User login
School-located influenza vaccination programs can be effective
School-located influenza vaccination (SLIV) increased seasonal influenza vaccination rates countywide and in both suburban and urban settings, a study found.
“Schools have a stake in influenza vaccination because immunization of schoolchildren can reduce absenteeism throughout the community. Nevertheless, only 6% of childhood influenza vaccinations occur at school. SLIV poses logistical challenges: obtaining parental consent, ordering and administering vaccine, and billing,” said Peter G. Szilagyi, MD, of Mattel Children’s Hospital, Los Angeles, and his associates.
From 2014 to 2015, 44 elementary schools were randomized in upstate New York in an organized cluster-randomized trial in which 19,776 children were eligible candidates. Seven percent of SLIV school students, 5% of suburban SLIV school students, and 9% of urban SLIV students were vaccinated at SLIV clinics. Children in SLIV schools had higher flu vaccination rates than did children in control schools countywide (54% vs. 47%, P less than .001) and in suburban (62% vs. 54%, P less than .001) and urban schools (44% vs. 39%; P less than .001).
SLIV did substitute for vaccination for urban settings serving more Vaccines for Children–covered students, but did not substitute for practice-based vaccination in the suburbs, where pediatricians often preorder influenza vaccine.
“SLIV, using Web-based consent, is a potential strategy to improve influenza vaccination coverage among large populations of children,” the researchers concluded.
Read the full story here: Pediatrics. 2016. doi: 10.1542/peds.2016-1746.
School-located influenza vaccination (SLIV) increased seasonal influenza vaccination rates countywide and in both suburban and urban settings, a study found.
“Schools have a stake in influenza vaccination because immunization of schoolchildren can reduce absenteeism throughout the community. Nevertheless, only 6% of childhood influenza vaccinations occur at school. SLIV poses logistical challenges: obtaining parental consent, ordering and administering vaccine, and billing,” said Peter G. Szilagyi, MD, of Mattel Children’s Hospital, Los Angeles, and his associates.
From 2014 to 2015, 44 elementary schools were randomized in upstate New York in an organized cluster-randomized trial in which 19,776 children were eligible candidates. Seven percent of SLIV school students, 5% of suburban SLIV school students, and 9% of urban SLIV students were vaccinated at SLIV clinics. Children in SLIV schools had higher flu vaccination rates than did children in control schools countywide (54% vs. 47%, P less than .001) and in suburban (62% vs. 54%, P less than .001) and urban schools (44% vs. 39%; P less than .001).
SLIV did substitute for vaccination for urban settings serving more Vaccines for Children–covered students, but did not substitute for practice-based vaccination in the suburbs, where pediatricians often preorder influenza vaccine.
“SLIV, using Web-based consent, is a potential strategy to improve influenza vaccination coverage among large populations of children,” the researchers concluded.
Read the full story here: Pediatrics. 2016. doi: 10.1542/peds.2016-1746.
School-located influenza vaccination (SLIV) increased seasonal influenza vaccination rates countywide and in both suburban and urban settings, a study found.
“Schools have a stake in influenza vaccination because immunization of schoolchildren can reduce absenteeism throughout the community. Nevertheless, only 6% of childhood influenza vaccinations occur at school. SLIV poses logistical challenges: obtaining parental consent, ordering and administering vaccine, and billing,” said Peter G. Szilagyi, MD, of Mattel Children’s Hospital, Los Angeles, and his associates.
From 2014 to 2015, 44 elementary schools were randomized in upstate New York in an organized cluster-randomized trial in which 19,776 children were eligible candidates. Seven percent of SLIV school students, 5% of suburban SLIV school students, and 9% of urban SLIV students were vaccinated at SLIV clinics. Children in SLIV schools had higher flu vaccination rates than did children in control schools countywide (54% vs. 47%, P less than .001) and in suburban (62% vs. 54%, P less than .001) and urban schools (44% vs. 39%; P less than .001).
SLIV did substitute for vaccination for urban settings serving more Vaccines for Children–covered students, but did not substitute for practice-based vaccination in the suburbs, where pediatricians often preorder influenza vaccine.
“SLIV, using Web-based consent, is a potential strategy to improve influenza vaccination coverage among large populations of children,” the researchers concluded.
Read the full story here: Pediatrics. 2016. doi: 10.1542/peds.2016-1746.
FROM PEDIATRICS
Low parental confidence in HPV vaccine stymies adolescent vaccination rates
More than a quarter of U.S. parents surveyed refused human papillomavirus (HPV) vaccination for their adolescents because of a lack of overall trust in adolescent vaccination programs and higher levels of perceived harm, a study found.
In an online survey of 1,484 U.S. parents, 28% of respondents reported they had refused the HPV vaccine on behalf of their children aged 11-17 years at least once. Another 8% responded they had elected to delay vaccination. The remaining two-thirds of respondents said they had neither refused nor delayed the vaccination, reported Melissa B. Gilkey, PhD, of Harvard Medical School, Boston, and her associates (Hum Vaccin Immunother. 2016. doi: 10.1080/21645515.2016.1247134).
Compared with parents who reported neither refusal nor delay, refusal was associated with lower confidence in adolescent vaccination (relative risk ratio = 0.66, 95% CI, 0.48-0.91), lower perceived HPV vaccine effectiveness (RRR = 0.68, 95% CI, 0.50-0.91), and higher perceived harms (RRR = 3.49, 95% CI, 2.65-4.60). Parents who reported delaying vaccination were more likely to endorse insufficient information as the reason (RRR = 1.76, 95% CI, 1.08-2.85). While 79% of parents who had delayed HPV vaccination said talking with a physician would help them with their decision, 61% of parents who refused the vaccination said it would. In addition, nearly half of parents who delayed vaccination said they did so out of a preference to wait until their children were older.
In adolescents whose parents had ever refused the vaccine, only 27% had received one HPV vaccine vs. 59% in those whose parents had elected to delay vaccination. Among adolescents whose parents responded they had neither refused nor delayed the vaccine, 56% had received one HPV vaccine.
Although the investigators did not find race, ethnicity, nor educational attainment were drivers of whether a parent chose to vaccinate, families with higher income levels tended to refuse the HPV vaccine more often than did other parents (RRR: 1.48, 95% confidence interval, 1.02-2.15).
Merck and the National Cancer Institute funded the study. Coauthor Noel T. Brewer, PhD, has received HPV vaccine-related grants from, or been on paid advisory boards for, Merck, GlaxoSmithKline, and Pfizer; he served on the National Vaccine Advisory Committee Working Group on HPV Vaccine and is chair of the National HPV Vaccination Roundtable.
More than a quarter of U.S. parents surveyed refused human papillomavirus (HPV) vaccination for their adolescents because of a lack of overall trust in adolescent vaccination programs and higher levels of perceived harm, a study found.
In an online survey of 1,484 U.S. parents, 28% of respondents reported they had refused the HPV vaccine on behalf of their children aged 11-17 years at least once. Another 8% responded they had elected to delay vaccination. The remaining two-thirds of respondents said they had neither refused nor delayed the vaccination, reported Melissa B. Gilkey, PhD, of Harvard Medical School, Boston, and her associates (Hum Vaccin Immunother. 2016. doi: 10.1080/21645515.2016.1247134).
Compared with parents who reported neither refusal nor delay, refusal was associated with lower confidence in adolescent vaccination (relative risk ratio = 0.66, 95% CI, 0.48-0.91), lower perceived HPV vaccine effectiveness (RRR = 0.68, 95% CI, 0.50-0.91), and higher perceived harms (RRR = 3.49, 95% CI, 2.65-4.60). Parents who reported delaying vaccination were more likely to endorse insufficient information as the reason (RRR = 1.76, 95% CI, 1.08-2.85). While 79% of parents who had delayed HPV vaccination said talking with a physician would help them with their decision, 61% of parents who refused the vaccination said it would. In addition, nearly half of parents who delayed vaccination said they did so out of a preference to wait until their children were older.
In adolescents whose parents had ever refused the vaccine, only 27% had received one HPV vaccine vs. 59% in those whose parents had elected to delay vaccination. Among adolescents whose parents responded they had neither refused nor delayed the vaccine, 56% had received one HPV vaccine.
Although the investigators did not find race, ethnicity, nor educational attainment were drivers of whether a parent chose to vaccinate, families with higher income levels tended to refuse the HPV vaccine more often than did other parents (RRR: 1.48, 95% confidence interval, 1.02-2.15).
Merck and the National Cancer Institute funded the study. Coauthor Noel T. Brewer, PhD, has received HPV vaccine-related grants from, or been on paid advisory boards for, Merck, GlaxoSmithKline, and Pfizer; he served on the National Vaccine Advisory Committee Working Group on HPV Vaccine and is chair of the National HPV Vaccination Roundtable.
More than a quarter of U.S. parents surveyed refused human papillomavirus (HPV) vaccination for their adolescents because of a lack of overall trust in adolescent vaccination programs and higher levels of perceived harm, a study found.
In an online survey of 1,484 U.S. parents, 28% of respondents reported they had refused the HPV vaccine on behalf of their children aged 11-17 years at least once. Another 8% responded they had elected to delay vaccination. The remaining two-thirds of respondents said they had neither refused nor delayed the vaccination, reported Melissa B. Gilkey, PhD, of Harvard Medical School, Boston, and her associates (Hum Vaccin Immunother. 2016. doi: 10.1080/21645515.2016.1247134).
Compared with parents who reported neither refusal nor delay, refusal was associated with lower confidence in adolescent vaccination (relative risk ratio = 0.66, 95% CI, 0.48-0.91), lower perceived HPV vaccine effectiveness (RRR = 0.68, 95% CI, 0.50-0.91), and higher perceived harms (RRR = 3.49, 95% CI, 2.65-4.60). Parents who reported delaying vaccination were more likely to endorse insufficient information as the reason (RRR = 1.76, 95% CI, 1.08-2.85). While 79% of parents who had delayed HPV vaccination said talking with a physician would help them with their decision, 61% of parents who refused the vaccination said it would. In addition, nearly half of parents who delayed vaccination said they did so out of a preference to wait until their children were older.
In adolescents whose parents had ever refused the vaccine, only 27% had received one HPV vaccine vs. 59% in those whose parents had elected to delay vaccination. Among adolescents whose parents responded they had neither refused nor delayed the vaccine, 56% had received one HPV vaccine.
Although the investigators did not find race, ethnicity, nor educational attainment were drivers of whether a parent chose to vaccinate, families with higher income levels tended to refuse the HPV vaccine more often than did other parents (RRR: 1.48, 95% confidence interval, 1.02-2.15).
Merck and the National Cancer Institute funded the study. Coauthor Noel T. Brewer, PhD, has received HPV vaccine-related grants from, or been on paid advisory boards for, Merck, GlaxoSmithKline, and Pfizer; he served on the National Vaccine Advisory Committee Working Group on HPV Vaccine and is chair of the National HPV Vaccination Roundtable.
Key clinical point:
Major finding: HPV vaccine refusal rate was 28% in parents of teens and preteens; the rate of vaccine delay was 8%.
Data source: Online survey conducted in 2014-2015 of 1,484 U.S. parents with children between ages of 11 and 17 years.
Disclosures: Merck and the National Cancer Institute funded the study. Coauthor Noel T. Brewer, PhD, has received HPV vaccine-related grants from, or been on paid advisory boards for, Merck, GlaxoSmithKline, and Pfizer; he served on the National Vaccine Advisory Committee Working Group on HPV Vaccine and is chair of the National HPV Vaccination Roundtable.
Roommates
The American Academy of Pediatrics has recently released a new policy for parents on safe sleep practices that in addition to the previous warnings about bed sharing and positioning includes the recommendation that an infant sleep in the same room as her parent for at least the first 6 months (Pediatrics. 2016 Oct;138[5]:e20162938). Apparently what prompted this new set of recommendations is the observation that deaths from sudden unexpected infant deaths (SUIDS) and sudden infant deaths (SIDS) has plateaued since the dramatic decline we witnessed in the 1990s following the Back-to-Sleep campaign.
Although the policy statement refers to “new research” that has become available since the last policy statement was released in 2011, I have had trouble finding convincing evidence in the references I reviewed to support the room sharing recommendation. In some studies, room sharing was the cultural norm, making it difficult to establish a control group. In one of the most frequently cited papers from New Zealand, the authors could not sort out the effects of prone sleeping and sleeping alone, and wonder whether both factors may be affecting risk “through a common mechanism” (Lancet. 1996 Jan 6;347[8993]:7-12).

For some, parents attempting to follow this recommendation may not be without its negative consequences. Sleeping like a baby is not the same as sleeping quietly. Infants often breathe in a pattern that includes long, anxiety-provoking pauses. The implication of this policy recommendation is that parents can prevent crib death by being more vigilant at night. Do we have enough evidence that this is indeed the case?
Most parents are already anxious, and none of them are getting enough sleep. I can envision that trying to follow this recommendation could aggravate both conditions for some parents. Sleep-deprived parents often are not as capable parents as they could be. And they certainly aren’t as happy as they could be. Postpartum depression compounded by sleep deprivation continues to be an underreported and inadequately managed condition that can have negative effects for the health of the child.
For some parents, room sharing is something they gravitate toward naturally, and it can help them deal with the anxiety of new parenthood. They may sleep better with their infant close by. But for others, the better solution to their own sleep deprivation lies in sleep training, a strategy that is very difficult, if not impossible, for parents who are sharing their bedroom with their infant.
As the authors of one of the most frequently quoted papers that supports room sharing have written, “the traditional habit of labeling one sleep arrangement as being superior to another without awareness of the family context is not only wrong but potentially harmful” (Paediatric Resp Review. 2005, Jun;6[2]:134-52).
I think the academy has gone too far or at least moved prematurely with its room sharing recommendation. For some families, room sharing is a better arrangement, for others it is not. It may well be that the plateau in crib deaths is telling us that we have reached the limits of our abilities to effect any further decline with our recommendations about sleep environments. But more research needs to be done.
On a more positive note, the new recommendation may force parents to reevaluate their habit of having a television in their bedroom. Will it be baby or TV in the bedroom? Unfortunately, I fear too many will opt to have both.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.” Email him at pdnews@frontlinemedcom.com.
The American Academy of Pediatrics has recently released a new policy for parents on safe sleep practices that in addition to the previous warnings about bed sharing and positioning includes the recommendation that an infant sleep in the same room as her parent for at least the first 6 months (Pediatrics. 2016 Oct;138[5]:e20162938). Apparently what prompted this new set of recommendations is the observation that deaths from sudden unexpected infant deaths (SUIDS) and sudden infant deaths (SIDS) has plateaued since the dramatic decline we witnessed in the 1990s following the Back-to-Sleep campaign.
Although the policy statement refers to “new research” that has become available since the last policy statement was released in 2011, I have had trouble finding convincing evidence in the references I reviewed to support the room sharing recommendation. In some studies, room sharing was the cultural norm, making it difficult to establish a control group. In one of the most frequently cited papers from New Zealand, the authors could not sort out the effects of prone sleeping and sleeping alone, and wonder whether both factors may be affecting risk “through a common mechanism” (Lancet. 1996 Jan 6;347[8993]:7-12).
For some, parents attempting to follow this recommendation may not be without its negative consequences. Sleeping like a baby is not the same as sleeping quietly. Infants often breathe in a pattern that includes long, anxiety-provoking pauses. The implication of this policy recommendation is that parents can prevent crib death by being more vigilant at night. Do we have enough evidence that this is indeed the case?
Most parents are already anxious, and none of them are getting enough sleep. I can envision that trying to follow this recommendation could aggravate both conditions for some parents. Sleep-deprived parents often are not as capable parents as they could be. And they certainly aren’t as happy as they could be. Postpartum depression compounded by sleep deprivation continues to be an underreported and inadequately managed condition that can have negative effects for the health of the child.
For some parents, room sharing is something they gravitate toward naturally, and it can help them deal with the anxiety of new parenthood. They may sleep better with their infant close by. But for others, the better solution to their own sleep deprivation lies in sleep training, a strategy that is very difficult, if not impossible, for parents who are sharing their bedroom with their infant.
As the authors of one of the most frequently quoted papers that supports room sharing have written, “the traditional habit of labeling one sleep arrangement as being superior to another without awareness of the family context is not only wrong but potentially harmful” (Paediatric Resp Review. 2005, Jun;6[2]:134-52).
I think the academy has gone too far or at least moved prematurely with its room sharing recommendation. For some families, room sharing is a better arrangement, for others it is not. It may well be that the plateau in crib deaths is telling us that we have reached the limits of our abilities to effect any further decline with our recommendations about sleep environments. But more research needs to be done.
On a more positive note, the new recommendation may force parents to reevaluate their habit of having a television in their bedroom. Will it be baby or TV in the bedroom? Unfortunately, I fear too many will opt to have both.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.” Email him at pdnews@frontlinemedcom.com.
The American Academy of Pediatrics has recently released a new policy for parents on safe sleep practices that in addition to the previous warnings about bed sharing and positioning includes the recommendation that an infant sleep in the same room as her parent for at least the first 6 months (Pediatrics. 2016 Oct;138[5]:e20162938). Apparently what prompted this new set of recommendations is the observation that deaths from sudden unexpected infant deaths (SUIDS) and sudden infant deaths (SIDS) has plateaued since the dramatic decline we witnessed in the 1990s following the Back-to-Sleep campaign.
Although the policy statement refers to “new research” that has become available since the last policy statement was released in 2011, I have had trouble finding convincing evidence in the references I reviewed to support the room sharing recommendation. In some studies, room sharing was the cultural norm, making it difficult to establish a control group. In one of the most frequently cited papers from New Zealand, the authors could not sort out the effects of prone sleeping and sleeping alone, and wonder whether both factors may be affecting risk “through a common mechanism” (Lancet. 1996 Jan 6;347[8993]:7-12).
For some, parents attempting to follow this recommendation may not be without its negative consequences. Sleeping like a baby is not the same as sleeping quietly. Infants often breathe in a pattern that includes long, anxiety-provoking pauses. The implication of this policy recommendation is that parents can prevent crib death by being more vigilant at night. Do we have enough evidence that this is indeed the case?
Most parents are already anxious, and none of them are getting enough sleep. I can envision that trying to follow this recommendation could aggravate both conditions for some parents. Sleep-deprived parents often are not as capable parents as they could be. And they certainly aren’t as happy as they could be. Postpartum depression compounded by sleep deprivation continues to be an underreported and inadequately managed condition that can have negative effects for the health of the child.
For some parents, room sharing is something they gravitate toward naturally, and it can help them deal with the anxiety of new parenthood. They may sleep better with their infant close by. But for others, the better solution to their own sleep deprivation lies in sleep training, a strategy that is very difficult, if not impossible, for parents who are sharing their bedroom with their infant.
As the authors of one of the most frequently quoted papers that supports room sharing have written, “the traditional habit of labeling one sleep arrangement as being superior to another without awareness of the family context is not only wrong but potentially harmful” (Paediatric Resp Review. 2005, Jun;6[2]:134-52).
I think the academy has gone too far or at least moved prematurely with its room sharing recommendation. For some families, room sharing is a better arrangement, for others it is not. It may well be that the plateau in crib deaths is telling us that we have reached the limits of our abilities to effect any further decline with our recommendations about sleep environments. But more research needs to be done.
On a more positive note, the new recommendation may force parents to reevaluate their habit of having a television in their bedroom. Will it be baby or TV in the bedroom? Unfortunately, I fear too many will opt to have both.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.” Email him at pdnews@frontlinemedcom.com.
Cancer type, age at time of diagnosis implicated in risk of CVD-related deaths
Survivorship data derived from a U.K. cancer registry make it possible to more closely pinpoint the risk of cardiovascular disease in patients treated for cancer as adolescents and young adults.
Researchers report that 6% of the 2,016 deaths occurring in 200,945 cancer survivors diagnosed between the ages of 15 and 39 years were directly related to cardiovascular disease. A multivariable Poisson regression analysis of data from the Teenage and Young Adult Cancer Survivor Study also showed that survivors who were diagnosed between the ages of 15 and 19 years had 4.2 times the risk (95% confidence interval, 3.4-5.2) of death from cardiovascular disease, compared with their peers in the general population. But for survivors who were aged 35-39 years when diagnosed, that risk decreased to 1.2 times (95% CI, 1.1-1.3) that of their general population peers (P less than .0001). The standardized mortality ratios and absolute excess risks for ischemic heart disease, valvular heart disease, and cardiomyopathy were similar (Circulation. 2016;134:1521-33).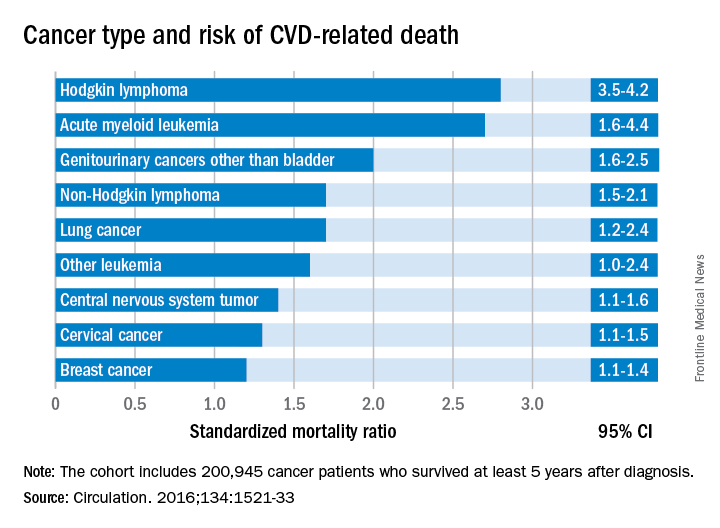
The findings should help clinicians craft more effective after-cancer care, according to Mike Hawkins, DPhil. “It helps them focus the most intensive follow-up care on those most at risk,” Dr. Hawkins, an epidemiology professor and director of the Centre for Childhood Cancer Survivor Studies at the University of Birmingham (England), said in a statement. “It is important for survivors because it empowers them by providing them with their long-term chances of a specific side effect of cancer treatment.”
The most significant relationship between cardiovascular disease and cancer occurred in those diagnosed with Hodgkin lymphoma, and at an earlier age. Overall, Hodgkin lymphoma survivors had a 3.8 times higher risk of cardiovascular disease–related death than their peers not diagnosed with any cancer. In those diagnosed at age 15-19 years, 6.9% had died from cardiovascular disease by age 55 years, compared with 2% of those who’d been diagnosed at age 35-39 years. Among these two age groups in the general population, fewer than 1% typically die from cardiovascular disease–related deaths. In Hodgkin lymphoma survivors aged 60 years or older, 27.5% of excess deaths were from cardiovascular disease.
Although not stratified by treatment, the study includes risk estimates for other cancers diagnosed in the teen and young adult years, stratified by the age at diagnosis, something the authors of the study noted is “a considerable advance on previous knowledge.”
Survivors of all age groups in the cohort diagnosed with a variety of cancers experienced a greater risk of death from heart disease, compared with their peers in the general population.
Survivorship data derived from a U.K. cancer registry make it possible to more closely pinpoint the risk of cardiovascular disease in patients treated for cancer as adolescents and young adults.
Researchers report that 6% of the 2,016 deaths occurring in 200,945 cancer survivors diagnosed between the ages of 15 and 39 years were directly related to cardiovascular disease. A multivariable Poisson regression analysis of data from the Teenage and Young Adult Cancer Survivor Study also showed that survivors who were diagnosed between the ages of 15 and 19 years had 4.2 times the risk (95% confidence interval, 3.4-5.2) of death from cardiovascular disease, compared with their peers in the general population. But for survivors who were aged 35-39 years when diagnosed, that risk decreased to 1.2 times (95% CI, 1.1-1.3) that of their general population peers (P less than .0001). The standardized mortality ratios and absolute excess risks for ischemic heart disease, valvular heart disease, and cardiomyopathy were similar (Circulation. 2016;134:1521-33).
The findings should help clinicians craft more effective after-cancer care, according to Mike Hawkins, DPhil. “It helps them focus the most intensive follow-up care on those most at risk,” Dr. Hawkins, an epidemiology professor and director of the Centre for Childhood Cancer Survivor Studies at the University of Birmingham (England), said in a statement. “It is important for survivors because it empowers them by providing them with their long-term chances of a specific side effect of cancer treatment.”
The most significant relationship between cardiovascular disease and cancer occurred in those diagnosed with Hodgkin lymphoma, and at an earlier age. Overall, Hodgkin lymphoma survivors had a 3.8 times higher risk of cardiovascular disease–related death than their peers not diagnosed with any cancer. In those diagnosed at age 15-19 years, 6.9% had died from cardiovascular disease by age 55 years, compared with 2% of those who’d been diagnosed at age 35-39 years. Among these two age groups in the general population, fewer than 1% typically die from cardiovascular disease–related deaths. In Hodgkin lymphoma survivors aged 60 years or older, 27.5% of excess deaths were from cardiovascular disease.
Although not stratified by treatment, the study includes risk estimates for other cancers diagnosed in the teen and young adult years, stratified by the age at diagnosis, something the authors of the study noted is “a considerable advance on previous knowledge.”
Survivors of all age groups in the cohort diagnosed with a variety of cancers experienced a greater risk of death from heart disease, compared with their peers in the general population.
Survivorship data derived from a U.K. cancer registry make it possible to more closely pinpoint the risk of cardiovascular disease in patients treated for cancer as adolescents and young adults.
Researchers report that 6% of the 2,016 deaths occurring in 200,945 cancer survivors diagnosed between the ages of 15 and 39 years were directly related to cardiovascular disease. A multivariable Poisson regression analysis of data from the Teenage and Young Adult Cancer Survivor Study also showed that survivors who were diagnosed between the ages of 15 and 19 years had 4.2 times the risk (95% confidence interval, 3.4-5.2) of death from cardiovascular disease, compared with their peers in the general population. But for survivors who were aged 35-39 years when diagnosed, that risk decreased to 1.2 times (95% CI, 1.1-1.3) that of their general population peers (P less than .0001). The standardized mortality ratios and absolute excess risks for ischemic heart disease, valvular heart disease, and cardiomyopathy were similar (Circulation. 2016;134:1521-33).
The findings should help clinicians craft more effective after-cancer care, according to Mike Hawkins, DPhil. “It helps them focus the most intensive follow-up care on those most at risk,” Dr. Hawkins, an epidemiology professor and director of the Centre for Childhood Cancer Survivor Studies at the University of Birmingham (England), said in a statement. “It is important for survivors because it empowers them by providing them with their long-term chances of a specific side effect of cancer treatment.”
The most significant relationship between cardiovascular disease and cancer occurred in those diagnosed with Hodgkin lymphoma, and at an earlier age. Overall, Hodgkin lymphoma survivors had a 3.8 times higher risk of cardiovascular disease–related death than their peers not diagnosed with any cancer. In those diagnosed at age 15-19 years, 6.9% had died from cardiovascular disease by age 55 years, compared with 2% of those who’d been diagnosed at age 35-39 years. Among these two age groups in the general population, fewer than 1% typically die from cardiovascular disease–related deaths. In Hodgkin lymphoma survivors aged 60 years or older, 27.5% of excess deaths were from cardiovascular disease.
Although not stratified by treatment, the study includes risk estimates for other cancers diagnosed in the teen and young adult years, stratified by the age at diagnosis, something the authors of the study noted is “a considerable advance on previous knowledge.”
Survivors of all age groups in the cohort diagnosed with a variety of cancers experienced a greater risk of death from heart disease, compared with their peers in the general population.
FROM CIRCULATION
Key clinical point:
Major finding: Cancer survivors who were diagnosed at age 15-19 years had 4.2 times the risk of death from cardiovascular disease than did their peers in the general population.
Data source: A U.K. cancer registry of 200,945 persons between 15 and 39 years at time of diagnosis.
Disclosures: This study was supported by the National Institute for Health Research in the United Kingdom. The authors had no relevant disclosures.
Vector may make gene therapy safer

Photo courtesy of Washington
State University Spokane
Using modified foamy retroviral vectors to deliver gene therapy may reduce the risk of genotoxicity, according to research published in Scientific Reports.
These vectors have demonstrated promise in vitro, and researchers believe they could be used to treat X-linked severe combined immunodeficiency, thalassemia, and other diseases.
“We’ve started to translate this in collaboration with other scientists and medical doctors into the clinic,” said study author Grant Trobridge, PhD, of Washington State University Spokane.
Dr Trobridge and his colleagues said they decided to pursue foamy retroviral vectors as a delivery system for gene therapy because these vectors are less likely than gammaretroviral vectors or lentiviral vectors to activate nearby genes, including proto-oncogenes.
Still, the researchers altered the foamy retroviral vectors to change how they interact with target stem cells in an attempt to ensure the vectors would insert themselves into safer parts of the genome.
The team said they were able to retarget the foamy retroviral vectors away from genes and into satellite regions enriched for trimethylated histone H3 at lysine 9 by modifying the foamy virus Gag and Pol proteins.
These retargeted foamy retroviral vectors integrated near genes significantly less often than unmodified foamy retroviral vectors (P<0.001).
The researchers also noted that their retargeted foamy retroviral vectors can be produced at clinically relevant titers, and engineered target cells are not needed. Any target cell can be used by using alternate foamy helper plasmids during vector production. ![]()
Photo courtesy of Washington
State University Spokane
Using modified foamy retroviral vectors to deliver gene therapy may reduce the risk of genotoxicity, according to research published in Scientific Reports.
These vectors have demonstrated promise in vitro, and researchers believe they could be used to treat X-linked severe combined immunodeficiency, thalassemia, and other diseases.
“We’ve started to translate this in collaboration with other scientists and medical doctors into the clinic,” said study author Grant Trobridge, PhD, of Washington State University Spokane.
Dr Trobridge and his colleagues said they decided to pursue foamy retroviral vectors as a delivery system for gene therapy because these vectors are less likely than gammaretroviral vectors or lentiviral vectors to activate nearby genes, including proto-oncogenes.
Still, the researchers altered the foamy retroviral vectors to change how they interact with target stem cells in an attempt to ensure the vectors would insert themselves into safer parts of the genome.
The team said they were able to retarget the foamy retroviral vectors away from genes and into satellite regions enriched for trimethylated histone H3 at lysine 9 by modifying the foamy virus Gag and Pol proteins.
These retargeted foamy retroviral vectors integrated near genes significantly less often than unmodified foamy retroviral vectors (P<0.001).
The researchers also noted that their retargeted foamy retroviral vectors can be produced at clinically relevant titers, and engineered target cells are not needed. Any target cell can be used by using alternate foamy helper plasmids during vector production. ![]()
Photo courtesy of Washington
State University Spokane
Using modified foamy retroviral vectors to deliver gene therapy may reduce the risk of genotoxicity, according to research published in Scientific Reports.
These vectors have demonstrated promise in vitro, and researchers believe they could be used to treat X-linked severe combined immunodeficiency, thalassemia, and other diseases.
“We’ve started to translate this in collaboration with other scientists and medical doctors into the clinic,” said study author Grant Trobridge, PhD, of Washington State University Spokane.
Dr Trobridge and his colleagues said they decided to pursue foamy retroviral vectors as a delivery system for gene therapy because these vectors are less likely than gammaretroviral vectors or lentiviral vectors to activate nearby genes, including proto-oncogenes.
Still, the researchers altered the foamy retroviral vectors to change how they interact with target stem cells in an attempt to ensure the vectors would insert themselves into safer parts of the genome.
The team said they were able to retarget the foamy retroviral vectors away from genes and into satellite regions enriched for trimethylated histone H3 at lysine 9 by modifying the foamy virus Gag and Pol proteins.
These retargeted foamy retroviral vectors integrated near genes significantly less often than unmodified foamy retroviral vectors (P<0.001).
The researchers also noted that their retargeted foamy retroviral vectors can be produced at clinically relevant titers, and engineered target cells are not needed. Any target cell can be used by using alternate foamy helper plasmids during vector production. ![]()
FDA: Etanercept first biologic approved for pediatric psoriasis
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.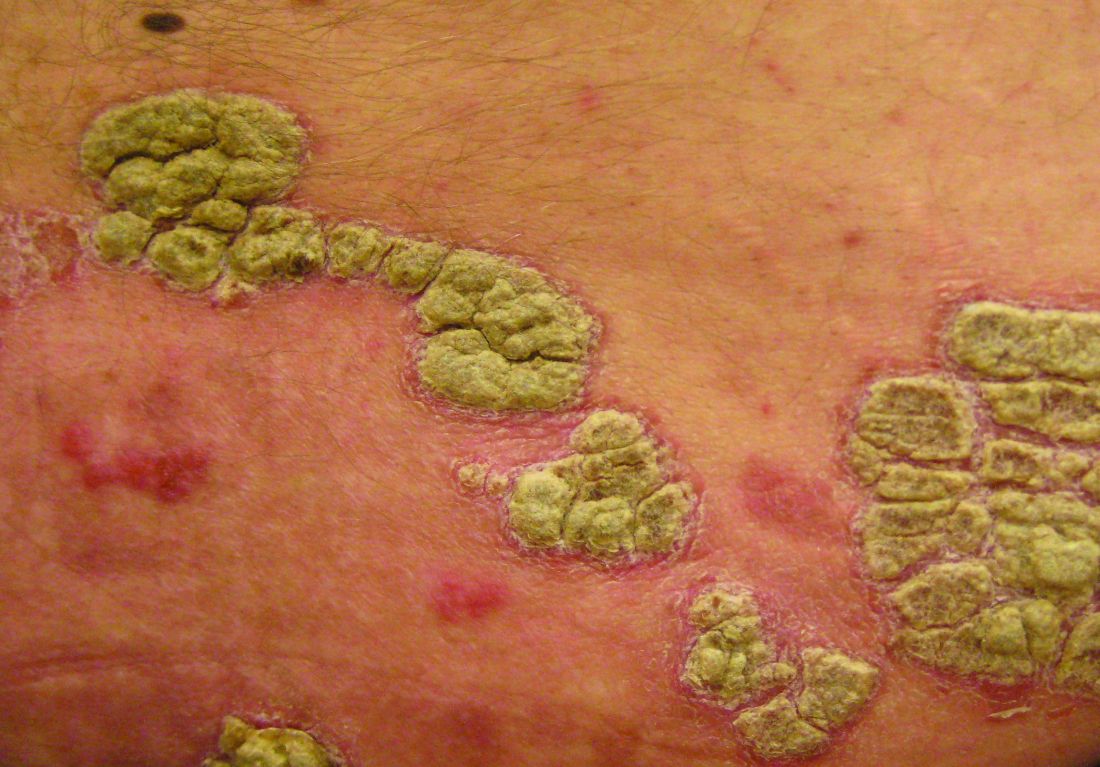
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.
FDA’s Woodcock: Give biosimilars a chance
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.

She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.

“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.
She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.
“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
BOSTON – Biosimilar drugs are not identical twins of original biologic agents, but there are very strong family ties, and the newcomer is expected to look and behave very much like its older relative, said Janet Woodcock, MD, director of the Center for Drug Evaluation and Research at the Food and Drug Administration.
Although biosimilars differ from generic, small-molecule drugs, concerns about the development of biosimilars mirrors the kerfuffle over generic drugs surrounding the passage of the Hatch-Waxman (Drug Price Competition and Patent Term Restoration) Act in 1984.
She noted that clinicians today are asking the same questions about biosimilars that were asked about generics three decades ago:
- Are biosimilars as effective and as safe as the originally licensed biopharmaceuticals?
- If pharmacists substitute biosimilars for prescribed biologics, will patients be adversely affected?
- Can biosimilars reduce the high cost of biologic therapy?
“In certain specialties, this skepticism has persisted to this very day,” she said.
ACA mandate
Biosimilars owe their existence in large measure to the Biologics Price Competition and Innovation Act of 2009 (BPCI), passed as a part of the Affordable Care Act and signed into law by President Obama in 2010.
The act created an abbreviated licensure pathway for biologic products that can be shown to be either biosimilar to or interchangeable with an FDA-licensed reference drug.
A biosimilar is defined as a biological product that is highly similar to the reference product “notwithstanding minor differences in clinically inactive components,” and with no clinically meaningful differences between it and the reference product in terms of purity, safety, and potency.
To be interchangeable, a biosimilar must be expected to produce the same clinical results as the reference drug in any given patient, and “for a product that is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the product and its reference product is not greater than the risk of using the reference product without such alternation or switch.”
The definition of interchangeability includes the understanding that the prescriber’s approval is not necessary for substitution of a biosimilar for its reference product.
“If we’re going to have that kind of switching, if they are going to be interchangeable, then we have to have a very high bar,” Dr. Woodcock said.
She added that “any pressure people are feeling to push their patients to biosimilars is from the reimbursement system. There is no non-prescriber switching allowed currently; however, that doesn’t say there isn’t pressure on prescribers to write a different prescription.”
Faster track and bridge to approval
The biosimilar development and approval requires only convincing demonstration of biosimilarity to an existing agent, rather than an independent finding of safety or effectiveness, and the purpose of clinical studies in this case is to address “residual uncertainties,” Dr. Woodcock said.
Drug developers and regulatory authorities alike “are having trouble getting their mind around this concept,” she said.
The FDA requires manufacturers to provide data in their biosimilar drug license applications demonstrating biosimilarity based on analytical studies, animal studies that include toxicity assessments, and one or more clinical studies that include information on immunogenicity and pharmacokinetics (PK) or pharmacodynamics (PD) that is sufficient to demonstrate the safety, purity, and potency of the candidate biosimilar.
The FDA is also allowing manufacturers to submit data from animal studies and specified clinical studies comparing a proposed biosimilar product with a product not licensed in the United States, “as long as we are convinced that the reference product is equivalent to the U.S. product,” Dr. Woodcock said.
This “analytical bridge” process was requested by manufacturers who began development of biosimilars in Europe. The European Medicines Agency approved a biosimilar process in 2005, and gave the nod to the first biosimilar agents to existing erythropoietin products in 2007.
Current and pending
As of early October 2016, 66 programs were enrolled in FDA’s Biosimilar Product Development Program, and CDER has received requests for meetings with manufacturers to discuss what tests and documents are required for the development of biosimilars to 20 different reference products.
The FDA is prohibited from publicly discussing the existence of a pending application unless it has been previously disclosed or acknowledged publicly with the manufacturer’s permission, Dr. Woodcock noted, but as of Oct. 10, 2016, seven companies have announced a total of 10 biologic license applications for biosimilars to etanercept (Enbrel), adalimumab (Humira), pegfilgrastim (Neulasta), epoetin alfa (Epogen/Procrit), filgrastim (Neupogen), and infliximab (Remicade).
The FDA has granted licenses to four biosimilars to date (the four-letter suffix is intended to differentiate biosimilars agents from other biosimilars to the same reference product):
- Zarxio (filgrastim-sndz).
- Inflectra (infliximab-dyyb).
- Erelzi (etanercept-szzs).
- Amjevita (adalimumab-atto).
Physician perspective
“Everything I have heard suggests that biosimilars will be useful, but the scientist in me is a skeptic,” commented Donald Massenburg, MD, PhD, a rheumatologist at Wheaton Franciscan Healthcare in Franklin, Wisc., in an interview.
“I feel better now after learning that current biosimilars are not considered interchangeable, meaning that there can’t be substitutions made without the treating physician’s consent,” he said.
He acknowledged that “I wouldn’t be that excited” if a specific biosimilar was approved for interchangeability and was given to his patients without his knowledge.
Dr. Massenburg pointed to data from the NOR-SWITCH trial comparing the biosimilar Remsima to the reference product Remicade for treatment of rheumatic diseases, psoriasis, and inflammatory bowel disease. The trial showed that Remsima was noninferior to the reference product.
“I would like to be able to say whether a patient should be switched to a biosimilar or not just because of that potential risk,” Dr. Massenburg said.
A rheumatologist in private practice in New England said that what’s really needed in rheumatology is not the availability of more drugs that act like other drugs, but innovative research into therapies with better targeted mechanism of action.
“We’ve been through the ‘me-too’ hype; we did that with nonsteroidal anti-inflammatory drugs,” said J. Scott Toder, MD, director of the Toder Rheumatology and Osteoporosis Center, Providence, R.I.
“I think we need to concentrate on innovative therapies, and we may be able to do something about the escalating price of the biologics on the market by creating drugs with new mechanism of action to actually increase competition and hopefully control prices. I don’t think that having multiple drugs with the same mechanism of action is in the best interest of our patients,” he said in an interview.
Dr. Woodcock, Dr. Massenburg, and Dr. Toder reported having no relevant disclosures.
EXPERT ANALYSIS FROM A BIOSIMILARS IN RHEUMATOLOGY SYMPOSIUM
ACR 2016 continues big buffet of basic and clinical science sessions
This year’s annual meeting of the American College of Rheumatology will feature cutting-edge research and results of studies that directly affect how attendees will manage patients once they are back in the clinical setting, according to both Richard Loeser, MD, program chair of the Annual Meeting Planning Committee (AMPC), and Gregory Gardner, MD, clinical subchair of the AMPC, who suggested special sessions of interest culled from the more than 450 sessions to be presented.
“It is an exciting time in rheumatology. Basic research is being translated into new therapies before our very eyes. Areas on the program this year that have translational potential include immunometabolism, blocking interleukin-1 (IL-1), T-cell receptor signaling, and meta-analysis of gene expression data. The meeting will also feature trials that refine and advance the management of rheumatologic diseases, including results on studies of new biologics,” Dr. Loeser said.

Hot sessions
Luke O’Neill, MD, will talk about immunometabolism Monday at 7:30 a.m. This session will explore a newly described connection between energy metabolism and the immune system and the link with inflammation.
Charles Dinarello, MD, will give the Philip Hensch Memorial Lecture Sunday at 8:30 a.m. on blocking IL-1 in inflammatory diseases. He will cover a host of diseases from gout to cancer, Dr. Loeser noted.
Another hot topic, T-cell receptor signaling in autoimmune diseases and the development of new therapies, will be discussed by Arthur Weiss, MD, Tuesday morning at 7:30 a.m.
Tuesday at 11:00 a.m., Peter Lipsky, MD, will tackle big data mining, presenting a meta-analysis of gene expression datasets to identify novel pathways and targets in systemic lupus erythematosus (SLE).
“SLE lags behind rheumatoid arthritis in therapeutic advances. A number of trials of biologics have failed in SLE, whereas they have been found effective in rheumatoid arthritis,” Dr. Loeser explained.
Clinical slant
Sunday’s Plenary session at 11:00 a.m. will feature several top-rated abstracts, among them results of a phase III study on tocilizumab in giant cell arteritis to be presented by John Stone, MD. “Tocilizumab is a major breakthrough as a steroid-sparing treatment for the most common form of vasculitis that affects older adults,” Dr. Loeser said.
At 2:30 p.m. on Sunday at The Great Debate, Paul Emery, MD, and Arthur Kavanaugh, MD, will tackle the very important clinical topic of “To Taper or Not to Taper? – Biologic DMARDs in Low Rheumatoid Arthritis Disease Activity.”
“People aren’t sure what to do. The fear with tapering is rebound, with the disease coming back even more forcefully. There is new evidence to suggest that tapering may be safe under certain circumstances. This session should inform attendees on how to make the decision to taper and on the best way to do it,” Dr. Loeser commented.
The Late-Breaking Abstract session on Tuesday at 4:30 p.m. will feature six clinical trials. Dr. Loeser singled out a study to be presented by Elaine Husni, MD, on “Vascular Safety of Celecoxib versus Ibuprofen or Naproxen” in more than 20,000 patients with osteoarthritis or rheumatoid arthritis.
“The fear is that COX-2 inhibitors have increased cardiovascular risk. The data from this study that will be presented at the meeting should answer the question of whether or not this is true in patients with arthritis,” Dr. Loeser explained.
Wednesday at 7:30 a.m. Candida Fratazzi, MD, will talk about “Emerging Biosimilars in Therapeutic Management,” a subject of great interest since they have the potential to be equally effective and less expensive than current biologics.
Two “bookends” of the meeting will frame the opening and closing. Sunday at 7:30 a.m., the “Year in Review” session will feature the best published studies on rheumatologic diseases from the past year, based on the judgment of two experts. Ingrid Lundberg, MD, will present the best clinical studies and Bruce Cronstein, MD, will present the best basic science studies. Wednesday at 7:30 a.m., John Cush, MD, and Dr. Kavanaugh will present the “Rheumatology Roundup” of the best abstracts and put them into context. “This session is usually quite entertaining,” Dr. Loeser said.

More sessions of clinical import
“In keeping with our meeting theme of fine-tuning our care of patients with rheumatic disease, I want to point out several sessions,” Dr. Gardner said.
Attendees interested in sessions on clinical applicability will have to choose between two different sessions Monday at 4:30 p.m.: one on dermatomyositis, a relatively rare but difficult-to-treat entity, and the other about treatment of the patient with rheumatoid arthritis when the patient is not well and suffering from comorbidities.
Monday at 8:30 a.m., an “Osteoporosis Update” will give listeners perspective on current and future therapies.
Sunday at 2:30 p.m., new guidelines for steroid-induced osteoporosis will be presented.
“Four or five sessions on the Tech Track will show rheumatologists how they can improve their practice by using technology,” Dr. Gardner said. “Several high-quality sessions are important to educators, including ‘Flipped Classroom, Technology, and Reflection’ [Monday at 12:30 p.m.] and ‘Year in Review’ [Sunday at 1:00 p.m.].”
Monday at 11:00 a.m., the Plenary session will feature Workforce Study results on how many rheumatologists will be needed in the year 2030, and in which geographic locations. This session will also include a discussion of the impact of part-time rheumatologists.
“Two sessions I am excited about are ‘Treat to Target in 2016,’ Tuesday at 4:30 p.m., and ‘Rheumatic Diseases in Native Americans,’ Sunday at 11:00 a.m.,” Dr. Gardner noted. “Concurrent abstract sessions throughout the meeting will feature discussions on new biologics, small molecules, and gene therapy.”
This year’s annual meeting of the American College of Rheumatology will feature cutting-edge research and results of studies that directly affect how attendees will manage patients once they are back in the clinical setting, according to both Richard Loeser, MD, program chair of the Annual Meeting Planning Committee (AMPC), and Gregory Gardner, MD, clinical subchair of the AMPC, who suggested special sessions of interest culled from the more than 450 sessions to be presented.
“It is an exciting time in rheumatology. Basic research is being translated into new therapies before our very eyes. Areas on the program this year that have translational potential include immunometabolism, blocking interleukin-1 (IL-1), T-cell receptor signaling, and meta-analysis of gene expression data. The meeting will also feature trials that refine and advance the management of rheumatologic diseases, including results on studies of new biologics,” Dr. Loeser said.
Hot sessions
Luke O’Neill, MD, will talk about immunometabolism Monday at 7:30 a.m. This session will explore a newly described connection between energy metabolism and the immune system and the link with inflammation.
Charles Dinarello, MD, will give the Philip Hensch Memorial Lecture Sunday at 8:30 a.m. on blocking IL-1 in inflammatory diseases. He will cover a host of diseases from gout to cancer, Dr. Loeser noted.
Another hot topic, T-cell receptor signaling in autoimmune diseases and the development of new therapies, will be discussed by Arthur Weiss, MD, Tuesday morning at 7:30 a.m.
Tuesday at 11:00 a.m., Peter Lipsky, MD, will tackle big data mining, presenting a meta-analysis of gene expression datasets to identify novel pathways and targets in systemic lupus erythematosus (SLE).
“SLE lags behind rheumatoid arthritis in therapeutic advances. A number of trials of biologics have failed in SLE, whereas they have been found effective in rheumatoid arthritis,” Dr. Loeser explained.
Clinical slant
Sunday’s Plenary session at 11:00 a.m. will feature several top-rated abstracts, among them results of a phase III study on tocilizumab in giant cell arteritis to be presented by John Stone, MD. “Tocilizumab is a major breakthrough as a steroid-sparing treatment for the most common form of vasculitis that affects older adults,” Dr. Loeser said.
At 2:30 p.m. on Sunday at The Great Debate, Paul Emery, MD, and Arthur Kavanaugh, MD, will tackle the very important clinical topic of “To Taper or Not to Taper? – Biologic DMARDs in Low Rheumatoid Arthritis Disease Activity.”
“People aren’t sure what to do. The fear with tapering is rebound, with the disease coming back even more forcefully. There is new evidence to suggest that tapering may be safe under certain circumstances. This session should inform attendees on how to make the decision to taper and on the best way to do it,” Dr. Loeser commented.
The Late-Breaking Abstract session on Tuesday at 4:30 p.m. will feature six clinical trials. Dr. Loeser singled out a study to be presented by Elaine Husni, MD, on “Vascular Safety of Celecoxib versus Ibuprofen or Naproxen” in more than 20,000 patients with osteoarthritis or rheumatoid arthritis.
“The fear is that COX-2 inhibitors have increased cardiovascular risk. The data from this study that will be presented at the meeting should answer the question of whether or not this is true in patients with arthritis,” Dr. Loeser explained.
Wednesday at 7:30 a.m. Candida Fratazzi, MD, will talk about “Emerging Biosimilars in Therapeutic Management,” a subject of great interest since they have the potential to be equally effective and less expensive than current biologics.
Two “bookends” of the meeting will frame the opening and closing. Sunday at 7:30 a.m., the “Year in Review” session will feature the best published studies on rheumatologic diseases from the past year, based on the judgment of two experts. Ingrid Lundberg, MD, will present the best clinical studies and Bruce Cronstein, MD, will present the best basic science studies. Wednesday at 7:30 a.m., John Cush, MD, and Dr. Kavanaugh will present the “Rheumatology Roundup” of the best abstracts and put them into context. “This session is usually quite entertaining,” Dr. Loeser said.
More sessions of clinical import
“In keeping with our meeting theme of fine-tuning our care of patients with rheumatic disease, I want to point out several sessions,” Dr. Gardner said.
Attendees interested in sessions on clinical applicability will have to choose between two different sessions Monday at 4:30 p.m.: one on dermatomyositis, a relatively rare but difficult-to-treat entity, and the other about treatment of the patient with rheumatoid arthritis when the patient is not well and suffering from comorbidities.
Monday at 8:30 a.m., an “Osteoporosis Update” will give listeners perspective on current and future therapies.
Sunday at 2:30 p.m., new guidelines for steroid-induced osteoporosis will be presented.
“Four or five sessions on the Tech Track will show rheumatologists how they can improve their practice by using technology,” Dr. Gardner said. “Several high-quality sessions are important to educators, including ‘Flipped Classroom, Technology, and Reflection’ [Monday at 12:30 p.m.] and ‘Year in Review’ [Sunday at 1:00 p.m.].”
Monday at 11:00 a.m., the Plenary session will feature Workforce Study results on how many rheumatologists will be needed in the year 2030, and in which geographic locations. This session will also include a discussion of the impact of part-time rheumatologists.
“Two sessions I am excited about are ‘Treat to Target in 2016,’ Tuesday at 4:30 p.m., and ‘Rheumatic Diseases in Native Americans,’ Sunday at 11:00 a.m.,” Dr. Gardner noted. “Concurrent abstract sessions throughout the meeting will feature discussions on new biologics, small molecules, and gene therapy.”
This year’s annual meeting of the American College of Rheumatology will feature cutting-edge research and results of studies that directly affect how attendees will manage patients once they are back in the clinical setting, according to both Richard Loeser, MD, program chair of the Annual Meeting Planning Committee (AMPC), and Gregory Gardner, MD, clinical subchair of the AMPC, who suggested special sessions of interest culled from the more than 450 sessions to be presented.
“It is an exciting time in rheumatology. Basic research is being translated into new therapies before our very eyes. Areas on the program this year that have translational potential include immunometabolism, blocking interleukin-1 (IL-1), T-cell receptor signaling, and meta-analysis of gene expression data. The meeting will also feature trials that refine and advance the management of rheumatologic diseases, including results on studies of new biologics,” Dr. Loeser said.
Hot sessions
Luke O’Neill, MD, will talk about immunometabolism Monday at 7:30 a.m. This session will explore a newly described connection between energy metabolism and the immune system and the link with inflammation.
Charles Dinarello, MD, will give the Philip Hensch Memorial Lecture Sunday at 8:30 a.m. on blocking IL-1 in inflammatory diseases. He will cover a host of diseases from gout to cancer, Dr. Loeser noted.
Another hot topic, T-cell receptor signaling in autoimmune diseases and the development of new therapies, will be discussed by Arthur Weiss, MD, Tuesday morning at 7:30 a.m.
Tuesday at 11:00 a.m., Peter Lipsky, MD, will tackle big data mining, presenting a meta-analysis of gene expression datasets to identify novel pathways and targets in systemic lupus erythematosus (SLE).
“SLE lags behind rheumatoid arthritis in therapeutic advances. A number of trials of biologics have failed in SLE, whereas they have been found effective in rheumatoid arthritis,” Dr. Loeser explained.
Clinical slant
Sunday’s Plenary session at 11:00 a.m. will feature several top-rated abstracts, among them results of a phase III study on tocilizumab in giant cell arteritis to be presented by John Stone, MD. “Tocilizumab is a major breakthrough as a steroid-sparing treatment for the most common form of vasculitis that affects older adults,” Dr. Loeser said.
At 2:30 p.m. on Sunday at The Great Debate, Paul Emery, MD, and Arthur Kavanaugh, MD, will tackle the very important clinical topic of “To Taper or Not to Taper? – Biologic DMARDs in Low Rheumatoid Arthritis Disease Activity.”
“People aren’t sure what to do. The fear with tapering is rebound, with the disease coming back even more forcefully. There is new evidence to suggest that tapering may be safe under certain circumstances. This session should inform attendees on how to make the decision to taper and on the best way to do it,” Dr. Loeser commented.
The Late-Breaking Abstract session on Tuesday at 4:30 p.m. will feature six clinical trials. Dr. Loeser singled out a study to be presented by Elaine Husni, MD, on “Vascular Safety of Celecoxib versus Ibuprofen or Naproxen” in more than 20,000 patients with osteoarthritis or rheumatoid arthritis.
“The fear is that COX-2 inhibitors have increased cardiovascular risk. The data from this study that will be presented at the meeting should answer the question of whether or not this is true in patients with arthritis,” Dr. Loeser explained.
Wednesday at 7:30 a.m. Candida Fratazzi, MD, will talk about “Emerging Biosimilars in Therapeutic Management,” a subject of great interest since they have the potential to be equally effective and less expensive than current biologics.
Two “bookends” of the meeting will frame the opening and closing. Sunday at 7:30 a.m., the “Year in Review” session will feature the best published studies on rheumatologic diseases from the past year, based on the judgment of two experts. Ingrid Lundberg, MD, will present the best clinical studies and Bruce Cronstein, MD, will present the best basic science studies. Wednesday at 7:30 a.m., John Cush, MD, and Dr. Kavanaugh will present the “Rheumatology Roundup” of the best abstracts and put them into context. “This session is usually quite entertaining,” Dr. Loeser said.
More sessions of clinical import
“In keeping with our meeting theme of fine-tuning our care of patients with rheumatic disease, I want to point out several sessions,” Dr. Gardner said.
Attendees interested in sessions on clinical applicability will have to choose between two different sessions Monday at 4:30 p.m.: one on dermatomyositis, a relatively rare but difficult-to-treat entity, and the other about treatment of the patient with rheumatoid arthritis when the patient is not well and suffering from comorbidities.
Monday at 8:30 a.m., an “Osteoporosis Update” will give listeners perspective on current and future therapies.
Sunday at 2:30 p.m., new guidelines for steroid-induced osteoporosis will be presented.
“Four or five sessions on the Tech Track will show rheumatologists how they can improve their practice by using technology,” Dr. Gardner said. “Several high-quality sessions are important to educators, including ‘Flipped Classroom, Technology, and Reflection’ [Monday at 12:30 p.m.] and ‘Year in Review’ [Sunday at 1:00 p.m.].”
Monday at 11:00 a.m., the Plenary session will feature Workforce Study results on how many rheumatologists will be needed in the year 2030, and in which geographic locations. This session will also include a discussion of the impact of part-time rheumatologists.
“Two sessions I am excited about are ‘Treat to Target in 2016,’ Tuesday at 4:30 p.m., and ‘Rheumatic Diseases in Native Americans,’ Sunday at 11:00 a.m.,” Dr. Gardner noted. “Concurrent abstract sessions throughout the meeting will feature discussions on new biologics, small molecules, and gene therapy.”
FROM THE ACR ANNUAL MEETING
Large increase seen in number of Zika-infected pregnancies
The 288 new cases of pregnant women in the United States with laboratory evidence of Zika infection reported for the week ending Oct. 27, 2016, represent the largest weekly increase so far this year, according to new data from the Centers for Disease Control and Prevention.
The 52 new cases in the 50 states and the District of Columbia and the 236 cases in the U.S. territories bring the totals for the year to 1,005 and 2,263, respectively, and to 3,268 for the United States as a whole, the CDC reported.
The week ending Oct. 27 also brought reports of two more infants born with Zika-related birth defects in the 50 states/D.C., but there were no reports of Zika-related pregnancy losses. So far in 2016, there have been 25 liveborn infants with Zika-related birth defects and five pregnancy losses in the states.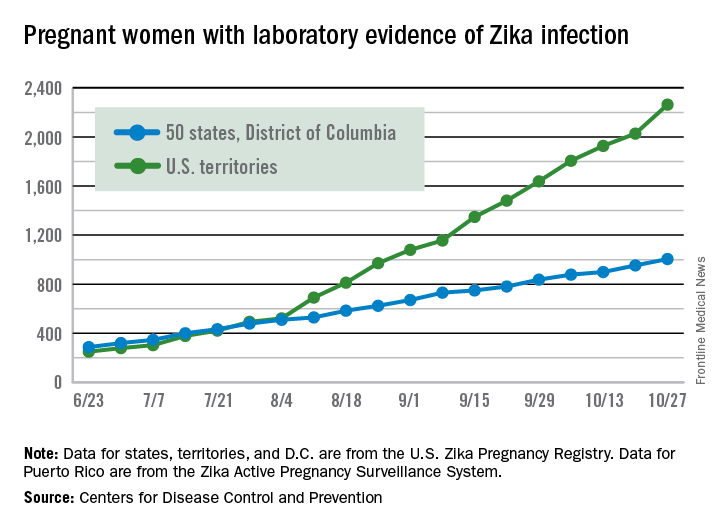
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The 288 new cases of pregnant women in the United States with laboratory evidence of Zika infection reported for the week ending Oct. 27, 2016, represent the largest weekly increase so far this year, according to new data from the Centers for Disease Control and Prevention.
The 52 new cases in the 50 states and the District of Columbia and the 236 cases in the U.S. territories bring the totals for the year to 1,005 and 2,263, respectively, and to 3,268 for the United States as a whole, the CDC reported.
The week ending Oct. 27 also brought reports of two more infants born with Zika-related birth defects in the 50 states/D.C., but there were no reports of Zika-related pregnancy losses. So far in 2016, there have been 25 liveborn infants with Zika-related birth defects and five pregnancy losses in the states.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The 288 new cases of pregnant women in the United States with laboratory evidence of Zika infection reported for the week ending Oct. 27, 2016, represent the largest weekly increase so far this year, according to new data from the Centers for Disease Control and Prevention.
The 52 new cases in the 50 states and the District of Columbia and the 236 cases in the U.S. territories bring the totals for the year to 1,005 and 2,263, respectively, and to 3,268 for the United States as a whole, the CDC reported.
The week ending Oct. 27 also brought reports of two more infants born with Zika-related birth defects in the 50 states/D.C., but there were no reports of Zika-related pregnancy losses. So far in 2016, there have been 25 liveborn infants with Zika-related birth defects and five pregnancy losses in the states.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
U.S. preterm birth rate rose slightly in 2015
The preterm birth rate in the United States for 2015 increased for the first time in 8 years, according to the March of Dimes.
The national preterm birth rate rose from 9.57% in 2014 to 9.63% last year, earning an overall grade of C on the March of Dimes 2016 Premature Birth Report Card.
The March of Dimes also ranked preterm births in the states by racial/ethnic disparity: Maine had the least disparity, followed by New Hampshire and Utah, while Hawaii had the greatest disparity, just ahead of Pennsylvania and Louisiana. Using an average of the 2012-2014 national preterm birth rates, the March of Dimes calculated that Asian/Pacific Islanders had the lowest preterm birth rate at 8.5%, compared with 9% for whites, 9.1% for Hispanics, 10.4% for American Indian/Alaska Natives, and 13.3% for blacks.
The report card shows “that there is an unfair burden of premature birth among specific racial and ethnic groups as well as geographic areas,” Jennifer L. Howse, PhD, president of the March of Dimes, said in a statement. “Babies in this country have different chances of surviving and thriving simply based on the circumstances of their birth.”
The preterm birth rate in the United States for 2015 increased for the first time in 8 years, according to the March of Dimes.
The national preterm birth rate rose from 9.57% in 2014 to 9.63% last year, earning an overall grade of C on the March of Dimes 2016 Premature Birth Report Card.
The March of Dimes also ranked preterm births in the states by racial/ethnic disparity: Maine had the least disparity, followed by New Hampshire and Utah, while Hawaii had the greatest disparity, just ahead of Pennsylvania and Louisiana. Using an average of the 2012-2014 national preterm birth rates, the March of Dimes calculated that Asian/Pacific Islanders had the lowest preterm birth rate at 8.5%, compared with 9% for whites, 9.1% for Hispanics, 10.4% for American Indian/Alaska Natives, and 13.3% for blacks.
The report card shows “that there is an unfair burden of premature birth among specific racial and ethnic groups as well as geographic areas,” Jennifer L. Howse, PhD, president of the March of Dimes, said in a statement. “Babies in this country have different chances of surviving and thriving simply based on the circumstances of their birth.”
The preterm birth rate in the United States for 2015 increased for the first time in 8 years, according to the March of Dimes.
The national preterm birth rate rose from 9.57% in 2014 to 9.63% last year, earning an overall grade of C on the March of Dimes 2016 Premature Birth Report Card.
The March of Dimes also ranked preterm births in the states by racial/ethnic disparity: Maine had the least disparity, followed by New Hampshire and Utah, while Hawaii had the greatest disparity, just ahead of Pennsylvania and Louisiana. Using an average of the 2012-2014 national preterm birth rates, the March of Dimes calculated that Asian/Pacific Islanders had the lowest preterm birth rate at 8.5%, compared with 9% for whites, 9.1% for Hispanics, 10.4% for American Indian/Alaska Natives, and 13.3% for blacks.
The report card shows “that there is an unfair burden of premature birth among specific racial and ethnic groups as well as geographic areas,” Jennifer L. Howse, PhD, president of the March of Dimes, said in a statement. “Babies in this country have different chances of surviving and thriving simply based on the circumstances of their birth.”