User login
Warfarin may decrease risk of cancer

New research suggests warfarin use may reduce the risk of cancer among people over the age of 50.
An observational study of more than 1 million people in Norway revealed that individuals who took warfarin had a lower incidence of cancers overall, as well as certain types of cancers, than people who did not take warfarin.
James B. Lorens, PhD, of the University of Bergen in Norway, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers analyzed data from the Norwegian National Registry, the Norwegian Prescription Database, and the Cancer Registry of Norway.
This included data for all individuals ages 52 to 82 who were living in Norway from January 1, 2006, through December 31, 2012, (n=1,256,725).
The researchers looked for cancer diagnoses in this group during the 7-year observation period. They compared the incidence of any new cancer (including certain cancer types) between warfarin users and non-users.
People were considered warfarin users if they had taken at least 6 months of a warfarin prescription and at least 2 years had elapsed from their first prescription to any cancer diagnosis.
Most study subjects were non-users (92.6%, n=1,163,783), but 7.4% (n=92,942) were warfarin users.
The researchers noted that warfarin users were older than non-users, with mean ages of 70.2 and 63.9, respectively. Warfarin users were also more likely to be male (61.7%; n=57,370) and non-users female (52.7%; n=613,803).
The incidence of cancer was 10.6% (n=132,687) in the entire study cohort, 9.4% (n=8754) among warfarin users, and 10.6% (n=123,933) among non-users. The most common cancer types were prostate, lung, colon, and breast.
The researchers found a significantly lower age- and sex-adjusted incidence rate ratio (IRR) for all cancers among warfarin users than nonusers. The IRR was 0.84 (95% CI, 0.82-0.86).
The IRR was also significantly lower among users than non-users for 3 of the 4 most common cancers. The IRR was 0.69 (95% CI, 0.65-0.72) for prostate cancer, 0.80 (95% CI, 0.75-0.86) for lung cancer, and 0.90 (95% CI, 0.82-1.00) for female breast cancer.
The researchers said there was no significant difference between users and non-users for colon cancer. The IRR was 0.99 (95% CI, 0.93-1.06).
The team also assessed hematologic malignancies. The IRRs, for users compared to non-users, were as follows:
- 0.99 (95% CI, 0.89-1.11) for leukemia
- 0.89 (95% CI, 0.71-1.11) for chronic lymphocytic leukemia
- 0.70 (95% CI, 0.51-0.98) for acute myeloid leukemia
- 0.92 (95% CI, 0.82-1.04) for lymphoma
- 0.66 (95% CI, 0.34-1.26) for Hodgkin lymphoma
- 0.92 (95% CI, 0.81-1.04) for non-Hodgkin lymphoma.
The researchers noted that they did not collect information on other medications subjects were taking or risk factors that might influence cancer development, and new cancers may have been cancer recurrences.
In addition, the team said the prescription of warfarin may be a marker for other healthcare factors that lead to cancer prevention.
Still, the researchers said warfarin appeared to be associated with reduced cancer risk in this study.
They therefore believe this finding could have implications for choosing anticoagulants, although additional research is needed. ![]()
New research suggests warfarin use may reduce the risk of cancer among people over the age of 50.
An observational study of more than 1 million people in Norway revealed that individuals who took warfarin had a lower incidence of cancers overall, as well as certain types of cancers, than people who did not take warfarin.
James B. Lorens, PhD, of the University of Bergen in Norway, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers analyzed data from the Norwegian National Registry, the Norwegian Prescription Database, and the Cancer Registry of Norway.
This included data for all individuals ages 52 to 82 who were living in Norway from January 1, 2006, through December 31, 2012, (n=1,256,725).
The researchers looked for cancer diagnoses in this group during the 7-year observation period. They compared the incidence of any new cancer (including certain cancer types) between warfarin users and non-users.
People were considered warfarin users if they had taken at least 6 months of a warfarin prescription and at least 2 years had elapsed from their first prescription to any cancer diagnosis.
Most study subjects were non-users (92.6%, n=1,163,783), but 7.4% (n=92,942) were warfarin users.
The researchers noted that warfarin users were older than non-users, with mean ages of 70.2 and 63.9, respectively. Warfarin users were also more likely to be male (61.7%; n=57,370) and non-users female (52.7%; n=613,803).
The incidence of cancer was 10.6% (n=132,687) in the entire study cohort, 9.4% (n=8754) among warfarin users, and 10.6% (n=123,933) among non-users. The most common cancer types were prostate, lung, colon, and breast.
The researchers found a significantly lower age- and sex-adjusted incidence rate ratio (IRR) for all cancers among warfarin users than nonusers. The IRR was 0.84 (95% CI, 0.82-0.86).
The IRR was also significantly lower among users than non-users for 3 of the 4 most common cancers. The IRR was 0.69 (95% CI, 0.65-0.72) for prostate cancer, 0.80 (95% CI, 0.75-0.86) for lung cancer, and 0.90 (95% CI, 0.82-1.00) for female breast cancer.
The researchers said there was no significant difference between users and non-users for colon cancer. The IRR was 0.99 (95% CI, 0.93-1.06).
The team also assessed hematologic malignancies. The IRRs, for users compared to non-users, were as follows:
- 0.99 (95% CI, 0.89-1.11) for leukemia
- 0.89 (95% CI, 0.71-1.11) for chronic lymphocytic leukemia
- 0.70 (95% CI, 0.51-0.98) for acute myeloid leukemia
- 0.92 (95% CI, 0.82-1.04) for lymphoma
- 0.66 (95% CI, 0.34-1.26) for Hodgkin lymphoma
- 0.92 (95% CI, 0.81-1.04) for non-Hodgkin lymphoma.
The researchers noted that they did not collect information on other medications subjects were taking or risk factors that might influence cancer development, and new cancers may have been cancer recurrences.
In addition, the team said the prescription of warfarin may be a marker for other healthcare factors that lead to cancer prevention.
Still, the researchers said warfarin appeared to be associated with reduced cancer risk in this study.
They therefore believe this finding could have implications for choosing anticoagulants, although additional research is needed. ![]()
New research suggests warfarin use may reduce the risk of cancer among people over the age of 50.
An observational study of more than 1 million people in Norway revealed that individuals who took warfarin had a lower incidence of cancers overall, as well as certain types of cancers, than people who did not take warfarin.
James B. Lorens, PhD, of the University of Bergen in Norway, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers analyzed data from the Norwegian National Registry, the Norwegian Prescription Database, and the Cancer Registry of Norway.
This included data for all individuals ages 52 to 82 who were living in Norway from January 1, 2006, through December 31, 2012, (n=1,256,725).
The researchers looked for cancer diagnoses in this group during the 7-year observation period. They compared the incidence of any new cancer (including certain cancer types) between warfarin users and non-users.
People were considered warfarin users if they had taken at least 6 months of a warfarin prescription and at least 2 years had elapsed from their first prescription to any cancer diagnosis.
Most study subjects were non-users (92.6%, n=1,163,783), but 7.4% (n=92,942) were warfarin users.
The researchers noted that warfarin users were older than non-users, with mean ages of 70.2 and 63.9, respectively. Warfarin users were also more likely to be male (61.7%; n=57,370) and non-users female (52.7%; n=613,803).
The incidence of cancer was 10.6% (n=132,687) in the entire study cohort, 9.4% (n=8754) among warfarin users, and 10.6% (n=123,933) among non-users. The most common cancer types were prostate, lung, colon, and breast.
The researchers found a significantly lower age- and sex-adjusted incidence rate ratio (IRR) for all cancers among warfarin users than nonusers. The IRR was 0.84 (95% CI, 0.82-0.86).
The IRR was also significantly lower among users than non-users for 3 of the 4 most common cancers. The IRR was 0.69 (95% CI, 0.65-0.72) for prostate cancer, 0.80 (95% CI, 0.75-0.86) for lung cancer, and 0.90 (95% CI, 0.82-1.00) for female breast cancer.
The researchers said there was no significant difference between users and non-users for colon cancer. The IRR was 0.99 (95% CI, 0.93-1.06).
The team also assessed hematologic malignancies. The IRRs, for users compared to non-users, were as follows:
- 0.99 (95% CI, 0.89-1.11) for leukemia
- 0.89 (95% CI, 0.71-1.11) for chronic lymphocytic leukemia
- 0.70 (95% CI, 0.51-0.98) for acute myeloid leukemia
- 0.92 (95% CI, 0.82-1.04) for lymphoma
- 0.66 (95% CI, 0.34-1.26) for Hodgkin lymphoma
- 0.92 (95% CI, 0.81-1.04) for non-Hodgkin lymphoma.
The researchers noted that they did not collect information on other medications subjects were taking or risk factors that might influence cancer development, and new cancers may have been cancer recurrences.
In addition, the team said the prescription of warfarin may be a marker for other healthcare factors that lead to cancer prevention.
Still, the researchers said warfarin appeared to be associated with reduced cancer risk in this study.
They therefore believe this finding could have implications for choosing anticoagulants, although additional research is needed. ![]()
FDA grants drug orphan designation for treatment of malaria

The US Food and Drug Administration (FDA) has granted orphan drug designation to artemisone, a product candidate for the treatment of malaria.
Artemisone is a synthetic derivative of the antimalarial drug artemisinin, which “has been optimized for potency, stability, and safety,” according to Artemis Therapeutics, Inc., the company developing artemisone.
The company said phase 2 trial data suggest artemisone is effective against Plasmodium falciparum malaria.
Ninety-five patients were enrolled in the trial, and they received a 2-day or 3-day course of artemisone. Patients also received a second antimalarial drug on the final day of artemisone treatment (in compliance with recommendations from the World Health Organization).
At 28 days, the cure rates were 100% in both the 2-day and 3-day course groups. However, parasite clearance time was 25% faster with the 2-day course.
Artemis Therapeutics, Inc. has not yet released safety data from this trial.
Phase 1 data suggested artemisone was well tolerated by healthy subjects. There were no serious adverse events in the trial and no clinically relevant changes in laboratory and vital parameters, according to researchers.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to artemisone, a product candidate for the treatment of malaria.
Artemisone is a synthetic derivative of the antimalarial drug artemisinin, which “has been optimized for potency, stability, and safety,” according to Artemis Therapeutics, Inc., the company developing artemisone.
The company said phase 2 trial data suggest artemisone is effective against Plasmodium falciparum malaria.
Ninety-five patients were enrolled in the trial, and they received a 2-day or 3-day course of artemisone. Patients also received a second antimalarial drug on the final day of artemisone treatment (in compliance with recommendations from the World Health Organization).
At 28 days, the cure rates were 100% in both the 2-day and 3-day course groups. However, parasite clearance time was 25% faster with the 2-day course.
Artemis Therapeutics, Inc. has not yet released safety data from this trial.
Phase 1 data suggested artemisone was well tolerated by healthy subjects. There were no serious adverse events in the trial and no clinically relevant changes in laboratory and vital parameters, according to researchers.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to artemisone, a product candidate for the treatment of malaria.
Artemisone is a synthetic derivative of the antimalarial drug artemisinin, which “has been optimized for potency, stability, and safety,” according to Artemis Therapeutics, Inc., the company developing artemisone.
The company said phase 2 trial data suggest artemisone is effective against Plasmodium falciparum malaria.
Ninety-five patients were enrolled in the trial, and they received a 2-day or 3-day course of artemisone. Patients also received a second antimalarial drug on the final day of artemisone treatment (in compliance with recommendations from the World Health Organization).
At 28 days, the cure rates were 100% in both the 2-day and 3-day course groups. However, parasite clearance time was 25% faster with the 2-day course.
Artemis Therapeutics, Inc. has not yet released safety data from this trial.
Phase 1 data suggested artemisone was well tolerated by healthy subjects. There were no serious adverse events in the trial and no clinically relevant changes in laboratory and vital parameters, according to researchers.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
EMA grants accelerated assessment to drug for AML

The European Medicines Agency’s Committee for Medicinal Products for Human Use has granted accelerated assessment to a marketing authorization application (MAA) for CPX-351 (Vyxeos™), a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
The MAA is for CPX-351 to treat adults with high-risk acute myeloid leukemia (AML), defined as therapy-related AML or AML with myelodysplasia-related changes.
Accelerated assessment is designed to reduce the review timeline for products of major interest for public health and therapeutic innovation.
“If approved, Vyxeos will become the first new chemotherapy treatment option specifically for European patients with therapy-related AML or AML with myelodysplasia-related changes,” said Karen Smith, MD, PhD, executive vice president, research and development and chief medical officer at Jazz Pharmaceuticals, the company developing and marketing CPX-351.
The MAA for CPX-351 is supported by clinical data from 5 studies, including a phase 3 study. Results from this study were presented at the 2016 ASCO Annual Meeting.
In this study, researchers compared CPX-351 to cytarabine and daunorubicin (7+3) in 309 patients, ages 60 to 75, with newly diagnosed, therapy-related AML or AML with myelodysplasia-related changes.
The complete response rate was 38% in the CPX-351 arm and 26% in the 7+3 arm (P=0.036).
The rate of hematopoietic stem cell transplant was 34% in the CPX-351 arm and 25% in the 7+3 arm.
The median overall survival was 9.6 months in the CPX-351 arm and 5.9 months in the 7+3 arm (P=0.005).
All-cause 30-day mortality was 6% in the CPX-351 arm and 11% in the 7+3 arm. Sixty-day mortality was 14% and 21%, respectively.
Six percent of patients in both arms had a fatal adverse event (AE) on treatment or within 30 days of therapy that was not in the setting of progressive disease.
The rate of AEs that led to discontinuation was 18% in the CPX-351 arm and 13% in the 7+3 arm. AEs leading to discontinuation in the CPX-351 arm included prolonged cytopenias, infection, cardiotoxicity, respiratory failure, hemorrhage, renal insufficiency, colitis, and generalized medical deterioration.
The most common AEs (incidence ≥ 25%) in the CPX-351 arm were hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The most common serious AEs (incidence ≥ 5%) in the CPX-351 arm were dyspnea, myocardial toxicity, sepsis, pneumonia, febrile neutropenia, bacteremia, and hemorrhage. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use has granted accelerated assessment to a marketing authorization application (MAA) for CPX-351 (Vyxeos™), a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
The MAA is for CPX-351 to treat adults with high-risk acute myeloid leukemia (AML), defined as therapy-related AML or AML with myelodysplasia-related changes.
Accelerated assessment is designed to reduce the review timeline for products of major interest for public health and therapeutic innovation.
“If approved, Vyxeos will become the first new chemotherapy treatment option specifically for European patients with therapy-related AML or AML with myelodysplasia-related changes,” said Karen Smith, MD, PhD, executive vice president, research and development and chief medical officer at Jazz Pharmaceuticals, the company developing and marketing CPX-351.
The MAA for CPX-351 is supported by clinical data from 5 studies, including a phase 3 study. Results from this study were presented at the 2016 ASCO Annual Meeting.
In this study, researchers compared CPX-351 to cytarabine and daunorubicin (7+3) in 309 patients, ages 60 to 75, with newly diagnosed, therapy-related AML or AML with myelodysplasia-related changes.
The complete response rate was 38% in the CPX-351 arm and 26% in the 7+3 arm (P=0.036).
The rate of hematopoietic stem cell transplant was 34% in the CPX-351 arm and 25% in the 7+3 arm.
The median overall survival was 9.6 months in the CPX-351 arm and 5.9 months in the 7+3 arm (P=0.005).
All-cause 30-day mortality was 6% in the CPX-351 arm and 11% in the 7+3 arm. Sixty-day mortality was 14% and 21%, respectively.
Six percent of patients in both arms had a fatal adverse event (AE) on treatment or within 30 days of therapy that was not in the setting of progressive disease.
The rate of AEs that led to discontinuation was 18% in the CPX-351 arm and 13% in the 7+3 arm. AEs leading to discontinuation in the CPX-351 arm included prolonged cytopenias, infection, cardiotoxicity, respiratory failure, hemorrhage, renal insufficiency, colitis, and generalized medical deterioration.
The most common AEs (incidence ≥ 25%) in the CPX-351 arm were hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The most common serious AEs (incidence ≥ 5%) in the CPX-351 arm were dyspnea, myocardial toxicity, sepsis, pneumonia, febrile neutropenia, bacteremia, and hemorrhage. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use has granted accelerated assessment to a marketing authorization application (MAA) for CPX-351 (Vyxeos™), a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
The MAA is for CPX-351 to treat adults with high-risk acute myeloid leukemia (AML), defined as therapy-related AML or AML with myelodysplasia-related changes.
Accelerated assessment is designed to reduce the review timeline for products of major interest for public health and therapeutic innovation.
“If approved, Vyxeos will become the first new chemotherapy treatment option specifically for European patients with therapy-related AML or AML with myelodysplasia-related changes,” said Karen Smith, MD, PhD, executive vice president, research and development and chief medical officer at Jazz Pharmaceuticals, the company developing and marketing CPX-351.
The MAA for CPX-351 is supported by clinical data from 5 studies, including a phase 3 study. Results from this study were presented at the 2016 ASCO Annual Meeting.
In this study, researchers compared CPX-351 to cytarabine and daunorubicin (7+3) in 309 patients, ages 60 to 75, with newly diagnosed, therapy-related AML or AML with myelodysplasia-related changes.
The complete response rate was 38% in the CPX-351 arm and 26% in the 7+3 arm (P=0.036).
The rate of hematopoietic stem cell transplant was 34% in the CPX-351 arm and 25% in the 7+3 arm.
The median overall survival was 9.6 months in the CPX-351 arm and 5.9 months in the 7+3 arm (P=0.005).
All-cause 30-day mortality was 6% in the CPX-351 arm and 11% in the 7+3 arm. Sixty-day mortality was 14% and 21%, respectively.
Six percent of patients in both arms had a fatal adverse event (AE) on treatment or within 30 days of therapy that was not in the setting of progressive disease.
The rate of AEs that led to discontinuation was 18% in the CPX-351 arm and 13% in the 7+3 arm. AEs leading to discontinuation in the CPX-351 arm included prolonged cytopenias, infection, cardiotoxicity, respiratory failure, hemorrhage, renal insufficiency, colitis, and generalized medical deterioration.
The most common AEs (incidence ≥ 25%) in the CPX-351 arm were hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The most common serious AEs (incidence ≥ 5%) in the CPX-351 arm were dyspnea, myocardial toxicity, sepsis, pneumonia, febrile neutropenia, bacteremia, and hemorrhage. ![]()
Drug receives orphan designation for treatment of PNH

The US Food and Drug Administration (FDA) has granted orphan drug designation to ACH-4471 for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
And the European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products has recommended the drug receive orphan status for the same indication in the European Economic Area.
ACH-4471 is a factor D inhibitor being developed by Achillion Pharmaceuticals, Inc.
In April, the company announced the initiation of a phase 2, three-month, dose-ranging trial with ACH-4471 for patients with untreated PNH (NCT03053102).
The primary objective of the trial is to assess the change from baseline in serum lactate dehydrogenase (LDH) levels. Secondary endpoints include changes in hemoglobin, PNH red blood cells, fatigue score (FACIT scale), changes in levels of complement pathway biomarkers such as Bb and factor D, pharmacokinetics, and safety.
The protocol allows for intra-patient dose-escalation. Patients initially receive 100 mg or 150 mg of ACH-4471 three times daily, and doses may be increased during the treatment period.
After patients complete 3 months of treatment and investigators have assessed safety and clinical benefit, patients may be enrolled in the long-term extension trial (NCT03181633).
To date, 200 mg three times daily has been the highest dose of ACH-4471 administered. And Achillion has collected data on 4 patients.
Two of the patients have completed the 3-month trial and entered the long-term extension trial. One patient continues to receive dosing in the 3-month trial, and the fourth patient voluntarily withdrew from the trial on day 41 for reasons unrelated to safety.
Thus far, ACH-4471 has produced clinically meaningful complement inhibition and demonstrated a favorable tolerability profile, with no reports of clinically meaningful increases in liver enzymes. ACH-4471 has improved LDH, hemoglobin, fatigue score, and other measures of response, including PNH clone size.
FDA’s orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
EMA’s orphan designation
The EMA’s orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. It also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to ACH-4471 for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
And the European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products has recommended the drug receive orphan status for the same indication in the European Economic Area.
ACH-4471 is a factor D inhibitor being developed by Achillion Pharmaceuticals, Inc.
In April, the company announced the initiation of a phase 2, three-month, dose-ranging trial with ACH-4471 for patients with untreated PNH (NCT03053102).
The primary objective of the trial is to assess the change from baseline in serum lactate dehydrogenase (LDH) levels. Secondary endpoints include changes in hemoglobin, PNH red blood cells, fatigue score (FACIT scale), changes in levels of complement pathway biomarkers such as Bb and factor D, pharmacokinetics, and safety.
The protocol allows for intra-patient dose-escalation. Patients initially receive 100 mg or 150 mg of ACH-4471 three times daily, and doses may be increased during the treatment period.
After patients complete 3 months of treatment and investigators have assessed safety and clinical benefit, patients may be enrolled in the long-term extension trial (NCT03181633).
To date, 200 mg three times daily has been the highest dose of ACH-4471 administered. And Achillion has collected data on 4 patients.
Two of the patients have completed the 3-month trial and entered the long-term extension trial. One patient continues to receive dosing in the 3-month trial, and the fourth patient voluntarily withdrew from the trial on day 41 for reasons unrelated to safety.
Thus far, ACH-4471 has produced clinically meaningful complement inhibition and demonstrated a favorable tolerability profile, with no reports of clinically meaningful increases in liver enzymes. ACH-4471 has improved LDH, hemoglobin, fatigue score, and other measures of response, including PNH clone size.
FDA’s orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
EMA’s orphan designation
The EMA’s orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. It also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to ACH-4471 for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
And the European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products has recommended the drug receive orphan status for the same indication in the European Economic Area.
ACH-4471 is a factor D inhibitor being developed by Achillion Pharmaceuticals, Inc.
In April, the company announced the initiation of a phase 2, three-month, dose-ranging trial with ACH-4471 for patients with untreated PNH (NCT03053102).
The primary objective of the trial is to assess the change from baseline in serum lactate dehydrogenase (LDH) levels. Secondary endpoints include changes in hemoglobin, PNH red blood cells, fatigue score (FACIT scale), changes in levels of complement pathway biomarkers such as Bb and factor D, pharmacokinetics, and safety.
The protocol allows for intra-patient dose-escalation. Patients initially receive 100 mg or 150 mg of ACH-4471 three times daily, and doses may be increased during the treatment period.
After patients complete 3 months of treatment and investigators have assessed safety and clinical benefit, patients may be enrolled in the long-term extension trial (NCT03181633).
To date, 200 mg three times daily has been the highest dose of ACH-4471 administered. And Achillion has collected data on 4 patients.
Two of the patients have completed the 3-month trial and entered the long-term extension trial. One patient continues to receive dosing in the 3-month trial, and the fourth patient voluntarily withdrew from the trial on day 41 for reasons unrelated to safety.
Thus far, ACH-4471 has produced clinically meaningful complement inhibition and demonstrated a favorable tolerability profile, with no reports of clinically meaningful increases in liver enzymes. ACH-4471 has improved LDH, hemoglobin, fatigue score, and other measures of response, including PNH clone size.
FDA’s orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
EMA’s orphan designation
The EMA’s orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. It also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
Shedding skin
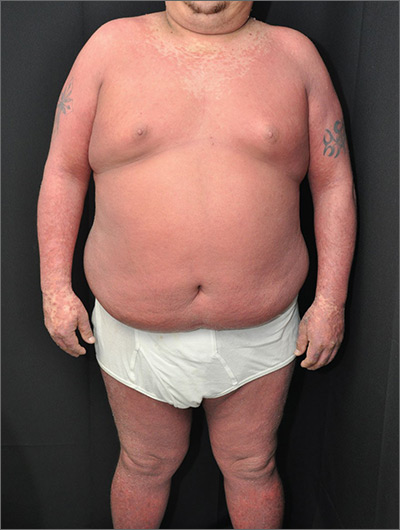
The physician made a diagnosis of erythroderma. The FP considered psoriasis, atopic dermatitis, or another, more rare disease as causes. Psoriasis was the most likely etiology due to the nail pitting and the fact that atopic dermatitis starts in childhood. A 4-mm punch biopsy (rush requested) was performed to confirm this.
Erythroderma is an uncommon condition that affects all age groups and is characterized by a generalized erythematous rash with associated scaling. It is generally a manifestation of another underlying dermatosis or systemic disorder. Erythroderma is associated with a range of morbidity, and can have life-threatening metabolic and cardiovascular complications. Therapy is usually focused on treating the underlying disease, as well as addressing the systemic complications.
In this patient’s case, the FP consulted a dermatology colleague (an FP with dermatology fellowship training). She recommended ordering the following labs in anticipation of oral cyclosporine therapy: complete blood count, chemistry panel, uric acid, magnesium level, QuantiFERON TB gold, hepatitis C antibody, and hepatitis B surface antigen, surface antibody, and core antibody.
The FP prescribed a one-pound jar of 0.1% triamcinolone ointment to be applied twice daily. He instructed the patient to apply the ointment to the red areas, cover the involved skin with wet pajamas or soft clothing, then apply a dry blanket over the wet layer. (The pajamas should be made wet with lukewarm water and wrung out so that they are not dripping.) The FP instructed the patient to sleep in this overnight and only remove it if he became chilled or was unable to sleep. The patient was also counseled to quit smoking and drinking.
The dermatology specialist agreed to see the patient in 2 days. At that time, the patient’s lab results were normal and he was feeling a bit better from the topical triamcinolone. The dermatology specialist started the patient on cyclosporine and he improved rapidly. At follow-up, the dermatology specialist discussed other systemic treatments and the need to taper off the cyclosporine. Fortunately, the patient had stopped smoking and drinking, and his blood pressure and labs remained normal.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Henderson D. Erythroderma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 915-921.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The physician made a diagnosis of erythroderma. The FP considered psoriasis, atopic dermatitis, or another, more rare disease as causes. Psoriasis was the most likely etiology due to the nail pitting and the fact that atopic dermatitis starts in childhood. A 4-mm punch biopsy (rush requested) was performed to confirm this.
Erythroderma is an uncommon condition that affects all age groups and is characterized by a generalized erythematous rash with associated scaling. It is generally a manifestation of another underlying dermatosis or systemic disorder. Erythroderma is associated with a range of morbidity, and can have life-threatening metabolic and cardiovascular complications. Therapy is usually focused on treating the underlying disease, as well as addressing the systemic complications.
In this patient’s case, the FP consulted a dermatology colleague (an FP with dermatology fellowship training). She recommended ordering the following labs in anticipation of oral cyclosporine therapy: complete blood count, chemistry panel, uric acid, magnesium level, QuantiFERON TB gold, hepatitis C antibody, and hepatitis B surface antigen, surface antibody, and core antibody.
The FP prescribed a one-pound jar of 0.1% triamcinolone ointment to be applied twice daily. He instructed the patient to apply the ointment to the red areas, cover the involved skin with wet pajamas or soft clothing, then apply a dry blanket over the wet layer. (The pajamas should be made wet with lukewarm water and wrung out so that they are not dripping.) The FP instructed the patient to sleep in this overnight and only remove it if he became chilled or was unable to sleep. The patient was also counseled to quit smoking and drinking.
The dermatology specialist agreed to see the patient in 2 days. At that time, the patient’s lab results were normal and he was feeling a bit better from the topical triamcinolone. The dermatology specialist started the patient on cyclosporine and he improved rapidly. At follow-up, the dermatology specialist discussed other systemic treatments and the need to taper off the cyclosporine. Fortunately, the patient had stopped smoking and drinking, and his blood pressure and labs remained normal.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Henderson D. Erythroderma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 915-921.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The physician made a diagnosis of erythroderma. The FP considered psoriasis, atopic dermatitis, or another, more rare disease as causes. Psoriasis was the most likely etiology due to the nail pitting and the fact that atopic dermatitis starts in childhood. A 4-mm punch biopsy (rush requested) was performed to confirm this.
Erythroderma is an uncommon condition that affects all age groups and is characterized by a generalized erythematous rash with associated scaling. It is generally a manifestation of another underlying dermatosis or systemic disorder. Erythroderma is associated with a range of morbidity, and can have life-threatening metabolic and cardiovascular complications. Therapy is usually focused on treating the underlying disease, as well as addressing the systemic complications.
In this patient’s case, the FP consulted a dermatology colleague (an FP with dermatology fellowship training). She recommended ordering the following labs in anticipation of oral cyclosporine therapy: complete blood count, chemistry panel, uric acid, magnesium level, QuantiFERON TB gold, hepatitis C antibody, and hepatitis B surface antigen, surface antibody, and core antibody.
The FP prescribed a one-pound jar of 0.1% triamcinolone ointment to be applied twice daily. He instructed the patient to apply the ointment to the red areas, cover the involved skin with wet pajamas or soft clothing, then apply a dry blanket over the wet layer. (The pajamas should be made wet with lukewarm water and wrung out so that they are not dripping.) The FP instructed the patient to sleep in this overnight and only remove it if he became chilled or was unable to sleep. The patient was also counseled to quit smoking and drinking.
The dermatology specialist agreed to see the patient in 2 days. At that time, the patient’s lab results were normal and he was feeling a bit better from the topical triamcinolone. The dermatology specialist started the patient on cyclosporine and he improved rapidly. At follow-up, the dermatology specialist discussed other systemic treatments and the need to taper off the cyclosporine. Fortunately, the patient had stopped smoking and drinking, and his blood pressure and labs remained normal.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Henderson D. Erythroderma. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill; 2013: 915-921.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
Scratching the Surface of the Problem
ANSWER
The least likely diagnosis is staph infection (choice “b”). By their very nature, staph infections are suppurative, often involving redness, swelling, pain, and purulent drainage. They can be chronic (eg, MRSA) but are more typically of acute onset. And they usually resolve on their own or with treatment; recall that this patient was treated for staph infection many times without any change.
She was also treated repeatedly for scabies (choice “a”), a condition that could certainly last 15 years. However, the lack of improvement with treatment, combined with the absence of suggestive signs, make this diagnosis improbable.
Mycosis fungoides (MF; choice “c”) is a type of T-cell lymphoma that often develops with itching and plaque formation over the course of years. It was ultimately ruled out by the biopsy results (as were scabies and staph infection).
What the biopsy did show was epidermal thickening—a characteristic sign of prurigo nodularis (choice “d”).
DISCUSSION
Prurigo nodularis is a localized form of neurodermatitis caused by picking and scratching. As with the classic form, the more the patient scratches, the more the lesions itch and multiply. In this case, biopsy of the larger plaque showed hypertrophic scarring.
The patient’s skin-picking habit likely developed during (and perhaps because of) her methamphetamine use. Long-term exposure to the itch-scratch-itch cycle can make treatment problematic. In this case, a class 4 topical steroid cream was prescribed, along with injection of several larger lesions with 10 mg/mL triamcinolone suspension.
It’s worth mentioning that for patients with this type of history, general lab testing (ie, complete blood count and complete metabolic panel) should be performed to rule out organic disease and other serious conditions (eg, renal or hepatic failure, leukemia). Fortunately, this patient’s results were reassuring on that front.
ANSWER
The least likely diagnosis is staph infection (choice “b”). By their very nature, staph infections are suppurative, often involving redness, swelling, pain, and purulent drainage. They can be chronic (eg, MRSA) but are more typically of acute onset. And they usually resolve on their own or with treatment; recall that this patient was treated for staph infection many times without any change.
She was also treated repeatedly for scabies (choice “a”), a condition that could certainly last 15 years. However, the lack of improvement with treatment, combined with the absence of suggestive signs, make this diagnosis improbable.
Mycosis fungoides (MF; choice “c”) is a type of T-cell lymphoma that often develops with itching and plaque formation over the course of years. It was ultimately ruled out by the biopsy results (as were scabies and staph infection).
What the biopsy did show was epidermal thickening—a characteristic sign of prurigo nodularis (choice “d”).
DISCUSSION
Prurigo nodularis is a localized form of neurodermatitis caused by picking and scratching. As with the classic form, the more the patient scratches, the more the lesions itch and multiply. In this case, biopsy of the larger plaque showed hypertrophic scarring.
The patient’s skin-picking habit likely developed during (and perhaps because of) her methamphetamine use. Long-term exposure to the itch-scratch-itch cycle can make treatment problematic. In this case, a class 4 topical steroid cream was prescribed, along with injection of several larger lesions with 10 mg/mL triamcinolone suspension.
It’s worth mentioning that for patients with this type of history, general lab testing (ie, complete blood count and complete metabolic panel) should be performed to rule out organic disease and other serious conditions (eg, renal or hepatic failure, leukemia). Fortunately, this patient’s results were reassuring on that front.
ANSWER
The least likely diagnosis is staph infection (choice “b”). By their very nature, staph infections are suppurative, often involving redness, swelling, pain, and purulent drainage. They can be chronic (eg, MRSA) but are more typically of acute onset. And they usually resolve on their own or with treatment; recall that this patient was treated for staph infection many times without any change.
She was also treated repeatedly for scabies (choice “a”), a condition that could certainly last 15 years. However, the lack of improvement with treatment, combined with the absence of suggestive signs, make this diagnosis improbable.
Mycosis fungoides (MF; choice “c”) is a type of T-cell lymphoma that often develops with itching and plaque formation over the course of years. It was ultimately ruled out by the biopsy results (as were scabies and staph infection).
What the biopsy did show was epidermal thickening—a characteristic sign of prurigo nodularis (choice “d”).
DISCUSSION
Prurigo nodularis is a localized form of neurodermatitis caused by picking and scratching. As with the classic form, the more the patient scratches, the more the lesions itch and multiply. In this case, biopsy of the larger plaque showed hypertrophic scarring.
The patient’s skin-picking habit likely developed during (and perhaps because of) her methamphetamine use. Long-term exposure to the itch-scratch-itch cycle can make treatment problematic. In this case, a class 4 topical steroid cream was prescribed, along with injection of several larger lesions with 10 mg/mL triamcinolone suspension.
It’s worth mentioning that for patients with this type of history, general lab testing (ie, complete blood count and complete metabolic panel) should be performed to rule out organic disease and other serious conditions (eg, renal or hepatic failure, leukemia). Fortunately, this patient’s results were reassuring on that front.
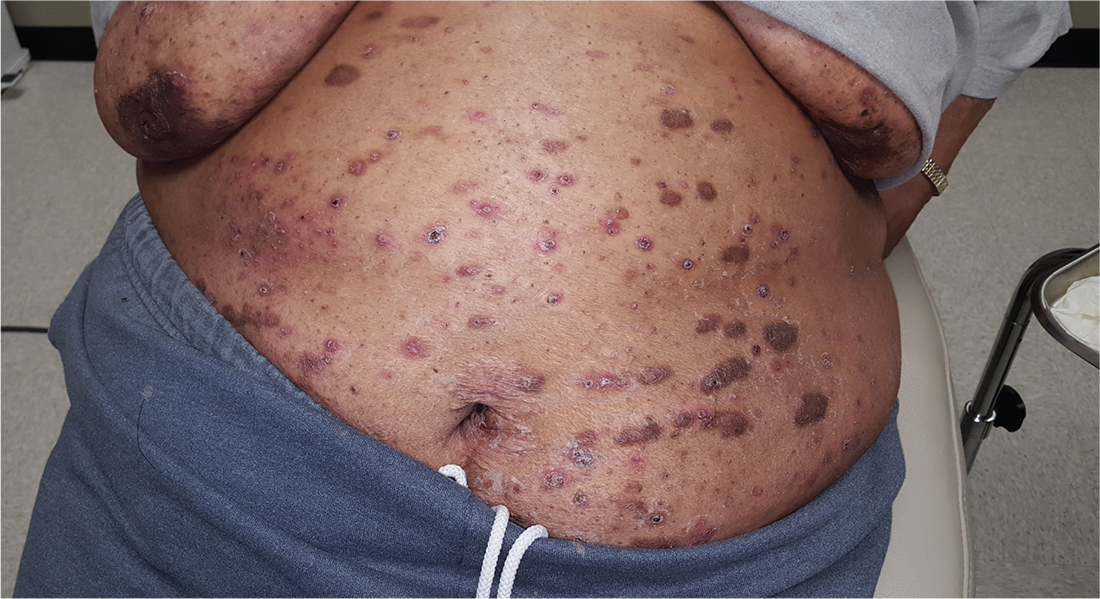
For at least 15 years, this 61-year-old African-American woman has had itchy lesions within arm’s reach. The patient says the problem manifested during a period in her life when she was addicted to several drugs, including methamphetamine. She complains bitterly about the itching and says it is difficult for her to leave the lesions alone.
No one else in her household is similarly affected. The patient has consulted a variety of health care providers, from primary care to dermatology and other specialties. She has received diagnoses of everything from scabies (for which she was treated, unsuccessfully) to pyoderma, bedbugs, and various forms of staph infection.
Examination reveals more than 100 lesions, mostly confined to her abdomen and lower chest, ranging from pinpoint to more than 3 cm. Many are excoriated, but most are purplish brown, oval plaques that somewhat match her type IV skin.
Her back, arms, and hands are spared. No lesions are seen between her fingers or on her volar wrists.
A biopsy is performed on two of the lesions—one small, the other larger.

FDA asked to approve add-on drug for eosinophilic COPD
GlaxoSmithKline asked the Food and Drug Administration to approve an interleuklin-5 antagonist as an add-on maintenance therapy for patients with eosinophilic chronic obstructive pulmonary disease (COPD).
The pharmaceutical and health care company is seeking approval of mepolizumab to be used specifically to treat COPD patients with an eosinophilic phenotype. The drug currently is indicated to treat patients aged 12 years or older with severe asthma and asthma with an eosinophilic phenotype and is sold under the name Nucala, according to a GlaxoSmithKline statement issued November 7.![]()
Headache, injection site reaction, back pain, and fatigue are the most common adverse reactions seen in patients who took mepolizumab during clinical trials.
Mepolizumab is not approved for the treatment of COPD anywhere in the world, and GlaxoSmithKline intends to also ask other countries’ regulatory authorities to allow this drug to be sold as a therapy for COPD.
GlaxoSmithKline asked the Food and Drug Administration to approve an interleuklin-5 antagonist as an add-on maintenance therapy for patients with eosinophilic chronic obstructive pulmonary disease (COPD).
The pharmaceutical and health care company is seeking approval of mepolizumab to be used specifically to treat COPD patients with an eosinophilic phenotype. The drug currently is indicated to treat patients aged 12 years or older with severe asthma and asthma with an eosinophilic phenotype and is sold under the name Nucala, according to a GlaxoSmithKline statement issued November 7.![]()
Headache, injection site reaction, back pain, and fatigue are the most common adverse reactions seen in patients who took mepolizumab during clinical trials.
Mepolizumab is not approved for the treatment of COPD anywhere in the world, and GlaxoSmithKline intends to also ask other countries’ regulatory authorities to allow this drug to be sold as a therapy for COPD.
GlaxoSmithKline asked the Food and Drug Administration to approve an interleuklin-5 antagonist as an add-on maintenance therapy for patients with eosinophilic chronic obstructive pulmonary disease (COPD).
The pharmaceutical and health care company is seeking approval of mepolizumab to be used specifically to treat COPD patients with an eosinophilic phenotype. The drug currently is indicated to treat patients aged 12 years or older with severe asthma and asthma with an eosinophilic phenotype and is sold under the name Nucala, according to a GlaxoSmithKline statement issued November 7.![]()
Headache, injection site reaction, back pain, and fatigue are the most common adverse reactions seen in patients who took mepolizumab during clinical trials.
Mepolizumab is not approved for the treatment of COPD anywhere in the world, and GlaxoSmithKline intends to also ask other countries’ regulatory authorities to allow this drug to be sold as a therapy for COPD.
Red cell age: No impact on mortality after transfusion
Critically ill patients who received transfusions of the freshest-available red cells had a mortality rate similar to that of patients who received standard-issue, oldest-available red cells, according to results from a large randomized trial.
In some earlier studies, transfusion of older red cells was linked to increased mortality for critically ill, surgical, and trauma patients. But the new results provide “strong evidence” that transfusing very fresh red cells rather than older red cells “provides no clinically meaningful benefits” in the critically ill population, reported D. James Cooper, MD, of Monash University, Melbourne, and his colleagues.
“Our results support the current international usual practice of transfusing patients with the oldest red cells available,” the researchers wrote in the report on the trial, known as TRANSFUSE (Standard Issue Transfusion versus Fresher Red-Cell Use in Intensive Care).
Red cells are stored up to 42 days and can undergo biochemical, structural, or metabolic changes during that time that “may cause harm,” the researchers wrote. However, blood banks typically issue the oldest compatible red cell units available to them, and it’s uncertain whether doing so increases mortality.
To see if the age of red cells impacted mortality, Dr. Cooper and has colleagues at 59 centers in five countries randomized 4,994 critically ill adults to receive the freshest-available or standard oldest-available red cells (N Engl J Med. 2017;377:1858-67).
At 90 days after transfusion, mortality was 24.8% in the group of patients receiving the freshest-available red cells, and 24.1% for the oldest-available group, or an absolute risk difference of just 0.7 percentage points (95% confidence interval, –1.7 to 3.1; P = .57).
“Among the many secondary outcomes tested, we noted a nominal difference in febrile nonhemolytic transfusion reactions that was small, and we are not sure of its clinical significance,” the researchers wrote.
The average duration of red cell storage was 11.8 days versus 22.4 days for the freshest-available and oldest-available groups, respectively.
The TRANSFUSE trial is not the first to suggest that age of red blood cells does not make a difference in mortality after transfusion. There were two earlier trials, ABLE (Age of Blood Evaluation) and INFORM (Informing Fresh versus Old Red Cell Management) that came to similar conclusions. However, the ABLE trial had a small sample size, and INFORM had “limited outcome data” including a low mortality rate “suggesting low illness severity,” the researchers noted.
“The lower in-hospital mortality in the ICU subgroup in the INFORM trial (13.0%) than that observed in our trial at 90 days (24.5%) is consistent with lower illness severity in the INFORM patients,” they wrote.
The study was funded by organizations including the Australian National Health and Medical Research Council. Dr. Cooper reported receiving consulting fees from Eustralis Pharmaceuticals that were paid to Monash University. No other potential conflicts of interest were reported.
Critically ill patients who received transfusions of the freshest-available red cells had a mortality rate similar to that of patients who received standard-issue, oldest-available red cells, according to results from a large randomized trial.
In some earlier studies, transfusion of older red cells was linked to increased mortality for critically ill, surgical, and trauma patients. But the new results provide “strong evidence” that transfusing very fresh red cells rather than older red cells “provides no clinically meaningful benefits” in the critically ill population, reported D. James Cooper, MD, of Monash University, Melbourne, and his colleagues.
“Our results support the current international usual practice of transfusing patients with the oldest red cells available,” the researchers wrote in the report on the trial, known as TRANSFUSE (Standard Issue Transfusion versus Fresher Red-Cell Use in Intensive Care).
Red cells are stored up to 42 days and can undergo biochemical, structural, or metabolic changes during that time that “may cause harm,” the researchers wrote. However, blood banks typically issue the oldest compatible red cell units available to them, and it’s uncertain whether doing so increases mortality.
To see if the age of red cells impacted mortality, Dr. Cooper and has colleagues at 59 centers in five countries randomized 4,994 critically ill adults to receive the freshest-available or standard oldest-available red cells (N Engl J Med. 2017;377:1858-67).
At 90 days after transfusion, mortality was 24.8% in the group of patients receiving the freshest-available red cells, and 24.1% for the oldest-available group, or an absolute risk difference of just 0.7 percentage points (95% confidence interval, –1.7 to 3.1; P = .57).
“Among the many secondary outcomes tested, we noted a nominal difference in febrile nonhemolytic transfusion reactions that was small, and we are not sure of its clinical significance,” the researchers wrote.
The average duration of red cell storage was 11.8 days versus 22.4 days for the freshest-available and oldest-available groups, respectively.
The TRANSFUSE trial is not the first to suggest that age of red blood cells does not make a difference in mortality after transfusion. There were two earlier trials, ABLE (Age of Blood Evaluation) and INFORM (Informing Fresh versus Old Red Cell Management) that came to similar conclusions. However, the ABLE trial had a small sample size, and INFORM had “limited outcome data” including a low mortality rate “suggesting low illness severity,” the researchers noted.
“The lower in-hospital mortality in the ICU subgroup in the INFORM trial (13.0%) than that observed in our trial at 90 days (24.5%) is consistent with lower illness severity in the INFORM patients,” they wrote.
The study was funded by organizations including the Australian National Health and Medical Research Council. Dr. Cooper reported receiving consulting fees from Eustralis Pharmaceuticals that were paid to Monash University. No other potential conflicts of interest were reported.
Critically ill patients who received transfusions of the freshest-available red cells had a mortality rate similar to that of patients who received standard-issue, oldest-available red cells, according to results from a large randomized trial.
In some earlier studies, transfusion of older red cells was linked to increased mortality for critically ill, surgical, and trauma patients. But the new results provide “strong evidence” that transfusing very fresh red cells rather than older red cells “provides no clinically meaningful benefits” in the critically ill population, reported D. James Cooper, MD, of Monash University, Melbourne, and his colleagues.
“Our results support the current international usual practice of transfusing patients with the oldest red cells available,” the researchers wrote in the report on the trial, known as TRANSFUSE (Standard Issue Transfusion versus Fresher Red-Cell Use in Intensive Care).
Red cells are stored up to 42 days and can undergo biochemical, structural, or metabolic changes during that time that “may cause harm,” the researchers wrote. However, blood banks typically issue the oldest compatible red cell units available to them, and it’s uncertain whether doing so increases mortality.
To see if the age of red cells impacted mortality, Dr. Cooper and has colleagues at 59 centers in five countries randomized 4,994 critically ill adults to receive the freshest-available or standard oldest-available red cells (N Engl J Med. 2017;377:1858-67).
At 90 days after transfusion, mortality was 24.8% in the group of patients receiving the freshest-available red cells, and 24.1% for the oldest-available group, or an absolute risk difference of just 0.7 percentage points (95% confidence interval, –1.7 to 3.1; P = .57).
“Among the many secondary outcomes tested, we noted a nominal difference in febrile nonhemolytic transfusion reactions that was small, and we are not sure of its clinical significance,” the researchers wrote.
The average duration of red cell storage was 11.8 days versus 22.4 days for the freshest-available and oldest-available groups, respectively.
The TRANSFUSE trial is not the first to suggest that age of red blood cells does not make a difference in mortality after transfusion. There were two earlier trials, ABLE (Age of Blood Evaluation) and INFORM (Informing Fresh versus Old Red Cell Management) that came to similar conclusions. However, the ABLE trial had a small sample size, and INFORM had “limited outcome data” including a low mortality rate “suggesting low illness severity,” the researchers noted.
“The lower in-hospital mortality in the ICU subgroup in the INFORM trial (13.0%) than that observed in our trial at 90 days (24.5%) is consistent with lower illness severity in the INFORM patients,” they wrote.
The study was funded by organizations including the Australian National Health and Medical Research Council. Dr. Cooper reported receiving consulting fees from Eustralis Pharmaceuticals that were paid to Monash University. No other potential conflicts of interest were reported.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point:
Major finding: Mortality at 90 days after transfusion was 24.8% in patients receiving the freshest-available red cells and 24.1% in patients receiving standard-issue, oldest-available red cells (P = 0.57).
Data source: An international, randomized, double-blind trial including nearly 5,000 critically ill adults at 59 centers in five countries.
Disclosures: The study was funded by organizations including the Australian National Health and Medical Research Council. Dr. Cooper reported receiving consulting fees from Eustralis Pharmaceuticals that were paid to Monash University. No other potential conflicts of interest were reported.
Breast cancer recurrence risk substantial after endocrine treatment
Women who stop adjuvant endocrine therapy after 5 years are still at substantial risk of distant recurrence over the next 15 years, even if their tumors were small, according to results of a recent meta-analysis of 88 clinical trials.
For women with T1N0 disease, the annual rate of distant recurrence was approximately 1% each year during 5-20 years, resulting in a cumulative risk of distant recurrence of 13%, authors of the meta-analysis reported (N Engl J Med. 2017 Nov. 8. doi: 10.1056/NEJMoa1701830).
Tumor diameter and nodal status was associated with the risk of distant recurrence during the later years and was approximately additive, with the risk increasing from 13% for T1N0 to 41% for T2N4–9 disease, wrote investigator Hongchao Pan, PhD, of the Nuffield Department of Population Health, University of Oxford, England, and his coauthors.
“Recognition of the magnitude of the long-term risks of ER-positive disease can help women and their health care professionals decide whether to extend therapy beyond 5 years and whether to persist if adverse events occur,” the authors wrote in the report.
The meta-analysis by Dr. Pan and his colleagues included 62,923 women with ER-positive breast cancer who were free of disease after 5 years of scheduled endocrine therapy.
They had hoped to identify a subgroup of women with a recurrence risks so small that the risk of additional side effects caused by extending endocrine therapy would outweigh any potential benefits of that additional treatment. However, the finding of measurable risk even in the women with T1N0 disease led them to recommend that extending endocrine therapy at least be considered for all patients.
“An absolute reduction of a few percentage points in the risk of distant metastases over the next 15 years might well be possible even for such low-risk women, with correspondingly greater absolute benefits for women with larger tumors or node-positive disease,” they wrote.
Whether reducing risk translates into improved survival remains to be seen.
As of now, “reliable trial evidence is not yet available” to confirm the clinical benefit of extending endocrine therapy beyond 5 years, the authors noted.
Cancer Research UK and others funded the study. Senior author Daniel F. Hayes, MD, reported grant support from Eli Lilly, Janssen Research & Development, Veridex, Puma, Pfizer, and AstraZeneca, among other disclosures. Full disclosures for all authors were provided on the NEJM website.
“This study reaffirms the potential for recurrences very late after the original diagnosis, an observation made with other datasets as well. This pattern of recurrence is most consistent with hormone-sensitive breast cancer,” William J. Gradishar, MD, said in an interview.

Dr. William J. Gradishar is the Betsy Bramsen Professor of Breast Oncology & professor of medicine at Northwestern University, Chicago.
“This study reaffirms the potential for recurrences very late after the original diagnosis, an observation made with other datasets as well. This pattern of recurrence is most consistent with hormone-sensitive breast cancer,” William J. Gradishar, MD, said in an interview.
Dr. William J. Gradishar is the Betsy Bramsen Professor of Breast Oncology & professor of medicine at Northwestern University, Chicago.
“This study reaffirms the potential for recurrences very late after the original diagnosis, an observation made with other datasets as well. This pattern of recurrence is most consistent with hormone-sensitive breast cancer,” William J. Gradishar, MD, said in an interview.
Dr. William J. Gradishar is the Betsy Bramsen Professor of Breast Oncology & professor of medicine at Northwestern University, Chicago.
Women who stop adjuvant endocrine therapy after 5 years are still at substantial risk of distant recurrence over the next 15 years, even if their tumors were small, according to results of a recent meta-analysis of 88 clinical trials.
For women with T1N0 disease, the annual rate of distant recurrence was approximately 1% each year during 5-20 years, resulting in a cumulative risk of distant recurrence of 13%, authors of the meta-analysis reported (N Engl J Med. 2017 Nov. 8. doi: 10.1056/NEJMoa1701830).
Tumor diameter and nodal status was associated with the risk of distant recurrence during the later years and was approximately additive, with the risk increasing from 13% for T1N0 to 41% for T2N4–9 disease, wrote investigator Hongchao Pan, PhD, of the Nuffield Department of Population Health, University of Oxford, England, and his coauthors.
“Recognition of the magnitude of the long-term risks of ER-positive disease can help women and their health care professionals decide whether to extend therapy beyond 5 years and whether to persist if adverse events occur,” the authors wrote in the report.
The meta-analysis by Dr. Pan and his colleagues included 62,923 women with ER-positive breast cancer who were free of disease after 5 years of scheduled endocrine therapy.
They had hoped to identify a subgroup of women with a recurrence risks so small that the risk of additional side effects caused by extending endocrine therapy would outweigh any potential benefits of that additional treatment. However, the finding of measurable risk even in the women with T1N0 disease led them to recommend that extending endocrine therapy at least be considered for all patients.
“An absolute reduction of a few percentage points in the risk of distant metastases over the next 15 years might well be possible even for such low-risk women, with correspondingly greater absolute benefits for women with larger tumors or node-positive disease,” they wrote.
Whether reducing risk translates into improved survival remains to be seen.
As of now, “reliable trial evidence is not yet available” to confirm the clinical benefit of extending endocrine therapy beyond 5 years, the authors noted.
Cancer Research UK and others funded the study. Senior author Daniel F. Hayes, MD, reported grant support from Eli Lilly, Janssen Research & Development, Veridex, Puma, Pfizer, and AstraZeneca, among other disclosures. Full disclosures for all authors were provided on the NEJM website.
Women who stop adjuvant endocrine therapy after 5 years are still at substantial risk of distant recurrence over the next 15 years, even if their tumors were small, according to results of a recent meta-analysis of 88 clinical trials.
For women with T1N0 disease, the annual rate of distant recurrence was approximately 1% each year during 5-20 years, resulting in a cumulative risk of distant recurrence of 13%, authors of the meta-analysis reported (N Engl J Med. 2017 Nov. 8. doi: 10.1056/NEJMoa1701830).
Tumor diameter and nodal status was associated with the risk of distant recurrence during the later years and was approximately additive, with the risk increasing from 13% for T1N0 to 41% for T2N4–9 disease, wrote investigator Hongchao Pan, PhD, of the Nuffield Department of Population Health, University of Oxford, England, and his coauthors.
“Recognition of the magnitude of the long-term risks of ER-positive disease can help women and their health care professionals decide whether to extend therapy beyond 5 years and whether to persist if adverse events occur,” the authors wrote in the report.
The meta-analysis by Dr. Pan and his colleagues included 62,923 women with ER-positive breast cancer who were free of disease after 5 years of scheduled endocrine therapy.
They had hoped to identify a subgroup of women with a recurrence risks so small that the risk of additional side effects caused by extending endocrine therapy would outweigh any potential benefits of that additional treatment. However, the finding of measurable risk even in the women with T1N0 disease led them to recommend that extending endocrine therapy at least be considered for all patients.
“An absolute reduction of a few percentage points in the risk of distant metastases over the next 15 years might well be possible even for such low-risk women, with correspondingly greater absolute benefits for women with larger tumors or node-positive disease,” they wrote.
Whether reducing risk translates into improved survival remains to be seen.
As of now, “reliable trial evidence is not yet available” to confirm the clinical benefit of extending endocrine therapy beyond 5 years, the authors noted.
Cancer Research UK and others funded the study. Senior author Daniel F. Hayes, MD, reported grant support from Eli Lilly, Janssen Research & Development, Veridex, Puma, Pfizer, and AstraZeneca, among other disclosures. Full disclosures for all authors were provided on the NEJM website.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Among women with early-stage, estrogen-receptor (ER)–positive breast cancer who stop adjuvant endocrine therapy after 5 years, distant recurrences happened at a steady rate over the ensuing 15 years.
Major finding: Distant recurrence risk ranged from 10% to 41%, depending on tumor diameter and nodal status (TN) and tumor grade.
Data source: A meta-analysis of 88 trials including 62,923 women with ER-positive breast cancer who were disease free after 5 years of scheduled endocrine therapy.
Disclosures: The study was funded by Cancer Research UK and others. Senior author Daniel F. Hayes, MD, reported grant support from Eli Lilly, Janssen Research & Development, Veridex, Puma, Pfizer, and AstraZeneca, among other disclosures. Full disclosures for all authors were provided on the NEJM website.
Neoantigen profiling predicts response to immunotherapy
In antitumor immunity and immunotherapy, quality and fitness count.
Specifically, the quality and fitness of neoantigens – tumor-specific mutated peptides on the surface of cancer cells – can influence a patient’s response to immune checkpoint inhibitors, and mathematical models of neoantigen fitness can serve as biomarkers for response to immunotherapy, according to investigators of two separate but related studies published in Nature.
In one study, Marta Łuksza, PhD, from the Simons Center for Systems Biology at the Institute for Advanced Study in Princeton, N.J., and colleagues propose a neoantigen fitness model that can predict tumor response to checkpoint blockade immunotherapy.
Importantly, low-fitness neoantigens identified by our method may be leveraged for developing novel immunotherapies,” they wrote (Nature. 2017 Nov 8. doi: 10.1038/nature24473).
In a related study, Vinod P. Balachandran, MD, from the David M. Rubinstein Center for Pancreatic Cancer Research at Memorial Sloan Kettering Cancer Center in New York and colleagues, including Dr. Łuksza and others, looked at T-cell antigens in long-term survivors of pancreatic cancer and identified specific neoantigens as T-cell targets.
“More broadly, we identify neoantigen quality as a biomarker for immunogenic tumors that may guide the application of immunotherapies,” Dr. Balachandran and colleagues wrote (Nature. 2017 Nov 8. doi: 10.1038/nature24462).
Proof of concept
The studies provide a proof of concept that mathematical modeling of tumor evolution and the interactions of tumors with the immune system may soon provide clinicians with valuable and actionable information about responses to immunotherapy, Benjamin Greenbaum, PhD, senior author on the study by Łuksza et al., and a coauthor on the pancreatic cancer study said in an interview.
“We’re trying to come up with measures that take into account what we think the underlying processes are and what lies behind therapy response, and that should lead to better predictive models associated with response in the future,” said Dr. Greenbaum, of the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai Medical Center, New York.
One of the key findings of the studies is that neoantigen quality – the ability of neoantigens to spark T-cell recognition – seems to be as or more important than neoantigen quantity for influencing immune responses during tumor evolution.
“The general logic behind the idea that mutational burden can be a good predictor of response is that the more mutations you have, the more likely that you have a neoantigen, a peptide generated by a tumor mutation, that elicits productive T-cell recognition. We tried to model that process that might lead to productive T-cell recognition, to assign a kind of number to every neoantigen to provide some estimate of how likely it was to undergo a productive process,” Dr. Greenbaum explained.
Melanoma and lung cancer survivors
In the study by Łuksza et al., the investigators created a mathematical fitness model that can predict how tumors respond to immunotherapy based on how neoantigens interact with the immune system and applied the model to data on three previously reported patient cohorts, including two groups of patients with malignant melanoma treated with a cytotoxic T-lymphocyte associated protein 4 (CTLA4) immune checkpoint such as ipilimumab (Yervoy), and one group of patients with non–small cell lung cancer treated with a programmed death-1 (PD-1) inhibitor (for example, nivolumab [Opdivo]).
They found that their proposed model is more accurate than genomic biomarkers for predicting how a specific tumor may respond to immunotherapy.
“Importantly, low-fitness neoantigens identified by our method may be leveraged for developing novel immunotherapies. By using an immune fitness model to study immunotherapy, we reveal broad similarities between the evolution of tumors and rapidly evolving pathogens,” they wrote.
Pancreatic cancer survivors
Fewer than 7% of patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) survive more than 5 years, despite the best surgical and medical therapy. But a few lucky patients are long-term survivors, and Dr. Balachandran and associates sought to examine what aspects of T-cell immunity contributed to their longevity.
Rather than relying on genomic analysis of tumor samples, however, they used a combination of genetic, immunohistochemical, and transcriptional immunoprofiling, as well as computational biophysics and function to identify T-cell antigens in the long-term survivors.
When they compared surgically resected patients matched by tumor stage, they found that tumors from those with a median overall survival (OS) of 6 years had a 3-fold greater density of CD8-positive T cells and a 12-fold greater density of cytolytic CD8-positive cells, as well as more mature dendritic cells, regulatory T cells, and macrophages, but decreased numbers of CD4-positive T cells, compared with patients with a more typical course of survival (median OS, 0.8 years). There were no differences between long- and short-term survivors in either B cells or major histocompatibility complex (MHC) class I–positive cells.
They then performed whole-exome sequencing on tumor samples to determine the frequency of neoantigens and found a median of 38 predicted neoantigens per tumor.
“Notably, patients with both the highest predicted neoantigen number and either the greatest CD3+, CD8+, or polyclonal T-cell repertoire, but neither alone, exhibited the longest survival,” they wrote.
When they looked for qualities of neoantigens responsible for promoting T-cell activation in the long-term survivors, they found that the tumors from the survivors, compared with others, were enriched in neoantigen qualities that could be described by a mathematical fitness model.
“Our results provide insight into the heterogeneous immunobiology of PDAC, a presumed poorly immunogenic and checkpoint blockade–refractory tumor, demonstrating that neoantigens may be T-cell targets in [long-term survivors]”, they wrote.
The investigators propose that immunity to neoantigens that are generated during the outgrowth of a primary tumor could at least partially explain the lower incidence of relapse and prolonged survival of a small minority of patients with pancreatic cancer.
“Our findings support the development of strategies to harness neoantigen-specific immunity to treat checkpoint blockade–refractory cancers, and the identification of immunogenic hot spots for directed neoantigen targeting,” they concluded.
The studies were supported by grants from Stand Up to Cancer, American Cancer Society, National Science Foundation, Lustgarten Foundation, Janssen Research & Development, the STARR Cancer Consortium, the Pershing Square Sohn Cancer Research Alliance, the National Institutes of Health, the V Foundation, Swim Across America, Ludwig Institute for Cancer Research, the Parker Institute for Cancer Immunotherapy, a National Cancer Institute Career Development Award, and a Memorial Sloan Kettering Cancer Center core grant. Dr. Łuksza and Dr. Greenbaum disclosed consulting for Merck. Dr. Balachandran disclosed research funding from Bristol-Myers Squibb.
In antitumor immunity and immunotherapy, quality and fitness count.
Specifically, the quality and fitness of neoantigens – tumor-specific mutated peptides on the surface of cancer cells – can influence a patient’s response to immune checkpoint inhibitors, and mathematical models of neoantigen fitness can serve as biomarkers for response to immunotherapy, according to investigators of two separate but related studies published in Nature.
In one study, Marta Łuksza, PhD, from the Simons Center for Systems Biology at the Institute for Advanced Study in Princeton, N.J., and colleagues propose a neoantigen fitness model that can predict tumor response to checkpoint blockade immunotherapy.
Importantly, low-fitness neoantigens identified by our method may be leveraged for developing novel immunotherapies,” they wrote (Nature. 2017 Nov 8. doi: 10.1038/nature24473).
In a related study, Vinod P. Balachandran, MD, from the David M. Rubinstein Center for Pancreatic Cancer Research at Memorial Sloan Kettering Cancer Center in New York and colleagues, including Dr. Łuksza and others, looked at T-cell antigens in long-term survivors of pancreatic cancer and identified specific neoantigens as T-cell targets.
“More broadly, we identify neoantigen quality as a biomarker for immunogenic tumors that may guide the application of immunotherapies,” Dr. Balachandran and colleagues wrote (Nature. 2017 Nov 8. doi: 10.1038/nature24462).
Proof of concept
The studies provide a proof of concept that mathematical modeling of tumor evolution and the interactions of tumors with the immune system may soon provide clinicians with valuable and actionable information about responses to immunotherapy, Benjamin Greenbaum, PhD, senior author on the study by Łuksza et al., and a coauthor on the pancreatic cancer study said in an interview.
“We’re trying to come up with measures that take into account what we think the underlying processes are and what lies behind therapy response, and that should lead to better predictive models associated with response in the future,” said Dr. Greenbaum, of the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai Medical Center, New York.
One of the key findings of the studies is that neoantigen quality – the ability of neoantigens to spark T-cell recognition – seems to be as or more important than neoantigen quantity for influencing immune responses during tumor evolution.
“The general logic behind the idea that mutational burden can be a good predictor of response is that the more mutations you have, the more likely that you have a neoantigen, a peptide generated by a tumor mutation, that elicits productive T-cell recognition. We tried to model that process that might lead to productive T-cell recognition, to assign a kind of number to every neoantigen to provide some estimate of how likely it was to undergo a productive process,” Dr. Greenbaum explained.
Melanoma and lung cancer survivors
In the study by Łuksza et al., the investigators created a mathematical fitness model that can predict how tumors respond to immunotherapy based on how neoantigens interact with the immune system and applied the model to data on three previously reported patient cohorts, including two groups of patients with malignant melanoma treated with a cytotoxic T-lymphocyte associated protein 4 (CTLA4) immune checkpoint such as ipilimumab (Yervoy), and one group of patients with non–small cell lung cancer treated with a programmed death-1 (PD-1) inhibitor (for example, nivolumab [Opdivo]).
They found that their proposed model is more accurate than genomic biomarkers for predicting how a specific tumor may respond to immunotherapy.
“Importantly, low-fitness neoantigens identified by our method may be leveraged for developing novel immunotherapies. By using an immune fitness model to study immunotherapy, we reveal broad similarities between the evolution of tumors and rapidly evolving pathogens,” they wrote.
Pancreatic cancer survivors
Fewer than 7% of patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) survive more than 5 years, despite the best surgical and medical therapy. But a few lucky patients are long-term survivors, and Dr. Balachandran and associates sought to examine what aspects of T-cell immunity contributed to their longevity.
Rather than relying on genomic analysis of tumor samples, however, they used a combination of genetic, immunohistochemical, and transcriptional immunoprofiling, as well as computational biophysics and function to identify T-cell antigens in the long-term survivors.
When they compared surgically resected patients matched by tumor stage, they found that tumors from those with a median overall survival (OS) of 6 years had a 3-fold greater density of CD8-positive T cells and a 12-fold greater density of cytolytic CD8-positive cells, as well as more mature dendritic cells, regulatory T cells, and macrophages, but decreased numbers of CD4-positive T cells, compared with patients with a more typical course of survival (median OS, 0.8 years). There were no differences between long- and short-term survivors in either B cells or major histocompatibility complex (MHC) class I–positive cells.
They then performed whole-exome sequencing on tumor samples to determine the frequency of neoantigens and found a median of 38 predicted neoantigens per tumor.
“Notably, patients with both the highest predicted neoantigen number and either the greatest CD3+, CD8+, or polyclonal T-cell repertoire, but neither alone, exhibited the longest survival,” they wrote.
When they looked for qualities of neoantigens responsible for promoting T-cell activation in the long-term survivors, they found that the tumors from the survivors, compared with others, were enriched in neoantigen qualities that could be described by a mathematical fitness model.
“Our results provide insight into the heterogeneous immunobiology of PDAC, a presumed poorly immunogenic and checkpoint blockade–refractory tumor, demonstrating that neoantigens may be T-cell targets in [long-term survivors]”, they wrote.
The investigators propose that immunity to neoantigens that are generated during the outgrowth of a primary tumor could at least partially explain the lower incidence of relapse and prolonged survival of a small minority of patients with pancreatic cancer.
“Our findings support the development of strategies to harness neoantigen-specific immunity to treat checkpoint blockade–refractory cancers, and the identification of immunogenic hot spots for directed neoantigen targeting,” they concluded.
The studies were supported by grants from Stand Up to Cancer, American Cancer Society, National Science Foundation, Lustgarten Foundation, Janssen Research & Development, the STARR Cancer Consortium, the Pershing Square Sohn Cancer Research Alliance, the National Institutes of Health, the V Foundation, Swim Across America, Ludwig Institute for Cancer Research, the Parker Institute for Cancer Immunotherapy, a National Cancer Institute Career Development Award, and a Memorial Sloan Kettering Cancer Center core grant. Dr. Łuksza and Dr. Greenbaum disclosed consulting for Merck. Dr. Balachandran disclosed research funding from Bristol-Myers Squibb.
In antitumor immunity and immunotherapy, quality and fitness count.
Specifically, the quality and fitness of neoantigens – tumor-specific mutated peptides on the surface of cancer cells – can influence a patient’s response to immune checkpoint inhibitors, and mathematical models of neoantigen fitness can serve as biomarkers for response to immunotherapy, according to investigators of two separate but related studies published in Nature.
In one study, Marta Łuksza, PhD, from the Simons Center for Systems Biology at the Institute for Advanced Study in Princeton, N.J., and colleagues propose a neoantigen fitness model that can predict tumor response to checkpoint blockade immunotherapy.
Importantly, low-fitness neoantigens identified by our method may be leveraged for developing novel immunotherapies,” they wrote (Nature. 2017 Nov 8. doi: 10.1038/nature24473).
In a related study, Vinod P. Balachandran, MD, from the David M. Rubinstein Center for Pancreatic Cancer Research at Memorial Sloan Kettering Cancer Center in New York and colleagues, including Dr. Łuksza and others, looked at T-cell antigens in long-term survivors of pancreatic cancer and identified specific neoantigens as T-cell targets.
“More broadly, we identify neoantigen quality as a biomarker for immunogenic tumors that may guide the application of immunotherapies,” Dr. Balachandran and colleagues wrote (Nature. 2017 Nov 8. doi: 10.1038/nature24462).
Proof of concept
The studies provide a proof of concept that mathematical modeling of tumor evolution and the interactions of tumors with the immune system may soon provide clinicians with valuable and actionable information about responses to immunotherapy, Benjamin Greenbaum, PhD, senior author on the study by Łuksza et al., and a coauthor on the pancreatic cancer study said in an interview.
“We’re trying to come up with measures that take into account what we think the underlying processes are and what lies behind therapy response, and that should lead to better predictive models associated with response in the future,” said Dr. Greenbaum, of the Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai Medical Center, New York.
One of the key findings of the studies is that neoantigen quality – the ability of neoantigens to spark T-cell recognition – seems to be as or more important than neoantigen quantity for influencing immune responses during tumor evolution.
“The general logic behind the idea that mutational burden can be a good predictor of response is that the more mutations you have, the more likely that you have a neoantigen, a peptide generated by a tumor mutation, that elicits productive T-cell recognition. We tried to model that process that might lead to productive T-cell recognition, to assign a kind of number to every neoantigen to provide some estimate of how likely it was to undergo a productive process,” Dr. Greenbaum explained.
Melanoma and lung cancer survivors
In the study by Łuksza et al., the investigators created a mathematical fitness model that can predict how tumors respond to immunotherapy based on how neoantigens interact with the immune system and applied the model to data on three previously reported patient cohorts, including two groups of patients with malignant melanoma treated with a cytotoxic T-lymphocyte associated protein 4 (CTLA4) immune checkpoint such as ipilimumab (Yervoy), and one group of patients with non–small cell lung cancer treated with a programmed death-1 (PD-1) inhibitor (for example, nivolumab [Opdivo]).
They found that their proposed model is more accurate than genomic biomarkers for predicting how a specific tumor may respond to immunotherapy.
“Importantly, low-fitness neoantigens identified by our method may be leveraged for developing novel immunotherapies. By using an immune fitness model to study immunotherapy, we reveal broad similarities between the evolution of tumors and rapidly evolving pathogens,” they wrote.
Pancreatic cancer survivors
Fewer than 7% of patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) survive more than 5 years, despite the best surgical and medical therapy. But a few lucky patients are long-term survivors, and Dr. Balachandran and associates sought to examine what aspects of T-cell immunity contributed to their longevity.
Rather than relying on genomic analysis of tumor samples, however, they used a combination of genetic, immunohistochemical, and transcriptional immunoprofiling, as well as computational biophysics and function to identify T-cell antigens in the long-term survivors.
When they compared surgically resected patients matched by tumor stage, they found that tumors from those with a median overall survival (OS) of 6 years had a 3-fold greater density of CD8-positive T cells and a 12-fold greater density of cytolytic CD8-positive cells, as well as more mature dendritic cells, regulatory T cells, and macrophages, but decreased numbers of CD4-positive T cells, compared with patients with a more typical course of survival (median OS, 0.8 years). There were no differences between long- and short-term survivors in either B cells or major histocompatibility complex (MHC) class I–positive cells.
They then performed whole-exome sequencing on tumor samples to determine the frequency of neoantigens and found a median of 38 predicted neoantigens per tumor.
“Notably, patients with both the highest predicted neoantigen number and either the greatest CD3+, CD8+, or polyclonal T-cell repertoire, but neither alone, exhibited the longest survival,” they wrote.
When they looked for qualities of neoantigens responsible for promoting T-cell activation in the long-term survivors, they found that the tumors from the survivors, compared with others, were enriched in neoantigen qualities that could be described by a mathematical fitness model.
“Our results provide insight into the heterogeneous immunobiology of PDAC, a presumed poorly immunogenic and checkpoint blockade–refractory tumor, demonstrating that neoantigens may be T-cell targets in [long-term survivors]”, they wrote.
The investigators propose that immunity to neoantigens that are generated during the outgrowth of a primary tumor could at least partially explain the lower incidence of relapse and prolonged survival of a small minority of patients with pancreatic cancer.
“Our findings support the development of strategies to harness neoantigen-specific immunity to treat checkpoint blockade–refractory cancers, and the identification of immunogenic hot spots for directed neoantigen targeting,” they concluded.
The studies were supported by grants from Stand Up to Cancer, American Cancer Society, National Science Foundation, Lustgarten Foundation, Janssen Research & Development, the STARR Cancer Consortium, the Pershing Square Sohn Cancer Research Alliance, the National Institutes of Health, the V Foundation, Swim Across America, Ludwig Institute for Cancer Research, the Parker Institute for Cancer Immunotherapy, a National Cancer Institute Career Development Award, and a Memorial Sloan Kettering Cancer Center core grant. Dr. Łuksza and Dr. Greenbaum disclosed consulting for Merck. Dr. Balachandran disclosed research funding from Bristol-Myers Squibb.
FROM NATURE
Key clinical point: Proof-of-concept studies show that mathematical modeling of neoantigens can be used to predict tumor responses to immune checkpoint inhibitors.
Major finding: Neoantigen quality may be a better biomarker for guiding immunotherapy than tumor genomic profiling.
Data source: Basic science reports focusing on neoantigens and their potential influence on tumor interactions with the immune system.
Disclosures: The studies were supported by grants from Stand Up to Cancer, American Cancer Society, National Science Foundation, Lustgarten Foundation, Janssen Research & Development, the STARR Cancer Consortium, the Pershing Square Sohn Cancer Research Alliance, the National Institutes of Health, the V Foundation, Swim Across America, Ludwig Institute for Cancer Research, the Parker Institute for Cancer Immunotherapy, a National Cancer Institute Career Development Award, and a Memorial Sloan Kettering Cancer Center core grant. Dr. Łuksza and Dr. Greenbaum disclosed consulting for Merck. Dr. Balachandran disclosed research funding from Bristol-Myers Squibb.