User login
Masquerading as Metastatic Carcinoma: A Diagnostic Challenge
Masquerading as Metastatic Carcinoma: A Diagnostic Challenge
Case Presentation
A 55-year-old White male Gulf War Marine veteran with a history of irritable bowel syndrome, chronic low back pain, anxiety, posttraumatic stress disorder (PTSD), and a 35 pack-year smoking history presented to the Edward Hines, Jr. Veterans Affairs Hospital primary care clinic with moderate abdominal pain for 1 week and altered bowel habits for several months. He reported mainly having diarrhea and more bloating than usual. The patient stated that he had experienced progressive fatigue over the past 6 months, along with new drenching night sweats and chills. He reported a 20-lb unintentional weight loss over the past 6 months. The patient noted no substance use or changes to his diet or exercise pattern. He had previously served in the US Marine Corps and was deployed to Okinawa, Camp Lejeune, Iraq, and Honduras during his 10-year military career.
This patient’s initial abdominal computed tomography (CT) revealed multiple prominent lymph nodes in the peripancreatic, periportal, retroperitoneal, bilateral iliac, and inguinal regions. Due to substantial lymphadenopathy and gastrointestinal complaints, he was evaluated by gastroenterology and underwent both esophagogastroduodenoscopy and colonoscopy. Results were notable for gastric mucosal inflammation and intestinal metaplasia, as well as tubular adenomas throughout the colon; however, there was no evidence of mass lesions.
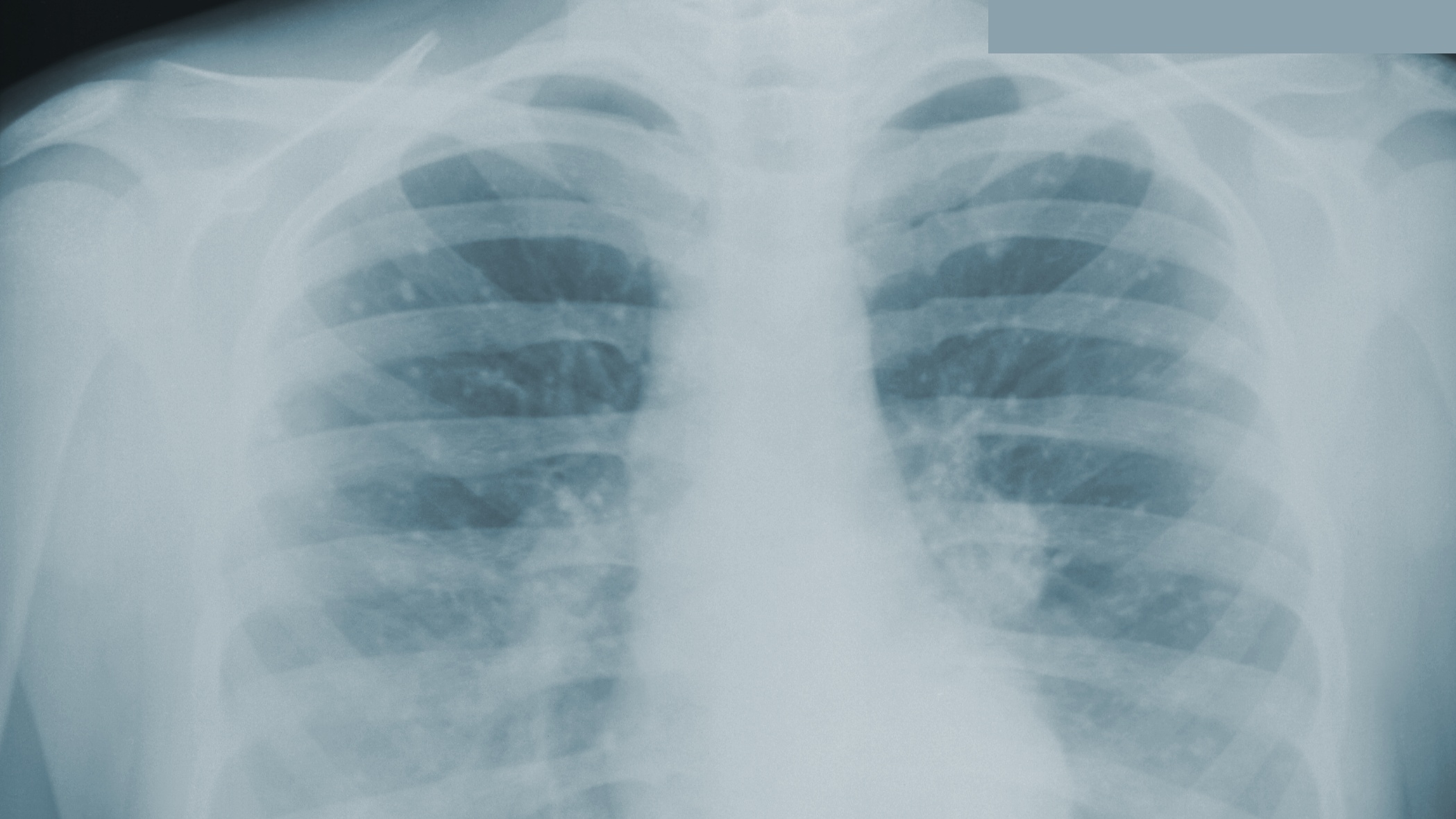
Due to ongoing systemic symptoms of unknown etiology, the patient underwent CT of the chest, revealing a 2.3-cm right lower lobe (RLL) pulmonary mass with right hilar lymphadenopathy. A positron emission tomography (PET) demonstrated hypermetabolic activity in the RLL nodule (standardized uptake value [SUV], 3.1) and adjacent lymph nodes (SUV, ≤ 5.8).
The patient was told that he likely had metastatic disease and was referred to the Hematology-Oncology service. He reported increased anxiety and insomnia. He also was referred to the Mental Health service, which started him on escitalopram 10 mg daily and hydroxyzine 25 mg daily as needed, and recommended therapy.
The Pulmonology service performed an endobronchial ultrasound (EBUS)-guided biopsy of mediastinal lymph nodes and the RLL nodule. Pathologic examination yielded necrotic debris and benign lymphoid tissue. All cultures and stains for fungal, mycobacterial, and bacterial organisms were negative.
The patient then underwent thoracic surgery for diagnostic wedge resection of the RLL. Histopathology revealed necrotizing granulomatous inflammation with fungal organisms morphologically consistent with Histoplasma. All microbiologic cultures remained negative. The patient’s urine and serum histoplasma antigen, serum cryptococcal antigen, β-D-glucan, QuantiFERON-TB Gold, and blastomycosis tests were negative. The patient started itraconazole 200 mg twice daily and experienced slow improvements in his symptoms with follow-up by the Infectious Disease service and decreased anxiety.
Discussion
This case highlights the diagnostic challenges associated with disseminated fungal infections, namely histoplasmosis, which can closely mimic malignancy in both clinical presentation and imaging.1-3 The patient presented with classic B symptoms, diffuse lymphadenopathy, and a suspicious lung nodule, all features highly suggestive of malignancy.
Classic “B symptoms” (eg, fever, drenching night sweats, and unintentional weight loss) are constitutional manifestations that occur in association with lymphoid malignancies, particularly Hodgkin lymphoma and certain non-Hodgkin lymphomas. Their presence is central to disease staging, prognosis, and therapeutic decision-making.4 The Ann Arbor staging system classifies systemic symptoms as B symptoms when any of the following are present: fever > 38 °C in the absence of infection, often intermittent; drenching night sweats requiring a change of bedclothes or sheets; or unintentional weight loss of > 10% of baseline body weight over 6 months.5
Although often associated with lymphomas, B symptoms are not pathognomonic. They may occur in chronic infections, such as tuberculosis and HIV infection, autoimmune diseases, and solid tumors.5,6 Thorough evaluation, including infectious and inflammatory workups, is essential before attributing these systemic manifestations to a neoplasm.
Histoplasma capsulatum, the causative agent of histoplasmosis, is endemic in certain parts of the United States, including the Ohio and Mississippi River valleys. However, the fungus is also found in the Middle East, Asia, Africa, and Central and South America.7 It is acquired via inhalation of soil contaminated with bird or bat droppings.1 The patient’s military service represents a potential environmental exposure risk. While immunocompetent individuals typically clear the infection, chronic progressive disseminated histoplasmosis can develop, particularly in patients with underlying pulmonary disease or subtle immunosuppression. 1,8
Negative fungal serologies and antigen tests, as seen in this patient, are not uncommon in subacute or localized disease and should not delay further diagnostic interventions when clinical suspicion remains high.2,9 Invasive procedures, including wedge resection, may be required to establish a definitive diagnosis.
EBUS-guided transbronchial needle aspiration (EBUS-TBNA) is a valuable minimally invasive technique for the evaluation of mediastinal and hilar lymphadenopathy. However, its diagnostic performance in cases of histoplasmosis is limited by several factors. First, low organism burden is a significant limitation. In immunocompetent individuals with subacute pulmonary histoplasmosis, the fungal load within lymph nodes or pulmonary tissue is often low, leading to negative fungal cultures specimens obtained using EBUS.10 Second, the diagnostic yield of EBUS-TBNA for identifying Histoplasma capsulatum is variable. Detection of fungal elements or successful culture may be inconsistent and is influenced by operator expertise, needle size, number of passes, and specimen handling and processing techniques. Third, overlapping cytopathologic findings can complicate interpretation. The granulomatous inflammation characteristic of histoplasmosis is nonspecific and may also be observed in other conditions such as tuberculosis, sarcoidosis, and certain lymphoproliferative disorders. Therefore, clinical and radiologic correlation, as well as adjunctive testing, are essential for accurate diagnosis. 11 Fourth, sample quality poses additional challenges. Differentiating inflammatory cells from neoplastic elements, managing poorly preserved specimens, and avoiding contamination from bronchial wall tissue can all affect diagnostic accuracy.12 Finally, inadequate or nondiagnostic sampling occurs in a subset of patients. Case series have reported nondiagnostic EBUS-TBNA results, necessitating reliance on complementary diagnostic modalities, including serologic assays, urine antigen testing, or surgical biopsy, to establish a definitive diagnosis.9
Necrotizing granulomas without confirmed microbiologic identification may still justify antifungal treatment if histopathologic findings are consistent with fungal infection, as in this patient.1,3 Itraconazole remains the first-line treatment, though adverse effects and treatment duration may impact adherence.3
The presentation of pulmonary nodules and diffuse lymphadenopathy in this patient was strongly suggestive of metastatic disease, especially given his smoking history. However, histoplasmosis can closely mimic metastatic cancer radiographically, leading to potential misdiagnosis.13 In endemic areas, histoplasmosis should be included in the differential for any patient with suspicious thoracic imaging.
In this case, the diagnosis of malignancy was prematurely communicated to the patient based solely on imaging, prior to histologic confirmation. This approach carries significant risk, including psychological distress, initiation of inappropriate treatments, and delayed antifungal therapy. A similar diagnostic dilemma was described by Wheat et al, who emphasized the need for tissue sampling in distinguishing fungal infections from malignancy.14 In the absence of tissue diagnosis, reliance on radiologic criteria alone can be misleading. PETs, which detect metabolic activity, are not specific and can show uptake in both neoplastic and inflammatory processes.15
This case reinforces the principle of diagnostic humility: no diagnosis, especially one as consequential as cancer, should be confirmed without histologic or microbiologic evidence. Multidisciplinary evaluation and appropriate use of tissue sampling are crucial to avoid harmful diagnostic errors.
Conclusions
In patients presenting with constitutional symptoms, diffuse lymphadenopathy, and pulmonary nodules, disseminated fungal infections such as histoplasmosis must remain in the differential diagnosis, even in the absence of positive fungal serologies.1,2 Tissue diagnosis through invasive procedures may be necessary, particularly when initial biopsies and cultures are inconclusive. 1 Early recognition and treatment are critical to avoid complications from progressive disseminated infection.3,8
- Wheat LJ, Azar MM, Bahr NC, et al. Histoplasmosis. Infect Dis Clin North Am. 2016;30:207-227.
- Fielding DI, et al. Diagnostic yield of EBUS-TBNA for infectious diseases. Respirology. 2012;17:876-882
- Hage CA, Azar MM, Bahr N, et al. Histoplasmosis: up-to-date evidence-based approach to diagnosis and management. Semin Respir Crit Care Med. 2015;36:729-745. doi:10.1055/s-0035-1563546
- Azar MM, Loyd JL, Relich RF, et al. Diagnostic challenges of pulmonary histoplasmosis: a case series and review of the literature. Respir Med Case Rep. 2019;27:100825. doi:10.1016/j.rmcr.2019.100825
- Hage CA, Ribes JA, Wengenack NL, et al. A multicenter evaluation of tests for diagnosis of histoplasmosis. Clin Infect Dis. 2011;53:448-454. doi:10.1093/cid/cir435
- Love C, Tomas MB, Tronco GG, et al. FDG PET of infection and inflammation. Radiographics. 2005;25:1357-1368. doi:10.1148/rg.255045122
- Nakajima T, Yasufuku K. The role of EBUS-TBNA in the diagnosis of infectious diseases. J Thorac Dis. 2013;5:S478- S482.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev. 2007;20:115-132. doi:10.1128/CMR.00027-06
- Gupta D, Dadhwal DS, Agarwal R, et al. Endobronchial ultrasound-guided transbronchial needle aspiration in mediastinal granulomatous diseases. Lung India. 2014;31:212-216.
- Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2024;74:215-235. doi:10.3322/caac.21892
- McKinsey DS, Spiegel RA, Hutwagner L, et al. Clinical and diagnostic features of patients with disseminated histoplasmosis. Am J Med. 1997;102:370-378. doi:10.1016/S0002-9343(97)00017-6
- Goodwin RA Jr, Shapiro JL, Thurman GH, et al. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore).1980;59:1-33. doi:10.1097/00005792-198001000-00001
- Azar MM, Hage CA. Laboratory diagnostics for histoplasmosis. J Clin Microbiol. 2017;55:1612-1620. doi:10.1128/JCM.00120-17
- Wheat LJ, Freifeld AG, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45:807-825. doi:10.1086/521259
- Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res. 1971;31:1860-1861.
Case Presentation
A 55-year-old White male Gulf War Marine veteran with a history of irritable bowel syndrome, chronic low back pain, anxiety, posttraumatic stress disorder (PTSD), and a 35 pack-year smoking history presented to the Edward Hines, Jr. Veterans Affairs Hospital primary care clinic with moderate abdominal pain for 1 week and altered bowel habits for several months. He reported mainly having diarrhea and more bloating than usual. The patient stated that he had experienced progressive fatigue over the past 6 months, along with new drenching night sweats and chills. He reported a 20-lb unintentional weight loss over the past 6 months. The patient noted no substance use or changes to his diet or exercise pattern. He had previously served in the US Marine Corps and was deployed to Okinawa, Camp Lejeune, Iraq, and Honduras during his 10-year military career.
This patient’s initial abdominal computed tomography (CT) revealed multiple prominent lymph nodes in the peripancreatic, periportal, retroperitoneal, bilateral iliac, and inguinal regions. Due to substantial lymphadenopathy and gastrointestinal complaints, he was evaluated by gastroenterology and underwent both esophagogastroduodenoscopy and colonoscopy. Results were notable for gastric mucosal inflammation and intestinal metaplasia, as well as tubular adenomas throughout the colon; however, there was no evidence of mass lesions.
Due to ongoing systemic symptoms of unknown etiology, the patient underwent CT of the chest, revealing a 2.3-cm right lower lobe (RLL) pulmonary mass with right hilar lymphadenopathy. A positron emission tomography (PET) demonstrated hypermetabolic activity in the RLL nodule (standardized uptake value [SUV], 3.1) and adjacent lymph nodes (SUV, ≤ 5.8).
The patient was told that he likely had metastatic disease and was referred to the Hematology-Oncology service. He reported increased anxiety and insomnia. He also was referred to the Mental Health service, which started him on escitalopram 10 mg daily and hydroxyzine 25 mg daily as needed, and recommended therapy.
The Pulmonology service performed an endobronchial ultrasound (EBUS)-guided biopsy of mediastinal lymph nodes and the RLL nodule. Pathologic examination yielded necrotic debris and benign lymphoid tissue. All cultures and stains for fungal, mycobacterial, and bacterial organisms were negative.
The patient then underwent thoracic surgery for diagnostic wedge resection of the RLL. Histopathology revealed necrotizing granulomatous inflammation with fungal organisms morphologically consistent with Histoplasma. All microbiologic cultures remained negative. The patient’s urine and serum histoplasma antigen, serum cryptococcal antigen, β-D-glucan, QuantiFERON-TB Gold, and blastomycosis tests were negative. The patient started itraconazole 200 mg twice daily and experienced slow improvements in his symptoms with follow-up by the Infectious Disease service and decreased anxiety.
Discussion
This case highlights the diagnostic challenges associated with disseminated fungal infections, namely histoplasmosis, which can closely mimic malignancy in both clinical presentation and imaging.1-3 The patient presented with classic B symptoms, diffuse lymphadenopathy, and a suspicious lung nodule, all features highly suggestive of malignancy.
Classic “B symptoms” (eg, fever, drenching night sweats, and unintentional weight loss) are constitutional manifestations that occur in association with lymphoid malignancies, particularly Hodgkin lymphoma and certain non-Hodgkin lymphomas. Their presence is central to disease staging, prognosis, and therapeutic decision-making.4 The Ann Arbor staging system classifies systemic symptoms as B symptoms when any of the following are present: fever > 38 °C in the absence of infection, often intermittent; drenching night sweats requiring a change of bedclothes or sheets; or unintentional weight loss of > 10% of baseline body weight over 6 months.5
Although often associated with lymphomas, B symptoms are not pathognomonic. They may occur in chronic infections, such as tuberculosis and HIV infection, autoimmune diseases, and solid tumors.5,6 Thorough evaluation, including infectious and inflammatory workups, is essential before attributing these systemic manifestations to a neoplasm.
Histoplasma capsulatum, the causative agent of histoplasmosis, is endemic in certain parts of the United States, including the Ohio and Mississippi River valleys. However, the fungus is also found in the Middle East, Asia, Africa, and Central and South America.7 It is acquired via inhalation of soil contaminated with bird or bat droppings.1 The patient’s military service represents a potential environmental exposure risk. While immunocompetent individuals typically clear the infection, chronic progressive disseminated histoplasmosis can develop, particularly in patients with underlying pulmonary disease or subtle immunosuppression. 1,8
Negative fungal serologies and antigen tests, as seen in this patient, are not uncommon in subacute or localized disease and should not delay further diagnostic interventions when clinical suspicion remains high.2,9 Invasive procedures, including wedge resection, may be required to establish a definitive diagnosis.
EBUS-guided transbronchial needle aspiration (EBUS-TBNA) is a valuable minimally invasive technique for the evaluation of mediastinal and hilar lymphadenopathy. However, its diagnostic performance in cases of histoplasmosis is limited by several factors. First, low organism burden is a significant limitation. In immunocompetent individuals with subacute pulmonary histoplasmosis, the fungal load within lymph nodes or pulmonary tissue is often low, leading to negative fungal cultures specimens obtained using EBUS.10 Second, the diagnostic yield of EBUS-TBNA for identifying Histoplasma capsulatum is variable. Detection of fungal elements or successful culture may be inconsistent and is influenced by operator expertise, needle size, number of passes, and specimen handling and processing techniques. Third, overlapping cytopathologic findings can complicate interpretation. The granulomatous inflammation characteristic of histoplasmosis is nonspecific and may also be observed in other conditions such as tuberculosis, sarcoidosis, and certain lymphoproliferative disorders. Therefore, clinical and radiologic correlation, as well as adjunctive testing, are essential for accurate diagnosis. 11 Fourth, sample quality poses additional challenges. Differentiating inflammatory cells from neoplastic elements, managing poorly preserved specimens, and avoiding contamination from bronchial wall tissue can all affect diagnostic accuracy.12 Finally, inadequate or nondiagnostic sampling occurs in a subset of patients. Case series have reported nondiagnostic EBUS-TBNA results, necessitating reliance on complementary diagnostic modalities, including serologic assays, urine antigen testing, or surgical biopsy, to establish a definitive diagnosis.9
Necrotizing granulomas without confirmed microbiologic identification may still justify antifungal treatment if histopathologic findings are consistent with fungal infection, as in this patient.1,3 Itraconazole remains the first-line treatment, though adverse effects and treatment duration may impact adherence.3
The presentation of pulmonary nodules and diffuse lymphadenopathy in this patient was strongly suggestive of metastatic disease, especially given his smoking history. However, histoplasmosis can closely mimic metastatic cancer radiographically, leading to potential misdiagnosis.13 In endemic areas, histoplasmosis should be included in the differential for any patient with suspicious thoracic imaging.
In this case, the diagnosis of malignancy was prematurely communicated to the patient based solely on imaging, prior to histologic confirmation. This approach carries significant risk, including psychological distress, initiation of inappropriate treatments, and delayed antifungal therapy. A similar diagnostic dilemma was described by Wheat et al, who emphasized the need for tissue sampling in distinguishing fungal infections from malignancy.14 In the absence of tissue diagnosis, reliance on radiologic criteria alone can be misleading. PETs, which detect metabolic activity, are not specific and can show uptake in both neoplastic and inflammatory processes.15
This case reinforces the principle of diagnostic humility: no diagnosis, especially one as consequential as cancer, should be confirmed without histologic or microbiologic evidence. Multidisciplinary evaluation and appropriate use of tissue sampling are crucial to avoid harmful diagnostic errors.
Conclusions
In patients presenting with constitutional symptoms, diffuse lymphadenopathy, and pulmonary nodules, disseminated fungal infections such as histoplasmosis must remain in the differential diagnosis, even in the absence of positive fungal serologies.1,2 Tissue diagnosis through invasive procedures may be necessary, particularly when initial biopsies and cultures are inconclusive. 1 Early recognition and treatment are critical to avoid complications from progressive disseminated infection.3,8
Case Presentation
A 55-year-old White male Gulf War Marine veteran with a history of irritable bowel syndrome, chronic low back pain, anxiety, posttraumatic stress disorder (PTSD), and a 35 pack-year smoking history presented to the Edward Hines, Jr. Veterans Affairs Hospital primary care clinic with moderate abdominal pain for 1 week and altered bowel habits for several months. He reported mainly having diarrhea and more bloating than usual. The patient stated that he had experienced progressive fatigue over the past 6 months, along with new drenching night sweats and chills. He reported a 20-lb unintentional weight loss over the past 6 months. The patient noted no substance use or changes to his diet or exercise pattern. He had previously served in the US Marine Corps and was deployed to Okinawa, Camp Lejeune, Iraq, and Honduras during his 10-year military career.
This patient’s initial abdominal computed tomography (CT) revealed multiple prominent lymph nodes in the peripancreatic, periportal, retroperitoneal, bilateral iliac, and inguinal regions. Due to substantial lymphadenopathy and gastrointestinal complaints, he was evaluated by gastroenterology and underwent both esophagogastroduodenoscopy and colonoscopy. Results were notable for gastric mucosal inflammation and intestinal metaplasia, as well as tubular adenomas throughout the colon; however, there was no evidence of mass lesions.
Due to ongoing systemic symptoms of unknown etiology, the patient underwent CT of the chest, revealing a 2.3-cm right lower lobe (RLL) pulmonary mass with right hilar lymphadenopathy. A positron emission tomography (PET) demonstrated hypermetabolic activity in the RLL nodule (standardized uptake value [SUV], 3.1) and adjacent lymph nodes (SUV, ≤ 5.8).
The patient was told that he likely had metastatic disease and was referred to the Hematology-Oncology service. He reported increased anxiety and insomnia. He also was referred to the Mental Health service, which started him on escitalopram 10 mg daily and hydroxyzine 25 mg daily as needed, and recommended therapy.
The Pulmonology service performed an endobronchial ultrasound (EBUS)-guided biopsy of mediastinal lymph nodes and the RLL nodule. Pathologic examination yielded necrotic debris and benign lymphoid tissue. All cultures and stains for fungal, mycobacterial, and bacterial organisms were negative.
The patient then underwent thoracic surgery for diagnostic wedge resection of the RLL. Histopathology revealed necrotizing granulomatous inflammation with fungal organisms morphologically consistent with Histoplasma. All microbiologic cultures remained negative. The patient’s urine and serum histoplasma antigen, serum cryptococcal antigen, β-D-glucan, QuantiFERON-TB Gold, and blastomycosis tests were negative. The patient started itraconazole 200 mg twice daily and experienced slow improvements in his symptoms with follow-up by the Infectious Disease service and decreased anxiety.
Discussion
This case highlights the diagnostic challenges associated with disseminated fungal infections, namely histoplasmosis, which can closely mimic malignancy in both clinical presentation and imaging.1-3 The patient presented with classic B symptoms, diffuse lymphadenopathy, and a suspicious lung nodule, all features highly suggestive of malignancy.
Classic “B symptoms” (eg, fever, drenching night sweats, and unintentional weight loss) are constitutional manifestations that occur in association with lymphoid malignancies, particularly Hodgkin lymphoma and certain non-Hodgkin lymphomas. Their presence is central to disease staging, prognosis, and therapeutic decision-making.4 The Ann Arbor staging system classifies systemic symptoms as B symptoms when any of the following are present: fever > 38 °C in the absence of infection, often intermittent; drenching night sweats requiring a change of bedclothes or sheets; or unintentional weight loss of > 10% of baseline body weight over 6 months.5
Although often associated with lymphomas, B symptoms are not pathognomonic. They may occur in chronic infections, such as tuberculosis and HIV infection, autoimmune diseases, and solid tumors.5,6 Thorough evaluation, including infectious and inflammatory workups, is essential before attributing these systemic manifestations to a neoplasm.
Histoplasma capsulatum, the causative agent of histoplasmosis, is endemic in certain parts of the United States, including the Ohio and Mississippi River valleys. However, the fungus is also found in the Middle East, Asia, Africa, and Central and South America.7 It is acquired via inhalation of soil contaminated with bird or bat droppings.1 The patient’s military service represents a potential environmental exposure risk. While immunocompetent individuals typically clear the infection, chronic progressive disseminated histoplasmosis can develop, particularly in patients with underlying pulmonary disease or subtle immunosuppression. 1,8
Negative fungal serologies and antigen tests, as seen in this patient, are not uncommon in subacute or localized disease and should not delay further diagnostic interventions when clinical suspicion remains high.2,9 Invasive procedures, including wedge resection, may be required to establish a definitive diagnosis.
EBUS-guided transbronchial needle aspiration (EBUS-TBNA) is a valuable minimally invasive technique for the evaluation of mediastinal and hilar lymphadenopathy. However, its diagnostic performance in cases of histoplasmosis is limited by several factors. First, low organism burden is a significant limitation. In immunocompetent individuals with subacute pulmonary histoplasmosis, the fungal load within lymph nodes or pulmonary tissue is often low, leading to negative fungal cultures specimens obtained using EBUS.10 Second, the diagnostic yield of EBUS-TBNA for identifying Histoplasma capsulatum is variable. Detection of fungal elements or successful culture may be inconsistent and is influenced by operator expertise, needle size, number of passes, and specimen handling and processing techniques. Third, overlapping cytopathologic findings can complicate interpretation. The granulomatous inflammation characteristic of histoplasmosis is nonspecific and may also be observed in other conditions such as tuberculosis, sarcoidosis, and certain lymphoproliferative disorders. Therefore, clinical and radiologic correlation, as well as adjunctive testing, are essential for accurate diagnosis. 11 Fourth, sample quality poses additional challenges. Differentiating inflammatory cells from neoplastic elements, managing poorly preserved specimens, and avoiding contamination from bronchial wall tissue can all affect diagnostic accuracy.12 Finally, inadequate or nondiagnostic sampling occurs in a subset of patients. Case series have reported nondiagnostic EBUS-TBNA results, necessitating reliance on complementary diagnostic modalities, including serologic assays, urine antigen testing, or surgical biopsy, to establish a definitive diagnosis.9
Necrotizing granulomas without confirmed microbiologic identification may still justify antifungal treatment if histopathologic findings are consistent with fungal infection, as in this patient.1,3 Itraconazole remains the first-line treatment, though adverse effects and treatment duration may impact adherence.3
The presentation of pulmonary nodules and diffuse lymphadenopathy in this patient was strongly suggestive of metastatic disease, especially given his smoking history. However, histoplasmosis can closely mimic metastatic cancer radiographically, leading to potential misdiagnosis.13 In endemic areas, histoplasmosis should be included in the differential for any patient with suspicious thoracic imaging.
In this case, the diagnosis of malignancy was prematurely communicated to the patient based solely on imaging, prior to histologic confirmation. This approach carries significant risk, including psychological distress, initiation of inappropriate treatments, and delayed antifungal therapy. A similar diagnostic dilemma was described by Wheat et al, who emphasized the need for tissue sampling in distinguishing fungal infections from malignancy.14 In the absence of tissue diagnosis, reliance on radiologic criteria alone can be misleading. PETs, which detect metabolic activity, are not specific and can show uptake in both neoplastic and inflammatory processes.15
This case reinforces the principle of diagnostic humility: no diagnosis, especially one as consequential as cancer, should be confirmed without histologic or microbiologic evidence. Multidisciplinary evaluation and appropriate use of tissue sampling are crucial to avoid harmful diagnostic errors.
Conclusions
In patients presenting with constitutional symptoms, diffuse lymphadenopathy, and pulmonary nodules, disseminated fungal infections such as histoplasmosis must remain in the differential diagnosis, even in the absence of positive fungal serologies.1,2 Tissue diagnosis through invasive procedures may be necessary, particularly when initial biopsies and cultures are inconclusive. 1 Early recognition and treatment are critical to avoid complications from progressive disseminated infection.3,8
- Wheat LJ, Azar MM, Bahr NC, et al. Histoplasmosis. Infect Dis Clin North Am. 2016;30:207-227.
- Fielding DI, et al. Diagnostic yield of EBUS-TBNA for infectious diseases. Respirology. 2012;17:876-882
- Hage CA, Azar MM, Bahr N, et al. Histoplasmosis: up-to-date evidence-based approach to diagnosis and management. Semin Respir Crit Care Med. 2015;36:729-745. doi:10.1055/s-0035-1563546
- Azar MM, Loyd JL, Relich RF, et al. Diagnostic challenges of pulmonary histoplasmosis: a case series and review of the literature. Respir Med Case Rep. 2019;27:100825. doi:10.1016/j.rmcr.2019.100825
- Hage CA, Ribes JA, Wengenack NL, et al. A multicenter evaluation of tests for diagnosis of histoplasmosis. Clin Infect Dis. 2011;53:448-454. doi:10.1093/cid/cir435
- Love C, Tomas MB, Tronco GG, et al. FDG PET of infection and inflammation. Radiographics. 2005;25:1357-1368. doi:10.1148/rg.255045122
- Nakajima T, Yasufuku K. The role of EBUS-TBNA in the diagnosis of infectious diseases. J Thorac Dis. 2013;5:S478- S482.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev. 2007;20:115-132. doi:10.1128/CMR.00027-06
- Gupta D, Dadhwal DS, Agarwal R, et al. Endobronchial ultrasound-guided transbronchial needle aspiration in mediastinal granulomatous diseases. Lung India. 2014;31:212-216.
- Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2024;74:215-235. doi:10.3322/caac.21892
- McKinsey DS, Spiegel RA, Hutwagner L, et al. Clinical and diagnostic features of patients with disseminated histoplasmosis. Am J Med. 1997;102:370-378. doi:10.1016/S0002-9343(97)00017-6
- Goodwin RA Jr, Shapiro JL, Thurman GH, et al. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore).1980;59:1-33. doi:10.1097/00005792-198001000-00001
- Azar MM, Hage CA. Laboratory diagnostics for histoplasmosis. J Clin Microbiol. 2017;55:1612-1620. doi:10.1128/JCM.00120-17
- Wheat LJ, Freifeld AG, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45:807-825. doi:10.1086/521259
- Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res. 1971;31:1860-1861.
- Wheat LJ, Azar MM, Bahr NC, et al. Histoplasmosis. Infect Dis Clin North Am. 2016;30:207-227.
- Fielding DI, et al. Diagnostic yield of EBUS-TBNA for infectious diseases. Respirology. 2012;17:876-882
- Hage CA, Azar MM, Bahr N, et al. Histoplasmosis: up-to-date evidence-based approach to diagnosis and management. Semin Respir Crit Care Med. 2015;36:729-745. doi:10.1055/s-0035-1563546
- Azar MM, Loyd JL, Relich RF, et al. Diagnostic challenges of pulmonary histoplasmosis: a case series and review of the literature. Respir Med Case Rep. 2019;27:100825. doi:10.1016/j.rmcr.2019.100825
- Hage CA, Ribes JA, Wengenack NL, et al. A multicenter evaluation of tests for diagnosis of histoplasmosis. Clin Infect Dis. 2011;53:448-454. doi:10.1093/cid/cir435
- Love C, Tomas MB, Tronco GG, et al. FDG PET of infection and inflammation. Radiographics. 2005;25:1357-1368. doi:10.1148/rg.255045122
- Nakajima T, Yasufuku K. The role of EBUS-TBNA in the diagnosis of infectious diseases. J Thorac Dis. 2013;5:S478- S482.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev. 2007;20:115-132. doi:10.1128/CMR.00027-06
- Gupta D, Dadhwal DS, Agarwal R, et al. Endobronchial ultrasound-guided transbronchial needle aspiration in mediastinal granulomatous diseases. Lung India. 2014;31:212-216.
- Shanbhag S, Ambinder RF. Hodgkin lymphoma: a review and update on recent progress. CA Cancer J Clin. 2024;74:215-235. doi:10.3322/caac.21892
- McKinsey DS, Spiegel RA, Hutwagner L, et al. Clinical and diagnostic features of patients with disseminated histoplasmosis. Am J Med. 1997;102:370-378. doi:10.1016/S0002-9343(97)00017-6
- Goodwin RA Jr, Shapiro JL, Thurman GH, et al. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore).1980;59:1-33. doi:10.1097/00005792-198001000-00001
- Azar MM, Hage CA. Laboratory diagnostics for histoplasmosis. J Clin Microbiol. 2017;55:1612-1620. doi:10.1128/JCM.00120-17
- Wheat LJ, Freifeld AG, Kleiman MB, et al. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. 2007;45:807-825. doi:10.1086/521259
- Carbone PP, Kaplan HS, Musshoff K, et al. Report of the Committee on Hodgkin’s Disease Staging Classification. Cancer Res. 1971;31:1860-1861.
Masquerading as Metastatic Carcinoma: A Diagnostic Challenge
Masquerading as Metastatic Carcinoma: A Diagnostic Challenge
Managing Requests for Medical Aid in Dying Within the Veterans Health Administration
Managing Requests for Medical Aid in Dying Within the Veterans Health Administration
Requests for medical aid in dying (MAID) within the Veterans Health Administration (VHA) present unique ethical, legal, and clinical challenges. MAID is a process in which a physician provides a terminally ill patient with the means to end their own life. It is expressly prohibited by federal law, including within the US Department of Veterans Affairs (VA), regardless of its legality at the state level.1 MAID is also prohibited within community care institutions funded by the VA. The American Medical Association, American Geriatrics Society, and American Academy of Hospice and Palliative Medicine have adopted neutral positions regarding MAID due to varying opinions among their respective members.2-4 VHA palliative care clinicians are trained to identify and honor preferences for care and alleviate physical and emotional distress, which may complicate the management of MAID requests. Veterans can request MAID due to their desire for autonomy and pain relief, but the VHA prohibits clinicians from honoring these specific preferences. The inability to help veterans achieve their care preferences conflicts with the core mission of palliative care to reduce suffering and respect end-of-life wishes.
This case report describes the management of a veteran who requested MAID while also exhibiting active suicidal ideation. The patient’s distress stemmed from fears of impending loss of autonomy and functional decline, factors frequently linked to requests for MAID in terminally ill patients.5,6 Addressing the veteran’s request for MAID required balancing respect for patient autonomy and concerns about future suffering with the VA mission to protect veterans from self-harm and provide mental health care for suicidal ideation. This case highlights the importance of nuanced clinical approaches, ethical reflection, and interdisciplinary collaboration in navigating such complex scenarios. Informed consent was obtained from the patient’s family and health care agent (HCA) to publish this report.
Case Presentation
A 73-year-old male veteran, with Parkinson disease (PD), diagnosed at age 52 years, was referred to palliative care following diagnosis of a glioblastoma multiforme (GBM). The patient also had a history of major depressive disorder (MDD), suicidal ideation (SI), benign prostatic hypertrophy, and migraines. He was divorced, had no children, and his only sibling (sister) was deceased. His brother-in-law served as his HCA.
The patient had many close friends in the community, was an architect by training, and was active in the removal of barriers and increasing access for people with disabilities. Since 2010, about 7 years before his PD diagnosis, the patient used psychiatry and psychology resources to treat MDD, functional decline, and SI. He was hospitalized in 2016 after self-administration of heroin. During the hospitalization the patient received a high risk for suicide label. He articulated a firm and long-standing belief in his right to die and shared plans to end his life when he experienced a significant decline in his independence and quality of life (QoL).
When diagnosed with PD, the patient shared that his QoL was of utmost importance. He was aware that he would have significant physical decline as PD progressed and felt like there would be a point when his QoL would not be acceptable. When that happened, he wanted to end his life by available means. He was followed closely by his VHA care team for physical and emotional distress.
When diagnosed with a GBM in 2023, the patient declined treatment and was referred to palliative care, which had sporadically treated him for PD-related distress prior to 2023. During his previous palliative care visits, the patient had discussed a desire to engage in MAID when his functional status declined. After the GBM diagnosis, he reported no acute intent to harm himself with heroin, but planned to travel to Vermont for MAID when he felt he no longer had an adequate QoL based on functional capability.
Pharmacologic and nonpharmacologic approaches were used to treat the patient’s pain. He reported significant benefit from biofeedback therapy provided by the VA Headache Center of Excellence. This work also reconnected him to meditation, which he used daily to relieve pain and distress. The patient managed head pain with nonpharmacologic and pharmacologic interventions for 6 months and reported satisfaction with his QoL.
After 6 months, imaging showed progression of the brain tumor, which was associated with more fatigue and memory decline. At that time, the patient was enrolled in home hospice and reported continued intent to pursue MAID in Vermont but had not taken steps toward carrying it out. The patient understood the VA could not assist him in pursuing MAID; however, his care team was able to assist him in sharing his preferences for care with his loved ones and health care power of attorney.
He experienced rapid functional and cognitive decline due to progression of the GBM and was admitted to the VA Connecticut Healthcare System (VACHS) acute care unit where he exhibited confusion and screened positive for delirium using the Confusion Assessment Method.7 His physical and cognitive deterioration was likely due to the progressive brain tumor, and the patient lacked the capacity to make complex medical decisions. Formal consent was obtained from his HCA to transfer him to inpatient hospice. Psychiatry followed the patient throughout.
After 4 weeks of hospice care, the patient had a witnessed suicide attempt while the nurse was assisting him in the bathroom. The patient attempted to use hospital pajamas to hang himself when he wrapped a hospital gown around his neck and stated he was trying to tie a knot. Due to his confusion and delirium, the patient was unable to express his reasoning for the suicide attempt. He was seen by the Psychiatry service, which determined that his suicide risk was low to intermediate. The Psychiatry service did not recommend a 1:1 safety sitter, but suggested medication changes. Levetiracetam was discontinued, and valproate 500 mg orally twice daily was initiated for seizure prevention.
The hospice team was informed of the suicide attempt and psychiatry recommendations. The suicide prevention team was also updated following this event and agreed with psychiatry recommendations. The patient continued to decline, was no longer able to get out of bed, and had minimal speech. The patient received comfort medications, including intravenous morphine 2 mg and lorazepam 0.5 mg as needed ≤ 4 times daily. He died 8 days later.
Discussion
Chronic medical illness has been associated with increased suicide risk.8-10 The increased risk of suicide in chronically ill patients has been described as having as a bidirectional relationship with MDD, with depression not only increasing the risk of chronic medical illness but new-onset chronic medical illness being associated with new onset depression.11,12 Chronic medical conditions are associated with numerous psychiatric disorders, and the presence of a comorbid psychiatric illness is associated with higher rates of hospitalization, emergency department visits, and increased health care costs.13 Research has found that the association between suicide risk and chronic medical illness remains even after accounting for comorbid mental health disorders.14 This has been postulated to be due to a multitude of interpersonal, behavioral, cognitive, and affective factors (eg, perceived burdensomeness, loneliness, stress, pain catastrophizing, self-criticism).15 Additionally, some researchers have questioned whether suicidality constitutes a distinct mental disorder.16
Patients with cancer are at increased risk for suicidal ideation (including passive death wishes) and suicide attempts.17,18 Recent data indicate that compared with the general population, there is an 85% increased risk of suicide mortality in patients with cancer.19 Studies show the incidence of suicide is greater for individuals with cancer compared with the general population, with standardized mortality ratios ranging from 1.4 to 5.7.20-22
Among patients with cancer, suicide risk is associated with several factors: worse prognosis, older age, male sex, living in a socioeconomically vulnerable environment, and increased communication about suicidal intent prior to death.23-25 Just as the prevalence of suicidal ideation in people with cancer varies widely, reported rates of suicidality in caregivers of patients with cancer range from 2.7% to 71%.17,26 A survey of health care workers indicated the following reasons patients with cancer may die by suicide or seek aid in assisted suicide: social isolation, pain, physical impairment, loss of autonomy and meaning, terminal illness, and psychic distress and desperation.27
As with cancer, patients with PD exhibit increased suicidal ideation compared with the general population.28,29 Two studies found the suicide rate in individuals with PD is about twice as high as it is in the general population.30,31 Among people with PD, male sex, younger age, initial onset of motor symptoms in the upper or both upper and lower extremities, history of depression or any psychiatric diagnosis, delusions, higher levodopa dosing, and urban residence have been clinically correlated with suicide. Jumping has been a frequent method of suicide.30,31
Some research has evaluated the perspectives of loved ones after a patient chooses MAID. A study in the Netherlands found that 92% of relatives surveyed believed that access to MAID improved QoL and reduced pain at the end of life.32 In another, family members of individuals who used MAID reported higher quality on items related to physical symptom control and preparedness for death, compared with individuals who did not pursue MAID or who requested but did not receive it. There were no differences on items assessing connectedness to their loved one, being unafraid of death, level of consciousness, or global quality of death items.33 Another study found no significant differences in depression rates, grief, or use of mental health services among Oregon families whose loved ones died using MAID compared with those who did not.34
The higher suicide rate among terminally ill patients highlights the complex issue of MAID and the right to die. It is important to differentiate between euthanasia and medically-assisted dying. Euthanasia is an act whereby a person other than the patient acts to cause death. In MAID, the patient is provided with a medication that they self-administer. Recent Gallup polls found that > 70% of Americans believe physicians should be “allowed by law to end the patient’s life by some painless means if the patient and his or her family request it.”35
It is important to acknowledge MAID in the context of chronic suicidality, like in the case described in this article. It is imperative not to dismiss reports of suicidality in this population. Ignoring reports of suicidal ideation may lead to decreased access to pharmacologic and nonpharmacologic interventions. It is also important to maintain a timeline of symptom occurrence and to differentiate between chronic suicidality and the desire to die associated with having a terminal illness. A thorough assessment is necessary to assess whether the patient’s decision stems from a calculated decision with preserved capacity or from underlying mental health conditions. Other factors that may lead the patient to a hastened death (ie, pain, poor psychosocial support, delirium, cognitive impairment, incomplete understanding of treatment/prognosis) need to be addressed prior to finalizing choices. In this case, an assessment was performed by psychiatry, psychology, social work, and chaplains to ensure comprehensive evaluation.
The VHA offers resources to assist individuals experiencing suicidal ideation, including suicide prevention coordinators who work directly with veterans and offer consultation to teams working with veterans at risk for suicide. Support for VHA clinicians who treat veterans considering MAID may help address any moral distress. In this case, the care team met early for overnight sign-out, had daily core hospice team meetings, as-needed safety huddles, and weekly care plan meetings to ensure maximal physical and emotional comfort for the patient. These meetings cultivate open, honest, and transparent discussions regarding any staff concerns or personal distress around the plan of care. The VACHS chief well-being officer was also available for all staff.
A systematic review of the impact of MAID on clinicians found that MAID legislation influenced emotional responses. For countries whose MAID legislation emphasized alleviation from pain in addition to terminal illness, clinicians reported more emotional reflection. Whereas, in countries where MAID legislation is stricter and can be applied solely for terminal illness, clinicians reported a stronger and more polarizing range of emotions.36 This highlights the potential influence of the context in which clinicians work on their emotional experience with MAID. Given that MAID is not permitted in the VA, staff members may experience heightened emotional responses. In a survey of US adults, there was an interest in using MAID but there were knowledge deficits regarding the process and legality.37
Legal aspects come into play as well with regards to MAID. Eligibility requires the patient be aged ≥ 18 years, be terminally ill with a prognosis of ≤ 6 months, have the capacity to make their own health care decision, and be able to self-administer the medication. States also may have residency restrictions. Special care and adequate education are needed, as having anyone but the patient administer the medication may be considered criminal. Furthermore, since MAID is not allowed federally, this creates further distress in VHA clinicians entrusted to minimizing pain for patients.
Strategies to support veterans given prohibition of MAID include: conversations about the patient’s values, clarifying reasons for request, assessing all domains of distress, affirming concerns with compassion and nonjudgment, addressing any pain using pharmacologic and nonpharmacologic interventions, providing education on other permissible options for end-of-life care, and consulting other specialties.38
End-of-life options permitted by the VA include withholding/ withdrawing life-sustaining treatments, palliative sedation, and voluntary stopping of eating and drinking.39 Given the complexities of MAID, the VHA should initiate discussions of MAID, educate clinicians on what they can and cannot do as federal employees, and establish committees to discuss approaches that could minimize pain for patients and clinician distress.
Conclusions
Caring for veterans who request MAID requires clinicians to navigate a complex intersection of ethical obligations, legal constraints, and patient preferences. Within the VHA, where MAID is prohibited, clinicians must balance respect for patient autonomy with adherence to VA regulations. Comprehensive assessment to identify sources of distress, interdisciplinary collaboration, and recognition of permissible alternatives that align with patients’ values are essential to provide effective end-of-life care at the VHA for individuals considering MAID. As requests for MAID continue to emerge in clinical practice, the VHA has an opportunity to strengthen clinician education, clarify institutional expectations, and promote supportive structures that reduce both patient suffering and clinician moral distress.
- Meisel A, Snyder L, Quill T; American College of Physicians-- American Society of Internal Medicine End-of-Life Care Consensus Panel. Seven legal barriers to end-of- life care: myths, realities, and grains of truth. JAMA. 2000;284:2495-2501. doi:10.1001/jama.284.19.2495
- Physician-Assisted Suicide. American Medical Association Code of Medical Ethics. 2025. Accessed May 6, 2026. https://code-medical-ethics.ama-assn.org/ethics-opinions /physician-assisted-suicide
- Youngner SJ, Thoman R. AGS survey actually supports engaged neutrality for physician-assisted death. J Am Geriatr Soc. 2020;68:2140-2141. doi:10.1111/jgs.16679
- Physician-Assisted Dying. American Academy of Hospice and Palliative Medicine. Updated 2007. Accessed May 6, 2026. https://aahpm.org/advocacy/where-we-stand/pad/
- Ganzini L, Goy ER, Dobscha SK. Why Oregon patients request assisted death: family members’ views. J Gen Intern Med. 2008;23:154-157. doi:10.1007/s11606-007-0476-x
- Pearlman RA, Hsu C, Starks H, et al. Motivations for physician-assisted suicide: patient and family voices. J Gen Intern Med. 2005;20:234-239. doi:10.1111/j.1525-1497.2005.40225.x
- Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941- 948. doi:10.7326/0003-4819-113-12-941
- Fässberg MM, Cheung G, Canetto SS, et al. A systematic review of physical illness, functional disability, and suicidal behaviour among older adults.
Aging Ment Health. 2016;20:166-194. doi:10.1080/13607863.2015.1083945 - Gürhan N, Bes¸er NG, Polat Ü, et al. Suicide risk and depression in individuals with chronic illness. Community Ment Health J. 2019;55:840-848. doi:10.1007/s10597-019-00388-7
- Kye SY, Park K. Suicidal ideation and suicidal attempts among adults with chronic diseases: a crosssectional study. Compr Psychiatry. 2017;73:160-167. doi:10.1016/j.comppsych.2016.12.001
- Patten SB. Long-term medical conditions and major depression in a Canadian population study at waves 1 and 2. J Affect Disord. 2001;63:35-41. doi:10.1016/s0165-0327(00)00186-5
- Van der Kooy K, van Hout H, Marwijk H, et al. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int J Geriatr Psychiatry. 2007;22:613- 626. doi:10.1002/gps.1723
- Sporinova B, Manns B, Tonelli M, et al. Association of mental health disorders with health care utilization and costs among adults with chronic disease. JAMA Netw Open. 2019;2:e199910. doi:10.1001/jamanetworkopen.2019.9910
- Ahmedani BK, Peterson EL, Hu Y, et al. Major physical health conditions and risk of suicide. Am J Prev Med. 2017;53:308-315. doi:10.1016/j.amepre.2017.04.001
- Rogers ML, Joiner TE, Shahar G. Suicidality in chronic illness: an overview of cognitive-affective and interpersonal factors. J Clin Psychol Med Settings. 2021;28:137-148. doi:10.1007/s10880-020-09749-x
- Sisti D, Mann JJ, Oquendo MA. Toward a distinct mental disorder—suicidal behavior. JAMA Psychiatry. 2020;77:661-662. doi:10.1001/jamapsychiatry.2020.0111
- Kolva E, Hoffecker L, Cox-Martin E. Suicidal ideation in patients with cancer: a systematic review of prevalence, risk factors, intervention and assessment. Palliat Support Care. 2020;18:206-219. doi:10.1017/S1478951519000610
- Zaorsky NG, Zhang Y, Tuanquin L, et al. Suicide among cancer patients. Nat Commun. 2019;10:207. doi:10.1038/s41467-018-08170-1
- Heinrich M, Hofmann L, Baurecht H, et al. Suicide risk and mortality among patients with cancer. Nat Med. 2022;28:852-859. doi:10.1038/s41591-022-01745-y
- Yousaf U, Christensen ML, Engholm G, et al. Suicides among Danish cancer patients 1971-1999. Br J Cancer. 2005;92:995-1000. doi:10.1038/sj.bjc.6602424
- Misono S, Weiss NS, Fann JR, et al. Incidence of suicide in persons with cancer. J Clin Oncol. 2008;26:4731-4738. doi:10.1200/JCO.2007.13.8941
- Björkenstam C, Edberg A, Ayoubi S, et al. Are cancer patients at higher suicide risk than the general population?. Scand J Public Health. 2005;33:208-214. doi:10.1080/14034940410019226
- Kinslow CJ, Kumar P, Olfson M, et al. Prognosis and risk of suicide after cancer diagnosis. Cancer. 2024;130:588-596. doi:10.1002/cncr.35118
- Men VY, Emery CR, Yip PSF. Characteristics of cancer patients who died by suicide: a quantitative study of 15-year coronial records. Psychooncology. 2021;30:1051-1058. doi:10.1002/pon.5634
- Abdel-Rahman O. Socioeconomic predictors of suicide risk among cancer patients in the United States: a population- based study. Cancer Epidemiol. 2019;63:101601. doi:10.1016/j.canep.2019.101601
- O’Dwyer ST, Janssens A, Sansom A, et al. Suicidality in family caregivers of people with long-term illnesses and disabilities: a scoping review. Compr Psychiatry. 2021;110:152261. doi:10.1016/j.comppsych.2021.152261
- Senf B, Maiwurm P, Fettel J. Attitudes and opinions towards suicidality in professionals working with oncology patients: results from an online survey. Support Care Cancer. 2022;30:1775-1786. doi:10.1007/s00520-021-06590-2
- Berardelli I, Belvisi D, Nardella A, et al. Suicide in Parkinson’s disease: a systematic review. CNS Neurol Disord Drug Targets. 2019;18:466-477. doi:10.2174/1871527318666190703093345
- Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Survey. Arch Gen Psychiatry. 1999;56:617-626. doi:10.1001/archpsyc.56.7.617
- Chen YY, Yu S, Hu YH, et al. Risk of suicide among patients with Parkinson disease. JAMA Psychiatry. 2021;78:293-301. doi:10.1001/jamapsychiatry.2020.4001
- Lee T, Lee HB, Ahn MH, et al. Increased suicide risk and clinical correlates of suicide among patients with Parkinson’s disease. Parkinsonism Relat Disord. 2016;32:102- 107. doi:10.1016/j.parkreldis.2016.09.006
- Georges JJ, Onwuteaka-Philipsen BD, Muller MT, et al. Relatives’ perspective on the terminally ill patients who died after euthanasia or physician-assisted suicide: a retrospective cross-sectional interview study in the Netherlands. Death Stud. 2007;31:1-15. doi:10.1080/07481180600985041
- Smith KA, Goy ER, Harvath TA, et al. Quality of death and dying in patients who request physician-assisted death. J Palliat Med. 2011;14:445-450. doi:10.1089/jpm.2010.0425
- Ganzini L, Goy ER, Dobscha SK, et al. Mental health outcomes of family members of Oregonians who request physician aid in dying. J Pain Symptom Manage. 2009;38:807-815. doi:10.1016/j.jpainsymman.2009.04.026
- Yi R. Most Americans favor legal euthanasia. Gallup. August 8, 2024. Accessed May 6, 2026. https://news.gallup .com/poll/648215/americans-favor-legal-euthanasia.aspx
- Dholakia SY, Bagheri A, Simpson A. Emotional impact on healthcare providers involved in medical assistance in dying (MAiD): a systematic review and qualitative meta-synthesis. BMJ Open. 2022;12:e058523. doi:10.1136/bmjopen-2021-058523
- Kozlov E, Luth EA, Nemeth S, et al. Knowl - edge of and preferences for medical aid in dying. JAMA Netw Open. 2025;8:e2461495. doi:10.1001/jamanetworkopen.2024.61495
- Geppert C; Veterans Administration National Center for Ethics in Health Care. Medical aid in dying in the VA. Presented at: VISN 1 Palliative Care Summit, September 2024.
- National Ethics Committee, Veterans Health Administration. The ethics of palliative sedation as a therapy of last resort. Am J Hosp Palliat Care. 2006;23:483-491. doi:10.1177/1049909106294883
Requests for medical aid in dying (MAID) within the Veterans Health Administration (VHA) present unique ethical, legal, and clinical challenges. MAID is a process in which a physician provides a terminally ill patient with the means to end their own life. It is expressly prohibited by federal law, including within the US Department of Veterans Affairs (VA), regardless of its legality at the state level.1 MAID is also prohibited within community care institutions funded by the VA. The American Medical Association, American Geriatrics Society, and American Academy of Hospice and Palliative Medicine have adopted neutral positions regarding MAID due to varying opinions among their respective members.2-4 VHA palliative care clinicians are trained to identify and honor preferences for care and alleviate physical and emotional distress, which may complicate the management of MAID requests. Veterans can request MAID due to their desire for autonomy and pain relief, but the VHA prohibits clinicians from honoring these specific preferences. The inability to help veterans achieve their care preferences conflicts with the core mission of palliative care to reduce suffering and respect end-of-life wishes.
This case report describes the management of a veteran who requested MAID while also exhibiting active suicidal ideation. The patient’s distress stemmed from fears of impending loss of autonomy and functional decline, factors frequently linked to requests for MAID in terminally ill patients.5,6 Addressing the veteran’s request for MAID required balancing respect for patient autonomy and concerns about future suffering with the VA mission to protect veterans from self-harm and provide mental health care for suicidal ideation. This case highlights the importance of nuanced clinical approaches, ethical reflection, and interdisciplinary collaboration in navigating such complex scenarios. Informed consent was obtained from the patient’s family and health care agent (HCA) to publish this report.
Case Presentation
A 73-year-old male veteran, with Parkinson disease (PD), diagnosed at age 52 years, was referred to palliative care following diagnosis of a glioblastoma multiforme (GBM). The patient also had a history of major depressive disorder (MDD), suicidal ideation (SI), benign prostatic hypertrophy, and migraines. He was divorced, had no children, and his only sibling (sister) was deceased. His brother-in-law served as his HCA.
The patient had many close friends in the community, was an architect by training, and was active in the removal of barriers and increasing access for people with disabilities. Since 2010, about 7 years before his PD diagnosis, the patient used psychiatry and psychology resources to treat MDD, functional decline, and SI. He was hospitalized in 2016 after self-administration of heroin. During the hospitalization the patient received a high risk for suicide label. He articulated a firm and long-standing belief in his right to die and shared plans to end his life when he experienced a significant decline in his independence and quality of life (QoL).
When diagnosed with PD, the patient shared that his QoL was of utmost importance. He was aware that he would have significant physical decline as PD progressed and felt like there would be a point when his QoL would not be acceptable. When that happened, he wanted to end his life by available means. He was followed closely by his VHA care team for physical and emotional distress.
When diagnosed with a GBM in 2023, the patient declined treatment and was referred to palliative care, which had sporadically treated him for PD-related distress prior to 2023. During his previous palliative care visits, the patient had discussed a desire to engage in MAID when his functional status declined. After the GBM diagnosis, he reported no acute intent to harm himself with heroin, but planned to travel to Vermont for MAID when he felt he no longer had an adequate QoL based on functional capability.
Pharmacologic and nonpharmacologic approaches were used to treat the patient’s pain. He reported significant benefit from biofeedback therapy provided by the VA Headache Center of Excellence. This work also reconnected him to meditation, which he used daily to relieve pain and distress. The patient managed head pain with nonpharmacologic and pharmacologic interventions for 6 months and reported satisfaction with his QoL.
After 6 months, imaging showed progression of the brain tumor, which was associated with more fatigue and memory decline. At that time, the patient was enrolled in home hospice and reported continued intent to pursue MAID in Vermont but had not taken steps toward carrying it out. The patient understood the VA could not assist him in pursuing MAID; however, his care team was able to assist him in sharing his preferences for care with his loved ones and health care power of attorney.
He experienced rapid functional and cognitive decline due to progression of the GBM and was admitted to the VA Connecticut Healthcare System (VACHS) acute care unit where he exhibited confusion and screened positive for delirium using the Confusion Assessment Method.7 His physical and cognitive deterioration was likely due to the progressive brain tumor, and the patient lacked the capacity to make complex medical decisions. Formal consent was obtained from his HCA to transfer him to inpatient hospice. Psychiatry followed the patient throughout.
After 4 weeks of hospice care, the patient had a witnessed suicide attempt while the nurse was assisting him in the bathroom. The patient attempted to use hospital pajamas to hang himself when he wrapped a hospital gown around his neck and stated he was trying to tie a knot. Due to his confusion and delirium, the patient was unable to express his reasoning for the suicide attempt. He was seen by the Psychiatry service, which determined that his suicide risk was low to intermediate. The Psychiatry service did not recommend a 1:1 safety sitter, but suggested medication changes. Levetiracetam was discontinued, and valproate 500 mg orally twice daily was initiated for seizure prevention.
The hospice team was informed of the suicide attempt and psychiatry recommendations. The suicide prevention team was also updated following this event and agreed with psychiatry recommendations. The patient continued to decline, was no longer able to get out of bed, and had minimal speech. The patient received comfort medications, including intravenous morphine 2 mg and lorazepam 0.5 mg as needed ≤ 4 times daily. He died 8 days later.
Discussion
Chronic medical illness has been associated with increased suicide risk.8-10 The increased risk of suicide in chronically ill patients has been described as having as a bidirectional relationship with MDD, with depression not only increasing the risk of chronic medical illness but new-onset chronic medical illness being associated with new onset depression.11,12 Chronic medical conditions are associated with numerous psychiatric disorders, and the presence of a comorbid psychiatric illness is associated with higher rates of hospitalization, emergency department visits, and increased health care costs.13 Research has found that the association between suicide risk and chronic medical illness remains even after accounting for comorbid mental health disorders.14 This has been postulated to be due to a multitude of interpersonal, behavioral, cognitive, and affective factors (eg, perceived burdensomeness, loneliness, stress, pain catastrophizing, self-criticism).15 Additionally, some researchers have questioned whether suicidality constitutes a distinct mental disorder.16
Patients with cancer are at increased risk for suicidal ideation (including passive death wishes) and suicide attempts.17,18 Recent data indicate that compared with the general population, there is an 85% increased risk of suicide mortality in patients with cancer.19 Studies show the incidence of suicide is greater for individuals with cancer compared with the general population, with standardized mortality ratios ranging from 1.4 to 5.7.20-22
Among patients with cancer, suicide risk is associated with several factors: worse prognosis, older age, male sex, living in a socioeconomically vulnerable environment, and increased communication about suicidal intent prior to death.23-25 Just as the prevalence of suicidal ideation in people with cancer varies widely, reported rates of suicidality in caregivers of patients with cancer range from 2.7% to 71%.17,26 A survey of health care workers indicated the following reasons patients with cancer may die by suicide or seek aid in assisted suicide: social isolation, pain, physical impairment, loss of autonomy and meaning, terminal illness, and psychic distress and desperation.27
As with cancer, patients with PD exhibit increased suicidal ideation compared with the general population.28,29 Two studies found the suicide rate in individuals with PD is about twice as high as it is in the general population.30,31 Among people with PD, male sex, younger age, initial onset of motor symptoms in the upper or both upper and lower extremities, history of depression or any psychiatric diagnosis, delusions, higher levodopa dosing, and urban residence have been clinically correlated with suicide. Jumping has been a frequent method of suicide.30,31
Some research has evaluated the perspectives of loved ones after a patient chooses MAID. A study in the Netherlands found that 92% of relatives surveyed believed that access to MAID improved QoL and reduced pain at the end of life.32 In another, family members of individuals who used MAID reported higher quality on items related to physical symptom control and preparedness for death, compared with individuals who did not pursue MAID or who requested but did not receive it. There were no differences on items assessing connectedness to their loved one, being unafraid of death, level of consciousness, or global quality of death items.33 Another study found no significant differences in depression rates, grief, or use of mental health services among Oregon families whose loved ones died using MAID compared with those who did not.34
The higher suicide rate among terminally ill patients highlights the complex issue of MAID and the right to die. It is important to differentiate between euthanasia and medically-assisted dying. Euthanasia is an act whereby a person other than the patient acts to cause death. In MAID, the patient is provided with a medication that they self-administer. Recent Gallup polls found that > 70% of Americans believe physicians should be “allowed by law to end the patient’s life by some painless means if the patient and his or her family request it.”35
It is important to acknowledge MAID in the context of chronic suicidality, like in the case described in this article. It is imperative not to dismiss reports of suicidality in this population. Ignoring reports of suicidal ideation may lead to decreased access to pharmacologic and nonpharmacologic interventions. It is also important to maintain a timeline of symptom occurrence and to differentiate between chronic suicidality and the desire to die associated with having a terminal illness. A thorough assessment is necessary to assess whether the patient’s decision stems from a calculated decision with preserved capacity or from underlying mental health conditions. Other factors that may lead the patient to a hastened death (ie, pain, poor psychosocial support, delirium, cognitive impairment, incomplete understanding of treatment/prognosis) need to be addressed prior to finalizing choices. In this case, an assessment was performed by psychiatry, psychology, social work, and chaplains to ensure comprehensive evaluation.
The VHA offers resources to assist individuals experiencing suicidal ideation, including suicide prevention coordinators who work directly with veterans and offer consultation to teams working with veterans at risk for suicide. Support for VHA clinicians who treat veterans considering MAID may help address any moral distress. In this case, the care team met early for overnight sign-out, had daily core hospice team meetings, as-needed safety huddles, and weekly care plan meetings to ensure maximal physical and emotional comfort for the patient. These meetings cultivate open, honest, and transparent discussions regarding any staff concerns or personal distress around the plan of care. The VACHS chief well-being officer was also available for all staff.
A systematic review of the impact of MAID on clinicians found that MAID legislation influenced emotional responses. For countries whose MAID legislation emphasized alleviation from pain in addition to terminal illness, clinicians reported more emotional reflection. Whereas, in countries where MAID legislation is stricter and can be applied solely for terminal illness, clinicians reported a stronger and more polarizing range of emotions.36 This highlights the potential influence of the context in which clinicians work on their emotional experience with MAID. Given that MAID is not permitted in the VA, staff members may experience heightened emotional responses. In a survey of US adults, there was an interest in using MAID but there were knowledge deficits regarding the process and legality.37
Legal aspects come into play as well with regards to MAID. Eligibility requires the patient be aged ≥ 18 years, be terminally ill with a prognosis of ≤ 6 months, have the capacity to make their own health care decision, and be able to self-administer the medication. States also may have residency restrictions. Special care and adequate education are needed, as having anyone but the patient administer the medication may be considered criminal. Furthermore, since MAID is not allowed federally, this creates further distress in VHA clinicians entrusted to minimizing pain for patients.
Strategies to support veterans given prohibition of MAID include: conversations about the patient’s values, clarifying reasons for request, assessing all domains of distress, affirming concerns with compassion and nonjudgment, addressing any pain using pharmacologic and nonpharmacologic interventions, providing education on other permissible options for end-of-life care, and consulting other specialties.38
End-of-life options permitted by the VA include withholding/ withdrawing life-sustaining treatments, palliative sedation, and voluntary stopping of eating and drinking.39 Given the complexities of MAID, the VHA should initiate discussions of MAID, educate clinicians on what they can and cannot do as federal employees, and establish committees to discuss approaches that could minimize pain for patients and clinician distress.
Conclusions
Caring for veterans who request MAID requires clinicians to navigate a complex intersection of ethical obligations, legal constraints, and patient preferences. Within the VHA, where MAID is prohibited, clinicians must balance respect for patient autonomy with adherence to VA regulations. Comprehensive assessment to identify sources of distress, interdisciplinary collaboration, and recognition of permissible alternatives that align with patients’ values are essential to provide effective end-of-life care at the VHA for individuals considering MAID. As requests for MAID continue to emerge in clinical practice, the VHA has an opportunity to strengthen clinician education, clarify institutional expectations, and promote supportive structures that reduce both patient suffering and clinician moral distress.
Requests for medical aid in dying (MAID) within the Veterans Health Administration (VHA) present unique ethical, legal, and clinical challenges. MAID is a process in which a physician provides a terminally ill patient with the means to end their own life. It is expressly prohibited by federal law, including within the US Department of Veterans Affairs (VA), regardless of its legality at the state level.1 MAID is also prohibited within community care institutions funded by the VA. The American Medical Association, American Geriatrics Society, and American Academy of Hospice and Palliative Medicine have adopted neutral positions regarding MAID due to varying opinions among their respective members.2-4 VHA palliative care clinicians are trained to identify and honor preferences for care and alleviate physical and emotional distress, which may complicate the management of MAID requests. Veterans can request MAID due to their desire for autonomy and pain relief, but the VHA prohibits clinicians from honoring these specific preferences. The inability to help veterans achieve their care preferences conflicts with the core mission of palliative care to reduce suffering and respect end-of-life wishes.
This case report describes the management of a veteran who requested MAID while also exhibiting active suicidal ideation. The patient’s distress stemmed from fears of impending loss of autonomy and functional decline, factors frequently linked to requests for MAID in terminally ill patients.5,6 Addressing the veteran’s request for MAID required balancing respect for patient autonomy and concerns about future suffering with the VA mission to protect veterans from self-harm and provide mental health care for suicidal ideation. This case highlights the importance of nuanced clinical approaches, ethical reflection, and interdisciplinary collaboration in navigating such complex scenarios. Informed consent was obtained from the patient’s family and health care agent (HCA) to publish this report.
Case Presentation
A 73-year-old male veteran, with Parkinson disease (PD), diagnosed at age 52 years, was referred to palliative care following diagnosis of a glioblastoma multiforme (GBM). The patient also had a history of major depressive disorder (MDD), suicidal ideation (SI), benign prostatic hypertrophy, and migraines. He was divorced, had no children, and his only sibling (sister) was deceased. His brother-in-law served as his HCA.
The patient had many close friends in the community, was an architect by training, and was active in the removal of barriers and increasing access for people with disabilities. Since 2010, about 7 years before his PD diagnosis, the patient used psychiatry and psychology resources to treat MDD, functional decline, and SI. He was hospitalized in 2016 after self-administration of heroin. During the hospitalization the patient received a high risk for suicide label. He articulated a firm and long-standing belief in his right to die and shared plans to end his life when he experienced a significant decline in his independence and quality of life (QoL).
When diagnosed with PD, the patient shared that his QoL was of utmost importance. He was aware that he would have significant physical decline as PD progressed and felt like there would be a point when his QoL would not be acceptable. When that happened, he wanted to end his life by available means. He was followed closely by his VHA care team for physical and emotional distress.
When diagnosed with a GBM in 2023, the patient declined treatment and was referred to palliative care, which had sporadically treated him for PD-related distress prior to 2023. During his previous palliative care visits, the patient had discussed a desire to engage in MAID when his functional status declined. After the GBM diagnosis, he reported no acute intent to harm himself with heroin, but planned to travel to Vermont for MAID when he felt he no longer had an adequate QoL based on functional capability.
Pharmacologic and nonpharmacologic approaches were used to treat the patient’s pain. He reported significant benefit from biofeedback therapy provided by the VA Headache Center of Excellence. This work also reconnected him to meditation, which he used daily to relieve pain and distress. The patient managed head pain with nonpharmacologic and pharmacologic interventions for 6 months and reported satisfaction with his QoL.
After 6 months, imaging showed progression of the brain tumor, which was associated with more fatigue and memory decline. At that time, the patient was enrolled in home hospice and reported continued intent to pursue MAID in Vermont but had not taken steps toward carrying it out. The patient understood the VA could not assist him in pursuing MAID; however, his care team was able to assist him in sharing his preferences for care with his loved ones and health care power of attorney.
He experienced rapid functional and cognitive decline due to progression of the GBM and was admitted to the VA Connecticut Healthcare System (VACHS) acute care unit where he exhibited confusion and screened positive for delirium using the Confusion Assessment Method.7 His physical and cognitive deterioration was likely due to the progressive brain tumor, and the patient lacked the capacity to make complex medical decisions. Formal consent was obtained from his HCA to transfer him to inpatient hospice. Psychiatry followed the patient throughout.
After 4 weeks of hospice care, the patient had a witnessed suicide attempt while the nurse was assisting him in the bathroom. The patient attempted to use hospital pajamas to hang himself when he wrapped a hospital gown around his neck and stated he was trying to tie a knot. Due to his confusion and delirium, the patient was unable to express his reasoning for the suicide attempt. He was seen by the Psychiatry service, which determined that his suicide risk was low to intermediate. The Psychiatry service did not recommend a 1:1 safety sitter, but suggested medication changes. Levetiracetam was discontinued, and valproate 500 mg orally twice daily was initiated for seizure prevention.
The hospice team was informed of the suicide attempt and psychiatry recommendations. The suicide prevention team was also updated following this event and agreed with psychiatry recommendations. The patient continued to decline, was no longer able to get out of bed, and had minimal speech. The patient received comfort medications, including intravenous morphine 2 mg and lorazepam 0.5 mg as needed ≤ 4 times daily. He died 8 days later.
Discussion
Chronic medical illness has been associated with increased suicide risk.8-10 The increased risk of suicide in chronically ill patients has been described as having as a bidirectional relationship with MDD, with depression not only increasing the risk of chronic medical illness but new-onset chronic medical illness being associated with new onset depression.11,12 Chronic medical conditions are associated with numerous psychiatric disorders, and the presence of a comorbid psychiatric illness is associated with higher rates of hospitalization, emergency department visits, and increased health care costs.13 Research has found that the association between suicide risk and chronic medical illness remains even after accounting for comorbid mental health disorders.14 This has been postulated to be due to a multitude of interpersonal, behavioral, cognitive, and affective factors (eg, perceived burdensomeness, loneliness, stress, pain catastrophizing, self-criticism).15 Additionally, some researchers have questioned whether suicidality constitutes a distinct mental disorder.16
Patients with cancer are at increased risk for suicidal ideation (including passive death wishes) and suicide attempts.17,18 Recent data indicate that compared with the general population, there is an 85% increased risk of suicide mortality in patients with cancer.19 Studies show the incidence of suicide is greater for individuals with cancer compared with the general population, with standardized mortality ratios ranging from 1.4 to 5.7.20-22
Among patients with cancer, suicide risk is associated with several factors: worse prognosis, older age, male sex, living in a socioeconomically vulnerable environment, and increased communication about suicidal intent prior to death.23-25 Just as the prevalence of suicidal ideation in people with cancer varies widely, reported rates of suicidality in caregivers of patients with cancer range from 2.7% to 71%.17,26 A survey of health care workers indicated the following reasons patients with cancer may die by suicide or seek aid in assisted suicide: social isolation, pain, physical impairment, loss of autonomy and meaning, terminal illness, and psychic distress and desperation.27
As with cancer, patients with PD exhibit increased suicidal ideation compared with the general population.28,29 Two studies found the suicide rate in individuals with PD is about twice as high as it is in the general population.30,31 Among people with PD, male sex, younger age, initial onset of motor symptoms in the upper or both upper and lower extremities, history of depression or any psychiatric diagnosis, delusions, higher levodopa dosing, and urban residence have been clinically correlated with suicide. Jumping has been a frequent method of suicide.30,31
Some research has evaluated the perspectives of loved ones after a patient chooses MAID. A study in the Netherlands found that 92% of relatives surveyed believed that access to MAID improved QoL and reduced pain at the end of life.32 In another, family members of individuals who used MAID reported higher quality on items related to physical symptom control and preparedness for death, compared with individuals who did not pursue MAID or who requested but did not receive it. There were no differences on items assessing connectedness to their loved one, being unafraid of death, level of consciousness, or global quality of death items.33 Another study found no significant differences in depression rates, grief, or use of mental health services among Oregon families whose loved ones died using MAID compared with those who did not.34
The higher suicide rate among terminally ill patients highlights the complex issue of MAID and the right to die. It is important to differentiate between euthanasia and medically-assisted dying. Euthanasia is an act whereby a person other than the patient acts to cause death. In MAID, the patient is provided with a medication that they self-administer. Recent Gallup polls found that > 70% of Americans believe physicians should be “allowed by law to end the patient’s life by some painless means if the patient and his or her family request it.”35
It is important to acknowledge MAID in the context of chronic suicidality, like in the case described in this article. It is imperative not to dismiss reports of suicidality in this population. Ignoring reports of suicidal ideation may lead to decreased access to pharmacologic and nonpharmacologic interventions. It is also important to maintain a timeline of symptom occurrence and to differentiate between chronic suicidality and the desire to die associated with having a terminal illness. A thorough assessment is necessary to assess whether the patient’s decision stems from a calculated decision with preserved capacity or from underlying mental health conditions. Other factors that may lead the patient to a hastened death (ie, pain, poor psychosocial support, delirium, cognitive impairment, incomplete understanding of treatment/prognosis) need to be addressed prior to finalizing choices. In this case, an assessment was performed by psychiatry, psychology, social work, and chaplains to ensure comprehensive evaluation.
The VHA offers resources to assist individuals experiencing suicidal ideation, including suicide prevention coordinators who work directly with veterans and offer consultation to teams working with veterans at risk for suicide. Support for VHA clinicians who treat veterans considering MAID may help address any moral distress. In this case, the care team met early for overnight sign-out, had daily core hospice team meetings, as-needed safety huddles, and weekly care plan meetings to ensure maximal physical and emotional comfort for the patient. These meetings cultivate open, honest, and transparent discussions regarding any staff concerns or personal distress around the plan of care. The VACHS chief well-being officer was also available for all staff.
A systematic review of the impact of MAID on clinicians found that MAID legislation influenced emotional responses. For countries whose MAID legislation emphasized alleviation from pain in addition to terminal illness, clinicians reported more emotional reflection. Whereas, in countries where MAID legislation is stricter and can be applied solely for terminal illness, clinicians reported a stronger and more polarizing range of emotions.36 This highlights the potential influence of the context in which clinicians work on their emotional experience with MAID. Given that MAID is not permitted in the VA, staff members may experience heightened emotional responses. In a survey of US adults, there was an interest in using MAID but there were knowledge deficits regarding the process and legality.37
Legal aspects come into play as well with regards to MAID. Eligibility requires the patient be aged ≥ 18 years, be terminally ill with a prognosis of ≤ 6 months, have the capacity to make their own health care decision, and be able to self-administer the medication. States also may have residency restrictions. Special care and adequate education are needed, as having anyone but the patient administer the medication may be considered criminal. Furthermore, since MAID is not allowed federally, this creates further distress in VHA clinicians entrusted to minimizing pain for patients.
Strategies to support veterans given prohibition of MAID include: conversations about the patient’s values, clarifying reasons for request, assessing all domains of distress, affirming concerns with compassion and nonjudgment, addressing any pain using pharmacologic and nonpharmacologic interventions, providing education on other permissible options for end-of-life care, and consulting other specialties.38
End-of-life options permitted by the VA include withholding/ withdrawing life-sustaining treatments, palliative sedation, and voluntary stopping of eating and drinking.39 Given the complexities of MAID, the VHA should initiate discussions of MAID, educate clinicians on what they can and cannot do as federal employees, and establish committees to discuss approaches that could minimize pain for patients and clinician distress.
Conclusions
Caring for veterans who request MAID requires clinicians to navigate a complex intersection of ethical obligations, legal constraints, and patient preferences. Within the VHA, where MAID is prohibited, clinicians must balance respect for patient autonomy with adherence to VA regulations. Comprehensive assessment to identify sources of distress, interdisciplinary collaboration, and recognition of permissible alternatives that align with patients’ values are essential to provide effective end-of-life care at the VHA for individuals considering MAID. As requests for MAID continue to emerge in clinical practice, the VHA has an opportunity to strengthen clinician education, clarify institutional expectations, and promote supportive structures that reduce both patient suffering and clinician moral distress.
- Meisel A, Snyder L, Quill T; American College of Physicians-- American Society of Internal Medicine End-of-Life Care Consensus Panel. Seven legal barriers to end-of- life care: myths, realities, and grains of truth. JAMA. 2000;284:2495-2501. doi:10.1001/jama.284.19.2495
- Physician-Assisted Suicide. American Medical Association Code of Medical Ethics. 2025. Accessed May 6, 2026. https://code-medical-ethics.ama-assn.org/ethics-opinions /physician-assisted-suicide
- Youngner SJ, Thoman R. AGS survey actually supports engaged neutrality for physician-assisted death. J Am Geriatr Soc. 2020;68:2140-2141. doi:10.1111/jgs.16679
- Physician-Assisted Dying. American Academy of Hospice and Palliative Medicine. Updated 2007. Accessed May 6, 2026. https://aahpm.org/advocacy/where-we-stand/pad/
- Ganzini L, Goy ER, Dobscha SK. Why Oregon patients request assisted death: family members’ views. J Gen Intern Med. 2008;23:154-157. doi:10.1007/s11606-007-0476-x
- Pearlman RA, Hsu C, Starks H, et al. Motivations for physician-assisted suicide: patient and family voices. J Gen Intern Med. 2005;20:234-239. doi:10.1111/j.1525-1497.2005.40225.x
- Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941- 948. doi:10.7326/0003-4819-113-12-941
- Fässberg MM, Cheung G, Canetto SS, et al. A systematic review of physical illness, functional disability, and suicidal behaviour among older adults.
Aging Ment Health. 2016;20:166-194. doi:10.1080/13607863.2015.1083945 - Gürhan N, Bes¸er NG, Polat Ü, et al. Suicide risk and depression in individuals with chronic illness. Community Ment Health J. 2019;55:840-848. doi:10.1007/s10597-019-00388-7
- Kye SY, Park K. Suicidal ideation and suicidal attempts among adults with chronic diseases: a crosssectional study. Compr Psychiatry. 2017;73:160-167. doi:10.1016/j.comppsych.2016.12.001
- Patten SB. Long-term medical conditions and major depression in a Canadian population study at waves 1 and 2. J Affect Disord. 2001;63:35-41. doi:10.1016/s0165-0327(00)00186-5
- Van der Kooy K, van Hout H, Marwijk H, et al. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int J Geriatr Psychiatry. 2007;22:613- 626. doi:10.1002/gps.1723
- Sporinova B, Manns B, Tonelli M, et al. Association of mental health disorders with health care utilization and costs among adults with chronic disease. JAMA Netw Open. 2019;2:e199910. doi:10.1001/jamanetworkopen.2019.9910
- Ahmedani BK, Peterson EL, Hu Y, et al. Major physical health conditions and risk of suicide. Am J Prev Med. 2017;53:308-315. doi:10.1016/j.amepre.2017.04.001
- Rogers ML, Joiner TE, Shahar G. Suicidality in chronic illness: an overview of cognitive-affective and interpersonal factors. J Clin Psychol Med Settings. 2021;28:137-148. doi:10.1007/s10880-020-09749-x
- Sisti D, Mann JJ, Oquendo MA. Toward a distinct mental disorder—suicidal behavior. JAMA Psychiatry. 2020;77:661-662. doi:10.1001/jamapsychiatry.2020.0111
- Kolva E, Hoffecker L, Cox-Martin E. Suicidal ideation in patients with cancer: a systematic review of prevalence, risk factors, intervention and assessment. Palliat Support Care. 2020;18:206-219. doi:10.1017/S1478951519000610
- Zaorsky NG, Zhang Y, Tuanquin L, et al. Suicide among cancer patients. Nat Commun. 2019;10:207. doi:10.1038/s41467-018-08170-1
- Heinrich M, Hofmann L, Baurecht H, et al. Suicide risk and mortality among patients with cancer. Nat Med. 2022;28:852-859. doi:10.1038/s41591-022-01745-y
- Yousaf U, Christensen ML, Engholm G, et al. Suicides among Danish cancer patients 1971-1999. Br J Cancer. 2005;92:995-1000. doi:10.1038/sj.bjc.6602424
- Misono S, Weiss NS, Fann JR, et al. Incidence of suicide in persons with cancer. J Clin Oncol. 2008;26:4731-4738. doi:10.1200/JCO.2007.13.8941
- Björkenstam C, Edberg A, Ayoubi S, et al. Are cancer patients at higher suicide risk than the general population?. Scand J Public Health. 2005;33:208-214. doi:10.1080/14034940410019226
- Kinslow CJ, Kumar P, Olfson M, et al. Prognosis and risk of suicide after cancer diagnosis. Cancer. 2024;130:588-596. doi:10.1002/cncr.35118
- Men VY, Emery CR, Yip PSF. Characteristics of cancer patients who died by suicide: a quantitative study of 15-year coronial records. Psychooncology. 2021;30:1051-1058. doi:10.1002/pon.5634
- Abdel-Rahman O. Socioeconomic predictors of suicide risk among cancer patients in the United States: a population- based study. Cancer Epidemiol. 2019;63:101601. doi:10.1016/j.canep.2019.101601
- O’Dwyer ST, Janssens A, Sansom A, et al. Suicidality in family caregivers of people with long-term illnesses and disabilities: a scoping review. Compr Psychiatry. 2021;110:152261. doi:10.1016/j.comppsych.2021.152261
- Senf B, Maiwurm P, Fettel J. Attitudes and opinions towards suicidality in professionals working with oncology patients: results from an online survey. Support Care Cancer. 2022;30:1775-1786. doi:10.1007/s00520-021-06590-2
- Berardelli I, Belvisi D, Nardella A, et al. Suicide in Parkinson’s disease: a systematic review. CNS Neurol Disord Drug Targets. 2019;18:466-477. doi:10.2174/1871527318666190703093345
- Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Survey. Arch Gen Psychiatry. 1999;56:617-626. doi:10.1001/archpsyc.56.7.617
- Chen YY, Yu S, Hu YH, et al. Risk of suicide among patients with Parkinson disease. JAMA Psychiatry. 2021;78:293-301. doi:10.1001/jamapsychiatry.2020.4001
- Lee T, Lee HB, Ahn MH, et al. Increased suicide risk and clinical correlates of suicide among patients with Parkinson’s disease. Parkinsonism Relat Disord. 2016;32:102- 107. doi:10.1016/j.parkreldis.2016.09.006
- Georges JJ, Onwuteaka-Philipsen BD, Muller MT, et al. Relatives’ perspective on the terminally ill patients who died after euthanasia or physician-assisted suicide: a retrospective cross-sectional interview study in the Netherlands. Death Stud. 2007;31:1-15. doi:10.1080/07481180600985041
- Smith KA, Goy ER, Harvath TA, et al. Quality of death and dying in patients who request physician-assisted death. J Palliat Med. 2011;14:445-450. doi:10.1089/jpm.2010.0425
- Ganzini L, Goy ER, Dobscha SK, et al. Mental health outcomes of family members of Oregonians who request physician aid in dying. J Pain Symptom Manage. 2009;38:807-815. doi:10.1016/j.jpainsymman.2009.04.026
- Yi R. Most Americans favor legal euthanasia. Gallup. August 8, 2024. Accessed May 6, 2026. https://news.gallup .com/poll/648215/americans-favor-legal-euthanasia.aspx
- Dholakia SY, Bagheri A, Simpson A. Emotional impact on healthcare providers involved in medical assistance in dying (MAiD): a systematic review and qualitative meta-synthesis. BMJ Open. 2022;12:e058523. doi:10.1136/bmjopen-2021-058523
- Kozlov E, Luth EA, Nemeth S, et al. Knowl - edge of and preferences for medical aid in dying. JAMA Netw Open. 2025;8:e2461495. doi:10.1001/jamanetworkopen.2024.61495
- Geppert C; Veterans Administration National Center for Ethics in Health Care. Medical aid in dying in the VA. Presented at: VISN 1 Palliative Care Summit, September 2024.
- National Ethics Committee, Veterans Health Administration. The ethics of palliative sedation as a therapy of last resort. Am J Hosp Palliat Care. 2006;23:483-491. doi:10.1177/1049909106294883
- Meisel A, Snyder L, Quill T; American College of Physicians-- American Society of Internal Medicine End-of-Life Care Consensus Panel. Seven legal barriers to end-of- life care: myths, realities, and grains of truth. JAMA. 2000;284:2495-2501. doi:10.1001/jama.284.19.2495
- Physician-Assisted Suicide. American Medical Association Code of Medical Ethics. 2025. Accessed May 6, 2026. https://code-medical-ethics.ama-assn.org/ethics-opinions /physician-assisted-suicide
- Youngner SJ, Thoman R. AGS survey actually supports engaged neutrality for physician-assisted death. J Am Geriatr Soc. 2020;68:2140-2141. doi:10.1111/jgs.16679
- Physician-Assisted Dying. American Academy of Hospice and Palliative Medicine. Updated 2007. Accessed May 6, 2026. https://aahpm.org/advocacy/where-we-stand/pad/
- Ganzini L, Goy ER, Dobscha SK. Why Oregon patients request assisted death: family members’ views. J Gen Intern Med. 2008;23:154-157. doi:10.1007/s11606-007-0476-x
- Pearlman RA, Hsu C, Starks H, et al. Motivations for physician-assisted suicide: patient and family voices. J Gen Intern Med. 2005;20:234-239. doi:10.1111/j.1525-1497.2005.40225.x
- Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the Confusion Assessment Method. A new method for detection of delirium. Ann Intern Med. 1990;113:941- 948. doi:10.7326/0003-4819-113-12-941
- Fässberg MM, Cheung G, Canetto SS, et al. A systematic review of physical illness, functional disability, and suicidal behaviour among older adults.
Aging Ment Health. 2016;20:166-194. doi:10.1080/13607863.2015.1083945 - Gürhan N, Bes¸er NG, Polat Ü, et al. Suicide risk and depression in individuals with chronic illness. Community Ment Health J. 2019;55:840-848. doi:10.1007/s10597-019-00388-7
- Kye SY, Park K. Suicidal ideation and suicidal attempts among adults with chronic diseases: a crosssectional study. Compr Psychiatry. 2017;73:160-167. doi:10.1016/j.comppsych.2016.12.001
- Patten SB. Long-term medical conditions and major depression in a Canadian population study at waves 1 and 2. J Affect Disord. 2001;63:35-41. doi:10.1016/s0165-0327(00)00186-5
- Van der Kooy K, van Hout H, Marwijk H, et al. Depression and the risk for cardiovascular diseases: systematic review and meta analysis. Int J Geriatr Psychiatry. 2007;22:613- 626. doi:10.1002/gps.1723
- Sporinova B, Manns B, Tonelli M, et al. Association of mental health disorders with health care utilization and costs among adults with chronic disease. JAMA Netw Open. 2019;2:e199910. doi:10.1001/jamanetworkopen.2019.9910
- Ahmedani BK, Peterson EL, Hu Y, et al. Major physical health conditions and risk of suicide. Am J Prev Med. 2017;53:308-315. doi:10.1016/j.amepre.2017.04.001
- Rogers ML, Joiner TE, Shahar G. Suicidality in chronic illness: an overview of cognitive-affective and interpersonal factors. J Clin Psychol Med Settings. 2021;28:137-148. doi:10.1007/s10880-020-09749-x
- Sisti D, Mann JJ, Oquendo MA. Toward a distinct mental disorder—suicidal behavior. JAMA Psychiatry. 2020;77:661-662. doi:10.1001/jamapsychiatry.2020.0111
- Kolva E, Hoffecker L, Cox-Martin E. Suicidal ideation in patients with cancer: a systematic review of prevalence, risk factors, intervention and assessment. Palliat Support Care. 2020;18:206-219. doi:10.1017/S1478951519000610
- Zaorsky NG, Zhang Y, Tuanquin L, et al. Suicide among cancer patients. Nat Commun. 2019;10:207. doi:10.1038/s41467-018-08170-1
- Heinrich M, Hofmann L, Baurecht H, et al. Suicide risk and mortality among patients with cancer. Nat Med. 2022;28:852-859. doi:10.1038/s41591-022-01745-y
- Yousaf U, Christensen ML, Engholm G, et al. Suicides among Danish cancer patients 1971-1999. Br J Cancer. 2005;92:995-1000. doi:10.1038/sj.bjc.6602424
- Misono S, Weiss NS, Fann JR, et al. Incidence of suicide in persons with cancer. J Clin Oncol. 2008;26:4731-4738. doi:10.1200/JCO.2007.13.8941
- Björkenstam C, Edberg A, Ayoubi S, et al. Are cancer patients at higher suicide risk than the general population?. Scand J Public Health. 2005;33:208-214. doi:10.1080/14034940410019226
- Kinslow CJ, Kumar P, Olfson M, et al. Prognosis and risk of suicide after cancer diagnosis. Cancer. 2024;130:588-596. doi:10.1002/cncr.35118
- Men VY, Emery CR, Yip PSF. Characteristics of cancer patients who died by suicide: a quantitative study of 15-year coronial records. Psychooncology. 2021;30:1051-1058. doi:10.1002/pon.5634
- Abdel-Rahman O. Socioeconomic predictors of suicide risk among cancer patients in the United States: a population- based study. Cancer Epidemiol. 2019;63:101601. doi:10.1016/j.canep.2019.101601
- O’Dwyer ST, Janssens A, Sansom A, et al. Suicidality in family caregivers of people with long-term illnesses and disabilities: a scoping review. Compr Psychiatry. 2021;110:152261. doi:10.1016/j.comppsych.2021.152261
- Senf B, Maiwurm P, Fettel J. Attitudes and opinions towards suicidality in professionals working with oncology patients: results from an online survey. Support Care Cancer. 2022;30:1775-1786. doi:10.1007/s00520-021-06590-2
- Berardelli I, Belvisi D, Nardella A, et al. Suicide in Parkinson’s disease: a systematic review. CNS Neurol Disord Drug Targets. 2019;18:466-477. doi:10.2174/1871527318666190703093345
- Kessler RC, Borges G, Walters EE. Prevalence of and risk factors for lifetime suicide attempts in the National Comorbidity Survey. Arch Gen Psychiatry. 1999;56:617-626. doi:10.1001/archpsyc.56.7.617
- Chen YY, Yu S, Hu YH, et al. Risk of suicide among patients with Parkinson disease. JAMA Psychiatry. 2021;78:293-301. doi:10.1001/jamapsychiatry.2020.4001
- Lee T, Lee HB, Ahn MH, et al. Increased suicide risk and clinical correlates of suicide among patients with Parkinson’s disease. Parkinsonism Relat Disord. 2016;32:102- 107. doi:10.1016/j.parkreldis.2016.09.006
- Georges JJ, Onwuteaka-Philipsen BD, Muller MT, et al. Relatives’ perspective on the terminally ill patients who died after euthanasia or physician-assisted suicide: a retrospective cross-sectional interview study in the Netherlands. Death Stud. 2007;31:1-15. doi:10.1080/07481180600985041
- Smith KA, Goy ER, Harvath TA, et al. Quality of death and dying in patients who request physician-assisted death. J Palliat Med. 2011;14:445-450. doi:10.1089/jpm.2010.0425
- Ganzini L, Goy ER, Dobscha SK, et al. Mental health outcomes of family members of Oregonians who request physician aid in dying. J Pain Symptom Manage. 2009;38:807-815. doi:10.1016/j.jpainsymman.2009.04.026
- Yi R. Most Americans favor legal euthanasia. Gallup. August 8, 2024. Accessed May 6, 2026. https://news.gallup .com/poll/648215/americans-favor-legal-euthanasia.aspx
- Dholakia SY, Bagheri A, Simpson A. Emotional impact on healthcare providers involved in medical assistance in dying (MAiD): a systematic review and qualitative meta-synthesis. BMJ Open. 2022;12:e058523. doi:10.1136/bmjopen-2021-058523
- Kozlov E, Luth EA, Nemeth S, et al. Knowl - edge of and preferences for medical aid in dying. JAMA Netw Open. 2025;8:e2461495. doi:10.1001/jamanetworkopen.2024.61495
- Geppert C; Veterans Administration National Center for Ethics in Health Care. Medical aid in dying in the VA. Presented at: VISN 1 Palliative Care Summit, September 2024.
- National Ethics Committee, Veterans Health Administration. The ethics of palliative sedation as a therapy of last resort. Am J Hosp Palliat Care. 2006;23:483-491. doi:10.1177/1049909106294883
Managing Requests for Medical Aid in Dying Within the Veterans Health Administration
Managing Requests for Medical Aid in Dying Within the Veterans Health Administration
Case Series of Patients With Cardiac Amyloidosis at VA New York Harbor Healthcare-Brooklyn
Case Series of Patients With Cardiac Amyloidosis at VA New York Harbor Healthcare-Brooklyn
Transthyretin amyloid cardiomyopathy (ATTR-CM) is caused by the misfolding of the TTR protein, which results in aggregation of amyloid fibrils that deposit in the myocardium and causes restrictive cardiomyopathy. Though it remains underdiagnosed, ATTR-CM is increasingly being recognized as a cause of heart failure in geriatric patients.1 There are 2 categories of ATTRCM: wild-type ATTR-CM (wtATTR-CM), in which there is no mutation in the TTR gene, and hereditary ATTR-CM (hATTR-CM), in which a mutation is present in the TTR gene. Research has shown that wtATTR-CM accounted for as many as 30% of cases of heart failure (HF) with preserved ejection fraction (HFpEF) in patients aged > 75 years.2 A significant percentage of the veteran patient population consists of older males. Given their age, these patients are at greater risk for ATTR diagnosis.3
Identifying red flags for patients within this population may allow clinicians to make earlier diagnoses and improve outcomes. A high index of suspicion is needed to diagnose ATTR because many early signs and symptoms are extracardiac, which leads to delayed diagnoses and worse outcomes. This article describes 8 cases of ATTR-CM within the US Department of Veterans Affairs (VA) New York Harbor Healthcare System-Brooklyn (VANYHHSB).
Methods
This retrospective case series was reviewed and approved by the VANYHHSB Institutional Review Board where it was conducted. Patients diagnosed with ATTR between 2017 and 2024 were identified using International Classification of Diseases, Tenth Revision codes. Eleven patients were identified; 3 were excluded due to insufficient medical records. The remaining 8 patient records were retrospectively reviewed and included.
Case 1
A 67-year-old male with a history of carpal tunnel syndrome (CTS) presented following a syncopal episode. Initial electrocardiogram (ECG) showed sinus rhythm, first-degree atrioventricular block, and a bifascicular block. Transthoracic echocardiogram (TTE) showed moderate asymmetric left ventricular (LV) hypertrophy (LVH) and biatrial enlargement with an ejection fraction (EF) > 55%. The patient was discharged with a loop recorder and an outpatient follow-up appointment scheduled. One month later, he presented with worsening dyspnea on exertion with clinical signs of hypervolemia. A repeat TTE showed global LV wall thickening, moderately reduced LV systolic function (EF 40%), and moderate pulmonary hypertension. Given these findings, the patient underwent cardiac magnetic resonance imaging (CMR), which suggested an infiltrative cardiomyopathy. Amyloid light-chain (AL) amyloidosis evaluation, technetium-99m (99mTC) pyrophosphate imaging, and a fat pad biopsy were unrevealing. An endomyocardial biopsy was performed with electron microscopy, which confirmed amyloidosis. Genetic testing was negative, and the patient began taking tafamidis. There were no later admissions for decompensated HF; however, the patient developed atrial fibrillation (AF) and an interval TTE demonstrated no improvement in his EF. He died at age 73 years.
Case 2
A 78-year-old male with a history of CTS presented with lightheadedness. Initial ECG showed rate-controlled AF and TTE revealed a moderately thickened LV wall with normal LV size, mild left atrial enlargement, and an EF of 65%. The patient was discharged with a scheduled outpatient CMR appointment; however, he defaulted from follow-up. Two years later, he presented with recurrent syncopal episodes and physical examination was consistent with hypervolemia. Repeat TTE revealed moderate LVH, biatrial enlargement, and an EF of 55%. An inpatient CMR was suggestive of cardiac amyloidosis, and a pyrophosphate scan was diagnostic for ATTR. The patient started taking tafamidis, but continued to have recurrent admissions for HF exacerbation. He died at age 81 years.
Case 3
A 71-year-old male with a history of CTS presented with exertional dyspnea. The initial ECG showed sinus rhythm with left atrial enlargement and left axis deviation. Subsequent TTE revealed severe LVH, mildly reduced LV cavity size, moderate to severe biatrial enlargement, and an EF of 25% to 30%. Outpatient 99mTC pyrophosphate imaging suggested cardiac amyloidosis, and laboratory testing showed no evidence of monoclonal proteins. The patient was started on tafamidis for ATTR. At 2-year follow-up, he had new AF and to date has had no further hospitalizations for acute decompensated HF.
Case 4
A 92-year-old male with a history of AF and bilateral CTS presented with lightheadedness. An ECG revealed AF with a slowed ventricular response. Subsequent Holter monitoring demonstrated pauses exceeding 3 seconds, and a permanent pacemaker was recommended. During his preoperative evaluation, TTE revealed severe concentric LVH with a speckled appearance of the myocardium, mild-tomoderate biatrial enlargement, and an EF of 50% to 55%. 99mTC pyrophosphate imaging was positive for amyloidosis and the patient started taking tafamidis. Recurrent hospital admissions for decompensated HF complicated his progression. The patient died at age 95 years.
Case 5
A 72-year-old male with a history of bilateral CTS and cervical spinal stenosis presented with dyspnea on exertion. An ECG revealed a normal sinus rhythm. A TTE found severely reduced systolic function with an EF ≤ 25%, mild concentric LVH, grade 3 diastolic dysfunction, mild-tomoderate biatrial enlargement, and moderate pulmonary hypertension (Figure 1). He was started on guideline-directed medical therapy (GDMT) for HF, which included sacubitril/valsartan, metoprolol succinate, and empagliflozin. The patient’s dyspnea improved, and a workup for nonischemic cardiomyopathy was initiated. 99mTC pyrophosphate imaging 1 year after his initial presentation was positive, leading to ATTR-CM diagnosis. The patient started taking tafamidis and he has since had a stable progression and continued to demonstrate good exercise tolerance with no hospitalizations. His most recent TTE indicated an EF of 40% to 45%.
(parasternal long axis view) of patient 5
demonstrating concentric left ventricular
hypertrophy with a speckled myocardium
and dilated left atrium.
Case 6
A 76-year-old male with a history of paroxysmal AF status after multiple ablations, bilateral CTS, and severe cervical spinal stenosis presented with dyspnea on exertion. The patient’s ECG showed normal sinus rhythm and left axis deviation with concern for left anterior hemiblock. A TTE revealed moderate LVH with a speckled appearance of the myocardium and grade 3 diastolic dysfunction with preserved EF. Before completing the workup for underlying cardiomyopathy, the patient underwent an interventional radiology procedure for an angiomyolipoma, and his postoperative course was complicated by pulmonary edema, requiring admission to the coronary care unit for diuresis. A repeat TTE revealed a reduced EF of 35%, and he was discharged on GDMT. No monoclonal protein was seen in the serum or urine. The patient’s progression was complicated by recurrent admissions for acute decompensated HF and supraventricular tachycardia despite being on amiodarone, which led to a delay in obtaining 99mTC pyrophosphate imaging. Due to hypotension, the patient was unable to tolerate GDMT. He eventually underwent 99mTC pyrophosphate imaging that confirmed ATTR-CM and was started on tafamidis. One year following initial presentation, the patient’s EF progressively declined to 20% to 25%, and he died shortly after discharge to subacute rehabilitation.
Case 7
A 95-year-old male with a history of longstanding persistent AF and bilateral CTS presented with dyspnea on exertion, bendopnea, and worsening bilateral pedal edema for a week. An ECG showed AF with a controlled ventricular response and low-voltage QRS waves (Figure 2). A TTE showed biatrial enlargement, LVH, and preserved EF > 55%. He started taking furosemide and was discharged with a diagnosis of HFpEF. The patient missed cardiology follow-up and presented 1 year later with decompensated HF. An amyloidosis workup was recommended, but due to intermittent periods of being lost to follow-up, the patient did not pursue this workup until 3 years after his initial presentation when 99mTC pyrophosphate imaging confirmed ATTR-CM. The patient declined tafamidis and continued to be followed by the cardiology team. His HF is managed with furosemide as needed due to intolerance to GDMT.
demonstrating atrial fibrillation with controlled
ventricular response and low-voltage QRS.
Case 8
A 74-year-old male with history of bilateral CTS presented with right-sided chest pain associated with shortness of breath, diaphoresis, dizziness, and worsening abdominal pain. He was found to have inferior wall myocardial infarction on ECG with later percutaneous coronary intervention to the left circumflex. His hospital course was complicated by decompensated HF (EF 45% to 50%) and AF with rapid ventricular response. He was treated and discharged with follow-up visits scheduled in the cardiology clinic. Multiple attempts were made to place him on GDMT; however, the patient was unable to tolerate these medications due to recurrent admissions for syncope. During a cardiology clinic visit 3 years after his initial presentation, an amyloidosis workup was initiated. 99mTC pyrophosphate imaging was positive for ATTR-CM, and he started taking tafamidis. Before this diagnosis, ECG indicated low-voltage QRS complexes. His progression has since been complicated by admissions for decompensated HF, recurrent episodes of AF requiring atrioventricular node ablation, and biventricular implantable cardioverter-defibrillator implantation after failed attempts at electrical cardioversion. He continued to follow up in the HF clinic.
Discussion
ATTR-CM is an underdiagnosed cause of cardiomyopathy, particularly in older adults. TTR is a transport protein produced in the liver, and misfolding can occur due to age-related instability of the wild-type protein (wtATTR) or pathologic variants in the TTR (hATTR). The misfolding leads to restrictive physiology, HF (often with preserved EF) arrhythmias, and conduction system disease.1 It is likely that the predominantly older male veteran population would be predisposed to wtATTR cardiomyopathy given that misfolding in the condition is believed to be age-related.
Current criteria for ATTR diagnosis require a combination of clinical suspicion, imaging, laboratory testing, and, in some cases, tissue biopsy confirmation and genetic testing.1,4,5 The diagnostic algorithm is as follows:
Clinical suspicion. Consider ATTR in patients with unexplained HFpEF, LV wall thickness ≥ 12 to 14 mm, discordance between ECG voltage and wall thickness, or associated extracardiac manifestations such as CTS, lumbar spinal stenosis, or peripheral/ autonomic neuropathy.
Exclusion of AL amyloidosis. Bone scintigraphy alone cannot distinguish between AL amyloidosis and ATTR, so patients with suspected amyloid cardiomyopathy should undergo serum and urine immunofixation electrophoresis and serum free light chain assay to rule out a monoclonal gammopathy as seen in AL amyloidosis.
Cardiac scintigraphy. Once AL amyloidosis is excluded, a positive 99mTC-labeled boneavid tracer image (such as a pyrophosphate scan) with grade 2 or grade 3 myocardial uptake is diagnostic of ATTR.
Tissue biopsy. If there is monoclonal gammopathy or equivocal imaging, tissue biopsy (eg, endomyocardial) with Congo red staining and amyloid typing by mass spectrometry or immunohistochemistry is necessary.
Genetic testing. Once ATTR is confirmed, genetic testing distinguishes hATTR from wtATTR, which impacts management and determines the need to screen family members.
Currently, there are 3 therapies approved by the US Food and Drug Administration (FDA) to treat ATTR-CM: tafamidis, acoramidis, and vutrisiran.6-8 Tafamidis and acoramidis stabilize the TTR tetramer, preventing amyloid formation. Vutrisiran uses RNA interference to silence the gene that produces TTR. Tafamidis has been found to improve cardiovascular outcomes in ATTR-CM. In the ATTR-ACT trial, it reduced all-cause mortality and cardiovascular hospitalizations in patients with ATTR-CM and New York Heart Association class I-III symptoms.6 There are other disease-modifying therapies, such the TTR gene silencers inotersen and patisiran; however, these are only FDA-approved for hATTR polyneuropathy and not for ATTRCM; ongoing trials are evaluating their cardiac efficacy.
The mean age of ATTR-CM diagnosis in the patients described in this case series was 79 years, which is older than the mean age of 74 years reported in prior research.4 All patients were male, and the most common presenting symptom was dyspnea on exertion. Table 1 outlines baseline characteristics and associated comorbidities of patients in this case series. The patients presented with many red-flag signs of ATTR-CM (Figure 3). Among them included syncope (4 of 8 patients), spinal stenosis (2 of 8 patients), arrhythmia (7 of 8 patients), heart failure (7 of 8 patients), and bilateral CTS (all patients).
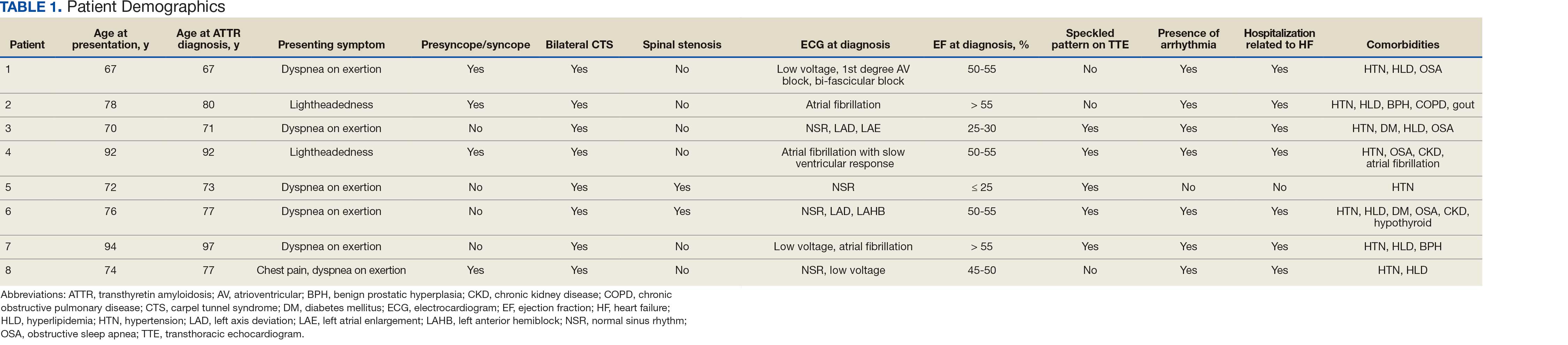
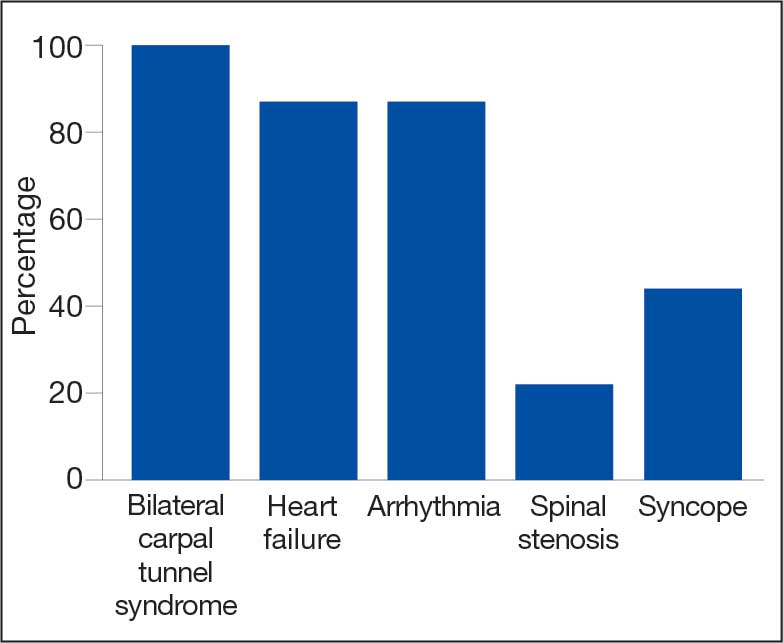
amyloidosis red-flag signs identified.
Bilateral CTS was diagnosed in all patients before their diagnosis of ATTR-CM. Patient 7 had a diagnosis of CTS 9 years before his ATTR-CM diagnosis, underscoring a subtle, yet important extracardiac sign that may increase clinical suspicion for ATTR-CM. Previous research found that the probability of having CTS is highest 5 to 9 years prior to the development of cardiomyopathy. Its presence is also a prognostic marker in ATTR, independent of cardiac involvement.9 The median interval between CTS diagnosis and cardiomyopathy diagnosis was 5 years in this series.
Although screening criteria for ATTR have been proposed, none have been incorporated into formal guidelines.10,11 We propose that a baseline ECG and screening TTE be obtained in any patient aged > 65 years with cardiac risk equivalents such as hypertension and diabetes, presenting with ≥ 1 extracardiac red-flag signs, such as bilateral CTS and spinal stenosis. This will likely facilitate an earlier diagnosis of ATTR-CM, leading to earlier treatment initiation and better patient outcomes. This initiative can be started in the primary care setting and facilitates early cardiology referral. This recommendation is based on literature supporting clinical patterns and observations the authors have made in clinical practice.
Obstructive sleep apnea (OSA) was a comorbidity present in 50% of the patients described in this case series. The literature describing this association is sparse; however, a prospective observational study reported that disorders of sleep inclusive of OSA are frequent in patients with cardiac amyloidosis.12 A theory behind this association is that amyloid deposits in the upper airway tissues lead to airway narrowing. 13 More research is needed to further assess the relationship between OSA and cardiac amyloidosis, particularly with respect to the timing of OSA prior to the development of cardiomyopathy as it may be a potential early sign for clinicians to acknowledge.
An important observation from this case series is the variability in the timing of tafamidis initiation relative to symptom onset and confirmed diagnosis of ATTR-CM (Table 2). Although tafamidis has been found to slow disease progression, several patients in this series began treatment at advanced stages or years after the onset of cardiac symptoms, potentially limiting its clinical benefit.
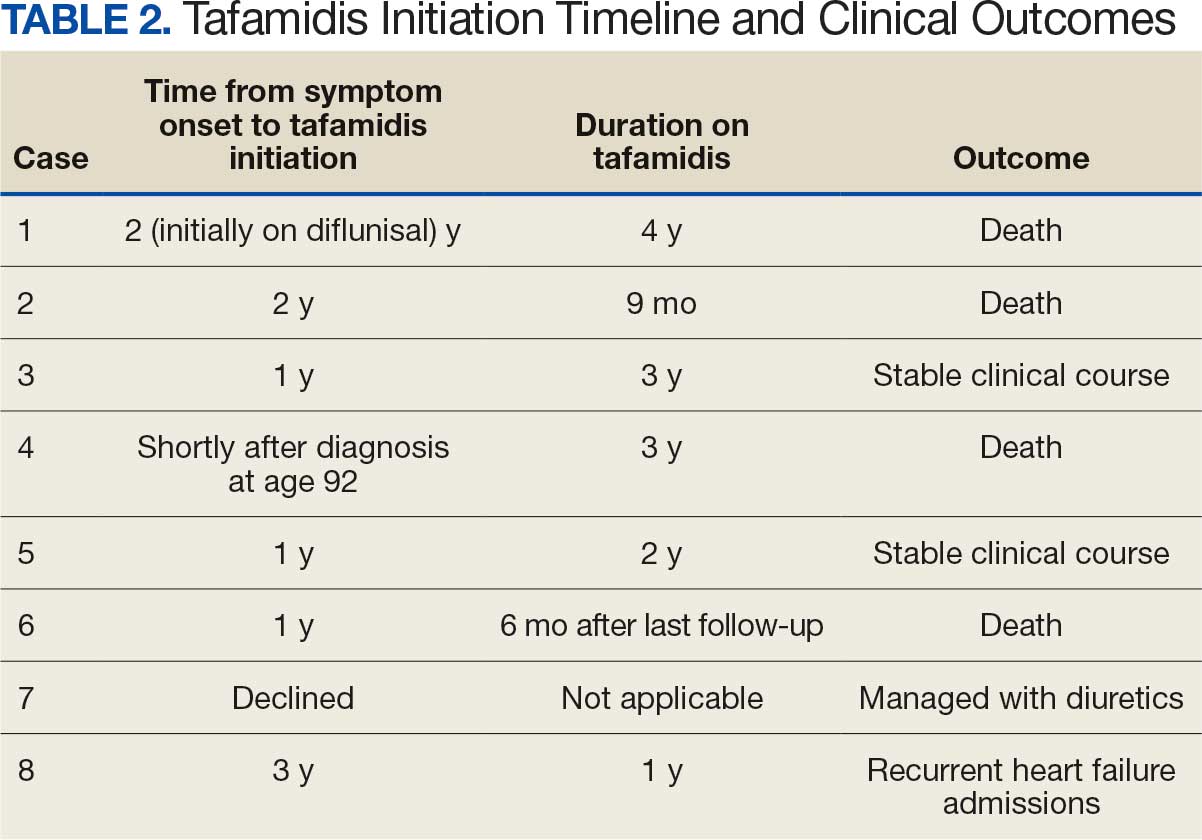
For example, patient 1 was diagnosed with ATTR 2 years before tafamidis became available on the market and was initially treated with diflunisal. Patients who started tafamidis earlier in the disease course (eg, patients 3 and 5) appeared to have better long-term outcomes, including an absence of heart failure hospitalizations after initiation. In contrast, patients with delayed treatment initiation (eg, patients 2, 6, and 8) experienced ongoing decompensations or early mortality. Patient 7 declined tafamidis, underscoring challenges in medication uptake among older adults. Randomized controlled trials are warranted to compare the effectiveness of tafamidis with other recently FDA-approved therapies, such as acoramidis and vutrisiran, particularly in terms of cardiovascular outcomes.
AF was the most common arrhythmia observed in these patients and may occur years before the development of HF symptoms in the setting of ATTR-CM. Due to inherent conduction system disease, AF in this population may have a controlled or slow ventricular response, as observed in patient 4. Patients with atrial fibrillation and cardiac amyloidosis should receive anticoagulation regardless of CHA2DS2- VASc score due to their high risk of intracardiac thrombus formation.4
Limitations
This case series lacked genetic testing following a confirmed ATTR-CM diagnosis. Although much of the treatment is the same regardless of the presence of a TTR mutation, knowing the specific subtype of ATTR-CM has implications for prognosis and for screening family members.
Conclusions
Following analysis of 8 patients diagnosed with ATTR, this case series could serve as a blueprint for research into ATTR in veterans. In clinical practice, following military service, veterans may not be routinely seen by an outpatient physician and may present with sequelae of advanced stages of ATTR. Early identification of red-flag symptoms can lead to a higher clinical suspicion, prompting early diagnostic evaluation and treatment initiation and ultimately mitigating adverse outcomes. Future research that includes genetic testing for those with confirmed ATTR-CM may prove useful as a foundation for detailed and informed discussions with patients and their families regarding prognosis and, if indicated, screening for family members.
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142:e7-e22. doi:10.1161/CIR.0000000000000792
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765-774. doi:10.1111/j.1532-5415.2011.03868.x
- Nativi-Nicolau J, Redd A, Kelly N, et al. Increasing number of amyloidosis diagnosis in the Veterans Affairs populations. J Card Fail. 2019;25:S96.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872-2891. doi:10.1016/j.jacc.2019.04.003
- Gillmore JD, Maurer, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404-2412. doi:10.1161/CIRCULATIONAHA.116.021612
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007-1016. doi:10.1056/NEJMoa1805689
- Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med. 2024;390:132-142. doi:10.1056/NEJMoa2305434
- Fontana M, Berk JL, Gillmore JD, et al. Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J Med. 2025;392:33-44. doi:10.1056/NEJMoa2409134
- Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22:507-515. doi:10.1002/ejhf.1742
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554-1568. doi:10.1093/eurheartj/ehab072
- Brito D, Albrecht FC, de Arenaza DP, et al. World Heart Federation consensus on transthyretin amyloidosis cardiomyopathy (ATTR-CM). Glob Heart. 2023;18:59. doi:10.5334/gh.1262
- Bodez D, Guellich A, Kharoubi M, et al. Prevalence, severity, and prognostic value of sleep apnea syndromes in cardiac amyloidosis. Sleep. 2016;39:1333-1341. doi:10.5665/sleep.5958
- Colaco B, Colaco C, Lipford M. A forgotten problem: sleep-disordered breathing in amyloidosis. Chest. 2016;150:1293A. doi:10.1016/j.chest.2016.08.1407
Transthyretin amyloid cardiomyopathy (ATTR-CM) is caused by the misfolding of the TTR protein, which results in aggregation of amyloid fibrils that deposit in the myocardium and causes restrictive cardiomyopathy. Though it remains underdiagnosed, ATTR-CM is increasingly being recognized as a cause of heart failure in geriatric patients.1 There are 2 categories of ATTRCM: wild-type ATTR-CM (wtATTR-CM), in which there is no mutation in the TTR gene, and hereditary ATTR-CM (hATTR-CM), in which a mutation is present in the TTR gene. Research has shown that wtATTR-CM accounted for as many as 30% of cases of heart failure (HF) with preserved ejection fraction (HFpEF) in patients aged > 75 years.2 A significant percentage of the veteran patient population consists of older males. Given their age, these patients are at greater risk for ATTR diagnosis.3
Identifying red flags for patients within this population may allow clinicians to make earlier diagnoses and improve outcomes. A high index of suspicion is needed to diagnose ATTR because many early signs and symptoms are extracardiac, which leads to delayed diagnoses and worse outcomes. This article describes 8 cases of ATTR-CM within the US Department of Veterans Affairs (VA) New York Harbor Healthcare System-Brooklyn (VANYHHSB).
Methods
This retrospective case series was reviewed and approved by the VANYHHSB Institutional Review Board where it was conducted. Patients diagnosed with ATTR between 2017 and 2024 were identified using International Classification of Diseases, Tenth Revision codes. Eleven patients were identified; 3 were excluded due to insufficient medical records. The remaining 8 patient records were retrospectively reviewed and included.
Case 1
A 67-year-old male with a history of carpal tunnel syndrome (CTS) presented following a syncopal episode. Initial electrocardiogram (ECG) showed sinus rhythm, first-degree atrioventricular block, and a bifascicular block. Transthoracic echocardiogram (TTE) showed moderate asymmetric left ventricular (LV) hypertrophy (LVH) and biatrial enlargement with an ejection fraction (EF) > 55%. The patient was discharged with a loop recorder and an outpatient follow-up appointment scheduled. One month later, he presented with worsening dyspnea on exertion with clinical signs of hypervolemia. A repeat TTE showed global LV wall thickening, moderately reduced LV systolic function (EF 40%), and moderate pulmonary hypertension. Given these findings, the patient underwent cardiac magnetic resonance imaging (CMR), which suggested an infiltrative cardiomyopathy. Amyloid light-chain (AL) amyloidosis evaluation, technetium-99m (99mTC) pyrophosphate imaging, and a fat pad biopsy were unrevealing. An endomyocardial biopsy was performed with electron microscopy, which confirmed amyloidosis. Genetic testing was negative, and the patient began taking tafamidis. There were no later admissions for decompensated HF; however, the patient developed atrial fibrillation (AF) and an interval TTE demonstrated no improvement in his EF. He died at age 73 years.
Case 2
A 78-year-old male with a history of CTS presented with lightheadedness. Initial ECG showed rate-controlled AF and TTE revealed a moderately thickened LV wall with normal LV size, mild left atrial enlargement, and an EF of 65%. The patient was discharged with a scheduled outpatient CMR appointment; however, he defaulted from follow-up. Two years later, he presented with recurrent syncopal episodes and physical examination was consistent with hypervolemia. Repeat TTE revealed moderate LVH, biatrial enlargement, and an EF of 55%. An inpatient CMR was suggestive of cardiac amyloidosis, and a pyrophosphate scan was diagnostic for ATTR. The patient started taking tafamidis, but continued to have recurrent admissions for HF exacerbation. He died at age 81 years.
Case 3
A 71-year-old male with a history of CTS presented with exertional dyspnea. The initial ECG showed sinus rhythm with left atrial enlargement and left axis deviation. Subsequent TTE revealed severe LVH, mildly reduced LV cavity size, moderate to severe biatrial enlargement, and an EF of 25% to 30%. Outpatient 99mTC pyrophosphate imaging suggested cardiac amyloidosis, and laboratory testing showed no evidence of monoclonal proteins. The patient was started on tafamidis for ATTR. At 2-year follow-up, he had new AF and to date has had no further hospitalizations for acute decompensated HF.
Case 4
A 92-year-old male with a history of AF and bilateral CTS presented with lightheadedness. An ECG revealed AF with a slowed ventricular response. Subsequent Holter monitoring demonstrated pauses exceeding 3 seconds, and a permanent pacemaker was recommended. During his preoperative evaluation, TTE revealed severe concentric LVH with a speckled appearance of the myocardium, mild-tomoderate biatrial enlargement, and an EF of 50% to 55%. 99mTC pyrophosphate imaging was positive for amyloidosis and the patient started taking tafamidis. Recurrent hospital admissions for decompensated HF complicated his progression. The patient died at age 95 years.
Case 5
A 72-year-old male with a history of bilateral CTS and cervical spinal stenosis presented with dyspnea on exertion. An ECG revealed a normal sinus rhythm. A TTE found severely reduced systolic function with an EF ≤ 25%, mild concentric LVH, grade 3 diastolic dysfunction, mild-tomoderate biatrial enlargement, and moderate pulmonary hypertension (Figure 1). He was started on guideline-directed medical therapy (GDMT) for HF, which included sacubitril/valsartan, metoprolol succinate, and empagliflozin. The patient’s dyspnea improved, and a workup for nonischemic cardiomyopathy was initiated. 99mTC pyrophosphate imaging 1 year after his initial presentation was positive, leading to ATTR-CM diagnosis. The patient started taking tafamidis and he has since had a stable progression and continued to demonstrate good exercise tolerance with no hospitalizations. His most recent TTE indicated an EF of 40% to 45%.
(parasternal long axis view) of patient 5
demonstrating concentric left ventricular
hypertrophy with a speckled myocardium
and dilated left atrium.
Case 6
A 76-year-old male with a history of paroxysmal AF status after multiple ablations, bilateral CTS, and severe cervical spinal stenosis presented with dyspnea on exertion. The patient’s ECG showed normal sinus rhythm and left axis deviation with concern for left anterior hemiblock. A TTE revealed moderate LVH with a speckled appearance of the myocardium and grade 3 diastolic dysfunction with preserved EF. Before completing the workup for underlying cardiomyopathy, the patient underwent an interventional radiology procedure for an angiomyolipoma, and his postoperative course was complicated by pulmonary edema, requiring admission to the coronary care unit for diuresis. A repeat TTE revealed a reduced EF of 35%, and he was discharged on GDMT. No monoclonal protein was seen in the serum or urine. The patient’s progression was complicated by recurrent admissions for acute decompensated HF and supraventricular tachycardia despite being on amiodarone, which led to a delay in obtaining 99mTC pyrophosphate imaging. Due to hypotension, the patient was unable to tolerate GDMT. He eventually underwent 99mTC pyrophosphate imaging that confirmed ATTR-CM and was started on tafamidis. One year following initial presentation, the patient’s EF progressively declined to 20% to 25%, and he died shortly after discharge to subacute rehabilitation.
Case 7
A 95-year-old male with a history of longstanding persistent AF and bilateral CTS presented with dyspnea on exertion, bendopnea, and worsening bilateral pedal edema for a week. An ECG showed AF with a controlled ventricular response and low-voltage QRS waves (Figure 2). A TTE showed biatrial enlargement, LVH, and preserved EF > 55%. He started taking furosemide and was discharged with a diagnosis of HFpEF. The patient missed cardiology follow-up and presented 1 year later with decompensated HF. An amyloidosis workup was recommended, but due to intermittent periods of being lost to follow-up, the patient did not pursue this workup until 3 years after his initial presentation when 99mTC pyrophosphate imaging confirmed ATTR-CM. The patient declined tafamidis and continued to be followed by the cardiology team. His HF is managed with furosemide as needed due to intolerance to GDMT.
demonstrating atrial fibrillation with controlled
ventricular response and low-voltage QRS.
Case 8
A 74-year-old male with history of bilateral CTS presented with right-sided chest pain associated with shortness of breath, diaphoresis, dizziness, and worsening abdominal pain. He was found to have inferior wall myocardial infarction on ECG with later percutaneous coronary intervention to the left circumflex. His hospital course was complicated by decompensated HF (EF 45% to 50%) and AF with rapid ventricular response. He was treated and discharged with follow-up visits scheduled in the cardiology clinic. Multiple attempts were made to place him on GDMT; however, the patient was unable to tolerate these medications due to recurrent admissions for syncope. During a cardiology clinic visit 3 years after his initial presentation, an amyloidosis workup was initiated. 99mTC pyrophosphate imaging was positive for ATTR-CM, and he started taking tafamidis. Before this diagnosis, ECG indicated low-voltage QRS complexes. His progression has since been complicated by admissions for decompensated HF, recurrent episodes of AF requiring atrioventricular node ablation, and biventricular implantable cardioverter-defibrillator implantation after failed attempts at electrical cardioversion. He continued to follow up in the HF clinic.
Discussion
ATTR-CM is an underdiagnosed cause of cardiomyopathy, particularly in older adults. TTR is a transport protein produced in the liver, and misfolding can occur due to age-related instability of the wild-type protein (wtATTR) or pathologic variants in the TTR (hATTR). The misfolding leads to restrictive physiology, HF (often with preserved EF) arrhythmias, and conduction system disease.1 It is likely that the predominantly older male veteran population would be predisposed to wtATTR cardiomyopathy given that misfolding in the condition is believed to be age-related.
Current criteria for ATTR diagnosis require a combination of clinical suspicion, imaging, laboratory testing, and, in some cases, tissue biopsy confirmation and genetic testing.1,4,5 The diagnostic algorithm is as follows:
Clinical suspicion. Consider ATTR in patients with unexplained HFpEF, LV wall thickness ≥ 12 to 14 mm, discordance between ECG voltage and wall thickness, or associated extracardiac manifestations such as CTS, lumbar spinal stenosis, or peripheral/ autonomic neuropathy.
Exclusion of AL amyloidosis. Bone scintigraphy alone cannot distinguish between AL amyloidosis and ATTR, so patients with suspected amyloid cardiomyopathy should undergo serum and urine immunofixation electrophoresis and serum free light chain assay to rule out a monoclonal gammopathy as seen in AL amyloidosis.
Cardiac scintigraphy. Once AL amyloidosis is excluded, a positive 99mTC-labeled boneavid tracer image (such as a pyrophosphate scan) with grade 2 or grade 3 myocardial uptake is diagnostic of ATTR.
Tissue biopsy. If there is monoclonal gammopathy or equivocal imaging, tissue biopsy (eg, endomyocardial) with Congo red staining and amyloid typing by mass spectrometry or immunohistochemistry is necessary.
Genetic testing. Once ATTR is confirmed, genetic testing distinguishes hATTR from wtATTR, which impacts management and determines the need to screen family members.
Currently, there are 3 therapies approved by the US Food and Drug Administration (FDA) to treat ATTR-CM: tafamidis, acoramidis, and vutrisiran.6-8 Tafamidis and acoramidis stabilize the TTR tetramer, preventing amyloid formation. Vutrisiran uses RNA interference to silence the gene that produces TTR. Tafamidis has been found to improve cardiovascular outcomes in ATTR-CM. In the ATTR-ACT trial, it reduced all-cause mortality and cardiovascular hospitalizations in patients with ATTR-CM and New York Heart Association class I-III symptoms.6 There are other disease-modifying therapies, such the TTR gene silencers inotersen and patisiran; however, these are only FDA-approved for hATTR polyneuropathy and not for ATTRCM; ongoing trials are evaluating their cardiac efficacy.
The mean age of ATTR-CM diagnosis in the patients described in this case series was 79 years, which is older than the mean age of 74 years reported in prior research.4 All patients were male, and the most common presenting symptom was dyspnea on exertion. Table 1 outlines baseline characteristics and associated comorbidities of patients in this case series. The patients presented with many red-flag signs of ATTR-CM (Figure 3). Among them included syncope (4 of 8 patients), spinal stenosis (2 of 8 patients), arrhythmia (7 of 8 patients), heart failure (7 of 8 patients), and bilateral CTS (all patients).
amyloidosis red-flag signs identified.
Bilateral CTS was diagnosed in all patients before their diagnosis of ATTR-CM. Patient 7 had a diagnosis of CTS 9 years before his ATTR-CM diagnosis, underscoring a subtle, yet important extracardiac sign that may increase clinical suspicion for ATTR-CM. Previous research found that the probability of having CTS is highest 5 to 9 years prior to the development of cardiomyopathy. Its presence is also a prognostic marker in ATTR, independent of cardiac involvement.9 The median interval between CTS diagnosis and cardiomyopathy diagnosis was 5 years in this series.
Although screening criteria for ATTR have been proposed, none have been incorporated into formal guidelines.10,11 We propose that a baseline ECG and screening TTE be obtained in any patient aged > 65 years with cardiac risk equivalents such as hypertension and diabetes, presenting with ≥ 1 extracardiac red-flag signs, such as bilateral CTS and spinal stenosis. This will likely facilitate an earlier diagnosis of ATTR-CM, leading to earlier treatment initiation and better patient outcomes. This initiative can be started in the primary care setting and facilitates early cardiology referral. This recommendation is based on literature supporting clinical patterns and observations the authors have made in clinical practice.
Obstructive sleep apnea (OSA) was a comorbidity present in 50% of the patients described in this case series. The literature describing this association is sparse; however, a prospective observational study reported that disorders of sleep inclusive of OSA are frequent in patients with cardiac amyloidosis.12 A theory behind this association is that amyloid deposits in the upper airway tissues lead to airway narrowing. 13 More research is needed to further assess the relationship between OSA and cardiac amyloidosis, particularly with respect to the timing of OSA prior to the development of cardiomyopathy as it may be a potential early sign for clinicians to acknowledge.
An important observation from this case series is the variability in the timing of tafamidis initiation relative to symptom onset and confirmed diagnosis of ATTR-CM (Table 2). Although tafamidis has been found to slow disease progression, several patients in this series began treatment at advanced stages or years after the onset of cardiac symptoms, potentially limiting its clinical benefit.
For example, patient 1 was diagnosed with ATTR 2 years before tafamidis became available on the market and was initially treated with diflunisal. Patients who started tafamidis earlier in the disease course (eg, patients 3 and 5) appeared to have better long-term outcomes, including an absence of heart failure hospitalizations after initiation. In contrast, patients with delayed treatment initiation (eg, patients 2, 6, and 8) experienced ongoing decompensations or early mortality. Patient 7 declined tafamidis, underscoring challenges in medication uptake among older adults. Randomized controlled trials are warranted to compare the effectiveness of tafamidis with other recently FDA-approved therapies, such as acoramidis and vutrisiran, particularly in terms of cardiovascular outcomes.
AF was the most common arrhythmia observed in these patients and may occur years before the development of HF symptoms in the setting of ATTR-CM. Due to inherent conduction system disease, AF in this population may have a controlled or slow ventricular response, as observed in patient 4. Patients with atrial fibrillation and cardiac amyloidosis should receive anticoagulation regardless of CHA2DS2- VASc score due to their high risk of intracardiac thrombus formation.4
Limitations
This case series lacked genetic testing following a confirmed ATTR-CM diagnosis. Although much of the treatment is the same regardless of the presence of a TTR mutation, knowing the specific subtype of ATTR-CM has implications for prognosis and for screening family members.
Conclusions
Following analysis of 8 patients diagnosed with ATTR, this case series could serve as a blueprint for research into ATTR in veterans. In clinical practice, following military service, veterans may not be routinely seen by an outpatient physician and may present with sequelae of advanced stages of ATTR. Early identification of red-flag symptoms can lead to a higher clinical suspicion, prompting early diagnostic evaluation and treatment initiation and ultimately mitigating adverse outcomes. Future research that includes genetic testing for those with confirmed ATTR-CM may prove useful as a foundation for detailed and informed discussions with patients and their families regarding prognosis and, if indicated, screening for family members.
Transthyretin amyloid cardiomyopathy (ATTR-CM) is caused by the misfolding of the TTR protein, which results in aggregation of amyloid fibrils that deposit in the myocardium and causes restrictive cardiomyopathy. Though it remains underdiagnosed, ATTR-CM is increasingly being recognized as a cause of heart failure in geriatric patients.1 There are 2 categories of ATTRCM: wild-type ATTR-CM (wtATTR-CM), in which there is no mutation in the TTR gene, and hereditary ATTR-CM (hATTR-CM), in which a mutation is present in the TTR gene. Research has shown that wtATTR-CM accounted for as many as 30% of cases of heart failure (HF) with preserved ejection fraction (HFpEF) in patients aged > 75 years.2 A significant percentage of the veteran patient population consists of older males. Given their age, these patients are at greater risk for ATTR diagnosis.3
Identifying red flags for patients within this population may allow clinicians to make earlier diagnoses and improve outcomes. A high index of suspicion is needed to diagnose ATTR because many early signs and symptoms are extracardiac, which leads to delayed diagnoses and worse outcomes. This article describes 8 cases of ATTR-CM within the US Department of Veterans Affairs (VA) New York Harbor Healthcare System-Brooklyn (VANYHHSB).
Methods
This retrospective case series was reviewed and approved by the VANYHHSB Institutional Review Board where it was conducted. Patients diagnosed with ATTR between 2017 and 2024 were identified using International Classification of Diseases, Tenth Revision codes. Eleven patients were identified; 3 were excluded due to insufficient medical records. The remaining 8 patient records were retrospectively reviewed and included.
Case 1
A 67-year-old male with a history of carpal tunnel syndrome (CTS) presented following a syncopal episode. Initial electrocardiogram (ECG) showed sinus rhythm, first-degree atrioventricular block, and a bifascicular block. Transthoracic echocardiogram (TTE) showed moderate asymmetric left ventricular (LV) hypertrophy (LVH) and biatrial enlargement with an ejection fraction (EF) > 55%. The patient was discharged with a loop recorder and an outpatient follow-up appointment scheduled. One month later, he presented with worsening dyspnea on exertion with clinical signs of hypervolemia. A repeat TTE showed global LV wall thickening, moderately reduced LV systolic function (EF 40%), and moderate pulmonary hypertension. Given these findings, the patient underwent cardiac magnetic resonance imaging (CMR), which suggested an infiltrative cardiomyopathy. Amyloid light-chain (AL) amyloidosis evaluation, technetium-99m (99mTC) pyrophosphate imaging, and a fat pad biopsy were unrevealing. An endomyocardial biopsy was performed with electron microscopy, which confirmed amyloidosis. Genetic testing was negative, and the patient began taking tafamidis. There were no later admissions for decompensated HF; however, the patient developed atrial fibrillation (AF) and an interval TTE demonstrated no improvement in his EF. He died at age 73 years.
Case 2
A 78-year-old male with a history of CTS presented with lightheadedness. Initial ECG showed rate-controlled AF and TTE revealed a moderately thickened LV wall with normal LV size, mild left atrial enlargement, and an EF of 65%. The patient was discharged with a scheduled outpatient CMR appointment; however, he defaulted from follow-up. Two years later, he presented with recurrent syncopal episodes and physical examination was consistent with hypervolemia. Repeat TTE revealed moderate LVH, biatrial enlargement, and an EF of 55%. An inpatient CMR was suggestive of cardiac amyloidosis, and a pyrophosphate scan was diagnostic for ATTR. The patient started taking tafamidis, but continued to have recurrent admissions for HF exacerbation. He died at age 81 years.
Case 3
A 71-year-old male with a history of CTS presented with exertional dyspnea. The initial ECG showed sinus rhythm with left atrial enlargement and left axis deviation. Subsequent TTE revealed severe LVH, mildly reduced LV cavity size, moderate to severe biatrial enlargement, and an EF of 25% to 30%. Outpatient 99mTC pyrophosphate imaging suggested cardiac amyloidosis, and laboratory testing showed no evidence of monoclonal proteins. The patient was started on tafamidis for ATTR. At 2-year follow-up, he had new AF and to date has had no further hospitalizations for acute decompensated HF.
Case 4
A 92-year-old male with a history of AF and bilateral CTS presented with lightheadedness. An ECG revealed AF with a slowed ventricular response. Subsequent Holter monitoring demonstrated pauses exceeding 3 seconds, and a permanent pacemaker was recommended. During his preoperative evaluation, TTE revealed severe concentric LVH with a speckled appearance of the myocardium, mild-tomoderate biatrial enlargement, and an EF of 50% to 55%. 99mTC pyrophosphate imaging was positive for amyloidosis and the patient started taking tafamidis. Recurrent hospital admissions for decompensated HF complicated his progression. The patient died at age 95 years.
Case 5
A 72-year-old male with a history of bilateral CTS and cervical spinal stenosis presented with dyspnea on exertion. An ECG revealed a normal sinus rhythm. A TTE found severely reduced systolic function with an EF ≤ 25%, mild concentric LVH, grade 3 diastolic dysfunction, mild-tomoderate biatrial enlargement, and moderate pulmonary hypertension (Figure 1). He was started on guideline-directed medical therapy (GDMT) for HF, which included sacubitril/valsartan, metoprolol succinate, and empagliflozin. The patient’s dyspnea improved, and a workup for nonischemic cardiomyopathy was initiated. 99mTC pyrophosphate imaging 1 year after his initial presentation was positive, leading to ATTR-CM diagnosis. The patient started taking tafamidis and he has since had a stable progression and continued to demonstrate good exercise tolerance with no hospitalizations. His most recent TTE indicated an EF of 40% to 45%.
(parasternal long axis view) of patient 5
demonstrating concentric left ventricular
hypertrophy with a speckled myocardium
and dilated left atrium.
Case 6
A 76-year-old male with a history of paroxysmal AF status after multiple ablations, bilateral CTS, and severe cervical spinal stenosis presented with dyspnea on exertion. The patient’s ECG showed normal sinus rhythm and left axis deviation with concern for left anterior hemiblock. A TTE revealed moderate LVH with a speckled appearance of the myocardium and grade 3 diastolic dysfunction with preserved EF. Before completing the workup for underlying cardiomyopathy, the patient underwent an interventional radiology procedure for an angiomyolipoma, and his postoperative course was complicated by pulmonary edema, requiring admission to the coronary care unit for diuresis. A repeat TTE revealed a reduced EF of 35%, and he was discharged on GDMT. No monoclonal protein was seen in the serum or urine. The patient’s progression was complicated by recurrent admissions for acute decompensated HF and supraventricular tachycardia despite being on amiodarone, which led to a delay in obtaining 99mTC pyrophosphate imaging. Due to hypotension, the patient was unable to tolerate GDMT. He eventually underwent 99mTC pyrophosphate imaging that confirmed ATTR-CM and was started on tafamidis. One year following initial presentation, the patient’s EF progressively declined to 20% to 25%, and he died shortly after discharge to subacute rehabilitation.
Case 7
A 95-year-old male with a history of longstanding persistent AF and bilateral CTS presented with dyspnea on exertion, bendopnea, and worsening bilateral pedal edema for a week. An ECG showed AF with a controlled ventricular response and low-voltage QRS waves (Figure 2). A TTE showed biatrial enlargement, LVH, and preserved EF > 55%. He started taking furosemide and was discharged with a diagnosis of HFpEF. The patient missed cardiology follow-up and presented 1 year later with decompensated HF. An amyloidosis workup was recommended, but due to intermittent periods of being lost to follow-up, the patient did not pursue this workup until 3 years after his initial presentation when 99mTC pyrophosphate imaging confirmed ATTR-CM. The patient declined tafamidis and continued to be followed by the cardiology team. His HF is managed with furosemide as needed due to intolerance to GDMT.
demonstrating atrial fibrillation with controlled
ventricular response and low-voltage QRS.
Case 8
A 74-year-old male with history of bilateral CTS presented with right-sided chest pain associated with shortness of breath, diaphoresis, dizziness, and worsening abdominal pain. He was found to have inferior wall myocardial infarction on ECG with later percutaneous coronary intervention to the left circumflex. His hospital course was complicated by decompensated HF (EF 45% to 50%) and AF with rapid ventricular response. He was treated and discharged with follow-up visits scheduled in the cardiology clinic. Multiple attempts were made to place him on GDMT; however, the patient was unable to tolerate these medications due to recurrent admissions for syncope. During a cardiology clinic visit 3 years after his initial presentation, an amyloidosis workup was initiated. 99mTC pyrophosphate imaging was positive for ATTR-CM, and he started taking tafamidis. Before this diagnosis, ECG indicated low-voltage QRS complexes. His progression has since been complicated by admissions for decompensated HF, recurrent episodes of AF requiring atrioventricular node ablation, and biventricular implantable cardioverter-defibrillator implantation after failed attempts at electrical cardioversion. He continued to follow up in the HF clinic.
Discussion
ATTR-CM is an underdiagnosed cause of cardiomyopathy, particularly in older adults. TTR is a transport protein produced in the liver, and misfolding can occur due to age-related instability of the wild-type protein (wtATTR) or pathologic variants in the TTR (hATTR). The misfolding leads to restrictive physiology, HF (often with preserved EF) arrhythmias, and conduction system disease.1 It is likely that the predominantly older male veteran population would be predisposed to wtATTR cardiomyopathy given that misfolding in the condition is believed to be age-related.
Current criteria for ATTR diagnosis require a combination of clinical suspicion, imaging, laboratory testing, and, in some cases, tissue biopsy confirmation and genetic testing.1,4,5 The diagnostic algorithm is as follows:
Clinical suspicion. Consider ATTR in patients with unexplained HFpEF, LV wall thickness ≥ 12 to 14 mm, discordance between ECG voltage and wall thickness, or associated extracardiac manifestations such as CTS, lumbar spinal stenosis, or peripheral/ autonomic neuropathy.
Exclusion of AL amyloidosis. Bone scintigraphy alone cannot distinguish between AL amyloidosis and ATTR, so patients with suspected amyloid cardiomyopathy should undergo serum and urine immunofixation electrophoresis and serum free light chain assay to rule out a monoclonal gammopathy as seen in AL amyloidosis.
Cardiac scintigraphy. Once AL amyloidosis is excluded, a positive 99mTC-labeled boneavid tracer image (such as a pyrophosphate scan) with grade 2 or grade 3 myocardial uptake is diagnostic of ATTR.
Tissue biopsy. If there is monoclonal gammopathy or equivocal imaging, tissue biopsy (eg, endomyocardial) with Congo red staining and amyloid typing by mass spectrometry or immunohistochemistry is necessary.
Genetic testing. Once ATTR is confirmed, genetic testing distinguishes hATTR from wtATTR, which impacts management and determines the need to screen family members.
Currently, there are 3 therapies approved by the US Food and Drug Administration (FDA) to treat ATTR-CM: tafamidis, acoramidis, and vutrisiran.6-8 Tafamidis and acoramidis stabilize the TTR tetramer, preventing amyloid formation. Vutrisiran uses RNA interference to silence the gene that produces TTR. Tafamidis has been found to improve cardiovascular outcomes in ATTR-CM. In the ATTR-ACT trial, it reduced all-cause mortality and cardiovascular hospitalizations in patients with ATTR-CM and New York Heart Association class I-III symptoms.6 There are other disease-modifying therapies, such the TTR gene silencers inotersen and patisiran; however, these are only FDA-approved for hATTR polyneuropathy and not for ATTRCM; ongoing trials are evaluating their cardiac efficacy.
The mean age of ATTR-CM diagnosis in the patients described in this case series was 79 years, which is older than the mean age of 74 years reported in prior research.4 All patients were male, and the most common presenting symptom was dyspnea on exertion. Table 1 outlines baseline characteristics and associated comorbidities of patients in this case series. The patients presented with many red-flag signs of ATTR-CM (Figure 3). Among them included syncope (4 of 8 patients), spinal stenosis (2 of 8 patients), arrhythmia (7 of 8 patients), heart failure (7 of 8 patients), and bilateral CTS (all patients).
amyloidosis red-flag signs identified.
Bilateral CTS was diagnosed in all patients before their diagnosis of ATTR-CM. Patient 7 had a diagnosis of CTS 9 years before his ATTR-CM diagnosis, underscoring a subtle, yet important extracardiac sign that may increase clinical suspicion for ATTR-CM. Previous research found that the probability of having CTS is highest 5 to 9 years prior to the development of cardiomyopathy. Its presence is also a prognostic marker in ATTR, independent of cardiac involvement.9 The median interval between CTS diagnosis and cardiomyopathy diagnosis was 5 years in this series.
Although screening criteria for ATTR have been proposed, none have been incorporated into formal guidelines.10,11 We propose that a baseline ECG and screening TTE be obtained in any patient aged > 65 years with cardiac risk equivalents such as hypertension and diabetes, presenting with ≥ 1 extracardiac red-flag signs, such as bilateral CTS and spinal stenosis. This will likely facilitate an earlier diagnosis of ATTR-CM, leading to earlier treatment initiation and better patient outcomes. This initiative can be started in the primary care setting and facilitates early cardiology referral. This recommendation is based on literature supporting clinical patterns and observations the authors have made in clinical practice.
Obstructive sleep apnea (OSA) was a comorbidity present in 50% of the patients described in this case series. The literature describing this association is sparse; however, a prospective observational study reported that disorders of sleep inclusive of OSA are frequent in patients with cardiac amyloidosis.12 A theory behind this association is that amyloid deposits in the upper airway tissues lead to airway narrowing. 13 More research is needed to further assess the relationship between OSA and cardiac amyloidosis, particularly with respect to the timing of OSA prior to the development of cardiomyopathy as it may be a potential early sign for clinicians to acknowledge.
An important observation from this case series is the variability in the timing of tafamidis initiation relative to symptom onset and confirmed diagnosis of ATTR-CM (Table 2). Although tafamidis has been found to slow disease progression, several patients in this series began treatment at advanced stages or years after the onset of cardiac symptoms, potentially limiting its clinical benefit.
For example, patient 1 was diagnosed with ATTR 2 years before tafamidis became available on the market and was initially treated with diflunisal. Patients who started tafamidis earlier in the disease course (eg, patients 3 and 5) appeared to have better long-term outcomes, including an absence of heart failure hospitalizations after initiation. In contrast, patients with delayed treatment initiation (eg, patients 2, 6, and 8) experienced ongoing decompensations or early mortality. Patient 7 declined tafamidis, underscoring challenges in medication uptake among older adults. Randomized controlled trials are warranted to compare the effectiveness of tafamidis with other recently FDA-approved therapies, such as acoramidis and vutrisiran, particularly in terms of cardiovascular outcomes.
AF was the most common arrhythmia observed in these patients and may occur years before the development of HF symptoms in the setting of ATTR-CM. Due to inherent conduction system disease, AF in this population may have a controlled or slow ventricular response, as observed in patient 4. Patients with atrial fibrillation and cardiac amyloidosis should receive anticoagulation regardless of CHA2DS2- VASc score due to their high risk of intracardiac thrombus formation.4
Limitations
This case series lacked genetic testing following a confirmed ATTR-CM diagnosis. Although much of the treatment is the same regardless of the presence of a TTR mutation, knowing the specific subtype of ATTR-CM has implications for prognosis and for screening family members.
Conclusions
Following analysis of 8 patients diagnosed with ATTR, this case series could serve as a blueprint for research into ATTR in veterans. In clinical practice, following military service, veterans may not be routinely seen by an outpatient physician and may present with sequelae of advanced stages of ATTR. Early identification of red-flag symptoms can lead to a higher clinical suspicion, prompting early diagnostic evaluation and treatment initiation and ultimately mitigating adverse outcomes. Future research that includes genetic testing for those with confirmed ATTR-CM may prove useful as a foundation for detailed and informed discussions with patients and their families regarding prognosis and, if indicated, screening for family members.
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142:e7-e22. doi:10.1161/CIR.0000000000000792
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765-774. doi:10.1111/j.1532-5415.2011.03868.x
- Nativi-Nicolau J, Redd A, Kelly N, et al. Increasing number of amyloidosis diagnosis in the Veterans Affairs populations. J Card Fail. 2019;25:S96.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872-2891. doi:10.1016/j.jacc.2019.04.003
- Gillmore JD, Maurer, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404-2412. doi:10.1161/CIRCULATIONAHA.116.021612
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007-1016. doi:10.1056/NEJMoa1805689
- Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med. 2024;390:132-142. doi:10.1056/NEJMoa2305434
- Fontana M, Berk JL, Gillmore JD, et al. Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J Med. 2025;392:33-44. doi:10.1056/NEJMoa2409134
- Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22:507-515. doi:10.1002/ejhf.1742
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554-1568. doi:10.1093/eurheartj/ehab072
- Brito D, Albrecht FC, de Arenaza DP, et al. World Heart Federation consensus on transthyretin amyloidosis cardiomyopathy (ATTR-CM). Glob Heart. 2023;18:59. doi:10.5334/gh.1262
- Bodez D, Guellich A, Kharoubi M, et al. Prevalence, severity, and prognostic value of sleep apnea syndromes in cardiac amyloidosis. Sleep. 2016;39:1333-1341. doi:10.5665/sleep.5958
- Colaco B, Colaco C, Lipford M. A forgotten problem: sleep-disordered breathing in amyloidosis. Chest. 2016;150:1293A. doi:10.1016/j.chest.2016.08.1407
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142:e7-e22. doi:10.1161/CIR.0000000000000792
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765-774. doi:10.1111/j.1532-5415.2011.03868.x
- Nativi-Nicolau J, Redd A, Kelly N, et al. Increasing number of amyloidosis diagnosis in the Veterans Affairs populations. J Card Fail. 2019;25:S96.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872-2891. doi:10.1016/j.jacc.2019.04.003
- Gillmore JD, Maurer, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404-2412. doi:10.1161/CIRCULATIONAHA.116.021612
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007-1016. doi:10.1056/NEJMoa1805689
- Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med. 2024;390:132-142. doi:10.1056/NEJMoa2305434
- Fontana M, Berk JL, Gillmore JD, et al. Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J Med. 2025;392:33-44. doi:10.1056/NEJMoa2409134
- Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22:507-515. doi:10.1002/ejhf.1742
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554-1568. doi:10.1093/eurheartj/ehab072
- Brito D, Albrecht FC, de Arenaza DP, et al. World Heart Federation consensus on transthyretin amyloidosis cardiomyopathy (ATTR-CM). Glob Heart. 2023;18:59. doi:10.5334/gh.1262
- Bodez D, Guellich A, Kharoubi M, et al. Prevalence, severity, and prognostic value of sleep apnea syndromes in cardiac amyloidosis. Sleep. 2016;39:1333-1341. doi:10.5665/sleep.5958
- Colaco B, Colaco C, Lipford M. A forgotten problem: sleep-disordered breathing in amyloidosis. Chest. 2016;150:1293A. doi:10.1016/j.chest.2016.08.1407
Case Series of Patients With Cardiac Amyloidosis at VA New York Harbor Healthcare-Brooklyn
Case Series of Patients With Cardiac Amyloidosis at VA New York Harbor Healthcare-Brooklyn
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
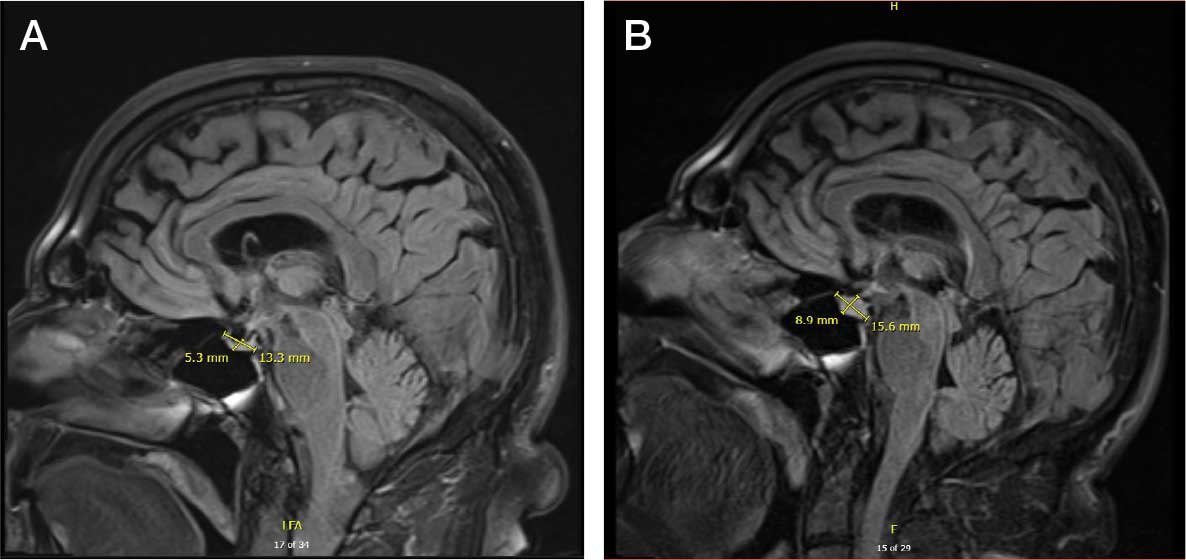
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.

No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
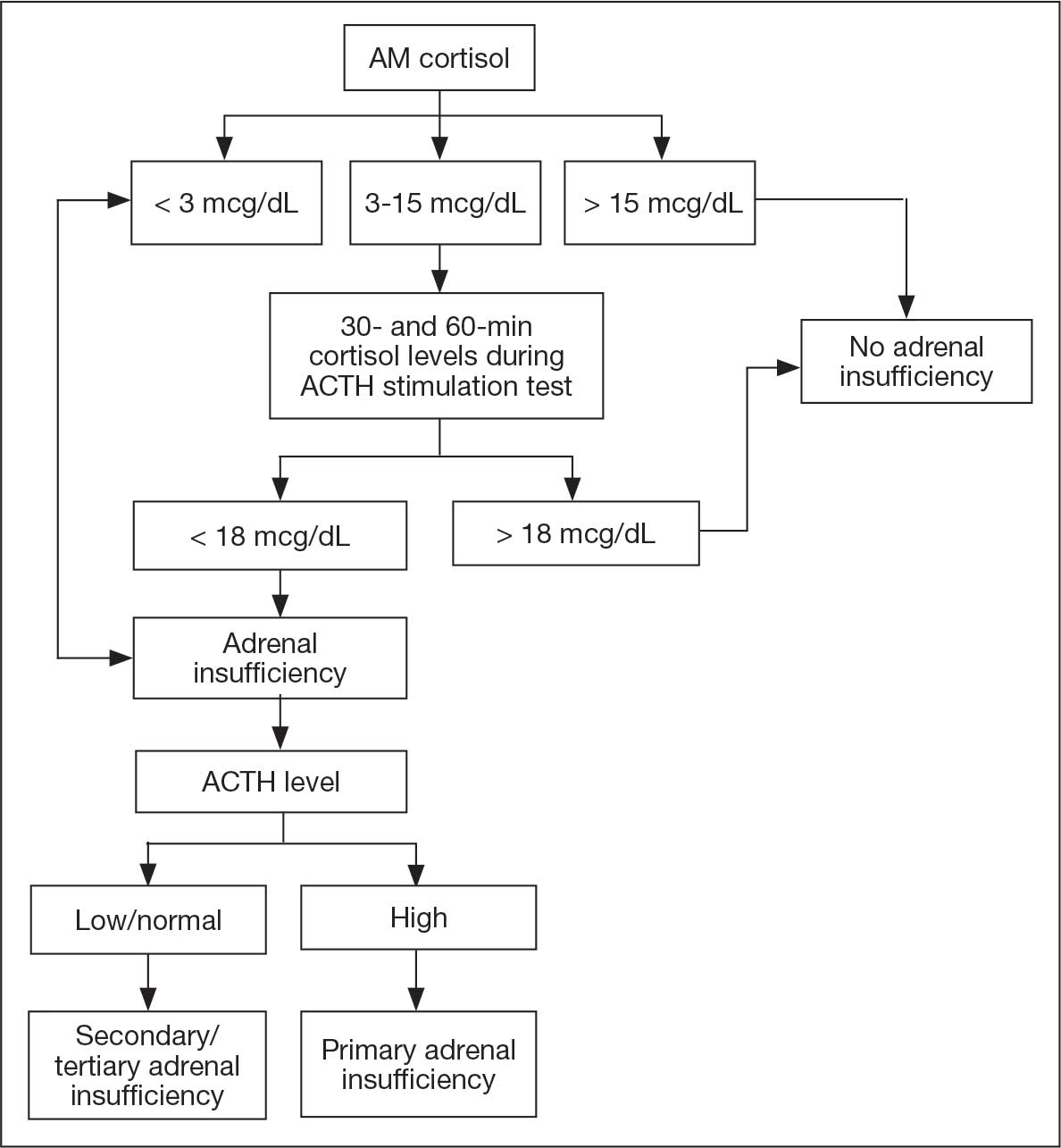
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
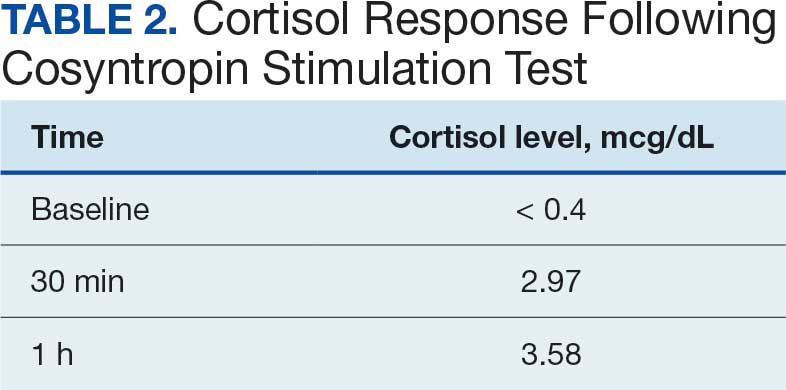
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.
No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.
No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
A Case of Birt-Hogg-Dubé Syndrome: A Rare but Essential Diagnosis to Consider
A Case of Birt-Hogg-Dubé Syndrome: A Rare but Essential Diagnosis to Consider
Birt-Hogg-Dubé syndrome (BHD) is an autosomal dominant disease that arises from loss-of-function mutations in the FLCN gene. FLCN encodes folliculin, which is presumed to function as a tumor suppressor, though its precise role is incompletely understood.1,2 BHD is characterized by multiple pulmonary cysts leading to recurrent spontaneous pneumothoraces, cutaneous lesions—specifically fibrofolliculomas—and an increased risk of renal malignancies. Diagnosing BHD is challenging due to the variable presentation of the disease. Some patients may only have cystic lung diseases, while others may not have characteristic skin lesions.3-5 It is important to maintain awareness of BHD, especially when the diagnosis dictates the need for genetic counseling.
Case Presentation
A male veteran in his 60s, who was a lifelong nonsmoker with a history of extensive bullous emphysema and recurrent pneumothoraces, presented to the Veterans Affairs Greater Los Angeles Healthcare System pulmonary clinic while transferring care from a separate institution.
According to the patient, the first pneumothorax episode occurred about 20 years before presentation, followed by a recurrence a few years later after he was diagnosed with emphysema. He underwent pleurodesis of the right lung during his service abroad. Another episode nearly a decade after the first pneumothorax necessitated pleurodesis of the left lung (Figure 1). The patient's family history revealed pulmonary cysts in 1 immediate family member but no history of renal tumors. Notably, his mother passed away at a young age due to tuberculosis.
On physical examination, numerous skin tags and acrochordons on the face were observed, which had been stable for > 30 years. Despite a slow decline in exercise capacity following pleurodesis, the patient could still walk multiple miles daily and climb 3 flights of stairs before needing to rest. Pulmonary function testing (PFT) showed a forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio of 0.84 with reduced FEV1, total lung capacity (TLC), and diffusion capacity for carbon monoxide (DLCO), indicating a mild restrictive ventilatory defect and reduced diffusing capacity.
Laboratory results revealed a normal α-1 antitrypsin level: 133 mg/dL (reference, 83-199 mg/dL), with a Pi*MS phenotype and undetectable antinuclear antibodies. The most recent chest computed tomography (CT) in 2019, displayed paraseptal and centrilobular emphysema, scattered blebs, and scarring consistent with prior pleurodesis procedures (Figure 2).
Genetic testing for the FLCN gene revealed heterozygous pathogenic mutation: c.1285del and p.His429Thrfs*39, which confirmed the diagnosis of BHD. A shave biopsy of a postauricular papular lesion confirmed a histologic pattern of fibrofolliculoma/trichodiscoma.
Follow-up and Outcomes
After confirmation of the BHD diagnosis, the patient was referred to genetic counseling and scheduled for annual magnetic resonance imaging (MRI) of the abdomen and pelvis to screen for renal malignancies. As the patient was able to establish care with a new long-term primary care practitioner in the outpatient setting, he continues regular follow-up visits in the pulmonary clinic with stable respiratory symptoms and no recurrent pneumothoraces thus far.
Discussion
Differential Diagnoses of Cystic Pulmonary Lesions
BHD is an important differential diagnosis to consider in the presentation of diffuse cystic lung diseases. Still, 2 other crucial considerations are pulmonary Langerhans cell histiocytosis (PLCH) and lymphangioleiomyomatosis (LAM), which occur at slightly higher frequencies than BHD.6
One of the first steps in radiographically evaluating cystic lung diseases is to characterize the cysts. The Fleischner Society defines true cysts as a “round parenchymal lucency or low-attenuating area with a well-defined interface with normal lung.”7 Mimics of cystic lesions may include cavitary lung lesions, thick-walled spaces within another area of mass, nodule, or consolidation. Another mimic is a pneumatocele, a pseudocyst that lacks epithelial lining and may be secondary to bacterial pneumonia, pneumocystis infections, trauma, or prior mechanical ventilation.8After characterizing true cysts, different patterns of cystic lesions can also be associated with specific diseases. Cysts in PLCH are commonly more uniform and round, whereas the cysts in LAM may be more irregularly shaped. 9 Cysts in BHD may be larger and predominantly located in basal and paramediastinal areas.4LAM is associated with tuberous sclerosis, which can also present with skin lesions (angiofibromas) and renal tumors (angiomyolipomas), thus creating a very similar picture to BHD. Therefore, tissue biopsies of skin lesions are essential as histopathology can identify characteristic fibrofolliculomas specific to BHD. While genetic testing would also strongly support the diagnosis of BHD, it is essential to note that negative genetic testing does not rule out BHD.4Lastly, lymphoid interstitial pneumonia (LIP) is another important consideration in the differential diagnosis of cystic lung diseases. LIP presents with not only perivascular cysts and centrilobular nodules but also diffuse ground-glass attenuation.10 In contrast to BHD, LIP is associated with autoimmune diseases such as Sjögren syndrome and infectious diseases such as HIV; thus, it may be differentiated from BHD by the presence of underlying disease processes and may warrant serologic testing for potential rheumatologic disorders.
Characteristics and Diagnostic Criteria
Cystic lung disease is the most common presentation of BHD. It presents in > 80% of cases and confers a 50-fold increase in the risk of spontaneous pneumothorax compared with the general population.4,11 Recurrent pneumothoraces are observed in about 25% to 30% of patients with BHD, typically occurring between the third and fifth decades of life and at significantly decreased rates after 50 years of age.12 A spontaneous pneumothorax might serve as the initial and perhaps the sole clinical presentation for some patients with BHD, but others may present with other respiratory symptoms such as cough and exertional dyspnea. PFT results may be normal or reveal a mild restrictive ventilatory defect and reduced DLCO, as reported in a few cases.6 The management of pulmonary complications primarily revolves around reducing the risk of pneumothoraces, which includes precautions such as avoiding positive pressure ventilation and air travel. Early pleurodesis with the first occurrence of a spontaneous pneumothorax is considered in some cases.13
The distinctive dermatologic features associated with BHD include multiple white papules primarily found on the nose and face. Pathologically, these manifestations have a range of histologic distinctions, from fibrofolliculomas to benign hamartomas of the hair follicles and trichodiscomas.5 The diagnostic criteria outlined by Menko et al note that confirmation of BHD requires the presence of either ≥ 5 pathologically confirmed fibrofolliculomas or trichodiscomas, a documented pathogenic FLCN gene mutation, or the fulfillment of 2 minor criteria. These minor criteria include the presence of multiple lung cysts, early-onset renal cancer, or a first-degree relative with BHD.5
Recurrent Pneumothoraces Management
After the first episode of spontaneous pneumothorax, early pleurodesis is indicated as the risk of recurrence can be as high as 75%.4,14 Specific pleurodesis modalities have shown promising results, such as total pleural covering with cellulose mesh. In a small retrospective review, cellulose mesh demonstrated a significant reduction in the recurrence rate of pneumothorax at 7.5 years for patients with BHD compared with partial covering.15 Apart from preventing further pneumothorax episodes in the affected lung, it is also important to highlight patient education and monitoring after initial pleurodesis, as the contralateral lung is also at risk. As demonstrated in this case, the patient had received pleurodesis of his right lung but experienced another pneumothorax of his contralateral lung a few years later.
Lastly, the patient was advised to avoid air travel altogether; however, current data may suggest that air travel may not be an absolute contraindication for patients with BHD. Although the literature on this topic is limited, a retrospective study by Johannesma et al involving 158 patients with BHD surveyed on pneumothorax occurrence after air travel indicated a calculated risk of 0.63% per flight. Notably, only 3 of 13 patients with BHD and recurrent pneumothoraces after travel had undergone pleurodesis in the past.16 Therefore, counseling patients on the potential risks of air travel and allowing essential flights while diligently monitoring for symptoms during and after travel may be a reasonable, patient-centered approach in contrast to a complete restriction on air travel.
Timing to Diagnosis
Diagnosing BHD is challenging and often delayed. In a 2022 study by Steinlein et al, the average delay in BHD diagnoses in their cohort was 9.3 years, with 4 patients also diagnosed with renal malignancy during the study period.17 The difficulty in diagnosis can be attributed to the heterogeneous presentation among affected family members, some of whom may exclusively exhibit pulmonary cystic lesions without dermatologic findings.
A lack of longitudinal care for this patient may have contributed to the diagnostic delay. The patient had pneumothorax events across separate care settings and locations, and due to employment-related relocations, he often re-established care at various health care systems. This case highlights the importance of continuity of care, especially in BHD, where monitoring for renal tumors is also essential to long-term management.17,18
Renal Tumor Monitoring
Finally, once BHD is diagnosed, one of the most important considerations is to begin routine monitoring for renal malignancies. Current recommendations advise starting lifelong renal cancer screening, even as early as age 20 years, with annual MRIs, as renal ultrasound may not be sufficiently sensitive to detect smaller lesions.19 The screening interval can be extended to every 2 years for patients without a family history of renal tumors or suspicious renal lesions. If tumors are found, then nephron-sparing surgery is recommended, given the potential for the development of chronic renal insufficiency in patients with BHD.20
Conclusions
BHD is a rare and complex syndrome in which early recognition and diagnosis play a pivotal role in preventing potentially severe complications such as renal malignancies. Suspicion of a genetic disorder, such as BHD, LAM, or PLCH, should arise in patients who experience spontaneous pneumothorax, especially in the presence of multiple cystic lesions or a family history of pneumothoraces. Early consideration of pleurodesis after the first spontaneous pneumothorax is advisable. The complex presentation of BHD contributes to the difficulty of diagnosis and may delay recognition, which can be exacerbated by variable continuity of care.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé-Syndrome. Nat Rev Urol. 2015;12:558-569. doi:10.1038/nrurol.2015.206
- Lim DHK, Rehal PK, Nahorski MS, et al. A new locus-specific database (LSDB) for mutations in the folliculin (FLCN) gene. Hum Mutat. 2010;31:E1043-1051. doi:10.1002/humu.21130
- Aivaz O, Berkman S, Middelton L, et al. Comedonal and cystic fibrofolliculomas in Birt-Hogg-Dube syndrome. JAMA Dermatology. 2015;151:770-774. doi:10.1001/jamadermatol.2015.0215
- Daccord C, Good JM, Morren MA, et al. Birt–Hogg–Dubé syndrome. Eur Respir Rev. 2020;29:200042. doi:10.1183/16000617.0042-2020
- Menko FH, van Steensel MA, Giraud S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. The Lancet Oncology. 2009;10:1199-1206. doi:10.1016/S1470-2045(09)70188-3
- Daccord C, Cottin V, Prévot G, et al. Lung function in Birt-Hogg-Dubé syndrome: a retrospective analysis of 96 patients. Orphanet J Rare Dis. 2020;15:120. doi:10.1186/s13023-020-01402-y
- Hansell DM, Bankier AA, MacMahon H, et al. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246:697-722. doi:10.1148/radiol.2462070712
- Jamil A, Kasi A. Pneumatocele. In: StatPearls. StatPearls Publishing; 2024. Accessed March 2, 2026. http://www.ncbi.nlm.nih.gov/books/NBK556146/
- Bhardwaj H, Bhardwaj B. Differentiating pulmonary lymphangioleiomyomatosis from pulmonary langerhans cell histiocytosis and Birt-Hogg-Dube syndrome. Lung India. 2013;30:372-373. doi:10.4103/0970-2113.120611
- Swigris JJ, Berry GJ, Raffin TA, et al. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122:2150-2164. doi:10.1378/chest.122.6.2150
- Zbar B, Alvord WG, Glenn G, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393-400.
- Sattler EC, Steinlein OK. Delayed diagnosis of Birt-Hogg-Dubé syndrome due to marked intrafamilial clinical variability: a case report. BMC Med Genet. 2018;19:45. doi:10.1186/s12881-018-0558-0
- Gupta N, Seyama K, McCormack FX. Pulmonary manifestations of Birt-Hogg-Dubé syndrome. Fam Cancer. 2013;12:387-396. doi:10.1007/s10689-013-9660-9
- Gupta N, Kopras EJ, Henske EP, et al. Spontaneous pneumothoraces in patients with Birt–Hogg–Dubé syndrome. Ann Am Thorac Soc. 2017;14:706-713. doi:10.1513/AnnalsATS.201611-886OC
- Mizobuchi T, Kurihara M, Ebana H, et al. A total pleural covering of absorbable cellulose mesh prevents pneumothorax recurrence in patients with Birt-Hogg-Dubé syndrome. Orphanet J Rare Dis. 2018;13:78. doi:10.1186/s13023-018-0790-x
- Johannesma PC, van de Beek I, van der Wel JWT, et al. Risk of spontaneous pneumothorax due to air travel and diving in patients with Birt–Hogg–Dubé syndrome. Springerplus. 2016;5:1506. doi:10.1186/s40064-016-3009-4
- Steinlein OK, Reithmair M, Syunyaeva Z, et al. Delayed diagnosis of Birt-Hogg-Dubé syndrome might be aggravated by gender bias. eClinicalMedicine. 2022;51:101572. doi:10.1016/j.eclinm.2022.101572
- Pereira Gray DJ, Sidaway-Lee K, White E, et al. Continuity of care with doctors—a matter of life and death? A systematic review of continuity of care and mortality. BMJ Open. 2018;8:e021161. doi:10.1136/bmjopen-2017-021161
- Sattler EC, Steinlein OK. GeneReviews Birt-Hogg-Dubé syndrome. January 30, 2020. Accessed March 2, 2026. https://www.ncbi.nlm.nih.gov/books/NBK1522/table
- Stamatakis L, Metwalli AR, Middelton LA, et al. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. 2013;12:397-402. doi:10.1007/s10689-013-9657-4
Birt-Hogg-Dubé syndrome (BHD) is an autosomal dominant disease that arises from loss-of-function mutations in the FLCN gene. FLCN encodes folliculin, which is presumed to function as a tumor suppressor, though its precise role is incompletely understood.1,2 BHD is characterized by multiple pulmonary cysts leading to recurrent spontaneous pneumothoraces, cutaneous lesions—specifically fibrofolliculomas—and an increased risk of renal malignancies. Diagnosing BHD is challenging due to the variable presentation of the disease. Some patients may only have cystic lung diseases, while others may not have characteristic skin lesions.3-5 It is important to maintain awareness of BHD, especially when the diagnosis dictates the need for genetic counseling.
Case Presentation
A male veteran in his 60s, who was a lifelong nonsmoker with a history of extensive bullous emphysema and recurrent pneumothoraces, presented to the Veterans Affairs Greater Los Angeles Healthcare System pulmonary clinic while transferring care from a separate institution.
According to the patient, the first pneumothorax episode occurred about 20 years before presentation, followed by a recurrence a few years later after he was diagnosed with emphysema. He underwent pleurodesis of the right lung during his service abroad. Another episode nearly a decade after the first pneumothorax necessitated pleurodesis of the left lung (Figure 1). The patient's family history revealed pulmonary cysts in 1 immediate family member but no history of renal tumors. Notably, his mother passed away at a young age due to tuberculosis.
On physical examination, numerous skin tags and acrochordons on the face were observed, which had been stable for > 30 years. Despite a slow decline in exercise capacity following pleurodesis, the patient could still walk multiple miles daily and climb 3 flights of stairs before needing to rest. Pulmonary function testing (PFT) showed a forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio of 0.84 with reduced FEV1, total lung capacity (TLC), and diffusion capacity for carbon monoxide (DLCO), indicating a mild restrictive ventilatory defect and reduced diffusing capacity.
Laboratory results revealed a normal α-1 antitrypsin level: 133 mg/dL (reference, 83-199 mg/dL), with a Pi*MS phenotype and undetectable antinuclear antibodies. The most recent chest computed tomography (CT) in 2019, displayed paraseptal and centrilobular emphysema, scattered blebs, and scarring consistent with prior pleurodesis procedures (Figure 2).
Genetic testing for the FLCN gene revealed heterozygous pathogenic mutation: c.1285del and p.His429Thrfs*39, which confirmed the diagnosis of BHD. A shave biopsy of a postauricular papular lesion confirmed a histologic pattern of fibrofolliculoma/trichodiscoma.
Follow-up and Outcomes
After confirmation of the BHD diagnosis, the patient was referred to genetic counseling and scheduled for annual magnetic resonance imaging (MRI) of the abdomen and pelvis to screen for renal malignancies. As the patient was able to establish care with a new long-term primary care practitioner in the outpatient setting, he continues regular follow-up visits in the pulmonary clinic with stable respiratory symptoms and no recurrent pneumothoraces thus far.
Discussion
Differential Diagnoses of Cystic Pulmonary Lesions
BHD is an important differential diagnosis to consider in the presentation of diffuse cystic lung diseases. Still, 2 other crucial considerations are pulmonary Langerhans cell histiocytosis (PLCH) and lymphangioleiomyomatosis (LAM), which occur at slightly higher frequencies than BHD.6
One of the first steps in radiographically evaluating cystic lung diseases is to characterize the cysts. The Fleischner Society defines true cysts as a “round parenchymal lucency or low-attenuating area with a well-defined interface with normal lung.”7 Mimics of cystic lesions may include cavitary lung lesions, thick-walled spaces within another area of mass, nodule, or consolidation. Another mimic is a pneumatocele, a pseudocyst that lacks epithelial lining and may be secondary to bacterial pneumonia, pneumocystis infections, trauma, or prior mechanical ventilation.8After characterizing true cysts, different patterns of cystic lesions can also be associated with specific diseases. Cysts in PLCH are commonly more uniform and round, whereas the cysts in LAM may be more irregularly shaped. 9 Cysts in BHD may be larger and predominantly located in basal and paramediastinal areas.4LAM is associated with tuberous sclerosis, which can also present with skin lesions (angiofibromas) and renal tumors (angiomyolipomas), thus creating a very similar picture to BHD. Therefore, tissue biopsies of skin lesions are essential as histopathology can identify characteristic fibrofolliculomas specific to BHD. While genetic testing would also strongly support the diagnosis of BHD, it is essential to note that negative genetic testing does not rule out BHD.4Lastly, lymphoid interstitial pneumonia (LIP) is another important consideration in the differential diagnosis of cystic lung diseases. LIP presents with not only perivascular cysts and centrilobular nodules but also diffuse ground-glass attenuation.10 In contrast to BHD, LIP is associated with autoimmune diseases such as Sjögren syndrome and infectious diseases such as HIV; thus, it may be differentiated from BHD by the presence of underlying disease processes and may warrant serologic testing for potential rheumatologic disorders.
Characteristics and Diagnostic Criteria
Cystic lung disease is the most common presentation of BHD. It presents in > 80% of cases and confers a 50-fold increase in the risk of spontaneous pneumothorax compared with the general population.4,11 Recurrent pneumothoraces are observed in about 25% to 30% of patients with BHD, typically occurring between the third and fifth decades of life and at significantly decreased rates after 50 years of age.12 A spontaneous pneumothorax might serve as the initial and perhaps the sole clinical presentation for some patients with BHD, but others may present with other respiratory symptoms such as cough and exertional dyspnea. PFT results may be normal or reveal a mild restrictive ventilatory defect and reduced DLCO, as reported in a few cases.6 The management of pulmonary complications primarily revolves around reducing the risk of pneumothoraces, which includes precautions such as avoiding positive pressure ventilation and air travel. Early pleurodesis with the first occurrence of a spontaneous pneumothorax is considered in some cases.13
The distinctive dermatologic features associated with BHD include multiple white papules primarily found on the nose and face. Pathologically, these manifestations have a range of histologic distinctions, from fibrofolliculomas to benign hamartomas of the hair follicles and trichodiscomas.5 The diagnostic criteria outlined by Menko et al note that confirmation of BHD requires the presence of either ≥ 5 pathologically confirmed fibrofolliculomas or trichodiscomas, a documented pathogenic FLCN gene mutation, or the fulfillment of 2 minor criteria. These minor criteria include the presence of multiple lung cysts, early-onset renal cancer, or a first-degree relative with BHD.5
Recurrent Pneumothoraces Management
After the first episode of spontaneous pneumothorax, early pleurodesis is indicated as the risk of recurrence can be as high as 75%.4,14 Specific pleurodesis modalities have shown promising results, such as total pleural covering with cellulose mesh. In a small retrospective review, cellulose mesh demonstrated a significant reduction in the recurrence rate of pneumothorax at 7.5 years for patients with BHD compared with partial covering.15 Apart from preventing further pneumothorax episodes in the affected lung, it is also important to highlight patient education and monitoring after initial pleurodesis, as the contralateral lung is also at risk. As demonstrated in this case, the patient had received pleurodesis of his right lung but experienced another pneumothorax of his contralateral lung a few years later.
Lastly, the patient was advised to avoid air travel altogether; however, current data may suggest that air travel may not be an absolute contraindication for patients with BHD. Although the literature on this topic is limited, a retrospective study by Johannesma et al involving 158 patients with BHD surveyed on pneumothorax occurrence after air travel indicated a calculated risk of 0.63% per flight. Notably, only 3 of 13 patients with BHD and recurrent pneumothoraces after travel had undergone pleurodesis in the past.16 Therefore, counseling patients on the potential risks of air travel and allowing essential flights while diligently monitoring for symptoms during and after travel may be a reasonable, patient-centered approach in contrast to a complete restriction on air travel.
Timing to Diagnosis
Diagnosing BHD is challenging and often delayed. In a 2022 study by Steinlein et al, the average delay in BHD diagnoses in their cohort was 9.3 years, with 4 patients also diagnosed with renal malignancy during the study period.17 The difficulty in diagnosis can be attributed to the heterogeneous presentation among affected family members, some of whom may exclusively exhibit pulmonary cystic lesions without dermatologic findings.
A lack of longitudinal care for this patient may have contributed to the diagnostic delay. The patient had pneumothorax events across separate care settings and locations, and due to employment-related relocations, he often re-established care at various health care systems. This case highlights the importance of continuity of care, especially in BHD, where monitoring for renal tumors is also essential to long-term management.17,18
Renal Tumor Monitoring
Finally, once BHD is diagnosed, one of the most important considerations is to begin routine monitoring for renal malignancies. Current recommendations advise starting lifelong renal cancer screening, even as early as age 20 years, with annual MRIs, as renal ultrasound may not be sufficiently sensitive to detect smaller lesions.19 The screening interval can be extended to every 2 years for patients without a family history of renal tumors or suspicious renal lesions. If tumors are found, then nephron-sparing surgery is recommended, given the potential for the development of chronic renal insufficiency in patients with BHD.20
Conclusions
BHD is a rare and complex syndrome in which early recognition and diagnosis play a pivotal role in preventing potentially severe complications such as renal malignancies. Suspicion of a genetic disorder, such as BHD, LAM, or PLCH, should arise in patients who experience spontaneous pneumothorax, especially in the presence of multiple cystic lesions or a family history of pneumothoraces. Early consideration of pleurodesis after the first spontaneous pneumothorax is advisable. The complex presentation of BHD contributes to the difficulty of diagnosis and may delay recognition, which can be exacerbated by variable continuity of care.
Birt-Hogg-Dubé syndrome (BHD) is an autosomal dominant disease that arises from loss-of-function mutations in the FLCN gene. FLCN encodes folliculin, which is presumed to function as a tumor suppressor, though its precise role is incompletely understood.1,2 BHD is characterized by multiple pulmonary cysts leading to recurrent spontaneous pneumothoraces, cutaneous lesions—specifically fibrofolliculomas—and an increased risk of renal malignancies. Diagnosing BHD is challenging due to the variable presentation of the disease. Some patients may only have cystic lung diseases, while others may not have characteristic skin lesions.3-5 It is important to maintain awareness of BHD, especially when the diagnosis dictates the need for genetic counseling.
Case Presentation
A male veteran in his 60s, who was a lifelong nonsmoker with a history of extensive bullous emphysema and recurrent pneumothoraces, presented to the Veterans Affairs Greater Los Angeles Healthcare System pulmonary clinic while transferring care from a separate institution.
According to the patient, the first pneumothorax episode occurred about 20 years before presentation, followed by a recurrence a few years later after he was diagnosed with emphysema. He underwent pleurodesis of the right lung during his service abroad. Another episode nearly a decade after the first pneumothorax necessitated pleurodesis of the left lung (Figure 1). The patient's family history revealed pulmonary cysts in 1 immediate family member but no history of renal tumors. Notably, his mother passed away at a young age due to tuberculosis.
On physical examination, numerous skin tags and acrochordons on the face were observed, which had been stable for > 30 years. Despite a slow decline in exercise capacity following pleurodesis, the patient could still walk multiple miles daily and climb 3 flights of stairs before needing to rest. Pulmonary function testing (PFT) showed a forced expiratory volume in 1 second (FEV1)/forced vital capacity ratio of 0.84 with reduced FEV1, total lung capacity (TLC), and diffusion capacity for carbon monoxide (DLCO), indicating a mild restrictive ventilatory defect and reduced diffusing capacity.
Laboratory results revealed a normal α-1 antitrypsin level: 133 mg/dL (reference, 83-199 mg/dL), with a Pi*MS phenotype and undetectable antinuclear antibodies. The most recent chest computed tomography (CT) in 2019, displayed paraseptal and centrilobular emphysema, scattered blebs, and scarring consistent with prior pleurodesis procedures (Figure 2).
Genetic testing for the FLCN gene revealed heterozygous pathogenic mutation: c.1285del and p.His429Thrfs*39, which confirmed the diagnosis of BHD. A shave biopsy of a postauricular papular lesion confirmed a histologic pattern of fibrofolliculoma/trichodiscoma.
Follow-up and Outcomes
After confirmation of the BHD diagnosis, the patient was referred to genetic counseling and scheduled for annual magnetic resonance imaging (MRI) of the abdomen and pelvis to screen for renal malignancies. As the patient was able to establish care with a new long-term primary care practitioner in the outpatient setting, he continues regular follow-up visits in the pulmonary clinic with stable respiratory symptoms and no recurrent pneumothoraces thus far.
Discussion
Differential Diagnoses of Cystic Pulmonary Lesions
BHD is an important differential diagnosis to consider in the presentation of diffuse cystic lung diseases. Still, 2 other crucial considerations are pulmonary Langerhans cell histiocytosis (PLCH) and lymphangioleiomyomatosis (LAM), which occur at slightly higher frequencies than BHD.6
One of the first steps in radiographically evaluating cystic lung diseases is to characterize the cysts. The Fleischner Society defines true cysts as a “round parenchymal lucency or low-attenuating area with a well-defined interface with normal lung.”7 Mimics of cystic lesions may include cavitary lung lesions, thick-walled spaces within another area of mass, nodule, or consolidation. Another mimic is a pneumatocele, a pseudocyst that lacks epithelial lining and may be secondary to bacterial pneumonia, pneumocystis infections, trauma, or prior mechanical ventilation.8After characterizing true cysts, different patterns of cystic lesions can also be associated with specific diseases. Cysts in PLCH are commonly more uniform and round, whereas the cysts in LAM may be more irregularly shaped. 9 Cysts in BHD may be larger and predominantly located in basal and paramediastinal areas.4LAM is associated with tuberous sclerosis, which can also present with skin lesions (angiofibromas) and renal tumors (angiomyolipomas), thus creating a very similar picture to BHD. Therefore, tissue biopsies of skin lesions are essential as histopathology can identify characteristic fibrofolliculomas specific to BHD. While genetic testing would also strongly support the diagnosis of BHD, it is essential to note that negative genetic testing does not rule out BHD.4Lastly, lymphoid interstitial pneumonia (LIP) is another important consideration in the differential diagnosis of cystic lung diseases. LIP presents with not only perivascular cysts and centrilobular nodules but also diffuse ground-glass attenuation.10 In contrast to BHD, LIP is associated with autoimmune diseases such as Sjögren syndrome and infectious diseases such as HIV; thus, it may be differentiated from BHD by the presence of underlying disease processes and may warrant serologic testing for potential rheumatologic disorders.
Characteristics and Diagnostic Criteria
Cystic lung disease is the most common presentation of BHD. It presents in > 80% of cases and confers a 50-fold increase in the risk of spontaneous pneumothorax compared with the general population.4,11 Recurrent pneumothoraces are observed in about 25% to 30% of patients with BHD, typically occurring between the third and fifth decades of life and at significantly decreased rates after 50 years of age.12 A spontaneous pneumothorax might serve as the initial and perhaps the sole clinical presentation for some patients with BHD, but others may present with other respiratory symptoms such as cough and exertional dyspnea. PFT results may be normal or reveal a mild restrictive ventilatory defect and reduced DLCO, as reported in a few cases.6 The management of pulmonary complications primarily revolves around reducing the risk of pneumothoraces, which includes precautions such as avoiding positive pressure ventilation and air travel. Early pleurodesis with the first occurrence of a spontaneous pneumothorax is considered in some cases.13
The distinctive dermatologic features associated with BHD include multiple white papules primarily found on the nose and face. Pathologically, these manifestations have a range of histologic distinctions, from fibrofolliculomas to benign hamartomas of the hair follicles and trichodiscomas.5 The diagnostic criteria outlined by Menko et al note that confirmation of BHD requires the presence of either ≥ 5 pathologically confirmed fibrofolliculomas or trichodiscomas, a documented pathogenic FLCN gene mutation, or the fulfillment of 2 minor criteria. These minor criteria include the presence of multiple lung cysts, early-onset renal cancer, or a first-degree relative with BHD.5
Recurrent Pneumothoraces Management
After the first episode of spontaneous pneumothorax, early pleurodesis is indicated as the risk of recurrence can be as high as 75%.4,14 Specific pleurodesis modalities have shown promising results, such as total pleural covering with cellulose mesh. In a small retrospective review, cellulose mesh demonstrated a significant reduction in the recurrence rate of pneumothorax at 7.5 years for patients with BHD compared with partial covering.15 Apart from preventing further pneumothorax episodes in the affected lung, it is also important to highlight patient education and monitoring after initial pleurodesis, as the contralateral lung is also at risk. As demonstrated in this case, the patient had received pleurodesis of his right lung but experienced another pneumothorax of his contralateral lung a few years later.
Lastly, the patient was advised to avoid air travel altogether; however, current data may suggest that air travel may not be an absolute contraindication for patients with BHD. Although the literature on this topic is limited, a retrospective study by Johannesma et al involving 158 patients with BHD surveyed on pneumothorax occurrence after air travel indicated a calculated risk of 0.63% per flight. Notably, only 3 of 13 patients with BHD and recurrent pneumothoraces after travel had undergone pleurodesis in the past.16 Therefore, counseling patients on the potential risks of air travel and allowing essential flights while diligently monitoring for symptoms during and after travel may be a reasonable, patient-centered approach in contrast to a complete restriction on air travel.
Timing to Diagnosis
Diagnosing BHD is challenging and often delayed. In a 2022 study by Steinlein et al, the average delay in BHD diagnoses in their cohort was 9.3 years, with 4 patients also diagnosed with renal malignancy during the study period.17 The difficulty in diagnosis can be attributed to the heterogeneous presentation among affected family members, some of whom may exclusively exhibit pulmonary cystic lesions without dermatologic findings.
A lack of longitudinal care for this patient may have contributed to the diagnostic delay. The patient had pneumothorax events across separate care settings and locations, and due to employment-related relocations, he often re-established care at various health care systems. This case highlights the importance of continuity of care, especially in BHD, where monitoring for renal tumors is also essential to long-term management.17,18
Renal Tumor Monitoring
Finally, once BHD is diagnosed, one of the most important considerations is to begin routine monitoring for renal malignancies. Current recommendations advise starting lifelong renal cancer screening, even as early as age 20 years, with annual MRIs, as renal ultrasound may not be sufficiently sensitive to detect smaller lesions.19 The screening interval can be extended to every 2 years for patients without a family history of renal tumors or suspicious renal lesions. If tumors are found, then nephron-sparing surgery is recommended, given the potential for the development of chronic renal insufficiency in patients with BHD.20
Conclusions
BHD is a rare and complex syndrome in which early recognition and diagnosis play a pivotal role in preventing potentially severe complications such as renal malignancies. Suspicion of a genetic disorder, such as BHD, LAM, or PLCH, should arise in patients who experience spontaneous pneumothorax, especially in the presence of multiple cystic lesions or a family history of pneumothoraces. Early consideration of pleurodesis after the first spontaneous pneumothorax is advisable. The complex presentation of BHD contributes to the difficulty of diagnosis and may delay recognition, which can be exacerbated by variable continuity of care.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé-Syndrome. Nat Rev Urol. 2015;12:558-569. doi:10.1038/nrurol.2015.206
- Lim DHK, Rehal PK, Nahorski MS, et al. A new locus-specific database (LSDB) for mutations in the folliculin (FLCN) gene. Hum Mutat. 2010;31:E1043-1051. doi:10.1002/humu.21130
- Aivaz O, Berkman S, Middelton L, et al. Comedonal and cystic fibrofolliculomas in Birt-Hogg-Dube syndrome. JAMA Dermatology. 2015;151:770-774. doi:10.1001/jamadermatol.2015.0215
- Daccord C, Good JM, Morren MA, et al. Birt–Hogg–Dubé syndrome. Eur Respir Rev. 2020;29:200042. doi:10.1183/16000617.0042-2020
- Menko FH, van Steensel MA, Giraud S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. The Lancet Oncology. 2009;10:1199-1206. doi:10.1016/S1470-2045(09)70188-3
- Daccord C, Cottin V, Prévot G, et al. Lung function in Birt-Hogg-Dubé syndrome: a retrospective analysis of 96 patients. Orphanet J Rare Dis. 2020;15:120. doi:10.1186/s13023-020-01402-y
- Hansell DM, Bankier AA, MacMahon H, et al. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246:697-722. doi:10.1148/radiol.2462070712
- Jamil A, Kasi A. Pneumatocele. In: StatPearls. StatPearls Publishing; 2024. Accessed March 2, 2026. http://www.ncbi.nlm.nih.gov/books/NBK556146/
- Bhardwaj H, Bhardwaj B. Differentiating pulmonary lymphangioleiomyomatosis from pulmonary langerhans cell histiocytosis and Birt-Hogg-Dube syndrome. Lung India. 2013;30:372-373. doi:10.4103/0970-2113.120611
- Swigris JJ, Berry GJ, Raffin TA, et al. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122:2150-2164. doi:10.1378/chest.122.6.2150
- Zbar B, Alvord WG, Glenn G, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393-400.
- Sattler EC, Steinlein OK. Delayed diagnosis of Birt-Hogg-Dubé syndrome due to marked intrafamilial clinical variability: a case report. BMC Med Genet. 2018;19:45. doi:10.1186/s12881-018-0558-0
- Gupta N, Seyama K, McCormack FX. Pulmonary manifestations of Birt-Hogg-Dubé syndrome. Fam Cancer. 2013;12:387-396. doi:10.1007/s10689-013-9660-9
- Gupta N, Kopras EJ, Henske EP, et al. Spontaneous pneumothoraces in patients with Birt–Hogg–Dubé syndrome. Ann Am Thorac Soc. 2017;14:706-713. doi:10.1513/AnnalsATS.201611-886OC
- Mizobuchi T, Kurihara M, Ebana H, et al. A total pleural covering of absorbable cellulose mesh prevents pneumothorax recurrence in patients with Birt-Hogg-Dubé syndrome. Orphanet J Rare Dis. 2018;13:78. doi:10.1186/s13023-018-0790-x
- Johannesma PC, van de Beek I, van der Wel JWT, et al. Risk of spontaneous pneumothorax due to air travel and diving in patients with Birt–Hogg–Dubé syndrome. Springerplus. 2016;5:1506. doi:10.1186/s40064-016-3009-4
- Steinlein OK, Reithmair M, Syunyaeva Z, et al. Delayed diagnosis of Birt-Hogg-Dubé syndrome might be aggravated by gender bias. eClinicalMedicine. 2022;51:101572. doi:10.1016/j.eclinm.2022.101572
- Pereira Gray DJ, Sidaway-Lee K, White E, et al. Continuity of care with doctors—a matter of life and death? A systematic review of continuity of care and mortality. BMJ Open. 2018;8:e021161. doi:10.1136/bmjopen-2017-021161
- Sattler EC, Steinlein OK. GeneReviews Birt-Hogg-Dubé syndrome. January 30, 2020. Accessed March 2, 2026. https://www.ncbi.nlm.nih.gov/books/NBK1522/table
- Stamatakis L, Metwalli AR, Middelton LA, et al. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. 2013;12:397-402. doi:10.1007/s10689-013-9657-4
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé-Syndrome. Nat Rev Urol. 2015;12:558-569. doi:10.1038/nrurol.2015.206
- Lim DHK, Rehal PK, Nahorski MS, et al. A new locus-specific database (LSDB) for mutations in the folliculin (FLCN) gene. Hum Mutat. 2010;31:E1043-1051. doi:10.1002/humu.21130
- Aivaz O, Berkman S, Middelton L, et al. Comedonal and cystic fibrofolliculomas in Birt-Hogg-Dube syndrome. JAMA Dermatology. 2015;151:770-774. doi:10.1001/jamadermatol.2015.0215
- Daccord C, Good JM, Morren MA, et al. Birt–Hogg–Dubé syndrome. Eur Respir Rev. 2020;29:200042. doi:10.1183/16000617.0042-2020
- Menko FH, van Steensel MA, Giraud S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. The Lancet Oncology. 2009;10:1199-1206. doi:10.1016/S1470-2045(09)70188-3
- Daccord C, Cottin V, Prévot G, et al. Lung function in Birt-Hogg-Dubé syndrome: a retrospective analysis of 96 patients. Orphanet J Rare Dis. 2020;15:120. doi:10.1186/s13023-020-01402-y
- Hansell DM, Bankier AA, MacMahon H, et al. Fleischner Society: glossary of terms for thoracic imaging. Radiology. 2008;246:697-722. doi:10.1148/radiol.2462070712
- Jamil A, Kasi A. Pneumatocele. In: StatPearls. StatPearls Publishing; 2024. Accessed March 2, 2026. http://www.ncbi.nlm.nih.gov/books/NBK556146/
- Bhardwaj H, Bhardwaj B. Differentiating pulmonary lymphangioleiomyomatosis from pulmonary langerhans cell histiocytosis and Birt-Hogg-Dube syndrome. Lung India. 2013;30:372-373. doi:10.4103/0970-2113.120611
- Swigris JJ, Berry GJ, Raffin TA, et al. Lymphoid interstitial pneumonia: a narrative review. Chest. 2002;122:2150-2164. doi:10.1378/chest.122.6.2150
- Zbar B, Alvord WG, Glenn G, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev. 2002;11:393-400.
- Sattler EC, Steinlein OK. Delayed diagnosis of Birt-Hogg-Dubé syndrome due to marked intrafamilial clinical variability: a case report. BMC Med Genet. 2018;19:45. doi:10.1186/s12881-018-0558-0
- Gupta N, Seyama K, McCormack FX. Pulmonary manifestations of Birt-Hogg-Dubé syndrome. Fam Cancer. 2013;12:387-396. doi:10.1007/s10689-013-9660-9
- Gupta N, Kopras EJ, Henske EP, et al. Spontaneous pneumothoraces in patients with Birt–Hogg–Dubé syndrome. Ann Am Thorac Soc. 2017;14:706-713. doi:10.1513/AnnalsATS.201611-886OC
- Mizobuchi T, Kurihara M, Ebana H, et al. A total pleural covering of absorbable cellulose mesh prevents pneumothorax recurrence in patients with Birt-Hogg-Dubé syndrome. Orphanet J Rare Dis. 2018;13:78. doi:10.1186/s13023-018-0790-x
- Johannesma PC, van de Beek I, van der Wel JWT, et al. Risk of spontaneous pneumothorax due to air travel and diving in patients with Birt–Hogg–Dubé syndrome. Springerplus. 2016;5:1506. doi:10.1186/s40064-016-3009-4
- Steinlein OK, Reithmair M, Syunyaeva Z, et al. Delayed diagnosis of Birt-Hogg-Dubé syndrome might be aggravated by gender bias. eClinicalMedicine. 2022;51:101572. doi:10.1016/j.eclinm.2022.101572
- Pereira Gray DJ, Sidaway-Lee K, White E, et al. Continuity of care with doctors—a matter of life and death? A systematic review of continuity of care and mortality. BMJ Open. 2018;8:e021161. doi:10.1136/bmjopen-2017-021161
- Sattler EC, Steinlein OK. GeneReviews Birt-Hogg-Dubé syndrome. January 30, 2020. Accessed March 2, 2026. https://www.ncbi.nlm.nih.gov/books/NBK1522/table
- Stamatakis L, Metwalli AR, Middelton LA, et al. Diagnosis and management of BHD-associated kidney cancer. Fam Cancer. 2013;12:397-402. doi:10.1007/s10689-013-9657-4
A Case of Birt-Hogg-Dubé Syndrome: A Rare but Essential Diagnosis to Consider
A Case of Birt-Hogg-Dubé Syndrome: A Rare but Essential Diagnosis to Consider
Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
Magnetic resonance image (MRI) contrast agents can induce profound complications, including gadolinium encephalopathy, kidney injury, gadolinium-associated plaques, and progressive systemic fibrosis, which can be fatal.1-10 About 50% of MRIs use gadolinium-based contrast (Gd3+), a toxic rare earth metal ion that enhances imaging but requires binding with pharmaceutical ligands to reduce toxicity and promote renal elimination (Figure 1). Despite these measures, Gd3+ can persist in the body, including the brain.11,12 Wastewater treatment fails to remove these agents, making Gd3+ a growing pollutant in water and the food chain.13-15 Because Gd3+ is a rare earth metal ion in the milieu intérieur, there is an urgent need to study its biological and long-term effects (Appendix 1).
Case Presentation
A 65-year-old Vietnam-era veteran presented to nephrology at the Raymond G. Murphy Veterans Affairs Medical Center (RGMVAMC) in Albuquerque, New Mexico, for evaluation of gadolinium-induced symptoms. His medical history included metabolic syndrome, hypertension, hyperlipidemia, hypogonadism, cervical spondylosis, and an elevated prostate-specific antigen, previously assessed with a contrast-enhanced MRI in 2019 (Gadobenic acid, 19 mL). Surgical history included cervical fusion and ankle hardware.
The patient had a scheduled MRI 25 days earlier, following an elevated prostate specific antigen test result, prompting urologic surveillance and concern for malignancy. In preparation for the contrast-enhanced MRI, his right arm was cannulated with a line primed with gadobenic acid contrast. Though the technician stated the infusion had not started, the patient’s symptoms began shortly after entry into the scanner, before any programmed pulse sequences. The patient experienced claustrophobia, diaphoresis, palpitations, xerostomia, dysgeusia, shortness of breath, and a sensation of heat in his groin, chest, “kidneys,” and lower back. The MRI was terminated prematurely in response to the patient’s acute symptomatology. The patient continued experiencing new symptoms intermittently during the following week, including lightheadedness, headaches, right clavicular pain, raspy voice, edema, and a sense of doom.
The patient presented to the RGMVAMC emergency department (ED) 8 days after the MRI with worsening symptoms and was hospitalized for 10 days. During this time, he was referred to nephrology for outpatient evaluation. While awaiting his nephrology appointment, the patient presented to the RGMVAMC ED 20 days after the initial episode with ongoing symptoms. “I thought I was dying,” he said. Laboratory results and a 12-lead electrocardiogram showed a finely static background, wide P waves (> 80 ms) with notching in lead II, sinusoidal P waves in V1, R transition in V2, RR’ in V2, ST flat in lead III, and sinus bradycardia (Table 1 and Appendix 2).
The patient’s medical and surgical histories were reviewed at the nephrology evaluation 25 days following the MRI. He reported that household water was sourced from a well and that he filtered his drinking water with a reverse osmosis system. He served in the US Army for 10 years as an engineer specializing in mechanical systems, power generation, and vehicles. Following Army retirement, the patient served in the US Air Force Reserves for 15 years, working as a crew chief in pneudraulics. The patient reported stopping tobacco use 1 year before and also reported regular use of a broad array of prescription medications and dietary supplements, including dexamethasone (4 mg twice daily), fluticasone nasal spray (50 mcg per nostril, twice daily), ibuprofen (400 mg twice daily, as needed), loratadine (10 mg daily), aspirin (81 mg daily), and metoprolol succinate (50 mg nightly). In addition, he reported consistent use of cholecalciferol (3000 IU daily), another supplemental vitamin D preparation, chelated magnesium glycinate (3 tablets daily for bone issues), turmeric (1 tablet daily), a multivitamin (Living Green Liquid Gel, daily), and a mega-B complex.
Physical examination revealed a well-nourished, tall man with hypertension (145/87 mmHg) and bilateral lower extremity edema. Oral examination showed poor dentition, including missing molars (#1-3, #14-16, #17-19, #30-31), with the anterior teeth replaced by bridges supported by dental implants. The review of systems was otherwise unremarkable, with nocturia noted before the consultation.
Serum and urine gadolinium testing, (Mayo Clinic Laboratories) revealed gadolinium levels of 0.3 mcg/24 h in the urine and 0.1 ng/mL in the serum. Nonzero values indicated detectable gadolinium, suggesting retention. The patient had a prior gadolinium exposure during a 2019 MRI (about 1340 days before) and suspected a repeat exposure on day 0, although the MRI technician stated that no contrast was administered. Given his elevated vitamin D levels, the patient was advised to minimize dietary supplements, particularly vitamin D, to avoid confounding symptoms. The plan included monitoring symptoms and a follow-up evaluation with repeat laboratory tests on day 116.
At the nephrology follow-up 4 months postexposure, the patient's symptoms had primarily abated, with a marked reduction in the previously noted metallic dysgeusia. Physical examination remained consistent with prior findings. He was afebrile (97.7 °F) with a blood pressure of 111/72 mmHg, a pulse of 63 beats per minute, and an oxygen saturation of 98% on ambient air. Laboratory analysis revealed serum and urine gadolinium levels below detectable thresholds (< 0.1 ng/mL and < 0.1 mcg/24 h). A 24-hour creatinine clearance, calculated from a urine volume of 1300 mL, measured at an optimal 106 mL/min, indicating preserved renal function (Tables 2 and 3). Of note, his 24-hour oxalate was above the reference range, with a urine pH below the reference range and a high supersaturation index for calcium oxalate.
Discussion
Use of enhanced MRI has increased in the Veterans Health Administration (Figure 2). A growing range of indications for enhanced procedures (eg, cardiac MRI) has contributed to this rise. The market has grown with new gadolinium-based contrast agents, such as gadopiclenol. However, reliance on untested assumptions about the safety of newer agents and need for robust clinical trials pose potential risks to patient safety.
Without prospective evidence, the American College of Radiology (ACR) classifies gadolinium-based contrast agents into 3 groups: Group 1, associated with the highest number of nephrogenic systemic fibrosis cases; Group 2, linked to few, if any, unconfounded cases; and Group 3, where data on nephrogenic systemic fibrosis risk have been limited. As of April 2024, the ACR reclassified Group 3 agents (Ablavar/Vasovist/Angiomark and Primovist/Eovist) into Group 2. Curiously, Vueway and Elucirem were approved in late 2022 and should clearly be categorized as Group 3 (Table 4).There were 19 cases of nephrogenic systemic fibrosis or similar manifestations, 8 of which were unconfounded by other factors. These patients had been exposed to gadobutrol, often combined with other agents. Gadobutrol—like other Group 2 agents—has been associated with nephrogenic systemic fibrosis.16,17 Despite US Food and Drug Administration (FDA) documentation of rising reports, many clinicians remain unaware that nephrogenic systemic fibrosis is increasingly linked to Group 2 agents classified by the ACR.18 While declines in reported cases of nephrogenic systemic fibrosis may suggest reduced incidence, this trend may reflect diminished clinical vigilance and underreporting, particularly given emerging evidence implicating even Group 2 gadolinium-based contrast agents in delayed and underrecognized presentations. This information has yet to permeate the medical community, particularly among nephrologists. Considering these cases, revisiting the ACR guidelines may be prudent.
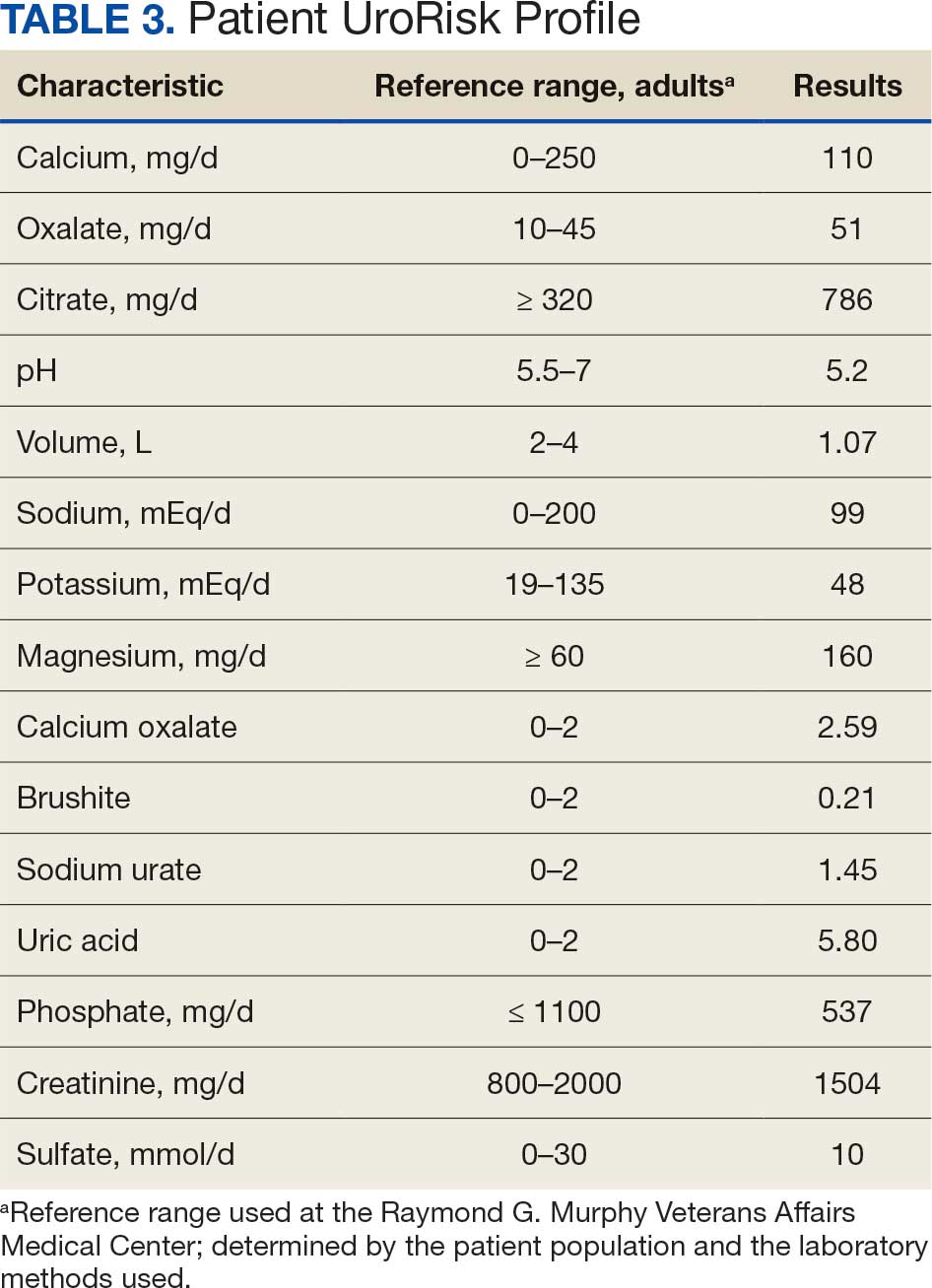
To address this growing concern, clinicians must adopt stricter vigilance and actively pursue updated information to mitigate patient risks tied to these contrast agents.
There exists an illusion of knowledge in disregarding the confounded exposures of MRI contrast agents. Ten distinct brands of contrast agents have been approved for clinical use. With repeated imaging, patients are often exposed to varying formulations of gadolinium-based agents. Yet investigators commonly discard these data points when assessing risk. By doing so, they assume—without evidence—that some formulations are inherently less likely to provoke adverse effects (AEs) than others. This untested presumption becomes perilous, especially given the limited understanding of the mechanisms underlying gadolinium-induced pathologies. As Aldous Huxley warned, “Facts do not cease to exist because they are ignored.”19
Gadolinium Persistence
Contrary to expectations, gadolinium persists in the body far longer than initially presumed. Symptoms associated with gadolinium exposure (SAGE) encapsulate the chronic, often enigmatic maladies tied to MRI contrast agents.20 The prolonged retention of this rare earth metal offers a compelling hypothesis for the etiology of SAGE. It has been hypothesized that Lewis base-rich metabolites increase susceptibility to gadolinium-based contrast agent complications.21
The blood and urine concentration elimination curves of gadolinium are exponential and categorized as fast, intermediate, and long-term.1 For urinary elimination, the function of the curves is exponential. The quantity of gadolinium in the urine at a time (t) after exposure (D[Gd](t)) is equal to the product of the amount of gadolinium in the sample (urine or blood) at the end of the fast elimination period (D[Gd](t0)) and the exponential decay with k being a rate constant.
To the authors’ knowledge, we are the only research team currently investigating the rate constant for the intermediate- and long-term phase gadolinium elimination. The Retention and Toxicity of Gadolinium-based Contrast Agents study was approved by the University of New Mexico Health Sciences Center Institutional Review Board on May 27, 2020 (IRB ID 19-660). The data for the patient in this case were compared with preliminary results for patients with exposure-to-measurement intervals < 100 days.
The patient in this case presented with detectable gadolinium levels in urine and serum shortly after an attempted contrast-enhanced MRI procedure (Figure 3). The presence of detectable gadolinium levels in the patient’s urine and serum suggests a likely exposure to a contrast agent about 27 days before his consultation. While the technician reported that no contrast was administered during the attempted MRI, it remains possible that a small amount was introduced during cannulation, potentially triggering the patient’s symptoms. Linear modeling of semilogarithmic plots for participants exposed to contrast agents within 100 days (urine: P = 1.8 × 10ˉ8, adjusted r² = 0.62; blood: P = .005, adjusted r² = 0.21) provided clearance rates (k values) for urine and blood. Extrapolating from these models to the presumed exposure date, the intercepts estimate that the patient received between 0.5% and 8% of a standard contrast dose.

MRI contrast agents can cause skin disease. Systemic fibrosis is considered one of the most severe AEs. Skin pathophysiology involving myeloid cells is driven by elevated levels of monocyte chemoattractant protein-1, which recruits circulating fibroblasts via the C-C chemokine receptor 2.22,23 This occurs alongside activation of NADPH oxidase Nox4.4,24,25 Intracellular gadolinium-rich nanoparticles likely serve as catalysts for this reactive cascade.2,18,22,26,27 These particles assemble around intracellular lipid droplets and ferrule them in spiculated rare earth-rich shells that compromise cellular architecture.2,18,21,22,26,27 Frequently sequestered within endosomal compartments, they disrupt vesicular integrity and threaten cellular homeostasis. Interference with degradative systems such as the endolysosomal axis perturbs energy-recycling pathways—an insidious disturbance, particularly in cells with high metabolic demand. Skin-related symptoms are among the most frequently reported AEs, according to the FDA AE reporting system.18
Studies indicate repeated exposure to MRI contrast agents can lead to permanent gadolinium retention in the brain and other vital organs. Intravenous (IV) contrast agents cross the blood-brain barrier rapidly, while intrathecal administration has been linked to significant and lasting neurologic effects.18
Gadolinium is chemically bound to pharmaceutical ligands to enhance renal clearance and reduce toxicity. However, available data from human samples suggest potential ligand exchanges with undefined physiologic substances. This exchange may facilitate gadolinium precipitation and accumulation within cells into spiculated nanoparticles. Transmission electron microscopy reveals the formation of unilamellar bodies associated with mitochondriopathy and cellular damage, particularly in renal proximal tubules.2,18,22,26,27 It is proposed that intracellular nanoparticle formation represents a key mechanism driving the systemic symptoms observed in patients.1,2,18, 22,26,27
Any hypothesis based on free soluble gadolinium—or concept derived from it—should be discarded. The high affinity of pharmaceutical ligands for gadolinium suggests that the cationic rare earth metal remains predominantly in a ligand-bound, soluble form. It is hypothesized that gadolinium undergoes ligand exchange with physiologic substances, directly leading to nanoparticle formation. Current data demonstrate gadolinium precipitation according to the Le Chatelier’s principle. Since precipitated gadolinium does not readily re-equilibrate with pharmaceutical ligands, repeated administration of different contrast agent brands may contribute to nanoparticle growth.26
Meanwhile, a growing number of patients are turning to chelation therapy, a largely untested treatment. The premise of chelation therapy is rooted in several unproven assumptions.18,21 First, it assumes that clinically significant amounts of gadolinium persist in compartments such as the extracellular space, where they can be effectively chelated and cleared. Second, it presumes that free gadolinium is the primary driver of chronic symptoms, an assertion that remains scientifically unsubstantiated. Finally, chelation proponents overlook the potential harm caused by depleting essential physiological metals during the process, assuming without evidence that the scant removal of gadolinium outweighs the risk of physiological mineral depletion.
These assumptions underpin an unproven remedy that demands critical scrutiny. Recent findings reveal that gadolinium deposits in the skin and kidney often take the form of intracellular nanoparticles, directly challenging the foundation of chelation therapy. Chelation advocates must demonstrate that these intracellular gadolinium deposits neither trigger cellular toxicity nor initiate a cytokine cascade. Chelation supporters must prove that the systemic response to these foreign particles is unrelated to the symptoms reported by patients. Until then, the validity of chelation therapy remains highly questionable.
The causality of the symptoms, mainly whether IV gadolinium was administered, was examined. The null hypothesis stated that the patient was not exposed to gadolinium. However, this hypothesis was contradicted by the detection of gadolinium in the serum and urine 27 days after the potential exposure.
Two plausible explanations exist for the nonzero gadolinium levels detected in the serum and urine. The first possibility is that minute quantities of gadolinium were introduced during cannulation, with the amount being sufficient to persist in measurable concentrations 27 days postexposure. The second possibility is that the gadolinium originated from an MRI contrast agent administered 4 years earlier. In this scenario, gadolinium stored in organ reservoirs such as bone, liver, or kidneys may have been mobilized into the extracellular fluid compartment due to the administration of high-dose steroids 20 days after the recent contrast-enhanced MRI procedure attempt. Coyte et al reported elevated gadolinium levels in the serum, cord blood, breast milk, and placenta of pregnant women with prior exposure to MRI contrast agents.28 These findings suggest that gadolinium, stored in organs such as bone may be remobilized by variables affecting bone remodeling (eg, high-dose steroids).
Significantly, the patient exhibited elevated urinary oxalate levels. Previous research has found that oxalic acid reacts rapidly with MRI contrast agents, forming digadolinium trioxalate. While the gadolinium-rich nanoparticles identified in tissues such as the skin and kidney (including the human kidney) are amorphous, these in vitro findings establish a proof-of-concept: the intracellular environment facilitates gadolinium dissociation from pharmaceutical chelates.
Furthermore, in vitro experiments show that proteins and lysosomal pH promote this dissociation, underscoring how human metabolic conditions—particularly oxalic acid concentration—may drive intracellular gadolinium deposition.
Patient Perspective
“They put something into my body that they cannot get out.” This stark realization underpins the patient’s profound concern about gadolinium-based contrast agents and their potential long-term effects. Reflecting on his experience, the patient expressed deep fears about the unknown future impacts: “I’m concerned about my kidneys, I’m concerned about my heart, and I’m concerned about my brain. I don’t know how this stuff is going to affect me in the future.”
He drew an unsettling parallel between gadolinium and heavy metals: “Heavy metal is poison. The body does not produce this kind of stuff on its own.” His reaction to the procedure left a lasting impression, prompting him to question the logic of using a substance that cannot be purged: “Why would you put something into someone’s body that you cannot extract? Nobody—nobody—should experience what I went through.”
The patient emphasized the lack of clear research on long-term outcomes, which compounds his anxiety: “If there was research that said, ‘Well, this is only going to affect these organs for this long,’ OK, I might be able to accept that. But there is no research like that. Nobody can tell me what’s going to happen in 5 years.”
Strengths and Limitations
A significant strength of this approach is the ability to track gadolinium elimination and symptom resolution over time, supported by unique access to intermediate and long-term clearance data from our ongoing research protocol. The investigators were equipped to back-extrapolate the exposure, which provided a rare opportunity to correlate gadolinium levels with clinical outcomes. The primary limitation is the lack of a defined clinical case definition for gadolinium toxicity and limited mechanistic understanding of SAGE, which hinders diagnosis and management.
Metabolites, proteins, and lipids rich in Lewis bases could initiate this process as substrates for intracellular gadolinium sedimentation. Future studies should investigate whether metabolic conditions such as oxalate burden or altered parathyroid hormone levels modulate gadolinium compartmentalization and tissue retention. If gadolinium-rich nanoparticle formation and accumulation disrupt cellular equilibrium, it underscores an urgent need to understand the implications of long-term gadolinium retention. The research team continues to gather evidence that the gadolinium cation remains chelated from the moment MRI contrast agents are administered through to the formation of intracellular nanoparticles. Retained gadolinium nanoparticles may act as a nidus, triggering cellular signaling cascades that lead to multisymptomatic illnesses. Intracellular and insoluble retained gadolinium challenges proponents of untested chelation therapies.
Conclusions
This case highlights emerging clinical and ethical concerns surrounding gadolinium-based contrast agent use. Clinicians may benefit from considering gadolinium retention as a contributor to persistent, unexplained symptoms—particularly in patients with recent imaging exposure. As contrast use continues to rise within federal health systems, regulatory and administrative stakeholders would do well to re-examine current safety frameworks. Informed consent should reflect what is known: gadolinium can remain in the body long after administration, potentially indefinitely. The long-term consequences of cumulative exposure remain poorly defined, but the presence of a lanthanide element in human tissue warrants greater attention from researchers and regulators alike. Interest in alternative imaging modalities and long-term safety monitoring would mark progress toward more transparent, accountable care.
Jackson DB, MacIntyre T, Duarte-Miramontes V, et al. Gadolinium deposition disease: a case report and the prevalence of enhanced MRI procedures within the Veterans Health Administration. Fed Pract. 2022;39:218-225. doi:10.12788/fp.0258
Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1:561-568.doi:10.34067/kid.0000272019
Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28:154-162. doi:10.1097/MNH.0000000000000475
Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311:F1-F11. doi:10.1152/ajprenal.00166.2016
Maramattom BV, Manno EM, Wijdicks EF, et al. Gadolinium encephalopathy in a patient with renal failure. Neurology. 2005;64:1276-1278.doi:10.1212/01.WNL.0000156805.45547.6E
Sam AD II, Morasch MD, Collins J, et al. Safety of gadolinium contrast angiography in patients with chronic renal insufficiency. J Vasc Surg. 2003;38:313-318. doi:10.1016/s0741-5214(03)00315-x
Schenker MP, Solomon JA, Roberts DA. Gadolinium arteriography complicated by acute pancreatitis and acute renal failure. J Vasc Interv Radiol. 2001;12:393. doi:10.1016/s1051-0443(07)61925-3
Gemery J, Idelson B, Reid S, et al. Acute renal failure after arteriography with a gadolinium-based contrast agent. AJR Am J Roentgenol. 1998;171:1277-1278. doi:10.2214/ajr.171.5.9798860
Akgun H, Gonlusen G, Cartwright J Jr, et al. Are gadolinium-based contrast media nephrotoxic? A renal biopsy study. Arch Pathol Lab Med. 2006;130:1354-1357. doi:10.5858/2006-130-1354-AGCMNA
Gathings RM, Reddy R, Santa Cruz D, et al. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151:316-319. doi:10.1001/jamadermatol.2014.2660
McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
Kanda T, Ishii K, Kawaguchi H, et al. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270(3):834-841. doi:10.1148/radiol.13131669
Schmidt K, Bau M, Merschel G, et al. Anthropogenic gadolinium in tap water and in tap water-based beverages from fast-food franchises in six major cities in Germany. Sci Total Environ. 2019;687:1401-1408. doi:10.1016/j.scitotenv.2019.07.075
Kulaksız S, Bau M. Anthropogenic gadolinium as a microcontaminant in tap water used as drinking water in urban areas and megacities. Appl Geochem. 2011;26:1877-1885.
Brunjes R, Hofmann T. Anthropogenic gadolinium in freshwater and drinking water systems. Water Res. 2020;182:115966. doi:10.1016/j.watres.2020.115966
Endrikat J, Gutberlet M, Hoffmann KT, et al. Clinical safety of gadobutrol: review of over 25 years of use exceeding 100 million administrations. Invest Radiol. 2024;59(9):605-613. doi:10.1097/RLI.0000000000001072
Elmholdt TR, Jørgensen B, Ramsing M, et al. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287. doi:10.1093/ndtplus/sfq028
Cunningham A, Kirk M, Hong E, et al. The safety of magnetic resonance imaging contrast agents. Front Toxicol. 2024;6:1376587. doi:10.3389/ftox.2024.1376587
Huxley A. Complete Essays. Volume II, 1926-1929. Chicago; 2000:227.
McDonald RJ, Weinreb JC, Davenport MS. Symptoms associated with gadolinium exposure (SAGE): a suggested term. Radiology. 2022;302(2):270-273. doi:10.1148/radiol.2021211349
Henderson IM, Benevidez AD, Mowry CD, et al. Precipitation of gadolinium from magnetic resonance imaging contrast agents may be the Brass tacks of toxicity. Magn Reson Imaging. 2025;119:110383. doi:10.1016/j.mri.2025.110383
Do C, Drel V, Tan C, et al. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143. doi:10.1016/j.jid.2019.03.1145
Drel VR, Tan C, Barnes JL, et al. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH oxidase 4 for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
DeAguero J, Howard T, Kusewitt D, et al. The onset of rare earth metallosis begins with renal gadolinium-rich nanoparticles from magnetic resonance imaging contrast agent exposure. Sci Rep. 2023;13(1):2025. doi:10.1038/s41598-023-28666-1
Do C, Ford B, Lee DY, et al. Gadolinium-based contrast agents: Stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
Coyte RM, Darrah T, Olesik J, et al. Gadolinium during human pregnancy following administration of gadolinium chelate before pregnancy. Birth Defects Res. 2023;115(14):1264-1273. doi:10.1002/bdr2.2209
Magnetic resonance image (MRI) contrast agents can induce profound complications, including gadolinium encephalopathy, kidney injury, gadolinium-associated plaques, and progressive systemic fibrosis, which can be fatal.1-10 About 50% of MRIs use gadolinium-based contrast (Gd3+), a toxic rare earth metal ion that enhances imaging but requires binding with pharmaceutical ligands to reduce toxicity and promote renal elimination (Figure 1). Despite these measures, Gd3+ can persist in the body, including the brain.11,12 Wastewater treatment fails to remove these agents, making Gd3+ a growing pollutant in water and the food chain.13-15 Because Gd3+ is a rare earth metal ion in the milieu intérieur, there is an urgent need to study its biological and long-term effects (Appendix 1).
Case Presentation
A 65-year-old Vietnam-era veteran presented to nephrology at the Raymond G. Murphy Veterans Affairs Medical Center (RGMVAMC) in Albuquerque, New Mexico, for evaluation of gadolinium-induced symptoms. His medical history included metabolic syndrome, hypertension, hyperlipidemia, hypogonadism, cervical spondylosis, and an elevated prostate-specific antigen, previously assessed with a contrast-enhanced MRI in 2019 (Gadobenic acid, 19 mL). Surgical history included cervical fusion and ankle hardware.
The patient had a scheduled MRI 25 days earlier, following an elevated prostate specific antigen test result, prompting urologic surveillance and concern for malignancy. In preparation for the contrast-enhanced MRI, his right arm was cannulated with a line primed with gadobenic acid contrast. Though the technician stated the infusion had not started, the patient’s symptoms began shortly after entry into the scanner, before any programmed pulse sequences. The patient experienced claustrophobia, diaphoresis, palpitations, xerostomia, dysgeusia, shortness of breath, and a sensation of heat in his groin, chest, “kidneys,” and lower back. The MRI was terminated prematurely in response to the patient’s acute symptomatology. The patient continued experiencing new symptoms intermittently during the following week, including lightheadedness, headaches, right clavicular pain, raspy voice, edema, and a sense of doom.
The patient presented to the RGMVAMC emergency department (ED) 8 days after the MRI with worsening symptoms and was hospitalized for 10 days. During this time, he was referred to nephrology for outpatient evaluation. While awaiting his nephrology appointment, the patient presented to the RGMVAMC ED 20 days after the initial episode with ongoing symptoms. “I thought I was dying,” he said. Laboratory results and a 12-lead electrocardiogram showed a finely static background, wide P waves (> 80 ms) with notching in lead II, sinusoidal P waves in V1, R transition in V2, RR’ in V2, ST flat in lead III, and sinus bradycardia (Table 1 and Appendix 2).
The patient’s medical and surgical histories were reviewed at the nephrology evaluation 25 days following the MRI. He reported that household water was sourced from a well and that he filtered his drinking water with a reverse osmosis system. He served in the US Army for 10 years as an engineer specializing in mechanical systems, power generation, and vehicles. Following Army retirement, the patient served in the US Air Force Reserves for 15 years, working as a crew chief in pneudraulics. The patient reported stopping tobacco use 1 year before and also reported regular use of a broad array of prescription medications and dietary supplements, including dexamethasone (4 mg twice daily), fluticasone nasal spray (50 mcg per nostril, twice daily), ibuprofen (400 mg twice daily, as needed), loratadine (10 mg daily), aspirin (81 mg daily), and metoprolol succinate (50 mg nightly). In addition, he reported consistent use of cholecalciferol (3000 IU daily), another supplemental vitamin D preparation, chelated magnesium glycinate (3 tablets daily for bone issues), turmeric (1 tablet daily), a multivitamin (Living Green Liquid Gel, daily), and a mega-B complex.
Physical examination revealed a well-nourished, tall man with hypertension (145/87 mmHg) and bilateral lower extremity edema. Oral examination showed poor dentition, including missing molars (#1-3, #14-16, #17-19, #30-31), with the anterior teeth replaced by bridges supported by dental implants. The review of systems was otherwise unremarkable, with nocturia noted before the consultation.
Serum and urine gadolinium testing, (Mayo Clinic Laboratories) revealed gadolinium levels of 0.3 mcg/24 h in the urine and 0.1 ng/mL in the serum. Nonzero values indicated detectable gadolinium, suggesting retention. The patient had a prior gadolinium exposure during a 2019 MRI (about 1340 days before) and suspected a repeat exposure on day 0, although the MRI technician stated that no contrast was administered. Given his elevated vitamin D levels, the patient was advised to minimize dietary supplements, particularly vitamin D, to avoid confounding symptoms. The plan included monitoring symptoms and a follow-up evaluation with repeat laboratory tests on day 116.
At the nephrology follow-up 4 months postexposure, the patient's symptoms had primarily abated, with a marked reduction in the previously noted metallic dysgeusia. Physical examination remained consistent with prior findings. He was afebrile (97.7 °F) with a blood pressure of 111/72 mmHg, a pulse of 63 beats per minute, and an oxygen saturation of 98% on ambient air. Laboratory analysis revealed serum and urine gadolinium levels below detectable thresholds (< 0.1 ng/mL and < 0.1 mcg/24 h). A 24-hour creatinine clearance, calculated from a urine volume of 1300 mL, measured at an optimal 106 mL/min, indicating preserved renal function (Tables 2 and 3). Of note, his 24-hour oxalate was above the reference range, with a urine pH below the reference range and a high supersaturation index for calcium oxalate.
Discussion
Use of enhanced MRI has increased in the Veterans Health Administration (Figure 2). A growing range of indications for enhanced procedures (eg, cardiac MRI) has contributed to this rise. The market has grown with new gadolinium-based contrast agents, such as gadopiclenol. However, reliance on untested assumptions about the safety of newer agents and need for robust clinical trials pose potential risks to patient safety.
Without prospective evidence, the American College of Radiology (ACR) classifies gadolinium-based contrast agents into 3 groups: Group 1, associated with the highest number of nephrogenic systemic fibrosis cases; Group 2, linked to few, if any, unconfounded cases; and Group 3, where data on nephrogenic systemic fibrosis risk have been limited. As of April 2024, the ACR reclassified Group 3 agents (Ablavar/Vasovist/Angiomark and Primovist/Eovist) into Group 2. Curiously, Vueway and Elucirem were approved in late 2022 and should clearly be categorized as Group 3 (Table 4).There were 19 cases of nephrogenic systemic fibrosis or similar manifestations, 8 of which were unconfounded by other factors. These patients had been exposed to gadobutrol, often combined with other agents. Gadobutrol—like other Group 2 agents—has been associated with nephrogenic systemic fibrosis.16,17 Despite US Food and Drug Administration (FDA) documentation of rising reports, many clinicians remain unaware that nephrogenic systemic fibrosis is increasingly linked to Group 2 agents classified by the ACR.18 While declines in reported cases of nephrogenic systemic fibrosis may suggest reduced incidence, this trend may reflect diminished clinical vigilance and underreporting, particularly given emerging evidence implicating even Group 2 gadolinium-based contrast agents in delayed and underrecognized presentations. This information has yet to permeate the medical community, particularly among nephrologists. Considering these cases, revisiting the ACR guidelines may be prudent.
To address this growing concern, clinicians must adopt stricter vigilance and actively pursue updated information to mitigate patient risks tied to these contrast agents.
There exists an illusion of knowledge in disregarding the confounded exposures of MRI contrast agents. Ten distinct brands of contrast agents have been approved for clinical use. With repeated imaging, patients are often exposed to varying formulations of gadolinium-based agents. Yet investigators commonly discard these data points when assessing risk. By doing so, they assume—without evidence—that some formulations are inherently less likely to provoke adverse effects (AEs) than others. This untested presumption becomes perilous, especially given the limited understanding of the mechanisms underlying gadolinium-induced pathologies. As Aldous Huxley warned, “Facts do not cease to exist because they are ignored.”19
Gadolinium Persistence
Contrary to expectations, gadolinium persists in the body far longer than initially presumed. Symptoms associated with gadolinium exposure (SAGE) encapsulate the chronic, often enigmatic maladies tied to MRI contrast agents.20 The prolonged retention of this rare earth metal offers a compelling hypothesis for the etiology of SAGE. It has been hypothesized that Lewis base-rich metabolites increase susceptibility to gadolinium-based contrast agent complications.21
The blood and urine concentration elimination curves of gadolinium are exponential and categorized as fast, intermediate, and long-term.1 For urinary elimination, the function of the curves is exponential. The quantity of gadolinium in the urine at a time (t) after exposure (D[Gd](t)) is equal to the product of the amount of gadolinium in the sample (urine or blood) at the end of the fast elimination period (D[Gd](t0)) and the exponential decay with k being a rate constant.
To the authors’ knowledge, we are the only research team currently investigating the rate constant for the intermediate- and long-term phase gadolinium elimination. The Retention and Toxicity of Gadolinium-based Contrast Agents study was approved by the University of New Mexico Health Sciences Center Institutional Review Board on May 27, 2020 (IRB ID 19-660). The data for the patient in this case were compared with preliminary results for patients with exposure-to-measurement intervals < 100 days.
The patient in this case presented with detectable gadolinium levels in urine and serum shortly after an attempted contrast-enhanced MRI procedure (Figure 3). The presence of detectable gadolinium levels in the patient’s urine and serum suggests a likely exposure to a contrast agent about 27 days before his consultation. While the technician reported that no contrast was administered during the attempted MRI, it remains possible that a small amount was introduced during cannulation, potentially triggering the patient’s symptoms. Linear modeling of semilogarithmic plots for participants exposed to contrast agents within 100 days (urine: P = 1.8 × 10ˉ8, adjusted r² = 0.62; blood: P = .005, adjusted r² = 0.21) provided clearance rates (k values) for urine and blood. Extrapolating from these models to the presumed exposure date, the intercepts estimate that the patient received between 0.5% and 8% of a standard contrast dose.
MRI contrast agents can cause skin disease. Systemic fibrosis is considered one of the most severe AEs. Skin pathophysiology involving myeloid cells is driven by elevated levels of monocyte chemoattractant protein-1, which recruits circulating fibroblasts via the C-C chemokine receptor 2.22,23 This occurs alongside activation of NADPH oxidase Nox4.4,24,25 Intracellular gadolinium-rich nanoparticles likely serve as catalysts for this reactive cascade.2,18,22,26,27 These particles assemble around intracellular lipid droplets and ferrule them in spiculated rare earth-rich shells that compromise cellular architecture.2,18,21,22,26,27 Frequently sequestered within endosomal compartments, they disrupt vesicular integrity and threaten cellular homeostasis. Interference with degradative systems such as the endolysosomal axis perturbs energy-recycling pathways—an insidious disturbance, particularly in cells with high metabolic demand. Skin-related symptoms are among the most frequently reported AEs, according to the FDA AE reporting system.18
Studies indicate repeated exposure to MRI contrast agents can lead to permanent gadolinium retention in the brain and other vital organs. Intravenous (IV) contrast agents cross the blood-brain barrier rapidly, while intrathecal administration has been linked to significant and lasting neurologic effects.18
Gadolinium is chemically bound to pharmaceutical ligands to enhance renal clearance and reduce toxicity. However, available data from human samples suggest potential ligand exchanges with undefined physiologic substances. This exchange may facilitate gadolinium precipitation and accumulation within cells into spiculated nanoparticles. Transmission electron microscopy reveals the formation of unilamellar bodies associated with mitochondriopathy and cellular damage, particularly in renal proximal tubules.2,18,22,26,27 It is proposed that intracellular nanoparticle formation represents a key mechanism driving the systemic symptoms observed in patients.1,2,18, 22,26,27
Any hypothesis based on free soluble gadolinium—or concept derived from it—should be discarded. The high affinity of pharmaceutical ligands for gadolinium suggests that the cationic rare earth metal remains predominantly in a ligand-bound, soluble form. It is hypothesized that gadolinium undergoes ligand exchange with physiologic substances, directly leading to nanoparticle formation. Current data demonstrate gadolinium precipitation according to the Le Chatelier’s principle. Since precipitated gadolinium does not readily re-equilibrate with pharmaceutical ligands, repeated administration of different contrast agent brands may contribute to nanoparticle growth.26
Meanwhile, a growing number of patients are turning to chelation therapy, a largely untested treatment. The premise of chelation therapy is rooted in several unproven assumptions.18,21 First, it assumes that clinically significant amounts of gadolinium persist in compartments such as the extracellular space, where they can be effectively chelated and cleared. Second, it presumes that free gadolinium is the primary driver of chronic symptoms, an assertion that remains scientifically unsubstantiated. Finally, chelation proponents overlook the potential harm caused by depleting essential physiological metals during the process, assuming without evidence that the scant removal of gadolinium outweighs the risk of physiological mineral depletion.
These assumptions underpin an unproven remedy that demands critical scrutiny. Recent findings reveal that gadolinium deposits in the skin and kidney often take the form of intracellular nanoparticles, directly challenging the foundation of chelation therapy. Chelation advocates must demonstrate that these intracellular gadolinium deposits neither trigger cellular toxicity nor initiate a cytokine cascade. Chelation supporters must prove that the systemic response to these foreign particles is unrelated to the symptoms reported by patients. Until then, the validity of chelation therapy remains highly questionable.
The causality of the symptoms, mainly whether IV gadolinium was administered, was examined. The null hypothesis stated that the patient was not exposed to gadolinium. However, this hypothesis was contradicted by the detection of gadolinium in the serum and urine 27 days after the potential exposure.
Two plausible explanations exist for the nonzero gadolinium levels detected in the serum and urine. The first possibility is that minute quantities of gadolinium were introduced during cannulation, with the amount being sufficient to persist in measurable concentrations 27 days postexposure. The second possibility is that the gadolinium originated from an MRI contrast agent administered 4 years earlier. In this scenario, gadolinium stored in organ reservoirs such as bone, liver, or kidneys may have been mobilized into the extracellular fluid compartment due to the administration of high-dose steroids 20 days after the recent contrast-enhanced MRI procedure attempt. Coyte et al reported elevated gadolinium levels in the serum, cord blood, breast milk, and placenta of pregnant women with prior exposure to MRI contrast agents.28 These findings suggest that gadolinium, stored in organs such as bone may be remobilized by variables affecting bone remodeling (eg, high-dose steroids).
Significantly, the patient exhibited elevated urinary oxalate levels. Previous research has found that oxalic acid reacts rapidly with MRI contrast agents, forming digadolinium trioxalate. While the gadolinium-rich nanoparticles identified in tissues such as the skin and kidney (including the human kidney) are amorphous, these in vitro findings establish a proof-of-concept: the intracellular environment facilitates gadolinium dissociation from pharmaceutical chelates.
Furthermore, in vitro experiments show that proteins and lysosomal pH promote this dissociation, underscoring how human metabolic conditions—particularly oxalic acid concentration—may drive intracellular gadolinium deposition.
Patient Perspective
“They put something into my body that they cannot get out.” This stark realization underpins the patient’s profound concern about gadolinium-based contrast agents and their potential long-term effects. Reflecting on his experience, the patient expressed deep fears about the unknown future impacts: “I’m concerned about my kidneys, I’m concerned about my heart, and I’m concerned about my brain. I don’t know how this stuff is going to affect me in the future.”
He drew an unsettling parallel between gadolinium and heavy metals: “Heavy metal is poison. The body does not produce this kind of stuff on its own.” His reaction to the procedure left a lasting impression, prompting him to question the logic of using a substance that cannot be purged: “Why would you put something into someone’s body that you cannot extract? Nobody—nobody—should experience what I went through.”
The patient emphasized the lack of clear research on long-term outcomes, which compounds his anxiety: “If there was research that said, ‘Well, this is only going to affect these organs for this long,’ OK, I might be able to accept that. But there is no research like that. Nobody can tell me what’s going to happen in 5 years.”
Strengths and Limitations
A significant strength of this approach is the ability to track gadolinium elimination and symptom resolution over time, supported by unique access to intermediate and long-term clearance data from our ongoing research protocol. The investigators were equipped to back-extrapolate the exposure, which provided a rare opportunity to correlate gadolinium levels with clinical outcomes. The primary limitation is the lack of a defined clinical case definition for gadolinium toxicity and limited mechanistic understanding of SAGE, which hinders diagnosis and management.
Metabolites, proteins, and lipids rich in Lewis bases could initiate this process as substrates for intracellular gadolinium sedimentation. Future studies should investigate whether metabolic conditions such as oxalate burden or altered parathyroid hormone levels modulate gadolinium compartmentalization and tissue retention. If gadolinium-rich nanoparticle formation and accumulation disrupt cellular equilibrium, it underscores an urgent need to understand the implications of long-term gadolinium retention. The research team continues to gather evidence that the gadolinium cation remains chelated from the moment MRI contrast agents are administered through to the formation of intracellular nanoparticles. Retained gadolinium nanoparticles may act as a nidus, triggering cellular signaling cascades that lead to multisymptomatic illnesses. Intracellular and insoluble retained gadolinium challenges proponents of untested chelation therapies.
Conclusions
This case highlights emerging clinical and ethical concerns surrounding gadolinium-based contrast agent use. Clinicians may benefit from considering gadolinium retention as a contributor to persistent, unexplained symptoms—particularly in patients with recent imaging exposure. As contrast use continues to rise within federal health systems, regulatory and administrative stakeholders would do well to re-examine current safety frameworks. Informed consent should reflect what is known: gadolinium can remain in the body long after administration, potentially indefinitely. The long-term consequences of cumulative exposure remain poorly defined, but the presence of a lanthanide element in human tissue warrants greater attention from researchers and regulators alike. Interest in alternative imaging modalities and long-term safety monitoring would mark progress toward more transparent, accountable care.
Magnetic resonance image (MRI) contrast agents can induce profound complications, including gadolinium encephalopathy, kidney injury, gadolinium-associated plaques, and progressive systemic fibrosis, which can be fatal.1-10 About 50% of MRIs use gadolinium-based contrast (Gd3+), a toxic rare earth metal ion that enhances imaging but requires binding with pharmaceutical ligands to reduce toxicity and promote renal elimination (Figure 1). Despite these measures, Gd3+ can persist in the body, including the brain.11,12 Wastewater treatment fails to remove these agents, making Gd3+ a growing pollutant in water and the food chain.13-15 Because Gd3+ is a rare earth metal ion in the milieu intérieur, there is an urgent need to study its biological and long-term effects (Appendix 1).
Case Presentation
A 65-year-old Vietnam-era veteran presented to nephrology at the Raymond G. Murphy Veterans Affairs Medical Center (RGMVAMC) in Albuquerque, New Mexico, for evaluation of gadolinium-induced symptoms. His medical history included metabolic syndrome, hypertension, hyperlipidemia, hypogonadism, cervical spondylosis, and an elevated prostate-specific antigen, previously assessed with a contrast-enhanced MRI in 2019 (Gadobenic acid, 19 mL). Surgical history included cervical fusion and ankle hardware.
The patient had a scheduled MRI 25 days earlier, following an elevated prostate specific antigen test result, prompting urologic surveillance and concern for malignancy. In preparation for the contrast-enhanced MRI, his right arm was cannulated with a line primed with gadobenic acid contrast. Though the technician stated the infusion had not started, the patient’s symptoms began shortly after entry into the scanner, before any programmed pulse sequences. The patient experienced claustrophobia, diaphoresis, palpitations, xerostomia, dysgeusia, shortness of breath, and a sensation of heat in his groin, chest, “kidneys,” and lower back. The MRI was terminated prematurely in response to the patient’s acute symptomatology. The patient continued experiencing new symptoms intermittently during the following week, including lightheadedness, headaches, right clavicular pain, raspy voice, edema, and a sense of doom.
The patient presented to the RGMVAMC emergency department (ED) 8 days after the MRI with worsening symptoms and was hospitalized for 10 days. During this time, he was referred to nephrology for outpatient evaluation. While awaiting his nephrology appointment, the patient presented to the RGMVAMC ED 20 days after the initial episode with ongoing symptoms. “I thought I was dying,” he said. Laboratory results and a 12-lead electrocardiogram showed a finely static background, wide P waves (> 80 ms) with notching in lead II, sinusoidal P waves in V1, R transition in V2, RR’ in V2, ST flat in lead III, and sinus bradycardia (Table 1 and Appendix 2).
The patient’s medical and surgical histories were reviewed at the nephrology evaluation 25 days following the MRI. He reported that household water was sourced from a well and that he filtered his drinking water with a reverse osmosis system. He served in the US Army for 10 years as an engineer specializing in mechanical systems, power generation, and vehicles. Following Army retirement, the patient served in the US Air Force Reserves for 15 years, working as a crew chief in pneudraulics. The patient reported stopping tobacco use 1 year before and also reported regular use of a broad array of prescription medications and dietary supplements, including dexamethasone (4 mg twice daily), fluticasone nasal spray (50 mcg per nostril, twice daily), ibuprofen (400 mg twice daily, as needed), loratadine (10 mg daily), aspirin (81 mg daily), and metoprolol succinate (50 mg nightly). In addition, he reported consistent use of cholecalciferol (3000 IU daily), another supplemental vitamin D preparation, chelated magnesium glycinate (3 tablets daily for bone issues), turmeric (1 tablet daily), a multivitamin (Living Green Liquid Gel, daily), and a mega-B complex.
Physical examination revealed a well-nourished, tall man with hypertension (145/87 mmHg) and bilateral lower extremity edema. Oral examination showed poor dentition, including missing molars (#1-3, #14-16, #17-19, #30-31), with the anterior teeth replaced by bridges supported by dental implants. The review of systems was otherwise unremarkable, with nocturia noted before the consultation.
Serum and urine gadolinium testing, (Mayo Clinic Laboratories) revealed gadolinium levels of 0.3 mcg/24 h in the urine and 0.1 ng/mL in the serum. Nonzero values indicated detectable gadolinium, suggesting retention. The patient had a prior gadolinium exposure during a 2019 MRI (about 1340 days before) and suspected a repeat exposure on day 0, although the MRI technician stated that no contrast was administered. Given his elevated vitamin D levels, the patient was advised to minimize dietary supplements, particularly vitamin D, to avoid confounding symptoms. The plan included monitoring symptoms and a follow-up evaluation with repeat laboratory tests on day 116.
At the nephrology follow-up 4 months postexposure, the patient's symptoms had primarily abated, with a marked reduction in the previously noted metallic dysgeusia. Physical examination remained consistent with prior findings. He was afebrile (97.7 °F) with a blood pressure of 111/72 mmHg, a pulse of 63 beats per minute, and an oxygen saturation of 98% on ambient air. Laboratory analysis revealed serum and urine gadolinium levels below detectable thresholds (< 0.1 ng/mL and < 0.1 mcg/24 h). A 24-hour creatinine clearance, calculated from a urine volume of 1300 mL, measured at an optimal 106 mL/min, indicating preserved renal function (Tables 2 and 3). Of note, his 24-hour oxalate was above the reference range, with a urine pH below the reference range and a high supersaturation index for calcium oxalate.
Discussion
Use of enhanced MRI has increased in the Veterans Health Administration (Figure 2). A growing range of indications for enhanced procedures (eg, cardiac MRI) has contributed to this rise. The market has grown with new gadolinium-based contrast agents, such as gadopiclenol. However, reliance on untested assumptions about the safety of newer agents and need for robust clinical trials pose potential risks to patient safety.
Without prospective evidence, the American College of Radiology (ACR) classifies gadolinium-based contrast agents into 3 groups: Group 1, associated with the highest number of nephrogenic systemic fibrosis cases; Group 2, linked to few, if any, unconfounded cases; and Group 3, where data on nephrogenic systemic fibrosis risk have been limited. As of April 2024, the ACR reclassified Group 3 agents (Ablavar/Vasovist/Angiomark and Primovist/Eovist) into Group 2. Curiously, Vueway and Elucirem were approved in late 2022 and should clearly be categorized as Group 3 (Table 4).There were 19 cases of nephrogenic systemic fibrosis or similar manifestations, 8 of which were unconfounded by other factors. These patients had been exposed to gadobutrol, often combined with other agents. Gadobutrol—like other Group 2 agents—has been associated with nephrogenic systemic fibrosis.16,17 Despite US Food and Drug Administration (FDA) documentation of rising reports, many clinicians remain unaware that nephrogenic systemic fibrosis is increasingly linked to Group 2 agents classified by the ACR.18 While declines in reported cases of nephrogenic systemic fibrosis may suggest reduced incidence, this trend may reflect diminished clinical vigilance and underreporting, particularly given emerging evidence implicating even Group 2 gadolinium-based contrast agents in delayed and underrecognized presentations. This information has yet to permeate the medical community, particularly among nephrologists. Considering these cases, revisiting the ACR guidelines may be prudent.
To address this growing concern, clinicians must adopt stricter vigilance and actively pursue updated information to mitigate patient risks tied to these contrast agents.
There exists an illusion of knowledge in disregarding the confounded exposures of MRI contrast agents. Ten distinct brands of contrast agents have been approved for clinical use. With repeated imaging, patients are often exposed to varying formulations of gadolinium-based agents. Yet investigators commonly discard these data points when assessing risk. By doing so, they assume—without evidence—that some formulations are inherently less likely to provoke adverse effects (AEs) than others. This untested presumption becomes perilous, especially given the limited understanding of the mechanisms underlying gadolinium-induced pathologies. As Aldous Huxley warned, “Facts do not cease to exist because they are ignored.”19
Gadolinium Persistence
Contrary to expectations, gadolinium persists in the body far longer than initially presumed. Symptoms associated with gadolinium exposure (SAGE) encapsulate the chronic, often enigmatic maladies tied to MRI contrast agents.20 The prolonged retention of this rare earth metal offers a compelling hypothesis for the etiology of SAGE. It has been hypothesized that Lewis base-rich metabolites increase susceptibility to gadolinium-based contrast agent complications.21
The blood and urine concentration elimination curves of gadolinium are exponential and categorized as fast, intermediate, and long-term.1 For urinary elimination, the function of the curves is exponential. The quantity of gadolinium in the urine at a time (t) after exposure (D[Gd](t)) is equal to the product of the amount of gadolinium in the sample (urine or blood) at the end of the fast elimination period (D[Gd](t0)) and the exponential decay with k being a rate constant.
To the authors’ knowledge, we are the only research team currently investigating the rate constant for the intermediate- and long-term phase gadolinium elimination. The Retention and Toxicity of Gadolinium-based Contrast Agents study was approved by the University of New Mexico Health Sciences Center Institutional Review Board on May 27, 2020 (IRB ID 19-660). The data for the patient in this case were compared with preliminary results for patients with exposure-to-measurement intervals < 100 days.
The patient in this case presented with detectable gadolinium levels in urine and serum shortly after an attempted contrast-enhanced MRI procedure (Figure 3). The presence of detectable gadolinium levels in the patient’s urine and serum suggests a likely exposure to a contrast agent about 27 days before his consultation. While the technician reported that no contrast was administered during the attempted MRI, it remains possible that a small amount was introduced during cannulation, potentially triggering the patient’s symptoms. Linear modeling of semilogarithmic plots for participants exposed to contrast agents within 100 days (urine: P = 1.8 × 10ˉ8, adjusted r² = 0.62; blood: P = .005, adjusted r² = 0.21) provided clearance rates (k values) for urine and blood. Extrapolating from these models to the presumed exposure date, the intercepts estimate that the patient received between 0.5% and 8% of a standard contrast dose.
MRI contrast agents can cause skin disease. Systemic fibrosis is considered one of the most severe AEs. Skin pathophysiology involving myeloid cells is driven by elevated levels of monocyte chemoattractant protein-1, which recruits circulating fibroblasts via the C-C chemokine receptor 2.22,23 This occurs alongside activation of NADPH oxidase Nox4.4,24,25 Intracellular gadolinium-rich nanoparticles likely serve as catalysts for this reactive cascade.2,18,22,26,27 These particles assemble around intracellular lipid droplets and ferrule them in spiculated rare earth-rich shells that compromise cellular architecture.2,18,21,22,26,27 Frequently sequestered within endosomal compartments, they disrupt vesicular integrity and threaten cellular homeostasis. Interference with degradative systems such as the endolysosomal axis perturbs energy-recycling pathways—an insidious disturbance, particularly in cells with high metabolic demand. Skin-related symptoms are among the most frequently reported AEs, according to the FDA AE reporting system.18
Studies indicate repeated exposure to MRI contrast agents can lead to permanent gadolinium retention in the brain and other vital organs. Intravenous (IV) contrast agents cross the blood-brain barrier rapidly, while intrathecal administration has been linked to significant and lasting neurologic effects.18
Gadolinium is chemically bound to pharmaceutical ligands to enhance renal clearance and reduce toxicity. However, available data from human samples suggest potential ligand exchanges with undefined physiologic substances. This exchange may facilitate gadolinium precipitation and accumulation within cells into spiculated nanoparticles. Transmission electron microscopy reveals the formation of unilamellar bodies associated with mitochondriopathy and cellular damage, particularly in renal proximal tubules.2,18,22,26,27 It is proposed that intracellular nanoparticle formation represents a key mechanism driving the systemic symptoms observed in patients.1,2,18, 22,26,27
Any hypothesis based on free soluble gadolinium—or concept derived from it—should be discarded. The high affinity of pharmaceutical ligands for gadolinium suggests that the cationic rare earth metal remains predominantly in a ligand-bound, soluble form. It is hypothesized that gadolinium undergoes ligand exchange with physiologic substances, directly leading to nanoparticle formation. Current data demonstrate gadolinium precipitation according to the Le Chatelier’s principle. Since precipitated gadolinium does not readily re-equilibrate with pharmaceutical ligands, repeated administration of different contrast agent brands may contribute to nanoparticle growth.26
Meanwhile, a growing number of patients are turning to chelation therapy, a largely untested treatment. The premise of chelation therapy is rooted in several unproven assumptions.18,21 First, it assumes that clinically significant amounts of gadolinium persist in compartments such as the extracellular space, where they can be effectively chelated and cleared. Second, it presumes that free gadolinium is the primary driver of chronic symptoms, an assertion that remains scientifically unsubstantiated. Finally, chelation proponents overlook the potential harm caused by depleting essential physiological metals during the process, assuming without evidence that the scant removal of gadolinium outweighs the risk of physiological mineral depletion.
These assumptions underpin an unproven remedy that demands critical scrutiny. Recent findings reveal that gadolinium deposits in the skin and kidney often take the form of intracellular nanoparticles, directly challenging the foundation of chelation therapy. Chelation advocates must demonstrate that these intracellular gadolinium deposits neither trigger cellular toxicity nor initiate a cytokine cascade. Chelation supporters must prove that the systemic response to these foreign particles is unrelated to the symptoms reported by patients. Until then, the validity of chelation therapy remains highly questionable.
The causality of the symptoms, mainly whether IV gadolinium was administered, was examined. The null hypothesis stated that the patient was not exposed to gadolinium. However, this hypothesis was contradicted by the detection of gadolinium in the serum and urine 27 days after the potential exposure.
Two plausible explanations exist for the nonzero gadolinium levels detected in the serum and urine. The first possibility is that minute quantities of gadolinium were introduced during cannulation, with the amount being sufficient to persist in measurable concentrations 27 days postexposure. The second possibility is that the gadolinium originated from an MRI contrast agent administered 4 years earlier. In this scenario, gadolinium stored in organ reservoirs such as bone, liver, or kidneys may have been mobilized into the extracellular fluid compartment due to the administration of high-dose steroids 20 days after the recent contrast-enhanced MRI procedure attempt. Coyte et al reported elevated gadolinium levels in the serum, cord blood, breast milk, and placenta of pregnant women with prior exposure to MRI contrast agents.28 These findings suggest that gadolinium, stored in organs such as bone may be remobilized by variables affecting bone remodeling (eg, high-dose steroids).
Significantly, the patient exhibited elevated urinary oxalate levels. Previous research has found that oxalic acid reacts rapidly with MRI contrast agents, forming digadolinium trioxalate. While the gadolinium-rich nanoparticles identified in tissues such as the skin and kidney (including the human kidney) are amorphous, these in vitro findings establish a proof-of-concept: the intracellular environment facilitates gadolinium dissociation from pharmaceutical chelates.
Furthermore, in vitro experiments show that proteins and lysosomal pH promote this dissociation, underscoring how human metabolic conditions—particularly oxalic acid concentration—may drive intracellular gadolinium deposition.
Patient Perspective
“They put something into my body that they cannot get out.” This stark realization underpins the patient’s profound concern about gadolinium-based contrast agents and their potential long-term effects. Reflecting on his experience, the patient expressed deep fears about the unknown future impacts: “I’m concerned about my kidneys, I’m concerned about my heart, and I’m concerned about my brain. I don’t know how this stuff is going to affect me in the future.”
He drew an unsettling parallel between gadolinium and heavy metals: “Heavy metal is poison. The body does not produce this kind of stuff on its own.” His reaction to the procedure left a lasting impression, prompting him to question the logic of using a substance that cannot be purged: “Why would you put something into someone’s body that you cannot extract? Nobody—nobody—should experience what I went through.”
The patient emphasized the lack of clear research on long-term outcomes, which compounds his anxiety: “If there was research that said, ‘Well, this is only going to affect these organs for this long,’ OK, I might be able to accept that. But there is no research like that. Nobody can tell me what’s going to happen in 5 years.”
Strengths and Limitations
A significant strength of this approach is the ability to track gadolinium elimination and symptom resolution over time, supported by unique access to intermediate and long-term clearance data from our ongoing research protocol. The investigators were equipped to back-extrapolate the exposure, which provided a rare opportunity to correlate gadolinium levels with clinical outcomes. The primary limitation is the lack of a defined clinical case definition for gadolinium toxicity and limited mechanistic understanding of SAGE, which hinders diagnosis and management.
Metabolites, proteins, and lipids rich in Lewis bases could initiate this process as substrates for intracellular gadolinium sedimentation. Future studies should investigate whether metabolic conditions such as oxalate burden or altered parathyroid hormone levels modulate gadolinium compartmentalization and tissue retention. If gadolinium-rich nanoparticle formation and accumulation disrupt cellular equilibrium, it underscores an urgent need to understand the implications of long-term gadolinium retention. The research team continues to gather evidence that the gadolinium cation remains chelated from the moment MRI contrast agents are administered through to the formation of intracellular nanoparticles. Retained gadolinium nanoparticles may act as a nidus, triggering cellular signaling cascades that lead to multisymptomatic illnesses. Intracellular and insoluble retained gadolinium challenges proponents of untested chelation therapies.
Conclusions
This case highlights emerging clinical and ethical concerns surrounding gadolinium-based contrast agent use. Clinicians may benefit from considering gadolinium retention as a contributor to persistent, unexplained symptoms—particularly in patients with recent imaging exposure. As contrast use continues to rise within federal health systems, regulatory and administrative stakeholders would do well to re-examine current safety frameworks. Informed consent should reflect what is known: gadolinium can remain in the body long after administration, potentially indefinitely. The long-term consequences of cumulative exposure remain poorly defined, but the presence of a lanthanide element in human tissue warrants greater attention from researchers and regulators alike. Interest in alternative imaging modalities and long-term safety monitoring would mark progress toward more transparent, accountable care.
Jackson DB, MacIntyre T, Duarte-Miramontes V, et al. Gadolinium deposition disease: a case report and the prevalence of enhanced MRI procedures within the Veterans Health Administration. Fed Pract. 2022;39:218-225. doi:10.12788/fp.0258
Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1:561-568.doi:10.34067/kid.0000272019
Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28:154-162. doi:10.1097/MNH.0000000000000475
Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311:F1-F11. doi:10.1152/ajprenal.00166.2016
Maramattom BV, Manno EM, Wijdicks EF, et al. Gadolinium encephalopathy in a patient with renal failure. Neurology. 2005;64:1276-1278.doi:10.1212/01.WNL.0000156805.45547.6E
Sam AD II, Morasch MD, Collins J, et al. Safety of gadolinium contrast angiography in patients with chronic renal insufficiency. J Vasc Surg. 2003;38:313-318. doi:10.1016/s0741-5214(03)00315-x
Schenker MP, Solomon JA, Roberts DA. Gadolinium arteriography complicated by acute pancreatitis and acute renal failure. J Vasc Interv Radiol. 2001;12:393. doi:10.1016/s1051-0443(07)61925-3
Gemery J, Idelson B, Reid S, et al. Acute renal failure after arteriography with a gadolinium-based contrast agent. AJR Am J Roentgenol. 1998;171:1277-1278. doi:10.2214/ajr.171.5.9798860
Akgun H, Gonlusen G, Cartwright J Jr, et al. Are gadolinium-based contrast media nephrotoxic? A renal biopsy study. Arch Pathol Lab Med. 2006;130:1354-1357. doi:10.5858/2006-130-1354-AGCMNA
Gathings RM, Reddy R, Santa Cruz D, et al. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151:316-319. doi:10.1001/jamadermatol.2014.2660
McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
Kanda T, Ishii K, Kawaguchi H, et al. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270(3):834-841. doi:10.1148/radiol.13131669
Schmidt K, Bau M, Merschel G, et al. Anthropogenic gadolinium in tap water and in tap water-based beverages from fast-food franchises in six major cities in Germany. Sci Total Environ. 2019;687:1401-1408. doi:10.1016/j.scitotenv.2019.07.075
Kulaksız S, Bau M. Anthropogenic gadolinium as a microcontaminant in tap water used as drinking water in urban areas and megacities. Appl Geochem. 2011;26:1877-1885.
Brunjes R, Hofmann T. Anthropogenic gadolinium in freshwater and drinking water systems. Water Res. 2020;182:115966. doi:10.1016/j.watres.2020.115966
Endrikat J, Gutberlet M, Hoffmann KT, et al. Clinical safety of gadobutrol: review of over 25 years of use exceeding 100 million administrations. Invest Radiol. 2024;59(9):605-613. doi:10.1097/RLI.0000000000001072
Elmholdt TR, Jørgensen B, Ramsing M, et al. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287. doi:10.1093/ndtplus/sfq028
Cunningham A, Kirk M, Hong E, et al. The safety of magnetic resonance imaging contrast agents. Front Toxicol. 2024;6:1376587. doi:10.3389/ftox.2024.1376587
Huxley A. Complete Essays. Volume II, 1926-1929. Chicago; 2000:227.
McDonald RJ, Weinreb JC, Davenport MS. Symptoms associated with gadolinium exposure (SAGE): a suggested term. Radiology. 2022;302(2):270-273. doi:10.1148/radiol.2021211349
Henderson IM, Benevidez AD, Mowry CD, et al. Precipitation of gadolinium from magnetic resonance imaging contrast agents may be the Brass tacks of toxicity. Magn Reson Imaging. 2025;119:110383. doi:10.1016/j.mri.2025.110383
Do C, Drel V, Tan C, et al. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143. doi:10.1016/j.jid.2019.03.1145
Drel VR, Tan C, Barnes JL, et al. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH oxidase 4 for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
DeAguero J, Howard T, Kusewitt D, et al. The onset of rare earth metallosis begins with renal gadolinium-rich nanoparticles from magnetic resonance imaging contrast agent exposure. Sci Rep. 2023;13(1):2025. doi:10.1038/s41598-023-28666-1
Do C, Ford B, Lee DY, et al. Gadolinium-based contrast agents: Stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
Coyte RM, Darrah T, Olesik J, et al. Gadolinium during human pregnancy following administration of gadolinium chelate before pregnancy. Birth Defects Res. 2023;115(14):1264-1273. doi:10.1002/bdr2.2209
Jackson DB, MacIntyre T, Duarte-Miramontes V, et al. Gadolinium deposition disease: a case report and the prevalence of enhanced MRI procedures within the Veterans Health Administration. Fed Pract. 2022;39:218-225. doi:10.12788/fp.0258
Do C, DeAguero J, Brearley A, et al. Gadolinium-based contrast agent use, their safety, and practice evolution. Kidney360. 2020;1:561-568.doi:10.34067/kid.0000272019
Leyba K, Wagner B. Gadolinium-based contrast agents: why nephrologists need to be concerned. Curr Opin Nephrol Hypertens. 2019;28:154-162. doi:10.1097/MNH.0000000000000475
Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;311:F1-F11. doi:10.1152/ajprenal.00166.2016
Maramattom BV, Manno EM, Wijdicks EF, et al. Gadolinium encephalopathy in a patient with renal failure. Neurology. 2005;64:1276-1278.doi:10.1212/01.WNL.0000156805.45547.6E
Sam AD II, Morasch MD, Collins J, et al. Safety of gadolinium contrast angiography in patients with chronic renal insufficiency. J Vasc Surg. 2003;38:313-318. doi:10.1016/s0741-5214(03)00315-x
Schenker MP, Solomon JA, Roberts DA. Gadolinium arteriography complicated by acute pancreatitis and acute renal failure. J Vasc Interv Radiol. 2001;12:393. doi:10.1016/s1051-0443(07)61925-3
Gemery J, Idelson B, Reid S, et al. Acute renal failure after arteriography with a gadolinium-based contrast agent. AJR Am J Roentgenol. 1998;171:1277-1278. doi:10.2214/ajr.171.5.9798860
Akgun H, Gonlusen G, Cartwright J Jr, et al. Are gadolinium-based contrast media nephrotoxic? A renal biopsy study. Arch Pathol Lab Med. 2006;130:1354-1357. doi:10.5858/2006-130-1354-AGCMNA
Gathings RM, Reddy R, Santa Cruz D, et al. Gadolinium-associated plaques: a new, distinctive clinical entity. JAMA Dermatol. 2015;151:316-319. doi:10.1001/jamadermatol.2014.2660
McDonald RJ, McDonald JS, Kallmes DF, et al. Gadolinium deposition in human brain tissues after contrast-enhanced MR imaging in adult patients without intracranial abnormalities. Radiology. 2017;285(2):546-554. doi:10.1148/radiol.2017161595
Kanda T, Ishii K, Kawaguchi H, et al. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270(3):834-841. doi:10.1148/radiol.13131669
Schmidt K, Bau M, Merschel G, et al. Anthropogenic gadolinium in tap water and in tap water-based beverages from fast-food franchises in six major cities in Germany. Sci Total Environ. 2019;687:1401-1408. doi:10.1016/j.scitotenv.2019.07.075
Kulaksız S, Bau M. Anthropogenic gadolinium as a microcontaminant in tap water used as drinking water in urban areas and megacities. Appl Geochem. 2011;26:1877-1885.
Brunjes R, Hofmann T. Anthropogenic gadolinium in freshwater and drinking water systems. Water Res. 2020;182:115966. doi:10.1016/j.watres.2020.115966
Endrikat J, Gutberlet M, Hoffmann KT, et al. Clinical safety of gadobutrol: review of over 25 years of use exceeding 100 million administrations. Invest Radiol. 2024;59(9):605-613. doi:10.1097/RLI.0000000000001072
Elmholdt TR, Jørgensen B, Ramsing M, et al. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287. doi:10.1093/ndtplus/sfq028
Cunningham A, Kirk M, Hong E, et al. The safety of magnetic resonance imaging contrast agents. Front Toxicol. 2024;6:1376587. doi:10.3389/ftox.2024.1376587
Huxley A. Complete Essays. Volume II, 1926-1929. Chicago; 2000:227.
McDonald RJ, Weinreb JC, Davenport MS. Symptoms associated with gadolinium exposure (SAGE): a suggested term. Radiology. 2022;302(2):270-273. doi:10.1148/radiol.2021211349
Henderson IM, Benevidez AD, Mowry CD, et al. Precipitation of gadolinium from magnetic resonance imaging contrast agents may be the Brass tacks of toxicity. Magn Reson Imaging. 2025;119:110383. doi:10.1016/j.mri.2025.110383
Do C, Drel V, Tan C, et al. Nephrogenic systemic fibrosis is mediated by myeloid C-C chemokine receptor 2. J Invest Dermatol. 2019;139(10):2134-2143. doi:10.1016/j.jid.2019.03.1145
Drel VR, Tan C, Barnes JL, et al. Centrality of bone marrow in the severity of gadolinium-based contrast-induced systemic fibrosis. FASEB J. 2016;30(9):3026-3038. doi:10.1096/fj.201500188R
Bruno F, DeAguero J, Do C, et al. Overlapping roles of NADPH oxidase 4 for diabetic and gadolinium-based contrast agent-induced systemic fibrosis. Am J Physiol Renal Physiol. 2021;320(4):F617-F627. doi:10.1152/ajprenal.00456.2020
Wagner B, Tan C, Barnes JL, et al. Nephrogenic systemic fibrosis: evidence for oxidative stress and bone marrow-derived fibrocytes in skin, liver, and heart lesions using a 5/6 nephrectomy rodent model. Am J Pathol. 2012;181(6):1941-1952. doi:10.1016/j.ajpath.2012.08.026
DeAguero J, Howard T, Kusewitt D, et al. The onset of rare earth metallosis begins with renal gadolinium-rich nanoparticles from magnetic resonance imaging contrast agent exposure. Sci Rep. 2023;13(1):2025. doi:10.1038/s41598-023-28666-1
Do C, Ford B, Lee DY, et al. Gadolinium-based contrast agents: Stimulators of myeloid-induced renal fibrosis and major metabolic disruptors. Toxicol Appl Pharmacol. 2019;375:32-45. doi:10.1016/j.taap.2019.05.009
Coyte RM, Darrah T, Olesik J, et al. Gadolinium during human pregnancy following administration of gadolinium chelate before pregnancy. Birth Defects Res. 2023;115(14):1264-1273. doi:10.1002/bdr2.2209
Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
Gadolinium Intermediate Elimination and Persistent Symptoms After Magnetic Resonance Imaging Contrast Agent Exposure
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
Tracheal deviation mostly occurs from mechanical compression of the trachea, and can be caused by a variety of clinical conditions, including trauma,¹ pharyngeal abscess,² neck hematoma,³ thyroid enlargement,4 and kyphoscoliosis.5 These conditions often result in lateral tracheal deviation, which can be associated with tracheal compression and reduction in tracheal caliber.
Anterior-posterior (A-P) tracheal deviation has rarely been reported. Kyphoscoliosis, scarring after a tracheostomy, or innominate vein compression are probable causes of A-P tracheal deviation and can be associated with tracheal narrowing and vascular fistula formation. This report describes a case of difficult endotracheal tube (ETT) advancement secondary to unexpected acute posterior tracheal deviation encountered during cardiopulmonary resuscitation (CPR). A waiver of patient consent was obtained from the Human Research Protection Program at the US Department of Veterans Affairs (VA) Puget Sound Health Care System.
Case Presentation
A 50-year-old male with a history of chronic cerebral venous sinus thrombosis and taking enoxaparin, presented to the emergency department for recurrent headaches. He experienced sudden cardiac arrest, and CPR in the form of chest compression and bag mask ventilation was immediately initiated. With the patient's head in an extended position and using a video laryngoscope, a Cormack–Lehane grade 1 view of the glottic opening was obtained and the trachea was intubated with an 8 mm (internal diameter) polyvinyl chloride ETT. Tracheal intubation was confirmed by utilizing continuous EtCO2 monitoring. The ETT was secured at 22 cm measured at the teeth.
After about 40 minutes of CPR, spontaneous circulation restarted and a portable A-P chest X-ray with the head in a neutral position indicated the ETT tip was at the level of the first rib (Figure 1). This finding, along with a persistent air leak, prompted blind advancement of the ETT to 26 cm at the teeth, but resistance to advancement was noted. A subsequent chest computed tomography (CT) with the head in a neutral position revealed the ETT remained inappropriately positioned with the tip measured 8.2 cm above the carina (Figure 2A). Concurrently, a sagittal CT view demonstrated significant posterior deviation of the mid and lower trachea. This deviation was determined to be the most likely cause of the difficulty encountered in advancing the ETT. No masses or lesions contributing to the acute tracheal angulation could be identified. Comparing CT imaging from 2 months prior, the trachea was of normal caliber and ordinarily aligned with the vertebral column (Figure 2B).
With the patient in Fowler position with the head midline, a flexible fiber-optic bronchoscopy was performed. Acute, almost 90-degree tracheal angulation was encountered and navigated by retroflexion of the flexible bronchoscope. Once the posterior tracheal wall was encountered, retroflexion was relaxed and the carina was visualized. The bronchoscope tip was placed near the carina, and the ETT was advanced over the fiber-optic bronchoscope to terminate 3 cm above the carina. A subsequent chest X-ray confirmed appropriate ETT position (Figure 3).
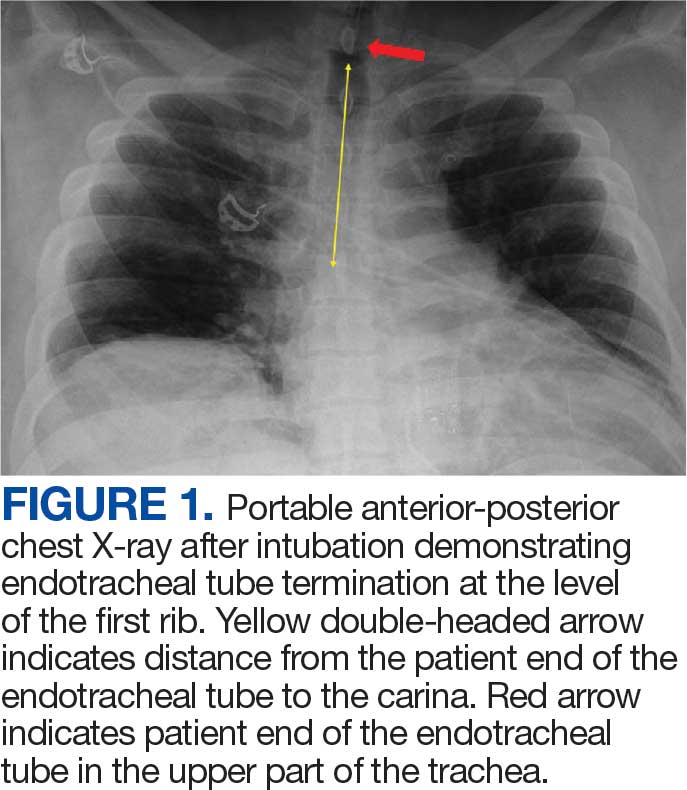
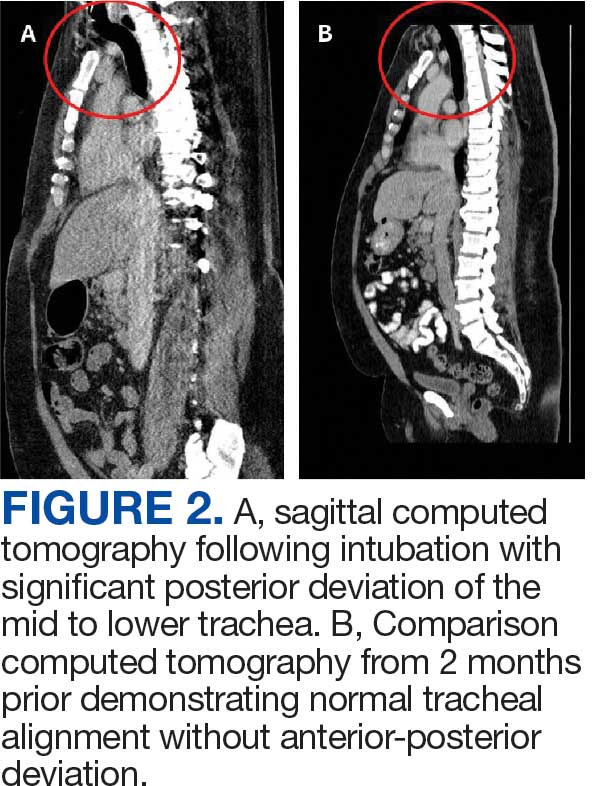
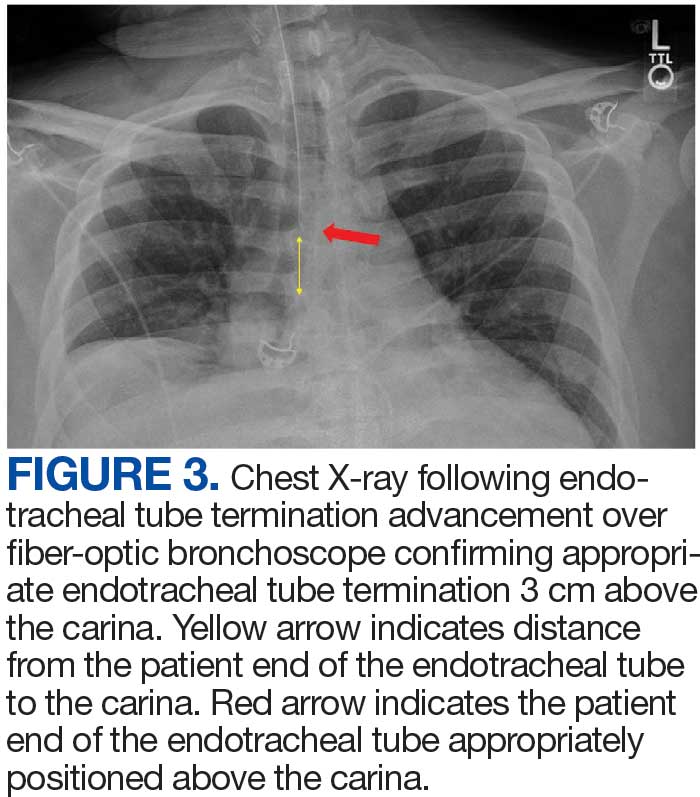
Discussion
Tracheal deviation in the A-P dimension resulting in difficult tracheal intubation has rarely been reported. Previous reports have described anatomical lesions contributing to similar tracheal deviation, such as retro-tracheal thyroid tissue, pronounced cervical lordosis, and severe kyphoscoliosis with destructive cervical fusion.5-8 In a study of the anatomical correlation of double lumen tube placement while using positron emission tomography CT, Cameron et al evaluated the size and angulation of the glottis and proximal trachea using calibrated CT measurements and an online digital protractor and note nearly perfect alignment of the pharynx and glottis.9 However, the trachea turned posteriorly relative to the glottis, resulting in an overall posterior angle of the proximal trachea compared to the glottis of 30.4 to 50.1 degrees, with no sex differences. The need to maneuver similar proximal tracheal angulation during endotracheal intubation has been reported as a cause of difficult intubation.10
In this case, the posterior angulation was not encountered in the proximal trachea but rather in the more distal trachea. The extreme A-P tracheal deviation was not associated with any identifiable masses or lesions. A CT performed 2 months prior demonstrated normal tracheal anatomy, and there was no interval history of neck trauma or tracheal obstruction suggestive of a likely cause for this deviation. This change in the patient’s tracheal anatomy was only discovered after CPR had been performed and as part of the workup for cardiac arrest. Iatrogenic injuries are known to occur during CPR. Common CPR-related airway injuries include tracheal mucosal injury from traumatic intubation and bony injuries to the chest wall from compressions.11 Laryngeal cartilage damage from intubation may also occur, but tracheal displacement following CPR has not been previously reported.11
This case of tracheal deviation is unlikely to be related to patient positioning, as the A-P deviation persisted in 3 separate head and neck alignments. First, during indirect laryngoscopy, performed in a standard sniffing position. Second, during the CT, performed in the supine position, with no head support. The acute A-P deviation seen in Figure 2 was clearly noted in this position. Lastly, flexible fiber-optic bronchoscopy was performed in a semiupright position with the head supported on a pillow. A-P deviation was encountered and navigated in this position during flexible fiber-optic guided ETT repositioning.
Using magnetic resonance imaging, alterations in the alignment of pharyngeal and tracheal axes have been described with changes in neck positioning; however, tracheal deviation has not been described with changes in head and neck alignment.12 Although the clinical presentation in this case was consistent with prior reports, we were unable to identify any previously reported anatomic cause for the tracheal deviation.5,6,8 Initial glottic visualization with a video laryngoscope was unremarkable, but resistance to sufficient ETT advancement past the vocal cords and a persistent air leak due to cuff herniation through the glottic opening was noticeable. The ETT was maneuvered to an appropriate position in the trachea using a flexible fiber-optic bronchoscope. The acute angulation of the trachea that was appreciated on bronchoscopy did not result in kinking of the ETT both initially and after in-situ thermosoftening of the polyvinyl chloride tube.13 Previously reported instances of A-P tracheal deviation have outlined the necessity of using alternative techniques to establish a patent airway, including the use of a laryngeal mask airway and a cuffless ETT with saline-soaked gauze packing.5,8 In 1 reported case, awake fiber-optic intubation was performed when difficult tracheal intubation was anticipated due to known A-P tracheal deviation.6
Failure of ETT advancement can be due to obstruction from the arytenoids and at the level of the vocal cords.14 When the ETT has been visualized to have traversed the vocal cords, tracheal A-P deviation should be considered as a cause of difficult ETT advancement. If an adequate endotracheal airway cannot be established, prompt consideration should be given to placement of a supraglottic airway. Early fiber-optic bronchoscopy should be used to establish the diagnosis and assist with proper ETT positioning.
Conclusions
This case illustrates the rare occurrence of A-P tracheal deviation leading to difficult intubation during CPR. The findings underscore the importance of considering A-P deviation as a potential cause of airway complications in emergency settings, especially in patients with previously normal tracheal anatomy. The successful use of flexible fiber-optic bronchoscopy in this case provides a valuable technique for addressing acute tracheal angulation. This report contributes to the limited literature on A-P tracheal deviation and serves as a reminder for clinicians to maintain a high index of suspicion for unusual airway challenges during critical interventions.
Creasy JD, Chiles C, Routh WD, et al. Overview of traumatic injury of the thoracic aorta. Radiogr Rev Publ Radiol Soc N Am Inc. 1997;17:27-45. doi:10.1148/radiographics.17.1.9017797
Yee AM, Christensen DN, Waterbrook AL, et al. Parapharyngeal abscess with tracheal deviation. Intern Emerg Med. 2017;12:1077-1078.doi:10.1007/s11739-017-1634-8
Querney J, Singh SI, Sebbag I. Tracheal deviation with phrenic nerve palsy after brachial plexus block. Anaesth Rep. 2021;9:41-43. doi:10.1002/anr3.12100
Geissler B, Wagner T, Dorn R, et al. Extensive sterile abscess in an invasive fibrous thyroiditis (Riedel’s thyroiditis) caused by an occlusive vasculitis. J Endocrinol Invest. 2001;24:111-115. doi:10.1007/BF03343824
Kim HJ, Choi YS, Park SH, et al. Difficult endotracheal intubation secondary to tracheal deviation and stenosis in a patient with severe kyphoscoliosis: a case report. Korean J Anesthesiol. 2016;69:386-389. doi:10.4097/kjae.2016.69.4.386
Crabb IJ. Anterior deviation of the trachea. Anaesthesia. 2001;56:284-286.doi:10.1046/j.1365-2044.2001.01918-17.x
De Cassai A, Boscolo A, Rose K, et al. Predictive parameters of difficult intubation in thyroid surgery: a meta-analysis. Minerva Anestesiol. 2020;86:317-326. doi:10.23736/S0375-9393.19.14127-2
Davies R. Difficult tracheal intubation secondary to a tracheal diverticulum and a 90 degree deviation in the trachea. Anaesthesia. 2000;55:923-925. doi:10.1046/j.1365-2044.2000.01664-18.x
Cameron RB, Peacock WJ, Chang XG, et al. Double lumen endobronchial tube intubation: lessons learned from anatomy. BMC Anesthesiol. 2024;24:150. doi:10.1186/s12871-024-02517-6
Walls RM, Samuels-Kalow M, Perkins A. A new maneuver for endotracheal tube insertion during difficult GlideScope intubation. J Emerg Med. 2010;39:86-88. doi:10.1016/j.jemermed.2009.11.005
Buschmann CT, Tsokos M. Frequent and rare complications of resuscitation attempts. Intensive Care Med. 2009;35:397-404. doi:10.1007/s00134-008-1255-9
Greenland KB, Edwards MJ, Hutton NJ, et al. Changes in airway configuration with different head and neck positions using magnetic resonance imaging of normal airways: a new concept with possible clinical applications. Br J Anaesth. 2010;105:683-690. doi:10.1093/bja/aeq239
Takasugi Y, Futagawa K, Umeda T, et al. Thermophysical Properties of Thermosoftening Nasotracheal Tubes. Anesth Prog. 2018;65:100-105. doi:10.2344/anpr-65-02-06
Phelan MP. Use of the endotracheal bougie introducer for difficult intubations. Am J Emerg Med. 2004;22:479-482. doi:10.1016/j.ajem.2004.07.017
Tracheal deviation mostly occurs from mechanical compression of the trachea, and can be caused by a variety of clinical conditions, including trauma,¹ pharyngeal abscess,² neck hematoma,³ thyroid enlargement,4 and kyphoscoliosis.5 These conditions often result in lateral tracheal deviation, which can be associated with tracheal compression and reduction in tracheal caliber.
Anterior-posterior (A-P) tracheal deviation has rarely been reported. Kyphoscoliosis, scarring after a tracheostomy, or innominate vein compression are probable causes of A-P tracheal deviation and can be associated with tracheal narrowing and vascular fistula formation. This report describes a case of difficult endotracheal tube (ETT) advancement secondary to unexpected acute posterior tracheal deviation encountered during cardiopulmonary resuscitation (CPR). A waiver of patient consent was obtained from the Human Research Protection Program at the US Department of Veterans Affairs (VA) Puget Sound Health Care System.
Case Presentation
A 50-year-old male with a history of chronic cerebral venous sinus thrombosis and taking enoxaparin, presented to the emergency department for recurrent headaches. He experienced sudden cardiac arrest, and CPR in the form of chest compression and bag mask ventilation was immediately initiated. With the patient's head in an extended position and using a video laryngoscope, a Cormack–Lehane grade 1 view of the glottic opening was obtained and the trachea was intubated with an 8 mm (internal diameter) polyvinyl chloride ETT. Tracheal intubation was confirmed by utilizing continuous EtCO2 monitoring. The ETT was secured at 22 cm measured at the teeth.
After about 40 minutes of CPR, spontaneous circulation restarted and a portable A-P chest X-ray with the head in a neutral position indicated the ETT tip was at the level of the first rib (Figure 1). This finding, along with a persistent air leak, prompted blind advancement of the ETT to 26 cm at the teeth, but resistance to advancement was noted. A subsequent chest computed tomography (CT) with the head in a neutral position revealed the ETT remained inappropriately positioned with the tip measured 8.2 cm above the carina (Figure 2A). Concurrently, a sagittal CT view demonstrated significant posterior deviation of the mid and lower trachea. This deviation was determined to be the most likely cause of the difficulty encountered in advancing the ETT. No masses or lesions contributing to the acute tracheal angulation could be identified. Comparing CT imaging from 2 months prior, the trachea was of normal caliber and ordinarily aligned with the vertebral column (Figure 2B).
With the patient in Fowler position with the head midline, a flexible fiber-optic bronchoscopy was performed. Acute, almost 90-degree tracheal angulation was encountered and navigated by retroflexion of the flexible bronchoscope. Once the posterior tracheal wall was encountered, retroflexion was relaxed and the carina was visualized. The bronchoscope tip was placed near the carina, and the ETT was advanced over the fiber-optic bronchoscope to terminate 3 cm above the carina. A subsequent chest X-ray confirmed appropriate ETT position (Figure 3).
Discussion
Tracheal deviation in the A-P dimension resulting in difficult tracheal intubation has rarely been reported. Previous reports have described anatomical lesions contributing to similar tracheal deviation, such as retro-tracheal thyroid tissue, pronounced cervical lordosis, and severe kyphoscoliosis with destructive cervical fusion.5-8 In a study of the anatomical correlation of double lumen tube placement while using positron emission tomography CT, Cameron et al evaluated the size and angulation of the glottis and proximal trachea using calibrated CT measurements and an online digital protractor and note nearly perfect alignment of the pharynx and glottis.9 However, the trachea turned posteriorly relative to the glottis, resulting in an overall posterior angle of the proximal trachea compared to the glottis of 30.4 to 50.1 degrees, with no sex differences. The need to maneuver similar proximal tracheal angulation during endotracheal intubation has been reported as a cause of difficult intubation.10
In this case, the posterior angulation was not encountered in the proximal trachea but rather in the more distal trachea. The extreme A-P tracheal deviation was not associated with any identifiable masses or lesions. A CT performed 2 months prior demonstrated normal tracheal anatomy, and there was no interval history of neck trauma or tracheal obstruction suggestive of a likely cause for this deviation. This change in the patient’s tracheal anatomy was only discovered after CPR had been performed and as part of the workup for cardiac arrest. Iatrogenic injuries are known to occur during CPR. Common CPR-related airway injuries include tracheal mucosal injury from traumatic intubation and bony injuries to the chest wall from compressions.11 Laryngeal cartilage damage from intubation may also occur, but tracheal displacement following CPR has not been previously reported.11
This case of tracheal deviation is unlikely to be related to patient positioning, as the A-P deviation persisted in 3 separate head and neck alignments. First, during indirect laryngoscopy, performed in a standard sniffing position. Second, during the CT, performed in the supine position, with no head support. The acute A-P deviation seen in Figure 2 was clearly noted in this position. Lastly, flexible fiber-optic bronchoscopy was performed in a semiupright position with the head supported on a pillow. A-P deviation was encountered and navigated in this position during flexible fiber-optic guided ETT repositioning.
Using magnetic resonance imaging, alterations in the alignment of pharyngeal and tracheal axes have been described with changes in neck positioning; however, tracheal deviation has not been described with changes in head and neck alignment.12 Although the clinical presentation in this case was consistent with prior reports, we were unable to identify any previously reported anatomic cause for the tracheal deviation.5,6,8 Initial glottic visualization with a video laryngoscope was unremarkable, but resistance to sufficient ETT advancement past the vocal cords and a persistent air leak due to cuff herniation through the glottic opening was noticeable. The ETT was maneuvered to an appropriate position in the trachea using a flexible fiber-optic bronchoscope. The acute angulation of the trachea that was appreciated on bronchoscopy did not result in kinking of the ETT both initially and after in-situ thermosoftening of the polyvinyl chloride tube.13 Previously reported instances of A-P tracheal deviation have outlined the necessity of using alternative techniques to establish a patent airway, including the use of a laryngeal mask airway and a cuffless ETT with saline-soaked gauze packing.5,8 In 1 reported case, awake fiber-optic intubation was performed when difficult tracheal intubation was anticipated due to known A-P tracheal deviation.6
Failure of ETT advancement can be due to obstruction from the arytenoids and at the level of the vocal cords.14 When the ETT has been visualized to have traversed the vocal cords, tracheal A-P deviation should be considered as a cause of difficult ETT advancement. If an adequate endotracheal airway cannot be established, prompt consideration should be given to placement of a supraglottic airway. Early fiber-optic bronchoscopy should be used to establish the diagnosis and assist with proper ETT positioning.
Conclusions
This case illustrates the rare occurrence of A-P tracheal deviation leading to difficult intubation during CPR. The findings underscore the importance of considering A-P deviation as a potential cause of airway complications in emergency settings, especially in patients with previously normal tracheal anatomy. The successful use of flexible fiber-optic bronchoscopy in this case provides a valuable technique for addressing acute tracheal angulation. This report contributes to the limited literature on A-P tracheal deviation and serves as a reminder for clinicians to maintain a high index of suspicion for unusual airway challenges during critical interventions.
Tracheal deviation mostly occurs from mechanical compression of the trachea, and can be caused by a variety of clinical conditions, including trauma,¹ pharyngeal abscess,² neck hematoma,³ thyroid enlargement,4 and kyphoscoliosis.5 These conditions often result in lateral tracheal deviation, which can be associated with tracheal compression and reduction in tracheal caliber.
Anterior-posterior (A-P) tracheal deviation has rarely been reported. Kyphoscoliosis, scarring after a tracheostomy, or innominate vein compression are probable causes of A-P tracheal deviation and can be associated with tracheal narrowing and vascular fistula formation. This report describes a case of difficult endotracheal tube (ETT) advancement secondary to unexpected acute posterior tracheal deviation encountered during cardiopulmonary resuscitation (CPR). A waiver of patient consent was obtained from the Human Research Protection Program at the US Department of Veterans Affairs (VA) Puget Sound Health Care System.
Case Presentation
A 50-year-old male with a history of chronic cerebral venous sinus thrombosis and taking enoxaparin, presented to the emergency department for recurrent headaches. He experienced sudden cardiac arrest, and CPR in the form of chest compression and bag mask ventilation was immediately initiated. With the patient's head in an extended position and using a video laryngoscope, a Cormack–Lehane grade 1 view of the glottic opening was obtained and the trachea was intubated with an 8 mm (internal diameter) polyvinyl chloride ETT. Tracheal intubation was confirmed by utilizing continuous EtCO2 monitoring. The ETT was secured at 22 cm measured at the teeth.
After about 40 minutes of CPR, spontaneous circulation restarted and a portable A-P chest X-ray with the head in a neutral position indicated the ETT tip was at the level of the first rib (Figure 1). This finding, along with a persistent air leak, prompted blind advancement of the ETT to 26 cm at the teeth, but resistance to advancement was noted. A subsequent chest computed tomography (CT) with the head in a neutral position revealed the ETT remained inappropriately positioned with the tip measured 8.2 cm above the carina (Figure 2A). Concurrently, a sagittal CT view demonstrated significant posterior deviation of the mid and lower trachea. This deviation was determined to be the most likely cause of the difficulty encountered in advancing the ETT. No masses or lesions contributing to the acute tracheal angulation could be identified. Comparing CT imaging from 2 months prior, the trachea was of normal caliber and ordinarily aligned with the vertebral column (Figure 2B).
With the patient in Fowler position with the head midline, a flexible fiber-optic bronchoscopy was performed. Acute, almost 90-degree tracheal angulation was encountered and navigated by retroflexion of the flexible bronchoscope. Once the posterior tracheal wall was encountered, retroflexion was relaxed and the carina was visualized. The bronchoscope tip was placed near the carina, and the ETT was advanced over the fiber-optic bronchoscope to terminate 3 cm above the carina. A subsequent chest X-ray confirmed appropriate ETT position (Figure 3).
Discussion
Tracheal deviation in the A-P dimension resulting in difficult tracheal intubation has rarely been reported. Previous reports have described anatomical lesions contributing to similar tracheal deviation, such as retro-tracheal thyroid tissue, pronounced cervical lordosis, and severe kyphoscoliosis with destructive cervical fusion.5-8 In a study of the anatomical correlation of double lumen tube placement while using positron emission tomography CT, Cameron et al evaluated the size and angulation of the glottis and proximal trachea using calibrated CT measurements and an online digital protractor and note nearly perfect alignment of the pharynx and glottis.9 However, the trachea turned posteriorly relative to the glottis, resulting in an overall posterior angle of the proximal trachea compared to the glottis of 30.4 to 50.1 degrees, with no sex differences. The need to maneuver similar proximal tracheal angulation during endotracheal intubation has been reported as a cause of difficult intubation.10
In this case, the posterior angulation was not encountered in the proximal trachea but rather in the more distal trachea. The extreme A-P tracheal deviation was not associated with any identifiable masses or lesions. A CT performed 2 months prior demonstrated normal tracheal anatomy, and there was no interval history of neck trauma or tracheal obstruction suggestive of a likely cause for this deviation. This change in the patient’s tracheal anatomy was only discovered after CPR had been performed and as part of the workup for cardiac arrest. Iatrogenic injuries are known to occur during CPR. Common CPR-related airway injuries include tracheal mucosal injury from traumatic intubation and bony injuries to the chest wall from compressions.11 Laryngeal cartilage damage from intubation may also occur, but tracheal displacement following CPR has not been previously reported.11
This case of tracheal deviation is unlikely to be related to patient positioning, as the A-P deviation persisted in 3 separate head and neck alignments. First, during indirect laryngoscopy, performed in a standard sniffing position. Second, during the CT, performed in the supine position, with no head support. The acute A-P deviation seen in Figure 2 was clearly noted in this position. Lastly, flexible fiber-optic bronchoscopy was performed in a semiupright position with the head supported on a pillow. A-P deviation was encountered and navigated in this position during flexible fiber-optic guided ETT repositioning.
Using magnetic resonance imaging, alterations in the alignment of pharyngeal and tracheal axes have been described with changes in neck positioning; however, tracheal deviation has not been described with changes in head and neck alignment.12 Although the clinical presentation in this case was consistent with prior reports, we were unable to identify any previously reported anatomic cause for the tracheal deviation.5,6,8 Initial glottic visualization with a video laryngoscope was unremarkable, but resistance to sufficient ETT advancement past the vocal cords and a persistent air leak due to cuff herniation through the glottic opening was noticeable. The ETT was maneuvered to an appropriate position in the trachea using a flexible fiber-optic bronchoscope. The acute angulation of the trachea that was appreciated on bronchoscopy did not result in kinking of the ETT both initially and after in-situ thermosoftening of the polyvinyl chloride tube.13 Previously reported instances of A-P tracheal deviation have outlined the necessity of using alternative techniques to establish a patent airway, including the use of a laryngeal mask airway and a cuffless ETT with saline-soaked gauze packing.5,8 In 1 reported case, awake fiber-optic intubation was performed when difficult tracheal intubation was anticipated due to known A-P tracheal deviation.6
Failure of ETT advancement can be due to obstruction from the arytenoids and at the level of the vocal cords.14 When the ETT has been visualized to have traversed the vocal cords, tracheal A-P deviation should be considered as a cause of difficult ETT advancement. If an adequate endotracheal airway cannot be established, prompt consideration should be given to placement of a supraglottic airway. Early fiber-optic bronchoscopy should be used to establish the diagnosis and assist with proper ETT positioning.
Conclusions
This case illustrates the rare occurrence of A-P tracheal deviation leading to difficult intubation during CPR. The findings underscore the importance of considering A-P deviation as a potential cause of airway complications in emergency settings, especially in patients with previously normal tracheal anatomy. The successful use of flexible fiber-optic bronchoscopy in this case provides a valuable technique for addressing acute tracheal angulation. This report contributes to the limited literature on A-P tracheal deviation and serves as a reminder for clinicians to maintain a high index of suspicion for unusual airway challenges during critical interventions.
Creasy JD, Chiles C, Routh WD, et al. Overview of traumatic injury of the thoracic aorta. Radiogr Rev Publ Radiol Soc N Am Inc. 1997;17:27-45. doi:10.1148/radiographics.17.1.9017797
Yee AM, Christensen DN, Waterbrook AL, et al. Parapharyngeal abscess with tracheal deviation. Intern Emerg Med. 2017;12:1077-1078.doi:10.1007/s11739-017-1634-8
Querney J, Singh SI, Sebbag I. Tracheal deviation with phrenic nerve palsy after brachial plexus block. Anaesth Rep. 2021;9:41-43. doi:10.1002/anr3.12100
Geissler B, Wagner T, Dorn R, et al. Extensive sterile abscess in an invasive fibrous thyroiditis (Riedel’s thyroiditis) caused by an occlusive vasculitis. J Endocrinol Invest. 2001;24:111-115. doi:10.1007/BF03343824
Kim HJ, Choi YS, Park SH, et al. Difficult endotracheal intubation secondary to tracheal deviation and stenosis in a patient with severe kyphoscoliosis: a case report. Korean J Anesthesiol. 2016;69:386-389. doi:10.4097/kjae.2016.69.4.386
Crabb IJ. Anterior deviation of the trachea. Anaesthesia. 2001;56:284-286.doi:10.1046/j.1365-2044.2001.01918-17.x
De Cassai A, Boscolo A, Rose K, et al. Predictive parameters of difficult intubation in thyroid surgery: a meta-analysis. Minerva Anestesiol. 2020;86:317-326. doi:10.23736/S0375-9393.19.14127-2
Davies R. Difficult tracheal intubation secondary to a tracheal diverticulum and a 90 degree deviation in the trachea. Anaesthesia. 2000;55:923-925. doi:10.1046/j.1365-2044.2000.01664-18.x
Cameron RB, Peacock WJ, Chang XG, et al. Double lumen endobronchial tube intubation: lessons learned from anatomy. BMC Anesthesiol. 2024;24:150. doi:10.1186/s12871-024-02517-6
Walls RM, Samuels-Kalow M, Perkins A. A new maneuver for endotracheal tube insertion during difficult GlideScope intubation. J Emerg Med. 2010;39:86-88. doi:10.1016/j.jemermed.2009.11.005
Buschmann CT, Tsokos M. Frequent and rare complications of resuscitation attempts. Intensive Care Med. 2009;35:397-404. doi:10.1007/s00134-008-1255-9
Greenland KB, Edwards MJ, Hutton NJ, et al. Changes in airway configuration with different head and neck positions using magnetic resonance imaging of normal airways: a new concept with possible clinical applications. Br J Anaesth. 2010;105:683-690. doi:10.1093/bja/aeq239
Takasugi Y, Futagawa K, Umeda T, et al. Thermophysical Properties of Thermosoftening Nasotracheal Tubes. Anesth Prog. 2018;65:100-105. doi:10.2344/anpr-65-02-06
Phelan MP. Use of the endotracheal bougie introducer for difficult intubations. Am J Emerg Med. 2004;22:479-482. doi:10.1016/j.ajem.2004.07.017
Creasy JD, Chiles C, Routh WD, et al. Overview of traumatic injury of the thoracic aorta. Radiogr Rev Publ Radiol Soc N Am Inc. 1997;17:27-45. doi:10.1148/radiographics.17.1.9017797
Yee AM, Christensen DN, Waterbrook AL, et al. Parapharyngeal abscess with tracheal deviation. Intern Emerg Med. 2017;12:1077-1078.doi:10.1007/s11739-017-1634-8
Querney J, Singh SI, Sebbag I. Tracheal deviation with phrenic nerve palsy after brachial plexus block. Anaesth Rep. 2021;9:41-43. doi:10.1002/anr3.12100
Geissler B, Wagner T, Dorn R, et al. Extensive sterile abscess in an invasive fibrous thyroiditis (Riedel’s thyroiditis) caused by an occlusive vasculitis. J Endocrinol Invest. 2001;24:111-115. doi:10.1007/BF03343824
Kim HJ, Choi YS, Park SH, et al. Difficult endotracheal intubation secondary to tracheal deviation and stenosis in a patient with severe kyphoscoliosis: a case report. Korean J Anesthesiol. 2016;69:386-389. doi:10.4097/kjae.2016.69.4.386
Crabb IJ. Anterior deviation of the trachea. Anaesthesia. 2001;56:284-286.doi:10.1046/j.1365-2044.2001.01918-17.x
De Cassai A, Boscolo A, Rose K, et al. Predictive parameters of difficult intubation in thyroid surgery: a meta-analysis. Minerva Anestesiol. 2020;86:317-326. doi:10.23736/S0375-9393.19.14127-2
Davies R. Difficult tracheal intubation secondary to a tracheal diverticulum and a 90 degree deviation in the trachea. Anaesthesia. 2000;55:923-925. doi:10.1046/j.1365-2044.2000.01664-18.x
Cameron RB, Peacock WJ, Chang XG, et al. Double lumen endobronchial tube intubation: lessons learned from anatomy. BMC Anesthesiol. 2024;24:150. doi:10.1186/s12871-024-02517-6
Walls RM, Samuels-Kalow M, Perkins A. A new maneuver for endotracheal tube insertion during difficult GlideScope intubation. J Emerg Med. 2010;39:86-88. doi:10.1016/j.jemermed.2009.11.005
Buschmann CT, Tsokos M. Frequent and rare complications of resuscitation attempts. Intensive Care Med. 2009;35:397-404. doi:10.1007/s00134-008-1255-9
Greenland KB, Edwards MJ, Hutton NJ, et al. Changes in airway configuration with different head and neck positions using magnetic resonance imaging of normal airways: a new concept with possible clinical applications. Br J Anaesth. 2010;105:683-690. doi:10.1093/bja/aeq239
Takasugi Y, Futagawa K, Umeda T, et al. Thermophysical Properties of Thermosoftening Nasotracheal Tubes. Anesth Prog. 2018;65:100-105. doi:10.2344/anpr-65-02-06
Phelan MP. Use of the endotracheal bougie introducer for difficult intubations. Am J Emerg Med. 2004;22:479-482. doi:10.1016/j.ajem.2004.07.017
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
A Case Report of Unanticipated Difficult Intubation Due to Posterior Tracheal Angulation
A Case Report on Bortezomib-Induced Hypotension: Rare Adverse Effect in Proteasome Inhibitor Therapy
Case Presentation
A 75-year-old man with chronic kidney disease, hypertension and diabetes mellitus presented with acute kidney injury (creatinine 5.2 from baseline 4.2) and a two-week history of increased urinary frequency. Labs revealed high anion gap metabolic acidosis, proteinuria, hematuria, pyuria, and acute on chronic anemia. He was diagnosed with kappa light chain nephropathy and multiple myeloma with 32% plasma cells on bone marrow biopsy. He began treatment with bortezomib, cyclophosphamide, and dexamethasone (Cy- BorD). Three days after cyclophosphamide and five days after bortezomib, the patient developed persistent hypotension with systolic BP in the 50s, unresponsive to fluids and Trendelenburg position. Due to end-stage renal disease with anuria, fluid resuscitation was limited. He required norepinephrine and was transferred to the ICU. Given instability, hemodialysis was deferred, and continuous renal replacement therapy was initiated. Shock evaluation included a CT abdomen showing enteritis versus ileus; however, infectious workup was negative. Cardiogenic shock was ruled out with a serial echocardiogram showing normal ejection fractions of 59-67% without significant valvular disease. The workup for adrenal insufficiency was negative. After the exclusion of other potential causes of shock, severe refractory hypotension was attributed to bortezomib toxicity.Hypotension is a known adverse effect of bortezomib. Orthostatic hypotension may occur in 8 to 9% of patients, and rarely, patients may experience heart failure, conduction disorders and arrhythmias, or cardiogenic shock. The pathologic mechanism of this toxicity is still poorly understood. Proposed mechanisms include direct endothelial toxicity as evidenced by thrombotic microangiopathy or impairment of sympathetic and parasympathetic nerve fibres. Most commonly, patients experience neurotoxicity, which may manifest as autonomic dysfunction or peripheral neuropathy. Cardiovascular complications are typically reversible. Our patient’s cardiac function remained within normal limits; therefore, his persistent hypotension was felt to be the result of direct toxicity from bortezomib rather than cardiogenic shock. Ultimately, blood pressure did improve, and vasopressors were discontinued. However, he continued to have orthostatic hypotension and continued to require supportive fludrocortisone, midodrine, and pyridostigmine. Goals of care have been discussed, and he wished to continue pursuing restorative care, with a plan for transition to carfilzomib versus daratumumab outpatient.
Case Presentation
A 75-year-old man with chronic kidney disease, hypertension and diabetes mellitus presented with acute kidney injury (creatinine 5.2 from baseline 4.2) and a two-week history of increased urinary frequency. Labs revealed high anion gap metabolic acidosis, proteinuria, hematuria, pyuria, and acute on chronic anemia. He was diagnosed with kappa light chain nephropathy and multiple myeloma with 32% plasma cells on bone marrow biopsy. He began treatment with bortezomib, cyclophosphamide, and dexamethasone (Cy- BorD). Three days after cyclophosphamide and five days after bortezomib, the patient developed persistent hypotension with systolic BP in the 50s, unresponsive to fluids and Trendelenburg position. Due to end-stage renal disease with anuria, fluid resuscitation was limited. He required norepinephrine and was transferred to the ICU. Given instability, hemodialysis was deferred, and continuous renal replacement therapy was initiated. Shock evaluation included a CT abdomen showing enteritis versus ileus; however, infectious workup was negative. Cardiogenic shock was ruled out with a serial echocardiogram showing normal ejection fractions of 59-67% without significant valvular disease. The workup for adrenal insufficiency was negative. After the exclusion of other potential causes of shock, severe refractory hypotension was attributed to bortezomib toxicity.Hypotension is a known adverse effect of bortezomib. Orthostatic hypotension may occur in 8 to 9% of patients, and rarely, patients may experience heart failure, conduction disorders and arrhythmias, or cardiogenic shock. The pathologic mechanism of this toxicity is still poorly understood. Proposed mechanisms include direct endothelial toxicity as evidenced by thrombotic microangiopathy or impairment of sympathetic and parasympathetic nerve fibres. Most commonly, patients experience neurotoxicity, which may manifest as autonomic dysfunction or peripheral neuropathy. Cardiovascular complications are typically reversible. Our patient’s cardiac function remained within normal limits; therefore, his persistent hypotension was felt to be the result of direct toxicity from bortezomib rather than cardiogenic shock. Ultimately, blood pressure did improve, and vasopressors were discontinued. However, he continued to have orthostatic hypotension and continued to require supportive fludrocortisone, midodrine, and pyridostigmine. Goals of care have been discussed, and he wished to continue pursuing restorative care, with a plan for transition to carfilzomib versus daratumumab outpatient.
Case Presentation
A 75-year-old man with chronic kidney disease, hypertension and diabetes mellitus presented with acute kidney injury (creatinine 5.2 from baseline 4.2) and a two-week history of increased urinary frequency. Labs revealed high anion gap metabolic acidosis, proteinuria, hematuria, pyuria, and acute on chronic anemia. He was diagnosed with kappa light chain nephropathy and multiple myeloma with 32% plasma cells on bone marrow biopsy. He began treatment with bortezomib, cyclophosphamide, and dexamethasone (Cy- BorD). Three days after cyclophosphamide and five days after bortezomib, the patient developed persistent hypotension with systolic BP in the 50s, unresponsive to fluids and Trendelenburg position. Due to end-stage renal disease with anuria, fluid resuscitation was limited. He required norepinephrine and was transferred to the ICU. Given instability, hemodialysis was deferred, and continuous renal replacement therapy was initiated. Shock evaluation included a CT abdomen showing enteritis versus ileus; however, infectious workup was negative. Cardiogenic shock was ruled out with a serial echocardiogram showing normal ejection fractions of 59-67% without significant valvular disease. The workup for adrenal insufficiency was negative. After the exclusion of other potential causes of shock, severe refractory hypotension was attributed to bortezomib toxicity.Hypotension is a known adverse effect of bortezomib. Orthostatic hypotension may occur in 8 to 9% of patients, and rarely, patients may experience heart failure, conduction disorders and arrhythmias, or cardiogenic shock. The pathologic mechanism of this toxicity is still poorly understood. Proposed mechanisms include direct endothelial toxicity as evidenced by thrombotic microangiopathy or impairment of sympathetic and parasympathetic nerve fibres. Most commonly, patients experience neurotoxicity, which may manifest as autonomic dysfunction or peripheral neuropathy. Cardiovascular complications are typically reversible. Our patient’s cardiac function remained within normal limits; therefore, his persistent hypotension was felt to be the result of direct toxicity from bortezomib rather than cardiogenic shock. Ultimately, blood pressure did improve, and vasopressors were discontinued. However, he continued to have orthostatic hypotension and continued to require supportive fludrocortisone, midodrine, and pyridostigmine. Goals of care have been discussed, and he wished to continue pursuing restorative care, with a plan for transition to carfilzomib versus daratumumab outpatient.
Diagnostic Challenges of Persistent Hypoglycemia in a Patient with Gastrointestinal Stromal Tumors
Background
Gastrointestinal stromal tumors (GISTs) are rare neoplasms of the gastrointestinal (GI) tract, accounting for approximately 1–2% of GI cancers. Hypoglycemia in patients with GIST is an uncommon and diagnostically challenging presentation, often involving a broad differential diagnosis. This case report explores the diagnostic difficulties encountered in managing persistent hypoglycemia in a patient with a history of advanced GIST.
Case Presentation
An 80-year-old male with a history of stage IV GIST, diagnosed in 2010, presented with persistent symptomatic hypoglycemia. His medical history included extensive abdominal disease, managed with multiple interventions: esophagogastrostomy, left lateral liver resection, a Whipple procedure, and Y-90 radioembolization. He received adjuvant imatinib therapy, which was discontinued in April 2024 due to significant adverse effects, including anasarca. In 2025, the patient developed progressive hypoglycemia, ultimately requiring continuous D10 infusion to maintain euglycemia, prompting an endocrinology evaluation. The initial diagnostic workup included cortisol, insulin, C-peptide levels, and IGF-1/IGF-2 ratio ruling out insulinoma, adrenal insufficiency, and GISTrelated paraneoplastic syndrome. Imaging studies, including PET and CT, showed no radiological evidence of recurrent GIST. Treatment with octreotide infusion resulted in minimal improvement, whereas daily corticosteroid therapy significantly alleviated the patient’s symptoms. The etiology of hypoglycemia remains elusive, with potential causes under consideration including Y-90 radioembolization-induced damage to glucagon-producing cells, immunotherapy-related adverse effects, or radiologically occult GIST. Insulin autoantibody testing is pending, and the case remains under active investigation, highlighting the diagnostic complexity of hypoglycemia in advanced GIST.
Discussion
Hypoglycemia in the context of GIST is a rare and poorly understood phenomenon. Potential mechanisms include paraneoplastic syndromes, such as non-islet cell tumor hypoglycemia (NICTH) mediated by IGF-2, or treatment-related effects, such as radiation-induced pancreatic or hepatic dysfunction. In this case, the absence of detectable IGF-2 abnormalities and negative imaging complicates the diagnosis. The lack of response to octreotide indicates that somatostatin receptor-mediated pathways may not be involved. The discontinuation of imatinib and prior Y-90 radioembolization further broadens the differential, as both could contribute to metabolic dysregulation.
Conclusions
This case illustrates the need for a systematic and multidisciplinary approach to evaluate hypoglycemia in patients with advanced GIST.
Background
Gastrointestinal stromal tumors (GISTs) are rare neoplasms of the gastrointestinal (GI) tract, accounting for approximately 1–2% of GI cancers. Hypoglycemia in patients with GIST is an uncommon and diagnostically challenging presentation, often involving a broad differential diagnosis. This case report explores the diagnostic difficulties encountered in managing persistent hypoglycemia in a patient with a history of advanced GIST.
Case Presentation
An 80-year-old male with a history of stage IV GIST, diagnosed in 2010, presented with persistent symptomatic hypoglycemia. His medical history included extensive abdominal disease, managed with multiple interventions: esophagogastrostomy, left lateral liver resection, a Whipple procedure, and Y-90 radioembolization. He received adjuvant imatinib therapy, which was discontinued in April 2024 due to significant adverse effects, including anasarca. In 2025, the patient developed progressive hypoglycemia, ultimately requiring continuous D10 infusion to maintain euglycemia, prompting an endocrinology evaluation. The initial diagnostic workup included cortisol, insulin, C-peptide levels, and IGF-1/IGF-2 ratio ruling out insulinoma, adrenal insufficiency, and GISTrelated paraneoplastic syndrome. Imaging studies, including PET and CT, showed no radiological evidence of recurrent GIST. Treatment with octreotide infusion resulted in minimal improvement, whereas daily corticosteroid therapy significantly alleviated the patient’s symptoms. The etiology of hypoglycemia remains elusive, with potential causes under consideration including Y-90 radioembolization-induced damage to glucagon-producing cells, immunotherapy-related adverse effects, or radiologically occult GIST. Insulin autoantibody testing is pending, and the case remains under active investigation, highlighting the diagnostic complexity of hypoglycemia in advanced GIST.
Discussion
Hypoglycemia in the context of GIST is a rare and poorly understood phenomenon. Potential mechanisms include paraneoplastic syndromes, such as non-islet cell tumor hypoglycemia (NICTH) mediated by IGF-2, or treatment-related effects, such as radiation-induced pancreatic or hepatic dysfunction. In this case, the absence of detectable IGF-2 abnormalities and negative imaging complicates the diagnosis. The lack of response to octreotide indicates that somatostatin receptor-mediated pathways may not be involved. The discontinuation of imatinib and prior Y-90 radioembolization further broadens the differential, as both could contribute to metabolic dysregulation.
Conclusions
This case illustrates the need for a systematic and multidisciplinary approach to evaluate hypoglycemia in patients with advanced GIST.
Background
Gastrointestinal stromal tumors (GISTs) are rare neoplasms of the gastrointestinal (GI) tract, accounting for approximately 1–2% of GI cancers. Hypoglycemia in patients with GIST is an uncommon and diagnostically challenging presentation, often involving a broad differential diagnosis. This case report explores the diagnostic difficulties encountered in managing persistent hypoglycemia in a patient with a history of advanced GIST.
Case Presentation
An 80-year-old male with a history of stage IV GIST, diagnosed in 2010, presented with persistent symptomatic hypoglycemia. His medical history included extensive abdominal disease, managed with multiple interventions: esophagogastrostomy, left lateral liver resection, a Whipple procedure, and Y-90 radioembolization. He received adjuvant imatinib therapy, which was discontinued in April 2024 due to significant adverse effects, including anasarca. In 2025, the patient developed progressive hypoglycemia, ultimately requiring continuous D10 infusion to maintain euglycemia, prompting an endocrinology evaluation. The initial diagnostic workup included cortisol, insulin, C-peptide levels, and IGF-1/IGF-2 ratio ruling out insulinoma, adrenal insufficiency, and GISTrelated paraneoplastic syndrome. Imaging studies, including PET and CT, showed no radiological evidence of recurrent GIST. Treatment with octreotide infusion resulted in minimal improvement, whereas daily corticosteroid therapy significantly alleviated the patient’s symptoms. The etiology of hypoglycemia remains elusive, with potential causes under consideration including Y-90 radioembolization-induced damage to glucagon-producing cells, immunotherapy-related adverse effects, or radiologically occult GIST. Insulin autoantibody testing is pending, and the case remains under active investigation, highlighting the diagnostic complexity of hypoglycemia in advanced GIST.
Discussion
Hypoglycemia in the context of GIST is a rare and poorly understood phenomenon. Potential mechanisms include paraneoplastic syndromes, such as non-islet cell tumor hypoglycemia (NICTH) mediated by IGF-2, or treatment-related effects, such as radiation-induced pancreatic or hepatic dysfunction. In this case, the absence of detectable IGF-2 abnormalities and negative imaging complicates the diagnosis. The lack of response to octreotide indicates that somatostatin receptor-mediated pathways may not be involved. The discontinuation of imatinib and prior Y-90 radioembolization further broadens the differential, as both could contribute to metabolic dysregulation.
Conclusions
This case illustrates the need for a systematic and multidisciplinary approach to evaluate hypoglycemia in patients with advanced GIST.
Checkpoint Inhibitor-Associated Optic Neuritis: A Rare irAE With Reversible Vision Loss
Background
Immune-related adverse events (irAEs) associated with checkpoint inhibitors can involve virtually any organ system. Optic neuritis is a rare but potentially reversible toxicity, with limited reports in the literature.
Case Presentation
A 57-year-old male with Stage IV poorly-differentiated neuroendocrine carcinoma presented with progressive bilateral vision loss following a near-complete response to four cycles of atezolizumab, carboplatin, and etoposide chemotherapy, and one cycle of maintenance atezolizumab. Symptoms began in the right eye and progressed to the left over 12 days. Neurological and ophthalmological evaluations included brain and orbital MRI, autoimmune panels, and infectious workup, all of which were unrevealing. The clinical picture remained consistent with isolated, immunemediated optic neuritis.
Discussion
High-dose intravenous methylprednisolone was initiated, resulting in gradual improvement and partial visual recovery by day four. An oral prednisone taper was prescribed for continued treatment. This is the second reported case of isolated optic neuritis associated with PD-L1 inhibitor therapy and the second with negative imaging findings. The rarity of this irAE and the absence of radiographic abnormalities may delay diagnosis and treatment.
Conclusions
Checkpoint-inhibitor-induced optic neuritis should be considered in patients with visual symptoms on immunotherapy, even in the setting of negative imaging. Early recognition and corticosteroid therapy are critical in preserving visual function.
Background
Immune-related adverse events (irAEs) associated with checkpoint inhibitors can involve virtually any organ system. Optic neuritis is a rare but potentially reversible toxicity, with limited reports in the literature.
Case Presentation
A 57-year-old male with Stage IV poorly-differentiated neuroendocrine carcinoma presented with progressive bilateral vision loss following a near-complete response to four cycles of atezolizumab, carboplatin, and etoposide chemotherapy, and one cycle of maintenance atezolizumab. Symptoms began in the right eye and progressed to the left over 12 days. Neurological and ophthalmological evaluations included brain and orbital MRI, autoimmune panels, and infectious workup, all of which were unrevealing. The clinical picture remained consistent with isolated, immunemediated optic neuritis.
Discussion
High-dose intravenous methylprednisolone was initiated, resulting in gradual improvement and partial visual recovery by day four. An oral prednisone taper was prescribed for continued treatment. This is the second reported case of isolated optic neuritis associated with PD-L1 inhibitor therapy and the second with negative imaging findings. The rarity of this irAE and the absence of radiographic abnormalities may delay diagnosis and treatment.
Conclusions
Checkpoint-inhibitor-induced optic neuritis should be considered in patients with visual symptoms on immunotherapy, even in the setting of negative imaging. Early recognition and corticosteroid therapy are critical in preserving visual function.
Background
Immune-related adverse events (irAEs) associated with checkpoint inhibitors can involve virtually any organ system. Optic neuritis is a rare but potentially reversible toxicity, with limited reports in the literature.
Case Presentation
A 57-year-old male with Stage IV poorly-differentiated neuroendocrine carcinoma presented with progressive bilateral vision loss following a near-complete response to four cycles of atezolizumab, carboplatin, and etoposide chemotherapy, and one cycle of maintenance atezolizumab. Symptoms began in the right eye and progressed to the left over 12 days. Neurological and ophthalmological evaluations included brain and orbital MRI, autoimmune panels, and infectious workup, all of which were unrevealing. The clinical picture remained consistent with isolated, immunemediated optic neuritis.
Discussion
High-dose intravenous methylprednisolone was initiated, resulting in gradual improvement and partial visual recovery by day four. An oral prednisone taper was prescribed for continued treatment. This is the second reported case of isolated optic neuritis associated with PD-L1 inhibitor therapy and the second with negative imaging findings. The rarity of this irAE and the absence of radiographic abnormalities may delay diagnosis and treatment.
Conclusions
Checkpoint-inhibitor-induced optic neuritis should be considered in patients with visual symptoms on immunotherapy, even in the setting of negative imaging. Early recognition and corticosteroid therapy are critical in preserving visual function.