User login
-
Oncology Mergers Are on the Rise. How Can Independent Practices Survive?
When he completed his fellowship at Fox Chase Cancer Center in Philadelphia, Moshe Chasky, MD, joined a small five-person practice that rented space from the city’s Jefferson Hospital in Philadelphia. The arrangement seemed to work well for the hospital and the small practice, which remained independent.
Within 10 years, the hospital sought to buy the practice, Alliance Cancer Specialists.
But the oncologists at Alliance did not want to join Jefferson.
The hospital eventually entered into an exclusive agreement with its own medical group to provide inpatient oncology/hematology services at three Jefferson Health–Northeast hospitals and stripped Dr. Chasky and his colleagues of their privileges at those facilities, Medscape Medical News reported last year.
said Jeff Patton, MD, CEO of OneOncology, a management services organization.
A 2020 report from the Community Oncology Alliance (COA), for instance, tracked mergers, acquisitions, and closures in the community oncology setting and found the number of practices acquired by hospitals, known as vertical integration, nearly tripled from 2010 to 2020.
“Some hospitals are pretty predatory in their approach,” Dr. Patton said. If hospitals have their own oncology program, “they’ll employ the referring doctors and then discourage them or prevent them from referring patients to our independent practices that are not owned by the hospital.”
Still, in the face of growing pressure to join hospitals, some community oncology practices are finding ways to survive and maintain their independence.
A Growing Trend
The latest data continue to show a clear trend: Consolidation in oncology is on the rise.
A 2024 study revealed that the pace of consolidation seems to be increasing.
The analysis found that, between 2015 and 2022, the number of medical oncologists increased by 14% and the number of medical oncologists per practice increased by 40%, while the number of practices decreased by 18%.
While about 44% of practices remain independent, the percentage of medical oncologists working in practices with more than 25 clinicians has increased from 34% in 2015 to 44% in 2022. By 2022, the largest 102 practices in the United States employed more than 40% of all medical oncologists.
“The rate of consolidation seems to be rapid,” study coauthor Parsa Erfani, MD, an internal medicine resident at Brigham & Women’s Hospital, Boston, explained.
Consolidation appears to breed more consolidation. The researchers found, for instance, that markets with greater hospital consolidation and more hospital beds per capita were more likely to undergo consolidation in oncology.
Consolidation may be higher in these markets “because hospitals or health systems are buying up oncology practices or conversely because oncology practices are merging to compete more effectively with larger hospitals in the area,” Dr. Erfani told this news organization.
Mergers among independent practices, known as horizontal integration, have also been on the rise, according to the 2020 COA report. These mergers can help counter pressures from hospitals seeking to acquire community practices as well as prevent practices and their clinics from closing.
Although Dr. Erfani’s research wasn’t designed to determine the factors behind consolidation, he and his colleagues point to the Affordable Care Act (ACA) and the federal 340B Drug Pricing Program as potential drivers of this trend.
The ACA encouraged consolidation as a way to improve efficiency and created the need for ever-larger information systems to collect and report quality data. But these data collection and reporting requirements have become increasingly difficult for smaller practices to take on.
The 340B Program, however, may be a bigger contributing factor to consolidation. Created in 1992, the 340B Program allows qualifying hospitals and clinics that treat low-income and uninsured patients to buy outpatient prescription drugs at a 25%-50% discount.
Hospitals seeking to capitalize on the margins possible under the 340B Program will “buy all the referring physicians in a market so that the medical oncology group is left with little choice but to sell to the hospital,” said Dr. Patton.
“Those 340B dollars are worth a lot to hospitals,” said David A. Eagle, MD, a hematologist/oncologist with New York Cancer & Blood Specialists and past president of COA. The program “creates an appetite for nonprofit hospitals to want to grow their medical oncology programs,” he told this news organization.
Declining Medicare reimbursement has also hit independent practices hard.
Over the past 15 years, compared with inflation, physicians have gotten “a pay rate decrease from Medicare,” said Dr. Patton. Payers have followed that lead and tried to cut pay for clinicians, especially those who do not have market share, he said. Paying them less is “disingenuous knowing that our costs of providing care are going up,” he said.
Less Access, Higher Costs, Worse Care?
Many studies have demonstrated that, when hospitals become behemoths in a given market, healthcare costs go up.
“There are robust data showing that consolidation increases healthcare costs by reducing competition, including in oncology,” wrote Dr. Erfani and colleagues.
Oncology practices that are owned by hospitals bill facility fees for outpatient chemotherapy treatment, adding another layer of cost, the researchers explained, citing a 2019 Health Economics study.
Another analysis, published in 2020, found that hospital prices for the top 37 infused cancer drugs averaged 86% more per unit than the price charged by physician offices. Hospital outpatient departments charged even more, on average, for drugs — 128% more for nivolumab and 428% more for fluorouracil, for instance.
In their 2024 analysis, Dr. Erfani and colleagues also found that increased hospital market concentration was associated with worse quality of care, across all assessed patient satisfaction measures, and may result in worse access to care as well.
Overall, these consolidation “trends have important implications for cancer care cost, quality, and access,” the authors concluded.
Navigating the Consolidation Trend
In the face of mounting pressure to join hospitals, community oncology practices have typically relied on horizontal mergers to maintain their independence. An increasing number of practices, however, are now turning to another strategy: Management services organizations.
According to some oncologists, a core benefit of joining a management services organization is their community practices can maintain autonomy, hold on to referrals, and benefit from access to a wider network of peers and recently approved treatments such as chimeric antigen receptor T-cell therapies.
In these arrangements, the management company also provides business assistance to practices, including help with billing and collection, payer negotiations, supply chain issues, and credentialing, as well as recruiting, hiring, and marketing.
These management organizations, which include American Oncology Network, Integrated Oncology Network, OneOncology, and Verdi Oncology, are, however, backed by private equity. According to a 2022 report, private equity–backed management organizations have ramped up arrangements with community oncology practices over the past few years — a trend that has concerned some experts.
The authors of a recent analysis in JAMA Internal Medicine explained that, although private equity involvement in physician practices may enable operational efficiencies, “critics point to potential conflicts of interest” and highlight concerns that patients “may face additional barriers to both accessibility and affordability of care.”
The difference, according to some oncologists, is their practices are not owned by the management services organization; instead, the practices enter contracts that outline the boundaries of the relationship and stipulate fees to the management organizations.
In 2020, Dr. Chasky’s practice, Alliance Cancer Specialists, joined The US Oncology Network, a management services organization wholly owned by McKesson. The organization provides the practice with capital and other resources, as well as access to the Sarah Cannon Research Institute, so patients can participate in clinical trials.
“We totally function as an independent practice,” said Dr. Chasky. “We make our own management decisions,” he said. For instance, if Alliance wants to hire a new clinician, US Oncology helps with the recruitment. “But at the end of the day, it’s our practice,” he said.
Davey Daniel, MD — whose community practice joined the management services organization OneOncology — has seen the benefits of being part of a larger network. For instance, bispecific therapies for leukemias, lymphomas, and multiple myeloma are typically administered at academic centers because of the risk for cytokine release syndrome.
However, physician leaders in the OneOncology network “came up with a playbook on how to do it safely” in the community setting, said Dr. Daniel. “It meant that we were adopting FDA newly approved therapies in a very short course.”
Being able to draw from a wider pool of expertise has had other advantages. Dr. Daniel can lean on pathologists and research scientists in the network for advice on targeted therapy use. “We’re actually bringing precision medicine expertise to the community,” Dr. Daniel said.
Dr. Chasky and Dr. Eagle, whose practice is also part of OneOncology, said that continuing to work in the community setting has allowed them greater flexibility.
Dr. Eagle explained that New York Cancer & Blood Specialists tries to offer patients an appointment within 2 days of a referral, and it allows walk-in visits.
Dr. Chasky leans into the flexibility by having staff stay late, when needed, to ensure that all patients are seen. “We’re there for our patients at all hours,” Dr. Chasky said, adding that often “you don’t have that flexibility when you work for a big hospital system.”
The bottom line is community oncology can still thrive, said Nick Ferreyros, managing director of COA, “as long as we have a healthy competitive ecosystem where [we] are valued and seen as an important part of our cancer care system.”
A version of this article first appeared on Medscape.com.
When he completed his fellowship at Fox Chase Cancer Center in Philadelphia, Moshe Chasky, MD, joined a small five-person practice that rented space from the city’s Jefferson Hospital in Philadelphia. The arrangement seemed to work well for the hospital and the small practice, which remained independent.
Within 10 years, the hospital sought to buy the practice, Alliance Cancer Specialists.
But the oncologists at Alliance did not want to join Jefferson.
The hospital eventually entered into an exclusive agreement with its own medical group to provide inpatient oncology/hematology services at three Jefferson Health–Northeast hospitals and stripped Dr. Chasky and his colleagues of their privileges at those facilities, Medscape Medical News reported last year.
said Jeff Patton, MD, CEO of OneOncology, a management services organization.
A 2020 report from the Community Oncology Alliance (COA), for instance, tracked mergers, acquisitions, and closures in the community oncology setting and found the number of practices acquired by hospitals, known as vertical integration, nearly tripled from 2010 to 2020.
“Some hospitals are pretty predatory in their approach,” Dr. Patton said. If hospitals have their own oncology program, “they’ll employ the referring doctors and then discourage them or prevent them from referring patients to our independent practices that are not owned by the hospital.”
Still, in the face of growing pressure to join hospitals, some community oncology practices are finding ways to survive and maintain their independence.
A Growing Trend
The latest data continue to show a clear trend: Consolidation in oncology is on the rise.
A 2024 study revealed that the pace of consolidation seems to be increasing.
The analysis found that, between 2015 and 2022, the number of medical oncologists increased by 14% and the number of medical oncologists per practice increased by 40%, while the number of practices decreased by 18%.
While about 44% of practices remain independent, the percentage of medical oncologists working in practices with more than 25 clinicians has increased from 34% in 2015 to 44% in 2022. By 2022, the largest 102 practices in the United States employed more than 40% of all medical oncologists.
“The rate of consolidation seems to be rapid,” study coauthor Parsa Erfani, MD, an internal medicine resident at Brigham & Women’s Hospital, Boston, explained.
Consolidation appears to breed more consolidation. The researchers found, for instance, that markets with greater hospital consolidation and more hospital beds per capita were more likely to undergo consolidation in oncology.
Consolidation may be higher in these markets “because hospitals or health systems are buying up oncology practices or conversely because oncology practices are merging to compete more effectively with larger hospitals in the area,” Dr. Erfani told this news organization.
Mergers among independent practices, known as horizontal integration, have also been on the rise, according to the 2020 COA report. These mergers can help counter pressures from hospitals seeking to acquire community practices as well as prevent practices and their clinics from closing.
Although Dr. Erfani’s research wasn’t designed to determine the factors behind consolidation, he and his colleagues point to the Affordable Care Act (ACA) and the federal 340B Drug Pricing Program as potential drivers of this trend.
The ACA encouraged consolidation as a way to improve efficiency and created the need for ever-larger information systems to collect and report quality data. But these data collection and reporting requirements have become increasingly difficult for smaller practices to take on.
The 340B Program, however, may be a bigger contributing factor to consolidation. Created in 1992, the 340B Program allows qualifying hospitals and clinics that treat low-income and uninsured patients to buy outpatient prescription drugs at a 25%-50% discount.
Hospitals seeking to capitalize on the margins possible under the 340B Program will “buy all the referring physicians in a market so that the medical oncology group is left with little choice but to sell to the hospital,” said Dr. Patton.
“Those 340B dollars are worth a lot to hospitals,” said David A. Eagle, MD, a hematologist/oncologist with New York Cancer & Blood Specialists and past president of COA. The program “creates an appetite for nonprofit hospitals to want to grow their medical oncology programs,” he told this news organization.
Declining Medicare reimbursement has also hit independent practices hard.
Over the past 15 years, compared with inflation, physicians have gotten “a pay rate decrease from Medicare,” said Dr. Patton. Payers have followed that lead and tried to cut pay for clinicians, especially those who do not have market share, he said. Paying them less is “disingenuous knowing that our costs of providing care are going up,” he said.
Less Access, Higher Costs, Worse Care?
Many studies have demonstrated that, when hospitals become behemoths in a given market, healthcare costs go up.
“There are robust data showing that consolidation increases healthcare costs by reducing competition, including in oncology,” wrote Dr. Erfani and colleagues.
Oncology practices that are owned by hospitals bill facility fees for outpatient chemotherapy treatment, adding another layer of cost, the researchers explained, citing a 2019 Health Economics study.
Another analysis, published in 2020, found that hospital prices for the top 37 infused cancer drugs averaged 86% more per unit than the price charged by physician offices. Hospital outpatient departments charged even more, on average, for drugs — 128% more for nivolumab and 428% more for fluorouracil, for instance.
In their 2024 analysis, Dr. Erfani and colleagues also found that increased hospital market concentration was associated with worse quality of care, across all assessed patient satisfaction measures, and may result in worse access to care as well.
Overall, these consolidation “trends have important implications for cancer care cost, quality, and access,” the authors concluded.
Navigating the Consolidation Trend
In the face of mounting pressure to join hospitals, community oncology practices have typically relied on horizontal mergers to maintain their independence. An increasing number of practices, however, are now turning to another strategy: Management services organizations.
According to some oncologists, a core benefit of joining a management services organization is their community practices can maintain autonomy, hold on to referrals, and benefit from access to a wider network of peers and recently approved treatments such as chimeric antigen receptor T-cell therapies.
In these arrangements, the management company also provides business assistance to practices, including help with billing and collection, payer negotiations, supply chain issues, and credentialing, as well as recruiting, hiring, and marketing.
These management organizations, which include American Oncology Network, Integrated Oncology Network, OneOncology, and Verdi Oncology, are, however, backed by private equity. According to a 2022 report, private equity–backed management organizations have ramped up arrangements with community oncology practices over the past few years — a trend that has concerned some experts.
The authors of a recent analysis in JAMA Internal Medicine explained that, although private equity involvement in physician practices may enable operational efficiencies, “critics point to potential conflicts of interest” and highlight concerns that patients “may face additional barriers to both accessibility and affordability of care.”
The difference, according to some oncologists, is their practices are not owned by the management services organization; instead, the practices enter contracts that outline the boundaries of the relationship and stipulate fees to the management organizations.
In 2020, Dr. Chasky’s practice, Alliance Cancer Specialists, joined The US Oncology Network, a management services organization wholly owned by McKesson. The organization provides the practice with capital and other resources, as well as access to the Sarah Cannon Research Institute, so patients can participate in clinical trials.
“We totally function as an independent practice,” said Dr. Chasky. “We make our own management decisions,” he said. For instance, if Alliance wants to hire a new clinician, US Oncology helps with the recruitment. “But at the end of the day, it’s our practice,” he said.
Davey Daniel, MD — whose community practice joined the management services organization OneOncology — has seen the benefits of being part of a larger network. For instance, bispecific therapies for leukemias, lymphomas, and multiple myeloma are typically administered at academic centers because of the risk for cytokine release syndrome.
However, physician leaders in the OneOncology network “came up with a playbook on how to do it safely” in the community setting, said Dr. Daniel. “It meant that we were adopting FDA newly approved therapies in a very short course.”
Being able to draw from a wider pool of expertise has had other advantages. Dr. Daniel can lean on pathologists and research scientists in the network for advice on targeted therapy use. “We’re actually bringing precision medicine expertise to the community,” Dr. Daniel said.
Dr. Chasky and Dr. Eagle, whose practice is also part of OneOncology, said that continuing to work in the community setting has allowed them greater flexibility.
Dr. Eagle explained that New York Cancer & Blood Specialists tries to offer patients an appointment within 2 days of a referral, and it allows walk-in visits.
Dr. Chasky leans into the flexibility by having staff stay late, when needed, to ensure that all patients are seen. “We’re there for our patients at all hours,” Dr. Chasky said, adding that often “you don’t have that flexibility when you work for a big hospital system.”
The bottom line is community oncology can still thrive, said Nick Ferreyros, managing director of COA, “as long as we have a healthy competitive ecosystem where [we] are valued and seen as an important part of our cancer care system.”
A version of this article first appeared on Medscape.com.
When he completed his fellowship at Fox Chase Cancer Center in Philadelphia, Moshe Chasky, MD, joined a small five-person practice that rented space from the city’s Jefferson Hospital in Philadelphia. The arrangement seemed to work well for the hospital and the small practice, which remained independent.
Within 10 years, the hospital sought to buy the practice, Alliance Cancer Specialists.
But the oncologists at Alliance did not want to join Jefferson.
The hospital eventually entered into an exclusive agreement with its own medical group to provide inpatient oncology/hematology services at three Jefferson Health–Northeast hospitals and stripped Dr. Chasky and his colleagues of their privileges at those facilities, Medscape Medical News reported last year.
said Jeff Patton, MD, CEO of OneOncology, a management services organization.
A 2020 report from the Community Oncology Alliance (COA), for instance, tracked mergers, acquisitions, and closures in the community oncology setting and found the number of practices acquired by hospitals, known as vertical integration, nearly tripled from 2010 to 2020.
“Some hospitals are pretty predatory in their approach,” Dr. Patton said. If hospitals have their own oncology program, “they’ll employ the referring doctors and then discourage them or prevent them from referring patients to our independent practices that are not owned by the hospital.”
Still, in the face of growing pressure to join hospitals, some community oncology practices are finding ways to survive and maintain their independence.
A Growing Trend
The latest data continue to show a clear trend: Consolidation in oncology is on the rise.
A 2024 study revealed that the pace of consolidation seems to be increasing.
The analysis found that, between 2015 and 2022, the number of medical oncologists increased by 14% and the number of medical oncologists per practice increased by 40%, while the number of practices decreased by 18%.
While about 44% of practices remain independent, the percentage of medical oncologists working in practices with more than 25 clinicians has increased from 34% in 2015 to 44% in 2022. By 2022, the largest 102 practices in the United States employed more than 40% of all medical oncologists.
“The rate of consolidation seems to be rapid,” study coauthor Parsa Erfani, MD, an internal medicine resident at Brigham & Women’s Hospital, Boston, explained.
Consolidation appears to breed more consolidation. The researchers found, for instance, that markets with greater hospital consolidation and more hospital beds per capita were more likely to undergo consolidation in oncology.
Consolidation may be higher in these markets “because hospitals or health systems are buying up oncology practices or conversely because oncology practices are merging to compete more effectively with larger hospitals in the area,” Dr. Erfani told this news organization.
Mergers among independent practices, known as horizontal integration, have also been on the rise, according to the 2020 COA report. These mergers can help counter pressures from hospitals seeking to acquire community practices as well as prevent practices and their clinics from closing.
Although Dr. Erfani’s research wasn’t designed to determine the factors behind consolidation, he and his colleagues point to the Affordable Care Act (ACA) and the federal 340B Drug Pricing Program as potential drivers of this trend.
The ACA encouraged consolidation as a way to improve efficiency and created the need for ever-larger information systems to collect and report quality data. But these data collection and reporting requirements have become increasingly difficult for smaller practices to take on.
The 340B Program, however, may be a bigger contributing factor to consolidation. Created in 1992, the 340B Program allows qualifying hospitals and clinics that treat low-income and uninsured patients to buy outpatient prescription drugs at a 25%-50% discount.
Hospitals seeking to capitalize on the margins possible under the 340B Program will “buy all the referring physicians in a market so that the medical oncology group is left with little choice but to sell to the hospital,” said Dr. Patton.
“Those 340B dollars are worth a lot to hospitals,” said David A. Eagle, MD, a hematologist/oncologist with New York Cancer & Blood Specialists and past president of COA. The program “creates an appetite for nonprofit hospitals to want to grow their medical oncology programs,” he told this news organization.
Declining Medicare reimbursement has also hit independent practices hard.
Over the past 15 years, compared with inflation, physicians have gotten “a pay rate decrease from Medicare,” said Dr. Patton. Payers have followed that lead and tried to cut pay for clinicians, especially those who do not have market share, he said. Paying them less is “disingenuous knowing that our costs of providing care are going up,” he said.
Less Access, Higher Costs, Worse Care?
Many studies have demonstrated that, when hospitals become behemoths in a given market, healthcare costs go up.
“There are robust data showing that consolidation increases healthcare costs by reducing competition, including in oncology,” wrote Dr. Erfani and colleagues.
Oncology practices that are owned by hospitals bill facility fees for outpatient chemotherapy treatment, adding another layer of cost, the researchers explained, citing a 2019 Health Economics study.
Another analysis, published in 2020, found that hospital prices for the top 37 infused cancer drugs averaged 86% more per unit than the price charged by physician offices. Hospital outpatient departments charged even more, on average, for drugs — 128% more for nivolumab and 428% more for fluorouracil, for instance.
In their 2024 analysis, Dr. Erfani and colleagues also found that increased hospital market concentration was associated with worse quality of care, across all assessed patient satisfaction measures, and may result in worse access to care as well.
Overall, these consolidation “trends have important implications for cancer care cost, quality, and access,” the authors concluded.
Navigating the Consolidation Trend
In the face of mounting pressure to join hospitals, community oncology practices have typically relied on horizontal mergers to maintain their independence. An increasing number of practices, however, are now turning to another strategy: Management services organizations.
According to some oncologists, a core benefit of joining a management services organization is their community practices can maintain autonomy, hold on to referrals, and benefit from access to a wider network of peers and recently approved treatments such as chimeric antigen receptor T-cell therapies.
In these arrangements, the management company also provides business assistance to practices, including help with billing and collection, payer negotiations, supply chain issues, and credentialing, as well as recruiting, hiring, and marketing.
These management organizations, which include American Oncology Network, Integrated Oncology Network, OneOncology, and Verdi Oncology, are, however, backed by private equity. According to a 2022 report, private equity–backed management organizations have ramped up arrangements with community oncology practices over the past few years — a trend that has concerned some experts.
The authors of a recent analysis in JAMA Internal Medicine explained that, although private equity involvement in physician practices may enable operational efficiencies, “critics point to potential conflicts of interest” and highlight concerns that patients “may face additional barriers to both accessibility and affordability of care.”
The difference, according to some oncologists, is their practices are not owned by the management services organization; instead, the practices enter contracts that outline the boundaries of the relationship and stipulate fees to the management organizations.
In 2020, Dr. Chasky’s practice, Alliance Cancer Specialists, joined The US Oncology Network, a management services organization wholly owned by McKesson. The organization provides the practice with capital and other resources, as well as access to the Sarah Cannon Research Institute, so patients can participate in clinical trials.
“We totally function as an independent practice,” said Dr. Chasky. “We make our own management decisions,” he said. For instance, if Alliance wants to hire a new clinician, US Oncology helps with the recruitment. “But at the end of the day, it’s our practice,” he said.
Davey Daniel, MD — whose community practice joined the management services organization OneOncology — has seen the benefits of being part of a larger network. For instance, bispecific therapies for leukemias, lymphomas, and multiple myeloma are typically administered at academic centers because of the risk for cytokine release syndrome.
However, physician leaders in the OneOncology network “came up with a playbook on how to do it safely” in the community setting, said Dr. Daniel. “It meant that we were adopting FDA newly approved therapies in a very short course.”
Being able to draw from a wider pool of expertise has had other advantages. Dr. Daniel can lean on pathologists and research scientists in the network for advice on targeted therapy use. “We’re actually bringing precision medicine expertise to the community,” Dr. Daniel said.
Dr. Chasky and Dr. Eagle, whose practice is also part of OneOncology, said that continuing to work in the community setting has allowed them greater flexibility.
Dr. Eagle explained that New York Cancer & Blood Specialists tries to offer patients an appointment within 2 days of a referral, and it allows walk-in visits.
Dr. Chasky leans into the flexibility by having staff stay late, when needed, to ensure that all patients are seen. “We’re there for our patients at all hours,” Dr. Chasky said, adding that often “you don’t have that flexibility when you work for a big hospital system.”
The bottom line is community oncology can still thrive, said Nick Ferreyros, managing director of COA, “as long as we have a healthy competitive ecosystem where [we] are valued and seen as an important part of our cancer care system.”
A version of this article first appeared on Medscape.com.
Medicare Advantage Plans Not Always Advantageous
While Medicare Advantage (MA) plans are marketed as providing more generous benefits than traditional Medicare (TM), differences in the financial burden between beneficiaries switching to MA and staying with TM, are minimal, a longitudinal cohort analysis found.
In fact, according to a study by Sungchul Park, PhD, a health economist at Korea University in Seoul, and colleagues, the estimated annual out-of-pocket spending when switching to MA was $168 higher than staying in TM. That amounted to a 10.5% relative increase based on baseline out-of-pocket spending of $1597 annually among switchers, ranging widely, however, from a $133 decrease to a $469 increase. And for some, MA enrollment was associated with a higher likelihood of catastrophic financial burden.
“Our findings contrast with the notion that MA’s apparently more generous health insurance benefits lead to financial savings for enrollees,” Dr. Park and associates wrote in Annals of Internal Medicine.
The study
The analysis looked at costs for 7054 TM stayers and 1544 TM-to-MA switchers from the 2014-2020 Medical Expenditure Panel Survey, focusing on a cohort in which 18% of TM-covered individuals in year 1 switched to MA in year 2.
Comparative financial outcome measures included individual healthcare costs (out-of-pocket spending/cost sharing), financial burden (high/catastrophic), and subjective financial hardship (difficulty paying medical bills).
Although the overall out-of-pocket differences for MA were minimal and amounted to less than 1% of total healthcare expenses, MA was associated with a greater financial burden in vulnerable, especially in low-income populations. For every 100 beneficiaries with family incomes below 200% of the federal poverty level, one to six more switchers faced a catastrophic financial burden, with their out-of-pocket costs consuming more than 40% of household income in the year after switching.
The gap between the perception of lower costs and reality may be caused by a substantially heavier cost-sharing burden for certain services in MA plans, Dr. Park and associates pointed out. While MA enrollees generally paid less in some studies than the Part A hospital deductible for TM for inpatient stays of 3 days, they were more likely to face higher cost sharing for stays exceeding 7 days
Furthermore, whereas TM covers home health services without cost sharing, some MA plans have copayments. In addition, out-of-network health services can cost more. MA enrollees paid an average of $9 more for mental health services than for other in-network services and often encountered limited access to in-network providers. According to a 2021 study, only 18.2% of mental health professionals, 34.4% of cardiologists, 50.0% of psychiatrists, and 57.9% of primary care providers were included in MA networks,
An accompanying editorial noted that private MA plans will reap $83 billion in overpayments from U.S. taxpayers this year, according to Congress’s Medicare Payment Advisory Commission.
And as the data from Dr. Park and colleagues reveal, switchers don’t get much financial protection, according to primary care physician and healthcare researcher Steffi J. Woolhandler, MD, MPH, and internist David U. Himmelstein, MD, both of City University of New York at Hunter College in New York City.
“Medicare Advantage looks good when you’re healthy and don’t need much care. But when you need coverage, it often fails, leaving you with big bills and narrow choices for care,” Dr. Woolhandler said in an interview.
So how do these findings square with insurers’ hard-sell claims and enrollees’ perceptions that MA cuts out-of-pocket costs? “The likeliest explanation is that MA insurers have structured their benefits to advantage low-cost (that is, profitable) enrollees and disadvantage those requiring expensive care,” the editorial commentators wrote. For beneficiaries on inexpensive medications, MA plans would be a financial win. “But for patients requiring expensive chemotherapies, the 20% coinsurance that most MA plans charge could be financially ruinous.”
Commenting on the study but not involved in it, David A. Lipschutz, JD, LLB, associate director of the Center for Medicare Advocacy in Washington, DC, called the study an important one that provides more evidence that significant overpayments to MA plans don’t translate to better financial protections for plan enrollees, particularly lower-income individuals. “While there has been some recent movement to hold plans more accountable for providing necessary care, much more impactful action by policymakers is required to mitigate the harms of the growing privatization of the Medicare program,” he said. “MA overpayments could be redistributed to traditional Medicare in order to enrich all Medicare beneficiaries instead of just insurance companies.”
This study was supported by the National Research Foundation of Korea. Dr. Park disclosed no competing interests. One study coauthor reported support from government and not-for-profit research-funding bodies. Editorialists Dr. Woolhandler and Dr. Himmelstein had no competing interests to declare. Dr. Lipschutz disclosed Medicare advocacy work.
While Medicare Advantage (MA) plans are marketed as providing more generous benefits than traditional Medicare (TM), differences in the financial burden between beneficiaries switching to MA and staying with TM, are minimal, a longitudinal cohort analysis found.
In fact, according to a study by Sungchul Park, PhD, a health economist at Korea University in Seoul, and colleagues, the estimated annual out-of-pocket spending when switching to MA was $168 higher than staying in TM. That amounted to a 10.5% relative increase based on baseline out-of-pocket spending of $1597 annually among switchers, ranging widely, however, from a $133 decrease to a $469 increase. And for some, MA enrollment was associated with a higher likelihood of catastrophic financial burden.
“Our findings contrast with the notion that MA’s apparently more generous health insurance benefits lead to financial savings for enrollees,” Dr. Park and associates wrote in Annals of Internal Medicine.
The study
The analysis looked at costs for 7054 TM stayers and 1544 TM-to-MA switchers from the 2014-2020 Medical Expenditure Panel Survey, focusing on a cohort in which 18% of TM-covered individuals in year 1 switched to MA in year 2.
Comparative financial outcome measures included individual healthcare costs (out-of-pocket spending/cost sharing), financial burden (high/catastrophic), and subjective financial hardship (difficulty paying medical bills).
Although the overall out-of-pocket differences for MA were minimal and amounted to less than 1% of total healthcare expenses, MA was associated with a greater financial burden in vulnerable, especially in low-income populations. For every 100 beneficiaries with family incomes below 200% of the federal poverty level, one to six more switchers faced a catastrophic financial burden, with their out-of-pocket costs consuming more than 40% of household income in the year after switching.
The gap between the perception of lower costs and reality may be caused by a substantially heavier cost-sharing burden for certain services in MA plans, Dr. Park and associates pointed out. While MA enrollees generally paid less in some studies than the Part A hospital deductible for TM for inpatient stays of 3 days, they were more likely to face higher cost sharing for stays exceeding 7 days
Furthermore, whereas TM covers home health services without cost sharing, some MA plans have copayments. In addition, out-of-network health services can cost more. MA enrollees paid an average of $9 more for mental health services than for other in-network services and often encountered limited access to in-network providers. According to a 2021 study, only 18.2% of mental health professionals, 34.4% of cardiologists, 50.0% of psychiatrists, and 57.9% of primary care providers were included in MA networks,
An accompanying editorial noted that private MA plans will reap $83 billion in overpayments from U.S. taxpayers this year, according to Congress’s Medicare Payment Advisory Commission.
And as the data from Dr. Park and colleagues reveal, switchers don’t get much financial protection, according to primary care physician and healthcare researcher Steffi J. Woolhandler, MD, MPH, and internist David U. Himmelstein, MD, both of City University of New York at Hunter College in New York City.
“Medicare Advantage looks good when you’re healthy and don’t need much care. But when you need coverage, it often fails, leaving you with big bills and narrow choices for care,” Dr. Woolhandler said in an interview.
So how do these findings square with insurers’ hard-sell claims and enrollees’ perceptions that MA cuts out-of-pocket costs? “The likeliest explanation is that MA insurers have structured their benefits to advantage low-cost (that is, profitable) enrollees and disadvantage those requiring expensive care,” the editorial commentators wrote. For beneficiaries on inexpensive medications, MA plans would be a financial win. “But for patients requiring expensive chemotherapies, the 20% coinsurance that most MA plans charge could be financially ruinous.”
Commenting on the study but not involved in it, David A. Lipschutz, JD, LLB, associate director of the Center for Medicare Advocacy in Washington, DC, called the study an important one that provides more evidence that significant overpayments to MA plans don’t translate to better financial protections for plan enrollees, particularly lower-income individuals. “While there has been some recent movement to hold plans more accountable for providing necessary care, much more impactful action by policymakers is required to mitigate the harms of the growing privatization of the Medicare program,” he said. “MA overpayments could be redistributed to traditional Medicare in order to enrich all Medicare beneficiaries instead of just insurance companies.”
This study was supported by the National Research Foundation of Korea. Dr. Park disclosed no competing interests. One study coauthor reported support from government and not-for-profit research-funding bodies. Editorialists Dr. Woolhandler and Dr. Himmelstein had no competing interests to declare. Dr. Lipschutz disclosed Medicare advocacy work.
While Medicare Advantage (MA) plans are marketed as providing more generous benefits than traditional Medicare (TM), differences in the financial burden between beneficiaries switching to MA and staying with TM, are minimal, a longitudinal cohort analysis found.
In fact, according to a study by Sungchul Park, PhD, a health economist at Korea University in Seoul, and colleagues, the estimated annual out-of-pocket spending when switching to MA was $168 higher than staying in TM. That amounted to a 10.5% relative increase based on baseline out-of-pocket spending of $1597 annually among switchers, ranging widely, however, from a $133 decrease to a $469 increase. And for some, MA enrollment was associated with a higher likelihood of catastrophic financial burden.
“Our findings contrast with the notion that MA’s apparently more generous health insurance benefits lead to financial savings for enrollees,” Dr. Park and associates wrote in Annals of Internal Medicine.
The study
The analysis looked at costs for 7054 TM stayers and 1544 TM-to-MA switchers from the 2014-2020 Medical Expenditure Panel Survey, focusing on a cohort in which 18% of TM-covered individuals in year 1 switched to MA in year 2.
Comparative financial outcome measures included individual healthcare costs (out-of-pocket spending/cost sharing), financial burden (high/catastrophic), and subjective financial hardship (difficulty paying medical bills).
Although the overall out-of-pocket differences for MA were minimal and amounted to less than 1% of total healthcare expenses, MA was associated with a greater financial burden in vulnerable, especially in low-income populations. For every 100 beneficiaries with family incomes below 200% of the federal poverty level, one to six more switchers faced a catastrophic financial burden, with their out-of-pocket costs consuming more than 40% of household income in the year after switching.
The gap between the perception of lower costs and reality may be caused by a substantially heavier cost-sharing burden for certain services in MA plans, Dr. Park and associates pointed out. While MA enrollees generally paid less in some studies than the Part A hospital deductible for TM for inpatient stays of 3 days, they were more likely to face higher cost sharing for stays exceeding 7 days
Furthermore, whereas TM covers home health services without cost sharing, some MA plans have copayments. In addition, out-of-network health services can cost more. MA enrollees paid an average of $9 more for mental health services than for other in-network services and often encountered limited access to in-network providers. According to a 2021 study, only 18.2% of mental health professionals, 34.4% of cardiologists, 50.0% of psychiatrists, and 57.9% of primary care providers were included in MA networks,
An accompanying editorial noted that private MA plans will reap $83 billion in overpayments from U.S. taxpayers this year, according to Congress’s Medicare Payment Advisory Commission.
And as the data from Dr. Park and colleagues reveal, switchers don’t get much financial protection, according to primary care physician and healthcare researcher Steffi J. Woolhandler, MD, MPH, and internist David U. Himmelstein, MD, both of City University of New York at Hunter College in New York City.
“Medicare Advantage looks good when you’re healthy and don’t need much care. But when you need coverage, it often fails, leaving you with big bills and narrow choices for care,” Dr. Woolhandler said in an interview.
So how do these findings square with insurers’ hard-sell claims and enrollees’ perceptions that MA cuts out-of-pocket costs? “The likeliest explanation is that MA insurers have structured their benefits to advantage low-cost (that is, profitable) enrollees and disadvantage those requiring expensive care,” the editorial commentators wrote. For beneficiaries on inexpensive medications, MA plans would be a financial win. “But for patients requiring expensive chemotherapies, the 20% coinsurance that most MA plans charge could be financially ruinous.”
Commenting on the study but not involved in it, David A. Lipschutz, JD, LLB, associate director of the Center for Medicare Advocacy in Washington, DC, called the study an important one that provides more evidence that significant overpayments to MA plans don’t translate to better financial protections for plan enrollees, particularly lower-income individuals. “While there has been some recent movement to hold plans more accountable for providing necessary care, much more impactful action by policymakers is required to mitigate the harms of the growing privatization of the Medicare program,” he said. “MA overpayments could be redistributed to traditional Medicare in order to enrich all Medicare beneficiaries instead of just insurance companies.”
This study was supported by the National Research Foundation of Korea. Dr. Park disclosed no competing interests. One study coauthor reported support from government and not-for-profit research-funding bodies. Editorialists Dr. Woolhandler and Dr. Himmelstein had no competing interests to declare. Dr. Lipschutz disclosed Medicare advocacy work.
FROM ANNALS OF INTERNAL MEDICINE
US Hospitals Prone to Cyberattacks Like One That Impacted Patient Care at Ascension, Experts Say
In the wake of a debilitating cyberattack against one of the nation’s largest health care systems, Marvin Ruckle, a nurse at an Ascension hospital in Wichita, Kansas, said he had a frightening experience: He nearly gave a baby “the wrong dose of narcotic” because of confusing paperwork.
A May 8 ransomware attack against Ascension, a Catholic health system with 140 hospitals in at least 10 states, locked providers out of systems that track and coordinate nearly every aspect of patient care. They include its systems for electronic health records, some phones, and ones “utilized to order certain tests, procedures and medications,” the company said in a May 9 statement.
More than a dozen doctors and nurses who work for the sprawling health system told Michigan Public and KFF Health News that patient care at its hospitals across the nation was compromised in the fallout of the cyberattack over the past several weeks. Clinicians working for hospitals in three states described harrowing lapses, including delayed or lost lab results, medication errors, and an absence of routine safety checks via technology to prevent potentially fatal mistakes.
Despite a precipitous rise in cyberattacks against the health sector in recent years, a weeks-long disruption of this magnitude is beyond what most health systems are prepared for, said John S. Clark, an associate chief pharmacy officer at the University of Michigan health system.
“I don’t believe that anyone is fully prepared,” he said. Most emergency management plans “are designed around long-term downtimes that are into one, two, or three days.”
Ascension in a public statement May 9 said its care teams were “trained for these kinds of disruptions,” but did not respond to questions in early June about whether it had prepared for longer periods of downtime. Ascension said June 14 it had restored access to electronic health records across its network, but that patient “medical records and other information collected between May 8” and when the service was restored “may be temporarily inaccessible as we work to update the portal with information collected during the system downtime.”
Ruckle said he “had no training” for the cyberattack.
Back to Paper
Lisa Watson, an intensive care unit nurse at Ascension Via Christi St. Francis hospital in Wichita, described her own close call. She said she nearly administered the wrong medication to a critically ill patient because she couldn’t scan it as she normally would. “My patient probably would have passed away had I not caught it,” she said.
Watson is no stranger to using paper for patients’ medical charts, saying she did so “for probably half of my career,” before electronic health records became ubiquitous in hospitals. What happened after the cyberattack was “by no means the same.”
“When we paper-charted, we had systems in place to get those orders to other departments in a timely manner,” she said, “and those have all gone away.”
Melissa LaRue, an ICU nurse at Ascension Saint Agnes Hospital in Baltimore, described a close call with “administering the wrong dosage” of a patient’s blood pressure medication. “Luckily,” she said, it was “triple-checked and remedied before that could happen. But I think the potential for harm is there when you have so much information and paperwork that you have to go through.”
Clinicians say their hospitals have relied on slapdash workarounds, using handwritten notes, faxes, sticky notes, and basic computer spreadsheets — many devised on the fly by doctors and nurses — to care for patients.
More than a dozen other nurses and doctors, some of them without union protections, at Ascension hospitals in Michigan recounted situations in which they say patient care was compromised. Those clinicians spoke on the condition that they not be named for fear of retaliation by their employer.
An Ascension hospital emergency room doctor in Detroit said a man on the city’s east side was given a dangerous narcotic intended for another patient because of a paperwork mix-up. As a result, the patient’s breathing slowed to the point that he had to be put on a ventilator. “We intubated him and we sent him to the ICU because he got the wrong medication.”
A nurse in a Michigan Ascension hospital ER said a woman with low blood sugar and “altered mental status” went into cardiac arrest and died after staff said they waited four hours for lab results they needed to determine how to treat her, but never received. “If I started having crushing chest pain in the middle of work and thought I was having a big one, I would grab someone to drive me down the street to another hospital,” the same ER nurse said.
Similar concerns reportedly led a travel nurse at an Ascension hospital in Indiana to quit. “I just want to warn those patients that are coming to any of the Ascension facilities that there will be delays in care. There is potential for error and for harm,” Justin Neisser told CBS4 in Indianapolis in May.
Several nurses and doctors at Ascension hospitals said they feared the errors they’ve witnessed since the cyberattack began could threaten their professional licenses. “This is how a RaDonda Vaught happens,” one nurse said, referring to the Tennessee nurse who was convicted of criminally negligent homicide in 2022 for a fatal drug error.
Reporters were not able to review records to verify clinicians’ claims because of privacy laws surrounding patients’ medical information that apply to health care professionals.
Ascension declined to answer questions about claims that care has been affected by the ransomware attack. “As we have made clear throughout this cyber attack which has impacted our system and our dedicated clinical providers, caring for our patients is our highest priority,” Sean Fitzpatrick, Ascension’s vice president of external communications, said via email on June 3. “We are confident that our care providers in our hospitals and facilities continue to provide quality medical care.”
The federal government requires hospitals to protect patients’ sensitive health data, according to cybersecurity experts. However, there are no federal requirements for hospitals to prevent or prepare for cyberattacks that could compromise their electronic systems.
Hospitals: ‘The No.1 Target of Ransomware’
“We’ve started to think about these as public health issues and disasters on the scale of earthquakes or hurricanes,” said Jeff Tully, a co-director of the Center for Healthcare Cybersecurity at the University of California-San Diego. “These types of cybersecurity incidents should be thought of as a matter of when, and not if.”
Josh Corman, a cybersecurity expert and advocate, said ransom crews regard hospitals as the perfect prey: “They have terrible security and they’ll pay. So almost immediately, hospitals went to the No. 1 target of ransomware.”
In 2023, the health sector experienced the largest share of ransomware attacks of 16 infrastructure sectors considered vital to national security or safety, according to an FBI report on internet crimes. In March, the federal Department of Health and Human Services said reported large breaches involving ransomware had jumped by 264% over the past five years.
A cyberattack this year on Change Healthcare, a unit of UnitedHealth Group’s Optum division that processes billions of health care transactions every year, crippled the business of providers, pharmacies, and hospitals.
In May, UnitedHealth Group CEO Andrew Witty told lawmakers the company paid a $22 million ransom as a result of the Change Healthcare attack — which occurred after hackers accessed a company portal that didn’t have multifactor authentication, a basic cybersecurity tool.
The Biden administration in recent months has pushed to bolster health care cybersecurity standards, but it’s not clear which new measures will be required.
In January, HHS nudged companies to improve email security, add multifactor authentication, and institute cybersecurity training and testing, among other voluntary measures. The Centers for Medicare & Medicaid Services is expected to release new requirements for hospitals, but the scope and timing are unclear. The same is true of an update HHS is expected to make to patient privacy regulations.
HHS said the voluntary measures “will inform the creation of new enforceable cybersecurity standards,” department spokesperson Jeff Nesbit said in a statement.
“The recent cyberattack at Ascension only underscores the need for everyone in the health care ecosystem to do their part to secure their systems and protect patients,” Nesbit said.
Meanwhile, lobbyists for the hospital industry contend cybersecurity mandates or penalties are misplaced and would curtail hospitals’ resources to fend off attacks.
“Hospitals and health systems are not the primary source of cyber risk exposure facing the health care sector,” the American Hospital Association, the largest lobbying group for U.S. hospitals, said in an April statement prepared for U.S. House lawmakers. Most large data breaches that hit hospitals in 2023 originated with third-party “business associates” or other health entities, including CMS itself, the AHA statement said.
Hospitals consolidating into large multistate health systems face increased risk of data breaches and ransomware attacks, according to one study. Ascension in 2022 was the third-largest hospital chain in the U.S. by number of beds, according to the most recent data from the federal Agency for Healthcare Research and Quality.
And while cybersecurity regulations can quickly become outdated, they can at least make it clear that if health systems fail to implement basic protections there “should be consequences for that,” Jim Bagian, a former director of the National Center for Patient Safety at the Veterans Health Administration, told Michigan Public’s Stateside.
Patients can pay the price when lapses occur. Those in hospital care face a greater likelihood of death during a cyberattack, according to researchers at the University of Minnesota School of Public Health.
Workers concerned about patient safety at Ascension hospitals in Michigan have called for the company to make changes.
“We implore Ascension to recognize the internal problems that continue to plague its hospitals, both publicly and transparently,” said Dina Carlisle, a nurse and the president of the OPEIU Local 40 union, which represents nurses at Ascension Providence Rochester. At least 125 staff members at that Ascension hospital have signed a petition asking administrators to temporarily reduce elective surgeries and nonemergency patient admissions, like under the protocols many hospitals adopted early in the covid-19 pandemic.
Watson, the Kansas ICU nurse, said in late May that nurses had urged management to bring in more nurses to help manage the workflow. “Everything that we say has fallen on deaf ears,” she said.
“It is very hard to be a nurse at Ascension right now,” Watson said in late May. “It is very hard to be a patient at Ascension right now.”
If you’re a patient or worker at an Ascension hospital and would like to tell KFF Health News about your experiences, click here to share your story with us.
KFF Health News is a national newsroom that produces in-depth journalism about health issues and is one of the core operating programs at KFF—an independent source of health policy research, polling, and journalism. Learn more about KFF.
In the wake of a debilitating cyberattack against one of the nation’s largest health care systems, Marvin Ruckle, a nurse at an Ascension hospital in Wichita, Kansas, said he had a frightening experience: He nearly gave a baby “the wrong dose of narcotic” because of confusing paperwork.
A May 8 ransomware attack against Ascension, a Catholic health system with 140 hospitals in at least 10 states, locked providers out of systems that track and coordinate nearly every aspect of patient care. They include its systems for electronic health records, some phones, and ones “utilized to order certain tests, procedures and medications,” the company said in a May 9 statement.
More than a dozen doctors and nurses who work for the sprawling health system told Michigan Public and KFF Health News that patient care at its hospitals across the nation was compromised in the fallout of the cyberattack over the past several weeks. Clinicians working for hospitals in three states described harrowing lapses, including delayed or lost lab results, medication errors, and an absence of routine safety checks via technology to prevent potentially fatal mistakes.
Despite a precipitous rise in cyberattacks against the health sector in recent years, a weeks-long disruption of this magnitude is beyond what most health systems are prepared for, said John S. Clark, an associate chief pharmacy officer at the University of Michigan health system.
“I don’t believe that anyone is fully prepared,” he said. Most emergency management plans “are designed around long-term downtimes that are into one, two, or three days.”
Ascension in a public statement May 9 said its care teams were “trained for these kinds of disruptions,” but did not respond to questions in early June about whether it had prepared for longer periods of downtime. Ascension said June 14 it had restored access to electronic health records across its network, but that patient “medical records and other information collected between May 8” and when the service was restored “may be temporarily inaccessible as we work to update the portal with information collected during the system downtime.”
Ruckle said he “had no training” for the cyberattack.
Back to Paper
Lisa Watson, an intensive care unit nurse at Ascension Via Christi St. Francis hospital in Wichita, described her own close call. She said she nearly administered the wrong medication to a critically ill patient because she couldn’t scan it as she normally would. “My patient probably would have passed away had I not caught it,” she said.
Watson is no stranger to using paper for patients’ medical charts, saying she did so “for probably half of my career,” before electronic health records became ubiquitous in hospitals. What happened after the cyberattack was “by no means the same.”
“When we paper-charted, we had systems in place to get those orders to other departments in a timely manner,” she said, “and those have all gone away.”
Melissa LaRue, an ICU nurse at Ascension Saint Agnes Hospital in Baltimore, described a close call with “administering the wrong dosage” of a patient’s blood pressure medication. “Luckily,” she said, it was “triple-checked and remedied before that could happen. But I think the potential for harm is there when you have so much information and paperwork that you have to go through.”
Clinicians say their hospitals have relied on slapdash workarounds, using handwritten notes, faxes, sticky notes, and basic computer spreadsheets — many devised on the fly by doctors and nurses — to care for patients.
More than a dozen other nurses and doctors, some of them without union protections, at Ascension hospitals in Michigan recounted situations in which they say patient care was compromised. Those clinicians spoke on the condition that they not be named for fear of retaliation by their employer.
An Ascension hospital emergency room doctor in Detroit said a man on the city’s east side was given a dangerous narcotic intended for another patient because of a paperwork mix-up. As a result, the patient’s breathing slowed to the point that he had to be put on a ventilator. “We intubated him and we sent him to the ICU because he got the wrong medication.”
A nurse in a Michigan Ascension hospital ER said a woman with low blood sugar and “altered mental status” went into cardiac arrest and died after staff said they waited four hours for lab results they needed to determine how to treat her, but never received. “If I started having crushing chest pain in the middle of work and thought I was having a big one, I would grab someone to drive me down the street to another hospital,” the same ER nurse said.
Similar concerns reportedly led a travel nurse at an Ascension hospital in Indiana to quit. “I just want to warn those patients that are coming to any of the Ascension facilities that there will be delays in care. There is potential for error and for harm,” Justin Neisser told CBS4 in Indianapolis in May.
Several nurses and doctors at Ascension hospitals said they feared the errors they’ve witnessed since the cyberattack began could threaten their professional licenses. “This is how a RaDonda Vaught happens,” one nurse said, referring to the Tennessee nurse who was convicted of criminally negligent homicide in 2022 for a fatal drug error.
Reporters were not able to review records to verify clinicians’ claims because of privacy laws surrounding patients’ medical information that apply to health care professionals.
Ascension declined to answer questions about claims that care has been affected by the ransomware attack. “As we have made clear throughout this cyber attack which has impacted our system and our dedicated clinical providers, caring for our patients is our highest priority,” Sean Fitzpatrick, Ascension’s vice president of external communications, said via email on June 3. “We are confident that our care providers in our hospitals and facilities continue to provide quality medical care.”
The federal government requires hospitals to protect patients’ sensitive health data, according to cybersecurity experts. However, there are no federal requirements for hospitals to prevent or prepare for cyberattacks that could compromise their electronic systems.
Hospitals: ‘The No.1 Target of Ransomware’
“We’ve started to think about these as public health issues and disasters on the scale of earthquakes or hurricanes,” said Jeff Tully, a co-director of the Center for Healthcare Cybersecurity at the University of California-San Diego. “These types of cybersecurity incidents should be thought of as a matter of when, and not if.”
Josh Corman, a cybersecurity expert and advocate, said ransom crews regard hospitals as the perfect prey: “They have terrible security and they’ll pay. So almost immediately, hospitals went to the No. 1 target of ransomware.”
In 2023, the health sector experienced the largest share of ransomware attacks of 16 infrastructure sectors considered vital to national security or safety, according to an FBI report on internet crimes. In March, the federal Department of Health and Human Services said reported large breaches involving ransomware had jumped by 264% over the past five years.
A cyberattack this year on Change Healthcare, a unit of UnitedHealth Group’s Optum division that processes billions of health care transactions every year, crippled the business of providers, pharmacies, and hospitals.
In May, UnitedHealth Group CEO Andrew Witty told lawmakers the company paid a $22 million ransom as a result of the Change Healthcare attack — which occurred after hackers accessed a company portal that didn’t have multifactor authentication, a basic cybersecurity tool.
The Biden administration in recent months has pushed to bolster health care cybersecurity standards, but it’s not clear which new measures will be required.
In January, HHS nudged companies to improve email security, add multifactor authentication, and institute cybersecurity training and testing, among other voluntary measures. The Centers for Medicare & Medicaid Services is expected to release new requirements for hospitals, but the scope and timing are unclear. The same is true of an update HHS is expected to make to patient privacy regulations.
HHS said the voluntary measures “will inform the creation of new enforceable cybersecurity standards,” department spokesperson Jeff Nesbit said in a statement.
“The recent cyberattack at Ascension only underscores the need for everyone in the health care ecosystem to do their part to secure their systems and protect patients,” Nesbit said.
Meanwhile, lobbyists for the hospital industry contend cybersecurity mandates or penalties are misplaced and would curtail hospitals’ resources to fend off attacks.
“Hospitals and health systems are not the primary source of cyber risk exposure facing the health care sector,” the American Hospital Association, the largest lobbying group for U.S. hospitals, said in an April statement prepared for U.S. House lawmakers. Most large data breaches that hit hospitals in 2023 originated with third-party “business associates” or other health entities, including CMS itself, the AHA statement said.
Hospitals consolidating into large multistate health systems face increased risk of data breaches and ransomware attacks, according to one study. Ascension in 2022 was the third-largest hospital chain in the U.S. by number of beds, according to the most recent data from the federal Agency for Healthcare Research and Quality.
And while cybersecurity regulations can quickly become outdated, they can at least make it clear that if health systems fail to implement basic protections there “should be consequences for that,” Jim Bagian, a former director of the National Center for Patient Safety at the Veterans Health Administration, told Michigan Public’s Stateside.
Patients can pay the price when lapses occur. Those in hospital care face a greater likelihood of death during a cyberattack, according to researchers at the University of Minnesota School of Public Health.
Workers concerned about patient safety at Ascension hospitals in Michigan have called for the company to make changes.
“We implore Ascension to recognize the internal problems that continue to plague its hospitals, both publicly and transparently,” said Dina Carlisle, a nurse and the president of the OPEIU Local 40 union, which represents nurses at Ascension Providence Rochester. At least 125 staff members at that Ascension hospital have signed a petition asking administrators to temporarily reduce elective surgeries and nonemergency patient admissions, like under the protocols many hospitals adopted early in the covid-19 pandemic.
Watson, the Kansas ICU nurse, said in late May that nurses had urged management to bring in more nurses to help manage the workflow. “Everything that we say has fallen on deaf ears,” she said.
“It is very hard to be a nurse at Ascension right now,” Watson said in late May. “It is very hard to be a patient at Ascension right now.”
If you’re a patient or worker at an Ascension hospital and would like to tell KFF Health News about your experiences, click here to share your story with us.
KFF Health News is a national newsroom that produces in-depth journalism about health issues and is one of the core operating programs at KFF—an independent source of health policy research, polling, and journalism. Learn more about KFF.
In the wake of a debilitating cyberattack against one of the nation’s largest health care systems, Marvin Ruckle, a nurse at an Ascension hospital in Wichita, Kansas, said he had a frightening experience: He nearly gave a baby “the wrong dose of narcotic” because of confusing paperwork.
A May 8 ransomware attack against Ascension, a Catholic health system with 140 hospitals in at least 10 states, locked providers out of systems that track and coordinate nearly every aspect of patient care. They include its systems for electronic health records, some phones, and ones “utilized to order certain tests, procedures and medications,” the company said in a May 9 statement.
More than a dozen doctors and nurses who work for the sprawling health system told Michigan Public and KFF Health News that patient care at its hospitals across the nation was compromised in the fallout of the cyberattack over the past several weeks. Clinicians working for hospitals in three states described harrowing lapses, including delayed or lost lab results, medication errors, and an absence of routine safety checks via technology to prevent potentially fatal mistakes.
Despite a precipitous rise in cyberattacks against the health sector in recent years, a weeks-long disruption of this magnitude is beyond what most health systems are prepared for, said John S. Clark, an associate chief pharmacy officer at the University of Michigan health system.
“I don’t believe that anyone is fully prepared,” he said. Most emergency management plans “are designed around long-term downtimes that are into one, two, or three days.”
Ascension in a public statement May 9 said its care teams were “trained for these kinds of disruptions,” but did not respond to questions in early June about whether it had prepared for longer periods of downtime. Ascension said June 14 it had restored access to electronic health records across its network, but that patient “medical records and other information collected between May 8” and when the service was restored “may be temporarily inaccessible as we work to update the portal with information collected during the system downtime.”
Ruckle said he “had no training” for the cyberattack.
Back to Paper
Lisa Watson, an intensive care unit nurse at Ascension Via Christi St. Francis hospital in Wichita, described her own close call. She said she nearly administered the wrong medication to a critically ill patient because she couldn’t scan it as she normally would. “My patient probably would have passed away had I not caught it,” she said.
Watson is no stranger to using paper for patients’ medical charts, saying she did so “for probably half of my career,” before electronic health records became ubiquitous in hospitals. What happened after the cyberattack was “by no means the same.”
“When we paper-charted, we had systems in place to get those orders to other departments in a timely manner,” she said, “and those have all gone away.”
Melissa LaRue, an ICU nurse at Ascension Saint Agnes Hospital in Baltimore, described a close call with “administering the wrong dosage” of a patient’s blood pressure medication. “Luckily,” she said, it was “triple-checked and remedied before that could happen. But I think the potential for harm is there when you have so much information and paperwork that you have to go through.”
Clinicians say their hospitals have relied on slapdash workarounds, using handwritten notes, faxes, sticky notes, and basic computer spreadsheets — many devised on the fly by doctors and nurses — to care for patients.
More than a dozen other nurses and doctors, some of them without union protections, at Ascension hospitals in Michigan recounted situations in which they say patient care was compromised. Those clinicians spoke on the condition that they not be named for fear of retaliation by their employer.
An Ascension hospital emergency room doctor in Detroit said a man on the city’s east side was given a dangerous narcotic intended for another patient because of a paperwork mix-up. As a result, the patient’s breathing slowed to the point that he had to be put on a ventilator. “We intubated him and we sent him to the ICU because he got the wrong medication.”
A nurse in a Michigan Ascension hospital ER said a woman with low blood sugar and “altered mental status” went into cardiac arrest and died after staff said they waited four hours for lab results they needed to determine how to treat her, but never received. “If I started having crushing chest pain in the middle of work and thought I was having a big one, I would grab someone to drive me down the street to another hospital,” the same ER nurse said.
Similar concerns reportedly led a travel nurse at an Ascension hospital in Indiana to quit. “I just want to warn those patients that are coming to any of the Ascension facilities that there will be delays in care. There is potential for error and for harm,” Justin Neisser told CBS4 in Indianapolis in May.
Several nurses and doctors at Ascension hospitals said they feared the errors they’ve witnessed since the cyberattack began could threaten their professional licenses. “This is how a RaDonda Vaught happens,” one nurse said, referring to the Tennessee nurse who was convicted of criminally negligent homicide in 2022 for a fatal drug error.
Reporters were not able to review records to verify clinicians’ claims because of privacy laws surrounding patients’ medical information that apply to health care professionals.
Ascension declined to answer questions about claims that care has been affected by the ransomware attack. “As we have made clear throughout this cyber attack which has impacted our system and our dedicated clinical providers, caring for our patients is our highest priority,” Sean Fitzpatrick, Ascension’s vice president of external communications, said via email on June 3. “We are confident that our care providers in our hospitals and facilities continue to provide quality medical care.”
The federal government requires hospitals to protect patients’ sensitive health data, according to cybersecurity experts. However, there are no federal requirements for hospitals to prevent or prepare for cyberattacks that could compromise their electronic systems.
Hospitals: ‘The No.1 Target of Ransomware’
“We’ve started to think about these as public health issues and disasters on the scale of earthquakes or hurricanes,” said Jeff Tully, a co-director of the Center for Healthcare Cybersecurity at the University of California-San Diego. “These types of cybersecurity incidents should be thought of as a matter of when, and not if.”
Josh Corman, a cybersecurity expert and advocate, said ransom crews regard hospitals as the perfect prey: “They have terrible security and they’ll pay. So almost immediately, hospitals went to the No. 1 target of ransomware.”
In 2023, the health sector experienced the largest share of ransomware attacks of 16 infrastructure sectors considered vital to national security or safety, according to an FBI report on internet crimes. In March, the federal Department of Health and Human Services said reported large breaches involving ransomware had jumped by 264% over the past five years.
A cyberattack this year on Change Healthcare, a unit of UnitedHealth Group’s Optum division that processes billions of health care transactions every year, crippled the business of providers, pharmacies, and hospitals.
In May, UnitedHealth Group CEO Andrew Witty told lawmakers the company paid a $22 million ransom as a result of the Change Healthcare attack — which occurred after hackers accessed a company portal that didn’t have multifactor authentication, a basic cybersecurity tool.
The Biden administration in recent months has pushed to bolster health care cybersecurity standards, but it’s not clear which new measures will be required.
In January, HHS nudged companies to improve email security, add multifactor authentication, and institute cybersecurity training and testing, among other voluntary measures. The Centers for Medicare & Medicaid Services is expected to release new requirements for hospitals, but the scope and timing are unclear. The same is true of an update HHS is expected to make to patient privacy regulations.
HHS said the voluntary measures “will inform the creation of new enforceable cybersecurity standards,” department spokesperson Jeff Nesbit said in a statement.
“The recent cyberattack at Ascension only underscores the need for everyone in the health care ecosystem to do their part to secure their systems and protect patients,” Nesbit said.
Meanwhile, lobbyists for the hospital industry contend cybersecurity mandates or penalties are misplaced and would curtail hospitals’ resources to fend off attacks.
“Hospitals and health systems are not the primary source of cyber risk exposure facing the health care sector,” the American Hospital Association, the largest lobbying group for U.S. hospitals, said in an April statement prepared for U.S. House lawmakers. Most large data breaches that hit hospitals in 2023 originated with third-party “business associates” or other health entities, including CMS itself, the AHA statement said.
Hospitals consolidating into large multistate health systems face increased risk of data breaches and ransomware attacks, according to one study. Ascension in 2022 was the third-largest hospital chain in the U.S. by number of beds, according to the most recent data from the federal Agency for Healthcare Research and Quality.
And while cybersecurity regulations can quickly become outdated, they can at least make it clear that if health systems fail to implement basic protections there “should be consequences for that,” Jim Bagian, a former director of the National Center for Patient Safety at the Veterans Health Administration, told Michigan Public’s Stateside.
Patients can pay the price when lapses occur. Those in hospital care face a greater likelihood of death during a cyberattack, according to researchers at the University of Minnesota School of Public Health.
Workers concerned about patient safety at Ascension hospitals in Michigan have called for the company to make changes.
“We implore Ascension to recognize the internal problems that continue to plague its hospitals, both publicly and transparently,” said Dina Carlisle, a nurse and the president of the OPEIU Local 40 union, which represents nurses at Ascension Providence Rochester. At least 125 staff members at that Ascension hospital have signed a petition asking administrators to temporarily reduce elective surgeries and nonemergency patient admissions, like under the protocols many hospitals adopted early in the covid-19 pandemic.
Watson, the Kansas ICU nurse, said in late May that nurses had urged management to bring in more nurses to help manage the workflow. “Everything that we say has fallen on deaf ears,” she said.
“It is very hard to be a nurse at Ascension right now,” Watson said in late May. “It is very hard to be a patient at Ascension right now.”
If you’re a patient or worker at an Ascension hospital and would like to tell KFF Health News about your experiences, click here to share your story with us.
KFF Health News is a national newsroom that produces in-depth journalism about health issues and is one of the core operating programs at KFF—an independent source of health policy research, polling, and journalism. Learn more about KFF.
DLBCL: Glofitamab Plus Chemo Boosts Survival
“Glofitamab is the first CD20xCD3 bispecific antibody to demonstrate an overall survival benefit in DLBCL in a randomized phase 3 trial,” said first author Jeremy Abramson, MD, of the Massachusetts General Hospital Cancer Center, Boston, Massachusetts, in a press briefing at the annual meeting of the European Hematology Association (EHA) in Madrid.
“These results support the use of glofitamab GemOx as new, off-the-shelf treatment for relapsed/refractory DLBCL in patients who are transplant ineligible in the second-line or later setting,” he said.
The findings are from the phase 3 STARGLO study involving 274 patients with R/R DLBCL who had previously been treated either with at least two prior lines of therapy, or—if only one prior line of therapy—were determined to be ineligible for autologous stem cell transplant (ASCT).
At a median follow-up of 21 months, those treated with glofitamab combined with GemOx had a significantly higher median overall survival of 25.5 months, compared with those treated with the standard of care of rituximab and GemOx (12.9 months; hazard ratio [HR], 0.62; P = .006).
“The results show a 38% lower risk of death with the glofitamab plus GemOx, compared with [the rituximab regimen],” Dr. Abramson said.
Secondary endpoints showed consistent benefits with the glofitamab regimen, with significant improvements in progression-free survival and complete remission.
Unmet Need for Accessible Therapies
Relapsed/refractory DLBCL, the most common form of non-Hodgkin lymphoma (NHL) in the United States, is an aggressive blood cancer. The standard second-line therapy is high-dose chemotherapy followed by ASCT. However, factors including older age or coexisting medical conditions can compromise response, and those who relapse or are refractory to subsequent therapies have poor outcomes.
“Relapsed DLBCL in the second-line setting or later continues to represent an area of medical need,” Dr. Abramson said.
While several CD20xCD3 bispecific antibody drugs are under development to address the need, glofitamab was the first off-the-shelf, fixed-duration bispecific antibody to receive accelerated approval from the US Food and Drug Administration (FDA) for the treatment of R/R DLBCL, specifically as monotherapy after two or more lines of systemic therapy.
That approval was based on results from a pivotal phase 1/2 study, which showed high rates of deep and durable complete remission with the monotherapy.
To further evaluate glofitamab in combination with GemOx, the authors conducted the multicenter, open-label STARGLO trial, which extended enrollment to patients with just one prior therapy if they were determined to be stem cell transplant ineligible. Patients were also required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2.
Patients were randomized 2:1 either to treatment with glofitamab combined with GemOx, involving 8 cycles, in addition to 4 cycles of glofitamab monotherapy (n = 183), or to the rituximab plus GemOx regimen in 8 cycles (n = 91).
Overall, 153 (55.8%) of patients had primary refractory disease and 166 (60.6%) were refractory to their last therapy. The median age was 68, and 37% had two or more lines of therapy, including some who had received chimeric antigen receptor T-cell (CAR T) therapy (8.8% in the glofitamab group and 7.1% in the rituximab group).
In addition to the significant overall survival benefit, the glofitamab regimen also showed significantly improved progression-free survival at a median follow-up of 16.1 months, as observed by IRC-assessed PFS, with a median progression-free survival rate of 13.8 months vs 3.6 months (HR, 0.40; P < .0001).
The complete remission rate was doubled with glofitamab-GemOx, with a rate of 58.5% vs 25.3%, respectively; P < .0001.
Similar results were observed in subgroups, including relapsed vs refractory patients and those treated as a second or third line of care.
The median number of cycles received was higher among those receiving glofitamab (11 vs four cycles).
Adverse event (AE) rates were higher with glofitamab vs rituximab, including grade 3-4 AEs (69.4 vs 36.4%), grade 5 AEs (8.3 vs 4.5%; primarily driven by an imbalance of COVID-19 AEs), and serious AEs (54.4 vs 17.0%; primarily cytokine release syndrome [CRS]).
CRS was the most frequently reported AE in the glofitamab group (grade 1: 31.4%; grade 2: 10.5%; and grade 3: 2.3%), and events consistent with immune effector cell–associated neurotoxicity syndrome were reported in four patients (2.3%), all of which were concurrent with CRS.
Other AEs were consistent with the known risks associated with the therapy regimens.
“We found that with glofitamab GemOx, the toxicities were manageable, and the most common toxicity of CRS was predominantly low-grade and occurred with step-up dosing in cycle one and was completely reversible,” Dr. Abramson said.
He noted that the higher rate of grade 5 AEs with glofitamab GemOx “was far outweighed by the survival benefit for disease control.”
Overall, “these findings represent the best outcomes observed in a phase 3 trial for relapsed/refractory diffuse large B-cell lymphoma patients who are considered transplant ineligible,” Dr. Abramson said in an interview.
Improved Accessibility Vs CAR-T Therapy
Among key developments in the treatment of R/R DLBCL has been the advent and approval of potentially highly effective CAR T-cell therapy, with the anti-CD19 CAR T cell isiocabtagene maraleucel also FDA approved in the non–transplant eligible DLBCL second-line setting.
Asked in the press briefing about the role of glofitamab GemOx in relation to CAR T cell’s significant benefits, Dr. Abramson underscored the important limitations in CAR T-cell accessibility.
“What I would say is a rising tide lifts all boats,” he responded. “It’s great to have multiple effective immunotherapy strategies.”
However, “CAR T cells of course are not available to most people in the US or worldwide,” he explained.
“They are more difficult to access, they require lymphodepleting chemotherapy, and so ultimately, the majority of patients who could potentially benefit from a CAR T cell probably don’t have access to them in the first place.”
He noted that “the appeal of a regimen like [glofitamab] is that it is an off-the-shelf, targeted immunotherapy combined with a well-tolerated chemotherapy backbone and should be more broadly accessible outside of just tertiary care centers in major cities.”
Long-Term Durability?
Looking ahead, Dr. Abramson noted that a key issue of focus is how long the encouraging results actually last.
“The major ongoing question with this trial is the long-term durability of remissions,” he said.
“Thus far, with a median of 21 months of follow-up for overall survival, the results are encouraging but longer follow-up is needed,” he added.
“Further trials are needed in a broader large B-cell lymphoma population as this trial was limited to DLBCL not otherwise specified, so did not include patients with transformed lymphoma, primary mediastinal B-cell lymphoma, high grade B-cell lymphoma, etc.”
Is Chemo Necessary?
Commenting on the findings, Jonathan W. Friedberg, MD, director of the Wilmot Cancer Institute, University of Rochester School of Medicine, in Rochester, New York, underscored that, “given the overall survival benefit, these findings are clearly clinically significant.”
Noting that “these results add to evidence of high activity of bispecific antibodies in this disease,” Dr. Friedberg speculated on the role of chemotherapy with the therapy.
“Indeed, an important question in this study is whether the addition of chemotherapy to glofitamab is necessary, as high response rates with durable responses in patients who achieve complete remission have been demonstrated with single agent bispecific antibody therapy,” he said.
With the durability of CAR T therapy shown in long-term follow-up of trials to exceed 5 years, Dr. Friedberg added that “it is not known how bispecific antibody therapy, with or without chemotherapy, compares to CAR T-cell therapy and how to sequence CAR T and bispecific antibody therapy.”
Dr. Friedberg agreed that longer-term results are needed get a clearer, fuller picture of the therapy’s effects.
“I have no doubt that the overall survival benefit will endure, but in DLBCL our goal should be cure, and whether glofitamab cures as many patients as CAR T-cell therapy is not currently known and will require further follow-up of this and other trials.”
The study was sponsored by F. Hoffman-La Roche Ltd. Dr. Abramson reported ties with AbbVie, ADC Therapeutics, AstraZeneca, BeiGene, BMS, Cellectar, Caribou Biosciences, Celgene, Genentech, Gilead, Incyte, Interius, Janssen, Lilly, Novartis, F. Hoffmann-La Roche Ltd, Seagen, and Takeda. Dr. Friedberg had no disclosures.
“Glofitamab is the first CD20xCD3 bispecific antibody to demonstrate an overall survival benefit in DLBCL in a randomized phase 3 trial,” said first author Jeremy Abramson, MD, of the Massachusetts General Hospital Cancer Center, Boston, Massachusetts, in a press briefing at the annual meeting of the European Hematology Association (EHA) in Madrid.
“These results support the use of glofitamab GemOx as new, off-the-shelf treatment for relapsed/refractory DLBCL in patients who are transplant ineligible in the second-line or later setting,” he said.
The findings are from the phase 3 STARGLO study involving 274 patients with R/R DLBCL who had previously been treated either with at least two prior lines of therapy, or—if only one prior line of therapy—were determined to be ineligible for autologous stem cell transplant (ASCT).
At a median follow-up of 21 months, those treated with glofitamab combined with GemOx had a significantly higher median overall survival of 25.5 months, compared with those treated with the standard of care of rituximab and GemOx (12.9 months; hazard ratio [HR], 0.62; P = .006).
“The results show a 38% lower risk of death with the glofitamab plus GemOx, compared with [the rituximab regimen],” Dr. Abramson said.
Secondary endpoints showed consistent benefits with the glofitamab regimen, with significant improvements in progression-free survival and complete remission.
Unmet Need for Accessible Therapies
Relapsed/refractory DLBCL, the most common form of non-Hodgkin lymphoma (NHL) in the United States, is an aggressive blood cancer. The standard second-line therapy is high-dose chemotherapy followed by ASCT. However, factors including older age or coexisting medical conditions can compromise response, and those who relapse or are refractory to subsequent therapies have poor outcomes.
“Relapsed DLBCL in the second-line setting or later continues to represent an area of medical need,” Dr. Abramson said.
While several CD20xCD3 bispecific antibody drugs are under development to address the need, glofitamab was the first off-the-shelf, fixed-duration bispecific antibody to receive accelerated approval from the US Food and Drug Administration (FDA) for the treatment of R/R DLBCL, specifically as monotherapy after two or more lines of systemic therapy.
That approval was based on results from a pivotal phase 1/2 study, which showed high rates of deep and durable complete remission with the monotherapy.
To further evaluate glofitamab in combination with GemOx, the authors conducted the multicenter, open-label STARGLO trial, which extended enrollment to patients with just one prior therapy if they were determined to be stem cell transplant ineligible. Patients were also required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2.
Patients were randomized 2:1 either to treatment with glofitamab combined with GemOx, involving 8 cycles, in addition to 4 cycles of glofitamab monotherapy (n = 183), or to the rituximab plus GemOx regimen in 8 cycles (n = 91).
Overall, 153 (55.8%) of patients had primary refractory disease and 166 (60.6%) were refractory to their last therapy. The median age was 68, and 37% had two or more lines of therapy, including some who had received chimeric antigen receptor T-cell (CAR T) therapy (8.8% in the glofitamab group and 7.1% in the rituximab group).
In addition to the significant overall survival benefit, the glofitamab regimen also showed significantly improved progression-free survival at a median follow-up of 16.1 months, as observed by IRC-assessed PFS, with a median progression-free survival rate of 13.8 months vs 3.6 months (HR, 0.40; P < .0001).
The complete remission rate was doubled with glofitamab-GemOx, with a rate of 58.5% vs 25.3%, respectively; P < .0001.
Similar results were observed in subgroups, including relapsed vs refractory patients and those treated as a second or third line of care.
The median number of cycles received was higher among those receiving glofitamab (11 vs four cycles).
Adverse event (AE) rates were higher with glofitamab vs rituximab, including grade 3-4 AEs (69.4 vs 36.4%), grade 5 AEs (8.3 vs 4.5%; primarily driven by an imbalance of COVID-19 AEs), and serious AEs (54.4 vs 17.0%; primarily cytokine release syndrome [CRS]).
CRS was the most frequently reported AE in the glofitamab group (grade 1: 31.4%; grade 2: 10.5%; and grade 3: 2.3%), and events consistent with immune effector cell–associated neurotoxicity syndrome were reported in four patients (2.3%), all of which were concurrent with CRS.
Other AEs were consistent with the known risks associated with the therapy regimens.
“We found that with glofitamab GemOx, the toxicities were manageable, and the most common toxicity of CRS was predominantly low-grade and occurred with step-up dosing in cycle one and was completely reversible,” Dr. Abramson said.
He noted that the higher rate of grade 5 AEs with glofitamab GemOx “was far outweighed by the survival benefit for disease control.”
Overall, “these findings represent the best outcomes observed in a phase 3 trial for relapsed/refractory diffuse large B-cell lymphoma patients who are considered transplant ineligible,” Dr. Abramson said in an interview.
Improved Accessibility Vs CAR-T Therapy
Among key developments in the treatment of R/R DLBCL has been the advent and approval of potentially highly effective CAR T-cell therapy, with the anti-CD19 CAR T cell isiocabtagene maraleucel also FDA approved in the non–transplant eligible DLBCL second-line setting.
Asked in the press briefing about the role of glofitamab GemOx in relation to CAR T cell’s significant benefits, Dr. Abramson underscored the important limitations in CAR T-cell accessibility.
“What I would say is a rising tide lifts all boats,” he responded. “It’s great to have multiple effective immunotherapy strategies.”
However, “CAR T cells of course are not available to most people in the US or worldwide,” he explained.
“They are more difficult to access, they require lymphodepleting chemotherapy, and so ultimately, the majority of patients who could potentially benefit from a CAR T cell probably don’t have access to them in the first place.”
He noted that “the appeal of a regimen like [glofitamab] is that it is an off-the-shelf, targeted immunotherapy combined with a well-tolerated chemotherapy backbone and should be more broadly accessible outside of just tertiary care centers in major cities.”
Long-Term Durability?
Looking ahead, Dr. Abramson noted that a key issue of focus is how long the encouraging results actually last.
“The major ongoing question with this trial is the long-term durability of remissions,” he said.
“Thus far, with a median of 21 months of follow-up for overall survival, the results are encouraging but longer follow-up is needed,” he added.
“Further trials are needed in a broader large B-cell lymphoma population as this trial was limited to DLBCL not otherwise specified, so did not include patients with transformed lymphoma, primary mediastinal B-cell lymphoma, high grade B-cell lymphoma, etc.”
Is Chemo Necessary?
Commenting on the findings, Jonathan W. Friedberg, MD, director of the Wilmot Cancer Institute, University of Rochester School of Medicine, in Rochester, New York, underscored that, “given the overall survival benefit, these findings are clearly clinically significant.”
Noting that “these results add to evidence of high activity of bispecific antibodies in this disease,” Dr. Friedberg speculated on the role of chemotherapy with the therapy.
“Indeed, an important question in this study is whether the addition of chemotherapy to glofitamab is necessary, as high response rates with durable responses in patients who achieve complete remission have been demonstrated with single agent bispecific antibody therapy,” he said.
With the durability of CAR T therapy shown in long-term follow-up of trials to exceed 5 years, Dr. Friedberg added that “it is not known how bispecific antibody therapy, with or without chemotherapy, compares to CAR T-cell therapy and how to sequence CAR T and bispecific antibody therapy.”
Dr. Friedberg agreed that longer-term results are needed get a clearer, fuller picture of the therapy’s effects.
“I have no doubt that the overall survival benefit will endure, but in DLBCL our goal should be cure, and whether glofitamab cures as many patients as CAR T-cell therapy is not currently known and will require further follow-up of this and other trials.”
The study was sponsored by F. Hoffman-La Roche Ltd. Dr. Abramson reported ties with AbbVie, ADC Therapeutics, AstraZeneca, BeiGene, BMS, Cellectar, Caribou Biosciences, Celgene, Genentech, Gilead, Incyte, Interius, Janssen, Lilly, Novartis, F. Hoffmann-La Roche Ltd, Seagen, and Takeda. Dr. Friedberg had no disclosures.
“Glofitamab is the first CD20xCD3 bispecific antibody to demonstrate an overall survival benefit in DLBCL in a randomized phase 3 trial,” said first author Jeremy Abramson, MD, of the Massachusetts General Hospital Cancer Center, Boston, Massachusetts, in a press briefing at the annual meeting of the European Hematology Association (EHA) in Madrid.
“These results support the use of glofitamab GemOx as new, off-the-shelf treatment for relapsed/refractory DLBCL in patients who are transplant ineligible in the second-line or later setting,” he said.
The findings are from the phase 3 STARGLO study involving 274 patients with R/R DLBCL who had previously been treated either with at least two prior lines of therapy, or—if only one prior line of therapy—were determined to be ineligible for autologous stem cell transplant (ASCT).
At a median follow-up of 21 months, those treated with glofitamab combined with GemOx had a significantly higher median overall survival of 25.5 months, compared with those treated with the standard of care of rituximab and GemOx (12.9 months; hazard ratio [HR], 0.62; P = .006).
“The results show a 38% lower risk of death with the glofitamab plus GemOx, compared with [the rituximab regimen],” Dr. Abramson said.
Secondary endpoints showed consistent benefits with the glofitamab regimen, with significant improvements in progression-free survival and complete remission.
Unmet Need for Accessible Therapies
Relapsed/refractory DLBCL, the most common form of non-Hodgkin lymphoma (NHL) in the United States, is an aggressive blood cancer. The standard second-line therapy is high-dose chemotherapy followed by ASCT. However, factors including older age or coexisting medical conditions can compromise response, and those who relapse or are refractory to subsequent therapies have poor outcomes.
“Relapsed DLBCL in the second-line setting or later continues to represent an area of medical need,” Dr. Abramson said.
While several CD20xCD3 bispecific antibody drugs are under development to address the need, glofitamab was the first off-the-shelf, fixed-duration bispecific antibody to receive accelerated approval from the US Food and Drug Administration (FDA) for the treatment of R/R DLBCL, specifically as monotherapy after two or more lines of systemic therapy.
That approval was based on results from a pivotal phase 1/2 study, which showed high rates of deep and durable complete remission with the monotherapy.
To further evaluate glofitamab in combination with GemOx, the authors conducted the multicenter, open-label STARGLO trial, which extended enrollment to patients with just one prior therapy if they were determined to be stem cell transplant ineligible. Patients were also required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2.
Patients were randomized 2:1 either to treatment with glofitamab combined with GemOx, involving 8 cycles, in addition to 4 cycles of glofitamab monotherapy (n = 183), or to the rituximab plus GemOx regimen in 8 cycles (n = 91).
Overall, 153 (55.8%) of patients had primary refractory disease and 166 (60.6%) were refractory to their last therapy. The median age was 68, and 37% had two or more lines of therapy, including some who had received chimeric antigen receptor T-cell (CAR T) therapy (8.8% in the glofitamab group and 7.1% in the rituximab group).
In addition to the significant overall survival benefit, the glofitamab regimen also showed significantly improved progression-free survival at a median follow-up of 16.1 months, as observed by IRC-assessed PFS, with a median progression-free survival rate of 13.8 months vs 3.6 months (HR, 0.40; P < .0001).
The complete remission rate was doubled with glofitamab-GemOx, with a rate of 58.5% vs 25.3%, respectively; P < .0001.
Similar results were observed in subgroups, including relapsed vs refractory patients and those treated as a second or third line of care.
The median number of cycles received was higher among those receiving glofitamab (11 vs four cycles).
Adverse event (AE) rates were higher with glofitamab vs rituximab, including grade 3-4 AEs (69.4 vs 36.4%), grade 5 AEs (8.3 vs 4.5%; primarily driven by an imbalance of COVID-19 AEs), and serious AEs (54.4 vs 17.0%; primarily cytokine release syndrome [CRS]).
CRS was the most frequently reported AE in the glofitamab group (grade 1: 31.4%; grade 2: 10.5%; and grade 3: 2.3%), and events consistent with immune effector cell–associated neurotoxicity syndrome were reported in four patients (2.3%), all of which were concurrent with CRS.
Other AEs were consistent with the known risks associated with the therapy regimens.
“We found that with glofitamab GemOx, the toxicities were manageable, and the most common toxicity of CRS was predominantly low-grade and occurred with step-up dosing in cycle one and was completely reversible,” Dr. Abramson said.
He noted that the higher rate of grade 5 AEs with glofitamab GemOx “was far outweighed by the survival benefit for disease control.”
Overall, “these findings represent the best outcomes observed in a phase 3 trial for relapsed/refractory diffuse large B-cell lymphoma patients who are considered transplant ineligible,” Dr. Abramson said in an interview.
Improved Accessibility Vs CAR-T Therapy
Among key developments in the treatment of R/R DLBCL has been the advent and approval of potentially highly effective CAR T-cell therapy, with the anti-CD19 CAR T cell isiocabtagene maraleucel also FDA approved in the non–transplant eligible DLBCL second-line setting.
Asked in the press briefing about the role of glofitamab GemOx in relation to CAR T cell’s significant benefits, Dr. Abramson underscored the important limitations in CAR T-cell accessibility.
“What I would say is a rising tide lifts all boats,” he responded. “It’s great to have multiple effective immunotherapy strategies.”
However, “CAR T cells of course are not available to most people in the US or worldwide,” he explained.
“They are more difficult to access, they require lymphodepleting chemotherapy, and so ultimately, the majority of patients who could potentially benefit from a CAR T cell probably don’t have access to them in the first place.”
He noted that “the appeal of a regimen like [glofitamab] is that it is an off-the-shelf, targeted immunotherapy combined with a well-tolerated chemotherapy backbone and should be more broadly accessible outside of just tertiary care centers in major cities.”
Long-Term Durability?
Looking ahead, Dr. Abramson noted that a key issue of focus is how long the encouraging results actually last.
“The major ongoing question with this trial is the long-term durability of remissions,” he said.
“Thus far, with a median of 21 months of follow-up for overall survival, the results are encouraging but longer follow-up is needed,” he added.
“Further trials are needed in a broader large B-cell lymphoma population as this trial was limited to DLBCL not otherwise specified, so did not include patients with transformed lymphoma, primary mediastinal B-cell lymphoma, high grade B-cell lymphoma, etc.”
Is Chemo Necessary?
Commenting on the findings, Jonathan W. Friedberg, MD, director of the Wilmot Cancer Institute, University of Rochester School of Medicine, in Rochester, New York, underscored that, “given the overall survival benefit, these findings are clearly clinically significant.”
Noting that “these results add to evidence of high activity of bispecific antibodies in this disease,” Dr. Friedberg speculated on the role of chemotherapy with the therapy.
“Indeed, an important question in this study is whether the addition of chemotherapy to glofitamab is necessary, as high response rates with durable responses in patients who achieve complete remission have been demonstrated with single agent bispecific antibody therapy,” he said.
With the durability of CAR T therapy shown in long-term follow-up of trials to exceed 5 years, Dr. Friedberg added that “it is not known how bispecific antibody therapy, with or without chemotherapy, compares to CAR T-cell therapy and how to sequence CAR T and bispecific antibody therapy.”
Dr. Friedberg agreed that longer-term results are needed get a clearer, fuller picture of the therapy’s effects.
“I have no doubt that the overall survival benefit will endure, but in DLBCL our goal should be cure, and whether glofitamab cures as many patients as CAR T-cell therapy is not currently known and will require further follow-up of this and other trials.”
The study was sponsored by F. Hoffman-La Roche Ltd. Dr. Abramson reported ties with AbbVie, ADC Therapeutics, AstraZeneca, BeiGene, BMS, Cellectar, Caribou Biosciences, Celgene, Genentech, Gilead, Incyte, Interius, Janssen, Lilly, Novartis, F. Hoffmann-La Roche Ltd, Seagen, and Takeda. Dr. Friedberg had no disclosures.
FROM EHA 2024
APL: Should Chemo-Free Regimen Become New Standard?
“First-line therapy with ATRA-ATO with two initial doses of idarubicin results in superior event-free survival, compared to conventional ATRA-chemotherapy in patients with high-risk APL,” said first author Uwe Platzbecker, MD, of the University Hospital Leipzig, department for hematology, cellular therapy, hemostaseology, and infectious diseases, in Leipzig, Germany, at the annual meeting of the European Hematology Association (EHA) in Madrid, Spain.
“We believe that the trial may support the implementation of this regimen as a new standard of care in all patients with high-risk APL,” he said.
In the treatment of low and intermediate risk APL, a subtype of acute myeloid leukemia (AML), the combination of ATRA and ATO has become standard since being shown in a pivotal 2013 study to be superior versus ATRA and chemotherapy. The approach is approved by the Food and Drug Administration in the treatment of adults with newly diagnosed low-risk APL.
Importantly, the improved survival with ATRA/ATO approach may result from “reduced severe hematologic toxicity together with similar antileukemic efficacy,” compared with the regimen that include chemotherapy, the authors of the 2013 study speculated.
However, the treatment regimen has not been evaluated in randomized trials in patients with high-risk APL, defined as having a white blood cell count of more than 10,000 cells per μL.
For those patients, the conventional treatment remains ATRA with a chemotherapy backbone, Dr. Platzbecker explained.
To evaluate if the improvements extend to high-risk APL patients without compromising safety, Dr. Platzbecker and colleagues conducted the open-label, prospective APOLLO trial, involving newly diagnosed high-risk APL who were enrolled between 2016 and 2022 at 143 sites in six European countries.
The patients were randomized into one of two groups: ATRA/ATO, involving treatment consisting of two doses of idarubicin (12 mg/m2) on days 1 and 3 at the time of induction therapy, in addition to ATO 0.15 mg/kg and ATRA 45 mg/m2, daily until complete remission, or the ATRA-chemotherapy arm, involving standard ATRA also with idarubicin induction, followed by three cycles of chemotherapy-based consolidation as well as 2 years of maintenance treatment.
While the study was prematurely discontinued in August 2022 because of COVID-19–related recruitment delays and expiration of the study drug, the maintenance and observational periods are ongoing.
Of 131 patients with high-risk APL who were evaluable for the outcome analysis, 68 were in the ATRA/ATO group and 63 in the ATRA-chemotherapy arm.
Overall, participants had a mean age of 46, 50% were female, their median Eastern Cooperative Oncology Group score was 1. Their median white blood cell count was 36 × 109/L, with 39% having a white blood cell count greater than 50 × 109/L.
Molecular resistance occurred in 1.7% in the ATRA/ATO arm vs 5.5% in the ATRA chemotherapy arm, which was not statistically significant (P = .268); however, the incidence of molecular relapse was much lower without chemotherapy, at 1.6% with ATRA/ATO vs 14% with ATRA and chemotherapy.
For the primary endpoint, with a median follow-up of 31 months, the 2-year rate of event-free survival those in the ATRA/ATO arm was 88% vs 70% in the ATRA plus chemotherapy regimen (P = .02). The 5-year event-free survival continued to favor ATRA-ATO (87% vs 55%; P = .0034).
The estimated 5-year overall survival was 93% vs 82% for ATRA/ATO vs ATRA-chemotherapy, respectively, which was not significantly different (P = .17).
There were no significant differences between the arms in complete response (93% with ATRA/ATO vs 91% with ATRA-chemotherapy; P = .65), and rates of early death (within the first 30 days) were also similar across arms, at 7% vs 10%, respectively.
Death while in complete remission occurred in zero patients in the ATRA/ATO arm and three in the ATRA chemotherapy arm.
In terms of toxicities, the ATRA/ATO group had significantly lower rates of hematologic toxicity versus ATRA-chemotherapy, including rates of thrombocytopenia grade 1-4 and neutropenia grade 3-4 (P < .001), while there were no significant differences between the groups in hepatic toxicities (11.8% and 14.3%, respectively; P = .08) or differentiation syndrome (1.5% vs 4.8%; P = .27).
QTc prolongation grade 3-4 occurred in 4.4 patients receiving ATRA/ATO, compared with 0 in the ATRA-chemotherapy group; however, Dr. Platzbecker said the cases had no clinical implications.
Asked to elaborate on the regimens’ toxicities in the press briefing, Dr. Platzbecker noted that “what is very important especially for patients, is [lower rates] of issues such as hair loss and constipation that are much less common with the ATRO/ATO regimen.”
“In addition, we know from the early experiences with this that younger patients are being cured by this regimen,” hence improving pregnancy prospects for women.
A take-home message from the overall results is that the ATRO/ATO regimen for high-risk APL patients should represent “a new treatment paradigm” that will “hopefully soon” be reflected in guideline recommendations, Dr. Platzbecker said in an interview.
Concerns Included Relapse, Differentiation Syndrome
Commenting on the research, Mikkael A. Sekeres, MD, explained that, while the “less is more” non-chemotherapy approach was adopted in widespread utilization in low-risk APL because of superior outcomes, a variety of concerns surrounded its use in high-risk patients.
“In high-risk patients, there were concerns that a durable response would be lower and that relapse would be higher for patients receiving ATRA and ATO than those receiving standard chemotherapy,” Dr. Sekeres, who is chief of the division of hematology, department of medicine, Sylvester Comprehensive Cancer Center, University of Miami, Miami, Florida, said in an interview.
“In addition, it was theoretically possible that patients receiving the differentiating agents ATRA and ATO could suffer higher rates of differentiation syndrome, which could contribute to early death,” he explained. “These fears were simply not realized in the trial.”
Caveats of the trial “include the relatively small sample size and that the trial was stopped prematurely due to low enrollment during the COVID pandemic,” he noted.
Another limitation was the median follow-up of about 2.5 years.
However, Dr. Sekeres said he agreed that, “with further follow-up and continued superiority of the idarubicin, ATRA, and ATO combination, this could become a new standard of care for high-risk patients with APL.”
Dr. Platzbecker’s disclosures include ties with Teva, BMS, Curis, Janssen, AbbVie, and Takeda. Dr. Sekeres had no disclosures.
“First-line therapy with ATRA-ATO with two initial doses of idarubicin results in superior event-free survival, compared to conventional ATRA-chemotherapy in patients with high-risk APL,” said first author Uwe Platzbecker, MD, of the University Hospital Leipzig, department for hematology, cellular therapy, hemostaseology, and infectious diseases, in Leipzig, Germany, at the annual meeting of the European Hematology Association (EHA) in Madrid, Spain.
“We believe that the trial may support the implementation of this regimen as a new standard of care in all patients with high-risk APL,” he said.
In the treatment of low and intermediate risk APL, a subtype of acute myeloid leukemia (AML), the combination of ATRA and ATO has become standard since being shown in a pivotal 2013 study to be superior versus ATRA and chemotherapy. The approach is approved by the Food and Drug Administration in the treatment of adults with newly diagnosed low-risk APL.
Importantly, the improved survival with ATRA/ATO approach may result from “reduced severe hematologic toxicity together with similar antileukemic efficacy,” compared with the regimen that include chemotherapy, the authors of the 2013 study speculated.
However, the treatment regimen has not been evaluated in randomized trials in patients with high-risk APL, defined as having a white blood cell count of more than 10,000 cells per μL.
For those patients, the conventional treatment remains ATRA with a chemotherapy backbone, Dr. Platzbecker explained.
To evaluate if the improvements extend to high-risk APL patients without compromising safety, Dr. Platzbecker and colleagues conducted the open-label, prospective APOLLO trial, involving newly diagnosed high-risk APL who were enrolled between 2016 and 2022 at 143 sites in six European countries.
The patients were randomized into one of two groups: ATRA/ATO, involving treatment consisting of two doses of idarubicin (12 mg/m2) on days 1 and 3 at the time of induction therapy, in addition to ATO 0.15 mg/kg and ATRA 45 mg/m2, daily until complete remission, or the ATRA-chemotherapy arm, involving standard ATRA also with idarubicin induction, followed by three cycles of chemotherapy-based consolidation as well as 2 years of maintenance treatment.
While the study was prematurely discontinued in August 2022 because of COVID-19–related recruitment delays and expiration of the study drug, the maintenance and observational periods are ongoing.
Of 131 patients with high-risk APL who were evaluable for the outcome analysis, 68 were in the ATRA/ATO group and 63 in the ATRA-chemotherapy arm.
Overall, participants had a mean age of 46, 50% were female, their median Eastern Cooperative Oncology Group score was 1. Their median white blood cell count was 36 × 109/L, with 39% having a white blood cell count greater than 50 × 109/L.
Molecular resistance occurred in 1.7% in the ATRA/ATO arm vs 5.5% in the ATRA chemotherapy arm, which was not statistically significant (P = .268); however, the incidence of molecular relapse was much lower without chemotherapy, at 1.6% with ATRA/ATO vs 14% with ATRA and chemotherapy.
For the primary endpoint, with a median follow-up of 31 months, the 2-year rate of event-free survival those in the ATRA/ATO arm was 88% vs 70% in the ATRA plus chemotherapy regimen (P = .02). The 5-year event-free survival continued to favor ATRA-ATO (87% vs 55%; P = .0034).
The estimated 5-year overall survival was 93% vs 82% for ATRA/ATO vs ATRA-chemotherapy, respectively, which was not significantly different (P = .17).
There were no significant differences between the arms in complete response (93% with ATRA/ATO vs 91% with ATRA-chemotherapy; P = .65), and rates of early death (within the first 30 days) were also similar across arms, at 7% vs 10%, respectively.
Death while in complete remission occurred in zero patients in the ATRA/ATO arm and three in the ATRA chemotherapy arm.
In terms of toxicities, the ATRA/ATO group had significantly lower rates of hematologic toxicity versus ATRA-chemotherapy, including rates of thrombocytopenia grade 1-4 and neutropenia grade 3-4 (P < .001), while there were no significant differences between the groups in hepatic toxicities (11.8% and 14.3%, respectively; P = .08) or differentiation syndrome (1.5% vs 4.8%; P = .27).
QTc prolongation grade 3-4 occurred in 4.4 patients receiving ATRA/ATO, compared with 0 in the ATRA-chemotherapy group; however, Dr. Platzbecker said the cases had no clinical implications.
Asked to elaborate on the regimens’ toxicities in the press briefing, Dr. Platzbecker noted that “what is very important especially for patients, is [lower rates] of issues such as hair loss and constipation that are much less common with the ATRO/ATO regimen.”
“In addition, we know from the early experiences with this that younger patients are being cured by this regimen,” hence improving pregnancy prospects for women.
A take-home message from the overall results is that the ATRO/ATO regimen for high-risk APL patients should represent “a new treatment paradigm” that will “hopefully soon” be reflected in guideline recommendations, Dr. Platzbecker said in an interview.
Concerns Included Relapse, Differentiation Syndrome
Commenting on the research, Mikkael A. Sekeres, MD, explained that, while the “less is more” non-chemotherapy approach was adopted in widespread utilization in low-risk APL because of superior outcomes, a variety of concerns surrounded its use in high-risk patients.
“In high-risk patients, there were concerns that a durable response would be lower and that relapse would be higher for patients receiving ATRA and ATO than those receiving standard chemotherapy,” Dr. Sekeres, who is chief of the division of hematology, department of medicine, Sylvester Comprehensive Cancer Center, University of Miami, Miami, Florida, said in an interview.
“In addition, it was theoretically possible that patients receiving the differentiating agents ATRA and ATO could suffer higher rates of differentiation syndrome, which could contribute to early death,” he explained. “These fears were simply not realized in the trial.”
Caveats of the trial “include the relatively small sample size and that the trial was stopped prematurely due to low enrollment during the COVID pandemic,” he noted.
Another limitation was the median follow-up of about 2.5 years.
However, Dr. Sekeres said he agreed that, “with further follow-up and continued superiority of the idarubicin, ATRA, and ATO combination, this could become a new standard of care for high-risk patients with APL.”
Dr. Platzbecker’s disclosures include ties with Teva, BMS, Curis, Janssen, AbbVie, and Takeda. Dr. Sekeres had no disclosures.
“First-line therapy with ATRA-ATO with two initial doses of idarubicin results in superior event-free survival, compared to conventional ATRA-chemotherapy in patients with high-risk APL,” said first author Uwe Platzbecker, MD, of the University Hospital Leipzig, department for hematology, cellular therapy, hemostaseology, and infectious diseases, in Leipzig, Germany, at the annual meeting of the European Hematology Association (EHA) in Madrid, Spain.
“We believe that the trial may support the implementation of this regimen as a new standard of care in all patients with high-risk APL,” he said.
In the treatment of low and intermediate risk APL, a subtype of acute myeloid leukemia (AML), the combination of ATRA and ATO has become standard since being shown in a pivotal 2013 study to be superior versus ATRA and chemotherapy. The approach is approved by the Food and Drug Administration in the treatment of adults with newly diagnosed low-risk APL.
Importantly, the improved survival with ATRA/ATO approach may result from “reduced severe hematologic toxicity together with similar antileukemic efficacy,” compared with the regimen that include chemotherapy, the authors of the 2013 study speculated.
However, the treatment regimen has not been evaluated in randomized trials in patients with high-risk APL, defined as having a white blood cell count of more than 10,000 cells per μL.
For those patients, the conventional treatment remains ATRA with a chemotherapy backbone, Dr. Platzbecker explained.
To evaluate if the improvements extend to high-risk APL patients without compromising safety, Dr. Platzbecker and colleagues conducted the open-label, prospective APOLLO trial, involving newly diagnosed high-risk APL who were enrolled between 2016 and 2022 at 143 sites in six European countries.
The patients were randomized into one of two groups: ATRA/ATO, involving treatment consisting of two doses of idarubicin (12 mg/m2) on days 1 and 3 at the time of induction therapy, in addition to ATO 0.15 mg/kg and ATRA 45 mg/m2, daily until complete remission, or the ATRA-chemotherapy arm, involving standard ATRA also with idarubicin induction, followed by three cycles of chemotherapy-based consolidation as well as 2 years of maintenance treatment.
While the study was prematurely discontinued in August 2022 because of COVID-19–related recruitment delays and expiration of the study drug, the maintenance and observational periods are ongoing.
Of 131 patients with high-risk APL who were evaluable for the outcome analysis, 68 were in the ATRA/ATO group and 63 in the ATRA-chemotherapy arm.
Overall, participants had a mean age of 46, 50% were female, their median Eastern Cooperative Oncology Group score was 1. Their median white blood cell count was 36 × 109/L, with 39% having a white blood cell count greater than 50 × 109/L.
Molecular resistance occurred in 1.7% in the ATRA/ATO arm vs 5.5% in the ATRA chemotherapy arm, which was not statistically significant (P = .268); however, the incidence of molecular relapse was much lower without chemotherapy, at 1.6% with ATRA/ATO vs 14% with ATRA and chemotherapy.
For the primary endpoint, with a median follow-up of 31 months, the 2-year rate of event-free survival those in the ATRA/ATO arm was 88% vs 70% in the ATRA plus chemotherapy regimen (P = .02). The 5-year event-free survival continued to favor ATRA-ATO (87% vs 55%; P = .0034).
The estimated 5-year overall survival was 93% vs 82% for ATRA/ATO vs ATRA-chemotherapy, respectively, which was not significantly different (P = .17).
There were no significant differences between the arms in complete response (93% with ATRA/ATO vs 91% with ATRA-chemotherapy; P = .65), and rates of early death (within the first 30 days) were also similar across arms, at 7% vs 10%, respectively.
Death while in complete remission occurred in zero patients in the ATRA/ATO arm and three in the ATRA chemotherapy arm.
In terms of toxicities, the ATRA/ATO group had significantly lower rates of hematologic toxicity versus ATRA-chemotherapy, including rates of thrombocytopenia grade 1-4 and neutropenia grade 3-4 (P < .001), while there were no significant differences between the groups in hepatic toxicities (11.8% and 14.3%, respectively; P = .08) or differentiation syndrome (1.5% vs 4.8%; P = .27).
QTc prolongation grade 3-4 occurred in 4.4 patients receiving ATRA/ATO, compared with 0 in the ATRA-chemotherapy group; however, Dr. Platzbecker said the cases had no clinical implications.
Asked to elaborate on the regimens’ toxicities in the press briefing, Dr. Platzbecker noted that “what is very important especially for patients, is [lower rates] of issues such as hair loss and constipation that are much less common with the ATRO/ATO regimen.”
“In addition, we know from the early experiences with this that younger patients are being cured by this regimen,” hence improving pregnancy prospects for women.
A take-home message from the overall results is that the ATRO/ATO regimen for high-risk APL patients should represent “a new treatment paradigm” that will “hopefully soon” be reflected in guideline recommendations, Dr. Platzbecker said in an interview.
Concerns Included Relapse, Differentiation Syndrome
Commenting on the research, Mikkael A. Sekeres, MD, explained that, while the “less is more” non-chemotherapy approach was adopted in widespread utilization in low-risk APL because of superior outcomes, a variety of concerns surrounded its use in high-risk patients.
“In high-risk patients, there were concerns that a durable response would be lower and that relapse would be higher for patients receiving ATRA and ATO than those receiving standard chemotherapy,” Dr. Sekeres, who is chief of the division of hematology, department of medicine, Sylvester Comprehensive Cancer Center, University of Miami, Miami, Florida, said in an interview.
“In addition, it was theoretically possible that patients receiving the differentiating agents ATRA and ATO could suffer higher rates of differentiation syndrome, which could contribute to early death,” he explained. “These fears were simply not realized in the trial.”
Caveats of the trial “include the relatively small sample size and that the trial was stopped prematurely due to low enrollment during the COVID pandemic,” he noted.
Another limitation was the median follow-up of about 2.5 years.
However, Dr. Sekeres said he agreed that, “with further follow-up and continued superiority of the idarubicin, ATRA, and ATO combination, this could become a new standard of care for high-risk patients with APL.”
Dr. Platzbecker’s disclosures include ties with Teva, BMS, Curis, Janssen, AbbVie, and Takeda. Dr. Sekeres had no disclosures.
FROM EHA 2024
VEXAS Syndrome: Study Highlights Cutaneous Symptoms
Additionally, the most common histologic findings include leukocytoclastic vasculitis, neutrophilic dermatosis, and perivascular dermatitis; different variants in the UBA1 gene are associated with specific skin manifestations.
Those are key findings from a cohort study of 112 patients with VEXAS published online in JAMA Dermatology. The study, conducted by researchers at the National Institutes of Health (NIH) and several other institutions, aimed to define the spectrum of cutaneous manifestations in VEXAS in association with genetic, histologic, and other clinical findings.

First described in 2020, VEXAS syndrome is an adult-onset multisystem disease that can pose a diagnostic challenge to clinicians, the study’s corresponding author, Edward W. Cowen, MD, MHSc, of the dermatology branch at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), said in an interview. The disease is caused by pathogenic variants in the UBA1 gene, located on the X chromosome. Affected individuals exhibit a wide range of manifestations, including cytopenia/myelodysplasia, multiorgan systemic inflammation, and cutaneous involvement.
“Patients may present to a variety of disease specialists depending on their symptoms and providers may not immediately consider a genetic etiology in an older individual,” Dr. Cowen said in an interview. “Although skin involvement occurs in more than 80% of patients, it is pleomorphic and may resemble a variety of other conditions such as vasculitis and Sweet syndrome.”
To better understand the cutaneous manifestations of VEXAS syndrome, the researchers evaluated data from 112 patients with VEXAS-defining genetic variants in the UBA1 gene between 2019 and 2023. Of the 112 patients, 73 underwent medical record review only, and 39 were prospectively evaluated at NIH. All but one of the patients were men, 94% were White individuals, and their mean age was 64 years. Skin involvement occurred in 83% of cases and was the most common presenting feature of VEXAS in 61% of cases.
Of the 64 histopathologic reports available from 60 patients, the main skin histopathologic findings were leukocytoclastic vasculitis in 23 patients (36%), neutrophilic dermatosis in 22 patients (34%), and perivascular dermatitis in 19 patients (30%). According to Dr. Cowen, one key histologic finding was a distinct pattern of “histiocytoid” dermal neutrophilic inflammation, which was present in 13 of 15 specimens (86%) that underwent central re-review. “This pattern can occasionally also be seen in patients with Sweet syndrome, unrelated to VEXAS, but was a hallmark feature found in the majority of skin biopsies of patients with VEXAS,” he said.
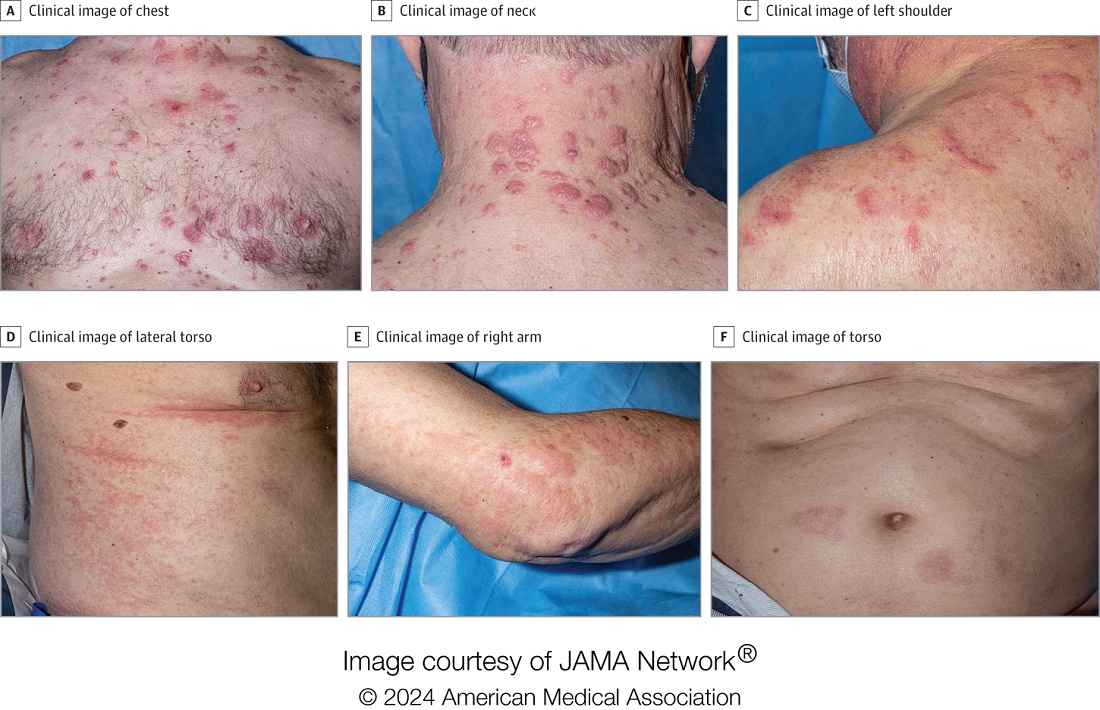
“Together with another pathologic finding, leukocytoclasia, these features can be useful clues to alert the pathologist to a potential diagnosis of VEXAS. This myeloid predominant pattern of skin inflammation was also most strongly associated with the leucine pathogenic variant of the UBA1 gene.” In contrast, cutaneous vasculitis was most strongly associated with the valine pathogenic variant of UBA1. “This is important because the valine variant has been previously independently linked to decreased survival,” he said.
In findings related to pathogenic genetic variants, the researchers observed that the p.Met41Leu variant was most frequently associated with neutrophilic dermal infiltrates in 14 of 17 patients (82%) with this variant and often resembled histiocytoid Sweet syndrome. In addition, the p.Met41Val variant was associated with vasculitic lesions in 11 of 20 patients (55%) with this variant and with a mixed leukocytic infiltrate in 17 of these 20 patients (85%).
Treatment Outcomes
In the realm of therapies, skin manifestations improved in 67 of 73 patients (92%) treated with oral prednisone, while treatment with the interleukin-1 receptor antagonist anakinra improved cutaneous disease in 9 of the 16 (56%) who received it. However, 12 (75%) of those who received anakinra developed severe injection-site reactions, including ulceration in two patients and abscess formation in one patient.
Dr. Cowen noted that VEXAS is associated with high mortality (22% in this cohort), and a high degree of suspicion is required to diagnose patients with VEXAS before significant end organ damage has occurred. “This diagnosis should be considered in all older male patients who present with neutrophilic dermatosis — particularly histiocytoid Sweet syndrome, vasculitis, or leukocytoclasia without vasculitis. Patients who appear to have isolated skin involvement may have cytopenias and acute phase reactants. Therefore, complete blood count with differential and ESR and CRP should be considered to investigate for macrocytosis, cytopenias, and systemic inflammation.”
He acknowledged certain limitations of the study, including the fact that many patients were first evaluated at the NIH after having disease symptoms for many months or years. “It is possible that patients with VEXAS referred to the NIH, either for genetic testing or in person evaluation, represent a population with more aggressive disease.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Connecticut, who was asked to comment on the study, emphasized the importance of the UBA1 mutation in the diagnosis of this complex syndrome. “Dermatologists should be aware of VEXAS syndrome as the majority of patients present with skin lesions, which can range from urticarial to Sweet syndrome–like to palpable purpura,” Dr. Ko said.
“Chondritis and periorbital edema, sometimes unilateral, are also associated. Histopathologic clues include a predominantly histiocytoid infiltrate,” she noted. In addition, “the prominent myxoid stroma around blood vessels and adnexal structures as a clue to VEXAS syndrome surprised me; I had not read that before.”
The study was supported by the Intramural Research Program of NIAMS. One of the study authors reported personal fees from Alexion, Novartis, and Sobi outside of the submitted work. No other disclosures were reported. Dr. Ko reported having no disclosures.
A version of this article appeared on Medscape.com .
Additionally, the most common histologic findings include leukocytoclastic vasculitis, neutrophilic dermatosis, and perivascular dermatitis; different variants in the UBA1 gene are associated with specific skin manifestations.
Those are key findings from a cohort study of 112 patients with VEXAS published online in JAMA Dermatology. The study, conducted by researchers at the National Institutes of Health (NIH) and several other institutions, aimed to define the spectrum of cutaneous manifestations in VEXAS in association with genetic, histologic, and other clinical findings.
First described in 2020, VEXAS syndrome is an adult-onset multisystem disease that can pose a diagnostic challenge to clinicians, the study’s corresponding author, Edward W. Cowen, MD, MHSc, of the dermatology branch at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), said in an interview. The disease is caused by pathogenic variants in the UBA1 gene, located on the X chromosome. Affected individuals exhibit a wide range of manifestations, including cytopenia/myelodysplasia, multiorgan systemic inflammation, and cutaneous involvement.
“Patients may present to a variety of disease specialists depending on their symptoms and providers may not immediately consider a genetic etiology in an older individual,” Dr. Cowen said in an interview. “Although skin involvement occurs in more than 80% of patients, it is pleomorphic and may resemble a variety of other conditions such as vasculitis and Sweet syndrome.”
To better understand the cutaneous manifestations of VEXAS syndrome, the researchers evaluated data from 112 patients with VEXAS-defining genetic variants in the UBA1 gene between 2019 and 2023. Of the 112 patients, 73 underwent medical record review only, and 39 were prospectively evaluated at NIH. All but one of the patients were men, 94% were White individuals, and their mean age was 64 years. Skin involvement occurred in 83% of cases and was the most common presenting feature of VEXAS in 61% of cases.
Of the 64 histopathologic reports available from 60 patients, the main skin histopathologic findings were leukocytoclastic vasculitis in 23 patients (36%), neutrophilic dermatosis in 22 patients (34%), and perivascular dermatitis in 19 patients (30%). According to Dr. Cowen, one key histologic finding was a distinct pattern of “histiocytoid” dermal neutrophilic inflammation, which was present in 13 of 15 specimens (86%) that underwent central re-review. “This pattern can occasionally also be seen in patients with Sweet syndrome, unrelated to VEXAS, but was a hallmark feature found in the majority of skin biopsies of patients with VEXAS,” he said.
“Together with another pathologic finding, leukocytoclasia, these features can be useful clues to alert the pathologist to a potential diagnosis of VEXAS. This myeloid predominant pattern of skin inflammation was also most strongly associated with the leucine pathogenic variant of the UBA1 gene.” In contrast, cutaneous vasculitis was most strongly associated with the valine pathogenic variant of UBA1. “This is important because the valine variant has been previously independently linked to decreased survival,” he said.
In findings related to pathogenic genetic variants, the researchers observed that the p.Met41Leu variant was most frequently associated with neutrophilic dermal infiltrates in 14 of 17 patients (82%) with this variant and often resembled histiocytoid Sweet syndrome. In addition, the p.Met41Val variant was associated with vasculitic lesions in 11 of 20 patients (55%) with this variant and with a mixed leukocytic infiltrate in 17 of these 20 patients (85%).
Treatment Outcomes
In the realm of therapies, skin manifestations improved in 67 of 73 patients (92%) treated with oral prednisone, while treatment with the interleukin-1 receptor antagonist anakinra improved cutaneous disease in 9 of the 16 (56%) who received it. However, 12 (75%) of those who received anakinra developed severe injection-site reactions, including ulceration in two patients and abscess formation in one patient.
Dr. Cowen noted that VEXAS is associated with high mortality (22% in this cohort), and a high degree of suspicion is required to diagnose patients with VEXAS before significant end organ damage has occurred. “This diagnosis should be considered in all older male patients who present with neutrophilic dermatosis — particularly histiocytoid Sweet syndrome, vasculitis, or leukocytoclasia without vasculitis. Patients who appear to have isolated skin involvement may have cytopenias and acute phase reactants. Therefore, complete blood count with differential and ESR and CRP should be considered to investigate for macrocytosis, cytopenias, and systemic inflammation.”
He acknowledged certain limitations of the study, including the fact that many patients were first evaluated at the NIH after having disease symptoms for many months or years. “It is possible that patients with VEXAS referred to the NIH, either for genetic testing or in person evaluation, represent a population with more aggressive disease.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Connecticut, who was asked to comment on the study, emphasized the importance of the UBA1 mutation in the diagnosis of this complex syndrome. “Dermatologists should be aware of VEXAS syndrome as the majority of patients present with skin lesions, which can range from urticarial to Sweet syndrome–like to palpable purpura,” Dr. Ko said.
“Chondritis and periorbital edema, sometimes unilateral, are also associated. Histopathologic clues include a predominantly histiocytoid infiltrate,” she noted. In addition, “the prominent myxoid stroma around blood vessels and adnexal structures as a clue to VEXAS syndrome surprised me; I had not read that before.”
The study was supported by the Intramural Research Program of NIAMS. One of the study authors reported personal fees from Alexion, Novartis, and Sobi outside of the submitted work. No other disclosures were reported. Dr. Ko reported having no disclosures.
A version of this article appeared on Medscape.com .
Additionally, the most common histologic findings include leukocytoclastic vasculitis, neutrophilic dermatosis, and perivascular dermatitis; different variants in the UBA1 gene are associated with specific skin manifestations.
Those are key findings from a cohort study of 112 patients with VEXAS published online in JAMA Dermatology. The study, conducted by researchers at the National Institutes of Health (NIH) and several other institutions, aimed to define the spectrum of cutaneous manifestations in VEXAS in association with genetic, histologic, and other clinical findings.
First described in 2020, VEXAS syndrome is an adult-onset multisystem disease that can pose a diagnostic challenge to clinicians, the study’s corresponding author, Edward W. Cowen, MD, MHSc, of the dermatology branch at the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), said in an interview. The disease is caused by pathogenic variants in the UBA1 gene, located on the X chromosome. Affected individuals exhibit a wide range of manifestations, including cytopenia/myelodysplasia, multiorgan systemic inflammation, and cutaneous involvement.
“Patients may present to a variety of disease specialists depending on their symptoms and providers may not immediately consider a genetic etiology in an older individual,” Dr. Cowen said in an interview. “Although skin involvement occurs in more than 80% of patients, it is pleomorphic and may resemble a variety of other conditions such as vasculitis and Sweet syndrome.”
To better understand the cutaneous manifestations of VEXAS syndrome, the researchers evaluated data from 112 patients with VEXAS-defining genetic variants in the UBA1 gene between 2019 and 2023. Of the 112 patients, 73 underwent medical record review only, and 39 were prospectively evaluated at NIH. All but one of the patients were men, 94% were White individuals, and their mean age was 64 years. Skin involvement occurred in 83% of cases and was the most common presenting feature of VEXAS in 61% of cases.
Of the 64 histopathologic reports available from 60 patients, the main skin histopathologic findings were leukocytoclastic vasculitis in 23 patients (36%), neutrophilic dermatosis in 22 patients (34%), and perivascular dermatitis in 19 patients (30%). According to Dr. Cowen, one key histologic finding was a distinct pattern of “histiocytoid” dermal neutrophilic inflammation, which was present in 13 of 15 specimens (86%) that underwent central re-review. “This pattern can occasionally also be seen in patients with Sweet syndrome, unrelated to VEXAS, but was a hallmark feature found in the majority of skin biopsies of patients with VEXAS,” he said.
“Together with another pathologic finding, leukocytoclasia, these features can be useful clues to alert the pathologist to a potential diagnosis of VEXAS. This myeloid predominant pattern of skin inflammation was also most strongly associated with the leucine pathogenic variant of the UBA1 gene.” In contrast, cutaneous vasculitis was most strongly associated with the valine pathogenic variant of UBA1. “This is important because the valine variant has been previously independently linked to decreased survival,” he said.
In findings related to pathogenic genetic variants, the researchers observed that the p.Met41Leu variant was most frequently associated with neutrophilic dermal infiltrates in 14 of 17 patients (82%) with this variant and often resembled histiocytoid Sweet syndrome. In addition, the p.Met41Val variant was associated with vasculitic lesions in 11 of 20 patients (55%) with this variant and with a mixed leukocytic infiltrate in 17 of these 20 patients (85%).
Treatment Outcomes
In the realm of therapies, skin manifestations improved in 67 of 73 patients (92%) treated with oral prednisone, while treatment with the interleukin-1 receptor antagonist anakinra improved cutaneous disease in 9 of the 16 (56%) who received it. However, 12 (75%) of those who received anakinra developed severe injection-site reactions, including ulceration in two patients and abscess formation in one patient.
Dr. Cowen noted that VEXAS is associated with high mortality (22% in this cohort), and a high degree of suspicion is required to diagnose patients with VEXAS before significant end organ damage has occurred. “This diagnosis should be considered in all older male patients who present with neutrophilic dermatosis — particularly histiocytoid Sweet syndrome, vasculitis, or leukocytoclasia without vasculitis. Patients who appear to have isolated skin involvement may have cytopenias and acute phase reactants. Therefore, complete blood count with differential and ESR and CRP should be considered to investigate for macrocytosis, cytopenias, and systemic inflammation.”
He acknowledged certain limitations of the study, including the fact that many patients were first evaluated at the NIH after having disease symptoms for many months or years. “It is possible that patients with VEXAS referred to the NIH, either for genetic testing or in person evaluation, represent a population with more aggressive disease.”
Christine Ko, MD, professor of dermatology and pathology at Yale University, New Haven, Connecticut, who was asked to comment on the study, emphasized the importance of the UBA1 mutation in the diagnosis of this complex syndrome. “Dermatologists should be aware of VEXAS syndrome as the majority of patients present with skin lesions, which can range from urticarial to Sweet syndrome–like to palpable purpura,” Dr. Ko said.
“Chondritis and periorbital edema, sometimes unilateral, are also associated. Histopathologic clues include a predominantly histiocytoid infiltrate,” she noted. In addition, “the prominent myxoid stroma around blood vessels and adnexal structures as a clue to VEXAS syndrome surprised me; I had not read that before.”
The study was supported by the Intramural Research Program of NIAMS. One of the study authors reported personal fees from Alexion, Novartis, and Sobi outside of the submitted work. No other disclosures were reported. Dr. Ko reported having no disclosures.
A version of this article appeared on Medscape.com .
FROM JAMA DERMATOLOGY
Doctors Endorsing Products on X May Not Disclose Company Ties
Lead author Aaron Mitchell, MD, MPH, a medical oncologist at Memorial Sloan Kettering Cancer Center in New York City, told this news organization that he and his colleagues undertook the study in part to see whether physicians were adhering to professional and industry guidelines regarding marketing communications.
The team reviewed posts by physicians on X during 2022, looking for key words that might indicate that the posts were intended as endorsements of a product. The researchers then delved into the Centers for Medicare and Medicaid Services Open Payments database to see how many of those identified as having endorsed a product were paid by the manufacturers.
What Dr. Mitchell found concerned him, he said.
Overall, the researchers identified 28 physician endorsers who received a total of $1.4 million from sponsors in 2022. Among these, 26 physicians (93%) received payments from the product’s manufacturer, totaling $713,976, and 24 physicians (86%) accepted payments related to the endorsed drug or device, totaling $492,098.
While most did disclose that the posts were sponsored — by adding the word “sponsored” or using #sponsored — nine physicians did not.
Although 28 physician endorsers represent a “small fraction” of the overall number of physicians who use X, each endorsement was ultimately posted dozens, if not hundreds of times, said Dr. Mitchell. In fact, he said he saw the same particular endorsement post every time he opened his X app for months.
Overall, Dr. Mitchell noted that it’s less about the fact that the endorsements are occurring on social media and more that there are these paid endorsements taking place at all.
Among the physician specialties promoting a product, urologists and oncologists dominated. Almost one third were urologists, and 57% were oncologists — six medical oncologists, six radiation oncologists, and four gynecologic oncologists. Of the remaining three physicians, two were internists and one was a pulmonary and critical care medicine specialist.
The authors tracked posts from physicians and industry accounts. Many of the posts on industry accounts were physician testimonials, usually videos. Almost half — 8 of 17 — of those testimonials did not disclose that the doctor was being paid by the manufacturer. In another case, a physician did not disclose that they were paid to endorse a white paper.
Fifteen promotional posts were for a Boston Scientific product, followed by six for GlaxoSmithKline, two for Eisai, two for Exelixis, and one each for AstraZeneca, Novartis, and Pfizer.
In general, Dr. Mitchell said, industry guidelines suggest that manufacturer-paid speakers or consultants should have well-regarded expertise in the area they are being asked to weigh in on, but most physician endorsers in the study were not key opinion leaders or experts.
The authors examined the paid endorsers’ H-index — a measure of academic productivity provided by Scopus. Overall, 19 of the 28 physicians had an H-index below 20, which is considered less accomplished, and 14 had no published research related to the endorsed product.
Ten received payments from manufacturers for research purposes, and only one received research payments related to the endorsed product ($224,577).
“Physicians’ participation in industry marketing raises questions regarding professionalism and their responsibilities as patient advocates,” the JAMA authors wrote.
The study was supported by grants from the National Cancer Institute. Dr. Mitchell reported no relevant financial relationships. Coauthors Samer Al Hadidi, MD, reported receiving personal fees from Pfizer, Sanofi, and Janssen during the conduct of the study, and Timothy S. Anderson, MD, reported receiving grants from the National Institute on Aging, the American Heart Association, and the American College of Cardiology, and receiving consulting fees from the American Medical Student Association. Dr. Anderson is also an associate editor of JAMA Internal Medicine.
A version of this article appeared on Medscape.com.
Lead author Aaron Mitchell, MD, MPH, a medical oncologist at Memorial Sloan Kettering Cancer Center in New York City, told this news organization that he and his colleagues undertook the study in part to see whether physicians were adhering to professional and industry guidelines regarding marketing communications.
The team reviewed posts by physicians on X during 2022, looking for key words that might indicate that the posts were intended as endorsements of a product. The researchers then delved into the Centers for Medicare and Medicaid Services Open Payments database to see how many of those identified as having endorsed a product were paid by the manufacturers.
What Dr. Mitchell found concerned him, he said.
Overall, the researchers identified 28 physician endorsers who received a total of $1.4 million from sponsors in 2022. Among these, 26 physicians (93%) received payments from the product’s manufacturer, totaling $713,976, and 24 physicians (86%) accepted payments related to the endorsed drug or device, totaling $492,098.
While most did disclose that the posts were sponsored — by adding the word “sponsored” or using #sponsored — nine physicians did not.
Although 28 physician endorsers represent a “small fraction” of the overall number of physicians who use X, each endorsement was ultimately posted dozens, if not hundreds of times, said Dr. Mitchell. In fact, he said he saw the same particular endorsement post every time he opened his X app for months.
Overall, Dr. Mitchell noted that it’s less about the fact that the endorsements are occurring on social media and more that there are these paid endorsements taking place at all.
Among the physician specialties promoting a product, urologists and oncologists dominated. Almost one third were urologists, and 57% were oncologists — six medical oncologists, six radiation oncologists, and four gynecologic oncologists. Of the remaining three physicians, two were internists and one was a pulmonary and critical care medicine specialist.
The authors tracked posts from physicians and industry accounts. Many of the posts on industry accounts were physician testimonials, usually videos. Almost half — 8 of 17 — of those testimonials did not disclose that the doctor was being paid by the manufacturer. In another case, a physician did not disclose that they were paid to endorse a white paper.
Fifteen promotional posts were for a Boston Scientific product, followed by six for GlaxoSmithKline, two for Eisai, two for Exelixis, and one each for AstraZeneca, Novartis, and Pfizer.
In general, Dr. Mitchell said, industry guidelines suggest that manufacturer-paid speakers or consultants should have well-regarded expertise in the area they are being asked to weigh in on, but most physician endorsers in the study were not key opinion leaders or experts.
The authors examined the paid endorsers’ H-index — a measure of academic productivity provided by Scopus. Overall, 19 of the 28 physicians had an H-index below 20, which is considered less accomplished, and 14 had no published research related to the endorsed product.
Ten received payments from manufacturers for research purposes, and only one received research payments related to the endorsed product ($224,577).
“Physicians’ participation in industry marketing raises questions regarding professionalism and their responsibilities as patient advocates,” the JAMA authors wrote.
The study was supported by grants from the National Cancer Institute. Dr. Mitchell reported no relevant financial relationships. Coauthors Samer Al Hadidi, MD, reported receiving personal fees from Pfizer, Sanofi, and Janssen during the conduct of the study, and Timothy S. Anderson, MD, reported receiving grants from the National Institute on Aging, the American Heart Association, and the American College of Cardiology, and receiving consulting fees from the American Medical Student Association. Dr. Anderson is also an associate editor of JAMA Internal Medicine.
A version of this article appeared on Medscape.com.
Lead author Aaron Mitchell, MD, MPH, a medical oncologist at Memorial Sloan Kettering Cancer Center in New York City, told this news organization that he and his colleagues undertook the study in part to see whether physicians were adhering to professional and industry guidelines regarding marketing communications.
The team reviewed posts by physicians on X during 2022, looking for key words that might indicate that the posts were intended as endorsements of a product. The researchers then delved into the Centers for Medicare and Medicaid Services Open Payments database to see how many of those identified as having endorsed a product were paid by the manufacturers.
What Dr. Mitchell found concerned him, he said.
Overall, the researchers identified 28 physician endorsers who received a total of $1.4 million from sponsors in 2022. Among these, 26 physicians (93%) received payments from the product’s manufacturer, totaling $713,976, and 24 physicians (86%) accepted payments related to the endorsed drug or device, totaling $492,098.
While most did disclose that the posts were sponsored — by adding the word “sponsored” or using #sponsored — nine physicians did not.
Although 28 physician endorsers represent a “small fraction” of the overall number of physicians who use X, each endorsement was ultimately posted dozens, if not hundreds of times, said Dr. Mitchell. In fact, he said he saw the same particular endorsement post every time he opened his X app for months.
Overall, Dr. Mitchell noted that it’s less about the fact that the endorsements are occurring on social media and more that there are these paid endorsements taking place at all.
Among the physician specialties promoting a product, urologists and oncologists dominated. Almost one third were urologists, and 57% were oncologists — six medical oncologists, six radiation oncologists, and four gynecologic oncologists. Of the remaining three physicians, two were internists and one was a pulmonary and critical care medicine specialist.
The authors tracked posts from physicians and industry accounts. Many of the posts on industry accounts were physician testimonials, usually videos. Almost half — 8 of 17 — of those testimonials did not disclose that the doctor was being paid by the manufacturer. In another case, a physician did not disclose that they were paid to endorse a white paper.
Fifteen promotional posts were for a Boston Scientific product, followed by six for GlaxoSmithKline, two for Eisai, two for Exelixis, and one each for AstraZeneca, Novartis, and Pfizer.
In general, Dr. Mitchell said, industry guidelines suggest that manufacturer-paid speakers or consultants should have well-regarded expertise in the area they are being asked to weigh in on, but most physician endorsers in the study were not key opinion leaders or experts.
The authors examined the paid endorsers’ H-index — a measure of academic productivity provided by Scopus. Overall, 19 of the 28 physicians had an H-index below 20, which is considered less accomplished, and 14 had no published research related to the endorsed product.
Ten received payments from manufacturers for research purposes, and only one received research payments related to the endorsed product ($224,577).
“Physicians’ participation in industry marketing raises questions regarding professionalism and their responsibilities as patient advocates,” the JAMA authors wrote.
The study was supported by grants from the National Cancer Institute. Dr. Mitchell reported no relevant financial relationships. Coauthors Samer Al Hadidi, MD, reported receiving personal fees from Pfizer, Sanofi, and Janssen during the conduct of the study, and Timothy S. Anderson, MD, reported receiving grants from the National Institute on Aging, the American Heart Association, and the American College of Cardiology, and receiving consulting fees from the American Medical Student Association. Dr. Anderson is also an associate editor of JAMA Internal Medicine.
A version of this article appeared on Medscape.com.
One Patient Changed This Oncologist’s View of Hope. Here’s How.
CHICAGO — Carlos, a 21-year-old, lay in a hospital bed, barely clinging to life. Following a stem cell transplant for leukemia, Carlos had developed a life-threatening case of graft-vs-host disease.
But Carlos’ mother had faith.
“I have hope things will get better,” she said, via interpreter, to Richard Leiter, MD, a palliative care doctor in training at that time.
“I hope they will,” Dr. Leiter told her.
“I should have stopped there,” said Dr. Leiter, recounting an early-career lesson on hope during the ASCO Voices session at the American Society of Clinical Oncology annual meeting. “But in my eagerness to show my attending and myself that I could handle this conversation, I kept going, mistakenly.”
“But none of us think they will,” Dr. Leiter continued.
Carlos’ mother looked Dr. Leiter in the eye. “You want him to die,” she said.
“I knew, even then, that she was right,” recalled Dr. Leiter, now a palliative care physician at Dana-Farber Cancer Institute and Brigham and Women’s Hospital and an assistant professor of medicine at Harvard Medical School, Boston.
Although there was nothing he could do to save Carlos, Dr. Leiter also couldn’t sit with the extreme suffering. “The pain was too great,” Dr. Leiter said. “I needed her to adopt our narrative that we had done everything we could to help him live, and now, we would do everything we could to help his death be a comfortable one.”
But looking back, Dr. Leiter realized, “How could we have asked her to accept what was fundamentally unacceptable, to comprehend the incomprehensible?”
The Importance of Hope
Alan B. Astrow, MD, said during an ASCO symposium on “The Art and Science of Hope.”
“How we think about hope directly influences patient care,” said Dr. Astrow, chief of hematology and medical oncology at NewYork-Presbyterian Brooklyn Methodist Hospital and a professor of clinical medicine at Weill Cornell Medicine in New York City.
Hope, whatever it turns out to be neurobiologically, is “very much a gift” that underlies human existence, he said.
Physicians have the capacity to restore or shatter a patient’s hopes, and those who come to understand the importance of hope will wish to extend the gift to others, Dr. Astrow said.
Asking patients about their hopes is the “golden question,” Steven Z. Pantilat, MD, said at the symposium. “When you think about the future, what do you hope for?”
Often, the answers reveal not only “things beyond a cure that matter tremendously to the patient but things that we can help with,” said Dr. Pantilat, professor and chief of the Division of Palliative Medicine at the University of California San Francisco.
Dr. Pantilat recalled a patient with advanced pancreatic cancer who wished to see her daughter’s wedding in 10 months. He knew that was unlikely, but the discussion led to another solution.
Her daughter moved the wedding to the ICU.
Hope can persist and uplift even in the darkest of times, and “as clinicians, we need to be in the true hope business,” he said.
While some patients may wish for a cure, others may want more time with family or comfort in the face of suffering. People can “hope for all the things that can still be, despite the fact that there’s a lot of things that can’t,” he said.
However, fear that a patient will hope for a cure, and that the difficult discussions to follow might destroy hope or lead to false hope, sometimes means physicians won’t begin the conversation.
“We want to be honest with our patients — compassionate and kind, but honest — when we talk about their hopes,” Dr. Pantilat explained. Sometimes that means he needs to tell patients, “I wish that could happen. I wish I had a treatment that could make your cancer go away, but unfortunately, I don’t. So let’s think about what else we can do to help you.”
Having these difficult discussions matters. The evidence, although limited, indicates that feeling hopeful can improve patients’ well-being and may even boost their cancer outcomes.
One recent study found, for instance, that patients who reported feeling more hopeful also had lower levels of depression and anxiety. Early research also suggests that greater levels of hope may have a hand in reducing inflammation in patients with ovarian cancer and could even improve survival in some patients with advanced cancer.
For Dr. Leiter, while these lessons came early in his career as a palliative care physician, they persist and influence his practice today.
“I know that I could not have prevented Carlos’ death. None of us could have, and none of us could have protected his mother from the unimaginable grief that will stay with her for the rest of her life,” he said. “But I could have made things just a little bit less difficult for her.
“I could have acted as her guide rather than her cross-examiner,” he continued, explaining that he now sees hope as “a generous collaborator” that can coexist with rising creatinine levels, failing livers, and fears about intubation.
“As clinicians, we can always find space to hope with our patients and their families,” he said. “So now, years later when I sit with a terrified and grieving family and they tell me they hope their loved one gets better, I remember Carlos’ mother’s eyes piercing mine ... and I know how to respond: ‘I hope so, too.’ And I do.”
A version of this article appeared on Medscape.com.
CHICAGO — Carlos, a 21-year-old, lay in a hospital bed, barely clinging to life. Following a stem cell transplant for leukemia, Carlos had developed a life-threatening case of graft-vs-host disease.
But Carlos’ mother had faith.
“I have hope things will get better,” she said, via interpreter, to Richard Leiter, MD, a palliative care doctor in training at that time.
“I hope they will,” Dr. Leiter told her.
“I should have stopped there,” said Dr. Leiter, recounting an early-career lesson on hope during the ASCO Voices session at the American Society of Clinical Oncology annual meeting. “But in my eagerness to show my attending and myself that I could handle this conversation, I kept going, mistakenly.”
“But none of us think they will,” Dr. Leiter continued.
Carlos’ mother looked Dr. Leiter in the eye. “You want him to die,” she said.
“I knew, even then, that she was right,” recalled Dr. Leiter, now a palliative care physician at Dana-Farber Cancer Institute and Brigham and Women’s Hospital and an assistant professor of medicine at Harvard Medical School, Boston.
Although there was nothing he could do to save Carlos, Dr. Leiter also couldn’t sit with the extreme suffering. “The pain was too great,” Dr. Leiter said. “I needed her to adopt our narrative that we had done everything we could to help him live, and now, we would do everything we could to help his death be a comfortable one.”
But looking back, Dr. Leiter realized, “How could we have asked her to accept what was fundamentally unacceptable, to comprehend the incomprehensible?”
The Importance of Hope
Alan B. Astrow, MD, said during an ASCO symposium on “The Art and Science of Hope.”
“How we think about hope directly influences patient care,” said Dr. Astrow, chief of hematology and medical oncology at NewYork-Presbyterian Brooklyn Methodist Hospital and a professor of clinical medicine at Weill Cornell Medicine in New York City.
Hope, whatever it turns out to be neurobiologically, is “very much a gift” that underlies human existence, he said.
Physicians have the capacity to restore or shatter a patient’s hopes, and those who come to understand the importance of hope will wish to extend the gift to others, Dr. Astrow said.
Asking patients about their hopes is the “golden question,” Steven Z. Pantilat, MD, said at the symposium. “When you think about the future, what do you hope for?”
Often, the answers reveal not only “things beyond a cure that matter tremendously to the patient but things that we can help with,” said Dr. Pantilat, professor and chief of the Division of Palliative Medicine at the University of California San Francisco.
Dr. Pantilat recalled a patient with advanced pancreatic cancer who wished to see her daughter’s wedding in 10 months. He knew that was unlikely, but the discussion led to another solution.
Her daughter moved the wedding to the ICU.
Hope can persist and uplift even in the darkest of times, and “as clinicians, we need to be in the true hope business,” he said.
While some patients may wish for a cure, others may want more time with family or comfort in the face of suffering. People can “hope for all the things that can still be, despite the fact that there’s a lot of things that can’t,” he said.
However, fear that a patient will hope for a cure, and that the difficult discussions to follow might destroy hope or lead to false hope, sometimes means physicians won’t begin the conversation.
“We want to be honest with our patients — compassionate and kind, but honest — when we talk about their hopes,” Dr. Pantilat explained. Sometimes that means he needs to tell patients, “I wish that could happen. I wish I had a treatment that could make your cancer go away, but unfortunately, I don’t. So let’s think about what else we can do to help you.”
Having these difficult discussions matters. The evidence, although limited, indicates that feeling hopeful can improve patients’ well-being and may even boost their cancer outcomes.
One recent study found, for instance, that patients who reported feeling more hopeful also had lower levels of depression and anxiety. Early research also suggests that greater levels of hope may have a hand in reducing inflammation in patients with ovarian cancer and could even improve survival in some patients with advanced cancer.
For Dr. Leiter, while these lessons came early in his career as a palliative care physician, they persist and influence his practice today.
“I know that I could not have prevented Carlos’ death. None of us could have, and none of us could have protected his mother from the unimaginable grief that will stay with her for the rest of her life,” he said. “But I could have made things just a little bit less difficult for her.
“I could have acted as her guide rather than her cross-examiner,” he continued, explaining that he now sees hope as “a generous collaborator” that can coexist with rising creatinine levels, failing livers, and fears about intubation.
“As clinicians, we can always find space to hope with our patients and their families,” he said. “So now, years later when I sit with a terrified and grieving family and they tell me they hope their loved one gets better, I remember Carlos’ mother’s eyes piercing mine ... and I know how to respond: ‘I hope so, too.’ And I do.”
A version of this article appeared on Medscape.com.
CHICAGO — Carlos, a 21-year-old, lay in a hospital bed, barely clinging to life. Following a stem cell transplant for leukemia, Carlos had developed a life-threatening case of graft-vs-host disease.
But Carlos’ mother had faith.
“I have hope things will get better,” she said, via interpreter, to Richard Leiter, MD, a palliative care doctor in training at that time.
“I hope they will,” Dr. Leiter told her.
“I should have stopped there,” said Dr. Leiter, recounting an early-career lesson on hope during the ASCO Voices session at the American Society of Clinical Oncology annual meeting. “But in my eagerness to show my attending and myself that I could handle this conversation, I kept going, mistakenly.”
“But none of us think they will,” Dr. Leiter continued.
Carlos’ mother looked Dr. Leiter in the eye. “You want him to die,” she said.
“I knew, even then, that she was right,” recalled Dr. Leiter, now a palliative care physician at Dana-Farber Cancer Institute and Brigham and Women’s Hospital and an assistant professor of medicine at Harvard Medical School, Boston.
Although there was nothing he could do to save Carlos, Dr. Leiter also couldn’t sit with the extreme suffering. “The pain was too great,” Dr. Leiter said. “I needed her to adopt our narrative that we had done everything we could to help him live, and now, we would do everything we could to help his death be a comfortable one.”
But looking back, Dr. Leiter realized, “How could we have asked her to accept what was fundamentally unacceptable, to comprehend the incomprehensible?”
The Importance of Hope
Alan B. Astrow, MD, said during an ASCO symposium on “The Art and Science of Hope.”
“How we think about hope directly influences patient care,” said Dr. Astrow, chief of hematology and medical oncology at NewYork-Presbyterian Brooklyn Methodist Hospital and a professor of clinical medicine at Weill Cornell Medicine in New York City.
Hope, whatever it turns out to be neurobiologically, is “very much a gift” that underlies human existence, he said.
Physicians have the capacity to restore or shatter a patient’s hopes, and those who come to understand the importance of hope will wish to extend the gift to others, Dr. Astrow said.
Asking patients about their hopes is the “golden question,” Steven Z. Pantilat, MD, said at the symposium. “When you think about the future, what do you hope for?”
Often, the answers reveal not only “things beyond a cure that matter tremendously to the patient but things that we can help with,” said Dr. Pantilat, professor and chief of the Division of Palliative Medicine at the University of California San Francisco.
Dr. Pantilat recalled a patient with advanced pancreatic cancer who wished to see her daughter’s wedding in 10 months. He knew that was unlikely, but the discussion led to another solution.
Her daughter moved the wedding to the ICU.
Hope can persist and uplift even in the darkest of times, and “as clinicians, we need to be in the true hope business,” he said.
While some patients may wish for a cure, others may want more time with family or comfort in the face of suffering. People can “hope for all the things that can still be, despite the fact that there’s a lot of things that can’t,” he said.
However, fear that a patient will hope for a cure, and that the difficult discussions to follow might destroy hope or lead to false hope, sometimes means physicians won’t begin the conversation.
“We want to be honest with our patients — compassionate and kind, but honest — when we talk about their hopes,” Dr. Pantilat explained. Sometimes that means he needs to tell patients, “I wish that could happen. I wish I had a treatment that could make your cancer go away, but unfortunately, I don’t. So let’s think about what else we can do to help you.”
Having these difficult discussions matters. The evidence, although limited, indicates that feeling hopeful can improve patients’ well-being and may even boost their cancer outcomes.
One recent study found, for instance, that patients who reported feeling more hopeful also had lower levels of depression and anxiety. Early research also suggests that greater levels of hope may have a hand in reducing inflammation in patients with ovarian cancer and could even improve survival in some patients with advanced cancer.
For Dr. Leiter, while these lessons came early in his career as a palliative care physician, they persist and influence his practice today.
“I know that I could not have prevented Carlos’ death. None of us could have, and none of us could have protected his mother from the unimaginable grief that will stay with her for the rest of her life,” he said. “But I could have made things just a little bit less difficult for her.
“I could have acted as her guide rather than her cross-examiner,” he continued, explaining that he now sees hope as “a generous collaborator” that can coexist with rising creatinine levels, failing livers, and fears about intubation.
“As clinicians, we can always find space to hope with our patients and their families,” he said. “So now, years later when I sit with a terrified and grieving family and they tell me they hope their loved one gets better, I remember Carlos’ mother’s eyes piercing mine ... and I know how to respond: ‘I hope so, too.’ And I do.”
A version of this article appeared on Medscape.com.
FROM ASCO 2024
DEA Training Mandate: 8 Hours of My Life I’d Like Back
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty. 
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes! 
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty.
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes!
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
It’s time to renew two of my three narcotic prescribing licenses. For the first time in my career, I’ve waffled on whether the financial outlay to the US Drug Enforcement Agency (DEA) is worth it.
At $888 each, I’ve considered letting two licenses lapse because I only work part-time in Montana. But several friends advised me to keep a “spare” in case I transfer to a new location.
I thought about just paying the fees until I could do a little more research, but there is no mechanism for a refund unless I die within the first year of the 3-year cycle, provide incorrect credit card digits, or accidentally duplicate payments.
The renewal fee is just part of the issue.
Mandatory 8-Hour Training
I also received an alert about the requirement for more “narcotics prescribing education” thanks to the Medication Access and Training Expansion Act (MATE).
The requirement seems counterintuitive because opioid prescribing has decreased for the 10th consecutive year, according to the AMA Overdose Epidemic Report. The continuing rise in overdose deaths is largely due to illegitimate manufacturing of synthetic opioids.
I’ve written zero outpatient narcotics prescriptions in the past 6 years, and I’ve written very few in my 33 years of practice. My use is limited to intravenous morphine for flash pulmonary edema or refractory angina, but unless you graduated from a training program within 5 years of the June 2023 mandate or are boarded in addiction medicine, there is no way to escape the 8-hour education requirement.
The problem is that these courses are never just 8 hours in duration. After signing up for one such CME course that cost $150, I was still dying of boredom and at risk for DVT 4 days later. That’s how long it took to sit through.
Instead of the 30 seconds it should have taken to review the simple instructions to deliver Narcan, there were scores of screens followed by juvenile quizlets and cartoons. All but about 2 hours out of the 4 days is now relegated to that category of “hours of my life that I can never get back.” Additionally, none of that mandatory “education” will change my prescribing habits one whit.
And beware the penalty.
Of course, I would always be truthful when asked to check the box on the DEA renewal application attesting to my having completed the required education. On the outside chance that you plan to check the yes box without completing the relevant courses, those found guilty of such false claims could be fined up to $250,000 and subject to “not more than four years in prison,” or both. Yikes!
Larry Houck, a former DEA investigator, explained that “[t]here are lot of people who are coming up for renewal and log on but still don’t know this is a requirement.” Neither ignorance nor complacency is an acceptable defense.
Changes Needed
The only good thing that came of those 4 long days of opioid education was a motivation to drive change in our current licensing and educational experience. Why not use this opportunity to reform the DEA-physician/prescriber relationship?
The educational requirements should be curtailed for those of us who do not provide outpatient narcotic prescriptions even if we use inpatient opioids. Meds with low abuse potential should be rescheduled to minimize who gets caught in the broad net of the education requirement.
We should reduce overregulation of the legitimate prescribers by lowering, instead of increasing, licensing fees. We should change to a single license number that covers every state. In this digital age, there is no legitimate excuse to prevent this from happening.
After all, the settlements from opioid manufacturers and distributors will in time total $50 billion. It seems that at least some of the responsibilities of the DEA could shift to states, cities, and towns.
My friend Siamak Karimian, MD, who provides locum services in multiple states, pays for seven active DEA licenses every 3 years. He pointed out the hypocrisy in the current regulatory system: “It’s funny that you can have only one DEA or state license and work for the government in all other states or territories with no limits, including the VA, Indian healthcare systems, or prison systems.”
All other prescribers require a separate DEA number for every state. Ultimately, you’d think tracking prescriptions for a single DEA number should be far simpler than tracking someone with seven.
Competent physicians not guilty of criminal overprescribing seem to be the last to be considered in nearly every healthcare endeavor these days. It would be refreshing if they would reduce our fees and prevent this waste of our time.
And while we are at it, perhaps a more fitting punishment is due for Richard Sackler and all the Purdue Pharma–affiliated family members. The Sacklers will pay out $6 billion in exchange for immunity against civil litigation. That doesn’t seem like much when they are worth $11 billion.
Perhaps they should be made to take an 8-hour course on opioid prescribing, annually and in perpetuity. Let’s see them complete a few quizlets and sit through screens of instruction on how to administer Naloxone. Of course, that would be a mild punishment for those who manufactured a drug that killed hundreds of thousands. But it would be a start.
Dr. Walton-Shirley, a clinical cardiologist in Nashville, Tennessee, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Beta Thalassemia: Pricey Gene Therapy Hits The Mark
With luck, maybe Ms. Ahmed’s son could follow in his aunt’s footsteps and get a stem cell transplant from a compatible family donor. But while little Yusuf Saeed has a twin sister of his own, she wasn’t a match. Without another treatment option, he’d face the prospect of a lifetime not only cut short but burdened by multiple monthly transfusions and severe limitations.
Then came glimpses of hope. One of Yusuf’s physicians at Cohen Children’s Medical Center in Long Island, New York, told Yusuf’s mother about a new kind of gene therapy on the horizon. But it took time to get FDA approval. Yusuf grew older, heading toward his teenage years, when regular transfusions would be a huge burden. “He’s turning 5 and 6, and there’s nothing,” Ms. Ahmed recalled, and the family worried.
Finally, the FDA approved the one-time treatment — betibeglogene autotemcel (beti-cel, Zynteglo) in 2022. By January 2024, the hospital was ready to treat Yusuf. At age 8, he became the first patient in the state of New York to undergo gene therapy for beta thalassemia.
A medical team infused Yusuf with his own stem cells, which had been genetically engineered to boost production of hemoglobin and prevent thalassemia’s devastating effects.
There are caveats about the treatment. It’s an extraordinarily expensive therapy that can be performed at only a few institutions. And it’s so brand new that caveats may not even have appeared yet. Yet, for kids like Yusuf, the gene therapy could transform a life.
“We feel like a weight has been lifted,” Ms. Ahmed said in an interview. “It’s something we’ve been waiting for.”
Anemia Becomes a Lifetime Threat
Among all genetic diseases, thalassemia stands alone. It’s the most common condition caused by a single gene, according to Hanny Al-Samkari, MD, a hematologist/clinical investigator at Massachusetts General Hospital and associate professor of medicine at Harvard Medical School, in Boston, Massachusetts.
Millions of people have the thalassemia trait, especially in southern Europe, the Middle East, southeast Asia, and Africa, Dr. Al-Samkari said. (Yusuf’s parents are from Pakistan.)
The trait, which appears to provide protection against malaria, may cause mild anemia in some cases but is otherwise harmless. However, a child born to parents with the same kind of trait has a high risk of developing alpha thalassemia or beta thalassemia. Like his aunt, Yusuf developed beta thalassemia, which is generally more severe. Yusuf’s bleeding disorder requires him to be transfusion-dependent.
In these patients, the disease disrupts the production of red blood cells in the bone marrow, Dr. Al-Samkari said. Hemoglobin levels can fall to 7 or 8 g/dL, compared with the normal levels of 12-16 g/dL in adults. “They’re chronically anemic, and that low hemoglobin that leads to things you associate with anemia: fatigue, reduced exercise tolerance, mind fog, challenges with work or school, and hypersomnolence.”
In addition, the bones become thinner and more brittle, he said, leading to fractures.
Transfusions are one treatment option, but they’re needed for a lifetime and cause their own problems, such as iron overload. Care of thalassemia patients “becomes quite complex and quite challenging for both families and medical institutions,” Alexis A. Thompson MD, MPH, chief of hematology at Children’s Hospital of Philadelphia, Pennsylvania, said in an interview.
Yusara Ahmed remembers her sister’s endless visits to the hospital after she was diagnosed at age 4. “We were all very traumatized by the hospital environment,” she said. But good news came in 2008, a few years later, when her sister was able to get a stem cell transplant from their brother.
But while stem cell transplants can be curative, most children don’t have a relative who can be a suitable match as a donor, Dr. Thompson said. Now, gene therapy offers another option, by turning a patient into his or her own matched donor.
Stem Cells Out, Stem Cells In
Last year, Yusuf went to Cohen Children’s Medical Center to donate stem cells, which were sent to a laboratory where they were genetically engineered to add copies of the beta-globin gene. Then, in January 2024, the modified stem cells were infused back into Yusuf after he underwent chemotherapy to make room for them in his bone marrow.
In April, a bald-headed Yusuf played with toy dinosaurs while his mother and clinicians met the media at a hospital press conference about his so-far-successful treatment. Early reports about the efficacy of the treatment suggest it may be the proverbial “game changer” for many of the estimated 100,000-plus people in the world who are diagnosed with transfusion-dependent beta thalassemia each year.
Over a median follow-up of 29.5 months, 20 of 22 patients treated with beti-cel no longer needed transfusions, according to a 2022 open-label phase 3 study published in the New England Journal of Medicine. Only one adverse event — thrombocytopenia in one patient — was considered both serious and related to the treatment, the industry-funded trial reported.
Costly Treatment Seems to Be Cost-Effective
As of 2022, gene therapy for transfusion-dependent beta thalassemia was listed as $2.8 million per treatment making it the most expensive single-treatment therapy ever approved in the United States. The price is “extraordinary,” said Dr. Thompson. “For some families, it gives them pause when they first hear about it.”
The hospital makes the case to insurers that covering the treatment is cost-effective in the long run, considering the high cost of traditional treatment, she said. “We’ve been very successful in getting coverage.”
In addition, the independent Institute for Clinical and Economic Review reported in 2022 that the treatment will be cost-effective at the “anticipated price of $2.1 million with an 80% payback option for patients who do not achieve and maintain transfusion independence over a 5-year period.”
Moving Forward, Clinicians Want to Reduce Complications
What’s next for transfusion-dependent beta thalassemia treatment? Earlier this year, the FDA approved a second gene therapy treatment called exagamglogene autotemcel (exa-cel, Casgevy). “We’re just beginning to evaluate individuals for the product, and we intend to make it available for families as well,” Dr. Thompson said.
In the bigger picture, she said gene therapy still has room for improvement. The need for chemotherapy is one target. According to her, it causes most of the complications related to gene therapy.
“Chemotherapy is a part of all gene therapies today because one has to make space in the bone marrow in order to have modified stem cells to come back to settle in and grow,” she said.
One strategy is to reduce the number of stem cells that are required for the therapy to work. “That would essentially eliminate the need for chemotherapy,” she said. “We’re not there yet.”
Another goal is to reduce the small risk of complications from gene therapy itself, she said. “Overall, though, this doesn’t detract us at all from being very excited about how well children are doing with the current approach. We’re very enthusiastic and very confident in recommending it to families.”
Back on Long Island, a Sense of Relief
Several months after his treatment, Yusuf is doing well. His hemoglobin levels are increasing, and his bone marrow has grown back, his mother said. He’s being home-schooled for the time being because he still faces a risk of infection. (Ms. Ahmed, a stay-at-home mom, has worked a teacher and mosque volunteer. Her husband runs a consumer electronics business.)
As Yusuf gets better, his parents hope they’ll soon be able to take a long trip back home to Pakistan to see relatives. They’ll be able to share their son with family along with something else: a sense of relief.
Dr. Al-Samkari discloses consulting for Agios. Dr. Thompson discloses research for Beam, Bluebird Bio, Editas, Novartis, and Novo Nordisk and consulting for Beam, Bluebird Bio, Editas, Roche, and Vertex.
With luck, maybe Ms. Ahmed’s son could follow in his aunt’s footsteps and get a stem cell transplant from a compatible family donor. But while little Yusuf Saeed has a twin sister of his own, she wasn’t a match. Without another treatment option, he’d face the prospect of a lifetime not only cut short but burdened by multiple monthly transfusions and severe limitations.
Then came glimpses of hope. One of Yusuf’s physicians at Cohen Children’s Medical Center in Long Island, New York, told Yusuf’s mother about a new kind of gene therapy on the horizon. But it took time to get FDA approval. Yusuf grew older, heading toward his teenage years, when regular transfusions would be a huge burden. “He’s turning 5 and 6, and there’s nothing,” Ms. Ahmed recalled, and the family worried.
Finally, the FDA approved the one-time treatment — betibeglogene autotemcel (beti-cel, Zynteglo) in 2022. By January 2024, the hospital was ready to treat Yusuf. At age 8, he became the first patient in the state of New York to undergo gene therapy for beta thalassemia.
A medical team infused Yusuf with his own stem cells, which had been genetically engineered to boost production of hemoglobin and prevent thalassemia’s devastating effects.
There are caveats about the treatment. It’s an extraordinarily expensive therapy that can be performed at only a few institutions. And it’s so brand new that caveats may not even have appeared yet. Yet, for kids like Yusuf, the gene therapy could transform a life.
“We feel like a weight has been lifted,” Ms. Ahmed said in an interview. “It’s something we’ve been waiting for.”
Anemia Becomes a Lifetime Threat
Among all genetic diseases, thalassemia stands alone. It’s the most common condition caused by a single gene, according to Hanny Al-Samkari, MD, a hematologist/clinical investigator at Massachusetts General Hospital and associate professor of medicine at Harvard Medical School, in Boston, Massachusetts.
Millions of people have the thalassemia trait, especially in southern Europe, the Middle East, southeast Asia, and Africa, Dr. Al-Samkari said. (Yusuf’s parents are from Pakistan.)
The trait, which appears to provide protection against malaria, may cause mild anemia in some cases but is otherwise harmless. However, a child born to parents with the same kind of trait has a high risk of developing alpha thalassemia or beta thalassemia. Like his aunt, Yusuf developed beta thalassemia, which is generally more severe. Yusuf’s bleeding disorder requires him to be transfusion-dependent.
In these patients, the disease disrupts the production of red blood cells in the bone marrow, Dr. Al-Samkari said. Hemoglobin levels can fall to 7 or 8 g/dL, compared with the normal levels of 12-16 g/dL in adults. “They’re chronically anemic, and that low hemoglobin that leads to things you associate with anemia: fatigue, reduced exercise tolerance, mind fog, challenges with work or school, and hypersomnolence.”
In addition, the bones become thinner and more brittle, he said, leading to fractures.
Transfusions are one treatment option, but they’re needed for a lifetime and cause their own problems, such as iron overload. Care of thalassemia patients “becomes quite complex and quite challenging for both families and medical institutions,” Alexis A. Thompson MD, MPH, chief of hematology at Children’s Hospital of Philadelphia, Pennsylvania, said in an interview.
Yusara Ahmed remembers her sister’s endless visits to the hospital after she was diagnosed at age 4. “We were all very traumatized by the hospital environment,” she said. But good news came in 2008, a few years later, when her sister was able to get a stem cell transplant from their brother.
But while stem cell transplants can be curative, most children don’t have a relative who can be a suitable match as a donor, Dr. Thompson said. Now, gene therapy offers another option, by turning a patient into his or her own matched donor.
Stem Cells Out, Stem Cells In
Last year, Yusuf went to Cohen Children’s Medical Center to donate stem cells, which were sent to a laboratory where they were genetically engineered to add copies of the beta-globin gene. Then, in January 2024, the modified stem cells were infused back into Yusuf after he underwent chemotherapy to make room for them in his bone marrow.
In April, a bald-headed Yusuf played with toy dinosaurs while his mother and clinicians met the media at a hospital press conference about his so-far-successful treatment. Early reports about the efficacy of the treatment suggest it may be the proverbial “game changer” for many of the estimated 100,000-plus people in the world who are diagnosed with transfusion-dependent beta thalassemia each year.
Over a median follow-up of 29.5 months, 20 of 22 patients treated with beti-cel no longer needed transfusions, according to a 2022 open-label phase 3 study published in the New England Journal of Medicine. Only one adverse event — thrombocytopenia in one patient — was considered both serious and related to the treatment, the industry-funded trial reported.
Costly Treatment Seems to Be Cost-Effective
As of 2022, gene therapy for transfusion-dependent beta thalassemia was listed as $2.8 million per treatment making it the most expensive single-treatment therapy ever approved in the United States. The price is “extraordinary,” said Dr. Thompson. “For some families, it gives them pause when they first hear about it.”
The hospital makes the case to insurers that covering the treatment is cost-effective in the long run, considering the high cost of traditional treatment, she said. “We’ve been very successful in getting coverage.”
In addition, the independent Institute for Clinical and Economic Review reported in 2022 that the treatment will be cost-effective at the “anticipated price of $2.1 million with an 80% payback option for patients who do not achieve and maintain transfusion independence over a 5-year period.”
Moving Forward, Clinicians Want to Reduce Complications
What’s next for transfusion-dependent beta thalassemia treatment? Earlier this year, the FDA approved a second gene therapy treatment called exagamglogene autotemcel (exa-cel, Casgevy). “We’re just beginning to evaluate individuals for the product, and we intend to make it available for families as well,” Dr. Thompson said.
In the bigger picture, she said gene therapy still has room for improvement. The need for chemotherapy is one target. According to her, it causes most of the complications related to gene therapy.
“Chemotherapy is a part of all gene therapies today because one has to make space in the bone marrow in order to have modified stem cells to come back to settle in and grow,” she said.
One strategy is to reduce the number of stem cells that are required for the therapy to work. “That would essentially eliminate the need for chemotherapy,” she said. “We’re not there yet.”
Another goal is to reduce the small risk of complications from gene therapy itself, she said. “Overall, though, this doesn’t detract us at all from being very excited about how well children are doing with the current approach. We’re very enthusiastic and very confident in recommending it to families.”
Back on Long Island, a Sense of Relief
Several months after his treatment, Yusuf is doing well. His hemoglobin levels are increasing, and his bone marrow has grown back, his mother said. He’s being home-schooled for the time being because he still faces a risk of infection. (Ms. Ahmed, a stay-at-home mom, has worked a teacher and mosque volunteer. Her husband runs a consumer electronics business.)
As Yusuf gets better, his parents hope they’ll soon be able to take a long trip back home to Pakistan to see relatives. They’ll be able to share their son with family along with something else: a sense of relief.
Dr. Al-Samkari discloses consulting for Agios. Dr. Thompson discloses research for Beam, Bluebird Bio, Editas, Novartis, and Novo Nordisk and consulting for Beam, Bluebird Bio, Editas, Roche, and Vertex.
With luck, maybe Ms. Ahmed’s son could follow in his aunt’s footsteps and get a stem cell transplant from a compatible family donor. But while little Yusuf Saeed has a twin sister of his own, she wasn’t a match. Without another treatment option, he’d face the prospect of a lifetime not only cut short but burdened by multiple monthly transfusions and severe limitations.
Then came glimpses of hope. One of Yusuf’s physicians at Cohen Children’s Medical Center in Long Island, New York, told Yusuf’s mother about a new kind of gene therapy on the horizon. But it took time to get FDA approval. Yusuf grew older, heading toward his teenage years, when regular transfusions would be a huge burden. “He’s turning 5 and 6, and there’s nothing,” Ms. Ahmed recalled, and the family worried.
Finally, the FDA approved the one-time treatment — betibeglogene autotemcel (beti-cel, Zynteglo) in 2022. By January 2024, the hospital was ready to treat Yusuf. At age 8, he became the first patient in the state of New York to undergo gene therapy for beta thalassemia.
A medical team infused Yusuf with his own stem cells, which had been genetically engineered to boost production of hemoglobin and prevent thalassemia’s devastating effects.
There are caveats about the treatment. It’s an extraordinarily expensive therapy that can be performed at only a few institutions. And it’s so brand new that caveats may not even have appeared yet. Yet, for kids like Yusuf, the gene therapy could transform a life.
“We feel like a weight has been lifted,” Ms. Ahmed said in an interview. “It’s something we’ve been waiting for.”
Anemia Becomes a Lifetime Threat
Among all genetic diseases, thalassemia stands alone. It’s the most common condition caused by a single gene, according to Hanny Al-Samkari, MD, a hematologist/clinical investigator at Massachusetts General Hospital and associate professor of medicine at Harvard Medical School, in Boston, Massachusetts.
Millions of people have the thalassemia trait, especially in southern Europe, the Middle East, southeast Asia, and Africa, Dr. Al-Samkari said. (Yusuf’s parents are from Pakistan.)
The trait, which appears to provide protection against malaria, may cause mild anemia in some cases but is otherwise harmless. However, a child born to parents with the same kind of trait has a high risk of developing alpha thalassemia or beta thalassemia. Like his aunt, Yusuf developed beta thalassemia, which is generally more severe. Yusuf’s bleeding disorder requires him to be transfusion-dependent.
In these patients, the disease disrupts the production of red blood cells in the bone marrow, Dr. Al-Samkari said. Hemoglobin levels can fall to 7 or 8 g/dL, compared with the normal levels of 12-16 g/dL in adults. “They’re chronically anemic, and that low hemoglobin that leads to things you associate with anemia: fatigue, reduced exercise tolerance, mind fog, challenges with work or school, and hypersomnolence.”
In addition, the bones become thinner and more brittle, he said, leading to fractures.
Transfusions are one treatment option, but they’re needed for a lifetime and cause their own problems, such as iron overload. Care of thalassemia patients “becomes quite complex and quite challenging for both families and medical institutions,” Alexis A. Thompson MD, MPH, chief of hematology at Children’s Hospital of Philadelphia, Pennsylvania, said in an interview.
Yusara Ahmed remembers her sister’s endless visits to the hospital after she was diagnosed at age 4. “We were all very traumatized by the hospital environment,” she said. But good news came in 2008, a few years later, when her sister was able to get a stem cell transplant from their brother.
But while stem cell transplants can be curative, most children don’t have a relative who can be a suitable match as a donor, Dr. Thompson said. Now, gene therapy offers another option, by turning a patient into his or her own matched donor.
Stem Cells Out, Stem Cells In
Last year, Yusuf went to Cohen Children’s Medical Center to donate stem cells, which were sent to a laboratory where they were genetically engineered to add copies of the beta-globin gene. Then, in January 2024, the modified stem cells were infused back into Yusuf after he underwent chemotherapy to make room for them in his bone marrow.
In April, a bald-headed Yusuf played with toy dinosaurs while his mother and clinicians met the media at a hospital press conference about his so-far-successful treatment. Early reports about the efficacy of the treatment suggest it may be the proverbial “game changer” for many of the estimated 100,000-plus people in the world who are diagnosed with transfusion-dependent beta thalassemia each year.
Over a median follow-up of 29.5 months, 20 of 22 patients treated with beti-cel no longer needed transfusions, according to a 2022 open-label phase 3 study published in the New England Journal of Medicine. Only one adverse event — thrombocytopenia in one patient — was considered both serious and related to the treatment, the industry-funded trial reported.
Costly Treatment Seems to Be Cost-Effective
As of 2022, gene therapy for transfusion-dependent beta thalassemia was listed as $2.8 million per treatment making it the most expensive single-treatment therapy ever approved in the United States. The price is “extraordinary,” said Dr. Thompson. “For some families, it gives them pause when they first hear about it.”
The hospital makes the case to insurers that covering the treatment is cost-effective in the long run, considering the high cost of traditional treatment, she said. “We’ve been very successful in getting coverage.”
In addition, the independent Institute for Clinical and Economic Review reported in 2022 that the treatment will be cost-effective at the “anticipated price of $2.1 million with an 80% payback option for patients who do not achieve and maintain transfusion independence over a 5-year period.”
Moving Forward, Clinicians Want to Reduce Complications
What’s next for transfusion-dependent beta thalassemia treatment? Earlier this year, the FDA approved a second gene therapy treatment called exagamglogene autotemcel (exa-cel, Casgevy). “We’re just beginning to evaluate individuals for the product, and we intend to make it available for families as well,” Dr. Thompson said.
In the bigger picture, she said gene therapy still has room for improvement. The need for chemotherapy is one target. According to her, it causes most of the complications related to gene therapy.
“Chemotherapy is a part of all gene therapies today because one has to make space in the bone marrow in order to have modified stem cells to come back to settle in and grow,” she said.
One strategy is to reduce the number of stem cells that are required for the therapy to work. “That would essentially eliminate the need for chemotherapy,” she said. “We’re not there yet.”
Another goal is to reduce the small risk of complications from gene therapy itself, she said. “Overall, though, this doesn’t detract us at all from being very excited about how well children are doing with the current approach. We’re very enthusiastic and very confident in recommending it to families.”
Back on Long Island, a Sense of Relief
Several months after his treatment, Yusuf is doing well. His hemoglobin levels are increasing, and his bone marrow has grown back, his mother said. He’s being home-schooled for the time being because he still faces a risk of infection. (Ms. Ahmed, a stay-at-home mom, has worked a teacher and mosque volunteer. Her husband runs a consumer electronics business.)
As Yusuf gets better, his parents hope they’ll soon be able to take a long trip back home to Pakistan to see relatives. They’ll be able to share their son with family along with something else: a sense of relief.
Dr. Al-Samkari discloses consulting for Agios. Dr. Thompson discloses research for Beam, Bluebird Bio, Editas, Novartis, and Novo Nordisk and consulting for Beam, Bluebird Bio, Editas, Roche, and Vertex.