User login
CMS proposes inpatient payment model for CAR T therapies
Physicians may finally have some clarity on payment for inpatient administration of two chimeric antigen receptor–T-cell therapies if a proposed rule from the Centers of Medicare & Medicaid Services becomes final.
The agency is seeking to assign ICD-10-PCS codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019. The agency is also considering the creation of a new Medicare Severity–Diagnosis Related Group (MS-DRG) code for procedures involving the use of chimeric antigen receptor (CAR) T-cell therapy drugs. The proposal was published in May in the Federal Register.
The proposal demonstrates that CMS is listening to physicians’ concerns about CAR T payments and working to provide a more reasonable framework, said Stephanie Farnia, director of health policy and strategic relations for the American Society for Blood and Marrow Transplantation.
“The primary point of significance is that CAR-T care episodes should be assigned to a specific MS-DRG in FY2019, which will give physicians a clearer sense of inpatient reimbursement in advance,” she said in an interview.
Uncertainty about inpatient payment for administration of the two approved CAR-T therapies have been a lingering concern of specialists using, or interested in using, the therapies. In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
However, physicians noted that even if the drugs are first administered in the outpatient setting, inpatient care is likely to occur with CAR T-cell therapies because some patients will need to be admitted in order to be monitored for serious side effects. In such cases, all payments will become part of the inpatient stay per CMS’ 3-day payment window rule.
In outlining the most recent payment proposal, CMS stated that its clinical advisers believe that patients receiving treatment with CAR T-cell therapy would have similar clinical characteristics and comorbidities as patients treated with autologous bone marrow transplant therapy, who are currently assigned to MS-DRG 016 “Autologous Bone Marrow Transplant with CC/MCC”. Therefore, CMS officials said they are suggesting ICD-10-PCS procedure codes XW033C3 and XW043C3 to pre-MDC MS-DRG 016. Additionally, the agency is proposing to revise the title of MS-DRG 016 from “Autologous Bone Marrow Transplant with CC/MCC” to “Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy.”
However, the agency emphasized that it invites public comment on alternative payment approaches for CAR T-cell therapies in the context of the pending, new technology add-on payment applications by the CAR-T drugmakers Novartis Pharmaceuticals and Kite Pharma. If approved in the final rule, the technology add-on payments would provide an additional and separate payment equivalent to up to 50% of the product cost plus the MS-DRG payment received for the episode of care.
Other items in the proposed rule include approximate base payments for hematopoietic stem cell transplantation for fiscal 2019, which starts Oct. 1, 2018. The approximate per unit payment is $5,498, which translates to a total base payment of $64,790 for allogeneic bone marrow transplant (MS-DRG 014) for instance. In an American Society for Blood and Marrow Transplantation blog, Ms. Farnia outlined the approximate base weights and estimated reimbursements calculated from the rule.
CMS did not make any changes to the payment model for allogeneic hematopoietic cell transplantation despite continued requests by physicians for donor acquisition charges. Ms. Farnia encourages doctors to express to CMS the importance of such reimbursement during the comment period.
Public comments on the inpatient payment proposal are due by June 25, 2018, and can be submitted at https://www.regulations.gov. Additionally, CMS will convene a meeting of the Medicare Evidence Development & Coverage Advisory Committee on August 22, 2018, to consider whether to issue a national coverage policy for CAR T-cell therapies that would set consistent parameters for patient access.
Physicians may finally have some clarity on payment for inpatient administration of two chimeric antigen receptor–T-cell therapies if a proposed rule from the Centers of Medicare & Medicaid Services becomes final.
The agency is seeking to assign ICD-10-PCS codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019. The agency is also considering the creation of a new Medicare Severity–Diagnosis Related Group (MS-DRG) code for procedures involving the use of chimeric antigen receptor (CAR) T-cell therapy drugs. The proposal was published in May in the Federal Register.
The proposal demonstrates that CMS is listening to physicians’ concerns about CAR T payments and working to provide a more reasonable framework, said Stephanie Farnia, director of health policy and strategic relations for the American Society for Blood and Marrow Transplantation.
“The primary point of significance is that CAR-T care episodes should be assigned to a specific MS-DRG in FY2019, which will give physicians a clearer sense of inpatient reimbursement in advance,” she said in an interview.
Uncertainty about inpatient payment for administration of the two approved CAR-T therapies have been a lingering concern of specialists using, or interested in using, the therapies. In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
However, physicians noted that even if the drugs are first administered in the outpatient setting, inpatient care is likely to occur with CAR T-cell therapies because some patients will need to be admitted in order to be monitored for serious side effects. In such cases, all payments will become part of the inpatient stay per CMS’ 3-day payment window rule.
In outlining the most recent payment proposal, CMS stated that its clinical advisers believe that patients receiving treatment with CAR T-cell therapy would have similar clinical characteristics and comorbidities as patients treated with autologous bone marrow transplant therapy, who are currently assigned to MS-DRG 016 “Autologous Bone Marrow Transplant with CC/MCC”. Therefore, CMS officials said they are suggesting ICD-10-PCS procedure codes XW033C3 and XW043C3 to pre-MDC MS-DRG 016. Additionally, the agency is proposing to revise the title of MS-DRG 016 from “Autologous Bone Marrow Transplant with CC/MCC” to “Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy.”
However, the agency emphasized that it invites public comment on alternative payment approaches for CAR T-cell therapies in the context of the pending, new technology add-on payment applications by the CAR-T drugmakers Novartis Pharmaceuticals and Kite Pharma. If approved in the final rule, the technology add-on payments would provide an additional and separate payment equivalent to up to 50% of the product cost plus the MS-DRG payment received for the episode of care.
Other items in the proposed rule include approximate base payments for hematopoietic stem cell transplantation for fiscal 2019, which starts Oct. 1, 2018. The approximate per unit payment is $5,498, which translates to a total base payment of $64,790 for allogeneic bone marrow transplant (MS-DRG 014) for instance. In an American Society for Blood and Marrow Transplantation blog, Ms. Farnia outlined the approximate base weights and estimated reimbursements calculated from the rule.
CMS did not make any changes to the payment model for allogeneic hematopoietic cell transplantation despite continued requests by physicians for donor acquisition charges. Ms. Farnia encourages doctors to express to CMS the importance of such reimbursement during the comment period.
Public comments on the inpatient payment proposal are due by June 25, 2018, and can be submitted at https://www.regulations.gov. Additionally, CMS will convene a meeting of the Medicare Evidence Development & Coverage Advisory Committee on August 22, 2018, to consider whether to issue a national coverage policy for CAR T-cell therapies that would set consistent parameters for patient access.
Physicians may finally have some clarity on payment for inpatient administration of two chimeric antigen receptor–T-cell therapies if a proposed rule from the Centers of Medicare & Medicaid Services becomes final.
The agency is seeking to assign ICD-10-PCS codes XW033C3 and XW043C3 to the use of axicabtagene ciloleucel (Yescarta) and tisagenlecleucel (Kymriah) in the inpatient setting for fiscal year 2019. The agency is also considering the creation of a new Medicare Severity–Diagnosis Related Group (MS-DRG) code for procedures involving the use of chimeric antigen receptor (CAR) T-cell therapy drugs. The proposal was published in May in the Federal Register.
The proposal demonstrates that CMS is listening to physicians’ concerns about CAR T payments and working to provide a more reasonable framework, said Stephanie Farnia, director of health policy and strategic relations for the American Society for Blood and Marrow Transplantation.
“The primary point of significance is that CAR-T care episodes should be assigned to a specific MS-DRG in FY2019, which will give physicians a clearer sense of inpatient reimbursement in advance,” she said in an interview.
Uncertainty about inpatient payment for administration of the two approved CAR-T therapies have been a lingering concern of specialists using, or interested in using, the therapies. In April 2018, CMS announced payment rates for outpatient administration of the two drugs, settling on $395,380 for axicabtagene ciloleucel and $500,839 for tisagenlecleucel. The two medications have list prices of $373,000 and $475,000, respectively.
However, physicians noted that even if the drugs are first administered in the outpatient setting, inpatient care is likely to occur with CAR T-cell therapies because some patients will need to be admitted in order to be monitored for serious side effects. In such cases, all payments will become part of the inpatient stay per CMS’ 3-day payment window rule.
In outlining the most recent payment proposal, CMS stated that its clinical advisers believe that patients receiving treatment with CAR T-cell therapy would have similar clinical characteristics and comorbidities as patients treated with autologous bone marrow transplant therapy, who are currently assigned to MS-DRG 016 “Autologous Bone Marrow Transplant with CC/MCC”. Therefore, CMS officials said they are suggesting ICD-10-PCS procedure codes XW033C3 and XW043C3 to pre-MDC MS-DRG 016. Additionally, the agency is proposing to revise the title of MS-DRG 016 from “Autologous Bone Marrow Transplant with CC/MCC” to “Autologous Bone Marrow Transplant with CC/MCC or T-cell Immunotherapy.”
However, the agency emphasized that it invites public comment on alternative payment approaches for CAR T-cell therapies in the context of the pending, new technology add-on payment applications by the CAR-T drugmakers Novartis Pharmaceuticals and Kite Pharma. If approved in the final rule, the technology add-on payments would provide an additional and separate payment equivalent to up to 50% of the product cost plus the MS-DRG payment received for the episode of care.
Other items in the proposed rule include approximate base payments for hematopoietic stem cell transplantation for fiscal 2019, which starts Oct. 1, 2018. The approximate per unit payment is $5,498, which translates to a total base payment of $64,790 for allogeneic bone marrow transplant (MS-DRG 014) for instance. In an American Society for Blood and Marrow Transplantation blog, Ms. Farnia outlined the approximate base weights and estimated reimbursements calculated from the rule.
CMS did not make any changes to the payment model for allogeneic hematopoietic cell transplantation despite continued requests by physicians for donor acquisition charges. Ms. Farnia encourages doctors to express to CMS the importance of such reimbursement during the comment period.
Public comments on the inpatient payment proposal are due by June 25, 2018, and can be submitted at https://www.regulations.gov. Additionally, CMS will convene a meeting of the Medicare Evidence Development & Coverage Advisory Committee on August 22, 2018, to consider whether to issue a national coverage policy for CAR T-cell therapies that would set consistent parameters for patient access.
Secondary Syphilis: An Atypical Presentation Complicated by a False Negative Rapid Plasma Reagin Test
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
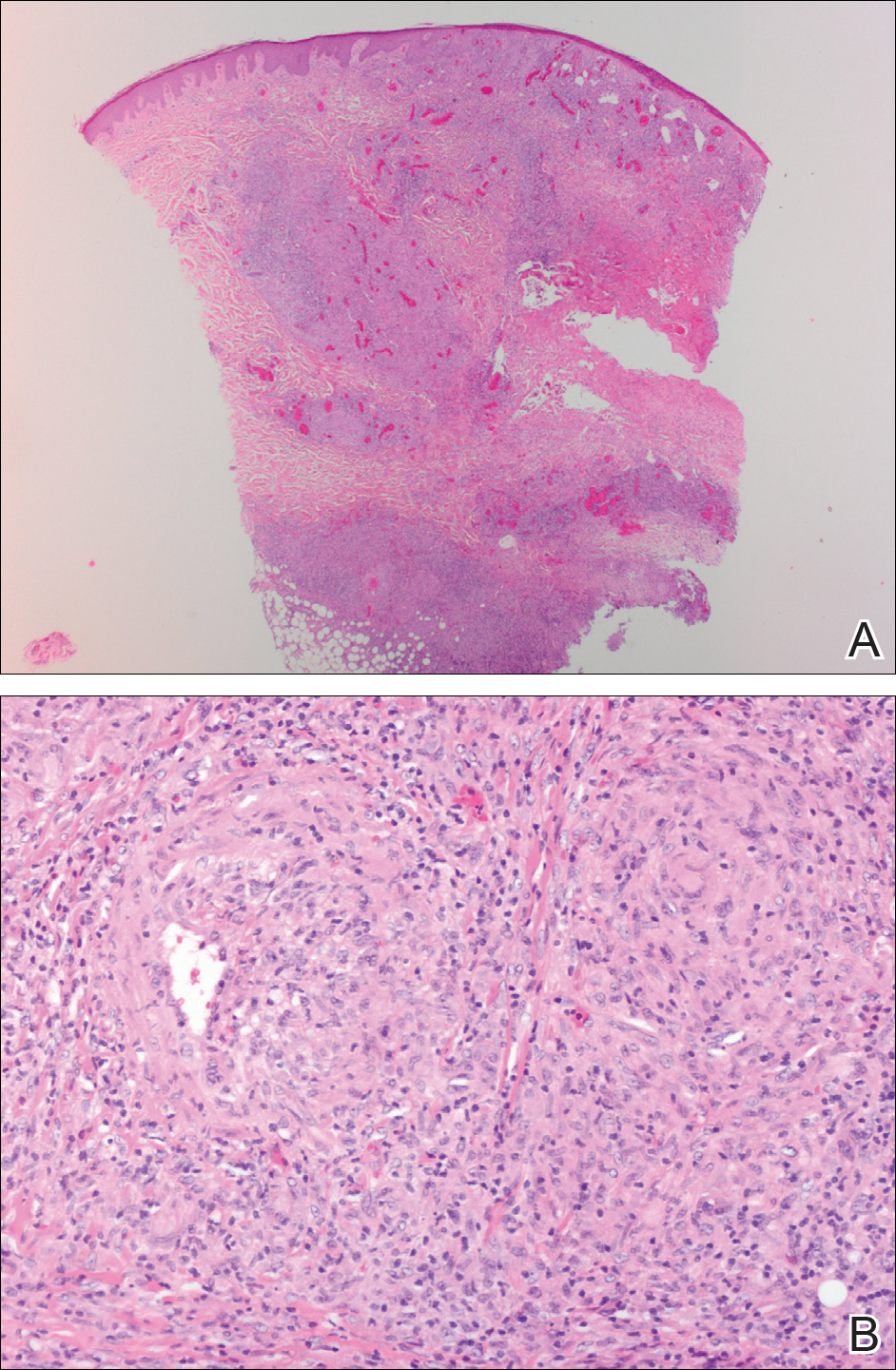
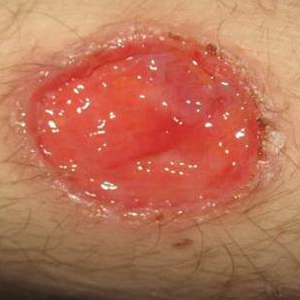
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.

Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.
Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.
Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
Practice Points
- Fluorescent treponemal antibody absorption testing more accurately detects syphilis than rapid plasma reagin (RPR).
- Rapid plasma reagin testing is more useful in monitoring serum response once treatment has been initiated.
- If only RPR is being performed at your institution, ensure the laboratory is performing serial dilutions to negate the prozone phenomenon.
Noninvasive Neurostimulator Shows Efficacy in Episodic Migraine
LOS ANGELES—Noninvasive vagus nerve stimulation (nVNS) is a rapidly effective, well-tolerated, and practical option for the acute treatment of episodic migraine, according to the results of PRESTO, a multicenter, randomized, sham-controlled, double-blind trial presented at the 70th Annual Meeting of the American Academy of Neurology. “There are multiple options for nVNS in the acute treatment of migraine,” said Cristina Tassorelli, MD, PhD, Professor of Neurology at the University of Pavia and Director of the Headache Science Centre at the Casimiro Mondino National Neurological Institute of Pavia in Italy. “It can be used alone because it is effective, it can be used in combination with drugs because we do not expect any significant interactions, and it is also indicated in patients who have developed medication overuse.”

Neuromodulation for Migraine
The aim of the PRESTO trial was to evaluate the efficacy, safety, and tolerability of gammaCore, an nVNS device, for the acute treatment of migraine. The handheld gammaCore device is already FDA cleared for the acute treatment of pain associated with episodic cluster headache and migraine headache in adults. PRESTO was the trial that supported the FDA clearance of gammaCore for migraine.
Following an observational period of four weeks, patients enrolled in PRESTO were randomized to nVNS or sham stimulation for four weeks or until five attacks were treated. Following this period, patients entered the open-label period for another four weeks or five additional attacks.
Low-intensity current, which induced a tingling sensation on the skin that was similar to the sensation of the active stimulation, was used for the sham stimulation. Patients were instructed to treat their attack early with two two-minute stimulations, one for each side of the neck. At 15 minutes, patients assessed the intensity of their pain. If pain was still present, another set of two stimulations was self-administered. A second assessment of pain occurred at 120 minutes, with the possibility of administering another stimulation. Rescue medication use before 120 minutes was considered to indicate treatment failure.
Patients with episodic migraine with or without aura were recruited from 10 Italian tertiary headache centers. A total of 285 patients were enrolled, and 248 randomized: 122 to the active arm and 126 to the sham arm. A total of 117 patients completed the double-blind period in the active arm, and 123 patients in the sham arm. “We had a few discontinuations in both arms, one of which was due to the device,” Dr. Tassorelli and colleagues noted. “Demographic and baseline characteristics show typical pictures of patients with episodic migraine,” Dr. Tassorelli said. Study subjects were mostly young, mostly female, and mostly experiencing migraine without aura. Subjects had a mean of five attacks of migraine per month, a mean of six days of headache per month, and a mean monthly intake of five acute migraine medications. One-third of the patients were on stable prophylactic medications. About 40% of the patients were having moderate pain when they treated their first attack. One-third of them were experiencing mild pain, while 23% were experiencing severe pain in the active group and 15% were having severe pain in the sham group.
Stimulation Reduced Pain
The primary end point of the study was pain freedom at 120 minutes for the first attack. The investigators found a significant difference between the two arms at 30 minutes that became more evident at 60 minutes. At 120 minutes, there was a difference, “but we lost the statistical significance,” Dr. Tassorelli reported. “However, a more refined, post hoc repeated-measure analysis showed that the group treated with the active stimulation had significantly more pain relief than the sham stimulation over the 120-minute period.”
One of the secondary outcome measures was headache
The rate of participants who had a therapeutic response for 50% of attacks or more at 120 minutes speaks to the consistency of the response. Almost half of the patients responded to nVNS at 120 minutes for 50% or more of all treated attacks—47.6% achieved mild or no pain at 120 minutes in the treatment group versus 32.3% in the sham group; 32.4% achieved pain freedom at 120 minutes in the treatment group versus 18.2% in the sham group.
“We did not have many adverse events in this study,” Dr. Tassorelli said. Adverse effects of nVNS, mainly application site discomfort, were infrequent, mild, and transitory. No serious adverse events were recorded, and none of the patients in the active treatment group discontinued treatment due to adverse events.
In summary, nVNS “proved superior to sham for pain freedom at 30 minutes and at 60 minutes, but not at 120 minutes, which was our primary end point,” said Dr. Tassorelli and colleagues. “However, repeated-measures analysis validated the primary end point, indicating the superiority of active stimulation over sham through 120 minutes. This study provides class one evidence for the efficacy of nVNS in the acute treatment of episodic migraine.”
This study was sponsored by electroCore.
—Glenn S. Williams
LOS ANGELES—Noninvasive vagus nerve stimulation (nVNS) is a rapidly effective, well-tolerated, and practical option for the acute treatment of episodic migraine, according to the results of PRESTO, a multicenter, randomized, sham-controlled, double-blind trial presented at the 70th Annual Meeting of the American Academy of Neurology. “There are multiple options for nVNS in the acute treatment of migraine,” said Cristina Tassorelli, MD, PhD, Professor of Neurology at the University of Pavia and Director of the Headache Science Centre at the Casimiro Mondino National Neurological Institute of Pavia in Italy. “It can be used alone because it is effective, it can be used in combination with drugs because we do not expect any significant interactions, and it is also indicated in patients who have developed medication overuse.”
Neuromodulation for Migraine
The aim of the PRESTO trial was to evaluate the efficacy, safety, and tolerability of gammaCore, an nVNS device, for the acute treatment of migraine. The handheld gammaCore device is already FDA cleared for the acute treatment of pain associated with episodic cluster headache and migraine headache in adults. PRESTO was the trial that supported the FDA clearance of gammaCore for migraine.
Following an observational period of four weeks, patients enrolled in PRESTO were randomized to nVNS or sham stimulation for four weeks or until five attacks were treated. Following this period, patients entered the open-label period for another four weeks or five additional attacks.
Low-intensity current, which induced a tingling sensation on the skin that was similar to the sensation of the active stimulation, was used for the sham stimulation. Patients were instructed to treat their attack early with two two-minute stimulations, one for each side of the neck. At 15 minutes, patients assessed the intensity of their pain. If pain was still present, another set of two stimulations was self-administered. A second assessment of pain occurred at 120 minutes, with the possibility of administering another stimulation. Rescue medication use before 120 minutes was considered to indicate treatment failure.
Patients with episodic migraine with or without aura were recruited from 10 Italian tertiary headache centers. A total of 285 patients were enrolled, and 248 randomized: 122 to the active arm and 126 to the sham arm. A total of 117 patients completed the double-blind period in the active arm, and 123 patients in the sham arm. “We had a few discontinuations in both arms, one of which was due to the device,” Dr. Tassorelli and colleagues noted. “Demographic and baseline characteristics show typical pictures of patients with episodic migraine,” Dr. Tassorelli said. Study subjects were mostly young, mostly female, and mostly experiencing migraine without aura. Subjects had a mean of five attacks of migraine per month, a mean of six days of headache per month, and a mean monthly intake of five acute migraine medications. One-third of the patients were on stable prophylactic medications. About 40% of the patients were having moderate pain when they treated their first attack. One-third of them were experiencing mild pain, while 23% were experiencing severe pain in the active group and 15% were having severe pain in the sham group.
Stimulation Reduced Pain
The primary end point of the study was pain freedom at 120 minutes for the first attack. The investigators found a significant difference between the two arms at 30 minutes that became more evident at 60 minutes. At 120 minutes, there was a difference, “but we lost the statistical significance,” Dr. Tassorelli reported. “However, a more refined, post hoc repeated-measure analysis showed that the group treated with the active stimulation had significantly more pain relief than the sham stimulation over the 120-minute period.”
One of the secondary outcome measures was headache
The rate of participants who had a therapeutic response for 50% of attacks or more at 120 minutes speaks to the consistency of the response. Almost half of the patients responded to nVNS at 120 minutes for 50% or more of all treated attacks—47.6% achieved mild or no pain at 120 minutes in the treatment group versus 32.3% in the sham group; 32.4% achieved pain freedom at 120 minutes in the treatment group versus 18.2% in the sham group.
“We did not have many adverse events in this study,” Dr. Tassorelli said. Adverse effects of nVNS, mainly application site discomfort, were infrequent, mild, and transitory. No serious adverse events were recorded, and none of the patients in the active treatment group discontinued treatment due to adverse events.
In summary, nVNS “proved superior to sham for pain freedom at 30 minutes and at 60 minutes, but not at 120 minutes, which was our primary end point,” said Dr. Tassorelli and colleagues. “However, repeated-measures analysis validated the primary end point, indicating the superiority of active stimulation over sham through 120 minutes. This study provides class one evidence for the efficacy of nVNS in the acute treatment of episodic migraine.”
This study was sponsored by electroCore.
—Glenn S. Williams
LOS ANGELES—Noninvasive vagus nerve stimulation (nVNS) is a rapidly effective, well-tolerated, and practical option for the acute treatment of episodic migraine, according to the results of PRESTO, a multicenter, randomized, sham-controlled, double-blind trial presented at the 70th Annual Meeting of the American Academy of Neurology. “There are multiple options for nVNS in the acute treatment of migraine,” said Cristina Tassorelli, MD, PhD, Professor of Neurology at the University of Pavia and Director of the Headache Science Centre at the Casimiro Mondino National Neurological Institute of Pavia in Italy. “It can be used alone because it is effective, it can be used in combination with drugs because we do not expect any significant interactions, and it is also indicated in patients who have developed medication overuse.”
Neuromodulation for Migraine
The aim of the PRESTO trial was to evaluate the efficacy, safety, and tolerability of gammaCore, an nVNS device, for the acute treatment of migraine. The handheld gammaCore device is already FDA cleared for the acute treatment of pain associated with episodic cluster headache and migraine headache in adults. PRESTO was the trial that supported the FDA clearance of gammaCore for migraine.
Following an observational period of four weeks, patients enrolled in PRESTO were randomized to nVNS or sham stimulation for four weeks or until five attacks were treated. Following this period, patients entered the open-label period for another four weeks or five additional attacks.
Low-intensity current, which induced a tingling sensation on the skin that was similar to the sensation of the active stimulation, was used for the sham stimulation. Patients were instructed to treat their attack early with two two-minute stimulations, one for each side of the neck. At 15 minutes, patients assessed the intensity of their pain. If pain was still present, another set of two stimulations was self-administered. A second assessment of pain occurred at 120 minutes, with the possibility of administering another stimulation. Rescue medication use before 120 minutes was considered to indicate treatment failure.
Patients with episodic migraine with or without aura were recruited from 10 Italian tertiary headache centers. A total of 285 patients were enrolled, and 248 randomized: 122 to the active arm and 126 to the sham arm. A total of 117 patients completed the double-blind period in the active arm, and 123 patients in the sham arm. “We had a few discontinuations in both arms, one of which was due to the device,” Dr. Tassorelli and colleagues noted. “Demographic and baseline characteristics show typical pictures of patients with episodic migraine,” Dr. Tassorelli said. Study subjects were mostly young, mostly female, and mostly experiencing migraine without aura. Subjects had a mean of five attacks of migraine per month, a mean of six days of headache per month, and a mean monthly intake of five acute migraine medications. One-third of the patients were on stable prophylactic medications. About 40% of the patients were having moderate pain when they treated their first attack. One-third of them were experiencing mild pain, while 23% were experiencing severe pain in the active group and 15% were having severe pain in the sham group.
Stimulation Reduced Pain
The primary end point of the study was pain freedom at 120 minutes for the first attack. The investigators found a significant difference between the two arms at 30 minutes that became more evident at 60 minutes. At 120 minutes, there was a difference, “but we lost the statistical significance,” Dr. Tassorelli reported. “However, a more refined, post hoc repeated-measure analysis showed that the group treated with the active stimulation had significantly more pain relief than the sham stimulation over the 120-minute period.”
One of the secondary outcome measures was headache
The rate of participants who had a therapeutic response for 50% of attacks or more at 120 minutes speaks to the consistency of the response. Almost half of the patients responded to nVNS at 120 minutes for 50% or more of all treated attacks—47.6% achieved mild or no pain at 120 minutes in the treatment group versus 32.3% in the sham group; 32.4% achieved pain freedom at 120 minutes in the treatment group versus 18.2% in the sham group.
“We did not have many adverse events in this study,” Dr. Tassorelli said. Adverse effects of nVNS, mainly application site discomfort, were infrequent, mild, and transitory. No serious adverse events were recorded, and none of the patients in the active treatment group discontinued treatment due to adverse events.
In summary, nVNS “proved superior to sham for pain freedom at 30 minutes and at 60 minutes, but not at 120 minutes, which was our primary end point,” said Dr. Tassorelli and colleagues. “However, repeated-measures analysis validated the primary end point, indicating the superiority of active stimulation over sham through 120 minutes. This study provides class one evidence for the efficacy of nVNS in the acute treatment of episodic migraine.”
This study was sponsored by electroCore.
—Glenn S. Williams
Blood Pressure Change Influences Outcome of Endovascular Therapy
LOS ANGELES—Decreases in blood pressure during endovascular therapy are common and associated with worse functional outcomes, according to research presented at the 70th Annual Meeting of the American Academy of Neurology.
Patients with acute ischemic stroke have impaired cerebral autoregulation, which raises the need for blood pressure management to avoid secondary neurologic injury. Neurologists have not reached a consensus on the optimal management of blood pressure during endovascular therapy, however.
Anson Wang, a postgraduate research associate at the Yale School of Medicine in New Haven, Connecticut, and colleagues prospectively enrolled into their study patients with acute ischemic stroke due to large-vessel occlusion who were scheduled to undergo endovascular therapy between 2014 and 2018 at Yale-New Haven Hospital. Nils Petersen, MD, MSc, Assistant Professor of Neurology at Yale School of Medicine, led the study. The researchers monitored admission mean arterial pressure (MAP) and intraprocedural MAP using a noninvasive blood pressure cuff or an intra-arterial catheter. They calculated change in MAP as the difference between admission MAP and lowest MAP during endovascular therapy. Sustained hypotension was measured as the area between admission MAP and continuous measurements of intraprocedural MAP (aMAP).

Mr. Wang and colleagues examined associations between blood pressure and functional outcome at discharge and 90 days, as measured using the modified Rankin Scale (mRS), using ordinal logistic regression. They defined an unfavorable outcome as an mRS score of 3 or more.
Most Patients Had Reductions in Blood Pressure
The investigators included 115 patients in the study. Participants’ mean age was 72, and mean NIH Stroke Scale (NIHSS) score was 18. Ninety-day outcomes were available for 78 patients. Admission MAP was 107 mm Hg. Approximately 89% of patients had MAP reductions during endovascular therapy (mean, 26 ± 21 mm Hg). Median reduction in MAP among patients with favorable outcomes was 18 mm Hg, compared with 34 mm Hg among patients with unfavorable outcomes.
After Mr. Wang and colleagues adjusted the data for age, gender, and admission NIHSS, change in MAP was independently associated with higher mRS scores at discharge and at 90 days. Every 10-mm Hg reduction in MAP from admission during endovascular therapy was associated with a 26% increase in the likelihood of an unfavorable functional outcome at discharge and a 32% increase in the likelihood of an unfavorable functional outcome at 90 days. The association between aMAP and outcome was highly significant at discharge and 90 days.
Blood Pressure Change May Affect Infarct Volume
To investigate how decreases in blood pressure could cause worse functional outcomes, Mr. Wang and colleagues measured MRI scans of 57 patients with a Thrombolysis in Cerebral Infarction (TICI) scale score of 2b or 3. The mean time between scans was 28 hours. They observed an association between larger final infarct volumes and higher drops in blood pressure. The median infarct volume in patients with a good outcome was approximately 12 cm3.
The limitations of the study include the retrospective data analysis and the lack of continuous blood pressure measurements during the period between arrival at the emergency department and presentation for surgery. Nevertheless, “these results underline the importance of blood pressure management during endovascular therapy and highlight the need for further investigation of active blood pressure management strategies to optimize clinical outcomes,” said Mr. Wang.
—Erik Greb
LOS ANGELES—Decreases in blood pressure during endovascular therapy are common and associated with worse functional outcomes, according to research presented at the 70th Annual Meeting of the American Academy of Neurology.
Patients with acute ischemic stroke have impaired cerebral autoregulation, which raises the need for blood pressure management to avoid secondary neurologic injury. Neurologists have not reached a consensus on the optimal management of blood pressure during endovascular therapy, however.
Anson Wang, a postgraduate research associate at the Yale School of Medicine in New Haven, Connecticut, and colleagues prospectively enrolled into their study patients with acute ischemic stroke due to large-vessel occlusion who were scheduled to undergo endovascular therapy between 2014 and 2018 at Yale-New Haven Hospital. Nils Petersen, MD, MSc, Assistant Professor of Neurology at Yale School of Medicine, led the study. The researchers monitored admission mean arterial pressure (MAP) and intraprocedural MAP using a noninvasive blood pressure cuff or an intra-arterial catheter. They calculated change in MAP as the difference between admission MAP and lowest MAP during endovascular therapy. Sustained hypotension was measured as the area between admission MAP and continuous measurements of intraprocedural MAP (aMAP).
Mr. Wang and colleagues examined associations between blood pressure and functional outcome at discharge and 90 days, as measured using the modified Rankin Scale (mRS), using ordinal logistic regression. They defined an unfavorable outcome as an mRS score of 3 or more.
Most Patients Had Reductions in Blood Pressure
The investigators included 115 patients in the study. Participants’ mean age was 72, and mean NIH Stroke Scale (NIHSS) score was 18. Ninety-day outcomes were available for 78 patients. Admission MAP was 107 mm Hg. Approximately 89% of patients had MAP reductions during endovascular therapy (mean, 26 ± 21 mm Hg). Median reduction in MAP among patients with favorable outcomes was 18 mm Hg, compared with 34 mm Hg among patients with unfavorable outcomes.
After Mr. Wang and colleagues adjusted the data for age, gender, and admission NIHSS, change in MAP was independently associated with higher mRS scores at discharge and at 90 days. Every 10-mm Hg reduction in MAP from admission during endovascular therapy was associated with a 26% increase in the likelihood of an unfavorable functional outcome at discharge and a 32% increase in the likelihood of an unfavorable functional outcome at 90 days. The association between aMAP and outcome was highly significant at discharge and 90 days.
Blood Pressure Change May Affect Infarct Volume
To investigate how decreases in blood pressure could cause worse functional outcomes, Mr. Wang and colleagues measured MRI scans of 57 patients with a Thrombolysis in Cerebral Infarction (TICI) scale score of 2b or 3. The mean time between scans was 28 hours. They observed an association between larger final infarct volumes and higher drops in blood pressure. The median infarct volume in patients with a good outcome was approximately 12 cm3.
The limitations of the study include the retrospective data analysis and the lack of continuous blood pressure measurements during the period between arrival at the emergency department and presentation for surgery. Nevertheless, “these results underline the importance of blood pressure management during endovascular therapy and highlight the need for further investigation of active blood pressure management strategies to optimize clinical outcomes,” said Mr. Wang.
—Erik Greb
LOS ANGELES—Decreases in blood pressure during endovascular therapy are common and associated with worse functional outcomes, according to research presented at the 70th Annual Meeting of the American Academy of Neurology.
Patients with acute ischemic stroke have impaired cerebral autoregulation, which raises the need for blood pressure management to avoid secondary neurologic injury. Neurologists have not reached a consensus on the optimal management of blood pressure during endovascular therapy, however.
Anson Wang, a postgraduate research associate at the Yale School of Medicine in New Haven, Connecticut, and colleagues prospectively enrolled into their study patients with acute ischemic stroke due to large-vessel occlusion who were scheduled to undergo endovascular therapy between 2014 and 2018 at Yale-New Haven Hospital. Nils Petersen, MD, MSc, Assistant Professor of Neurology at Yale School of Medicine, led the study. The researchers monitored admission mean arterial pressure (MAP) and intraprocedural MAP using a noninvasive blood pressure cuff or an intra-arterial catheter. They calculated change in MAP as the difference between admission MAP and lowest MAP during endovascular therapy. Sustained hypotension was measured as the area between admission MAP and continuous measurements of intraprocedural MAP (aMAP).
Mr. Wang and colleagues examined associations between blood pressure and functional outcome at discharge and 90 days, as measured using the modified Rankin Scale (mRS), using ordinal logistic regression. They defined an unfavorable outcome as an mRS score of 3 or more.
Most Patients Had Reductions in Blood Pressure
The investigators included 115 patients in the study. Participants’ mean age was 72, and mean NIH Stroke Scale (NIHSS) score was 18. Ninety-day outcomes were available for 78 patients. Admission MAP was 107 mm Hg. Approximately 89% of patients had MAP reductions during endovascular therapy (mean, 26 ± 21 mm Hg). Median reduction in MAP among patients with favorable outcomes was 18 mm Hg, compared with 34 mm Hg among patients with unfavorable outcomes.
After Mr. Wang and colleagues adjusted the data for age, gender, and admission NIHSS, change in MAP was independently associated with higher mRS scores at discharge and at 90 days. Every 10-mm Hg reduction in MAP from admission during endovascular therapy was associated with a 26% increase in the likelihood of an unfavorable functional outcome at discharge and a 32% increase in the likelihood of an unfavorable functional outcome at 90 days. The association between aMAP and outcome was highly significant at discharge and 90 days.
Blood Pressure Change May Affect Infarct Volume
To investigate how decreases in blood pressure could cause worse functional outcomes, Mr. Wang and colleagues measured MRI scans of 57 patients with a Thrombolysis in Cerebral Infarction (TICI) scale score of 2b or 3. The mean time between scans was 28 hours. They observed an association between larger final infarct volumes and higher drops in blood pressure. The median infarct volume in patients with a good outcome was approximately 12 cm3.
The limitations of the study include the retrospective data analysis and the lack of continuous blood pressure measurements during the period between arrival at the emergency department and presentation for surgery. Nevertheless, “these results underline the importance of blood pressure management during endovascular therapy and highlight the need for further investigation of active blood pressure management strategies to optimize clinical outcomes,” said Mr. Wang.
—Erik Greb
Rehabilitation in MS is hot topic at CMSC 2018
This year’s annual meeting of the Consortium of Multiple Sclerosis Centers in Nashville, Tenn., will feature an intense focus on rehabilitation in MS. The topic is often a priority for patients but not necessarily at top of mind for health care providers, said rehabilitation therapist Patty Bobryk, secretary of the CMSC.
“This year, some of the Rehab Track topics include addressing respiratory issues in MS, exploring the impact of visual impairments on rehab, and recommending the proper orthotics, as well as discussing treatments that focus on mind-body understanding,” said Ms. Bobryk of MS Comprehensive Care Center of Central Florida, Orlando. She serves as cochair of the International Organization of MS Rehabilitation Therapists.

Rehabilitation in MS is a controversial topic. Two years ago, the CMSC endorsed a statement criticizing a 2015 report by the American Academy of Neurology that found limited evidence supporting MS rehabilitation (Neurology. 2015 Nov 24;85[21]:1896-903). The statement, also supported by the International Organization of MS Nurses and International Organization of MS Rehabilitation Therapists, declared that the AAN report “presents an incomplete review of the evidence published” (Neurol Clin Pract. 2016 Dec;6[6]:475-9).
However, the statement acknowledged that “larger studies with better research methodologies and higher-quality evidence are needed in rehabilitation.”
More recently, a 2017 systematic review of systematic reviews of rehabilitation in MS found that “strong” evidence only exists “for physical therapy for improved activity and participation, and for exercise-based educational programs for the reduction of patient-reported fatigue.” The review of reviews also found there’s “ ‘moderate’ evidence for multidisciplinary rehabilitation for longer-term gains at the levels of activity (disability) and participation, for cognitive-behavior therapy for the treatment of depression, and for information-provision interventions for improved patient knowledge” (Arch Phys Med Rehabil. 2017 Feb;98[2]:353-67).
In an interview, Ms. Bobryk said the MS rehabilitation community is evolving toward “a better understanding of neuroplasticity and the impact that rehab can have on maintaining and improving functional skills.”
In addition, she said, “more emphasis is being placed on early intervention and preventing the secondary effects of the disease versus providing only compensatory strategies.”
Highlights of this year’s rehab sessions at the CMSC conference include a 3-hour course on imbalance in MS that will “provide an intensive review of the anatomy of the balance system and educate the audience on evaluation and treatment interventions specific to retraining the balance system,” Ms. Bobryk said.
Another highlight: A half-day course that will “provide the rationale for including rehabilitation in a comprehensive model of care for people with severe MS,” she said. “Unique interventions such as the use of functional electrical stimulation cycling and body weight-supported treadmill training will be discussed.”
In the big picture, “clinicians who are working in the field of MS realize the benefits of rehab, but more education is needed to spread the word to the broader scope of health care providers,” Ms. Bobryk said. “Patients are often the ones that bring up rehab to their physicians to initiate a referral.”
This year’s annual meeting of the Consortium of Multiple Sclerosis Centers in Nashville, Tenn., will feature an intense focus on rehabilitation in MS. The topic is often a priority for patients but not necessarily at top of mind for health care providers, said rehabilitation therapist Patty Bobryk, secretary of the CMSC.
“This year, some of the Rehab Track topics include addressing respiratory issues in MS, exploring the impact of visual impairments on rehab, and recommending the proper orthotics, as well as discussing treatments that focus on mind-body understanding,” said Ms. Bobryk of MS Comprehensive Care Center of Central Florida, Orlando. She serves as cochair of the International Organization of MS Rehabilitation Therapists.
Rehabilitation in MS is a controversial topic. Two years ago, the CMSC endorsed a statement criticizing a 2015 report by the American Academy of Neurology that found limited evidence supporting MS rehabilitation (Neurology. 2015 Nov 24;85[21]:1896-903). The statement, also supported by the International Organization of MS Nurses and International Organization of MS Rehabilitation Therapists, declared that the AAN report “presents an incomplete review of the evidence published” (Neurol Clin Pract. 2016 Dec;6[6]:475-9).
However, the statement acknowledged that “larger studies with better research methodologies and higher-quality evidence are needed in rehabilitation.”
More recently, a 2017 systematic review of systematic reviews of rehabilitation in MS found that “strong” evidence only exists “for physical therapy for improved activity and participation, and for exercise-based educational programs for the reduction of patient-reported fatigue.” The review of reviews also found there’s “ ‘moderate’ evidence for multidisciplinary rehabilitation for longer-term gains at the levels of activity (disability) and participation, for cognitive-behavior therapy for the treatment of depression, and for information-provision interventions for improved patient knowledge” (Arch Phys Med Rehabil. 2017 Feb;98[2]:353-67).
In an interview, Ms. Bobryk said the MS rehabilitation community is evolving toward “a better understanding of neuroplasticity and the impact that rehab can have on maintaining and improving functional skills.”
In addition, she said, “more emphasis is being placed on early intervention and preventing the secondary effects of the disease versus providing only compensatory strategies.”
Highlights of this year’s rehab sessions at the CMSC conference include a 3-hour course on imbalance in MS that will “provide an intensive review of the anatomy of the balance system and educate the audience on evaluation and treatment interventions specific to retraining the balance system,” Ms. Bobryk said.
Another highlight: A half-day course that will “provide the rationale for including rehabilitation in a comprehensive model of care for people with severe MS,” she said. “Unique interventions such as the use of functional electrical stimulation cycling and body weight-supported treadmill training will be discussed.”
In the big picture, “clinicians who are working in the field of MS realize the benefits of rehab, but more education is needed to spread the word to the broader scope of health care providers,” Ms. Bobryk said. “Patients are often the ones that bring up rehab to their physicians to initiate a referral.”
This year’s annual meeting of the Consortium of Multiple Sclerosis Centers in Nashville, Tenn., will feature an intense focus on rehabilitation in MS. The topic is often a priority for patients but not necessarily at top of mind for health care providers, said rehabilitation therapist Patty Bobryk, secretary of the CMSC.
“This year, some of the Rehab Track topics include addressing respiratory issues in MS, exploring the impact of visual impairments on rehab, and recommending the proper orthotics, as well as discussing treatments that focus on mind-body understanding,” said Ms. Bobryk of MS Comprehensive Care Center of Central Florida, Orlando. She serves as cochair of the International Organization of MS Rehabilitation Therapists.
Rehabilitation in MS is a controversial topic. Two years ago, the CMSC endorsed a statement criticizing a 2015 report by the American Academy of Neurology that found limited evidence supporting MS rehabilitation (Neurology. 2015 Nov 24;85[21]:1896-903). The statement, also supported by the International Organization of MS Nurses and International Organization of MS Rehabilitation Therapists, declared that the AAN report “presents an incomplete review of the evidence published” (Neurol Clin Pract. 2016 Dec;6[6]:475-9).
However, the statement acknowledged that “larger studies with better research methodologies and higher-quality evidence are needed in rehabilitation.”
More recently, a 2017 systematic review of systematic reviews of rehabilitation in MS found that “strong” evidence only exists “for physical therapy for improved activity and participation, and for exercise-based educational programs for the reduction of patient-reported fatigue.” The review of reviews also found there’s “ ‘moderate’ evidence for multidisciplinary rehabilitation for longer-term gains at the levels of activity (disability) and participation, for cognitive-behavior therapy for the treatment of depression, and for information-provision interventions for improved patient knowledge” (Arch Phys Med Rehabil. 2017 Feb;98[2]:353-67).
In an interview, Ms. Bobryk said the MS rehabilitation community is evolving toward “a better understanding of neuroplasticity and the impact that rehab can have on maintaining and improving functional skills.”
In addition, she said, “more emphasis is being placed on early intervention and preventing the secondary effects of the disease versus providing only compensatory strategies.”
Highlights of this year’s rehab sessions at the CMSC conference include a 3-hour course on imbalance in MS that will “provide an intensive review of the anatomy of the balance system and educate the audience on evaluation and treatment interventions specific to retraining the balance system,” Ms. Bobryk said.
Another highlight: A half-day course that will “provide the rationale for including rehabilitation in a comprehensive model of care for people with severe MS,” she said. “Unique interventions such as the use of functional electrical stimulation cycling and body weight-supported treadmill training will be discussed.”
In the big picture, “clinicians who are working in the field of MS realize the benefits of rehab, but more education is needed to spread the word to the broader scope of health care providers,” Ms. Bobryk said. “Patients are often the ones that bring up rehab to their physicians to initiate a referral.”
Wide variety of MS topics on tap at CMSC 2018
More than 2,000 members of the multiple sclerosis care, advocacy, research, and patient communities will gather in Nashville, Tenn., May 30–June 2 for the annual meeting of the Consortium of Multiple Sclerosis Centers.
Dozens of topics will be discussed, ranging from complementary/alternative therapies, ethics, and neuropsychiatry to neuroimmunology and disease models, relapse management, and self-care. Clinicians also will tackle sensitive topics such as suicide, depression, and cognitive impairment.
“Accredited continuing education will be offered for MDs, registered nurses, pharmacists, occupational therapists, physical therapists, social workers, and psychologists,” said Gary Cutter, PhD, president of the CMSC. “Our offerings include beginner courses, advanced science sessions, rehab and mental health tracks, platform and poster sessions, and roundtables.”

“We’ll discuss meds for progressive forms of MS as well as targeted therapies based on new information from genetics,” Dr. Cutter said. Participants will also gain insight from registries and other data sources, he added.
Lecture topics will include the use of computerized screening for cognitive dysfunction in the MS clinic and new research into MS pathology.
Other sessions will explore the use of cannabis, infusion therapies, respiratory enhancement, and a new class of medications for blocking lipid metabolism. Rehabilitation will also be a major focus.
One session will examine MS in patients before conception, during pregnancy, and in the postpartum period. Another session will explore suicide in MS and discuss how clinics can identify and help patients at risk.
The annual CMSC conference stands apart because it’s a “relatively unique meeting where the entire MS treatment team, researchers, and persons with MS can interact and discuss the complex issues in MS care,” Dr. Cutter said. “Much of this takes place outside of the formal program but can still have major impact on all of us. The enthusiasm of the young attendees is always amazing. They are supported, involved, and encouraged.”
More than 2,000 members of the multiple sclerosis care, advocacy, research, and patient communities will gather in Nashville, Tenn., May 30–June 2 for the annual meeting of the Consortium of Multiple Sclerosis Centers.
Dozens of topics will be discussed, ranging from complementary/alternative therapies, ethics, and neuropsychiatry to neuroimmunology and disease models, relapse management, and self-care. Clinicians also will tackle sensitive topics such as suicide, depression, and cognitive impairment.
“Accredited continuing education will be offered for MDs, registered nurses, pharmacists, occupational therapists, physical therapists, social workers, and psychologists,” said Gary Cutter, PhD, president of the CMSC. “Our offerings include beginner courses, advanced science sessions, rehab and mental health tracks, platform and poster sessions, and roundtables.”
“We’ll discuss meds for progressive forms of MS as well as targeted therapies based on new information from genetics,” Dr. Cutter said. Participants will also gain insight from registries and other data sources, he added.
Lecture topics will include the use of computerized screening for cognitive dysfunction in the MS clinic and new research into MS pathology.
Other sessions will explore the use of cannabis, infusion therapies, respiratory enhancement, and a new class of medications for blocking lipid metabolism. Rehabilitation will also be a major focus.
One session will examine MS in patients before conception, during pregnancy, and in the postpartum period. Another session will explore suicide in MS and discuss how clinics can identify and help patients at risk.
The annual CMSC conference stands apart because it’s a “relatively unique meeting where the entire MS treatment team, researchers, and persons with MS can interact and discuss the complex issues in MS care,” Dr. Cutter said. “Much of this takes place outside of the formal program but can still have major impact on all of us. The enthusiasm of the young attendees is always amazing. They are supported, involved, and encouraged.”
More than 2,000 members of the multiple sclerosis care, advocacy, research, and patient communities will gather in Nashville, Tenn., May 30–June 2 for the annual meeting of the Consortium of Multiple Sclerosis Centers.
Dozens of topics will be discussed, ranging from complementary/alternative therapies, ethics, and neuropsychiatry to neuroimmunology and disease models, relapse management, and self-care. Clinicians also will tackle sensitive topics such as suicide, depression, and cognitive impairment.
“Accredited continuing education will be offered for MDs, registered nurses, pharmacists, occupational therapists, physical therapists, social workers, and psychologists,” said Gary Cutter, PhD, president of the CMSC. “Our offerings include beginner courses, advanced science sessions, rehab and mental health tracks, platform and poster sessions, and roundtables.”
“We’ll discuss meds for progressive forms of MS as well as targeted therapies based on new information from genetics,” Dr. Cutter said. Participants will also gain insight from registries and other data sources, he added.
Lecture topics will include the use of computerized screening for cognitive dysfunction in the MS clinic and new research into MS pathology.
Other sessions will explore the use of cannabis, infusion therapies, respiratory enhancement, and a new class of medications for blocking lipid metabolism. Rehabilitation will also be a major focus.
One session will examine MS in patients before conception, during pregnancy, and in the postpartum period. Another session will explore suicide in MS and discuss how clinics can identify and help patients at risk.
The annual CMSC conference stands apart because it’s a “relatively unique meeting where the entire MS treatment team, researchers, and persons with MS can interact and discuss the complex issues in MS care,” Dr. Cutter said. “Much of this takes place outside of the formal program but can still have major impact on all of us. The enthusiasm of the young attendees is always amazing. They are supported, involved, and encouraged.”
LAAC in nonvalvular AF provides stroke protection comparable to warfarin
Background: Because thrombi typically form in the left atrial appendage, LAAC may be an alternative to chronic oral anticoagulation in nonvalvular atrial fibrillation. Two prior randomized controlled trials compared outcomes in patients treated with LAAC with outcomes with warfarin. PROTECT AF trial showed noninferiority of LAAC to warfarin but noted high procedural complication rates. Subsequently, PREVAIL trial failed to demonstrate noninferiority, although complication rates were low overall and similar in both groups. However, longer-term follow-up data were lacking.
Study design: Patient-level meta-analysis of two prospective randomized trials.
Setting: Fifty-nine centers in the United States and Europe (PROTECT AF trial) and 41 centers in the United States (PREVAIL trial).
Synopsis: Meta-analysis of 5-year follow-up data from 1,114 adult patients with atrial fibrillation, most with CHADS2 score greater than or equal to 2 , randomized to receive LAAC or warfarin showed similar frequency of the composite endpoint of stroke, systemic embolism, or cardiovascular/unexplained death (hazard ratio, 0.820; P = .27). Subgroup analysis showed no significant difference in outcomes by patient subset, including CHADS2 or HAS-BLED scores. While the rate of ischemic stroke was similar between groups, the rates of hemorrhagic and disabling/fatal stroke were significantly lower with LAAC (HR, 0.20; P = .0022 and HR, 0.45; P = .034, respectively). All-cause and cardiovascular mortality also were significantly lower with LAAC (HR, 0.73; P = .035 and HR, 0.59; P = .027, respectively), likely because of lower incidence of hemorrhagic stroke.

These data cannot be generalized to patients who have an absolute contraindication to anticoagulation, as these patients were excluded. Further, these trials were conducted before widespread clinical use of novel oral anticoagulants, and LAAC has not yet been compared with these anticoagulants.
Bottom line: In patients with nonvalvular atrial fibrillation, LAAC with the Watchman device provides all-stroke prevention comparable with that of warfarin, and is associated with significantly lower rates of hemorrhagic stroke, disabling or fatal stroke, and mortality.
Citation: Reddy VY et al. 5-year outcomes after left atrial appendage closure: From the PREVAIL and PROTECT AF trials. J Am Coll Cardiol. 2017;70(24):2964-75.
Dr. Indovina is a hospitalist at Denver Health Medical Center and an assistant professor of medicine at the University of Colorado at Denver, Aurora.
Background: Because thrombi typically form in the left atrial appendage, LAAC may be an alternative to chronic oral anticoagulation in nonvalvular atrial fibrillation. Two prior randomized controlled trials compared outcomes in patients treated with LAAC with outcomes with warfarin. PROTECT AF trial showed noninferiority of LAAC to warfarin but noted high procedural complication rates. Subsequently, PREVAIL trial failed to demonstrate noninferiority, although complication rates were low overall and similar in both groups. However, longer-term follow-up data were lacking.
Study design: Patient-level meta-analysis of two prospective randomized trials.
Setting: Fifty-nine centers in the United States and Europe (PROTECT AF trial) and 41 centers in the United States (PREVAIL trial).
Synopsis: Meta-analysis of 5-year follow-up data from 1,114 adult patients with atrial fibrillation, most with CHADS2 score greater than or equal to 2 , randomized to receive LAAC or warfarin showed similar frequency of the composite endpoint of stroke, systemic embolism, or cardiovascular/unexplained death (hazard ratio, 0.820; P = .27). Subgroup analysis showed no significant difference in outcomes by patient subset, including CHADS2 or HAS-BLED scores. While the rate of ischemic stroke was similar between groups, the rates of hemorrhagic and disabling/fatal stroke were significantly lower with LAAC (HR, 0.20; P = .0022 and HR, 0.45; P = .034, respectively). All-cause and cardiovascular mortality also were significantly lower with LAAC (HR, 0.73; P = .035 and HR, 0.59; P = .027, respectively), likely because of lower incidence of hemorrhagic stroke.
These data cannot be generalized to patients who have an absolute contraindication to anticoagulation, as these patients were excluded. Further, these trials were conducted before widespread clinical use of novel oral anticoagulants, and LAAC has not yet been compared with these anticoagulants.
Bottom line: In patients with nonvalvular atrial fibrillation, LAAC with the Watchman device provides all-stroke prevention comparable with that of warfarin, and is associated with significantly lower rates of hemorrhagic stroke, disabling or fatal stroke, and mortality.
Citation: Reddy VY et al. 5-year outcomes after left atrial appendage closure: From the PREVAIL and PROTECT AF trials. J Am Coll Cardiol. 2017;70(24):2964-75.
Dr. Indovina is a hospitalist at Denver Health Medical Center and an assistant professor of medicine at the University of Colorado at Denver, Aurora.
Background: Because thrombi typically form in the left atrial appendage, LAAC may be an alternative to chronic oral anticoagulation in nonvalvular atrial fibrillation. Two prior randomized controlled trials compared outcomes in patients treated with LAAC with outcomes with warfarin. PROTECT AF trial showed noninferiority of LAAC to warfarin but noted high procedural complication rates. Subsequently, PREVAIL trial failed to demonstrate noninferiority, although complication rates were low overall and similar in both groups. However, longer-term follow-up data were lacking.
Study design: Patient-level meta-analysis of two prospective randomized trials.
Setting: Fifty-nine centers in the United States and Europe (PROTECT AF trial) and 41 centers in the United States (PREVAIL trial).
Synopsis: Meta-analysis of 5-year follow-up data from 1,114 adult patients with atrial fibrillation, most with CHADS2 score greater than or equal to 2 , randomized to receive LAAC or warfarin showed similar frequency of the composite endpoint of stroke, systemic embolism, or cardiovascular/unexplained death (hazard ratio, 0.820; P = .27). Subgroup analysis showed no significant difference in outcomes by patient subset, including CHADS2 or HAS-BLED scores. While the rate of ischemic stroke was similar between groups, the rates of hemorrhagic and disabling/fatal stroke were significantly lower with LAAC (HR, 0.20; P = .0022 and HR, 0.45; P = .034, respectively). All-cause and cardiovascular mortality also were significantly lower with LAAC (HR, 0.73; P = .035 and HR, 0.59; P = .027, respectively), likely because of lower incidence of hemorrhagic stroke.
These data cannot be generalized to patients who have an absolute contraindication to anticoagulation, as these patients were excluded. Further, these trials were conducted before widespread clinical use of novel oral anticoagulants, and LAAC has not yet been compared with these anticoagulants.
Bottom line: In patients with nonvalvular atrial fibrillation, LAAC with the Watchman device provides all-stroke prevention comparable with that of warfarin, and is associated with significantly lower rates of hemorrhagic stroke, disabling or fatal stroke, and mortality.
Citation: Reddy VY et al. 5-year outcomes after left atrial appendage closure: From the PREVAIL and PROTECT AF trials. J Am Coll Cardiol. 2017;70(24):2964-75.
Dr. Indovina is a hospitalist at Denver Health Medical Center and an assistant professor of medicine at the University of Colorado at Denver, Aurora.
Screening for brain mets could improve quality of life for some with breast cancer
Despite having more extensive metastases at presentation, breast cancer patients had outcomes after brain-directed therapy similar to those of lung cancer patients, results of a retrospective, single-center study show.
The breast cancer patients had larger and more numerous brain metastases compared with the non-small-cell lung cancer (NSCLC) patients, according to study results published in JAMA Oncology.
However, median survival was not statistically different between groups, at 1.45 years for the breast cancer patients and 1.09 years for NSCLC patients (P = .06), wrote Daniel N. Cagney, MD, of Dana-Farber/Brigham and Women’s Cancer Center, Harvard Medical School, Boston, and his coauthors.
“This finding suggests that intracranial disease in patients with breast cancer was not more aggressive or resistant to treatment, but rather was diagnosed at a later stage,” noted Dr. Cagney and his colleagues.
They described a retrospective analysis of 349 patients with breast cancer and 659 patients with NSCLC, all treated between 2000 and 2015 at Dana-Farber/Brigham and Women’s Cancer Center.
Median metastasis diameter at presentation was 17 mm for the breast cancer patients, compared with 14 mm for the lung cancer patients (P less than .001). Breast cancer patients were significantly more likely to be symptomatic, have seizures, harbor brainstem involvement, and have leptomeningeal disease at the time of diagnosis, the researchers wrote.
“After initial brain-directed therapy, no significant differences in recurrence or treatment-based intracranial outcomes were found between the two groups,” they noted. However, neurological death was seen in 37.3% of the breast cancer group versus 19.9% of the lung cancer group (P less than .001).
Dr. Cagney and his coauthors said they conducted the study to identify the potential value of brain-directed MRI screening in breast cancer, which they said is “important given the impact of neurological compromise on quality of life.”
Brain metastases are common in some subsets of breast cancer patients, yet National Comprehensive Cancer Network guidelines do not recommend brain-directed screening in breast cancer, “a recommendation that is based only on expert consensus given the lack of definitive or prospective studies on this issue,” they wrote.
In light of their findings, the investigators suggest that brain-directed MRI screening is important for breast cancer patients who present with potential for intracranial involvement.
“Early identification of intracranial disease facilitates less invasive or less toxic approaches, such as stereotactic radiosurgery or careful use of promising systemic agents, rather than [whole brain radiation therapy] or neurosurgical resection.” they wrote.
In this study, whole brain radiation therapy was more common in the breast cancer group (59.9% versus 42.9% for the lung cancer group; P less than .001), the investigators noted.
Dr. Cagney and colleagues had no conflicts of interest to report.
SOURCE: Cagney DN et al. JAMA Oncol. 2018 May 17. doi: 10.1001/jamaoncol.2018.0813.
Despite having more extensive metastases at presentation, breast cancer patients had outcomes after brain-directed therapy similar to those of lung cancer patients, results of a retrospective, single-center study show.
The breast cancer patients had larger and more numerous brain metastases compared with the non-small-cell lung cancer (NSCLC) patients, according to study results published in JAMA Oncology.
However, median survival was not statistically different between groups, at 1.45 years for the breast cancer patients and 1.09 years for NSCLC patients (P = .06), wrote Daniel N. Cagney, MD, of Dana-Farber/Brigham and Women’s Cancer Center, Harvard Medical School, Boston, and his coauthors.
“This finding suggests that intracranial disease in patients with breast cancer was not more aggressive or resistant to treatment, but rather was diagnosed at a later stage,” noted Dr. Cagney and his colleagues.
They described a retrospective analysis of 349 patients with breast cancer and 659 patients with NSCLC, all treated between 2000 and 2015 at Dana-Farber/Brigham and Women’s Cancer Center.
Median metastasis diameter at presentation was 17 mm for the breast cancer patients, compared with 14 mm for the lung cancer patients (P less than .001). Breast cancer patients were significantly more likely to be symptomatic, have seizures, harbor brainstem involvement, and have leptomeningeal disease at the time of diagnosis, the researchers wrote.
“After initial brain-directed therapy, no significant differences in recurrence or treatment-based intracranial outcomes were found between the two groups,” they noted. However, neurological death was seen in 37.3% of the breast cancer group versus 19.9% of the lung cancer group (P less than .001).
Dr. Cagney and his coauthors said they conducted the study to identify the potential value of brain-directed MRI screening in breast cancer, which they said is “important given the impact of neurological compromise on quality of life.”
Brain metastases are common in some subsets of breast cancer patients, yet National Comprehensive Cancer Network guidelines do not recommend brain-directed screening in breast cancer, “a recommendation that is based only on expert consensus given the lack of definitive or prospective studies on this issue,” they wrote.
In light of their findings, the investigators suggest that brain-directed MRI screening is important for breast cancer patients who present with potential for intracranial involvement.
“Early identification of intracranial disease facilitates less invasive or less toxic approaches, such as stereotactic radiosurgery or careful use of promising systemic agents, rather than [whole brain radiation therapy] or neurosurgical resection.” they wrote.
In this study, whole brain radiation therapy was more common in the breast cancer group (59.9% versus 42.9% for the lung cancer group; P less than .001), the investigators noted.
Dr. Cagney and colleagues had no conflicts of interest to report.
SOURCE: Cagney DN et al. JAMA Oncol. 2018 May 17. doi: 10.1001/jamaoncol.2018.0813.
Despite having more extensive metastases at presentation, breast cancer patients had outcomes after brain-directed therapy similar to those of lung cancer patients, results of a retrospective, single-center study show.
The breast cancer patients had larger and more numerous brain metastases compared with the non-small-cell lung cancer (NSCLC) patients, according to study results published in JAMA Oncology.
However, median survival was not statistically different between groups, at 1.45 years for the breast cancer patients and 1.09 years for NSCLC patients (P = .06), wrote Daniel N. Cagney, MD, of Dana-Farber/Brigham and Women’s Cancer Center, Harvard Medical School, Boston, and his coauthors.
“This finding suggests that intracranial disease in patients with breast cancer was not more aggressive or resistant to treatment, but rather was diagnosed at a later stage,” noted Dr. Cagney and his colleagues.
They described a retrospective analysis of 349 patients with breast cancer and 659 patients with NSCLC, all treated between 2000 and 2015 at Dana-Farber/Brigham and Women’s Cancer Center.
Median metastasis diameter at presentation was 17 mm for the breast cancer patients, compared with 14 mm for the lung cancer patients (P less than .001). Breast cancer patients were significantly more likely to be symptomatic, have seizures, harbor brainstem involvement, and have leptomeningeal disease at the time of diagnosis, the researchers wrote.
“After initial brain-directed therapy, no significant differences in recurrence or treatment-based intracranial outcomes were found between the two groups,” they noted. However, neurological death was seen in 37.3% of the breast cancer group versus 19.9% of the lung cancer group (P less than .001).
Dr. Cagney and his coauthors said they conducted the study to identify the potential value of brain-directed MRI screening in breast cancer, which they said is “important given the impact of neurological compromise on quality of life.”
Brain metastases are common in some subsets of breast cancer patients, yet National Comprehensive Cancer Network guidelines do not recommend brain-directed screening in breast cancer, “a recommendation that is based only on expert consensus given the lack of definitive or prospective studies on this issue,” they wrote.
In light of their findings, the investigators suggest that brain-directed MRI screening is important for breast cancer patients who present with potential for intracranial involvement.
“Early identification of intracranial disease facilitates less invasive or less toxic approaches, such as stereotactic radiosurgery or careful use of promising systemic agents, rather than [whole brain radiation therapy] or neurosurgical resection.” they wrote.
In this study, whole brain radiation therapy was more common in the breast cancer group (59.9% versus 42.9% for the lung cancer group; P less than .001), the investigators noted.
Dr. Cagney and colleagues had no conflicts of interest to report.
SOURCE: Cagney DN et al. JAMA Oncol. 2018 May 17. doi: 10.1001/jamaoncol.2018.0813.
FROM JAMA ONCOLOGY
Key clinical point: Breast cancer patients presented with larger and more numerous brain metastases compared with non–small-cell lung cancer patients, but after brain-directed therapy, there were no differences in outcomes between groups.
Major finding: Median survival was 1.45 years for breast cancer patients and 1.09 for NSCLC patients.
Study details: A retrospective analysis of 349 patients with breast cancer and 659 patients with NSCLC treated between 2000 and 2015 at Dana-Farber/Brigham and Women’s Cancer Center.
Disclosures: The authors reported no conflicts of interest.
Source: Cagney DN et al. JAMA Oncol. 2018 May 17. doi: 10.1001/jamaoncol.2018.0813.
The ‘Other’ Risks of High Blood Pressure
Other cardiovascular disease outcomes, such as kidney disease and dementia, are less known than the “tradional” outcomes, such as heart attack, heart failure, and stroke. In a Centers for Disease Control and Prevention (CDC) study of 4,166 adults, only 38.5% of those with self-reported hypertension were aware of the risk of kidney disease, versus 24.8% of normotensive adults. Awareness of the risk of dementia was “markedly low”: 8.7% and 7.9%, respectively.
The researchers found “notable” socioeconomic and racial/ethnic differences in awareness of risks. For example, high-income respondents had greater awareness of the association between uncontrolled hypertension and kidney disease, stroke, and dementia, when compared with low-income respondents. Non-Hispanic whites had greater awareness of risk than non-Hispanic blacks (who have a higher prevalence of hypertension and uncontrolled hypertension).
Nearly 35 million people have uncontrolled hypertension, the researchers note. To educate more diverse patient populations about the risks, they recommend expanding current initiatives such as the “Mind Your Risks” program (https://mindyourrisks.nih.gov), promoted by the National Institute of Neurological Disorders and Stroke; and the “Measure Up/Pressure Down (www.measureuppressuredown.com) program, sponsored by the American Medical Group Association.
Other cardiovascular disease outcomes, such as kidney disease and dementia, are less known than the “tradional” outcomes, such as heart attack, heart failure, and stroke. In a Centers for Disease Control and Prevention (CDC) study of 4,166 adults, only 38.5% of those with self-reported hypertension were aware of the risk of kidney disease, versus 24.8% of normotensive adults. Awareness of the risk of dementia was “markedly low”: 8.7% and 7.9%, respectively.
The researchers found “notable” socioeconomic and racial/ethnic differences in awareness of risks. For example, high-income respondents had greater awareness of the association between uncontrolled hypertension and kidney disease, stroke, and dementia, when compared with low-income respondents. Non-Hispanic whites had greater awareness of risk than non-Hispanic blacks (who have a higher prevalence of hypertension and uncontrolled hypertension).
Nearly 35 million people have uncontrolled hypertension, the researchers note. To educate more diverse patient populations about the risks, they recommend expanding current initiatives such as the “Mind Your Risks” program (https://mindyourrisks.nih.gov), promoted by the National Institute of Neurological Disorders and Stroke; and the “Measure Up/Pressure Down (www.measureuppressuredown.com) program, sponsored by the American Medical Group Association.
Other cardiovascular disease outcomes, such as kidney disease and dementia, are less known than the “tradional” outcomes, such as heart attack, heart failure, and stroke. In a Centers for Disease Control and Prevention (CDC) study of 4,166 adults, only 38.5% of those with self-reported hypertension were aware of the risk of kidney disease, versus 24.8% of normotensive adults. Awareness of the risk of dementia was “markedly low”: 8.7% and 7.9%, respectively.
The researchers found “notable” socioeconomic and racial/ethnic differences in awareness of risks. For example, high-income respondents had greater awareness of the association between uncontrolled hypertension and kidney disease, stroke, and dementia, when compared with low-income respondents. Non-Hispanic whites had greater awareness of risk than non-Hispanic blacks (who have a higher prevalence of hypertension and uncontrolled hypertension).
Nearly 35 million people have uncontrolled hypertension, the researchers note. To educate more diverse patient populations about the risks, they recommend expanding current initiatives such as the “Mind Your Risks” program (https://mindyourrisks.nih.gov), promoted by the National Institute of Neurological Disorders and Stroke; and the “Measure Up/Pressure Down (www.measureuppressuredown.com) program, sponsored by the American Medical Group Association.
Perioperative rVWF alone sufficient for some VWD patients
GLASGOW—Recombinant von Willebrand factor (rVWF) alone can be sufficient as perioperative management for some patients with severe von Willebrand disease (VWD), according to researchers.
In a phase 3 study, 10 of 15 patients were able to achieve hemostatic efficacy ratings of “good” or “excellent” when receiving only rVWF before, during, and/or after surgery.
The remaining 5 patients also achieved favorable hemostatic efficacy ratings, but they received recombinant factor VIII (FVIII) as well.
These results were presented at the World Federation of Hemophilia (WFH) 2018 World Congress (abstract W-MP-63 [749]). The research was sponsored by Shire, the company marketing rVWF as Vonvendi.
“There is an unmet clinical need for those living with von Willebrand disease, as they face a heightened risk of bleeding during surgery,” said study investigator Flora Peyvandi, MD, PhD, of the University of Milan in Italy.
“People with von Willebrand disease lack proper function or quantity of von Willebrand factor, and some also have a secondary factor VIII deficiency. In this study, recombinant von Willebrand factor was administered to replace the insufficient or dysfunctional von Willebrand factor, allowing the body to naturally replenish FVIII in most patients. These study results demonstrate clinical promise as physicians were able to tailor treatment based on each patient’s individual need for one or both factor therapies.”
The study included 15 adults with severe VWD who were undergoing elective surgical procedures. Ten patients were undergoing major surgery, 4 minor, and 1 oral surgery.
The patients’ median age was 40 (range, 20-70), and 8 were female. Most (n=8) had type 3 VWD, 3 had type 1, 2 had type 2A, 1 had 2B, and 1 had 2M.
At baseline, the mean endogenous FVIII level (FVIII:C) was 16.4 IU/dL, and the mean VWF ristocetin cofactor (VWF:Rco) was 10.6 IU/dL.
The patients received rVWF at 40 to 60 IU/kg VWF:RCo intravenously 12 to 24 hours before surgery to allow FVIII:C levels to increase to at least 30 IU/dL for minor or oral surgery or to at least 60 IU/dL for major surgery, within 3 hours before surgery.
If the desired levels were achieved, rVWF could be given alone. If the levels were not achieved, patients would receive rFVIII as well, within 1 to 2 hours before surgery. Patients were monitored for 14 days after surgery.
Results
All 15 patients had overall/intraoperative hemostatic efficacy ratings of “excellent” (as good as or better than expected) or “good” (probably as good as expected).
The patients received a median of 6 (range, 2 to 15) rVWF infusions at a median dose of 55 IU/kg (range, 36.1 to 59.9). Most patients (n=11) did not receive rVWF every day. For some, infusions were separated by 2 to 9 days.
Ten patients received rVWF alone, 12 did not receive any preoperative FVIII, and 2 did not receive rVWF postoperatively.
Most rVWF infusions (89.4%, 93/104) were given alone, and 70% (7/10) of the major surgeries were performed with rVWF alone.
The researchers said that, with rVWF alone, patients had hemostatically effective levels of FVIII:C as early as 6 hours after surgery, and this was sustained for 72 to 96 hours.
There were 5 patients who received rVWF with rFVIII. Of the 11 infusions these patients received, 9 were given when FVIII:C levels were above 60 IU/dL.
Three patients received rVWF with rFVIII 1 hour before major surgery—total hip replacement, molar extraction, and left ankle prosthesis. However, 2 of these patients had FVIII:C levels above 60 IU/dL.
The patient undergoing a molar extraction received rVWF with rFVIII 6 times after surgery. In 5 cases, the patient’s FVIII:C levels were 110 to 152 IU/dL. In the remaining case, the FVIII:C level was 23 IU/dL.
Two patients received rVWF with rFVIII for minor surgery. One patient undergoing a tooth extraction received rVWF with rFVIII intraoperatively when the FVIII:C level was 72 IU/dL.
The other patient received rVWF with rFVIII after radioisotope synovectomy when the FVIII:C level was 73 IU/dL. This patient received a postoperative dose of rVWF alone as well.
One patient tested positive for binding antibodies to VWF, and 1 patient developed deep vein thrombosis 3 days after total hip replacement while receiving rVWF.
GLASGOW—Recombinant von Willebrand factor (rVWF) alone can be sufficient as perioperative management for some patients with severe von Willebrand disease (VWD), according to researchers.
In a phase 3 study, 10 of 15 patients were able to achieve hemostatic efficacy ratings of “good” or “excellent” when receiving only rVWF before, during, and/or after surgery.
The remaining 5 patients also achieved favorable hemostatic efficacy ratings, but they received recombinant factor VIII (FVIII) as well.
These results were presented at the World Federation of Hemophilia (WFH) 2018 World Congress (abstract W-MP-63 [749]). The research was sponsored by Shire, the company marketing rVWF as Vonvendi.
“There is an unmet clinical need for those living with von Willebrand disease, as they face a heightened risk of bleeding during surgery,” said study investigator Flora Peyvandi, MD, PhD, of the University of Milan in Italy.
“People with von Willebrand disease lack proper function or quantity of von Willebrand factor, and some also have a secondary factor VIII deficiency. In this study, recombinant von Willebrand factor was administered to replace the insufficient or dysfunctional von Willebrand factor, allowing the body to naturally replenish FVIII in most patients. These study results demonstrate clinical promise as physicians were able to tailor treatment based on each patient’s individual need for one or both factor therapies.”
The study included 15 adults with severe VWD who were undergoing elective surgical procedures. Ten patients were undergoing major surgery, 4 minor, and 1 oral surgery.
The patients’ median age was 40 (range, 20-70), and 8 were female. Most (n=8) had type 3 VWD, 3 had type 1, 2 had type 2A, 1 had 2B, and 1 had 2M.
At baseline, the mean endogenous FVIII level (FVIII:C) was 16.4 IU/dL, and the mean VWF ristocetin cofactor (VWF:Rco) was 10.6 IU/dL.
The patients received rVWF at 40 to 60 IU/kg VWF:RCo intravenously 12 to 24 hours before surgery to allow FVIII:C levels to increase to at least 30 IU/dL for minor or oral surgery or to at least 60 IU/dL for major surgery, within 3 hours before surgery.
If the desired levels were achieved, rVWF could be given alone. If the levels were not achieved, patients would receive rFVIII as well, within 1 to 2 hours before surgery. Patients were monitored for 14 days after surgery.
Results
All 15 patients had overall/intraoperative hemostatic efficacy ratings of “excellent” (as good as or better than expected) or “good” (probably as good as expected).
The patients received a median of 6 (range, 2 to 15) rVWF infusions at a median dose of 55 IU/kg (range, 36.1 to 59.9). Most patients (n=11) did not receive rVWF every day. For some, infusions were separated by 2 to 9 days.
Ten patients received rVWF alone, 12 did not receive any preoperative FVIII, and 2 did not receive rVWF postoperatively.
Most rVWF infusions (89.4%, 93/104) were given alone, and 70% (7/10) of the major surgeries were performed with rVWF alone.
The researchers said that, with rVWF alone, patients had hemostatically effective levels of FVIII:C as early as 6 hours after surgery, and this was sustained for 72 to 96 hours.
There were 5 patients who received rVWF with rFVIII. Of the 11 infusions these patients received, 9 were given when FVIII:C levels were above 60 IU/dL.
Three patients received rVWF with rFVIII 1 hour before major surgery—total hip replacement, molar extraction, and left ankle prosthesis. However, 2 of these patients had FVIII:C levels above 60 IU/dL.
The patient undergoing a molar extraction received rVWF with rFVIII 6 times after surgery. In 5 cases, the patient’s FVIII:C levels were 110 to 152 IU/dL. In the remaining case, the FVIII:C level was 23 IU/dL.
Two patients received rVWF with rFVIII for minor surgery. One patient undergoing a tooth extraction received rVWF with rFVIII intraoperatively when the FVIII:C level was 72 IU/dL.
The other patient received rVWF with rFVIII after radioisotope synovectomy when the FVIII:C level was 73 IU/dL. This patient received a postoperative dose of rVWF alone as well.
One patient tested positive for binding antibodies to VWF, and 1 patient developed deep vein thrombosis 3 days after total hip replacement while receiving rVWF.
GLASGOW—Recombinant von Willebrand factor (rVWF) alone can be sufficient as perioperative management for some patients with severe von Willebrand disease (VWD), according to researchers.
In a phase 3 study, 10 of 15 patients were able to achieve hemostatic efficacy ratings of “good” or “excellent” when receiving only rVWF before, during, and/or after surgery.
The remaining 5 patients also achieved favorable hemostatic efficacy ratings, but they received recombinant factor VIII (FVIII) as well.
These results were presented at the World Federation of Hemophilia (WFH) 2018 World Congress (abstract W-MP-63 [749]). The research was sponsored by Shire, the company marketing rVWF as Vonvendi.
“There is an unmet clinical need for those living with von Willebrand disease, as they face a heightened risk of bleeding during surgery,” said study investigator Flora Peyvandi, MD, PhD, of the University of Milan in Italy.
“People with von Willebrand disease lack proper function or quantity of von Willebrand factor, and some also have a secondary factor VIII deficiency. In this study, recombinant von Willebrand factor was administered to replace the insufficient or dysfunctional von Willebrand factor, allowing the body to naturally replenish FVIII in most patients. These study results demonstrate clinical promise as physicians were able to tailor treatment based on each patient’s individual need for one or both factor therapies.”
The study included 15 adults with severe VWD who were undergoing elective surgical procedures. Ten patients were undergoing major surgery, 4 minor, and 1 oral surgery.
The patients’ median age was 40 (range, 20-70), and 8 were female. Most (n=8) had type 3 VWD, 3 had type 1, 2 had type 2A, 1 had 2B, and 1 had 2M.
At baseline, the mean endogenous FVIII level (FVIII:C) was 16.4 IU/dL, and the mean VWF ristocetin cofactor (VWF:Rco) was 10.6 IU/dL.
The patients received rVWF at 40 to 60 IU/kg VWF:RCo intravenously 12 to 24 hours before surgery to allow FVIII:C levels to increase to at least 30 IU/dL for minor or oral surgery or to at least 60 IU/dL for major surgery, within 3 hours before surgery.
If the desired levels were achieved, rVWF could be given alone. If the levels were not achieved, patients would receive rFVIII as well, within 1 to 2 hours before surgery. Patients were monitored for 14 days after surgery.
Results
All 15 patients had overall/intraoperative hemostatic efficacy ratings of “excellent” (as good as or better than expected) or “good” (probably as good as expected).
The patients received a median of 6 (range, 2 to 15) rVWF infusions at a median dose of 55 IU/kg (range, 36.1 to 59.9). Most patients (n=11) did not receive rVWF every day. For some, infusions were separated by 2 to 9 days.
Ten patients received rVWF alone, 12 did not receive any preoperative FVIII, and 2 did not receive rVWF postoperatively.
Most rVWF infusions (89.4%, 93/104) were given alone, and 70% (7/10) of the major surgeries were performed with rVWF alone.
The researchers said that, with rVWF alone, patients had hemostatically effective levels of FVIII:C as early as 6 hours after surgery, and this was sustained for 72 to 96 hours.
There were 5 patients who received rVWF with rFVIII. Of the 11 infusions these patients received, 9 were given when FVIII:C levels were above 60 IU/dL.
Three patients received rVWF with rFVIII 1 hour before major surgery—total hip replacement, molar extraction, and left ankle prosthesis. However, 2 of these patients had FVIII:C levels above 60 IU/dL.
The patient undergoing a molar extraction received rVWF with rFVIII 6 times after surgery. In 5 cases, the patient’s FVIII:C levels were 110 to 152 IU/dL. In the remaining case, the FVIII:C level was 23 IU/dL.
Two patients received rVWF with rFVIII for minor surgery. One patient undergoing a tooth extraction received rVWF with rFVIII intraoperatively when the FVIII:C level was 72 IU/dL.
The other patient received rVWF with rFVIII after radioisotope synovectomy when the FVIII:C level was 73 IU/dL. This patient received a postoperative dose of rVWF alone as well.
One patient tested positive for binding antibodies to VWF, and 1 patient developed deep vein thrombosis 3 days after total hip replacement while receiving rVWF.