User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Nail Biopsy: 6 Techniques to Biopsy the Nail Matrix
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
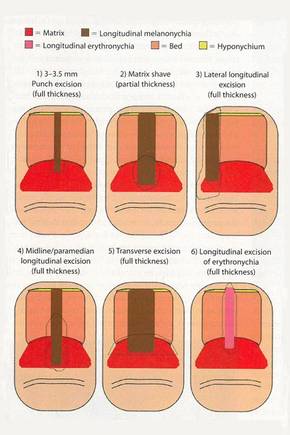
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
Nail matrix biopsies are performed to confirm a diagnosis or surgically remove a skin lesion that is affecting the growth of the nail plate. The procedure may be used to identify:
- Inflammatory conditions such as nail psoriasis and lichen planus
- Benign tumors
- Solitary melanonychia
- Squamous cell carcinoma (SCC)
- Other nail disorders
Nail biopsy can lead to complications such as bleeding, infection, or scarring. Postoperative scarring can cause permanent nail splitting, dystrophy, or both.
In a Cosmetic Dermatology article, “Matrix Biopsy of Longitudinal Melanonychia and Longitudinal Erythronychia: A Step-by-Step Approach,” Drs. Siobhan C. Collins and Nathaniel J. Jellinek review 6 techniques used to biopsy the nail matrix.
- Punch excision
- Matrix shave
- Lateral longitudinal excision
- Midline/paramedian longitudinal excision
- Transverse excision
- Longitudinal excision of erythronychia
In the setting of longitudinal melanonychia (to diagnose nail melanoma or SCC) and longitudinal erythronychia (to diagnose SCC and rarely amelanotic melanoma or basal cell carcinoma), the techniques they describe accomplish 3 fundamental goals of nail surgery:
- Obtain adequate tissue via an excisional biopsy to make an accurate diagnosis and avoid sampling error
- Avoid unnecessary trauma to surrounding nail tissues by the judicious use of partial plate avulsions whenever feasible
- Avoid unnecessary postoperative nail scarring whenever possible
Dermatologists must be confident when performing nail biopsies and the techniques discussed by the authors will help approach nail surgery with more certainty.
At the 73rd Annual Meeting of the American Academy of Dermatology, Dr. Jellinek provides a hands-on approach to nail surgery. On Saturday, March 21, he will provide tips for nail surgeries at the “Medical and Surgical Management of Nail Disorders” lecture.
For more information, read the Collins and Jellinek article from Cosmetic Dermatology.
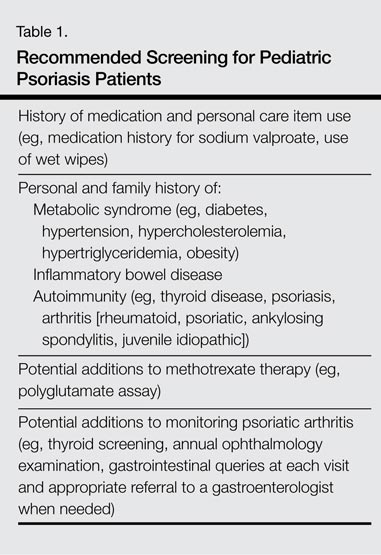
Update on Pediatric Psoriasis
Psoriasis affects 2% to 4% of the US population, with approximately one-third of cases beginning in childhood. The understanding of pediatric psoriasis has developed at a far slower pace than adult disease, with limitations in care including few medications that are approved by the US Food and Drug Administration for pediatric and adolescent use. Recently, a stable fixed-combination dose of calcipo-triene 0.005%–betamethasone dipropionate 0.064% topical suspension was approved for treatment of plaque psoriasis of the scalp in patients aged 12 to 17 years, which hopefully will lead a trend in psoriasis medication approval for children and teenagers.1 Based on a PubMed search of articles indexed for MEDLINE using the search terms pediatric psoriasis, psoriasis, and strep that were published from April 2012 to April 2014, this article reviews newer data to address the issues that surround pediatric psoriasis and to provide an update on prior review articles on pediatric psoriasis.2-5 This article reviews some of the newer literature on clinical presentation and comorbidities in pediatric psoriasis.5 Based on these recent findings, additional screenings including review of obesity parameters are recommended for pediatric patients with psoriasis (Table 1).
Update on Disease Manifestations, Associations, and Comorbidities
Disease Manifestations
A 2013 multicenter study delineated the clinical features of pediatric psoriasis.6 The study was conducted at 8 geographically diverse dermatology clinics in the United States to delineate the clinical manifestations of pediatric psoriasis. In an assessment of 181 participants aged 5 to 17 years, the investigators sought to determine the frequency of disease sites, severity, and guttate disease. Over a period of approximately 2 years, 43.1% of participants were determined to have mild disease and 56.9% had severe disease. Family history of psoriasis was present in 51.4% of participants, with first-degree relatives affected in 59.8% of cases. Scalp involvement at some time was noted in 79.0% of participants, and nail disease was noted in 55% of boys and 29% of girls. Guttate psoriasis was noted in 30% of participants, with more cases in the severe range (35.9%) versus the mild range (21.8%). Additionally, 22.1% of participants had a precipitating streptococcal infection, with the association being more common in pediatric patients with guttate psoriasis than plaque psoriasis.6 This study highlighted that pediatric psoriasis has a genetic basis, is frequently guttate in nature, commonly affects the nails, shows a trend toward being classified as severe, and may be triggered by streptococcal infections.
Streptococcal Infection
Pediatric psoriasis may be triggered or flared by Streptococcus pyogenes (group A β-hemolytic streptococci) infections, specifically β-hemolytic streptococci groups A, C, and G that have streptococcal M protein,2,3,7 and this tendency can be associated with HLA-Cw6 or guttate psoriasis. Newer data have elucidated the role of streptococcal throat infections in psoriasis. Given that streptococcal throat infections are most common in school-aged children, these studies suggest a putative mechanism in pediatric psoriasis for triggering streptococcal infections, which would need to be confirmed in future studies, specifically in pediatric psoriasis patients.
It has been shown that T cells in psoriasis patients recognize common streptococcal M proteins and keratin determinants.7 Ferran et al8 recently demonstrated activation of circulating cutaneous lymphocyte–associated antigen (CLA)+ T cells but not CLA- memory T cells in 27 psoriasis patients (ages not specified) when mixed with streptococcal throat extracts, causing production of IL-17, IP-10, IL-22, and IFN-γ; activation was not found in 6 healthy control patients. Antistreptolysin O levels were correlated with the messenger RNA up- regulation for IL-17, IP-10, IL-22, and IFN-γ, and also correlated with psoriasis area and severity index score in psoriasis patients. In this same study, injection of the activated culture supernatant into mouse skin caused epidermal hyperkeratosis and activation of nonlesional epidermal cells from psoriatic patients. This study thereby delineated some of the potential pathways of the streptococcal induction of psoriasis and psoriatic flares in childhood8; however, confirmation is needed through further study of pediatric psoriatic lymphocyte activity.
Differential Diagnosis
Additions to the extensive differential list have been cited in the recent literature. The differential diagnosis of pediatric psoriasis now includes sodium valproate–induced psoriasiform drug eruption9 and allergic contact dermatitis to methylchloroisothiazolinone and methylisothiazolinone, which are present in many sanitizing hand and diaper wipes and has been reported to cause psoriasiform dermatitis in a periorificial or perineal distribution.10 Clinicians should inquire about the use of these wipes, as caregivers rarely suspect this agent to be causative of the eruption.
Psoriatic Arthritis
Previously, psoriasis and psoriatic arthritis have been linked to autoimmune thyroid disease in adults.11 A study of the Childhood Arthritis & Rheumatology Research Alliance (CARRA) registry showed that family history of psoriasis, autoimmune thyroiditis, Crohn disease, and ankylosing spondylitis in a first-degree relative has been linked to juvenile idiopathic arthritis, highlighting that pediatric psoriasis can be genetically linked or associated with multiple autoimmune conditions and vice versa.12
Obesity, Metabolic Syndrome, and Cardiovascular Risks
Obesity is associated with pediatric psoriasis as highlighted in a growing body of recent literature.13 Excess adiposity as manifested by body mass index in the 85th percentile or greater (37.9% of 155 pediatric psoriasis patients vs 20.5% of 42 controls) and excess central adiposity as manifested by excess waist circumference and increased waist-to-height ratios are more common in pediatric patients with psoriasis than in controls.14
Obesity may be a trigger or associated with increased disease activity in pediatric psoriasis patients. Excess overall adiposity correlates with more severe disease. Obesity parameters may correlate with the onset of psoriasis and with disease severity. In fact, the odds of obesity may be higher in childhood than in adults.14,15 A 2011 report of pediatric psoriasis patients aged 10 to 17 years (n=12) and wart controls (n=6)(mean age, 13.2 and 13.5 years, respectively) demonstrated that 4 of 12 patients with psoriasis and 0 of 6 patients with warts met criteria for metabolic syndrome as defined by 3 of the following: (1) triglycerides greater than or equal to 100 mg/dL; (2) high-density lipoprotein cholesterol less than 50 mg/dL in females and less than 5 mg/dL in males; (3) fasting blood glucose levels greater than or equal to 110 mg/dL, (4) waist circumference greater than the 75th percentile for age and sex; and (5) systolic or diastolic blood pressure greater than the 90th percentile for age, sex, and height.16 These studies highlight that obesity and metabolic syndrome are of concern in pediatric psoriasis patients; however, the best management approach using diet and weight interventions has yet to be identified.
Adiposity may precede the onset of psoriasis. A recent cohort of 27 pediatric psoriasis patients reported that the average age at onset of psoriasis was 8.7 years and the average age at onset of obesity was 4.1 years.15 In this study, 93% (25/27) of patients had adiposity preceding their psoriasis by 2 or more years. It is unclear if this is nature or nurture, as 48% (13/27) of patients had a family history of obesity, 41% (11/27) had a family history of psoriasis, and 48% (13/27) had a family history of hyperlipidemia.15 Therefore, obesity may be cultivated in some psoriatic families. The issue of household influences on diet and obesity needs to be addressed if successful weight management is to be achieved in future studies of pediatric psoriasis.
Cardiovascular risks in the pediatric psoriasis population are the subject of ongoing assessment but will likely mimic studies of adult psoriasis patients when reviewed longitudinally.16 Weight loss and healthy lifestyle interventions likely are beneficial to long-term health, but there is a lack of published data addressing dietary modification as a disease modifier for long-term care of pediatric psoriasis.
Anxiety and Depression
Anxiety and depression have been noted in adults with chronic skin diseases. A recent study assessed 118 patients and caregivers of pediatric patients with atopic dermatitis (n=50), psoriasis (n=25), or vitiligo (n=43) using the Children’s Dermatology Life Quality Index, the Hamilton Anxiety Scale, and the Beck Depression Inventory.17 Anxiety and depression were found in 36% of caregivers of pediatric psoriasis patients and depression was found in 36% of pediatric psoriasis patients, highlighting the need for interventions on a personal and family level to improve quality of life. As a comparator, anxiety was more prevalent in vitiligo caregivers (42%), but depression was only found in 26% of caregivers in the same group. Extent of disease (25%–75% body surface area affected) correlated with both depression and anxiety in the caregivers of pediatric patients with psoriasis as well as with anxiety in caregivers of pediatric patients with increased visible surface area of vitiligo.17 Parental anxiety has been reported at times to be linked to corticosteroid phobia, or corticophobia, which may interfere with disease therapy, as topical corticosteroids are considered the mainstay of therapy in childhood disease.18 Coordinating care with caregivers and addressing their concerns about the safety of medications should be integral to the pediatric psoriasis visit.
Pustular Psoriasis
Pustular psoriasis can be seen in any age group. Researchers recently have attempted to delineate the features and successful management of this severe subset of pediatric psoriasis patients. Twenty-four pediatric pustular psoriasis cases reviewed by Posso-De Los Rios et al19 revealed that 92% (22/24) had generalized and 8% (2/24) had limited acral disease. The mean (standard deviation) age at onset of pediatric pustular psoriasis was 6.3 (4.9) years. Half of the reported cases required more than one intervention. Treatment with acitretin, cyclosporine, and methotrexate was effective, but the investigators identified that there is a true dearth of evidence-based therapeutics in pediatric pustular psoriasis and much rebound with discontinuation.19 Although the subset of pediatric pustular psoriasis is rare, study of evidence-based intervention is needed.
Therapy
Recent reviews of pediatric and adolescent psoriasis highlight the paucity of therapeutic information for these patient populations. Investigators typically focus on topical therapies as the basis of treatment,20 as well as the addition of phototherapy in mild to moderate plaque or guttate psoriasis and biologic or systemic agents in moderate to severe flares of plaque, erythrodermic, or pustular psoriasis.21 Further studies are needed to identify evidence-based therapeutic paradigms for pediatric psoriasis and to pinpoint therapies associated with the best quality of life in patients and their caregivers.
Tumor Necrosis Factor α Inhibitors
Safety and efficacy of etanercept for juvenile idiopathic arthritis including oligoarthritis, enthesitis-related arthritis, and psoriatic arthritis recently was reviewed by Windschall et al22 using data from the German pediatric Biologika in der Kinderrheumatologie registry. Juvenile Arthritis Disease Activity Score 10 improved from baseline for 127 pediatric patients with psoriatic arthritis in 3 to 24 months (mean [standard deviation], 14.7 [6.4], 5.0 [4.6], 5.3 [6.4] at baseline, 3 months, and 24 months, respectively). Overall side effects were relatively higher in the psoriatic arthritis group; the rate of serious (relative risk, 1.39 [0.95-2.03; P=.08]) and nonserious (relative risk, 1.18 [1.02-1.35; P=.03]) adverse events also was elevated. Uveitis risk was greatest in the psoriatic arthritis group and the number of associated cases of inflammatory bowel disease outnumbered those seen in other forms of arthritis. The investigators concluded that monitoring for extra-articular immunopathies should be conducted in pediatric patients with psoriatic arthritis who are undergoing etanercept therapy.22
Tumor necrosis factor α (TNF-α) inhibitors have been associated with triggering psoriasiform dermatitis in pediatric patients treated for inflammatory bowel disease. A Finnish study of infliximab side effects in pediatric patients with inflammatory bowel disease (n=84; Crohn disease: n=64) demonstrated that almost half (47.6% [40/84]) of the participants presented with chronic skin reactions, 23% of which were severe in nature.23 Psoriasiform lesions of the scalp and ears were most common, followed by the periorificial area, genitals, trunk, and extremities. Rare association with HLA-Cw*0602 genotype was noted. Skin manifestations did not correlate with gut inflammation (as determined by fecal calprotectin levels). Discontinuation of therapy rarely was required.23 Other studies also have highlighted this side effect, suggesting an incidence of 2.7% in adults with colitis treated with TNF-α inhibitors24 and 10.5% in pediatric patients with Crohn disease.25 In a study by Sherlock et al,25 pediatric patients with Crohn disease developing psoriasis following infliximab therapy were more likely to be homozygous for specific polymorphisms in the IL-23R gene (rs10489628, rs10789229, and rs1343151).
Methotrexate
For pediatric patients who are being treated with methotrexate, the polyglutamate assay recently has been reported to be helpful in identifying patients needing a dose escalation.26 Higher numbers on the polyglutamate assay are associated with superior response to methotrexate therapy. Doses can be increased after 12 weeks in patients with low assays.26
IL-23
The safety of IL-23 blockade in pediatric psoriasis patients has not yet been established, but data from adult cases have implicated the IL-17 and IL-23 pathways in psoriasis/psoriatic arthritis, including an association with IL-23R polymorphisms27 and increases in soluble IL-20 and IL-22 associated with disease severity and an association of IL-17 levels with activity on the psoriasis area and severity index scores.28 The data are more limited for pediatric cases. Pediatric patients with inflammatory bowel disease who have an IL-23R polymorphism appear to be susceptible to psoriatic flares while on TNF-α inhibitor therapy,25 which suggests that the IL-23 blockade may be of benefit for some pediatric patients with psoriasis or psoriatic arthritis.
Conclusion
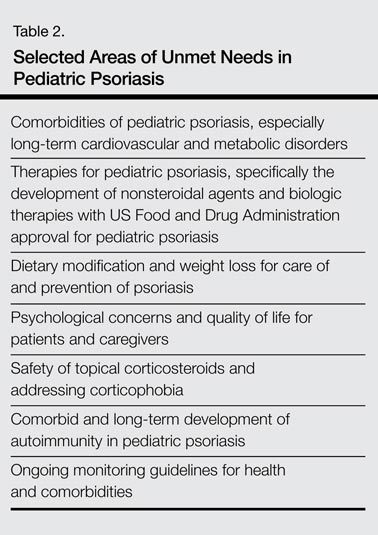
Pediatric psoriasis and psoriatic arthritis have now been identified as being part of the autoimmune spectrum and are associated with metabolic syndrome, including obesity and excess central adiposity, similar to their adult variants. An overview of potential unmet needs in pediatric psoriasis is included in Table 2. These unmet needs include further delineation of diet and weight modification in the care and prevention of psoriasis; expansion of therapeutic trials and US Food and Drug Administration–approved medications for children with psoriasis, especially severe variants such as extensive plaque and pustular disease; and development of guidelines for ongoing monitoring of children with psoriasis. The role of therapeutic interventions and weight management on long-term disease course remains to be shown in extended clinical trials. Despite the great advancements in psoriatic care, knowledge gaps remain in pediatric psoriasis that will need to be addressed in the future.
1. Taclonex Expanded Indication. OptumRx Web site. https://www.optumrx.com/vgnpreview/HCP/Assets/RxNews/Clinical%20Updates_Taclonex_2014-1003.pdf. Published August 29, 2014. Accessed January 28, 2015.
2. Silverberg NB. Update on pediatric psoriasis, part 1: clinical features and demographics. Cutis. 2010;86:118-124.
3. Silverberg NB. Update on pediatric psoriasis, part 2: therapeutic management. Cutis. 2010;86:172-176.
4. Cather JC. Psoriasis in children and women: addressing some special needs. Semin Cutan Med Surg. 2014;33(2 suppl 2):S42-S44.
5. Khorsand K, Sidbury R. Recent advances in pediatric dermatology. Arch Dis Child. 2014;99:944-948.
6. Mercy K, Kwasny M, Cordoro KM, et al. Clinical manifestations of pediatric psoriasis: results of a multicenter study in the United States. Pediatr Dermatol. 2013;30:424-428.
7. Gudjonsson JE, Thorarinsson AM, Sigurgeirsson B, et al. Streptococcal throat infections and exacerbation of chronic plaque psoriasis: a prospective study. Br J Dermatol. 2003;149:530-534.
8. Ferran M, Galván AB, Rincón C, et al. Streptococcus induces circulating CLA(+) memory T-cell-dependent epidermal cell activation in psoriasis. J Invest Dermatol. 2013;133:999-1007.
9. Gul Mert G, Incecik F, Gunasti S, et al. Psoriasiform drug eruption associated with sodium valproate [published online ahead of print November 13, 2013]. Case Rep Pediatr. 2013;2013:823469.
10. Chang MW, Nakrani R. Six children with allergic contact dermatitis to methylisothiazolinone in wet wipes (baby wipes). Pediatrics. 2014;133:e434-e438.
11. Gul U, Gonul M, Kaya I, et al. Autoimmune thyroid disorders in patients with psoriasis. Eur J Dermatol. 2009;19:221-223.
12. Prahalad S, McCracken C, Ponder L, et al. A120: Familial autoimmunity in the CARRA registry. Arthritis Rheumatol. 2014;66(suppl 11):S157.
13. Mercy KM, Paller AS. The relationship between obesity and psoriasis in the pediatric population: implications and future directions. Cutis. 2013;92:107-109.
14. Paller AS, Mercy K, Kwasny MJ, et al. Association of pediatric psoriasis severity with excess and central adiposity: an international cross-sectional study. JAMA Dermatol. 2013;149:166-176.
15. Becker L, Tom WL, Eshagh K, et al. Excess adiposity preceding pediatric psoriasis. JAMA Dermatol. 2014;150:573-574.
16. Volf EM, Levine DE, Michelon MA, et al. Assessor-blinded study of the metabolic syndrome and surrogate markers of increased cardiovascular risk in children with moderate-to-severe psoriasis compared with age-matched population of children with warts. J Drugs Dermatol. 2011;10:900-901.
17. Manzoni AP, Weber MB, Nagatomi AR, et al. Assessing depression and anxiety in the caregivers of pediatric patients with chronic skin disorders. An Bras Dermatol. 2013;88:894-899.
18. Belloni Fortina A, Neri L. Topical steroids and corticophobia. G Ital Dermatol Venereol. 2013;148:651-654.
19. Posso-De Los Rios CJ, Pope E, Lara-Corrales I. A systematic review of systemic medications for pustular psoriasis in pediatrics. Pediatr Dermatol. 2014;31:430-439.
20. Tollefson MM. Diagnosis and management of psoriasis in children. Pediatr Clin North Am. 2014;61:261-277.
21. Fotiadou C, Lazaridou E, Ioannides D. Management of psoriasis in adolescence. Adolesc Health Med Ther. 2014;5:25-34.
22. Windschall D, Müller T, Becker I, et al. Safety and efficacy of etanercept in children with the JIA categories extended oligoarthritis, enthesitis-related arthritis and psoriasis arthritis [published online ahead of print July 18, 2014]. Clin Rheumatol. 2015;34:61-69.
23. Mälkönen T, Wikström A, Heiskanen K, et al. Skin reactions during anti-TNFa therapy for pediatric inflammatory bowel disease: a 2-year prospective study. Inflamm Bowel Dis. 2014;20:1309-1315.
24. Afzali A, Wheat CL, Hu JK, et al. The association of psoriasiform rash with anti-tumor necrosis factor (anti-TNF) therapy in inflammatory bowel disease: a single academic center case series. J Crohns Colitis. 2014;8:480-488.
25. Sherlock ME, Walters T, Tabbers MM, et al. Infliximab-induced psoriasis and psoriasiform skin lesions in pediatric Crohn disease and a potential association with IL-23 receptor polymorphisms. J Pediatr Gastroenterol Nutr. 2013;56:512-518.
26. Rahman SI, Siegfried E, Flanagan KH, et al. The methotrexate polyglutamate assay supports the efficacy of methotrexate for severe inflammatory skin disease in children. J Am Acad Dermatol. 2014;70:252-256.
27. Suzuki E, Mellins ED, Gershwin ME, et al. The IL-23/IL-17 axis in psoriatic arthritis. Autoimmun Rev. 2014;13:496-502.
28. Michalak-Stoma A, Bartosi´nska J, Kowal M, et al. Serum levels of selected Th17 and Th22 cytokines in psoriatic patients. Dis Markers. 2013;35:625-631.
Psoriasis affects 2% to 4% of the US population, with approximately one-third of cases beginning in childhood. The understanding of pediatric psoriasis has developed at a far slower pace than adult disease, with limitations in care including few medications that are approved by the US Food and Drug Administration for pediatric and adolescent use. Recently, a stable fixed-combination dose of calcipo-triene 0.005%–betamethasone dipropionate 0.064% topical suspension was approved for treatment of plaque psoriasis of the scalp in patients aged 12 to 17 years, which hopefully will lead a trend in psoriasis medication approval for children and teenagers.1 Based on a PubMed search of articles indexed for MEDLINE using the search terms pediatric psoriasis, psoriasis, and strep that were published from April 2012 to April 2014, this article reviews newer data to address the issues that surround pediatric psoriasis and to provide an update on prior review articles on pediatric psoriasis.2-5 This article reviews some of the newer literature on clinical presentation and comorbidities in pediatric psoriasis.5 Based on these recent findings, additional screenings including review of obesity parameters are recommended for pediatric patients with psoriasis (Table 1).
Update on Disease Manifestations, Associations, and Comorbidities
Disease Manifestations
A 2013 multicenter study delineated the clinical features of pediatric psoriasis.6 The study was conducted at 8 geographically diverse dermatology clinics in the United States to delineate the clinical manifestations of pediatric psoriasis. In an assessment of 181 participants aged 5 to 17 years, the investigators sought to determine the frequency of disease sites, severity, and guttate disease. Over a period of approximately 2 years, 43.1% of participants were determined to have mild disease and 56.9% had severe disease. Family history of psoriasis was present in 51.4% of participants, with first-degree relatives affected in 59.8% of cases. Scalp involvement at some time was noted in 79.0% of participants, and nail disease was noted in 55% of boys and 29% of girls. Guttate psoriasis was noted in 30% of participants, with more cases in the severe range (35.9%) versus the mild range (21.8%). Additionally, 22.1% of participants had a precipitating streptococcal infection, with the association being more common in pediatric patients with guttate psoriasis than plaque psoriasis.6 This study highlighted that pediatric psoriasis has a genetic basis, is frequently guttate in nature, commonly affects the nails, shows a trend toward being classified as severe, and may be triggered by streptococcal infections.
Streptococcal Infection
Pediatric psoriasis may be triggered or flared by Streptococcus pyogenes (group A β-hemolytic streptococci) infections, specifically β-hemolytic streptococci groups A, C, and G that have streptococcal M protein,2,3,7 and this tendency can be associated with HLA-Cw6 or guttate psoriasis. Newer data have elucidated the role of streptococcal throat infections in psoriasis. Given that streptococcal throat infections are most common in school-aged children, these studies suggest a putative mechanism in pediatric psoriasis for triggering streptococcal infections, which would need to be confirmed in future studies, specifically in pediatric psoriasis patients.
It has been shown that T cells in psoriasis patients recognize common streptococcal M proteins and keratin determinants.7 Ferran et al8 recently demonstrated activation of circulating cutaneous lymphocyte–associated antigen (CLA)+ T cells but not CLA- memory T cells in 27 psoriasis patients (ages not specified) when mixed with streptococcal throat extracts, causing production of IL-17, IP-10, IL-22, and IFN-γ; activation was not found in 6 healthy control patients. Antistreptolysin O levels were correlated with the messenger RNA up- regulation for IL-17, IP-10, IL-22, and IFN-γ, and also correlated with psoriasis area and severity index score in psoriasis patients. In this same study, injection of the activated culture supernatant into mouse skin caused epidermal hyperkeratosis and activation of nonlesional epidermal cells from psoriatic patients. This study thereby delineated some of the potential pathways of the streptococcal induction of psoriasis and psoriatic flares in childhood8; however, confirmation is needed through further study of pediatric psoriatic lymphocyte activity.
Differential Diagnosis
Additions to the extensive differential list have been cited in the recent literature. The differential diagnosis of pediatric psoriasis now includes sodium valproate–induced psoriasiform drug eruption9 and allergic contact dermatitis to methylchloroisothiazolinone and methylisothiazolinone, which are present in many sanitizing hand and diaper wipes and has been reported to cause psoriasiform dermatitis in a periorificial or perineal distribution.10 Clinicians should inquire about the use of these wipes, as caregivers rarely suspect this agent to be causative of the eruption.
Psoriatic Arthritis
Previously, psoriasis and psoriatic arthritis have been linked to autoimmune thyroid disease in adults.11 A study of the Childhood Arthritis & Rheumatology Research Alliance (CARRA) registry showed that family history of psoriasis, autoimmune thyroiditis, Crohn disease, and ankylosing spondylitis in a first-degree relative has been linked to juvenile idiopathic arthritis, highlighting that pediatric psoriasis can be genetically linked or associated with multiple autoimmune conditions and vice versa.12
Obesity, Metabolic Syndrome, and Cardiovascular Risks
Obesity is associated with pediatric psoriasis as highlighted in a growing body of recent literature.13 Excess adiposity as manifested by body mass index in the 85th percentile or greater (37.9% of 155 pediatric psoriasis patients vs 20.5% of 42 controls) and excess central adiposity as manifested by excess waist circumference and increased waist-to-height ratios are more common in pediatric patients with psoriasis than in controls.14
Obesity may be a trigger or associated with increased disease activity in pediatric psoriasis patients. Excess overall adiposity correlates with more severe disease. Obesity parameters may correlate with the onset of psoriasis and with disease severity. In fact, the odds of obesity may be higher in childhood than in adults.14,15 A 2011 report of pediatric psoriasis patients aged 10 to 17 years (n=12) and wart controls (n=6)(mean age, 13.2 and 13.5 years, respectively) demonstrated that 4 of 12 patients with psoriasis and 0 of 6 patients with warts met criteria for metabolic syndrome as defined by 3 of the following: (1) triglycerides greater than or equal to 100 mg/dL; (2) high-density lipoprotein cholesterol less than 50 mg/dL in females and less than 5 mg/dL in males; (3) fasting blood glucose levels greater than or equal to 110 mg/dL, (4) waist circumference greater than the 75th percentile for age and sex; and (5) systolic or diastolic blood pressure greater than the 90th percentile for age, sex, and height.16 These studies highlight that obesity and metabolic syndrome are of concern in pediatric psoriasis patients; however, the best management approach using diet and weight interventions has yet to be identified.
Adiposity may precede the onset of psoriasis. A recent cohort of 27 pediatric psoriasis patients reported that the average age at onset of psoriasis was 8.7 years and the average age at onset of obesity was 4.1 years.15 In this study, 93% (25/27) of patients had adiposity preceding their psoriasis by 2 or more years. It is unclear if this is nature or nurture, as 48% (13/27) of patients had a family history of obesity, 41% (11/27) had a family history of psoriasis, and 48% (13/27) had a family history of hyperlipidemia.15 Therefore, obesity may be cultivated in some psoriatic families. The issue of household influences on diet and obesity needs to be addressed if successful weight management is to be achieved in future studies of pediatric psoriasis.
Cardiovascular risks in the pediatric psoriasis population are the subject of ongoing assessment but will likely mimic studies of adult psoriasis patients when reviewed longitudinally.16 Weight loss and healthy lifestyle interventions likely are beneficial to long-term health, but there is a lack of published data addressing dietary modification as a disease modifier for long-term care of pediatric psoriasis.
Anxiety and Depression
Anxiety and depression have been noted in adults with chronic skin diseases. A recent study assessed 118 patients and caregivers of pediatric patients with atopic dermatitis (n=50), psoriasis (n=25), or vitiligo (n=43) using the Children’s Dermatology Life Quality Index, the Hamilton Anxiety Scale, and the Beck Depression Inventory.17 Anxiety and depression were found in 36% of caregivers of pediatric psoriasis patients and depression was found in 36% of pediatric psoriasis patients, highlighting the need for interventions on a personal and family level to improve quality of life. As a comparator, anxiety was more prevalent in vitiligo caregivers (42%), but depression was only found in 26% of caregivers in the same group. Extent of disease (25%–75% body surface area affected) correlated with both depression and anxiety in the caregivers of pediatric patients with psoriasis as well as with anxiety in caregivers of pediatric patients with increased visible surface area of vitiligo.17 Parental anxiety has been reported at times to be linked to corticosteroid phobia, or corticophobia, which may interfere with disease therapy, as topical corticosteroids are considered the mainstay of therapy in childhood disease.18 Coordinating care with caregivers and addressing their concerns about the safety of medications should be integral to the pediatric psoriasis visit.
Pustular Psoriasis
Pustular psoriasis can be seen in any age group. Researchers recently have attempted to delineate the features and successful management of this severe subset of pediatric psoriasis patients. Twenty-four pediatric pustular psoriasis cases reviewed by Posso-De Los Rios et al19 revealed that 92% (22/24) had generalized and 8% (2/24) had limited acral disease. The mean (standard deviation) age at onset of pediatric pustular psoriasis was 6.3 (4.9) years. Half of the reported cases required more than one intervention. Treatment with acitretin, cyclosporine, and methotrexate was effective, but the investigators identified that there is a true dearth of evidence-based therapeutics in pediatric pustular psoriasis and much rebound with discontinuation.19 Although the subset of pediatric pustular psoriasis is rare, study of evidence-based intervention is needed.
Therapy
Recent reviews of pediatric and adolescent psoriasis highlight the paucity of therapeutic information for these patient populations. Investigators typically focus on topical therapies as the basis of treatment,20 as well as the addition of phototherapy in mild to moderate plaque or guttate psoriasis and biologic or systemic agents in moderate to severe flares of plaque, erythrodermic, or pustular psoriasis.21 Further studies are needed to identify evidence-based therapeutic paradigms for pediatric psoriasis and to pinpoint therapies associated with the best quality of life in patients and their caregivers.
Tumor Necrosis Factor α Inhibitors
Safety and efficacy of etanercept for juvenile idiopathic arthritis including oligoarthritis, enthesitis-related arthritis, and psoriatic arthritis recently was reviewed by Windschall et al22 using data from the German pediatric Biologika in der Kinderrheumatologie registry. Juvenile Arthritis Disease Activity Score 10 improved from baseline for 127 pediatric patients with psoriatic arthritis in 3 to 24 months (mean [standard deviation], 14.7 [6.4], 5.0 [4.6], 5.3 [6.4] at baseline, 3 months, and 24 months, respectively). Overall side effects were relatively higher in the psoriatic arthritis group; the rate of serious (relative risk, 1.39 [0.95-2.03; P=.08]) and nonserious (relative risk, 1.18 [1.02-1.35; P=.03]) adverse events also was elevated. Uveitis risk was greatest in the psoriatic arthritis group and the number of associated cases of inflammatory bowel disease outnumbered those seen in other forms of arthritis. The investigators concluded that monitoring for extra-articular immunopathies should be conducted in pediatric patients with psoriatic arthritis who are undergoing etanercept therapy.22
Tumor necrosis factor α (TNF-α) inhibitors have been associated with triggering psoriasiform dermatitis in pediatric patients treated for inflammatory bowel disease. A Finnish study of infliximab side effects in pediatric patients with inflammatory bowel disease (n=84; Crohn disease: n=64) demonstrated that almost half (47.6% [40/84]) of the participants presented with chronic skin reactions, 23% of which were severe in nature.23 Psoriasiform lesions of the scalp and ears were most common, followed by the periorificial area, genitals, trunk, and extremities. Rare association with HLA-Cw*0602 genotype was noted. Skin manifestations did not correlate with gut inflammation (as determined by fecal calprotectin levels). Discontinuation of therapy rarely was required.23 Other studies also have highlighted this side effect, suggesting an incidence of 2.7% in adults with colitis treated with TNF-α inhibitors24 and 10.5% in pediatric patients with Crohn disease.25 In a study by Sherlock et al,25 pediatric patients with Crohn disease developing psoriasis following infliximab therapy were more likely to be homozygous for specific polymorphisms in the IL-23R gene (rs10489628, rs10789229, and rs1343151).
Methotrexate
For pediatric patients who are being treated with methotrexate, the polyglutamate assay recently has been reported to be helpful in identifying patients needing a dose escalation.26 Higher numbers on the polyglutamate assay are associated with superior response to methotrexate therapy. Doses can be increased after 12 weeks in patients with low assays.26
IL-23
The safety of IL-23 blockade in pediatric psoriasis patients has not yet been established, but data from adult cases have implicated the IL-17 and IL-23 pathways in psoriasis/psoriatic arthritis, including an association with IL-23R polymorphisms27 and increases in soluble IL-20 and IL-22 associated with disease severity and an association of IL-17 levels with activity on the psoriasis area and severity index scores.28 The data are more limited for pediatric cases. Pediatric patients with inflammatory bowel disease who have an IL-23R polymorphism appear to be susceptible to psoriatic flares while on TNF-α inhibitor therapy,25 which suggests that the IL-23 blockade may be of benefit for some pediatric patients with psoriasis or psoriatic arthritis.
Conclusion
Pediatric psoriasis and psoriatic arthritis have now been identified as being part of the autoimmune spectrum and are associated with metabolic syndrome, including obesity and excess central adiposity, similar to their adult variants. An overview of potential unmet needs in pediatric psoriasis is included in Table 2. These unmet needs include further delineation of diet and weight modification in the care and prevention of psoriasis; expansion of therapeutic trials and US Food and Drug Administration–approved medications for children with psoriasis, especially severe variants such as extensive plaque and pustular disease; and development of guidelines for ongoing monitoring of children with psoriasis. The role of therapeutic interventions and weight management on long-term disease course remains to be shown in extended clinical trials. Despite the great advancements in psoriatic care, knowledge gaps remain in pediatric psoriasis that will need to be addressed in the future.
Psoriasis affects 2% to 4% of the US population, with approximately one-third of cases beginning in childhood. The understanding of pediatric psoriasis has developed at a far slower pace than adult disease, with limitations in care including few medications that are approved by the US Food and Drug Administration for pediatric and adolescent use. Recently, a stable fixed-combination dose of calcipo-triene 0.005%–betamethasone dipropionate 0.064% topical suspension was approved for treatment of plaque psoriasis of the scalp in patients aged 12 to 17 years, which hopefully will lead a trend in psoriasis medication approval for children and teenagers.1 Based on a PubMed search of articles indexed for MEDLINE using the search terms pediatric psoriasis, psoriasis, and strep that were published from April 2012 to April 2014, this article reviews newer data to address the issues that surround pediatric psoriasis and to provide an update on prior review articles on pediatric psoriasis.2-5 This article reviews some of the newer literature on clinical presentation and comorbidities in pediatric psoriasis.5 Based on these recent findings, additional screenings including review of obesity parameters are recommended for pediatric patients with psoriasis (Table 1).
Update on Disease Manifestations, Associations, and Comorbidities
Disease Manifestations
A 2013 multicenter study delineated the clinical features of pediatric psoriasis.6 The study was conducted at 8 geographically diverse dermatology clinics in the United States to delineate the clinical manifestations of pediatric psoriasis. In an assessment of 181 participants aged 5 to 17 years, the investigators sought to determine the frequency of disease sites, severity, and guttate disease. Over a period of approximately 2 years, 43.1% of participants were determined to have mild disease and 56.9% had severe disease. Family history of psoriasis was present in 51.4% of participants, with first-degree relatives affected in 59.8% of cases. Scalp involvement at some time was noted in 79.0% of participants, and nail disease was noted in 55% of boys and 29% of girls. Guttate psoriasis was noted in 30% of participants, with more cases in the severe range (35.9%) versus the mild range (21.8%). Additionally, 22.1% of participants had a precipitating streptococcal infection, with the association being more common in pediatric patients with guttate psoriasis than plaque psoriasis.6 This study highlighted that pediatric psoriasis has a genetic basis, is frequently guttate in nature, commonly affects the nails, shows a trend toward being classified as severe, and may be triggered by streptococcal infections.
Streptococcal Infection
Pediatric psoriasis may be triggered or flared by Streptococcus pyogenes (group A β-hemolytic streptococci) infections, specifically β-hemolytic streptococci groups A, C, and G that have streptococcal M protein,2,3,7 and this tendency can be associated with HLA-Cw6 or guttate psoriasis. Newer data have elucidated the role of streptococcal throat infections in psoriasis. Given that streptococcal throat infections are most common in school-aged children, these studies suggest a putative mechanism in pediatric psoriasis for triggering streptococcal infections, which would need to be confirmed in future studies, specifically in pediatric psoriasis patients.
It has been shown that T cells in psoriasis patients recognize common streptococcal M proteins and keratin determinants.7 Ferran et al8 recently demonstrated activation of circulating cutaneous lymphocyte–associated antigen (CLA)+ T cells but not CLA- memory T cells in 27 psoriasis patients (ages not specified) when mixed with streptococcal throat extracts, causing production of IL-17, IP-10, IL-22, and IFN-γ; activation was not found in 6 healthy control patients. Antistreptolysin O levels were correlated with the messenger RNA up- regulation for IL-17, IP-10, IL-22, and IFN-γ, and also correlated with psoriasis area and severity index score in psoriasis patients. In this same study, injection of the activated culture supernatant into mouse skin caused epidermal hyperkeratosis and activation of nonlesional epidermal cells from psoriatic patients. This study thereby delineated some of the potential pathways of the streptococcal induction of psoriasis and psoriatic flares in childhood8; however, confirmation is needed through further study of pediatric psoriatic lymphocyte activity.
Differential Diagnosis
Additions to the extensive differential list have been cited in the recent literature. The differential diagnosis of pediatric psoriasis now includes sodium valproate–induced psoriasiform drug eruption9 and allergic contact dermatitis to methylchloroisothiazolinone and methylisothiazolinone, which are present in many sanitizing hand and diaper wipes and has been reported to cause psoriasiform dermatitis in a periorificial or perineal distribution.10 Clinicians should inquire about the use of these wipes, as caregivers rarely suspect this agent to be causative of the eruption.
Psoriatic Arthritis
Previously, psoriasis and psoriatic arthritis have been linked to autoimmune thyroid disease in adults.11 A study of the Childhood Arthritis & Rheumatology Research Alliance (CARRA) registry showed that family history of psoriasis, autoimmune thyroiditis, Crohn disease, and ankylosing spondylitis in a first-degree relative has been linked to juvenile idiopathic arthritis, highlighting that pediatric psoriasis can be genetically linked or associated with multiple autoimmune conditions and vice versa.12
Obesity, Metabolic Syndrome, and Cardiovascular Risks
Obesity is associated with pediatric psoriasis as highlighted in a growing body of recent literature.13 Excess adiposity as manifested by body mass index in the 85th percentile or greater (37.9% of 155 pediatric psoriasis patients vs 20.5% of 42 controls) and excess central adiposity as manifested by excess waist circumference and increased waist-to-height ratios are more common in pediatric patients with psoriasis than in controls.14
Obesity may be a trigger or associated with increased disease activity in pediatric psoriasis patients. Excess overall adiposity correlates with more severe disease. Obesity parameters may correlate with the onset of psoriasis and with disease severity. In fact, the odds of obesity may be higher in childhood than in adults.14,15 A 2011 report of pediatric psoriasis patients aged 10 to 17 years (n=12) and wart controls (n=6)(mean age, 13.2 and 13.5 years, respectively) demonstrated that 4 of 12 patients with psoriasis and 0 of 6 patients with warts met criteria for metabolic syndrome as defined by 3 of the following: (1) triglycerides greater than or equal to 100 mg/dL; (2) high-density lipoprotein cholesterol less than 50 mg/dL in females and less than 5 mg/dL in males; (3) fasting blood glucose levels greater than or equal to 110 mg/dL, (4) waist circumference greater than the 75th percentile for age and sex; and (5) systolic or diastolic blood pressure greater than the 90th percentile for age, sex, and height.16 These studies highlight that obesity and metabolic syndrome are of concern in pediatric psoriasis patients; however, the best management approach using diet and weight interventions has yet to be identified.
Adiposity may precede the onset of psoriasis. A recent cohort of 27 pediatric psoriasis patients reported that the average age at onset of psoriasis was 8.7 years and the average age at onset of obesity was 4.1 years.15 In this study, 93% (25/27) of patients had adiposity preceding their psoriasis by 2 or more years. It is unclear if this is nature or nurture, as 48% (13/27) of patients had a family history of obesity, 41% (11/27) had a family history of psoriasis, and 48% (13/27) had a family history of hyperlipidemia.15 Therefore, obesity may be cultivated in some psoriatic families. The issue of household influences on diet and obesity needs to be addressed if successful weight management is to be achieved in future studies of pediatric psoriasis.
Cardiovascular risks in the pediatric psoriasis population are the subject of ongoing assessment but will likely mimic studies of adult psoriasis patients when reviewed longitudinally.16 Weight loss and healthy lifestyle interventions likely are beneficial to long-term health, but there is a lack of published data addressing dietary modification as a disease modifier for long-term care of pediatric psoriasis.
Anxiety and Depression
Anxiety and depression have been noted in adults with chronic skin diseases. A recent study assessed 118 patients and caregivers of pediatric patients with atopic dermatitis (n=50), psoriasis (n=25), or vitiligo (n=43) using the Children’s Dermatology Life Quality Index, the Hamilton Anxiety Scale, and the Beck Depression Inventory.17 Anxiety and depression were found in 36% of caregivers of pediatric psoriasis patients and depression was found in 36% of pediatric psoriasis patients, highlighting the need for interventions on a personal and family level to improve quality of life. As a comparator, anxiety was more prevalent in vitiligo caregivers (42%), but depression was only found in 26% of caregivers in the same group. Extent of disease (25%–75% body surface area affected) correlated with both depression and anxiety in the caregivers of pediatric patients with psoriasis as well as with anxiety in caregivers of pediatric patients with increased visible surface area of vitiligo.17 Parental anxiety has been reported at times to be linked to corticosteroid phobia, or corticophobia, which may interfere with disease therapy, as topical corticosteroids are considered the mainstay of therapy in childhood disease.18 Coordinating care with caregivers and addressing their concerns about the safety of medications should be integral to the pediatric psoriasis visit.
Pustular Psoriasis
Pustular psoriasis can be seen in any age group. Researchers recently have attempted to delineate the features and successful management of this severe subset of pediatric psoriasis patients. Twenty-four pediatric pustular psoriasis cases reviewed by Posso-De Los Rios et al19 revealed that 92% (22/24) had generalized and 8% (2/24) had limited acral disease. The mean (standard deviation) age at onset of pediatric pustular psoriasis was 6.3 (4.9) years. Half of the reported cases required more than one intervention. Treatment with acitretin, cyclosporine, and methotrexate was effective, but the investigators identified that there is a true dearth of evidence-based therapeutics in pediatric pustular psoriasis and much rebound with discontinuation.19 Although the subset of pediatric pustular psoriasis is rare, study of evidence-based intervention is needed.
Therapy
Recent reviews of pediatric and adolescent psoriasis highlight the paucity of therapeutic information for these patient populations. Investigators typically focus on topical therapies as the basis of treatment,20 as well as the addition of phototherapy in mild to moderate plaque or guttate psoriasis and biologic or systemic agents in moderate to severe flares of plaque, erythrodermic, or pustular psoriasis.21 Further studies are needed to identify evidence-based therapeutic paradigms for pediatric psoriasis and to pinpoint therapies associated with the best quality of life in patients and their caregivers.
Tumor Necrosis Factor α Inhibitors
Safety and efficacy of etanercept for juvenile idiopathic arthritis including oligoarthritis, enthesitis-related arthritis, and psoriatic arthritis recently was reviewed by Windschall et al22 using data from the German pediatric Biologika in der Kinderrheumatologie registry. Juvenile Arthritis Disease Activity Score 10 improved from baseline for 127 pediatric patients with psoriatic arthritis in 3 to 24 months (mean [standard deviation], 14.7 [6.4], 5.0 [4.6], 5.3 [6.4] at baseline, 3 months, and 24 months, respectively). Overall side effects were relatively higher in the psoriatic arthritis group; the rate of serious (relative risk, 1.39 [0.95-2.03; P=.08]) and nonserious (relative risk, 1.18 [1.02-1.35; P=.03]) adverse events also was elevated. Uveitis risk was greatest in the psoriatic arthritis group and the number of associated cases of inflammatory bowel disease outnumbered those seen in other forms of arthritis. The investigators concluded that monitoring for extra-articular immunopathies should be conducted in pediatric patients with psoriatic arthritis who are undergoing etanercept therapy.22
Tumor necrosis factor α (TNF-α) inhibitors have been associated with triggering psoriasiform dermatitis in pediatric patients treated for inflammatory bowel disease. A Finnish study of infliximab side effects in pediatric patients with inflammatory bowel disease (n=84; Crohn disease: n=64) demonstrated that almost half (47.6% [40/84]) of the participants presented with chronic skin reactions, 23% of which were severe in nature.23 Psoriasiform lesions of the scalp and ears were most common, followed by the periorificial area, genitals, trunk, and extremities. Rare association with HLA-Cw*0602 genotype was noted. Skin manifestations did not correlate with gut inflammation (as determined by fecal calprotectin levels). Discontinuation of therapy rarely was required.23 Other studies also have highlighted this side effect, suggesting an incidence of 2.7% in adults with colitis treated with TNF-α inhibitors24 and 10.5% in pediatric patients with Crohn disease.25 In a study by Sherlock et al,25 pediatric patients with Crohn disease developing psoriasis following infliximab therapy were more likely to be homozygous for specific polymorphisms in the IL-23R gene (rs10489628, rs10789229, and rs1343151).
Methotrexate
For pediatric patients who are being treated with methotrexate, the polyglutamate assay recently has been reported to be helpful in identifying patients needing a dose escalation.26 Higher numbers on the polyglutamate assay are associated with superior response to methotrexate therapy. Doses can be increased after 12 weeks in patients with low assays.26
IL-23
The safety of IL-23 blockade in pediatric psoriasis patients has not yet been established, but data from adult cases have implicated the IL-17 and IL-23 pathways in psoriasis/psoriatic arthritis, including an association with IL-23R polymorphisms27 and increases in soluble IL-20 and IL-22 associated with disease severity and an association of IL-17 levels with activity on the psoriasis area and severity index scores.28 The data are more limited for pediatric cases. Pediatric patients with inflammatory bowel disease who have an IL-23R polymorphism appear to be susceptible to psoriatic flares while on TNF-α inhibitor therapy,25 which suggests that the IL-23 blockade may be of benefit for some pediatric patients with psoriasis or psoriatic arthritis.
Conclusion
Pediatric psoriasis and psoriatic arthritis have now been identified as being part of the autoimmune spectrum and are associated with metabolic syndrome, including obesity and excess central adiposity, similar to their adult variants. An overview of potential unmet needs in pediatric psoriasis is included in Table 2. These unmet needs include further delineation of diet and weight modification in the care and prevention of psoriasis; expansion of therapeutic trials and US Food and Drug Administration–approved medications for children with psoriasis, especially severe variants such as extensive plaque and pustular disease; and development of guidelines for ongoing monitoring of children with psoriasis. The role of therapeutic interventions and weight management on long-term disease course remains to be shown in extended clinical trials. Despite the great advancements in psoriatic care, knowledge gaps remain in pediatric psoriasis that will need to be addressed in the future.
1. Taclonex Expanded Indication. OptumRx Web site. https://www.optumrx.com/vgnpreview/HCP/Assets/RxNews/Clinical%20Updates_Taclonex_2014-1003.pdf. Published August 29, 2014. Accessed January 28, 2015.
2. Silverberg NB. Update on pediatric psoriasis, part 1: clinical features and demographics. Cutis. 2010;86:118-124.
3. Silverberg NB. Update on pediatric psoriasis, part 2: therapeutic management. Cutis. 2010;86:172-176.
4. Cather JC. Psoriasis in children and women: addressing some special needs. Semin Cutan Med Surg. 2014;33(2 suppl 2):S42-S44.
5. Khorsand K, Sidbury R. Recent advances in pediatric dermatology. Arch Dis Child. 2014;99:944-948.
6. Mercy K, Kwasny M, Cordoro KM, et al. Clinical manifestations of pediatric psoriasis: results of a multicenter study in the United States. Pediatr Dermatol. 2013;30:424-428.
7. Gudjonsson JE, Thorarinsson AM, Sigurgeirsson B, et al. Streptococcal throat infections and exacerbation of chronic plaque psoriasis: a prospective study. Br J Dermatol. 2003;149:530-534.
8. Ferran M, Galván AB, Rincón C, et al. Streptococcus induces circulating CLA(+) memory T-cell-dependent epidermal cell activation in psoriasis. J Invest Dermatol. 2013;133:999-1007.
9. Gul Mert G, Incecik F, Gunasti S, et al. Psoriasiform drug eruption associated with sodium valproate [published online ahead of print November 13, 2013]. Case Rep Pediatr. 2013;2013:823469.
10. Chang MW, Nakrani R. Six children with allergic contact dermatitis to methylisothiazolinone in wet wipes (baby wipes). Pediatrics. 2014;133:e434-e438.
11. Gul U, Gonul M, Kaya I, et al. Autoimmune thyroid disorders in patients with psoriasis. Eur J Dermatol. 2009;19:221-223.
12. Prahalad S, McCracken C, Ponder L, et al. A120: Familial autoimmunity in the CARRA registry. Arthritis Rheumatol. 2014;66(suppl 11):S157.
13. Mercy KM, Paller AS. The relationship between obesity and psoriasis in the pediatric population: implications and future directions. Cutis. 2013;92:107-109.
14. Paller AS, Mercy K, Kwasny MJ, et al. Association of pediatric psoriasis severity with excess and central adiposity: an international cross-sectional study. JAMA Dermatol. 2013;149:166-176.
15. Becker L, Tom WL, Eshagh K, et al. Excess adiposity preceding pediatric psoriasis. JAMA Dermatol. 2014;150:573-574.
16. Volf EM, Levine DE, Michelon MA, et al. Assessor-blinded study of the metabolic syndrome and surrogate markers of increased cardiovascular risk in children with moderate-to-severe psoriasis compared with age-matched population of children with warts. J Drugs Dermatol. 2011;10:900-901.
17. Manzoni AP, Weber MB, Nagatomi AR, et al. Assessing depression and anxiety in the caregivers of pediatric patients with chronic skin disorders. An Bras Dermatol. 2013;88:894-899.
18. Belloni Fortina A, Neri L. Topical steroids and corticophobia. G Ital Dermatol Venereol. 2013;148:651-654.
19. Posso-De Los Rios CJ, Pope E, Lara-Corrales I. A systematic review of systemic medications for pustular psoriasis in pediatrics. Pediatr Dermatol. 2014;31:430-439.
20. Tollefson MM. Diagnosis and management of psoriasis in children. Pediatr Clin North Am. 2014;61:261-277.
21. Fotiadou C, Lazaridou E, Ioannides D. Management of psoriasis in adolescence. Adolesc Health Med Ther. 2014;5:25-34.
22. Windschall D, Müller T, Becker I, et al. Safety and efficacy of etanercept in children with the JIA categories extended oligoarthritis, enthesitis-related arthritis and psoriasis arthritis [published online ahead of print July 18, 2014]. Clin Rheumatol. 2015;34:61-69.
23. Mälkönen T, Wikström A, Heiskanen K, et al. Skin reactions during anti-TNFa therapy for pediatric inflammatory bowel disease: a 2-year prospective study. Inflamm Bowel Dis. 2014;20:1309-1315.
24. Afzali A, Wheat CL, Hu JK, et al. The association of psoriasiform rash with anti-tumor necrosis factor (anti-TNF) therapy in inflammatory bowel disease: a single academic center case series. J Crohns Colitis. 2014;8:480-488.
25. Sherlock ME, Walters T, Tabbers MM, et al. Infliximab-induced psoriasis and psoriasiform skin lesions in pediatric Crohn disease and a potential association with IL-23 receptor polymorphisms. J Pediatr Gastroenterol Nutr. 2013;56:512-518.
26. Rahman SI, Siegfried E, Flanagan KH, et al. The methotrexate polyglutamate assay supports the efficacy of methotrexate for severe inflammatory skin disease in children. J Am Acad Dermatol. 2014;70:252-256.
27. Suzuki E, Mellins ED, Gershwin ME, et al. The IL-23/IL-17 axis in psoriatic arthritis. Autoimmun Rev. 2014;13:496-502.
28. Michalak-Stoma A, Bartosi´nska J, Kowal M, et al. Serum levels of selected Th17 and Th22 cytokines in psoriatic patients. Dis Markers. 2013;35:625-631.
1. Taclonex Expanded Indication. OptumRx Web site. https://www.optumrx.com/vgnpreview/HCP/Assets/RxNews/Clinical%20Updates_Taclonex_2014-1003.pdf. Published August 29, 2014. Accessed January 28, 2015.
2. Silverberg NB. Update on pediatric psoriasis, part 1: clinical features and demographics. Cutis. 2010;86:118-124.
3. Silverberg NB. Update on pediatric psoriasis, part 2: therapeutic management. Cutis. 2010;86:172-176.
4. Cather JC. Psoriasis in children and women: addressing some special needs. Semin Cutan Med Surg. 2014;33(2 suppl 2):S42-S44.
5. Khorsand K, Sidbury R. Recent advances in pediatric dermatology. Arch Dis Child. 2014;99:944-948.
6. Mercy K, Kwasny M, Cordoro KM, et al. Clinical manifestations of pediatric psoriasis: results of a multicenter study in the United States. Pediatr Dermatol. 2013;30:424-428.
7. Gudjonsson JE, Thorarinsson AM, Sigurgeirsson B, et al. Streptococcal throat infections and exacerbation of chronic plaque psoriasis: a prospective study. Br J Dermatol. 2003;149:530-534.
8. Ferran M, Galván AB, Rincón C, et al. Streptococcus induces circulating CLA(+) memory T-cell-dependent epidermal cell activation in psoriasis. J Invest Dermatol. 2013;133:999-1007.
9. Gul Mert G, Incecik F, Gunasti S, et al. Psoriasiform drug eruption associated with sodium valproate [published online ahead of print November 13, 2013]. Case Rep Pediatr. 2013;2013:823469.
10. Chang MW, Nakrani R. Six children with allergic contact dermatitis to methylisothiazolinone in wet wipes (baby wipes). Pediatrics. 2014;133:e434-e438.
11. Gul U, Gonul M, Kaya I, et al. Autoimmune thyroid disorders in patients with psoriasis. Eur J Dermatol. 2009;19:221-223.
12. Prahalad S, McCracken C, Ponder L, et al. A120: Familial autoimmunity in the CARRA registry. Arthritis Rheumatol. 2014;66(suppl 11):S157.
13. Mercy KM, Paller AS. The relationship between obesity and psoriasis in the pediatric population: implications and future directions. Cutis. 2013;92:107-109.
14. Paller AS, Mercy K, Kwasny MJ, et al. Association of pediatric psoriasis severity with excess and central adiposity: an international cross-sectional study. JAMA Dermatol. 2013;149:166-176.
15. Becker L, Tom WL, Eshagh K, et al. Excess adiposity preceding pediatric psoriasis. JAMA Dermatol. 2014;150:573-574.
16. Volf EM, Levine DE, Michelon MA, et al. Assessor-blinded study of the metabolic syndrome and surrogate markers of increased cardiovascular risk in children with moderate-to-severe psoriasis compared with age-matched population of children with warts. J Drugs Dermatol. 2011;10:900-901.
17. Manzoni AP, Weber MB, Nagatomi AR, et al. Assessing depression and anxiety in the caregivers of pediatric patients with chronic skin disorders. An Bras Dermatol. 2013;88:894-899.
18. Belloni Fortina A, Neri L. Topical steroids and corticophobia. G Ital Dermatol Venereol. 2013;148:651-654.
19. Posso-De Los Rios CJ, Pope E, Lara-Corrales I. A systematic review of systemic medications for pustular psoriasis in pediatrics. Pediatr Dermatol. 2014;31:430-439.
20. Tollefson MM. Diagnosis and management of psoriasis in children. Pediatr Clin North Am. 2014;61:261-277.
21. Fotiadou C, Lazaridou E, Ioannides D. Management of psoriasis in adolescence. Adolesc Health Med Ther. 2014;5:25-34.
22. Windschall D, Müller T, Becker I, et al. Safety and efficacy of etanercept in children with the JIA categories extended oligoarthritis, enthesitis-related arthritis and psoriasis arthritis [published online ahead of print July 18, 2014]. Clin Rheumatol. 2015;34:61-69.
23. Mälkönen T, Wikström A, Heiskanen K, et al. Skin reactions during anti-TNFa therapy for pediatric inflammatory bowel disease: a 2-year prospective study. Inflamm Bowel Dis. 2014;20:1309-1315.
24. Afzali A, Wheat CL, Hu JK, et al. The association of psoriasiform rash with anti-tumor necrosis factor (anti-TNF) therapy in inflammatory bowel disease: a single academic center case series. J Crohns Colitis. 2014;8:480-488.
25. Sherlock ME, Walters T, Tabbers MM, et al. Infliximab-induced psoriasis and psoriasiform skin lesions in pediatric Crohn disease and a potential association with IL-23 receptor polymorphisms. J Pediatr Gastroenterol Nutr. 2013;56:512-518.
26. Rahman SI, Siegfried E, Flanagan KH, et al. The methotrexate polyglutamate assay supports the efficacy of methotrexate for severe inflammatory skin disease in children. J Am Acad Dermatol. 2014;70:252-256.
27. Suzuki E, Mellins ED, Gershwin ME, et al. The IL-23/IL-17 axis in psoriatic arthritis. Autoimmun Rev. 2014;13:496-502.
28. Michalak-Stoma A, Bartosi´nska J, Kowal M, et al. Serum levels of selected Th17 and Th22 cytokines in psoriatic patients. Dis Markers. 2013;35:625-631.
Practice Points
- The majority of children with psoriasis have severe disease, scalp involvement, and a family history.
- Pediatric psoriasis is associated with metabolic syndrome, especially obesity.
- Anxiety and depression may be noted in children with psoriasis as well as their caregivers.

What Is Your Diagnosis? Extramammary Paget Disease

A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
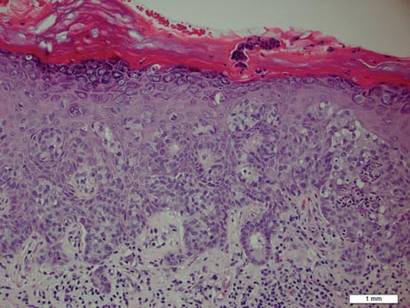
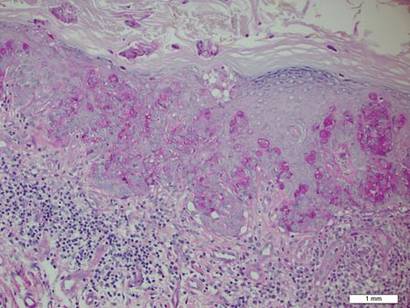
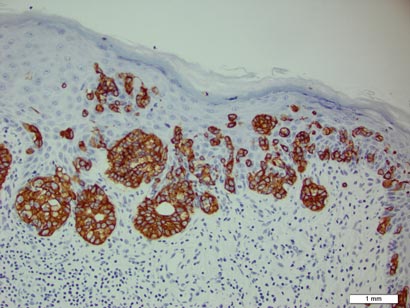
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
A 70-year-old man presented with a nonpruritic erythematous scaly plaque in the left suprapubic region of 6 months’ duration that had failed to respond to terbinafine cream 1% after 1 month of treatment of suspected tinea cruris. His medical history was remarkable for hypertension, hyperlipidemia, chronic obstructive pulmonary disease, benign prostatic hyperplasia, an abdominal aortic aneurysm, alcohol dependence, tobacco use disorder, and unintentional weight loss of 15 lb over the last year.
The Diagnosis: Extramammary Paget Disease
A biopsy of the plaque revealed an intraepidermal proliferation of large cells with abundant clear cytoplasm and large vesicular nuclei distributed throughout the epidermis (Figure 1). The neoplastic cells stained positive for both periodic acid–Schiff stain (Figure 2) and CK7 (Figure 3). Chemistry and liver function panel, urine analysis, carcinoembryonic antigen levels, and prostate-specific antigen levels were within reference range. A complete blood cell count revealed mild megaloblastic anemia. Subsequent computed tomography of the chest, abdomen, and pelvis revealed an abdominal aortic aneurysm and prostatic enlargement without any evidence of potential malignancies. Colonoscopy revealed multiple hyperplastic polyps and a tubular adenoma. Cystoscopy was normal, except for evidence of prostate enlargement. Urine cytology was unremarkable. The patient was referred for excision of the lesion with Mohs micrographic surgery. Follow-up was recommended every 3 months for the first 2 years following surgery and every 6 months thereafter to monitor for recurrence or secondary neoplasms.
|
Sir James Paget first described mammary Paget disease of the nipple in 1874 in his report of 15 women with skin eruptions of the nipple and areola and subsequent carcinoma of the underlying breast.1 Paget also described a patient with a similar eruption on the glans penis and Crocker2 described extramammary Paget disease (EMPD) of the scrotum and penis in 1889. The principle difference between mammary Paget disease and EMPD is the anatomic location.
Extramammary Paget disease is a rare condition that typically affects patients aged 50 to 80 years and is more common in women and white-skinned races.3 Extramammary Paget disease frequently targets cutaneous sites that are rich in apocrine glands. The most commonly affected site is the vulva followed by perineal, perianal, scrotal, and penile skin. Less commonly, the axillae, buttocks, thighs, eyelids, and external auditory canals may be affected.4
Patients with EMPD typically present with well-demarcated, nonresolving, erythematous and eczematous plaques that may have associated crusting, scaling, papillomatous excrescences, lichenification, ulceration, or bleeding. The most common symptom is pruritus, followed by burning, irritation, pain, and tenderness.5 Ten percent of patients are asymptomatic. The average interval between symptom onset and diagnosis is 2 years.5
Histopathology reveals diffusely infiltrating, irregular, neoplastic Paget cells within the epidermis that are large and vacuolated with abundant pale bluish cytoplasm and large vesicular nuclei, which may be centrally or laterally compressed. The cells may be distributed singly or in groups as strands, nests, or glandular patterns within the lower epidermis, rete ridges, and adnexal structures. Hyperkeratosis, acanthosis, and parakeratosis may also be present. Paget cells stain for immunohistochemical markers of apocrine and eccrine derivation including low-molecular-weight cytokeratins, gross cystic disease fluid protein 15, periodic acid–Schiff stain, and carcinoembryonic antigen.5 Perrotto et al6 studied 98 specimens from 61 patients and found that CK7 was positive in all EMPD specimens, while CK20 and gross cystic disease fluid protein 15 were positive in large subsets of both primary and secondary EMPD. Cases of EMPD secondary to anorectal adenocarcinoma were largely ERBB2 (formerly HER2/neu) negative and CDX2 positive.6
Diagnosis of EMPD should be followed by a thorough investigation for underlying carcinomas. In a review of 197 cases of EMPD, 24% of patients with EMPD had an associated underlying in situ or invasive adnexal apocrine carcinoma, which was associated with a higher mortality rate than in patients without this underlying malignancy. Additionally, 12% of EMPD patients had an associated underlying internal malignancy.7 These malignancies may include carcinomas of the urethra, bladder, vagina, cervix, endometrium, prostate, colon, and rectum. Perianal EMPD has a higher frequency of associated malignancies than vulvar EMPD.5 The location of EMPD is related to the location of the underlying malignancy; for example, perianal EMPD is associated with colorectal adenocarcinomas, and EMPD of the penis, scrotum, and groin is associated with genitourinary malignancies. Investigations to search for associated malignancies in patients with EMPD may include pelvic ultrasonography and/or magnetic resonance imaging, hysteroscopy, colonoscopy, sigmoidoscopy, cystoscopy, intravenous pyelogram, mammogram, and/or chest radiograph.
The most effective treatment of EMPD is margin-controlled surgical excision. High local recurrence rates may be due to irregular margins, multicentricity, and the tendency of EMPD to involve clinically normal-appearing skin. Hendi et al8 noted that EMPD may actually be unifocal with subclinical fingerlike projections extending beyond the main body of the tumor, requiring CK7 immunostaining for visualization to ensure complete margin control. The recurrence rate after standard surgical excision is 33% to 60%. The recurrence rate after excision via Mohs micrographic surgery is 16% for primary EMPD and 50% for recurrent EMPD.9 Other treatment modalities include radiotherapy, topical chemotherapy with 5-fluorouracil or imiquimod, and photodynamic therapy.10-13 Combined systemic chemotherapy with trastuzumab and paclitaxel can be considered for the treatment of ERBB2-positive EMPD.14
For patients with chronic genital or perianal lesions that are unresponsive to treatment, dermatologists should maintain a high index of suspicion for EMPD. If a patient is diagnosed with EMPD, a full-body skin examination should be performed with palpation of all lymph nodes. Imaging studies directed at the anatomic location of the involved skin should be utilized to search for an underlying internal malignancy.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
1. Paget J. On disease of the mammary areola preceding cancer of the mammary gland. St Bartholomew Hosp Rep. 1874;10:87-89.
2. Crocker H. Paget’s disease affecting the scrotum and penis. Trans Pathol Soc Lond. 1889;40:187-191.
3. Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
4. Heymann WR. Extramammary Paget’s disease. Clin Dermatol. 1993;11:83-87.5. Shepherd V, Davidson EJ, Davies-Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
6. Perrotto J, Abbott JJ, Ceilley RI, et al. The role of immunohistochemistry in discriminating primary from secondary extramammary Paget disease. Am J Dermatopathol. 2010;32:137-143.
7. Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
8. Hendi A, Perdikis G, Snow JL. Unifocality of extramammary Paget disease. J Am Acad Dermatol. 2008;59:811-813.
9. Hendi A, Brodland DG, Zitelli JA. Extramammary Paget’s disease: surgical treatment with mohs micrographic surgery. J Am Acad Dermatol. 2004;51:767-773.10. Zampogna JC, Flowers FP, Roth WI, et al. Treatment of primary limited cutaneous extramammary Paget’s disease with topical imiquimod monotherapy: two case reports. J Am Acad Dermatol. 2002;47:S229-S235.
11. Beleznay KM, Levesque MA, Gill S. Response to 5-fluorouracil in metastatic extramammary Paget disease of the scrotum presenting as pancytopenia and back pain. Curr Oncol. 2009;16:81-83.
12. Kitagawa KH, Bogner P, Zeitouni NC. Photodynamic therapy with methyl-aminolevulinate for the treatment of double extramammary Paget’s disease. Dermatol Surg. 2011;37:1043-1046.
13. Hata M, Omura M, Koike I, et al. Role of radiotherapy as curative treatment of extramammary Paget’s disease. Int J Radiat Oncol Biol Phys. 2011;80:47-54.
14. Takahagi S, Noda H, Kamegashira A, et al. Metastatic extramammary Paget’s disease treated with paclitaxel and trastuzumab combination chemotherapy. J Dermatol. 2009;36:457-461.
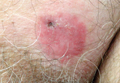
Onchocerciasis
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
 |  |
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4

Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
 |  |
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
| |
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4
Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
| |
The larvae of Onchocerca volvulus, a nematode that is most commonly found in tropical Africa, Yemen, Central America, and South America, are transmitted by flies of the genus Simulium that breed near fast-flowing rivers.1 The flies bite the host and transmit the larvae, and the larvae then mature into adults within the skin and subcutis, forming nodules that typically are not painful. The worms may reside within the skin for years and produce microfilariae, which can migrate and cause visual impairment, blindness, or a pruritic papular rash.1
The nematode produces a nodule within the dermis or subcutis with surrounding fibrous tissue and a mixed inflammatory infiltrate with eosinophils (Figure 1). In some cases, microfilariae can be seen within the lymphatics or within the uteri of the worms.1 Male and female worms typically are present and have a corrugated cuticle with a thin underlying layer of striated muscle. The females have paired uteri, which usually contain microfilariae2 (Figure 2).
| |
Dirofilaria repens also is a nematode that produces a subcutaneous nodule with an inflammatory reaction. This worm typically has a thick cuticle with longitudinal ridges, long thick muscle, and lateral cords.3 Additionally, because humans are not the usual host, Dirofilaria species do not complete their lifecycle and typically are not gravid, unlike Onchocerca species.
Myiasis is the presence of fly larvae within the skin. The larvae demonstrate a thick hyaline cuticle with pigmented brown-yellow spikes (Figure 3). There is a thick muscular layer under the cuticle and a tubular tracheal system containing vertical striations. The digestive system has an epithelial lining with prominent vessels. Adipose tissue with granulated cytoplasm, prominent nuclei, and coarse chromatin also are present.4
Scabies mites (Figure 4), ova, and scybala are present within the stratum corneum. A mixed inflammatory infiltrate also can be present.1 Tungiasis is caused by burrowing fleas and typically occurs on acral skin; therefore, it is more frequently found in the superficial portion of the skin. Erythrocytes usually are present in the gastrointestinal tract, and the females usually are gravid.2 A surrounding mixed inflammatory infiltrate is present, and necrosis also can occur (Figure 5).1
| |
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
1. Weedon D. Weedon’s Skin Pathology. 3rd ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2010.
2. Elston DM, Ferringer T. Dermatopathology: Requisites in Dermatology. Edinburgh, Scotland: Saunders Elsevier; 2008.
3. Tzanetou K, Gasteratos S, Pantazopoulou A, et al. Subcutaneous dirofilariasis caused by Dirofilaria repens in Greece: a case report. J Cutan Pathol. 2009;36:892-895.
4. Fernandez-Flores A, Saeb-Lima M. Pulse granuloma of the lip: morphologic clues in its differential diagnosis. J Cutan Pathol. 2014;41:394-399.
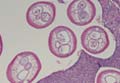
What’s Eating You? Cutaneous Larva Migrans
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
  |
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
|
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
|
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
Practice Points
- Classic cutaneous larva migrans (CLM) presents with a unilateral, serpiginous, pruritic eruption on the hands, feet, or buttocks following direct contact with sand or soil that is contaminated with Ancylostoma braziliense or Ancylostoma caninum.
- Atypical presentations of CLM include bilateral distribution; folliculitis and urticarial plaques; prolonged cases lasting up to 1 year; and Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia.
- Cutaneous larva migrans is self-limited, but treatment often is necessary due to intense pruritus. Treatment options include a single oral dose of albendazole or ivermectin, topical thiabendazole, and prolonged courses of oral albendazole in cases complicated by Löffler syndrome.
First Refusal
Treatment success in psoriasis, as in any dermatologic condition, is dependent on many factors. The willingness of patients to follow our suggested therapeutic plans certainly is one of the most important components of this process.
Halioua et al1 analyzed the issue of treatment refusal, which they defined as “a patient actively refusing to take treatment despite physician recommendations,” among psoriasis patients. Treatment refusal is a more complex phenomenon than nonadherence, as it requires an affirmative act that goes beyond more passive acts of not filling prescriptions, taking a medication sporadically, or forgetting to take a medication. Their objective was to investigate refusal of topical treatments by patients living with psoriasis in France as well as the factors that influence such refusal.1
The authors evaluated responses to an Internet study.1 Responses from participants who refused topical therapy (n=50) were compared to individuals who successfully applied topical treatment (n=205). Individuals receiving phototherapy, biologic therapy, and oral treatment were not included in the analysis. Spearman rank correlations completed by Fisher exact tests and Student t tests were performed.1
The researchers found that objective aspects of psoriasis, including comorbidities, localization of lesions, and symptoms associated with psoriasis, were not significant predictors of treatment refusal. The factors that did appear to influence refusal related more to patient perception of disease and its treatment.1
First, treatment refusal was defined by patient attitude toward treatment. In the treatment refusal group, significantly fewer participants reported believing that psoriasis can be managed (20.0% vs 38.5%; P<.01), and significantly more participants in the treatment refusal group reported believing that topical psoriasis treatments never work (58.0% vs 27.5%; odds ratio, 2.09; P<.0001). Additionally, significantly fewer participants in the treatment refusal group were willing to stay on prescription medications long-term (30.0% vs 77.6%; P<.001), and significantly more patients in the treatment refusal group believed that all creams (prescription or over-the-counter) work the same (54.0% vs 31.7%; odds ratio, 1.07; P=.003).1
The physician-patient relationship also influenced refusal. In the treatment refusal group, 60% of participants reported no longer consulting physicians for psoriasis treatment. The main reasons for cessation of medical care were lack of improvement of psoriasis (40%) and feeling that the physician did not take psoriasis seriously (20%). In the treatment acceptance group, only 10% of participants no longer consulted physicians.1 Among participants who continued to consult their physician (40% for the treatment refusal group and 90.2% for the treatment acceptance group), significantly fewer participants in the treatment refusal group reported that they were substantially helped by their physician (50.0% vs 73.0%; P=.03) and that they always followed physician recommendations (65.0% vs 85.4%; P=.02). Additionally, significantly fewer participants in the treatment refusal group considered that their physician took the time to listen to what he/she had to say (65.0% vs 85.9%; P=.02) and that their physician had provided clear instructions on how to utilize the treatment (65.0% vs 83.2%; P=.046).1
Therefore, treatment refusal is an important factor to be considered in the management of psoriasis. The findings of this study indicate possible strategies to reduce patient refusal. For example, enhanced education about the therapeutic options for psoriasis and their benefits could counter negative perceptions about these therapies. It also appears that increased focus on the physician-patient relationship may have a positive impact in this area.
Reference
1. Halioua B, Maury Le Breton A, de Fontaubert A, et al. Treatment refusal among patients with psoriasis [published online ahead of print]. J Dermatolog Treat. 2015;2:1-5.
Treatment success in psoriasis, as in any dermatologic condition, is dependent on many factors. The willingness of patients to follow our suggested therapeutic plans certainly is one of the most important components of this process.
Halioua et al1 analyzed the issue of treatment refusal, which they defined as “a patient actively refusing to take treatment despite physician recommendations,” among psoriasis patients. Treatment refusal is a more complex phenomenon than nonadherence, as it requires an affirmative act that goes beyond more passive acts of not filling prescriptions, taking a medication sporadically, or forgetting to take a medication. Their objective was to investigate refusal of topical treatments by patients living with psoriasis in France as well as the factors that influence such refusal.1
The authors evaluated responses to an Internet study.1 Responses from participants who refused topical therapy (n=50) were compared to individuals who successfully applied topical treatment (n=205). Individuals receiving phototherapy, biologic therapy, and oral treatment were not included in the analysis. Spearman rank correlations completed by Fisher exact tests and Student t tests were performed.1
The researchers found that objective aspects of psoriasis, including comorbidities, localization of lesions, and symptoms associated with psoriasis, were not significant predictors of treatment refusal. The factors that did appear to influence refusal related more to patient perception of disease and its treatment.1
First, treatment refusal was defined by patient attitude toward treatment. In the treatment refusal group, significantly fewer participants reported believing that psoriasis can be managed (20.0% vs 38.5%; P<.01), and significantly more participants in the treatment refusal group reported believing that topical psoriasis treatments never work (58.0% vs 27.5%; odds ratio, 2.09; P<.0001). Additionally, significantly fewer participants in the treatment refusal group were willing to stay on prescription medications long-term (30.0% vs 77.6%; P<.001), and significantly more patients in the treatment refusal group believed that all creams (prescription or over-the-counter) work the same (54.0% vs 31.7%; odds ratio, 1.07; P=.003).1
The physician-patient relationship also influenced refusal. In the treatment refusal group, 60% of participants reported no longer consulting physicians for psoriasis treatment. The main reasons for cessation of medical care were lack of improvement of psoriasis (40%) and feeling that the physician did not take psoriasis seriously (20%). In the treatment acceptance group, only 10% of participants no longer consulted physicians.1 Among participants who continued to consult their physician (40% for the treatment refusal group and 90.2% for the treatment acceptance group), significantly fewer participants in the treatment refusal group reported that they were substantially helped by their physician (50.0% vs 73.0%; P=.03) and that they always followed physician recommendations (65.0% vs 85.4%; P=.02). Additionally, significantly fewer participants in the treatment refusal group considered that their physician took the time to listen to what he/she had to say (65.0% vs 85.9%; P=.02) and that their physician had provided clear instructions on how to utilize the treatment (65.0% vs 83.2%; P=.046).1
Therefore, treatment refusal is an important factor to be considered in the management of psoriasis. The findings of this study indicate possible strategies to reduce patient refusal. For example, enhanced education about the therapeutic options for psoriasis and their benefits could counter negative perceptions about these therapies. It also appears that increased focus on the physician-patient relationship may have a positive impact in this area.
Treatment success in psoriasis, as in any dermatologic condition, is dependent on many factors. The willingness of patients to follow our suggested therapeutic plans certainly is one of the most important components of this process.
Halioua et al1 analyzed the issue of treatment refusal, which they defined as “a patient actively refusing to take treatment despite physician recommendations,” among psoriasis patients. Treatment refusal is a more complex phenomenon than nonadherence, as it requires an affirmative act that goes beyond more passive acts of not filling prescriptions, taking a medication sporadically, or forgetting to take a medication. Their objective was to investigate refusal of topical treatments by patients living with psoriasis in France as well as the factors that influence such refusal.1
The authors evaluated responses to an Internet study.1 Responses from participants who refused topical therapy (n=50) were compared to individuals who successfully applied topical treatment (n=205). Individuals receiving phototherapy, biologic therapy, and oral treatment were not included in the analysis. Spearman rank correlations completed by Fisher exact tests and Student t tests were performed.1
The researchers found that objective aspects of psoriasis, including comorbidities, localization of lesions, and symptoms associated with psoriasis, were not significant predictors of treatment refusal. The factors that did appear to influence refusal related more to patient perception of disease and its treatment.1
First, treatment refusal was defined by patient attitude toward treatment. In the treatment refusal group, significantly fewer participants reported believing that psoriasis can be managed (20.0% vs 38.5%; P<.01), and significantly more participants in the treatment refusal group reported believing that topical psoriasis treatments never work (58.0% vs 27.5%; odds ratio, 2.09; P<.0001). Additionally, significantly fewer participants in the treatment refusal group were willing to stay on prescription medications long-term (30.0% vs 77.6%; P<.001), and significantly more patients in the treatment refusal group believed that all creams (prescription or over-the-counter) work the same (54.0% vs 31.7%; odds ratio, 1.07; P=.003).1
The physician-patient relationship also influenced refusal. In the treatment refusal group, 60% of participants reported no longer consulting physicians for psoriasis treatment. The main reasons for cessation of medical care were lack of improvement of psoriasis (40%) and feeling that the physician did not take psoriasis seriously (20%). In the treatment acceptance group, only 10% of participants no longer consulted physicians.1 Among participants who continued to consult their physician (40% for the treatment refusal group and 90.2% for the treatment acceptance group), significantly fewer participants in the treatment refusal group reported that they were substantially helped by their physician (50.0% vs 73.0%; P=.03) and that they always followed physician recommendations (65.0% vs 85.4%; P=.02). Additionally, significantly fewer participants in the treatment refusal group considered that their physician took the time to listen to what he/she had to say (65.0% vs 85.9%; P=.02) and that their physician had provided clear instructions on how to utilize the treatment (65.0% vs 83.2%; P=.046).1
Therefore, treatment refusal is an important factor to be considered in the management of psoriasis. The findings of this study indicate possible strategies to reduce patient refusal. For example, enhanced education about the therapeutic options for psoriasis and their benefits could counter negative perceptions about these therapies. It also appears that increased focus on the physician-patient relationship may have a positive impact in this area.
Reference
1. Halioua B, Maury Le Breton A, de Fontaubert A, et al. Treatment refusal among patients with psoriasis [published online ahead of print]. J Dermatolog Treat. 2015;2:1-5.
Reference
1. Halioua B, Maury Le Breton A, de Fontaubert A, et al. Treatment refusal among patients with psoriasis [published online ahead of print]. J Dermatolog Treat. 2015;2:1-5.
A Case of Morfan Syndrome
To the Editor:
A 17-year-old adolescent girl presented to our clinic for evaluation of diffuse acanthosis nigricans (AN) that had been present since 7 years of age. The patient had a history of hypothyroidism, insulin resistance, ovarian cysts, and developmental delay. On examination, she presented with thick and verrucous plaques of AN involving the neck, abdomen, trunk, arms, and legs (Figure). The intertriginous areas were affected the most. The examination also was notable for dysmorphic facies and an endomorphic body habitus. Both parents denied similar health problems in their family and were normal in appearance. Although the patient was receiving metformin treatment for insulin resistance, she had not undergone any prior workup to identify a unifying syndromic cause for her physical and biochemical findings. A review of the literature showed a 1993 case report of a 5-year-old boy with mental retardation, body overgrowth, remarkable facies, and AN, which was termed Morfan syndrome.1 Because of the similarity in features, we believe that our patient’s presentation fits this syndrome. This report represents the second documented case of Morfan syndrome according to a PubMed search of articles indexed for MEDLINE using the search term Morfan. Additional searches using the terms acanthosis nigricans and syndrome also failed to identify any reports describing patients with a similar constellation of findings.
|
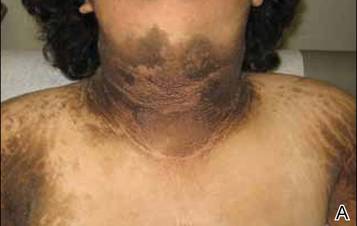

First described by Santi Unna and Monatsh Pollitzer in 1890, AN is a common dermatosis that is characterized by thick, hyperpigmented, and verrucous plaques.2,3 Although most common in symmetric distribution on flexural and intertriginous areas, AN also may involve mucosal surfaces.4 Acanthosis nigricans is associated with multiple etiologic factors, including direct autosomal transmission, genetic abnormalities, medications, malignancy, and endocrine imbalance.2 However, the diffuse generalized form of AN is almost always found in the context of malignancy or genetic syndromes.5 Historically, most attention on AN has focused on its eruptive form (so-called malignant AN), which is usually associated with internal malignancy but also may result from benign pituitary adenomas.6 Although the precise mechanism for paraneoplastic AN is still a matter of debate, it is likely the result of overactive growth factors, such as transforming growth factor α, epidermal growth factor, and α melanocyte-stimulating hormone.7 Generalized AN also has been associated with genetic abnormalities. Multiple genetic mutations have been associated with AN, including the genes coding for the insulin receptor, fibroblast growth receptors 2 and 3, lamin A/C, and seipin.8-12 Acanthosis nigricans has been described as a feature in other genetic syndromes but may represent an incidental finding.4 In 1976, Kahn et al13 linked AN with insulin resistance, which subsequently shifted the focus on AN’s link with obesity and as a precursor of type 2 diabetes mellitus.6,14 In these cases, it is hypothesized that excess insulin activates the insulinlike growth factor 1 receptor to stimulate keratinocyte and dermal fibroblast proliferation. It is likely that hyperinsulinemia also induces cellular proliferation indirectly through other pathways.15,16 Compared with paraneoplastic and syndromic causes, AN secondary to hyperinsulinemia and obesity does not tend to be generalized but instead is proportionate to the degree of metabolic derangement.17
Because our patient’s AN presentation was diffuse, long-standing, and occurred in the context of morphologic abnormalities, it was consistent with a syndrome. Based on the constellation of findings, we believe our patient fit the rare markers indicating Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome. As additional cases are reported, we hope that further biochemical, physical, and genetic studies may be pursued to better identify the syndrome and elucidate its pathogenesis.
1. Seemanová E, Rüdiger HW, Dreyer M. Morfan: a new syndrome characterized by mental retardation, pre- and postnatal overgrowth, remarkable face and acanthosis nigricans in 5-year-old boy. Am J Med Genet. 1993;45:525-528.
2. Rendon MI, Cruz PD Jr, Sontheimer RD, et al. Acanthosis nigricans: a cutaneous marker of tissue resistance to insulin. J Am Acad Dermatol. 1989;21:461-469.
3. Unna PG, Morris M, Besnier E, eds. International Atlas of Rare Skin Diseases. London, England: HK Lewis & Co; 1890.
4. Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol. 1994;31:1-19.
5. Inamdar AC, Palit A. Generalized acanthosis nigricans in childhood. Pediatr Dermatol. 2004;21:277-279.
6. Cruz PD, Hud JA. Excess insulin binding to insulin-like growth factor receptors: proposed mechanism for acanthosis nigricans. J Invest Dermatol. 1992;98:82-85.
7. Krawczyk M, Mykała-Cies´la J, Kołodziej-Jaskuła A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
8. Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 1995;11:462-464.
9. Moller DE, Cohen O, Yamaguchi Y, et al. Prevalence of mutations in the insulin receptor gene in subjects with features of the type A syndrome of insulin resistance. Diabetes. 1994;43:247-255.
10. Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nature Genet. 1996;13:492-494.
11. Anderson JL, Khan M, David WS, et al. Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21-22. Am J Med Genet. 1992;82:161-165.
12. Simha V, Agarwal AK, Aronin PA, et al. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet. 2008;146A:2318-2326.
13. Kahn CR, Flier JS, Bar RS, et al. the syndromes of insulin resistance and acanthosis nigricans. insulin-receptor disorders in man. N Engl J Med. 1976;294:739-745.
14. Brickman WWJ, Huang J, Silverman BL, et al. Acanthosis nigricans identifies youth at high risk for metabolic abnormalities. J Pediatr. 2010;156:87-92.
15. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. insulin-like growth factors.
N Engl J Med. 1997;336:633-640.
16. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818-835.
17. Berk DR, Spector EB, Bayliss SJ. Familial acanthosis nigricans due to K650T FGFR3 mutation. Arch Dermatol. 2007;143:1153-1156.
To the Editor:
A 17-year-old adolescent girl presented to our clinic for evaluation of diffuse acanthosis nigricans (AN) that had been present since 7 years of age. The patient had a history of hypothyroidism, insulin resistance, ovarian cysts, and developmental delay. On examination, she presented with thick and verrucous plaques of AN involving the neck, abdomen, trunk, arms, and legs (Figure). The intertriginous areas were affected the most. The examination also was notable for dysmorphic facies and an endomorphic body habitus. Both parents denied similar health problems in their family and were normal in appearance. Although the patient was receiving metformin treatment for insulin resistance, she had not undergone any prior workup to identify a unifying syndromic cause for her physical and biochemical findings. A review of the literature showed a 1993 case report of a 5-year-old boy with mental retardation, body overgrowth, remarkable facies, and AN, which was termed Morfan syndrome.1 Because of the similarity in features, we believe that our patient’s presentation fits this syndrome. This report represents the second documented case of Morfan syndrome according to a PubMed search of articles indexed for MEDLINE using the search term Morfan. Additional searches using the terms acanthosis nigricans and syndrome also failed to identify any reports describing patients with a similar constellation of findings.
|
First described by Santi Unna and Monatsh Pollitzer in 1890, AN is a common dermatosis that is characterized by thick, hyperpigmented, and verrucous plaques.2,3 Although most common in symmetric distribution on flexural and intertriginous areas, AN also may involve mucosal surfaces.4 Acanthosis nigricans is associated with multiple etiologic factors, including direct autosomal transmission, genetic abnormalities, medications, malignancy, and endocrine imbalance.2 However, the diffuse generalized form of AN is almost always found in the context of malignancy or genetic syndromes.5 Historically, most attention on AN has focused on its eruptive form (so-called malignant AN), which is usually associated with internal malignancy but also may result from benign pituitary adenomas.6 Although the precise mechanism for paraneoplastic AN is still a matter of debate, it is likely the result of overactive growth factors, such as transforming growth factor α, epidermal growth factor, and α melanocyte-stimulating hormone.7 Generalized AN also has been associated with genetic abnormalities. Multiple genetic mutations have been associated with AN, including the genes coding for the insulin receptor, fibroblast growth receptors 2 and 3, lamin A/C, and seipin.8-12 Acanthosis nigricans has been described as a feature in other genetic syndromes but may represent an incidental finding.4 In 1976, Kahn et al13 linked AN with insulin resistance, which subsequently shifted the focus on AN’s link with obesity and as a precursor of type 2 diabetes mellitus.6,14 In these cases, it is hypothesized that excess insulin activates the insulinlike growth factor 1 receptor to stimulate keratinocyte and dermal fibroblast proliferation. It is likely that hyperinsulinemia also induces cellular proliferation indirectly through other pathways.15,16 Compared with paraneoplastic and syndromic causes, AN secondary to hyperinsulinemia and obesity does not tend to be generalized but instead is proportionate to the degree of metabolic derangement.17
Because our patient’s AN presentation was diffuse, long-standing, and occurred in the context of morphologic abnormalities, it was consistent with a syndrome. Based on the constellation of findings, we believe our patient fit the rare markers indicating Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome. As additional cases are reported, we hope that further biochemical, physical, and genetic studies may be pursued to better identify the syndrome and elucidate its pathogenesis.
To the Editor:
A 17-year-old adolescent girl presented to our clinic for evaluation of diffuse acanthosis nigricans (AN) that had been present since 7 years of age. The patient had a history of hypothyroidism, insulin resistance, ovarian cysts, and developmental delay. On examination, she presented with thick and verrucous plaques of AN involving the neck, abdomen, trunk, arms, and legs (Figure). The intertriginous areas were affected the most. The examination also was notable for dysmorphic facies and an endomorphic body habitus. Both parents denied similar health problems in their family and were normal in appearance. Although the patient was receiving metformin treatment for insulin resistance, she had not undergone any prior workup to identify a unifying syndromic cause for her physical and biochemical findings. A review of the literature showed a 1993 case report of a 5-year-old boy with mental retardation, body overgrowth, remarkable facies, and AN, which was termed Morfan syndrome.1 Because of the similarity in features, we believe that our patient’s presentation fits this syndrome. This report represents the second documented case of Morfan syndrome according to a PubMed search of articles indexed for MEDLINE using the search term Morfan. Additional searches using the terms acanthosis nigricans and syndrome also failed to identify any reports describing patients with a similar constellation of findings.
|
First described by Santi Unna and Monatsh Pollitzer in 1890, AN is a common dermatosis that is characterized by thick, hyperpigmented, and verrucous plaques.2,3 Although most common in symmetric distribution on flexural and intertriginous areas, AN also may involve mucosal surfaces.4 Acanthosis nigricans is associated with multiple etiologic factors, including direct autosomal transmission, genetic abnormalities, medications, malignancy, and endocrine imbalance.2 However, the diffuse generalized form of AN is almost always found in the context of malignancy or genetic syndromes.5 Historically, most attention on AN has focused on its eruptive form (so-called malignant AN), which is usually associated with internal malignancy but also may result from benign pituitary adenomas.6 Although the precise mechanism for paraneoplastic AN is still a matter of debate, it is likely the result of overactive growth factors, such as transforming growth factor α, epidermal growth factor, and α melanocyte-stimulating hormone.7 Generalized AN also has been associated with genetic abnormalities. Multiple genetic mutations have been associated with AN, including the genes coding for the insulin receptor, fibroblast growth receptors 2 and 3, lamin A/C, and seipin.8-12 Acanthosis nigricans has been described as a feature in other genetic syndromes but may represent an incidental finding.4 In 1976, Kahn et al13 linked AN with insulin resistance, which subsequently shifted the focus on AN’s link with obesity and as a precursor of type 2 diabetes mellitus.6,14 In these cases, it is hypothesized that excess insulin activates the insulinlike growth factor 1 receptor to stimulate keratinocyte and dermal fibroblast proliferation. It is likely that hyperinsulinemia also induces cellular proliferation indirectly through other pathways.15,16 Compared with paraneoplastic and syndromic causes, AN secondary to hyperinsulinemia and obesity does not tend to be generalized but instead is proportionate to the degree of metabolic derangement.17
Because our patient’s AN presentation was diffuse, long-standing, and occurred in the context of morphologic abnormalities, it was consistent with a syndrome. Based on the constellation of findings, we believe our patient fit the rare markers indicating Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome. As additional cases are reported, we hope that further biochemical, physical, and genetic studies may be pursued to better identify the syndrome and elucidate its pathogenesis.
1. Seemanová E, Rüdiger HW, Dreyer M. Morfan: a new syndrome characterized by mental retardation, pre- and postnatal overgrowth, remarkable face and acanthosis nigricans in 5-year-old boy. Am J Med Genet. 1993;45:525-528.
2. Rendon MI, Cruz PD Jr, Sontheimer RD, et al. Acanthosis nigricans: a cutaneous marker of tissue resistance to insulin. J Am Acad Dermatol. 1989;21:461-469.
3. Unna PG, Morris M, Besnier E, eds. International Atlas of Rare Skin Diseases. London, England: HK Lewis & Co; 1890.
4. Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol. 1994;31:1-19.
5. Inamdar AC, Palit A. Generalized acanthosis nigricans in childhood. Pediatr Dermatol. 2004;21:277-279.
6. Cruz PD, Hud JA. Excess insulin binding to insulin-like growth factor receptors: proposed mechanism for acanthosis nigricans. J Invest Dermatol. 1992;98:82-85.
7. Krawczyk M, Mykała-Cies´la J, Kołodziej-Jaskuła A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
8. Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 1995;11:462-464.
9. Moller DE, Cohen O, Yamaguchi Y, et al. Prevalence of mutations in the insulin receptor gene in subjects with features of the type A syndrome of insulin resistance. Diabetes. 1994;43:247-255.
10. Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nature Genet. 1996;13:492-494.
11. Anderson JL, Khan M, David WS, et al. Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21-22. Am J Med Genet. 1992;82:161-165.
12. Simha V, Agarwal AK, Aronin PA, et al. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet. 2008;146A:2318-2326.
13. Kahn CR, Flier JS, Bar RS, et al. the syndromes of insulin resistance and acanthosis nigricans. insulin-receptor disorders in man. N Engl J Med. 1976;294:739-745.
14. Brickman WWJ, Huang J, Silverman BL, et al. Acanthosis nigricans identifies youth at high risk for metabolic abnormalities. J Pediatr. 2010;156:87-92.
15. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. insulin-like growth factors.
N Engl J Med. 1997;336:633-640.
16. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818-835.
17. Berk DR, Spector EB, Bayliss SJ. Familial acanthosis nigricans due to K650T FGFR3 mutation. Arch Dermatol. 2007;143:1153-1156.
1. Seemanová E, Rüdiger HW, Dreyer M. Morfan: a new syndrome characterized by mental retardation, pre- and postnatal overgrowth, remarkable face and acanthosis nigricans in 5-year-old boy. Am J Med Genet. 1993;45:525-528.
2. Rendon MI, Cruz PD Jr, Sontheimer RD, et al. Acanthosis nigricans: a cutaneous marker of tissue resistance to insulin. J Am Acad Dermatol. 1989;21:461-469.
3. Unna PG, Morris M, Besnier E, eds. International Atlas of Rare Skin Diseases. London, England: HK Lewis & Co; 1890.
4. Schwartz RA. Acanthosis nigricans. J Am Acad Dermatol. 1994;31:1-19.
5. Inamdar AC, Palit A. Generalized acanthosis nigricans in childhood. Pediatr Dermatol. 2004;21:277-279.
6. Cruz PD, Hud JA. Excess insulin binding to insulin-like growth factor receptors: proposed mechanism for acanthosis nigricans. J Invest Dermatol. 1992;98:82-85.
7. Krawczyk M, Mykała-Cies´la J, Kołodziej-Jaskuła A. Acanthosis nigricans as a paraneoplastic syndrome. case reports and review of literature. Pol Arch Med Wewn. 2009;119:180-183.
8. Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nature Genet. 1995;11:462-464.
9. Moller DE, Cohen O, Yamaguchi Y, et al. Prevalence of mutations in the insulin receptor gene in subjects with features of the type A syndrome of insulin resistance. Diabetes. 1994;43:247-255.
10. Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nature Genet. 1996;13:492-494.
11. Anderson JL, Khan M, David WS, et al. Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21-22. Am J Med Genet. 1992;82:161-165.
12. Simha V, Agarwal AK, Aronin PA, et al. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet. 2008;146A:2318-2326.
13. Kahn CR, Flier JS, Bar RS, et al. the syndromes of insulin resistance and acanthosis nigricans. insulin-receptor disorders in man. N Engl J Med. 1976;294:739-745.
14. Brickman WWJ, Huang J, Silverman BL, et al. Acanthosis nigricans identifies youth at high risk for metabolic abnormalities. J Pediatr. 2010;156:87-92.
15. Le Roith D. Seminars in medicine of the Beth Israel Deaconess Medical Center. insulin-like growth factors.
N Engl J Med. 1997;336:633-640.
16. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev. 2001;22:818-835.
17. Berk DR, Spector EB, Bayliss SJ. Familial acanthosis nigricans due to K650T FGFR3 mutation. Arch Dermatol. 2007;143:1153-1156.

Plasmapheresis in Refractory Pemphigus Vulgaris: Revisiting an Old Treatment Modality Used in Synchrony With Pulse Cyclophosphamide
To the Editor:
Pemphigus vulgaris is an uncommon autoimmune blistering dermatosis characterized by painful mucocutaneous erosions. It can be a life-threatening condition if left untreated. The autoimmune process is mediated by autoantibodies against the keratinocyte surface antigens desmoglein 1 and 3.1 Therapy is directed at lowering autoantibody levels with systemic corticosteroids and immunosuppressive agents. Use of these agents often is limited by collateral adverse effects.2 Refractory disease may occur despite the use of high-dose corticosteroids or a combination of other immunosuppressants. The level of these pathogenic autoantibodies generally parallels the extent of disease activity, and removing them with plasmapheresis followed by immunosuppression should result in therapeutic response.3 We report a case of refractory pemphigus vulgaris that was controlled with plasmapheresis used in synchrony with pulse cyclophosphamide.
A 48-year-old Chinese man first presented with mucocutaneous erosions 2 years ago, and a diagnosis of pemphigus vulgaris was confirmed based on typical histologic and immunofluorescence features. Histologic features included suprabasal acantholysis with an intraepidermal blister as well as basal keratinocytes attached to the dermal papillae and present along the entire dermoepidermal junction (Figure 1). Direct immunofluorescence demonstrated intercellular deposits of IgG and complements in the lower epidermis, and indirect immunofluorescence showed the presence of the pathogenic pemphigus autoantibodies. The patient was initially treated with prednisolone (up to 1 mg/kg daily) and mycophenolate mofetil (1 g twice daily) for 6 months with moderate disease response. Two months later he experienced a disease flare that was triggered by sun exposure and concomitant herpes simplex virus infection. He achieved moderate disease control with acyclovir, 3 days of intravenous immunoglobulin, and combination prednisolone and azathioprine. There was no other relevant medical history. For the last year, the patient received continuous prednisolone (varying doses 0.5–1 mg/kg daily), concomitant azathioprine (up to 3 mg/kg daily), and long-term prophylactic acyclovir, but he continued to have residual crusted erosions over the scalp and face (best score of 25 points based on the autoimmune bullous skin disorder intensity score [ABSIS] ranging from 0–150 points4). He was admitted at the current presentation with another, more severe disease flare with extensive painful erosions over the trunk, arms, legs, face, and scalp (80% body surface area involvement and ABSIS score of 120 points)(Figure 2)4 that occurred after azathioprine was temporarily ceased for 1 week due to transaminitis, and despite a temporary increment in prednisolone dose. There was, however, no significant oral mucosal involvement. The desmoglein 1 and 3 antibody levels were elevated at more than 300 U/mL and 186 U/mL, respectively (>20 U/mL indicates positivity). A 3-day course of pulse intravenous methylprednisolone (10 mg/kg) failed to achieve clinical improvement or reduction of antibody titers. The use of various immunosuppressive agents was limited by persistent transaminitis and transient leukopenia.
|
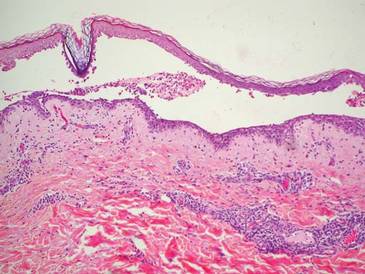
Because of remarkable morbidity, the patient underwent interim plasmapheresis for rapid disease control. Plasmapheresis was carried out through a pheresible central venous catheter. One plasma volume exchange was done each session, which was 5 L for the patient’s body weight and hematocrit. Equal volume of colloid comprising 2.5 L of fresh frozen plasma and 2.5 L of 5% albumin was used for replacement. Plasma exchange was performed with a cell separator by discontinuous flow centrifugation with 4% acid citrate dextrose as an anticoagulant. For each session of plasmapheresis, 16 cycles of exchange (each processing approximately 300 mL of blood) was carried out, the entire process lasting for 4 hours. The coagulation and biochemical profile was checked after each session of plasmapheresis and corrected when necessary. The patient underwent 9 sessions of plasmapheresis over a 3-week period, synchronized with pulse intravenous cyclophosphamide (15 mg/kg) immediately after completion of the plasmapheresis sessions, resulting in a remarkable decrease in pathogenic antibody titers to near undetectable levels and clinical improvement (Figure 3). The extensive erosions gradually healed with good reepithelialization, and there was a notable reduction in the ABSIS score to 12 points. He received 3 more monthly treatments with pulse intravenous cyclophosphamide (15 mg/kg) and is currently maintained on oral cyclophosphamide (2 mg/kg daily) and low-dose prednisolone (0.3 mg/kg daily). There was no subsequent disease relapse at 6-month follow-up, with the ABSIS score maintained at 5 points, and no increase in pathogenic autoantibody titers. The patient subsequently was lost to follow-up.
Patients with severe disease or refractory cases of pemphigus vulgaris that have been maintained on unacceptably high doses of corticosteroids or immunosuppressants that cannot be tapered without a disease flare may develop remarkable adverse effects, both from medications and from long-term immunosuppression.2 Our case illustrates the short-term benefit of plasmapheresis combined with immunosuppressants resulting in rapid disease control.
Plasmapheresis involves the selective removal of pathogenic materials from the circulation to achieve therapeutic effect, followed by appropriate replacement fluids. Treating pemphigus vulgaris with plasmapheresis was introduced in 1978 based on the rationale of removing pathogenic autoantibodies from the circulation.3,5 Using desmoglein enzyme-linked immunosorbent assay, it has been shown that one centrifugal plasmapheresis procedure eliminates approximately 15% of the IgG autoantibodies from the whole body.6 An average of 5 plasmapheresis sessions on alternate days usually is required to deplete the levels of pathogenic autoantibodies to near undetectable levels.7 Our case required 9 plasmapheresis sessions over 3 weeks to achieve good therapeutic response.
It seems that using plasmapheresis to treat pemphigus vulgaris has fallen out of favor due to its inability to prevent the antibody rebound occurring during weeks 1 and 2 posttreatment. Because of a feedback mechanism, a massive antibody depletion by plasmapheresis triggers a rebound synthesis of more autoantibodies by pathogenic B cells to titers comparable to or higher than those before plasmapheresis.8 The use of plasmapheresis should be supported by immunosuppressive therapy to prevent antibody feedback rebound. Due to the advent of available immunosuppressive agents in recent years, there is a resurgence in the successful use of this old treatment modality combined with immunosuppressive therapy in managing refractory pemphigus vulgaris.7,8 At present there is no clear data to support the use of one immunosuppressant versus another, but our case supports the use of pulse intravenous cyclophosphamide, as documented in other reports.7,9 The success of immunosuppressive agents at reducing antibody levels depends on the timing (immediately after plasmapheresis) as well as individual responsiveness to the immunosuppressant.7
Our armamentarium of therapies for refractory pemphigus vulgaris continues to evolve. A more selective method of removing antibodies by extracorporeal immunoadsorption has the benefit of higher removal rates and reduced inadvertent loss of other plasma components.10 The combination of protein A immunoadsorption with rituximab, a monoclonal anti-CD20 antibody that induces B-cell depletion, also has been shown to induce rapid and durable remission in refractory cases.11
Our case shows that plasmapheresis can be a useful alternative or adjunctive intervention in pemphigus vulgaris that is not responding to conventional therapy or in cases when steroids or immunosuppressants are contraindicated. There is a definite role for such therapeutic plasma exchanges in the rapid control of potentially life-threatening disease. Its benefits are optimized when used in synchrony with immunosuppressants immediately following plasmapheresis to prevent rebound effect of antibody depletion.
1. Udey MC, Stanley JR. Pemphigus–disease of antidesmosomal autoimmunity. JAMA. 1999;282:572-576.
2. Huilgol SC, Black MM. Management of the immunobullous disorders. II. pemphigus. Clin Exp Dermatol. 1995;20:283-293.
3. Cotterill JA, Barker DJ, Millard LG. Plasma exchange in the treatment of pemphigus vulgaris. Br J Dermatol. 1978;98:243.
4. Pfutze M, Niedermeier A, Hertl M, et al. Introducing a novel Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) in pemphigus [published online ahead of print February 27, 2007]. Eur J Dermatol. 2007;17:4-11.
5. Ruocco V, Rossi A, Argenziano G, et al. Pathogenicity of the intercellular antibodies of pemphigus their periodic removal from the circulation by plasmapheresis. Br J Dermatol. 1978;98:237-241.
6. Nagasaka T, Fujii Y, Ishida A, et al. Evaluating efficacy of plasmapheresis for patients with pemphigus using desmoglein enzyme-linked immunosorbent assay [published online ahead of print January 30, 2008]. Br J Dermatol. 2008;158:685-690.
7. Turner MS, Sutton D, Sauder DN. The use of plasmapheresis and immunosuppression in the treatment of pemphigus vulgaris. J Am Acad Dermatol. 2000;43:1058-1064.
8. Roujeau JC, Andre C, Joneau Fabre M, et al. Plasma exchange in pemphigus. uncontrolled study of ten patients. Arch Dermatol. 1983;119:215-221.
9. Euler HH, Löffler H, Christophers E. Synchronization of plasmapheresis and pulse cyclophosphamide therapy in pemphigus vulgaris. Arch Dermatol. 1987;123:1205-1210.
10. Lüftl M, Stauber A, Mainka A, et al. Successful removal of pathogenic autoantibodies in pemphigus by immunoadsorption with a tryptophan-linked polyvinylalcohol adsorber. Br J Dermatol. 2003;149:598-605.
11. Shimanovich I, Nitschke M, Rose C, et al. Treatment of severe pemphigus with protein A immunoadsorption, rituximab and intravenous immunoglobulins. Br J Dermatol. 2008;158:382-388.
To the Editor:
Pemphigus vulgaris is an uncommon autoimmune blistering dermatosis characterized by painful mucocutaneous erosions. It can be a life-threatening condition if left untreated. The autoimmune process is mediated by autoantibodies against the keratinocyte surface antigens desmoglein 1 and 3.1 Therapy is directed at lowering autoantibody levels with systemic corticosteroids and immunosuppressive agents. Use of these agents often is limited by collateral adverse effects.2 Refractory disease may occur despite the use of high-dose corticosteroids or a combination of other immunosuppressants. The level of these pathogenic autoantibodies generally parallels the extent of disease activity, and removing them with plasmapheresis followed by immunosuppression should result in therapeutic response.3 We report a case of refractory pemphigus vulgaris that was controlled with plasmapheresis used in synchrony with pulse cyclophosphamide.
A 48-year-old Chinese man first presented with mucocutaneous erosions 2 years ago, and a diagnosis of pemphigus vulgaris was confirmed based on typical histologic and immunofluorescence features. Histologic features included suprabasal acantholysis with an intraepidermal blister as well as basal keratinocytes attached to the dermal papillae and present along the entire dermoepidermal junction (Figure 1). Direct immunofluorescence demonstrated intercellular deposits of IgG and complements in the lower epidermis, and indirect immunofluorescence showed the presence of the pathogenic pemphigus autoantibodies. The patient was initially treated with prednisolone (up to 1 mg/kg daily) and mycophenolate mofetil (1 g twice daily) for 6 months with moderate disease response. Two months later he experienced a disease flare that was triggered by sun exposure and concomitant herpes simplex virus infection. He achieved moderate disease control with acyclovir, 3 days of intravenous immunoglobulin, and combination prednisolone and azathioprine. There was no other relevant medical history. For the last year, the patient received continuous prednisolone (varying doses 0.5–1 mg/kg daily), concomitant azathioprine (up to 3 mg/kg daily), and long-term prophylactic acyclovir, but he continued to have residual crusted erosions over the scalp and face (best score of 25 points based on the autoimmune bullous skin disorder intensity score [ABSIS] ranging from 0–150 points4). He was admitted at the current presentation with another, more severe disease flare with extensive painful erosions over the trunk, arms, legs, face, and scalp (80% body surface area involvement and ABSIS score of 120 points)(Figure 2)4 that occurred after azathioprine was temporarily ceased for 1 week due to transaminitis, and despite a temporary increment in prednisolone dose. There was, however, no significant oral mucosal involvement. The desmoglein 1 and 3 antibody levels were elevated at more than 300 U/mL and 186 U/mL, respectively (>20 U/mL indicates positivity). A 3-day course of pulse intravenous methylprednisolone (10 mg/kg) failed to achieve clinical improvement or reduction of antibody titers. The use of various immunosuppressive agents was limited by persistent transaminitis and transient leukopenia.
|
Because of remarkable morbidity, the patient underwent interim plasmapheresis for rapid disease control. Plasmapheresis was carried out through a pheresible central venous catheter. One plasma volume exchange was done each session, which was 5 L for the patient’s body weight and hematocrit. Equal volume of colloid comprising 2.5 L of fresh frozen plasma and 2.5 L of 5% albumin was used for replacement. Plasma exchange was performed with a cell separator by discontinuous flow centrifugation with 4% acid citrate dextrose as an anticoagulant. For each session of plasmapheresis, 16 cycles of exchange (each processing approximately 300 mL of blood) was carried out, the entire process lasting for 4 hours. The coagulation and biochemical profile was checked after each session of plasmapheresis and corrected when necessary. The patient underwent 9 sessions of plasmapheresis over a 3-week period, synchronized with pulse intravenous cyclophosphamide (15 mg/kg) immediately after completion of the plasmapheresis sessions, resulting in a remarkable decrease in pathogenic antibody titers to near undetectable levels and clinical improvement (Figure 3). The extensive erosions gradually healed with good reepithelialization, and there was a notable reduction in the ABSIS score to 12 points. He received 3 more monthly treatments with pulse intravenous cyclophosphamide (15 mg/kg) and is currently maintained on oral cyclophosphamide (2 mg/kg daily) and low-dose prednisolone (0.3 mg/kg daily). There was no subsequent disease relapse at 6-month follow-up, with the ABSIS score maintained at 5 points, and no increase in pathogenic autoantibody titers. The patient subsequently was lost to follow-up.
Patients with severe disease or refractory cases of pemphigus vulgaris that have been maintained on unacceptably high doses of corticosteroids or immunosuppressants that cannot be tapered without a disease flare may develop remarkable adverse effects, both from medications and from long-term immunosuppression.2 Our case illustrates the short-term benefit of plasmapheresis combined with immunosuppressants resulting in rapid disease control.
Plasmapheresis involves the selective removal of pathogenic materials from the circulation to achieve therapeutic effect, followed by appropriate replacement fluids. Treating pemphigus vulgaris with plasmapheresis was introduced in 1978 based on the rationale of removing pathogenic autoantibodies from the circulation.3,5 Using desmoglein enzyme-linked immunosorbent assay, it has been shown that one centrifugal plasmapheresis procedure eliminates approximately 15% of the IgG autoantibodies from the whole body.6 An average of 5 plasmapheresis sessions on alternate days usually is required to deplete the levels of pathogenic autoantibodies to near undetectable levels.7 Our case required 9 plasmapheresis sessions over 3 weeks to achieve good therapeutic response.
It seems that using plasmapheresis to treat pemphigus vulgaris has fallen out of favor due to its inability to prevent the antibody rebound occurring during weeks 1 and 2 posttreatment. Because of a feedback mechanism, a massive antibody depletion by plasmapheresis triggers a rebound synthesis of more autoantibodies by pathogenic B cells to titers comparable to or higher than those before plasmapheresis.8 The use of plasmapheresis should be supported by immunosuppressive therapy to prevent antibody feedback rebound. Due to the advent of available immunosuppressive agents in recent years, there is a resurgence in the successful use of this old treatment modality combined with immunosuppressive therapy in managing refractory pemphigus vulgaris.7,8 At present there is no clear data to support the use of one immunosuppressant versus another, but our case supports the use of pulse intravenous cyclophosphamide, as documented in other reports.7,9 The success of immunosuppressive agents at reducing antibody levels depends on the timing (immediately after plasmapheresis) as well as individual responsiveness to the immunosuppressant.7
Our armamentarium of therapies for refractory pemphigus vulgaris continues to evolve. A more selective method of removing antibodies by extracorporeal immunoadsorption has the benefit of higher removal rates and reduced inadvertent loss of other plasma components.10 The combination of protein A immunoadsorption with rituximab, a monoclonal anti-CD20 antibody that induces B-cell depletion, also has been shown to induce rapid and durable remission in refractory cases.11
Our case shows that plasmapheresis can be a useful alternative or adjunctive intervention in pemphigus vulgaris that is not responding to conventional therapy or in cases when steroids or immunosuppressants are contraindicated. There is a definite role for such therapeutic plasma exchanges in the rapid control of potentially life-threatening disease. Its benefits are optimized when used in synchrony with immunosuppressants immediately following plasmapheresis to prevent rebound effect of antibody depletion.
To the Editor:
Pemphigus vulgaris is an uncommon autoimmune blistering dermatosis characterized by painful mucocutaneous erosions. It can be a life-threatening condition if left untreated. The autoimmune process is mediated by autoantibodies against the keratinocyte surface antigens desmoglein 1 and 3.1 Therapy is directed at lowering autoantibody levels with systemic corticosteroids and immunosuppressive agents. Use of these agents often is limited by collateral adverse effects.2 Refractory disease may occur despite the use of high-dose corticosteroids or a combination of other immunosuppressants. The level of these pathogenic autoantibodies generally parallels the extent of disease activity, and removing them with plasmapheresis followed by immunosuppression should result in therapeutic response.3 We report a case of refractory pemphigus vulgaris that was controlled with plasmapheresis used in synchrony with pulse cyclophosphamide.
A 48-year-old Chinese man first presented with mucocutaneous erosions 2 years ago, and a diagnosis of pemphigus vulgaris was confirmed based on typical histologic and immunofluorescence features. Histologic features included suprabasal acantholysis with an intraepidermal blister as well as basal keratinocytes attached to the dermal papillae and present along the entire dermoepidermal junction (Figure 1). Direct immunofluorescence demonstrated intercellular deposits of IgG and complements in the lower epidermis, and indirect immunofluorescence showed the presence of the pathogenic pemphigus autoantibodies. The patient was initially treated with prednisolone (up to 1 mg/kg daily) and mycophenolate mofetil (1 g twice daily) for 6 months with moderate disease response. Two months later he experienced a disease flare that was triggered by sun exposure and concomitant herpes simplex virus infection. He achieved moderate disease control with acyclovir, 3 days of intravenous immunoglobulin, and combination prednisolone and azathioprine. There was no other relevant medical history. For the last year, the patient received continuous prednisolone (varying doses 0.5–1 mg/kg daily), concomitant azathioprine (up to 3 mg/kg daily), and long-term prophylactic acyclovir, but he continued to have residual crusted erosions over the scalp and face (best score of 25 points based on the autoimmune bullous skin disorder intensity score [ABSIS] ranging from 0–150 points4). He was admitted at the current presentation with another, more severe disease flare with extensive painful erosions over the trunk, arms, legs, face, and scalp (80% body surface area involvement and ABSIS score of 120 points)(Figure 2)4 that occurred after azathioprine was temporarily ceased for 1 week due to transaminitis, and despite a temporary increment in prednisolone dose. There was, however, no significant oral mucosal involvement. The desmoglein 1 and 3 antibody levels were elevated at more than 300 U/mL and 186 U/mL, respectively (>20 U/mL indicates positivity). A 3-day course of pulse intravenous methylprednisolone (10 mg/kg) failed to achieve clinical improvement or reduction of antibody titers. The use of various immunosuppressive agents was limited by persistent transaminitis and transient leukopenia.
|
Because of remarkable morbidity, the patient underwent interim plasmapheresis for rapid disease control. Plasmapheresis was carried out through a pheresible central venous catheter. One plasma volume exchange was done each session, which was 5 L for the patient’s body weight and hematocrit. Equal volume of colloid comprising 2.5 L of fresh frozen plasma and 2.5 L of 5% albumin was used for replacement. Plasma exchange was performed with a cell separator by discontinuous flow centrifugation with 4% acid citrate dextrose as an anticoagulant. For each session of plasmapheresis, 16 cycles of exchange (each processing approximately 300 mL of blood) was carried out, the entire process lasting for 4 hours. The coagulation and biochemical profile was checked after each session of plasmapheresis and corrected when necessary. The patient underwent 9 sessions of plasmapheresis over a 3-week period, synchronized with pulse intravenous cyclophosphamide (15 mg/kg) immediately after completion of the plasmapheresis sessions, resulting in a remarkable decrease in pathogenic antibody titers to near undetectable levels and clinical improvement (Figure 3). The extensive erosions gradually healed with good reepithelialization, and there was a notable reduction in the ABSIS score to 12 points. He received 3 more monthly treatments with pulse intravenous cyclophosphamide (15 mg/kg) and is currently maintained on oral cyclophosphamide (2 mg/kg daily) and low-dose prednisolone (0.3 mg/kg daily). There was no subsequent disease relapse at 6-month follow-up, with the ABSIS score maintained at 5 points, and no increase in pathogenic autoantibody titers. The patient subsequently was lost to follow-up.
Patients with severe disease or refractory cases of pemphigus vulgaris that have been maintained on unacceptably high doses of corticosteroids or immunosuppressants that cannot be tapered without a disease flare may develop remarkable adverse effects, both from medications and from long-term immunosuppression.2 Our case illustrates the short-term benefit of plasmapheresis combined with immunosuppressants resulting in rapid disease control.
Plasmapheresis involves the selective removal of pathogenic materials from the circulation to achieve therapeutic effect, followed by appropriate replacement fluids. Treating pemphigus vulgaris with plasmapheresis was introduced in 1978 based on the rationale of removing pathogenic autoantibodies from the circulation.3,5 Using desmoglein enzyme-linked immunosorbent assay, it has been shown that one centrifugal plasmapheresis procedure eliminates approximately 15% of the IgG autoantibodies from the whole body.6 An average of 5 plasmapheresis sessions on alternate days usually is required to deplete the levels of pathogenic autoantibodies to near undetectable levels.7 Our case required 9 plasmapheresis sessions over 3 weeks to achieve good therapeutic response.
It seems that using plasmapheresis to treat pemphigus vulgaris has fallen out of favor due to its inability to prevent the antibody rebound occurring during weeks 1 and 2 posttreatment. Because of a feedback mechanism, a massive antibody depletion by plasmapheresis triggers a rebound synthesis of more autoantibodies by pathogenic B cells to titers comparable to or higher than those before plasmapheresis.8 The use of plasmapheresis should be supported by immunosuppressive therapy to prevent antibody feedback rebound. Due to the advent of available immunosuppressive agents in recent years, there is a resurgence in the successful use of this old treatment modality combined with immunosuppressive therapy in managing refractory pemphigus vulgaris.7,8 At present there is no clear data to support the use of one immunosuppressant versus another, but our case supports the use of pulse intravenous cyclophosphamide, as documented in other reports.7,9 The success of immunosuppressive agents at reducing antibody levels depends on the timing (immediately after plasmapheresis) as well as individual responsiveness to the immunosuppressant.7
Our armamentarium of therapies for refractory pemphigus vulgaris continues to evolve. A more selective method of removing antibodies by extracorporeal immunoadsorption has the benefit of higher removal rates and reduced inadvertent loss of other plasma components.10 The combination of protein A immunoadsorption with rituximab, a monoclonal anti-CD20 antibody that induces B-cell depletion, also has been shown to induce rapid and durable remission in refractory cases.11
Our case shows that plasmapheresis can be a useful alternative or adjunctive intervention in pemphigus vulgaris that is not responding to conventional therapy or in cases when steroids or immunosuppressants are contraindicated. There is a definite role for such therapeutic plasma exchanges in the rapid control of potentially life-threatening disease. Its benefits are optimized when used in synchrony with immunosuppressants immediately following plasmapheresis to prevent rebound effect of antibody depletion.
1. Udey MC, Stanley JR. Pemphigus–disease of antidesmosomal autoimmunity. JAMA. 1999;282:572-576.
2. Huilgol SC, Black MM. Management of the immunobullous disorders. II. pemphigus. Clin Exp Dermatol. 1995;20:283-293.
3. Cotterill JA, Barker DJ, Millard LG. Plasma exchange in the treatment of pemphigus vulgaris. Br J Dermatol. 1978;98:243.
4. Pfutze M, Niedermeier A, Hertl M, et al. Introducing a novel Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) in pemphigus [published online ahead of print February 27, 2007]. Eur J Dermatol. 2007;17:4-11.
5. Ruocco V, Rossi A, Argenziano G, et al. Pathogenicity of the intercellular antibodies of pemphigus their periodic removal from the circulation by plasmapheresis. Br J Dermatol. 1978;98:237-241.
6. Nagasaka T, Fujii Y, Ishida A, et al. Evaluating efficacy of plasmapheresis for patients with pemphigus using desmoglein enzyme-linked immunosorbent assay [published online ahead of print January 30, 2008]. Br J Dermatol. 2008;158:685-690.
7. Turner MS, Sutton D, Sauder DN. The use of plasmapheresis and immunosuppression in the treatment of pemphigus vulgaris. J Am Acad Dermatol. 2000;43:1058-1064.
8. Roujeau JC, Andre C, Joneau Fabre M, et al. Plasma exchange in pemphigus. uncontrolled study of ten patients. Arch Dermatol. 1983;119:215-221.
9. Euler HH, Löffler H, Christophers E. Synchronization of plasmapheresis and pulse cyclophosphamide therapy in pemphigus vulgaris. Arch Dermatol. 1987;123:1205-1210.
10. Lüftl M, Stauber A, Mainka A, et al. Successful removal of pathogenic autoantibodies in pemphigus by immunoadsorption with a tryptophan-linked polyvinylalcohol adsorber. Br J Dermatol. 2003;149:598-605.
11. Shimanovich I, Nitschke M, Rose C, et al. Treatment of severe pemphigus with protein A immunoadsorption, rituximab and intravenous immunoglobulins. Br J Dermatol. 2008;158:382-388.
1. Udey MC, Stanley JR. Pemphigus–disease of antidesmosomal autoimmunity. JAMA. 1999;282:572-576.
2. Huilgol SC, Black MM. Management of the immunobullous disorders. II. pemphigus. Clin Exp Dermatol. 1995;20:283-293.
3. Cotterill JA, Barker DJ, Millard LG. Plasma exchange in the treatment of pemphigus vulgaris. Br J Dermatol. 1978;98:243.
4. Pfutze M, Niedermeier A, Hertl M, et al. Introducing a novel Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) in pemphigus [published online ahead of print February 27, 2007]. Eur J Dermatol. 2007;17:4-11.
5. Ruocco V, Rossi A, Argenziano G, et al. Pathogenicity of the intercellular antibodies of pemphigus their periodic removal from the circulation by plasmapheresis. Br J Dermatol. 1978;98:237-241.
6. Nagasaka T, Fujii Y, Ishida A, et al. Evaluating efficacy of plasmapheresis for patients with pemphigus using desmoglein enzyme-linked immunosorbent assay [published online ahead of print January 30, 2008]. Br J Dermatol. 2008;158:685-690.
7. Turner MS, Sutton D, Sauder DN. The use of plasmapheresis and immunosuppression in the treatment of pemphigus vulgaris. J Am Acad Dermatol. 2000;43:1058-1064.
8. Roujeau JC, Andre C, Joneau Fabre M, et al. Plasma exchange in pemphigus. uncontrolled study of ten patients. Arch Dermatol. 1983;119:215-221.
9. Euler HH, Löffler H, Christophers E. Synchronization of plasmapheresis and pulse cyclophosphamide therapy in pemphigus vulgaris. Arch Dermatol. 1987;123:1205-1210.
10. Lüftl M, Stauber A, Mainka A, et al. Successful removal of pathogenic autoantibodies in pemphigus by immunoadsorption with a tryptophan-linked polyvinylalcohol adsorber. Br J Dermatol. 2003;149:598-605.
11. Shimanovich I, Nitschke M, Rose C, et al. Treatment of severe pemphigus with protein A immunoadsorption, rituximab and intravenous immunoglobulins. Br J Dermatol. 2008;158:382-388.
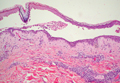
Bluish Red Verrucous Lesions on the Leg
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
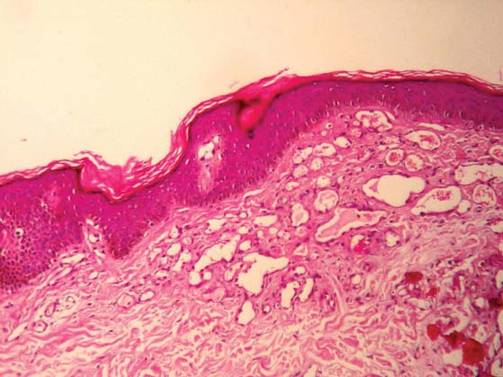
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
The Diagnosis: Linear Verrucous Hemangioma
Verrucous hemangioma is a rare congenital vascular malformation of the cutaneous and subcutaneous tissues. Although almost invariably present at birth, it may appear later in childhood or even in adulthood. Lesions commonly are found on the legs and may be linear, multiple, and disseminated or sometimes confined to the digits. In the early phase of evolution, the lesions are nonkeratotic, soft, blue-red plaques, but they gradually become increasingly hyperkeratotic.1 Linear verrucous hemangioma is even more rare with few published reports.
In 1937, Halter2 first used the term verrucous hemangioma to describe a 16-year-old adolescent boy who presented with a linear purpuric cluster of plaques extending from the right buttock to the toes. Imperial and Helwig3 later described it as a distinct entity. Since then, similar lesions have been described using a variety of names such as angiokeratoma circumscriptum neviforme, angiokeratoma circumscriptum, angiokeratoma corporis neviforme, keratotic hemangioma, nevus vascularis unius lateralis, and nevus keratoangiomatosus.3 Therefore, the exact incidence is difficult to determine.
Lesions generally are noted at birth or in early childhood and are often located on the lower extremities. The early lesions are bluish red in color. Secondary infection is a frequent complication, resulting in reactive papillomatosis and hyperkeratosis; thus, the older lesions acquire a verrucous or warty surface.3 Clinically, they may resemble angiokeratoma, lym-phangioma circumscriptum, verrucous epidermal nevus, verrucous cancer, or even malignant melanoma. Lesions initially resemble port-wine stains and later may become soft, bluish red vascular swellings that tend to grow in size and become verrucous.1
The histologic appearance closely resembles angiokeratoma, as both lesions show vascular spaces beneath a papillomatous and hyperkeratotic epidermis.4 However, in contrast to angiokeratoma, the vascular spaces in verrucous hemangioma also involve the lower dermis and subcutaneous tissues.
Although cases of linear verrucous hemangioma have been reported, its distribution along the lines of Blaschko is rare.5,6 It has been proposed that these lesions may actually be following dermatomal patterns or that the linear arrangement represents genetic mosaicism.5 In our case, the lesion started as a small plaque in childhood and gradually spread linearly to the buttock (Figure 1). A biopsy was taken from the lesion on the right buttock, which showed numerous dilated capillaries in the dermis (Figure 2).
|
Verrucous hemangiomas are best treated by excision. Larger lesions will need grafting. There is a tendency for recurrence to occur unless excision is complete.1 Yang and Ohara7 reported 14 patients with small localized lesions that were cured by 1 session of surgery without recurrence; 9 patients with wider and more extensive lesions required combination therapy in several stages for optimal results.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
1. Atherton DJ, Moss C. Naevi and other developmental defects. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. London, England: Blackwell Scientific Publications; 2004:15-60.
2. Halter K. Haemangioma verrucosum mit Osteoatrophie. Dermatol Z. 1937;75:271-279.
3. Imperial R, Helwig EB. Verrucous hemangioma: a clinicopathological study of 21 cases. Arch Dermatol. 1967;96:247-253.
4. Calduch L, Ortega C, Navarro V, et al. Verrucous hemangioma: Report of two cases and review of the literature. Pediatr Dermatol. 2000;17:213-217.
5. Wentscher U, Happle R. Linear verrucous hemangioma. J Am Acad Dermatol. 2000;42:516-518.
6. Jain VK, Aggarwal K, Jain S. Linear verrucous hemangioma on the leg. Indian J Dermatol Venereol Leprol. 2008;74:656-658.
7. Yang CH, Ohara K. Successful surgical treatment of verrucous hemangioma: a combined approach. Dermatol Surg. 2002;28:913-919.
An 18-year-old man presented with a history of multiple bluish verrucous lesions over the right leg. A small nodule on the lateral aspect of the right ankle was present at birth; it increased in size and number and gradually extended to the right buttock. He had recurrent bleeding and infection over the lesions. No other remarkable comorbidities were noted. Dermatologic examination revealed multiple well-circumscribed, bluish red, verrucous lesions distributed linearly along the lateral aspect of the leg. The surface of the lesions was verruciform and showed crusting at places. The second and third toes on the right foot were involved. On the buttock, multiple well-defined, bluish red plaques were present. Both limbs were of equal length.
Eyes on Ivermectin
In December 2014, the US Food and Drug Administration approved ivermectin cream 1% (Soolantra) for the treatment of inflammatory lesions of rosacea. This approval follows several safety and efficacy trials, particularly a phase 3 investigator-blinded, parallel-group study, published online in the British Journal of Dermatology on September 16, 2014, comparing once-daily application of ivermectin cream 1% and twice-daily metronidazole cream 0.75% in 962 patients with papulopustular rosacea over 16 weeks. Ivermectin showed more favorable local tolerability and significant reduction in lesion count versus metronidazole (83% vs 73.7%; P<.001) starting at week 3 and persisting throughout the study.
What’s the issue?
How many patients do you encounter each week who apply metronidazole topical treatments for years with little objective evidence of rosacea improvement? These data suggest that topical ivermectin may be a slightly more effective and tolerable alternative for papulopustular rosacea than the long-standing but modestly efficacious gold standard. With the recent approval of brimonidine gel for the erythematous component of rosacea, it is groundbreaking to introduce novel topical mechanisms into our rosacea prescription armamentarium as we attempt to elucidate the disease’s complex pathophysiology. What is your experience with this topical, and how do you think it will fit into your prescribing routines for rosacea?
In December 2014, the US Food and Drug Administration approved ivermectin cream 1% (Soolantra) for the treatment of inflammatory lesions of rosacea. This approval follows several safety and efficacy trials, particularly a phase 3 investigator-blinded, parallel-group study, published online in the British Journal of Dermatology on September 16, 2014, comparing once-daily application of ivermectin cream 1% and twice-daily metronidazole cream 0.75% in 962 patients with papulopustular rosacea over 16 weeks. Ivermectin showed more favorable local tolerability and significant reduction in lesion count versus metronidazole (83% vs 73.7%; P<.001) starting at week 3 and persisting throughout the study.
What’s the issue?
How many patients do you encounter each week who apply metronidazole topical treatments for years with little objective evidence of rosacea improvement? These data suggest that topical ivermectin may be a slightly more effective and tolerable alternative for papulopustular rosacea than the long-standing but modestly efficacious gold standard. With the recent approval of brimonidine gel for the erythematous component of rosacea, it is groundbreaking to introduce novel topical mechanisms into our rosacea prescription armamentarium as we attempt to elucidate the disease’s complex pathophysiology. What is your experience with this topical, and how do you think it will fit into your prescribing routines for rosacea?
In December 2014, the US Food and Drug Administration approved ivermectin cream 1% (Soolantra) for the treatment of inflammatory lesions of rosacea. This approval follows several safety and efficacy trials, particularly a phase 3 investigator-blinded, parallel-group study, published online in the British Journal of Dermatology on September 16, 2014, comparing once-daily application of ivermectin cream 1% and twice-daily metronidazole cream 0.75% in 962 patients with papulopustular rosacea over 16 weeks. Ivermectin showed more favorable local tolerability and significant reduction in lesion count versus metronidazole (83% vs 73.7%; P<.001) starting at week 3 and persisting throughout the study.
What’s the issue?
How many patients do you encounter each week who apply metronidazole topical treatments for years with little objective evidence of rosacea improvement? These data suggest that topical ivermectin may be a slightly more effective and tolerable alternative for papulopustular rosacea than the long-standing but modestly efficacious gold standard. With the recent approval of brimonidine gel for the erythematous component of rosacea, it is groundbreaking to introduce novel topical mechanisms into our rosacea prescription armamentarium as we attempt to elucidate the disease’s complex pathophysiology. What is your experience with this topical, and how do you think it will fit into your prescribing routines for rosacea?
