User login
Manufacturer releases new reprocessing instructions for TJF-Q180V duodenoscope
Olympus, the manufacturer of the TJF-Q180V duodenoscope, has issued new, validated instructions for reprocessing this particular model, as part of the response to recent reports of a possible association between multidrug-resistant bacterial infections and improperly processed duodenoscopes, according to the Food and Drug Administration.
The new instructions, which replace the manual reprocessing instructions included in the original labeling, and validation data have been reviewed by the FDA as part of its ongoing review of the device. The agency “recommends that any facilities that are using Olympus’ TJF-Q180V duodenoscope train staff on the new instructions and implement them as soon as possible,” according to an FDA statement. The instructions are provided in letters sent by Olympus to health care and other facilities that use this particular model.
“Key changes” have been made to the procedures for precleaning, manual cleaning, and manual high-level disinfection reprocessing procedures, the FDA said.
The TJF-Q180 V duodenoscope was the model used in four patients who had undergone an endoscopic retrograde cholangiopancreatography (ERCP) procedure between August 2014 and January 2015 with the same duodenoscope at Cedars-Sinai Medical Center in Los Angeles and had been infected with carbapenem-resistant Enterobacteriaceae (CRE). This outbreak was announced by the medical center in early March in a statement that said the infections occurred “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes” recommended in instructions provided by Olympus and the FDA.
In February, the FDA first announced that the agency had received reports of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures with duodenoscopes, despite proper cleaning and disinfection of the devices and that the “complex design of ERCP endoscopes (also called duodenoscopes) may impede effective reprocessing.”
In the latest statement, the FDA said it “is closely monitoring the possible association between reprocessed duodenoscopes and the transmission of infectious agents,” including multidrug-resistant bacterial infections caused by CRE. If they are not properly reprocessed, the statement adds, “residual body fluids and organic debris may remain in microscopic crevices of the device following an attempted cleaning and high-level disinfection. If these residual fluids contain microbial contamination, subsequent patients may be exposed to serious infections.”
Adverse events associated with duodenoscopes should be reported to the FDA’s MedWatch Program at 800-332-1088 or www.accessdata.fda.gov/scripts/medwatch.
Olympus, the manufacturer of the TJF-Q180V duodenoscope, has issued new, validated instructions for reprocessing this particular model, as part of the response to recent reports of a possible association between multidrug-resistant bacterial infections and improperly processed duodenoscopes, according to the Food and Drug Administration.
The new instructions, which replace the manual reprocessing instructions included in the original labeling, and validation data have been reviewed by the FDA as part of its ongoing review of the device. The agency “recommends that any facilities that are using Olympus’ TJF-Q180V duodenoscope train staff on the new instructions and implement them as soon as possible,” according to an FDA statement. The instructions are provided in letters sent by Olympus to health care and other facilities that use this particular model.
“Key changes” have been made to the procedures for precleaning, manual cleaning, and manual high-level disinfection reprocessing procedures, the FDA said.
The TJF-Q180 V duodenoscope was the model used in four patients who had undergone an endoscopic retrograde cholangiopancreatography (ERCP) procedure between August 2014 and January 2015 with the same duodenoscope at Cedars-Sinai Medical Center in Los Angeles and had been infected with carbapenem-resistant Enterobacteriaceae (CRE). This outbreak was announced by the medical center in early March in a statement that said the infections occurred “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes” recommended in instructions provided by Olympus and the FDA.
In February, the FDA first announced that the agency had received reports of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures with duodenoscopes, despite proper cleaning and disinfection of the devices and that the “complex design of ERCP endoscopes (also called duodenoscopes) may impede effective reprocessing.”
In the latest statement, the FDA said it “is closely monitoring the possible association between reprocessed duodenoscopes and the transmission of infectious agents,” including multidrug-resistant bacterial infections caused by CRE. If they are not properly reprocessed, the statement adds, “residual body fluids and organic debris may remain in microscopic crevices of the device following an attempted cleaning and high-level disinfection. If these residual fluids contain microbial contamination, subsequent patients may be exposed to serious infections.”
Adverse events associated with duodenoscopes should be reported to the FDA’s MedWatch Program at 800-332-1088 or www.accessdata.fda.gov/scripts/medwatch.
Olympus, the manufacturer of the TJF-Q180V duodenoscope, has issued new, validated instructions for reprocessing this particular model, as part of the response to recent reports of a possible association between multidrug-resistant bacterial infections and improperly processed duodenoscopes, according to the Food and Drug Administration.
The new instructions, which replace the manual reprocessing instructions included in the original labeling, and validation data have been reviewed by the FDA as part of its ongoing review of the device. The agency “recommends that any facilities that are using Olympus’ TJF-Q180V duodenoscope train staff on the new instructions and implement them as soon as possible,” according to an FDA statement. The instructions are provided in letters sent by Olympus to health care and other facilities that use this particular model.
“Key changes” have been made to the procedures for precleaning, manual cleaning, and manual high-level disinfection reprocessing procedures, the FDA said.
The TJF-Q180 V duodenoscope was the model used in four patients who had undergone an endoscopic retrograde cholangiopancreatography (ERCP) procedure between August 2014 and January 2015 with the same duodenoscope at Cedars-Sinai Medical Center in Los Angeles and had been infected with carbapenem-resistant Enterobacteriaceae (CRE). This outbreak was announced by the medical center in early March in a statement that said the infections occurred “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes” recommended in instructions provided by Olympus and the FDA.
In February, the FDA first announced that the agency had received reports of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures with duodenoscopes, despite proper cleaning and disinfection of the devices and that the “complex design of ERCP endoscopes (also called duodenoscopes) may impede effective reprocessing.”
In the latest statement, the FDA said it “is closely monitoring the possible association between reprocessed duodenoscopes and the transmission of infectious agents,” including multidrug-resistant bacterial infections caused by CRE. If they are not properly reprocessed, the statement adds, “residual body fluids and organic debris may remain in microscopic crevices of the device following an attempted cleaning and high-level disinfection. If these residual fluids contain microbial contamination, subsequent patients may be exposed to serious infections.”
Adverse events associated with duodenoscopes should be reported to the FDA’s MedWatch Program at 800-332-1088 or www.accessdata.fda.gov/scripts/medwatch.
Waning antibody levels seen in children who received meningococcal B vaccine as infants
A study that followed up children aged 5 years who had received a meningococcal group B vaccine as infants in an earlier study determined that protective antibody levels against the strains covered by the vaccine dropped over time, with variations depending on the strains and vaccine dose schedules, the investigators reported.
The phase II, open-label study evaluated serum levels of protective antibodies to the strains included in the serogroup B meningococcal vaccine manufactured by Novartis in 50 infants, 18-20 months after they had received the last dose. The results “provide important new information about how the persistence, at 5 years of age, of bactericidal activity induced by administration of 4CMenB vaccine differs between test strains and with different vaccination schedules,” wrote Dr. Fiona McQuaid of the Oxford Vaccine Group, Oxford University, and her coauthors. The study was published online March 23 in the CMAJ (2015 [doi:10.1503/cmaj.141200]).
The authors pointed out that there are limited data on the persistence of antibody responses after toddler and infant immunization and after booster doses, but this information “will be important for guiding the implementation” by the United Kingdom Joint Committee on Vaccination and Immunization recommendation that the 4CMenB vaccine be included in the routine U.K. immunization schedule.
Among the 16 children who had received the vaccine at 2, 4, 6, 12, and 40 months, 44%-88% had antibodies against strains in the vaccine that were protective, which varied by strains, at age 5 years. Among the five children who received the vaccine at 12, 40, and 42 months, 80%-100% had protective antibodies against vaccine strains. Among the 29 children who had received the vaccine at 40 and 42 months, 31%-100% had protective antibodies against vaccine strains. “Waning of antibody titres was observed for those immunized as infants, but the extent of waning varied by strain,” the authors observed.
The most common adverse events after the first and second doses were injection site pain and erythema; the rate of fever was low.
Although the study had limitations, including the small sample size, the data “give an early indication that protection against serogroup B meningococcal disease after infant immunization is unlikely to persist into adolescence, when the second peak of incidence occurs,” the authors wrote.
“The steep decline in disease incidence after the first year of life means that schedules involving vaccination at 40 and 42 months and at 60 and 62 months may not have a substantial effect on the number of cases and are therefore less likely to be used if the incidence of disease remains at its current low levels,” they noted, but added that their data “will prove useful in planning for a future epidemic or for countries planning catch-up campaigns.”
The Novartis vaccine (Bexsero) and Pfizer’s meningococcal group B vaccine, Trumenba, which are approved in Europe and other countries, were recently approved in the United States. At a meeting in February, the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices recommended serogroup B meningococcal vaccination for several groups at increased risk for serogroup B disease, including students during outbreaks at college campuses. Broader use of these vaccines in adolescents and college students is on the agenda of the next ACIP meeting in June 2015.
Dr. McQuaid and her coauthors pointed out that, in Canada, meningococcal B vaccination is recommended for those in high-risk groups, and that introducing the vaccine as a routine vaccine in the United Kingdom “provides an ideal opportunity to assess the effect of this vaccine in a real-world setting and will guide the implementation of 4CMenB vaccination in Canada and worldwide.”
In an interview, Dr. McQuaid noted that the vaccine is licensed for use in the United States for people aged 10-25 years, while this study focused on children immunized as infants and at 5 years of age and does not necessarily apply to older age groups.
In Europe, the vaccine is licensed from 2 months of age, “and these data show that children immunized at 5 years of age have a good immunological response 1 month after two doses of the vaccine, which is important in the context of use during outbreaks.”
“The reactogenicity data will also help physicians inform parents of what to expect after vaccination,” she said, adding that, if the vaccine is introduced routinely into the United Kingdom, as recommended by the U.K. Joint Committee on Vaccination and Immunization,” this will provide a great deal of important information on how the vaccine works in a real-world setting.”
A study that followed up children aged 5 years who had received a meningococcal group B vaccine as infants in an earlier study determined that protective antibody levels against the strains covered by the vaccine dropped over time, with variations depending on the strains and vaccine dose schedules, the investigators reported.
The phase II, open-label study evaluated serum levels of protective antibodies to the strains included in the serogroup B meningococcal vaccine manufactured by Novartis in 50 infants, 18-20 months after they had received the last dose. The results “provide important new information about how the persistence, at 5 years of age, of bactericidal activity induced by administration of 4CMenB vaccine differs between test strains and with different vaccination schedules,” wrote Dr. Fiona McQuaid of the Oxford Vaccine Group, Oxford University, and her coauthors. The study was published online March 23 in the CMAJ (2015 [doi:10.1503/cmaj.141200]).
The authors pointed out that there are limited data on the persistence of antibody responses after toddler and infant immunization and after booster doses, but this information “will be important for guiding the implementation” by the United Kingdom Joint Committee on Vaccination and Immunization recommendation that the 4CMenB vaccine be included in the routine U.K. immunization schedule.
Among the 16 children who had received the vaccine at 2, 4, 6, 12, and 40 months, 44%-88% had antibodies against strains in the vaccine that were protective, which varied by strains, at age 5 years. Among the five children who received the vaccine at 12, 40, and 42 months, 80%-100% had protective antibodies against vaccine strains. Among the 29 children who had received the vaccine at 40 and 42 months, 31%-100% had protective antibodies against vaccine strains. “Waning of antibody titres was observed for those immunized as infants, but the extent of waning varied by strain,” the authors observed.
The most common adverse events after the first and second doses were injection site pain and erythema; the rate of fever was low.
Although the study had limitations, including the small sample size, the data “give an early indication that protection against serogroup B meningococcal disease after infant immunization is unlikely to persist into adolescence, when the second peak of incidence occurs,” the authors wrote.
“The steep decline in disease incidence after the first year of life means that schedules involving vaccination at 40 and 42 months and at 60 and 62 months may not have a substantial effect on the number of cases and are therefore less likely to be used if the incidence of disease remains at its current low levels,” they noted, but added that their data “will prove useful in planning for a future epidemic or for countries planning catch-up campaigns.”
The Novartis vaccine (Bexsero) and Pfizer’s meningococcal group B vaccine, Trumenba, which are approved in Europe and other countries, were recently approved in the United States. At a meeting in February, the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices recommended serogroup B meningococcal vaccination for several groups at increased risk for serogroup B disease, including students during outbreaks at college campuses. Broader use of these vaccines in adolescents and college students is on the agenda of the next ACIP meeting in June 2015.
Dr. McQuaid and her coauthors pointed out that, in Canada, meningococcal B vaccination is recommended for those in high-risk groups, and that introducing the vaccine as a routine vaccine in the United Kingdom “provides an ideal opportunity to assess the effect of this vaccine in a real-world setting and will guide the implementation of 4CMenB vaccination in Canada and worldwide.”
In an interview, Dr. McQuaid noted that the vaccine is licensed for use in the United States for people aged 10-25 years, while this study focused on children immunized as infants and at 5 years of age and does not necessarily apply to older age groups.
In Europe, the vaccine is licensed from 2 months of age, “and these data show that children immunized at 5 years of age have a good immunological response 1 month after two doses of the vaccine, which is important in the context of use during outbreaks.”
“The reactogenicity data will also help physicians inform parents of what to expect after vaccination,” she said, adding that, if the vaccine is introduced routinely into the United Kingdom, as recommended by the U.K. Joint Committee on Vaccination and Immunization,” this will provide a great deal of important information on how the vaccine works in a real-world setting.”
A study that followed up children aged 5 years who had received a meningococcal group B vaccine as infants in an earlier study determined that protective antibody levels against the strains covered by the vaccine dropped over time, with variations depending on the strains and vaccine dose schedules, the investigators reported.
The phase II, open-label study evaluated serum levels of protective antibodies to the strains included in the serogroup B meningococcal vaccine manufactured by Novartis in 50 infants, 18-20 months after they had received the last dose. The results “provide important new information about how the persistence, at 5 years of age, of bactericidal activity induced by administration of 4CMenB vaccine differs between test strains and with different vaccination schedules,” wrote Dr. Fiona McQuaid of the Oxford Vaccine Group, Oxford University, and her coauthors. The study was published online March 23 in the CMAJ (2015 [doi:10.1503/cmaj.141200]).
The authors pointed out that there are limited data on the persistence of antibody responses after toddler and infant immunization and after booster doses, but this information “will be important for guiding the implementation” by the United Kingdom Joint Committee on Vaccination and Immunization recommendation that the 4CMenB vaccine be included in the routine U.K. immunization schedule.
Among the 16 children who had received the vaccine at 2, 4, 6, 12, and 40 months, 44%-88% had antibodies against strains in the vaccine that were protective, which varied by strains, at age 5 years. Among the five children who received the vaccine at 12, 40, and 42 months, 80%-100% had protective antibodies against vaccine strains. Among the 29 children who had received the vaccine at 40 and 42 months, 31%-100% had protective antibodies against vaccine strains. “Waning of antibody titres was observed for those immunized as infants, but the extent of waning varied by strain,” the authors observed.
The most common adverse events after the first and second doses were injection site pain and erythema; the rate of fever was low.
Although the study had limitations, including the small sample size, the data “give an early indication that protection against serogroup B meningococcal disease after infant immunization is unlikely to persist into adolescence, when the second peak of incidence occurs,” the authors wrote.
“The steep decline in disease incidence after the first year of life means that schedules involving vaccination at 40 and 42 months and at 60 and 62 months may not have a substantial effect on the number of cases and are therefore less likely to be used if the incidence of disease remains at its current low levels,” they noted, but added that their data “will prove useful in planning for a future epidemic or for countries planning catch-up campaigns.”
The Novartis vaccine (Bexsero) and Pfizer’s meningococcal group B vaccine, Trumenba, which are approved in Europe and other countries, were recently approved in the United States. At a meeting in February, the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices recommended serogroup B meningococcal vaccination for several groups at increased risk for serogroup B disease, including students during outbreaks at college campuses. Broader use of these vaccines in adolescents and college students is on the agenda of the next ACIP meeting in June 2015.
Dr. McQuaid and her coauthors pointed out that, in Canada, meningococcal B vaccination is recommended for those in high-risk groups, and that introducing the vaccine as a routine vaccine in the United Kingdom “provides an ideal opportunity to assess the effect of this vaccine in a real-world setting and will guide the implementation of 4CMenB vaccination in Canada and worldwide.”
In an interview, Dr. McQuaid noted that the vaccine is licensed for use in the United States for people aged 10-25 years, while this study focused on children immunized as infants and at 5 years of age and does not necessarily apply to older age groups.
In Europe, the vaccine is licensed from 2 months of age, “and these data show that children immunized at 5 years of age have a good immunological response 1 month after two doses of the vaccine, which is important in the context of use during outbreaks.”
“The reactogenicity data will also help physicians inform parents of what to expect after vaccination,” she said, adding that, if the vaccine is introduced routinely into the United Kingdom, as recommended by the U.K. Joint Committee on Vaccination and Immunization,” this will provide a great deal of important information on how the vaccine works in a real-world setting.”
FROM THE CMAJ
Key clinical point: A study evaluatingantibody titers to meningococcal B strains in 5-year-olds immunized as infants provide data that should be helpful in determining how to schedule meningococcal B vaccination.
Major finding: At age 5 years, 18-20 months after the last dose, protective antibody levels against strains in the meningococcal B vaccine the children had received as infants ranged from 31% to 100%, depending on the strain and schedule.
Data source: A phase II, open-label extension study at one center in the United Kingdom evaluated the levels of protective antibodies in 50 5-year-old children who had received a meningococcal B vaccine at different schedules as infants in another study, 18-20 months after they received their last dose.
Disclosures: The study was sponsored by Novartis, manufacturer of the vaccine. Author disclosures include being an investigator for studies funded by vaccine manufacturers, including Novartis and GlaxoSmithkline, and serving as consultants and advisors for those manufacturers. Three authors are Novartis employees.
DTaP-IPV vaccine approved for children aged 4-6 years
A quadrivalent vaccine has been approved for active immunization against diphtheria, tetanus, pertussis, and poliomyelitis in children aged 4-6 years in the United States, the manufacturer, Sanofi Pasteur, announced March 25.
The approved indication is for use in children in this age group “as a fifth dose in the diphtheria, tetanus, pertussis vaccination (DTaP) series, and as a fourth or fifth dose in the inactivated poliovirus vaccination (IPV) series, in children who have received 4 doses of Pentacel and/or Daptacel vaccine,” according to the prescribing information. The vaccine will be marketed as Quadracel (diphtheria and tetanus toxoids and acellular pertussis adsorbed and inactivated Poliovirus).

Pentacel (diphtheria and tetanus toxoids and acellular pertussis adsorbed, inactivated poliovirus and haemophilus b conjugate [tetanus toxoid conjugate]) and Daptacel (diphtheria and tetanus toxoids and acellular pertussis vaccine adsorbed) also are manufactured by Sanofi Pasteur.
The company statement announcing approval of Quadracel said that the Food and Drug Administration approval is based on a phase III randomized controlled study of children aged 4-6 years who had been vaccinated with the Daptacel and/or Pentacel vaccines. The study compared the safety and immunogenicity of the Quadracel vaccine with Daptacel (DTaP) and IPOL (IPV) and determined that the safety and immunogenicity profiles were similar, according to the company.
Another DTaP-IPV vaccine, Kinrix (diphtheria and tetanus toxoids and acellular pertussis adsorbed and inactivated poliovirus vaccine), was approved in 2008.
A quadrivalent vaccine has been approved for active immunization against diphtheria, tetanus, pertussis, and poliomyelitis in children aged 4-6 years in the United States, the manufacturer, Sanofi Pasteur, announced March 25.
The approved indication is for use in children in this age group “as a fifth dose in the diphtheria, tetanus, pertussis vaccination (DTaP) series, and as a fourth or fifth dose in the inactivated poliovirus vaccination (IPV) series, in children who have received 4 doses of Pentacel and/or Daptacel vaccine,” according to the prescribing information. The vaccine will be marketed as Quadracel (diphtheria and tetanus toxoids and acellular pertussis adsorbed and inactivated Poliovirus).
Pentacel (diphtheria and tetanus toxoids and acellular pertussis adsorbed, inactivated poliovirus and haemophilus b conjugate [tetanus toxoid conjugate]) and Daptacel (diphtheria and tetanus toxoids and acellular pertussis vaccine adsorbed) also are manufactured by Sanofi Pasteur.
The company statement announcing approval of Quadracel said that the Food and Drug Administration approval is based on a phase III randomized controlled study of children aged 4-6 years who had been vaccinated with the Daptacel and/or Pentacel vaccines. The study compared the safety and immunogenicity of the Quadracel vaccine with Daptacel (DTaP) and IPOL (IPV) and determined that the safety and immunogenicity profiles were similar, according to the company.
Another DTaP-IPV vaccine, Kinrix (diphtheria and tetanus toxoids and acellular pertussis adsorbed and inactivated poliovirus vaccine), was approved in 2008.
A quadrivalent vaccine has been approved for active immunization against diphtheria, tetanus, pertussis, and poliomyelitis in children aged 4-6 years in the United States, the manufacturer, Sanofi Pasteur, announced March 25.
The approved indication is for use in children in this age group “as a fifth dose in the diphtheria, tetanus, pertussis vaccination (DTaP) series, and as a fourth or fifth dose in the inactivated poliovirus vaccination (IPV) series, in children who have received 4 doses of Pentacel and/or Daptacel vaccine,” according to the prescribing information. The vaccine will be marketed as Quadracel (diphtheria and tetanus toxoids and acellular pertussis adsorbed and inactivated Poliovirus).
Pentacel (diphtheria and tetanus toxoids and acellular pertussis adsorbed, inactivated poliovirus and haemophilus b conjugate [tetanus toxoid conjugate]) and Daptacel (diphtheria and tetanus toxoids and acellular pertussis vaccine adsorbed) also are manufactured by Sanofi Pasteur.
The company statement announcing approval of Quadracel said that the Food and Drug Administration approval is based on a phase III randomized controlled study of children aged 4-6 years who had been vaccinated with the Daptacel and/or Pentacel vaccines. The study compared the safety and immunogenicity of the Quadracel vaccine with Daptacel (DTaP) and IPOL (IPV) and determined that the safety and immunogenicity profiles were similar, according to the company.
Another DTaP-IPV vaccine, Kinrix (diphtheria and tetanus toxoids and acellular pertussis adsorbed and inactivated poliovirus vaccine), was approved in 2008.

FDA approves another VEGF inhibitor for diabetic retinopathy in patients with DME
Aflibercept, a vascular endothelial growth factor (VEGF) inhibitor administered by intravitreal injection, is now approved for treating diabetic retinopathy in patients with diabetic macular edema (DME), the Food and Drug Administration announced on March 25.
Aflibercept was previously approved for treating wet (neovascular) age-related macular degeneration, and for treating DME and macular edema secondary to retinal vein occlusions, “both of which cause fluid to leak into the macula resulting in blurred vision,” according to the FDA statement announcing approval. The first five injections of aflibercept are administered once a month, followed by one injection every 2 months. Aflibercept is marketed as Eylea by Regeneron Pharmaceuticals; it was initially approved in 2011.
“Today’s approval gives patients with diabetic retinopathy and diabetic macular edema another therapy to treat this vision-impairing complication,” Dr. Edward Cox, director of the office of antimicrobial products in the FDA’s Center for Drug Evaluation and Research, said in the statement. In February, the FDA approved ranibizumab (Lucentis), another VEGF inhibitor, for the same indication.
Approval of aflibercept was based on two phase III studies of 679 patients with diabetic retinopathy and DME, who were randomized to treatment with aflibercept or macular laser photocoagulation. At 2 years, those treated with aflibercept “showed significant improvement in the severity of their diabetic retinopathy, compared to patients who did not receive Eylea,” according to the FDA statement. Conjunctival hemorrhage, eye pain, cataracts, vitreous floaters, increased intraocular pressure, and vitreous detachment were among the most common adverse events (reported by at least 5% of treated patients) associated with treatment. Endophthalmitis and retinal detachments were among the serious adverse events associated with treatment.
The two studies are the VISTA-DME and VIVID-DME trials, according to the Regeneron statement announcing approval.
Adverse events associated with aflibercept should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
Aflibercept, a vascular endothelial growth factor (VEGF) inhibitor administered by intravitreal injection, is now approved for treating diabetic retinopathy in patients with diabetic macular edema (DME), the Food and Drug Administration announced on March 25.
Aflibercept was previously approved for treating wet (neovascular) age-related macular degeneration, and for treating DME and macular edema secondary to retinal vein occlusions, “both of which cause fluid to leak into the macula resulting in blurred vision,” according to the FDA statement announcing approval. The first five injections of aflibercept are administered once a month, followed by one injection every 2 months. Aflibercept is marketed as Eylea by Regeneron Pharmaceuticals; it was initially approved in 2011.
“Today’s approval gives patients with diabetic retinopathy and diabetic macular edema another therapy to treat this vision-impairing complication,” Dr. Edward Cox, director of the office of antimicrobial products in the FDA’s Center for Drug Evaluation and Research, said in the statement. In February, the FDA approved ranibizumab (Lucentis), another VEGF inhibitor, for the same indication.
Approval of aflibercept was based on two phase III studies of 679 patients with diabetic retinopathy and DME, who were randomized to treatment with aflibercept or macular laser photocoagulation. At 2 years, those treated with aflibercept “showed significant improvement in the severity of their diabetic retinopathy, compared to patients who did not receive Eylea,” according to the FDA statement. Conjunctival hemorrhage, eye pain, cataracts, vitreous floaters, increased intraocular pressure, and vitreous detachment were among the most common adverse events (reported by at least 5% of treated patients) associated with treatment. Endophthalmitis and retinal detachments were among the serious adverse events associated with treatment.
The two studies are the VISTA-DME and VIVID-DME trials, according to the Regeneron statement announcing approval.
Adverse events associated with aflibercept should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
Aflibercept, a vascular endothelial growth factor (VEGF) inhibitor administered by intravitreal injection, is now approved for treating diabetic retinopathy in patients with diabetic macular edema (DME), the Food and Drug Administration announced on March 25.
Aflibercept was previously approved for treating wet (neovascular) age-related macular degeneration, and for treating DME and macular edema secondary to retinal vein occlusions, “both of which cause fluid to leak into the macula resulting in blurred vision,” according to the FDA statement announcing approval. The first five injections of aflibercept are administered once a month, followed by one injection every 2 months. Aflibercept is marketed as Eylea by Regeneron Pharmaceuticals; it was initially approved in 2011.
“Today’s approval gives patients with diabetic retinopathy and diabetic macular edema another therapy to treat this vision-impairing complication,” Dr. Edward Cox, director of the office of antimicrobial products in the FDA’s Center for Drug Evaluation and Research, said in the statement. In February, the FDA approved ranibizumab (Lucentis), another VEGF inhibitor, for the same indication.
Approval of aflibercept was based on two phase III studies of 679 patients with diabetic retinopathy and DME, who were randomized to treatment with aflibercept or macular laser photocoagulation. At 2 years, those treated with aflibercept “showed significant improvement in the severity of their diabetic retinopathy, compared to patients who did not receive Eylea,” according to the FDA statement. Conjunctival hemorrhage, eye pain, cataracts, vitreous floaters, increased intraocular pressure, and vitreous detachment were among the most common adverse events (reported by at least 5% of treated patients) associated with treatment. Endophthalmitis and retinal detachments were among the serious adverse events associated with treatment.
The two studies are the VISTA-DME and VIVID-DME trials, according to the Regeneron statement announcing approval.
Adverse events associated with aflibercept should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
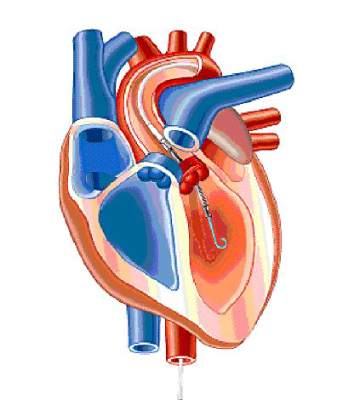
FDA approves miniature heart pump for use during high risk PCI
A miniature heart pump has been approved by the Food and Drug Administration to “help certain patients maintain stable heart function and circulation during certain high-risk percutaneous coronary intervention (HRPCI) procedures,” the agency has announced.
The Impella 2.5 System, manufactured by Abiomed, is “intended for temporary use by patients with severe symptomatic CAD [coronary artery disease] and diminished (but stable) heart function who are undergoing HRPCI but are not candidates for surgical coronary bypass treatment,” according to the FDA’s statement.
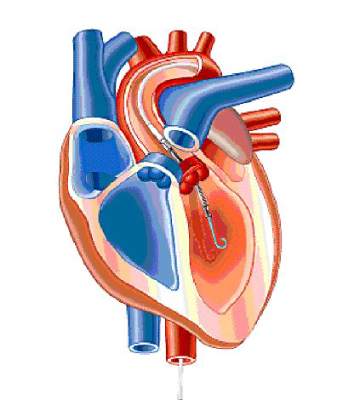
“Use of the Impella 2.5 System is intended to prevent episodes of unstable heart function, including unstable blood pressure and poor circulation, in patients who are at high risk for its occurrence,” Dr. William Maisel, acting director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, said in the statement.
Approval was based on the PROTECT II study and observational data from the USpella Registry.
“The overall data provided evidence that, for patients with severe CAD and diminished heart function, the temporary circulatory support provided by the Impella 2.5 System during an HRPCI procedure may allow a longer and more thorough procedure by preventing episodes of hemodynamic instability ... due to temporary abnormalities in heart function,” the FDA statement said. In addition, “fewer later adverse events” such as the need for repeat HRPCI procedures, “may occur in patients undergoing HRPCI with the pump compared to patients undergoing HRPCI with an intra-aortic balloon pump,” according to the FDA.
The FDA statement also noted that the system can be used as an alternative to the intra-aortic balloon pump “without significantly increasing the safety risks of the HRPCI procedure.”
As a postmarketing requirement, the manufacturer will conduct a single arm study of the device in high-risk PCI patients, according to the company’s statement announcing approval.
The wording of the approved indication is as follows, according to Abiomed: “The Impella 2.5 is a temporary (less than or equal to 6 hours) ventricular support device indicated for use during high-risk PCI performed in elective or urgent hemodynamically stable patients with severe coronary artery disease and depressed left ventricular ejection fraction, when a heart team, including a cardiac surgeon, has determined high-risk PCI is the appropriate therapeutic option. Use of the Impella 2.5 in these patients may prevent hemodynamic instability that may occur during planned temporary coronary occlusions and may reduce peri- and postprocedural adverse events.”
A miniature heart pump has been approved by the Food and Drug Administration to “help certain patients maintain stable heart function and circulation during certain high-risk percutaneous coronary intervention (HRPCI) procedures,” the agency has announced.
The Impella 2.5 System, manufactured by Abiomed, is “intended for temporary use by patients with severe symptomatic CAD [coronary artery disease] and diminished (but stable) heart function who are undergoing HRPCI but are not candidates for surgical coronary bypass treatment,” according to the FDA’s statement.
“Use of the Impella 2.5 System is intended to prevent episodes of unstable heart function, including unstable blood pressure and poor circulation, in patients who are at high risk for its occurrence,” Dr. William Maisel, acting director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, said in the statement.
Approval was based on the PROTECT II study and observational data from the USpella Registry.
“The overall data provided evidence that, for patients with severe CAD and diminished heart function, the temporary circulatory support provided by the Impella 2.5 System during an HRPCI procedure may allow a longer and more thorough procedure by preventing episodes of hemodynamic instability ... due to temporary abnormalities in heart function,” the FDA statement said. In addition, “fewer later adverse events” such as the need for repeat HRPCI procedures, “may occur in patients undergoing HRPCI with the pump compared to patients undergoing HRPCI with an intra-aortic balloon pump,” according to the FDA.
The FDA statement also noted that the system can be used as an alternative to the intra-aortic balloon pump “without significantly increasing the safety risks of the HRPCI procedure.”
As a postmarketing requirement, the manufacturer will conduct a single arm study of the device in high-risk PCI patients, according to the company’s statement announcing approval.
The wording of the approved indication is as follows, according to Abiomed: “The Impella 2.5 is a temporary (less than or equal to 6 hours) ventricular support device indicated for use during high-risk PCI performed in elective or urgent hemodynamically stable patients with severe coronary artery disease and depressed left ventricular ejection fraction, when a heart team, including a cardiac surgeon, has determined high-risk PCI is the appropriate therapeutic option. Use of the Impella 2.5 in these patients may prevent hemodynamic instability that may occur during planned temporary coronary occlusions and may reduce peri- and postprocedural adverse events.”
A miniature heart pump has been approved by the Food and Drug Administration to “help certain patients maintain stable heart function and circulation during certain high-risk percutaneous coronary intervention (HRPCI) procedures,” the agency has announced.
The Impella 2.5 System, manufactured by Abiomed, is “intended for temporary use by patients with severe symptomatic CAD [coronary artery disease] and diminished (but stable) heart function who are undergoing HRPCI but are not candidates for surgical coronary bypass treatment,” according to the FDA’s statement.
“Use of the Impella 2.5 System is intended to prevent episodes of unstable heart function, including unstable blood pressure and poor circulation, in patients who are at high risk for its occurrence,” Dr. William Maisel, acting director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, said in the statement.
Approval was based on the PROTECT II study and observational data from the USpella Registry.
“The overall data provided evidence that, for patients with severe CAD and diminished heart function, the temporary circulatory support provided by the Impella 2.5 System during an HRPCI procedure may allow a longer and more thorough procedure by preventing episodes of hemodynamic instability ... due to temporary abnormalities in heart function,” the FDA statement said. In addition, “fewer later adverse events” such as the need for repeat HRPCI procedures, “may occur in patients undergoing HRPCI with the pump compared to patients undergoing HRPCI with an intra-aortic balloon pump,” according to the FDA.
The FDA statement also noted that the system can be used as an alternative to the intra-aortic balloon pump “without significantly increasing the safety risks of the HRPCI procedure.”
As a postmarketing requirement, the manufacturer will conduct a single arm study of the device in high-risk PCI patients, according to the company’s statement announcing approval.
The wording of the approved indication is as follows, according to Abiomed: “The Impella 2.5 is a temporary (less than or equal to 6 hours) ventricular support device indicated for use during high-risk PCI performed in elective or urgent hemodynamically stable patients with severe coronary artery disease and depressed left ventricular ejection fraction, when a heart team, including a cardiac surgeon, has determined high-risk PCI is the appropriate therapeutic option. Use of the Impella 2.5 in these patients may prevent hemodynamic instability that may occur during planned temporary coronary occlusions and may reduce peri- and postprocedural adverse events.”
FDA: Avoid Using Amiodarone With Some Hepatitis C Antivirals
Taking the antiarrhythmic drug amiodarone with the hepatitis C antiviral drugs ledipasvir and sofosbuvir, or with sofosbuvir plus another direct-acting antiviral drug, has been associated with cases of symptomatic bradycardia – including a fatal cardiac arrest – according to the Food and Drug Administration.
Because of the reports, the antiviral drugs’ labels now recommend against using amiodarone with those hepatitis C drugs.
An FDA statement issued March 24 described the bradycardia cases as “serious and life-threatening.” Gilead Sciences markets the ledipasvir and sofosbuvir combination as Harvoni and markets sofosbuvir as Sovaldi to treat chronic hepatitis C virus (HCV) infection.
Gilead issued a “Dear Health Care Provider” letter that provides further details of the cases. There have been nine postmarketing reports of symptomatic bradycardia in patients who were taking amiodarone with Harvoni; amiodarone with Sovaldi plus another hepatitis C antiviral drug, simeprevir (Olysio); or amiodarone with an investigational hepatitis C antiviral drug, daclatasvir.
Of those cases, six occurred with in the first 24 hours of starting treatment with the antivirals, and three cases occurred within the first 2-12 days after antiviral therapy was started. A pacemaker was needed in three cases, and one case was a fatal cardiac arrest.
In three cases, a “rechallenge with HCV treatment in the setting of continued amiodarone therapy resulted in recurrence of symptomatic bradycardia,” according to the Gilead letter.
The effect of coadministration on the blood levels of the antiviral drugs is not known, nor is the mechanism behind the cardiac effect.
The labeling of the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni) now includes a section on “serious symptomatic bradycardia” when coadministered with amiodarone, and says that coadministration is not recommended. The label adds that if a patient on amiodarone or Harvoni has no other alternative than to take that combination, patients should be counseled about the bradycardia risk.
Cardiac monitoring is recommended for inpatients during the first 48 hours the patient is taking the drugs, “after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.”
The label notes that amiodarone has a long half-life, so cardiac monitoring is still necessary if the patient discontinues amiodarone just before starting treatment with Harvoni. Similar labeling changes have been made to the Sovaldi label.
Adverse events associated with Harvoni or Sovaldi should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/Safety/MedWatch/.
Taking the antiarrhythmic drug amiodarone with the hepatitis C antiviral drugs ledipasvir and sofosbuvir, or with sofosbuvir plus another direct-acting antiviral drug, has been associated with cases of symptomatic bradycardia – including a fatal cardiac arrest – according to the Food and Drug Administration.
Because of the reports, the antiviral drugs’ labels now recommend against using amiodarone with those hepatitis C drugs.
An FDA statement issued March 24 described the bradycardia cases as “serious and life-threatening.” Gilead Sciences markets the ledipasvir and sofosbuvir combination as Harvoni and markets sofosbuvir as Sovaldi to treat chronic hepatitis C virus (HCV) infection.
Gilead issued a “Dear Health Care Provider” letter that provides further details of the cases. There have been nine postmarketing reports of symptomatic bradycardia in patients who were taking amiodarone with Harvoni; amiodarone with Sovaldi plus another hepatitis C antiviral drug, simeprevir (Olysio); or amiodarone with an investigational hepatitis C antiviral drug, daclatasvir.
Of those cases, six occurred with in the first 24 hours of starting treatment with the antivirals, and three cases occurred within the first 2-12 days after antiviral therapy was started. A pacemaker was needed in three cases, and one case was a fatal cardiac arrest.
In three cases, a “rechallenge with HCV treatment in the setting of continued amiodarone therapy resulted in recurrence of symptomatic bradycardia,” according to the Gilead letter.
The effect of coadministration on the blood levels of the antiviral drugs is not known, nor is the mechanism behind the cardiac effect.
The labeling of the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni) now includes a section on “serious symptomatic bradycardia” when coadministered with amiodarone, and says that coadministration is not recommended. The label adds that if a patient on amiodarone or Harvoni has no other alternative than to take that combination, patients should be counseled about the bradycardia risk.
Cardiac monitoring is recommended for inpatients during the first 48 hours the patient is taking the drugs, “after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.”
The label notes that amiodarone has a long half-life, so cardiac monitoring is still necessary if the patient discontinues amiodarone just before starting treatment with Harvoni. Similar labeling changes have been made to the Sovaldi label.
Adverse events associated with Harvoni or Sovaldi should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/Safety/MedWatch/.
Taking the antiarrhythmic drug amiodarone with the hepatitis C antiviral drugs ledipasvir and sofosbuvir, or with sofosbuvir plus another direct-acting antiviral drug, has been associated with cases of symptomatic bradycardia – including a fatal cardiac arrest – according to the Food and Drug Administration.
Because of the reports, the antiviral drugs’ labels now recommend against using amiodarone with those hepatitis C drugs.
An FDA statement issued March 24 described the bradycardia cases as “serious and life-threatening.” Gilead Sciences markets the ledipasvir and sofosbuvir combination as Harvoni and markets sofosbuvir as Sovaldi to treat chronic hepatitis C virus (HCV) infection.
Gilead issued a “Dear Health Care Provider” letter that provides further details of the cases. There have been nine postmarketing reports of symptomatic bradycardia in patients who were taking amiodarone with Harvoni; amiodarone with Sovaldi plus another hepatitis C antiviral drug, simeprevir (Olysio); or amiodarone with an investigational hepatitis C antiviral drug, daclatasvir.
Of those cases, six occurred with in the first 24 hours of starting treatment with the antivirals, and three cases occurred within the first 2-12 days after antiviral therapy was started. A pacemaker was needed in three cases, and one case was a fatal cardiac arrest.
In three cases, a “rechallenge with HCV treatment in the setting of continued amiodarone therapy resulted in recurrence of symptomatic bradycardia,” according to the Gilead letter.
The effect of coadministration on the blood levels of the antiviral drugs is not known, nor is the mechanism behind the cardiac effect.
The labeling of the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni) now includes a section on “serious symptomatic bradycardia” when coadministered with amiodarone, and says that coadministration is not recommended. The label adds that if a patient on amiodarone or Harvoni has no other alternative than to take that combination, patients should be counseled about the bradycardia risk.
Cardiac monitoring is recommended for inpatients during the first 48 hours the patient is taking the drugs, “after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.”
The label notes that amiodarone has a long half-life, so cardiac monitoring is still necessary if the patient discontinues amiodarone just before starting treatment with Harvoni. Similar labeling changes have been made to the Sovaldi label.
Adverse events associated with Harvoni or Sovaldi should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/Safety/MedWatch/.
FDA: Avoid using amiodarone with some hepatitis C antivirals
Taking the antiarrythmic drug amiodarone with the hepatitis C antiviral drugs ledipasvir and sofosbuvir, or with sofosbuvir plus another direct-acting antiviral drug, has been associated with cases of symptomatic bradycardia – including a fatal cardiac arrest – according to the Food and Drug Administration.
Because of the reports, the antiviral drugs’ labels now recommend against using amiodarone with those hepatitis C drugs.
An FDA statement issued March 24 described the bradycardia cases as “serious and life-threatening.” Gilead Sciences markets the ledipasvir and sofosbuvir combination as Harvoni and markets sofosbuvir as Sovaldi to treat chronic hepatitis C virus (HCV) infection.
Gilead issued a “Dear Health Care Provider” letter that provides further details of the cases. There have been nine postmarketing reports of symptomatic bradycardia in patients who were taking amiodarone with Harvoni; amiodarone with Sovaldi plus another hepatitis C antiviral drug, simeprevir (Olysio); or amiodarone with an investigational hepatitis C antiviral drug, daclatasvir.
Of those cases, six occurred with in the first 24 hours of starting treatment with the antivirals, and three cases occurred within the first 2-12 days after antiviral therapy was started. A pacemaker was needed in three cases, and one case was a fatal cardiac arrest.
In three cases, a “rechallenge with HCV treatment in the setting of continued amiodarone therapy resulted in recurrence of symptomatic bradycardia,” according to the Gilead letter.
The effect of coadministration on the blood levels of the antiviral drugs is not known, nor is the mechanism behind the cardiac effect.
The labeling of the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni) now includes a section on “serious symptomatic bradycardia” when coadministered with amiodarone, and says that coadministration is not recommended. The label adds that if a patient on amiodarone or Harvoni has no other alternative than to take that combination, patients should be counseled about the bradycardia risk.
Cardiac monitoring is recommended for inpatients during the first 48 hours the patient is taking the drugs, “after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.”
The label notes that amiodarone has a long half-life, so cardiac monitoring is still necessary if the patient discontinues amiodarone just before starting treatment with Harvoni. Similar labeling changes have been made to the Sovaldi label.
Adverse events associated with Harvoni or Sovaldi should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/Safety/MedWatch/.
Taking the antiarrythmic drug amiodarone with the hepatitis C antiviral drugs ledipasvir and sofosbuvir, or with sofosbuvir plus another direct-acting antiviral drug, has been associated with cases of symptomatic bradycardia – including a fatal cardiac arrest – according to the Food and Drug Administration.
Because of the reports, the antiviral drugs’ labels now recommend against using amiodarone with those hepatitis C drugs.
An FDA statement issued March 24 described the bradycardia cases as “serious and life-threatening.” Gilead Sciences markets the ledipasvir and sofosbuvir combination as Harvoni and markets sofosbuvir as Sovaldi to treat chronic hepatitis C virus (HCV) infection.
Gilead issued a “Dear Health Care Provider” letter that provides further details of the cases. There have been nine postmarketing reports of symptomatic bradycardia in patients who were taking amiodarone with Harvoni; amiodarone with Sovaldi plus another hepatitis C antiviral drug, simeprevir (Olysio); or amiodarone with an investigational hepatitis C antiviral drug, daclatasvir.
Of those cases, six occurred with in the first 24 hours of starting treatment with the antivirals, and three cases occurred within the first 2-12 days after antiviral therapy was started. A pacemaker was needed in three cases, and one case was a fatal cardiac arrest.
In three cases, a “rechallenge with HCV treatment in the setting of continued amiodarone therapy resulted in recurrence of symptomatic bradycardia,” according to the Gilead letter.
The effect of coadministration on the blood levels of the antiviral drugs is not known, nor is the mechanism behind the cardiac effect.
The labeling of the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni) now includes a section on “serious symptomatic bradycardia” when coadministered with amiodarone, and says that coadministration is not recommended. The label adds that if a patient on amiodarone or Harvoni has no other alternative than to take that combination, patients should be counseled about the bradycardia risk.
Cardiac monitoring is recommended for inpatients during the first 48 hours the patient is taking the drugs, “after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.”
The label notes that amiodarone has a long half-life, so cardiac monitoring is still necessary if the patient discontinues amiodarone just before starting treatment with Harvoni. Similar labeling changes have been made to the Sovaldi label.
Adverse events associated with Harvoni or Sovaldi should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/Safety/MedWatch/.
Taking the antiarrythmic drug amiodarone with the hepatitis C antiviral drugs ledipasvir and sofosbuvir, or with sofosbuvir plus another direct-acting antiviral drug, has been associated with cases of symptomatic bradycardia – including a fatal cardiac arrest – according to the Food and Drug Administration.
Because of the reports, the antiviral drugs’ labels now recommend against using amiodarone with those hepatitis C drugs.
An FDA statement issued March 24 described the bradycardia cases as “serious and life-threatening.” Gilead Sciences markets the ledipasvir and sofosbuvir combination as Harvoni and markets sofosbuvir as Sovaldi to treat chronic hepatitis C virus (HCV) infection.
Gilead issued a “Dear Health Care Provider” letter that provides further details of the cases. There have been nine postmarketing reports of symptomatic bradycardia in patients who were taking amiodarone with Harvoni; amiodarone with Sovaldi plus another hepatitis C antiviral drug, simeprevir (Olysio); or amiodarone with an investigational hepatitis C antiviral drug, daclatasvir.
Of those cases, six occurred with in the first 24 hours of starting treatment with the antivirals, and three cases occurred within the first 2-12 days after antiviral therapy was started. A pacemaker was needed in three cases, and one case was a fatal cardiac arrest.
In three cases, a “rechallenge with HCV treatment in the setting of continued amiodarone therapy resulted in recurrence of symptomatic bradycardia,” according to the Gilead letter.
The effect of coadministration on the blood levels of the antiviral drugs is not known, nor is the mechanism behind the cardiac effect.
The labeling of the fixed-dose combination of ledipasvir and sofosbuvir (Harvoni) now includes a section on “serious symptomatic bradycardia” when coadministered with amiodarone, and says that coadministration is not recommended. The label adds that if a patient on amiodarone or Harvoni has no other alternative than to take that combination, patients should be counseled about the bradycardia risk.
Cardiac monitoring is recommended for inpatients during the first 48 hours the patient is taking the drugs, “after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.”
The label notes that amiodarone has a long half-life, so cardiac monitoring is still necessary if the patient discontinues amiodarone just before starting treatment with Harvoni. Similar labeling changes have been made to the Sovaldi label.
Adverse events associated with Harvoni or Sovaldi should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/Safety/MedWatch/.
FDA calls study of unexplained olanzapine deaths ‘inconclusive’
The Food and Drug Administration’s review of a study that looked into the unexplained deaths of two patients days after receiving injections of the long-acting injectable formulation of the atypical antipsychotic olanzapine are “inconclusive,” and no labeling changes are currently recommended, the agency has announced.
“We are unable to exclude the possibility that the deaths were caused by rapid, but delayed, entry of the drug into the bloodstream following intramuscular injection,” according to the drug safety communication issued March 23 by the FDA.
Patients who receive olanzapine pamoate must be monitored for 3 hours after an injection because of the risk of post-injection delirium sedation (PDSS) associated with the injection. These patients, however, died 3 to 4 days after having received appropriate IM doses of olanzapine pamoate (Zyprexa Relprevv) and were found to have very high levels of the drug. The deaths and subsequent investigation were announced by the FDA in 2013.
The March 23 statement said an animal study, conducted by olanzapine manufacturer Eli Lilly at the FDA’s request to determine whether an IM injection could result in “postmortem redistribution” of olanzapine pamoate, “suggested that much of the drug level increase could have occurred after death, a finding that could explain the extremely high blood levels” in the two patients.
Based on its review, the FDA is not recommending any changes to the prescribing information of olanzapine pamoate. However, the agency is advising health care professionals to continue to follow the Risk Evaluation and Mitigation Strategy in place for this drug, which includes mandatory enrollment of patients, prescribers, health care facilities, and pharmacies in the Zyprexa Relprevv Patient Care program. Patients “should not stop receiving treatment without first talking to their health care professionals,” the statement adds.
The REMS requirements include continuous monitoring of patients for 3 hours after the injection, which must be administered at a REMS-certified health care facility that has quick access to emergency response services. A medication guide explaining these and other risks associated with the drug are provided to patients.
The increased risk for severe sedation, including coma, and/or delirium after each olanzapine pamoate injection, also is described in a boxed warning in the prescribing information.
Adverse events associated with Zyprexa Relprevv should be reported to the FDA’s MedWatch program at 800-332-1088 or https://www.accessdata.fda.gov/scripts/medwatch/
The Food and Drug Administration’s review of a study that looked into the unexplained deaths of two patients days after receiving injections of the long-acting injectable formulation of the atypical antipsychotic olanzapine are “inconclusive,” and no labeling changes are currently recommended, the agency has announced.
“We are unable to exclude the possibility that the deaths were caused by rapid, but delayed, entry of the drug into the bloodstream following intramuscular injection,” according to the drug safety communication issued March 23 by the FDA.
Patients who receive olanzapine pamoate must be monitored for 3 hours after an injection because of the risk of post-injection delirium sedation (PDSS) associated with the injection. These patients, however, died 3 to 4 days after having received appropriate IM doses of olanzapine pamoate (Zyprexa Relprevv) and were found to have very high levels of the drug. The deaths and subsequent investigation were announced by the FDA in 2013.
The March 23 statement said an animal study, conducted by olanzapine manufacturer Eli Lilly at the FDA’s request to determine whether an IM injection could result in “postmortem redistribution” of olanzapine pamoate, “suggested that much of the drug level increase could have occurred after death, a finding that could explain the extremely high blood levels” in the two patients.
Based on its review, the FDA is not recommending any changes to the prescribing information of olanzapine pamoate. However, the agency is advising health care professionals to continue to follow the Risk Evaluation and Mitigation Strategy in place for this drug, which includes mandatory enrollment of patients, prescribers, health care facilities, and pharmacies in the Zyprexa Relprevv Patient Care program. Patients “should not stop receiving treatment without first talking to their health care professionals,” the statement adds.
The REMS requirements include continuous monitoring of patients for 3 hours after the injection, which must be administered at a REMS-certified health care facility that has quick access to emergency response services. A medication guide explaining these and other risks associated with the drug are provided to patients.
The increased risk for severe sedation, including coma, and/or delirium after each olanzapine pamoate injection, also is described in a boxed warning in the prescribing information.
Adverse events associated with Zyprexa Relprevv should be reported to the FDA’s MedWatch program at 800-332-1088 or https://www.accessdata.fda.gov/scripts/medwatch/
The Food and Drug Administration’s review of a study that looked into the unexplained deaths of two patients days after receiving injections of the long-acting injectable formulation of the atypical antipsychotic olanzapine are “inconclusive,” and no labeling changes are currently recommended, the agency has announced.
“We are unable to exclude the possibility that the deaths were caused by rapid, but delayed, entry of the drug into the bloodstream following intramuscular injection,” according to the drug safety communication issued March 23 by the FDA.
Patients who receive olanzapine pamoate must be monitored for 3 hours after an injection because of the risk of post-injection delirium sedation (PDSS) associated with the injection. These patients, however, died 3 to 4 days after having received appropriate IM doses of olanzapine pamoate (Zyprexa Relprevv) and were found to have very high levels of the drug. The deaths and subsequent investigation were announced by the FDA in 2013.
The March 23 statement said an animal study, conducted by olanzapine manufacturer Eli Lilly at the FDA’s request to determine whether an IM injection could result in “postmortem redistribution” of olanzapine pamoate, “suggested that much of the drug level increase could have occurred after death, a finding that could explain the extremely high blood levels” in the two patients.
Based on its review, the FDA is not recommending any changes to the prescribing information of olanzapine pamoate. However, the agency is advising health care professionals to continue to follow the Risk Evaluation and Mitigation Strategy in place for this drug, which includes mandatory enrollment of patients, prescribers, health care facilities, and pharmacies in the Zyprexa Relprevv Patient Care program. Patients “should not stop receiving treatment without first talking to their health care professionals,” the statement adds.
The REMS requirements include continuous monitoring of patients for 3 hours after the injection, which must be administered at a REMS-certified health care facility that has quick access to emergency response services. A medication guide explaining these and other risks associated with the drug are provided to patients.
The increased risk for severe sedation, including coma, and/or delirium after each olanzapine pamoate injection, also is described in a boxed warning in the prescribing information.
Adverse events associated with Zyprexa Relprevv should be reported to the FDA’s MedWatch program at 800-332-1088 or https://www.accessdata.fda.gov/scripts/medwatch/
ADA and EASD Recommend Improvements in Monitoring Insulin Pump Safety
Improved reporting of adverse events associated with insulin pumps and increased funding of registries and clinical studies that evaluate the safety of these devices are among the recommendations included in a statement by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD).
Insulin pumps “appear to provide clinically important and increasing benefits for people with diabetes,” leading to their acceptance by the international diabetes community. However, “there are associated challenges, including potential risks to users,” according to the diabetes technology working group of the ADA and the EASD.
The two organizations reviewed the current approach to the evaluation of the clinical safety of insulin pumps, with the aim of considering “how health care professionals, pump manufacturers, regulatory authorities, and policymakers can best ensure the safety of new and long-standing users of insulin pumps, as the technology continues to develop,” according to the statement, which is in the April issue of Diabetes Care (2015;38:1-7 [doi:10.2337/dc15-0168).
While the rapidly evolving technology in diabetes treatment is “certainly a good thing, we don’t have very good postmarketing surveillance for devices such as insulin pumps, particularly in Europe where manufacturers often introduce products prior to releasing them in the United States,” Dr. Anne Peters, director of the University of Southern California Clinical Diabetes Program, Los Angeles, and one of the statement’s authors, said in a press release issued by the ADA. “We need to make sure we have sufficient data about how the devices are working once they hit the market, so that we can support patients by helping them understand how to prevent errors in using them.”
The working group reviewed evidence from published studies, regulatory authorities, manufacturers of pumps, and other sources. Among the conclusions is that there is a need for a more “standardized and transparent approach to identifying, reporting, and cataloging” adverse event reports, which would help patients and health care providers understand the risks of insulin pump therapy more clearly.
The statement refers to limitations of the postmarketing adverse event reporting systems for devices in the United States and in Europe, MAUDE (Manufacturer and User Facility Device Experience) and EUDAMED (European Databank on Medical Devices). For example, EUDAMED data are not accessible to the public and the MAUDE database is difficult to search.
The statement recommends specific actions that should be undertaken by five groups: regulators, pump manufacturers, professional societies, research funding bodies, and health care teams. For their part, health care teams should “encourage and support pump users under their care to report all” adverse events, and should provide structured training and regular updates for their patients who use pumps, based on standards set by national and international guidelines,
Professional societies should “provide updated evidence-based guidelines on indications for insulin pump therapy,” and “set standards for levels of staffing and skills” for health care teams.
Patient registries have collected information on metabolic control of patients. They should expand their scope to include more data on adverse events associated with insulin pumps and other information, according to the statement. More funding should be provided for trials that evaluate clinically relevant issues, and manufacturer-funded trials should be conducted by “independent investigators to minimize bias and credibility.”
The authors estimated that there could be 750,000 to 1 million pump users worldwide and that most pump users have type 1 diabetes. But a more accurate estimate would be useful to more reliably calculate the rates of malfunctioning pumps and human errors, they added.
Members of the working group did not receive honoraria for writing the manuscript or attending related meetings; their travel costs were paid by the EASD and ADA. Most of the members work with industry, and disclosures included having served as advisers or consultants to companies that manufacture diagnostic or therapeutic products for diabetes. Dr. Peters disclosed having served as a consultant for Medtronic/MiniMed, and having served as a consultant and/or speaker for non–device-related companies.
Improved reporting of adverse events associated with insulin pumps and increased funding of registries and clinical studies that evaluate the safety of these devices are among the recommendations included in a statement by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD).
Insulin pumps “appear to provide clinically important and increasing benefits for people with diabetes,” leading to their acceptance by the international diabetes community. However, “there are associated challenges, including potential risks to users,” according to the diabetes technology working group of the ADA and the EASD.
The two organizations reviewed the current approach to the evaluation of the clinical safety of insulin pumps, with the aim of considering “how health care professionals, pump manufacturers, regulatory authorities, and policymakers can best ensure the safety of new and long-standing users of insulin pumps, as the technology continues to develop,” according to the statement, which is in the April issue of Diabetes Care (2015;38:1-7 [doi:10.2337/dc15-0168).
While the rapidly evolving technology in diabetes treatment is “certainly a good thing, we don’t have very good postmarketing surveillance for devices such as insulin pumps, particularly in Europe where manufacturers often introduce products prior to releasing them in the United States,” Dr. Anne Peters, director of the University of Southern California Clinical Diabetes Program, Los Angeles, and one of the statement’s authors, said in a press release issued by the ADA. “We need to make sure we have sufficient data about how the devices are working once they hit the market, so that we can support patients by helping them understand how to prevent errors in using them.”
The working group reviewed evidence from published studies, regulatory authorities, manufacturers of pumps, and other sources. Among the conclusions is that there is a need for a more “standardized and transparent approach to identifying, reporting, and cataloging” adverse event reports, which would help patients and health care providers understand the risks of insulin pump therapy more clearly.
The statement refers to limitations of the postmarketing adverse event reporting systems for devices in the United States and in Europe, MAUDE (Manufacturer and User Facility Device Experience) and EUDAMED (European Databank on Medical Devices). For example, EUDAMED data are not accessible to the public and the MAUDE database is difficult to search.
The statement recommends specific actions that should be undertaken by five groups: regulators, pump manufacturers, professional societies, research funding bodies, and health care teams. For their part, health care teams should “encourage and support pump users under their care to report all” adverse events, and should provide structured training and regular updates for their patients who use pumps, based on standards set by national and international guidelines,
Professional societies should “provide updated evidence-based guidelines on indications for insulin pump therapy,” and “set standards for levels of staffing and skills” for health care teams.
Patient registries have collected information on metabolic control of patients. They should expand their scope to include more data on adverse events associated with insulin pumps and other information, according to the statement. More funding should be provided for trials that evaluate clinically relevant issues, and manufacturer-funded trials should be conducted by “independent investigators to minimize bias and credibility.”
The authors estimated that there could be 750,000 to 1 million pump users worldwide and that most pump users have type 1 diabetes. But a more accurate estimate would be useful to more reliably calculate the rates of malfunctioning pumps and human errors, they added.
Members of the working group did not receive honoraria for writing the manuscript or attending related meetings; their travel costs were paid by the EASD and ADA. Most of the members work with industry, and disclosures included having served as advisers or consultants to companies that manufacture diagnostic or therapeutic products for diabetes. Dr. Peters disclosed having served as a consultant for Medtronic/MiniMed, and having served as a consultant and/or speaker for non–device-related companies.
Improved reporting of adverse events associated with insulin pumps and increased funding of registries and clinical studies that evaluate the safety of these devices are among the recommendations included in a statement by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD).
Insulin pumps “appear to provide clinically important and increasing benefits for people with diabetes,” leading to their acceptance by the international diabetes community. However, “there are associated challenges, including potential risks to users,” according to the diabetes technology working group of the ADA and the EASD.
The two organizations reviewed the current approach to the evaluation of the clinical safety of insulin pumps, with the aim of considering “how health care professionals, pump manufacturers, regulatory authorities, and policymakers can best ensure the safety of new and long-standing users of insulin pumps, as the technology continues to develop,” according to the statement, which is in the April issue of Diabetes Care (2015;38:1-7 [doi:10.2337/dc15-0168).
While the rapidly evolving technology in diabetes treatment is “certainly a good thing, we don’t have very good postmarketing surveillance for devices such as insulin pumps, particularly in Europe where manufacturers often introduce products prior to releasing them in the United States,” Dr. Anne Peters, director of the University of Southern California Clinical Diabetes Program, Los Angeles, and one of the statement’s authors, said in a press release issued by the ADA. “We need to make sure we have sufficient data about how the devices are working once they hit the market, so that we can support patients by helping them understand how to prevent errors in using them.”
The working group reviewed evidence from published studies, regulatory authorities, manufacturers of pumps, and other sources. Among the conclusions is that there is a need for a more “standardized and transparent approach to identifying, reporting, and cataloging” adverse event reports, which would help patients and health care providers understand the risks of insulin pump therapy more clearly.
The statement refers to limitations of the postmarketing adverse event reporting systems for devices in the United States and in Europe, MAUDE (Manufacturer and User Facility Device Experience) and EUDAMED (European Databank on Medical Devices). For example, EUDAMED data are not accessible to the public and the MAUDE database is difficult to search.
The statement recommends specific actions that should be undertaken by five groups: regulators, pump manufacturers, professional societies, research funding bodies, and health care teams. For their part, health care teams should “encourage and support pump users under their care to report all” adverse events, and should provide structured training and regular updates for their patients who use pumps, based on standards set by national and international guidelines,
Professional societies should “provide updated evidence-based guidelines on indications for insulin pump therapy,” and “set standards for levels of staffing and skills” for health care teams.
Patient registries have collected information on metabolic control of patients. They should expand their scope to include more data on adverse events associated with insulin pumps and other information, according to the statement. More funding should be provided for trials that evaluate clinically relevant issues, and manufacturer-funded trials should be conducted by “independent investigators to minimize bias and credibility.”
The authors estimated that there could be 750,000 to 1 million pump users worldwide and that most pump users have type 1 diabetes. But a more accurate estimate would be useful to more reliably calculate the rates of malfunctioning pumps and human errors, they added.
Members of the working group did not receive honoraria for writing the manuscript or attending related meetings; their travel costs were paid by the EASD and ADA. Most of the members work with industry, and disclosures included having served as advisers or consultants to companies that manufacture diagnostic or therapeutic products for diabetes. Dr. Peters disclosed having served as a consultant for Medtronic/MiniMed, and having served as a consultant and/or speaker for non–device-related companies.
FDA panel backs once-daily ICS/LABA combo for adults, not adolescents
GAITHERSBURG, MD.– A Food and Drug Administration advisory panel supported approval of a fluticasone and vilanterol combination to treat asthma in adults – but the majority voted against approval for adolescents, citing a need for more safety and effectiveness data in that younger population.
GlaxoSmithKline has proposed that two fixed-dose combinations, 100 mcg or 200 mcg of the inhaled corticosteroid (ICS) fluticasone with 25 mcg of the long-acting beta-agonist (LABA) vilanterol in a dry powder inhaler, be approved for maintenance treatment of asthma in patients age 12 years and older, administered once per day.
The FDA approved the 100 mcg/25 mcg dose in 2013 to treat chronic obstructive pulmonary disease; GSK markets it as Breo Ellipta. To earn an asthma indication, the company submitted data from four studies, including a large safety study that addressed safety concerns associated with LABAs in the treatment of asthma.
At the March 19 joint meeting of the FDA’s Pulmonary-Allergy Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, the panels voted 16-4 that the efficacy and safety data supported the approval of both proposed doses in adults.
Many panelists noted the benefit of having a once-daily inhaled treatment to treat asthma, which could improve compliance. Most agreed that the efficacy data in adults provided a substantial amount of evidence that the two doses had a “clinically meaningful benefit” in adults, with beneficial effects on lung function and exacerbation rates.
But the panelists voted 19-1 that the data did not support approval in adolescents, aged 12-17 years. Members expressed uncertainty about safety in that age group, which was studied in the same trials as adults.
In addition, study data indicated that the combination’s beneficial effects were no better than fluticasone alone, panelists said. They also noted a numerical imbalance in hospitalizations among adolescents in a large safety study, and they pointed to the availability of other treatments for that age group.
Panelists voting no also said the efficacy results in adolescents were inconsistent, with “no clear trends in the positive direction,” as one panelist noted. Furthermore, there was no evidence that the combination of fluticasone and vilanterol at either dose was more effective than the ICS alone on forced expiratory volume in 1 second or asthma exacerbations.
Panelists also cautioned that the positive results in adults, who made up the majority of patients enrolled in the studies, could not be extrapolated to adolescents. Thus, there was a need for a separate safety and efficacy study in adolescents.
“At least in the adults, it offers something, perhaps the additional benefit of better compliance,” said the panel chair, Dr. Erik Swenson, professor of medicine and physiology, pulmonary and critical care medicine, University of Washington, Seattle. The combination also was superior at both doses at improving lung function and reducing exacerbations.
But among patients age 12-17 years, “there seems to be no obvious superiority and possibly inferiority against just fluticasone alone,” Dr. Swenson said.
Considering that results of large, ongoing LABA safety trials may soon become available, “I don’t feel comfortable adding a new LABA to the mix right now,” said Dr. Judith Kramer, professor emerita of medicine, Duke University, Durham, N.C., who voted against approval both for pediatric and adult patients.
Because it is taken once a day, the combination’s main advantage would be adherence, Dr. Kramer added. But “it’s not adding any major new therapeutic benefit, and there is the safety concern in the adolescents, and there may well be off-label use.”
Given longstanding concerns about serious asthma-related events associated with LABAs – particularly in pediatric and black patients – the FDA has required manufacturers to conduct postmarketing safety studies evaluating the risks of LABAs when added to an ICS.
Those studies include a GSK study of more than 11,000 patients aged 12 years and older evaluating the safety of Advair, a combination of fluticasone and the LABA salmeterol. Results from that study could be available in early 2016.
For the fluticasone-vilanterol asthma indication, GSK submitted the results of two 12-week and one 24-week lung function studies, enrolling a total of about 2,200 patients whose mean age was 44 years (about 8% were aged 12-17 years).
Another study evaluated the time to first asthma exacerbation in more than 2,000 patients, which included almost 300 patients aged 12-17 years. In that study, the risk of asthma exacerbations was reduced by about 20% among those on the combination, compared with those on fluticasone alone. There were no intubations or deaths from asthma exacerbations.
But in a subgroup analysis of patients aged 12-17 years, conducted by the FDA, the risk of asthma exacerbations was higher among those treated with fluticasone-vilanterol, compared with patients 18 years and older.
In addition, an FDA meta-analysis of asthma-related serious adverse events in the four studies found a numerical imbalance in hospitalizations among adolescents: four hospitalizations among those on the combination drug, but none on fluticasone alone.
If approved, fluticasone-vilanterol would be the first once-daily inhaled ICS/LABA combination treatment for asthma. Fluticasone (100 mcg and 200 mcg) is approved for treating asthma. Unlike the LABAs salmeterol and formoterol, however, vilanterol is not approved for use as a single agent. Vilanterol (as part of the combination product) would be the first new LABA approved for asthma in 15 years.
The FDA usually follows the recommendations of its advisory panels; a decision is expected by April 30. Panelists had no disclosures.
GAITHERSBURG, MD.– A Food and Drug Administration advisory panel supported approval of a fluticasone and vilanterol combination to treat asthma in adults – but the majority voted against approval for adolescents, citing a need for more safety and effectiveness data in that younger population.
GlaxoSmithKline has proposed that two fixed-dose combinations, 100 mcg or 200 mcg of the inhaled corticosteroid (ICS) fluticasone with 25 mcg of the long-acting beta-agonist (LABA) vilanterol in a dry powder inhaler, be approved for maintenance treatment of asthma in patients age 12 years and older, administered once per day.
The FDA approved the 100 mcg/25 mcg dose in 2013 to treat chronic obstructive pulmonary disease; GSK markets it as Breo Ellipta. To earn an asthma indication, the company submitted data from four studies, including a large safety study that addressed safety concerns associated with LABAs in the treatment of asthma.
At the March 19 joint meeting of the FDA’s Pulmonary-Allergy Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, the panels voted 16-4 that the efficacy and safety data supported the approval of both proposed doses in adults.
Many panelists noted the benefit of having a once-daily inhaled treatment to treat asthma, which could improve compliance. Most agreed that the efficacy data in adults provided a substantial amount of evidence that the two doses had a “clinically meaningful benefit” in adults, with beneficial effects on lung function and exacerbation rates.
But the panelists voted 19-1 that the data did not support approval in adolescents, aged 12-17 years. Members expressed uncertainty about safety in that age group, which was studied in the same trials as adults.
In addition, study data indicated that the combination’s beneficial effects were no better than fluticasone alone, panelists said. They also noted a numerical imbalance in hospitalizations among adolescents in a large safety study, and they pointed to the availability of other treatments for that age group.
Panelists voting no also said the efficacy results in adolescents were inconsistent, with “no clear trends in the positive direction,” as one panelist noted. Furthermore, there was no evidence that the combination of fluticasone and vilanterol at either dose was more effective than the ICS alone on forced expiratory volume in 1 second or asthma exacerbations.
Panelists also cautioned that the positive results in adults, who made up the majority of patients enrolled in the studies, could not be extrapolated to adolescents. Thus, there was a need for a separate safety and efficacy study in adolescents.
“At least in the adults, it offers something, perhaps the additional benefit of better compliance,” said the panel chair, Dr. Erik Swenson, professor of medicine and physiology, pulmonary and critical care medicine, University of Washington, Seattle. The combination also was superior at both doses at improving lung function and reducing exacerbations.
But among patients age 12-17 years, “there seems to be no obvious superiority and possibly inferiority against just fluticasone alone,” Dr. Swenson said.
Considering that results of large, ongoing LABA safety trials may soon become available, “I don’t feel comfortable adding a new LABA to the mix right now,” said Dr. Judith Kramer, professor emerita of medicine, Duke University, Durham, N.C., who voted against approval both for pediatric and adult patients.
Because it is taken once a day, the combination’s main advantage would be adherence, Dr. Kramer added. But “it’s not adding any major new therapeutic benefit, and there is the safety concern in the adolescents, and there may well be off-label use.”
Given longstanding concerns about serious asthma-related events associated with LABAs – particularly in pediatric and black patients – the FDA has required manufacturers to conduct postmarketing safety studies evaluating the risks of LABAs when added to an ICS.
Those studies include a GSK study of more than 11,000 patients aged 12 years and older evaluating the safety of Advair, a combination of fluticasone and the LABA salmeterol. Results from that study could be available in early 2016.
For the fluticasone-vilanterol asthma indication, GSK submitted the results of two 12-week and one 24-week lung function studies, enrolling a total of about 2,200 patients whose mean age was 44 years (about 8% were aged 12-17 years).
Another study evaluated the time to first asthma exacerbation in more than 2,000 patients, which included almost 300 patients aged 12-17 years. In that study, the risk of asthma exacerbations was reduced by about 20% among those on the combination, compared with those on fluticasone alone. There were no intubations or deaths from asthma exacerbations.
But in a subgroup analysis of patients aged 12-17 years, conducted by the FDA, the risk of asthma exacerbations was higher among those treated with fluticasone-vilanterol, compared with patients 18 years and older.
In addition, an FDA meta-analysis of asthma-related serious adverse events in the four studies found a numerical imbalance in hospitalizations among adolescents: four hospitalizations among those on the combination drug, but none on fluticasone alone.
If approved, fluticasone-vilanterol would be the first once-daily inhaled ICS/LABA combination treatment for asthma. Fluticasone (100 mcg and 200 mcg) is approved for treating asthma. Unlike the LABAs salmeterol and formoterol, however, vilanterol is not approved for use as a single agent. Vilanterol (as part of the combination product) would be the first new LABA approved for asthma in 15 years.
The FDA usually follows the recommendations of its advisory panels; a decision is expected by April 30. Panelists had no disclosures.
GAITHERSBURG, MD.– A Food and Drug Administration advisory panel supported approval of a fluticasone and vilanterol combination to treat asthma in adults – but the majority voted against approval for adolescents, citing a need for more safety and effectiveness data in that younger population.
GlaxoSmithKline has proposed that two fixed-dose combinations, 100 mcg or 200 mcg of the inhaled corticosteroid (ICS) fluticasone with 25 mcg of the long-acting beta-agonist (LABA) vilanterol in a dry powder inhaler, be approved for maintenance treatment of asthma in patients age 12 years and older, administered once per day.
The FDA approved the 100 mcg/25 mcg dose in 2013 to treat chronic obstructive pulmonary disease; GSK markets it as Breo Ellipta. To earn an asthma indication, the company submitted data from four studies, including a large safety study that addressed safety concerns associated with LABAs in the treatment of asthma.
At the March 19 joint meeting of the FDA’s Pulmonary-Allergy Drugs Advisory Committee and Drug Safety and Risk Management Advisory Committee, the panels voted 16-4 that the efficacy and safety data supported the approval of both proposed doses in adults.
Many panelists noted the benefit of having a once-daily inhaled treatment to treat asthma, which could improve compliance. Most agreed that the efficacy data in adults provided a substantial amount of evidence that the two doses had a “clinically meaningful benefit” in adults, with beneficial effects on lung function and exacerbation rates.
But the panelists voted 19-1 that the data did not support approval in adolescents, aged 12-17 years. Members expressed uncertainty about safety in that age group, which was studied in the same trials as adults.
In addition, study data indicated that the combination’s beneficial effects were no better than fluticasone alone, panelists said. They also noted a numerical imbalance in hospitalizations among adolescents in a large safety study, and they pointed to the availability of other treatments for that age group.
Panelists voting no also said the efficacy results in adolescents were inconsistent, with “no clear trends in the positive direction,” as one panelist noted. Furthermore, there was no evidence that the combination of fluticasone and vilanterol at either dose was more effective than the ICS alone on forced expiratory volume in 1 second or asthma exacerbations.
Panelists also cautioned that the positive results in adults, who made up the majority of patients enrolled in the studies, could not be extrapolated to adolescents. Thus, there was a need for a separate safety and efficacy study in adolescents.
“At least in the adults, it offers something, perhaps the additional benefit of better compliance,” said the panel chair, Dr. Erik Swenson, professor of medicine and physiology, pulmonary and critical care medicine, University of Washington, Seattle. The combination also was superior at both doses at improving lung function and reducing exacerbations.
But among patients age 12-17 years, “there seems to be no obvious superiority and possibly inferiority against just fluticasone alone,” Dr. Swenson said.
Considering that results of large, ongoing LABA safety trials may soon become available, “I don’t feel comfortable adding a new LABA to the mix right now,” said Dr. Judith Kramer, professor emerita of medicine, Duke University, Durham, N.C., who voted against approval both for pediatric and adult patients.
Because it is taken once a day, the combination’s main advantage would be adherence, Dr. Kramer added. But “it’s not adding any major new therapeutic benefit, and there is the safety concern in the adolescents, and there may well be off-label use.”
Given longstanding concerns about serious asthma-related events associated with LABAs – particularly in pediatric and black patients – the FDA has required manufacturers to conduct postmarketing safety studies evaluating the risks of LABAs when added to an ICS.
Those studies include a GSK study of more than 11,000 patients aged 12 years and older evaluating the safety of Advair, a combination of fluticasone and the LABA salmeterol. Results from that study could be available in early 2016.
For the fluticasone-vilanterol asthma indication, GSK submitted the results of two 12-week and one 24-week lung function studies, enrolling a total of about 2,200 patients whose mean age was 44 years (about 8% were aged 12-17 years).
Another study evaluated the time to first asthma exacerbation in more than 2,000 patients, which included almost 300 patients aged 12-17 years. In that study, the risk of asthma exacerbations was reduced by about 20% among those on the combination, compared with those on fluticasone alone. There were no intubations or deaths from asthma exacerbations.
But in a subgroup analysis of patients aged 12-17 years, conducted by the FDA, the risk of asthma exacerbations was higher among those treated with fluticasone-vilanterol, compared with patients 18 years and older.
In addition, an FDA meta-analysis of asthma-related serious adverse events in the four studies found a numerical imbalance in hospitalizations among adolescents: four hospitalizations among those on the combination drug, but none on fluticasone alone.
If approved, fluticasone-vilanterol would be the first once-daily inhaled ICS/LABA combination treatment for asthma. Fluticasone (100 mcg and 200 mcg) is approved for treating asthma. Unlike the LABAs salmeterol and formoterol, however, vilanterol is not approved for use as a single agent. Vilanterol (as part of the combination product) would be the first new LABA approved for asthma in 15 years.
The FDA usually follows the recommendations of its advisory panels; a decision is expected by April 30. Panelists had no disclosures.
AT AN FDA ADVISORY COMMITTEE MEETING