User login
Case Series of Patients With Cardiac Amyloidosis at VA New York Harbor Healthcare-Brooklyn
Transthyretin amyloid cardiomyopathy (ATTR-CM) is caused by the misfolding of the TTR protein, which results in aggregation of amyloid fibrils that deposit in the myocardium and causes restrictive cardiomyopathy. Though it remains underdiagnosed, ATTR-CM is increasingly being recognized as a cause of heart failure in geriatric patients.1 There are 2 categories of ATTRCM: wild-type ATTR-CM (wtATTR-CM), in which there is no mutation in the TTR gene, and hereditary ATTR-CM (hATTR-CM), in which a mutation is present in the TTR gene. Research has shown that wtATTR-CM accounted for as many as 30% of cases of heart failure (HF) with preserved ejection fraction (HFpEF) in patients aged > 75 years.2 A significant percentage of the veteran patient population consists of older males. Given their age, these patients are at greater risk for ATTR diagnosis.3
Identifying red flags for patients within this population may allow clinicians to make earlier diagnoses and improve outcomes. A high index of suspicion is needed to diagnose ATTR because many early signs and symptoms are extracardiac, which leads to delayed diagnoses and worse outcomes. This article describes 8 cases of ATTR-CM within the US Department of Veterans Affairs (VA) New York Harbor Healthcare System-Brooklyn (VANYHHSB).
Methods
This retrospective case series was reviewed and approved by the VANYHHSB Institutional Review Board where it was conducted. Patients diagnosed with ATTR between 2017 and 2024 were identified using International Classification of Diseases, Tenth Revision codes. Eleven patients were identified; 3 were excluded due to insufficient medical records. The remaining 8 patient records were retrospectively reviewed and included.
Case 1
A 67-year-old male with a history of carpal tunnel syndrome (CTS) presented following a syncopal episode. Initial electrocardiogram (ECG) showed sinus rhythm, first-degree atrioventricular block, and a bifascicular block. Transthoracic echocardiogram (TTE) showed moderate asymmetric left ventricular (LV) hypertrophy (LVH) and biatrial enlargement with an ejection fraction (EF) > 55%. The patient was discharged with a loop recorder and an outpatient follow-up appointment scheduled. One month later, he presented with worsening dyspnea on exertion with clinical signs of hypervolemia. A repeat TTE showed global LV wall thickening, moderately reduced LV systolic function (EF 40%), and moderate pulmonary hypertension. Given these findings, the patient underwent cardiac magnetic resonance imaging (CMR), which suggested an infiltrative cardiomyopathy. Amyloid light-chain (AL) amyloidosis evaluation, technetium-99m (99mTC) pyrophosphate imaging, and a fat pad biopsy were unrevealing. An endomyocardial biopsy was performed with electron microscopy, which confirmed amyloidosis. Genetic testing was negative, and the patient began taking tafamidis. There were no later admissions for decompensated HF; however, the patient developed atrial fibrillation (AF) and an interval TTE demonstrated no improvement in his EF. He died at age 73 years.
Case 2
A 78-year-old male with a history of CTS presented with lightheadedness. Initial ECG showed rate-controlled AF and TTE revealed a moderately thickened LV wall with normal LV size, mild left atrial enlargement, and an EF of 65%. The patient was discharged with a scheduled outpatient CMR appointment; however, he defaulted from follow-up. Two years later, he presented with recurrent syncopal episodes and physical examination was consistent with hypervolemia. Repeat TTE revealed moderate LVH, biatrial enlargement, and an EF of 55%. An inpatient CMR was suggestive of cardiac amyloidosis, and a pyrophosphate scan was diagnostic for ATTR. The patient started taking tafamidis, but continued to have recurrent admissions for HF exacerbation. He died at age 81 years.
Case 3
A 71-year-old male with a history of CTS presented with exertional dyspnea. The initial ECG showed sinus rhythm with left atrial enlargement and left axis deviation. Subsequent TTE revealed severe LVH, mildly reduced LV cavity size, moderate to severe biatrial enlargement, and an EF of 25% to 30%. Outpatient 99mTC pyrophosphate imaging suggested cardiac amyloidosis, and laboratory testing showed no evidence of monoclonal proteins. The patient was started on tafamidis for ATTR. At 2-year follow-up, he had new AF and to date has had no further hospitalizations for acute decompensated HF.
Case 4
A 92-year-old male with a history of AF and bilateral CTS presented with lightheadedness. An ECG revealed AF with a slowed ventricular response. Subsequent Holter monitoring demonstrated pauses exceeding 3 seconds, and a permanent pacemaker was recommended. During his preoperative evaluation, TTE revealed severe concentric LVH with a speckled appearance of the myocardium, mild-tomoderate biatrial enlargement, and an EF of 50% to 55%. 99mTC pyrophosphate imaging was positive for amyloidosis and the patient started taking tafamidis. Recurrent hospital admissions for decompensated HF complicated his progression. The patient died at age 95 years.
Case 5
A 72-year-old male with a history of bilateral CTS and cervical spinal stenosis presented with dyspnea on exertion. An ECG revealed a normal sinus rhythm. A TTE found severely reduced systolic function with an EF ≤ 25%, mild concentric LVH, grade 3 diastolic dysfunction, mild-tomoderate biatrial enlargement, and moderate pulmonary hypertension (Figure 1). He was started on guideline-directed medical therapy (GDMT) for HF, which included sacubitril/valsartan, metoprolol succinate, and empagliflozin. The patient’s dyspnea improved, and a workup for nonischemic cardiomyopathy was initiated. 99mTC pyrophosphate imaging 1 year after his initial presentation was positive, leading to ATTR-CM diagnosis. The patient started taking tafamidis and he has since had a stable progression and continued to demonstrate good exercise tolerance with no hospitalizations. His most recent TTE indicated an EF of 40% to 45%.
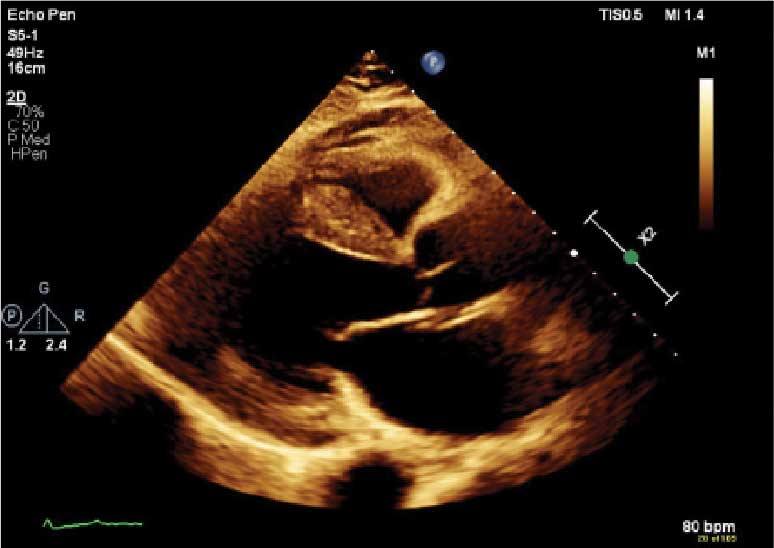
(parasternal long axis view) of patient 5
demonstrating concentric left ventricular
hypertrophy with a speckled myocardium
and dilated left atrium.
Case 6
A 76-year-old male with a history of paroxysmal AF status after multiple ablations, bilateral CTS, and severe cervical spinal stenosis presented with dyspnea on exertion. The patient’s ECG showed normal sinus rhythm and left axis deviation with concern for left anterior hemiblock. A TTE revealed moderate LVH with a speckled appearance of the myocardium and grade 3 diastolic dysfunction with preserved EF. Before completing the workup for underlying cardiomyopathy, the patient underwent an interventional radiology procedure for an angiomyolipoma, and his postoperative course was complicated by pulmonary edema, requiring admission to the coronary care unit for diuresis. A repeat TTE revealed a reduced EF of 35%, and he was discharged on GDMT. No monoclonal protein was seen in the serum or urine. The patient’s progression was complicated by recurrent admissions for acute decompensated HF and supraventricular tachycardia despite being on amiodarone, which led to a delay in obtaining 99mTC pyrophosphate imaging. Due to hypotension, the patient was unable to tolerate GDMT. He eventually underwent 99mTC pyrophosphate imaging that confirmed ATTR-CM and was started on tafamidis. One year following initial presentation, the patient’s EF progressively declined to 20% to 25%, and he died shortly after discharge to subacute rehabilitation.
Case 7
A 95-year-old male with a history of longstanding persistent AF and bilateral CTS presented with dyspnea on exertion, bendopnea, and worsening bilateral pedal edema for a week. An ECG showed AF with a controlled ventricular response and low-voltage QRS waves (Figure 2). A TTE showed biatrial enlargement, LVH, and preserved EF > 55%. He started taking furosemide and was discharged with a diagnosis of HFpEF. The patient missed cardiology follow-up and presented 1 year later with decompensated HF. An amyloidosis workup was recommended, but due to intermittent periods of being lost to follow-up, the patient did not pursue this workup until 3 years after his initial presentation when 99mTC pyrophosphate imaging confirmed ATTR-CM. The patient declined tafamidis and continued to be followed by the cardiology team. His HF is managed with furosemide as needed due to intolerance to GDMT.
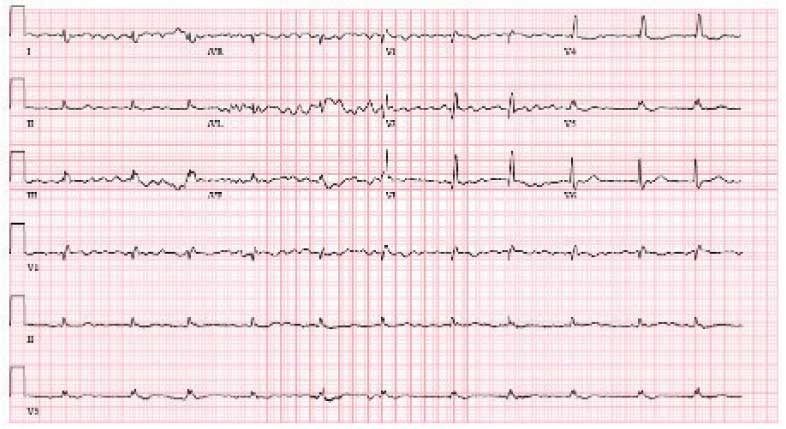
demonstrating atrial fibrillation with controlled
ventricular response and low-voltage QRS.
Case 8
A 74-year-old male with history of bilateral CTS presented with right-sided chest pain associated with shortness of breath, diaphoresis, dizziness, and worsening abdominal pain. He was found to have inferior wall myocardial infarction on ECG with later percutaneous coronary intervention to the left circumflex. His hospital course was complicated by decompensated HF (EF 45% to 50%) and AF with rapid ventricular response. He was treated and discharged with follow-up visits scheduled in the cardiology clinic. Multiple attempts were made to place him on GDMT; however, the patient was unable to tolerate these medications due to recurrent admissions for syncope. During a cardiology clinic visit 3 years after his initial presentation, an amyloidosis workup was initiated. 99mTC pyrophosphate imaging was positive for ATTR-CM, and he started taking tafamidis. Before this diagnosis, ECG indicated low-voltage QRS complexes. His progression has since been complicated by admissions for decompensated HF, recurrent episodes of AF requiring atrioventricular node ablation, and biventricular implantable cardioverter-defibrillator implantation after failed attempts at electrical cardioversion. He continued to follow up in the HF clinic.
Discussion
ATTR-CM is an underdiagnosed cause of cardiomyopathy, particularly in older adults. TTR is a transport protein produced in the liver, and misfolding can occur due to age-related instability of the wild-type protein (wtATTR) or pathologic variants in the TTR (hATTR). The misfolding leads to restrictive physiology, HF (often with preserved EF) arrhythmias, and conduction system disease.1 It is likely that the predominantly older male veteran population would be predisposed to wtATTR cardiomyopathy given that misfolding in the condition is believed to be age-related.
Current criteria for ATTR diagnosis require a combination of clinical suspicion, imaging, laboratory testing, and, in some cases, tissue biopsy confirmation and genetic testing.1,4,5 The diagnostic algorithm is as follows:
Clinical suspicion. Consider ATTR in patients with unexplained HFpEF, LV wall thickness ≥ 12 to 14 mm, discordance between ECG voltage and wall thickness, or associated extracardiac manifestations such as CTS, lumbar spinal stenosis, or peripheral/ autonomic neuropathy.
Exclusion of AL amyloidosis. Bone scintigraphy alone cannot distinguish between AL amyloidosis and ATTR, so patients with suspected amyloid cardiomyopathy should undergo serum and urine immunofixation electrophoresis and serum free light chain assay to rule out a monoclonal gammopathy as seen in AL amyloidosis.
Cardiac scintigraphy. Once AL amyloidosis is excluded, a positive 99mTC-labeled boneavid tracer image (such as a pyrophosphate scan) with grade 2 or grade 3 myocardial uptake is diagnostic of ATTR.
Tissue biopsy. If there is monoclonal gammopathy or equivocal imaging, tissue biopsy (eg, endomyocardial) with Congo red staining and amyloid typing by mass spectrometry or immunohistochemistry is necessary.
Genetic testing. Once ATTR is confirmed, genetic testing distinguishes hATTR from wtATTR, which impacts management and determines the need to screen family members.
Currently, there are 3 therapies approved by the US Food and Drug Administration (FDA) to treat ATTR-CM: tafamidis, acoramidis, and vutrisiran.6-8 Tafamidis and acoramidis stabilize the TTR tetramer, preventing amyloid formation. Vutrisiran uses RNA interference to silence the gene that produces TTR. Tafamidis has been found to improve cardiovascular outcomes in ATTR-CM. In the ATTR-ACT trial, it reduced all-cause mortality and cardiovascular hospitalizations in patients with ATTR-CM and New York Heart Association class I-III symptoms.6 There are other disease-modifying therapies, such the TTR gene silencers inotersen and patisiran; however, these are only FDA-approved for hATTR polyneuropathy and not for ATTRCM; ongoing trials are evaluating their cardiac efficacy.
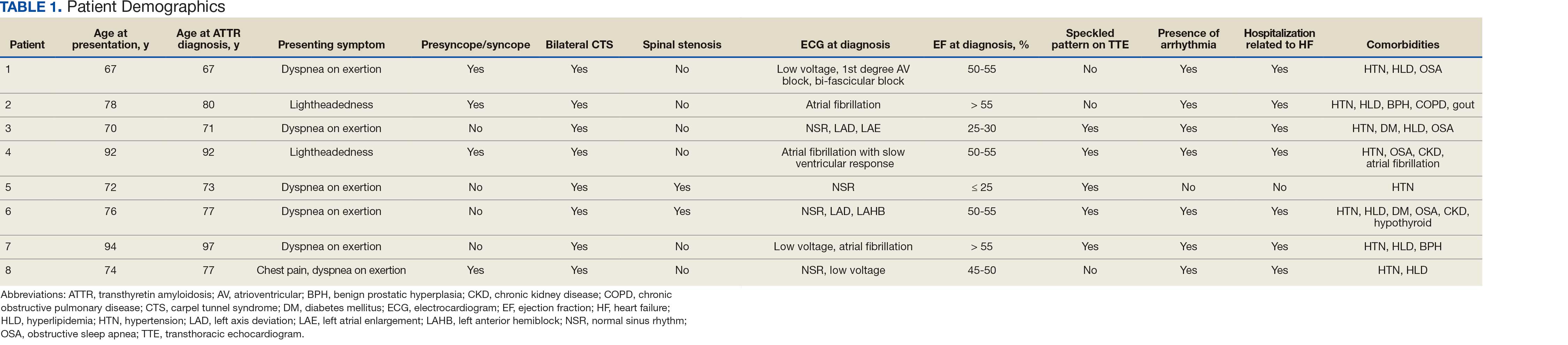
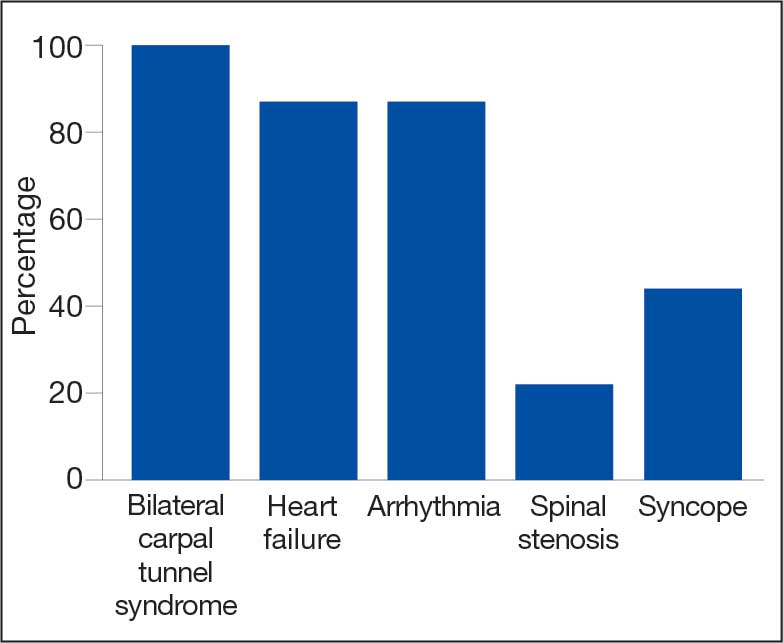
The mean age of ATTR-CM diagnosis in the patients described in this case series was 79 years, which is older than the mean age of 74 years reported in prior research.4 All patients were male, and the most common presenting symptom was dyspnea on exertion. Table 1 outlines baseline characteristics and associated comorbidities of patients in this case series. The patients presented with many red-flag signs of ATTR-CM (Figure 3). Among them included syncope (4 of 8 patients), spinal stenosis (2 of 8 patients), arrhythmia (7 of 8 patients), heart failure (7 of 8 patients), and bilateral CTS (all patients).
amyloidosis red-flag signs identified.
Bilateral CTS was diagnosed in all patients before their diagnosis of ATTR-CM. Patient 7 had a diagnosis of CTS 9 years before his ATTR-CM diagnosis, underscoring a subtle, yet important extracardiac sign that may increase clinical suspicion for ATTR-CM. Previous research found that the probability of having CTS is highest 5 to 9 years prior to the development of cardiomyopathy. Its presence is also a prognostic marker in ATTR, independent of cardiac involvement.9 The median interval between CTS diagnosis and cardiomyopathy diagnosis was 5 years in this series.
Although screening criteria for ATTR have been proposed, none have been incorporated into formal guidelines.10,11 We propose that a baseline ECG and screening TTE be obtained in any patient aged > 65 years with cardiac risk equivalents such as hypertension and diabetes, presenting with ≥ 1 extracardiac red-flag signs, such as bilateral CTS and spinal stenosis. This will likely facilitate an earlier diagnosis of ATTR-CM, leading to earlier treatment initiation and better patient outcomes. This initiative can be started in the primary care setting and facilitates early cardiology referral. This recommendation is based on literature supporting clinical patterns and observations the authors have made in clinical practice.
Obstructive sleep apnea (OSA) was a comorbidity present in 50% of the patients described in this case series. The literature describing this association is sparse; however, a prospective observational study reported that disorders of sleep inclusive of OSA are frequent in patients with cardiac amyloidosis.12 A theory behind this association is that amyloid deposits in the upper airway tissues lead to airway narrowing. 13 More research is needed to further assess the relationship between OSA and cardiac amyloidosis, particularly with respect to the timing of OSA prior to the development of cardiomyopathy as it may be a potential early sign for clinicians to acknowledge.
An important observation from this case series is the variability in the timing of tafamidis initiation relative to symptom onset and confirmed diagnosis of ATTR-CM (Table 2). Although tafamidis has been found to slow disease progression, several patients in this series began treatment at advanced stages or years after the onset of cardiac symptoms, potentially limiting its clinical benefit.
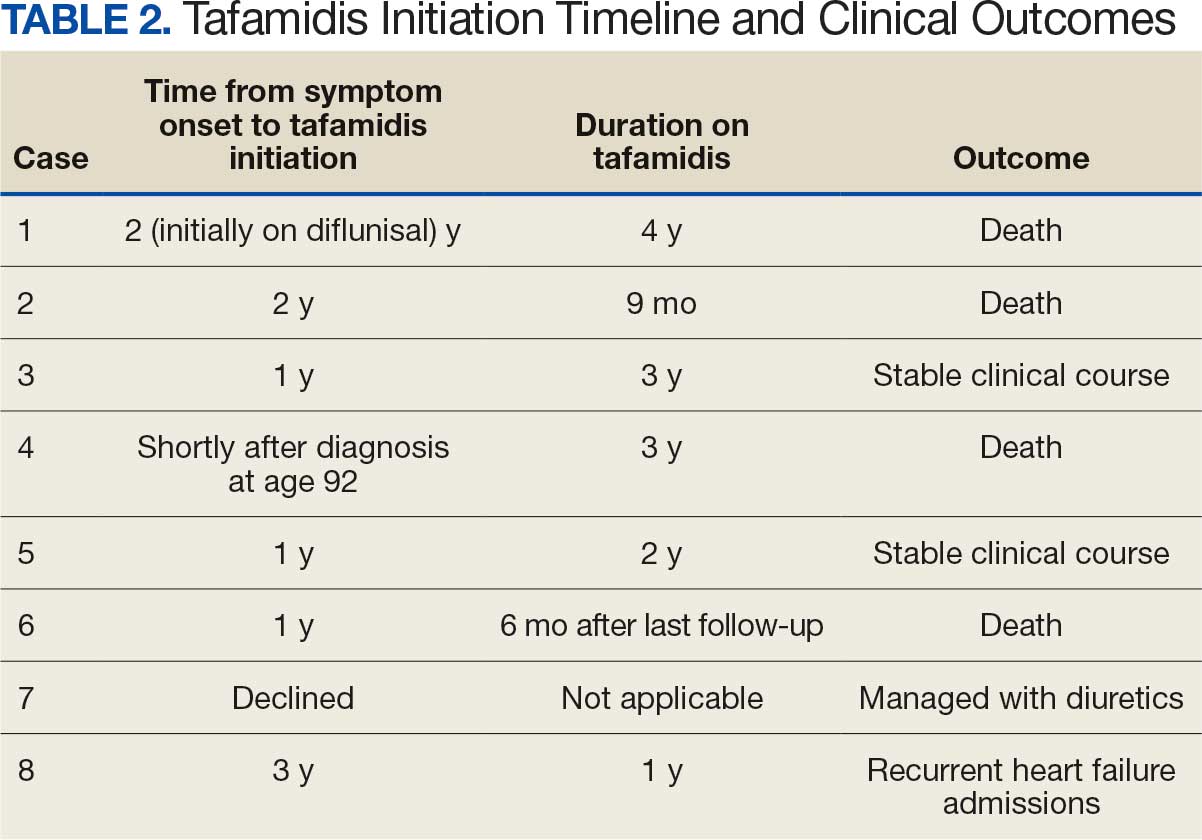
For example, patient 1 was diagnosed with ATTR 2 years before tafamidis became available on the market and was initially treated with diflunisal. Patients who started tafamidis earlier in the disease course (eg, patients 3 and 5) appeared to have better long-term outcomes, including an absence of heart failure hospitalizations after initiation. In contrast, patients with delayed treatment initiation (eg, patients 2, 6, and 8) experienced ongoing decompensations or early mortality. Patient 7 declined tafamidis, underscoring challenges in medication uptake among older adults. Randomized controlled trials are warranted to compare the effectiveness of tafamidis with other recently FDA-approved therapies, such as acoramidis and vutrisiran, particularly in terms of cardiovascular outcomes.
AF was the most common arrhythmia observed in these patients and may occur years before the development of HF symptoms in the setting of ATTR-CM. Due to inherent conduction system disease, AF in this population may have a controlled or slow ventricular response, as observed in patient 4. Patients with atrial fibrillation and cardiac amyloidosis should receive anticoagulation regardless of CHA2DS2- VASc score due to their high risk of intracardiac thrombus formation.4
Limitations
This case series lacked genetic testing following a confirmed ATTR-CM diagnosis. Although much of the treatment is the same regardless of the presence of a TTR mutation, knowing the specific subtype of ATTR-CM has implications for prognosis and for screening family members.
Conclusions
Following analysis of 8 patients diagnosed with ATTR, this case series could serve as a blueprint for research into ATTR in veterans. In clinical practice, following military service, veterans may not be routinely seen by an outpatient physician and may present with sequelae of advanced stages of ATTR. Early identification of red-flag symptoms can lead to a higher clinical suspicion, prompting early diagnostic evaluation and treatment initiation and ultimately mitigating adverse outcomes. Future research that includes genetic testing for those with confirmed ATTR-CM may prove useful as a foundation for detailed and informed discussions with patients and their families regarding prognosis and, if indicated, screening for family members.
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142:e7-e22. doi:10.1161/CIR.0000000000000792
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765-774. doi:10.1111/j.1532-5415.2011.03868.x
- Nativi-Nicolau J, Redd A, Kelly N, et al. Increasing number of amyloidosis diagnosis in the Veterans Affairs populations. J Card Fail. 2019;25:S96.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872-2891. doi:10.1016/j.jacc.2019.04.003
- Gillmore JD, Maurer, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404-2412. doi:10.1161/CIRCULATIONAHA.116.021612
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007-1016. doi:10.1056/NEJMoa1805689
- Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med. 2024;390:132-142. doi:10.1056/NEJMoa2305434
- Fontana M, Berk JL, Gillmore JD, et al. Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J Med. 2025;392:33-44. doi:10.1056/NEJMoa2409134
- Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22:507-515. doi:10.1002/ejhf.1742
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554-1568. doi:10.1093/eurheartj/ehab072
- Brito D, Albrecht FC, de Arenaza DP, et al. World Heart Federation consensus on transthyretin amyloidosis cardiomyopathy (ATTR-CM). Glob Heart. 2023;18:59. doi:10.5334/gh.1262
- Bodez D, Guellich A, Kharoubi M, et al. Prevalence, severity, and prognostic value of sleep apnea syndromes in cardiac amyloidosis. Sleep. 2016;39:1333-1341. doi:10.5665/sleep.5958
- Colaco B, Colaco C, Lipford M. A forgotten problem: sleep-disordered breathing in amyloidosis. Chest. 2016;150:1293A. doi:10.1016/j.chest.2016.08.1407
Transthyretin amyloid cardiomyopathy (ATTR-CM) is caused by the misfolding of the TTR protein, which results in aggregation of amyloid fibrils that deposit in the myocardium and causes restrictive cardiomyopathy. Though it remains underdiagnosed, ATTR-CM is increasingly being recognized as a cause of heart failure in geriatric patients.1 There are 2 categories of ATTRCM: wild-type ATTR-CM (wtATTR-CM), in which there is no mutation in the TTR gene, and hereditary ATTR-CM (hATTR-CM), in which a mutation is present in the TTR gene. Research has shown that wtATTR-CM accounted for as many as 30% of cases of heart failure (HF) with preserved ejection fraction (HFpEF) in patients aged > 75 years.2 A significant percentage of the veteran patient population consists of older males. Given their age, these patients are at greater risk for ATTR diagnosis.3
Identifying red flags for patients within this population may allow clinicians to make earlier diagnoses and improve outcomes. A high index of suspicion is needed to diagnose ATTR because many early signs and symptoms are extracardiac, which leads to delayed diagnoses and worse outcomes. This article describes 8 cases of ATTR-CM within the US Department of Veterans Affairs (VA) New York Harbor Healthcare System-Brooklyn (VANYHHSB).
Methods
This retrospective case series was reviewed and approved by the VANYHHSB Institutional Review Board where it was conducted. Patients diagnosed with ATTR between 2017 and 2024 were identified using International Classification of Diseases, Tenth Revision codes. Eleven patients were identified; 3 were excluded due to insufficient medical records. The remaining 8 patient records were retrospectively reviewed and included.
Case 1
A 67-year-old male with a history of carpal tunnel syndrome (CTS) presented following a syncopal episode. Initial electrocardiogram (ECG) showed sinus rhythm, first-degree atrioventricular block, and a bifascicular block. Transthoracic echocardiogram (TTE) showed moderate asymmetric left ventricular (LV) hypertrophy (LVH) and biatrial enlargement with an ejection fraction (EF) > 55%. The patient was discharged with a loop recorder and an outpatient follow-up appointment scheduled. One month later, he presented with worsening dyspnea on exertion with clinical signs of hypervolemia. A repeat TTE showed global LV wall thickening, moderately reduced LV systolic function (EF 40%), and moderate pulmonary hypertension. Given these findings, the patient underwent cardiac magnetic resonance imaging (CMR), which suggested an infiltrative cardiomyopathy. Amyloid light-chain (AL) amyloidosis evaluation, technetium-99m (99mTC) pyrophosphate imaging, and a fat pad biopsy were unrevealing. An endomyocardial biopsy was performed with electron microscopy, which confirmed amyloidosis. Genetic testing was negative, and the patient began taking tafamidis. There were no later admissions for decompensated HF; however, the patient developed atrial fibrillation (AF) and an interval TTE demonstrated no improvement in his EF. He died at age 73 years.
Case 2
A 78-year-old male with a history of CTS presented with lightheadedness. Initial ECG showed rate-controlled AF and TTE revealed a moderately thickened LV wall with normal LV size, mild left atrial enlargement, and an EF of 65%. The patient was discharged with a scheduled outpatient CMR appointment; however, he defaulted from follow-up. Two years later, he presented with recurrent syncopal episodes and physical examination was consistent with hypervolemia. Repeat TTE revealed moderate LVH, biatrial enlargement, and an EF of 55%. An inpatient CMR was suggestive of cardiac amyloidosis, and a pyrophosphate scan was diagnostic for ATTR. The patient started taking tafamidis, but continued to have recurrent admissions for HF exacerbation. He died at age 81 years.
Case 3
A 71-year-old male with a history of CTS presented with exertional dyspnea. The initial ECG showed sinus rhythm with left atrial enlargement and left axis deviation. Subsequent TTE revealed severe LVH, mildly reduced LV cavity size, moderate to severe biatrial enlargement, and an EF of 25% to 30%. Outpatient 99mTC pyrophosphate imaging suggested cardiac amyloidosis, and laboratory testing showed no evidence of monoclonal proteins. The patient was started on tafamidis for ATTR. At 2-year follow-up, he had new AF and to date has had no further hospitalizations for acute decompensated HF.
Case 4
A 92-year-old male with a history of AF and bilateral CTS presented with lightheadedness. An ECG revealed AF with a slowed ventricular response. Subsequent Holter monitoring demonstrated pauses exceeding 3 seconds, and a permanent pacemaker was recommended. During his preoperative evaluation, TTE revealed severe concentric LVH with a speckled appearance of the myocardium, mild-tomoderate biatrial enlargement, and an EF of 50% to 55%. 99mTC pyrophosphate imaging was positive for amyloidosis and the patient started taking tafamidis. Recurrent hospital admissions for decompensated HF complicated his progression. The patient died at age 95 years.
Case 5
A 72-year-old male with a history of bilateral CTS and cervical spinal stenosis presented with dyspnea on exertion. An ECG revealed a normal sinus rhythm. A TTE found severely reduced systolic function with an EF ≤ 25%, mild concentric LVH, grade 3 diastolic dysfunction, mild-tomoderate biatrial enlargement, and moderate pulmonary hypertension (Figure 1). He was started on guideline-directed medical therapy (GDMT) for HF, which included sacubitril/valsartan, metoprolol succinate, and empagliflozin. The patient’s dyspnea improved, and a workup for nonischemic cardiomyopathy was initiated. 99mTC pyrophosphate imaging 1 year after his initial presentation was positive, leading to ATTR-CM diagnosis. The patient started taking tafamidis and he has since had a stable progression and continued to demonstrate good exercise tolerance with no hospitalizations. His most recent TTE indicated an EF of 40% to 45%.
(parasternal long axis view) of patient 5
demonstrating concentric left ventricular
hypertrophy with a speckled myocardium
and dilated left atrium.
Case 6
A 76-year-old male with a history of paroxysmal AF status after multiple ablations, bilateral CTS, and severe cervical spinal stenosis presented with dyspnea on exertion. The patient’s ECG showed normal sinus rhythm and left axis deviation with concern for left anterior hemiblock. A TTE revealed moderate LVH with a speckled appearance of the myocardium and grade 3 diastolic dysfunction with preserved EF. Before completing the workup for underlying cardiomyopathy, the patient underwent an interventional radiology procedure for an angiomyolipoma, and his postoperative course was complicated by pulmonary edema, requiring admission to the coronary care unit for diuresis. A repeat TTE revealed a reduced EF of 35%, and he was discharged on GDMT. No monoclonal protein was seen in the serum or urine. The patient’s progression was complicated by recurrent admissions for acute decompensated HF and supraventricular tachycardia despite being on amiodarone, which led to a delay in obtaining 99mTC pyrophosphate imaging. Due to hypotension, the patient was unable to tolerate GDMT. He eventually underwent 99mTC pyrophosphate imaging that confirmed ATTR-CM and was started on tafamidis. One year following initial presentation, the patient’s EF progressively declined to 20% to 25%, and he died shortly after discharge to subacute rehabilitation.
Case 7
A 95-year-old male with a history of longstanding persistent AF and bilateral CTS presented with dyspnea on exertion, bendopnea, and worsening bilateral pedal edema for a week. An ECG showed AF with a controlled ventricular response and low-voltage QRS waves (Figure 2). A TTE showed biatrial enlargement, LVH, and preserved EF > 55%. He started taking furosemide and was discharged with a diagnosis of HFpEF. The patient missed cardiology follow-up and presented 1 year later with decompensated HF. An amyloidosis workup was recommended, but due to intermittent periods of being lost to follow-up, the patient did not pursue this workup until 3 years after his initial presentation when 99mTC pyrophosphate imaging confirmed ATTR-CM. The patient declined tafamidis and continued to be followed by the cardiology team. His HF is managed with furosemide as needed due to intolerance to GDMT.
demonstrating atrial fibrillation with controlled
ventricular response and low-voltage QRS.
Case 8
A 74-year-old male with history of bilateral CTS presented with right-sided chest pain associated with shortness of breath, diaphoresis, dizziness, and worsening abdominal pain. He was found to have inferior wall myocardial infarction on ECG with later percutaneous coronary intervention to the left circumflex. His hospital course was complicated by decompensated HF (EF 45% to 50%) and AF with rapid ventricular response. He was treated and discharged with follow-up visits scheduled in the cardiology clinic. Multiple attempts were made to place him on GDMT; however, the patient was unable to tolerate these medications due to recurrent admissions for syncope. During a cardiology clinic visit 3 years after his initial presentation, an amyloidosis workup was initiated. 99mTC pyrophosphate imaging was positive for ATTR-CM, and he started taking tafamidis. Before this diagnosis, ECG indicated low-voltage QRS complexes. His progression has since been complicated by admissions for decompensated HF, recurrent episodes of AF requiring atrioventricular node ablation, and biventricular implantable cardioverter-defibrillator implantation after failed attempts at electrical cardioversion. He continued to follow up in the HF clinic.
Discussion
ATTR-CM is an underdiagnosed cause of cardiomyopathy, particularly in older adults. TTR is a transport protein produced in the liver, and misfolding can occur due to age-related instability of the wild-type protein (wtATTR) or pathologic variants in the TTR (hATTR). The misfolding leads to restrictive physiology, HF (often with preserved EF) arrhythmias, and conduction system disease.1 It is likely that the predominantly older male veteran population would be predisposed to wtATTR cardiomyopathy given that misfolding in the condition is believed to be age-related.
Current criteria for ATTR diagnosis require a combination of clinical suspicion, imaging, laboratory testing, and, in some cases, tissue biopsy confirmation and genetic testing.1,4,5 The diagnostic algorithm is as follows:
Clinical suspicion. Consider ATTR in patients with unexplained HFpEF, LV wall thickness ≥ 12 to 14 mm, discordance between ECG voltage and wall thickness, or associated extracardiac manifestations such as CTS, lumbar spinal stenosis, or peripheral/ autonomic neuropathy.
Exclusion of AL amyloidosis. Bone scintigraphy alone cannot distinguish between AL amyloidosis and ATTR, so patients with suspected amyloid cardiomyopathy should undergo serum and urine immunofixation electrophoresis and serum free light chain assay to rule out a monoclonal gammopathy as seen in AL amyloidosis.
Cardiac scintigraphy. Once AL amyloidosis is excluded, a positive 99mTC-labeled boneavid tracer image (such as a pyrophosphate scan) with grade 2 or grade 3 myocardial uptake is diagnostic of ATTR.
Tissue biopsy. If there is monoclonal gammopathy or equivocal imaging, tissue biopsy (eg, endomyocardial) with Congo red staining and amyloid typing by mass spectrometry or immunohistochemistry is necessary.
Genetic testing. Once ATTR is confirmed, genetic testing distinguishes hATTR from wtATTR, which impacts management and determines the need to screen family members.
Currently, there are 3 therapies approved by the US Food and Drug Administration (FDA) to treat ATTR-CM: tafamidis, acoramidis, and vutrisiran.6-8 Tafamidis and acoramidis stabilize the TTR tetramer, preventing amyloid formation. Vutrisiran uses RNA interference to silence the gene that produces TTR. Tafamidis has been found to improve cardiovascular outcomes in ATTR-CM. In the ATTR-ACT trial, it reduced all-cause mortality and cardiovascular hospitalizations in patients with ATTR-CM and New York Heart Association class I-III symptoms.6 There are other disease-modifying therapies, such the TTR gene silencers inotersen and patisiran; however, these are only FDA-approved for hATTR polyneuropathy and not for ATTRCM; ongoing trials are evaluating their cardiac efficacy.
The mean age of ATTR-CM diagnosis in the patients described in this case series was 79 years, which is older than the mean age of 74 years reported in prior research.4 All patients were male, and the most common presenting symptom was dyspnea on exertion. Table 1 outlines baseline characteristics and associated comorbidities of patients in this case series. The patients presented with many red-flag signs of ATTR-CM (Figure 3). Among them included syncope (4 of 8 patients), spinal stenosis (2 of 8 patients), arrhythmia (7 of 8 patients), heart failure (7 of 8 patients), and bilateral CTS (all patients).
amyloidosis red-flag signs identified.
Bilateral CTS was diagnosed in all patients before their diagnosis of ATTR-CM. Patient 7 had a diagnosis of CTS 9 years before his ATTR-CM diagnosis, underscoring a subtle, yet important extracardiac sign that may increase clinical suspicion for ATTR-CM. Previous research found that the probability of having CTS is highest 5 to 9 years prior to the development of cardiomyopathy. Its presence is also a prognostic marker in ATTR, independent of cardiac involvement.9 The median interval between CTS diagnosis and cardiomyopathy diagnosis was 5 years in this series.
Although screening criteria for ATTR have been proposed, none have been incorporated into formal guidelines.10,11 We propose that a baseline ECG and screening TTE be obtained in any patient aged > 65 years with cardiac risk equivalents such as hypertension and diabetes, presenting with ≥ 1 extracardiac red-flag signs, such as bilateral CTS and spinal stenosis. This will likely facilitate an earlier diagnosis of ATTR-CM, leading to earlier treatment initiation and better patient outcomes. This initiative can be started in the primary care setting and facilitates early cardiology referral. This recommendation is based on literature supporting clinical patterns and observations the authors have made in clinical practice.
Obstructive sleep apnea (OSA) was a comorbidity present in 50% of the patients described in this case series. The literature describing this association is sparse; however, a prospective observational study reported that disorders of sleep inclusive of OSA are frequent in patients with cardiac amyloidosis.12 A theory behind this association is that amyloid deposits in the upper airway tissues lead to airway narrowing. 13 More research is needed to further assess the relationship between OSA and cardiac amyloidosis, particularly with respect to the timing of OSA prior to the development of cardiomyopathy as it may be a potential early sign for clinicians to acknowledge.
An important observation from this case series is the variability in the timing of tafamidis initiation relative to symptom onset and confirmed diagnosis of ATTR-CM (Table 2). Although tafamidis has been found to slow disease progression, several patients in this series began treatment at advanced stages or years after the onset of cardiac symptoms, potentially limiting its clinical benefit.
For example, patient 1 was diagnosed with ATTR 2 years before tafamidis became available on the market and was initially treated with diflunisal. Patients who started tafamidis earlier in the disease course (eg, patients 3 and 5) appeared to have better long-term outcomes, including an absence of heart failure hospitalizations after initiation. In contrast, patients with delayed treatment initiation (eg, patients 2, 6, and 8) experienced ongoing decompensations or early mortality. Patient 7 declined tafamidis, underscoring challenges in medication uptake among older adults. Randomized controlled trials are warranted to compare the effectiveness of tafamidis with other recently FDA-approved therapies, such as acoramidis and vutrisiran, particularly in terms of cardiovascular outcomes.
AF was the most common arrhythmia observed in these patients and may occur years before the development of HF symptoms in the setting of ATTR-CM. Due to inherent conduction system disease, AF in this population may have a controlled or slow ventricular response, as observed in patient 4. Patients with atrial fibrillation and cardiac amyloidosis should receive anticoagulation regardless of CHA2DS2- VASc score due to their high risk of intracardiac thrombus formation.4
Limitations
This case series lacked genetic testing following a confirmed ATTR-CM diagnosis. Although much of the treatment is the same regardless of the presence of a TTR mutation, knowing the specific subtype of ATTR-CM has implications for prognosis and for screening family members.
Conclusions
Following analysis of 8 patients diagnosed with ATTR, this case series could serve as a blueprint for research into ATTR in veterans. In clinical practice, following military service, veterans may not be routinely seen by an outpatient physician and may present with sequelae of advanced stages of ATTR. Early identification of red-flag symptoms can lead to a higher clinical suspicion, prompting early diagnostic evaluation and treatment initiation and ultimately mitigating adverse outcomes. Future research that includes genetic testing for those with confirmed ATTR-CM may prove useful as a foundation for detailed and informed discussions with patients and their families regarding prognosis and, if indicated, screening for family members.
Transthyretin amyloid cardiomyopathy (ATTR-CM) is caused by the misfolding of the TTR protein, which results in aggregation of amyloid fibrils that deposit in the myocardium and causes restrictive cardiomyopathy. Though it remains underdiagnosed, ATTR-CM is increasingly being recognized as a cause of heart failure in geriatric patients.1 There are 2 categories of ATTRCM: wild-type ATTR-CM (wtATTR-CM), in which there is no mutation in the TTR gene, and hereditary ATTR-CM (hATTR-CM), in which a mutation is present in the TTR gene. Research has shown that wtATTR-CM accounted for as many as 30% of cases of heart failure (HF) with preserved ejection fraction (HFpEF) in patients aged > 75 years.2 A significant percentage of the veteran patient population consists of older males. Given their age, these patients are at greater risk for ATTR diagnosis.3
Identifying red flags for patients within this population may allow clinicians to make earlier diagnoses and improve outcomes. A high index of suspicion is needed to diagnose ATTR because many early signs and symptoms are extracardiac, which leads to delayed diagnoses and worse outcomes. This article describes 8 cases of ATTR-CM within the US Department of Veterans Affairs (VA) New York Harbor Healthcare System-Brooklyn (VANYHHSB).
Methods
This retrospective case series was reviewed and approved by the VANYHHSB Institutional Review Board where it was conducted. Patients diagnosed with ATTR between 2017 and 2024 were identified using International Classification of Diseases, Tenth Revision codes. Eleven patients were identified; 3 were excluded due to insufficient medical records. The remaining 8 patient records were retrospectively reviewed and included.
Case 1
A 67-year-old male with a history of carpal tunnel syndrome (CTS) presented following a syncopal episode. Initial electrocardiogram (ECG) showed sinus rhythm, first-degree atrioventricular block, and a bifascicular block. Transthoracic echocardiogram (TTE) showed moderate asymmetric left ventricular (LV) hypertrophy (LVH) and biatrial enlargement with an ejection fraction (EF) > 55%. The patient was discharged with a loop recorder and an outpatient follow-up appointment scheduled. One month later, he presented with worsening dyspnea on exertion with clinical signs of hypervolemia. A repeat TTE showed global LV wall thickening, moderately reduced LV systolic function (EF 40%), and moderate pulmonary hypertension. Given these findings, the patient underwent cardiac magnetic resonance imaging (CMR), which suggested an infiltrative cardiomyopathy. Amyloid light-chain (AL) amyloidosis evaluation, technetium-99m (99mTC) pyrophosphate imaging, and a fat pad biopsy were unrevealing. An endomyocardial biopsy was performed with electron microscopy, which confirmed amyloidosis. Genetic testing was negative, and the patient began taking tafamidis. There were no later admissions for decompensated HF; however, the patient developed atrial fibrillation (AF) and an interval TTE demonstrated no improvement in his EF. He died at age 73 years.
Case 2
A 78-year-old male with a history of CTS presented with lightheadedness. Initial ECG showed rate-controlled AF and TTE revealed a moderately thickened LV wall with normal LV size, mild left atrial enlargement, and an EF of 65%. The patient was discharged with a scheduled outpatient CMR appointment; however, he defaulted from follow-up. Two years later, he presented with recurrent syncopal episodes and physical examination was consistent with hypervolemia. Repeat TTE revealed moderate LVH, biatrial enlargement, and an EF of 55%. An inpatient CMR was suggestive of cardiac amyloidosis, and a pyrophosphate scan was diagnostic for ATTR. The patient started taking tafamidis, but continued to have recurrent admissions for HF exacerbation. He died at age 81 years.
Case 3
A 71-year-old male with a history of CTS presented with exertional dyspnea. The initial ECG showed sinus rhythm with left atrial enlargement and left axis deviation. Subsequent TTE revealed severe LVH, mildly reduced LV cavity size, moderate to severe biatrial enlargement, and an EF of 25% to 30%. Outpatient 99mTC pyrophosphate imaging suggested cardiac amyloidosis, and laboratory testing showed no evidence of monoclonal proteins. The patient was started on tafamidis for ATTR. At 2-year follow-up, he had new AF and to date has had no further hospitalizations for acute decompensated HF.
Case 4
A 92-year-old male with a history of AF and bilateral CTS presented with lightheadedness. An ECG revealed AF with a slowed ventricular response. Subsequent Holter monitoring demonstrated pauses exceeding 3 seconds, and a permanent pacemaker was recommended. During his preoperative evaluation, TTE revealed severe concentric LVH with a speckled appearance of the myocardium, mild-tomoderate biatrial enlargement, and an EF of 50% to 55%. 99mTC pyrophosphate imaging was positive for amyloidosis and the patient started taking tafamidis. Recurrent hospital admissions for decompensated HF complicated his progression. The patient died at age 95 years.
Case 5
A 72-year-old male with a history of bilateral CTS and cervical spinal stenosis presented with dyspnea on exertion. An ECG revealed a normal sinus rhythm. A TTE found severely reduced systolic function with an EF ≤ 25%, mild concentric LVH, grade 3 diastolic dysfunction, mild-tomoderate biatrial enlargement, and moderate pulmonary hypertension (Figure 1). He was started on guideline-directed medical therapy (GDMT) for HF, which included sacubitril/valsartan, metoprolol succinate, and empagliflozin. The patient’s dyspnea improved, and a workup for nonischemic cardiomyopathy was initiated. 99mTC pyrophosphate imaging 1 year after his initial presentation was positive, leading to ATTR-CM diagnosis. The patient started taking tafamidis and he has since had a stable progression and continued to demonstrate good exercise tolerance with no hospitalizations. His most recent TTE indicated an EF of 40% to 45%.
(parasternal long axis view) of patient 5
demonstrating concentric left ventricular
hypertrophy with a speckled myocardium
and dilated left atrium.
Case 6
A 76-year-old male with a history of paroxysmal AF status after multiple ablations, bilateral CTS, and severe cervical spinal stenosis presented with dyspnea on exertion. The patient’s ECG showed normal sinus rhythm and left axis deviation with concern for left anterior hemiblock. A TTE revealed moderate LVH with a speckled appearance of the myocardium and grade 3 diastolic dysfunction with preserved EF. Before completing the workup for underlying cardiomyopathy, the patient underwent an interventional radiology procedure for an angiomyolipoma, and his postoperative course was complicated by pulmonary edema, requiring admission to the coronary care unit for diuresis. A repeat TTE revealed a reduced EF of 35%, and he was discharged on GDMT. No monoclonal protein was seen in the serum or urine. The patient’s progression was complicated by recurrent admissions for acute decompensated HF and supraventricular tachycardia despite being on amiodarone, which led to a delay in obtaining 99mTC pyrophosphate imaging. Due to hypotension, the patient was unable to tolerate GDMT. He eventually underwent 99mTC pyrophosphate imaging that confirmed ATTR-CM and was started on tafamidis. One year following initial presentation, the patient’s EF progressively declined to 20% to 25%, and he died shortly after discharge to subacute rehabilitation.
Case 7
A 95-year-old male with a history of longstanding persistent AF and bilateral CTS presented with dyspnea on exertion, bendopnea, and worsening bilateral pedal edema for a week. An ECG showed AF with a controlled ventricular response and low-voltage QRS waves (Figure 2). A TTE showed biatrial enlargement, LVH, and preserved EF > 55%. He started taking furosemide and was discharged with a diagnosis of HFpEF. The patient missed cardiology follow-up and presented 1 year later with decompensated HF. An amyloidosis workup was recommended, but due to intermittent periods of being lost to follow-up, the patient did not pursue this workup until 3 years after his initial presentation when 99mTC pyrophosphate imaging confirmed ATTR-CM. The patient declined tafamidis and continued to be followed by the cardiology team. His HF is managed with furosemide as needed due to intolerance to GDMT.
demonstrating atrial fibrillation with controlled
ventricular response and low-voltage QRS.
Case 8
A 74-year-old male with history of bilateral CTS presented with right-sided chest pain associated with shortness of breath, diaphoresis, dizziness, and worsening abdominal pain. He was found to have inferior wall myocardial infarction on ECG with later percutaneous coronary intervention to the left circumflex. His hospital course was complicated by decompensated HF (EF 45% to 50%) and AF with rapid ventricular response. He was treated and discharged with follow-up visits scheduled in the cardiology clinic. Multiple attempts were made to place him on GDMT; however, the patient was unable to tolerate these medications due to recurrent admissions for syncope. During a cardiology clinic visit 3 years after his initial presentation, an amyloidosis workup was initiated. 99mTC pyrophosphate imaging was positive for ATTR-CM, and he started taking tafamidis. Before this diagnosis, ECG indicated low-voltage QRS complexes. His progression has since been complicated by admissions for decompensated HF, recurrent episodes of AF requiring atrioventricular node ablation, and biventricular implantable cardioverter-defibrillator implantation after failed attempts at electrical cardioversion. He continued to follow up in the HF clinic.
Discussion
ATTR-CM is an underdiagnosed cause of cardiomyopathy, particularly in older adults. TTR is a transport protein produced in the liver, and misfolding can occur due to age-related instability of the wild-type protein (wtATTR) or pathologic variants in the TTR (hATTR). The misfolding leads to restrictive physiology, HF (often with preserved EF) arrhythmias, and conduction system disease.1 It is likely that the predominantly older male veteran population would be predisposed to wtATTR cardiomyopathy given that misfolding in the condition is believed to be age-related.
Current criteria for ATTR diagnosis require a combination of clinical suspicion, imaging, laboratory testing, and, in some cases, tissue biopsy confirmation and genetic testing.1,4,5 The diagnostic algorithm is as follows:
Clinical suspicion. Consider ATTR in patients with unexplained HFpEF, LV wall thickness ≥ 12 to 14 mm, discordance between ECG voltage and wall thickness, or associated extracardiac manifestations such as CTS, lumbar spinal stenosis, or peripheral/ autonomic neuropathy.
Exclusion of AL amyloidosis. Bone scintigraphy alone cannot distinguish between AL amyloidosis and ATTR, so patients with suspected amyloid cardiomyopathy should undergo serum and urine immunofixation electrophoresis and serum free light chain assay to rule out a monoclonal gammopathy as seen in AL amyloidosis.
Cardiac scintigraphy. Once AL amyloidosis is excluded, a positive 99mTC-labeled boneavid tracer image (such as a pyrophosphate scan) with grade 2 or grade 3 myocardial uptake is diagnostic of ATTR.
Tissue biopsy. If there is monoclonal gammopathy or equivocal imaging, tissue biopsy (eg, endomyocardial) with Congo red staining and amyloid typing by mass spectrometry or immunohistochemistry is necessary.
Genetic testing. Once ATTR is confirmed, genetic testing distinguishes hATTR from wtATTR, which impacts management and determines the need to screen family members.
Currently, there are 3 therapies approved by the US Food and Drug Administration (FDA) to treat ATTR-CM: tafamidis, acoramidis, and vutrisiran.6-8 Tafamidis and acoramidis stabilize the TTR tetramer, preventing amyloid formation. Vutrisiran uses RNA interference to silence the gene that produces TTR. Tafamidis has been found to improve cardiovascular outcomes in ATTR-CM. In the ATTR-ACT trial, it reduced all-cause mortality and cardiovascular hospitalizations in patients with ATTR-CM and New York Heart Association class I-III symptoms.6 There are other disease-modifying therapies, such the TTR gene silencers inotersen and patisiran; however, these are only FDA-approved for hATTR polyneuropathy and not for ATTRCM; ongoing trials are evaluating their cardiac efficacy.
The mean age of ATTR-CM diagnosis in the patients described in this case series was 79 years, which is older than the mean age of 74 years reported in prior research.4 All patients were male, and the most common presenting symptom was dyspnea on exertion. Table 1 outlines baseline characteristics and associated comorbidities of patients in this case series. The patients presented with many red-flag signs of ATTR-CM (Figure 3). Among them included syncope (4 of 8 patients), spinal stenosis (2 of 8 patients), arrhythmia (7 of 8 patients), heart failure (7 of 8 patients), and bilateral CTS (all patients).
amyloidosis red-flag signs identified.
Bilateral CTS was diagnosed in all patients before their diagnosis of ATTR-CM. Patient 7 had a diagnosis of CTS 9 years before his ATTR-CM diagnosis, underscoring a subtle, yet important extracardiac sign that may increase clinical suspicion for ATTR-CM. Previous research found that the probability of having CTS is highest 5 to 9 years prior to the development of cardiomyopathy. Its presence is also a prognostic marker in ATTR, independent of cardiac involvement.9 The median interval between CTS diagnosis and cardiomyopathy diagnosis was 5 years in this series.
Although screening criteria for ATTR have been proposed, none have been incorporated into formal guidelines.10,11 We propose that a baseline ECG and screening TTE be obtained in any patient aged > 65 years with cardiac risk equivalents such as hypertension and diabetes, presenting with ≥ 1 extracardiac red-flag signs, such as bilateral CTS and spinal stenosis. This will likely facilitate an earlier diagnosis of ATTR-CM, leading to earlier treatment initiation and better patient outcomes. This initiative can be started in the primary care setting and facilitates early cardiology referral. This recommendation is based on literature supporting clinical patterns and observations the authors have made in clinical practice.
Obstructive sleep apnea (OSA) was a comorbidity present in 50% of the patients described in this case series. The literature describing this association is sparse; however, a prospective observational study reported that disorders of sleep inclusive of OSA are frequent in patients with cardiac amyloidosis.12 A theory behind this association is that amyloid deposits in the upper airway tissues lead to airway narrowing. 13 More research is needed to further assess the relationship between OSA and cardiac amyloidosis, particularly with respect to the timing of OSA prior to the development of cardiomyopathy as it may be a potential early sign for clinicians to acknowledge.
An important observation from this case series is the variability in the timing of tafamidis initiation relative to symptom onset and confirmed diagnosis of ATTR-CM (Table 2). Although tafamidis has been found to slow disease progression, several patients in this series began treatment at advanced stages or years after the onset of cardiac symptoms, potentially limiting its clinical benefit.
For example, patient 1 was diagnosed with ATTR 2 years before tafamidis became available on the market and was initially treated with diflunisal. Patients who started tafamidis earlier in the disease course (eg, patients 3 and 5) appeared to have better long-term outcomes, including an absence of heart failure hospitalizations after initiation. In contrast, patients with delayed treatment initiation (eg, patients 2, 6, and 8) experienced ongoing decompensations or early mortality. Patient 7 declined tafamidis, underscoring challenges in medication uptake among older adults. Randomized controlled trials are warranted to compare the effectiveness of tafamidis with other recently FDA-approved therapies, such as acoramidis and vutrisiran, particularly in terms of cardiovascular outcomes.
AF was the most common arrhythmia observed in these patients and may occur years before the development of HF symptoms in the setting of ATTR-CM. Due to inherent conduction system disease, AF in this population may have a controlled or slow ventricular response, as observed in patient 4. Patients with atrial fibrillation and cardiac amyloidosis should receive anticoagulation regardless of CHA2DS2- VASc score due to their high risk of intracardiac thrombus formation.4
Limitations
This case series lacked genetic testing following a confirmed ATTR-CM diagnosis. Although much of the treatment is the same regardless of the presence of a TTR mutation, knowing the specific subtype of ATTR-CM has implications for prognosis and for screening family members.
Conclusions
Following analysis of 8 patients diagnosed with ATTR, this case series could serve as a blueprint for research into ATTR in veterans. In clinical practice, following military service, veterans may not be routinely seen by an outpatient physician and may present with sequelae of advanced stages of ATTR. Early identification of red-flag symptoms can lead to a higher clinical suspicion, prompting early diagnostic evaluation and treatment initiation and ultimately mitigating adverse outcomes. Future research that includes genetic testing for those with confirmed ATTR-CM may prove useful as a foundation for detailed and informed discussions with patients and their families regarding prognosis and, if indicated, screening for family members.
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142:e7-e22. doi:10.1161/CIR.0000000000000792
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765-774. doi:10.1111/j.1532-5415.2011.03868.x
- Nativi-Nicolau J, Redd A, Kelly N, et al. Increasing number of amyloidosis diagnosis in the Veterans Affairs populations. J Card Fail. 2019;25:S96.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872-2891. doi:10.1016/j.jacc.2019.04.003
- Gillmore JD, Maurer, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404-2412. doi:10.1161/CIRCULATIONAHA.116.021612
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007-1016. doi:10.1056/NEJMoa1805689
- Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med. 2024;390:132-142. doi:10.1056/NEJMoa2305434
- Fontana M, Berk JL, Gillmore JD, et al. Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J Med. 2025;392:33-44. doi:10.1056/NEJMoa2409134
- Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22:507-515. doi:10.1002/ejhf.1742
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554-1568. doi:10.1093/eurheartj/ehab072
- Brito D, Albrecht FC, de Arenaza DP, et al. World Heart Federation consensus on transthyretin amyloidosis cardiomyopathy (ATTR-CM). Glob Heart. 2023;18:59. doi:10.5334/gh.1262
- Bodez D, Guellich A, Kharoubi M, et al. Prevalence, severity, and prognostic value of sleep apnea syndromes in cardiac amyloidosis. Sleep. 2016;39:1333-1341. doi:10.5665/sleep.5958
- Colaco B, Colaco C, Lipford M. A forgotten problem: sleep-disordered breathing in amyloidosis. Chest. 2016;150:1293A. doi:10.1016/j.chest.2016.08.1407
- Kittleson MM, Maurer MS, Ambardekar AV, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142:e7-e22. doi:10.1161/CIR.0000000000000792
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765-774. doi:10.1111/j.1532-5415.2011.03868.x
- Nativi-Nicolau J, Redd A, Kelly N, et al. Increasing number of amyloidosis diagnosis in the Veterans Affairs populations. J Card Fail. 2019;25:S96.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872-2891. doi:10.1016/j.jacc.2019.04.003
- Gillmore JD, Maurer, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404-2412. doi:10.1161/CIRCULATIONAHA.116.021612
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007-1016. doi:10.1056/NEJMoa1805689
- Gillmore JD, Judge DP, Cappelli F, et al. Efficacy and safety of acoramidis in transthyretin amyloid cardiomyopathy. N Engl J Med. 2024;390:132-142. doi:10.1056/NEJMoa2305434
- Fontana M, Berk JL, Gillmore JD, et al. Vutrisiran in patients with transthyretin amyloidosis with cardiomyopathy. N Engl J Med. 2025;392:33-44. doi:10.1056/NEJMoa2409134
- Milandri A, Farioli A, Gagliardi C, et al. Carpal tunnel syndrome in cardiac amyloidosis: implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur J Heart Fail. 2020;22:507-515. doi:10.1002/ejhf.1742
- Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2021;42:1554-1568. doi:10.1093/eurheartj/ehab072
- Brito D, Albrecht FC, de Arenaza DP, et al. World Heart Federation consensus on transthyretin amyloidosis cardiomyopathy (ATTR-CM). Glob Heart. 2023;18:59. doi:10.5334/gh.1262
- Bodez D, Guellich A, Kharoubi M, et al. Prevalence, severity, and prognostic value of sleep apnea syndromes in cardiac amyloidosis. Sleep. 2016;39:1333-1341. doi:10.5665/sleep.5958
- Colaco B, Colaco C, Lipford M. A forgotten problem: sleep-disordered breathing in amyloidosis. Chest. 2016;150:1293A. doi:10.1016/j.chest.2016.08.1407
Case Series of Patients With Cardiac Amyloidosis at VA New York Harbor Healthcare-Brooklyn
Case Series of Patients With Cardiac Amyloidosis at VA New York Harbor Healthcare-Brooklyn