User login
ADA and EASD recommend improvements in monitoring insulin pump safety
Improved reporting of adverse events associated with insulin pumps and increased funding of registries and clinical studies that evaluate the safety of these devices are among the recommendations included in a statement by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD).
Insulin pumps “appear to provide clinically important and increasing benefits for people with diabetes,” leading to their acceptance by the international diabetes community. However, “there are associated challenges, including potential risks to users,” according to the diabetes technology working group of the ADA and the EASD.
The two organizations reviewed the current approach to the evaluation of the clinical safety of insulin pumps, with the aim of considering “how health care professionals, pump manufacturers, regulatory authorities, and policymakers can best ensure the safety of new and long-standing users of insulin pumps, as the technology continues to develop,” according to the statement, which is in the April issue of Diabetes Care (2015;38:1-7 [doi:10.2337/dc15-0168).
While the rapidly evolving technology in diabetes treatment is “certainly a good thing, we don’t have very good postmarketing surveillance for devices such as insulin pumps, particularly in Europe where manufacturers often introduce products prior to releasing them in the United States,” Dr. Anne Peters, director of the University of Southern California Clinical Diabetes Program, Los Angeles, and one of the statement’s authors, said in a press release issued by the ADA. “We need to make sure we have sufficient data about how the devices are working once they hit the market, so that we can support patients by helping them understand how to prevent errors in using them.”
The working group reviewed evidence from published studies, regulatory authorities, manufacturers of pumps, and other sources. Among the conclusions is that there is a need for a more “standardized and transparent approach to identifying, reporting, and cataloging” adverse event reports, which would help patients and health care providers understand the risks of insulin pump therapy more clearly.
The statement refers to limitations of the postmarketing adverse event reporting systems for devices in the United States and in Europe, MAUDE (Manufacturer and User Facility Device Experience) and EUDAMED (European Databank on Medical Devices). For example, EUDAMED data are not accessible to the public and the MAUDE database is difficult to search.
The statement recommends specific actions that should be undertaken by five groups: regulators, pump manufacturers, professional societies, research funding bodies, and health care teams. For their part, health care teams should “encourage and support pump users under their care to report all” adverse events, and should provide structured training and regular updates for their patients who use pumps, based on standards set by national and international guidelines,
Professional societies should “provide updated evidence-based guidelines on indications for insulin pump therapy,” and “set standards for levels of staffing and skills” for health care teams.
Patient registries have collected information on metabolic control of patients. They should expand their scope to include more data on adverse events associated with insulin pumps and other information, according to the statement. More funding should be provided for trials that evaluate clinically relevant issues, and manufacturer-funded trials should be conducted by “independent investigators to minimize bias and credibility.”
The authors estimated that there could be 750,000 to 1 million pump users worldwide and that most pump users have type 1 diabetes. But a more accurate estimate would be useful to more reliably calculate the rates of malfunctioning pumps and human errors, they added.
Members of the working group did not receive honoraria for writing the manuscript or attending related meetings; their travel costs were paid by the EASD and ADA. Most of the members work with industry, and disclosures included having served as advisers or consultants to companies that manufacture diagnostic or therapeutic products for diabetes. Dr. Peters disclosed having served as a consultant for Medtronic/MiniMed, and having served as a consultant and/or speaker for non–device-related companies.
Improved reporting of adverse events associated with insulin pumps and increased funding of registries and clinical studies that evaluate the safety of these devices are among the recommendations included in a statement by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD).
Insulin pumps “appear to provide clinically important and increasing benefits for people with diabetes,” leading to their acceptance by the international diabetes community. However, “there are associated challenges, including potential risks to users,” according to the diabetes technology working group of the ADA and the EASD.
The two organizations reviewed the current approach to the evaluation of the clinical safety of insulin pumps, with the aim of considering “how health care professionals, pump manufacturers, regulatory authorities, and policymakers can best ensure the safety of new and long-standing users of insulin pumps, as the technology continues to develop,” according to the statement, which is in the April issue of Diabetes Care (2015;38:1-7 [doi:10.2337/dc15-0168).
While the rapidly evolving technology in diabetes treatment is “certainly a good thing, we don’t have very good postmarketing surveillance for devices such as insulin pumps, particularly in Europe where manufacturers often introduce products prior to releasing them in the United States,” Dr. Anne Peters, director of the University of Southern California Clinical Diabetes Program, Los Angeles, and one of the statement’s authors, said in a press release issued by the ADA. “We need to make sure we have sufficient data about how the devices are working once they hit the market, so that we can support patients by helping them understand how to prevent errors in using them.”
The working group reviewed evidence from published studies, regulatory authorities, manufacturers of pumps, and other sources. Among the conclusions is that there is a need for a more “standardized and transparent approach to identifying, reporting, and cataloging” adverse event reports, which would help patients and health care providers understand the risks of insulin pump therapy more clearly.
The statement refers to limitations of the postmarketing adverse event reporting systems for devices in the United States and in Europe, MAUDE (Manufacturer and User Facility Device Experience) and EUDAMED (European Databank on Medical Devices). For example, EUDAMED data are not accessible to the public and the MAUDE database is difficult to search.
The statement recommends specific actions that should be undertaken by five groups: regulators, pump manufacturers, professional societies, research funding bodies, and health care teams. For their part, health care teams should “encourage and support pump users under their care to report all” adverse events, and should provide structured training and regular updates for their patients who use pumps, based on standards set by national and international guidelines,
Professional societies should “provide updated evidence-based guidelines on indications for insulin pump therapy,” and “set standards for levels of staffing and skills” for health care teams.
Patient registries have collected information on metabolic control of patients. They should expand their scope to include more data on adverse events associated with insulin pumps and other information, according to the statement. More funding should be provided for trials that evaluate clinically relevant issues, and manufacturer-funded trials should be conducted by “independent investigators to minimize bias and credibility.”
The authors estimated that there could be 750,000 to 1 million pump users worldwide and that most pump users have type 1 diabetes. But a more accurate estimate would be useful to more reliably calculate the rates of malfunctioning pumps and human errors, they added.
Members of the working group did not receive honoraria for writing the manuscript or attending related meetings; their travel costs were paid by the EASD and ADA. Most of the members work with industry, and disclosures included having served as advisers or consultants to companies that manufacture diagnostic or therapeutic products for diabetes. Dr. Peters disclosed having served as a consultant for Medtronic/MiniMed, and having served as a consultant and/or speaker for non–device-related companies.
Improved reporting of adverse events associated with insulin pumps and increased funding of registries and clinical studies that evaluate the safety of these devices are among the recommendations included in a statement by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD).
Insulin pumps “appear to provide clinically important and increasing benefits for people with diabetes,” leading to their acceptance by the international diabetes community. However, “there are associated challenges, including potential risks to users,” according to the diabetes technology working group of the ADA and the EASD.
The two organizations reviewed the current approach to the evaluation of the clinical safety of insulin pumps, with the aim of considering “how health care professionals, pump manufacturers, regulatory authorities, and policymakers can best ensure the safety of new and long-standing users of insulin pumps, as the technology continues to develop,” according to the statement, which is in the April issue of Diabetes Care (2015;38:1-7 [doi:10.2337/dc15-0168).
While the rapidly evolving technology in diabetes treatment is “certainly a good thing, we don’t have very good postmarketing surveillance for devices such as insulin pumps, particularly in Europe where manufacturers often introduce products prior to releasing them in the United States,” Dr. Anne Peters, director of the University of Southern California Clinical Diabetes Program, Los Angeles, and one of the statement’s authors, said in a press release issued by the ADA. “We need to make sure we have sufficient data about how the devices are working once they hit the market, so that we can support patients by helping them understand how to prevent errors in using them.”
The working group reviewed evidence from published studies, regulatory authorities, manufacturers of pumps, and other sources. Among the conclusions is that there is a need for a more “standardized and transparent approach to identifying, reporting, and cataloging” adverse event reports, which would help patients and health care providers understand the risks of insulin pump therapy more clearly.
The statement refers to limitations of the postmarketing adverse event reporting systems for devices in the United States and in Europe, MAUDE (Manufacturer and User Facility Device Experience) and EUDAMED (European Databank on Medical Devices). For example, EUDAMED data are not accessible to the public and the MAUDE database is difficult to search.
The statement recommends specific actions that should be undertaken by five groups: regulators, pump manufacturers, professional societies, research funding bodies, and health care teams. For their part, health care teams should “encourage and support pump users under their care to report all” adverse events, and should provide structured training and regular updates for their patients who use pumps, based on standards set by national and international guidelines,
Professional societies should “provide updated evidence-based guidelines on indications for insulin pump therapy,” and “set standards for levels of staffing and skills” for health care teams.
Patient registries have collected information on metabolic control of patients. They should expand their scope to include more data on adverse events associated with insulin pumps and other information, according to the statement. More funding should be provided for trials that evaluate clinically relevant issues, and manufacturer-funded trials should be conducted by “independent investigators to minimize bias and credibility.”
The authors estimated that there could be 750,000 to 1 million pump users worldwide and that most pump users have type 1 diabetes. But a more accurate estimate would be useful to more reliably calculate the rates of malfunctioning pumps and human errors, they added.
Members of the working group did not receive honoraria for writing the manuscript or attending related meetings; their travel costs were paid by the EASD and ADA. Most of the members work with industry, and disclosures included having served as advisers or consultants to companies that manufacture diagnostic or therapeutic products for diabetes. Dr. Peters disclosed having served as a consultant for Medtronic/MiniMed, and having served as a consultant and/or speaker for non–device-related companies.
FROM DIABETES CARE
FDA approves cholic acid for rare metabolic conditions
The Food and Drug Administration approved cholic acid capsules for treatment of children aged 3 weeks and older and adults with bile acid synthesis disorders due to single enzyme defects. It is the first drug approved for this indication, the agency announced.
The bile acid also was approved as adjunctive treatment of peroxisomal disorders including Zellweger spectrum disorders in patients who exhibit manifestations of liver disease, steatorrhea, or complications from decreased fat soluble vitamin absorption. Cholic acid will be marketed as Cholbam by Asklepion Pharmaceuticals, the FDA said in its statement.
Approval was based on uncontrolled studies, which evaluated treatment of patients, mostly children, over a period of 18 years. In the study of 62 patients with bile acid synthesis disorders due to single enzyme defects, 64% of patients with evaluable data responded with improvements in baseline liver function tests and weight; two-thirds of patients survived longer than 3 years. In the study of cholic acid for the treatment of peroxisomal disorder in 31 children, improvements in baseline liver function tests and weight were noted in 46%; 42% of patients survived longer than 3 years.
Diarrhea was the most common adverse event associated with treatment. “The use of Cholbam should be carefully monitored by an experienced hepatologist or pediatric gastroenterologist, and treatment discontinued in patients developing worsening liver function,” according to the FDA statement.
The manufacturer is required to conduct a postapproval observational study to evaluate the long-term safety of cholic acid.
The prescribing information is available at www.accessdata.fda.gov/drugsatfda_docs/label/2015/205750s000lbl.pdf.
The Food and Drug Administration approved cholic acid capsules for treatment of children aged 3 weeks and older and adults with bile acid synthesis disorders due to single enzyme defects. It is the first drug approved for this indication, the agency announced.
The bile acid also was approved as adjunctive treatment of peroxisomal disorders including Zellweger spectrum disorders in patients who exhibit manifestations of liver disease, steatorrhea, or complications from decreased fat soluble vitamin absorption. Cholic acid will be marketed as Cholbam by Asklepion Pharmaceuticals, the FDA said in its statement.
Approval was based on uncontrolled studies, which evaluated treatment of patients, mostly children, over a period of 18 years. In the study of 62 patients with bile acid synthesis disorders due to single enzyme defects, 64% of patients with evaluable data responded with improvements in baseline liver function tests and weight; two-thirds of patients survived longer than 3 years. In the study of cholic acid for the treatment of peroxisomal disorder in 31 children, improvements in baseline liver function tests and weight were noted in 46%; 42% of patients survived longer than 3 years.
Diarrhea was the most common adverse event associated with treatment. “The use of Cholbam should be carefully monitored by an experienced hepatologist or pediatric gastroenterologist, and treatment discontinued in patients developing worsening liver function,” according to the FDA statement.
The manufacturer is required to conduct a postapproval observational study to evaluate the long-term safety of cholic acid.
The prescribing information is available at www.accessdata.fda.gov/drugsatfda_docs/label/2015/205750s000lbl.pdf.
The Food and Drug Administration approved cholic acid capsules for treatment of children aged 3 weeks and older and adults with bile acid synthesis disorders due to single enzyme defects. It is the first drug approved for this indication, the agency announced.
The bile acid also was approved as adjunctive treatment of peroxisomal disorders including Zellweger spectrum disorders in patients who exhibit manifestations of liver disease, steatorrhea, or complications from decreased fat soluble vitamin absorption. Cholic acid will be marketed as Cholbam by Asklepion Pharmaceuticals, the FDA said in its statement.
Approval was based on uncontrolled studies, which evaluated treatment of patients, mostly children, over a period of 18 years. In the study of 62 patients with bile acid synthesis disorders due to single enzyme defects, 64% of patients with evaluable data responded with improvements in baseline liver function tests and weight; two-thirds of patients survived longer than 3 years. In the study of cholic acid for the treatment of peroxisomal disorder in 31 children, improvements in baseline liver function tests and weight were noted in 46%; 42% of patients survived longer than 3 years.
Diarrhea was the most common adverse event associated with treatment. “The use of Cholbam should be carefully monitored by an experienced hepatologist or pediatric gastroenterologist, and treatment discontinued in patients developing worsening liver function,” according to the FDA statement.
The manufacturer is required to conduct a postapproval observational study to evaluate the long-term safety of cholic acid.
The prescribing information is available at www.accessdata.fda.gov/drugsatfda_docs/label/2015/205750s000lbl.pdf.
FDA guidance focuses on infections with reusable devices, including duodenoscopes
Recommendations to manufacturers about improving the safety of reusable medical devices and an upcoming advisory committee meeting on duodenoscope-associated infections are two efforts recently announced by the Food and Drug Administration that address the risks associated with reusable devices.
A final guidance document for industry on reprocessing reusable medical devices includes recommendations “aimed at helping device manufacturers develop safer reusable devices, especially those devices that pose a greater risk of infection,” according to the March 12 announcement. Also included in the guidance are criteria that should be met in instructions for reprocessing reusable devices, “to ensure users understand and correctly follow the reprocessing instructions,” and recommendations that manufacturers should consider “reprocessing challenges” at the early stages of the design of such devices.
The same announcement said that in mid-May, the FDA was convening a 2-day meeting of the agency’s Gastroenterology and Urology Devices Panel to discuss the recent reports of infections associated with the use of duodenoscopes in endoscopic retrograde cholangiopancreatography (ERCP) procedures in U.S. hospitals.
The announcement was issued less than a month after the agency alerted health care professionals and the public about the association with duodenoscopes and the transmission of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures, despite proper cleaning and disinfection of the devices. Between January 2013 and December 2014, the agency received 75 medical device adverse event reports for about 135 patients in the United States “relating to possible microbial transmission from reprocessed duodenoscopes,” according to the safety communication issued by the FDA on Feb. 19.
These reports and cases described in the medical literature have occurred even when manufacturer instructions for cleaning and sterilization were followed.
“Although the complex design of duodenoscopes improves the efficiency and effectiveness of ERCP, it causes challenges for cleaning and high-level disinfection,” according to the statement, which pointed out that it can be difficult to access some parts of the duodenoscopes when they are cleaned. Problems include the “elevator” mechanism at the tip of the duodenoscope, which should be manually brushed, but a brush may not be able to reach microscopic crevices in this mechanism and “residual body fluids and organic debris may remain in these crevices after cleaning and disinfection,” possibly exposing patients to serious infections if the fluids are contaminated with microbes.
The infections reported include carbapenem-resistant Enterobacteriaceae (CRE), according to the first FDA statement, which did not mention whether any of the reports were fatal.
But on Feb. 18, the UCLA Health System announced that CRE may have been transmitted to seven patients during ERCP procedures, and may have contributed to the death of two of the patients. The two devices implicated in these cases are no longer used and the medical center has started to use a decontamination process “that goes above and beyond manufacturer and national standards” for the devices, the statement said. More than 100 patients who had an ERCP between October 2014 and January 2015 at UCLA have been notified they may have been infected with CRE.
The FDA statement includes recommendations for facilities and staff that reprocess duodenoscopes, for patients, and for health care professionals. One recommendation is to take a duodenoscope out of service if there is any suspicion it may be linked to a multidrug-resistant infection in a patient who has undergone ERCP.
In early March, another outbreak was reported at Cedars-Sinai Medical Center in Los Angeles, which announced that four patients who had undergone an ERCP procedure between August 2014 and January 2015 with the same duodenoscope had been infected with CRE, “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes recommended in instructions provided by the manufacturer (Olympus Corporation) and the FDA.” This duodenoscope was the TJF-Q180 V model, a Cedars-Sinai spokesperson confirmed.
This particular duodenoscope has not yet been cleared for marketing, but has been used commercially, according to an FDA statement March 4 updating the duodenoscope-associated infection issue. The statement said that there was “no evidence” that the lack of clearance was associated with infections, and that the reported infections were associated with duodenoscopes from all three manufacturers of the devices used in the United States. In addition, the FDA statement noted that if the TJF-Q180 V duodenoscope was removed from the market, there may not be enough duodenoscopes to meet “the clinical demand in the United States of approximately 500,000 procedures per year.”
At the advisory panel meeting May 14-15, the FDA will ask the expert panel to discuss and make recommendations on various issues, including approaches that ensure patient safety during ERCP procedures and the effectiveness of the cleaning, disinfection, and sterilization procedures for duodenoscopes.
The FDA is asking health care professionals to report any infections possibly related to ERCP duodenoscopes to the manufacturers and the FDA’s MedWatch program.
The Centers for Disease Control and Prevention has provided an interim protocol for health care facilities, with information on monitoring for bacterial contamination of duodenoscopes after reprocessing and other reprocessing issues.
Recommendations to manufacturers about improving the safety of reusable medical devices and an upcoming advisory committee meeting on duodenoscope-associated infections are two efforts recently announced by the Food and Drug Administration that address the risks associated with reusable devices.
A final guidance document for industry on reprocessing reusable medical devices includes recommendations “aimed at helping device manufacturers develop safer reusable devices, especially those devices that pose a greater risk of infection,” according to the March 12 announcement. Also included in the guidance are criteria that should be met in instructions for reprocessing reusable devices, “to ensure users understand and correctly follow the reprocessing instructions,” and recommendations that manufacturers should consider “reprocessing challenges” at the early stages of the design of such devices.
The same announcement said that in mid-May, the FDA was convening a 2-day meeting of the agency’s Gastroenterology and Urology Devices Panel to discuss the recent reports of infections associated with the use of duodenoscopes in endoscopic retrograde cholangiopancreatography (ERCP) procedures in U.S. hospitals.
The announcement was issued less than a month after the agency alerted health care professionals and the public about the association with duodenoscopes and the transmission of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures, despite proper cleaning and disinfection of the devices. Between January 2013 and December 2014, the agency received 75 medical device adverse event reports for about 135 patients in the United States “relating to possible microbial transmission from reprocessed duodenoscopes,” according to the safety communication issued by the FDA on Feb. 19.
These reports and cases described in the medical literature have occurred even when manufacturer instructions for cleaning and sterilization were followed.
“Although the complex design of duodenoscopes improves the efficiency and effectiveness of ERCP, it causes challenges for cleaning and high-level disinfection,” according to the statement, which pointed out that it can be difficult to access some parts of the duodenoscopes when they are cleaned. Problems include the “elevator” mechanism at the tip of the duodenoscope, which should be manually brushed, but a brush may not be able to reach microscopic crevices in this mechanism and “residual body fluids and organic debris may remain in these crevices after cleaning and disinfection,” possibly exposing patients to serious infections if the fluids are contaminated with microbes.
The infections reported include carbapenem-resistant Enterobacteriaceae (CRE), according to the first FDA statement, which did not mention whether any of the reports were fatal.
But on Feb. 18, the UCLA Health System announced that CRE may have been transmitted to seven patients during ERCP procedures, and may have contributed to the death of two of the patients. The two devices implicated in these cases are no longer used and the medical center has started to use a decontamination process “that goes above and beyond manufacturer and national standards” for the devices, the statement said. More than 100 patients who had an ERCP between October 2014 and January 2015 at UCLA have been notified they may have been infected with CRE.
The FDA statement includes recommendations for facilities and staff that reprocess duodenoscopes, for patients, and for health care professionals. One recommendation is to take a duodenoscope out of service if there is any suspicion it may be linked to a multidrug-resistant infection in a patient who has undergone ERCP.
In early March, another outbreak was reported at Cedars-Sinai Medical Center in Los Angeles, which announced that four patients who had undergone an ERCP procedure between August 2014 and January 2015 with the same duodenoscope had been infected with CRE, “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes recommended in instructions provided by the manufacturer (Olympus Corporation) and the FDA.” This duodenoscope was the TJF-Q180 V model, a Cedars-Sinai spokesperson confirmed.
This particular duodenoscope has not yet been cleared for marketing, but has been used commercially, according to an FDA statement March 4 updating the duodenoscope-associated infection issue. The statement said that there was “no evidence” that the lack of clearance was associated with infections, and that the reported infections were associated with duodenoscopes from all three manufacturers of the devices used in the United States. In addition, the FDA statement noted that if the TJF-Q180 V duodenoscope was removed from the market, there may not be enough duodenoscopes to meet “the clinical demand in the United States of approximately 500,000 procedures per year.”
At the advisory panel meeting May 14-15, the FDA will ask the expert panel to discuss and make recommendations on various issues, including approaches that ensure patient safety during ERCP procedures and the effectiveness of the cleaning, disinfection, and sterilization procedures for duodenoscopes.
The FDA is asking health care professionals to report any infections possibly related to ERCP duodenoscopes to the manufacturers and the FDA’s MedWatch program.
The Centers for Disease Control and Prevention has provided an interim protocol for health care facilities, with information on monitoring for bacterial contamination of duodenoscopes after reprocessing and other reprocessing issues.
Recommendations to manufacturers about improving the safety of reusable medical devices and an upcoming advisory committee meeting on duodenoscope-associated infections are two efforts recently announced by the Food and Drug Administration that address the risks associated with reusable devices.
A final guidance document for industry on reprocessing reusable medical devices includes recommendations “aimed at helping device manufacturers develop safer reusable devices, especially those devices that pose a greater risk of infection,” according to the March 12 announcement. Also included in the guidance are criteria that should be met in instructions for reprocessing reusable devices, “to ensure users understand and correctly follow the reprocessing instructions,” and recommendations that manufacturers should consider “reprocessing challenges” at the early stages of the design of such devices.
The same announcement said that in mid-May, the FDA was convening a 2-day meeting of the agency’s Gastroenterology and Urology Devices Panel to discuss the recent reports of infections associated with the use of duodenoscopes in endoscopic retrograde cholangiopancreatography (ERCP) procedures in U.S. hospitals.
The announcement was issued less than a month after the agency alerted health care professionals and the public about the association with duodenoscopes and the transmission of multidrug-resistant bacterial infections in patients who had undergone ERCP procedures, despite proper cleaning and disinfection of the devices. Between January 2013 and December 2014, the agency received 75 medical device adverse event reports for about 135 patients in the United States “relating to possible microbial transmission from reprocessed duodenoscopes,” according to the safety communication issued by the FDA on Feb. 19.
These reports and cases described in the medical literature have occurred even when manufacturer instructions for cleaning and sterilization were followed.
“Although the complex design of duodenoscopes improves the efficiency and effectiveness of ERCP, it causes challenges for cleaning and high-level disinfection,” according to the statement, which pointed out that it can be difficult to access some parts of the duodenoscopes when they are cleaned. Problems include the “elevator” mechanism at the tip of the duodenoscope, which should be manually brushed, but a brush may not be able to reach microscopic crevices in this mechanism and “residual body fluids and organic debris may remain in these crevices after cleaning and disinfection,” possibly exposing patients to serious infections if the fluids are contaminated with microbes.
The infections reported include carbapenem-resistant Enterobacteriaceae (CRE), according to the first FDA statement, which did not mention whether any of the reports were fatal.
But on Feb. 18, the UCLA Health System announced that CRE may have been transmitted to seven patients during ERCP procedures, and may have contributed to the death of two of the patients. The two devices implicated in these cases are no longer used and the medical center has started to use a decontamination process “that goes above and beyond manufacturer and national standards” for the devices, the statement said. More than 100 patients who had an ERCP between October 2014 and January 2015 at UCLA have been notified they may have been infected with CRE.
The FDA statement includes recommendations for facilities and staff that reprocess duodenoscopes, for patients, and for health care professionals. One recommendation is to take a duodenoscope out of service if there is any suspicion it may be linked to a multidrug-resistant infection in a patient who has undergone ERCP.
In early March, another outbreak was reported at Cedars-Sinai Medical Center in Los Angeles, which announced that four patients who had undergone an ERCP procedure between August 2014 and January 2015 with the same duodenoscope had been infected with CRE, “despite the fact that Cedars-Sinai meticulously followed the disinfection procedure for duodenoscopes recommended in instructions provided by the manufacturer (Olympus Corporation) and the FDA.” This duodenoscope was the TJF-Q180 V model, a Cedars-Sinai spokesperson confirmed.
This particular duodenoscope has not yet been cleared for marketing, but has been used commercially, according to an FDA statement March 4 updating the duodenoscope-associated infection issue. The statement said that there was “no evidence” that the lack of clearance was associated with infections, and that the reported infections were associated with duodenoscopes from all three manufacturers of the devices used in the United States. In addition, the FDA statement noted that if the TJF-Q180 V duodenoscope was removed from the market, there may not be enough duodenoscopes to meet “the clinical demand in the United States of approximately 500,000 procedures per year.”
At the advisory panel meeting May 14-15, the FDA will ask the expert panel to discuss and make recommendations on various issues, including approaches that ensure patient safety during ERCP procedures and the effectiveness of the cleaning, disinfection, and sterilization procedures for duodenoscopes.
The FDA is asking health care professionals to report any infections possibly related to ERCP duodenoscopes to the manufacturers and the FDA’s MedWatch program.
The Centers for Disease Control and Prevention has provided an interim protocol for health care facilities, with information on monitoring for bacterial contamination of duodenoscopes after reprocessing and other reprocessing issues.
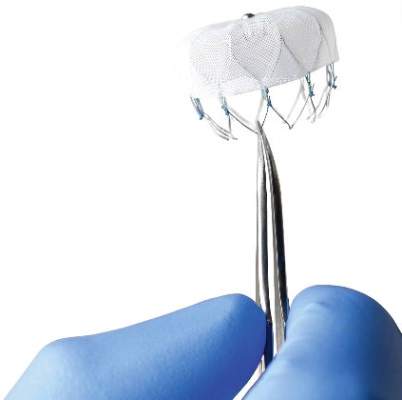
Novel Watchman device approved as warfarin alternative in atrial fib
The Watchman left atrial appendage (LAA) closure device has been approved in the United States as an alternative to warfarin for patients with nonvalvular atrial fibrillation, for a narrower indication than the one submitted for approval to the Food and Drug Administration.
The device is a percutaneously delivered permanent cardiac implant placed in the LAA to prevent the embolization of thrombi formed in the LAA, and is manufactured by Boston Scientific. The FDA approved the Watchman for reducing the risk of thromboembolism from the LAA in patients with nonvalvular atrial fibrillation “who are at increased risk for stroke and systemic embolism based on CHADS2 or CHA2DS2-VASc scores, are deemed by their physicians to be suitable for warfarin; and have an appropriate rationale to seek a nonpharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device, compared to warfarin,” according to a statement issued by the company on March 13.
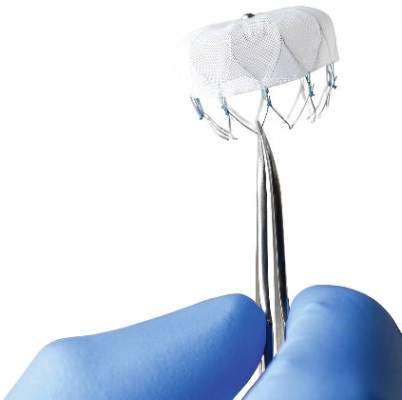
The approved indication is worded differently than the proposed indication that was submitted to the FDA for approval and discussed at an FDA panel meeting in October, to “prevent thromboembolism from the left atrial appendage.” The changes in the indication include the replacement of “prevent” with “reduce the risk” of thromboembolism, and the addition of the following qualifiers: In patients who “are deemed by their physicians to be suitable for warfarin,” and who have “an appropriate rationale to seek a nonpharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device compared to warfarin.”
“These changes were made to more accurately reflect the appropriate patient population for this device,” according to an FDA spokesperson.
At a meeting in October 2014, the FDA’s Circulatory System Devices Panel voted 6-5 with one abstention that the benefits of the device outweighed its risks for the proposed indication, but several panelists who voted no said they would support approval of a second-line indication. In addition, panelists voting on both sides of this question said that the indication was too broad and should be revised to describe the device as a second-line alternative to warfarin, making clear it is not appropriate for all warfarin-eligible patients. (At the meeting, the panel unanimously agreed that there was “reasonable assurance” that the device was safe for use in this population.)
At the first advisory panel meeting on the device, in December 2013, the panel voted 13-1 to recommend approval, based on data from the PREVAIL and PROTECT-AF studies, which compared the device to chronic warfarin, and information from the Continued Access to PREVAIL (CAP2) registry.PREVAIL compared implantation of the device – with 45 days of warfarin plus 81 mg of aspirin for 45 days, followed by 325 mg of aspirin and 75 mg of clopidogrel through 6 months, followed by 325 mg of aspirin a day indefinitely – to chronic warfarin.
The October meeting was convened by the FDA to review longer follow-up data from PREVAIL, which found additional cases of ischemic strokes in the Watchman group and none in the warfarin-treated group.
The Watchman device has been available outside of the United States since 2009, is registered in 75 countries, and has been used to treat more than 10,000 patients, according to Boston Scientific.
The Watchman left atrial appendage (LAA) closure device has been approved in the United States as an alternative to warfarin for patients with nonvalvular atrial fibrillation, for a narrower indication than the one submitted for approval to the Food and Drug Administration.
The device is a percutaneously delivered permanent cardiac implant placed in the LAA to prevent the embolization of thrombi formed in the LAA, and is manufactured by Boston Scientific. The FDA approved the Watchman for reducing the risk of thromboembolism from the LAA in patients with nonvalvular atrial fibrillation “who are at increased risk for stroke and systemic embolism based on CHADS2 or CHA2DS2-VASc scores, are deemed by their physicians to be suitable for warfarin; and have an appropriate rationale to seek a nonpharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device, compared to warfarin,” according to a statement issued by the company on March 13.
The approved indication is worded differently than the proposed indication that was submitted to the FDA for approval and discussed at an FDA panel meeting in October, to “prevent thromboembolism from the left atrial appendage.” The changes in the indication include the replacement of “prevent” with “reduce the risk” of thromboembolism, and the addition of the following qualifiers: In patients who “are deemed by their physicians to be suitable for warfarin,” and who have “an appropriate rationale to seek a nonpharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device compared to warfarin.”
“These changes were made to more accurately reflect the appropriate patient population for this device,” according to an FDA spokesperson.
At a meeting in October 2014, the FDA’s Circulatory System Devices Panel voted 6-5 with one abstention that the benefits of the device outweighed its risks for the proposed indication, but several panelists who voted no said they would support approval of a second-line indication. In addition, panelists voting on both sides of this question said that the indication was too broad and should be revised to describe the device as a second-line alternative to warfarin, making clear it is not appropriate for all warfarin-eligible patients. (At the meeting, the panel unanimously agreed that there was “reasonable assurance” that the device was safe for use in this population.)
At the first advisory panel meeting on the device, in December 2013, the panel voted 13-1 to recommend approval, based on data from the PREVAIL and PROTECT-AF studies, which compared the device to chronic warfarin, and information from the Continued Access to PREVAIL (CAP2) registry.PREVAIL compared implantation of the device – with 45 days of warfarin plus 81 mg of aspirin for 45 days, followed by 325 mg of aspirin and 75 mg of clopidogrel through 6 months, followed by 325 mg of aspirin a day indefinitely – to chronic warfarin.
The October meeting was convened by the FDA to review longer follow-up data from PREVAIL, which found additional cases of ischemic strokes in the Watchman group and none in the warfarin-treated group.
The Watchman device has been available outside of the United States since 2009, is registered in 75 countries, and has been used to treat more than 10,000 patients, according to Boston Scientific.
The Watchman left atrial appendage (LAA) closure device has been approved in the United States as an alternative to warfarin for patients with nonvalvular atrial fibrillation, for a narrower indication than the one submitted for approval to the Food and Drug Administration.
The device is a percutaneously delivered permanent cardiac implant placed in the LAA to prevent the embolization of thrombi formed in the LAA, and is manufactured by Boston Scientific. The FDA approved the Watchman for reducing the risk of thromboembolism from the LAA in patients with nonvalvular atrial fibrillation “who are at increased risk for stroke and systemic embolism based on CHADS2 or CHA2DS2-VASc scores, are deemed by their physicians to be suitable for warfarin; and have an appropriate rationale to seek a nonpharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device, compared to warfarin,” according to a statement issued by the company on March 13.
The approved indication is worded differently than the proposed indication that was submitted to the FDA for approval and discussed at an FDA panel meeting in October, to “prevent thromboembolism from the left atrial appendage.” The changes in the indication include the replacement of “prevent” with “reduce the risk” of thromboembolism, and the addition of the following qualifiers: In patients who “are deemed by their physicians to be suitable for warfarin,” and who have “an appropriate rationale to seek a nonpharmacologic alternative to warfarin, taking into account the safety and effectiveness of the device compared to warfarin.”
“These changes were made to more accurately reflect the appropriate patient population for this device,” according to an FDA spokesperson.
At a meeting in October 2014, the FDA’s Circulatory System Devices Panel voted 6-5 with one abstention that the benefits of the device outweighed its risks for the proposed indication, but several panelists who voted no said they would support approval of a second-line indication. In addition, panelists voting on both sides of this question said that the indication was too broad and should be revised to describe the device as a second-line alternative to warfarin, making clear it is not appropriate for all warfarin-eligible patients. (At the meeting, the panel unanimously agreed that there was “reasonable assurance” that the device was safe for use in this population.)
At the first advisory panel meeting on the device, in December 2013, the panel voted 13-1 to recommend approval, based on data from the PREVAIL and PROTECT-AF studies, which compared the device to chronic warfarin, and information from the Continued Access to PREVAIL (CAP2) registry.PREVAIL compared implantation of the device – with 45 days of warfarin plus 81 mg of aspirin for 45 days, followed by 325 mg of aspirin and 75 mg of clopidogrel through 6 months, followed by 325 mg of aspirin a day indefinitely – to chronic warfarin.
The October meeting was convened by the FDA to review longer follow-up data from PREVAIL, which found additional cases of ischemic strokes in the Watchman group and none in the warfarin-treated group.
The Watchman device has been available outside of the United States since 2009, is registered in 75 countries, and has been used to treat more than 10,000 patients, according to Boston Scientific.
FDA Panel Backs Injectable Treatment For Chin Fat
SILVER SPRING, MD – A less-invasive alternative to liposuction and other cosmetic treatments for submental fat will likely become available in the United States in the wake of a Food and Drug Administration advisory panel’s unanimous support for approval of deoxycholic acid as an injection into subcutaneous fat under the chin.
At a meeting on March 9, the FDA’s dermatologic and ophthalmic drugs advisory committee voted 17-0 that the efficacy and safety data on 1% deoxycholic acid (DCA) injection supported approval for the proposed indication, “the improvement in the appearance of moderate to severe convexity or fullness associated with submental fat” in adults.
The product is a synthetic version of naturally occurring DCA, an endogenous secondary bile acid that “serves to emulsify and solubilize dietary fat,” and for cosmetic use, “disrupts cell membranes of adipocytes, causing destruction of fat cells,” according to the manufacturer, Kythera Biopharmaceuticals.
The regimen proposed by Kythera is up to six treatment sessions with 4-week intervals between sessions. Each session would involve up to 50 injections, with 0.2 mL of DCA solution per injection, administered subcutaneously using a grid to control spacing of injections. The product would be the first drug approved for treatment of submental fat.
A pair of phase III studies including about 1,000 adults (median age, 48-49 years) randomized patients to treatment with DCA or placebo saline injections of 6 or fewer treatments, one month apart. The effects were evaluated with validated Clinician-Reported and a Patient-Reported Submental Fat Rating Scales (CR-SMFRS and PR-SMFRS), which Kythera developed in consultation with the FDA. On the CR-SMFRS, a 5-point rating scale ranged from 0 (no localized submental fat evident) to 4 (extreme submental convexity).
Most patients enrolled were rated as 2 (moderate, with prominent localized submental fat) or 3 (severe, with marked localized submental fat); the company is not seeking an indication for extreme submental convexity. On the PR-SMFRS, patients had five options to check off, based on how much fat they thought they had under their chin when looking in a mirror, ranging from “no chin fat at all” to a “very large amount of fat.”
The coprimary efficacy endpoints were those who had at least a 2-grade improvement on both scales, or at least a 1-grade improvement in both scales, 12 weeks after the first treatment.
Continue for study results >>
In the two studies, 70% and 66% of those treated with DCA achieved at least a 1-grade improvement in both scales, vs.19% and 22%, respectively, in the placebo group. The difference was statistically significant. In addition, 13% and 19% of those treated with DCA in the two studies had at least a 2-grade improvement in both scales at that time, vs. 0-3% of those in the placebo group, also statistically significant differences.
One issue raised at the meeting was that the scales were somewhat subjective. A more objective secondary endpoint used MRI volumetric measurements to evaluate results in 449 patients from both studies; this measure determined that 40%-46% of those treated with DCA achieved at least a 10% reduction in MRI volume of submental fat, vs. 5% of those on placebo.
Most patients in the placebo and DCA groups experienced adverse events that were mostly mild and were related to the injections. The rate of moderate adverse events was 17% in the DCA-treated patients and 10% in the placebo group. Injection site reactions more common in the DCA group vs. placebo included pain (70% vs. 31%), numbness (66% vs. 6%), edema (60% vs. 29%), and swelling (33% vs. 16%). There were 11 cases of dysphagia, all of which resolved, except for 1 patient who withdrew from the study and was not available for follow-up. There have been no cases of glandular injuries, including salivary or thyroid injuries, according to the company.
In a safety database of almost 2,000 patients, including 1,050 treated with DCA, the rate of marginal mandibular nerve injuries has been 4% in those treated with DCA. The nerve injuries lasted a median of 45 days (1-298 days) and all resolved. The rate of dysphagia was 2% among those on DCA , lasting a median of 3 days, vs. fewer than 1% among those on placebo, according to the FDA reviewers. One patient did not resolve, but the company noted that this patient dropped out of the study and was not followed. There were no cases of anaphylactic reactions; injection site urticaria was rare and resolved.
In addition, the potential risk of marginal mandibular nerve injuries, issues raised during the meeting included the concerns that the treatment would be used by clinicians who were not adequately trained to provide the treatment or not adequately familiar with the anatomy of the treated area. Other concerns include potential use of the product in areas that include the periorbitum, as well as off-label use of large amounts in different parts of the body, such as the abdomen or thighs. The company said that the product will not be provided to clinicians who have not gone through the planned training program and that it was better suited to small areas of the body.
While there are some questions that still need to be answered and concerns about off-label use and the need for proper training, the panel agreed this was safe and effective if used properly with appropriate training and appropriate precautions, said the panel chair, Dr. Lynn A. Drake of the department of dermatology, Massachusetts General Hospital, Boston.
DCA should not be used off label, a concern that could be at least partially addressed in the label, and “without appropriate training, this product could be misused and mishandled and cause damage,” she added. In addition, because of some “very novel” aspects to this treatment, postmarketing data are needed, addressing questions that include whether it causes scarring that could affect surgery and other interventions in the future, she said.
The FDA usually follows the recommendations of its advisory panels. None of the panel members had relevant disclosures.
SILVER SPRING, MD – A less-invasive alternative to liposuction and other cosmetic treatments for submental fat will likely become available in the United States in the wake of a Food and Drug Administration advisory panel’s unanimous support for approval of deoxycholic acid as an injection into subcutaneous fat under the chin.
At a meeting on March 9, the FDA’s dermatologic and ophthalmic drugs advisory committee voted 17-0 that the efficacy and safety data on 1% deoxycholic acid (DCA) injection supported approval for the proposed indication, “the improvement in the appearance of moderate to severe convexity or fullness associated with submental fat” in adults.
The product is a synthetic version of naturally occurring DCA, an endogenous secondary bile acid that “serves to emulsify and solubilize dietary fat,” and for cosmetic use, “disrupts cell membranes of adipocytes, causing destruction of fat cells,” according to the manufacturer, Kythera Biopharmaceuticals.
The regimen proposed by Kythera is up to six treatment sessions with 4-week intervals between sessions. Each session would involve up to 50 injections, with 0.2 mL of DCA solution per injection, administered subcutaneously using a grid to control spacing of injections. The product would be the first drug approved for treatment of submental fat.
A pair of phase III studies including about 1,000 adults (median age, 48-49 years) randomized patients to treatment with DCA or placebo saline injections of 6 or fewer treatments, one month apart. The effects were evaluated with validated Clinician-Reported and a Patient-Reported Submental Fat Rating Scales (CR-SMFRS and PR-SMFRS), which Kythera developed in consultation with the FDA. On the CR-SMFRS, a 5-point rating scale ranged from 0 (no localized submental fat evident) to 4 (extreme submental convexity).
Most patients enrolled were rated as 2 (moderate, with prominent localized submental fat) or 3 (severe, with marked localized submental fat); the company is not seeking an indication for extreme submental convexity. On the PR-SMFRS, patients had five options to check off, based on how much fat they thought they had under their chin when looking in a mirror, ranging from “no chin fat at all” to a “very large amount of fat.”
The coprimary efficacy endpoints were those who had at least a 2-grade improvement on both scales, or at least a 1-grade improvement in both scales, 12 weeks after the first treatment.
Continue for study results >>
In the two studies, 70% and 66% of those treated with DCA achieved at least a 1-grade improvement in both scales, vs.19% and 22%, respectively, in the placebo group. The difference was statistically significant. In addition, 13% and 19% of those treated with DCA in the two studies had at least a 2-grade improvement in both scales at that time, vs. 0-3% of those in the placebo group, also statistically significant differences.
One issue raised at the meeting was that the scales were somewhat subjective. A more objective secondary endpoint used MRI volumetric measurements to evaluate results in 449 patients from both studies; this measure determined that 40%-46% of those treated with DCA achieved at least a 10% reduction in MRI volume of submental fat, vs. 5% of those on placebo.
Most patients in the placebo and DCA groups experienced adverse events that were mostly mild and were related to the injections. The rate of moderate adverse events was 17% in the DCA-treated patients and 10% in the placebo group. Injection site reactions more common in the DCA group vs. placebo included pain (70% vs. 31%), numbness (66% vs. 6%), edema (60% vs. 29%), and swelling (33% vs. 16%). There were 11 cases of dysphagia, all of which resolved, except for 1 patient who withdrew from the study and was not available for follow-up. There have been no cases of glandular injuries, including salivary or thyroid injuries, according to the company.
In a safety database of almost 2,000 patients, including 1,050 treated with DCA, the rate of marginal mandibular nerve injuries has been 4% in those treated with DCA. The nerve injuries lasted a median of 45 days (1-298 days) and all resolved. The rate of dysphagia was 2% among those on DCA , lasting a median of 3 days, vs. fewer than 1% among those on placebo, according to the FDA reviewers. One patient did not resolve, but the company noted that this patient dropped out of the study and was not followed. There were no cases of anaphylactic reactions; injection site urticaria was rare and resolved.
In addition, the potential risk of marginal mandibular nerve injuries, issues raised during the meeting included the concerns that the treatment would be used by clinicians who were not adequately trained to provide the treatment or not adequately familiar with the anatomy of the treated area. Other concerns include potential use of the product in areas that include the periorbitum, as well as off-label use of large amounts in different parts of the body, such as the abdomen or thighs. The company said that the product will not be provided to clinicians who have not gone through the planned training program and that it was better suited to small areas of the body.
While there are some questions that still need to be answered and concerns about off-label use and the need for proper training, the panel agreed this was safe and effective if used properly with appropriate training and appropriate precautions, said the panel chair, Dr. Lynn A. Drake of the department of dermatology, Massachusetts General Hospital, Boston.
DCA should not be used off label, a concern that could be at least partially addressed in the label, and “without appropriate training, this product could be misused and mishandled and cause damage,” she added. In addition, because of some “very novel” aspects to this treatment, postmarketing data are needed, addressing questions that include whether it causes scarring that could affect surgery and other interventions in the future, she said.
The FDA usually follows the recommendations of its advisory panels. None of the panel members had relevant disclosures.
SILVER SPRING, MD – A less-invasive alternative to liposuction and other cosmetic treatments for submental fat will likely become available in the United States in the wake of a Food and Drug Administration advisory panel’s unanimous support for approval of deoxycholic acid as an injection into subcutaneous fat under the chin.
At a meeting on March 9, the FDA’s dermatologic and ophthalmic drugs advisory committee voted 17-0 that the efficacy and safety data on 1% deoxycholic acid (DCA) injection supported approval for the proposed indication, “the improvement in the appearance of moderate to severe convexity or fullness associated with submental fat” in adults.
The product is a synthetic version of naturally occurring DCA, an endogenous secondary bile acid that “serves to emulsify and solubilize dietary fat,” and for cosmetic use, “disrupts cell membranes of adipocytes, causing destruction of fat cells,” according to the manufacturer, Kythera Biopharmaceuticals.
The regimen proposed by Kythera is up to six treatment sessions with 4-week intervals between sessions. Each session would involve up to 50 injections, with 0.2 mL of DCA solution per injection, administered subcutaneously using a grid to control spacing of injections. The product would be the first drug approved for treatment of submental fat.
A pair of phase III studies including about 1,000 adults (median age, 48-49 years) randomized patients to treatment with DCA or placebo saline injections of 6 or fewer treatments, one month apart. The effects were evaluated with validated Clinician-Reported and a Patient-Reported Submental Fat Rating Scales (CR-SMFRS and PR-SMFRS), which Kythera developed in consultation with the FDA. On the CR-SMFRS, a 5-point rating scale ranged from 0 (no localized submental fat evident) to 4 (extreme submental convexity).
Most patients enrolled were rated as 2 (moderate, with prominent localized submental fat) or 3 (severe, with marked localized submental fat); the company is not seeking an indication for extreme submental convexity. On the PR-SMFRS, patients had five options to check off, based on how much fat they thought they had under their chin when looking in a mirror, ranging from “no chin fat at all” to a “very large amount of fat.”
The coprimary efficacy endpoints were those who had at least a 2-grade improvement on both scales, or at least a 1-grade improvement in both scales, 12 weeks after the first treatment.
Continue for study results >>
In the two studies, 70% and 66% of those treated with DCA achieved at least a 1-grade improvement in both scales, vs.19% and 22%, respectively, in the placebo group. The difference was statistically significant. In addition, 13% and 19% of those treated with DCA in the two studies had at least a 2-grade improvement in both scales at that time, vs. 0-3% of those in the placebo group, also statistically significant differences.
One issue raised at the meeting was that the scales were somewhat subjective. A more objective secondary endpoint used MRI volumetric measurements to evaluate results in 449 patients from both studies; this measure determined that 40%-46% of those treated with DCA achieved at least a 10% reduction in MRI volume of submental fat, vs. 5% of those on placebo.
Most patients in the placebo and DCA groups experienced adverse events that were mostly mild and were related to the injections. The rate of moderate adverse events was 17% in the DCA-treated patients and 10% in the placebo group. Injection site reactions more common in the DCA group vs. placebo included pain (70% vs. 31%), numbness (66% vs. 6%), edema (60% vs. 29%), and swelling (33% vs. 16%). There were 11 cases of dysphagia, all of which resolved, except for 1 patient who withdrew from the study and was not available for follow-up. There have been no cases of glandular injuries, including salivary or thyroid injuries, according to the company.
In a safety database of almost 2,000 patients, including 1,050 treated with DCA, the rate of marginal mandibular nerve injuries has been 4% in those treated with DCA. The nerve injuries lasted a median of 45 days (1-298 days) and all resolved. The rate of dysphagia was 2% among those on DCA , lasting a median of 3 days, vs. fewer than 1% among those on placebo, according to the FDA reviewers. One patient did not resolve, but the company noted that this patient dropped out of the study and was not followed. There were no cases of anaphylactic reactions; injection site urticaria was rare and resolved.
In addition, the potential risk of marginal mandibular nerve injuries, issues raised during the meeting included the concerns that the treatment would be used by clinicians who were not adequately trained to provide the treatment or not adequately familiar with the anatomy of the treated area. Other concerns include potential use of the product in areas that include the periorbitum, as well as off-label use of large amounts in different parts of the body, such as the abdomen or thighs. The company said that the product will not be provided to clinicians who have not gone through the planned training program and that it was better suited to small areas of the body.
While there are some questions that still need to be answered and concerns about off-label use and the need for proper training, the panel agreed this was safe and effective if used properly with appropriate training and appropriate precautions, said the panel chair, Dr. Lynn A. Drake of the department of dermatology, Massachusetts General Hospital, Boston.
DCA should not be used off label, a concern that could be at least partially addressed in the label, and “without appropriate training, this product could be misused and mishandled and cause damage,” she added. In addition, because of some “very novel” aspects to this treatment, postmarketing data are needed, addressing questions that include whether it causes scarring that could affect surgery and other interventions in the future, she said.
The FDA usually follows the recommendations of its advisory panels. None of the panel members had relevant disclosures.
AT AN FDA ADVISORY COMMITTEE MEETING
FDA panel backs injectable treatment for chin fat
SILVER SPRING, MD – A less-invasive alternative to liposuction and other cosmetic treatments for submental fat will likely become available in the United States in the wake of a Food and Drug Administration advisory panel’s unanimous support for approval of deoxycholic acid as an injection into subcutaneous fat under the chin.
At a meeting on March 9, the FDA’s dermatologic and ophthalmic drugs advisory committee voted 17-0 that the efficacy and safety data on 1% deoxycholic acid (DCA) injection supported approval for the proposed indication, “the improvement in the appearance of moderate to severe convexity or fullness associated with submental fat” in adults.
The product is a synthetic version of naturally occurring DCA, an endogenous secondary bile acid that “serves to emulsify and solubilize dietary fat,” and for cosmetic use, “disrupts cell membranes of adipocytes, causing destruction of fat cells,” according to the manufacturer, Kythera Biopharmaceuticals.
The regimen proposed by Kythera is up to six treatment sessions with 4-week intervals between sessions. Each session would involve up to 50 injections, with 0.2 mL of DCA solution per injection, administered subcutaneously using a grid to control spacing of injections. The product would be the first drug approved for treatment of submental fat.
A pair of phase III studies including about 1,000 adults (median age, 48-49 years) randomized patients to treatment with DCA or placebo saline injections of 6 or fewer treatments, one month apart. The effects were evaluated with validated Clinician-Reported and a Patient-Reported Submental Fat Rating Scales (CR-SMFRS and PR-SMFRS), which Kythera developed in consultation with the FDA. On the CR-SMFRS, a 5-point rating scale ranged from 0 (no localized submental fat evident) to 4 (extreme submental convexity).
Most patients enrolled were rated as 2 (moderate, with prominent localized submental fat) or 3 (severe, with marked localized submental fat); the company is not seeking an indication for extreme submental convexity. On the PR-SMFRS, patients had five options to check off, based on how much fat they thought they had under their chin when looking in a mirror, ranging from “no chin fat at all” to a “very large amount of fat.”
The coprimary efficacy endpoints were those who had at least a 2-grade improvement on both scales, or at least a 1-grade improvement in both scales, 12 weeks after the first treatment.
In the two studies, 70% and 66% of those treated with DCA achieved at least a 1-grade improvement in both scales, vs.19% and 22%, respectively, in the placebo group. The difference was statistically significant. In addition, 13% and 19% of those treated with DCA in the two studies had at least a 2-grade improvement in both scales at that time, vs. 0-3% of those in the placebo group, also statistically significant differences.
One issue raised at the meeting was that the scales were somewhat subjective. A more objective secondary endpoint used MRI volumetric measurements to evaluate results in 449 patients from both studies; this measure determined that 40%-46% of those treated with DCA achieved at least a 10% reduction in MRI volume of submental fat, vs. 5% of those on placebo.
Most patients in the placebo and DCA groups experienced adverse events that were mostly mild and were related to the injections. The rate of moderate adverse events was 17% in the DCA-treated patients and 10% in the placebo group. Injection site reactions more common in the DCA group vs. placebo included pain (70% vs. 31%), numbness (66% vs. 6%), edema (60% vs. 29%), and swelling (33% vs. 16%). There were 11 cases of dysphagia, all of which resolved, except for 1 patient who withdrew from the study and was not available for follow-up. There have been no cases of glandular injuries, including salivary or thyroid injuries, according to the company.
In a safety database of almost 2,000 patients, including 1,050 treated with DCA, the rate of marginal mandibular nerve injuries has been 4% in those treated with DCA. The nerve injuries lasted a median of 45 days (1-298 days) and all resolved. The rate of dysphagia was 2% among those on DCA , lasting a median of 3 days, vs. fewer than 1% among those on placebo, according to the FDA reviewers. One patient did not resolve, but the company noted that this patient dropped out of the study and was not followed. There were no cases of anaphylactic reactions; injection site urticaria was rare and resolved.
In addition, the potential risk of marginal mandibular nerve injuries, issues raised during the meeting included the concerns that the treatment would be used by clinicians who were not adequately trained to provide the treatment or not adequately familiar with the anatomy of the treated area. Other concerns include potential use of the product in areas that include the periorbitum, as well as off-label use of large amounts in different parts of the body, such as the abdomen or thighs. The company said that the product will not be provided to clinicians who have not gone through the planned training program and that it was better suited to small areas of the body.
While there are some questions that still need to be answered and concerns about off-label use and the need for proper training, the panel agreed this was safe and effective if used properly with appropriate training and appropriate precautions, said the panel chair, Dr. Lynn A. Drake of the department of dermatology, Massachusetts General Hospital, Boston.
DCA should not be used off label, a concern that could be at least partially addressed in the label, and “without appropriate training, this product could be misused and mishandled and cause damage,” she added. In addition, because of some “very novel” aspects to this treatment, postmarketing data are needed, addressing questions that include whether it causes scarring that could affect surgery and other interventions in the future, she said.
The FDA usually follows the recommendations of its advisory panels. None of the panel members had relevant disclosures.
SILVER SPRING, MD – A less-invasive alternative to liposuction and other cosmetic treatments for submental fat will likely become available in the United States in the wake of a Food and Drug Administration advisory panel’s unanimous support for approval of deoxycholic acid as an injection into subcutaneous fat under the chin.
At a meeting on March 9, the FDA’s dermatologic and ophthalmic drugs advisory committee voted 17-0 that the efficacy and safety data on 1% deoxycholic acid (DCA) injection supported approval for the proposed indication, “the improvement in the appearance of moderate to severe convexity or fullness associated with submental fat” in adults.
The product is a synthetic version of naturally occurring DCA, an endogenous secondary bile acid that “serves to emulsify and solubilize dietary fat,” and for cosmetic use, “disrupts cell membranes of adipocytes, causing destruction of fat cells,” according to the manufacturer, Kythera Biopharmaceuticals.
The regimen proposed by Kythera is up to six treatment sessions with 4-week intervals between sessions. Each session would involve up to 50 injections, with 0.2 mL of DCA solution per injection, administered subcutaneously using a grid to control spacing of injections. The product would be the first drug approved for treatment of submental fat.
A pair of phase III studies including about 1,000 adults (median age, 48-49 years) randomized patients to treatment with DCA or placebo saline injections of 6 or fewer treatments, one month apart. The effects were evaluated with validated Clinician-Reported and a Patient-Reported Submental Fat Rating Scales (CR-SMFRS and PR-SMFRS), which Kythera developed in consultation with the FDA. On the CR-SMFRS, a 5-point rating scale ranged from 0 (no localized submental fat evident) to 4 (extreme submental convexity).
Most patients enrolled were rated as 2 (moderate, with prominent localized submental fat) or 3 (severe, with marked localized submental fat); the company is not seeking an indication for extreme submental convexity. On the PR-SMFRS, patients had five options to check off, based on how much fat they thought they had under their chin when looking in a mirror, ranging from “no chin fat at all” to a “very large amount of fat.”
The coprimary efficacy endpoints were those who had at least a 2-grade improvement on both scales, or at least a 1-grade improvement in both scales, 12 weeks after the first treatment.
In the two studies, 70% and 66% of those treated with DCA achieved at least a 1-grade improvement in both scales, vs.19% and 22%, respectively, in the placebo group. The difference was statistically significant. In addition, 13% and 19% of those treated with DCA in the two studies had at least a 2-grade improvement in both scales at that time, vs. 0-3% of those in the placebo group, also statistically significant differences.
One issue raised at the meeting was that the scales were somewhat subjective. A more objective secondary endpoint used MRI volumetric measurements to evaluate results in 449 patients from both studies; this measure determined that 40%-46% of those treated with DCA achieved at least a 10% reduction in MRI volume of submental fat, vs. 5% of those on placebo.
Most patients in the placebo and DCA groups experienced adverse events that were mostly mild and were related to the injections. The rate of moderate adverse events was 17% in the DCA-treated patients and 10% in the placebo group. Injection site reactions more common in the DCA group vs. placebo included pain (70% vs. 31%), numbness (66% vs. 6%), edema (60% vs. 29%), and swelling (33% vs. 16%). There were 11 cases of dysphagia, all of which resolved, except for 1 patient who withdrew from the study and was not available for follow-up. There have been no cases of glandular injuries, including salivary or thyroid injuries, according to the company.
In a safety database of almost 2,000 patients, including 1,050 treated with DCA, the rate of marginal mandibular nerve injuries has been 4% in those treated with DCA. The nerve injuries lasted a median of 45 days (1-298 days) and all resolved. The rate of dysphagia was 2% among those on DCA , lasting a median of 3 days, vs. fewer than 1% among those on placebo, according to the FDA reviewers. One patient did not resolve, but the company noted that this patient dropped out of the study and was not followed. There were no cases of anaphylactic reactions; injection site urticaria was rare and resolved.
In addition, the potential risk of marginal mandibular nerve injuries, issues raised during the meeting included the concerns that the treatment would be used by clinicians who were not adequately trained to provide the treatment or not adequately familiar with the anatomy of the treated area. Other concerns include potential use of the product in areas that include the periorbitum, as well as off-label use of large amounts in different parts of the body, such as the abdomen or thighs. The company said that the product will not be provided to clinicians who have not gone through the planned training program and that it was better suited to small areas of the body.
While there are some questions that still need to be answered and concerns about off-label use and the need for proper training, the panel agreed this was safe and effective if used properly with appropriate training and appropriate precautions, said the panel chair, Dr. Lynn A. Drake of the department of dermatology, Massachusetts General Hospital, Boston.
DCA should not be used off label, a concern that could be at least partially addressed in the label, and “without appropriate training, this product could be misused and mishandled and cause damage,” she added. In addition, because of some “very novel” aspects to this treatment, postmarketing data are needed, addressing questions that include whether it causes scarring that could affect surgery and other interventions in the future, she said.
The FDA usually follows the recommendations of its advisory panels. None of the panel members had relevant disclosures.
SILVER SPRING, MD – A less-invasive alternative to liposuction and other cosmetic treatments for submental fat will likely become available in the United States in the wake of a Food and Drug Administration advisory panel’s unanimous support for approval of deoxycholic acid as an injection into subcutaneous fat under the chin.
At a meeting on March 9, the FDA’s dermatologic and ophthalmic drugs advisory committee voted 17-0 that the efficacy and safety data on 1% deoxycholic acid (DCA) injection supported approval for the proposed indication, “the improvement in the appearance of moderate to severe convexity or fullness associated with submental fat” in adults.
The product is a synthetic version of naturally occurring DCA, an endogenous secondary bile acid that “serves to emulsify and solubilize dietary fat,” and for cosmetic use, “disrupts cell membranes of adipocytes, causing destruction of fat cells,” according to the manufacturer, Kythera Biopharmaceuticals.
The regimen proposed by Kythera is up to six treatment sessions with 4-week intervals between sessions. Each session would involve up to 50 injections, with 0.2 mL of DCA solution per injection, administered subcutaneously using a grid to control spacing of injections. The product would be the first drug approved for treatment of submental fat.
A pair of phase III studies including about 1,000 adults (median age, 48-49 years) randomized patients to treatment with DCA or placebo saline injections of 6 or fewer treatments, one month apart. The effects were evaluated with validated Clinician-Reported and a Patient-Reported Submental Fat Rating Scales (CR-SMFRS and PR-SMFRS), which Kythera developed in consultation with the FDA. On the CR-SMFRS, a 5-point rating scale ranged from 0 (no localized submental fat evident) to 4 (extreme submental convexity).
Most patients enrolled were rated as 2 (moderate, with prominent localized submental fat) or 3 (severe, with marked localized submental fat); the company is not seeking an indication for extreme submental convexity. On the PR-SMFRS, patients had five options to check off, based on how much fat they thought they had under their chin when looking in a mirror, ranging from “no chin fat at all” to a “very large amount of fat.”
The coprimary efficacy endpoints were those who had at least a 2-grade improvement on both scales, or at least a 1-grade improvement in both scales, 12 weeks after the first treatment.
In the two studies, 70% and 66% of those treated with DCA achieved at least a 1-grade improvement in both scales, vs.19% and 22%, respectively, in the placebo group. The difference was statistically significant. In addition, 13% and 19% of those treated with DCA in the two studies had at least a 2-grade improvement in both scales at that time, vs. 0-3% of those in the placebo group, also statistically significant differences.
One issue raised at the meeting was that the scales were somewhat subjective. A more objective secondary endpoint used MRI volumetric measurements to evaluate results in 449 patients from both studies; this measure determined that 40%-46% of those treated with DCA achieved at least a 10% reduction in MRI volume of submental fat, vs. 5% of those on placebo.
Most patients in the placebo and DCA groups experienced adverse events that were mostly mild and were related to the injections. The rate of moderate adverse events was 17% in the DCA-treated patients and 10% in the placebo group. Injection site reactions more common in the DCA group vs. placebo included pain (70% vs. 31%), numbness (66% vs. 6%), edema (60% vs. 29%), and swelling (33% vs. 16%). There were 11 cases of dysphagia, all of which resolved, except for 1 patient who withdrew from the study and was not available for follow-up. There have been no cases of glandular injuries, including salivary or thyroid injuries, according to the company.
In a safety database of almost 2,000 patients, including 1,050 treated with DCA, the rate of marginal mandibular nerve injuries has been 4% in those treated with DCA. The nerve injuries lasted a median of 45 days (1-298 days) and all resolved. The rate of dysphagia was 2% among those on DCA , lasting a median of 3 days, vs. fewer than 1% among those on placebo, according to the FDA reviewers. One patient did not resolve, but the company noted that this patient dropped out of the study and was not followed. There were no cases of anaphylactic reactions; injection site urticaria was rare and resolved.
In addition, the potential risk of marginal mandibular nerve injuries, issues raised during the meeting included the concerns that the treatment would be used by clinicians who were not adequately trained to provide the treatment or not adequately familiar with the anatomy of the treated area. Other concerns include potential use of the product in areas that include the periorbitum, as well as off-label use of large amounts in different parts of the body, such as the abdomen or thighs. The company said that the product will not be provided to clinicians who have not gone through the planned training program and that it was better suited to small areas of the body.
While there are some questions that still need to be answered and concerns about off-label use and the need for proper training, the panel agreed this was safe and effective if used properly with appropriate training and appropriate precautions, said the panel chair, Dr. Lynn A. Drake of the department of dermatology, Massachusetts General Hospital, Boston.
DCA should not be used off label, a concern that could be at least partially addressed in the label, and “without appropriate training, this product could be misused and mishandled and cause damage,” she added. In addition, because of some “very novel” aspects to this treatment, postmarketing data are needed, addressing questions that include whether it causes scarring that could affect surgery and other interventions in the future, she said.
The FDA usually follows the recommendations of its advisory panels. None of the panel members had relevant disclosures.
AT AN FDA ADVISORY COMMITTEE MEETING
FDA approves new antifungal for aspergillosis and mucormycosis
Isavuconazonium sulfate, an azole antifungal drug, has been approved to treat invasive aspergillosis and invasive mucormycosis, “rare but serious infections,” the Food and Drug Administration announced on March 6.
Available in both intravenous and capsule formulations, the drug will be marketed as Cresemba by Astellas Pharma US.
Approval for the aspergillosis indication was based on a study of 516 patients randomized to treatment with isavuconazonium or voriconazole, which is approved for treating invasive aspergillosis and is the standard of care; approval of the mucormycosis indication was based on a single-arm study of 37 patients, which compared treatment results with “the natural disease progression associated with untreated mucormycosis,” according to the FDA statement announcing the approval.
“Both studies showed Cresemba was safe and effective in treating these serious fungal infections,” which most often affect immunocompromised patients, the statement said.
At a meeting in January, the FDA’s Anti-Infective Drugs Advisory Committee recommended approval of both indications, although they were more ambivalent about the mucormycosis indication because of the small amount of data in those patients. One of the manufacturer’s postmarketing commitments is to establish a registry “to collect and analyze clinical efficacy-related outcome data on patients treated with isavuconazonium sulfate who have invasive mucormycosis or infection with non–fumigatus aspergillus species,” according to the FDA’s approval letter.
The most common side effects associated with isavuconazonium include nausea, vomiting, diarrhea, headache, elevated liver chemistry tests, hypokalemia, constipation, dyspnea, cough, peripheral edema and back pain. Serious adverse events include liver problems, infusion reactions and severe allergic and skin reactions. The IV formulation comes in a powder formulation that is reconstituted for intravenous administration, and when administered intravenously, an in-line filter should be used because of the possibility that insoluble particulate may be formed after reconstitution, according to the prescribing information.
Serious adverse events associated with isavuconazonium should be reported to the FDA’s MedWatch program online or at 800-332-0178.
Isavuconazonium sulfate, an azole antifungal drug, has been approved to treat invasive aspergillosis and invasive mucormycosis, “rare but serious infections,” the Food and Drug Administration announced on March 6.
Available in both intravenous and capsule formulations, the drug will be marketed as Cresemba by Astellas Pharma US.
Approval for the aspergillosis indication was based on a study of 516 patients randomized to treatment with isavuconazonium or voriconazole, which is approved for treating invasive aspergillosis and is the standard of care; approval of the mucormycosis indication was based on a single-arm study of 37 patients, which compared treatment results with “the natural disease progression associated with untreated mucormycosis,” according to the FDA statement announcing the approval.
“Both studies showed Cresemba was safe and effective in treating these serious fungal infections,” which most often affect immunocompromised patients, the statement said.
At a meeting in January, the FDA’s Anti-Infective Drugs Advisory Committee recommended approval of both indications, although they were more ambivalent about the mucormycosis indication because of the small amount of data in those patients. One of the manufacturer’s postmarketing commitments is to establish a registry “to collect and analyze clinical efficacy-related outcome data on patients treated with isavuconazonium sulfate who have invasive mucormycosis or infection with non–fumigatus aspergillus species,” according to the FDA’s approval letter.
The most common side effects associated with isavuconazonium include nausea, vomiting, diarrhea, headache, elevated liver chemistry tests, hypokalemia, constipation, dyspnea, cough, peripheral edema and back pain. Serious adverse events include liver problems, infusion reactions and severe allergic and skin reactions. The IV formulation comes in a powder formulation that is reconstituted for intravenous administration, and when administered intravenously, an in-line filter should be used because of the possibility that insoluble particulate may be formed after reconstitution, according to the prescribing information.
Serious adverse events associated with isavuconazonium should be reported to the FDA’s MedWatch program online or at 800-332-0178.
Isavuconazonium sulfate, an azole antifungal drug, has been approved to treat invasive aspergillosis and invasive mucormycosis, “rare but serious infections,” the Food and Drug Administration announced on March 6.
Available in both intravenous and capsule formulations, the drug will be marketed as Cresemba by Astellas Pharma US.
Approval for the aspergillosis indication was based on a study of 516 patients randomized to treatment with isavuconazonium or voriconazole, which is approved for treating invasive aspergillosis and is the standard of care; approval of the mucormycosis indication was based on a single-arm study of 37 patients, which compared treatment results with “the natural disease progression associated with untreated mucormycosis,” according to the FDA statement announcing the approval.
“Both studies showed Cresemba was safe and effective in treating these serious fungal infections,” which most often affect immunocompromised patients, the statement said.
At a meeting in January, the FDA’s Anti-Infective Drugs Advisory Committee recommended approval of both indications, although they were more ambivalent about the mucormycosis indication because of the small amount of data in those patients. One of the manufacturer’s postmarketing commitments is to establish a registry “to collect and analyze clinical efficacy-related outcome data on patients treated with isavuconazonium sulfate who have invasive mucormycosis or infection with non–fumigatus aspergillus species,” according to the FDA’s approval letter.
The most common side effects associated with isavuconazonium include nausea, vomiting, diarrhea, headache, elevated liver chemistry tests, hypokalemia, constipation, dyspnea, cough, peripheral edema and back pain. Serious adverse events include liver problems, infusion reactions and severe allergic and skin reactions. The IV formulation comes in a powder formulation that is reconstituted for intravenous administration, and when administered intravenously, an in-line filter should be used because of the possibility that insoluble particulate may be formed after reconstitution, according to the prescribing information.
Serious adverse events associated with isavuconazonium should be reported to the FDA’s MedWatch program online or at 800-332-0178.
FDA’s mobile app provides real-time drug shortage information
A mobile app that provides up-to-date information on drug shortages has been launched by the Food and Drug Administration, the agency has announced.
The mobile application is “specifically designed to speed public access to valuable information about drug shortages,” namely, current and resolved drug shortages and discontinuations of drug shortages, according to the FDA statement. Clinicians and others can search by a drug’s generic name, active ingredient, or therapeutic category, and also can use the app to report a drug shortage or a supply issue to the FDA.

The app “is an innovative tool that will offer easier and faster access to important drug shortage information,” Capt. Valerie Jensen, R.Ph., associate director of the drug shortages program in the FDA’s Center for Drug Evaluation and Research, said in the statement. “The FDA understands that health care professionals and pharmacists need real-time information about drug shortages to make treatment decisions,” she added.
The app is free and can be downloaded via iTunes for Apple devices and the Google Play store for Android devices, searching for “FDA Drug Shortages.”
More information on drug shortages is available on the FDA’s web site at http://www.accessdata.fda.gov/scripts/drugshortages/default.cfm.
A mobile app that provides up-to-date information on drug shortages has been launched by the Food and Drug Administration, the agency has announced.
The mobile application is “specifically designed to speed public access to valuable information about drug shortages,” namely, current and resolved drug shortages and discontinuations of drug shortages, according to the FDA statement. Clinicians and others can search by a drug’s generic name, active ingredient, or therapeutic category, and also can use the app to report a drug shortage or a supply issue to the FDA.
The app “is an innovative tool that will offer easier and faster access to important drug shortage information,” Capt. Valerie Jensen, R.Ph., associate director of the drug shortages program in the FDA’s Center for Drug Evaluation and Research, said in the statement. “The FDA understands that health care professionals and pharmacists need real-time information about drug shortages to make treatment decisions,” she added.
The app is free and can be downloaded via iTunes for Apple devices and the Google Play store for Android devices, searching for “FDA Drug Shortages.”
More information on drug shortages is available on the FDA’s web site at http://www.accessdata.fda.gov/scripts/drugshortages/default.cfm.
A mobile app that provides up-to-date information on drug shortages has been launched by the Food and Drug Administration, the agency has announced.
The mobile application is “specifically designed to speed public access to valuable information about drug shortages,” namely, current and resolved drug shortages and discontinuations of drug shortages, according to the FDA statement. Clinicians and others can search by a drug’s generic name, active ingredient, or therapeutic category, and also can use the app to report a drug shortage or a supply issue to the FDA.
The app “is an innovative tool that will offer easier and faster access to important drug shortage information,” Capt. Valerie Jensen, R.Ph., associate director of the drug shortages program in the FDA’s Center for Drug Evaluation and Research, said in the statement. “The FDA understands that health care professionals and pharmacists need real-time information about drug shortages to make treatment decisions,” she added.
The app is free and can be downloaded via iTunes for Apple devices and the Google Play store for Android devices, searching for “FDA Drug Shortages.”
More information on drug shortages is available on the FDA’s web site at http://www.accessdata.fda.gov/scripts/drugshortages/default.cfm.
FROM THE FDA

FDA Panel Recommends Two New Strains for 2015-2016 Influenza Vaccines
SILVER SPRING, MD.– Two components of the trivalent and quadrivalent influenza vaccines used during the current season should be replaced for the 2015-2016 vaccine, including the influenza A(H3N2) component, a Food and Drug Administration advisory panel has stated.
During the current season, most of the influenza activity in the United States has been due to influenza A(H3N2), and more than two-thirds of the A(H3N2) viruses tested at the Centers for Disease Control and Prevention have “drifted” from the A (H3N2) strain included in the current vaccines, reducing their effectiveness.
The FDA’s Vaccines and Related Biologicals Products Advisory Committee voted at a meeting March 4 to recommend that the following viruses be used for the 2015-2016 trivalent vaccine: an A/California/7/2009 (H1N1)pdm09-like virus; an A/Switzerland/9715293/2013 (H3N2)-like virus; and a B/Phuket/3073/2013-like virus (B/Yamagata lineage). The A(H3N2) strain and the B/Yamagata lineage strain would replace the strains in the current vaccine.
The committee recommended a B/Brisbane/60/2008-like virus (B/Victoria lineage) for the second influenza B strain in the quadrivalent vaccine, which is included in the current quadrivalent vaccine. The panel votes separately on the strains; all votes were unanimous, except for the vote on the B/Yamagata lineage strain in the trivalent vaccine, which was supported by a 14-1 vote.
The FDA panel’s recommendation is the same as the recommendation made recently by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. Every year, the FDA panel meets at this time and considers the WHO recommendation, as well as information that includes influenza surveillance and epidemiology data in North America and worldwide.
This season has been “moderately severe” and started about 4 weeks earlier than average, peaking in late December and early January, according to Dr. Lisa Grohskopf of the epidemiology & prevention branch in the CDC’s influenza division.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups. As of Feb. 21, the preliminary estimate of hospitalizations in this age group was 51.7 cases per 100,000 people, compared with about 27 per 100,000 during the last season. This is the highest rate recorded for this age group since surveillance began during the 2005-2006 season, she added.
To date, there have been 92 pediatric deaths associated with influenza, compared with 109 reported during the 2013-2014 season, 171 during 2012-2013, and 37 during 2011-2012.
When asked why the hospitalization rate has been so high among the elderly, Dr. Grohskopf said that A(H3N2)-predominant seasons tend to be associated with more severe disease, and vaccine efficacy this season was reduced.
The data for the pediatric deaths are incomplete, but most of these cases have been associated with influenza A, and the subtypes tested have been A(H3N2). Whether they represent drifted strains is not yet known, she said. The children’s vaccination status also is not yet known, but historically about 85%-90% of influenza-associated deaths in children are in those who were not vaccinated, she noted.
Updated estimates of the current vaccine effectiveness against influenza A (H3N2) viruses, provided by the CDC on Feb. 26, is 18%. For all influenza viruses overall, estimated effectiveness is 19%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 19%, according to the update. During seasons when the vaccine is a good match for circulating viruses, vaccine effectiveness is in the 60% range, according to speakers at the FDA panel meeting.
The FDA usually follows the recommendations of its panel members. None of the panelists had disclosures.
SILVER SPRING, MD.– Two components of the trivalent and quadrivalent influenza vaccines used during the current season should be replaced for the 2015-2016 vaccine, including the influenza A(H3N2) component, a Food and Drug Administration advisory panel has stated.
During the current season, most of the influenza activity in the United States has been due to influenza A(H3N2), and more than two-thirds of the A(H3N2) viruses tested at the Centers for Disease Control and Prevention have “drifted” from the A (H3N2) strain included in the current vaccines, reducing their effectiveness.
The FDA’s Vaccines and Related Biologicals Products Advisory Committee voted at a meeting March 4 to recommend that the following viruses be used for the 2015-2016 trivalent vaccine: an A/California/7/2009 (H1N1)pdm09-like virus; an A/Switzerland/9715293/2013 (H3N2)-like virus; and a B/Phuket/3073/2013-like virus (B/Yamagata lineage). The A(H3N2) strain and the B/Yamagata lineage strain would replace the strains in the current vaccine.
The committee recommended a B/Brisbane/60/2008-like virus (B/Victoria lineage) for the second influenza B strain in the quadrivalent vaccine, which is included in the current quadrivalent vaccine. The panel votes separately on the strains; all votes were unanimous, except for the vote on the B/Yamagata lineage strain in the trivalent vaccine, which was supported by a 14-1 vote.
The FDA panel’s recommendation is the same as the recommendation made recently by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. Every year, the FDA panel meets at this time and considers the WHO recommendation, as well as information that includes influenza surveillance and epidemiology data in North America and worldwide.
This season has been “moderately severe” and started about 4 weeks earlier than average, peaking in late December and early January, according to Dr. Lisa Grohskopf of the epidemiology & prevention branch in the CDC’s influenza division.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups. As of Feb. 21, the preliminary estimate of hospitalizations in this age group was 51.7 cases per 100,000 people, compared with about 27 per 100,000 during the last season. This is the highest rate recorded for this age group since surveillance began during the 2005-2006 season, she added.
To date, there have been 92 pediatric deaths associated with influenza, compared with 109 reported during the 2013-2014 season, 171 during 2012-2013, and 37 during 2011-2012.
When asked why the hospitalization rate has been so high among the elderly, Dr. Grohskopf said that A(H3N2)-predominant seasons tend to be associated with more severe disease, and vaccine efficacy this season was reduced.
The data for the pediatric deaths are incomplete, but most of these cases have been associated with influenza A, and the subtypes tested have been A(H3N2). Whether they represent drifted strains is not yet known, she said. The children’s vaccination status also is not yet known, but historically about 85%-90% of influenza-associated deaths in children are in those who were not vaccinated, she noted.
Updated estimates of the current vaccine effectiveness against influenza A (H3N2) viruses, provided by the CDC on Feb. 26, is 18%. For all influenza viruses overall, estimated effectiveness is 19%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 19%, according to the update. During seasons when the vaccine is a good match for circulating viruses, vaccine effectiveness is in the 60% range, according to speakers at the FDA panel meeting.
The FDA usually follows the recommendations of its panel members. None of the panelists had disclosures.
SILVER SPRING, MD.– Two components of the trivalent and quadrivalent influenza vaccines used during the current season should be replaced for the 2015-2016 vaccine, including the influenza A(H3N2) component, a Food and Drug Administration advisory panel has stated.
During the current season, most of the influenza activity in the United States has been due to influenza A(H3N2), and more than two-thirds of the A(H3N2) viruses tested at the Centers for Disease Control and Prevention have “drifted” from the A (H3N2) strain included in the current vaccines, reducing their effectiveness.
The FDA’s Vaccines and Related Biologicals Products Advisory Committee voted at a meeting March 4 to recommend that the following viruses be used for the 2015-2016 trivalent vaccine: an A/California/7/2009 (H1N1)pdm09-like virus; an A/Switzerland/9715293/2013 (H3N2)-like virus; and a B/Phuket/3073/2013-like virus (B/Yamagata lineage). The A(H3N2) strain and the B/Yamagata lineage strain would replace the strains in the current vaccine.
The committee recommended a B/Brisbane/60/2008-like virus (B/Victoria lineage) for the second influenza B strain in the quadrivalent vaccine, which is included in the current quadrivalent vaccine. The panel votes separately on the strains; all votes were unanimous, except for the vote on the B/Yamagata lineage strain in the trivalent vaccine, which was supported by a 14-1 vote.
The FDA panel’s recommendation is the same as the recommendation made recently by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. Every year, the FDA panel meets at this time and considers the WHO recommendation, as well as information that includes influenza surveillance and epidemiology data in North America and worldwide.
This season has been “moderately severe” and started about 4 weeks earlier than average, peaking in late December and early January, according to Dr. Lisa Grohskopf of the epidemiology & prevention branch in the CDC’s influenza division.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups. As of Feb. 21, the preliminary estimate of hospitalizations in this age group was 51.7 cases per 100,000 people, compared with about 27 per 100,000 during the last season. This is the highest rate recorded for this age group since surveillance began during the 2005-2006 season, she added.
To date, there have been 92 pediatric deaths associated with influenza, compared with 109 reported during the 2013-2014 season, 171 during 2012-2013, and 37 during 2011-2012.
When asked why the hospitalization rate has been so high among the elderly, Dr. Grohskopf said that A(H3N2)-predominant seasons tend to be associated with more severe disease, and vaccine efficacy this season was reduced.
The data for the pediatric deaths are incomplete, but most of these cases have been associated with influenza A, and the subtypes tested have been A(H3N2). Whether they represent drifted strains is not yet known, she said. The children’s vaccination status also is not yet known, but historically about 85%-90% of influenza-associated deaths in children are in those who were not vaccinated, she noted.
Updated estimates of the current vaccine effectiveness against influenza A (H3N2) viruses, provided by the CDC on Feb. 26, is 18%. For all influenza viruses overall, estimated effectiveness is 19%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 19%, according to the update. During seasons when the vaccine is a good match for circulating viruses, vaccine effectiveness is in the 60% range, according to speakers at the FDA panel meeting.
The FDA usually follows the recommendations of its panel members. None of the panelists had disclosures.
FDA panel recommends two new strains for 2015-2016 influenza vaccines
SILVER SPRING, MD.– Two components of the trivalent and quadrivalent influenza vaccines used during the current season should be replaced for the 2015-2016 vaccine, including the influenza A(H3N2) component, a Food and Drug Administration advisory panel has stated.
During the current season, most of the influenza activity in the United States has been due to influenza A(H3N2), and more than two-thirds of the A(H3N2) viruses tested at the Centers for Disease Control and Prevention have “drifted” from the A (H3N2) strain included in the current vaccines, reducing their effectiveness.
The FDA’s Vaccines and Related Biologicals Products Advisory Committee voted at a meeting March 4 to recommend that the following viruses be used for the 2015-2016 trivalent vaccine: an A/California/7/2009 (H1N1)pdm09-like virus; an A/Switzerland/9715293/2013 (H3N2)-like virus; and a B/Phuket/3073/2013-like virus (B/Yamagata lineage). The A(H3N2) strain and the B/Yamagata lineage strain would replace the strains in the current vaccine.
The committee recommended a B/Brisbane/60/2008-like virus (B/Victoria lineage) for the second influenza B strain in the quadrivalent vaccine, which is included in the current quadrivalent vaccine. The panel votes separately on the strains; all votes were unanimous, except for the vote on the B/Yamagata lineage strain in the trivalent vaccine, which was supported by a 14-1 vote.
The FDA panel’s recommendation is the same as the recommendation made recently by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. Every year, the FDA panel meets at this time and considers the WHO recommendation, as well as information that includes influenza surveillance and epidemiology data in North America and worldwide.
This season has been “moderately severe” and started about 4 weeks earlier than average, peaking in late December and early January, according to Dr. Lisa Grohskopf of the epidemiology & prevention branch in the CDC’s influenza division.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups. As of Feb. 21, the preliminary estimate of hospitalizations in this age group was 51.7 cases per 100,000 people, compared with about 27 per 100,000 during the last season. This is the highest rate recorded for this age group since surveillance began during the 2005-2006 season, she added.
To date, there have been 92 pediatric deaths associated with influenza, compared with 109 reported during the 2013-2014 season, 171 during 2012-2013, and 37 during 2011-2012.
When asked why the hospitalization rate has been so high among the elderly, Dr. Grohskopf said that A(H3N2)-predominant seasons tend to be associated with more severe disease, and vaccine efficacy this season was reduced.
The data for the pediatric deaths are incomplete, but most of these cases have been associated with influenza A, and the subtypes tested have been A(H3N2). Whether they represent drifted strains is not yet known, she said. The children’s vaccination status also is not yet known, but historically about 85%-90% of influenza-associated deaths in children are in those who were not vaccinated, she noted.
Updated estimates of the current vaccine effectiveness against influenza A (H3N2) viruses, provided by the CDC on Feb. 26, is 18%. For all influenza viruses overall, estimated effectiveness is 19%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 19%, according to the update. During seasons when the vaccine is a good match for circulating viruses, vaccine effectiveness is in the 60% range, according to speakers at the FDA panel meeting.
The FDA usually follows the recommendations of its panel members. None of the panelists had disclosures.
SILVER SPRING, MD.– Two components of the trivalent and quadrivalent influenza vaccines used during the current season should be replaced for the 2015-2016 vaccine, including the influenza A(H3N2) component, a Food and Drug Administration advisory panel has stated.
During the current season, most of the influenza activity in the United States has been due to influenza A(H3N2), and more than two-thirds of the A(H3N2) viruses tested at the Centers for Disease Control and Prevention have “drifted” from the A (H3N2) strain included in the current vaccines, reducing their effectiveness.
The FDA’s Vaccines and Related Biologicals Products Advisory Committee voted at a meeting March 4 to recommend that the following viruses be used for the 2015-2016 trivalent vaccine: an A/California/7/2009 (H1N1)pdm09-like virus; an A/Switzerland/9715293/2013 (H3N2)-like virus; and a B/Phuket/3073/2013-like virus (B/Yamagata lineage). The A(H3N2) strain and the B/Yamagata lineage strain would replace the strains in the current vaccine.
The committee recommended a B/Brisbane/60/2008-like virus (B/Victoria lineage) for the second influenza B strain in the quadrivalent vaccine, which is included in the current quadrivalent vaccine. The panel votes separately on the strains; all votes were unanimous, except for the vote on the B/Yamagata lineage strain in the trivalent vaccine, which was supported by a 14-1 vote.
The FDA panel’s recommendation is the same as the recommendation made recently by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. Every year, the FDA panel meets at this time and considers the WHO recommendation, as well as information that includes influenza surveillance and epidemiology data in North America and worldwide.
This season has been “moderately severe” and started about 4 weeks earlier than average, peaking in late December and early January, according to Dr. Lisa Grohskopf of the epidemiology & prevention branch in the CDC’s influenza division.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups. As of Feb. 21, the preliminary estimate of hospitalizations in this age group was 51.7 cases per 100,000 people, compared with about 27 per 100,000 during the last season. This is the highest rate recorded for this age group since surveillance began during the 2005-2006 season, she added.
To date, there have been 92 pediatric deaths associated with influenza, compared with 109 reported during the 2013-2014 season, 171 during 2012-2013, and 37 during 2011-2012.
When asked why the hospitalization rate has been so high among the elderly, Dr. Grohskopf said that A(H3N2)-predominant seasons tend to be associated with more severe disease, and vaccine efficacy this season was reduced.
The data for the pediatric deaths are incomplete, but most of these cases have been associated with influenza A, and the subtypes tested have been A(H3N2). Whether they represent drifted strains is not yet known, she said. The children’s vaccination status also is not yet known, but historically about 85%-90% of influenza-associated deaths in children are in those who were not vaccinated, she noted.
Updated estimates of the current vaccine effectiveness against influenza A (H3N2) viruses, provided by the CDC on Feb. 26, is 18%. For all influenza viruses overall, estimated effectiveness is 19%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 19%, according to the update. During seasons when the vaccine is a good match for circulating viruses, vaccine effectiveness is in the 60% range, according to speakers at the FDA panel meeting.
The FDA usually follows the recommendations of its panel members. None of the panelists had disclosures.
SILVER SPRING, MD.– Two components of the trivalent and quadrivalent influenza vaccines used during the current season should be replaced for the 2015-2016 vaccine, including the influenza A(H3N2) component, a Food and Drug Administration advisory panel has stated.
During the current season, most of the influenza activity in the United States has been due to influenza A(H3N2), and more than two-thirds of the A(H3N2) viruses tested at the Centers for Disease Control and Prevention have “drifted” from the A (H3N2) strain included in the current vaccines, reducing their effectiveness.
The FDA’s Vaccines and Related Biologicals Products Advisory Committee voted at a meeting March 4 to recommend that the following viruses be used for the 2015-2016 trivalent vaccine: an A/California/7/2009 (H1N1)pdm09-like virus; an A/Switzerland/9715293/2013 (H3N2)-like virus; and a B/Phuket/3073/2013-like virus (B/Yamagata lineage). The A(H3N2) strain and the B/Yamagata lineage strain would replace the strains in the current vaccine.
The committee recommended a B/Brisbane/60/2008-like virus (B/Victoria lineage) for the second influenza B strain in the quadrivalent vaccine, which is included in the current quadrivalent vaccine. The panel votes separately on the strains; all votes were unanimous, except for the vote on the B/Yamagata lineage strain in the trivalent vaccine, which was supported by a 14-1 vote.
The FDA panel’s recommendation is the same as the recommendation made recently by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. Every year, the FDA panel meets at this time and considers the WHO recommendation, as well as information that includes influenza surveillance and epidemiology data in North America and worldwide.
This season has been “moderately severe” and started about 4 weeks earlier than average, peaking in late December and early January, according to Dr. Lisa Grohskopf of the epidemiology & prevention branch in the CDC’s influenza division.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups. As of Feb. 21, the preliminary estimate of hospitalizations in this age group was 51.7 cases per 100,000 people, compared with about 27 per 100,000 during the last season. This is the highest rate recorded for this age group since surveillance began during the 2005-2006 season, she added.
To date, there have been 92 pediatric deaths associated with influenza, compared with 109 reported during the 2013-2014 season, 171 during 2012-2013, and 37 during 2011-2012.
When asked why the hospitalization rate has been so high among the elderly, Dr. Grohskopf said that A(H3N2)-predominant seasons tend to be associated with more severe disease, and vaccine efficacy this season was reduced.
The data for the pediatric deaths are incomplete, but most of these cases have been associated with influenza A, and the subtypes tested have been A(H3N2). Whether they represent drifted strains is not yet known, she said. The children’s vaccination status also is not yet known, but historically about 85%-90% of influenza-associated deaths in children are in those who were not vaccinated, she noted.
Updated estimates of the current vaccine effectiveness against influenza A (H3N2) viruses, provided by the CDC on Feb. 26, is 18%. For all influenza viruses overall, estimated effectiveness is 19%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 19%, according to the update. During seasons when the vaccine is a good match for circulating viruses, vaccine effectiveness is in the 60% range, according to speakers at the FDA panel meeting.
The FDA usually follows the recommendations of its panel members. None of the panelists had disclosures.
AT AN FDA ADVISORY COMMITTEE MEETING