must be tailored to the patient.
Conclusion
Overall, steatocystomas are a relatively rare pathology, with a limited consensus on their etiology and management. This review summarizes the current knowledge on the condition to support clinicians in diagnosis and management, ranging from watchful waiting to surgical removal. By individualizing treatment plans, clinicians ultimately can optimize outcomes in patients with steatocystomas.
- Santana CN, Pereira DD, Lisboa AP, et al. Steatocystoma multiplex suppurativa: case report of a rare condition. An Bras Dermatol. 2016;91(5 suppl 1):51-53.
- Atzori L, Zanniello R, Pilloni L, et al. Steatocystoma multiplex suppurativa associated with hidradenitis suppurativa successfully treated with adalimumab. J Eur Acad Dermatol Venereol. 2019;33(Suppl 6):42-44.
- Jamieson WA. Case of numerous cutaneous cysts scattered over the body. Edinb Med J. 1873;19:223-225.
- Kamra HT, Gadgil PA, Ovhal AG, et al. Steatocystoma multiplex-a rare genetic disorder: a case report and review of the literature. J Clin Diagn Res. 2013;7:166-168.
- Brownstein MH. Steatocystoma simplex. A solitary steatocystoma. Arch Dermatol. 1982;118:409-411.
- McDonald RM, Reed WB. Natal teeth and steatocystoma multiplex complicated by hidradenitis suppurativa. A new syndrome. Arch Dermatol. 1976;112:1132-1134.
- Plewig G, Wolff HH, Braun-Falco O. Steatocystoma multiplex: anatomic reevaluation, electron microscopy, and autoradiography. Arch Dermatol. 1982;272:363-380.
- Fletcher J, Posso-De Los Rios C, Jambrosic J, A, et al. Coexistence of hidradenitis suppurativa and steatocystoma multiplex: is it a new variant of hidradenitis suppurativa? J Cutan Med Surg. 2021;25:586-590.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Yang L, Zhang S, Wang G. Keratin 17 in disease pathogenesis: from cancer to dermatoses. J Pathol. 2019;247:158-165.
- Shamloul G, Khachemoune A. An updated review of the sebaceous gland and its role in health and diseases Part 1: embryology, evolution, structure, and function of sebaceous glands. Dermatol Ther. 2021;34:e14695.
- Del Rosso JQ, Kircik LH, Stein Gold L, et al. Androgens, androgen receptors, and the skin: from the laboratory to the clinic with emphasis on clinical and therapeutic implications. J Drugs Dermatol. 2020;19:30-35.
- Porras Fimbres DC, Wolfe SA, Kelley CE. Proliferation of steatocystomas in 2 transgender men. JAAD Case Rep. 2022;26:70-72.
- Marasca C, Megna M, Donnarumma M, et al. A case of steatocystoma multiplex in a psoriatic patient during treatment with anti-IL-12/23. Skin Appendage Disord. 2020;6:309-311.
- Gordon Spratt EA, Kaplan J, Patel RR, et al. Steatocystoma. Dermatol Online J. 2013;19:20721.
- Sharma A, Agrawal S, Dhurat R, et al. An unusual case of facial steatocystoma multiplex: a clinicopathologic and dermoscopic report. Dermatopathology (Basel). 2018;5:58-63.
- Rahman MH, Islam MS, Ansari NP. Atypical steatocystoma multiplex with calcification. ISRN Dermatol. 2011;2011:381901.
- Beyer AV, Vossmann D. Steatocystoma multiplex. Article in German. Hautarzt. 1996;47:469-471.
- Yanagi T, Matsumura T. Steatocystoma multiplex presenting as acral subcutaneous nodules. Acta Derm Venereol. 2006;86:374-375.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Ferrandiz C, Peyri J. Steatocystoma multiplex. Article in Spanish. Med Cutan Ibero Lat Am. 1984;12:173-176.
- Alotaibi L, Alsaif M, Alhumidi A, et al. Steatocystoma multiplex suppurativa: a case with unusual giant cysts over the scalp and neck. Case Rep Dermatol. 2019;11:71-76.
- Kim SJ, Park HJ, Oh ST, et al. A case of steatocystoma multiplex limited to scalp. Ann Dermatol. 2009;21:106-109.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Tomková H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Patokar AS, Holani AR, Khandait GH, et al. Eruptive vellus hair cysts: an underdiagnosed entity. Int J Trichology. 2022;14:31-33.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5 Pt 2):876-878.
- Yoon H, Kang Y, Park H, et al. Sonographic appearance of steatocystoma: an analysis of 14 pathologically confirmed lesions. Taehan Yongsang Uihakhoe Chi. 2021;82:382-392.
- Varshney M, Aziz M, Maheshwari V, et al. Steatocystoma multiplex. BMJ Case Rep. 2011;2011:bcr0420114165.
- Tsai MH, Hsiao YP, Lin WL, et al. Steatocystoma multiplex as initial impression of non-small cell lung cancer with complete response to gefitinib. Chin J Cancer Res. 2014;26:E5-E9.
- Zussino M, Nazzaro G, Moltrasio C, et al. Coexistence of steatocystoma multiplex and hidradenitis suppurativa: assessment of this unique association by means of ultrasonography and color Doppler. Skin Res Technol. 2019;25:877-880.
- Whittle C, Silva-Hirschberg C, Loyola K, et al. Ultrasonographic spectrum of cutaneous cysts with stratified squamous epithelium in pediatric dermatology: pictorial essay. J Ultrasound Med. 2023;42:923-930.
- Arceu M, Martinez G, Alfaro D, et al. Ultrasound morphologic features of steatocystoma multiplex with clinical correlation. J Ultrasound Med. 2020;39:2255-2260.
- Reick-Mitrisin V, Reddy A, Shah BA. A breast imaging case of steatocystoma multiplex: a rare condition involving multiple anatomic regions. Cureus. 2022;14:E27756.
- Yoon H, Kang Y, Park H, et al. Sonographic appearance of steatocystoma: an analysis of 14 pathologically confirmed lesions. Taehan Yongsang Uihakhoe Chi. 2021;82:382-392.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sharma A, Agrawal S, Dhurat R, et al. An unusual case of facial steatocystoma multiplex: a clinicopathologic and dermoscopic report. Dermatopathology (Basel). 2018;5:58-63.
- Moritz DL, Silverman RA. Steatocystoma multiplex treated with isotretinoin: a delayed response. Cutis. 1988;42:437-439.
- Schwartz JL, Goldsmith LA. Steatocystoma multiplex suppurativum: treatment with isotretinoin. Cutis. 1984;34:149-153.
- Kim SJ, Park HJ, Oh ST, et al. A case of steatocystoma multiplex limited to the scalp. Ann Dermatol. 2009;21:106-109.
- Fekete GL, Fekete JE. Steatocystoma multiplex generalisata partially suppurativa--case report. Acta Dermatovenerol Croat. 2010;18:114-119.
- Choudhary S, Koley S, Salodkar A. A modified surgical technique for steatocystoma multiplex. J Cutan Aesthet Surg. 2010;3:25-28.
- Kaya TI, Ikizoglu G, Kokturk A, et al. A simple surgical technique for the treatment of steatocystoma multiplex. Int J Dermatol. 2001;40:785-788.
- Oertel YC, Scott DM. Cytologic-pathologic correlations: fine needle aspiration of three cases of steatocystoma multiplex. Ann Diagn Pathol. 1998;2:318-320.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.
- Lee SJ, Choe YS, Park BC, et al. The vein hook successfully used for eradication of steatocystoma multiplex. Dermatol Surg. 2007;33:82-84.
- Bettes PSL, Lopes SL, Prestes MA, et al. Treatment of a facial variant of the multiple steatocystoma with skin graft: case report. Rev Bras Cir Plást. 1998;13:31-36
- Düzova AN, Sentürk GB. Suggestion for the treatment of steatocystoma multiplex located exclusively on the face. Int J Dermatol. 2004;43:60-62. doi:10.1111/j.1365-4632.2004.02068.x
- Choudhary S, Koley S, Salodkar A. A modified surgical technique for steatocystoma multiplex. J Cutan Aesthet Surg. 2010;3:25-28.
- Kaya TI, Ikizoglu G, Kokturk A, et al. A simple surgical technique for the treatment of steatocystoma multiplex. Int J Dermatol. 2001;40:785-788.
- Bakkour W, Madan V. Carbon dioxide laser perforation and extirpation of steatocystoma multiplex. Dermatol Surg. 2014;40:658-662.
- Mumcuog?lu CT, Gurel MS, Kiremitci U, et al. Er: yag laser therapy for steatocystoma multiplex. Indian J Dermatol. 2010;55:300-301.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7 Pt 1):1104-1106.
- Cheon DU, Ko JY. 1927-nm fiber-optic diode laser: a novel therapeutic option for facial steatocystoma multiplex. J Cosmet Dermatol. 2019;18:1326-1329.
- Kim KT, Sun H, Chung EH. Comparison of complete surgical excision and minimally invasive excision using CO2 laser for removal of epidermal cysts on the face. Arch Craniofac Surg. 2019;20:84-88.
- Kassira S, Korta DZ, de Feraudy S, et al. Fractionated ablative carbon dioxide laser treatment of steatocystoma multiplex. J Cosmet Laser Ther. 2016;18:364-366.
- Dixit N, Sardana K, Paliwal P. The rationale of ideal pulse duration and pulse interval in the treatment of steatocystoma multiplex using the carbon dioxide laser in a super-pulse mode as opposedto the ultra-pulse mode. Indian J Dermatol Venereol Leprol. 2020;86:454-456.
Steatocystomas are small sebum-filled cysts that typically manifest in the dermis and originate from sebaceous follicles. Although commonly asymptomatic, these lesions can manifest with pruritus or become infected, predisposing patients to further complications.1 Steatocystomas can manifest as single (steatocystoma simplex [SS]) or numerous (steatocystoma multiplex [SM]) lesions; the lesions also can spontaneously rupture with characteristics that resemble hidradenitis suppurativa (HS)(steatocystoma multiplex suppurativa [SMS]).1,2
Steatocystomas are relatively rare, and there is limited consensus in the published literature on the etiology and management of this condition. In this article, we present a comprehensive review of steatocystomas in the current literature. We highlight important features to consider when making the diagnosis and also offer recommendations for best-practice treatment.
Historical Background
Although not explicitly identified by name, the first documentation of steatocystomas is a case report published in 1873. In this account, the author described a patient who presented with approximately 250 flesh-colored dermal cysts across the body that varied in size.3 In 1899, the term steatocystoma multiple—derived from Greek roots meaning “fatty bag”—was first used.4
In 1982, almost a century later, Brownstein5 reported some of the earliest cases of SS. This solitary subtype is identical to SM on a microscopic level; however, unlike SM, this variant occurs as a single lesion that typically forms in adulthood and in the absence of family history. Other benign adnexal tumors (eg, pilomatricomas, pilar cysts, and sebaceous hyperplasias) also can manifest as either solitary or multiple lesions.
In 1976, McDonald and Reed6 reported the first known cases of patients with both SM and HS. At the time, the co-occurrence of these conditions was viewed as coincidental, but there were postulations of a shared inflammatory process and hereditary link6; it was not until 1982 that the term steatocystoma multiplex suppurativum was coined to describe this variant.7 Although rare, there have been multiple documented instances of SMS since. It has been suggested that the convergence of these conditions may indicate a shared follicular proliferation defect.8 Ongoing investigation is warranted to explain the underlying pathogenesis of this unique variant.
Epidemiology
The available epidemiologic data primarily relate to SM, the most common steatocystoma variant. Nevertheless, SM is a relatively rare condition, and the exact incidence and prevalence remain unknown.8,9 Steatocystomas typically manifest in the first and second decades of life and have been observed in patients of both sexes, with studies demonstrating no notable sex bias.4,9
Etiology and Pathophysiology
Steatocystomas can occur sporadically or may be inherited as an autosomal-dominant condition.4 Typically, SS tends to manifest as an isolated occurrence without any inherent genetic predisposition.5 Alternatively, SM may develop sporadically or be associated with a mutation in the keratin 17 gene (KRT17).4 Steatocystoma multiplex also has been associated with at least 4 different missense mutations, including N92H, R94H, and R94C, located on the long (q) arm of chromosome 17.4,10-12
The keratin 17 gene is responsible for encoding the keratin 17 protein, a type I intermediate filament predominantly synthesized in the basal cells of epithelial tissue. This fibrous structural protein can regulate many processes, including inflammation and cell proliferation, and is found in regions such as the sebaceous glands, hair follicles, and eccrine sweat glands. Overexpression of KRT17 has been suggested in other cutaneous conditions, most notably psoriasis.12 Despite KRT17’s many roles, it remains unclear why SM typically manifests with a myriad of sebum-containing cysts as the primary symptom.12 Continued investigation into the genetic underpinnings of SM and the keratin 17 protein is necessary to further elucidate a more comprehensive understanding of this condition.
Hormonal influences have been suggested as a potential trigger for steatocystoma growth.4,13 This condition is associated with dysfunction of the sebaceous glands, and, correspondingly, the incidence of disease is highest in pubertal patients, in whom androgen levels and sebum production are elevated.4,13,14 Two cases of transgender men taking testosterone therapy presenting with steatocystomas provide additional clinical support for this association.15
Additionally, the use of immunomodulatory agents, such as ustekinumab (anti–interleukin 12/interleukin 23), has been shown to trigger SM. It is predicted that the reduced expression of certain interferons and interleukins may lead to downstream consequences in the keratin 17 pathway and lead to SM lesion formation in genetically susceptible individuals.16 Targeting these potential causes in the future may prove efficacious in the secondary prevention of familial SM manifestation or exacerbations.
Mutations in the KRT17 gene also have been implicated in pachyonychia congenita type 2 (PC-2).4 Marked by extensive systemic hyperkeratosis, PC-2 has been observed to coincide with SM in certain patients.4,5 Interestingly, the location of the KRT17 mutations are identical in both PC-2 and SM.4 Although most individuals with hereditary SM do not exhibit the characteristic features of PC-2, mild nail and dental abnormalities have been observed in some SM cases.4,10 This relationship suggests that SM may be a less severe variant of PC-2 or part of a complex polygenetic spectrum of disease.10 Further research is imperative to determine the exact nature and extent of the relationship between these conditions.
Clinical Manifestations
Steatocystomas are flesh-colored subcutaneous cysts that range in size from less than 3 mm to larger than 3 cm in diameter (Figure). They form within a single pilosebaceous unit and typically display firm attachment due to their origination in the dermis.2,7,17 Steatocystomas generally contain lipid material, and less frequently, keratin and hair shafts, distinguishing them as the only “true” sebaceous cysts.18 Their color can range from flesh-toned to yellow, with reports of occasional dark-blue shades and calcifications.19,20 Steatocystomas can persist indefinitely, and they usually are asymptomatic.
Diagnosis of steatocystoma is confirmed by biopsy.4 Steatocystomas are characterized by a dermal cyst lined by stratified squamous cell epithelium (eFigures 1 and 2).21 Classically they feature flattened sebaceous lobules, multinucleated giant cells, and abortive hair follicles. The lining of these cysts is marked by lymphocytic infiltrate and a dense, wrinkled, eosinophilic keratin cuticle that replaces the granular layer.22 The cyst maintains an epidermal connection through a follicular infundibulum characterized by clumps of keratinocytes, sebocytes, corneocytes, and/or hair follicles.7 Aspirated contents reveal crystalline structures and anucleate squamous cells upon microscopic analysis. That being said, variable histologic findings of steatocystomas have been described.23
Steatocystoma simplex, as the name implies, classifies a single isolated steatocystoma. This subtype exhibits similar histopathologic and clinical features to the other subtypes of steatocystomas. Notably, SS is not associated with a genetic mutation and is not an inherited condition within families.5 Steatocystoma multiplex manifests with many steatocystomas, often distributed widely across the body.3,4 The chest, axillae, and groin are the most common locations; however, these cysts can manifest on the face, back, abdomen, and extremities.4,18-22 Rare occurrences of SM limited to the face, scalp, and distal extremities have been documented.18,21,24,25 Due to the possibility of an autosomal-dominant inheritance, it is advisable to take a comprehensive family history in patients for whom SM is in the differential.17
Steatocystoma multiplex—especially familial variants—has been shown to develop in conjunction with other dermatologic conditions, including eruptive vellus hair (EVH) cysts, persistent infantile milia, and epidermoid/dermoid cysts.26 While some investigators regard these as separate entities due to their varied genetic etiology, it has been suggested that these conditions may be related and that the diagnosis is determined by the location of cyst origin along the sebaceous ducts.26,27 Other dermatologic conditions and lesions that frequently manifest comorbidly with SM include hidrocystomas, syringomas, pilonidal cysts, lichen planus, nodulocystic acne, trichotillomania, trichoblastomas, trichoepithelioma, HS, keratoacanthomas, acrokeratosis verruciformis of Hopf, and embryonal hair formation. Steatocystoma multiplex, manifesting comorbidly with dental and orofacial malformations (eg, partial noneruption of secondary teeth, natal and defective teeth, and bilateral preauricular sinuses) has been classified as SM natal teeth syndrome.6
Steatocystoma multiplex suppurativa is a rare and serious variant of SM characterized by inflammation, cyst rupture, sinus tract formation, and scarring.24 Patients with SMS typically have multiple intact SM cysts, which can aid in differentiation from HS.2,24 Steatocystoma multiplex suppurativa is associated with more complications than SS and SM, including cyst perforation, development of purulent and/or foul-smelling discharge, infection, scarring, pain, and overall discomfort.2
Given its rarity and the potential manifestations that overlap with other conditions, steatocystomas easily can be misdiagnosed. In some clinical instances, EVHs may share similar characteristics with SM; however, certain distinguishing features exist, including a central tuft of protruding hairs and different expressed contents, such as the vellus hair shafts, from the cyst’s lumen.28 Furthermore, histologic examination of EVHs reveals epidermoid keratinization of the lining as well as a lack of sebaceous glands within the wall.28,29 Other similar conditions include epidermoid cysts, pilar cysts, lipomas, epidermal inclusion cysts, dermoid cysts, sebaceous hyperplasia, folliculitis, xanthomas, neurofibromatosis, and syringomas.30 Occasionally, SMS can be mistaken for HS or acne conglobata, and SM lesions with a facial distribution can mimic acne vulgaris.1,31 These conditions should be excluded before a diagnosis of SS, SM, or SMS is made.
Importantly, SM is visually indistinguishable from subcutaneous metastasis on physical examination, and there are reports of oncologic conditions (eg, pulmonary adenocarcinoma metastasized to the skin) being mistaken for SS or SM.32 Therefore, a thorough clinical examination, histopathologic analysis, and potential use of other imaging modalities such as ultrasonography (US) are needed to ensure an accurate diagnosis.
Ultrasonography has demonstrated utility in diagnosing steatocystomas.33-35 Steatocystomas have incidentally been found on routine mammograms and can demonstrate well-defined circular nodules with radiolucent characteristics and a thin radiodense outline.33,36 Homogeneous hypoechoic nodules within the dermis without posterior acoustic features generally are observed (eFigure 3).33,37 In patients declining biopsy, US may be useful in further characterization of an unknown lesion. Color Doppler US can be used to distinguish SMS from HS. Specifically, SM typically exhibits an absence of Doppler signaling due to a lack of vascularity, providing a helpful diagnostic clue for the SMS variant.33
Management and Treatment Options
There is no established standard treatment for steatocystomas; therefore, the approach to management is contingent on clinical presentation and patient preferences. Various medical, surgical, and laser management options are available, each with its own advantages and limitations. Treatment of SM is difficult due to the large number of lesions.38 In many cases, continued observation is a viable treatment option, as most SS and SM lesions are asymptomatic; however, cosmetic concerns can be debilitating for patients with SM and may warrant intervention.39 More extensive medical and surgical management often are necessary in SMS due to associated morbidity. Discussing options and goals as well as setting realistic expectations with the patient are essential in determining the optimal approach.
Medical Management—In medical literature, oral isotretinoin (13-cis-retinoic acid) has been the mainstay of therapy for steatocystoma, as its effect on the size and activity of sebaceous glands is hypothesized to decrease disease activity.38,40 Interventional studies and case reports have exhibited varying degrees of effectiveness.1,38-41 Some reports depict a reduction in the formation of new lesions and a decrease in the size of pre-existing lesions, some show mild delayed therapeutic efficacy, and others suggest exacerbation of the condition.1,38-41 This outcome variability is attributed to isotretinoin’s preferential efficacy in treating inflammatory lesions.40,42
Tetracycline derivatives and intralesional steroid injections also have been employed with some efficacy in patients with focal inflammatory SM and SMS.43 There is limited evidence on the long-term outcomes of these interventions, and intralesional injections often are not recommended in conditions such as SM, in which there are many lesions present.
Surgical Management—Minimally invasive surgical procedures including drainage and resections have been used with varying efficacy in SS and SM. Typically, a 2- to 3-mm incision or sharp-tipped cautery is employed to puncture the cyst. Alternatively, radiofrequency probes with a 2.4-MHz frequency setting have been used to minimize incision size.44 The contents then are expressed with manual pressure or forceps, and the cyst sac is extracted using forceps and/or a vein hook (eFigure 4).44,45 The specific surgical techniques and their respective advantages and limitations are summarized in the eTable. Reported advantages and limitations of surgical techniques are derived from information provided by the authors of steatocystoma case reports, which are based on observations of a very limited sample size.
Laser Treatment—Various laser modalities have been used in the management of steatocystomas, including carbon dioxide lasers, erbium-doped yttrium aluminum garnet lasers, 1450-nm diode plus 1550-nm fractionated erbium-doped fiber lasers, and 1927-nm diode lasers.54,55-57 These lasers are used to perforate the cyst before extirpation and have displayed advantages in minimizing scar length.58 The super-pulse mode of carbon dioxide lasers demonstrates efficacy with minimal scarring and recurrence, and this mode is preferred to minimize thermal damage.54,59 Furthermore, this modality can be especially useful in patients whose condition is refractory to other noninvasive options.59 Similarly, the erbium-doped yttrium aluminum garnet laser was well tolerated with no complications noted.55 The 1927-nm diode laser also displayed good outcomes as well as no recurrence.57 With laser use, it is important to note that multiple treatments are needed to see optimal outcomes.54 Moreover, laser settings must be carefully considered, especially in patients with Fitzpatrick skin type III or higher, and topical anti-inflammatory agents should be considered posttreatment to minimize complications.54,59,60
Recommendations
For management of SS, we recommend conservative therapy of watchful observation, as scarring or postinflammatory pigment change may be brought on by medical or surgical therapy; however, if SS is cosmetically bothersome, laser or surgical excision can be done (eFigure 4).4,43-53 It is important to counsel the patient on risks/benefits. For SM, watchful observation also is indicated; however, systemic therapies aimed at prevention may be the most efficacious by limiting disease progression, and oral tetracycline or isotretinoin may be tried.4 Tetracyclines have the risk for photosensitivity and are teratogenic, while isotretinoin is extremely teratogenic, requires laboratory monitoring and regular pregnancy tests in women, and often causes substantial mucosal dryness. If lesions are bothersome or refractory to these therapies, intralesional steroids or surgical/laser procedures can be tried throughout multiple visits.43-53 For SMS, systemic therapies frequently are recommended. The risks of systemic tetracycline and isotretinoin therapies must be discussed. Patients with treatment-refractory SMS may require surgical excision or deroofing of sinus tracts.43-53 This management is similar to that of HS and must be tailored to the patient.
Conclusion
Overall, steatocystomas are a relatively rare pathology, with a limited consensus on their etiology and management. This review summarizes the current knowledge on the condition to support clinicians in diagnosis and management, ranging from watchful waiting to surgical removal. By individualizing treatment plans, clinicians ultimately can optimize outcomes in patients with steatocystomas.
Steatocystomas are small sebum-filled cysts that typically manifest in the dermis and originate from sebaceous follicles. Although commonly asymptomatic, these lesions can manifest with pruritus or become infected, predisposing patients to further complications.1 Steatocystomas can manifest as single (steatocystoma simplex [SS]) or numerous (steatocystoma multiplex [SM]) lesions; the lesions also can spontaneously rupture with characteristics that resemble hidradenitis suppurativa (HS)(steatocystoma multiplex suppurativa [SMS]).1,2
Steatocystomas are relatively rare, and there is limited consensus in the published literature on the etiology and management of this condition. In this article, we present a comprehensive review of steatocystomas in the current literature. We highlight important features to consider when making the diagnosis and also offer recommendations for best-practice treatment.
Historical Background
Although not explicitly identified by name, the first documentation of steatocystomas is a case report published in 1873. In this account, the author described a patient who presented with approximately 250 flesh-colored dermal cysts across the body that varied in size.3 In 1899, the term steatocystoma multiple—derived from Greek roots meaning “fatty bag”—was first used.4
In 1982, almost a century later, Brownstein5 reported some of the earliest cases of SS. This solitary subtype is identical to SM on a microscopic level; however, unlike SM, this variant occurs as a single lesion that typically forms in adulthood and in the absence of family history. Other benign adnexal tumors (eg, pilomatricomas, pilar cysts, and sebaceous hyperplasias) also can manifest as either solitary or multiple lesions.
In 1976, McDonald and Reed6 reported the first known cases of patients with both SM and HS. At the time, the co-occurrence of these conditions was viewed as coincidental, but there were postulations of a shared inflammatory process and hereditary link6; it was not until 1982 that the term steatocystoma multiplex suppurativum was coined to describe this variant.7 Although rare, there have been multiple documented instances of SMS since. It has been suggested that the convergence of these conditions may indicate a shared follicular proliferation defect.8 Ongoing investigation is warranted to explain the underlying pathogenesis of this unique variant.
Epidemiology
The available epidemiologic data primarily relate to SM, the most common steatocystoma variant. Nevertheless, SM is a relatively rare condition, and the exact incidence and prevalence remain unknown.8,9 Steatocystomas typically manifest in the first and second decades of life and have been observed in patients of both sexes, with studies demonstrating no notable sex bias.4,9
Etiology and Pathophysiology
Steatocystomas can occur sporadically or may be inherited as an autosomal-dominant condition.4 Typically, SS tends to manifest as an isolated occurrence without any inherent genetic predisposition.5 Alternatively, SM may develop sporadically or be associated with a mutation in the keratin 17 gene (KRT17).4 Steatocystoma multiplex also has been associated with at least 4 different missense mutations, including N92H, R94H, and R94C, located on the long (q) arm of chromosome 17.4,10-12
The keratin 17 gene is responsible for encoding the keratin 17 protein, a type I intermediate filament predominantly synthesized in the basal cells of epithelial tissue. This fibrous structural protein can regulate many processes, including inflammation and cell proliferation, and is found in regions such as the sebaceous glands, hair follicles, and eccrine sweat glands. Overexpression of KRT17 has been suggested in other cutaneous conditions, most notably psoriasis.12 Despite KRT17’s many roles, it remains unclear why SM typically manifests with a myriad of sebum-containing cysts as the primary symptom.12 Continued investigation into the genetic underpinnings of SM and the keratin 17 protein is necessary to further elucidate a more comprehensive understanding of this condition.
Hormonal influences have been suggested as a potential trigger for steatocystoma growth.4,13 This condition is associated with dysfunction of the sebaceous glands, and, correspondingly, the incidence of disease is highest in pubertal patients, in whom androgen levels and sebum production are elevated.4,13,14 Two cases of transgender men taking testosterone therapy presenting with steatocystomas provide additional clinical support for this association.15
Additionally, the use of immunomodulatory agents, such as ustekinumab (anti–interleukin 12/interleukin 23), has been shown to trigger SM. It is predicted that the reduced expression of certain interferons and interleukins may lead to downstream consequences in the keratin 17 pathway and lead to SM lesion formation in genetically susceptible individuals.16 Targeting these potential causes in the future may prove efficacious in the secondary prevention of familial SM manifestation or exacerbations.
Mutations in the KRT17 gene also have been implicated in pachyonychia congenita type 2 (PC-2).4 Marked by extensive systemic hyperkeratosis, PC-2 has been observed to coincide with SM in certain patients.4,5 Interestingly, the location of the KRT17 mutations are identical in both PC-2 and SM.4 Although most individuals with hereditary SM do not exhibit the characteristic features of PC-2, mild nail and dental abnormalities have been observed in some SM cases.4,10 This relationship suggests that SM may be a less severe variant of PC-2 or part of a complex polygenetic spectrum of disease.10 Further research is imperative to determine the exact nature and extent of the relationship between these conditions.
Clinical Manifestations
Steatocystomas are flesh-colored subcutaneous cysts that range in size from less than 3 mm to larger than 3 cm in diameter (Figure). They form within a single pilosebaceous unit and typically display firm attachment due to their origination in the dermis.2,7,17 Steatocystomas generally contain lipid material, and less frequently, keratin and hair shafts, distinguishing them as the only “true” sebaceous cysts.18 Their color can range from flesh-toned to yellow, with reports of occasional dark-blue shades and calcifications.19,20 Steatocystomas can persist indefinitely, and they usually are asymptomatic.
Diagnosis of steatocystoma is confirmed by biopsy.4 Steatocystomas are characterized by a dermal cyst lined by stratified squamous cell epithelium (eFigures 1 and 2).21 Classically they feature flattened sebaceous lobules, multinucleated giant cells, and abortive hair follicles. The lining of these cysts is marked by lymphocytic infiltrate and a dense, wrinkled, eosinophilic keratin cuticle that replaces the granular layer.22 The cyst maintains an epidermal connection through a follicular infundibulum characterized by clumps of keratinocytes, sebocytes, corneocytes, and/or hair follicles.7 Aspirated contents reveal crystalline structures and anucleate squamous cells upon microscopic analysis. That being said, variable histologic findings of steatocystomas have been described.23
Steatocystoma simplex, as the name implies, classifies a single isolated steatocystoma. This subtype exhibits similar histopathologic and clinical features to the other subtypes of steatocystomas. Notably, SS is not associated with a genetic mutation and is not an inherited condition within families.5 Steatocystoma multiplex manifests with many steatocystomas, often distributed widely across the body.3,4 The chest, axillae, and groin are the most common locations; however, these cysts can manifest on the face, back, abdomen, and extremities.4,18-22 Rare occurrences of SM limited to the face, scalp, and distal extremities have been documented.18,21,24,25 Due to the possibility of an autosomal-dominant inheritance, it is advisable to take a comprehensive family history in patients for whom SM is in the differential.17
Steatocystoma multiplex—especially familial variants—has been shown to develop in conjunction with other dermatologic conditions, including eruptive vellus hair (EVH) cysts, persistent infantile milia, and epidermoid/dermoid cysts.26 While some investigators regard these as separate entities due to their varied genetic etiology, it has been suggested that these conditions may be related and that the diagnosis is determined by the location of cyst origin along the sebaceous ducts.26,27 Other dermatologic conditions and lesions that frequently manifest comorbidly with SM include hidrocystomas, syringomas, pilonidal cysts, lichen planus, nodulocystic acne, trichotillomania, trichoblastomas, trichoepithelioma, HS, keratoacanthomas, acrokeratosis verruciformis of Hopf, and embryonal hair formation. Steatocystoma multiplex, manifesting comorbidly with dental and orofacial malformations (eg, partial noneruption of secondary teeth, natal and defective teeth, and bilateral preauricular sinuses) has been classified as SM natal teeth syndrome.6
Steatocystoma multiplex suppurativa is a rare and serious variant of SM characterized by inflammation, cyst rupture, sinus tract formation, and scarring.24 Patients with SMS typically have multiple intact SM cysts, which can aid in differentiation from HS.2,24 Steatocystoma multiplex suppurativa is associated with more complications than SS and SM, including cyst perforation, development of purulent and/or foul-smelling discharge, infection, scarring, pain, and overall discomfort.2
Given its rarity and the potential manifestations that overlap with other conditions, steatocystomas easily can be misdiagnosed. In some clinical instances, EVHs may share similar characteristics with SM; however, certain distinguishing features exist, including a central tuft of protruding hairs and different expressed contents, such as the vellus hair shafts, from the cyst’s lumen.28 Furthermore, histologic examination of EVHs reveals epidermoid keratinization of the lining as well as a lack of sebaceous glands within the wall.28,29 Other similar conditions include epidermoid cysts, pilar cysts, lipomas, epidermal inclusion cysts, dermoid cysts, sebaceous hyperplasia, folliculitis, xanthomas, neurofibromatosis, and syringomas.30 Occasionally, SMS can be mistaken for HS or acne conglobata, and SM lesions with a facial distribution can mimic acne vulgaris.1,31 These conditions should be excluded before a diagnosis of SS, SM, or SMS is made.
Importantly, SM is visually indistinguishable from subcutaneous metastasis on physical examination, and there are reports of oncologic conditions (eg, pulmonary adenocarcinoma metastasized to the skin) being mistaken for SS or SM.32 Therefore, a thorough clinical examination, histopathologic analysis, and potential use of other imaging modalities such as ultrasonography (US) are needed to ensure an accurate diagnosis.
Ultrasonography has demonstrated utility in diagnosing steatocystomas.33-35 Steatocystomas have incidentally been found on routine mammograms and can demonstrate well-defined circular nodules with radiolucent characteristics and a thin radiodense outline.33,36 Homogeneous hypoechoic nodules within the dermis without posterior acoustic features generally are observed (eFigure 3).33,37 In patients declining biopsy, US may be useful in further characterization of an unknown lesion. Color Doppler US can be used to distinguish SMS from HS. Specifically, SM typically exhibits an absence of Doppler signaling due to a lack of vascularity, providing a helpful diagnostic clue for the SMS variant.33
Management and Treatment Options
There is no established standard treatment for steatocystomas; therefore, the approach to management is contingent on clinical presentation and patient preferences. Various medical, surgical, and laser management options are available, each with its own advantages and limitations. Treatment of SM is difficult due to the large number of lesions.38 In many cases, continued observation is a viable treatment option, as most SS and SM lesions are asymptomatic; however, cosmetic concerns can be debilitating for patients with SM and may warrant intervention.39 More extensive medical and surgical management often are necessary in SMS due to associated morbidity. Discussing options and goals as well as setting realistic expectations with the patient are essential in determining the optimal approach.
Medical Management—In medical literature, oral isotretinoin (13-cis-retinoic acid) has been the mainstay of therapy for steatocystoma, as its effect on the size and activity of sebaceous glands is hypothesized to decrease disease activity.38,40 Interventional studies and case reports have exhibited varying degrees of effectiveness.1,38-41 Some reports depict a reduction in the formation of new lesions and a decrease in the size of pre-existing lesions, some show mild delayed therapeutic efficacy, and others suggest exacerbation of the condition.1,38-41 This outcome variability is attributed to isotretinoin’s preferential efficacy in treating inflammatory lesions.40,42
Tetracycline derivatives and intralesional steroid injections also have been employed with some efficacy in patients with focal inflammatory SM and SMS.43 There is limited evidence on the long-term outcomes of these interventions, and intralesional injections often are not recommended in conditions such as SM, in which there are many lesions present.
Surgical Management—Minimally invasive surgical procedures including drainage and resections have been used with varying efficacy in SS and SM. Typically, a 2- to 3-mm incision or sharp-tipped cautery is employed to puncture the cyst. Alternatively, radiofrequency probes with a 2.4-MHz frequency setting have been used to minimize incision size.44 The contents then are expressed with manual pressure or forceps, and the cyst sac is extracted using forceps and/or a vein hook (eFigure 4).44,45 The specific surgical techniques and their respective advantages and limitations are summarized in the eTable. Reported advantages and limitations of surgical techniques are derived from information provided by the authors of steatocystoma case reports, which are based on observations of a very limited sample size.
Laser Treatment—Various laser modalities have been used in the management of steatocystomas, including carbon dioxide lasers, erbium-doped yttrium aluminum garnet lasers, 1450-nm diode plus 1550-nm fractionated erbium-doped fiber lasers, and 1927-nm diode lasers.54,55-57 These lasers are used to perforate the cyst before extirpation and have displayed advantages in minimizing scar length.58 The super-pulse mode of carbon dioxide lasers demonstrates efficacy with minimal scarring and recurrence, and this mode is preferred to minimize thermal damage.54,59 Furthermore, this modality can be especially useful in patients whose condition is refractory to other noninvasive options.59 Similarly, the erbium-doped yttrium aluminum garnet laser was well tolerated with no complications noted.55 The 1927-nm diode laser also displayed good outcomes as well as no recurrence.57 With laser use, it is important to note that multiple treatments are needed to see optimal outcomes.54 Moreover, laser settings must be carefully considered, especially in patients with Fitzpatrick skin type III or higher, and topical anti-inflammatory agents should be considered posttreatment to minimize complications.54,59,60
Recommendations
For management of SS, we recommend conservative therapy of watchful observation, as scarring or postinflammatory pigment change may be brought on by medical or surgical therapy; however, if SS is cosmetically bothersome, laser or surgical excision can be done (eFigure 4).4,43-53 It is important to counsel the patient on risks/benefits. For SM, watchful observation also is indicated; however, systemic therapies aimed at prevention may be the most efficacious by limiting disease progression, and oral tetracycline or isotretinoin may be tried.4 Tetracyclines have the risk for photosensitivity and are teratogenic, while isotretinoin is extremely teratogenic, requires laboratory monitoring and regular pregnancy tests in women, and often causes substantial mucosal dryness. If lesions are bothersome or refractory to these therapies, intralesional steroids or surgical/laser procedures can be tried throughout multiple visits.43-53 For SMS, systemic therapies frequently are recommended. The risks of systemic tetracycline and isotretinoin therapies must be discussed. Patients with treatment-refractory SMS may require surgical excision or deroofing of sinus tracts.43-53 This management is similar to that of HS and must be tailored to the patient.
Conclusion
Overall, steatocystomas are a relatively rare pathology, with a limited consensus on their etiology and management. This review summarizes the current knowledge on the condition to support clinicians in diagnosis and management, ranging from watchful waiting to surgical removal. By individualizing treatment plans, clinicians ultimately can optimize outcomes in patients with steatocystomas.
- Santana CN, Pereira DD, Lisboa AP, et al. Steatocystoma multiplex suppurativa: case report of a rare condition. An Bras Dermatol. 2016;91(5 suppl 1):51-53.
- Atzori L, Zanniello R, Pilloni L, et al. Steatocystoma multiplex suppurativa associated with hidradenitis suppurativa successfully treated with adalimumab. J Eur Acad Dermatol Venereol. 2019;33(Suppl 6):42-44.
- Jamieson WA. Case of numerous cutaneous cysts scattered over the body. Edinb Med J. 1873;19:223-225.
- Kamra HT, Gadgil PA, Ovhal AG, et al. Steatocystoma multiplex-a rare genetic disorder: a case report and review of the literature. J Clin Diagn Res. 2013;7:166-168.
- Brownstein MH. Steatocystoma simplex. A solitary steatocystoma. Arch Dermatol. 1982;118:409-411.
- McDonald RM, Reed WB. Natal teeth and steatocystoma multiplex complicated by hidradenitis suppurativa. A new syndrome. Arch Dermatol. 1976;112:1132-1134.
- Plewig G, Wolff HH, Braun-Falco O. Steatocystoma multiplex: anatomic reevaluation, electron microscopy, and autoradiography. Arch Dermatol. 1982;272:363-380.
- Fletcher J, Posso-De Los Rios C, Jambrosic J, A, et al. Coexistence of hidradenitis suppurativa and steatocystoma multiplex: is it a new variant of hidradenitis suppurativa? J Cutan Med Surg. 2021;25:586-590.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Yang L, Zhang S, Wang G. Keratin 17 in disease pathogenesis: from cancer to dermatoses. J Pathol. 2019;247:158-165.
- Shamloul G, Khachemoune A. An updated review of the sebaceous gland and its role in health and diseases Part 1: embryology, evolution, structure, and function of sebaceous glands. Dermatol Ther. 2021;34:e14695.
- Del Rosso JQ, Kircik LH, Stein Gold L, et al. Androgens, androgen receptors, and the skin: from the laboratory to the clinic with emphasis on clinical and therapeutic implications. J Drugs Dermatol. 2020;19:30-35.
- Porras Fimbres DC, Wolfe SA, Kelley CE. Proliferation of steatocystomas in 2 transgender men. JAAD Case Rep. 2022;26:70-72.
- Marasca C, Megna M, Donnarumma M, et al. A case of steatocystoma multiplex in a psoriatic patient during treatment with anti-IL-12/23. Skin Appendage Disord. 2020;6:309-311.
- Gordon Spratt EA, Kaplan J, Patel RR, et al. Steatocystoma. Dermatol Online J. 2013;19:20721.
- Sharma A, Agrawal S, Dhurat R, et al. An unusual case of facial steatocystoma multiplex: a clinicopathologic and dermoscopic report. Dermatopathology (Basel). 2018;5:58-63.
- Rahman MH, Islam MS, Ansari NP. Atypical steatocystoma multiplex with calcification. ISRN Dermatol. 2011;2011:381901.
- Beyer AV, Vossmann D. Steatocystoma multiplex. Article in German. Hautarzt. 1996;47:469-471.
- Yanagi T, Matsumura T. Steatocystoma multiplex presenting as acral subcutaneous nodules. Acta Derm Venereol. 2006;86:374-375.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Ferrandiz C, Peyri J. Steatocystoma multiplex. Article in Spanish. Med Cutan Ibero Lat Am. 1984;12:173-176.
- Alotaibi L, Alsaif M, Alhumidi A, et al. Steatocystoma multiplex suppurativa: a case with unusual giant cysts over the scalp and neck. Case Rep Dermatol. 2019;11:71-76.
- Kim SJ, Park HJ, Oh ST, et al. A case of steatocystoma multiplex limited to scalp. Ann Dermatol. 2009;21:106-109.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Tomková H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Patokar AS, Holani AR, Khandait GH, et al. Eruptive vellus hair cysts: an underdiagnosed entity. Int J Trichology. 2022;14:31-33.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5 Pt 2):876-878.
- Yoon H, Kang Y, Park H, et al. Sonographic appearance of steatocystoma: an analysis of 14 pathologically confirmed lesions. Taehan Yongsang Uihakhoe Chi. 2021;82:382-392.
- Varshney M, Aziz M, Maheshwari V, et al. Steatocystoma multiplex. BMJ Case Rep. 2011;2011:bcr0420114165.
- Tsai MH, Hsiao YP, Lin WL, et al. Steatocystoma multiplex as initial impression of non-small cell lung cancer with complete response to gefitinib. Chin J Cancer Res. 2014;26:E5-E9.
- Zussino M, Nazzaro G, Moltrasio C, et al. Coexistence of steatocystoma multiplex and hidradenitis suppurativa: assessment of this unique association by means of ultrasonography and color Doppler. Skin Res Technol. 2019;25:877-880.
- Whittle C, Silva-Hirschberg C, Loyola K, et al. Ultrasonographic spectrum of cutaneous cysts with stratified squamous epithelium in pediatric dermatology: pictorial essay. J Ultrasound Med. 2023;42:923-930.
- Arceu M, Martinez G, Alfaro D, et al. Ultrasound morphologic features of steatocystoma multiplex with clinical correlation. J Ultrasound Med. 2020;39:2255-2260.
- Reick-Mitrisin V, Reddy A, Shah BA. A breast imaging case of steatocystoma multiplex: a rare condition involving multiple anatomic regions. Cureus. 2022;14:E27756.
- Yoon H, Kang Y, Park H, et al. Sonographic appearance of steatocystoma: an analysis of 14 pathologically confirmed lesions. Taehan Yongsang Uihakhoe Chi. 2021;82:382-392.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sharma A, Agrawal S, Dhurat R, et al. An unusual case of facial steatocystoma multiplex: a clinicopathologic and dermoscopic report. Dermatopathology (Basel). 2018;5:58-63.
- Moritz DL, Silverman RA. Steatocystoma multiplex treated with isotretinoin: a delayed response. Cutis. 1988;42:437-439.
- Schwartz JL, Goldsmith LA. Steatocystoma multiplex suppurativum: treatment with isotretinoin. Cutis. 1984;34:149-153.
- Kim SJ, Park HJ, Oh ST, et al. A case of steatocystoma multiplex limited to the scalp. Ann Dermatol. 2009;21:106-109.
- Fekete GL, Fekete JE. Steatocystoma multiplex generalisata partially suppurativa--case report. Acta Dermatovenerol Croat. 2010;18:114-119.
- Choudhary S, Koley S, Salodkar A. A modified surgical technique for steatocystoma multiplex. J Cutan Aesthet Surg. 2010;3:25-28.
- Kaya TI, Ikizoglu G, Kokturk A, et al. A simple surgical technique for the treatment of steatocystoma multiplex. Int J Dermatol. 2001;40:785-788.
- Oertel YC, Scott DM. Cytologic-pathologic correlations: fine needle aspiration of three cases of steatocystoma multiplex. Ann Diagn Pathol. 1998;2:318-320.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.
- Lee SJ, Choe YS, Park BC, et al. The vein hook successfully used for eradication of steatocystoma multiplex. Dermatol Surg. 2007;33:82-84.
- Bettes PSL, Lopes SL, Prestes MA, et al. Treatment of a facial variant of the multiple steatocystoma with skin graft: case report. Rev Bras Cir Plást. 1998;13:31-36
- Düzova AN, Sentürk GB. Suggestion for the treatment of steatocystoma multiplex located exclusively on the face. Int J Dermatol. 2004;43:60-62. doi:10.1111/j.1365-4632.2004.02068.x
- Choudhary S, Koley S, Salodkar A. A modified surgical technique for steatocystoma multiplex. J Cutan Aesthet Surg. 2010;3:25-28.
- Kaya TI, Ikizoglu G, Kokturk A, et al. A simple surgical technique for the treatment of steatocystoma multiplex. Int J Dermatol. 2001;40:785-788.
- Bakkour W, Madan V. Carbon dioxide laser perforation and extirpation of steatocystoma multiplex. Dermatol Surg. 2014;40:658-662.
- Mumcuog?lu CT, Gurel MS, Kiremitci U, et al. Er: yag laser therapy for steatocystoma multiplex. Indian J Dermatol. 2010;55:300-301.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7 Pt 1):1104-1106.
- Cheon DU, Ko JY. 1927-nm fiber-optic diode laser: a novel therapeutic option for facial steatocystoma multiplex. J Cosmet Dermatol. 2019;18:1326-1329.
- Kim KT, Sun H, Chung EH. Comparison of complete surgical excision and minimally invasive excision using CO2 laser for removal of epidermal cysts on the face. Arch Craniofac Surg. 2019;20:84-88.
- Kassira S, Korta DZ, de Feraudy S, et al. Fractionated ablative carbon dioxide laser treatment of steatocystoma multiplex. J Cosmet Laser Ther. 2016;18:364-366.
- Dixit N, Sardana K, Paliwal P. The rationale of ideal pulse duration and pulse interval in the treatment of steatocystoma multiplex using the carbon dioxide laser in a super-pulse mode as opposedto the ultra-pulse mode. Indian J Dermatol Venereol Leprol. 2020;86:454-456.
- Santana CN, Pereira DD, Lisboa AP, et al. Steatocystoma multiplex suppurativa: case report of a rare condition. An Bras Dermatol. 2016;91(5 suppl 1):51-53.
- Atzori L, Zanniello R, Pilloni L, et al. Steatocystoma multiplex suppurativa associated with hidradenitis suppurativa successfully treated with adalimumab. J Eur Acad Dermatol Venereol. 2019;33(Suppl 6):42-44.
- Jamieson WA. Case of numerous cutaneous cysts scattered over the body. Edinb Med J. 1873;19:223-225.
- Kamra HT, Gadgil PA, Ovhal AG, et al. Steatocystoma multiplex-a rare genetic disorder: a case report and review of the literature. J Clin Diagn Res. 2013;7:166-168.
- Brownstein MH. Steatocystoma simplex. A solitary steatocystoma. Arch Dermatol. 1982;118:409-411.
- McDonald RM, Reed WB. Natal teeth and steatocystoma multiplex complicated by hidradenitis suppurativa. A new syndrome. Arch Dermatol. 1976;112:1132-1134.
- Plewig G, Wolff HH, Braun-Falco O. Steatocystoma multiplex: anatomic reevaluation, electron microscopy, and autoradiography. Arch Dermatol. 1982;272:363-380.
- Fletcher J, Posso-De Los Rios C, Jambrosic J, A, et al. Coexistence of hidradenitis suppurativa and steatocystoma multiplex: is it a new variant of hidradenitis suppurativa? J Cutan Med Surg. 2021;25:586-590.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Covello SP, Smith FJ, Sillevis Smitt JH, et al. Keratin 17 mutations cause either steatocystoma multiplex or pachyonychia congenita type 2. Br J Dermatol. 1998;139:475-480.
- Liu Q, Wu W, Lu J, et al. Steatocystoma multiplex is associated with the R94C mutation in the KRTl7 gene. Mol Med Rep. 2015;12:5072-5076.
- Yang L, Zhang S, Wang G. Keratin 17 in disease pathogenesis: from cancer to dermatoses. J Pathol. 2019;247:158-165.
- Shamloul G, Khachemoune A. An updated review of the sebaceous gland and its role in health and diseases Part 1: embryology, evolution, structure, and function of sebaceous glands. Dermatol Ther. 2021;34:e14695.
- Del Rosso JQ, Kircik LH, Stein Gold L, et al. Androgens, androgen receptors, and the skin: from the laboratory to the clinic with emphasis on clinical and therapeutic implications. J Drugs Dermatol. 2020;19:30-35.
- Porras Fimbres DC, Wolfe SA, Kelley CE. Proliferation of steatocystomas in 2 transgender men. JAAD Case Rep. 2022;26:70-72.
- Marasca C, Megna M, Donnarumma M, et al. A case of steatocystoma multiplex in a psoriatic patient during treatment with anti-IL-12/23. Skin Appendage Disord. 2020;6:309-311.
- Gordon Spratt EA, Kaplan J, Patel RR, et al. Steatocystoma. Dermatol Online J. 2013;19:20721.
- Sharma A, Agrawal S, Dhurat R, et al. An unusual case of facial steatocystoma multiplex: a clinicopathologic and dermoscopic report. Dermatopathology (Basel). 2018;5:58-63.
- Rahman MH, Islam MS, Ansari NP. Atypical steatocystoma multiplex with calcification. ISRN Dermatol. 2011;2011:381901.
- Beyer AV, Vossmann D. Steatocystoma multiplex. Article in German. Hautarzt. 1996;47:469-471.
- Yanagi T, Matsumura T. Steatocystoma multiplex presenting as acral subcutaneous nodules. Acta Derm Venereol. 2006;86:374-375.
- Marzano AV, Tavecchio S, Balice Y, et al. Acral subcutaneous steatocystoma multiplex: a distinct subtype of the disease? Australas J Dermatol. 2012;53:198-201.
- Ferrandiz C, Peyri J. Steatocystoma multiplex. Article in Spanish. Med Cutan Ibero Lat Am. 1984;12:173-176.
- Alotaibi L, Alsaif M, Alhumidi A, et al. Steatocystoma multiplex suppurativa: a case with unusual giant cysts over the scalp and neck. Case Rep Dermatol. 2019;11:71-76.
- Kim SJ, Park HJ, Oh ST, et al. A case of steatocystoma multiplex limited to scalp. Ann Dermatol. 2009;21:106-109.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Tomková H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Patokar AS, Holani AR, Khandait GH, et al. Eruptive vellus hair cysts: an underdiagnosed entity. Int J Trichology. 2022;14:31-33.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5 Pt 2):876-878.
- Yoon H, Kang Y, Park H, et al. Sonographic appearance of steatocystoma: an analysis of 14 pathologically confirmed lesions. Taehan Yongsang Uihakhoe Chi. 2021;82:382-392.
- Varshney M, Aziz M, Maheshwari V, et al. Steatocystoma multiplex. BMJ Case Rep. 2011;2011:bcr0420114165.
- Tsai MH, Hsiao YP, Lin WL, et al. Steatocystoma multiplex as initial impression of non-small cell lung cancer with complete response to gefitinib. Chin J Cancer Res. 2014;26:E5-E9.
- Zussino M, Nazzaro G, Moltrasio C, et al. Coexistence of steatocystoma multiplex and hidradenitis suppurativa: assessment of this unique association by means of ultrasonography and color Doppler. Skin Res Technol. 2019;25:877-880.
- Whittle C, Silva-Hirschberg C, Loyola K, et al. Ultrasonographic spectrum of cutaneous cysts with stratified squamous epithelium in pediatric dermatology: pictorial essay. J Ultrasound Med. 2023;42:923-930.
- Arceu M, Martinez G, Alfaro D, et al. Ultrasound morphologic features of steatocystoma multiplex with clinical correlation. J Ultrasound Med. 2020;39:2255-2260.
- Reick-Mitrisin V, Reddy A, Shah BA. A breast imaging case of steatocystoma multiplex: a rare condition involving multiple anatomic regions. Cureus. 2022;14:E27756.
- Yoon H, Kang Y, Park H, et al. Sonographic appearance of steatocystoma: an analysis of 14 pathologically confirmed lesions. Taehan Yongsang Uihakhoe Chi. 2021;82:382-392.
- Apaydin R, Bilen N, Bayramgurler D, et al. Steatocystoma multiplex suppurativum: oral isotretinoin treatment combined with cryotherapy. Australas J Dermatol. 2000;41:98-100.
- Sharma A, Agrawal S, Dhurat R, et al. An unusual case of facial steatocystoma multiplex: a clinicopathologic and dermoscopic report. Dermatopathology (Basel). 2018;5:58-63.
- Moritz DL, Silverman RA. Steatocystoma multiplex treated with isotretinoin: a delayed response. Cutis. 1988;42:437-439.
- Schwartz JL, Goldsmith LA. Steatocystoma multiplex suppurativum: treatment with isotretinoin. Cutis. 1984;34:149-153.
- Kim SJ, Park HJ, Oh ST, et al. A case of steatocystoma multiplex limited to the scalp. Ann Dermatol. 2009;21:106-109.
- Fekete GL, Fekete JE. Steatocystoma multiplex generalisata partially suppurativa--case report. Acta Dermatovenerol Croat. 2010;18:114-119.
- Choudhary S, Koley S, Salodkar A. A modified surgical technique for steatocystoma multiplex. J Cutan Aesthet Surg. 2010;3:25-28.
- Kaya TI, Ikizoglu G, Kokturk A, et al. A simple surgical technique for the treatment of steatocystoma multiplex. Int J Dermatol. 2001;40:785-788.
- Oertel YC, Scott DM. Cytologic-pathologic correlations: fine needle aspiration of three cases of steatocystoma multiplex. Ann Diagn Pathol. 1998;2:318-320.
- Egbert BM, Price NM, Segal RJ. Steatocystoma multiplex. Report of a florid case and a review. Arch Dermatol. 1979;115:334-335.
- Adams BB, Mutasim DF, Nordlund JJ. Steatocystoma multiplex: a quick removal technique. Cutis. 1999;64:127-130.
- Lee SJ, Choe YS, Park BC, et al. The vein hook successfully used for eradication of steatocystoma multiplex. Dermatol Surg. 2007;33:82-84.
- Bettes PSL, Lopes SL, Prestes MA, et al. Treatment of a facial variant of the multiple steatocystoma with skin graft: case report. Rev Bras Cir Plást. 1998;13:31-36
- Düzova AN, Sentürk GB. Suggestion for the treatment of steatocystoma multiplex located exclusively on the face. Int J Dermatol. 2004;43:60-62. doi:10.1111/j.1365-4632.2004.02068.x
- Choudhary S, Koley S, Salodkar A. A modified surgical technique for steatocystoma multiplex. J Cutan Aesthet Surg. 2010;3:25-28.
- Kaya TI, Ikizoglu G, Kokturk A, et al. A simple surgical technique for the treatment of steatocystoma multiplex. Int J Dermatol. 2001;40:785-788.
- Bakkour W, Madan V. Carbon dioxide laser perforation and extirpation of steatocystoma multiplex. Dermatol Surg. 2014;40:658-662.
- Mumcuog?lu CT, Gurel MS, Kiremitci U, et al. Er: yag laser therapy for steatocystoma multiplex. Indian J Dermatol. 2010;55:300-301.
- Moody MN, Landau JM, Goldberg LH, et al. 1,450-nm diode laser in combination with the 1550-nm fractionated erbium-doped fiber laser for the treatment of steatocystoma multiplex: a case report. Dermatol Surg. 2012;38(7 Pt 1):1104-1106.
- Cheon DU, Ko JY. 1927-nm fiber-optic diode laser: a novel therapeutic option for facial steatocystoma multiplex. J Cosmet Dermatol. 2019;18:1326-1329.
- Kim KT, Sun H, Chung EH. Comparison of complete surgical excision and minimally invasive excision using CO2 laser for removal of epidermal cysts on the face. Arch Craniofac Surg. 2019;20:84-88.
- Kassira S, Korta DZ, de Feraudy S, et al. Fractionated ablative carbon dioxide laser treatment of steatocystoma multiplex. J Cosmet Laser Ther. 2016;18:364-366.
- Dixit N, Sardana K, Paliwal P. The rationale of ideal pulse duration and pulse interval in the treatment of steatocystoma multiplex using the carbon dioxide laser in a super-pulse mode as opposedto the ultra-pulse mode. Indian J Dermatol Venereol Leprol. 2020;86:454-456.
Steatocystomas: Update on Clinical Manifestations, Diagnosis, and Management
Steatocystomas: Update on Clinical Manifestations, Diagnosis, and Management
Practice Points
- Steatocystomas, which manifest as single or multiple flesh-colored subcutaneous cysts ranging from less than 3 mm to more than 3 cm, typically are asymptomatic and can persist indefinitely.
- Treatment options for steatocystomas include oral isotretinoin, tetracycline derivatives, and intralesional steroid injections. Minimally invasive procedures such as drainage and resection also are available, employing techniques such as blade incision, radiofrequency probes, and laser treatments to minimize scarring and recurrence.
- Conservative therapies such as watchful waiting are recommended for the simplex and multiplex variants, while more aggressive management such as surgical removal is recommended for the multiplex suppurativa variant due to its elevated risk for complications.

Operational Risk Management in Dermatologic Procedures
Operational Risk Management in Dermatologic Procedures
Operational risk management (ORM) refers to the systematic identification and assessment of daily operational risks within an organization designed to mitigate negative financial, reputational, and safety outcomes while maximizing efficiency and achievement of objectives.1 Operational risk management is indispensable to modern military operations, optimizing mission readiness while minimizing complications and personnel morbidity. Application of ORM in medicine holds considerable promise due to the emphasis on precise and efficient decision-making in high-stakes environments, where the margin for error is minimal. In this article, we propose integrating ORM principles into dermatologic surgery to enhance patient-centered care through improved counseling, risk assessment, and procedural outcomes.
Principles and Processes of ORM
The ORM framework is built on 4 fundamental principles: accept risk when benefits outweigh the cost, accept no unnecessary risk, anticipate and manage risk by planning, and make risk decisions at the right level.2 These principles form the foundation of the ORM’s systematic 5-step approach to identify hazards, assess hazards, make risk decisions, implement controls, and supervise. Key to the ORM process is the use of risk assessment codes and the risk assessment matrix to quantify and prioritize risks. Risk assessment codes are numerical values assigned to hazards based on their assessed severity and probability. The risk assessment matrix is a tool that plots the severity of a hazard against its probability. By locating a hazard on the matrix, users can visualize its risk level in terms of severity and probability. Building and using the risk assessment matrix begins with determining severity by assessing the potential impact of a hazard and categorizing it into levels (catastrophic, critical, moderate, or negligible). Next, probability is determined by evaluating the likelihood of occurrence (frequent, likely, occasional, seldom, or unlikely). Finally, the severity and probability are combined to assign a risk assessment code, which indicates the risk level and helps visualize criticality. Systematically applying these principles and processes enables users to make informed decisions that balance mission objectives with safety.
Proposed Framework for ORM in Dermatology Surgery
Current risk mitigation in dermatologic surgery includes strict medication oversight, sterilization protocols, and photography to prevent wrong-site surgeries. Preoperative risk assessment through conducting a thorough patient history is vital, considering factors such as pregnancy, allergies, bleeding history, cardiac devices, and keloid propensity, all of which impact surgical outcomes.3-5 After gathering the patient’s history, dermatologists determine appropriateness for surgery and its inherent risks, typically via an informed consent process outlining the diagnosis and procedure purpose as well as a list of risks, benefits, and alternatives, including forgoing treatment.
Importantly, the standard process for dermatologic risk evaluation often lacks a comprehensive systematic approach seen in other higher-risk surgical fields. For example, general surgeons frequently utilize risk assessment calculators such as the one developed by the American College of Surgeons’ National Surgical Quality Improvement Program to estimate surgical complications.6 While specific guidelines exist for evaluating factors such as hypertension or anticoagulant use, no single tool synthesizes all patient risk factors for a unified assessment. Therefore, we propose integrating ORM as a structured decision-making process that offers a more consistent means for dermatologists to evaluate, synthesize, categorize, and present risks to patients. Our proposed process includes translating military mishap severity into a framework that helps patients better understand decisions about their health care when using ORM (eTable 1). The proposed process also provides dermatologists with a systematic, proactive, and iterative approach to assessing risks that allows them to consistently qualify medical decisions (eTable 2).


Patients often struggle to understand surgical risk severity, including overestimating the risks of routine minor procedures or underestimating the risks of more intensive procedures.7,8 Incorporating ORM into patient communication mirrors the provider’s process but uses patient-friendly terminology—it is discussion based and integrates patient preferences and tolerances (eTable 2). These steps often occur informally in dermatologic counseling; however, an organized structured approach, especially using a visual aid such as a risk assessment matrix, enhances patient comprehension, recall, and satisfaction.9
Practical Scenarios
Integrating ORM into dermatologic surgery is a proactive iterative process for both provider decision-making and patient communication. Leveraging a risk assessment matrix as a visual aid allows for clear identification, evaluation, and mitigation of hazards, fostering collaborative choices with regard to the treatment approach. Here we provide 2 case scenarios highlighting how ORM and the risk assessment matrix can be used in the management of a complex patient with a lesion in a high-risk location as well as to address patient anxiety and comorbidities. It is important to note that the way the matrices are completed in the examples provided may differ compared to other providers. The purpose of ORM is not to dictate risk categories but to serve as a tool for providers to take their own experiences and knowledge of the patient to guide their decision-making and counseling processes.
Case Scenario 1—An elderly man with a history of diabetes, cardiovascular accident, coronary artery bypass grafting, and multiple squamous cell carcinoma excisions presents for evaluation of a 1-cm squamous cell carcinoma in situ on the left leg. His current medications include an anticoagulant and antihypertensives.
In this scenario, the provider would apply ORM by identifying and assessing hazards, making risk decisions, implementing controls, and supervising care.
General hazards for excision on the leg include bleeding, infection, scarring, pain, delayed healing, activity limitations, and possible further procedures. Before the visit, the provider should prepare baseline risk matrices for 2 potential treatment options: wide local excision and electrodessication and curettage. For example, surgical bleeding may be assessed as negligible severity and almost certain probability for a general excision.
Next, the provider would incorporate the patient’s unique history in the risk matrices (eFigures 1 and 2). The patient’s use of an anticoagulant indicates a bleeding risk; therefore, the provider may shift the severity to minimal clinical concern, understanding the need for enhanced perioperative management. The history of diabetes also has a considerable impact on wound healing, so the provider might elevate the probability of delayed wound healing from rare to unlikely and the severity from moderate to severe. The prior cardiovascular accident also raises concerns about mobility and activity limitations during recovery, which could be escalated from minimal to moderate clinical concern if postoperative limitations on ambulation increase the risk for new clots. Based on this internal assessment, the provider identifies which risks are elevated and require further attention and discussion with the patient, helping tailor the counseling approach and potential treatment plan. The provider should begin to consider initial control measures such as coordinating anticoagulant management, ensuring diabetes is well controlled, and planning for postoperative ambulation support.
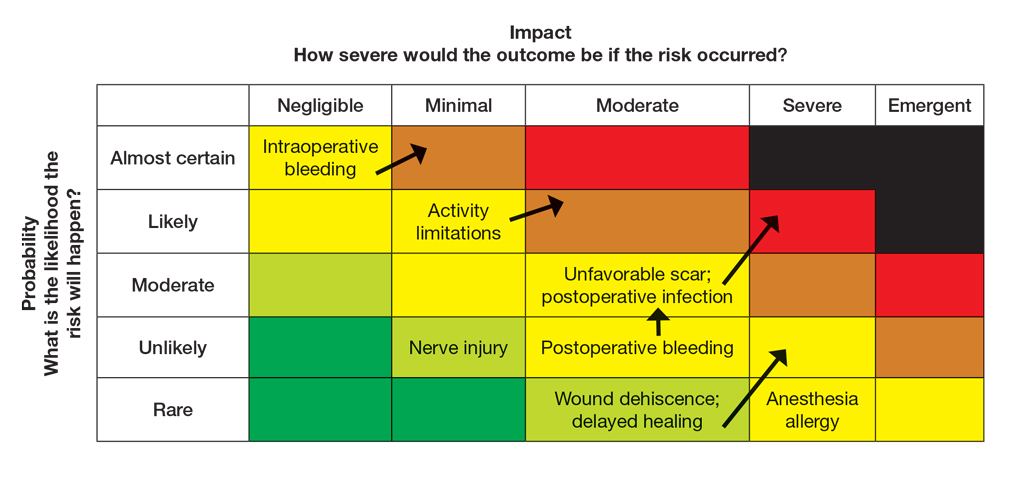
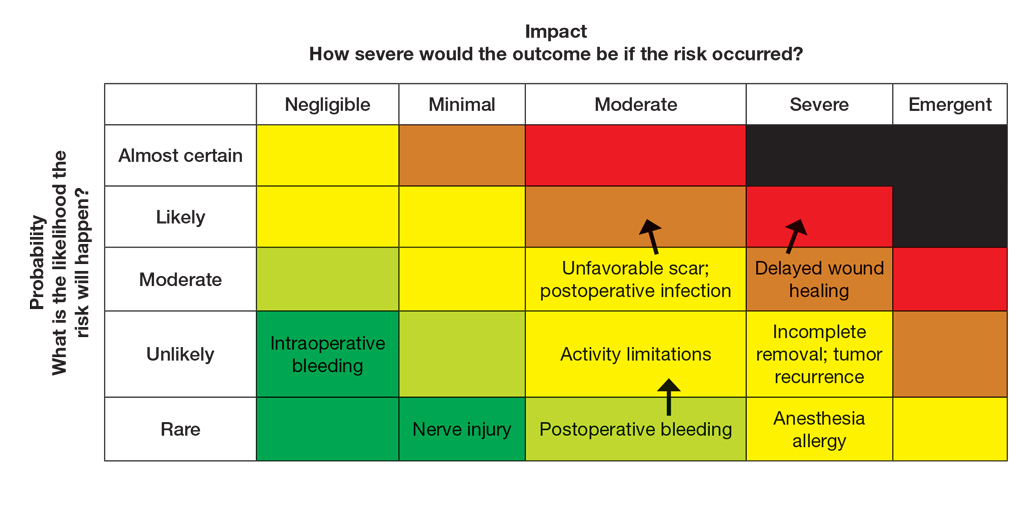
Once the provider has conducted the internal assessment, the ORM matrices become powerful tools for shared decision-making with the patient. The provider can walk the patient through the procedures and their common risks and then explain how their individual situation modifies the risks. The visual and explicit upgrade on the matrices allows the patient to clearly see how unique factors influence their personal risk profile, moving beyond a generic list of complications. The provider then should engage the patient in a discussion about their risk tolerance, which is crucial for mutual agreement on whether to proceed with treatment and, if so, which procedure is most appropriate given the patient’s comfort level with their individualized risk profile. Then the provider should reinforce the proactive steps planned to mitigate the identified risks to provide assurance and reinforce the collaborative approach to safety.
Finally, throughout the preoperative and postoperative phases, the provider should continuously monitor the patient’s condition and the effectiveness of the control measures, adjusting the plan as needed.
In this scenario, both the provider and the patient participated in the risk assessment, with the provider completing the assessment before the visit and presenting it to the patient or performing the assessment in real time with the patient present to explain the reasoning behind assignment of risk based on each procedure and the patient’s unique risk factors.
Case Scenario 2—A 38-year-old woman with a history of hypertension and procedural anxiety presents for evaluation of a biopsy-proven basal cell carcinoma on the nasal ala. The patient is taking diltiazem for hypertension and is compliant with her medication. Her blood pressure at the current visit is 148/96 mm Hg, which she attributes to white coat syndrome. Mohs micrographic surgery generally is the gold standard treatment for this case.
The provider’s ORM process, conducted either before or in real time during the visit, would begin with identification and assessment of the hazards. For Mohs surgery on the nasal ala, common hazards would include scarring, pain, infection, bleeding, and potential cosmetic distortion. Unique to this patient are the procedural anxiety and hypertension.
To populate the risk assessment matrix (eFigure 3), the provider would first map the baseline risks of Mohs surgery, which include considerable scarring as a moderate clinical concern but a seldom probability. Because the patient’s procedural anxiety directly increases the probability of intraoperative distress or elevated blood pressure during the procedure, the provider might assess patient distress/anxiety as a moderate clinical concern with a likely probability. While the patient’s blood pressure is controlled, the white coat syndrome raises the probability of hypertensive urgency/emergency during surgery; this might be elevated from unlikely to occasional or likely probability, and severity might increase from minimal to moderate due to its potential impact on procedural safety. The provider should consider strategies to address these elevated risks during the consultation. Then, as part of preprocedure planning, the provider should consider discussing anxiolytics, emphasizing medication compliance, and ensuring a calm environment for the patient’s surgery.
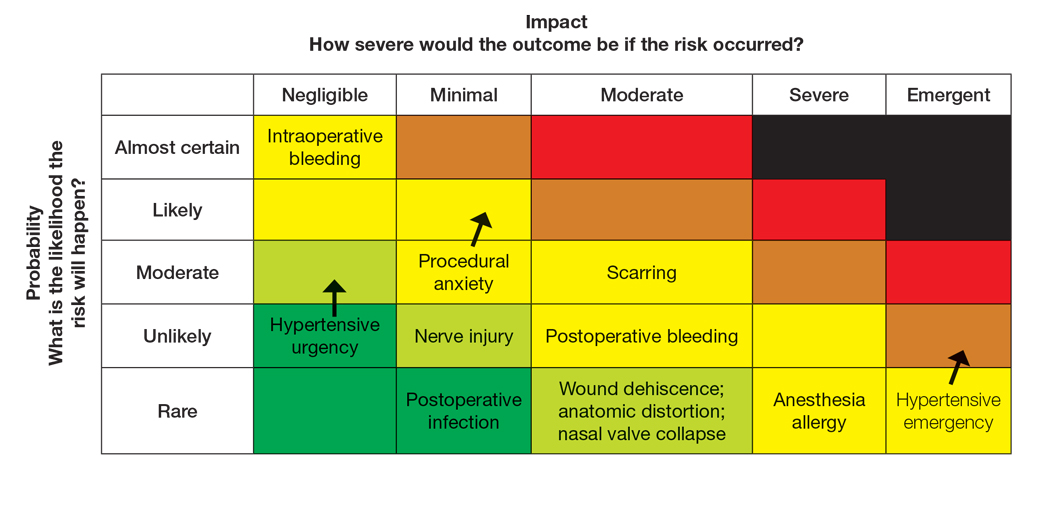
For this patient, the risk assessment matrix becomes a powerful tool to address fears and proactively manage her unique risk factors. To start the counseling process, the provider should explain the procedure, its benefits, and potential adverse effects. Then, the patient’s individualized risks can be visualized using the matrix, which also is an opportunity for reassurance, as it can alleviate patient fears by contextualizing rare but impactful outcomes.9
Now the provider can assess the patient’s risk tolerance. This discussion ensures that the patient’s comfort level and preferences are central to the treatment decision, even for a gold-standard procedure such as Mohs surgery. By listening and responding to the patient’s input, the provider can build trust and discuss strategies that can help control for some risk factors.
Finally, the provider would re-evaluate throughout the procedure by continuously monitoring the patient’s anxiety and vital signs. The provider should also be ready to adjust pain management or employ anxiety-reduction techniques.
Final Thoughts
Reviewing the risk assessment matrix can be an effective way to nonjudgmentally discuss a patient’s unique risk factors and provide a complete understanding of the planned treatment or procedure. It conveys to the patient that, as the provider, you are taking their health seriously when considering treatment options and can be a means to build patient rapport and trust. This approach mirrors risk communication strategies long employed in military operational planning, where transparency and structured risk evaluation are essential to maintaining mission readiness and unit cohesion.
- The OR Society. The history of OR. The OR Society. Published 2023.
- Naval Postgraduate School. ORM: operational risk management. Accessed September 12, 2025. https://nps.edu/web/safety/orm
- Smith C, Srivastava D, Nijhawan RI. Optimizing patient safety in dermatologic surgery. Dermatol Clin. 2019;37:319-328.
- Minkis K, Whittington A, Alam M. Dermatologic surgery emergencies: complications caused by systemic reactions, high-energy systems, and trauma. J Am Acad Dermatol. 2016;75:265-284.
- Pomerantz RG, Lee DA, Siegel DM. Risk assessment in surgical patients: balancing iatrogenic risks and benefits. Clin Dermatol. 2011;29:669-677.
- Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aid and informed consent tool for patients and surgeons. J Am Coll Surgeons. 2013;217:833-842.
- Lloyd AJ. The extent of patients’ understanding of the risk of treatments. BMJ Qual Saf. 2001;10:i14-i18.
- Falagas ME, Korbila IP, Giannopoulou KP, et al. Informed consent: how much and what do patients understand? Am J Surg. 2009;198:420-435.
- Cohen SM, Baimas-George M, Ponce C, et al. Is a picture worth a thousand words? a scoping review of the impact of visual aids on patients undergoing surgery. J Surg Educ. 2024;81:1276-1292.
Operational risk management (ORM) refers to the systematic identification and assessment of daily operational risks within an organization designed to mitigate negative financial, reputational, and safety outcomes while maximizing efficiency and achievement of objectives.1 Operational risk management is indispensable to modern military operations, optimizing mission readiness while minimizing complications and personnel morbidity. Application of ORM in medicine holds considerable promise due to the emphasis on precise and efficient decision-making in high-stakes environments, where the margin for error is minimal. In this article, we propose integrating ORM principles into dermatologic surgery to enhance patient-centered care through improved counseling, risk assessment, and procedural outcomes.
Principles and Processes of ORM
The ORM framework is built on 4 fundamental principles: accept risk when benefits outweigh the cost, accept no unnecessary risk, anticipate and manage risk by planning, and make risk decisions at the right level.2 These principles form the foundation of the ORM’s systematic 5-step approach to identify hazards, assess hazards, make risk decisions, implement controls, and supervise. Key to the ORM process is the use of risk assessment codes and the risk assessment matrix to quantify and prioritize risks. Risk assessment codes are numerical values assigned to hazards based on their assessed severity and probability. The risk assessment matrix is a tool that plots the severity of a hazard against its probability. By locating a hazard on the matrix, users can visualize its risk level in terms of severity and probability. Building and using the risk assessment matrix begins with determining severity by assessing the potential impact of a hazard and categorizing it into levels (catastrophic, critical, moderate, or negligible). Next, probability is determined by evaluating the likelihood of occurrence (frequent, likely, occasional, seldom, or unlikely). Finally, the severity and probability are combined to assign a risk assessment code, which indicates the risk level and helps visualize criticality. Systematically applying these principles and processes enables users to make informed decisions that balance mission objectives with safety.
Proposed Framework for ORM in Dermatology Surgery
Current risk mitigation in dermatologic surgery includes strict medication oversight, sterilization protocols, and photography to prevent wrong-site surgeries. Preoperative risk assessment through conducting a thorough patient history is vital, considering factors such as pregnancy, allergies, bleeding history, cardiac devices, and keloid propensity, all of which impact surgical outcomes.3-5 After gathering the patient’s history, dermatologists determine appropriateness for surgery and its inherent risks, typically via an informed consent process outlining the diagnosis and procedure purpose as well as a list of risks, benefits, and alternatives, including forgoing treatment.
Importantly, the standard process for dermatologic risk evaluation often lacks a comprehensive systematic approach seen in other higher-risk surgical fields. For example, general surgeons frequently utilize risk assessment calculators such as the one developed by the American College of Surgeons’ National Surgical Quality Improvement Program to estimate surgical complications.6 While specific guidelines exist for evaluating factors such as hypertension or anticoagulant use, no single tool synthesizes all patient risk factors for a unified assessment. Therefore, we propose integrating ORM as a structured decision-making process that offers a more consistent means for dermatologists to evaluate, synthesize, categorize, and present risks to patients. Our proposed process includes translating military mishap severity into a framework that helps patients better understand decisions about their health care when using ORM (eTable 1). The proposed process also provides dermatologists with a systematic, proactive, and iterative approach to assessing risks that allows them to consistently qualify medical decisions (eTable 2).
Patients often struggle to understand surgical risk severity, including overestimating the risks of routine minor procedures or underestimating the risks of more intensive procedures.7,8 Incorporating ORM into patient communication mirrors the provider’s process but uses patient-friendly terminology—it is discussion based and integrates patient preferences and tolerances (eTable 2). These steps often occur informally in dermatologic counseling; however, an organized structured approach, especially using a visual aid such as a risk assessment matrix, enhances patient comprehension, recall, and satisfaction.9
Practical Scenarios
Integrating ORM into dermatologic surgery is a proactive iterative process for both provider decision-making and patient communication. Leveraging a risk assessment matrix as a visual aid allows for clear identification, evaluation, and mitigation of hazards, fostering collaborative choices with regard to the treatment approach. Here we provide 2 case scenarios highlighting how ORM and the risk assessment matrix can be used in the management of a complex patient with a lesion in a high-risk location as well as to address patient anxiety and comorbidities. It is important to note that the way the matrices are completed in the examples provided may differ compared to other providers. The purpose of ORM is not to dictate risk categories but to serve as a tool for providers to take their own experiences and knowledge of the patient to guide their decision-making and counseling processes.
Case Scenario 1—An elderly man with a history of diabetes, cardiovascular accident, coronary artery bypass grafting, and multiple squamous cell carcinoma excisions presents for evaluation of a 1-cm squamous cell carcinoma in situ on the left leg. His current medications include an anticoagulant and antihypertensives.
In this scenario, the provider would apply ORM by identifying and assessing hazards, making risk decisions, implementing controls, and supervising care.
General hazards for excision on the leg include bleeding, infection, scarring, pain, delayed healing, activity limitations, and possible further procedures. Before the visit, the provider should prepare baseline risk matrices for 2 potential treatment options: wide local excision and electrodessication and curettage. For example, surgical bleeding may be assessed as negligible severity and almost certain probability for a general excision.
Next, the provider would incorporate the patient’s unique history in the risk matrices (eFigures 1 and 2). The patient’s use of an anticoagulant indicates a bleeding risk; therefore, the provider may shift the severity to minimal clinical concern, understanding the need for enhanced perioperative management. The history of diabetes also has a considerable impact on wound healing, so the provider might elevate the probability of delayed wound healing from rare to unlikely and the severity from moderate to severe. The prior cardiovascular accident also raises concerns about mobility and activity limitations during recovery, which could be escalated from minimal to moderate clinical concern if postoperative limitations on ambulation increase the risk for new clots. Based on this internal assessment, the provider identifies which risks are elevated and require further attention and discussion with the patient, helping tailor the counseling approach and potential treatment plan. The provider should begin to consider initial control measures such as coordinating anticoagulant management, ensuring diabetes is well controlled, and planning for postoperative ambulation support.
Once the provider has conducted the internal assessment, the ORM matrices become powerful tools for shared decision-making with the patient. The provider can walk the patient through the procedures and their common risks and then explain how their individual situation modifies the risks. The visual and explicit upgrade on the matrices allows the patient to clearly see how unique factors influence their personal risk profile, moving beyond a generic list of complications. The provider then should engage the patient in a discussion about their risk tolerance, which is crucial for mutual agreement on whether to proceed with treatment and, if so, which procedure is most appropriate given the patient’s comfort level with their individualized risk profile. Then the provider should reinforce the proactive steps planned to mitigate the identified risks to provide assurance and reinforce the collaborative approach to safety.
Finally, throughout the preoperative and postoperative phases, the provider should continuously monitor the patient’s condition and the effectiveness of the control measures, adjusting the plan as needed.
In this scenario, both the provider and the patient participated in the risk assessment, with the provider completing the assessment before the visit and presenting it to the patient or performing the assessment in real time with the patient present to explain the reasoning behind assignment of risk based on each procedure and the patient’s unique risk factors.
Case Scenario 2—A 38-year-old woman with a history of hypertension and procedural anxiety presents for evaluation of a biopsy-proven basal cell carcinoma on the nasal ala. The patient is taking diltiazem for hypertension and is compliant with her medication. Her blood pressure at the current visit is 148/96 mm Hg, which she attributes to white coat syndrome. Mohs micrographic surgery generally is the gold standard treatment for this case.
The provider’s ORM process, conducted either before or in real time during the visit, would begin with identification and assessment of the hazards. For Mohs surgery on the nasal ala, common hazards would include scarring, pain, infection, bleeding, and potential cosmetic distortion. Unique to this patient are the procedural anxiety and hypertension.
To populate the risk assessment matrix (eFigure 3), the provider would first map the baseline risks of Mohs surgery, which include considerable scarring as a moderate clinical concern but a seldom probability. Because the patient’s procedural anxiety directly increases the probability of intraoperative distress or elevated blood pressure during the procedure, the provider might assess patient distress/anxiety as a moderate clinical concern with a likely probability. While the patient’s blood pressure is controlled, the white coat syndrome raises the probability of hypertensive urgency/emergency during surgery; this might be elevated from unlikely to occasional or likely probability, and severity might increase from minimal to moderate due to its potential impact on procedural safety. The provider should consider strategies to address these elevated risks during the consultation. Then, as part of preprocedure planning, the provider should consider discussing anxiolytics, emphasizing medication compliance, and ensuring a calm environment for the patient’s surgery.
For this patient, the risk assessment matrix becomes a powerful tool to address fears and proactively manage her unique risk factors. To start the counseling process, the provider should explain the procedure, its benefits, and potential adverse effects. Then, the patient’s individualized risks can be visualized using the matrix, which also is an opportunity for reassurance, as it can alleviate patient fears by contextualizing rare but impactful outcomes.9
Now the provider can assess the patient’s risk tolerance. This discussion ensures that the patient’s comfort level and preferences are central to the treatment decision, even for a gold-standard procedure such as Mohs surgery. By listening and responding to the patient’s input, the provider can build trust and discuss strategies that can help control for some risk factors.
Finally, the provider would re-evaluate throughout the procedure by continuously monitoring the patient’s anxiety and vital signs. The provider should also be ready to adjust pain management or employ anxiety-reduction techniques.
Final Thoughts
Reviewing the risk assessment matrix can be an effective way to nonjudgmentally discuss a patient’s unique risk factors and provide a complete understanding of the planned treatment or procedure. It conveys to the patient that, as the provider, you are taking their health seriously when considering treatment options and can be a means to build patient rapport and trust. This approach mirrors risk communication strategies long employed in military operational planning, where transparency and structured risk evaluation are essential to maintaining mission readiness and unit cohesion.
Operational risk management (ORM) refers to the systematic identification and assessment of daily operational risks within an organization designed to mitigate negative financial, reputational, and safety outcomes while maximizing efficiency and achievement of objectives.1 Operational risk management is indispensable to modern military operations, optimizing mission readiness while minimizing complications and personnel morbidity. Application of ORM in medicine holds considerable promise due to the emphasis on precise and efficient decision-making in high-stakes environments, where the margin for error is minimal. In this article, we propose integrating ORM principles into dermatologic surgery to enhance patient-centered care through improved counseling, risk assessment, and procedural outcomes.
Principles and Processes of ORM
The ORM framework is built on 4 fundamental principles: accept risk when benefits outweigh the cost, accept no unnecessary risk, anticipate and manage risk by planning, and make risk decisions at the right level.2 These principles form the foundation of the ORM’s systematic 5-step approach to identify hazards, assess hazards, make risk decisions, implement controls, and supervise. Key to the ORM process is the use of risk assessment codes and the risk assessment matrix to quantify and prioritize risks. Risk assessment codes are numerical values assigned to hazards based on their assessed severity and probability. The risk assessment matrix is a tool that plots the severity of a hazard against its probability. By locating a hazard on the matrix, users can visualize its risk level in terms of severity and probability. Building and using the risk assessment matrix begins with determining severity by assessing the potential impact of a hazard and categorizing it into levels (catastrophic, critical, moderate, or negligible). Next, probability is determined by evaluating the likelihood of occurrence (frequent, likely, occasional, seldom, or unlikely). Finally, the severity and probability are combined to assign a risk assessment code, which indicates the risk level and helps visualize criticality. Systematically applying these principles and processes enables users to make informed decisions that balance mission objectives with safety.
Proposed Framework for ORM in Dermatology Surgery
Current risk mitigation in dermatologic surgery includes strict medication oversight, sterilization protocols, and photography to prevent wrong-site surgeries. Preoperative risk assessment through conducting a thorough patient history is vital, considering factors such as pregnancy, allergies, bleeding history, cardiac devices, and keloid propensity, all of which impact surgical outcomes.3-5 After gathering the patient’s history, dermatologists determine appropriateness for surgery and its inherent risks, typically via an informed consent process outlining the diagnosis and procedure purpose as well as a list of risks, benefits, and alternatives, including forgoing treatment.
Importantly, the standard process for dermatologic risk evaluation often lacks a comprehensive systematic approach seen in other higher-risk surgical fields. For example, general surgeons frequently utilize risk assessment calculators such as the one developed by the American College of Surgeons’ National Surgical Quality Improvement Program to estimate surgical complications.6 While specific guidelines exist for evaluating factors such as hypertension or anticoagulant use, no single tool synthesizes all patient risk factors for a unified assessment. Therefore, we propose integrating ORM as a structured decision-making process that offers a more consistent means for dermatologists to evaluate, synthesize, categorize, and present risks to patients. Our proposed process includes translating military mishap severity into a framework that helps patients better understand decisions about their health care when using ORM (eTable 1). The proposed process also provides dermatologists with a systematic, proactive, and iterative approach to assessing risks that allows them to consistently qualify medical decisions (eTable 2).
Patients often struggle to understand surgical risk severity, including overestimating the risks of routine minor procedures or underestimating the risks of more intensive procedures.7,8 Incorporating ORM into patient communication mirrors the provider’s process but uses patient-friendly terminology—it is discussion based and integrates patient preferences and tolerances (eTable 2). These steps often occur informally in dermatologic counseling; however, an organized structured approach, especially using a visual aid such as a risk assessment matrix, enhances patient comprehension, recall, and satisfaction.9
Practical Scenarios
Integrating ORM into dermatologic surgery is a proactive iterative process for both provider decision-making and patient communication. Leveraging a risk assessment matrix as a visual aid allows for clear identification, evaluation, and mitigation of hazards, fostering collaborative choices with regard to the treatment approach. Here we provide 2 case scenarios highlighting how ORM and the risk assessment matrix can be used in the management of a complex patient with a lesion in a high-risk location as well as to address patient anxiety and comorbidities. It is important to note that the way the matrices are completed in the examples provided may differ compared to other providers. The purpose of ORM is not to dictate risk categories but to serve as a tool for providers to take their own experiences and knowledge of the patient to guide their decision-making and counseling processes.
Case Scenario 1—An elderly man with a history of diabetes, cardiovascular accident, coronary artery bypass grafting, and multiple squamous cell carcinoma excisions presents for evaluation of a 1-cm squamous cell carcinoma in situ on the left leg. His current medications include an anticoagulant and antihypertensives.
In this scenario, the provider would apply ORM by identifying and assessing hazards, making risk decisions, implementing controls, and supervising care.
General hazards for excision on the leg include bleeding, infection, scarring, pain, delayed healing, activity limitations, and possible further procedures. Before the visit, the provider should prepare baseline risk matrices for 2 potential treatment options: wide local excision and electrodessication and curettage. For example, surgical bleeding may be assessed as negligible severity and almost certain probability for a general excision.
Next, the provider would incorporate the patient’s unique history in the risk matrices (eFigures 1 and 2). The patient’s use of an anticoagulant indicates a bleeding risk; therefore, the provider may shift the severity to minimal clinical concern, understanding the need for enhanced perioperative management. The history of diabetes also has a considerable impact on wound healing, so the provider might elevate the probability of delayed wound healing from rare to unlikely and the severity from moderate to severe. The prior cardiovascular accident also raises concerns about mobility and activity limitations during recovery, which could be escalated from minimal to moderate clinical concern if postoperative limitations on ambulation increase the risk for new clots. Based on this internal assessment, the provider identifies which risks are elevated and require further attention and discussion with the patient, helping tailor the counseling approach and potential treatment plan. The provider should begin to consider initial control measures such as coordinating anticoagulant management, ensuring diabetes is well controlled, and planning for postoperative ambulation support.
Once the provider has conducted the internal assessment, the ORM matrices become powerful tools for shared decision-making with the patient. The provider can walk the patient through the procedures and their common risks and then explain how their individual situation modifies the risks. The visual and explicit upgrade on the matrices allows the patient to clearly see how unique factors influence their personal risk profile, moving beyond a generic list of complications. The provider then should engage the patient in a discussion about their risk tolerance, which is crucial for mutual agreement on whether to proceed with treatment and, if so, which procedure is most appropriate given the patient’s comfort level with their individualized risk profile. Then the provider should reinforce the proactive steps planned to mitigate the identified risks to provide assurance and reinforce the collaborative approach to safety.
Finally, throughout the preoperative and postoperative phases, the provider should continuously monitor the patient’s condition and the effectiveness of the control measures, adjusting the plan as needed.
In this scenario, both the provider and the patient participated in the risk assessment, with the provider completing the assessment before the visit and presenting it to the patient or performing the assessment in real time with the patient present to explain the reasoning behind assignment of risk based on each procedure and the patient’s unique risk factors.
Case Scenario 2—A 38-year-old woman with a history of hypertension and procedural anxiety presents for evaluation of a biopsy-proven basal cell carcinoma on the nasal ala. The patient is taking diltiazem for hypertension and is compliant with her medication. Her blood pressure at the current visit is 148/96 mm Hg, which she attributes to white coat syndrome. Mohs micrographic surgery generally is the gold standard treatment for this case.
The provider’s ORM process, conducted either before or in real time during the visit, would begin with identification and assessment of the hazards. For Mohs surgery on the nasal ala, common hazards would include scarring, pain, infection, bleeding, and potential cosmetic distortion. Unique to this patient are the procedural anxiety and hypertension.
To populate the risk assessment matrix (eFigure 3), the provider would first map the baseline risks of Mohs surgery, which include considerable scarring as a moderate clinical concern but a seldom probability. Because the patient’s procedural anxiety directly increases the probability of intraoperative distress or elevated blood pressure during the procedure, the provider might assess patient distress/anxiety as a moderate clinical concern with a likely probability. While the patient’s blood pressure is controlled, the white coat syndrome raises the probability of hypertensive urgency/emergency during surgery; this might be elevated from unlikely to occasional or likely probability, and severity might increase from minimal to moderate due to its potential impact on procedural safety. The provider should consider strategies to address these elevated risks during the consultation. Then, as part of preprocedure planning, the provider should consider discussing anxiolytics, emphasizing medication compliance, and ensuring a calm environment for the patient’s surgery.
For this patient, the risk assessment matrix becomes a powerful tool to address fears and proactively manage her unique risk factors. To start the counseling process, the provider should explain the procedure, its benefits, and potential adverse effects. Then, the patient’s individualized risks can be visualized using the matrix, which also is an opportunity for reassurance, as it can alleviate patient fears by contextualizing rare but impactful outcomes.9
Now the provider can assess the patient’s risk tolerance. This discussion ensures that the patient’s comfort level and preferences are central to the treatment decision, even for a gold-standard procedure such as Mohs surgery. By listening and responding to the patient’s input, the provider can build trust and discuss strategies that can help control for some risk factors.
Finally, the provider would re-evaluate throughout the procedure by continuously monitoring the patient’s anxiety and vital signs. The provider should also be ready to adjust pain management or employ anxiety-reduction techniques.
Final Thoughts
Reviewing the risk assessment matrix can be an effective way to nonjudgmentally discuss a patient’s unique risk factors and provide a complete understanding of the planned treatment or procedure. It conveys to the patient that, as the provider, you are taking their health seriously when considering treatment options and can be a means to build patient rapport and trust. This approach mirrors risk communication strategies long employed in military operational planning, where transparency and structured risk evaluation are essential to maintaining mission readiness and unit cohesion.
- The OR Society. The history of OR. The OR Society. Published 2023.
- Naval Postgraduate School. ORM: operational risk management. Accessed September 12, 2025. https://nps.edu/web/safety/orm
- Smith C, Srivastava D, Nijhawan RI. Optimizing patient safety in dermatologic surgery. Dermatol Clin. 2019;37:319-328.
- Minkis K, Whittington A, Alam M. Dermatologic surgery emergencies: complications caused by systemic reactions, high-energy systems, and trauma. J Am Acad Dermatol. 2016;75:265-284.
- Pomerantz RG, Lee DA, Siegel DM. Risk assessment in surgical patients: balancing iatrogenic risks and benefits. Clin Dermatol. 2011;29:669-677.
- Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aid and informed consent tool for patients and surgeons. J Am Coll Surgeons. 2013;217:833-842.
- Lloyd AJ. The extent of patients’ understanding of the risk of treatments. BMJ Qual Saf. 2001;10:i14-i18.
- Falagas ME, Korbila IP, Giannopoulou KP, et al. Informed consent: how much and what do patients understand? Am J Surg. 2009;198:420-435.
- Cohen SM, Baimas-George M, Ponce C, et al. Is a picture worth a thousand words? a scoping review of the impact of visual aids on patients undergoing surgery. J Surg Educ. 2024;81:1276-1292.
- The OR Society. The history of OR. The OR Society. Published 2023.
- Naval Postgraduate School. ORM: operational risk management. Accessed September 12, 2025. https://nps.edu/web/safety/orm
- Smith C, Srivastava D, Nijhawan RI. Optimizing patient safety in dermatologic surgery. Dermatol Clin. 2019;37:319-328.
- Minkis K, Whittington A, Alam M. Dermatologic surgery emergencies: complications caused by systemic reactions, high-energy systems, and trauma. J Am Acad Dermatol. 2016;75:265-284.
- Pomerantz RG, Lee DA, Siegel DM. Risk assessment in surgical patients: balancing iatrogenic risks and benefits. Clin Dermatol. 2011;29:669-677.
- Bilimoria KY, Liu Y, Paruch JL, et al. Development and evaluation of the universal ACS NSQIP surgical risk calculator: a decision aid and informed consent tool for patients and surgeons. J Am Coll Surgeons. 2013;217:833-842.
- Lloyd AJ. The extent of patients’ understanding of the risk of treatments. BMJ Qual Saf. 2001;10:i14-i18.
- Falagas ME, Korbila IP, Giannopoulou KP, et al. Informed consent: how much and what do patients understand? Am J Surg. 2009;198:420-435.
- Cohen SM, Baimas-George M, Ponce C, et al. Is a picture worth a thousand words? a scoping review of the impact of visual aids on patients undergoing surgery. J Surg Educ. 2024;81:1276-1292.
Operational Risk Management in Dermatologic Procedures
Operational Risk Management in Dermatologic Procedures

Dermoscopic Documentation of a No-see-um Bite
Dermoscopic Documentation of a No-see-um Bite
Biting midges, commonly known as no-see-ums, are true flies (order Diptera) and members of the Ceratopogonidae family. Regionally, they are known as punkies in the Northeast, pinyon gnats in the Southwest, moose flies in Canada, and sand gnats in Georgia, among other names.1 There are 6206 species found worldwide except for Antarctica.2 The 3 genera of greatest importance to human and livestock health in the United States are Culicoides, Leptoconops, and Forcipomyia.1 Forty-seven species of the genus Culicoides are known to be present in Florida.3 Species belonging to the genus Leptoconops also are present in coastal areas of southeast Florida as well as in the tropics, subtropics, and Caribbean.3 In the United States, biting midges primarily are a nuisance; the major medical issue associated with Culicoides insects are allergic reactions to their bites. Even though no-see-ums are not known to transmit disease in humans, they have an impact on other animal species in the United States as biting pests and vectors of disease-causing pathogens.1 Biting midges pose quite a nuisance for the proper enjoyment of outdoor spaces in the southeastern United States.
Characteristics
Morphologically, no-see-ums are gray flies measuring 1 to 3 mm in length (eFigure 1). Adults have 2 wings with distinctive patterns, large compound eyes, a thorax that extends slightly over the head, an abdomen with 9 segments, and antennae with 15 segments (eFigure 2).1,3,4 Females have modified mouth parts including mandibles that lacerate the skin during feeding, which is mainly on blood from vertebrate hosts (primarily mammals but also birds, reptiles, and amphibians).1,4 They also can feed on invertebrate hosts. Both male and female no-see-ums feed on nectar, but adult females require a blood meal to develop their eggs.2 Their life cycle progresses in stages from egg to larva to pupa to adult. Larval habitats include salt marshes, swamps, shores of streams and ponds, water-holding plants, rotting fruit, and saturated wood- and manure-enriched soil. Adults can live 2 to 7 weeks. They are weak fliers, particularly in windy conditions.1


In Florida, no-see-ums are more active during the rainy months of May to October but are active year-round in the southeastern United States and the Gulf Coast from Florida to West Texas. They are active throughout the United States in the warmer months of June and July.5 Their peak feeding activity occurs at dawn and dusk, but different species of biting midges such as Leptoconops and Culicoides also can feed during daylight hours and at night, respectively.1,6,7
Case Report
One of the authors (M.J.S.), a healthy 54-year-old man with no remarkable medical history or current use of medications, documented the natural progression of a no-see-um bite by sitting in an outdoor Florida space at 8:00

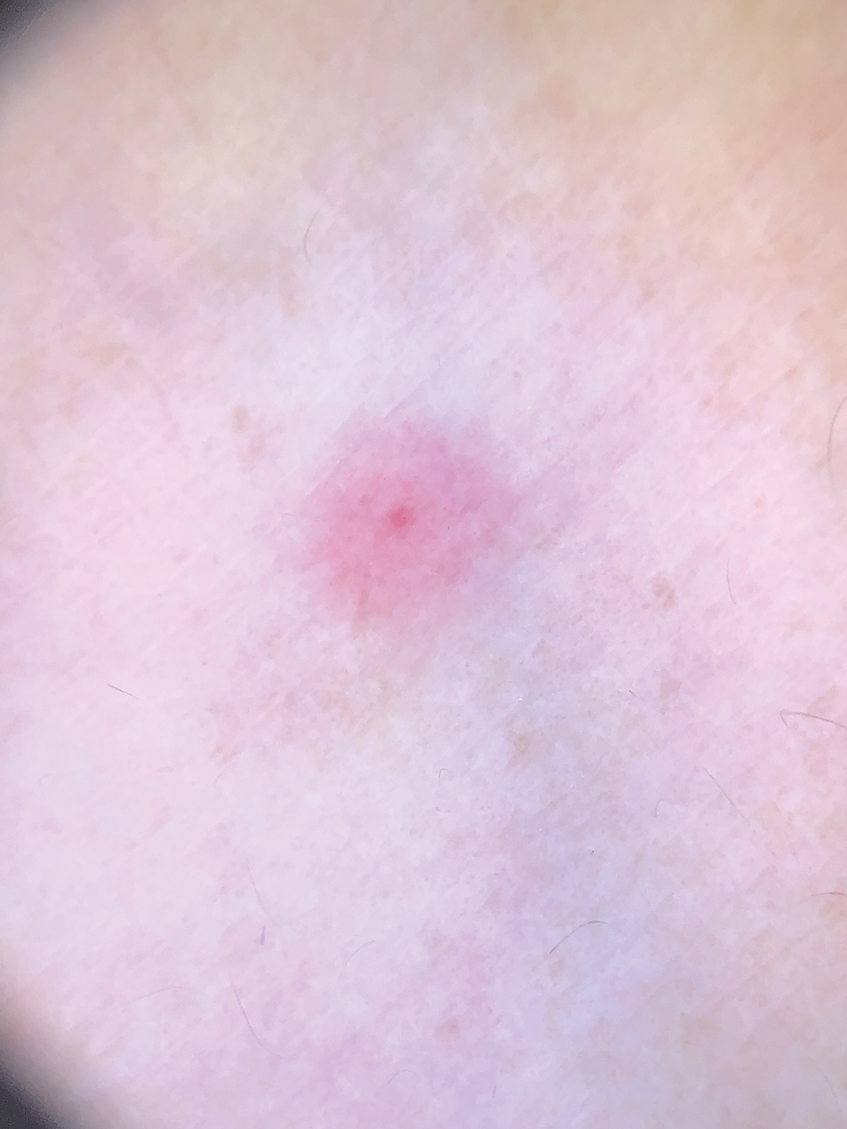
Clinical Manifestations
Although no-see-ums are not known to transmit disease in the United States, they are important biting pests that can affect tourism and prevent enjoyment of outdoor spaces and activities.2 The bite reactions on the host can range from wheal-like lesions to papules measuring 2 to 3 mm (at times with overlying vesicles) to nodules up to 1 cm in diameter.8 In our reported case, the small wheals disappeared within hours, but pruritic papules have been described to last from weeks to months. Published histopathologic correlation of biopsied indurated papules within 3 days of bite occurrence have revealed a superficial infiltrate composed of lymphocytes and histiocytes, while eosinophils were found in the deeper dermis and subcutaneous fat. Within 2 weeks, as the lesions aged, the infiltrate contained a smaller percentage of eosinophils and predominantly was present in only the superficial dermis.8 Delayed-type hypersensitivity reactions including pustules and bullous lesions also have been described.9,10 Host immune reaction to the saliva introduced during the bite dictates the severity of the response, and lesions may become secondarily infected due to scratching.11
Management Recommendations
Management consists of cleaning the bite site with soap and water to prevent infection, applying cold compresses or ice packs to relieve the intense itch, and avoiding scratching.11 Application of over-the-counter calamine lotion or hydrocortisone cream can relieve itch, and mid- to high-potency topical corticosteroids also can be prescribed for 1 to 2 weeks for more intense bite reactions in conjunction with oral antihistamines. Topical or oral antibiotics may be indicated if redness and swelling progress at the bite site or if breaks in the skin become secondarily infected.
Final Thoughts
Because of the wide-ranging habitats of no-see-ums, eradication programs using insecticides have been inefficient or environmentally suboptimal. Emptying all standing water in outdoor spaces will reduce the number of no-see-ums. Avoidance of the outdoors at dawn and dusk when no-see-ums are most active is helpful, as well as protecting exposed skin by wearing long-sleeved shirts and long pants when outside. Insect repellents containing DEET (N-N-diethyl-meta-toluamide) or picaridin can offer additional protection on the remaining exposed skin. Oil of lemon eucalyptus, or active compound p-menthane-3,8-diol, has been shown to be effective against no-see-ums. Use of DEET should be avoided in children younger than 2 years and p-menthane-3,8-diol in those younger than 3 years. Picaridin is safe for use in children.12 Citronella oil is ineffective. Installing window and patio screens with a mesh size less than 16 can prevent no-see-ums from passing through the netting but will restrict air flow.3 Turning off porch lights also is helpful, as no-see-ums are attracted to light sources.6 Since no-see-ums are weak flyers, setting ceiling or window fans at high speeds can minimize exposure; similarly, being outdoors on a windy day may decrease the likelihood of being bitten. Ultimately, the best remedy for a bite is to prevent them from happening.
- Hill CA, MacDonald JF. Biting midges: biology and public health risk. Purdue University. Published July 2013. Accessed September 3, 2025. http://extension.entm.purdue.edu/publichealth/insects/bitingmidge.html
- Borkent A, Dominiak P. Catalog of the biting midges of the world (Diptera: Ceratopogonidae). Zootaxa. 2020;4787:1-377.
- Connelly CR. Biting midges, no-see-ums Culicoides spp. (Insecta: Diptera: Ceratopogonidae). University of Florida publication #EENY 349. Published August 2, 2022. Accessed September 3, 2025. https://edis.ifas.ufl.edu/publication/IN626
- Mullen GR, Murphree CS. Biting midges (Ceratopogonidae). In: Mullen GR, Durden LA, eds. Medical and Veterinary Entomology. 3rd ed. Academic Press; 2019:213-236.
- Best Bee Brothers. No-see-um seasonality range map & season information. Published March 4, 2022. Accessed September 3, 2025. https://bestbeebrothers.com/blogs/blog/no-see-um-season
- Biology Insights. Is there a season for no see ums in Florida? Published August 28, 2025. Accessed September 16, 2025. https://biologyinsights.com/is-there-a-season-for-no-see-ums-in-florida/
- Burris S. Florida no see ums: how to navigate the woes of no see ums in Florida. The Bug Agenda. Published February 2, 2022. Accessed September 3, 2025. https://thebugagenda.com/no-see-ums-in-florida/
- Steffen C. Clinical and histopathologic correlation of midge bites. Arch Dermatol. 1981;117:785-787.
- Krakowski AC, Ho B. Arthropod assault from biting midges. J Pediatr. 2013;163:298.
- Maves RC, Reaves EJ, Martin GJ. Images in clinical tropical medicine: bullous leg lesions caused by Culicoides midges after travel in the Amazon basin. Am J Trop Med Hyg. 2010;83:447.
- Swank B. How long do no-see-ums live? Pest Source. Updated March 17, 2025. Accessed September 3, 2025. https://pestsource.com/no-see-um/lifespan/
- Nguyen QD, Vu MN, Herbert AA. Insect repellents: an updated review for the clinician. J Am Acad Dermatol. 2023;88:123-130.
Biting midges, commonly known as no-see-ums, are true flies (order Diptera) and members of the Ceratopogonidae family. Regionally, they are known as punkies in the Northeast, pinyon gnats in the Southwest, moose flies in Canada, and sand gnats in Georgia, among other names.1 There are 6206 species found worldwide except for Antarctica.2 The 3 genera of greatest importance to human and livestock health in the United States are Culicoides, Leptoconops, and Forcipomyia.1 Forty-seven species of the genus Culicoides are known to be present in Florida.3 Species belonging to the genus Leptoconops also are present in coastal areas of southeast Florida as well as in the tropics, subtropics, and Caribbean.3 In the United States, biting midges primarily are a nuisance; the major medical issue associated with Culicoides insects are allergic reactions to their bites. Even though no-see-ums are not known to transmit disease in humans, they have an impact on other animal species in the United States as biting pests and vectors of disease-causing pathogens.1 Biting midges pose quite a nuisance for the proper enjoyment of outdoor spaces in the southeastern United States.
Characteristics
Morphologically, no-see-ums are gray flies measuring 1 to 3 mm in length (eFigure 1). Adults have 2 wings with distinctive patterns, large compound eyes, a thorax that extends slightly over the head, an abdomen with 9 segments, and antennae with 15 segments (eFigure 2).1,3,4 Females have modified mouth parts including mandibles that lacerate the skin during feeding, which is mainly on blood from vertebrate hosts (primarily mammals but also birds, reptiles, and amphibians).1,4 They also can feed on invertebrate hosts. Both male and female no-see-ums feed on nectar, but adult females require a blood meal to develop their eggs.2 Their life cycle progresses in stages from egg to larva to pupa to adult. Larval habitats include salt marshes, swamps, shores of streams and ponds, water-holding plants, rotting fruit, and saturated wood- and manure-enriched soil. Adults can live 2 to 7 weeks. They are weak fliers, particularly in windy conditions.1
In Florida, no-see-ums are more active during the rainy months of May to October but are active year-round in the southeastern United States and the Gulf Coast from Florida to West Texas. They are active throughout the United States in the warmer months of June and July.5 Their peak feeding activity occurs at dawn and dusk, but different species of biting midges such as Leptoconops and Culicoides also can feed during daylight hours and at night, respectively.1,6,7
Case Report
One of the authors (M.J.S.), a healthy 54-year-old man with no remarkable medical history or current use of medications, documented the natural progression of a no-see-um bite by sitting in an outdoor Florida space at 8:00
Clinical Manifestations
Although no-see-ums are not known to transmit disease in the United States, they are important biting pests that can affect tourism and prevent enjoyment of outdoor spaces and activities.2 The bite reactions on the host can range from wheal-like lesions to papules measuring 2 to 3 mm (at times with overlying vesicles) to nodules up to 1 cm in diameter.8 In our reported case, the small wheals disappeared within hours, but pruritic papules have been described to last from weeks to months. Published histopathologic correlation of biopsied indurated papules within 3 days of bite occurrence have revealed a superficial infiltrate composed of lymphocytes and histiocytes, while eosinophils were found in the deeper dermis and subcutaneous fat. Within 2 weeks, as the lesions aged, the infiltrate contained a smaller percentage of eosinophils and predominantly was present in only the superficial dermis.8 Delayed-type hypersensitivity reactions including pustules and bullous lesions also have been described.9,10 Host immune reaction to the saliva introduced during the bite dictates the severity of the response, and lesions may become secondarily infected due to scratching.11
Management Recommendations
Management consists of cleaning the bite site with soap and water to prevent infection, applying cold compresses or ice packs to relieve the intense itch, and avoiding scratching.11 Application of over-the-counter calamine lotion or hydrocortisone cream can relieve itch, and mid- to high-potency topical corticosteroids also can be prescribed for 1 to 2 weeks for more intense bite reactions in conjunction with oral antihistamines. Topical or oral antibiotics may be indicated if redness and swelling progress at the bite site or if breaks in the skin become secondarily infected.
Final Thoughts
Because of the wide-ranging habitats of no-see-ums, eradication programs using insecticides have been inefficient or environmentally suboptimal. Emptying all standing water in outdoor spaces will reduce the number of no-see-ums. Avoidance of the outdoors at dawn and dusk when no-see-ums are most active is helpful, as well as protecting exposed skin by wearing long-sleeved shirts and long pants when outside. Insect repellents containing DEET (N-N-diethyl-meta-toluamide) or picaridin can offer additional protection on the remaining exposed skin. Oil of lemon eucalyptus, or active compound p-menthane-3,8-diol, has been shown to be effective against no-see-ums. Use of DEET should be avoided in children younger than 2 years and p-menthane-3,8-diol in those younger than 3 years. Picaridin is safe for use in children.12 Citronella oil is ineffective. Installing window and patio screens with a mesh size less than 16 can prevent no-see-ums from passing through the netting but will restrict air flow.3 Turning off porch lights also is helpful, as no-see-ums are attracted to light sources.6 Since no-see-ums are weak flyers, setting ceiling or window fans at high speeds can minimize exposure; similarly, being outdoors on a windy day may decrease the likelihood of being bitten. Ultimately, the best remedy for a bite is to prevent them from happening.
Biting midges, commonly known as no-see-ums, are true flies (order Diptera) and members of the Ceratopogonidae family. Regionally, they are known as punkies in the Northeast, pinyon gnats in the Southwest, moose flies in Canada, and sand gnats in Georgia, among other names.1 There are 6206 species found worldwide except for Antarctica.2 The 3 genera of greatest importance to human and livestock health in the United States are Culicoides, Leptoconops, and Forcipomyia.1 Forty-seven species of the genus Culicoides are known to be present in Florida.3 Species belonging to the genus Leptoconops also are present in coastal areas of southeast Florida as well as in the tropics, subtropics, and Caribbean.3 In the United States, biting midges primarily are a nuisance; the major medical issue associated with Culicoides insects are allergic reactions to their bites. Even though no-see-ums are not known to transmit disease in humans, they have an impact on other animal species in the United States as biting pests and vectors of disease-causing pathogens.1 Biting midges pose quite a nuisance for the proper enjoyment of outdoor spaces in the southeastern United States.
Characteristics
Morphologically, no-see-ums are gray flies measuring 1 to 3 mm in length (eFigure 1). Adults have 2 wings with distinctive patterns, large compound eyes, a thorax that extends slightly over the head, an abdomen with 9 segments, and antennae with 15 segments (eFigure 2).1,3,4 Females have modified mouth parts including mandibles that lacerate the skin during feeding, which is mainly on blood from vertebrate hosts (primarily mammals but also birds, reptiles, and amphibians).1,4 They also can feed on invertebrate hosts. Both male and female no-see-ums feed on nectar, but adult females require a blood meal to develop their eggs.2 Their life cycle progresses in stages from egg to larva to pupa to adult. Larval habitats include salt marshes, swamps, shores of streams and ponds, water-holding plants, rotting fruit, and saturated wood- and manure-enriched soil. Adults can live 2 to 7 weeks. They are weak fliers, particularly in windy conditions.1
In Florida, no-see-ums are more active during the rainy months of May to October but are active year-round in the southeastern United States and the Gulf Coast from Florida to West Texas. They are active throughout the United States in the warmer months of June and July.5 Their peak feeding activity occurs at dawn and dusk, but different species of biting midges such as Leptoconops and Culicoides also can feed during daylight hours and at night, respectively.1,6,7
Case Report
One of the authors (M.J.S.), a healthy 54-year-old man with no remarkable medical history or current use of medications, documented the natural progression of a no-see-um bite by sitting in an outdoor Florida space at 8:00
Clinical Manifestations
Although no-see-ums are not known to transmit disease in the United States, they are important biting pests that can affect tourism and prevent enjoyment of outdoor spaces and activities.2 The bite reactions on the host can range from wheal-like lesions to papules measuring 2 to 3 mm (at times with overlying vesicles) to nodules up to 1 cm in diameter.8 In our reported case, the small wheals disappeared within hours, but pruritic papules have been described to last from weeks to months. Published histopathologic correlation of biopsied indurated papules within 3 days of bite occurrence have revealed a superficial infiltrate composed of lymphocytes and histiocytes, while eosinophils were found in the deeper dermis and subcutaneous fat. Within 2 weeks, as the lesions aged, the infiltrate contained a smaller percentage of eosinophils and predominantly was present in only the superficial dermis.8 Delayed-type hypersensitivity reactions including pustules and bullous lesions also have been described.9,10 Host immune reaction to the saliva introduced during the bite dictates the severity of the response, and lesions may become secondarily infected due to scratching.11
Management Recommendations
Management consists of cleaning the bite site with soap and water to prevent infection, applying cold compresses or ice packs to relieve the intense itch, and avoiding scratching.11 Application of over-the-counter calamine lotion or hydrocortisone cream can relieve itch, and mid- to high-potency topical corticosteroids also can be prescribed for 1 to 2 weeks for more intense bite reactions in conjunction with oral antihistamines. Topical or oral antibiotics may be indicated if redness and swelling progress at the bite site or if breaks in the skin become secondarily infected.
Final Thoughts
Because of the wide-ranging habitats of no-see-ums, eradication programs using insecticides have been inefficient or environmentally suboptimal. Emptying all standing water in outdoor spaces will reduce the number of no-see-ums. Avoidance of the outdoors at dawn and dusk when no-see-ums are most active is helpful, as well as protecting exposed skin by wearing long-sleeved shirts and long pants when outside. Insect repellents containing DEET (N-N-diethyl-meta-toluamide) or picaridin can offer additional protection on the remaining exposed skin. Oil of lemon eucalyptus, or active compound p-menthane-3,8-diol, has been shown to be effective against no-see-ums. Use of DEET should be avoided in children younger than 2 years and p-menthane-3,8-diol in those younger than 3 years. Picaridin is safe for use in children.12 Citronella oil is ineffective. Installing window and patio screens with a mesh size less than 16 can prevent no-see-ums from passing through the netting but will restrict air flow.3 Turning off porch lights also is helpful, as no-see-ums are attracted to light sources.6 Since no-see-ums are weak flyers, setting ceiling or window fans at high speeds can minimize exposure; similarly, being outdoors on a windy day may decrease the likelihood of being bitten. Ultimately, the best remedy for a bite is to prevent them from happening.
- Hill CA, MacDonald JF. Biting midges: biology and public health risk. Purdue University. Published July 2013. Accessed September 3, 2025. http://extension.entm.purdue.edu/publichealth/insects/bitingmidge.html
- Borkent A, Dominiak P. Catalog of the biting midges of the world (Diptera: Ceratopogonidae). Zootaxa. 2020;4787:1-377.
- Connelly CR. Biting midges, no-see-ums Culicoides spp. (Insecta: Diptera: Ceratopogonidae). University of Florida publication #EENY 349. Published August 2, 2022. Accessed September 3, 2025. https://edis.ifas.ufl.edu/publication/IN626
- Mullen GR, Murphree CS. Biting midges (Ceratopogonidae). In: Mullen GR, Durden LA, eds. Medical and Veterinary Entomology. 3rd ed. Academic Press; 2019:213-236.
- Best Bee Brothers. No-see-um seasonality range map & season information. Published March 4, 2022. Accessed September 3, 2025. https://bestbeebrothers.com/blogs/blog/no-see-um-season
- Biology Insights. Is there a season for no see ums in Florida? Published August 28, 2025. Accessed September 16, 2025. https://biologyinsights.com/is-there-a-season-for-no-see-ums-in-florida/
- Burris S. Florida no see ums: how to navigate the woes of no see ums in Florida. The Bug Agenda. Published February 2, 2022. Accessed September 3, 2025. https://thebugagenda.com/no-see-ums-in-florida/
- Steffen C. Clinical and histopathologic correlation of midge bites. Arch Dermatol. 1981;117:785-787.
- Krakowski AC, Ho B. Arthropod assault from biting midges. J Pediatr. 2013;163:298.
- Maves RC, Reaves EJ, Martin GJ. Images in clinical tropical medicine: bullous leg lesions caused by Culicoides midges after travel in the Amazon basin. Am J Trop Med Hyg. 2010;83:447.
- Swank B. How long do no-see-ums live? Pest Source. Updated March 17, 2025. Accessed September 3, 2025. https://pestsource.com/no-see-um/lifespan/
- Nguyen QD, Vu MN, Herbert AA. Insect repellents: an updated review for the clinician. J Am Acad Dermatol. 2023;88:123-130.
- Hill CA, MacDonald JF. Biting midges: biology and public health risk. Purdue University. Published July 2013. Accessed September 3, 2025. http://extension.entm.purdue.edu/publichealth/insects/bitingmidge.html
- Borkent A, Dominiak P. Catalog of the biting midges of the world (Diptera: Ceratopogonidae). Zootaxa. 2020;4787:1-377.
- Connelly CR. Biting midges, no-see-ums Culicoides spp. (Insecta: Diptera: Ceratopogonidae). University of Florida publication #EENY 349. Published August 2, 2022. Accessed September 3, 2025. https://edis.ifas.ufl.edu/publication/IN626
- Mullen GR, Murphree CS. Biting midges (Ceratopogonidae). In: Mullen GR, Durden LA, eds. Medical and Veterinary Entomology. 3rd ed. Academic Press; 2019:213-236.
- Best Bee Brothers. No-see-um seasonality range map & season information. Published March 4, 2022. Accessed September 3, 2025. https://bestbeebrothers.com/blogs/blog/no-see-um-season
- Biology Insights. Is there a season for no see ums in Florida? Published August 28, 2025. Accessed September 16, 2025. https://biologyinsights.com/is-there-a-season-for-no-see-ums-in-florida/
- Burris S. Florida no see ums: how to navigate the woes of no see ums in Florida. The Bug Agenda. Published February 2, 2022. Accessed September 3, 2025. https://thebugagenda.com/no-see-ums-in-florida/
- Steffen C. Clinical and histopathologic correlation of midge bites. Arch Dermatol. 1981;117:785-787.
- Krakowski AC, Ho B. Arthropod assault from biting midges. J Pediatr. 2013;163:298.
- Maves RC, Reaves EJ, Martin GJ. Images in clinical tropical medicine: bullous leg lesions caused by Culicoides midges after travel in the Amazon basin. Am J Trop Med Hyg. 2010;83:447.
- Swank B. How long do no-see-ums live? Pest Source. Updated March 17, 2025. Accessed September 3, 2025. https://pestsource.com/no-see-um/lifespan/
- Nguyen QD, Vu MN, Herbert AA. Insect repellents: an updated review for the clinician. J Am Acad Dermatol. 2023;88:123-130.
Dermoscopic Documentation of a No-see-um Bite
Dermoscopic Documentation of a No-see-um Bite
Practice Points
- Biting midges, commonly known as no-see-ums, are extremely small flies whose bites can cause a burning sensation, mild pain, and reactions ranging from small wheals to intensely pruritic papules.
- Medical management of no-see-um bites is based on the severity of the skin reaction.
