User login
VIDEO: Expert picks top studies in Alzheimer’s, migraine, and stroke at AAN
WASHINGTON – A promising early Alzheimer’s treatment, a new finding in the brains of migraineurs, and a practice-changing stroke treatment study are the top picks for clinical research being presented at the annual meeting of the American Academy of Neurology, according to Dr. Natalia Rost, vice chair of the AAN Science Committee.
The first is an early-phase clinical study evaluating a new monoclonal antibody in 166 people with prodromal or mild Alzheimer’s disease. The second study evaluated the association between pain threshold and cortical thickness in patients with migraines. The third is a presentation of the MR CLEAN study results from the Netherlands, which compared standard of care versus standard of care plus mechanical embolectomy in 500 patients with acute stroke – a study that Dr. Rost, a stroke neurologist, described in this video interview as “game-changing.” Dr. Rost is associate professor of neurology at Harvard Medical School and associate director of acute stroke services at Massachusetts General Hospital, both in Boston.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WASHINGTON – A promising early Alzheimer’s treatment, a new finding in the brains of migraineurs, and a practice-changing stroke treatment study are the top picks for clinical research being presented at the annual meeting of the American Academy of Neurology, according to Dr. Natalia Rost, vice chair of the AAN Science Committee.
The first is an early-phase clinical study evaluating a new monoclonal antibody in 166 people with prodromal or mild Alzheimer’s disease. The second study evaluated the association between pain threshold and cortical thickness in patients with migraines. The third is a presentation of the MR CLEAN study results from the Netherlands, which compared standard of care versus standard of care plus mechanical embolectomy in 500 patients with acute stroke – a study that Dr. Rost, a stroke neurologist, described in this video interview as “game-changing.” Dr. Rost is associate professor of neurology at Harvard Medical School and associate director of acute stroke services at Massachusetts General Hospital, both in Boston.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WASHINGTON – A promising early Alzheimer’s treatment, a new finding in the brains of migraineurs, and a practice-changing stroke treatment study are the top picks for clinical research being presented at the annual meeting of the American Academy of Neurology, according to Dr. Natalia Rost, vice chair of the AAN Science Committee.
The first is an early-phase clinical study evaluating a new monoclonal antibody in 166 people with prodromal or mild Alzheimer’s disease. The second study evaluated the association between pain threshold and cortical thickness in patients with migraines. The third is a presentation of the MR CLEAN study results from the Netherlands, which compared standard of care versus standard of care plus mechanical embolectomy in 500 patients with acute stroke – a study that Dr. Rost, a stroke neurologist, described in this video interview as “game-changing.” Dr. Rost is associate professor of neurology at Harvard Medical School and associate director of acute stroke services at Massachusetts General Hospital, both in Boston.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT THE AAN 2015 ANNUAL MEETING

FDA moves to alert physicians, public to counterfeit Botox
Counterfeit Botox has been found in the United States and could be in offices and clinics across the country, the Food and Drug Administration announced on April 17.
The counterfeit Botox “may have been sold” to medical clinics and physicians’ offices; the source is an unlicensed supplier that is not authorized to ship or distribute drug products in this country. “The counterfeit products are considered unsafe and should not be used. FDA cannot confirm that the manufacture, quality, storage, and handling of these suspect products follow U.S. standards,” according to the FDA statement, which notes that the agency is not aware of any adverse events resulting from the use of the counterfeit product.
“Currently, there is no indication that Allergan’s FDA-approved version is at risk, and the genuine product should be considered safe and effective for its intended and approved use,” the FDA statement said.
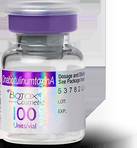
Features that can help identify the counterfeit product include a missing lot number on the vial. And instead of “OnabotulinumtoxinA” on the vial and outer carton (which is printed on the label and outer carton of the FDA-approved Botox manufactured by Allergan), the counterfeit product has “Botulinum Toxin Type A” on the outer carton and vial.

The counterfeit product also does not have anything written after the “LOT: MFG: EXP:” section of the outer carton.
The Allergan website also has information on how to identify counterfeit Botox, which includes a list of authorized distributors. In addition, a list of the state agencies that license wholesale prescription drug distributors is available on the FDA’s website.
The FDA is asking anybody who is aware of any Botox product thought to be counterfeit to contact the agency’s Office of Criminal Investigations (OCI) at 800-551-3989, www.accessdata.fda.gov/scripts/email/oc/oci/contact.cfm, or by e-mail at DrugSupplyChainIntegrity@fda.hhs.gov.
Adverse events thought to be related to counterfeit Botox should be reported to the FDA’s MedWatch program at800-332-1088, or https://www.accessdata.fda.gov/scripts/medwatch/.
Counterfeit Botox has been found in the United States and could be in offices and clinics across the country, the Food and Drug Administration announced on April 17.
The counterfeit Botox “may have been sold” to medical clinics and physicians’ offices; the source is an unlicensed supplier that is not authorized to ship or distribute drug products in this country. “The counterfeit products are considered unsafe and should not be used. FDA cannot confirm that the manufacture, quality, storage, and handling of these suspect products follow U.S. standards,” according to the FDA statement, which notes that the agency is not aware of any adverse events resulting from the use of the counterfeit product.
“Currently, there is no indication that Allergan’s FDA-approved version is at risk, and the genuine product should be considered safe and effective for its intended and approved use,” the FDA statement said.
Features that can help identify the counterfeit product include a missing lot number on the vial. And instead of “OnabotulinumtoxinA” on the vial and outer carton (which is printed on the label and outer carton of the FDA-approved Botox manufactured by Allergan), the counterfeit product has “Botulinum Toxin Type A” on the outer carton and vial.
The counterfeit product also does not have anything written after the “LOT: MFG: EXP:” section of the outer carton.
The Allergan website also has information on how to identify counterfeit Botox, which includes a list of authorized distributors. In addition, a list of the state agencies that license wholesale prescription drug distributors is available on the FDA’s website.
The FDA is asking anybody who is aware of any Botox product thought to be counterfeit to contact the agency’s Office of Criminal Investigations (OCI) at 800-551-3989, www.accessdata.fda.gov/scripts/email/oc/oci/contact.cfm, or by e-mail at DrugSupplyChainIntegrity@fda.hhs.gov.
Adverse events thought to be related to counterfeit Botox should be reported to the FDA’s MedWatch program at800-332-1088, or https://www.accessdata.fda.gov/scripts/medwatch/.
Counterfeit Botox has been found in the United States and could be in offices and clinics across the country, the Food and Drug Administration announced on April 17.
The counterfeit Botox “may have been sold” to medical clinics and physicians’ offices; the source is an unlicensed supplier that is not authorized to ship or distribute drug products in this country. “The counterfeit products are considered unsafe and should not be used. FDA cannot confirm that the manufacture, quality, storage, and handling of these suspect products follow U.S. standards,” according to the FDA statement, which notes that the agency is not aware of any adverse events resulting from the use of the counterfeit product.
“Currently, there is no indication that Allergan’s FDA-approved version is at risk, and the genuine product should be considered safe and effective for its intended and approved use,” the FDA statement said.
Features that can help identify the counterfeit product include a missing lot number on the vial. And instead of “OnabotulinumtoxinA” on the vial and outer carton (which is printed on the label and outer carton of the FDA-approved Botox manufactured by Allergan), the counterfeit product has “Botulinum Toxin Type A” on the outer carton and vial.
The counterfeit product also does not have anything written after the “LOT: MFG: EXP:” section of the outer carton.
The Allergan website also has information on how to identify counterfeit Botox, which includes a list of authorized distributors. In addition, a list of the state agencies that license wholesale prescription drug distributors is available on the FDA’s website.
The FDA is asking anybody who is aware of any Botox product thought to be counterfeit to contact the agency’s Office of Criminal Investigations (OCI) at 800-551-3989, www.accessdata.fda.gov/scripts/email/oc/oci/contact.cfm, or by e-mail at DrugSupplyChainIntegrity@fda.hhs.gov.
Adverse events thought to be related to counterfeit Botox should be reported to the FDA’s MedWatch program at800-332-1088, or https://www.accessdata.fda.gov/scripts/medwatch/.
FROM THE FDA
FDA approves first generic version of MS drug glatiramer acetate
The first generic formulation of the multiple sclerosis drug glatiramer acetate has been approved by the Food and Drug Administration, despite efforts by Copaxone manufacturer Teva to delay the approval of a generic version of the drug.
But it is unclear when the generic will be available.
The approved generic, in a 20 mg/mL daily injection, is manufactured by Sandoz, the FDA announced in a statement issued on April 16. The statement was accompanied by a letter from the FDA denying Teva’s citizen’s petition requesting that the FDA not approve generic versions of the drug “unless and until” certain conditions were met, including the conduct of extensive studies showing generic versions are bioequivalent to Copaxone. This was the eighth petition submitted by Teva, according to the FDA’s letter.
The FDA’s statement announcing the approval said that the agency “applies the same rigorous and reliable standards to evaluate all generic drug products. As needed, the agency requires appropriate information to demonstrate sameness for complex active ingredients, such as glatiramer acetate. For this approval, FDA scientists established a thorough scientific approach for demonstrating active ingredient sameness that takes into consideration the complexity of glatiramer acetate.”
Before approval of this product, “given its complexity, we reviewed additional information to make sure that the generic product is as safe and effective as the brand name product,” Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said in the FDA’s statement.
In a statement, Sandoz said it will market the generic form as Glatopa. A spokesperson said that the company cannot comment on plans for launching Glatopa.
In January, the U.S. Supreme Court overturned a federal circuit court’s judgment that had said that a patent for Copaxone 20 mg/mL was invalid because Teva’s listing of the molecular weight of its active ingredient as “5 to 9 kilodaltons” was fatally indefinite, sending it back to the U.S. Court of Appeals for the Federal Circuit for further review.
“Given the pending decision from the U.S. Court of Appeals for the Federal Circuit, any companies that launch a generic version of Copaxone would do so at risk,” a Teva spokesperson said in response to a request for a comment.
Approved in 1996, glatiramer acetate is indicated for relapsing forms of MS; the 20 mg/mL dose is administered once a day. Teva also markets a 40 mg/mL dose that is administered three times a week.
The first generic formulation of the multiple sclerosis drug glatiramer acetate has been approved by the Food and Drug Administration, despite efforts by Copaxone manufacturer Teva to delay the approval of a generic version of the drug.
But it is unclear when the generic will be available.
The approved generic, in a 20 mg/mL daily injection, is manufactured by Sandoz, the FDA announced in a statement issued on April 16. The statement was accompanied by a letter from the FDA denying Teva’s citizen’s petition requesting that the FDA not approve generic versions of the drug “unless and until” certain conditions were met, including the conduct of extensive studies showing generic versions are bioequivalent to Copaxone. This was the eighth petition submitted by Teva, according to the FDA’s letter.
The FDA’s statement announcing the approval said that the agency “applies the same rigorous and reliable standards to evaluate all generic drug products. As needed, the agency requires appropriate information to demonstrate sameness for complex active ingredients, such as glatiramer acetate. For this approval, FDA scientists established a thorough scientific approach for demonstrating active ingredient sameness that takes into consideration the complexity of glatiramer acetate.”
Before approval of this product, “given its complexity, we reviewed additional information to make sure that the generic product is as safe and effective as the brand name product,” Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said in the FDA’s statement.
In a statement, Sandoz said it will market the generic form as Glatopa. A spokesperson said that the company cannot comment on plans for launching Glatopa.
In January, the U.S. Supreme Court overturned a federal circuit court’s judgment that had said that a patent for Copaxone 20 mg/mL was invalid because Teva’s listing of the molecular weight of its active ingredient as “5 to 9 kilodaltons” was fatally indefinite, sending it back to the U.S. Court of Appeals for the Federal Circuit for further review.
“Given the pending decision from the U.S. Court of Appeals for the Federal Circuit, any companies that launch a generic version of Copaxone would do so at risk,” a Teva spokesperson said in response to a request for a comment.
Approved in 1996, glatiramer acetate is indicated for relapsing forms of MS; the 20 mg/mL dose is administered once a day. Teva also markets a 40 mg/mL dose that is administered three times a week.
The first generic formulation of the multiple sclerosis drug glatiramer acetate has been approved by the Food and Drug Administration, despite efforts by Copaxone manufacturer Teva to delay the approval of a generic version of the drug.
But it is unclear when the generic will be available.
The approved generic, in a 20 mg/mL daily injection, is manufactured by Sandoz, the FDA announced in a statement issued on April 16. The statement was accompanied by a letter from the FDA denying Teva’s citizen’s petition requesting that the FDA not approve generic versions of the drug “unless and until” certain conditions were met, including the conduct of extensive studies showing generic versions are bioequivalent to Copaxone. This was the eighth petition submitted by Teva, according to the FDA’s letter.
The FDA’s statement announcing the approval said that the agency “applies the same rigorous and reliable standards to evaluate all generic drug products. As needed, the agency requires appropriate information to demonstrate sameness for complex active ingredients, such as glatiramer acetate. For this approval, FDA scientists established a thorough scientific approach for demonstrating active ingredient sameness that takes into consideration the complexity of glatiramer acetate.”
Before approval of this product, “given its complexity, we reviewed additional information to make sure that the generic product is as safe and effective as the brand name product,” Dr. Janet Woodcock, director of the FDA’s Center for Drug Evaluation and Research, said in the FDA’s statement.
In a statement, Sandoz said it will market the generic form as Glatopa. A spokesperson said that the company cannot comment on plans for launching Glatopa.
In January, the U.S. Supreme Court overturned a federal circuit court’s judgment that had said that a patent for Copaxone 20 mg/mL was invalid because Teva’s listing of the molecular weight of its active ingredient as “5 to 9 kilodaltons” was fatally indefinite, sending it back to the U.S. Court of Appeals for the Federal Circuit for further review.
“Given the pending decision from the U.S. Court of Appeals for the Federal Circuit, any companies that launch a generic version of Copaxone would do so at risk,” a Teva spokesperson said in response to a request for a comment.
Approved in 1996, glatiramer acetate is indicated for relapsing forms of MS; the 20 mg/mL dose is administered once a day. Teva also markets a 40 mg/mL dose that is administered three times a week.
FDA panel cautiously backs approval of short-acting IV antiplatelet drug
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported approval of cangrelor, a short-acting, intravenous antiplatelet drug, for use in patients undergoing percutaneous coronary intervention, with precautions that it not be used widely or indiscriminately.
At a meeting on April 15, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 9-2, with one abstention, that cangrelor should be approved as an adjunct to PCI “for reducing the periprocedural thrombotic events, such as MI, stent thrombosis, and ischemia-driven revascularization.” The proposed indication includes the statement that use is for patients undergoing PCI “who have not received an oral P2Y12 inhibitor prior to the PCI procedure and in whom oral therapy with P2Y12 inhibitors is not feasible or desirable.”
Cangrelor, a P2Y12 receptor inhibitor, has a half-life of 3-6 minutes, and platelet function completely returns to normal within 1 hour of stopping the infusion, according to the product’s sponsor, the Medicines Company.
The vote was based on analyses of revised endpoints of the data from the CHAMPION PHOENIX study, which compared cangrelor to clopidogrel in more than 11,000 patients undergoing PCI. The same panel voted 7-2 against approval in February 2014, for reasons that included doubts about the clinical consequences of some of the endpoints of the trial.
At the request of the FDA, the company reanalyzed the data, dropping intraprocedural stent thrombosis and MIs that did not meet the SCAI (Society for Cardiovascular Angiography Interventions) criteria for a clinically relevant MI from the primary endpoint. At 48 hours, 1.4% of those in the cangrelor arm met this revised endpoint, vs. 2.1% of those in the clopidogrel arm, for an odds ratio of 0.69 (P = .011). Most of the panel agreed that these analyses showed the drug had a beneficial effect.
Deaths and serious adverse events were similar between cangrelor and clopidogrel, but bleeding events were more common among patients treated with cangrelor, according to the company. A risk-benefit analysis, which took into account bleeding events, provided by the FDA concluded that the “benefit of cangrelor is small, but the risk is smaller.”
“There’s a great responsibility that this gets used appropriately and [is] not overused,” said Dr. James de Lemos, who voted against approval at the last meeting but voted for approval at this meeting. Dr. de Lemos, distinguished chair in cardiology and professor of medicine at the University of Texas Southwestern Medical Center, Dallas, said that his concerns about the robustness of the primary endpoint that had been “mostly addressed” by the FDA’s and company’s analyses.
However, he added that he remained concerned by the very narrow clinical benefit of the drug "relative to the risks and relative to what I worry will be the potential use of the drug.”
”What I would hope is that this drug is used very selectively and very narrowly in circumstances where P2Y12 inhibitors cannot be used appropriately,” he commented. “If it is used broadly and indiscriminately, it will not only be expensive but it [also] will expose low-risk patients to unnecessary bleeding, some of which can be quite substantial.”
The FDA usually follows the recommendations of its advisory panels. The members of the panel had no relevant disclosures.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported approval of cangrelor, a short-acting, intravenous antiplatelet drug, for use in patients undergoing percutaneous coronary intervention, with precautions that it not be used widely or indiscriminately.
At a meeting on April 15, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 9-2, with one abstention, that cangrelor should be approved as an adjunct to PCI “for reducing the periprocedural thrombotic events, such as MI, stent thrombosis, and ischemia-driven revascularization.” The proposed indication includes the statement that use is for patients undergoing PCI “who have not received an oral P2Y12 inhibitor prior to the PCI procedure and in whom oral therapy with P2Y12 inhibitors is not feasible or desirable.”
Cangrelor, a P2Y12 receptor inhibitor, has a half-life of 3-6 minutes, and platelet function completely returns to normal within 1 hour of stopping the infusion, according to the product’s sponsor, the Medicines Company.
The vote was based on analyses of revised endpoints of the data from the CHAMPION PHOENIX study, which compared cangrelor to clopidogrel in more than 11,000 patients undergoing PCI. The same panel voted 7-2 against approval in February 2014, for reasons that included doubts about the clinical consequences of some of the endpoints of the trial.
At the request of the FDA, the company reanalyzed the data, dropping intraprocedural stent thrombosis and MIs that did not meet the SCAI (Society for Cardiovascular Angiography Interventions) criteria for a clinically relevant MI from the primary endpoint. At 48 hours, 1.4% of those in the cangrelor arm met this revised endpoint, vs. 2.1% of those in the clopidogrel arm, for an odds ratio of 0.69 (P = .011). Most of the panel agreed that these analyses showed the drug had a beneficial effect.
Deaths and serious adverse events were similar between cangrelor and clopidogrel, but bleeding events were more common among patients treated with cangrelor, according to the company. A risk-benefit analysis, which took into account bleeding events, provided by the FDA concluded that the “benefit of cangrelor is small, but the risk is smaller.”
“There’s a great responsibility that this gets used appropriately and [is] not overused,” said Dr. James de Lemos, who voted against approval at the last meeting but voted for approval at this meeting. Dr. de Lemos, distinguished chair in cardiology and professor of medicine at the University of Texas Southwestern Medical Center, Dallas, said that his concerns about the robustness of the primary endpoint that had been “mostly addressed” by the FDA’s and company’s analyses.
However, he added that he remained concerned by the very narrow clinical benefit of the drug "relative to the risks and relative to what I worry will be the potential use of the drug.”
”What I would hope is that this drug is used very selectively and very narrowly in circumstances where P2Y12 inhibitors cannot be used appropriately,” he commented. “If it is used broadly and indiscriminately, it will not only be expensive but it [also] will expose low-risk patients to unnecessary bleeding, some of which can be quite substantial.”
The FDA usually follows the recommendations of its advisory panels. The members of the panel had no relevant disclosures.
SILVER SPRING, MD. – The majority of a Food and Drug Administration advisory panel supported approval of cangrelor, a short-acting, intravenous antiplatelet drug, for use in patients undergoing percutaneous coronary intervention, with precautions that it not be used widely or indiscriminately.
At a meeting on April 15, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 9-2, with one abstention, that cangrelor should be approved as an adjunct to PCI “for reducing the periprocedural thrombotic events, such as MI, stent thrombosis, and ischemia-driven revascularization.” The proposed indication includes the statement that use is for patients undergoing PCI “who have not received an oral P2Y12 inhibitor prior to the PCI procedure and in whom oral therapy with P2Y12 inhibitors is not feasible or desirable.”
Cangrelor, a P2Y12 receptor inhibitor, has a half-life of 3-6 minutes, and platelet function completely returns to normal within 1 hour of stopping the infusion, according to the product’s sponsor, the Medicines Company.
The vote was based on analyses of revised endpoints of the data from the CHAMPION PHOENIX study, which compared cangrelor to clopidogrel in more than 11,000 patients undergoing PCI. The same panel voted 7-2 against approval in February 2014, for reasons that included doubts about the clinical consequences of some of the endpoints of the trial.
At the request of the FDA, the company reanalyzed the data, dropping intraprocedural stent thrombosis and MIs that did not meet the SCAI (Society for Cardiovascular Angiography Interventions) criteria for a clinically relevant MI from the primary endpoint. At 48 hours, 1.4% of those in the cangrelor arm met this revised endpoint, vs. 2.1% of those in the clopidogrel arm, for an odds ratio of 0.69 (P = .011). Most of the panel agreed that these analyses showed the drug had a beneficial effect.
Deaths and serious adverse events were similar between cangrelor and clopidogrel, but bleeding events were more common among patients treated with cangrelor, according to the company. A risk-benefit analysis, which took into account bleeding events, provided by the FDA concluded that the “benefit of cangrelor is small, but the risk is smaller.”
“There’s a great responsibility that this gets used appropriately and [is] not overused,” said Dr. James de Lemos, who voted against approval at the last meeting but voted for approval at this meeting. Dr. de Lemos, distinguished chair in cardiology and professor of medicine at the University of Texas Southwestern Medical Center, Dallas, said that his concerns about the robustness of the primary endpoint that had been “mostly addressed” by the FDA’s and company’s analyses.
However, he added that he remained concerned by the very narrow clinical benefit of the drug "relative to the risks and relative to what I worry will be the potential use of the drug.”
”What I would hope is that this drug is used very selectively and very narrowly in circumstances where P2Y12 inhibitors cannot be used appropriately,” he commented. “If it is used broadly and indiscriminately, it will not only be expensive but it [also] will expose low-risk patients to unnecessary bleeding, some of which can be quite substantial.”
The FDA usually follows the recommendations of its advisory panels. The members of the panel had no relevant disclosures.
AT AN FDA ADVISORY COMMITTEE MEETING
Controversial technique appeals to women with mitochondrial diseases
WASHINGTON– Despite ethical issues raised by mitochondrial replacement therapy and uncertainties about the potential risks, women with serious mitochondrial diseases appear willing to accept these unknowns for the chance to prevent transmission of the diseases to their biological children.
Mitochondrial replacement therapy (MRT), which was recently granted approval in the United Kingdom, involves removing the nuclear DNA from the egg or fertilized egg of a woman with mitochondrial abnormalities and inserting it into a donor egg with normal mitochondria, after the nuclear DNA of the donor egg has been removed.
An MRT technique that entails removing the mitochondrial DNA from the intended mother’s egg is being evaluated at two centers in the United States, the University of Oregon, Eugene, where it has been performed in macaque monkeys, and the New York Stem Cell Foundation.
The technique, also known as mitochondrial donation, mitochondrial transfer, or mitochondrial DNA replacement, has generated significant controversy, with critics warning that it will lead to “germline enhancements,” eugenics, and designer babies.
But at a 2-day meeting sponsored by the Institute of Medicine (IOM), researchers presented survey data showing that for women who are carriers of these mutations, the ethical concerns are secondary.
In a survey of 92 women who were known carriers of a mitochondrial DNA mutation or were at risk of carrying a mutation, there was universal concern about transmitting mitochondrial DNA mutations to their children, and “overwhelming support and widespread interest” in the clinical use of MRT, particularly among women considering having children at the time of the survey, according to Dr. Michio Hirano, professor of neurology at Columbia University, which collaborates with the New York Stem Cell Foundation on this research.
Almost 80% of the women said they had considered not having children because of the transmission risk and almost three-quarters of those who already had children said they would have considered not having children if they had known they carried the mutation.
Carrying a mutation “has a profound effect on what these women think about their progeny and the risk of transmitting disease,” said Dr. Hirano, who is codirector of the North American Mitochondrial Disease Consortium.
Almost all of the 21 women who were considering having children at the time of the survey said that having a biological child was “very” or “somewhat” important to them, and almost all said they would be interested in the option of MRT. Dr. Hirano, noted that preimplantation genetic diagnosis (PGD), an option often raised as an alternative to MRT, is not always reliable.
Kirah Fasano, a 32-year-old with chronic progressive external ophthalmoplegia, who spoke at the IOM meeting, said that because of her type of mitochondrial disease, PGD was not an option, and after several failed attempts with in-vitro fertilization using her own eggs, she and her husband opted for an egg donor.
She now has a healthy baby, but MRT “is exactly what we would have been looking for,” she said. “I would do it today if it were available,” even after hearing the potential downsides of the technology, she added.
Heather Ward, who has MELAS (mitochondrial encephalopathy, lactic acidosis, and strokelike episodes) and whose daughter was diagnosed with the disease at 18 months, acknowledged that children do bear risk with new techniques such as MRT. But she said that risk must be weighed against the benefits of potentially preventing neurodegenerative mitochondrial diseases in those children.
“It’s very important, if we have the option, to be able to give mothers with MELAS the option to have healthy children. We have the technology. We need to study it,” she said. “And I think it’s a huge disservice to women who have this disease to not give them that option.”
But another meeting participant, Marcy Darnovsky, Ph.D., said that there are two applications of these technologies – human reproductive cloning and human germline modification – which “are so dangerous from a safety and a social standpoint, that they should be taken off the table.”
These genetic techniques should not be evaluated as a medical treatment, where the medical risks are weighed against the medical benefits, because they do not provide a medical benefit but “a very real social benefit,” the ability to have a genetically related child, said Dr. Darnofsky, executive director of the Center for Genetics and Society, in Berkeley, Calif. While the desire of parents should be taken seriously, “they don’t trump everything else,” she added.
Other topics discussed during the meeting included whether egg and sperm donors view themselves as parents, the importance of a direct genetic connection to one’s child, and the concept of procreative liberty. The participants also discussed the unknown risks of manipulating the mitochondria, how to follow-up with children born as a result of MRT, and the risks to young egg donors paid to donate.
The IOM, which is studying the ethical and social policy considerations associated with mitochondrial replacement therapy at the request of the Food and Drug Administration, will hold several more public meetings before issuing a consensus report in early 2016. The regulation of human cells used in therapies that involve the transfer of genetic material “by means other than the union of gamete nuclei” is regulated by the FDA.
WASHINGTON– Despite ethical issues raised by mitochondrial replacement therapy and uncertainties about the potential risks, women with serious mitochondrial diseases appear willing to accept these unknowns for the chance to prevent transmission of the diseases to their biological children.
Mitochondrial replacement therapy (MRT), which was recently granted approval in the United Kingdom, involves removing the nuclear DNA from the egg or fertilized egg of a woman with mitochondrial abnormalities and inserting it into a donor egg with normal mitochondria, after the nuclear DNA of the donor egg has been removed.
An MRT technique that entails removing the mitochondrial DNA from the intended mother’s egg is being evaluated at two centers in the United States, the University of Oregon, Eugene, where it has been performed in macaque monkeys, and the New York Stem Cell Foundation.
The technique, also known as mitochondrial donation, mitochondrial transfer, or mitochondrial DNA replacement, has generated significant controversy, with critics warning that it will lead to “germline enhancements,” eugenics, and designer babies.
But at a 2-day meeting sponsored by the Institute of Medicine (IOM), researchers presented survey data showing that for women who are carriers of these mutations, the ethical concerns are secondary.
In a survey of 92 women who were known carriers of a mitochondrial DNA mutation or were at risk of carrying a mutation, there was universal concern about transmitting mitochondrial DNA mutations to their children, and “overwhelming support and widespread interest” in the clinical use of MRT, particularly among women considering having children at the time of the survey, according to Dr. Michio Hirano, professor of neurology at Columbia University, which collaborates with the New York Stem Cell Foundation on this research.
Almost 80% of the women said they had considered not having children because of the transmission risk and almost three-quarters of those who already had children said they would have considered not having children if they had known they carried the mutation.
Carrying a mutation “has a profound effect on what these women think about their progeny and the risk of transmitting disease,” said Dr. Hirano, who is codirector of the North American Mitochondrial Disease Consortium.
Almost all of the 21 women who were considering having children at the time of the survey said that having a biological child was “very” or “somewhat” important to them, and almost all said they would be interested in the option of MRT. Dr. Hirano, noted that preimplantation genetic diagnosis (PGD), an option often raised as an alternative to MRT, is not always reliable.
Kirah Fasano, a 32-year-old with chronic progressive external ophthalmoplegia, who spoke at the IOM meeting, said that because of her type of mitochondrial disease, PGD was not an option, and after several failed attempts with in-vitro fertilization using her own eggs, she and her husband opted for an egg donor.
She now has a healthy baby, but MRT “is exactly what we would have been looking for,” she said. “I would do it today if it were available,” even after hearing the potential downsides of the technology, she added.
Heather Ward, who has MELAS (mitochondrial encephalopathy, lactic acidosis, and strokelike episodes) and whose daughter was diagnosed with the disease at 18 months, acknowledged that children do bear risk with new techniques such as MRT. But she said that risk must be weighed against the benefits of potentially preventing neurodegenerative mitochondrial diseases in those children.
“It’s very important, if we have the option, to be able to give mothers with MELAS the option to have healthy children. We have the technology. We need to study it,” she said. “And I think it’s a huge disservice to women who have this disease to not give them that option.”
But another meeting participant, Marcy Darnovsky, Ph.D., said that there are two applications of these technologies – human reproductive cloning and human germline modification – which “are so dangerous from a safety and a social standpoint, that they should be taken off the table.”
These genetic techniques should not be evaluated as a medical treatment, where the medical risks are weighed against the medical benefits, because they do not provide a medical benefit but “a very real social benefit,” the ability to have a genetically related child, said Dr. Darnofsky, executive director of the Center for Genetics and Society, in Berkeley, Calif. While the desire of parents should be taken seriously, “they don’t trump everything else,” she added.
Other topics discussed during the meeting included whether egg and sperm donors view themselves as parents, the importance of a direct genetic connection to one’s child, and the concept of procreative liberty. The participants also discussed the unknown risks of manipulating the mitochondria, how to follow-up with children born as a result of MRT, and the risks to young egg donors paid to donate.
The IOM, which is studying the ethical and social policy considerations associated with mitochondrial replacement therapy at the request of the Food and Drug Administration, will hold several more public meetings before issuing a consensus report in early 2016. The regulation of human cells used in therapies that involve the transfer of genetic material “by means other than the union of gamete nuclei” is regulated by the FDA.
WASHINGTON– Despite ethical issues raised by mitochondrial replacement therapy and uncertainties about the potential risks, women with serious mitochondrial diseases appear willing to accept these unknowns for the chance to prevent transmission of the diseases to their biological children.
Mitochondrial replacement therapy (MRT), which was recently granted approval in the United Kingdom, involves removing the nuclear DNA from the egg or fertilized egg of a woman with mitochondrial abnormalities and inserting it into a donor egg with normal mitochondria, after the nuclear DNA of the donor egg has been removed.
An MRT technique that entails removing the mitochondrial DNA from the intended mother’s egg is being evaluated at two centers in the United States, the University of Oregon, Eugene, where it has been performed in macaque monkeys, and the New York Stem Cell Foundation.
The technique, also known as mitochondrial donation, mitochondrial transfer, or mitochondrial DNA replacement, has generated significant controversy, with critics warning that it will lead to “germline enhancements,” eugenics, and designer babies.
But at a 2-day meeting sponsored by the Institute of Medicine (IOM), researchers presented survey data showing that for women who are carriers of these mutations, the ethical concerns are secondary.
In a survey of 92 women who were known carriers of a mitochondrial DNA mutation or were at risk of carrying a mutation, there was universal concern about transmitting mitochondrial DNA mutations to their children, and “overwhelming support and widespread interest” in the clinical use of MRT, particularly among women considering having children at the time of the survey, according to Dr. Michio Hirano, professor of neurology at Columbia University, which collaborates with the New York Stem Cell Foundation on this research.
Almost 80% of the women said they had considered not having children because of the transmission risk and almost three-quarters of those who already had children said they would have considered not having children if they had known they carried the mutation.
Carrying a mutation “has a profound effect on what these women think about their progeny and the risk of transmitting disease,” said Dr. Hirano, who is codirector of the North American Mitochondrial Disease Consortium.
Almost all of the 21 women who were considering having children at the time of the survey said that having a biological child was “very” or “somewhat” important to them, and almost all said they would be interested in the option of MRT. Dr. Hirano, noted that preimplantation genetic diagnosis (PGD), an option often raised as an alternative to MRT, is not always reliable.
Kirah Fasano, a 32-year-old with chronic progressive external ophthalmoplegia, who spoke at the IOM meeting, said that because of her type of mitochondrial disease, PGD was not an option, and after several failed attempts with in-vitro fertilization using her own eggs, she and her husband opted for an egg donor.
She now has a healthy baby, but MRT “is exactly what we would have been looking for,” she said. “I would do it today if it were available,” even after hearing the potential downsides of the technology, she added.
Heather Ward, who has MELAS (mitochondrial encephalopathy, lactic acidosis, and strokelike episodes) and whose daughter was diagnosed with the disease at 18 months, acknowledged that children do bear risk with new techniques such as MRT. But she said that risk must be weighed against the benefits of potentially preventing neurodegenerative mitochondrial diseases in those children.
“It’s very important, if we have the option, to be able to give mothers with MELAS the option to have healthy children. We have the technology. We need to study it,” she said. “And I think it’s a huge disservice to women who have this disease to not give them that option.”
But another meeting participant, Marcy Darnovsky, Ph.D., said that there are two applications of these technologies – human reproductive cloning and human germline modification – which “are so dangerous from a safety and a social standpoint, that they should be taken off the table.”
These genetic techniques should not be evaluated as a medical treatment, where the medical risks are weighed against the medical benefits, because they do not provide a medical benefit but “a very real social benefit,” the ability to have a genetically related child, said Dr. Darnofsky, executive director of the Center for Genetics and Society, in Berkeley, Calif. While the desire of parents should be taken seriously, “they don’t trump everything else,” she added.
Other topics discussed during the meeting included whether egg and sperm donors view themselves as parents, the importance of a direct genetic connection to one’s child, and the concept of procreative liberty. The participants also discussed the unknown risks of manipulating the mitochondria, how to follow-up with children born as a result of MRT, and the risks to young egg donors paid to donate.
The IOM, which is studying the ethical and social policy considerations associated with mitochondrial replacement therapy at the request of the Food and Drug Administration, will hold several more public meetings before issuing a consensus report in early 2016. The regulation of human cells used in therapies that involve the transfer of genetic material “by means other than the union of gamete nuclei” is regulated by the FDA.
AT A MEETING OF THE INSTITUTE OF MEDICINE
Alogliptin CV risk acceptable, FDA panel agrees
SILVER SPRING, MD.– Alogliptin’s cardiovascular risk profile is acceptably safe for high-risk patients with type 2 diabetes, a Food and Drug Administration advisory panel has unanimously agreed.
At a meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee on April 14, panelists reviewed the results of the results of a large cardiovascular outcomes study of the dipeptidyl peptidase-4 (DPP-4) inhibitor, the Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care (EXAMINE) study, an international, randomized, double-blind, placebo-controlled trial of about 5,400 people with type 2 diabetes and established cardiovascular disease.
The FDA is requiring CV outcomes trials for all type 2 diabetes drugs, including those in development, and issued a guidance for industry for these trials in December 2008, which states that the trials should evaluate the major adverse cardiovascular event (MACE) incidence in patients at increased risk for CVD and that a 30% or greater excess CV risk over placebo should be excluded to meet the criteria for acceptable CV safety.
Alogliptin, marketed as Nesina by Takeda Pharmaceutical USA, was approved in January 2013. It also is combined with metformin (Kazano) and with pioglitazone (Oseni). The EXAMINE study, which was underway at the time of approval, enrolled people who had an acute coronary syndrome event (acute MI or unstable angina requiring hospitalization) within 15-90 days of enrollment.
Over a median follow-up of 1.5 years, there was no increase in the MACE composite endpoint of CV death, nonfatal MI, and nonfatal stroke among those on alogliptin (11.3%), compared with those on placebo (11.8%), for a hazard ratio of 0.96 – meeting the FDA criteria for CV safety (N. Engl. J. Med. 2013;369:1327-35).
Some of the issues raised with the study included a lower-than-planned proportion of patients enrolled in the United States and Canada – less than 16% of the total – and a subgroup analysis showing that, in the North American patients, the MACE rate among those on alogliptin was elevated compared with placebo (HR, 1.28). The FDA reviewers said that the North American population had some characteristics that might explain this difference.
Another finding discussed was the higher number of hospitalizations for heart failure in patients on alogliptin (106) than for those on placebo (89), for a rate of 2.6 vs. 2.3 cases per 100 patient-years (HR, 1.19), according to the FDA. In the CV outcomes study of another DPP-4 inhibitor, saxagliptin, which the panel reviewed earlier in the day, there was a similar signal for an increased risk of heart failure hospitalizations associated with the drug.
Panelists, however, said they were not convinced that the geographic differences represented a real effect, which they said could be due to chance and did not affect the overall results – although they would have preferred to have more people from the United States enrolled in the study. Panelists also were not overly concerned about the risk of heart failure, but they pointed out that the risk of heart failure risk should be monitored in patients on the drug because of the signal seen with saxagliptin. Several panelists said that they suspected that heart failure may turn out to be a class effect. CV outcomes trials for the two other approved DPP-4 inhibitors – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing.
“It’s clear that the trial achieved its primary objective in a manner consistent with the 2008 guidelines,” said one of the panelists, Dr. Thomas Wang, director of the division of cardiovascular medicine at Vanderbilt University, Nashville, Tenn. Although there were some limitations of the study, such as the low proportion of U.S. participants and the relatively short duration of follow-up, “these limitations do not come close to negating the conclusion” regarding the cardiovascular risk profile, he commented.
In another vote, 13 of the 16 panelists said that the new safety information should be added to the alogliptin prescribing information. “This trial represented a huge effort and we know a lot about the outcomes related to this drug now,” and that information should be included in the label, said one of the panelists, Dr. Susan Heckbert, professor in the department of epidemiology, University of Washington, Seattle.
Three panelists voted not to add any information to the label. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest
SILVER SPRING, MD.– Alogliptin’s cardiovascular risk profile is acceptably safe for high-risk patients with type 2 diabetes, a Food and Drug Administration advisory panel has unanimously agreed.
At a meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee on April 14, panelists reviewed the results of the results of a large cardiovascular outcomes study of the dipeptidyl peptidase-4 (DPP-4) inhibitor, the Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care (EXAMINE) study, an international, randomized, double-blind, placebo-controlled trial of about 5,400 people with type 2 diabetes and established cardiovascular disease.
The FDA is requiring CV outcomes trials for all type 2 diabetes drugs, including those in development, and issued a guidance for industry for these trials in December 2008, which states that the trials should evaluate the major adverse cardiovascular event (MACE) incidence in patients at increased risk for CVD and that a 30% or greater excess CV risk over placebo should be excluded to meet the criteria for acceptable CV safety.
Alogliptin, marketed as Nesina by Takeda Pharmaceutical USA, was approved in January 2013. It also is combined with metformin (Kazano) and with pioglitazone (Oseni). The EXAMINE study, which was underway at the time of approval, enrolled people who had an acute coronary syndrome event (acute MI or unstable angina requiring hospitalization) within 15-90 days of enrollment.
Over a median follow-up of 1.5 years, there was no increase in the MACE composite endpoint of CV death, nonfatal MI, and nonfatal stroke among those on alogliptin (11.3%), compared with those on placebo (11.8%), for a hazard ratio of 0.96 – meeting the FDA criteria for CV safety (N. Engl. J. Med. 2013;369:1327-35).
Some of the issues raised with the study included a lower-than-planned proportion of patients enrolled in the United States and Canada – less than 16% of the total – and a subgroup analysis showing that, in the North American patients, the MACE rate among those on alogliptin was elevated compared with placebo (HR, 1.28). The FDA reviewers said that the North American population had some characteristics that might explain this difference.
Another finding discussed was the higher number of hospitalizations for heart failure in patients on alogliptin (106) than for those on placebo (89), for a rate of 2.6 vs. 2.3 cases per 100 patient-years (HR, 1.19), according to the FDA. In the CV outcomes study of another DPP-4 inhibitor, saxagliptin, which the panel reviewed earlier in the day, there was a similar signal for an increased risk of heart failure hospitalizations associated with the drug.
Panelists, however, said they were not convinced that the geographic differences represented a real effect, which they said could be due to chance and did not affect the overall results – although they would have preferred to have more people from the United States enrolled in the study. Panelists also were not overly concerned about the risk of heart failure, but they pointed out that the risk of heart failure risk should be monitored in patients on the drug because of the signal seen with saxagliptin. Several panelists said that they suspected that heart failure may turn out to be a class effect. CV outcomes trials for the two other approved DPP-4 inhibitors – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing.
“It’s clear that the trial achieved its primary objective in a manner consistent with the 2008 guidelines,” said one of the panelists, Dr. Thomas Wang, director of the division of cardiovascular medicine at Vanderbilt University, Nashville, Tenn. Although there were some limitations of the study, such as the low proportion of U.S. participants and the relatively short duration of follow-up, “these limitations do not come close to negating the conclusion” regarding the cardiovascular risk profile, he commented.
In another vote, 13 of the 16 panelists said that the new safety information should be added to the alogliptin prescribing information. “This trial represented a huge effort and we know a lot about the outcomes related to this drug now,” and that information should be included in the label, said one of the panelists, Dr. Susan Heckbert, professor in the department of epidemiology, University of Washington, Seattle.
Three panelists voted not to add any information to the label. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest
SILVER SPRING, MD.– Alogliptin’s cardiovascular risk profile is acceptably safe for high-risk patients with type 2 diabetes, a Food and Drug Administration advisory panel has unanimously agreed.
At a meeting of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee on April 14, panelists reviewed the results of the results of a large cardiovascular outcomes study of the dipeptidyl peptidase-4 (DPP-4) inhibitor, the Examination of Cardiovascular Outcomes with Alogliptin versus Standard of Care (EXAMINE) study, an international, randomized, double-blind, placebo-controlled trial of about 5,400 people with type 2 diabetes and established cardiovascular disease.
The FDA is requiring CV outcomes trials for all type 2 diabetes drugs, including those in development, and issued a guidance for industry for these trials in December 2008, which states that the trials should evaluate the major adverse cardiovascular event (MACE) incidence in patients at increased risk for CVD and that a 30% or greater excess CV risk over placebo should be excluded to meet the criteria for acceptable CV safety.
Alogliptin, marketed as Nesina by Takeda Pharmaceutical USA, was approved in January 2013. It also is combined with metformin (Kazano) and with pioglitazone (Oseni). The EXAMINE study, which was underway at the time of approval, enrolled people who had an acute coronary syndrome event (acute MI or unstable angina requiring hospitalization) within 15-90 days of enrollment.
Over a median follow-up of 1.5 years, there was no increase in the MACE composite endpoint of CV death, nonfatal MI, and nonfatal stroke among those on alogliptin (11.3%), compared with those on placebo (11.8%), for a hazard ratio of 0.96 – meeting the FDA criteria for CV safety (N. Engl. J. Med. 2013;369:1327-35).
Some of the issues raised with the study included a lower-than-planned proportion of patients enrolled in the United States and Canada – less than 16% of the total – and a subgroup analysis showing that, in the North American patients, the MACE rate among those on alogliptin was elevated compared with placebo (HR, 1.28). The FDA reviewers said that the North American population had some characteristics that might explain this difference.
Another finding discussed was the higher number of hospitalizations for heart failure in patients on alogliptin (106) than for those on placebo (89), for a rate of 2.6 vs. 2.3 cases per 100 patient-years (HR, 1.19), according to the FDA. In the CV outcomes study of another DPP-4 inhibitor, saxagliptin, which the panel reviewed earlier in the day, there was a similar signal for an increased risk of heart failure hospitalizations associated with the drug.
Panelists, however, said they were not convinced that the geographic differences represented a real effect, which they said could be due to chance and did not affect the overall results – although they would have preferred to have more people from the United States enrolled in the study. Panelists also were not overly concerned about the risk of heart failure, but they pointed out that the risk of heart failure risk should be monitored in patients on the drug because of the signal seen with saxagliptin. Several panelists said that they suspected that heart failure may turn out to be a class effect. CV outcomes trials for the two other approved DPP-4 inhibitors – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing.
“It’s clear that the trial achieved its primary objective in a manner consistent with the 2008 guidelines,” said one of the panelists, Dr. Thomas Wang, director of the division of cardiovascular medicine at Vanderbilt University, Nashville, Tenn. Although there were some limitations of the study, such as the low proportion of U.S. participants and the relatively short duration of follow-up, “these limitations do not come close to negating the conclusion” regarding the cardiovascular risk profile, he commented.
In another vote, 13 of the 16 panelists said that the new safety information should be added to the alogliptin prescribing information. “This trial represented a huge effort and we know a lot about the outcomes related to this drug now,” and that information should be included in the label, said one of the panelists, Dr. Susan Heckbert, professor in the department of epidemiology, University of Washington, Seattle.
Three panelists voted not to add any information to the label. The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest
AT AN FDA ADVISORY COMMITTEE MEETING
FDA panel reassured by saxagliptin’s CV safety data
SILVER SPRING, MD.– Results of a large postmarketing study evaluating the cardiovascular safety of saxagliptin in a high-risk population provided reassuring evidence about the drug’s overall cardiovascular risk, although a signal for heart failure hospitalizations associated with the drug was a concern, according to a Food and Drug Administration advisory panel.
At a meeting on April 14, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1, with one abstention, that the results of the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus) trial showed that the use of saxagliptin in patients with type 2 diabetes had an acceptable cardiovascular risk profile. However, the panelists were concerned about the signal for an increased risk of hospitalizations for heart failure associated with saxagliptin in the study, which they agreed should be studied further. Nearly all of the panelists also voted that the safety data, including the heart failure finding and an imbalance in all-cause mortality among those on saxagliptin arm of the study, be added to the drug’s prescribing information.

Approved in 2009, saxagliptin, a dipeptidyl peptidase–4 (DPP4) inhibitor, is indicated “as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus in multiple clinical settings” and is marketed as Onglyza by AstraZeneca. A fixed-dose combination of saxagliptin with extended-release metformin (Kombiglyze XR) was approved in 2010. The FDA panel votes apply to both products.
The FDA is requiring that manufacturers conduct CV outcomes trials for all type 2 diabetes drugs in patients at high risk for cardiovascular disease (CVD), and in December 2008, issued a guidance document for industry, outlining the requirements for these studies, which included showing that the CVD risk, based on a major adverse cardiovascular event (MACE) endpoint, is not increased by more than 30% compared to placebo.
AstraZeneca conducted SAVOR, a prospective, randomized, double-blind, placebo-controlled study of nearly 16,500 people with type 2 diabetes, who had or were at risk of CVD, comparing saxagliptin to placebo, on top of standard treatment (N. Engl. J. Med. 2013;369:1317-26). After a median follow-up of about 2 years, the incidence of MACE (a composite of cardiovascular death, nonfatal MI, and nonfatal ischemic stroke) was identical in both groups, at 7.4%, with a hazard ratio of 1.0, meeting the FDA criteria for cardiovascular safety as outlined in the guidance, according to the company. Saxagliptin was not superior to placebo in reducing the MACE events, another primary objective of the study.
Unexpectedly, hospitalization for heart failure (HF), a component of the secondary composite endpoint, was increased among those treated with saxagliptin (hazard ratio, 1.27). There was also a “numerical imbalance” in all-cause mortality between groups, with 17 deaths in the saxagliptin patients vs. 11 in the placebo group (HR, 1.11).
However, deaths caused by heart failure were balanced between those on saxagliptin and those on placebo, and no possible mechanism for the HF finding could be identified, according to AstraZeneca, which is planning a mechanistic study to evaluate the effects of saxagliptin on volume, neurohormonal changes, and cardiac function. The company has proposed that the HF finding be addressed by adding this information to the prescribing information.
The cardiovascular safety results were reassuring, said Dr. Morris Schambelan, a panelist and professor emeritus of medicine in the division of endocrinology at the University of California, San Francisco, but like others on the committee, added that he believes the heart failure signal was real and that providing clinicians with information in the drug’s label to help predict those at higher risk in would be helpful.
Several panelists were somewhat concerned about the all-cause mortality finding, which is a strong endpoint, and therefore cannot be ruled out, they said. But they also pointed out that follow-up was relatively short for a drug that is used long term and that the results should be applied cautiously to patients at lower risk than were those enrolled in the trial.
The FDA usually follows the recommendations of its advisory panel members. Panelists had no relevant disclosures.
CV outcomes trials for other approved DPP4 inhibitors approved for type 2 diabetes – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing. The panel review of the CV outcomes trial for another DPP4 inhibitor approved, alogliptin (Nesina), followed the meeting on saxagliptin.
SILVER SPRING, MD.– Results of a large postmarketing study evaluating the cardiovascular safety of saxagliptin in a high-risk population provided reassuring evidence about the drug’s overall cardiovascular risk, although a signal for heart failure hospitalizations associated with the drug was a concern, according to a Food and Drug Administration advisory panel.
At a meeting on April 14, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1, with one abstention, that the results of the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus) trial showed that the use of saxagliptin in patients with type 2 diabetes had an acceptable cardiovascular risk profile. However, the panelists were concerned about the signal for an increased risk of hospitalizations for heart failure associated with saxagliptin in the study, which they agreed should be studied further. Nearly all of the panelists also voted that the safety data, including the heart failure finding and an imbalance in all-cause mortality among those on saxagliptin arm of the study, be added to the drug’s prescribing information.
Approved in 2009, saxagliptin, a dipeptidyl peptidase–4 (DPP4) inhibitor, is indicated “as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus in multiple clinical settings” and is marketed as Onglyza by AstraZeneca. A fixed-dose combination of saxagliptin with extended-release metformin (Kombiglyze XR) was approved in 2010. The FDA panel votes apply to both products.
The FDA is requiring that manufacturers conduct CV outcomes trials for all type 2 diabetes drugs in patients at high risk for cardiovascular disease (CVD), and in December 2008, issued a guidance document for industry, outlining the requirements for these studies, which included showing that the CVD risk, based on a major adverse cardiovascular event (MACE) endpoint, is not increased by more than 30% compared to placebo.
AstraZeneca conducted SAVOR, a prospective, randomized, double-blind, placebo-controlled study of nearly 16,500 people with type 2 diabetes, who had or were at risk of CVD, comparing saxagliptin to placebo, on top of standard treatment (N. Engl. J. Med. 2013;369:1317-26). After a median follow-up of about 2 years, the incidence of MACE (a composite of cardiovascular death, nonfatal MI, and nonfatal ischemic stroke) was identical in both groups, at 7.4%, with a hazard ratio of 1.0, meeting the FDA criteria for cardiovascular safety as outlined in the guidance, according to the company. Saxagliptin was not superior to placebo in reducing the MACE events, another primary objective of the study.
Unexpectedly, hospitalization for heart failure (HF), a component of the secondary composite endpoint, was increased among those treated with saxagliptin (hazard ratio, 1.27). There was also a “numerical imbalance” in all-cause mortality between groups, with 17 deaths in the saxagliptin patients vs. 11 in the placebo group (HR, 1.11).
However, deaths caused by heart failure were balanced between those on saxagliptin and those on placebo, and no possible mechanism for the HF finding could be identified, according to AstraZeneca, which is planning a mechanistic study to evaluate the effects of saxagliptin on volume, neurohormonal changes, and cardiac function. The company has proposed that the HF finding be addressed by adding this information to the prescribing information.
The cardiovascular safety results were reassuring, said Dr. Morris Schambelan, a panelist and professor emeritus of medicine in the division of endocrinology at the University of California, San Francisco, but like others on the committee, added that he believes the heart failure signal was real and that providing clinicians with information in the drug’s label to help predict those at higher risk in would be helpful.
Several panelists were somewhat concerned about the all-cause mortality finding, which is a strong endpoint, and therefore cannot be ruled out, they said. But they also pointed out that follow-up was relatively short for a drug that is used long term and that the results should be applied cautiously to patients at lower risk than were those enrolled in the trial.
The FDA usually follows the recommendations of its advisory panel members. Panelists had no relevant disclosures.
CV outcomes trials for other approved DPP4 inhibitors approved for type 2 diabetes – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing. The panel review of the CV outcomes trial for another DPP4 inhibitor approved, alogliptin (Nesina), followed the meeting on saxagliptin.
SILVER SPRING, MD.– Results of a large postmarketing study evaluating the cardiovascular safety of saxagliptin in a high-risk population provided reassuring evidence about the drug’s overall cardiovascular risk, although a signal for heart failure hospitalizations associated with the drug was a concern, according to a Food and Drug Administration advisory panel.
At a meeting on April 14, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted 13-1, with one abstention, that the results of the SAVOR (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients With Diabetes Mellitus) trial showed that the use of saxagliptin in patients with type 2 diabetes had an acceptable cardiovascular risk profile. However, the panelists were concerned about the signal for an increased risk of hospitalizations for heart failure associated with saxagliptin in the study, which they agreed should be studied further. Nearly all of the panelists also voted that the safety data, including the heart failure finding and an imbalance in all-cause mortality among those on saxagliptin arm of the study, be added to the drug’s prescribing information.
Approved in 2009, saxagliptin, a dipeptidyl peptidase–4 (DPP4) inhibitor, is indicated “as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus in multiple clinical settings” and is marketed as Onglyza by AstraZeneca. A fixed-dose combination of saxagliptin with extended-release metformin (Kombiglyze XR) was approved in 2010. The FDA panel votes apply to both products.
The FDA is requiring that manufacturers conduct CV outcomes trials for all type 2 diabetes drugs in patients at high risk for cardiovascular disease (CVD), and in December 2008, issued a guidance document for industry, outlining the requirements for these studies, which included showing that the CVD risk, based on a major adverse cardiovascular event (MACE) endpoint, is not increased by more than 30% compared to placebo.
AstraZeneca conducted SAVOR, a prospective, randomized, double-blind, placebo-controlled study of nearly 16,500 people with type 2 diabetes, who had or were at risk of CVD, comparing saxagliptin to placebo, on top of standard treatment (N. Engl. J. Med. 2013;369:1317-26). After a median follow-up of about 2 years, the incidence of MACE (a composite of cardiovascular death, nonfatal MI, and nonfatal ischemic stroke) was identical in both groups, at 7.4%, with a hazard ratio of 1.0, meeting the FDA criteria for cardiovascular safety as outlined in the guidance, according to the company. Saxagliptin was not superior to placebo in reducing the MACE events, another primary objective of the study.
Unexpectedly, hospitalization for heart failure (HF), a component of the secondary composite endpoint, was increased among those treated with saxagliptin (hazard ratio, 1.27). There was also a “numerical imbalance” in all-cause mortality between groups, with 17 deaths in the saxagliptin patients vs. 11 in the placebo group (HR, 1.11).
However, deaths caused by heart failure were balanced between those on saxagliptin and those on placebo, and no possible mechanism for the HF finding could be identified, according to AstraZeneca, which is planning a mechanistic study to evaluate the effects of saxagliptin on volume, neurohormonal changes, and cardiac function. The company has proposed that the HF finding be addressed by adding this information to the prescribing information.
The cardiovascular safety results were reassuring, said Dr. Morris Schambelan, a panelist and professor emeritus of medicine in the division of endocrinology at the University of California, San Francisco, but like others on the committee, added that he believes the heart failure signal was real and that providing clinicians with information in the drug’s label to help predict those at higher risk in would be helpful.
Several panelists were somewhat concerned about the all-cause mortality finding, which is a strong endpoint, and therefore cannot be ruled out, they said. But they also pointed out that follow-up was relatively short for a drug that is used long term and that the results should be applied cautiously to patients at lower risk than were those enrolled in the trial.
The FDA usually follows the recommendations of its advisory panel members. Panelists had no relevant disclosures.
CV outcomes trials for other approved DPP4 inhibitors approved for type 2 diabetes – sitagliptin (Januvia) and linagliptin (Tradjenta) – are ongoing. The panel review of the CV outcomes trial for another DPP4 inhibitor approved, alogliptin (Nesina), followed the meeting on saxagliptin.
AT AN FDA ADVISORY COMMITTEE MEETING

Several genetic variants linked to salivary gland cancer risk
Several single nucleotide polymorphisms were found to be significantly associated with salivary gland carcinoma in a genomewide association study, Li Xu, Ph.D., and associates at the University of Texas MD Anderson Cancer Center, Houston, reported online in Cancer.
The results of what they believe is the first such study conducted to identify common genetic variants associated with salivary gland carcinoma (SGC) need to be confirmed, but indicate that these single nucleotide polymorphisms (SNPs) could be further evaluated as possible screening tools for the rare cancer, they wrote (Cancer 2015 March 30 [doi:10.1002/cncr.29381]).
The study involved genotyping analyses of 309 cases of SGC in patients (mean age, 54 years) who had been treated at MD Anderson from September 2001 through February 2014; and 535 controls (mean age, 51 years), patients with no cancer.
Five SNPs were found to be associated with SGC risk. “The finding that the five novel SNPs associated with SGC risk are coding SNPs with functional potential, the finding that the genetic effects were considerable,” and the finding that the two SNPs with the strongest SGC risk were relatively rare among controls, “support that these five SNPs may be good candidate SNPs for SGC screening and prevention,” the authors concluded. “These findings support the existence of genetic heterogeneity between histological subtypes of SGC and provide a set of candidate SNPs and genes worthy of in-depth evaluation in future studies,” they added.
The small size of the study was among the limitations, and the results need to be confirmed in another study that also analyzes the function of the SNPs identified in the study, they noted.
In the United States, salivary gland carcinomas are rare, and account for about 0.3% of all malignancies, with an annual incidence of about 1 case per 100,000 people, Dr. Xu and associates said. A small proportion of SGCs are associated with high-dose ionizing radiation, the only well-documented environmental factor associated with the cancer, “suggesting a genetic role in the development of SGC,” they pointed out.
The authors had no disclosures. The study was partly supported by funds from the University of Texas MD Anderson Cancer Center, tge National Institutes of Health grants, and a Cancer Center Support grant.
Several single nucleotide polymorphisms were found to be significantly associated with salivary gland carcinoma in a genomewide association study, Li Xu, Ph.D., and associates at the University of Texas MD Anderson Cancer Center, Houston, reported online in Cancer.
The results of what they believe is the first such study conducted to identify common genetic variants associated with salivary gland carcinoma (SGC) need to be confirmed, but indicate that these single nucleotide polymorphisms (SNPs) could be further evaluated as possible screening tools for the rare cancer, they wrote (Cancer 2015 March 30 [doi:10.1002/cncr.29381]).
The study involved genotyping analyses of 309 cases of SGC in patients (mean age, 54 years) who had been treated at MD Anderson from September 2001 through February 2014; and 535 controls (mean age, 51 years), patients with no cancer.
Five SNPs were found to be associated with SGC risk. “The finding that the five novel SNPs associated with SGC risk are coding SNPs with functional potential, the finding that the genetic effects were considerable,” and the finding that the two SNPs with the strongest SGC risk were relatively rare among controls, “support that these five SNPs may be good candidate SNPs for SGC screening and prevention,” the authors concluded. “These findings support the existence of genetic heterogeneity between histological subtypes of SGC and provide a set of candidate SNPs and genes worthy of in-depth evaluation in future studies,” they added.
The small size of the study was among the limitations, and the results need to be confirmed in another study that also analyzes the function of the SNPs identified in the study, they noted.
In the United States, salivary gland carcinomas are rare, and account for about 0.3% of all malignancies, with an annual incidence of about 1 case per 100,000 people, Dr. Xu and associates said. A small proportion of SGCs are associated with high-dose ionizing radiation, the only well-documented environmental factor associated with the cancer, “suggesting a genetic role in the development of SGC,” they pointed out.
The authors had no disclosures. The study was partly supported by funds from the University of Texas MD Anderson Cancer Center, tge National Institutes of Health grants, and a Cancer Center Support grant.
Several single nucleotide polymorphisms were found to be significantly associated with salivary gland carcinoma in a genomewide association study, Li Xu, Ph.D., and associates at the University of Texas MD Anderson Cancer Center, Houston, reported online in Cancer.
The results of what they believe is the first such study conducted to identify common genetic variants associated with salivary gland carcinoma (SGC) need to be confirmed, but indicate that these single nucleotide polymorphisms (SNPs) could be further evaluated as possible screening tools for the rare cancer, they wrote (Cancer 2015 March 30 [doi:10.1002/cncr.29381]).
The study involved genotyping analyses of 309 cases of SGC in patients (mean age, 54 years) who had been treated at MD Anderson from September 2001 through February 2014; and 535 controls (mean age, 51 years), patients with no cancer.
Five SNPs were found to be associated with SGC risk. “The finding that the five novel SNPs associated with SGC risk are coding SNPs with functional potential, the finding that the genetic effects were considerable,” and the finding that the two SNPs with the strongest SGC risk were relatively rare among controls, “support that these five SNPs may be good candidate SNPs for SGC screening and prevention,” the authors concluded. “These findings support the existence of genetic heterogeneity between histological subtypes of SGC and provide a set of candidate SNPs and genes worthy of in-depth evaluation in future studies,” they added.
The small size of the study was among the limitations, and the results need to be confirmed in another study that also analyzes the function of the SNPs identified in the study, they noted.
In the United States, salivary gland carcinomas are rare, and account for about 0.3% of all malignancies, with an annual incidence of about 1 case per 100,000 people, Dr. Xu and associates said. A small proportion of SGCs are associated with high-dose ionizing radiation, the only well-documented environmental factor associated with the cancer, “suggesting a genetic role in the development of SGC,” they pointed out.
The authors had no disclosures. The study was partly supported by funds from the University of Texas MD Anderson Cancer Center, tge National Institutes of Health grants, and a Cancer Center Support grant.
Key clinical point: The identification of several gene variants associated with salivary gland carcinoma could eventually lead to screening tests for people at high risk for developing this rare cancer.
Major finding: Five different single nucleotide polymorphisms (SNPs) were significantly associated with salivary gland carcinomas.
Data source: A case-control genomewide association study looked for associations with salivary gland carcinomas, comparing results in 309 patients with salivary gland carcinoma and 535 controls with no cancer.
Disclosures: The authors had no disclosures. The study was partly supported by funds from the University of Texas MD Anderson Cancer Center, grants from the National Institutes of Health, and a Cancer Center Support grant.
FDA strengthens allergy warning for IV anemia therapy ferumoxytol
The warning about potentially life-threatening allergic reactions with the intravenous anemia drug ferumoxytol is being strengthened to reflect cases of allergic reactions, including fatalities, associated with this drug since approval, the Food and Drug Administration announced on March 30.
Information about the risk of potentially life-threatening allergic reactions was included in the warnings and precautions section of the prescribing information for ferumoxytol (Feraheme) when it was approved in 2009, and this risk is associated with all IV iron therapies. Since approval of ferumoxytol, however, “serious reactions, including deaths, have occurred despite the proper use of therapies to treat these reactions and emergency resuscitation measures,” according to the FDA’s statement, a drug safety communication.
Ways to reduce this risk have been identified and “health care professionals should follow the new recommendations in the drug label,” which includes the new contraindication, a “strong recommendation against use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product,” the statement said.
Other recommendations included in the revised prescribing information include instructions to administer diluted ferumoxytol in an IV infusion administered over at least 15 minutes and to never administer it as an undiluted solution; to closely monitor patients for signs and symptoms of serious allergic reactions, which includes monitoring pulse and blood pressure during administration and for at least 30 minutes after each infusion; and to “carefully consider the potential risks and benefits” of the drug in elderly patients and in patients who have multiple drug allergies, two groups of patients who may be at increased risk for serious reactions.
Between June 2009 and June 30, 2014, 79 reports of anaphylactic reactions associated with ferumoxytol were identified in people from aged 19-96 years in the FDA Adverse Event Reporting System database, of which 18 were fatal, “despite immediate medical intervention and emergency resuscitation attempts. Almost half were reported with the first dose and 60 cases – about 75% – were reported to have started during the infusion or within 5 minutes after it had been fully infused.
Of the 79 cases, 34 (43%) were reported in people with a history of drug allergy; and 24% had a history of multiple drug allergies. Cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing were among the common symptoms described.
Serious adverse events associated with ferumoxytol or other IV iron therapies should be reported to the FDA’s MedWatch program at 800-332-1099 or on-line at https://www.accessdata.fda.gov/scripts/medwatch/.
The warning about potentially life-threatening allergic reactions with the intravenous anemia drug ferumoxytol is being strengthened to reflect cases of allergic reactions, including fatalities, associated with this drug since approval, the Food and Drug Administration announced on March 30.
Information about the risk of potentially life-threatening allergic reactions was included in the warnings and precautions section of the prescribing information for ferumoxytol (Feraheme) when it was approved in 2009, and this risk is associated with all IV iron therapies. Since approval of ferumoxytol, however, “serious reactions, including deaths, have occurred despite the proper use of therapies to treat these reactions and emergency resuscitation measures,” according to the FDA’s statement, a drug safety communication.
Ways to reduce this risk have been identified and “health care professionals should follow the new recommendations in the drug label,” which includes the new contraindication, a “strong recommendation against use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product,” the statement said.
Other recommendations included in the revised prescribing information include instructions to administer diluted ferumoxytol in an IV infusion administered over at least 15 minutes and to never administer it as an undiluted solution; to closely monitor patients for signs and symptoms of serious allergic reactions, which includes monitoring pulse and blood pressure during administration and for at least 30 minutes after each infusion; and to “carefully consider the potential risks and benefits” of the drug in elderly patients and in patients who have multiple drug allergies, two groups of patients who may be at increased risk for serious reactions.
Between June 2009 and June 30, 2014, 79 reports of anaphylactic reactions associated with ferumoxytol were identified in people from aged 19-96 years in the FDA Adverse Event Reporting System database, of which 18 were fatal, “despite immediate medical intervention and emergency resuscitation attempts. Almost half were reported with the first dose and 60 cases – about 75% – were reported to have started during the infusion or within 5 minutes after it had been fully infused.
Of the 79 cases, 34 (43%) were reported in people with a history of drug allergy; and 24% had a history of multiple drug allergies. Cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing were among the common symptoms described.
Serious adverse events associated with ferumoxytol or other IV iron therapies should be reported to the FDA’s MedWatch program at 800-332-1099 or on-line at https://www.accessdata.fda.gov/scripts/medwatch/.
The warning about potentially life-threatening allergic reactions with the intravenous anemia drug ferumoxytol is being strengthened to reflect cases of allergic reactions, including fatalities, associated with this drug since approval, the Food and Drug Administration announced on March 30.
Information about the risk of potentially life-threatening allergic reactions was included in the warnings and precautions section of the prescribing information for ferumoxytol (Feraheme) when it was approved in 2009, and this risk is associated with all IV iron therapies. Since approval of ferumoxytol, however, “serious reactions, including deaths, have occurred despite the proper use of therapies to treat these reactions and emergency resuscitation measures,” according to the FDA’s statement, a drug safety communication.
Ways to reduce this risk have been identified and “health care professionals should follow the new recommendations in the drug label,” which includes the new contraindication, a “strong recommendation against use of Feraheme in patients who have had an allergic reaction to any intravenous (IV) iron replacement product,” the statement said.
Other recommendations included in the revised prescribing information include instructions to administer diluted ferumoxytol in an IV infusion administered over at least 15 minutes and to never administer it as an undiluted solution; to closely monitor patients for signs and symptoms of serious allergic reactions, which includes monitoring pulse and blood pressure during administration and for at least 30 minutes after each infusion; and to “carefully consider the potential risks and benefits” of the drug in elderly patients and in patients who have multiple drug allergies, two groups of patients who may be at increased risk for serious reactions.
Between June 2009 and June 30, 2014, 79 reports of anaphylactic reactions associated with ferumoxytol were identified in people from aged 19-96 years in the FDA Adverse Event Reporting System database, of which 18 were fatal, “despite immediate medical intervention and emergency resuscitation attempts. Almost half were reported with the first dose and 60 cases – about 75% – were reported to have started during the infusion or within 5 minutes after it had been fully infused.
Of the 79 cases, 34 (43%) were reported in people with a history of drug allergy; and 24% had a history of multiple drug allergies. Cardiac arrest, hypotension, dyspnea, nausea, vomiting, and flushing were among the common symptoms described.
Serious adverse events associated with ferumoxytol or other IV iron therapies should be reported to the FDA’s MedWatch program at 800-332-1099 or on-line at https://www.accessdata.fda.gov/scripts/medwatch/.
Manufacturer releases new reprocessing instructions for TJF-Q180V duodenoscope
Olympus, the manufacturer of the TJF-Q180V duodenoscope, has issued new, validated instructions for reprocessing this particular model, as part of the response to recent reports of a possible association between multidrug-resistant bacterial infections and improperly processed duodenoscopes, according to the Food and Drug Administration.
The new instructions, which replace the manual reprocessing instructions included in the original labeling, and validation data have been reviewed by the FDA as part of its ongoing review of the device. The agency “recommends that any facilities that are using Olympus’ TJF-Q180V duodenoscope train staff on the new instructions and implement them as soon as possible,” according to an FDA statement. The instructions are provided in letters sent by Olympus to health care and other facilities that use this particular model.
“Key changes” have been made to the procedures for precleaning, manual cleaning, and manual high-level disinfection reprocessing procedures, the FDA said.
The TJF-Q180 V duodenoscope was the model used in four patients who had undergone an endoscopic retrograde cholangiopancreatography (ERCP) procedure between August 2014 and January 2015 with the same duodenoscope at Cedars-Sinai Medical Center in Los Angeles and had been infected with carbapenem-resistant Enterobacteriaceae (CRE). This outbreak was announced by the medical center in early March in a statement that said the infections occurred “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes” recommended in instructions provided by Olympus and the FDA.
In February, the FDA first announced that the agency had received reports of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures with duodenoscopes, despite proper cleaning and disinfection of the devices and that the “complex design of ERCP endoscopes (also called duodenoscopes) may impede effective reprocessing.”
In the latest statement, the FDA said it “is closely monitoring the possible association between reprocessed duodenoscopes and the transmission of infectious agents,” including multidrug-resistant bacterial infections caused by CRE. If they are not properly reprocessed, the statement adds, “residual body fluids and organic debris may remain in microscopic crevices of the device following an attempted cleaning and high-level disinfection. If these residual fluids contain microbial contamination, subsequent patients may be exposed to serious infections.”
Adverse events associated with duodenoscopes should be reported to the FDA’s MedWatch Program at 800-332-1088 or www.accessdata.fda.gov/scripts/medwatch.
AGA Resource
Read more about AGA’s efforts and recommendations to stop duodenoscope infections here.
Olympus, the manufacturer of the TJF-Q180V duodenoscope, has issued new, validated instructions for reprocessing this particular model, as part of the response to recent reports of a possible association between multidrug-resistant bacterial infections and improperly processed duodenoscopes, according to the Food and Drug Administration.
The new instructions, which replace the manual reprocessing instructions included in the original labeling, and validation data have been reviewed by the FDA as part of its ongoing review of the device. The agency “recommends that any facilities that are using Olympus’ TJF-Q180V duodenoscope train staff on the new instructions and implement them as soon as possible,” according to an FDA statement. The instructions are provided in letters sent by Olympus to health care and other facilities that use this particular model.
“Key changes” have been made to the procedures for precleaning, manual cleaning, and manual high-level disinfection reprocessing procedures, the FDA said.
The TJF-Q180 V duodenoscope was the model used in four patients who had undergone an endoscopic retrograde cholangiopancreatography (ERCP) procedure between August 2014 and January 2015 with the same duodenoscope at Cedars-Sinai Medical Center in Los Angeles and had been infected with carbapenem-resistant Enterobacteriaceae (CRE). This outbreak was announced by the medical center in early March in a statement that said the infections occurred “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes” recommended in instructions provided by Olympus and the FDA.
In February, the FDA first announced that the agency had received reports of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures with duodenoscopes, despite proper cleaning and disinfection of the devices and that the “complex design of ERCP endoscopes (also called duodenoscopes) may impede effective reprocessing.”
In the latest statement, the FDA said it “is closely monitoring the possible association between reprocessed duodenoscopes and the transmission of infectious agents,” including multidrug-resistant bacterial infections caused by CRE. If they are not properly reprocessed, the statement adds, “residual body fluids and organic debris may remain in microscopic crevices of the device following an attempted cleaning and high-level disinfection. If these residual fluids contain microbial contamination, subsequent patients may be exposed to serious infections.”
Adverse events associated with duodenoscopes should be reported to the FDA’s MedWatch Program at 800-332-1088 or www.accessdata.fda.gov/scripts/medwatch.
AGA Resource
Read more about AGA’s efforts and recommendations to stop duodenoscope infections here.
Olympus, the manufacturer of the TJF-Q180V duodenoscope, has issued new, validated instructions for reprocessing this particular model, as part of the response to recent reports of a possible association between multidrug-resistant bacterial infections and improperly processed duodenoscopes, according to the Food and Drug Administration.
The new instructions, which replace the manual reprocessing instructions included in the original labeling, and validation data have been reviewed by the FDA as part of its ongoing review of the device. The agency “recommends that any facilities that are using Olympus’ TJF-Q180V duodenoscope train staff on the new instructions and implement them as soon as possible,” according to an FDA statement. The instructions are provided in letters sent by Olympus to health care and other facilities that use this particular model.
“Key changes” have been made to the procedures for precleaning, manual cleaning, and manual high-level disinfection reprocessing procedures, the FDA said.
The TJF-Q180 V duodenoscope was the model used in four patients who had undergone an endoscopic retrograde cholangiopancreatography (ERCP) procedure between August 2014 and January 2015 with the same duodenoscope at Cedars-Sinai Medical Center in Los Angeles and had been infected with carbapenem-resistant Enterobacteriaceae (CRE). This outbreak was announced by the medical center in early March in a statement that said the infections occurred “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes” recommended in instructions provided by Olympus and the FDA.
In February, the FDA first announced that the agency had received reports of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures with duodenoscopes, despite proper cleaning and disinfection of the devices and that the “complex design of ERCP endoscopes (also called duodenoscopes) may impede effective reprocessing.”
In the latest statement, the FDA said it “is closely monitoring the possible association between reprocessed duodenoscopes and the transmission of infectious agents,” including multidrug-resistant bacterial infections caused by CRE. If they are not properly reprocessed, the statement adds, “residual body fluids and organic debris may remain in microscopic crevices of the device following an attempted cleaning and high-level disinfection. If these residual fluids contain microbial contamination, subsequent patients may be exposed to serious infections.”
Adverse events associated with duodenoscopes should be reported to the FDA’s MedWatch Program at 800-332-1088 or www.accessdata.fda.gov/scripts/medwatch.
AGA Resource
Read more about AGA’s efforts and recommendations to stop duodenoscope infections here.