User login
Theme
medstat_cc
mclcc
Unpublish
Altmetric
Click for Credit Button Label
Click For Credit
DSM Affiliated
Display in offset block
Enable Disqus
Publication Type
News
Slot System
Featured Buckets
Disable Sticky Ads
Featured Buckets Admin
Show Ads on this Publication's Homepage
Consolidated Pub
Show Article Page Numbers on TOC
Use larger logo size
Off
publication_blueconic_enabled
Off
Disable Adhesion on Publication
Off
Disable Facebook Pixel from Publication
Exclude this publication from publication selection on articles and quiz
Challenge Center
Disable Inline Native ads
Medchallenges
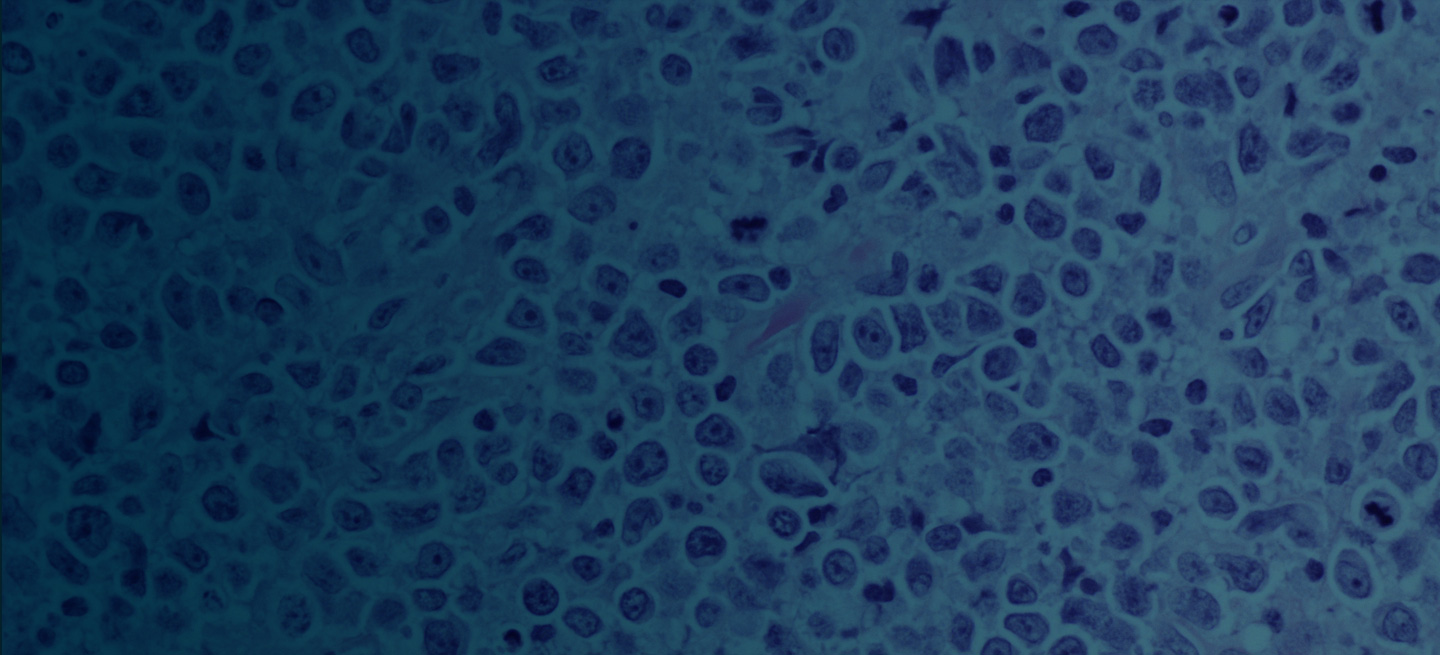
Leukemic Non-Nodal MCL
MCL Workup
MCL Management
MCL Genetics
Challenge Center Topic
Challenge Center Slideshow


Medchallenge Read More Link
https://app.medchallenge.net/select-opponent/mantle-cell-lymphoma
Supporter Name /ID
Pirtobrutinib [ 5829 ]
Activity Salesforce Deliverable ID
343187.3
Activity ID
94960
Product Name
Challenge Center
Product ID
121
Challenge Center Slideshow Media