User login
VIDEO: New topical acne therapies will target sebum
WAIKOLOA, HAWAII – Three new approaches to topical treatment of acne are on the horizon, and they all share a common foe: sebum.
“One exciting new avenue for topical therapy are drugs that actually target the production of sebum,” explained Dr. Linda F. Stein Gold, director of dermatology research at Henry Ford Health System, Detroit. “For the first time, we have a drug that potentially targets sebum with a topical mechanism. In the past, we’ve only been able to do that with oral therapy.”
In an interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation, Dr. Stein Gold discussed three topical, sebum-focused drugs in clinical trials and outlined their differing mechanisms of action.
SDEF and this news organization are owned by the same parent company.
WAIKOLOA, HAWAII – Three new approaches to topical treatment of acne are on the horizon, and they all share a common foe: sebum.
“One exciting new avenue for topical therapy are drugs that actually target the production of sebum,” explained Dr. Linda F. Stein Gold, director of dermatology research at Henry Ford Health System, Detroit. “For the first time, we have a drug that potentially targets sebum with a topical mechanism. In the past, we’ve only been able to do that with oral therapy.”
In an interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation, Dr. Stein Gold discussed three topical, sebum-focused drugs in clinical trials and outlined their differing mechanisms of action.
SDEF and this news organization are owned by the same parent company.
WAIKOLOA, HAWAII – Three new approaches to topical treatment of acne are on the horizon, and they all share a common foe: sebum.
“One exciting new avenue for topical therapy are drugs that actually target the production of sebum,” explained Dr. Linda F. Stein Gold, director of dermatology research at Henry Ford Health System, Detroit. “For the first time, we have a drug that potentially targets sebum with a topical mechanism. In the past, we’ve only been able to do that with oral therapy.”
In an interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation, Dr. Stein Gold discussed three topical, sebum-focused drugs in clinical trials and outlined their differing mechanisms of action.
SDEF and this news organization are owned by the same parent company.
AT SDEF HAWAII DERMATOLOGY SEMINAR

VIDEO: What’s new on atopic dermatitis drugs and cancer concerns?
WAIKOLOA, HAWAII – Topical calcineurin inhibitors’ boxed warnings give many patients and physicians pause over cancer concerns – but a new database analysis may put some minds at ease about the drugs’ use for atopic dermatitis.
“Pimecrolimus and tacrolimus are given topically, not internally – very little absorption occurs. So, it was hoped that ... we wouldn’t see cancer increases in these patients,” explained Dr. Joseph F. Fowler Jr., clinical professor of dermatology at the University of Louisville (Ky.). “And in fact, that’s exactly what was shown in this large study.”
In an interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation, Dr. Fowler discussed the data from new research examining cancer incidence and calcineurin inhibitor use.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAIKOLOA, HAWAII – Topical calcineurin inhibitors’ boxed warnings give many patients and physicians pause over cancer concerns – but a new database analysis may put some minds at ease about the drugs’ use for atopic dermatitis.
“Pimecrolimus and tacrolimus are given topically, not internally – very little absorption occurs. So, it was hoped that ... we wouldn’t see cancer increases in these patients,” explained Dr. Joseph F. Fowler Jr., clinical professor of dermatology at the University of Louisville (Ky.). “And in fact, that’s exactly what was shown in this large study.”
In an interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation, Dr. Fowler discussed the data from new research examining cancer incidence and calcineurin inhibitor use.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAIKOLOA, HAWAII – Topical calcineurin inhibitors’ boxed warnings give many patients and physicians pause over cancer concerns – but a new database analysis may put some minds at ease about the drugs’ use for atopic dermatitis.
“Pimecrolimus and tacrolimus are given topically, not internally – very little absorption occurs. So, it was hoped that ... we wouldn’t see cancer increases in these patients,” explained Dr. Joseph F. Fowler Jr., clinical professor of dermatology at the University of Louisville (Ky.). “And in fact, that’s exactly what was shown in this large study.”
In an interview at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation, Dr. Fowler discussed the data from new research examining cancer incidence and calcineurin inhibitor use.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SDEF HAWAII DERMATOLOGY SEMINAR

Accumulated victimization tied to depression, PTSD in LGBT youth
Lesbian, gay, bisexual, and transgender youth who report a history of increasing or high levels of victimization are at increased risk for major depressive disorder and posttraumatic stress disorder, a longitudinal study shows.
Results from the study, which is ongoing, show that people who identify as lesbian, gay, bisexual, and transgender (LGBT) are at higher risk for other mental disorders also, such as anxiety and suicide, than are their heterosexual or cisgender counterparts. Further, a history of harassment, victimization, or discrimination proved a predictor of mental health disorders.

The researchers, led by Brian Mustanski, Ph.D., sought to understand the trajectories of victimization and how they affect the development of depression and PTSD in LGBT youth. Initial recruitment included 248 youth. The mean age of the participants was 18.7 years old, and 54.7% were black. Eligibility was met in 234 participants (53% female), and after the seventh wave of data collection, 206 participants (83.1%) remained in the study (Am J Public Health. 2016 Jan 21. doi: 10.2105/AJPH.2015.302976), reported Dr. Mustanski and his colleagues, of the department of medical social sciences at Northwestern University, Chicago.
LGBT victimization was measured using a 10-item questionnaire, and depression and PTSD were assessed with psychiatric interviews at baseline and at the seventh wave of data collection. At the 48-month follow-up, female participants were more likely to complete the interview than were male participants (odds ratio, 2.01; 95% confidence interval, 1.12-4.51; P less than .05).
The researchers were able to identify 4 classes of victimization. Class 1 accounted for 65.4% of the sample and reported low levels of victimization that declined over time (P less than .01). Class 2 accounted for 10.3% of the sample and reported moderate victimization that increased over time (P less than .01). Class 3 included 5.1% of the sample and reported high levels of victimization that remained steady (P = .87). Class 4 included 19.2% of the sample and reported high levels of victimization that declined over time (P less than .01).
Between the classes, significant differences were not found by race or age, and participants in the class 1 group included a higher proportion of females.
Participants in class 2 (OR, 5.54; 95% CI, 1.94-15.82; P less than .001) and class 3 (OR, 4.23; 95% CI, 1.15-15.48; P less than .05) were found to be at higher risk for depression at follow-up than participants in class 1. Likewise, a higher risk for PTSD was found in participants in class 2 (OR, 9.37; 95% CI, 2.76-31.88; P less than .001) and class 3 (OR, 8.66; 95% CI, 1.93-39; P less than .01), compared with class 1. Class 4, however, was associated with a higher risk of PTSD (OR, 4.19; 95% CI, 1.39-12.63; P less than .01) but not depression.
“Our results highlight that it is not only isolated experiences of victimization that affect mental health (which has been the focus of much previous research),” the authors wrote, “but instead, the accumulation of these stressors that exacerbates mental health problems.”
The National Institutes of Mental Health, the American Foundation for Suicide Prevention, the William T. Grant Foundation, and the David Bohnett Foundation funded the study.
Lesbian, gay, bisexual, and transgender youth who report a history of increasing or high levels of victimization are at increased risk for major depressive disorder and posttraumatic stress disorder, a longitudinal study shows.
Results from the study, which is ongoing, show that people who identify as lesbian, gay, bisexual, and transgender (LGBT) are at higher risk for other mental disorders also, such as anxiety and suicide, than are their heterosexual or cisgender counterparts. Further, a history of harassment, victimization, or discrimination proved a predictor of mental health disorders.
The researchers, led by Brian Mustanski, Ph.D., sought to understand the trajectories of victimization and how they affect the development of depression and PTSD in LGBT youth. Initial recruitment included 248 youth. The mean age of the participants was 18.7 years old, and 54.7% were black. Eligibility was met in 234 participants (53% female), and after the seventh wave of data collection, 206 participants (83.1%) remained in the study (Am J Public Health. 2016 Jan 21. doi: 10.2105/AJPH.2015.302976), reported Dr. Mustanski and his colleagues, of the department of medical social sciences at Northwestern University, Chicago.
LGBT victimization was measured using a 10-item questionnaire, and depression and PTSD were assessed with psychiatric interviews at baseline and at the seventh wave of data collection. At the 48-month follow-up, female participants were more likely to complete the interview than were male participants (odds ratio, 2.01; 95% confidence interval, 1.12-4.51; P less than .05).
The researchers were able to identify 4 classes of victimization. Class 1 accounted for 65.4% of the sample and reported low levels of victimization that declined over time (P less than .01). Class 2 accounted for 10.3% of the sample and reported moderate victimization that increased over time (P less than .01). Class 3 included 5.1% of the sample and reported high levels of victimization that remained steady (P = .87). Class 4 included 19.2% of the sample and reported high levels of victimization that declined over time (P less than .01).
Between the classes, significant differences were not found by race or age, and participants in the class 1 group included a higher proportion of females.
Participants in class 2 (OR, 5.54; 95% CI, 1.94-15.82; P less than .001) and class 3 (OR, 4.23; 95% CI, 1.15-15.48; P less than .05) were found to be at higher risk for depression at follow-up than participants in class 1. Likewise, a higher risk for PTSD was found in participants in class 2 (OR, 9.37; 95% CI, 2.76-31.88; P less than .001) and class 3 (OR, 8.66; 95% CI, 1.93-39; P less than .01), compared with class 1. Class 4, however, was associated with a higher risk of PTSD (OR, 4.19; 95% CI, 1.39-12.63; P less than .01) but not depression.
“Our results highlight that it is not only isolated experiences of victimization that affect mental health (which has been the focus of much previous research),” the authors wrote, “but instead, the accumulation of these stressors that exacerbates mental health problems.”
The National Institutes of Mental Health, the American Foundation for Suicide Prevention, the William T. Grant Foundation, and the David Bohnett Foundation funded the study.
Lesbian, gay, bisexual, and transgender youth who report a history of increasing or high levels of victimization are at increased risk for major depressive disorder and posttraumatic stress disorder, a longitudinal study shows.
Results from the study, which is ongoing, show that people who identify as lesbian, gay, bisexual, and transgender (LGBT) are at higher risk for other mental disorders also, such as anxiety and suicide, than are their heterosexual or cisgender counterparts. Further, a history of harassment, victimization, or discrimination proved a predictor of mental health disorders.
The researchers, led by Brian Mustanski, Ph.D., sought to understand the trajectories of victimization and how they affect the development of depression and PTSD in LGBT youth. Initial recruitment included 248 youth. The mean age of the participants was 18.7 years old, and 54.7% were black. Eligibility was met in 234 participants (53% female), and after the seventh wave of data collection, 206 participants (83.1%) remained in the study (Am J Public Health. 2016 Jan 21. doi: 10.2105/AJPH.2015.302976), reported Dr. Mustanski and his colleagues, of the department of medical social sciences at Northwestern University, Chicago.
LGBT victimization was measured using a 10-item questionnaire, and depression and PTSD were assessed with psychiatric interviews at baseline and at the seventh wave of data collection. At the 48-month follow-up, female participants were more likely to complete the interview than were male participants (odds ratio, 2.01; 95% confidence interval, 1.12-4.51; P less than .05).
The researchers were able to identify 4 classes of victimization. Class 1 accounted for 65.4% of the sample and reported low levels of victimization that declined over time (P less than .01). Class 2 accounted for 10.3% of the sample and reported moderate victimization that increased over time (P less than .01). Class 3 included 5.1% of the sample and reported high levels of victimization that remained steady (P = .87). Class 4 included 19.2% of the sample and reported high levels of victimization that declined over time (P less than .01).
Between the classes, significant differences were not found by race or age, and participants in the class 1 group included a higher proportion of females.
Participants in class 2 (OR, 5.54; 95% CI, 1.94-15.82; P less than .001) and class 3 (OR, 4.23; 95% CI, 1.15-15.48; P less than .05) were found to be at higher risk for depression at follow-up than participants in class 1. Likewise, a higher risk for PTSD was found in participants in class 2 (OR, 9.37; 95% CI, 2.76-31.88; P less than .001) and class 3 (OR, 8.66; 95% CI, 1.93-39; P less than .01), compared with class 1. Class 4, however, was associated with a higher risk of PTSD (OR, 4.19; 95% CI, 1.39-12.63; P less than .01) but not depression.
“Our results highlight that it is not only isolated experiences of victimization that affect mental health (which has been the focus of much previous research),” the authors wrote, “but instead, the accumulation of these stressors that exacerbates mental health problems.”
The National Institutes of Mental Health, the American Foundation for Suicide Prevention, the William T. Grant Foundation, and the David Bohnett Foundation funded the study.
FROM THE AMERICAN JOURNAL OF PUBLIC HEALTH
Key clinical point: Understanding the experiences of LGBT youth and trajectories of victimization can lead to interventions that might decrease their risk of developing mental health problems such as major depressive disorder and posttraumatic stress disorder.
Major finding: Participants who experience moderate, increasing victimization and those in the high, steady victimization categories were found to be at higher risk for depression and PTSD, compared with those who had low, decreasing victimization.
Data source: LGBT victimization was measured using a 10-item questionnaire. Depression and PTSD were assessed with psychiatric interviews at baseline and at the seventh wave of data collection.
Disclosures: The National Institutes of Mental Health, the American Foundation for Suicide Prevention, the William T. Grant Foundation, and the David Bohnett Foundation funded the study.

Why so many pertussis outbreaks despite acellular pertussis vaccine? A call to action
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.

aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.
aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.
There has been a justified re-examination of acellular pertussis vaccine (aP)1,2 in light of the multiple large outbreaks of pertussis since 2000, particularly the two large California outbreaks in 2010 and 2014.
Lessons learned: aP protection is less durable than originally thought, and much pertussis is not in infants, but in the school-age and adolescent populations.
aP appears to produce reasonable protection (approximately 84% overall) for infants and preschool children, plus a much improved adverse effect profile, compared with whole cell pertussis vaccine (WCP), which provided approximately 94% protection.1 This 10% difference in aP versus WCP, however, means that herd immunity is more difficult to attain. The accepted pertussis immunization rate needed to provide herd immunity is 92%-94%. Because our current tools (DTaP and Tdap) provide only 84% protection at least in infants and preschoolers, even 100% uptake may leave us 6% to 8% short of the threshold for complete herd immunity.
The California outbreak data from school-age and teenage populations show protection rates drop each year post aP booster. That means that by the fourth year after the last dose, protection is less than 10%. So despite a Tdap dose at 11- to 12-years-of-age, protection gaps occur in 8-to 10-year-olds and 14- to 18-year-olds. These vulnerable periods in older children add to aP’s 84% vs. WCP’s 94% protection for those greater than 3 years of age, explaining more frequent pertussis outbreaks as the pool of WCP-immunized children among older populations decreased.
But before we place all blame on switching to aP, consider that we can now confirm more pertussis infections with molecular assays than was possible with culture and fluorescent assay testing in the WCP era. So improved testing sensitivity means more reports of minimally symptomatic cases that may have been missed before. So WCP, if still used today, might not show 94% protection either.
Additionally, aPs rely heavily on pertactin as a target antigen,3 likely less than WCP, given that WCP contained all pertussis antigens rather than just the 3-5 purified antigens in aPs. So the emergence of pertactin-altered pertussis strains could disproportionately affect protection from aP, compared with WCP.
There seem to be no quick fixes to preventing outbreaks using aPs as our vaccine. One suggestion by the authors of the California outbreak report is to use aP mostly to terminate outbreaks rather than routinely in late childhood. My concern is that if we do not continue routine use in 4-to 6-year-olds, 10-to 11-year-olds, and in early adulthood, the vulnerable proportion of the population during outbreaks would be larger, making outbreaks more difficult to terminate. So continuing to produce some protection, albeit short-lived, with current schedules of aP vaccines seems important.
Also remember that T cells, particularly TH 17 pertussis-specific cells, may be as important as pertussis antibody. Therefore, crafting pertussis vaccines that yield improved antibody plus T cell responses is the current goal. Disease and WCP seem to elicit more T-17 response than aP. One method to craft a better vaccine is to use antigen blends that differ from those in the current vaccines, such as antigens derived from circulating pertussis strains instead of the standard laboratory strain. Another option is to use current antigens but with more potent adjuvants. Such vaccines are likely 5 years away.
But we need to have reasonable expectations for pertussis vaccines. Pertussis infection begins in respiratory epithelium. Many of the most obvious signs and symptoms are due to destruction of ciliated respiratory epithelium plus increased tenacity/volume of secretions. Can a parenterally administered vaccine that induces mostly serum antibody protect against infection of epithelium where antibody concentrations are likely 10% or less than in serum? The short answer is – likely not. We should expect neither aP nor WCP to consistently protect against pertussis infection, but it does seem reasonable to expect aP to reduce disease severity. Preventing infections awaits a vaccine that induces surface IgA. Mucosally administered vaccines produce surface IgA – for example, rotavirus vaccine – but no mucosal pertussis vaccine appears imminent.
A key question is whether our most vulnerable populations, young children, have increased morbidity and mortality. Data from the California suggest an increase but mostly in infants under 6 months of age, the group not old enough to benefit from even the most effective of infant vaccines. Protecting young infants depends on vaccine administered prenatally to mothers. The over-representation of the Hispanic infants among fatalities shows a population on which to focus with maternal immunization. Hopefully, the recent universal TdaP recommendation in pregnancy will help when maternal immunization is higher than current approximately 50% rates.4
Despite the problems, it seems clear that we must continue to use current aP vaccines according to the current schedules, attempting to get as close to 100% uptake as possible. While the current, nearly 10% unimmunized rates add to the likelihood that we are losing complete herd immunity, partial herd immunity is better than no herd immunity.
Expectations: There will be ongoing outbreaks. Continue to be alert for signs of pertussis. They are often less obvious in older patients, and may be as subtle as more than 2 weeks of persistent cough. During outbreaks, we may be called upon to give aP doses at intervals shorter than the usual schedule.
Our responsibility: Do not become discouraged or lose enthusiasm for aP, but explain to parents that because aP is less reactogenic, it produces less protection and is less durable, particularly in school-age children. But please emphasize that modest protection is best in the youngest and modest protection of older children is better than none. Emphasize that the adverse effect profile of current aPs puts the harm/benefit balance heavily in favor of aP.
Bottom line: We can hopefully do better than the current 88% to 92% rate of aP vaccine uptake. We need to get as close to 100% uptake as possible until new vaccines or new strategies become available.
1. Clin Infect Dis. 2016 Feb 7; doi: 10.1093/cid/ciw051.
2. Pediatrics. 2016 Feb 5; doi: 10.1542/peds.2015-3326.
3. Expert Rev Vaccines. 2007 Feb;6(1):47-56.
4. Vaccine. 2016 Feb 10;34(7):968-73.
Dr. Harrison is professor of pediatrics and pediatric infectious diseases at Children’s Mercy Hospitals and Clinics, Kansas City, Mo. He disclosed that his institution received grant support for a study on hexavalent infant vaccine containing pertussis from GlaxoSmithKline, and he was the local primary investigator.*
*Correction, 2/17/2016: An earlier version of this article incompletely stated Dr. Harrison's disclosure information.

Binge drinking in adolescents
Binge drinking is a common problem in adolescents as well as adults. In a 2013 survey, 60 million Americans, representing 22% of the population, 12 years of age and older engaged in binge drinking in the previous month. Among those 12-20 years of age, 14%, or one in seven, reported binge drinking. Depending on the survey, between one-third and one-half of all high school students currently drink alcohol. Among young people who drink, a higher proportion drink heavily than among adult drinkers. For teenagers who drink, 28%-60% report binge drinking. Among high school seniors, 1 in 10 report drinking more than 10 drinks in a row. The predominant liquor consumed by 13- to 20-year-olds is vodka (44% ), whereas beer is consumed by less than one-third of respondents. Underage drinkers typically get alcohol from adults of legal drinking age, and frequently drink in their own home or that of a friend. Clearly, this is an important, underappreciated problem that warrants clinical attention.
Background
Binge drinking is defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) as the pattern of drinking required to bring the blood alcohol concentration (BAC) to 0.08% or greater. For adolescents, the required amount of alcohol to achieve the same BAC is thought to be less than the amount required for adults. Binge drinking for adults has traditionally been defined as more than five drinks over a 2-hour period for men or more than four drinks over 2 hours for women. Different cutoffs have been suggested for youth in several studies but were not clearly defined for this analysis.

Implications
Alcohol consumption in adolescents is associated with adverse events, and this is only exacerbated by binge drinking. These events include, but are not limited to, engaging in higher-risk behaviors, such as impaired driving or being the passenger in a vehicle with an impaired driver; an increased risk of suicide or attempted suicide; and an increased risk of nonautomobile accidents that can lead to severe injury or drowning. Adolescents who start drinking before the age of 15 years are four times as likely to develop alcohol dependence as people who start drinking after 20 years of age. In addition, adolescents who engage in binge drinking have an increased rate of high-risk sexual activity, which may result in a sexually transmitted infection as well as an unplanned pregnancy. Adolescents who continue to imbibe during pregnancy risk the development of fetal alcohol spectrum disorder. Finally, adolescent binge drinkers are subject to the immediate effects of alcohol, including hangover, blackout, and alcohol poisoning.
Guidance for the provider
The AAP recommends screening every adolescent for substance abuse, but if time constraints exist, alcohol abuse only (Pediatrics. 2015;136[3]:e718-26). The AAP has set up specific screening questions that are based upon the age group of the patient:
For elementary school students (ages 9-11) the following questions are appropriate:
• Do you have any friends who drank beer, wine, or any alcoholic drink in the past year?
• How about you? Have you ever had more than a few sips of beer, wine, or any drink with alcohol?
For patients in middle school (ages 11-14):
• Do you have any friends who drank beer, wine, or any alcohol-containing drink in the past year?
• How about you? Over the past year, how many days have you had where you’ve had more than a few sips of beer, wine, or any other alcohol?
For patients in high school (ages 14-18):
• Over the past year, how many days have you had where you’ve had more than a few sips of beer, wine, or any other alcohol?
• If your friends drink, how many drinks do they usually drink on an occasion?
The above questions help to risk stratify patients into three groups: low, moderate, and high risk. The guidelines do not make a recommendation on how to use these questions to risk stratify the patients. Low-risk patients receive brief counseling. Moderate-risk patients receive more intensive counseling as well as motivational interviewing. Motivational interviewing delivered over the course of one or more sessions has been found to be effective in reducing consumption of alcohol. Lastly, the high-risk group is given motivational interviewing, as well as possible referral.
The bottom line
Alcohol abuse in adolescents can have significant detrimental effects, both short and long term. The AAP recommends screening every adolescent for alcohol abuse and, if time permits, for other drug abuse as well, in order to prevent the morbidity and mortality associated with these substances. Interventions can range from brief counseling to motivational interviewing of adolescents at risk and finally referral to substance abuse specialists in the highest-risk groups
Dr. Skolnik is associate director of the family medicine residency program at Abington (Pa.) Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia. Dr. Kiriakov is a second-year resident in the Abington-Jefferson Family Medicine Residency Program in Abington.
Binge drinking is a common problem in adolescents as well as adults. In a 2013 survey, 60 million Americans, representing 22% of the population, 12 years of age and older engaged in binge drinking in the previous month. Among those 12-20 years of age, 14%, or one in seven, reported binge drinking. Depending on the survey, between one-third and one-half of all high school students currently drink alcohol. Among young people who drink, a higher proportion drink heavily than among adult drinkers. For teenagers who drink, 28%-60% report binge drinking. Among high school seniors, 1 in 10 report drinking more than 10 drinks in a row. The predominant liquor consumed by 13- to 20-year-olds is vodka (44% ), whereas beer is consumed by less than one-third of respondents. Underage drinkers typically get alcohol from adults of legal drinking age, and frequently drink in their own home or that of a friend. Clearly, this is an important, underappreciated problem that warrants clinical attention.
Background
Binge drinking is defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) as the pattern of drinking required to bring the blood alcohol concentration (BAC) to 0.08% or greater. For adolescents, the required amount of alcohol to achieve the same BAC is thought to be less than the amount required for adults. Binge drinking for adults has traditionally been defined as more than five drinks over a 2-hour period for men or more than four drinks over 2 hours for women. Different cutoffs have been suggested for youth in several studies but were not clearly defined for this analysis.
Implications
Alcohol consumption in adolescents is associated with adverse events, and this is only exacerbated by binge drinking. These events include, but are not limited to, engaging in higher-risk behaviors, such as impaired driving or being the passenger in a vehicle with an impaired driver; an increased risk of suicide or attempted suicide; and an increased risk of nonautomobile accidents that can lead to severe injury or drowning. Adolescents who start drinking before the age of 15 years are four times as likely to develop alcohol dependence as people who start drinking after 20 years of age. In addition, adolescents who engage in binge drinking have an increased rate of high-risk sexual activity, which may result in a sexually transmitted infection as well as an unplanned pregnancy. Adolescents who continue to imbibe during pregnancy risk the development of fetal alcohol spectrum disorder. Finally, adolescent binge drinkers are subject to the immediate effects of alcohol, including hangover, blackout, and alcohol poisoning.
Guidance for the provider
The AAP recommends screening every adolescent for substance abuse, but if time constraints exist, alcohol abuse only (Pediatrics. 2015;136[3]:e718-26). The AAP has set up specific screening questions that are based upon the age group of the patient:
For elementary school students (ages 9-11) the following questions are appropriate:
• Do you have any friends who drank beer, wine, or any alcoholic drink in the past year?
• How about you? Have you ever had more than a few sips of beer, wine, or any drink with alcohol?
For patients in middle school (ages 11-14):
• Do you have any friends who drank beer, wine, or any alcohol-containing drink in the past year?
• How about you? Over the past year, how many days have you had where you’ve had more than a few sips of beer, wine, or any other alcohol?
For patients in high school (ages 14-18):
• Over the past year, how many days have you had where you’ve had more than a few sips of beer, wine, or any other alcohol?
• If your friends drink, how many drinks do they usually drink on an occasion?
The above questions help to risk stratify patients into three groups: low, moderate, and high risk. The guidelines do not make a recommendation on how to use these questions to risk stratify the patients. Low-risk patients receive brief counseling. Moderate-risk patients receive more intensive counseling as well as motivational interviewing. Motivational interviewing delivered over the course of one or more sessions has been found to be effective in reducing consumption of alcohol. Lastly, the high-risk group is given motivational interviewing, as well as possible referral.
The bottom line
Alcohol abuse in adolescents can have significant detrimental effects, both short and long term. The AAP recommends screening every adolescent for alcohol abuse and, if time permits, for other drug abuse as well, in order to prevent the morbidity and mortality associated with these substances. Interventions can range from brief counseling to motivational interviewing of adolescents at risk and finally referral to substance abuse specialists in the highest-risk groups
Dr. Skolnik is associate director of the family medicine residency program at Abington (Pa.) Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia. Dr. Kiriakov is a second-year resident in the Abington-Jefferson Family Medicine Residency Program in Abington.
Binge drinking is a common problem in adolescents as well as adults. In a 2013 survey, 60 million Americans, representing 22% of the population, 12 years of age and older engaged in binge drinking in the previous month. Among those 12-20 years of age, 14%, or one in seven, reported binge drinking. Depending on the survey, between one-third and one-half of all high school students currently drink alcohol. Among young people who drink, a higher proportion drink heavily than among adult drinkers. For teenagers who drink, 28%-60% report binge drinking. Among high school seniors, 1 in 10 report drinking more than 10 drinks in a row. The predominant liquor consumed by 13- to 20-year-olds is vodka (44% ), whereas beer is consumed by less than one-third of respondents. Underage drinkers typically get alcohol from adults of legal drinking age, and frequently drink in their own home or that of a friend. Clearly, this is an important, underappreciated problem that warrants clinical attention.
Background
Binge drinking is defined by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) as the pattern of drinking required to bring the blood alcohol concentration (BAC) to 0.08% or greater. For adolescents, the required amount of alcohol to achieve the same BAC is thought to be less than the amount required for adults. Binge drinking for adults has traditionally been defined as more than five drinks over a 2-hour period for men or more than four drinks over 2 hours for women. Different cutoffs have been suggested for youth in several studies but were not clearly defined for this analysis.
Implications
Alcohol consumption in adolescents is associated with adverse events, and this is only exacerbated by binge drinking. These events include, but are not limited to, engaging in higher-risk behaviors, such as impaired driving or being the passenger in a vehicle with an impaired driver; an increased risk of suicide or attempted suicide; and an increased risk of nonautomobile accidents that can lead to severe injury or drowning. Adolescents who start drinking before the age of 15 years are four times as likely to develop alcohol dependence as people who start drinking after 20 years of age. In addition, adolescents who engage in binge drinking have an increased rate of high-risk sexual activity, which may result in a sexually transmitted infection as well as an unplanned pregnancy. Adolescents who continue to imbibe during pregnancy risk the development of fetal alcohol spectrum disorder. Finally, adolescent binge drinkers are subject to the immediate effects of alcohol, including hangover, blackout, and alcohol poisoning.
Guidance for the provider
The AAP recommends screening every adolescent for substance abuse, but if time constraints exist, alcohol abuse only (Pediatrics. 2015;136[3]:e718-26). The AAP has set up specific screening questions that are based upon the age group of the patient:
For elementary school students (ages 9-11) the following questions are appropriate:
• Do you have any friends who drank beer, wine, or any alcoholic drink in the past year?
• How about you? Have you ever had more than a few sips of beer, wine, or any drink with alcohol?
For patients in middle school (ages 11-14):
• Do you have any friends who drank beer, wine, or any alcohol-containing drink in the past year?
• How about you? Over the past year, how many days have you had where you’ve had more than a few sips of beer, wine, or any other alcohol?
For patients in high school (ages 14-18):
• Over the past year, how many days have you had where you’ve had more than a few sips of beer, wine, or any other alcohol?
• If your friends drink, how many drinks do they usually drink on an occasion?
The above questions help to risk stratify patients into three groups: low, moderate, and high risk. The guidelines do not make a recommendation on how to use these questions to risk stratify the patients. Low-risk patients receive brief counseling. Moderate-risk patients receive more intensive counseling as well as motivational interviewing. Motivational interviewing delivered over the course of one or more sessions has been found to be effective in reducing consumption of alcohol. Lastly, the high-risk group is given motivational interviewing, as well as possible referral.
The bottom line
Alcohol abuse in adolescents can have significant detrimental effects, both short and long term. The AAP recommends screening every adolescent for alcohol abuse and, if time permits, for other drug abuse as well, in order to prevent the morbidity and mortality associated with these substances. Interventions can range from brief counseling to motivational interviewing of adolescents at risk and finally referral to substance abuse specialists in the highest-risk groups
Dr. Skolnik is associate director of the family medicine residency program at Abington (Pa.) Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia. Dr. Kiriakov is a second-year resident in the Abington-Jefferson Family Medicine Residency Program in Abington.

Piebaldism in Children
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
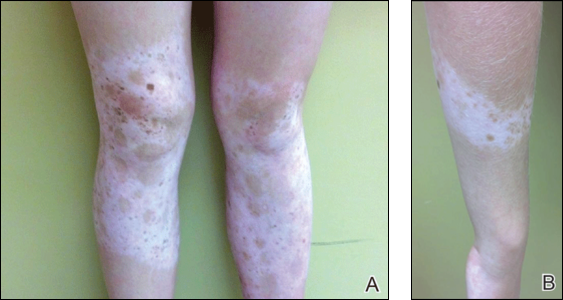
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Practice Points
- Poliosis circumscripta (or white forelock) is commonly the only manifestation of piebaldism in children.
- Affected areas of leukoderma in piebaldism are classically distributed on the central forehead, anterior trunk, and mid extremities.
- The presence of congenital leukoderma should prompt a thorough skin examination and review of the patient’s medical history for evidence of ocular, auditory, and/or neurologic abnormalities.
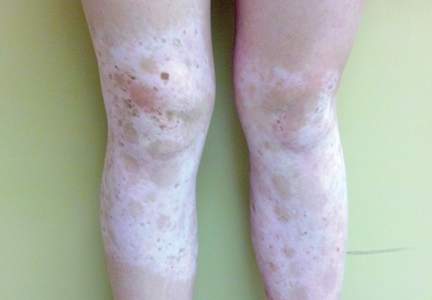
Universal autism screening lacks sufficient evidence, says USPSTF
Evidence is insufficient to assess whether universal screening for autism spectrum disorder (ASD) is in the best interest of public health, a federal task force has concluded.
The U.S. Preventive Services Task Force issued an “I statement” for insufficient evidence of whether there is a preponderance of either benefit or harm in screening a child for ASD when no previous alarms have been raised by that child’s parents or physicians. Instead, the task force recommends clinicians use their best judgment on a case-by-case basis. The full statement was published online in JAMA (doi: 10.1001/jama.2016.0081).

Guidance from other medical and pediatric organizations is mixed. The American Academy of Pediatrics recommends screening children between ages 18 and 24 months, while the American Academy of Family Physicians agrees evidence is insufficient to say one way or the other. The American Academy of Child and Adolescent Psychiatry (J Am Acad Child Adolesc Psychiatry. 2014;53[2]:237-57) recommends all young children receive routine psychiatric assessments, including questions about ASD symptoms.
After reviewing 26 randomly assigned controlled studies of early intensive behavioral and developmental interventions for ASD in this cohort, the task force could see benefit only in the treatment of older children who had been flagged for the disorder by family members, clinicians, or teachers.
Citing a lack of studies focused on the clinical outcomes of children identified with ASD through screening, as well as treatment studies that were small and featured children clinically referred rather than detected through screening, the task force called on researchers to fill the gap.
“Large, good quality, randomized clinical trials of treatment that enroll young children with ASD identified through screening and that report patient-centered outcomes are critical to understanding the effects of screening. Treatment could be compared with a wait-list control, less intense treatment, or an alternative treatment,” task force members wrote.
Agreeing with the USPSTF’s decision, Dr. Michael Silverstein of Boston University and Dr. Jenny Radesky of the University of Michigan, Ann Arbor, wrote in an accompanying editorial, that the report, “presents an opportunity to motivate better, more relevant research” and demonstrates an understanding of the real-world complexities of primary care. “The USPSTF embraced this issue in all its complexity … and other stakeholders should do the same,” they wrote.
For clinicians seeking a screening tool, two screens the task force identified as useful were the M-CHAT/F and the M-CHAT-R/F (Modified Checklist for Autism in Toddlers, Revised With Follow-Up). Both use parent-rated scales that can lead to a follow-up interview and, if necessary, referral for diagnosis.
The U.S. Centers for Disease Control and Prevention estimated in 2010 that 1 in 68 children were affected by ASD, a 23% increase from 2008. The spike is as yet unattributable to any specific cause, according to the CDC.
Dr. Silverstein and Dr. Radesky did not report any relevant disclosures.
On Twitter @whitneymcknight
“Although studies that directly compare the long-term outcomes of screened vs. nonscreened children in the general population are needed, there is ample evidence that justifies the continued practice of universal autism screening as recommended by the American Academy of Pediatrics,” Geraldine Dawson, Ph.D., wrote in an editorial accompanying the U.S. Preventive Services Task Force’s declaration that there is insufficient evidence to recommend the practice one way or the other.
“Studies indicate that available screening tools do identify children with ASD who would have been otherwise missed and children who begin intervention at an earlier age have improved outcomes,” Dr. Dawson, who directs the Duke Center for Autism and Brain Development in Durham, N.C., wrote.
“The current guidelines that encourage universal autism screening offer the best chance for individuals with ASD to reach their full potential and lead productive lives. We should, therefore, stay the course while continuing to advance our knowledge about the full impact” of autism screening, she said.
Dr. Dawson is on the scientific advisory boards of Janssen Research and Development, Roche Pharmaceuticals, Akili, and Progenity.
“Although studies that directly compare the long-term outcomes of screened vs. nonscreened children in the general population are needed, there is ample evidence that justifies the continued practice of universal autism screening as recommended by the American Academy of Pediatrics,” Geraldine Dawson, Ph.D., wrote in an editorial accompanying the U.S. Preventive Services Task Force’s declaration that there is insufficient evidence to recommend the practice one way or the other.
“Studies indicate that available screening tools do identify children with ASD who would have been otherwise missed and children who begin intervention at an earlier age have improved outcomes,” Dr. Dawson, who directs the Duke Center for Autism and Brain Development in Durham, N.C., wrote.
“The current guidelines that encourage universal autism screening offer the best chance for individuals with ASD to reach their full potential and lead productive lives. We should, therefore, stay the course while continuing to advance our knowledge about the full impact” of autism screening, she said.
Dr. Dawson is on the scientific advisory boards of Janssen Research and Development, Roche Pharmaceuticals, Akili, and Progenity.
“Although studies that directly compare the long-term outcomes of screened vs. nonscreened children in the general population are needed, there is ample evidence that justifies the continued practice of universal autism screening as recommended by the American Academy of Pediatrics,” Geraldine Dawson, Ph.D., wrote in an editorial accompanying the U.S. Preventive Services Task Force’s declaration that there is insufficient evidence to recommend the practice one way or the other.
“Studies indicate that available screening tools do identify children with ASD who would have been otherwise missed and children who begin intervention at an earlier age have improved outcomes,” Dr. Dawson, who directs the Duke Center for Autism and Brain Development in Durham, N.C., wrote.
“The current guidelines that encourage universal autism screening offer the best chance for individuals with ASD to reach their full potential and lead productive lives. We should, therefore, stay the course while continuing to advance our knowledge about the full impact” of autism screening, she said.
Dr. Dawson is on the scientific advisory boards of Janssen Research and Development, Roche Pharmaceuticals, Akili, and Progenity.
Evidence is insufficient to assess whether universal screening for autism spectrum disorder (ASD) is in the best interest of public health, a federal task force has concluded.
The U.S. Preventive Services Task Force issued an “I statement” for insufficient evidence of whether there is a preponderance of either benefit or harm in screening a child for ASD when no previous alarms have been raised by that child’s parents or physicians. Instead, the task force recommends clinicians use their best judgment on a case-by-case basis. The full statement was published online in JAMA (doi: 10.1001/jama.2016.0081).
Guidance from other medical and pediatric organizations is mixed. The American Academy of Pediatrics recommends screening children between ages 18 and 24 months, while the American Academy of Family Physicians agrees evidence is insufficient to say one way or the other. The American Academy of Child and Adolescent Psychiatry (J Am Acad Child Adolesc Psychiatry. 2014;53[2]:237-57) recommends all young children receive routine psychiatric assessments, including questions about ASD symptoms.
After reviewing 26 randomly assigned controlled studies of early intensive behavioral and developmental interventions for ASD in this cohort, the task force could see benefit only in the treatment of older children who had been flagged for the disorder by family members, clinicians, or teachers.
Citing a lack of studies focused on the clinical outcomes of children identified with ASD through screening, as well as treatment studies that were small and featured children clinically referred rather than detected through screening, the task force called on researchers to fill the gap.
“Large, good quality, randomized clinical trials of treatment that enroll young children with ASD identified through screening and that report patient-centered outcomes are critical to understanding the effects of screening. Treatment could be compared with a wait-list control, less intense treatment, or an alternative treatment,” task force members wrote.
Agreeing with the USPSTF’s decision, Dr. Michael Silverstein of Boston University and Dr. Jenny Radesky of the University of Michigan, Ann Arbor, wrote in an accompanying editorial, that the report, “presents an opportunity to motivate better, more relevant research” and demonstrates an understanding of the real-world complexities of primary care. “The USPSTF embraced this issue in all its complexity … and other stakeholders should do the same,” they wrote.
For clinicians seeking a screening tool, two screens the task force identified as useful were the M-CHAT/F and the M-CHAT-R/F (Modified Checklist for Autism in Toddlers, Revised With Follow-Up). Both use parent-rated scales that can lead to a follow-up interview and, if necessary, referral for diagnosis.
The U.S. Centers for Disease Control and Prevention estimated in 2010 that 1 in 68 children were affected by ASD, a 23% increase from 2008. The spike is as yet unattributable to any specific cause, according to the CDC.
Dr. Silverstein and Dr. Radesky did not report any relevant disclosures.
On Twitter @whitneymcknight
Evidence is insufficient to assess whether universal screening for autism spectrum disorder (ASD) is in the best interest of public health, a federal task force has concluded.
The U.S. Preventive Services Task Force issued an “I statement” for insufficient evidence of whether there is a preponderance of either benefit or harm in screening a child for ASD when no previous alarms have been raised by that child’s parents or physicians. Instead, the task force recommends clinicians use their best judgment on a case-by-case basis. The full statement was published online in JAMA (doi: 10.1001/jama.2016.0081).
Guidance from other medical and pediatric organizations is mixed. The American Academy of Pediatrics recommends screening children between ages 18 and 24 months, while the American Academy of Family Physicians agrees evidence is insufficient to say one way or the other. The American Academy of Child and Adolescent Psychiatry (J Am Acad Child Adolesc Psychiatry. 2014;53[2]:237-57) recommends all young children receive routine psychiatric assessments, including questions about ASD symptoms.
After reviewing 26 randomly assigned controlled studies of early intensive behavioral and developmental interventions for ASD in this cohort, the task force could see benefit only in the treatment of older children who had been flagged for the disorder by family members, clinicians, or teachers.
Citing a lack of studies focused on the clinical outcomes of children identified with ASD through screening, as well as treatment studies that were small and featured children clinically referred rather than detected through screening, the task force called on researchers to fill the gap.
“Large, good quality, randomized clinical trials of treatment that enroll young children with ASD identified through screening and that report patient-centered outcomes are critical to understanding the effects of screening. Treatment could be compared with a wait-list control, less intense treatment, or an alternative treatment,” task force members wrote.
Agreeing with the USPSTF’s decision, Dr. Michael Silverstein of Boston University and Dr. Jenny Radesky of the University of Michigan, Ann Arbor, wrote in an accompanying editorial, that the report, “presents an opportunity to motivate better, more relevant research” and demonstrates an understanding of the real-world complexities of primary care. “The USPSTF embraced this issue in all its complexity … and other stakeholders should do the same,” they wrote.
For clinicians seeking a screening tool, two screens the task force identified as useful were the M-CHAT/F and the M-CHAT-R/F (Modified Checklist for Autism in Toddlers, Revised With Follow-Up). Both use parent-rated scales that can lead to a follow-up interview and, if necessary, referral for diagnosis.
The U.S. Centers for Disease Control and Prevention estimated in 2010 that 1 in 68 children were affected by ASD, a 23% increase from 2008. The spike is as yet unattributable to any specific cause, according to the CDC.
Dr. Silverstein and Dr. Radesky did not report any relevant disclosures.
On Twitter @whitneymcknight
FROM JAMA

SDEF: Have higher degree of suspicion for pediatric allergic contact dermatitis
Allergic contact dermatitis is often missed in pediatric patients who present with eczema-like skin eruptions, in part because less is known about ACD in children than in adults.
“Often when we see a child with dermatitis, we automatically think of atopic dermatitis, but we should also consider the possibility of allergic contact dermatitis,” Dr. Joseph F. Fowler Jr. said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.

A 2012 cohort study of 349 children between 0 and 15 years indicated that even very young children who were patch tested for common allergens had at least one positive result. Investigators found that nearly three-quarters of children studied tested positive for at least one allergen, typically nickel, other metals, fragrance, or preservatives (Dermatitis. 2012 Nov-Dec;23[6]:275-80).
“This is very similar to what we see in the adult population,” said Dr. Fowler, clinical professor of dermatology at the University of Louisville (Ky.). “Other studies in recent years have borne this out.”
Dr. Fowler suggested having a high degree of suspicion for ACD, especially when pediatric patients present with:
• Chronic, difficult to control atopic condition, as this could indicate a systemic reaction.
• Localized or facial dermatitis, as this could indicate the point of contact with an allergen.
• Scattered, generalized dermatitis, which also could represent systemic allergic contact dermatitis.
• Dermatitis that worsens, despite otherwise adequate treatment regimen.
• Reactions following contact with metals, fragrances, topical components, such as preservatives or neomycin.
“In these situations, patch testing will help determine that an allergen is implicated,” Dr. Fowler said.
In children with eczema, Dr. Fowler recommended patch testing when the eczema is not in the typical areas such as behind the knees or elbows, or if it started in typical areas and then spread elsewhere, especially in children around 5 years old.
“The moral of the story is that kids can be allergic to the same things as adults, even though we have less about this in the literature,” Dr. Fowler. “Skin testing or blood testing for food allergies, unless very strongly positive, usually aren’t helpful in the management of the atopic individual. Patch test more and prick test less.”
Dr. Fowler disclosed a number of relationships with companies in the dermatology space. SDEF and this news organization are owned by the same parent company.
On Twitter @whitneymcknight
Allergic contact dermatitis is often missed in pediatric patients who present with eczema-like skin eruptions, in part because less is known about ACD in children than in adults.
“Often when we see a child with dermatitis, we automatically think of atopic dermatitis, but we should also consider the possibility of allergic contact dermatitis,” Dr. Joseph F. Fowler Jr. said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
A 2012 cohort study of 349 children between 0 and 15 years indicated that even very young children who were patch tested for common allergens had at least one positive result. Investigators found that nearly three-quarters of children studied tested positive for at least one allergen, typically nickel, other metals, fragrance, or preservatives (Dermatitis. 2012 Nov-Dec;23[6]:275-80).
“This is very similar to what we see in the adult population,” said Dr. Fowler, clinical professor of dermatology at the University of Louisville (Ky.). “Other studies in recent years have borne this out.”
Dr. Fowler suggested having a high degree of suspicion for ACD, especially when pediatric patients present with:
• Chronic, difficult to control atopic condition, as this could indicate a systemic reaction.
• Localized or facial dermatitis, as this could indicate the point of contact with an allergen.
• Scattered, generalized dermatitis, which also could represent systemic allergic contact dermatitis.
• Dermatitis that worsens, despite otherwise adequate treatment regimen.
• Reactions following contact with metals, fragrances, topical components, such as preservatives or neomycin.
“In these situations, patch testing will help determine that an allergen is implicated,” Dr. Fowler said.
In children with eczema, Dr. Fowler recommended patch testing when the eczema is not in the typical areas such as behind the knees or elbows, or if it started in typical areas and then spread elsewhere, especially in children around 5 years old.
“The moral of the story is that kids can be allergic to the same things as adults, even though we have less about this in the literature,” Dr. Fowler. “Skin testing or blood testing for food allergies, unless very strongly positive, usually aren’t helpful in the management of the atopic individual. Patch test more and prick test less.”
Dr. Fowler disclosed a number of relationships with companies in the dermatology space. SDEF and this news organization are owned by the same parent company.
On Twitter @whitneymcknight
Allergic contact dermatitis is often missed in pediatric patients who present with eczema-like skin eruptions, in part because less is known about ACD in children than in adults.
“Often when we see a child with dermatitis, we automatically think of atopic dermatitis, but we should also consider the possibility of allergic contact dermatitis,” Dr. Joseph F. Fowler Jr. said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
A 2012 cohort study of 349 children between 0 and 15 years indicated that even very young children who were patch tested for common allergens had at least one positive result. Investigators found that nearly three-quarters of children studied tested positive for at least one allergen, typically nickel, other metals, fragrance, or preservatives (Dermatitis. 2012 Nov-Dec;23[6]:275-80).
“This is very similar to what we see in the adult population,” said Dr. Fowler, clinical professor of dermatology at the University of Louisville (Ky.). “Other studies in recent years have borne this out.”
Dr. Fowler suggested having a high degree of suspicion for ACD, especially when pediatric patients present with:
• Chronic, difficult to control atopic condition, as this could indicate a systemic reaction.
• Localized or facial dermatitis, as this could indicate the point of contact with an allergen.
• Scattered, generalized dermatitis, which also could represent systemic allergic contact dermatitis.
• Dermatitis that worsens, despite otherwise adequate treatment regimen.
• Reactions following contact with metals, fragrances, topical components, such as preservatives or neomycin.
“In these situations, patch testing will help determine that an allergen is implicated,” Dr. Fowler said.
In children with eczema, Dr. Fowler recommended patch testing when the eczema is not in the typical areas such as behind the knees or elbows, or if it started in typical areas and then spread elsewhere, especially in children around 5 years old.
“The moral of the story is that kids can be allergic to the same things as adults, even though we have less about this in the literature,” Dr. Fowler. “Skin testing or blood testing for food allergies, unless very strongly positive, usually aren’t helpful in the management of the atopic individual. Patch test more and prick test less.”
Dr. Fowler disclosed a number of relationships with companies in the dermatology space. SDEF and this news organization are owned by the same parent company.
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR

Lichen Striatus
Lichen striatus (LS) is a benign, uncommon, self-limited, linear inflammatory skin disorder that primarily affects children up to 15 years of age, most commonly around 2 to 3 years of age, and is seen more frequently in girls.1 It presents with a sudden eruption of asymptomatic small, flat-topped, lichenoid, scaly papules in a linear array on a single extremity. The lesions may be erythematous, flesh colored, or hypopigmented.1,2 Multiple lesions appear over days to weeks and coalesce into linear plaques in a continuous or interrupted pattern along the lines of Blaschko, indicating possible somatic mosaicism.1 Although typically asymptomatic, it may be pruritic. Most cases spontaneously resolve within 1 year.3 Recurrences are unusual. Digital involvement may result in onycholysis, longitudinal ridging, splitting, and nail loss.1 The underlying cause of LS may be an abnormal immunologic reaction or genetic predisposition that is precipitated by some trigger such as a viral infection, trauma, hypersensitivity reaction, vaccine, seasonal variation, medication, or pregnancy.1,2 An association with atopy has been described. Treatment is not necessary but options include topical steroids, topical retinoids, and topical calcineurin inhibitors.2
Histologically, findings in LS are somewhat variable but typically show a combination of spongiotic and lichenoid interface dermatitis with a perivascular and periadnexal lymphocytic infiltrate (Figure 1). Epidermal changes include intercellular and intracellular edema, focal spongiosis, lymphocytic exocytosis, parakeratosis, patchy hyperkeratosis, and keratinocyte necrosis (Figure 2A).1,3 The epidermis is normal or slightly acanthotic, and dyskeratotic keratinocytes can be found in the granular and horny layers or at the dermoepidermal junction.2 The lymphohistiocytic infiltrate in the superficial and deep dermis surrounds vascular plexuses and cutaneous adnexa such as eccrine glands and hair follicles.1 Perivascular lymphoid aggregates and eccrine coil involvement are particularly distinctive of LS (Figure 2B).4 Pigment incontinence also may be seen.
Another condition that distributes linearly along the lines of Blaschko is linear epidermolytic hyperkeratosis (EHK). Similar to LS, histology shows hyperkeratosis, focal parakeratosis, and acanthosis of the epidermis.5 However, EHK shows epidermolysis, acantholysis, and perinuclear vacuolization in spinous and granular layers (Figure 3).5 The lack of perivascular and periadnexal inflammation also can help differentiate EHK from LS.
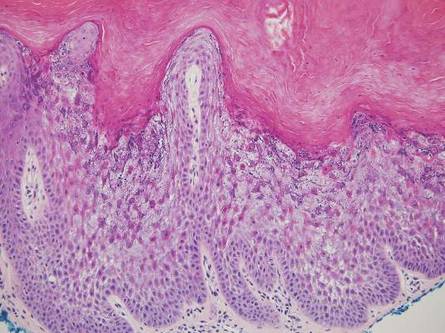
Linear lichen planus (LLP), similar to LS, histologically shows a lichenoid lymphocytic bandlike infiltrate obscuring the dermoepidermal junction, vacuolization of the basal cell layer, and pigment incontinence.1,2 Although LS and LLP can have histologic overlap, the absence of adnexal or perieccrine lymphocytic inflammation can help distinguish the two.3 The histopathologic changes of intercellular edema or mild spongiosis, exocytosis, and parakeratosis present in LS also are typically absent in LLP. Linear lichen planus characteristically consists of wedge-shaped hypergranulosis and irregular acanthosis with saw-toothed rete ridges (Figure 4).2 In addition, lobular eosinophilic deposits known as cytoid or Civatte bodies representing degenerated keratinocytes can be visualized at the dermoepidermal junction in LLP.2 Immunofluorescence will highlight Civatte bodies with IgM, IgG, and C3, also helping to differentiate these 2 conditions.1
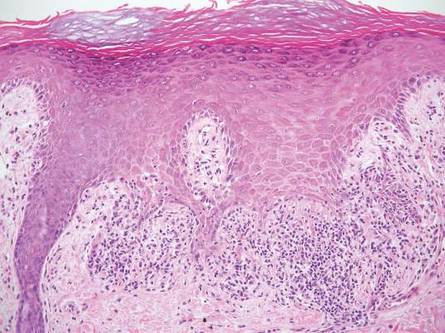
Linear porokeratosis can be mistaken for the linear lesion of LS. Both entities may reveal perivascular lymphocytes in the dermis, and porokeratosis can be lichenoid in the central portion of the lesion.6 However, porokeratosis is unique in that it contains a cornoid lamella, characterized by a thin column of tightly packed parakeratotic cells extending from an invagination of the epidermis through the adjacent stratum corneum (Figure 5).6 Beneath the cornoid lamella, the granular layer is either absent or markedly attenuated, and pyknotic keratinocytes with perinuclear edema are present in the spinous layer.6 The epidermis in the central portion of the porokeratotic lesion may be normal, hyperplastic, or atrophic with effacement of rete ridges.
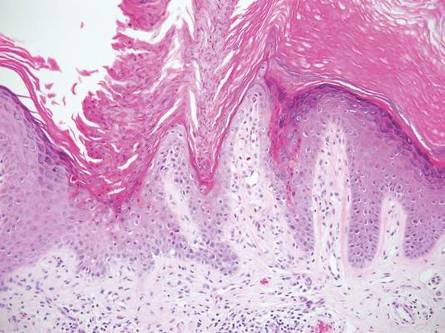
Similar to LS, linear psoriasis follows lines of Blaschko clinically. However, it is distinguished by its characteristic psoriatic epidermal changes as well as its lack of lichenoid or perieccrine inflammation.3 Typical findings in linear psoriasis include hyperkeratosis, confluent parakeratosis with entrapped neutrophilic microabscesses, acanthosis with regular elongation of rete ridges, intraepidermal neutrophils, thinned suprapapillary plates, dilated capillaries in the tips of the dermal papillae, and a chronic dermal inflammatory infiltrate (Figure 6).4
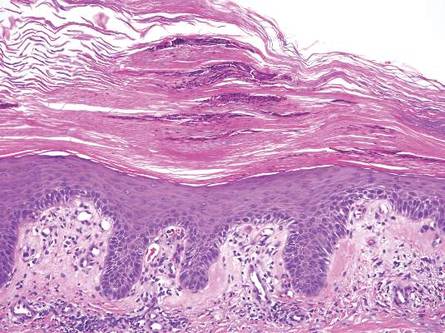
- Wang WL, Lazar A. Lichenoid and interface dermatitis. In: Calonje E, Brenn T, Lazar A, et al, eds. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier/Saunders; 2011:219-258.
- Shiohara T, Kano Y. Lichen planus and lichenoid dermatoses. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:183-202.
- Zhang Y, McNutt NS. Lichen striatus. histological, immunohistochemical, and ultrastructural study of 37 cases. J Cutan Pathol. 2001;28:65-71.
- Johnson M, Walker D, Galloway W, et al. Interface dermatitis along Blaschko’s lines. J Cutan Pathol. 2014;41:950-954.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Requena L, Requena C, Cockerell C. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:1795-1815.
Lichen striatus (LS) is a benign, uncommon, self-limited, linear inflammatory skin disorder that primarily affects children up to 15 years of age, most commonly around 2 to 3 years of age, and is seen more frequently in girls.1 It presents with a sudden eruption of asymptomatic small, flat-topped, lichenoid, scaly papules in a linear array on a single extremity. The lesions may be erythematous, flesh colored, or hypopigmented.1,2 Multiple lesions appear over days to weeks and coalesce into linear plaques in a continuous or interrupted pattern along the lines of Blaschko, indicating possible somatic mosaicism.1 Although typically asymptomatic, it may be pruritic. Most cases spontaneously resolve within 1 year.3 Recurrences are unusual. Digital involvement may result in onycholysis, longitudinal ridging, splitting, and nail loss.1 The underlying cause of LS may be an abnormal immunologic reaction or genetic predisposition that is precipitated by some trigger such as a viral infection, trauma, hypersensitivity reaction, vaccine, seasonal variation, medication, or pregnancy.1,2 An association with atopy has been described. Treatment is not necessary but options include topical steroids, topical retinoids, and topical calcineurin inhibitors.2
Histologically, findings in LS are somewhat variable but typically show a combination of spongiotic and lichenoid interface dermatitis with a perivascular and periadnexal lymphocytic infiltrate (Figure 1). Epidermal changes include intercellular and intracellular edema, focal spongiosis, lymphocytic exocytosis, parakeratosis, patchy hyperkeratosis, and keratinocyte necrosis (Figure 2A).1,3 The epidermis is normal or slightly acanthotic, and dyskeratotic keratinocytes can be found in the granular and horny layers or at the dermoepidermal junction.2 The lymphohistiocytic infiltrate in the superficial and deep dermis surrounds vascular plexuses and cutaneous adnexa such as eccrine glands and hair follicles.1 Perivascular lymphoid aggregates and eccrine coil involvement are particularly distinctive of LS (Figure 2B).4 Pigment incontinence also may be seen.
Another condition that distributes linearly along the lines of Blaschko is linear epidermolytic hyperkeratosis (EHK). Similar to LS, histology shows hyperkeratosis, focal parakeratosis, and acanthosis of the epidermis.5 However, EHK shows epidermolysis, acantholysis, and perinuclear vacuolization in spinous and granular layers (Figure 3).5 The lack of perivascular and periadnexal inflammation also can help differentiate EHK from LS.
Linear lichen planus (LLP), similar to LS, histologically shows a lichenoid lymphocytic bandlike infiltrate obscuring the dermoepidermal junction, vacuolization of the basal cell layer, and pigment incontinence.1,2 Although LS and LLP can have histologic overlap, the absence of adnexal or perieccrine lymphocytic inflammation can help distinguish the two.3 The histopathologic changes of intercellular edema or mild spongiosis, exocytosis, and parakeratosis present in LS also are typically absent in LLP. Linear lichen planus characteristically consists of wedge-shaped hypergranulosis and irregular acanthosis with saw-toothed rete ridges (Figure 4).2 In addition, lobular eosinophilic deposits known as cytoid or Civatte bodies representing degenerated keratinocytes can be visualized at the dermoepidermal junction in LLP.2 Immunofluorescence will highlight Civatte bodies with IgM, IgG, and C3, also helping to differentiate these 2 conditions.1
Linear porokeratosis can be mistaken for the linear lesion of LS. Both entities may reveal perivascular lymphocytes in the dermis, and porokeratosis can be lichenoid in the central portion of the lesion.6 However, porokeratosis is unique in that it contains a cornoid lamella, characterized by a thin column of tightly packed parakeratotic cells extending from an invagination of the epidermis through the adjacent stratum corneum (Figure 5).6 Beneath the cornoid lamella, the granular layer is either absent or markedly attenuated, and pyknotic keratinocytes with perinuclear edema are present in the spinous layer.6 The epidermis in the central portion of the porokeratotic lesion may be normal, hyperplastic, or atrophic with effacement of rete ridges.
Similar to LS, linear psoriasis follows lines of Blaschko clinically. However, it is distinguished by its characteristic psoriatic epidermal changes as well as its lack of lichenoid or perieccrine inflammation.3 Typical findings in linear psoriasis include hyperkeratosis, confluent parakeratosis with entrapped neutrophilic microabscesses, acanthosis with regular elongation of rete ridges, intraepidermal neutrophils, thinned suprapapillary plates, dilated capillaries in the tips of the dermal papillae, and a chronic dermal inflammatory infiltrate (Figure 6).4
Lichen striatus (LS) is a benign, uncommon, self-limited, linear inflammatory skin disorder that primarily affects children up to 15 years of age, most commonly around 2 to 3 years of age, and is seen more frequently in girls.1 It presents with a sudden eruption of asymptomatic small, flat-topped, lichenoid, scaly papules in a linear array on a single extremity. The lesions may be erythematous, flesh colored, or hypopigmented.1,2 Multiple lesions appear over days to weeks and coalesce into linear plaques in a continuous or interrupted pattern along the lines of Blaschko, indicating possible somatic mosaicism.1 Although typically asymptomatic, it may be pruritic. Most cases spontaneously resolve within 1 year.3 Recurrences are unusual. Digital involvement may result in onycholysis, longitudinal ridging, splitting, and nail loss.1 The underlying cause of LS may be an abnormal immunologic reaction or genetic predisposition that is precipitated by some trigger such as a viral infection, trauma, hypersensitivity reaction, vaccine, seasonal variation, medication, or pregnancy.1,2 An association with atopy has been described. Treatment is not necessary but options include topical steroids, topical retinoids, and topical calcineurin inhibitors.2
Histologically, findings in LS are somewhat variable but typically show a combination of spongiotic and lichenoid interface dermatitis with a perivascular and periadnexal lymphocytic infiltrate (Figure 1). Epidermal changes include intercellular and intracellular edema, focal spongiosis, lymphocytic exocytosis, parakeratosis, patchy hyperkeratosis, and keratinocyte necrosis (Figure 2A).1,3 The epidermis is normal or slightly acanthotic, and dyskeratotic keratinocytes can be found in the granular and horny layers or at the dermoepidermal junction.2 The lymphohistiocytic infiltrate in the superficial and deep dermis surrounds vascular plexuses and cutaneous adnexa such as eccrine glands and hair follicles.1 Perivascular lymphoid aggregates and eccrine coil involvement are particularly distinctive of LS (Figure 2B).4 Pigment incontinence also may be seen.
Another condition that distributes linearly along the lines of Blaschko is linear epidermolytic hyperkeratosis (EHK). Similar to LS, histology shows hyperkeratosis, focal parakeratosis, and acanthosis of the epidermis.5 However, EHK shows epidermolysis, acantholysis, and perinuclear vacuolization in spinous and granular layers (Figure 3).5 The lack of perivascular and periadnexal inflammation also can help differentiate EHK from LS.
Linear lichen planus (LLP), similar to LS, histologically shows a lichenoid lymphocytic bandlike infiltrate obscuring the dermoepidermal junction, vacuolization of the basal cell layer, and pigment incontinence.1,2 Although LS and LLP can have histologic overlap, the absence of adnexal or perieccrine lymphocytic inflammation can help distinguish the two.3 The histopathologic changes of intercellular edema or mild spongiosis, exocytosis, and parakeratosis present in LS also are typically absent in LLP. Linear lichen planus characteristically consists of wedge-shaped hypergranulosis and irregular acanthosis with saw-toothed rete ridges (Figure 4).2 In addition, lobular eosinophilic deposits known as cytoid or Civatte bodies representing degenerated keratinocytes can be visualized at the dermoepidermal junction in LLP.2 Immunofluorescence will highlight Civatte bodies with IgM, IgG, and C3, also helping to differentiate these 2 conditions.1
Linear porokeratosis can be mistaken for the linear lesion of LS. Both entities may reveal perivascular lymphocytes in the dermis, and porokeratosis can be lichenoid in the central portion of the lesion.6 However, porokeratosis is unique in that it contains a cornoid lamella, characterized by a thin column of tightly packed parakeratotic cells extending from an invagination of the epidermis through the adjacent stratum corneum (Figure 5).6 Beneath the cornoid lamella, the granular layer is either absent or markedly attenuated, and pyknotic keratinocytes with perinuclear edema are present in the spinous layer.6 The epidermis in the central portion of the porokeratotic lesion may be normal, hyperplastic, or atrophic with effacement of rete ridges.
Similar to LS, linear psoriasis follows lines of Blaschko clinically. However, it is distinguished by its characteristic psoriatic epidermal changes as well as its lack of lichenoid or perieccrine inflammation.3 Typical findings in linear psoriasis include hyperkeratosis, confluent parakeratosis with entrapped neutrophilic microabscesses, acanthosis with regular elongation of rete ridges, intraepidermal neutrophils, thinned suprapapillary plates, dilated capillaries in the tips of the dermal papillae, and a chronic dermal inflammatory infiltrate (Figure 6).4
- Wang WL, Lazar A. Lichenoid and interface dermatitis. In: Calonje E, Brenn T, Lazar A, et al, eds. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier/Saunders; 2011:219-258.
- Shiohara T, Kano Y. Lichen planus and lichenoid dermatoses. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:183-202.
- Zhang Y, McNutt NS. Lichen striatus. histological, immunohistochemical, and ultrastructural study of 37 cases. J Cutan Pathol. 2001;28:65-71.
- Johnson M, Walker D, Galloway W, et al. Interface dermatitis along Blaschko’s lines. J Cutan Pathol. 2014;41:950-954.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Requena L, Requena C, Cockerell C. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:1795-1815.
- Wang WL, Lazar A. Lichenoid and interface dermatitis. In: Calonje E, Brenn T, Lazar A, et al, eds. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier/Saunders; 2011:219-258.
- Shiohara T, Kano Y. Lichen planus and lichenoid dermatoses. In: Bolognia J, Jorizzo J, Schaffer J, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:183-202.
- Zhang Y, McNutt NS. Lichen striatus. histological, immunohistochemical, and ultrastructural study of 37 cases. J Cutan Pathol. 2001;28:65-71.
- Johnson M, Walker D, Galloway W, et al. Interface dermatitis along Blaschko’s lines. J Cutan Pathol. 2014;41:950-954.
- Kumar P, Kumar R, Kumar Mandal RK, et al. Systematized linear epidermolytic hyperkeratosis. Dermatol Online J. 2014;20:21248.
- Requena L, Requena C, Cockerell C. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. Philadelphia, PA: Elsevier/Saunders; 2012:1795-1815.
Chemo linked to long-term learning problems in ALL survivors

Pediatric acute lymphoblastic leukemia (ALL) patients treated with chemotherapy alone are at risk for attention and learning problems that persist after treatment ends, according to a study published in the Journal of Clinical Oncology.
Researchers said this study is the largest assessment to date of neurocognitive outcomes in pediatric ALL survivors treated with intensive chemotherapy alone rather than in combination with cranial radiation.
“This is an important contribution to the literature because the smaller size and design of previous studies made examining the impact of treatment difficult,” said study author Lisa Jacola, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The findings underscore the need for neurocognitive and academic screening to be included as part of routine survivorship care for all pediatric ALL survivors.”
The study included patients enrolled in the St. Jude Total Therapy Study XV. They underwent neurocognitive assessments at the beginning of induction (n=142), end of maintenance (n=243), and 2 years after completing treatment (n=211).
The subjects completed standardized tests of overall intelligence, attention, learning, and academic performance. In addition, parents and other caregivers rated the subjects’ attention, learning, and behavior.
Two years after treatment completion, subjects performed as expected for their age (compared to national data) on measures of overall intelligence, learning, and memory. However, study subjects had significant attention deficits and a significantly greater frequency of learning problems (all P≤0.005).
The risk of such problems was greatest for survivors who were less than 5 years old when diagnosed with ALL and for those who received more intensive chemotherapy.
Specifically, the younger subjects had a greater risk of difficulties with attention, learning, working memory, and processing speed (all P≤0.05). And subjects who received higher-intensity, CNS-directed chemotherapy had a greater risk of difficulties in attention, processing speed, and academics (all P≤0.01).
Researchers also found that subjects with attention problems at the end of therapy had lower academic scores 2 years later (all P≤0.05).
“These findings provide additional evidence that neurocognitive functioning has improved in survivors of childhood ALL since cranial irradiation was replaced with intensified chemotherapy,” Dr Jacola said.
“But we also show these young people are at an elevated risk for attention problems that have real-world consequences, particularly for learning and school performance. Attention is a building block for learning, and, in this study, attention difficulties predicted academic problems later. If we know attention problems seen at the end of therapy continue and contribute to academic problems, then our goal is to intervene earlier to reduce or prevent such difficulties.” ![]()
Pediatric acute lymphoblastic leukemia (ALL) patients treated with chemotherapy alone are at risk for attention and learning problems that persist after treatment ends, according to a study published in the Journal of Clinical Oncology.
Researchers said this study is the largest assessment to date of neurocognitive outcomes in pediatric ALL survivors treated with intensive chemotherapy alone rather than in combination with cranial radiation.
“This is an important contribution to the literature because the smaller size and design of previous studies made examining the impact of treatment difficult,” said study author Lisa Jacola, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The findings underscore the need for neurocognitive and academic screening to be included as part of routine survivorship care for all pediatric ALL survivors.”
The study included patients enrolled in the St. Jude Total Therapy Study XV. They underwent neurocognitive assessments at the beginning of induction (n=142), end of maintenance (n=243), and 2 years after completing treatment (n=211).
The subjects completed standardized tests of overall intelligence, attention, learning, and academic performance. In addition, parents and other caregivers rated the subjects’ attention, learning, and behavior.
Two years after treatment completion, subjects performed as expected for their age (compared to national data) on measures of overall intelligence, learning, and memory. However, study subjects had significant attention deficits and a significantly greater frequency of learning problems (all P≤0.005).
The risk of such problems was greatest for survivors who were less than 5 years old when diagnosed with ALL and for those who received more intensive chemotherapy.
Specifically, the younger subjects had a greater risk of difficulties with attention, learning, working memory, and processing speed (all P≤0.05). And subjects who received higher-intensity, CNS-directed chemotherapy had a greater risk of difficulties in attention, processing speed, and academics (all P≤0.01).
Researchers also found that subjects with attention problems at the end of therapy had lower academic scores 2 years later (all P≤0.05).
“These findings provide additional evidence that neurocognitive functioning has improved in survivors of childhood ALL since cranial irradiation was replaced with intensified chemotherapy,” Dr Jacola said.
“But we also show these young people are at an elevated risk for attention problems that have real-world consequences, particularly for learning and school performance. Attention is a building block for learning, and, in this study, attention difficulties predicted academic problems later. If we know attention problems seen at the end of therapy continue and contribute to academic problems, then our goal is to intervene earlier to reduce or prevent such difficulties.” ![]()
Pediatric acute lymphoblastic leukemia (ALL) patients treated with chemotherapy alone are at risk for attention and learning problems that persist after treatment ends, according to a study published in the Journal of Clinical Oncology.
Researchers said this study is the largest assessment to date of neurocognitive outcomes in pediatric ALL survivors treated with intensive chemotherapy alone rather than in combination with cranial radiation.
“This is an important contribution to the literature because the smaller size and design of previous studies made examining the impact of treatment difficult,” said study author Lisa Jacola, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The findings underscore the need for neurocognitive and academic screening to be included as part of routine survivorship care for all pediatric ALL survivors.”
The study included patients enrolled in the St. Jude Total Therapy Study XV. They underwent neurocognitive assessments at the beginning of induction (n=142), end of maintenance (n=243), and 2 years after completing treatment (n=211).
The subjects completed standardized tests of overall intelligence, attention, learning, and academic performance. In addition, parents and other caregivers rated the subjects’ attention, learning, and behavior.
Two years after treatment completion, subjects performed as expected for their age (compared to national data) on measures of overall intelligence, learning, and memory. However, study subjects had significant attention deficits and a significantly greater frequency of learning problems (all P≤0.005).
The risk of such problems was greatest for survivors who were less than 5 years old when diagnosed with ALL and for those who received more intensive chemotherapy.
Specifically, the younger subjects had a greater risk of difficulties with attention, learning, working memory, and processing speed (all P≤0.05). And subjects who received higher-intensity, CNS-directed chemotherapy had a greater risk of difficulties in attention, processing speed, and academics (all P≤0.01).
Researchers also found that subjects with attention problems at the end of therapy had lower academic scores 2 years later (all P≤0.05).
“These findings provide additional evidence that neurocognitive functioning has improved in survivors of childhood ALL since cranial irradiation was replaced with intensified chemotherapy,” Dr Jacola said.
“But we also show these young people are at an elevated risk for attention problems that have real-world consequences, particularly for learning and school performance. Attention is a building block for learning, and, in this study, attention difficulties predicted academic problems later. If we know attention problems seen at the end of therapy continue and contribute to academic problems, then our goal is to intervene earlier to reduce or prevent such difficulties.” ![]()