User login
VIDEO: Gene profiling could signal start of personalized medicine in RA
PARIS – A set of genetic polymorphisms is beginning to allow researchers to predict which patients with rheumatoid arthritis will have a severe disease course, as well as determine their response to treatment and risk of death.
Changes in amino acids at positions 71 and 74 of the HLA-DRB1 gene, which are a part of the "shared epitope" that is already known to increase genetic susceptibility for rheumatoid arthritis, as well as a new polymorphism at position 11 of the HLA-DRB1 gene that is outside the shared epitope, are key to this effort. These polymorphisms predicted the radiologic outcome of rheumatoid arthritis patients, response to anti-tumor necrosis factor therapy, and mortality in an analysis of blood samples from three independent multicenter, prospective cohort studies. The three polymorphisms defined 16 haplotypes whose effects on RA susceptibility range from protective to increasing risk and were perfectly correlated with the observed levels of disease susceptibility.
Further studies will be necessary to validate the associations observed with the sets of polymorphisms, said Dr. Sebastien Viatte, first author of the study and a research fellow at the Centre for Musculoskeletal Research at the University of Manchester (England). Nonetheless, the results are an important step in showing that "genetics can be used to predict disease outcomes and is ... likely to enter the clinic within 5-10 years," he said in a video interview at the annual European Congress of Rheumatology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
PARIS – A set of genetic polymorphisms is beginning to allow researchers to predict which patients with rheumatoid arthritis will have a severe disease course, as well as determine their response to treatment and risk of death.
Changes in amino acids at positions 71 and 74 of the HLA-DRB1 gene, which are a part of the "shared epitope" that is already known to increase genetic susceptibility for rheumatoid arthritis, as well as a new polymorphism at position 11 of the HLA-DRB1 gene that is outside the shared epitope, are key to this effort. These polymorphisms predicted the radiologic outcome of rheumatoid arthritis patients, response to anti-tumor necrosis factor therapy, and mortality in an analysis of blood samples from three independent multicenter, prospective cohort studies. The three polymorphisms defined 16 haplotypes whose effects on RA susceptibility range from protective to increasing risk and were perfectly correlated with the observed levels of disease susceptibility.
Further studies will be necessary to validate the associations observed with the sets of polymorphisms, said Dr. Sebastien Viatte, first author of the study and a research fellow at the Centre for Musculoskeletal Research at the University of Manchester (England). Nonetheless, the results are an important step in showing that "genetics can be used to predict disease outcomes and is ... likely to enter the clinic within 5-10 years," he said in a video interview at the annual European Congress of Rheumatology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
PARIS – A set of genetic polymorphisms is beginning to allow researchers to predict which patients with rheumatoid arthritis will have a severe disease course, as well as determine their response to treatment and risk of death.
Changes in amino acids at positions 71 and 74 of the HLA-DRB1 gene, which are a part of the "shared epitope" that is already known to increase genetic susceptibility for rheumatoid arthritis, as well as a new polymorphism at position 11 of the HLA-DRB1 gene that is outside the shared epitope, are key to this effort. These polymorphisms predicted the radiologic outcome of rheumatoid arthritis patients, response to anti-tumor necrosis factor therapy, and mortality in an analysis of blood samples from three independent multicenter, prospective cohort studies. The three polymorphisms defined 16 haplotypes whose effects on RA susceptibility range from protective to increasing risk and were perfectly correlated with the observed levels of disease susceptibility.
Further studies will be necessary to validate the associations observed with the sets of polymorphisms, said Dr. Sebastien Viatte, first author of the study and a research fellow at the Centre for Musculoskeletal Research at the University of Manchester (England). Nonetheless, the results are an important step in showing that "genetics can be used to predict disease outcomes and is ... likely to enter the clinic within 5-10 years," he said in a video interview at the annual European Congress of Rheumatology.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE EULAR CONGRESS 2014

Tofacitinib bests methotrexate for RA
Tofacitinib was superior to methotrexate at reducing the signs and symptoms of rheumatoid arthritis, improving physical function, and slowing the progression of joint damage in a 2-year, phase III clinical trial sponsored by Pfizer.
The results of the study indicate that tofacitinib "can be more effective clinically, functionally, and radiographically than methotrexate in patients with rheumatoid arthritis who have not previously received methotrexate," Dr. Eun-Bong Lee of Seoul (South Korea) National University and his associates reported June 19 in the New England Journal of Medicine.
While nonbiologic or biologic disease-modifying antirheumatic drugs (DMARDs) in combination with methotrexate have been shown to be superior to methotrexate alone, other studies have not shown superiority of a biologic DMARD alone vs. methotrexate at times extending to 1 year or longer, they noted.
Tofacitinib (Xeljanz), a targeted, small-molecule Janus kinase inhibitor, was approved by the Food and Drug Administration in November 2012 for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to methotrexate.
In the trial conducted at 151 medical centers worldwide, 956 adults with active, moderate to severe RA who had never received methotrexate or tofacitinib were randomly assigned to receive 5 mg of oral tofacitinib twice daily (373 patients), 10 mg of oral tofacitinib twice daily (397 patients), or methotrexate (186 patients) for 2 years. The mean duration of RA at entry into the study was approximately 3 years, the investigators wrote.
At 6, 12, and 24 months, there was no decline in van der Heijde modified total Sharp score (a radiographic measure of structural joint damage), as well as on measures of joint erosion and joint-space narrowing, in a significantly greater proportion of patients who were taking tofacitinib than in those who were taking methotrexate. In addition, 25.5% of the patients taking 5 mg of tofacitinib and 37.7% of those taking 10 mg of tofacitinib achieved an American College of Rheumatology (ACR) 70 response (at least a 70% reduction in the number of tender and swollen joints plus equivalent improvement in three to five ACR core measures), compared with only 12.0% of patients in the methotrexate group.
Rates of remission and of low disease activity at 6, 12, and 24 months also were significantly higher in the two tofacitinib groups than in the methotrexate group, the investigators reported (N. Engl. J. Med. 2014;370:2377-86).
However, "the benefits of tofacitinib need to be considered in the context of the risks of adverse events," the authors wrote. As expected, the drug was associated with declines in neutrophil and lymphocyte counts, and with increases in lipid, aminotransferase, and creatinine levels. Herpes zoster infections developed in 4% of the two tofacitinib groups, compared with only 1.1% of the methotrexate group. *Five confirmed cases of cancer (non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, prostate cancer, Burkitt’s B-cell lymphoma, and colon cancer) and one case of adrenal adenoma with unknown malignancy status developed in the tofacitinib group, compared with one confirmed case of cancer (gastric) in the methotrexate group.
This study was funded by Pfizer, which also collected and analyzed the data and assisted with writing the report. Dr. Lee reported receiving consulting fees from Pfizer; Dr. Lee’s associates reported ties to numerous industry sources.
*Correction, 7/1/2014: An earlier version of this story misstated the total number of confirmed cancers in the tofacitinib groups.
Tofacitinib was superior to methotrexate at reducing the signs and symptoms of rheumatoid arthritis, improving physical function, and slowing the progression of joint damage in a 2-year, phase III clinical trial sponsored by Pfizer.
The results of the study indicate that tofacitinib "can be more effective clinically, functionally, and radiographically than methotrexate in patients with rheumatoid arthritis who have not previously received methotrexate," Dr. Eun-Bong Lee of Seoul (South Korea) National University and his associates reported June 19 in the New England Journal of Medicine.
While nonbiologic or biologic disease-modifying antirheumatic drugs (DMARDs) in combination with methotrexate have been shown to be superior to methotrexate alone, other studies have not shown superiority of a biologic DMARD alone vs. methotrexate at times extending to 1 year or longer, they noted.
Tofacitinib (Xeljanz), a targeted, small-molecule Janus kinase inhibitor, was approved by the Food and Drug Administration in November 2012 for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to methotrexate.
In the trial conducted at 151 medical centers worldwide, 956 adults with active, moderate to severe RA who had never received methotrexate or tofacitinib were randomly assigned to receive 5 mg of oral tofacitinib twice daily (373 patients), 10 mg of oral tofacitinib twice daily (397 patients), or methotrexate (186 patients) for 2 years. The mean duration of RA at entry into the study was approximately 3 years, the investigators wrote.
At 6, 12, and 24 months, there was no decline in van der Heijde modified total Sharp score (a radiographic measure of structural joint damage), as well as on measures of joint erosion and joint-space narrowing, in a significantly greater proportion of patients who were taking tofacitinib than in those who were taking methotrexate. In addition, 25.5% of the patients taking 5 mg of tofacitinib and 37.7% of those taking 10 mg of tofacitinib achieved an American College of Rheumatology (ACR) 70 response (at least a 70% reduction in the number of tender and swollen joints plus equivalent improvement in three to five ACR core measures), compared with only 12.0% of patients in the methotrexate group.
Rates of remission and of low disease activity at 6, 12, and 24 months also were significantly higher in the two tofacitinib groups than in the methotrexate group, the investigators reported (N. Engl. J. Med. 2014;370:2377-86).
However, "the benefits of tofacitinib need to be considered in the context of the risks of adverse events," the authors wrote. As expected, the drug was associated with declines in neutrophil and lymphocyte counts, and with increases in lipid, aminotransferase, and creatinine levels. Herpes zoster infections developed in 4% of the two tofacitinib groups, compared with only 1.1% of the methotrexate group. *Five confirmed cases of cancer (non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, prostate cancer, Burkitt’s B-cell lymphoma, and colon cancer) and one case of adrenal adenoma with unknown malignancy status developed in the tofacitinib group, compared with one confirmed case of cancer (gastric) in the methotrexate group.
This study was funded by Pfizer, which also collected and analyzed the data and assisted with writing the report. Dr. Lee reported receiving consulting fees from Pfizer; Dr. Lee’s associates reported ties to numerous industry sources.
*Correction, 7/1/2014: An earlier version of this story misstated the total number of confirmed cancers in the tofacitinib groups.
Tofacitinib was superior to methotrexate at reducing the signs and symptoms of rheumatoid arthritis, improving physical function, and slowing the progression of joint damage in a 2-year, phase III clinical trial sponsored by Pfizer.
The results of the study indicate that tofacitinib "can be more effective clinically, functionally, and radiographically than methotrexate in patients with rheumatoid arthritis who have not previously received methotrexate," Dr. Eun-Bong Lee of Seoul (South Korea) National University and his associates reported June 19 in the New England Journal of Medicine.
While nonbiologic or biologic disease-modifying antirheumatic drugs (DMARDs) in combination with methotrexate have been shown to be superior to methotrexate alone, other studies have not shown superiority of a biologic DMARD alone vs. methotrexate at times extending to 1 year or longer, they noted.
Tofacitinib (Xeljanz), a targeted, small-molecule Janus kinase inhibitor, was approved by the Food and Drug Administration in November 2012 for the treatment of adults with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to methotrexate.
In the trial conducted at 151 medical centers worldwide, 956 adults with active, moderate to severe RA who had never received methotrexate or tofacitinib were randomly assigned to receive 5 mg of oral tofacitinib twice daily (373 patients), 10 mg of oral tofacitinib twice daily (397 patients), or methotrexate (186 patients) for 2 years. The mean duration of RA at entry into the study was approximately 3 years, the investigators wrote.
At 6, 12, and 24 months, there was no decline in van der Heijde modified total Sharp score (a radiographic measure of structural joint damage), as well as on measures of joint erosion and joint-space narrowing, in a significantly greater proportion of patients who were taking tofacitinib than in those who were taking methotrexate. In addition, 25.5% of the patients taking 5 mg of tofacitinib and 37.7% of those taking 10 mg of tofacitinib achieved an American College of Rheumatology (ACR) 70 response (at least a 70% reduction in the number of tender and swollen joints plus equivalent improvement in three to five ACR core measures), compared with only 12.0% of patients in the methotrexate group.
Rates of remission and of low disease activity at 6, 12, and 24 months also were significantly higher in the two tofacitinib groups than in the methotrexate group, the investigators reported (N. Engl. J. Med. 2014;370:2377-86).
However, "the benefits of tofacitinib need to be considered in the context of the risks of adverse events," the authors wrote. As expected, the drug was associated with declines in neutrophil and lymphocyte counts, and with increases in lipid, aminotransferase, and creatinine levels. Herpes zoster infections developed in 4% of the two tofacitinib groups, compared with only 1.1% of the methotrexate group. *Five confirmed cases of cancer (non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, prostate cancer, Burkitt’s B-cell lymphoma, and colon cancer) and one case of adrenal adenoma with unknown malignancy status developed in the tofacitinib group, compared with one confirmed case of cancer (gastric) in the methotrexate group.
This study was funded by Pfizer, which also collected and analyzed the data and assisted with writing the report. Dr. Lee reported receiving consulting fees from Pfizer; Dr. Lee’s associates reported ties to numerous industry sources.
*Correction, 7/1/2014: An earlier version of this story misstated the total number of confirmed cancers in the tofacitinib groups.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Tofacitinib monotherapy provided better outcomes at 2 years than did methotrexate alone in patients who previously had not been treated with either drug.
Major finding: 25.5% of the patients taking 5 mg of tofacitinib twice daily and 37.7% of those taking 10 mg of tofacitinib twice daily achieved an ACR 70 response, compared with only 12.0% of patients in the methotrexate group.
Data source: An international, randomized, double-blind phase III trial performed at 151 medical centers involving 956 adults with active RA who were treated with either tofacitinib or methotrexate for 2 years.
Disclosures: This study was funded by Pfizer, which also collected and analyzed the data and assisted with writing the report. Dr. Lee reported receiving consulting fees from Pfizer.
International RA risk tool for CVD comes closer to reality
An international collaboration of experts has developed a cardiovascular risk calculator specifically for rheumatoid arthritis patients that has the potential to become part of routine rheumatology clinical practice in many parts of the world.
The ATACC-RA (A TransAtlantic Cardiovascular Risk Calculator for Rheumatoid Arthritis) consortium will be constructed from data collected at 13 rheumatology centers in 10 countries, Elke Arts said at the annual European Congress of Rheumatology.
The current version, which is still undergoing validation, contains data from eight rheumatology centers in seven countries (Greece, the Netherlands, Norway, South Africa, Sweden, the United Kingdom, and the United States), said Ms. Arts, a rheumatology researcher at Radboud University Medical Centre in Nijmegen, the Netherlands.
So far, ATACC-RA contains pooled data from 3,176 RA patients without cardiovascular disease. Individual data were collected on CV risk factors and outcomes and then combined with RA-specific information, such as disease duration, seropositivity, rheumatoid factor and/or anti-citrullinated protein antibodies, 28-joint Disease Activity Score (DAS28), and acute phase reactants.
During an average of almost 8 years of follow-up comprising 24,733 patient-years, 314 patients developed cardiovascular disease (CVD).
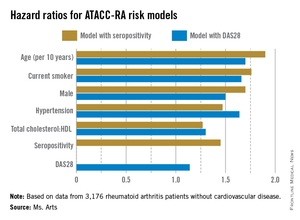
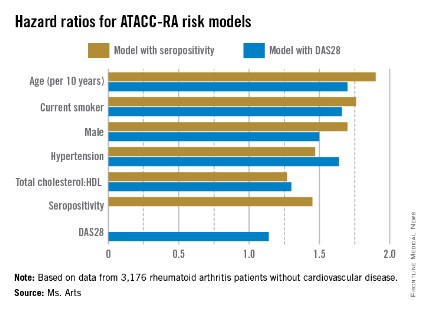
Two possible models came out of multivariable risk score modeling for predicting the RA-specific 10-year risk of CVD. Each incorporated the traditional risk factors of age, sex, current smoking, presence of hypertension, and ratio of total cholesterol to HDL cholesterol, but also either seropositivity or DAS28. The consortium specifically chose to assess potential risk factors that would be easily available to the health professional regardless of the setting (for example, primary, secondary, and tertiary care, and even private practice).
The models that included seropositivity or DAS28 both demonstrated good discrimination and calibration, when compared with the Framingham or SCORE (Systematic Coronary Risk Evaluation) algorithms. The models also showed good concordance with each other (see chart).

"By pooling data from many centers, it appears possible to develop an RA-specific CVD risk algorithm which is more accurate at predicting CVD in people with RA than the currently available risk algorithms, which have been developed for the general population," members of the consortium wrote in response to e-mailed questions.
The consortium is now working to validate the ATACC-RA calculator, with the ultimate aim of validating it in completely independent cohorts. The continued growth of the consortium will help make this a reality within the next couple of years. The end result may be a risk calculator that can be adapted to account for the underlying risk of each patient population.
Once the algorithm is validated, the consortium hopes it will become part of routine rheumatology clinical practice, much the same as the Framingham or SCORE algorithms are used currently in the general population, but more specifically applied to patients with RA.
"The models proved to be quite robust," Ms. Arts said. "If this holds true through the additional analysis and external validation, we may have one that will be applicable to a wide variety of patients all over the world."
Once fully validated, the investigators plan to produce the tool in a user-friendly application for computers or even smartphones.
The investigators had no conflicts of interest to declare.
An international collaboration of experts has developed a cardiovascular risk calculator specifically for rheumatoid arthritis patients that has the potential to become part of routine rheumatology clinical practice in many parts of the world.
The ATACC-RA (A TransAtlantic Cardiovascular Risk Calculator for Rheumatoid Arthritis) consortium will be constructed from data collected at 13 rheumatology centers in 10 countries, Elke Arts said at the annual European Congress of Rheumatology.
The current version, which is still undergoing validation, contains data from eight rheumatology centers in seven countries (Greece, the Netherlands, Norway, South Africa, Sweden, the United Kingdom, and the United States), said Ms. Arts, a rheumatology researcher at Radboud University Medical Centre in Nijmegen, the Netherlands.
So far, ATACC-RA contains pooled data from 3,176 RA patients without cardiovascular disease. Individual data were collected on CV risk factors and outcomes and then combined with RA-specific information, such as disease duration, seropositivity, rheumatoid factor and/or anti-citrullinated protein antibodies, 28-joint Disease Activity Score (DAS28), and acute phase reactants.
During an average of almost 8 years of follow-up comprising 24,733 patient-years, 314 patients developed cardiovascular disease (CVD).
Two possible models came out of multivariable risk score modeling for predicting the RA-specific 10-year risk of CVD. Each incorporated the traditional risk factors of age, sex, current smoking, presence of hypertension, and ratio of total cholesterol to HDL cholesterol, but also either seropositivity or DAS28. The consortium specifically chose to assess potential risk factors that would be easily available to the health professional regardless of the setting (for example, primary, secondary, and tertiary care, and even private practice).
The models that included seropositivity or DAS28 both demonstrated good discrimination and calibration, when compared with the Framingham or SCORE (Systematic Coronary Risk Evaluation) algorithms. The models also showed good concordance with each other (see chart).
"By pooling data from many centers, it appears possible to develop an RA-specific CVD risk algorithm which is more accurate at predicting CVD in people with RA than the currently available risk algorithms, which have been developed for the general population," members of the consortium wrote in response to e-mailed questions.
The consortium is now working to validate the ATACC-RA calculator, with the ultimate aim of validating it in completely independent cohorts. The continued growth of the consortium will help make this a reality within the next couple of years. The end result may be a risk calculator that can be adapted to account for the underlying risk of each patient population.
Once the algorithm is validated, the consortium hopes it will become part of routine rheumatology clinical practice, much the same as the Framingham or SCORE algorithms are used currently in the general population, but more specifically applied to patients with RA.
"The models proved to be quite robust," Ms. Arts said. "If this holds true through the additional analysis and external validation, we may have one that will be applicable to a wide variety of patients all over the world."
Once fully validated, the investigators plan to produce the tool in a user-friendly application for computers or even smartphones.
The investigators had no conflicts of interest to declare.
An international collaboration of experts has developed a cardiovascular risk calculator specifically for rheumatoid arthritis patients that has the potential to become part of routine rheumatology clinical practice in many parts of the world.
The ATACC-RA (A TransAtlantic Cardiovascular Risk Calculator for Rheumatoid Arthritis) consortium will be constructed from data collected at 13 rheumatology centers in 10 countries, Elke Arts said at the annual European Congress of Rheumatology.
The current version, which is still undergoing validation, contains data from eight rheumatology centers in seven countries (Greece, the Netherlands, Norway, South Africa, Sweden, the United Kingdom, and the United States), said Ms. Arts, a rheumatology researcher at Radboud University Medical Centre in Nijmegen, the Netherlands.
So far, ATACC-RA contains pooled data from 3,176 RA patients without cardiovascular disease. Individual data were collected on CV risk factors and outcomes and then combined with RA-specific information, such as disease duration, seropositivity, rheumatoid factor and/or anti-citrullinated protein antibodies, 28-joint Disease Activity Score (DAS28), and acute phase reactants.
During an average of almost 8 years of follow-up comprising 24,733 patient-years, 314 patients developed cardiovascular disease (CVD).
Two possible models came out of multivariable risk score modeling for predicting the RA-specific 10-year risk of CVD. Each incorporated the traditional risk factors of age, sex, current smoking, presence of hypertension, and ratio of total cholesterol to HDL cholesterol, but also either seropositivity or DAS28. The consortium specifically chose to assess potential risk factors that would be easily available to the health professional regardless of the setting (for example, primary, secondary, and tertiary care, and even private practice).
The models that included seropositivity or DAS28 both demonstrated good discrimination and calibration, when compared with the Framingham or SCORE (Systematic Coronary Risk Evaluation) algorithms. The models also showed good concordance with each other (see chart).
"By pooling data from many centers, it appears possible to develop an RA-specific CVD risk algorithm which is more accurate at predicting CVD in people with RA than the currently available risk algorithms, which have been developed for the general population," members of the consortium wrote in response to e-mailed questions.
The consortium is now working to validate the ATACC-RA calculator, with the ultimate aim of validating it in completely independent cohorts. The continued growth of the consortium will help make this a reality within the next couple of years. The end result may be a risk calculator that can be adapted to account for the underlying risk of each patient population.
Once the algorithm is validated, the consortium hopes it will become part of routine rheumatology clinical practice, much the same as the Framingham or SCORE algorithms are used currently in the general population, but more specifically applied to patients with RA.
"The models proved to be quite robust," Ms. Arts said. "If this holds true through the additional analysis and external validation, we may have one that will be applicable to a wide variety of patients all over the world."
Once fully validated, the investigators plan to produce the tool in a user-friendly application for computers or even smartphones.
The investigators had no conflicts of interest to declare.
FROM THE EULAR CONGRESS 2014
Key clinical point: An RA-specific calculator for 10-year CVD risk may soon be available.
Major finding: The risk factors in models that used seropositivity or DAS28 had similar hazard ratios contributing to CVD risk.
Data source: Pooled data from 3,176 RA patients without CVD who were followed for the development of CVD for a mean of nearly 8 years.
Disclosures: The investigators had no conflicts of interest to declare.
Sarilumab shown safe, effective for RA in phase III trial
PARIS – Rheumatoid arthritis patients who received the interleukin-6–blocking drug sarilumab plus methotrexate had significantly better short-term and 1-year outcomes than did patients on methotrexate alone in a phase III trial with 1,197 patients.
Sarilumab treatment also was relatively safe, with no new safety concerns arising from the study, Dr. Mark Genovese said at the annual European Congress of Rheumatology.

"Both sarilumab-dose groups showed a statistically significant improvement, compared to the placebo group, thereby demonstrating that, in this study, sarilumab plus methotrexate improved the signs and symptoms of rheumatoid arthritis [RA], improved physical function, and inhibited progression of joint damage in adult patients with active RA and inadequate response to methotrexate. Importantly, in this first phase III study with this agent, we did not see any unique safety issues," Dr. Genovese said in an interview.
The SARIL-RA-MOBILITY trial is the first study to finish from a panel of four phase III studies of sarilumab in patients with RA. The three other phase III trials currently in progress have enrolled a total of about 1,400 patients. A submission to the Food and Drug Administration for marketing approval of sarilumab for patients with RA will await completion of these three additional studies, said Dr. Genovese, professor of medicine and cochief of the division of immunology and rheumatology at Stanford (Calif.) University.
The trial enrolled patients with RA and an inadequate response to methotrexate alone at more than 200 centers in more than 30 countries, including the United States. The study randomized patients to treatment with methotrexate plus 150 mg sarilumab administered as a subcutaneous injection every 2 weeks, 200 mg injected every 2 weeks, or placebo.
The study had three primary efficacy endpoints:
• The percentage of patients achieving an American College of Rheumatology 20 response after 24 weeks of treatment, which was achieved by 58% of 400 patients who received 150 mg sarilumab, 66% of 399 patients who received the 200-mg dose, and 33% of the 398 patients who received placebo injections.
• The average change from baseline in the Health Assessment Questionnaire disability index after 16 weeks of treatment, which fell by 0.53 in patients who received the 150-mg dose of sarilumab, by 0.55 in those who received 200-mg sarilumab injections, and by 0.29 in those who received placebo.
• The average change from baseline in the van der Heijde–modified total Sharp score after 52 weeks of treatment, which increased by 0.90 among patients on the lower sarilumab dose, increased by 0.25 in patients on the higher dose, and increased by 2.78 among patients who received placebo.
For all three endpoints, the differences between each of the sarilumab groups and the placebo controls were statistically significant as well as clinically meaningful. "We are encouraged by the potential of sarilumab to offer a new alternative in the treatment armamentarium for patients suffering from RA," Dr. Genovese said. When the two companies developing sarilumab submit their approval application, they will likely initially seek indications for treating adult patients with moderately to severely active RA who had an inadequate response to disease-modifying antirheumatic drugs (DMARDs) or tumor necrosis factor–alpha inhibitors, he predicted.
Serious adverse events during 52 weeks of follow-up occurred in 23 placebo patients, 38 patients on the lower sarilumab dose, and 46 on the higher dose. The most common serious adverse event was infection, which occurred in 10 patients on placebo, 11 patients on low-dose sarilumab, and 17 on the higher dose. No serious infections occurred in patients whose neutrophil level fell below 1,000/mcL.
"The number of serious adverse events thus far remains low, and it will be important to see the integrated safety data across the phase III program before making any judgments regarding the ideal dose and the benefit-to-safety risk ratio of any single dose," Dr. Genovese said.
"Among patients treated with sarilumab, a dose-dependent decrease in mean neutrophil counts was observed. Serious infections were not associated with grades 3 and 4 neutropenia in this study. Increases in mean LDL cholesterol level and transaminases were observed. These safety findings were consistent with those observed in prior investigational studies with sarilumab. The choice of doses will ultimately depend on the integrated safety data across the program and the performance of each dose across a broad range of patient populations," he said.
Sarilumab is the first agent tested from the class of interleukin (IL)-6 blockers. "IL-6 is a key proinflammatory cytokine that impacts inflammation, bone, and metabolism," Dr. Genovese noted.
"Blockade of IL-6 should provide significant improvements in all these areas. This study has demonstrated that inhibition of IL-6 can reduce the signs and symptoms of RA and help to stop the destruction of joints in patients with RA. Additional phase III studies of sarilumab are underway and should provide additional evidence that anti–IL-6 receptor therapy with sarilumab may be a future option for RA patients. It is possibly too early to say if there are any special advantages or opportunities from the strategy of IL-6 blockage, compared with any other class of biologic DMARD," he said.
The trial was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.
On Twitter @mitchelzoler
PARIS – Rheumatoid arthritis patients who received the interleukin-6–blocking drug sarilumab plus methotrexate had significantly better short-term and 1-year outcomes than did patients on methotrexate alone in a phase III trial with 1,197 patients.
Sarilumab treatment also was relatively safe, with no new safety concerns arising from the study, Dr. Mark Genovese said at the annual European Congress of Rheumatology.
"Both sarilumab-dose groups showed a statistically significant improvement, compared to the placebo group, thereby demonstrating that, in this study, sarilumab plus methotrexate improved the signs and symptoms of rheumatoid arthritis [RA], improved physical function, and inhibited progression of joint damage in adult patients with active RA and inadequate response to methotrexate. Importantly, in this first phase III study with this agent, we did not see any unique safety issues," Dr. Genovese said in an interview.
The SARIL-RA-MOBILITY trial is the first study to finish from a panel of four phase III studies of sarilumab in patients with RA. The three other phase III trials currently in progress have enrolled a total of about 1,400 patients. A submission to the Food and Drug Administration for marketing approval of sarilumab for patients with RA will await completion of these three additional studies, said Dr. Genovese, professor of medicine and cochief of the division of immunology and rheumatology at Stanford (Calif.) University.
The trial enrolled patients with RA and an inadequate response to methotrexate alone at more than 200 centers in more than 30 countries, including the United States. The study randomized patients to treatment with methotrexate plus 150 mg sarilumab administered as a subcutaneous injection every 2 weeks, 200 mg injected every 2 weeks, or placebo.
The study had three primary efficacy endpoints:
• The percentage of patients achieving an American College of Rheumatology 20 response after 24 weeks of treatment, which was achieved by 58% of 400 patients who received 150 mg sarilumab, 66% of 399 patients who received the 200-mg dose, and 33% of the 398 patients who received placebo injections.
• The average change from baseline in the Health Assessment Questionnaire disability index after 16 weeks of treatment, which fell by 0.53 in patients who received the 150-mg dose of sarilumab, by 0.55 in those who received 200-mg sarilumab injections, and by 0.29 in those who received placebo.
• The average change from baseline in the van der Heijde–modified total Sharp score after 52 weeks of treatment, which increased by 0.90 among patients on the lower sarilumab dose, increased by 0.25 in patients on the higher dose, and increased by 2.78 among patients who received placebo.
For all three endpoints, the differences between each of the sarilumab groups and the placebo controls were statistically significant as well as clinically meaningful. "We are encouraged by the potential of sarilumab to offer a new alternative in the treatment armamentarium for patients suffering from RA," Dr. Genovese said. When the two companies developing sarilumab submit their approval application, they will likely initially seek indications for treating adult patients with moderately to severely active RA who had an inadequate response to disease-modifying antirheumatic drugs (DMARDs) or tumor necrosis factor–alpha inhibitors, he predicted.
Serious adverse events during 52 weeks of follow-up occurred in 23 placebo patients, 38 patients on the lower sarilumab dose, and 46 on the higher dose. The most common serious adverse event was infection, which occurred in 10 patients on placebo, 11 patients on low-dose sarilumab, and 17 on the higher dose. No serious infections occurred in patients whose neutrophil level fell below 1,000/mcL.
"The number of serious adverse events thus far remains low, and it will be important to see the integrated safety data across the phase III program before making any judgments regarding the ideal dose and the benefit-to-safety risk ratio of any single dose," Dr. Genovese said.
"Among patients treated with sarilumab, a dose-dependent decrease in mean neutrophil counts was observed. Serious infections were not associated with grades 3 and 4 neutropenia in this study. Increases in mean LDL cholesterol level and transaminases were observed. These safety findings were consistent with those observed in prior investigational studies with sarilumab. The choice of doses will ultimately depend on the integrated safety data across the program and the performance of each dose across a broad range of patient populations," he said.
Sarilumab is the first agent tested from the class of interleukin (IL)-6 blockers. "IL-6 is a key proinflammatory cytokine that impacts inflammation, bone, and metabolism," Dr. Genovese noted.
"Blockade of IL-6 should provide significant improvements in all these areas. This study has demonstrated that inhibition of IL-6 can reduce the signs and symptoms of RA and help to stop the destruction of joints in patients with RA. Additional phase III studies of sarilumab are underway and should provide additional evidence that anti–IL-6 receptor therapy with sarilumab may be a future option for RA patients. It is possibly too early to say if there are any special advantages or opportunities from the strategy of IL-6 blockage, compared with any other class of biologic DMARD," he said.
The trial was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.
On Twitter @mitchelzoler
PARIS – Rheumatoid arthritis patients who received the interleukin-6–blocking drug sarilumab plus methotrexate had significantly better short-term and 1-year outcomes than did patients on methotrexate alone in a phase III trial with 1,197 patients.
Sarilumab treatment also was relatively safe, with no new safety concerns arising from the study, Dr. Mark Genovese said at the annual European Congress of Rheumatology.
"Both sarilumab-dose groups showed a statistically significant improvement, compared to the placebo group, thereby demonstrating that, in this study, sarilumab plus methotrexate improved the signs and symptoms of rheumatoid arthritis [RA], improved physical function, and inhibited progression of joint damage in adult patients with active RA and inadequate response to methotrexate. Importantly, in this first phase III study with this agent, we did not see any unique safety issues," Dr. Genovese said in an interview.
The SARIL-RA-MOBILITY trial is the first study to finish from a panel of four phase III studies of sarilumab in patients with RA. The three other phase III trials currently in progress have enrolled a total of about 1,400 patients. A submission to the Food and Drug Administration for marketing approval of sarilumab for patients with RA will await completion of these three additional studies, said Dr. Genovese, professor of medicine and cochief of the division of immunology and rheumatology at Stanford (Calif.) University.
The trial enrolled patients with RA and an inadequate response to methotrexate alone at more than 200 centers in more than 30 countries, including the United States. The study randomized patients to treatment with methotrexate plus 150 mg sarilumab administered as a subcutaneous injection every 2 weeks, 200 mg injected every 2 weeks, or placebo.
The study had three primary efficacy endpoints:
• The percentage of patients achieving an American College of Rheumatology 20 response after 24 weeks of treatment, which was achieved by 58% of 400 patients who received 150 mg sarilumab, 66% of 399 patients who received the 200-mg dose, and 33% of the 398 patients who received placebo injections.
• The average change from baseline in the Health Assessment Questionnaire disability index after 16 weeks of treatment, which fell by 0.53 in patients who received the 150-mg dose of sarilumab, by 0.55 in those who received 200-mg sarilumab injections, and by 0.29 in those who received placebo.
• The average change from baseline in the van der Heijde–modified total Sharp score after 52 weeks of treatment, which increased by 0.90 among patients on the lower sarilumab dose, increased by 0.25 in patients on the higher dose, and increased by 2.78 among patients who received placebo.
For all three endpoints, the differences between each of the sarilumab groups and the placebo controls were statistically significant as well as clinically meaningful. "We are encouraged by the potential of sarilumab to offer a new alternative in the treatment armamentarium for patients suffering from RA," Dr. Genovese said. When the two companies developing sarilumab submit their approval application, they will likely initially seek indications for treating adult patients with moderately to severely active RA who had an inadequate response to disease-modifying antirheumatic drugs (DMARDs) or tumor necrosis factor–alpha inhibitors, he predicted.
Serious adverse events during 52 weeks of follow-up occurred in 23 placebo patients, 38 patients on the lower sarilumab dose, and 46 on the higher dose. The most common serious adverse event was infection, which occurred in 10 patients on placebo, 11 patients on low-dose sarilumab, and 17 on the higher dose. No serious infections occurred in patients whose neutrophil level fell below 1,000/mcL.
"The number of serious adverse events thus far remains low, and it will be important to see the integrated safety data across the phase III program before making any judgments regarding the ideal dose and the benefit-to-safety risk ratio of any single dose," Dr. Genovese said.
"Among patients treated with sarilumab, a dose-dependent decrease in mean neutrophil counts was observed. Serious infections were not associated with grades 3 and 4 neutropenia in this study. Increases in mean LDL cholesterol level and transaminases were observed. These safety findings were consistent with those observed in prior investigational studies with sarilumab. The choice of doses will ultimately depend on the integrated safety data across the program and the performance of each dose across a broad range of patient populations," he said.
Sarilumab is the first agent tested from the class of interleukin (IL)-6 blockers. "IL-6 is a key proinflammatory cytokine that impacts inflammation, bone, and metabolism," Dr. Genovese noted.
"Blockade of IL-6 should provide significant improvements in all these areas. This study has demonstrated that inhibition of IL-6 can reduce the signs and symptoms of RA and help to stop the destruction of joints in patients with RA. Additional phase III studies of sarilumab are underway and should provide additional evidence that anti–IL-6 receptor therapy with sarilumab may be a future option for RA patients. It is possibly too early to say if there are any special advantages or opportunities from the strategy of IL-6 blockage, compared with any other class of biologic DMARD," he said.
The trial was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.
On Twitter @mitchelzoler
AT THE EULAR CONGRESS 2014
Key clinical point: Treatment with the interleukin-6–blocking drug sarilumab showed efficacy and relative safety in the first reported results from a phase III trial.
Major finding: After 24 weeks, 58%-66% of patients on sarilumab had an ACR 20 response, compared with 33% of placebo patients.
Data source: The SARIL-RA-MOBILITY trial, which enrolled 1,197 patients at more than 200 international centers.
Disclosures: SARIL-RA-MOBILITY was sponsored by Sanofi and Regeneron, the companies developing the drug. Dr. Genovese said that he has been a consultant to and received research support from Sanofi. Several coauthors are employees of Sanofi or Regeneron.

Biosimilars match infliximab, etanercept for RA treatment
*PARIS - Two different biosimilar drugs, one a mimic of infliximab, the other modeled on etanercept, closely matched the efficacy and safety of their brand-name counterparts in a pair of separate, randomized, controlled studies.
In one study, treatment with infliximab and a biosimilar agent showed virtually identical efficacy and safety performance at several time points during the first 16 weeks of treatment of 189 patients with rheumatoid arthritis in a head-to-head, randomized comparison done at 23 centers in India.
In the second study, an agent produced to match etanercept showed very similar efficacy and safety to the branded formulation in a total of 233 randomized patients treated for 24 weeks at several centers in Korea.
The second study run in India was the "first clinical trial of a biosimilar infliximab to demonstrate and report kinetics of response to treatment at multiple time points prior to the plateau phase," which starts at about weeks 16, Dr. Jonathan Kay said at the annual European Congress of Rheumatology. It provides "convincing evidence of therapeutic equivalence," and also serves as a "new paradigm for comparative effectiveness testing of biosimilars," said Dr. Kay, a professor of medicine at the University of Massachusetts in Worcester.

Dr. Kay and his colleagues randomized 62 patients with active rheumatoid arthritis (RA) on stable doses of methotrexate to infliximab (Remicade) and 127 patients to BOW015, the biosimilar under study. They measured the proportion of responders at 2, 6, 14, and 16 weeks, with the study’s primary endpoint the percentage of patients showing an American College of Rheumatology 20 (ACR20) response at 16 weeks. The proportion of responders at each of these four time points was virtually identical, Dr. Kay reported. At 16 weeks, the percent of ACR20 responders was 86% in the infliximab arm and 90% in the biosimilar arm. The percent of ACR50 and ACR70 responders was also virtually the same in both treatment arms at 16 weeks, and the percent of ACR20 responders was virtually identical in the two treatment arms at weeks 2, 6, and 14.
The safety profiles of the two drugs also showed no statistically significant differences for any parameter measured except for the incidence of skin disorders, which occurred in one patient treated with the biosimilar and four patients treated with infliximab, a statistically significant difference.
At the same session, Dr. Sang-Cheol Bae of the Hanyang University Hospital for Rheumatic Diseases, Seoul, South Korea, presented findings from a randomized, controlled trial that compared the etanercept biosimilar HD203 with etanercept (Enbrel). Dr. Bae and his colleagues randomized 115 patients with active rheumatoid arthritis to HD203 and 118 patients to etanercept, both in combination with methotrexate, with the primary endpoint set as the proportion of patients achieving ACR20 by week 24. No statistically significant differences were seen between the patient groups for this primary endpoint, and equivalence in efficacy was demonstrated within predefined margins. The percent of ACR20 responders at 24 weeks was 79% in the biosimilar arm and 76% with etanercept.

Secondary analyses showed that the ACR50 responses occurred in 65% of the patients on the biosimilar at 24 weeks and in 53% of patients on etanercept, a statistically significant difference; and after 48 weeks of treatment, the ACR50 rate was 68% in the biosimilar arm and 55% with etanercept, also a statistically significant difference. The two study groups showed no significant difference in the rate of ACR70 responders after 24 or 48 weeks, and also no statistically significant difference in the rate of ACR20 responders after 48 weeks.
The results also showed that the biosimilar was "well tolerated," with a safety profile "comparable" with etanercept, Dr. Bae said.
Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.
* This story was updated 6/13/14.
*PARIS - Two different biosimilar drugs, one a mimic of infliximab, the other modeled on etanercept, closely matched the efficacy and safety of their brand-name counterparts in a pair of separate, randomized, controlled studies.
In one study, treatment with infliximab and a biosimilar agent showed virtually identical efficacy and safety performance at several time points during the first 16 weeks of treatment of 189 patients with rheumatoid arthritis in a head-to-head, randomized comparison done at 23 centers in India.
In the second study, an agent produced to match etanercept showed very similar efficacy and safety to the branded formulation in a total of 233 randomized patients treated for 24 weeks at several centers in Korea.
The second study run in India was the "first clinical trial of a biosimilar infliximab to demonstrate and report kinetics of response to treatment at multiple time points prior to the plateau phase," which starts at about weeks 16, Dr. Jonathan Kay said at the annual European Congress of Rheumatology. It provides "convincing evidence of therapeutic equivalence," and also serves as a "new paradigm for comparative effectiveness testing of biosimilars," said Dr. Kay, a professor of medicine at the University of Massachusetts in Worcester.
Dr. Kay and his colleagues randomized 62 patients with active rheumatoid arthritis (RA) on stable doses of methotrexate to infliximab (Remicade) and 127 patients to BOW015, the biosimilar under study. They measured the proportion of responders at 2, 6, 14, and 16 weeks, with the study’s primary endpoint the percentage of patients showing an American College of Rheumatology 20 (ACR20) response at 16 weeks. The proportion of responders at each of these four time points was virtually identical, Dr. Kay reported. At 16 weeks, the percent of ACR20 responders was 86% in the infliximab arm and 90% in the biosimilar arm. The percent of ACR50 and ACR70 responders was also virtually the same in both treatment arms at 16 weeks, and the percent of ACR20 responders was virtually identical in the two treatment arms at weeks 2, 6, and 14.
The safety profiles of the two drugs also showed no statistically significant differences for any parameter measured except for the incidence of skin disorders, which occurred in one patient treated with the biosimilar and four patients treated with infliximab, a statistically significant difference.
At the same session, Dr. Sang-Cheol Bae of the Hanyang University Hospital for Rheumatic Diseases, Seoul, South Korea, presented findings from a randomized, controlled trial that compared the etanercept biosimilar HD203 with etanercept (Enbrel). Dr. Bae and his colleagues randomized 115 patients with active rheumatoid arthritis to HD203 and 118 patients to etanercept, both in combination with methotrexate, with the primary endpoint set as the proportion of patients achieving ACR20 by week 24. No statistically significant differences were seen between the patient groups for this primary endpoint, and equivalence in efficacy was demonstrated within predefined margins. The percent of ACR20 responders at 24 weeks was 79% in the biosimilar arm and 76% with etanercept.
Secondary analyses showed that the ACR50 responses occurred in 65% of the patients on the biosimilar at 24 weeks and in 53% of patients on etanercept, a statistically significant difference; and after 48 weeks of treatment, the ACR50 rate was 68% in the biosimilar arm and 55% with etanercept, also a statistically significant difference. The two study groups showed no significant difference in the rate of ACR70 responders after 24 or 48 weeks, and also no statistically significant difference in the rate of ACR20 responders after 48 weeks.
The results also showed that the biosimilar was "well tolerated," with a safety profile "comparable" with etanercept, Dr. Bae said.
Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.
* This story was updated 6/13/14.
*PARIS - Two different biosimilar drugs, one a mimic of infliximab, the other modeled on etanercept, closely matched the efficacy and safety of their brand-name counterparts in a pair of separate, randomized, controlled studies.
In one study, treatment with infliximab and a biosimilar agent showed virtually identical efficacy and safety performance at several time points during the first 16 weeks of treatment of 189 patients with rheumatoid arthritis in a head-to-head, randomized comparison done at 23 centers in India.
In the second study, an agent produced to match etanercept showed very similar efficacy and safety to the branded formulation in a total of 233 randomized patients treated for 24 weeks at several centers in Korea.
The second study run in India was the "first clinical trial of a biosimilar infliximab to demonstrate and report kinetics of response to treatment at multiple time points prior to the plateau phase," which starts at about weeks 16, Dr. Jonathan Kay said at the annual European Congress of Rheumatology. It provides "convincing evidence of therapeutic equivalence," and also serves as a "new paradigm for comparative effectiveness testing of biosimilars," said Dr. Kay, a professor of medicine at the University of Massachusetts in Worcester.
Dr. Kay and his colleagues randomized 62 patients with active rheumatoid arthritis (RA) on stable doses of methotrexate to infliximab (Remicade) and 127 patients to BOW015, the biosimilar under study. They measured the proportion of responders at 2, 6, 14, and 16 weeks, with the study’s primary endpoint the percentage of patients showing an American College of Rheumatology 20 (ACR20) response at 16 weeks. The proportion of responders at each of these four time points was virtually identical, Dr. Kay reported. At 16 weeks, the percent of ACR20 responders was 86% in the infliximab arm and 90% in the biosimilar arm. The percent of ACR50 and ACR70 responders was also virtually the same in both treatment arms at 16 weeks, and the percent of ACR20 responders was virtually identical in the two treatment arms at weeks 2, 6, and 14.
The safety profiles of the two drugs also showed no statistically significant differences for any parameter measured except for the incidence of skin disorders, which occurred in one patient treated with the biosimilar and four patients treated with infliximab, a statistically significant difference.
At the same session, Dr. Sang-Cheol Bae of the Hanyang University Hospital for Rheumatic Diseases, Seoul, South Korea, presented findings from a randomized, controlled trial that compared the etanercept biosimilar HD203 with etanercept (Enbrel). Dr. Bae and his colleagues randomized 115 patients with active rheumatoid arthritis to HD203 and 118 patients to etanercept, both in combination with methotrexate, with the primary endpoint set as the proportion of patients achieving ACR20 by week 24. No statistically significant differences were seen between the patient groups for this primary endpoint, and equivalence in efficacy was demonstrated within predefined margins. The percent of ACR20 responders at 24 weeks was 79% in the biosimilar arm and 76% with etanercept.
Secondary analyses showed that the ACR50 responses occurred in 65% of the patients on the biosimilar at 24 weeks and in 53% of patients on etanercept, a statistically significant difference; and after 48 weeks of treatment, the ACR50 rate was 68% in the biosimilar arm and 55% with etanercept, also a statistically significant difference. The two study groups showed no significant difference in the rate of ACR70 responders after 24 or 48 weeks, and also no statistically significant difference in the rate of ACR20 responders after 48 weeks.
The results also showed that the biosimilar was "well tolerated," with a safety profile "comparable" with etanercept, Dr. Bae said.
Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.
* This story was updated 6/13/14.
AT THE EULAR CONGRESS 2014
Key clinical point: Two biosimilar agents showed nearly identical efficacy, safety to
infliximab and etanercept in a pair of randomized trials.
Major finding: An ACR20 response was seen in 86% of the infliximab arm and 90% in the biosimilar arm in the first study and 79% in the biosimilar arm and 76% with etanercept in the second study.
Data source: Two randomized, controlled studies, one with 189 patients, the other with 233 patients.
Disclosures: Dr. Kay has received research funding and/or consulting income from AbbVie, Amgen, Bristol-Myers Squibb, Eli Lilly, Epirus, Genentech, Hospira, Janssen, Pfizer, Roche, and UCB. Dr. Bae acknowledged receiving consulting income and/or research support from Abbott, Bristol-Myers Squibb, Eisai, GlaxoSmithKline, Hanwha Chemical, Merck Serono, MSD, and Pfizer.

Naloxegol cut opioid-associated constipation without impairing pain relief
Treatment with opioid receptor-antagonist naloxegol significantly improved opioid-associated constipation, compared with placebo, without affecting pain scores or daily opioid requirements, according to data from two identical double-blind studies.
Outpatients with noncancer pain who were given 25 mg of naloxegol showed a significantly shorter time to first spontaneous bowel movement after treatment, compared with those given placebo – a median time of 5.9 hours and 12 hours in the two studies, compared with 35.8 hours and 37.2 hours with placebo, according to a study published online June 4 in the New England Journal of Medicine.
Treatment with naloxegol was also associated with a significantly greater number of spontaneous bowel movements over the course of the 12-week study period, compared with placebo, and an increase in the mean number of days per week with one or more spontaneous bowel movements.
The Food and Drug Administration is currently considering whether to approve naloxegol; the agency is expected to decide by Sept. 16, 2014.
The two phase III randomized, controlled studies were nearly identical in size – one including 652 individuals with opioid-induced constipation, and the other including 700 – and entirely identical in design: Participants were randomized to receive either 25 mg or 12.5 mg of naloxegol daily, or placebo.
"In both studies, naloxegol at a dose of 25 mg was associated with an increased rate of response (10-15 percentage points higher than the response with placebo) over a period of 12 weeks," wrote Dr. William D. Chey of the University of Michigan Health System, Ann Arbor, and his colleagues.
The higher dose of naloxegol also was associated with more significant improvements in severity of straining, stool consistency, and the frequency of days with complete, spontaneous bowel movements.
Naloxegol’s benefits were even greater among individuals who had previously failed to respond to laxatives before study enrollment, which accounted for 71% of participants, a prespecified subgroup analysis showed (N. Engl. J. Med. 2014 June 4 [doi:10.1056/NEJMoa1310246]).
"In clinical practice, osmotic and stimulant laxatives are likely to be used before more expensive prescription medications," the researchers wrote. "Thus, the finding that naloxegol proved beneficial in patients who had persistent symptoms of opioid-induced constipation despite using standard laxatives is of potential importance."
There were some dose-related side effects observed in the naloxegol group, including abdominal pain, nausea, diarrhea, and vomiting, occurring soon after initiation of treatment. But most of these effects were mild to moderate.
There had been concern about potential cardiovascular side effects, which had been observed previously with alvimopan, another peripherally-acting mu-opioid antagonist. However the incidence of major cardiovascular events was rare and similar across both active and placebo groups.
Researchers found no significant interaction between naloxegol treatment and daily opioid dose, and there also were no significant differences in the mean change from baseline in pain scores.
Around half of the patients enrolled were taking opioids for back pain, while other reasons included arthritis, joint pain, or fibromyalgia. On average, participants had been taking opioids for 3.65 years.
AstraZeneca supported the study, and the authors declared a range of grants, consultancies, and other financial relationships with several pharmaceutical companies, including AstraZeneca.
Treatment with opioid receptor-antagonist naloxegol significantly improved opioid-associated constipation, compared with placebo, without affecting pain scores or daily opioid requirements, according to data from two identical double-blind studies.
Outpatients with noncancer pain who were given 25 mg of naloxegol showed a significantly shorter time to first spontaneous bowel movement after treatment, compared with those given placebo – a median time of 5.9 hours and 12 hours in the two studies, compared with 35.8 hours and 37.2 hours with placebo, according to a study published online June 4 in the New England Journal of Medicine.
Treatment with naloxegol was also associated with a significantly greater number of spontaneous bowel movements over the course of the 12-week study period, compared with placebo, and an increase in the mean number of days per week with one or more spontaneous bowel movements.
The Food and Drug Administration is currently considering whether to approve naloxegol; the agency is expected to decide by Sept. 16, 2014.
The two phase III randomized, controlled studies were nearly identical in size – one including 652 individuals with opioid-induced constipation, and the other including 700 – and entirely identical in design: Participants were randomized to receive either 25 mg or 12.5 mg of naloxegol daily, or placebo.
"In both studies, naloxegol at a dose of 25 mg was associated with an increased rate of response (10-15 percentage points higher than the response with placebo) over a period of 12 weeks," wrote Dr. William D. Chey of the University of Michigan Health System, Ann Arbor, and his colleagues.
The higher dose of naloxegol also was associated with more significant improvements in severity of straining, stool consistency, and the frequency of days with complete, spontaneous bowel movements.
Naloxegol’s benefits were even greater among individuals who had previously failed to respond to laxatives before study enrollment, which accounted for 71% of participants, a prespecified subgroup analysis showed (N. Engl. J. Med. 2014 June 4 [doi:10.1056/NEJMoa1310246]).
"In clinical practice, osmotic and stimulant laxatives are likely to be used before more expensive prescription medications," the researchers wrote. "Thus, the finding that naloxegol proved beneficial in patients who had persistent symptoms of opioid-induced constipation despite using standard laxatives is of potential importance."
There were some dose-related side effects observed in the naloxegol group, including abdominal pain, nausea, diarrhea, and vomiting, occurring soon after initiation of treatment. But most of these effects were mild to moderate.
There had been concern about potential cardiovascular side effects, which had been observed previously with alvimopan, another peripherally-acting mu-opioid antagonist. However the incidence of major cardiovascular events was rare and similar across both active and placebo groups.
Researchers found no significant interaction between naloxegol treatment and daily opioid dose, and there also were no significant differences in the mean change from baseline in pain scores.
Around half of the patients enrolled were taking opioids for back pain, while other reasons included arthritis, joint pain, or fibromyalgia. On average, participants had been taking opioids for 3.65 years.
AstraZeneca supported the study, and the authors declared a range of grants, consultancies, and other financial relationships with several pharmaceutical companies, including AstraZeneca.
Treatment with opioid receptor-antagonist naloxegol significantly improved opioid-associated constipation, compared with placebo, without affecting pain scores or daily opioid requirements, according to data from two identical double-blind studies.
Outpatients with noncancer pain who were given 25 mg of naloxegol showed a significantly shorter time to first spontaneous bowel movement after treatment, compared with those given placebo – a median time of 5.9 hours and 12 hours in the two studies, compared with 35.8 hours and 37.2 hours with placebo, according to a study published online June 4 in the New England Journal of Medicine.
Treatment with naloxegol was also associated with a significantly greater number of spontaneous bowel movements over the course of the 12-week study period, compared with placebo, and an increase in the mean number of days per week with one or more spontaneous bowel movements.
The Food and Drug Administration is currently considering whether to approve naloxegol; the agency is expected to decide by Sept. 16, 2014.
The two phase III randomized, controlled studies were nearly identical in size – one including 652 individuals with opioid-induced constipation, and the other including 700 – and entirely identical in design: Participants were randomized to receive either 25 mg or 12.5 mg of naloxegol daily, or placebo.
"In both studies, naloxegol at a dose of 25 mg was associated with an increased rate of response (10-15 percentage points higher than the response with placebo) over a period of 12 weeks," wrote Dr. William D. Chey of the University of Michigan Health System, Ann Arbor, and his colleagues.
The higher dose of naloxegol also was associated with more significant improvements in severity of straining, stool consistency, and the frequency of days with complete, spontaneous bowel movements.
Naloxegol’s benefits were even greater among individuals who had previously failed to respond to laxatives before study enrollment, which accounted for 71% of participants, a prespecified subgroup analysis showed (N. Engl. J. Med. 2014 June 4 [doi:10.1056/NEJMoa1310246]).
"In clinical practice, osmotic and stimulant laxatives are likely to be used before more expensive prescription medications," the researchers wrote. "Thus, the finding that naloxegol proved beneficial in patients who had persistent symptoms of opioid-induced constipation despite using standard laxatives is of potential importance."
There were some dose-related side effects observed in the naloxegol group, including abdominal pain, nausea, diarrhea, and vomiting, occurring soon after initiation of treatment. But most of these effects were mild to moderate.
There had been concern about potential cardiovascular side effects, which had been observed previously with alvimopan, another peripherally-acting mu-opioid antagonist. However the incidence of major cardiovascular events was rare and similar across both active and placebo groups.
Researchers found no significant interaction between naloxegol treatment and daily opioid dose, and there also were no significant differences in the mean change from baseline in pain scores.
Around half of the patients enrolled were taking opioids for back pain, while other reasons included arthritis, joint pain, or fibromyalgia. On average, participants had been taking opioids for 3.65 years.
AstraZeneca supported the study, and the authors declared a range of grants, consultancies, and other financial relationships with several pharmaceutical companies, including AstraZeneca.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Naloxegol improves opioid-induced constipation without affecting pain relief.
Major finding: Treatment with naloxegol was associated with a significantly shorter time to first spontaneous bowel movement, a significantly greater number of spontaneous bowel movements, and an increase in the mean number of days per week with one or more spontaneous bowel movements, compared with placebo.
Data source: Two identical 12-week, phase III, double-blind, randomized, controlled trials in a total of 1,352 patients with opioid-associated constipation.
Disclosures: AstraZeneca supported the study, and the authors declared a range of grants, consultancies, and other financial relationships with several pharmaceutical companies including AstraZeneca.
FDA approves blood test for membranous glomerulonephritis
The Food and Drug Administration has approved a noninvasive test to determine whether a chronic kidney disease is caused by an autoimmune disease or another cause such as infection.
The EUROIMMUN Anti- PLA2R IFA blood test detects an antibody that is specific to primary membranous glomerulonephritis (pMGN). MGN, a chronic kidney disease, damages the glomeruli; it can lead to kidney failure and transplant. Symptoms include swelling, hypercholesterolemia, hypertension, and an increased predisposition to blood clots.
The condition mostly affects white men. It occurs in 2 of every 10,000 people and is more common after age 40, according to the National Library of Medicine. Risk factors include cancers, especially lung and colon cancer; exposure to toxins, including gold and mercury; infections, including hepatitis B, malaria, syphilis, and endocarditis; certain medications, including penicillamine, trimethadione, and skin-lightening creams; and systemic lupus erythematosus, rheumatoid arthritis, Graves’ disease, and other autoimmune disorders.
"Treatment of MGN depends on the underlying cause of the disease," said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health, in a statement. "This test can help patients get a timely diagnosis for their MGN and aid with earlier treatment."
Test manufacturer EUROIMMUN US submitted data that compared 275 blood samples from patients with presumed pMGN, with 285 samples from patients diagnosed with other kidney diseases including secondary MGN (sMGN) and autoimmune diseases. The test detected pMGN in 77% of the presumed pMGN samples, and gave a false-positive result in less than 1% of the other samples.
The diagnostic test helped distinguish pMGN from sMGN in most of the patients.
The FDA said that the test should not be used alone to diagnose pMGN, but that patients’ symptoms and other laboratory test results should also be considered. A kidney biopsy is required for confirmation, according to the FDA.
On Twitter @aliciaault
The Food and Drug Administration has approved a noninvasive test to determine whether a chronic kidney disease is caused by an autoimmune disease or another cause such as infection.
The EUROIMMUN Anti- PLA2R IFA blood test detects an antibody that is specific to primary membranous glomerulonephritis (pMGN). MGN, a chronic kidney disease, damages the glomeruli; it can lead to kidney failure and transplant. Symptoms include swelling, hypercholesterolemia, hypertension, and an increased predisposition to blood clots.
The condition mostly affects white men. It occurs in 2 of every 10,000 people and is more common after age 40, according to the National Library of Medicine. Risk factors include cancers, especially lung and colon cancer; exposure to toxins, including gold and mercury; infections, including hepatitis B, malaria, syphilis, and endocarditis; certain medications, including penicillamine, trimethadione, and skin-lightening creams; and systemic lupus erythematosus, rheumatoid arthritis, Graves’ disease, and other autoimmune disorders.
"Treatment of MGN depends on the underlying cause of the disease," said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health, in a statement. "This test can help patients get a timely diagnosis for their MGN and aid with earlier treatment."
Test manufacturer EUROIMMUN US submitted data that compared 275 blood samples from patients with presumed pMGN, with 285 samples from patients diagnosed with other kidney diseases including secondary MGN (sMGN) and autoimmune diseases. The test detected pMGN in 77% of the presumed pMGN samples, and gave a false-positive result in less than 1% of the other samples.
The diagnostic test helped distinguish pMGN from sMGN in most of the patients.
The FDA said that the test should not be used alone to diagnose pMGN, but that patients’ symptoms and other laboratory test results should also be considered. A kidney biopsy is required for confirmation, according to the FDA.
On Twitter @aliciaault
The Food and Drug Administration has approved a noninvasive test to determine whether a chronic kidney disease is caused by an autoimmune disease or another cause such as infection.
The EUROIMMUN Anti- PLA2R IFA blood test detects an antibody that is specific to primary membranous glomerulonephritis (pMGN). MGN, a chronic kidney disease, damages the glomeruli; it can lead to kidney failure and transplant. Symptoms include swelling, hypercholesterolemia, hypertension, and an increased predisposition to blood clots.
The condition mostly affects white men. It occurs in 2 of every 10,000 people and is more common after age 40, according to the National Library of Medicine. Risk factors include cancers, especially lung and colon cancer; exposure to toxins, including gold and mercury; infections, including hepatitis B, malaria, syphilis, and endocarditis; certain medications, including penicillamine, trimethadione, and skin-lightening creams; and systemic lupus erythematosus, rheumatoid arthritis, Graves’ disease, and other autoimmune disorders.
"Treatment of MGN depends on the underlying cause of the disease," said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health at the FDA’s Center for Devices and Radiological Health, in a statement. "This test can help patients get a timely diagnosis for their MGN and aid with earlier treatment."
Test manufacturer EUROIMMUN US submitted data that compared 275 blood samples from patients with presumed pMGN, with 285 samples from patients diagnosed with other kidney diseases including secondary MGN (sMGN) and autoimmune diseases. The test detected pMGN in 77% of the presumed pMGN samples, and gave a false-positive result in less than 1% of the other samples.
The diagnostic test helped distinguish pMGN from sMGN in most of the patients.
The FDA said that the test should not be used alone to diagnose pMGN, but that patients’ symptoms and other laboratory test results should also be considered. A kidney biopsy is required for confirmation, according to the FDA.
On Twitter @aliciaault
RA’s heterogeneity poses challenge to ‘personalized medicine’
DESTIN, FLA. – Personalized medicine – picking the right patient for the right drug – is a hot topic in medicine in general, but the approach is challenging for rheumatoid arthritis because of its heterogeneity, according to Dr. Arthur Kavanaugh.
"How do we get to personalized medicine? Do we have a biomarker for any of our treatments? Well, ‘biomarker’ is sort of a generic term. We actually don’t want a biomarker – anything is a biomarker. What we want are surrogate markers. We want something that is so strong, that we can measure that and predict what’s going to happen," Dr. Kavanaugh, professor of medicine and director of the Center for Innovative Therapy at the University of California, San Diego, said at the annual Congress of Clinical Rheumatology.

He used the example of CD4 count in HIV infection and dual-energy x-ray absorptiometry scans for fragility fractures as useful surrogate markers.
"Some people would say ‘cholesterol for atherosclerotic disease.’ Well, you don’t care about the cholesterol – you care about whether you’re going to have a heart attack. So a surrogate marker is a much higher standard," he explained.
In rheumatology, there is a lack of such surrogate markers.
"We have lots of biomarkers, but there’s really nothing that’s a surrogate marker in rheumatology that will allow us to pick one treatment over another. It’s something we would love to see, and it’s been looked at in a number of ways," he said.
For example, the genomic approach was expected to be the answer.
"For those of us who were around before the human genome was sequenced – that was going to be the answer. Go to the doctor, swab your cheek, turn that in to the nurse, walk out with a prescription based on your genes – that was going to be it. It hasn’t worked out that way; humans are complicated," he said.
Proteomics, glycomics, immunomics – all kinds of things have been looked at, Dr. Kavanaugh pointed out.
For awhile, geneticists were promising that if they could just get enough samples, they would be able to figure out which patients should get which drug.
"Well, they kind of failed, and now I think a very interesting article that just came out in Nature is just sort of them surrendering and saying, ‘OK, what we’ve done now is take the treatments you know work, and we will figure out which genes predict that,’" he said.
The paper is a meta-analysis of genome-wide association studies, and it looks at how RA genetics can contribute to drug discovery (Nature 2014;506: 376-81).
The authors found about 100 loci that seemed to be informative, Dr. Kavanaugh said. "It’s very interesting, because ... a lot of the loci map to therapies that we have now."
So maybe this backwards "bedside-to-bench" approach will prove useful for providing more personalized medicine.
Epigenetics might also have promise for advancing personalized medicine for RA.
"It’s fascinating. We don’t know so much about it, but of course they are heritable alterations that don’t change the DNA sequence," he said.
Examples include DNA methylation, micro-RNAs, which affect the expression of genes, and histone modification.
These can be inherited, and while the concept opens up some politically charged and socioeconomic issues with respect to how epigenetic modifications might affect an individual, it’s an area that may have some relevance in personalized medicine, Dr. Kavanaugh said.
In some ways, the progress with personalized medicine in RA has been hampered by the results in other fields.
"There’s been so much progress in oncology, but they have more monogenic disease," he said, explaining that a single abnormality can predict whether a drug will work in 0% or 80% of cases. "That’s personalized medicine, but I don’t think we have that for rheumatic disease. It would be great if we did, but I don’t think we’re there yet."
In the rheumatic diseases, it may be that things have to be considered in a different, more complex way, by using combinations of biomarkers, for example.
In a poster presented at EULAR in 2013, based on the ADACTA study, he and his colleagues reported that none of a number of single biomarkers predicted response, but that patterns of biomarkers – in this case serum markers that correlated with immunologic phenotypes in the synovium from previous studies – appeared to have predictive value.
"Of course, this is predicting the past ... we have to see if this kind of stuff holds up. We would all love to have it. This is the next challenge – using it in a more rational way so that we can get our patients to the best possible place," he said.
Dr. Kavanaugh reported having no relevant financial disclosures.
DESTIN, FLA. – Personalized medicine – picking the right patient for the right drug – is a hot topic in medicine in general, but the approach is challenging for rheumatoid arthritis because of its heterogeneity, according to Dr. Arthur Kavanaugh.
"How do we get to personalized medicine? Do we have a biomarker for any of our treatments? Well, ‘biomarker’ is sort of a generic term. We actually don’t want a biomarker – anything is a biomarker. What we want are surrogate markers. We want something that is so strong, that we can measure that and predict what’s going to happen," Dr. Kavanaugh, professor of medicine and director of the Center for Innovative Therapy at the University of California, San Diego, said at the annual Congress of Clinical Rheumatology.
He used the example of CD4 count in HIV infection and dual-energy x-ray absorptiometry scans for fragility fractures as useful surrogate markers.
"Some people would say ‘cholesterol for atherosclerotic disease.’ Well, you don’t care about the cholesterol – you care about whether you’re going to have a heart attack. So a surrogate marker is a much higher standard," he explained.
In rheumatology, there is a lack of such surrogate markers.
"We have lots of biomarkers, but there’s really nothing that’s a surrogate marker in rheumatology that will allow us to pick one treatment over another. It’s something we would love to see, and it’s been looked at in a number of ways," he said.
For example, the genomic approach was expected to be the answer.
"For those of us who were around before the human genome was sequenced – that was going to be the answer. Go to the doctor, swab your cheek, turn that in to the nurse, walk out with a prescription based on your genes – that was going to be it. It hasn’t worked out that way; humans are complicated," he said.
Proteomics, glycomics, immunomics – all kinds of things have been looked at, Dr. Kavanaugh pointed out.
For awhile, geneticists were promising that if they could just get enough samples, they would be able to figure out which patients should get which drug.
"Well, they kind of failed, and now I think a very interesting article that just came out in Nature is just sort of them surrendering and saying, ‘OK, what we’ve done now is take the treatments you know work, and we will figure out which genes predict that,’" he said.
The paper is a meta-analysis of genome-wide association studies, and it looks at how RA genetics can contribute to drug discovery (Nature 2014;506: 376-81).
The authors found about 100 loci that seemed to be informative, Dr. Kavanaugh said. "It’s very interesting, because ... a lot of the loci map to therapies that we have now."
So maybe this backwards "bedside-to-bench" approach will prove useful for providing more personalized medicine.
Epigenetics might also have promise for advancing personalized medicine for RA.
"It’s fascinating. We don’t know so much about it, but of course they are heritable alterations that don’t change the DNA sequence," he said.
Examples include DNA methylation, micro-RNAs, which affect the expression of genes, and histone modification.
These can be inherited, and while the concept opens up some politically charged and socioeconomic issues with respect to how epigenetic modifications might affect an individual, it’s an area that may have some relevance in personalized medicine, Dr. Kavanaugh said.
In some ways, the progress with personalized medicine in RA has been hampered by the results in other fields.
"There’s been so much progress in oncology, but they have more monogenic disease," he said, explaining that a single abnormality can predict whether a drug will work in 0% or 80% of cases. "That’s personalized medicine, but I don’t think we have that for rheumatic disease. It would be great if we did, but I don’t think we’re there yet."
In the rheumatic diseases, it may be that things have to be considered in a different, more complex way, by using combinations of biomarkers, for example.
In a poster presented at EULAR in 2013, based on the ADACTA study, he and his colleagues reported that none of a number of single biomarkers predicted response, but that patterns of biomarkers – in this case serum markers that correlated with immunologic phenotypes in the synovium from previous studies – appeared to have predictive value.
"Of course, this is predicting the past ... we have to see if this kind of stuff holds up. We would all love to have it. This is the next challenge – using it in a more rational way so that we can get our patients to the best possible place," he said.
Dr. Kavanaugh reported having no relevant financial disclosures.
DESTIN, FLA. – Personalized medicine – picking the right patient for the right drug – is a hot topic in medicine in general, but the approach is challenging for rheumatoid arthritis because of its heterogeneity, according to Dr. Arthur Kavanaugh.
"How do we get to personalized medicine? Do we have a biomarker for any of our treatments? Well, ‘biomarker’ is sort of a generic term. We actually don’t want a biomarker – anything is a biomarker. What we want are surrogate markers. We want something that is so strong, that we can measure that and predict what’s going to happen," Dr. Kavanaugh, professor of medicine and director of the Center for Innovative Therapy at the University of California, San Diego, said at the annual Congress of Clinical Rheumatology.
He used the example of CD4 count in HIV infection and dual-energy x-ray absorptiometry scans for fragility fractures as useful surrogate markers.
"Some people would say ‘cholesterol for atherosclerotic disease.’ Well, you don’t care about the cholesterol – you care about whether you’re going to have a heart attack. So a surrogate marker is a much higher standard," he explained.
In rheumatology, there is a lack of such surrogate markers.
"We have lots of biomarkers, but there’s really nothing that’s a surrogate marker in rheumatology that will allow us to pick one treatment over another. It’s something we would love to see, and it’s been looked at in a number of ways," he said.
For example, the genomic approach was expected to be the answer.
"For those of us who were around before the human genome was sequenced – that was going to be the answer. Go to the doctor, swab your cheek, turn that in to the nurse, walk out with a prescription based on your genes – that was going to be it. It hasn’t worked out that way; humans are complicated," he said.
Proteomics, glycomics, immunomics – all kinds of things have been looked at, Dr. Kavanaugh pointed out.
For awhile, geneticists were promising that if they could just get enough samples, they would be able to figure out which patients should get which drug.
"Well, they kind of failed, and now I think a very interesting article that just came out in Nature is just sort of them surrendering and saying, ‘OK, what we’ve done now is take the treatments you know work, and we will figure out which genes predict that,’" he said.
The paper is a meta-analysis of genome-wide association studies, and it looks at how RA genetics can contribute to drug discovery (Nature 2014;506: 376-81).
The authors found about 100 loci that seemed to be informative, Dr. Kavanaugh said. "It’s very interesting, because ... a lot of the loci map to therapies that we have now."
So maybe this backwards "bedside-to-bench" approach will prove useful for providing more personalized medicine.
Epigenetics might also have promise for advancing personalized medicine for RA.
"It’s fascinating. We don’t know so much about it, but of course they are heritable alterations that don’t change the DNA sequence," he said.
Examples include DNA methylation, micro-RNAs, which affect the expression of genes, and histone modification.
These can be inherited, and while the concept opens up some politically charged and socioeconomic issues with respect to how epigenetic modifications might affect an individual, it’s an area that may have some relevance in personalized medicine, Dr. Kavanaugh said.
In some ways, the progress with personalized medicine in RA has been hampered by the results in other fields.
"There’s been so much progress in oncology, but they have more monogenic disease," he said, explaining that a single abnormality can predict whether a drug will work in 0% or 80% of cases. "That’s personalized medicine, but I don’t think we have that for rheumatic disease. It would be great if we did, but I don’t think we’re there yet."
In the rheumatic diseases, it may be that things have to be considered in a different, more complex way, by using combinations of biomarkers, for example.
In a poster presented at EULAR in 2013, based on the ADACTA study, he and his colleagues reported that none of a number of single biomarkers predicted response, but that patterns of biomarkers – in this case serum markers that correlated with immunologic phenotypes in the synovium from previous studies – appeared to have predictive value.
"Of course, this is predicting the past ... we have to see if this kind of stuff holds up. We would all love to have it. This is the next challenge – using it in a more rational way so that we can get our patients to the best possible place," he said.
Dr. Kavanaugh reported having no relevant financial disclosures.
EXPERT ANALYSIS FROM CCR 14

Triple therapy topped monotherapy in early rheumatoid arthritis
In arithmetic and newly diagnosed rheumatoid arthritis alike, three is greater than one.
Initial triple disease-modifying therapy with either of two glucocorticoid (GC) bridging therapies proved quicker in attaining treatment goals and required fewer treatment intensifications for patients with very early RA than did initial methotrexate monotherapy with oral GC therapy, according to the results of a randomized, comparative trial.
Several previous clinical trials concluded that initial combination therapy had better clinical efficacy than monotherapy. However, most rheumatologists have not incorporated this into daily practice, according to Dr. Pascal Hendrik Pieter de Jong of the University Medical Center, Rotterdam, the Netherlands, and his colleagues.
Previous combination-therapy studies have usually been disregarded, the researchers noted, because the trials were biased by the use of GCs. So, Dr. de Jong and his colleagues compared the benefits of the 1-year clinical efficacy of initial triple disease-modifying antirheumatic drugs to the benefits of initial methotrexate monotherapy unbiased by the use of GC therapy, either oral or intramuscular.
Primary outcomes of the study were the area under the curve of the Health Assessment Questionnaire (to assess functional ability) and Disease Activity Score (to assess disease state), and the proportion of patients with radiographic progression, the investigators reported online May 1 in Annals of the Rheumatic Diseases (Ann. Rheum. Dis. May 1, 2014 [doi: 10.1136/annrheumdis-2013-204788]).
The study population comprised the 281 patients with a high probability of progress to persistent arthritis (greater than 70%) who were in the Rotterdam Early Arthritis Cohort (tReach) trial. All patients were over age 18, with arthritis in one or more joints and symptom duration of less than 1 year; 68% of the patients were women.
Patients were randomized to one of three arms:
1) initial triple disease-modifying therapy (iTDT) of methotrexate plus sulfasalazine and hydroxychloroquine, with GCs administered intramuscularly,
2) iTDT with an oral GC tapering scheme, or
3) initial methotrexate monotherapy (iMM) with oral GCs similar to the second treatment arm.
Concurrent treatment with nonsteroidal anti-inflammatory drugs and intra-articular injections (maximum of two per 3 months) was allowed.
The largest difference in disease activity states between the treatment arms was seen after 3 months, although that effect gradually diminished to nonsignificance by 12 months. Similarly, there were no significant differences in functional ability at 12 months.
After 3 months, 40% fewer biological agents were prescribed in the iTDT group than in the iMM group, and that difference persisted over time. However, disease activity, functional ability, and radiographic progression were not different between the two treatments after 12 months, regardless of GC bridging therapies. Early initiation of biological agents in the iMM group might have prevented or delayed the radiographic progression, the researchers suggested.
There were no differences in serious adverse events seen in the treatment arms, but the proportion of patients with medication adjustments due to adverse events was significantly higher in the iTDT (65%), compared with the iMM group (45%). No differences were seen between the two GC bridging therapies. Gastrointestinal complaints (56%) and fatigue (36%) were the two most commonly reported adverse events.
"We found no differences in disease activity, functional ability, and radiographic progression after 12 months of treatment owing to our treat-to-target approach (intensifying treatment until the target is reached)," the investigators observed. "Therefore, it is not the endpoint, but progress towards the endpoint, which matters most."
In addition, delaying the implementation of biological agents with iTDT treatment resulted in "reducing costs enormously," according to the researchers, although the cost-utility analysis of the tREACH trial has to reconfirm this, they added.
"We think that future research should focus on developing a more personalized treatment approach, in which differentiation between patients who would thrive on iMM and those who need iTDT might be a first step," they said.
"Before choosing the initial treatment strategy, rheumatologists should be aware of the benefits and the risks, but additionally, known prognostic factors and the patient’s wish should be taken into account. One single intramuscular GC injection and a low-dose, oral GC tapering scheme would be sufficient as bridging therapy," the researchers concluded.
The research was supported by an unrestricted grant from Pfizer, which had no involvement in the study design, data collection, analysis, or publication of the results. Data management was sponsored by the Dutch Arthritis Foundation. The authors reported that they had no competing interests.
Initial triple disease-modifying therapy, glucocorticoid, early RA, methotrexate monotherapy with oral GC therapy, initial combination therapy, Dr. Pascal Hendrik Pieter de Jong, Health Assessment Questionnaire, Disease Activity Score, radiographic progression, Annals of the Rheumatic Diseases, Rotterdam Early Arthritis Cohort (tReach) trial,
In arithmetic and newly diagnosed rheumatoid arthritis alike, three is greater than one.
Initial triple disease-modifying therapy with either of two glucocorticoid (GC) bridging therapies proved quicker in attaining treatment goals and required fewer treatment intensifications for patients with very early RA than did initial methotrexate monotherapy with oral GC therapy, according to the results of a randomized, comparative trial.
Several previous clinical trials concluded that initial combination therapy had better clinical efficacy than monotherapy. However, most rheumatologists have not incorporated this into daily practice, according to Dr. Pascal Hendrik Pieter de Jong of the University Medical Center, Rotterdam, the Netherlands, and his colleagues.
Previous combination-therapy studies have usually been disregarded, the researchers noted, because the trials were biased by the use of GCs. So, Dr. de Jong and his colleagues compared the benefits of the 1-year clinical efficacy of initial triple disease-modifying antirheumatic drugs to the benefits of initial methotrexate monotherapy unbiased by the use of GC therapy, either oral or intramuscular.
Primary outcomes of the study were the area under the curve of the Health Assessment Questionnaire (to assess functional ability) and Disease Activity Score (to assess disease state), and the proportion of patients with radiographic progression, the investigators reported online May 1 in Annals of the Rheumatic Diseases (Ann. Rheum. Dis. May 1, 2014 [doi: 10.1136/annrheumdis-2013-204788]).
The study population comprised the 281 patients with a high probability of progress to persistent arthritis (greater than 70%) who were in the Rotterdam Early Arthritis Cohort (tReach) trial. All patients were over age 18, with arthritis in one or more joints and symptom duration of less than 1 year; 68% of the patients were women.
Patients were randomized to one of three arms:
1) initial triple disease-modifying therapy (iTDT) of methotrexate plus sulfasalazine and hydroxychloroquine, with GCs administered intramuscularly,
2) iTDT with an oral GC tapering scheme, or
3) initial methotrexate monotherapy (iMM) with oral GCs similar to the second treatment arm.
Concurrent treatment with nonsteroidal anti-inflammatory drugs and intra-articular injections (maximum of two per 3 months) was allowed.
The largest difference in disease activity states between the treatment arms was seen after 3 months, although that effect gradually diminished to nonsignificance by 12 months. Similarly, there were no significant differences in functional ability at 12 months.
After 3 months, 40% fewer biological agents were prescribed in the iTDT group than in the iMM group, and that difference persisted over time. However, disease activity, functional ability, and radiographic progression were not different between the two treatments after 12 months, regardless of GC bridging therapies. Early initiation of biological agents in the iMM group might have prevented or delayed the radiographic progression, the researchers suggested.
There were no differences in serious adverse events seen in the treatment arms, but the proportion of patients with medication adjustments due to adverse events was significantly higher in the iTDT (65%), compared with the iMM group (45%). No differences were seen between the two GC bridging therapies. Gastrointestinal complaints (56%) and fatigue (36%) were the two most commonly reported adverse events.
"We found no differences in disease activity, functional ability, and radiographic progression after 12 months of treatment owing to our treat-to-target approach (intensifying treatment until the target is reached)," the investigators observed. "Therefore, it is not the endpoint, but progress towards the endpoint, which matters most."
In addition, delaying the implementation of biological agents with iTDT treatment resulted in "reducing costs enormously," according to the researchers, although the cost-utility analysis of the tREACH trial has to reconfirm this, they added.
"We think that future research should focus on developing a more personalized treatment approach, in which differentiation between patients who would thrive on iMM and those who need iTDT might be a first step," they said.
"Before choosing the initial treatment strategy, rheumatologists should be aware of the benefits and the risks, but additionally, known prognostic factors and the patient’s wish should be taken into account. One single intramuscular GC injection and a low-dose, oral GC tapering scheme would be sufficient as bridging therapy," the researchers concluded.
The research was supported by an unrestricted grant from Pfizer, which had no involvement in the study design, data collection, analysis, or publication of the results. Data management was sponsored by the Dutch Arthritis Foundation. The authors reported that they had no competing interests.
In arithmetic and newly diagnosed rheumatoid arthritis alike, three is greater than one.
Initial triple disease-modifying therapy with either of two glucocorticoid (GC) bridging therapies proved quicker in attaining treatment goals and required fewer treatment intensifications for patients with very early RA than did initial methotrexate monotherapy with oral GC therapy, according to the results of a randomized, comparative trial.
Several previous clinical trials concluded that initial combination therapy had better clinical efficacy than monotherapy. However, most rheumatologists have not incorporated this into daily practice, according to Dr. Pascal Hendrik Pieter de Jong of the University Medical Center, Rotterdam, the Netherlands, and his colleagues.
Previous combination-therapy studies have usually been disregarded, the researchers noted, because the trials were biased by the use of GCs. So, Dr. de Jong and his colleagues compared the benefits of the 1-year clinical efficacy of initial triple disease-modifying antirheumatic drugs to the benefits of initial methotrexate monotherapy unbiased by the use of GC therapy, either oral or intramuscular.
Primary outcomes of the study were the area under the curve of the Health Assessment Questionnaire (to assess functional ability) and Disease Activity Score (to assess disease state), and the proportion of patients with radiographic progression, the investigators reported online May 1 in Annals of the Rheumatic Diseases (Ann. Rheum. Dis. May 1, 2014 [doi: 10.1136/annrheumdis-2013-204788]).
The study population comprised the 281 patients with a high probability of progress to persistent arthritis (greater than 70%) who were in the Rotterdam Early Arthritis Cohort (tReach) trial. All patients were over age 18, with arthritis in one or more joints and symptom duration of less than 1 year; 68% of the patients were women.
Patients were randomized to one of three arms:
1) initial triple disease-modifying therapy (iTDT) of methotrexate plus sulfasalazine and hydroxychloroquine, with GCs administered intramuscularly,
2) iTDT with an oral GC tapering scheme, or
3) initial methotrexate monotherapy (iMM) with oral GCs similar to the second treatment arm.
Concurrent treatment with nonsteroidal anti-inflammatory drugs and intra-articular injections (maximum of two per 3 months) was allowed.
The largest difference in disease activity states between the treatment arms was seen after 3 months, although that effect gradually diminished to nonsignificance by 12 months. Similarly, there were no significant differences in functional ability at 12 months.
After 3 months, 40% fewer biological agents were prescribed in the iTDT group than in the iMM group, and that difference persisted over time. However, disease activity, functional ability, and radiographic progression were not different between the two treatments after 12 months, regardless of GC bridging therapies. Early initiation of biological agents in the iMM group might have prevented or delayed the radiographic progression, the researchers suggested.
There were no differences in serious adverse events seen in the treatment arms, but the proportion of patients with medication adjustments due to adverse events was significantly higher in the iTDT (65%), compared with the iMM group (45%). No differences were seen between the two GC bridging therapies. Gastrointestinal complaints (56%) and fatigue (36%) were the two most commonly reported adverse events.
"We found no differences in disease activity, functional ability, and radiographic progression after 12 months of treatment owing to our treat-to-target approach (intensifying treatment until the target is reached)," the investigators observed. "Therefore, it is not the endpoint, but progress towards the endpoint, which matters most."
In addition, delaying the implementation of biological agents with iTDT treatment resulted in "reducing costs enormously," according to the researchers, although the cost-utility analysis of the tREACH trial has to reconfirm this, they added.
"We think that future research should focus on developing a more personalized treatment approach, in which differentiation between patients who would thrive on iMM and those who need iTDT might be a first step," they said.
"Before choosing the initial treatment strategy, rheumatologists should be aware of the benefits and the risks, but additionally, known prognostic factors and the patient’s wish should be taken into account. One single intramuscular GC injection and a low-dose, oral GC tapering scheme would be sufficient as bridging therapy," the researchers concluded.
The research was supported by an unrestricted grant from Pfizer, which had no involvement in the study design, data collection, analysis, or publication of the results. Data management was sponsored by the Dutch Arthritis Foundation. The authors reported that they had no competing interests.
Initial triple disease-modifying therapy, glucocorticoid, early RA, methotrexate monotherapy with oral GC therapy, initial combination therapy, Dr. Pascal Hendrik Pieter de Jong, Health Assessment Questionnaire, Disease Activity Score, radiographic progression, Annals of the Rheumatic Diseases, Rotterdam Early Arthritis Cohort (tReach) trial,
Initial triple disease-modifying therapy, glucocorticoid, early RA, methotrexate monotherapy with oral GC therapy, initial combination therapy, Dr. Pascal Hendrik Pieter de Jong, Health Assessment Questionnaire, Disease Activity Score, radiographic progression, Annals of the Rheumatic Diseases, Rotterdam Early Arthritis Cohort (tReach) trial,
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Start patients with newly diagnosed rheumatoid arthritis on triple disease-modifying therapy rather than methotrexate monotherapy.
Major finding: Triple therapy resulted in 40% fewer treatment intensifications than did monotherapy, but it required more medication adjustments because of adverse events. No differences in radiographic progression were seen.
Data source: The report summarized 1-year data from the tREACH trial, a randomized, single-blinded, three-arm clinical efficacy study with 281 patients.
Disclosures: The research was supported by an unrestricted grant from Pfizer, which had no involvement in the study design, data collection, analysis, or publication of the results. Data management was sponsored by the Dutch Arthritis Foundation. The authors reported that they had no competing interests.
Celebrex cost high in United States, low in Canada
North America is home to the most expensive and least expensive Celebrex, according to the International Federation of Health Plans’ 2013 Comparative Price Report.
The average price for a 1-month supply of Celebrex (celecoxib) was $225 in the United States last year, compared with $51 in Canada, the IFHP reported. The other countries in the survey were the Netherlands ($112), England ($112), Switzerland ($138), and Spain ($164).
Celebrex is approved in the United States for the treatment of osteoarthritis, rheumatoid arthritis, juvenile rheumatoid arthritis in patients 2 years and older, ankylosing spondylitis, acute pain, and primary dysmenorrhea.
The IFHP comprises more than 100 member companies in 25 countries. For the survey, the price for each country was submitted by participating member plans. Some prices are drawn from the public sector, some from the private, and some from both. U.S. averages were calculated from more than 100 million claims in the Truven MarketScan Research databases.
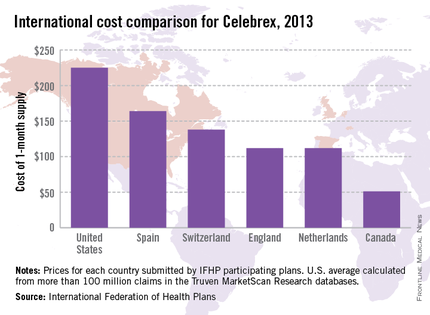
North America is home to the most expensive and least expensive Celebrex, according to the International Federation of Health Plans’ 2013 Comparative Price Report.
The average price for a 1-month supply of Celebrex (celecoxib) was $225 in the United States last year, compared with $51 in Canada, the IFHP reported. The other countries in the survey were the Netherlands ($112), England ($112), Switzerland ($138), and Spain ($164).
Celebrex is approved in the United States for the treatment of osteoarthritis, rheumatoid arthritis, juvenile rheumatoid arthritis in patients 2 years and older, ankylosing spondylitis, acute pain, and primary dysmenorrhea.
The IFHP comprises more than 100 member companies in 25 countries. For the survey, the price for each country was submitted by participating member plans. Some prices are drawn from the public sector, some from the private, and some from both. U.S. averages were calculated from more than 100 million claims in the Truven MarketScan Research databases.
North America is home to the most expensive and least expensive Celebrex, according to the International Federation of Health Plans’ 2013 Comparative Price Report.
The average price for a 1-month supply of Celebrex (celecoxib) was $225 in the United States last year, compared with $51 in Canada, the IFHP reported. The other countries in the survey were the Netherlands ($112), England ($112), Switzerland ($138), and Spain ($164).
Celebrex is approved in the United States for the treatment of osteoarthritis, rheumatoid arthritis, juvenile rheumatoid arthritis in patients 2 years and older, ankylosing spondylitis, acute pain, and primary dysmenorrhea.
The IFHP comprises more than 100 member companies in 25 countries. For the survey, the price for each country was submitted by participating member plans. Some prices are drawn from the public sector, some from the private, and some from both. U.S. averages were calculated from more than 100 million claims in the Truven MarketScan Research databases.