User login
Infections increase risk of idiopathic VTE
Infection and infection sites have been found to be associated with a significant increased risk of venous thromboembolism, according to results of a population-based, matched, case-control analysis of medical records covering the 13-year period 1988-2000.
Dr. Kevin P. Cohoon and his colleagues at the Mayo Clinic, Rochester, Minn., developed models using conditional logistic regression analysis to stratify the risk associated with specific infections and infection sites.
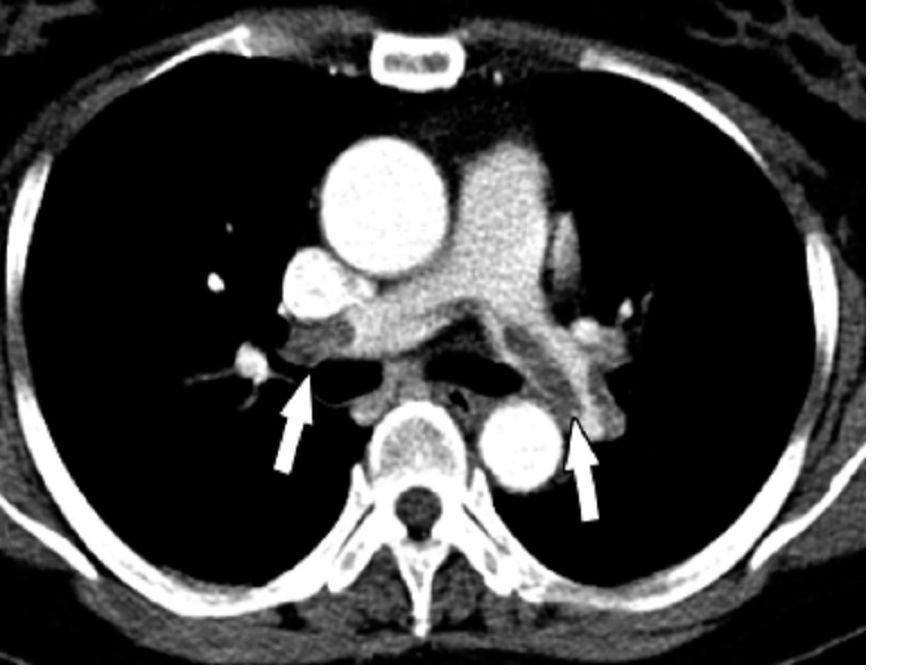
Five hundred thirteen (39.4%) cases and 189 (12.7%) controls had an infection within the previous 92 days (odds ratio, 4.5; P less than .0001). Known VTE risk factors and potentially confounding variables were used in the adjusted univariate and multivariate models, as reported in the American Journal of Medicine (2017. doi: 10.1016/j.amjmed.2017.09.015).
Dr. Cohoon and his colleagues reported that univariate analysis showed “most infection sites were strongly associated with venous thromboembolism” and the adjusted multivariate model resulted in 2.4-fold (P less than .0001) higher odds for VTE incidence, compared with uninfected controls.
Adjusted multivariate analysis ranked the odds of VTE according to specific infections. Dr. Cohoon and his colleagues reported that this modeling showed that the “highest magnitude of risk, compared with no infection, was imparted by intra-abdominal infection (OR, 18) followed by oral infection (OR, 12), systematic blood stream infection (OR, 11), lower respiratory infection such as pneumonia (OR, 3.6), and symptomatic urinary tract infection (OR, 2.2).”
The researchers concluded that their findings may allow for further refinement of inpatient VTE risk-prediction models such as the Padua prediction score and “future studies are required to assess the utility of venous thromboembolism prophylaxis among outpatients with high venous thromboembolism risk infections.”
The authors reported that they had no conflicts of interest.
Infection and infection sites have been found to be associated with a significant increased risk of venous thromboembolism, according to results of a population-based, matched, case-control analysis of medical records covering the 13-year period 1988-2000.
Dr. Kevin P. Cohoon and his colleagues at the Mayo Clinic, Rochester, Minn., developed models using conditional logistic regression analysis to stratify the risk associated with specific infections and infection sites.
Five hundred thirteen (39.4%) cases and 189 (12.7%) controls had an infection within the previous 92 days (odds ratio, 4.5; P less than .0001). Known VTE risk factors and potentially confounding variables were used in the adjusted univariate and multivariate models, as reported in the American Journal of Medicine (2017. doi: 10.1016/j.amjmed.2017.09.015).
Dr. Cohoon and his colleagues reported that univariate analysis showed “most infection sites were strongly associated with venous thromboembolism” and the adjusted multivariate model resulted in 2.4-fold (P less than .0001) higher odds for VTE incidence, compared with uninfected controls.
Adjusted multivariate analysis ranked the odds of VTE according to specific infections. Dr. Cohoon and his colleagues reported that this modeling showed that the “highest magnitude of risk, compared with no infection, was imparted by intra-abdominal infection (OR, 18) followed by oral infection (OR, 12), systematic blood stream infection (OR, 11), lower respiratory infection such as pneumonia (OR, 3.6), and symptomatic urinary tract infection (OR, 2.2).”
The researchers concluded that their findings may allow for further refinement of inpatient VTE risk-prediction models such as the Padua prediction score and “future studies are required to assess the utility of venous thromboembolism prophylaxis among outpatients with high venous thromboembolism risk infections.”
The authors reported that they had no conflicts of interest.
Infection and infection sites have been found to be associated with a significant increased risk of venous thromboembolism, according to results of a population-based, matched, case-control analysis of medical records covering the 13-year period 1988-2000.
Dr. Kevin P. Cohoon and his colleagues at the Mayo Clinic, Rochester, Minn., developed models using conditional logistic regression analysis to stratify the risk associated with specific infections and infection sites.
Five hundred thirteen (39.4%) cases and 189 (12.7%) controls had an infection within the previous 92 days (odds ratio, 4.5; P less than .0001). Known VTE risk factors and potentially confounding variables were used in the adjusted univariate and multivariate models, as reported in the American Journal of Medicine (2017. doi: 10.1016/j.amjmed.2017.09.015).
Dr. Cohoon and his colleagues reported that univariate analysis showed “most infection sites were strongly associated with venous thromboembolism” and the adjusted multivariate model resulted in 2.4-fold (P less than .0001) higher odds for VTE incidence, compared with uninfected controls.
Adjusted multivariate analysis ranked the odds of VTE according to specific infections. Dr. Cohoon and his colleagues reported that this modeling showed that the “highest magnitude of risk, compared with no infection, was imparted by intra-abdominal infection (OR, 18) followed by oral infection (OR, 12), systematic blood stream infection (OR, 11), lower respiratory infection such as pneumonia (OR, 3.6), and symptomatic urinary tract infection (OR, 2.2).”
The researchers concluded that their findings may allow for further refinement of inpatient VTE risk-prediction models such as the Padua prediction score and “future studies are required to assess the utility of venous thromboembolism prophylaxis among outpatients with high venous thromboembolism risk infections.”
The authors reported that they had no conflicts of interest.
FROM THE AMERICAN JOURNAL OF MEDICINE
Key clinical point:
Major finding: A significantly greater number of patients with infections developed VTE as compared with uninfected controls (OR, 4.5; P less than .0001).
Data source: Study was a retrospective database analysis of 1,303 VTE patients and 1,494 paired controls.
Disclosures: The authors reported that they had no conflicts of interest.
Online Help for People With Alcohol Use Disorder
In any given year, < 10% of people diagnosed with alcohol use disorder receive treatment, and many do not receive the type of care that best fits their needs. Two reasons may be that they don’t know where to turn for help, or they may not know that they have more treatment options beyond a mutual help group or long-term residential rehabilitation facility.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) has developed a new online tool that may help. The “comprehensive, yet easy-to-use” Alcohol Treatment Navigator was developed “to help address the alcohol ‘treatment gap,’” said NIAAA Director George Koob, PhD. It’s based on decades of scientific research into clinical interventions and health services with input from patients, providers, and researchers.
The Navigator includes an overview of alcohol use disorder and a description of professionally led treatment options. It also gives step-by-step instructions for searching online directories of treatment providers, 10 questions to ask a provider, and signs of quality to listen for. A downloadable tool kit helps organize and simplify the search process.
In any given year, < 10% of people diagnosed with alcohol use disorder receive treatment, and many do not receive the type of care that best fits their needs. Two reasons may be that they don’t know where to turn for help, or they may not know that they have more treatment options beyond a mutual help group or long-term residential rehabilitation facility.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) has developed a new online tool that may help. The “comprehensive, yet easy-to-use” Alcohol Treatment Navigator was developed “to help address the alcohol ‘treatment gap,’” said NIAAA Director George Koob, PhD. It’s based on decades of scientific research into clinical interventions and health services with input from patients, providers, and researchers.
The Navigator includes an overview of alcohol use disorder and a description of professionally led treatment options. It also gives step-by-step instructions for searching online directories of treatment providers, 10 questions to ask a provider, and signs of quality to listen for. A downloadable tool kit helps organize and simplify the search process.
In any given year, < 10% of people diagnosed with alcohol use disorder receive treatment, and many do not receive the type of care that best fits their needs. Two reasons may be that they don’t know where to turn for help, or they may not know that they have more treatment options beyond a mutual help group or long-term residential rehabilitation facility.
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) has developed a new online tool that may help. The “comprehensive, yet easy-to-use” Alcohol Treatment Navigator was developed “to help address the alcohol ‘treatment gap,’” said NIAAA Director George Koob, PhD. It’s based on decades of scientific research into clinical interventions and health services with input from patients, providers, and researchers.
The Navigator includes an overview of alcohol use disorder and a description of professionally led treatment options. It also gives step-by-step instructions for searching online directories of treatment providers, 10 questions to ask a provider, and signs of quality to listen for. A downloadable tool kit helps organize and simplify the search process.
Is MRD ready for prime time in multiple myeloma?

NEW YORK, NY—Speakers faced off over the issue of minimal residual disease (MRD) testing in multiple myeloma (MM) at Lymphoma & Myeloma 2017.
Ola Landgren, MD, PhD, of Weill Cornell Medicine in New York, New York, said, “it’s really a necessary and logical step forward to look at MRD.”
On the other hand, Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts, took the clinicians’ perspective and suggested that, at this point, “we’re not yet ready to apply it to everyday practice.”
“[P]atients who have a complete response (CR) and are MRD negative have longer progression-free survival (PFS),” Dr Landgren pointed out, “and there are indications that their overall survival (OS) is better than in those patients who are just CR and MRD positive.”
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena,” Dr Richardson contended. “The question for me, as a clinician, in my clinic, is ‘Do I apply it to everyday practice?’ And I would simply suggest to you, at this point, we’re not ready for that.”
Yes—MRD is ready for prime time
Dr Landgren based his argument on 2 meta-analyses published in 2016 and 2017 that outline the importance of MRD status in newly diagnosed MM patients.
The first analysis (Landgren et al 2016) showed that MRD negativity was associated with better PFS (hazard ratio [HR]=0.35] and OS (HR=0.48) than MRD positivity.
“So using more simple language,” Dr Landgren said, “this means that MRD negativity reduces the risk of progression by 65%, and it also reduces the risk of dying by 52%.”
The second analysis (Munshi et al 2017) also associated MRD-negative status with superior survival outcomes for both PFS (HR=0.41) and OS (HR=0.57).
As further confirmation of the importance of MRD status, the International Myeloma Working Group last year published response definitions that include MRD negativity at a sensitivity of 1 in 105 cells or higher as the deepest level of treatment response in MM.
Dr Landgren drew on additional studies to support routine MRD testing in patient care.
The IFM Study Group found that, in newly diagnosed patients treated with lenalidomide, bortezomib, and dexamethasone followed by 1 year of lenalidomide maintenance, patients who received a subsequent transplant achieved superior outcomes compared to non-transplanted patients, in terms of CR (58% vs 46%) and 3-year PFS (61% vs 48%).
However, in patients who were MRD negative in both arms, the PFS rates were very similar, Dr Landgren said. And in terms of 3-year OS, there was no difference, at 88% in both arms.
The experience with daratumumab in relapsed/refractory patients exhibited a similar pattern.
The phase 3 POLLUX trial first showed that adding daratumumab to lenalidomide and dexamethasone was superior to lenalidomide and dexamethasone only, with a PFS at 18 months of 78% and 52%, respectively. This amounted to a 63% reduction in the risk of disease progression.
Investigators then took one more step forward, Dr Landgren said, and looked at MRD.
At a sensitivity of 10-5, almost 25% of patients on the 3-drug regimen were MRD negative, “which is kind of amazing,” Dr Landgren said. “This is a very big step forward.”
“If you break down the results by MRD status, which is not the primary endpoint of the study, you see very similar patterns for PFS for MRD negative patients in each of the 2 arms,” he continued.
This raises the question of whether attaining MRD negativity is more important than the treatment modality.
MRD negativity has implications for speeding drug approvals, developing more sensitive assays, and future treatment management, Dr Landgren said.
No—MRD is not ready for prime time
Dr Richardson acknowledged that MRD assessment is important. However, he pointed out a number of caveats regarding how MRD assessment would be applied in clinical practice to support his position.
“I’d simply suggest to you that, in day-to-day practice, the definition [of MRD] is somewhat fluid,” he said. “And it varies, obviously, between diseases and technology used.”
For most malignancies, Dr Richardson said, 109 to 1010 malignant cells are undetectable with conventional methods. These may or may not lead to a full clinical relapse within months or even years.
Using a sensitive technique to determine the presence of MRD could permit analysis of treatments that induce a greater depth of response or identify patients at risk of early relapse who need further treatment.
Dr Richardson enumerated hematologic malignancies that utilize MRD as secondary endpoints—acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, follicular lymphoma, and mantle cell lymphoma.
In chronic myeloid leukemia, MRD is used as a primary endpoint that dictates practice.
“And I would applaud the field in that area because, obviously, molecular response accepted as an endpoint by FDA for second-generation TKIs has been a bedrock of that approval process, and it now applies in clinical practice,” Dr Richardson said.
“Obviously, that’s where we’d like to be, but I’d suggest to you, just again, with a certain amount of moderation and a certain amount of caution, that we may not be quite there yet.”
Dr Richardson suggested that MRD assessment in MM is less advanced than in leukemia and lymphoma.
“[W]e are currently at the point where MRD assessments are clearly secondary endpoints, an important research tool,” he said.
Some “remarkable combination therapies,” he added, have abrogated some of the “extraordinary genetic complexity” in MM.
“The critical point here, though, is that, while we’re more successful in terms of these triplets and quadruplets and now with the introduction of monoclonal antibodies and similar approaches, we’re able to throw a bigger net around the disease,” Dr Richardson said.
“We’re not able to eradicate it completely, and cure remains, in myeloma, frankly, evasive. And I think that’s a critical point.”
Dr Richardson reviewed various strategies for molecular response monitoring, from flow cytometry to polymerase chain reaction and next-generation sequencing, noting that there is variance in applicability and sensitivity.
For example, the limits of detection among 91 labs ranged from 0.10% to 0.001%.
Dr Richardson returned to the “very robust” meta-analysis by Munshi and colleagues discussed by Dr Landgren.
While the authors’ analysis demonstrated that MRD is predictive of both longer PFS and OS, they concluded that the evidence supported MRD as an endpoint and research tool in clinical trials.
“So I would humbly suggest perhaps it’s not ready for clinical prime time yet,” Dr Richardson said.
He also referred to the IFM Study Group trial described by Dr Landgren, calling it a “critical forward effort.”
“[W]hat’s so interesting is that there was no difference in overall survival,” Dr Richardson said. “Now, that’s a very important point as we soberly look at these data and judge what they mean for each patient.”
And so Dr Richardson stood by his assessment that MRD is not yet a standard of care but may be one day. ![]()
NEW YORK, NY—Speakers faced off over the issue of minimal residual disease (MRD) testing in multiple myeloma (MM) at Lymphoma & Myeloma 2017.
Ola Landgren, MD, PhD, of Weill Cornell Medicine in New York, New York, said, “it’s really a necessary and logical step forward to look at MRD.”
On the other hand, Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts, took the clinicians’ perspective and suggested that, at this point, “we’re not yet ready to apply it to everyday practice.”
“[P]atients who have a complete response (CR) and are MRD negative have longer progression-free survival (PFS),” Dr Landgren pointed out, “and there are indications that their overall survival (OS) is better than in those patients who are just CR and MRD positive.”
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena,” Dr Richardson contended. “The question for me, as a clinician, in my clinic, is ‘Do I apply it to everyday practice?’ And I would simply suggest to you, at this point, we’re not ready for that.”
Yes—MRD is ready for prime time
Dr Landgren based his argument on 2 meta-analyses published in 2016 and 2017 that outline the importance of MRD status in newly diagnosed MM patients.
The first analysis (Landgren et al 2016) showed that MRD negativity was associated with better PFS (hazard ratio [HR]=0.35] and OS (HR=0.48) than MRD positivity.
“So using more simple language,” Dr Landgren said, “this means that MRD negativity reduces the risk of progression by 65%, and it also reduces the risk of dying by 52%.”
The second analysis (Munshi et al 2017) also associated MRD-negative status with superior survival outcomes for both PFS (HR=0.41) and OS (HR=0.57).
As further confirmation of the importance of MRD status, the International Myeloma Working Group last year published response definitions that include MRD negativity at a sensitivity of 1 in 105 cells or higher as the deepest level of treatment response in MM.
Dr Landgren drew on additional studies to support routine MRD testing in patient care.
The IFM Study Group found that, in newly diagnosed patients treated with lenalidomide, bortezomib, and dexamethasone followed by 1 year of lenalidomide maintenance, patients who received a subsequent transplant achieved superior outcomes compared to non-transplanted patients, in terms of CR (58% vs 46%) and 3-year PFS (61% vs 48%).
However, in patients who were MRD negative in both arms, the PFS rates were very similar, Dr Landgren said. And in terms of 3-year OS, there was no difference, at 88% in both arms.
The experience with daratumumab in relapsed/refractory patients exhibited a similar pattern.
The phase 3 POLLUX trial first showed that adding daratumumab to lenalidomide and dexamethasone was superior to lenalidomide and dexamethasone only, with a PFS at 18 months of 78% and 52%, respectively. This amounted to a 63% reduction in the risk of disease progression.
Investigators then took one more step forward, Dr Landgren said, and looked at MRD.
At a sensitivity of 10-5, almost 25% of patients on the 3-drug regimen were MRD negative, “which is kind of amazing,” Dr Landgren said. “This is a very big step forward.”
“If you break down the results by MRD status, which is not the primary endpoint of the study, you see very similar patterns for PFS for MRD negative patients in each of the 2 arms,” he continued.
This raises the question of whether attaining MRD negativity is more important than the treatment modality.
MRD negativity has implications for speeding drug approvals, developing more sensitive assays, and future treatment management, Dr Landgren said.
No—MRD is not ready for prime time
Dr Richardson acknowledged that MRD assessment is important. However, he pointed out a number of caveats regarding how MRD assessment would be applied in clinical practice to support his position.
“I’d simply suggest to you that, in day-to-day practice, the definition [of MRD] is somewhat fluid,” he said. “And it varies, obviously, between diseases and technology used.”
For most malignancies, Dr Richardson said, 109 to 1010 malignant cells are undetectable with conventional methods. These may or may not lead to a full clinical relapse within months or even years.
Using a sensitive technique to determine the presence of MRD could permit analysis of treatments that induce a greater depth of response or identify patients at risk of early relapse who need further treatment.
Dr Richardson enumerated hematologic malignancies that utilize MRD as secondary endpoints—acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, follicular lymphoma, and mantle cell lymphoma.
In chronic myeloid leukemia, MRD is used as a primary endpoint that dictates practice.
“And I would applaud the field in that area because, obviously, molecular response accepted as an endpoint by FDA for second-generation TKIs has been a bedrock of that approval process, and it now applies in clinical practice,” Dr Richardson said.
“Obviously, that’s where we’d like to be, but I’d suggest to you, just again, with a certain amount of moderation and a certain amount of caution, that we may not be quite there yet.”
Dr Richardson suggested that MRD assessment in MM is less advanced than in leukemia and lymphoma.
“[W]e are currently at the point where MRD assessments are clearly secondary endpoints, an important research tool,” he said.
Some “remarkable combination therapies,” he added, have abrogated some of the “extraordinary genetic complexity” in MM.
“The critical point here, though, is that, while we’re more successful in terms of these triplets and quadruplets and now with the introduction of monoclonal antibodies and similar approaches, we’re able to throw a bigger net around the disease,” Dr Richardson said.
“We’re not able to eradicate it completely, and cure remains, in myeloma, frankly, evasive. And I think that’s a critical point.”
Dr Richardson reviewed various strategies for molecular response monitoring, from flow cytometry to polymerase chain reaction and next-generation sequencing, noting that there is variance in applicability and sensitivity.
For example, the limits of detection among 91 labs ranged from 0.10% to 0.001%.
Dr Richardson returned to the “very robust” meta-analysis by Munshi and colleagues discussed by Dr Landgren.
While the authors’ analysis demonstrated that MRD is predictive of both longer PFS and OS, they concluded that the evidence supported MRD as an endpoint and research tool in clinical trials.
“So I would humbly suggest perhaps it’s not ready for clinical prime time yet,” Dr Richardson said.
He also referred to the IFM Study Group trial described by Dr Landgren, calling it a “critical forward effort.”
“[W]hat’s so interesting is that there was no difference in overall survival,” Dr Richardson said. “Now, that’s a very important point as we soberly look at these data and judge what they mean for each patient.”
And so Dr Richardson stood by his assessment that MRD is not yet a standard of care but may be one day. ![]()
NEW YORK, NY—Speakers faced off over the issue of minimal residual disease (MRD) testing in multiple myeloma (MM) at Lymphoma & Myeloma 2017.
Ola Landgren, MD, PhD, of Weill Cornell Medicine in New York, New York, said, “it’s really a necessary and logical step forward to look at MRD.”
On the other hand, Paul Richardson, MD, of Dana-Farber Cancer Institute in Boston, Massachusetts, took the clinicians’ perspective and suggested that, at this point, “we’re not yet ready to apply it to everyday practice.”
“[P]atients who have a complete response (CR) and are MRD negative have longer progression-free survival (PFS),” Dr Landgren pointed out, “and there are indications that their overall survival (OS) is better than in those patients who are just CR and MRD positive.”
“My position on this is that MRD testing is absolutely ready for prime time in the research and regulatory arena,” Dr Richardson contended. “The question for me, as a clinician, in my clinic, is ‘Do I apply it to everyday practice?’ And I would simply suggest to you, at this point, we’re not ready for that.”
Yes—MRD is ready for prime time
Dr Landgren based his argument on 2 meta-analyses published in 2016 and 2017 that outline the importance of MRD status in newly diagnosed MM patients.
The first analysis (Landgren et al 2016) showed that MRD negativity was associated with better PFS (hazard ratio [HR]=0.35] and OS (HR=0.48) than MRD positivity.
“So using more simple language,” Dr Landgren said, “this means that MRD negativity reduces the risk of progression by 65%, and it also reduces the risk of dying by 52%.”
The second analysis (Munshi et al 2017) also associated MRD-negative status with superior survival outcomes for both PFS (HR=0.41) and OS (HR=0.57).
As further confirmation of the importance of MRD status, the International Myeloma Working Group last year published response definitions that include MRD negativity at a sensitivity of 1 in 105 cells or higher as the deepest level of treatment response in MM.
Dr Landgren drew on additional studies to support routine MRD testing in patient care.
The IFM Study Group found that, in newly diagnosed patients treated with lenalidomide, bortezomib, and dexamethasone followed by 1 year of lenalidomide maintenance, patients who received a subsequent transplant achieved superior outcomes compared to non-transplanted patients, in terms of CR (58% vs 46%) and 3-year PFS (61% vs 48%).
However, in patients who were MRD negative in both arms, the PFS rates were very similar, Dr Landgren said. And in terms of 3-year OS, there was no difference, at 88% in both arms.
The experience with daratumumab in relapsed/refractory patients exhibited a similar pattern.
The phase 3 POLLUX trial first showed that adding daratumumab to lenalidomide and dexamethasone was superior to lenalidomide and dexamethasone only, with a PFS at 18 months of 78% and 52%, respectively. This amounted to a 63% reduction in the risk of disease progression.
Investigators then took one more step forward, Dr Landgren said, and looked at MRD.
At a sensitivity of 10-5, almost 25% of patients on the 3-drug regimen were MRD negative, “which is kind of amazing,” Dr Landgren said. “This is a very big step forward.”
“If you break down the results by MRD status, which is not the primary endpoint of the study, you see very similar patterns for PFS for MRD negative patients in each of the 2 arms,” he continued.
This raises the question of whether attaining MRD negativity is more important than the treatment modality.
MRD negativity has implications for speeding drug approvals, developing more sensitive assays, and future treatment management, Dr Landgren said.
No—MRD is not ready for prime time
Dr Richardson acknowledged that MRD assessment is important. However, he pointed out a number of caveats regarding how MRD assessment would be applied in clinical practice to support his position.
“I’d simply suggest to you that, in day-to-day practice, the definition [of MRD] is somewhat fluid,” he said. “And it varies, obviously, between diseases and technology used.”
For most malignancies, Dr Richardson said, 109 to 1010 malignant cells are undetectable with conventional methods. These may or may not lead to a full clinical relapse within months or even years.
Using a sensitive technique to determine the presence of MRD could permit analysis of treatments that induce a greater depth of response or identify patients at risk of early relapse who need further treatment.
Dr Richardson enumerated hematologic malignancies that utilize MRD as secondary endpoints—acute lymphoblastic leukemia, acute myeloid leukemia, acute promyelocytic leukemia, chronic lymphocytic leukemia, follicular lymphoma, and mantle cell lymphoma.
In chronic myeloid leukemia, MRD is used as a primary endpoint that dictates practice.
“And I would applaud the field in that area because, obviously, molecular response accepted as an endpoint by FDA for second-generation TKIs has been a bedrock of that approval process, and it now applies in clinical practice,” Dr Richardson said.
“Obviously, that’s where we’d like to be, but I’d suggest to you, just again, with a certain amount of moderation and a certain amount of caution, that we may not be quite there yet.”
Dr Richardson suggested that MRD assessment in MM is less advanced than in leukemia and lymphoma.
“[W]e are currently at the point where MRD assessments are clearly secondary endpoints, an important research tool,” he said.
Some “remarkable combination therapies,” he added, have abrogated some of the “extraordinary genetic complexity” in MM.
“The critical point here, though, is that, while we’re more successful in terms of these triplets and quadruplets and now with the introduction of monoclonal antibodies and similar approaches, we’re able to throw a bigger net around the disease,” Dr Richardson said.
“We’re not able to eradicate it completely, and cure remains, in myeloma, frankly, evasive. And I think that’s a critical point.”
Dr Richardson reviewed various strategies for molecular response monitoring, from flow cytometry to polymerase chain reaction and next-generation sequencing, noting that there is variance in applicability and sensitivity.
For example, the limits of detection among 91 labs ranged from 0.10% to 0.001%.
Dr Richardson returned to the “very robust” meta-analysis by Munshi and colleagues discussed by Dr Landgren.
While the authors’ analysis demonstrated that MRD is predictive of both longer PFS and OS, they concluded that the evidence supported MRD as an endpoint and research tool in clinical trials.
“So I would humbly suggest perhaps it’s not ready for clinical prime time yet,” Dr Richardson said.
He also referred to the IFM Study Group trial described by Dr Landgren, calling it a “critical forward effort.”
“[W]hat’s so interesting is that there was no difference in overall survival,” Dr Richardson said. “Now, that’s a very important point as we soberly look at these data and judge what they mean for each patient.”
And so Dr Richardson stood by his assessment that MRD is not yet a standard of care but may be one day. ![]()
Ferric citrate approved to treat iron-deficiency anemia

The US Food and Drug Administration (FDA) has approved ferric citrate (Auryxia) to treat iron-deficiency anemia in adults with chronic kidney disease (CKD) who are not on dialysis.
Ferric citrate was originally approved by the FDA in September 2014 for the control of serum phosphorus levels in patients with CKD who require dialysis.
The full prescribing information for the drug is available at www.Auryxia.com.
“We are pleased with the broad indication permitted by the FDA, as a first-line treatment option for adults with iron-deficiency anemia and chronic kidney disease not on dialysis,” said John Neylan, MD, senior vice president and chief medical officer of Keryx Biopharmaceuticals, Inc., the company marketing ferric citrate.
“Physicians and their patients now have a new treatment option to help manage a serious complication of this complex disease.”
The new approval of ferric citrate was based on results from a 24-week, placebo-controlled, phase 3 trial. Results from this trial were published in the Journal of the American Society of Nephrology in January.
The trial enrolled 234 adults with stage 3-5, non-dialysis-dependent CKD and iron-deficiency anemia. Patients had hemoglobin levels between 9.0 g/dL and 11.5 g/dL and were intolerant to or had an inadequate response to prior treatment with oral iron supplements.
The starting dose of ferric citrate was 3 tablets per day, taken with meals. The mean dose was 5 tablets per day. Patients were not allowed to receive any intravenous or oral iron or erythropoiesis-stimulating agents.
Significantly more patients in the ferric citrate arm than the placebo arm had increases in hemoglobin levels of at least 1 g/dL at any point during the trial’s 16-week efficacy period—52.1% (61/117) and 19.1% (22/115), respectively (P<0.001).
Likewise, significantly more patients in the ferric citrate arm than the placebo arm had a sustained increase in hemoglobin of at least 0.75 g/dL over any 4-week period during the trial—48.7% (n=57) and 14.8% (n=17), respectively (P<0.001).
Serious adverse events occurred in 12.0% of patients in the ferric citrate arm and 11.2% of patients in the placebo arm. There were 2 treatment-emergent deaths in the ferric citrate arm (and none in the placebo arm), but they were not considered drug-related.
The most common (≥5%) treatment-emergent adverse events in patients who received ferric citrate were diarrhea (20.5%), constipation (18.8%), discolored feces (14.5%), nausea (11.1%), abdominal pain (6.0%), and hyperkalemia (6.8%). ![]()
The US Food and Drug Administration (FDA) has approved ferric citrate (Auryxia) to treat iron-deficiency anemia in adults with chronic kidney disease (CKD) who are not on dialysis.
Ferric citrate was originally approved by the FDA in September 2014 for the control of serum phosphorus levels in patients with CKD who require dialysis.
The full prescribing information for the drug is available at www.Auryxia.com.
“We are pleased with the broad indication permitted by the FDA, as a first-line treatment option for adults with iron-deficiency anemia and chronic kidney disease not on dialysis,” said John Neylan, MD, senior vice president and chief medical officer of Keryx Biopharmaceuticals, Inc., the company marketing ferric citrate.
“Physicians and their patients now have a new treatment option to help manage a serious complication of this complex disease.”
The new approval of ferric citrate was based on results from a 24-week, placebo-controlled, phase 3 trial. Results from this trial were published in the Journal of the American Society of Nephrology in January.
The trial enrolled 234 adults with stage 3-5, non-dialysis-dependent CKD and iron-deficiency anemia. Patients had hemoglobin levels between 9.0 g/dL and 11.5 g/dL and were intolerant to or had an inadequate response to prior treatment with oral iron supplements.
The starting dose of ferric citrate was 3 tablets per day, taken with meals. The mean dose was 5 tablets per day. Patients were not allowed to receive any intravenous or oral iron or erythropoiesis-stimulating agents.
Significantly more patients in the ferric citrate arm than the placebo arm had increases in hemoglobin levels of at least 1 g/dL at any point during the trial’s 16-week efficacy period—52.1% (61/117) and 19.1% (22/115), respectively (P<0.001).
Likewise, significantly more patients in the ferric citrate arm than the placebo arm had a sustained increase in hemoglobin of at least 0.75 g/dL over any 4-week period during the trial—48.7% (n=57) and 14.8% (n=17), respectively (P<0.001).
Serious adverse events occurred in 12.0% of patients in the ferric citrate arm and 11.2% of patients in the placebo arm. There were 2 treatment-emergent deaths in the ferric citrate arm (and none in the placebo arm), but they were not considered drug-related.
The most common (≥5%) treatment-emergent adverse events in patients who received ferric citrate were diarrhea (20.5%), constipation (18.8%), discolored feces (14.5%), nausea (11.1%), abdominal pain (6.0%), and hyperkalemia (6.8%). ![]()
The US Food and Drug Administration (FDA) has approved ferric citrate (Auryxia) to treat iron-deficiency anemia in adults with chronic kidney disease (CKD) who are not on dialysis.
Ferric citrate was originally approved by the FDA in September 2014 for the control of serum phosphorus levels in patients with CKD who require dialysis.
The full prescribing information for the drug is available at www.Auryxia.com.
“We are pleased with the broad indication permitted by the FDA, as a first-line treatment option for adults with iron-deficiency anemia and chronic kidney disease not on dialysis,” said John Neylan, MD, senior vice president and chief medical officer of Keryx Biopharmaceuticals, Inc., the company marketing ferric citrate.
“Physicians and their patients now have a new treatment option to help manage a serious complication of this complex disease.”
The new approval of ferric citrate was based on results from a 24-week, placebo-controlled, phase 3 trial. Results from this trial were published in the Journal of the American Society of Nephrology in January.
The trial enrolled 234 adults with stage 3-5, non-dialysis-dependent CKD and iron-deficiency anemia. Patients had hemoglobin levels between 9.0 g/dL and 11.5 g/dL and were intolerant to or had an inadequate response to prior treatment with oral iron supplements.
The starting dose of ferric citrate was 3 tablets per day, taken with meals. The mean dose was 5 tablets per day. Patients were not allowed to receive any intravenous or oral iron or erythropoiesis-stimulating agents.
Significantly more patients in the ferric citrate arm than the placebo arm had increases in hemoglobin levels of at least 1 g/dL at any point during the trial’s 16-week efficacy period—52.1% (61/117) and 19.1% (22/115), respectively (P<0.001).
Likewise, significantly more patients in the ferric citrate arm than the placebo arm had a sustained increase in hemoglobin of at least 0.75 g/dL over any 4-week period during the trial—48.7% (n=57) and 14.8% (n=17), respectively (P<0.001).
Serious adverse events occurred in 12.0% of patients in the ferric citrate arm and 11.2% of patients in the placebo arm. There were 2 treatment-emergent deaths in the ferric citrate arm (and none in the placebo arm), but they were not considered drug-related.
The most common (≥5%) treatment-emergent adverse events in patients who received ferric citrate were diarrhea (20.5%), constipation (18.8%), discolored feces (14.5%), nausea (11.1%), abdominal pain (6.0%), and hyperkalemia (6.8%). ![]()
FDA lifts hold on trials of universal CAR T-cell therapy

The US Food and Drug Administration (FDA) has lifted the full clinical hold on 2 phase 1 studies of UCART123, an allogeneic chimeric antigen receptor (CAR) T-cell therapy targeting CD123.
One of these studies was designed for patients with acute myeloid leukemia (AML), and the other was designed for patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The hold meant no new subjects could be enrolled in either trial, and there could be no further dosing of subjects who were already enrolled.
The hold was placed in September because the first patient treated in the BPDCN trial died. The patient developed grade 2 cytokine release syndrome (CRS) and a grade 3 lung infection. This was followed by grade 4 capillary leak syndrome and grade 5 CRS.
The first patient treated in the AML trial also developed grade 4 capillary leak syndrome and grade 3 CRS, but both resolved.
Now, the FDA has lifted the hold on the trials because Cellectis, the company developing UCART123, agreed to implement the following main revisions to phase 1 UCART123 protocols:
- Decrease the cohort dose level to 6.25 x 104 UCART123 cells/kg
- Decrease the cyclophosphamide dose of the lymphodepleting regimen to 750 mg/m²/day over 3 days, with a maximum daily dose of 1.33 grams
- Include specific criteria at Day 0, the day of UCART123 infusion, such as no new uncontrolled infection after receipt of lymphodepletion, afebrile, off all but replacement dose of corticosteroids, and no organ dysfunction since eligibility screening
- Ensure the next 3 patients to be treated in each protocol will be under the age of 65
- Ensure that enrollment will be staggered across the UCART123 protocols; at least 28 days should elapse between the enrollments of 2 patients across the 2 studies.
Cellectis is currently working with investigators and clinical sites to obtain internal review board approval on the revised protocols and resume patient enrollment. ![]()
The US Food and Drug Administration (FDA) has lifted the full clinical hold on 2 phase 1 studies of UCART123, an allogeneic chimeric antigen receptor (CAR) T-cell therapy targeting CD123.
One of these studies was designed for patients with acute myeloid leukemia (AML), and the other was designed for patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The hold meant no new subjects could be enrolled in either trial, and there could be no further dosing of subjects who were already enrolled.
The hold was placed in September because the first patient treated in the BPDCN trial died. The patient developed grade 2 cytokine release syndrome (CRS) and a grade 3 lung infection. This was followed by grade 4 capillary leak syndrome and grade 5 CRS.
The first patient treated in the AML trial also developed grade 4 capillary leak syndrome and grade 3 CRS, but both resolved.
Now, the FDA has lifted the hold on the trials because Cellectis, the company developing UCART123, agreed to implement the following main revisions to phase 1 UCART123 protocols:
- Decrease the cohort dose level to 6.25 x 104 UCART123 cells/kg
- Decrease the cyclophosphamide dose of the lymphodepleting regimen to 750 mg/m²/day over 3 days, with a maximum daily dose of 1.33 grams
- Include specific criteria at Day 0, the day of UCART123 infusion, such as no new uncontrolled infection after receipt of lymphodepletion, afebrile, off all but replacement dose of corticosteroids, and no organ dysfunction since eligibility screening
- Ensure the next 3 patients to be treated in each protocol will be under the age of 65
- Ensure that enrollment will be staggered across the UCART123 protocols; at least 28 days should elapse between the enrollments of 2 patients across the 2 studies.
Cellectis is currently working with investigators and clinical sites to obtain internal review board approval on the revised protocols and resume patient enrollment. ![]()
The US Food and Drug Administration (FDA) has lifted the full clinical hold on 2 phase 1 studies of UCART123, an allogeneic chimeric antigen receptor (CAR) T-cell therapy targeting CD123.
One of these studies was designed for patients with acute myeloid leukemia (AML), and the other was designed for patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN).
The hold meant no new subjects could be enrolled in either trial, and there could be no further dosing of subjects who were already enrolled.
The hold was placed in September because the first patient treated in the BPDCN trial died. The patient developed grade 2 cytokine release syndrome (CRS) and a grade 3 lung infection. This was followed by grade 4 capillary leak syndrome and grade 5 CRS.
The first patient treated in the AML trial also developed grade 4 capillary leak syndrome and grade 3 CRS, but both resolved.
Now, the FDA has lifted the hold on the trials because Cellectis, the company developing UCART123, agreed to implement the following main revisions to phase 1 UCART123 protocols:
- Decrease the cohort dose level to 6.25 x 104 UCART123 cells/kg
- Decrease the cyclophosphamide dose of the lymphodepleting regimen to 750 mg/m²/day over 3 days, with a maximum daily dose of 1.33 grams
- Include specific criteria at Day 0, the day of UCART123 infusion, such as no new uncontrolled infection after receipt of lymphodepletion, afebrile, off all but replacement dose of corticosteroids, and no organ dysfunction since eligibility screening
- Ensure the next 3 patients to be treated in each protocol will be under the age of 65
- Ensure that enrollment will be staggered across the UCART123 protocols; at least 28 days should elapse between the enrollments of 2 patients across the 2 studies.
Cellectis is currently working with investigators and clinical sites to obtain internal review board approval on the revised protocols and resume patient enrollment. ![]()
From 5K to … the End of the Driveway
ANSWER
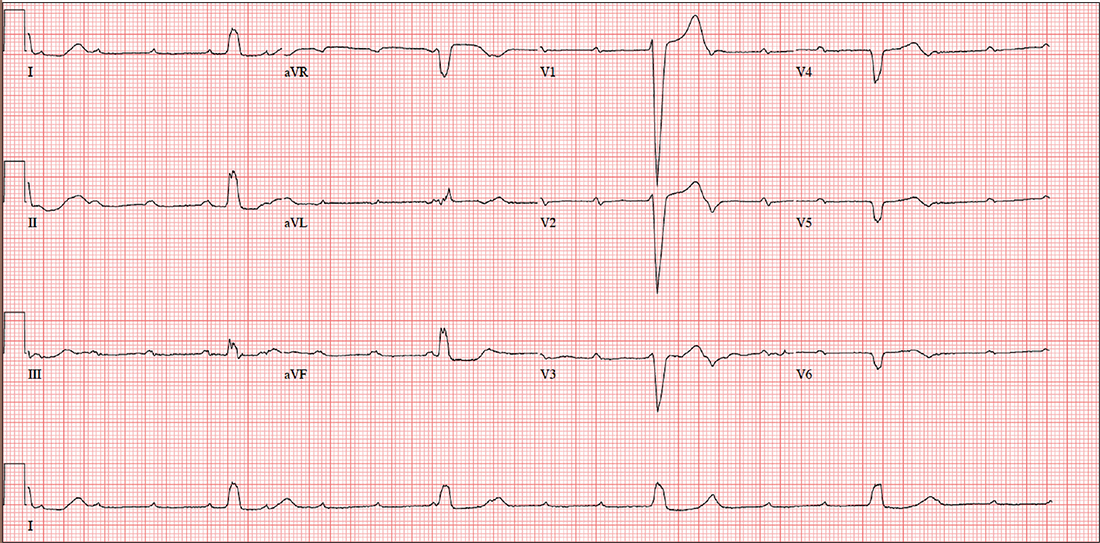
The correct interpretation includes sinus tachycardia with complete heart block and an idioventricular rhythm. Careful review of this ECG confirms complete atrioventricular dissociation, which is indicative of complete heart block.
Sinus tachycardia is indicated by a consistent P-P interval at a rate of 110 beats/min, idioventricular rhythm with a regular (but not normal) rate, and prolonged QRS interval of 148 ms. In this patient’s case, the tachycardia was presumed to be due to his upper respiratory infection. He underwent permanent pacemaker placement and resumed his normal activities without restriction.
ANSWER
The correct interpretation includes sinus tachycardia with complete heart block and an idioventricular rhythm. Careful review of this ECG confirms complete atrioventricular dissociation, which is indicative of complete heart block.
Sinus tachycardia is indicated by a consistent P-P interval at a rate of 110 beats/min, idioventricular rhythm with a regular (but not normal) rate, and prolonged QRS interval of 148 ms. In this patient’s case, the tachycardia was presumed to be due to his upper respiratory infection. He underwent permanent pacemaker placement and resumed his normal activities without restriction.
ANSWER
The correct interpretation includes sinus tachycardia with complete heart block and an idioventricular rhythm. Careful review of this ECG confirms complete atrioventricular dissociation, which is indicative of complete heart block.
Sinus tachycardia is indicated by a consistent P-P interval at a rate of 110 beats/min, idioventricular rhythm with a regular (but not normal) rate, and prolonged QRS interval of 148 ms. In this patient’s case, the tachycardia was presumed to be due to his upper respiratory infection. He underwent permanent pacemaker placement and resumed his normal activities without restriction.
Until three weeks ago, this 74-year-old man walked three miles every day without difficulty. But now, shortness of breath forces him to stop walking before he even reaches the end of his driveway. He denies chest pain (at rest or on exertion), palpitations, dyspnea at rest, and paroxysmal nocturnal dyspnea. There have been no recent weight changes.
Four years ago, he was diagnosed with coronary artery disease after experiencing chest pain at rest. A coronary angiography revealed stenosis in the proximal right coronary artery and the second obtuse marginal branch of his circumflex coronary artery; drug-eluting stents were placed, and he has had no further symptoms. An echocardiogram performed at a routine clinic visit six months ago showed mild aortic valve sclerosis, a left ventricular ejection fraction of 64%, and no regional wall motion abnormalities.
Surgical history is also remarkable for an open reduction and stabilization of a right high ankle fracture 10 years ago. Medical history includes type 2 diabetes and hyperlipidemia; treatment has normalized his A1C and his lipid panel.
His current medication list includes metoprolol, isosorbide dinitrate, metformin, and atorvastatin. He has no known drug allergies. He is unaware of any medical issues with his parents or grandparents.
The patient has two adult sons who live abroad and visit once a year. He was married for 53 years but lost his wife to lung cancer three years ago; her diagnosis prompted him to quit his long-term smoking habit. He consumes alcohol socially, having “one or two beers with friends on the weekends,” but denies current or previous use of marijuana or nonprescribed medications.
Review of systems is remarkable for a three-day history of upper respiratory infection with cough and rhinitis. He denies any change in bowel or bladder function.
Vital signs include a blood pressure of 104/54 mm Hg; pulse, 30 beats/min; respiratory rate, 16 breaths/min-1; and temperature, 99.4°F. His weight is 169 lb and his height, 70 in.
On physical exam, you note a thin, healthy-looking male in no acute distress. Pertinent findings include internally inflamed nares and oropharynx, a few scattered rales in both lower lung fields that clear with coughing, and no wheezing. There are no palpable lymph nodes in the head or neck.
Cardiac exam reveals a regular rhythm of 30 beats/min with no murmurs, rubs, or extra heart sounds. The abdomen is soft and nontender. Peripheral pulses are strong and palpable in both upper and lower extremities, and there is no peripheral edema. The neurologic exam is intact, without evidence of diabetic neuropathy.
Given the slow heart rate observed on physical exam, an ECG is ordered. It shows a ventricular rate of 29 beats/min; no discernable PR interval; QRS duration, 148 ms; QT/QTc interval, 584/405 ms; P axis, 64°; R axis, 55°; and T axis, 83°. What is your interpretation?
Apple pie and ...
How do you feel about apple pie? Is it a concept that evokes a positive feeling for you? Even if you prefer pumpkin or blueberry? Although your attitude toward apple pie may be relevant as we approach the holidays, is it a topic worthy of discussion in a publication devoted to pediatrics?
Certainly not, but what about motherhood? How do you feel about motherhood? As someone who is devoting his or her professional energies to the health of children, you must have formed some opinions about motherhood. Although your patients are children, it is their parents – and more often their mothers – with whom you communicate, particularly in the first several years of life. 
You may never have been asked that question in exactly that way before, but I suspect you have thought about it both professionally and personally. You may have considered the answer as you were deciding if, when, and how you were going to return to work after maternity leave. Or you may have been forced to consider the question in formulating an opinion in a case of contested child custody.
An opinion piece in the Wall Street Journal (“The Politicization of Motherhood,” by James Taranto, Oct. 27, 2017) suggests that how you answer my question about the biological necessity of motherhood will determine your position on one of our nation’s political divides. The article focuses on Erica Komisar, who has written a book in which she lays out evidence from the fields of neuroscience, psychology, and epigenetics supporting her view that a mother is biologically equipped to provide for the emotional development of her child (“Being There: Why Prioritizing Motherhood in the First Three Years Matters,” New York: TarcherPerigee, 2017).

I haven’t read Ms. Komisar’s book, nor am I aware of the studies she cites, but reading the article prompted me to think a bit more deeply regarding how I feel about motherhood. I guess I always have felt that there is something special that a mother can provide her children, particularly during the first 3 years of life. I don’t know whether there is a neurobiological basis for this special something, but if it is missing, the child’s emotional development can suffer. Are there situations where another person(s) can provide a substitute for this special maternal sauce? Of course, but it doesn’t always work as well as the real thing. And not every mother has an adequate amount of that certain maternal something.
As pediatricians, we are faced with two challenges. The first is to help families cope with situations in which that special maternal ingredient is absent or in short supply. Our second challenge is to help mothers who believe there is something special they can offer their children but feel guilty because, for whatever reason, they can’t be there to provide it.
I am interested to hear how you feel about motherhood ... and apple pie.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Email him at pdnews@frontlinemedcom.com.
How do you feel about apple pie? Is it a concept that evokes a positive feeling for you? Even if you prefer pumpkin or blueberry? Although your attitude toward apple pie may be relevant as we approach the holidays, is it a topic worthy of discussion in a publication devoted to pediatrics?
Certainly not, but what about motherhood? How do you feel about motherhood? As someone who is devoting his or her professional energies to the health of children, you must have formed some opinions about motherhood. Although your patients are children, it is their parents – and more often their mothers – with whom you communicate, particularly in the first several years of life.
You may never have been asked that question in exactly that way before, but I suspect you have thought about it both professionally and personally. You may have considered the answer as you were deciding if, when, and how you were going to return to work after maternity leave. Or you may have been forced to consider the question in formulating an opinion in a case of contested child custody.
An opinion piece in the Wall Street Journal (“The Politicization of Motherhood,” by James Taranto, Oct. 27, 2017) suggests that how you answer my question about the biological necessity of motherhood will determine your position on one of our nation’s political divides. The article focuses on Erica Komisar, who has written a book in which she lays out evidence from the fields of neuroscience, psychology, and epigenetics supporting her view that a mother is biologically equipped to provide for the emotional development of her child (“Being There: Why Prioritizing Motherhood in the First Three Years Matters,” New York: TarcherPerigee, 2017).
I haven’t read Ms. Komisar’s book, nor am I aware of the studies she cites, but reading the article prompted me to think a bit more deeply regarding how I feel about motherhood. I guess I always have felt that there is something special that a mother can provide her children, particularly during the first 3 years of life. I don’t know whether there is a neurobiological basis for this special something, but if it is missing, the child’s emotional development can suffer. Are there situations where another person(s) can provide a substitute for this special maternal sauce? Of course, but it doesn’t always work as well as the real thing. And not every mother has an adequate amount of that certain maternal something.
As pediatricians, we are faced with two challenges. The first is to help families cope with situations in which that special maternal ingredient is absent or in short supply. Our second challenge is to help mothers who believe there is something special they can offer their children but feel guilty because, for whatever reason, they can’t be there to provide it.
I am interested to hear how you feel about motherhood ... and apple pie.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Email him at pdnews@frontlinemedcom.com.
How do you feel about apple pie? Is it a concept that evokes a positive feeling for you? Even if you prefer pumpkin or blueberry? Although your attitude toward apple pie may be relevant as we approach the holidays, is it a topic worthy of discussion in a publication devoted to pediatrics?
Certainly not, but what about motherhood? How do you feel about motherhood? As someone who is devoting his or her professional energies to the health of children, you must have formed some opinions about motherhood. Although your patients are children, it is their parents – and more often their mothers – with whom you communicate, particularly in the first several years of life.
You may never have been asked that question in exactly that way before, but I suspect you have thought about it both professionally and personally. You may have considered the answer as you were deciding if, when, and how you were going to return to work after maternity leave. Or you may have been forced to consider the question in formulating an opinion in a case of contested child custody.
An opinion piece in the Wall Street Journal (“The Politicization of Motherhood,” by James Taranto, Oct. 27, 2017) suggests that how you answer my question about the biological necessity of motherhood will determine your position on one of our nation’s political divides. The article focuses on Erica Komisar, who has written a book in which she lays out evidence from the fields of neuroscience, psychology, and epigenetics supporting her view that a mother is biologically equipped to provide for the emotional development of her child (“Being There: Why Prioritizing Motherhood in the First Three Years Matters,” New York: TarcherPerigee, 2017).
I haven’t read Ms. Komisar’s book, nor am I aware of the studies she cites, but reading the article prompted me to think a bit more deeply regarding how I feel about motherhood. I guess I always have felt that there is something special that a mother can provide her children, particularly during the first 3 years of life. I don’t know whether there is a neurobiological basis for this special something, but if it is missing, the child’s emotional development can suffer. Are there situations where another person(s) can provide a substitute for this special maternal sauce? Of course, but it doesn’t always work as well as the real thing. And not every mother has an adequate amount of that certain maternal something.
As pediatricians, we are faced with two challenges. The first is to help families cope with situations in which that special maternal ingredient is absent or in short supply. Our second challenge is to help mothers who believe there is something special they can offer their children but feel guilty because, for whatever reason, they can’t be there to provide it.
I am interested to hear how you feel about motherhood ... and apple pie.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Email him at pdnews@frontlinemedcom.com.
Use of opioids, SSRIs linked to increased fracture risk in RA
SAN DIEGO – The use of selective serotonin reuptake inhibitors and opioids was associated with an increased osteoporotic fracture risk in patients with rheumatoid arthritis, results from an analysis of national data showed.
“Osteoporotic fractures are one of the important causes of disability, health-related costs, and mortality in RA, with substantially higher complication and mortality rates than the general population,” study author Gulsen Ozen, MD, said in an interview prior to the annual meeting of the American College of Rheumatology. “Given the burden of osteoporotic fractures and the suboptimal osteoporosis care, identifying the factors associated with fracture risk in RA patients is of paramount importance.”

During a median follow-up of nearly 6 years, 863 patients (7.8%) sustained osteoporotic fractures. Compared with patients who did not develop fractures, those who did were significantly older, had higher disease duration and activity, glucocorticoid use, comorbidity and FRAX, a fracture risk assessment tool, scores at baseline. After adjusting for sociodemographics, comorbidities, body mass index, fracture risk by FRAX, and RA severity measures, the researchers found a significant risk of osteoporotic fractures with use of opioids of any strength (weak agents, hazard ratio, 1.45; strong agents, HR, 1.79; P less than .001 for both), SSRI use (HR, 1.35; P = .003), and glucocorticoid use of 3 months or longer at a dose of at least 7.5 mg per day (HR, 1.74; P less than .05). Osteoporotic fracture risk increase started even after 1-30 days of opioid use (HR, 1.96; P less than .001), whereas SSRI-associated risk increase started after 3 months of use (HR, 1.42; P = .054). No significant association with fracture risk was observed with the use of other disease-modifying antirheumatic drugs, statins, antidepressants, proton pump inhibitors, NSAIDs, anticonvulsants, and antipsychotics.
“One of the first surprising findings was that almost 40% of the RA patients older than 40 years of age were at least once exposed to opioid analgesics,” said Dr. Ozen, who is a research fellow in the division of immunology and rheumatology at the medical center. “Another surprising finding was that even very short-term (1-30 days) use of opioids was associated with increased fracture risk.” She went on to note that careful and regular reviewing of patient medications “is an essential part of the RA patient care, as the use of medications not indicated anymore brings harm rather than a benefit. The most well-known example for this is glucocorticoid use. This is valid for all medications, too. Therefore, we hope that our findings provide more awareness about osteoporotic fractures and associated risk factors in RA patients.”
She acknowledged certain limitations of the study, including its observational design. “Additionally, fracture and the level of the trauma in our cohort were reported by patients,” she said. “Therefore, there might be some misclassification of fractures as osteoporotic fractures. Lastly, we did not have detailed data regarding fall risk, which might explain the associations we observed with opioids and potentially, SSRIs.”
Dr. Ozen reported having no disclosures.
SAN DIEGO – The use of selective serotonin reuptake inhibitors and opioids was associated with an increased osteoporotic fracture risk in patients with rheumatoid arthritis, results from an analysis of national data showed.
“Osteoporotic fractures are one of the important causes of disability, health-related costs, and mortality in RA, with substantially higher complication and mortality rates than the general population,” study author Gulsen Ozen, MD, said in an interview prior to the annual meeting of the American College of Rheumatology. “Given the burden of osteoporotic fractures and the suboptimal osteoporosis care, identifying the factors associated with fracture risk in RA patients is of paramount importance.”
During a median follow-up of nearly 6 years, 863 patients (7.8%) sustained osteoporotic fractures. Compared with patients who did not develop fractures, those who did were significantly older, had higher disease duration and activity, glucocorticoid use, comorbidity and FRAX, a fracture risk assessment tool, scores at baseline. After adjusting for sociodemographics, comorbidities, body mass index, fracture risk by FRAX, and RA severity measures, the researchers found a significant risk of osteoporotic fractures with use of opioids of any strength (weak agents, hazard ratio, 1.45; strong agents, HR, 1.79; P less than .001 for both), SSRI use (HR, 1.35; P = .003), and glucocorticoid use of 3 months or longer at a dose of at least 7.5 mg per day (HR, 1.74; P less than .05). Osteoporotic fracture risk increase started even after 1-30 days of opioid use (HR, 1.96; P less than .001), whereas SSRI-associated risk increase started after 3 months of use (HR, 1.42; P = .054). No significant association with fracture risk was observed with the use of other disease-modifying antirheumatic drugs, statins, antidepressants, proton pump inhibitors, NSAIDs, anticonvulsants, and antipsychotics.
“One of the first surprising findings was that almost 40% of the RA patients older than 40 years of age were at least once exposed to opioid analgesics,” said Dr. Ozen, who is a research fellow in the division of immunology and rheumatology at the medical center. “Another surprising finding was that even very short-term (1-30 days) use of opioids was associated with increased fracture risk.” She went on to note that careful and regular reviewing of patient medications “is an essential part of the RA patient care, as the use of medications not indicated anymore brings harm rather than a benefit. The most well-known example for this is glucocorticoid use. This is valid for all medications, too. Therefore, we hope that our findings provide more awareness about osteoporotic fractures and associated risk factors in RA patients.”
She acknowledged certain limitations of the study, including its observational design. “Additionally, fracture and the level of the trauma in our cohort were reported by patients,” she said. “Therefore, there might be some misclassification of fractures as osteoporotic fractures. Lastly, we did not have detailed data regarding fall risk, which might explain the associations we observed with opioids and potentially, SSRIs.”
Dr. Ozen reported having no disclosures.
SAN DIEGO – The use of selective serotonin reuptake inhibitors and opioids was associated with an increased osteoporotic fracture risk in patients with rheumatoid arthritis, results from an analysis of national data showed.
“Osteoporotic fractures are one of the important causes of disability, health-related costs, and mortality in RA, with substantially higher complication and mortality rates than the general population,” study author Gulsen Ozen, MD, said in an interview prior to the annual meeting of the American College of Rheumatology. “Given the burden of osteoporotic fractures and the suboptimal osteoporosis care, identifying the factors associated with fracture risk in RA patients is of paramount importance.”
During a median follow-up of nearly 6 years, 863 patients (7.8%) sustained osteoporotic fractures. Compared with patients who did not develop fractures, those who did were significantly older, had higher disease duration and activity, glucocorticoid use, comorbidity and FRAX, a fracture risk assessment tool, scores at baseline. After adjusting for sociodemographics, comorbidities, body mass index, fracture risk by FRAX, and RA severity measures, the researchers found a significant risk of osteoporotic fractures with use of opioids of any strength (weak agents, hazard ratio, 1.45; strong agents, HR, 1.79; P less than .001 for both), SSRI use (HR, 1.35; P = .003), and glucocorticoid use of 3 months or longer at a dose of at least 7.5 mg per day (HR, 1.74; P less than .05). Osteoporotic fracture risk increase started even after 1-30 days of opioid use (HR, 1.96; P less than .001), whereas SSRI-associated risk increase started after 3 months of use (HR, 1.42; P = .054). No significant association with fracture risk was observed with the use of other disease-modifying antirheumatic drugs, statins, antidepressants, proton pump inhibitors, NSAIDs, anticonvulsants, and antipsychotics.
“One of the first surprising findings was that almost 40% of the RA patients older than 40 years of age were at least once exposed to opioid analgesics,” said Dr. Ozen, who is a research fellow in the division of immunology and rheumatology at the medical center. “Another surprising finding was that even very short-term (1-30 days) use of opioids was associated with increased fracture risk.” She went on to note that careful and regular reviewing of patient medications “is an essential part of the RA patient care, as the use of medications not indicated anymore brings harm rather than a benefit. The most well-known example for this is glucocorticoid use. This is valid for all medications, too. Therefore, we hope that our findings provide more awareness about osteoporotic fractures and associated risk factors in RA patients.”
She acknowledged certain limitations of the study, including its observational design. “Additionally, fracture and the level of the trauma in our cohort were reported by patients,” she said. “Therefore, there might be some misclassification of fractures as osteoporotic fractures. Lastly, we did not have detailed data regarding fall risk, which might explain the associations we observed with opioids and potentially, SSRIs.”
Dr. Ozen reported having no disclosures.
AT ACR 2017
Key clinical point: When managing with opioids, even in the short-term, clinicians should be aware of the fracture risk.
Major finding: In patients with RA, concomitant use of selective serotonin reuptake inhibitors was associated with an increased risk of osteoporotic fracture (HR, 1.35; P = .003), as was opioid use (HR, 1.45 and HR, 1.79) for weak and strong agents, respectively; P less than .001 for both).
Study details: An observational study of 11,049 patients from the National Data Bank for Rheumatic Diseases.
Disclosures: Dr. Ozen reported having no disclosures.
Clinical and Radiographic Outcomes of Total Shoulder Arthroplasty With a Hybrid Dual-Radii Glenoid Component
Take-Home Points
- The authors have developed a total shoulder glenoid prosthesis that conforms with the humeral head in its center and is nonconforming on its peripheral edge.
- All clinical survey and range of motion parameters demonstrated statistically significant improvements at final follow-up.
- Only 3 shoulders (1.7%) required revision surgery.
- Eighty-six (63%) of 136 shoulders demonstrated no radiographic evidence of glenoid loosening.
- This is the first and largest study that evaluates the clinical and radiographic outcomes of this hybrid shoulder prosthesis.
Fixation of the glenoid component is the limiting factor in modern total shoulder arthroplasty (TSA). Glenoid loosening, the most common long-term complication, necessitates revision in up to 12% of patients.1-4 By contrast, humeral component loosening is relatively uncommon, affecting as few as 0.34% of patients.5 Multiple long-term studies have found consistently high rates (45%-93%) of radiolucencies around the glenoid component.3,6,7 Although their clinical significance has been debated, radiolucencies around the glenoid component raise concern about progressive loss of fixation.
Since TSA was introduced in the 1970s, complications with the glenoid component have been addressed with 2 different designs: conforming (congruent) and nonconforming. In a congruent articulation, the radii of curvature of the glenoid and humeral head components are identical, whereas they differ in a nonconforming model. Joint conformity is inversely related to glenohumeral translation.8 Neer’s original TSA was made congruent in order to limit translation and maximize the contact area. However, this design results in edge loading and a so-called rocking-horse phenomenon, which may lead to glenoid loosening.9-13 Surgeons therefore have increasingly turned to nonconforming implants. In the nonconforming design, the radius of curvature of the humeral head is smaller than that of the glenoid. Although this design may reduce edge loading,14 it allows more translation and reduces the relative contact area of the glenohumeral joint. As a result, more contact stress is transmitted to the glenoid component, leading to polyethylene deformation and wear.15,16
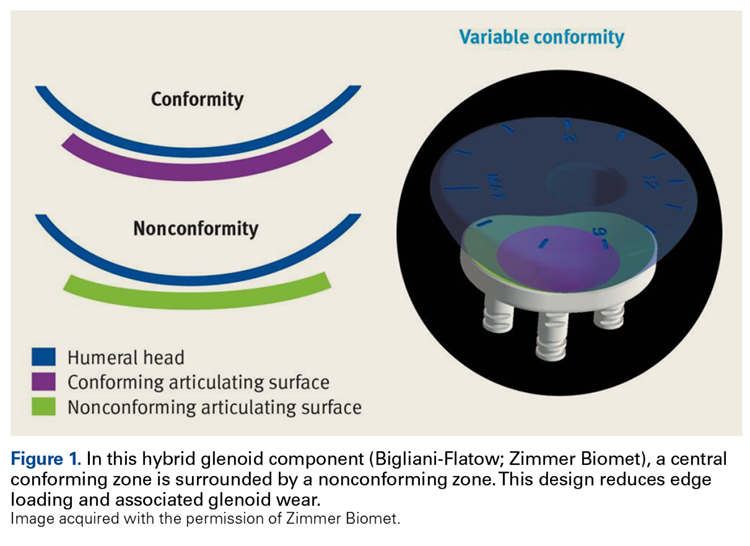
Dual radii of curvature are designed to augment joint stability without increasing component wear. Biomechanical data have indicated that edge loading is not increased by having a central conforming region added to a nonconforming model.17 The clinical value of this prosthesis, however, has not been determined. Therefore, we conducted a study to describe the intermediate-term clinical and radiographic outcomes of TSAs that use a novel hybrid glenoid component.
Materials and Methods
This study was approved (protocol AAAD3473) by the Institutional Review Board of Columbia University and was conducted in compliance with Health Insurance Portability and Accountability Act (HIPAA) regulations.
Patient Selection
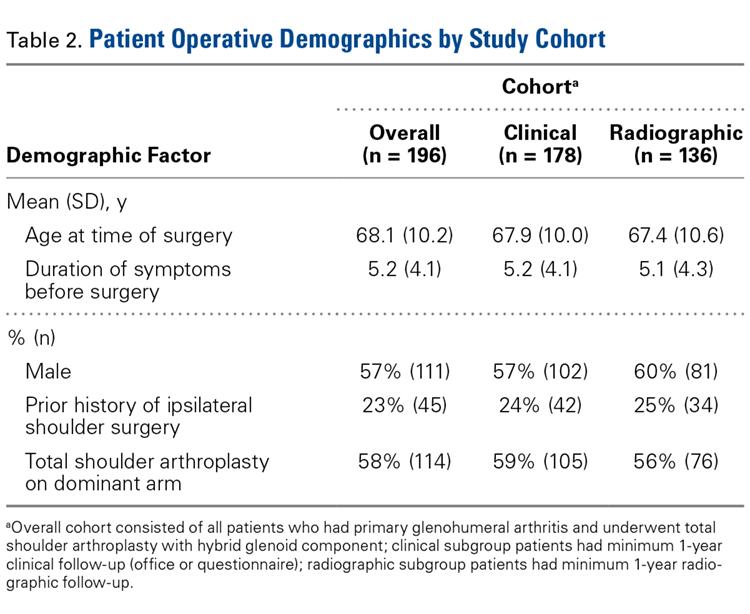
At Columbia University Medical Center, Dr. Bigliani performed 196 TSAs with a hybrid glenoid component (Bigliani-Flatow; Zimmer Biomet) in 169 patients between September 1998 and November 2007. All patients had received a diagnosis of primary glenohumeral arthritis as defined by Neer.18 Patients with previous surgery such as rotator cuff repair or subacromial decompression were included in our review, and patients with a nonprimary form of arthritis, such as rheumatoid, posttraumatic, or post-capsulorrhaphy arthritis, were excluded.
Operative Technique
For all surgeries, Dr. Bigliani performed a subscapularis tenotomy with regional anesthesia and a standard deltopectoral approach. A partial anterior capsulectomy was performed to increase the glenoid’s visibility. The inferior labrum was removed with a needle-tip bovie while the axillary nerve was being protected with a metal finger or narrow Darrach retractor. After reaming and trialing, the final glenoid component was cemented into place. Cement was placed only in the peg or keel holes and pressurized twice before final implantation. Of the 196 glenoid components, 168 (86%) were pegged and 28 (14%) keeled; in addition,190 of these components were all-polyethylene, whereas 6 had trabecular-metal backing. All glenoid components incorporated the hybrid design of dual radii of curvature. After the glenoid was cemented, the final humeral component was placed in 30° of retroversion. Whenever posterior wear was found, retroversion was reduced by 5° to 10°. The humeral prosthesis was cemented in cases (104/196, 53%) of poor bone quality or a large canal.
After surgery, the patient’s sling was fitted with an abduction pillow and a swathe, to be worn the first 24 hours, and the arm was passively ranged. Patients typically were discharged on postoperative day 2. Then, for 2 weeks, they followed an assisted passive range of motion (ROM) protocol, with limited external rotation, for promotion of subscapularis healing.
Clinical Outcomes
Dr. Bigliani assessed preoperative ROM in all planes. During initial evaluation, patients completed a questionnaire that consisted of the 36-Item Short Form Health Survey19,20 (SF-36) and the American Shoulder and Elbow Surgeons21 (ASES) and Simple Shoulder Test22 (SST) surveys. Postoperative clinical data were collected from office follow-up visits, survey questionnaires, or both. Postoperative office data included ROM, subscapularis integrity testing (belly-press or lift-off), and any complications. Patients with <1 year of office follow-up were excluded. In addition, the same survey questionnaire that was used before surgery was mailed to all patients after surgery; then, for anyone who did not respond by mail, we attempted contact by telephone. Neer criteria were based on patients’ subjective assessment of each arm on a 3-point Likert scale (1 = very satisfied, 2 = satisfied, 3 = dissatisfied). Patients were also asked about any specific complications or revision operations since their index procedure.
Physical examination and office follow-up data were obtained for 129 patients (148/196 shoulders, 76% follow-up) at a mean of 3.7 years (range 1.0-10.2 years) after surgery. Surveys were completed by 117 patients (139/196 shoulders, 71% follow-up) at a mean of 5.1 years (range, 1.6-11.2 years) after surgery. Only 15 patients had neither 1 year of office follow-up nor a completed questionnaire. The remaining 154 patients (178/196 shoulders, 91% follow-up) had clinical follow-up with office, mail, or telephone questionnaire at a mean of 4.8 years (range, 1.0-11.2 years) after surgery. This cohort of patients was used to determine rates of surgical revisions, subscapularis tears, dislocations, and other complications.
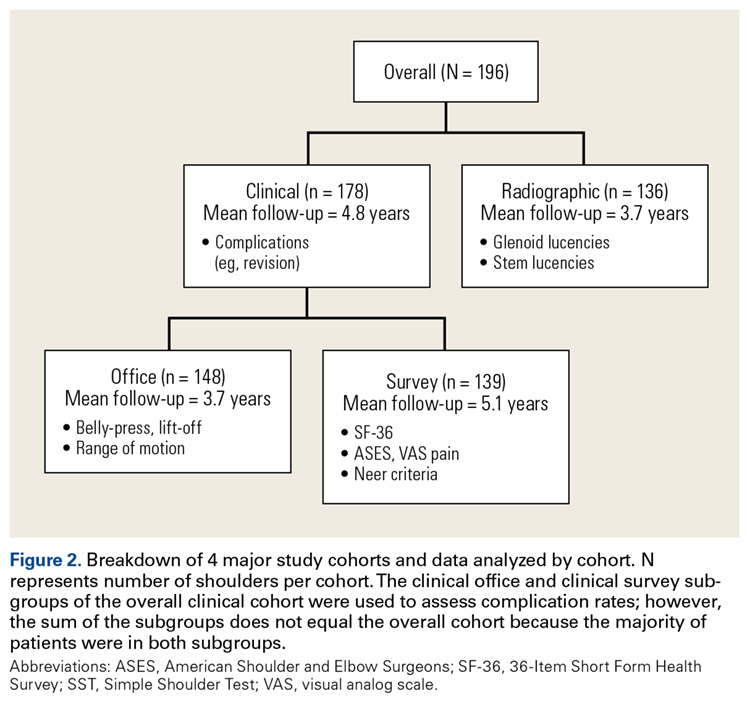
Radiographic Outcomes
Patients were included in the radiographic analysis if they had a shoulder radiograph at least 1 year after surgery. One hundred nineteen patients (136/196 shoulders, 69% follow-up) had radiographic follow-up at a mean of 3.7 years (range, 1.0-9.4 years) after surgery.
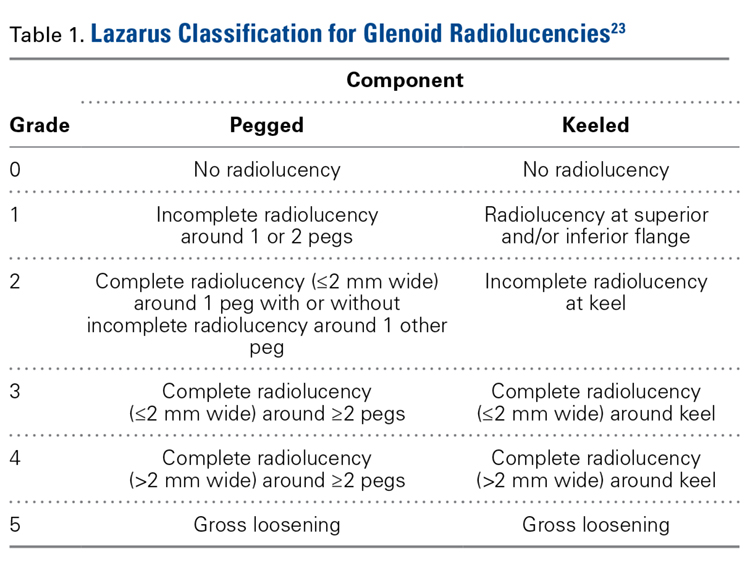
Statistical Analysis
Statistical analysis was performed with Stata Version 10.0. Paired t tests were used to compare preoperative and postoperative numerical data, including ROM and survey scores. We calculated 95% confidence intervals (CIs) and set statistical significance at P < .05. For qualitative measures, the Fisher exact test was used. Survivorship analysis was performed according to the Kaplan-Meier method, with right-censored data for no event or missing data.25
Results
Clinical Analysis of Demographics
In demographics, the clinical and radiographic patient subgroups were similar to each other and to the overall study population (Table 2). Of 196 patients overall, 16 (8%) had a concomitant rotator cuff repair, and 27 (14%) underwent staged bilateral shoulder arthroplasties.
Clinical Analysis of ROM and Survey Scores
Operative shoulder ROM in forward elevation, external rotation at side, external rotation in abduction, and internal rotation all showed statistically significant (P < .001) improvement from before surgery to after surgery. Over 3.7 years, mean (SD) forward elevation improved from 107.3° (34.8°) to 159.0° (29.4°), external rotation at side improved from 20.4° (16.7°) to 49.4° (11.3°), and external rotation in abduction improved from 53.7° (24.3°) to 84.7° (9.1°). Internal rotation improved from a mean (SD) vertebral level of S1 (6.0 levels) to T9 (3.7 levels).
All validated survey scores also showed statistically significant (P < .001) improvement from before surgery to after surgery. Over 5.1 years, mean (SD) SF-36 scores improved from 64.9 (13.4) to 73.6 (17.1), ASES scores improved from 41.1 (22.5) to 82.7 (17.7), SST scores improved from 3.9 (2.8) to 9.7 (2.2), and visual analog scale pain scores improved from 5.6 (3.2) to 1.4 (2.1). Of 139 patients with follow-up, 130 (93.5%) were either satisfied or very satisfied with their TSA, and only 119 (86%) were either satisfied or very satisfied with the nonoperative shoulder.
Clinical Analysis of Postoperative Complications
Of the 178 shoulders evaluated for complications, 3 (1.7%) underwent revision surgery. Mean time to revision was 2.3 years (range, 1.5-3.9 years). Two revisions involved the glenoid component, and the third involved the humerus. In one of the glenoid cases, a 77-year-old woman fell and sustained a fracture at the base of the trabecular metal glenoid pegs; her component was revised to an all-polyethylene component, and she had no further complications. In the other glenoid case, a 73-year-old man’s all-polyethylene component loosened after 2 years and was revised to a trabecular metal implant, which loosened as well and was later converted to a hemiarthroplasty. In the humeral case, a 33-year-old man had his 4-year-old index TSA revised to a cemented stem and had no further complications.
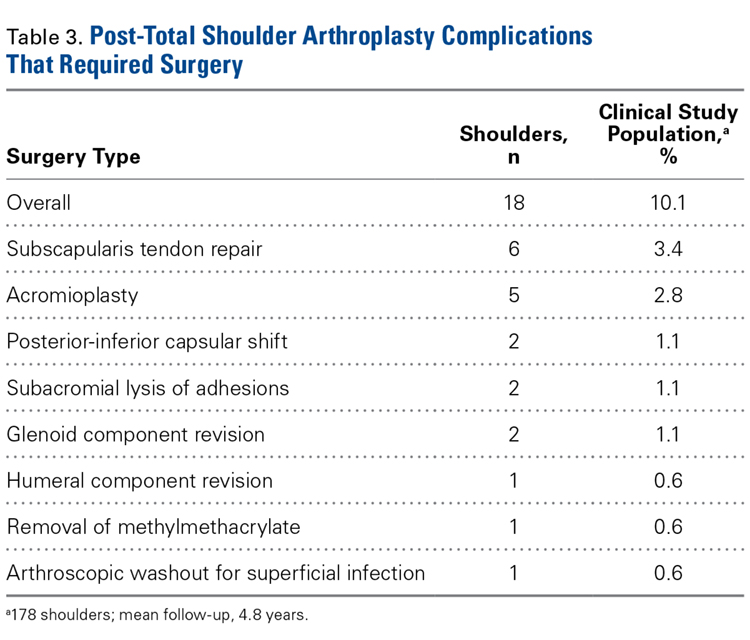
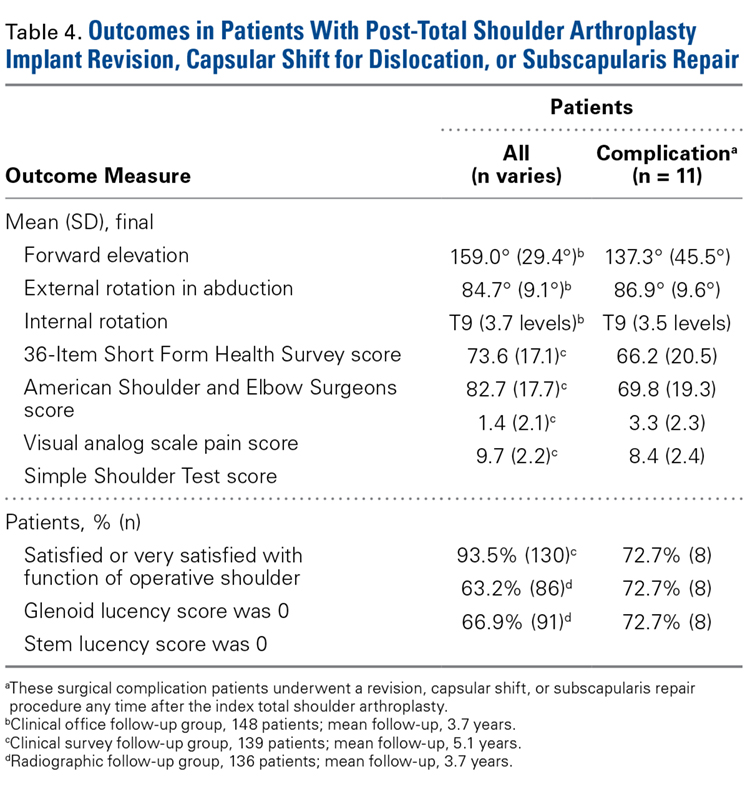
Table 4 compares the clinical and radiographic outcomes of patients who required subscapularis repair, capsular shift, or implant revision with the outcomes of all other study patients, and Figure 3 shows Kaplan-Meier survivorship.
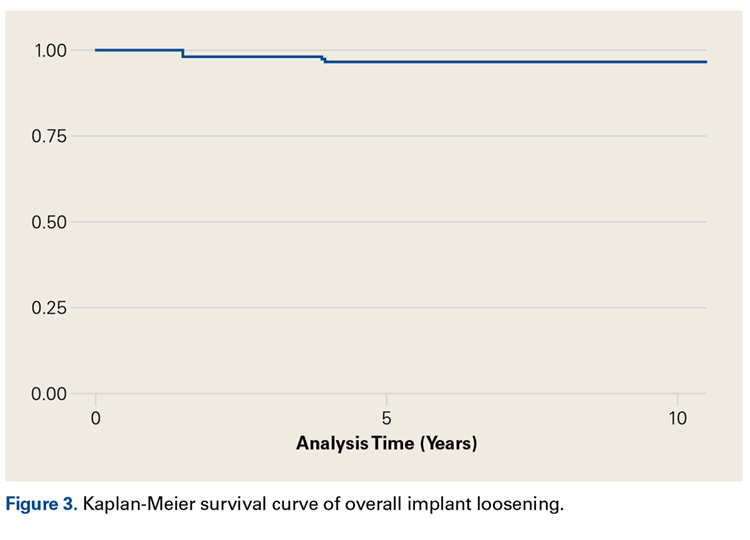
Postoperative Radiographic Analysis
Glenoid Component. At a mean of 3.7 years (minimum, 1 year) after surgery, 86 (63%) of 136 radiographically evaluated shoulders showed no glenoid lucencies; the other 50 (37%) showed ≥1 lucency. Of the 136 shoulders, 33 (24%) had a Lazarus score of 1, 15 (11%) had a score of 2, and only 2 (2%) had a score of 3. None of the shoulders had a score of 4 or 5.
Humeral Component. Of the 136 shoulders, 91 (67%) showed no lucencies in any of the 8 humeral stem zones; the other 45 (33%) showed 1 to 3 lucencies. Thirty (22%) of the 136 shoulders had 1 stem lucency zone, 8 (6%) had 2, and 3 (2%) had 3. None of the shoulders had >3 periprosthetic zones with lucent lines.
Discussion
In this article, we describe a hybrid glenoid TSA component with dual radii of curvature. Its central portion is congruent with the humeral head, and its peripheral portion is noncongruent and larger. The most significant finding of our study is the low rate (1.1%) of glenoid component revision 4.8 years after surgery. This rate is the lowest that has been reported in a study of ≥100 patients. Overall implant survival appeared as an almost flat Kaplan-Meir curve. We attribute this low revision rate to improved biomechanics with the hybrid glenoid design.
Symptomatic glenoid component loosening is the most common TSA complication.1,26-28 In a review of 73 Neer TSAs, Cofield7 found glenoid radiolucencies in 71% of patients 3.8 years after surgery. Radiographic evidence of loosening, defined as component migration, or tilt, or a circumferential lucency 1.5 mm thick, was present in another 11% of patients, and 4.1% developed symptomatic loosening that required glenoid revision. In a study with 12.2-year follow-up, Torchia and colleagues3 found rates of 84% for glenoid radiolucencies, 44% for radiographic loosening, and 5.6% for symptomatic loosening that required revision. In a systematic review of studies with follow-up of ≥10 years, Bohsali and colleagues27 found similar lucency and radiographic loosening rates and a 7% glenoid revision rate. These data suggest glenoid radiolucencies may progress to component loosening.
Degree of joint congruence is a key factor in glenoid loosening. Neer’s congruent design increases the contact area with concentric loading and reduces glenohumeral translation, which leads to reduced polyethylene wear and improved joint stability. In extreme arm positions, however, humeral head subluxation results in edge loading and a glenoid rocking-horse effect.9-13,17,29-31 Conversely, nonconforming implants allow increased glenohumeral translation without edge loading,14 though they also reduce the relative glenohumeral contact area and thus transmit more contact stress to the glenoid.16,17 A hybrid glenoid component with central conforming and peripheral nonconforming zones may reduce the rocking-horse effect while maximizing ROM and joint stability. Wang and colleagues32 studied the biomechanical properties of this glenoid design and found that the addition of a central conforming region did not increase edge loading.
Additional results from our study support the efficacy of a hybrid glenoid component. Patients’ clinical outcomes improved significantly. At 5.1 years after surgery, 93.5% of patients were satisfied or very satisfied with their procedure and reported less satisfaction (86%) with the nonoperative shoulder. Also significant was the reduced number of radiolucencies. At 3.7 years after surgery, the overall percentage of shoulders with ≥1 glenoid radiolucency was 37%, considerably lower than the 82% reported by Cofield7 and the rates in more recent studies.3,16,33-36 Of the 178 shoulders in our study, 10 (5.6%) had subscapularis tears, and 6 (3.4%) of 178 had these tears surgically repaired. This 3.4% compares favorably with the 5.9% (of 119 patients) found by Miller and colleagues37 28 months after surgery. Of our 178 shoulders, 27 (15.2%) had clinically significant postoperative complications; 18 (10.1%) of the 178 had these complications surgically treated, and 9 (5.1%) had them managed nonoperatively. Bohsali and colleagues27 systematically reviewed 33 TSA studies and found a slightly higher complication rate (16.3%) 5.3 years after surgery. Furthermore, in our study, the 11 patients who underwent revision, capsular shift, or subscapularis repair had final outcomes comparable to those of the rest of our study population.
Our study had several potential weaknesses. First, its minimum clinical and radiographic follow-up was 1 year, whereas most long-term TSA series set a minimum of 2 years. We used 1 year because this was the first clinical study of the hybrid glenoid component design, and we wanted to maximize its sample size by reporting on intermediate-length outcomes. Even so, 93% (166/178) of our clinical patients and 83% (113/136) of our radiographic patients have had ≥2 years of follow-up, and we continue to follow all study patients for long-term outcomes. Another weakness of the study was its lack of a uniform group of patients with all the office, survey, complications, and radiographic data. Our retrospective study design made it difficult to obtain such a group without significantly reducing the sample size, so we divided patients into 4 data groups. A third potential weakness was the study’s variable method for collecting complications data. Rates of complications in the 178 shoulders were calculated from either office evaluation or patient self-report by mail or telephone. This data collection method is subject to recall bias, but mail and telephone contact was needed so the study would capture the large number of patients who had traveled to our institution for their surgery or had since moved away. Fourth, belly-press and lift-off tests were used in part to assess subscapularis function, but recent literature suggests post-TSA subscapularis assessment can be unreliable.38 These tests may be positive in up to two-thirds of patients after 2 years.39 Fifth, the generalizability of our findings to diagnoses such as rheumatoid and posttraumatic arthritis is limited. We had to restrict the study to patients with primary glenohumeral arthritis in order to minimize confounders.
This study’s main strength is its description of the clinical and radiographic outcomes of using a single prosthetic system in operations performed by a single surgeon in a large number of patients. This was the first and largest study evaluating the clinical and radiographic outcomes of this hybrid glenoid implant. Excluding patients with nonprimary arthritis allowed us to minimize potential confounding factors that affect patient outcomes. In conclusion, our study results showed the favorable clinical and radiographic outcomes of TSAs that have a hybrid glenoid component with dual radii of curvature. At a mean of 3.7 years after surgery, 63% of patients had no glenoid lucencies, and, at a mean of 4.8 years, only 1.7% of patients required revision. We continue to follow these patients to obtain long-term results of this innovative prosthesis.
1. Rodosky MW, Bigliani LU. Indications for glenoid resurfacing in shoulder arthroplasty. J Shoulder Elbow Surg. 1996;5(3):231-248.
2. Boyd AD Jr, Thomas WH, Scott RD, Sledge CB, Thornhill TS. Total shoulder arthroplasty versus hemiarthroplasty. Indications for glenoid resurfacing. J Arthroplasty. 1990;5(4):329-336.
3. Torchia ME, Cofield RH, Settergren CR. Total shoulder arthroplasty with the Neer prosthesis: long-term results. J Shoulder Elbow Surg. 1997;6(6):495-505.
4. Iannotti JP, Norris TR. Influence of preoperative factors on outcome of shoulder arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2003;85(2):251-258.
5. Cofield RH. Degenerative and arthritic problems of the glenohumeral joint. In: Rockwood CA, Matsen FA, eds. The Shoulder. Philadelphia, PA: Saunders; 1990:740-745.
6. Neer CS 2nd, Watson KC, Stanton FJ. Recent experience in total shoulder replacement. J Bone Joint Surg Am. 1982;64(3):319-337.
7. Cofield RH. Total shoulder arthroplasty with the Neer prosthesis. J Bone Joint Surg Am. 1984;66(6):899-906.
8. Karduna AR, Williams GR, Williams JL, Iannotti JP. Kinematics of the glenohumeral joint: influences of muscle forces, ligamentous constraints, and articular geometry. J Orthop Res. 1996;14(6):986-993.
9. Karduna AR, Williams GR, Iannotti JP, Williams JL. Total shoulder arthroplasty biomechanics: a study of the forces and strains at the glenoid component. J Biomech Eng. 1998;120(1):92-99.
10. Karduna AR, Williams GR, Williams JL, Iannotti JP. Glenohumeral joint translations before and after total shoulder arthroplasty. A study in cadavera. J Bone Joint Surg Am. 1997;79(8):1166-1174.
11. Matsen FA 3rd, Clinton J, Lynch J, Bertelsen A, Richardson ML. Glenoid component failure in total shoulder arthroplasty. J Bone Joint Surg Am. 2008;90(4):885-896.
12. Franklin JL, Barrett WP, Jackins SE, Matsen FA 3rd. Glenoid loosening in total shoulder arthroplasty. Association with rotator cuff deficiency. J Arthroplasty. 1988;3(1):39-46.
13. Barrett WP, Franklin JL, Jackins SE, Wyss CR, Matsen FA 3rd. Total shoulder arthroplasty. J Bone Joint Surg Am. 1987;69(6):865-872.
14. Harryman DT, Sidles JA, Harris SL, Lippitt SB, Matsen FA 3rd. The effect of articular conformity and the size of the humeral head component on laxity and motion after glenohumeral arthroplasty. A study in cadavera. J Bone Joint Surg Am. 1995;77(4):555-563.
15. Flatow EL. Prosthetic design considerations in total shoulder arthroplasty. Semin Arthroplasty. 1995;6(4):233-244.
16. Klimkiewicz JJ, Iannotti JP, Rubash HE, Shanbhag AS. Aseptic loosening of the humeral component in total shoulder arthroplasty. J Shoulder Elbow Surg. 1998;7(4):422-426.
17. Wang VM, Krishnan R, Ugwonali OF, Flatow EL, Bigliani LU, Ateshian GA. Biomechanical evaluation of a novel glenoid design in total shoulder arthroplasty. J Shoulder Elbow Surg. 2005;14(1 suppl S):129S-140S.
18. Neer CS 2nd. Replacement arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 1974;56(1):1-13.
19. Boorman RS, Kopjar B, Fehringer E, Churchill RS, Smith K, Matsen FA 3rd. The effect of total shoulder arthroplasty on self-assessed health status is comparable to that of total hip arthroplasty and coronary artery bypass grafting. J Shoulder Elbow Surg. 2003;12(2):158-163.
20. Patel AA, Donegan D, Albert T. The 36-Item Short Form. J Am Acad Orthop Surg. 2007;15(2):126-134.
21. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3(6):347-352.
22. Wright RW, Baumgarten KM. Shoulder outcomes measures. J Am Acad Orthop Surg. 2010;18(7):436-444.
23. Lazarus MD, Jensen KL, Southworth C, Matsen FA 3rd. The radiographic evaluation of keeled and pegged glenoid component insertion. J Bone Joint Surg Am. 2002;84(7):1174-1182.
24. Sperling JW, Cofield RH, O’Driscoll SW, Torchia ME, Rowland CM. Radiographic assessment of ingrowth total shoulder arthroplasty. J Shoulder Elbow Surg. 2000;9(6):507-513.
25. Dinse GE, Lagakos SW. Nonparametric estimation of lifetime and disease onset distributions from incomplete observations. Biometrics. 1982;38(4):921-932.
26. Baumgarten KM, Lashgari CJ, Yamaguchi K. Glenoid resurfacing in shoulder arthroplasty: indications and contraindications. Instr Course Lect. 2004;53:3-11.
27. Bohsali KI, Wirth MA, Rockwood CA Jr. Complications of total shoulder arthroplasty. J Bone Joint Surg Am. 2006;88(10):2279-2292.
28. Wirth MA, Rockwood CA Jr. Complications of total shoulder-replacement arthroplasty. J Bone Joint Surg Am. 1996;78(4):603-616.
29. Poppen NK, Walker PS. Normal and abnormal motion of the shoulder. J Bone Joint Surg Am. 1976;58(2):195-201.
30. Cotton RE, Rideout DF. Tears of the humeral rotator cuff; a radiological and pathological necropsy survey. J Bone Joint Surg Br. 1964;46:314-328.
31. Bigliani LU, Kelkar R, Flatow EL, Pollock RG, Mow VC. Glenohumeral stability. Biomechanical properties of passive and active stabilizers. Clin Orthop Relat Res. 1996;(330):13-30.
32. Wang VM, Sugalski MT, Levine WN, Pawluk RJ, Mow VC, Bigliani LU. Comparison of glenohumeral mechanics following a capsular shift and anterior tightening. J Bone Joint Surg Am. 2005;87(6):1312-1322.
33. Young A, Walch G, Boileau P, et al. A multicentre study of the long-term results of using a flat-back polyethylene glenoid component in shoulder replacement for primary osteoarthritis. J Bone Joint Surg Br. 2011;93(2):210-216.
34. Khan A, Bunker TD, Kitson JB. Clinical and radiological follow-up of the Aequalis third-generation cemented total shoulder replacement: a minimum ten-year study. J Bone Joint Surg Br. 2009;91(12):1594-1600.
35. Walch G, Edwards TB, Boulahia A, Boileau P, Mole D, Adeleine P. The influence of glenohumeral prosthetic mismatch on glenoid radiolucent lines: results of a multicenter study. J Bone Joint Surg Am. 2002;84(12):2186-2191.
36. Bartelt R, Sperling JW, Schleck CD, Cofield RH. Shoulder arthroplasty in patients aged fifty-five years or younger with osteoarthritis. J Shoulder Elbow Surg. 2011;20(1):123-130.
37. Miller BS, Joseph TA, Noonan TJ, Horan MP, Hawkins RJ. Rupture of the subscapularis tendon after shoulder arthroplasty: diagnosis, treatment, and outcome. J Shoulder Elbow Surg. 2005;14(5):492-496.
38. Armstrong A, Lashgari C, Teefey S, Menendez J, Yamaguchi K, Galatz LM. Ultrasound evaluation and clinical correlation of subscapularis repair after total shoulder arthroplasty. J Shoulder Elbow Surg. 2006;15(5):541-548.
39. Miller SL, Hazrati Y, Klepps S, Chiang A, Flatow EL. Loss of subscapularis function after total shoulder replacement: a seldom recognized problem. J Shoulder Elbow Surg. 2003;12(1):29-34.
Take-Home Points
- The authors have developed a total shoulder glenoid prosthesis that conforms with the humeral head in its center and is nonconforming on its peripheral edge.
- All clinical survey and range of motion parameters demonstrated statistically significant improvements at final follow-up.
- Only 3 shoulders (1.7%) required revision surgery.
- Eighty-six (63%) of 136 shoulders demonstrated no radiographic evidence of glenoid loosening.
- This is the first and largest study that evaluates the clinical and radiographic outcomes of this hybrid shoulder prosthesis.
Fixation of the glenoid component is the limiting factor in modern total shoulder arthroplasty (TSA). Glenoid loosening, the most common long-term complication, necessitates revision in up to 12% of patients.1-4 By contrast, humeral component loosening is relatively uncommon, affecting as few as 0.34% of patients.5 Multiple long-term studies have found consistently high rates (45%-93%) of radiolucencies around the glenoid component.3,6,7 Although their clinical significance has been debated, radiolucencies around the glenoid component raise concern about progressive loss of fixation.
Since TSA was introduced in the 1970s, complications with the glenoid component have been addressed with 2 different designs: conforming (congruent) and nonconforming. In a congruent articulation, the radii of curvature of the glenoid and humeral head components are identical, whereas they differ in a nonconforming model. Joint conformity is inversely related to glenohumeral translation.8 Neer’s original TSA was made congruent in order to limit translation and maximize the contact area. However, this design results in edge loading and a so-called rocking-horse phenomenon, which may lead to glenoid loosening.9-13 Surgeons therefore have increasingly turned to nonconforming implants. In the nonconforming design, the radius of curvature of the humeral head is smaller than that of the glenoid. Although this design may reduce edge loading,14 it allows more translation and reduces the relative contact area of the glenohumeral joint. As a result, more contact stress is transmitted to the glenoid component, leading to polyethylene deformation and wear.15,16
Dual radii of curvature are designed to augment joint stability without increasing component wear. Biomechanical data have indicated that edge loading is not increased by having a central conforming region added to a nonconforming model.17 The clinical value of this prosthesis, however, has not been determined. Therefore, we conducted a study to describe the intermediate-term clinical and radiographic outcomes of TSAs that use a novel hybrid glenoid component.
Materials and Methods
This study was approved (protocol AAAD3473) by the Institutional Review Board of Columbia University and was conducted in compliance with Health Insurance Portability and Accountability Act (HIPAA) regulations.
Patient Selection
At Columbia University Medical Center, Dr. Bigliani performed 196 TSAs with a hybrid glenoid component (Bigliani-Flatow; Zimmer Biomet) in 169 patients between September 1998 and November 2007. All patients had received a diagnosis of primary glenohumeral arthritis as defined by Neer.18 Patients with previous surgery such as rotator cuff repair or subacromial decompression were included in our review, and patients with a nonprimary form of arthritis, such as rheumatoid, posttraumatic, or post-capsulorrhaphy arthritis, were excluded.
Operative Technique
For all surgeries, Dr. Bigliani performed a subscapularis tenotomy with regional anesthesia and a standard deltopectoral approach. A partial anterior capsulectomy was performed to increase the glenoid’s visibility. The inferior labrum was removed with a needle-tip bovie while the axillary nerve was being protected with a metal finger or narrow Darrach retractor. After reaming and trialing, the final glenoid component was cemented into place. Cement was placed only in the peg or keel holes and pressurized twice before final implantation. Of the 196 glenoid components, 168 (86%) were pegged and 28 (14%) keeled; in addition,190 of these components were all-polyethylene, whereas 6 had trabecular-metal backing. All glenoid components incorporated the hybrid design of dual radii of curvature. After the glenoid was cemented, the final humeral component was placed in 30° of retroversion. Whenever posterior wear was found, retroversion was reduced by 5° to 10°. The humeral prosthesis was cemented in cases (104/196, 53%) of poor bone quality or a large canal.
After surgery, the patient’s sling was fitted with an abduction pillow and a swathe, to be worn the first 24 hours, and the arm was passively ranged. Patients typically were discharged on postoperative day 2. Then, for 2 weeks, they followed an assisted passive range of motion (ROM) protocol, with limited external rotation, for promotion of subscapularis healing.
Clinical Outcomes
Dr. Bigliani assessed preoperative ROM in all planes. During initial evaluation, patients completed a questionnaire that consisted of the 36-Item Short Form Health Survey19,20 (SF-36) and the American Shoulder and Elbow Surgeons21 (ASES) and Simple Shoulder Test22 (SST) surveys. Postoperative clinical data were collected from office follow-up visits, survey questionnaires, or both. Postoperative office data included ROM, subscapularis integrity testing (belly-press or lift-off), and any complications. Patients with <1 year of office follow-up were excluded. In addition, the same survey questionnaire that was used before surgery was mailed to all patients after surgery; then, for anyone who did not respond by mail, we attempted contact by telephone. Neer criteria were based on patients’ subjective assessment of each arm on a 3-point Likert scale (1 = very satisfied, 2 = satisfied, 3 = dissatisfied). Patients were also asked about any specific complications or revision operations since their index procedure.
Physical examination and office follow-up data were obtained for 129 patients (148/196 shoulders, 76% follow-up) at a mean of 3.7 years (range 1.0-10.2 years) after surgery. Surveys were completed by 117 patients (139/196 shoulders, 71% follow-up) at a mean of 5.1 years (range, 1.6-11.2 years) after surgery. Only 15 patients had neither 1 year of office follow-up nor a completed questionnaire. The remaining 154 patients (178/196 shoulders, 91% follow-up) had clinical follow-up with office, mail, or telephone questionnaire at a mean of 4.8 years (range, 1.0-11.2 years) after surgery. This cohort of patients was used to determine rates of surgical revisions, subscapularis tears, dislocations, and other complications.
Radiographic Outcomes
Patients were included in the radiographic analysis if they had a shoulder radiograph at least 1 year after surgery. One hundred nineteen patients (136/196 shoulders, 69% follow-up) had radiographic follow-up at a mean of 3.7 years (range, 1.0-9.4 years) after surgery.
Statistical Analysis
Statistical analysis was performed with Stata Version 10.0. Paired t tests were used to compare preoperative and postoperative numerical data, including ROM and survey scores. We calculated 95% confidence intervals (CIs) and set statistical significance at P < .05. For qualitative measures, the Fisher exact test was used. Survivorship analysis was performed according to the Kaplan-Meier method, with right-censored data for no event or missing data.25
Results
Clinical Analysis of Demographics
In demographics, the clinical and radiographic patient subgroups were similar to each other and to the overall study population (Table 2). Of 196 patients overall, 16 (8%) had a concomitant rotator cuff repair, and 27 (14%) underwent staged bilateral shoulder arthroplasties.
Clinical Analysis of ROM and Survey Scores
Operative shoulder ROM in forward elevation, external rotation at side, external rotation in abduction, and internal rotation all showed statistically significant (P < .001) improvement from before surgery to after surgery. Over 3.7 years, mean (SD) forward elevation improved from 107.3° (34.8°) to 159.0° (29.4°), external rotation at side improved from 20.4° (16.7°) to 49.4° (11.3°), and external rotation in abduction improved from 53.7° (24.3°) to 84.7° (9.1°). Internal rotation improved from a mean (SD) vertebral level of S1 (6.0 levels) to T9 (3.7 levels).
All validated survey scores also showed statistically significant (P < .001) improvement from before surgery to after surgery. Over 5.1 years, mean (SD) SF-36 scores improved from 64.9 (13.4) to 73.6 (17.1), ASES scores improved from 41.1 (22.5) to 82.7 (17.7), SST scores improved from 3.9 (2.8) to 9.7 (2.2), and visual analog scale pain scores improved from 5.6 (3.2) to 1.4 (2.1). Of 139 patients with follow-up, 130 (93.5%) were either satisfied or very satisfied with their TSA, and only 119 (86%) were either satisfied or very satisfied with the nonoperative shoulder.
Clinical Analysis of Postoperative Complications
Of the 178 shoulders evaluated for complications, 3 (1.7%) underwent revision surgery. Mean time to revision was 2.3 years (range, 1.5-3.9 years). Two revisions involved the glenoid component, and the third involved the humerus. In one of the glenoid cases, a 77-year-old woman fell and sustained a fracture at the base of the trabecular metal glenoid pegs; her component was revised to an all-polyethylene component, and she had no further complications. In the other glenoid case, a 73-year-old man’s all-polyethylene component loosened after 2 years and was revised to a trabecular metal implant, which loosened as well and was later converted to a hemiarthroplasty. In the humeral case, a 33-year-old man had his 4-year-old index TSA revised to a cemented stem and had no further complications.
Table 4 compares the clinical and radiographic outcomes of patients who required subscapularis repair, capsular shift, or implant revision with the outcomes of all other study patients, and Figure 3 shows Kaplan-Meier survivorship.
Postoperative Radiographic Analysis
Glenoid Component. At a mean of 3.7 years (minimum, 1 year) after surgery, 86 (63%) of 136 radiographically evaluated shoulders showed no glenoid lucencies; the other 50 (37%) showed ≥1 lucency. Of the 136 shoulders, 33 (24%) had a Lazarus score of 1, 15 (11%) had a score of 2, and only 2 (2%) had a score of 3. None of the shoulders had a score of 4 or 5.
Humeral Component. Of the 136 shoulders, 91 (67%) showed no lucencies in any of the 8 humeral stem zones; the other 45 (33%) showed 1 to 3 lucencies. Thirty (22%) of the 136 shoulders had 1 stem lucency zone, 8 (6%) had 2, and 3 (2%) had 3. None of the shoulders had >3 periprosthetic zones with lucent lines.
Discussion
In this article, we describe a hybrid glenoid TSA component with dual radii of curvature. Its central portion is congruent with the humeral head, and its peripheral portion is noncongruent and larger. The most significant finding of our study is the low rate (1.1%) of glenoid component revision 4.8 years after surgery. This rate is the lowest that has been reported in a study of ≥100 patients. Overall implant survival appeared as an almost flat Kaplan-Meir curve. We attribute this low revision rate to improved biomechanics with the hybrid glenoid design.
Symptomatic glenoid component loosening is the most common TSA complication.1,26-28 In a review of 73 Neer TSAs, Cofield7 found glenoid radiolucencies in 71% of patients 3.8 years after surgery. Radiographic evidence of loosening, defined as component migration, or tilt, or a circumferential lucency 1.5 mm thick, was present in another 11% of patients, and 4.1% developed symptomatic loosening that required glenoid revision. In a study with 12.2-year follow-up, Torchia and colleagues3 found rates of 84% for glenoid radiolucencies, 44% for radiographic loosening, and 5.6% for symptomatic loosening that required revision. In a systematic review of studies with follow-up of ≥10 years, Bohsali and colleagues27 found similar lucency and radiographic loosening rates and a 7% glenoid revision rate. These data suggest glenoid radiolucencies may progress to component loosening.
Degree of joint congruence is a key factor in glenoid loosening. Neer’s congruent design increases the contact area with concentric loading and reduces glenohumeral translation, which leads to reduced polyethylene wear and improved joint stability. In extreme arm positions, however, humeral head subluxation results in edge loading and a glenoid rocking-horse effect.9-13,17,29-31 Conversely, nonconforming implants allow increased glenohumeral translation without edge loading,14 though they also reduce the relative glenohumeral contact area and thus transmit more contact stress to the glenoid.16,17 A hybrid glenoid component with central conforming and peripheral nonconforming zones may reduce the rocking-horse effect while maximizing ROM and joint stability. Wang and colleagues32 studied the biomechanical properties of this glenoid design and found that the addition of a central conforming region did not increase edge loading.
Additional results from our study support the efficacy of a hybrid glenoid component. Patients’ clinical outcomes improved significantly. At 5.1 years after surgery, 93.5% of patients were satisfied or very satisfied with their procedure and reported less satisfaction (86%) with the nonoperative shoulder. Also significant was the reduced number of radiolucencies. At 3.7 years after surgery, the overall percentage of shoulders with ≥1 glenoid radiolucency was 37%, considerably lower than the 82% reported by Cofield7 and the rates in more recent studies.3,16,33-36 Of the 178 shoulders in our study, 10 (5.6%) had subscapularis tears, and 6 (3.4%) of 178 had these tears surgically repaired. This 3.4% compares favorably with the 5.9% (of 119 patients) found by Miller and colleagues37 28 months after surgery. Of our 178 shoulders, 27 (15.2%) had clinically significant postoperative complications; 18 (10.1%) of the 178 had these complications surgically treated, and 9 (5.1%) had them managed nonoperatively. Bohsali and colleagues27 systematically reviewed 33 TSA studies and found a slightly higher complication rate (16.3%) 5.3 years after surgery. Furthermore, in our study, the 11 patients who underwent revision, capsular shift, or subscapularis repair had final outcomes comparable to those of the rest of our study population.
Our study had several potential weaknesses. First, its minimum clinical and radiographic follow-up was 1 year, whereas most long-term TSA series set a minimum of 2 years. We used 1 year because this was the first clinical study of the hybrid glenoid component design, and we wanted to maximize its sample size by reporting on intermediate-length outcomes. Even so, 93% (166/178) of our clinical patients and 83% (113/136) of our radiographic patients have had ≥2 years of follow-up, and we continue to follow all study patients for long-term outcomes. Another weakness of the study was its lack of a uniform group of patients with all the office, survey, complications, and radiographic data. Our retrospective study design made it difficult to obtain such a group without significantly reducing the sample size, so we divided patients into 4 data groups. A third potential weakness was the study’s variable method for collecting complications data. Rates of complications in the 178 shoulders were calculated from either office evaluation or patient self-report by mail or telephone. This data collection method is subject to recall bias, but mail and telephone contact was needed so the study would capture the large number of patients who had traveled to our institution for their surgery or had since moved away. Fourth, belly-press and lift-off tests were used in part to assess subscapularis function, but recent literature suggests post-TSA subscapularis assessment can be unreliable.38 These tests may be positive in up to two-thirds of patients after 2 years.39 Fifth, the generalizability of our findings to diagnoses such as rheumatoid and posttraumatic arthritis is limited. We had to restrict the study to patients with primary glenohumeral arthritis in order to minimize confounders.
This study’s main strength is its description of the clinical and radiographic outcomes of using a single prosthetic system in operations performed by a single surgeon in a large number of patients. This was the first and largest study evaluating the clinical and radiographic outcomes of this hybrid glenoid implant. Excluding patients with nonprimary arthritis allowed us to minimize potential confounding factors that affect patient outcomes. In conclusion, our study results showed the favorable clinical and radiographic outcomes of TSAs that have a hybrid glenoid component with dual radii of curvature. At a mean of 3.7 years after surgery, 63% of patients had no glenoid lucencies, and, at a mean of 4.8 years, only 1.7% of patients required revision. We continue to follow these patients to obtain long-term results of this innovative prosthesis.
Take-Home Points
- The authors have developed a total shoulder glenoid prosthesis that conforms with the humeral head in its center and is nonconforming on its peripheral edge.
- All clinical survey and range of motion parameters demonstrated statistically significant improvements at final follow-up.
- Only 3 shoulders (1.7%) required revision surgery.
- Eighty-six (63%) of 136 shoulders demonstrated no radiographic evidence of glenoid loosening.
- This is the first and largest study that evaluates the clinical and radiographic outcomes of this hybrid shoulder prosthesis.
Fixation of the glenoid component is the limiting factor in modern total shoulder arthroplasty (TSA). Glenoid loosening, the most common long-term complication, necessitates revision in up to 12% of patients.1-4 By contrast, humeral component loosening is relatively uncommon, affecting as few as 0.34% of patients.5 Multiple long-term studies have found consistently high rates (45%-93%) of radiolucencies around the glenoid component.3,6,7 Although their clinical significance has been debated, radiolucencies around the glenoid component raise concern about progressive loss of fixation.
Since TSA was introduced in the 1970s, complications with the glenoid component have been addressed with 2 different designs: conforming (congruent) and nonconforming. In a congruent articulation, the radii of curvature of the glenoid and humeral head components are identical, whereas they differ in a nonconforming model. Joint conformity is inversely related to glenohumeral translation.8 Neer’s original TSA was made congruent in order to limit translation and maximize the contact area. However, this design results in edge loading and a so-called rocking-horse phenomenon, which may lead to glenoid loosening.9-13 Surgeons therefore have increasingly turned to nonconforming implants. In the nonconforming design, the radius of curvature of the humeral head is smaller than that of the glenoid. Although this design may reduce edge loading,14 it allows more translation and reduces the relative contact area of the glenohumeral joint. As a result, more contact stress is transmitted to the glenoid component, leading to polyethylene deformation and wear.15,16
Dual radii of curvature are designed to augment joint stability without increasing component wear. Biomechanical data have indicated that edge loading is not increased by having a central conforming region added to a nonconforming model.17 The clinical value of this prosthesis, however, has not been determined. Therefore, we conducted a study to describe the intermediate-term clinical and radiographic outcomes of TSAs that use a novel hybrid glenoid component.
Materials and Methods
This study was approved (protocol AAAD3473) by the Institutional Review Board of Columbia University and was conducted in compliance with Health Insurance Portability and Accountability Act (HIPAA) regulations.
Patient Selection
At Columbia University Medical Center, Dr. Bigliani performed 196 TSAs with a hybrid glenoid component (Bigliani-Flatow; Zimmer Biomet) in 169 patients between September 1998 and November 2007. All patients had received a diagnosis of primary glenohumeral arthritis as defined by Neer.18 Patients with previous surgery such as rotator cuff repair or subacromial decompression were included in our review, and patients with a nonprimary form of arthritis, such as rheumatoid, posttraumatic, or post-capsulorrhaphy arthritis, were excluded.
Operative Technique
For all surgeries, Dr. Bigliani performed a subscapularis tenotomy with regional anesthesia and a standard deltopectoral approach. A partial anterior capsulectomy was performed to increase the glenoid’s visibility. The inferior labrum was removed with a needle-tip bovie while the axillary nerve was being protected with a metal finger or narrow Darrach retractor. After reaming and trialing, the final glenoid component was cemented into place. Cement was placed only in the peg or keel holes and pressurized twice before final implantation. Of the 196 glenoid components, 168 (86%) were pegged and 28 (14%) keeled; in addition,190 of these components were all-polyethylene, whereas 6 had trabecular-metal backing. All glenoid components incorporated the hybrid design of dual radii of curvature. After the glenoid was cemented, the final humeral component was placed in 30° of retroversion. Whenever posterior wear was found, retroversion was reduced by 5° to 10°. The humeral prosthesis was cemented in cases (104/196, 53%) of poor bone quality or a large canal.
After surgery, the patient’s sling was fitted with an abduction pillow and a swathe, to be worn the first 24 hours, and the arm was passively ranged. Patients typically were discharged on postoperative day 2. Then, for 2 weeks, they followed an assisted passive range of motion (ROM) protocol, with limited external rotation, for promotion of subscapularis healing.
Clinical Outcomes
Dr. Bigliani assessed preoperative ROM in all planes. During initial evaluation, patients completed a questionnaire that consisted of the 36-Item Short Form Health Survey19,20 (SF-36) and the American Shoulder and Elbow Surgeons21 (ASES) and Simple Shoulder Test22 (SST) surveys. Postoperative clinical data were collected from office follow-up visits, survey questionnaires, or both. Postoperative office data included ROM, subscapularis integrity testing (belly-press or lift-off), and any complications. Patients with <1 year of office follow-up were excluded. In addition, the same survey questionnaire that was used before surgery was mailed to all patients after surgery; then, for anyone who did not respond by mail, we attempted contact by telephone. Neer criteria were based on patients’ subjective assessment of each arm on a 3-point Likert scale (1 = very satisfied, 2 = satisfied, 3 = dissatisfied). Patients were also asked about any specific complications or revision operations since their index procedure.
Physical examination and office follow-up data were obtained for 129 patients (148/196 shoulders, 76% follow-up) at a mean of 3.7 years (range 1.0-10.2 years) after surgery. Surveys were completed by 117 patients (139/196 shoulders, 71% follow-up) at a mean of 5.1 years (range, 1.6-11.2 years) after surgery. Only 15 patients had neither 1 year of office follow-up nor a completed questionnaire. The remaining 154 patients (178/196 shoulders, 91% follow-up) had clinical follow-up with office, mail, or telephone questionnaire at a mean of 4.8 years (range, 1.0-11.2 years) after surgery. This cohort of patients was used to determine rates of surgical revisions, subscapularis tears, dislocations, and other complications.
Radiographic Outcomes
Patients were included in the radiographic analysis if they had a shoulder radiograph at least 1 year after surgery. One hundred nineteen patients (136/196 shoulders, 69% follow-up) had radiographic follow-up at a mean of 3.7 years (range, 1.0-9.4 years) after surgery.
Statistical Analysis
Statistical analysis was performed with Stata Version 10.0. Paired t tests were used to compare preoperative and postoperative numerical data, including ROM and survey scores. We calculated 95% confidence intervals (CIs) and set statistical significance at P < .05. For qualitative measures, the Fisher exact test was used. Survivorship analysis was performed according to the Kaplan-Meier method, with right-censored data for no event or missing data.25
Results
Clinical Analysis of Demographics
In demographics, the clinical and radiographic patient subgroups were similar to each other and to the overall study population (Table 2). Of 196 patients overall, 16 (8%) had a concomitant rotator cuff repair, and 27 (14%) underwent staged bilateral shoulder arthroplasties.
Clinical Analysis of ROM and Survey Scores
Operative shoulder ROM in forward elevation, external rotation at side, external rotation in abduction, and internal rotation all showed statistically significant (P < .001) improvement from before surgery to after surgery. Over 3.7 years, mean (SD) forward elevation improved from 107.3° (34.8°) to 159.0° (29.4°), external rotation at side improved from 20.4° (16.7°) to 49.4° (11.3°), and external rotation in abduction improved from 53.7° (24.3°) to 84.7° (9.1°). Internal rotation improved from a mean (SD) vertebral level of S1 (6.0 levels) to T9 (3.7 levels).
All validated survey scores also showed statistically significant (P < .001) improvement from before surgery to after surgery. Over 5.1 years, mean (SD) SF-36 scores improved from 64.9 (13.4) to 73.6 (17.1), ASES scores improved from 41.1 (22.5) to 82.7 (17.7), SST scores improved from 3.9 (2.8) to 9.7 (2.2), and visual analog scale pain scores improved from 5.6 (3.2) to 1.4 (2.1). Of 139 patients with follow-up, 130 (93.5%) were either satisfied or very satisfied with their TSA, and only 119 (86%) were either satisfied or very satisfied with the nonoperative shoulder.
Clinical Analysis of Postoperative Complications
Of the 178 shoulders evaluated for complications, 3 (1.7%) underwent revision surgery. Mean time to revision was 2.3 years (range, 1.5-3.9 years). Two revisions involved the glenoid component, and the third involved the humerus. In one of the glenoid cases, a 77-year-old woman fell and sustained a fracture at the base of the trabecular metal glenoid pegs; her component was revised to an all-polyethylene component, and she had no further complications. In the other glenoid case, a 73-year-old man’s all-polyethylene component loosened after 2 years and was revised to a trabecular metal implant, which loosened as well and was later converted to a hemiarthroplasty. In the humeral case, a 33-year-old man had his 4-year-old index TSA revised to a cemented stem and had no further complications.
Table 4 compares the clinical and radiographic outcomes of patients who required subscapularis repair, capsular shift, or implant revision with the outcomes of all other study patients, and Figure 3 shows Kaplan-Meier survivorship.
Postoperative Radiographic Analysis
Glenoid Component. At a mean of 3.7 years (minimum, 1 year) after surgery, 86 (63%) of 136 radiographically evaluated shoulders showed no glenoid lucencies; the other 50 (37%) showed ≥1 lucency. Of the 136 shoulders, 33 (24%) had a Lazarus score of 1, 15 (11%) had a score of 2, and only 2 (2%) had a score of 3. None of the shoulders had a score of 4 or 5.
Humeral Component. Of the 136 shoulders, 91 (67%) showed no lucencies in any of the 8 humeral stem zones; the other 45 (33%) showed 1 to 3 lucencies. Thirty (22%) of the 136 shoulders had 1 stem lucency zone, 8 (6%) had 2, and 3 (2%) had 3. None of the shoulders had >3 periprosthetic zones with lucent lines.
Discussion
In this article, we describe a hybrid glenoid TSA component with dual radii of curvature. Its central portion is congruent with the humeral head, and its peripheral portion is noncongruent and larger. The most significant finding of our study is the low rate (1.1%) of glenoid component revision 4.8 years after surgery. This rate is the lowest that has been reported in a study of ≥100 patients. Overall implant survival appeared as an almost flat Kaplan-Meir curve. We attribute this low revision rate to improved biomechanics with the hybrid glenoid design.
Symptomatic glenoid component loosening is the most common TSA complication.1,26-28 In a review of 73 Neer TSAs, Cofield7 found glenoid radiolucencies in 71% of patients 3.8 years after surgery. Radiographic evidence of loosening, defined as component migration, or tilt, or a circumferential lucency 1.5 mm thick, was present in another 11% of patients, and 4.1% developed symptomatic loosening that required glenoid revision. In a study with 12.2-year follow-up, Torchia and colleagues3 found rates of 84% for glenoid radiolucencies, 44% for radiographic loosening, and 5.6% for symptomatic loosening that required revision. In a systematic review of studies with follow-up of ≥10 years, Bohsali and colleagues27 found similar lucency and radiographic loosening rates and a 7% glenoid revision rate. These data suggest glenoid radiolucencies may progress to component loosening.
Degree of joint congruence is a key factor in glenoid loosening. Neer’s congruent design increases the contact area with concentric loading and reduces glenohumeral translation, which leads to reduced polyethylene wear and improved joint stability. In extreme arm positions, however, humeral head subluxation results in edge loading and a glenoid rocking-horse effect.9-13,17,29-31 Conversely, nonconforming implants allow increased glenohumeral translation without edge loading,14 though they also reduce the relative glenohumeral contact area and thus transmit more contact stress to the glenoid.16,17 A hybrid glenoid component with central conforming and peripheral nonconforming zones may reduce the rocking-horse effect while maximizing ROM and joint stability. Wang and colleagues32 studied the biomechanical properties of this glenoid design and found that the addition of a central conforming region did not increase edge loading.
Additional results from our study support the efficacy of a hybrid glenoid component. Patients’ clinical outcomes improved significantly. At 5.1 years after surgery, 93.5% of patients were satisfied or very satisfied with their procedure and reported less satisfaction (86%) with the nonoperative shoulder. Also significant was the reduced number of radiolucencies. At 3.7 years after surgery, the overall percentage of shoulders with ≥1 glenoid radiolucency was 37%, considerably lower than the 82% reported by Cofield7 and the rates in more recent studies.3,16,33-36 Of the 178 shoulders in our study, 10 (5.6%) had subscapularis tears, and 6 (3.4%) of 178 had these tears surgically repaired. This 3.4% compares favorably with the 5.9% (of 119 patients) found by Miller and colleagues37 28 months after surgery. Of our 178 shoulders, 27 (15.2%) had clinically significant postoperative complications; 18 (10.1%) of the 178 had these complications surgically treated, and 9 (5.1%) had them managed nonoperatively. Bohsali and colleagues27 systematically reviewed 33 TSA studies and found a slightly higher complication rate (16.3%) 5.3 years after surgery. Furthermore, in our study, the 11 patients who underwent revision, capsular shift, or subscapularis repair had final outcomes comparable to those of the rest of our study population.
Our study had several potential weaknesses. First, its minimum clinical and radiographic follow-up was 1 year, whereas most long-term TSA series set a minimum of 2 years. We used 1 year because this was the first clinical study of the hybrid glenoid component design, and we wanted to maximize its sample size by reporting on intermediate-length outcomes. Even so, 93% (166/178) of our clinical patients and 83% (113/136) of our radiographic patients have had ≥2 years of follow-up, and we continue to follow all study patients for long-term outcomes. Another weakness of the study was its lack of a uniform group of patients with all the office, survey, complications, and radiographic data. Our retrospective study design made it difficult to obtain such a group without significantly reducing the sample size, so we divided patients into 4 data groups. A third potential weakness was the study’s variable method for collecting complications data. Rates of complications in the 178 shoulders were calculated from either office evaluation or patient self-report by mail or telephone. This data collection method is subject to recall bias, but mail and telephone contact was needed so the study would capture the large number of patients who had traveled to our institution for their surgery or had since moved away. Fourth, belly-press and lift-off tests were used in part to assess subscapularis function, but recent literature suggests post-TSA subscapularis assessment can be unreliable.38 These tests may be positive in up to two-thirds of patients after 2 years.39 Fifth, the generalizability of our findings to diagnoses such as rheumatoid and posttraumatic arthritis is limited. We had to restrict the study to patients with primary glenohumeral arthritis in order to minimize confounders.
This study’s main strength is its description of the clinical and radiographic outcomes of using a single prosthetic system in operations performed by a single surgeon in a large number of patients. This was the first and largest study evaluating the clinical and radiographic outcomes of this hybrid glenoid implant. Excluding patients with nonprimary arthritis allowed us to minimize potential confounding factors that affect patient outcomes. In conclusion, our study results showed the favorable clinical and radiographic outcomes of TSAs that have a hybrid glenoid component with dual radii of curvature. At a mean of 3.7 years after surgery, 63% of patients had no glenoid lucencies, and, at a mean of 4.8 years, only 1.7% of patients required revision. We continue to follow these patients to obtain long-term results of this innovative prosthesis.
1. Rodosky MW, Bigliani LU. Indications for glenoid resurfacing in shoulder arthroplasty. J Shoulder Elbow Surg. 1996;5(3):231-248.
2. Boyd AD Jr, Thomas WH, Scott RD, Sledge CB, Thornhill TS. Total shoulder arthroplasty versus hemiarthroplasty. Indications for glenoid resurfacing. J Arthroplasty. 1990;5(4):329-336.
3. Torchia ME, Cofield RH, Settergren CR. Total shoulder arthroplasty with the Neer prosthesis: long-term results. J Shoulder Elbow Surg. 1997;6(6):495-505.
4. Iannotti JP, Norris TR. Influence of preoperative factors on outcome of shoulder arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2003;85(2):251-258.
5. Cofield RH. Degenerative and arthritic problems of the glenohumeral joint. In: Rockwood CA, Matsen FA, eds. The Shoulder. Philadelphia, PA: Saunders; 1990:740-745.
6. Neer CS 2nd, Watson KC, Stanton FJ. Recent experience in total shoulder replacement. J Bone Joint Surg Am. 1982;64(3):319-337.
7. Cofield RH. Total shoulder arthroplasty with the Neer prosthesis. J Bone Joint Surg Am. 1984;66(6):899-906.
8. Karduna AR, Williams GR, Williams JL, Iannotti JP. Kinematics of the glenohumeral joint: influences of muscle forces, ligamentous constraints, and articular geometry. J Orthop Res. 1996;14(6):986-993.
9. Karduna AR, Williams GR, Iannotti JP, Williams JL. Total shoulder arthroplasty biomechanics: a study of the forces and strains at the glenoid component. J Biomech Eng. 1998;120(1):92-99.
10. Karduna AR, Williams GR, Williams JL, Iannotti JP. Glenohumeral joint translations before and after total shoulder arthroplasty. A study in cadavera. J Bone Joint Surg Am. 1997;79(8):1166-1174.
11. Matsen FA 3rd, Clinton J, Lynch J, Bertelsen A, Richardson ML. Glenoid component failure in total shoulder arthroplasty. J Bone Joint Surg Am. 2008;90(4):885-896.
12. Franklin JL, Barrett WP, Jackins SE, Matsen FA 3rd. Glenoid loosening in total shoulder arthroplasty. Association with rotator cuff deficiency. J Arthroplasty. 1988;3(1):39-46.
13. Barrett WP, Franklin JL, Jackins SE, Wyss CR, Matsen FA 3rd. Total shoulder arthroplasty. J Bone Joint Surg Am. 1987;69(6):865-872.
14. Harryman DT, Sidles JA, Harris SL, Lippitt SB, Matsen FA 3rd. The effect of articular conformity and the size of the humeral head component on laxity and motion after glenohumeral arthroplasty. A study in cadavera. J Bone Joint Surg Am. 1995;77(4):555-563.
15. Flatow EL. Prosthetic design considerations in total shoulder arthroplasty. Semin Arthroplasty. 1995;6(4):233-244.
16. Klimkiewicz JJ, Iannotti JP, Rubash HE, Shanbhag AS. Aseptic loosening of the humeral component in total shoulder arthroplasty. J Shoulder Elbow Surg. 1998;7(4):422-426.
17. Wang VM, Krishnan R, Ugwonali OF, Flatow EL, Bigliani LU, Ateshian GA. Biomechanical evaluation of a novel glenoid design in total shoulder arthroplasty. J Shoulder Elbow Surg. 2005;14(1 suppl S):129S-140S.
18. Neer CS 2nd. Replacement arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 1974;56(1):1-13.
19. Boorman RS, Kopjar B, Fehringer E, Churchill RS, Smith K, Matsen FA 3rd. The effect of total shoulder arthroplasty on self-assessed health status is comparable to that of total hip arthroplasty and coronary artery bypass grafting. J Shoulder Elbow Surg. 2003;12(2):158-163.
20. Patel AA, Donegan D, Albert T. The 36-Item Short Form. J Am Acad Orthop Surg. 2007;15(2):126-134.
21. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3(6):347-352.
22. Wright RW, Baumgarten KM. Shoulder outcomes measures. J Am Acad Orthop Surg. 2010;18(7):436-444.
23. Lazarus MD, Jensen KL, Southworth C, Matsen FA 3rd. The radiographic evaluation of keeled and pegged glenoid component insertion. J Bone Joint Surg Am. 2002;84(7):1174-1182.
24. Sperling JW, Cofield RH, O’Driscoll SW, Torchia ME, Rowland CM. Radiographic assessment of ingrowth total shoulder arthroplasty. J Shoulder Elbow Surg. 2000;9(6):507-513.
25. Dinse GE, Lagakos SW. Nonparametric estimation of lifetime and disease onset distributions from incomplete observations. Biometrics. 1982;38(4):921-932.
26. Baumgarten KM, Lashgari CJ, Yamaguchi K. Glenoid resurfacing in shoulder arthroplasty: indications and contraindications. Instr Course Lect. 2004;53:3-11.
27. Bohsali KI, Wirth MA, Rockwood CA Jr. Complications of total shoulder arthroplasty. J Bone Joint Surg Am. 2006;88(10):2279-2292.
28. Wirth MA, Rockwood CA Jr. Complications of total shoulder-replacement arthroplasty. J Bone Joint Surg Am. 1996;78(4):603-616.
29. Poppen NK, Walker PS. Normal and abnormal motion of the shoulder. J Bone Joint Surg Am. 1976;58(2):195-201.
30. Cotton RE, Rideout DF. Tears of the humeral rotator cuff; a radiological and pathological necropsy survey. J Bone Joint Surg Br. 1964;46:314-328.
31. Bigliani LU, Kelkar R, Flatow EL, Pollock RG, Mow VC. Glenohumeral stability. Biomechanical properties of passive and active stabilizers. Clin Orthop Relat Res. 1996;(330):13-30.
32. Wang VM, Sugalski MT, Levine WN, Pawluk RJ, Mow VC, Bigliani LU. Comparison of glenohumeral mechanics following a capsular shift and anterior tightening. J Bone Joint Surg Am. 2005;87(6):1312-1322.
33. Young A, Walch G, Boileau P, et al. A multicentre study of the long-term results of using a flat-back polyethylene glenoid component in shoulder replacement for primary osteoarthritis. J Bone Joint Surg Br. 2011;93(2):210-216.
34. Khan A, Bunker TD, Kitson JB. Clinical and radiological follow-up of the Aequalis third-generation cemented total shoulder replacement: a minimum ten-year study. J Bone Joint Surg Br. 2009;91(12):1594-1600.
35. Walch G, Edwards TB, Boulahia A, Boileau P, Mole D, Adeleine P. The influence of glenohumeral prosthetic mismatch on glenoid radiolucent lines: results of a multicenter study. J Bone Joint Surg Am. 2002;84(12):2186-2191.
36. Bartelt R, Sperling JW, Schleck CD, Cofield RH. Shoulder arthroplasty in patients aged fifty-five years or younger with osteoarthritis. J Shoulder Elbow Surg. 2011;20(1):123-130.
37. Miller BS, Joseph TA, Noonan TJ, Horan MP, Hawkins RJ. Rupture of the subscapularis tendon after shoulder arthroplasty: diagnosis, treatment, and outcome. J Shoulder Elbow Surg. 2005;14(5):492-496.
38. Armstrong A, Lashgari C, Teefey S, Menendez J, Yamaguchi K, Galatz LM. Ultrasound evaluation and clinical correlation of subscapularis repair after total shoulder arthroplasty. J Shoulder Elbow Surg. 2006;15(5):541-548.
39. Miller SL, Hazrati Y, Klepps S, Chiang A, Flatow EL. Loss of subscapularis function after total shoulder replacement: a seldom recognized problem. J Shoulder Elbow Surg. 2003;12(1):29-34.
1. Rodosky MW, Bigliani LU. Indications for glenoid resurfacing in shoulder arthroplasty. J Shoulder Elbow Surg. 1996;5(3):231-248.
2. Boyd AD Jr, Thomas WH, Scott RD, Sledge CB, Thornhill TS. Total shoulder arthroplasty versus hemiarthroplasty. Indications for glenoid resurfacing. J Arthroplasty. 1990;5(4):329-336.
3. Torchia ME, Cofield RH, Settergren CR. Total shoulder arthroplasty with the Neer prosthesis: long-term results. J Shoulder Elbow Surg. 1997;6(6):495-505.
4. Iannotti JP, Norris TR. Influence of preoperative factors on outcome of shoulder arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 2003;85(2):251-258.
5. Cofield RH. Degenerative and arthritic problems of the glenohumeral joint. In: Rockwood CA, Matsen FA, eds. The Shoulder. Philadelphia, PA: Saunders; 1990:740-745.
6. Neer CS 2nd, Watson KC, Stanton FJ. Recent experience in total shoulder replacement. J Bone Joint Surg Am. 1982;64(3):319-337.
7. Cofield RH. Total shoulder arthroplasty with the Neer prosthesis. J Bone Joint Surg Am. 1984;66(6):899-906.
8. Karduna AR, Williams GR, Williams JL, Iannotti JP. Kinematics of the glenohumeral joint: influences of muscle forces, ligamentous constraints, and articular geometry. J Orthop Res. 1996;14(6):986-993.
9. Karduna AR, Williams GR, Iannotti JP, Williams JL. Total shoulder arthroplasty biomechanics: a study of the forces and strains at the glenoid component. J Biomech Eng. 1998;120(1):92-99.
10. Karduna AR, Williams GR, Williams JL, Iannotti JP. Glenohumeral joint translations before and after total shoulder arthroplasty. A study in cadavera. J Bone Joint Surg Am. 1997;79(8):1166-1174.
11. Matsen FA 3rd, Clinton J, Lynch J, Bertelsen A, Richardson ML. Glenoid component failure in total shoulder arthroplasty. J Bone Joint Surg Am. 2008;90(4):885-896.
12. Franklin JL, Barrett WP, Jackins SE, Matsen FA 3rd. Glenoid loosening in total shoulder arthroplasty. Association with rotator cuff deficiency. J Arthroplasty. 1988;3(1):39-46.
13. Barrett WP, Franklin JL, Jackins SE, Wyss CR, Matsen FA 3rd. Total shoulder arthroplasty. J Bone Joint Surg Am. 1987;69(6):865-872.
14. Harryman DT, Sidles JA, Harris SL, Lippitt SB, Matsen FA 3rd. The effect of articular conformity and the size of the humeral head component on laxity and motion after glenohumeral arthroplasty. A study in cadavera. J Bone Joint Surg Am. 1995;77(4):555-563.
15. Flatow EL. Prosthetic design considerations in total shoulder arthroplasty. Semin Arthroplasty. 1995;6(4):233-244.
16. Klimkiewicz JJ, Iannotti JP, Rubash HE, Shanbhag AS. Aseptic loosening of the humeral component in total shoulder arthroplasty. J Shoulder Elbow Surg. 1998;7(4):422-426.
17. Wang VM, Krishnan R, Ugwonali OF, Flatow EL, Bigliani LU, Ateshian GA. Biomechanical evaluation of a novel glenoid design in total shoulder arthroplasty. J Shoulder Elbow Surg. 2005;14(1 suppl S):129S-140S.
18. Neer CS 2nd. Replacement arthroplasty for glenohumeral osteoarthritis. J Bone Joint Surg Am. 1974;56(1):1-13.
19. Boorman RS, Kopjar B, Fehringer E, Churchill RS, Smith K, Matsen FA 3rd. The effect of total shoulder arthroplasty on self-assessed health status is comparable to that of total hip arthroplasty and coronary artery bypass grafting. J Shoulder Elbow Surg. 2003;12(2):158-163.
20. Patel AA, Donegan D, Albert T. The 36-Item Short Form. J Am Acad Orthop Surg. 2007;15(2):126-134.
21. Richards RR, An KN, Bigliani LU, et al. A standardized method for the assessment of shoulder function. J Shoulder Elbow Surg. 1994;3(6):347-352.
22. Wright RW, Baumgarten KM. Shoulder outcomes measures. J Am Acad Orthop Surg. 2010;18(7):436-444.
23. Lazarus MD, Jensen KL, Southworth C, Matsen FA 3rd. The radiographic evaluation of keeled and pegged glenoid component insertion. J Bone Joint Surg Am. 2002;84(7):1174-1182.
24. Sperling JW, Cofield RH, O’Driscoll SW, Torchia ME, Rowland CM. Radiographic assessment of ingrowth total shoulder arthroplasty. J Shoulder Elbow Surg. 2000;9(6):507-513.
25. Dinse GE, Lagakos SW. Nonparametric estimation of lifetime and disease onset distributions from incomplete observations. Biometrics. 1982;38(4):921-932.
26. Baumgarten KM, Lashgari CJ, Yamaguchi K. Glenoid resurfacing in shoulder arthroplasty: indications and contraindications. Instr Course Lect. 2004;53:3-11.
27. Bohsali KI, Wirth MA, Rockwood CA Jr. Complications of total shoulder arthroplasty. J Bone Joint Surg Am. 2006;88(10):2279-2292.
28. Wirth MA, Rockwood CA Jr. Complications of total shoulder-replacement arthroplasty. J Bone Joint Surg Am. 1996;78(4):603-616.
29. Poppen NK, Walker PS. Normal and abnormal motion of the shoulder. J Bone Joint Surg Am. 1976;58(2):195-201.
30. Cotton RE, Rideout DF. Tears of the humeral rotator cuff; a radiological and pathological necropsy survey. J Bone Joint Surg Br. 1964;46:314-328.
31. Bigliani LU, Kelkar R, Flatow EL, Pollock RG, Mow VC. Glenohumeral stability. Biomechanical properties of passive and active stabilizers. Clin Orthop Relat Res. 1996;(330):13-30.
32. Wang VM, Sugalski MT, Levine WN, Pawluk RJ, Mow VC, Bigliani LU. Comparison of glenohumeral mechanics following a capsular shift and anterior tightening. J Bone Joint Surg Am. 2005;87(6):1312-1322.
33. Young A, Walch G, Boileau P, et al. A multicentre study of the long-term results of using a flat-back polyethylene glenoid component in shoulder replacement for primary osteoarthritis. J Bone Joint Surg Br. 2011;93(2):210-216.
34. Khan A, Bunker TD, Kitson JB. Clinical and radiological follow-up of the Aequalis third-generation cemented total shoulder replacement: a minimum ten-year study. J Bone Joint Surg Br. 2009;91(12):1594-1600.
35. Walch G, Edwards TB, Boulahia A, Boileau P, Mole D, Adeleine P. The influence of glenohumeral prosthetic mismatch on glenoid radiolucent lines: results of a multicenter study. J Bone Joint Surg Am. 2002;84(12):2186-2191.
36. Bartelt R, Sperling JW, Schleck CD, Cofield RH. Shoulder arthroplasty in patients aged fifty-five years or younger with osteoarthritis. J Shoulder Elbow Surg. 2011;20(1):123-130.
37. Miller BS, Joseph TA, Noonan TJ, Horan MP, Hawkins RJ. Rupture of the subscapularis tendon after shoulder arthroplasty: diagnosis, treatment, and outcome. J Shoulder Elbow Surg. 2005;14(5):492-496.
38. Armstrong A, Lashgari C, Teefey S, Menendez J, Yamaguchi K, Galatz LM. Ultrasound evaluation and clinical correlation of subscapularis repair after total shoulder arthroplasty. J Shoulder Elbow Surg. 2006;15(5):541-548.
39. Miller SL, Hazrati Y, Klepps S, Chiang A, Flatow EL. Loss of subscapularis function after total shoulder replacement: a seldom recognized problem. J Shoulder Elbow Surg. 2003;12(1):29-34.
Immunization information systems show progress over recent years
From 2013 to 2016, all U.S. immunization information systems showed progress in bidirectional information exchange with EHRs, said Neil Murthy, MD, and his associates at the National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta.
Across all 55 jurisdictions in 49 states and six cities in the United States using immunization information systems (IIS), 106% of U.S. births were registered in IIS in 2016, which is an increase from 102% in 2013; percentages may exceed 100%, because a child who is born in one state but who lives in a different state might be recorded in both IISs.
Bidirectional exchange of data with EHRs is an important part of an IIS. In 2016, 91% of jurisdictions had an IIS that used a platform-independent messaging system that received vaccination histories from providers and returned acknowledgment messages, compared with 87% in 2013, the investigators said.
“Clinical Decision Support (CDS) functionalities enable providers to evaluate the validity of vaccine doses administered to patients and forecast future vaccines that will be needed, based on recommendations developed by the Advisory Committee on Immunization Practices,” Dr. Murthy and his associates said. In 2016, 58% of the 55 jurisdictions sent a vaccine forecast to another system, compared with 31% in 2013.
In 2016, 95% of the 55 jurisdictions gave a “predefined, automatic report on immunization coverage by provider site,” compared with 89% of jurisdictions in 2013.
“IISs are integral components of routine clinical practice and public health surveillance for immunization,” Dr. Murthy and his associates said. “Availability of more complete IIS data also offers many benefits to health care providers and public health practitioners, including consolidating patients’ vaccination histories, identifying undervaccinated subgroups, and forecasting the needs of individual patients for recommended vaccines.”
Read more in Morbidity and Mortality Weekly Report (2017 Nov 3;66[43]:1178-81).
From 2013 to 2016, all U.S. immunization information systems showed progress in bidirectional information exchange with EHRs, said Neil Murthy, MD, and his associates at the National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta.
Across all 55 jurisdictions in 49 states and six cities in the United States using immunization information systems (IIS), 106% of U.S. births were registered in IIS in 2016, which is an increase from 102% in 2013; percentages may exceed 100%, because a child who is born in one state but who lives in a different state might be recorded in both IISs.
Bidirectional exchange of data with EHRs is an important part of an IIS. In 2016, 91% of jurisdictions had an IIS that used a platform-independent messaging system that received vaccination histories from providers and returned acknowledgment messages, compared with 87% in 2013, the investigators said.
“Clinical Decision Support (CDS) functionalities enable providers to evaluate the validity of vaccine doses administered to patients and forecast future vaccines that will be needed, based on recommendations developed by the Advisory Committee on Immunization Practices,” Dr. Murthy and his associates said. In 2016, 58% of the 55 jurisdictions sent a vaccine forecast to another system, compared with 31% in 2013.
In 2016, 95% of the 55 jurisdictions gave a “predefined, automatic report on immunization coverage by provider site,” compared with 89% of jurisdictions in 2013.
“IISs are integral components of routine clinical practice and public health surveillance for immunization,” Dr. Murthy and his associates said. “Availability of more complete IIS data also offers many benefits to health care providers and public health practitioners, including consolidating patients’ vaccination histories, identifying undervaccinated subgroups, and forecasting the needs of individual patients for recommended vaccines.”
Read more in Morbidity and Mortality Weekly Report (2017 Nov 3;66[43]:1178-81).
From 2013 to 2016, all U.S. immunization information systems showed progress in bidirectional information exchange with EHRs, said Neil Murthy, MD, and his associates at the National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta.
Across all 55 jurisdictions in 49 states and six cities in the United States using immunization information systems (IIS), 106% of U.S. births were registered in IIS in 2016, which is an increase from 102% in 2013; percentages may exceed 100%, because a child who is born in one state but who lives in a different state might be recorded in both IISs.
Bidirectional exchange of data with EHRs is an important part of an IIS. In 2016, 91% of jurisdictions had an IIS that used a platform-independent messaging system that received vaccination histories from providers and returned acknowledgment messages, compared with 87% in 2013, the investigators said.
“Clinical Decision Support (CDS) functionalities enable providers to evaluate the validity of vaccine doses administered to patients and forecast future vaccines that will be needed, based on recommendations developed by the Advisory Committee on Immunization Practices,” Dr. Murthy and his associates said. In 2016, 58% of the 55 jurisdictions sent a vaccine forecast to another system, compared with 31% in 2013.
In 2016, 95% of the 55 jurisdictions gave a “predefined, automatic report on immunization coverage by provider site,” compared with 89% of jurisdictions in 2013.
“IISs are integral components of routine clinical practice and public health surveillance for immunization,” Dr. Murthy and his associates said. “Availability of more complete IIS data also offers many benefits to health care providers and public health practitioners, including consolidating patients’ vaccination histories, identifying undervaccinated subgroups, and forecasting the needs of individual patients for recommended vaccines.”
Read more in Morbidity and Mortality Weekly Report (2017 Nov 3;66[43]:1178-81).
FROM MMWR