User login
Study eyes fracture risk in pediatric patients taking antiepileptic drugs
HOUSTON – The incidence of fractures in pediatric patients taking antiepileptic medication stands at an estimated 1.1%, according to results from a 4-year period at a children’s hospital.
“Understanding the prevalence of fractures in pediatric patients on an AED [antiepileptic drug] will allow clinicians to weigh the risk versus benefit of therapy,” researchers led by Shannon DiCarlo, MD, wrote in an abstract presented during a poster session at the annual meeting of the American Epilepsy Society. “Recognizing a risk of fractures with AEDs will permit clinicians to provide appropriate supportive care and monitoring from the initiation of therapy.”
Dr. DiCarlo of Texas Children’s Hospital, Houston, and her associates went on to note that adults with epilepsy have a twofold to sixfold greater risk of experiencing fractures, compared with the general population, and that fractures secondary to seizures “are a major concern in pediatrics.” In fact, one survey of 404 pediatric neurologists found that only 41% of respondents were aware of the association between AEDs and reduced bone mass (Arch Neurol. 2001;58[9]:1369-74).
In an effort to evaluate the prevalence of fractures in pediatric patients on an antiepileptic drug, the researchers conducted a cohort study of 10,153 patients younger than 18 years of age who received an AED at Texas Children’s Hospital from 2011 to 2014. Half of the study population were female, and the most common concomitant disease was epilepsy (52.6%), followed by cerebral palsy (8.3%), epilepsy plus cerebral palsy (6.9%), osteoporosis (0.2%), and osteopenia (0.3%). In all, 113 patients (1.1%) experienced a fracture while on an antiepileptic drug, and the mean time from initiation of an AED to time of fracture was 1.6 years. Patients on enzyme-inducing AEDs were two times more likely to experience a fracture, while those with cerebral palsy and epilepsy were three times more likely to experience a fracture. Proton pump inhibitors and corticosteroids were the most common concomitant drugs. The researchers also found that less than 10% of patients were on calcium or vitamin D supplementation.
“Vigilant monitoring should be employed for at-risk patients,” the researchers concluded. “Regular monitoring of calcium and vitamin D levels may be warranted; further studies are needed to evaluate the roll of prophylactic calcium and vitamin D supplements.”
The researchers reported having no relevant financial disclosures.
HOUSTON – The incidence of fractures in pediatric patients taking antiepileptic medication stands at an estimated 1.1%, according to results from a 4-year period at a children’s hospital.
“Understanding the prevalence of fractures in pediatric patients on an AED [antiepileptic drug] will allow clinicians to weigh the risk versus benefit of therapy,” researchers led by Shannon DiCarlo, MD, wrote in an abstract presented during a poster session at the annual meeting of the American Epilepsy Society. “Recognizing a risk of fractures with AEDs will permit clinicians to provide appropriate supportive care and monitoring from the initiation of therapy.”
Dr. DiCarlo of Texas Children’s Hospital, Houston, and her associates went on to note that adults with epilepsy have a twofold to sixfold greater risk of experiencing fractures, compared with the general population, and that fractures secondary to seizures “are a major concern in pediatrics.” In fact, one survey of 404 pediatric neurologists found that only 41% of respondents were aware of the association between AEDs and reduced bone mass (Arch Neurol. 2001;58[9]:1369-74).
In an effort to evaluate the prevalence of fractures in pediatric patients on an antiepileptic drug, the researchers conducted a cohort study of 10,153 patients younger than 18 years of age who received an AED at Texas Children’s Hospital from 2011 to 2014. Half of the study population were female, and the most common concomitant disease was epilepsy (52.6%), followed by cerebral palsy (8.3%), epilepsy plus cerebral palsy (6.9%), osteoporosis (0.2%), and osteopenia (0.3%). In all, 113 patients (1.1%) experienced a fracture while on an antiepileptic drug, and the mean time from initiation of an AED to time of fracture was 1.6 years. Patients on enzyme-inducing AEDs were two times more likely to experience a fracture, while those with cerebral palsy and epilepsy were three times more likely to experience a fracture. Proton pump inhibitors and corticosteroids were the most common concomitant drugs. The researchers also found that less than 10% of patients were on calcium or vitamin D supplementation.
“Vigilant monitoring should be employed for at-risk patients,” the researchers concluded. “Regular monitoring of calcium and vitamin D levels may be warranted; further studies are needed to evaluate the roll of prophylactic calcium and vitamin D supplements.”
The researchers reported having no relevant financial disclosures.
HOUSTON – The incidence of fractures in pediatric patients taking antiepileptic medication stands at an estimated 1.1%, according to results from a 4-year period at a children’s hospital.
“Understanding the prevalence of fractures in pediatric patients on an AED [antiepileptic drug] will allow clinicians to weigh the risk versus benefit of therapy,” researchers led by Shannon DiCarlo, MD, wrote in an abstract presented during a poster session at the annual meeting of the American Epilepsy Society. “Recognizing a risk of fractures with AEDs will permit clinicians to provide appropriate supportive care and monitoring from the initiation of therapy.”
Dr. DiCarlo of Texas Children’s Hospital, Houston, and her associates went on to note that adults with epilepsy have a twofold to sixfold greater risk of experiencing fractures, compared with the general population, and that fractures secondary to seizures “are a major concern in pediatrics.” In fact, one survey of 404 pediatric neurologists found that only 41% of respondents were aware of the association between AEDs and reduced bone mass (Arch Neurol. 2001;58[9]:1369-74).
In an effort to evaluate the prevalence of fractures in pediatric patients on an antiepileptic drug, the researchers conducted a cohort study of 10,153 patients younger than 18 years of age who received an AED at Texas Children’s Hospital from 2011 to 2014. Half of the study population were female, and the most common concomitant disease was epilepsy (52.6%), followed by cerebral palsy (8.3%), epilepsy plus cerebral palsy (6.9%), osteoporosis (0.2%), and osteopenia (0.3%). In all, 113 patients (1.1%) experienced a fracture while on an antiepileptic drug, and the mean time from initiation of an AED to time of fracture was 1.6 years. Patients on enzyme-inducing AEDs were two times more likely to experience a fracture, while those with cerebral palsy and epilepsy were three times more likely to experience a fracture. Proton pump inhibitors and corticosteroids were the most common concomitant drugs. The researchers also found that less than 10% of patients were on calcium or vitamin D supplementation.
“Vigilant monitoring should be employed for at-risk patients,” the researchers concluded. “Regular monitoring of calcium and vitamin D levels may be warranted; further studies are needed to evaluate the roll of prophylactic calcium and vitamin D supplements.”
The researchers reported having no relevant financial disclosures.
Key clinical point:
Major finding: In all, 113 patients (1.1%) experienced a fracture while on an antiepileptic drug, and the mean time from initiation of AED to time of fracture was 1.6 years.
Data source: A cohort study of 10,153 patients younger than 18 years of age who received an AED at Texas Children’s Hospital from 2011 to 2014.
Disclosures: The researchers reported having no relevant financial disclosures.
Which maternal beta-blockers boost SGA risk?
NEW ORLEANS – The use of labetalol or atenolol in pregnancy is associated with significantly increased risk of having a small-for-gestational-age (SGA) baby; metoprolol and propranolol are not.
And none of these four beta-blockers are associated with increased risk of congenital cardiac anomalies, Angie Ng, MD, reported at the American Heart Association scientific sessions.

Overall, the average birth weight for babies whose mothers were on a beta-blocker was 2,996 g, significantly less than the 3,353 g in 374,391 controls who weren’t exposed to beta-blockers during pregnancy. But beta-blockers are not a monolithic class of drugs; their pharmacokinetics and physical properties differ. And so did their associated incidence of SGA, according to Dr. Ng of Kaiser Permanente Los Angeles.
The rate of SGA below the 10th percentile was 17.6% in the 3,357 women on labetalol during pregnancy and the same in the 638 women on atenolol. In contrast, the SGA rates in women on metoprolol or propranolol – 10.8% and 10.3%, respectively – weren’t significantly different from the 8.7% incidence in controls.
To deal with the possibility of confounding by indication, Dr. Ng and her coinvestigators performed a multivariate analysis adjusted for maternal age, white race, body mass index, gestational age, diabetes, hypertension, arrhythmias, dyslipidemia, and renal insufficiency. The resultant adjusted risk of having an SGA baby was 2.9-fold greater in women on labetalol and 2.4-fold greater in those on atenolol than in controls. Women on the other two beta-blockers faced no increased risk.
The incidence of congenital cardiac anomalies was 5.1% in women exposed to beta-blockers in pregnancy and 1.9% in controls who weren’t. The most commonly diagnosed anomalies – patent ductus arteriosus, atrial septal defect, and ventricular septal defect – were two- to threefold more frequent in the setting of maternal beta-blocker exposure. However, in a multivariate analysis the use of any beta-blocker was no longer associated with significantly elevated risk of congenital cardiac anomalies.
“This suggests that the initial association we see in the unadjusted analysis is likely due to confounders and not due to the beta-blocker exposure,” Dr. Ng said.
Labetalol and atenolol were prescribed during pregnancy most often for hypertension, while metoprolol and propranolol were typically prescribed to control arrhythmias.
Previous reports by other investigators have yielded conflicting results as to whether maternal beta-blocker therapy is associated with increased risk of SGA. A major limitation of those studies was that they examined beta-blockers as a class rather than assessing the impact of specific agents, according to Dr. Ng.
She reported having no financial conflicts of interest regarding her study, which was conducted free of commercial support.
NEW ORLEANS – The use of labetalol or atenolol in pregnancy is associated with significantly increased risk of having a small-for-gestational-age (SGA) baby; metoprolol and propranolol are not.
And none of these four beta-blockers are associated with increased risk of congenital cardiac anomalies, Angie Ng, MD, reported at the American Heart Association scientific sessions.
Overall, the average birth weight for babies whose mothers were on a beta-blocker was 2,996 g, significantly less than the 3,353 g in 374,391 controls who weren’t exposed to beta-blockers during pregnancy. But beta-blockers are not a monolithic class of drugs; their pharmacokinetics and physical properties differ. And so did their associated incidence of SGA, according to Dr. Ng of Kaiser Permanente Los Angeles.
The rate of SGA below the 10th percentile was 17.6% in the 3,357 women on labetalol during pregnancy and the same in the 638 women on atenolol. In contrast, the SGA rates in women on metoprolol or propranolol – 10.8% and 10.3%, respectively – weren’t significantly different from the 8.7% incidence in controls.
To deal with the possibility of confounding by indication, Dr. Ng and her coinvestigators performed a multivariate analysis adjusted for maternal age, white race, body mass index, gestational age, diabetes, hypertension, arrhythmias, dyslipidemia, and renal insufficiency. The resultant adjusted risk of having an SGA baby was 2.9-fold greater in women on labetalol and 2.4-fold greater in those on atenolol than in controls. Women on the other two beta-blockers faced no increased risk.
The incidence of congenital cardiac anomalies was 5.1% in women exposed to beta-blockers in pregnancy and 1.9% in controls who weren’t. The most commonly diagnosed anomalies – patent ductus arteriosus, atrial septal defect, and ventricular septal defect – were two- to threefold more frequent in the setting of maternal beta-blocker exposure. However, in a multivariate analysis the use of any beta-blocker was no longer associated with significantly elevated risk of congenital cardiac anomalies.
“This suggests that the initial association we see in the unadjusted analysis is likely due to confounders and not due to the beta-blocker exposure,” Dr. Ng said.
Labetalol and atenolol were prescribed during pregnancy most often for hypertension, while metoprolol and propranolol were typically prescribed to control arrhythmias.
Previous reports by other investigators have yielded conflicting results as to whether maternal beta-blocker therapy is associated with increased risk of SGA. A major limitation of those studies was that they examined beta-blockers as a class rather than assessing the impact of specific agents, according to Dr. Ng.
She reported having no financial conflicts of interest regarding her study, which was conducted free of commercial support.
NEW ORLEANS – The use of labetalol or atenolol in pregnancy is associated with significantly increased risk of having a small-for-gestational-age (SGA) baby; metoprolol and propranolol are not.
And none of these four beta-blockers are associated with increased risk of congenital cardiac anomalies, Angie Ng, MD, reported at the American Heart Association scientific sessions.
Overall, the average birth weight for babies whose mothers were on a beta-blocker was 2,996 g, significantly less than the 3,353 g in 374,391 controls who weren’t exposed to beta-blockers during pregnancy. But beta-blockers are not a monolithic class of drugs; their pharmacokinetics and physical properties differ. And so did their associated incidence of SGA, according to Dr. Ng of Kaiser Permanente Los Angeles.
The rate of SGA below the 10th percentile was 17.6% in the 3,357 women on labetalol during pregnancy and the same in the 638 women on atenolol. In contrast, the SGA rates in women on metoprolol or propranolol – 10.8% and 10.3%, respectively – weren’t significantly different from the 8.7% incidence in controls.
To deal with the possibility of confounding by indication, Dr. Ng and her coinvestigators performed a multivariate analysis adjusted for maternal age, white race, body mass index, gestational age, diabetes, hypertension, arrhythmias, dyslipidemia, and renal insufficiency. The resultant adjusted risk of having an SGA baby was 2.9-fold greater in women on labetalol and 2.4-fold greater in those on atenolol than in controls. Women on the other two beta-blockers faced no increased risk.
The incidence of congenital cardiac anomalies was 5.1% in women exposed to beta-blockers in pregnancy and 1.9% in controls who weren’t. The most commonly diagnosed anomalies – patent ductus arteriosus, atrial septal defect, and ventricular septal defect – were two- to threefold more frequent in the setting of maternal beta-blocker exposure. However, in a multivariate analysis the use of any beta-blocker was no longer associated with significantly elevated risk of congenital cardiac anomalies.
“This suggests that the initial association we see in the unadjusted analysis is likely due to confounders and not due to the beta-blocker exposure,” Dr. Ng said.
Labetalol and atenolol were prescribed during pregnancy most often for hypertension, while metoprolol and propranolol were typically prescribed to control arrhythmias.
Previous reports by other investigators have yielded conflicting results as to whether maternal beta-blocker therapy is associated with increased risk of SGA. A major limitation of those studies was that they examined beta-blockers as a class rather than assessing the impact of specific agents, according to Dr. Ng.
She reported having no financial conflicts of interest regarding her study, which was conducted free of commercial support.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: Women on labetalol or atenolol during pregnancy had a 17.6% incidence of small-for-gestational-age babies, a rate more than twice that in women not exposed to a beta-blocker during pregnancy.
Data source: This was a retrospective study of fetal outcomes in nearly 380,000 pregnant women, 4,847 of whom were on beta-blocker therapy during their pregnancy.
Disclosures: The presenter reported having no financial conflicts of interest regarding her study, which was conducted free of commercial support.
Infantile spasms diagnosis, treatment too often delayed
HOUSTON – The appropriate evaluation and treatment of infantile spasms often comes after significant delays that are largely attributable to lack of recognition of the condition, according to recent survey results.
“Parental suspicions that ‘something was wrong’ were often discounted by health care providers, and survey respondents frequently reported that health care providers (including neurologists) were unfamiliar with infantile spasms [IS],” wrote Johnson Lay and his collaborators, in presenting results of the recently-completed Assessment of Symptoms and Specialists in Infantile Spasms (ASSIST) study at the annual meeting of the American Epilepsy Society. It’s critically important not to discount parental concerns, senior author Shaun Hussain, MD, said in an interview about the study, which surveyed parents of children with IS: “Sometimes we get in the habit of reassuring patients too much.”

On average, a pediatrician will see two cases of infantile spasms in a career of caring for children, Dr. Hussain said. These characteristically brief episodes last just a second or so, and frequently come in clusters, during or after which crying is common. Though the classic presentation of IS is a brief drop of the head with accompanying elevation of the shoulders – and perhaps a lifting of the arms – there’s wide variation in presentation. “Infantile spasms can present in a lot of different ways; babies haven’t read the neurology textbooks,” Dr. Hussain said. A curated collection of videos showing the variety of infantile spasms is available on the Infantile Spasms Project website.
Mr. Lay and his collaborators surveyed the parents of 100 children with IS in order to understand and describe their experiences in having their child receive the correct diagnosis and treatment. Also, they sought to determine whether socioeconomic factors affected prompt access to care. Dr. Lay completed the study with Dr. Hussain and other coauthors after graduating from UCLA in 2015.
The study, presented during a poster session at the meeting, measured the time between the parents’ first identification of IS and their contact with an “effective provider.” A provider was deemed effective if he or she correctly diagnosed IS and prescribed a first-line therapy: adrenocorticotropin-releasing hormone (ACTH), corticosteroids, or vigabatrin (Sabril).
Of the 100 children whose families were surveyed, 29% were seen by an effective provider within a week of the onset of IS. The median time from the onset of IS to the patient’s being seen by an effective provider was 24.5 days, with a wide range seen in the population surveyed (interquartile range, 0-110.5 days). This exceeds the 7 days that has been identified as the goal cumulative latency to treatment, Mr. Lay and his coauthors said.
The median age of onset of IS was 6.6 months in the study population, 45% of whom were female.
A total of 64 patients were first seen for IS concerns by a provider who was not a neurologist. Of those, the cumulative latency to seeing the first effective provider was over 100 days, with some patients’ wait time stretching to over 200 days.
The survey asked about socioeconomic factors, including income, health insurance status, proximity to specialty care, ethnicity, and parental language preference. Of these, only a non-English parental language preference was associated with increased time to seeing an effective provider (hazard ratio, 0.37; 95% confidence interval, 0.20-0.68; P = .002).
“These results suggest that a simple lack of awareness of infantile spasms among health care providers may be responsible for potentially catastrophic delays in IS care,” wrote Mr. Lay and his coauthors. Dr. Hussain concurred: “The clear driver of treatment delay is the failure of health care providers to recognize the appearance, importance, and urgency of infantile spasms. Delay in diagnosis and treatment of infantile spasms is common, costly – to both patients and the health care system – and almost entirely preventable.”
The Elsie and Isaac Fogelman Endowment, the Hughes Family Foundation, and the UCLA Children’s Discovery and Innovation Institute funded the study. Dr. Hussain has received research support from and been on the advisory boards of several pharmaceutical companies. Mr. Lay reported no disclosures.
koakes@frontlinemedcom.com
On Twitter @karioakes
HOUSTON – The appropriate evaluation and treatment of infantile spasms often comes after significant delays that are largely attributable to lack of recognition of the condition, according to recent survey results.
“Parental suspicions that ‘something was wrong’ were often discounted by health care providers, and survey respondents frequently reported that health care providers (including neurologists) were unfamiliar with infantile spasms [IS],” wrote Johnson Lay and his collaborators, in presenting results of the recently-completed Assessment of Symptoms and Specialists in Infantile Spasms (ASSIST) study at the annual meeting of the American Epilepsy Society. It’s critically important not to discount parental concerns, senior author Shaun Hussain, MD, said in an interview about the study, which surveyed parents of children with IS: “Sometimes we get in the habit of reassuring patients too much.”
On average, a pediatrician will see two cases of infantile spasms in a career of caring for children, Dr. Hussain said. These characteristically brief episodes last just a second or so, and frequently come in clusters, during or after which crying is common. Though the classic presentation of IS is a brief drop of the head with accompanying elevation of the shoulders – and perhaps a lifting of the arms – there’s wide variation in presentation. “Infantile spasms can present in a lot of different ways; babies haven’t read the neurology textbooks,” Dr. Hussain said. A curated collection of videos showing the variety of infantile spasms is available on the Infantile Spasms Project website.
Mr. Lay and his collaborators surveyed the parents of 100 children with IS in order to understand and describe their experiences in having their child receive the correct diagnosis and treatment. Also, they sought to determine whether socioeconomic factors affected prompt access to care. Dr. Lay completed the study with Dr. Hussain and other coauthors after graduating from UCLA in 2015.
The study, presented during a poster session at the meeting, measured the time between the parents’ first identification of IS and their contact with an “effective provider.” A provider was deemed effective if he or she correctly diagnosed IS and prescribed a first-line therapy: adrenocorticotropin-releasing hormone (ACTH), corticosteroids, or vigabatrin (Sabril).
Of the 100 children whose families were surveyed, 29% were seen by an effective provider within a week of the onset of IS. The median time from the onset of IS to the patient’s being seen by an effective provider was 24.5 days, with a wide range seen in the population surveyed (interquartile range, 0-110.5 days). This exceeds the 7 days that has been identified as the goal cumulative latency to treatment, Mr. Lay and his coauthors said.
The median age of onset of IS was 6.6 months in the study population, 45% of whom were female.
A total of 64 patients were first seen for IS concerns by a provider who was not a neurologist. Of those, the cumulative latency to seeing the first effective provider was over 100 days, with some patients’ wait time stretching to over 200 days.
The survey asked about socioeconomic factors, including income, health insurance status, proximity to specialty care, ethnicity, and parental language preference. Of these, only a non-English parental language preference was associated with increased time to seeing an effective provider (hazard ratio, 0.37; 95% confidence interval, 0.20-0.68; P = .002).
“These results suggest that a simple lack of awareness of infantile spasms among health care providers may be responsible for potentially catastrophic delays in IS care,” wrote Mr. Lay and his coauthors. Dr. Hussain concurred: “The clear driver of treatment delay is the failure of health care providers to recognize the appearance, importance, and urgency of infantile spasms. Delay in diagnosis and treatment of infantile spasms is common, costly – to both patients and the health care system – and almost entirely preventable.”
The Elsie and Isaac Fogelman Endowment, the Hughes Family Foundation, and the UCLA Children’s Discovery and Innovation Institute funded the study. Dr. Hussain has received research support from and been on the advisory boards of several pharmaceutical companies. Mr. Lay reported no disclosures.
koakes@frontlinemedcom.com
On Twitter @karioakes
HOUSTON – The appropriate evaluation and treatment of infantile spasms often comes after significant delays that are largely attributable to lack of recognition of the condition, according to recent survey results.
“Parental suspicions that ‘something was wrong’ were often discounted by health care providers, and survey respondents frequently reported that health care providers (including neurologists) were unfamiliar with infantile spasms [IS],” wrote Johnson Lay and his collaborators, in presenting results of the recently-completed Assessment of Symptoms and Specialists in Infantile Spasms (ASSIST) study at the annual meeting of the American Epilepsy Society. It’s critically important not to discount parental concerns, senior author Shaun Hussain, MD, said in an interview about the study, which surveyed parents of children with IS: “Sometimes we get in the habit of reassuring patients too much.”
On average, a pediatrician will see two cases of infantile spasms in a career of caring for children, Dr. Hussain said. These characteristically brief episodes last just a second or so, and frequently come in clusters, during or after which crying is common. Though the classic presentation of IS is a brief drop of the head with accompanying elevation of the shoulders – and perhaps a lifting of the arms – there’s wide variation in presentation. “Infantile spasms can present in a lot of different ways; babies haven’t read the neurology textbooks,” Dr. Hussain said. A curated collection of videos showing the variety of infantile spasms is available on the Infantile Spasms Project website.
Mr. Lay and his collaborators surveyed the parents of 100 children with IS in order to understand and describe their experiences in having their child receive the correct diagnosis and treatment. Also, they sought to determine whether socioeconomic factors affected prompt access to care. Dr. Lay completed the study with Dr. Hussain and other coauthors after graduating from UCLA in 2015.
The study, presented during a poster session at the meeting, measured the time between the parents’ first identification of IS and their contact with an “effective provider.” A provider was deemed effective if he or she correctly diagnosed IS and prescribed a first-line therapy: adrenocorticotropin-releasing hormone (ACTH), corticosteroids, or vigabatrin (Sabril).
Of the 100 children whose families were surveyed, 29% were seen by an effective provider within a week of the onset of IS. The median time from the onset of IS to the patient’s being seen by an effective provider was 24.5 days, with a wide range seen in the population surveyed (interquartile range, 0-110.5 days). This exceeds the 7 days that has been identified as the goal cumulative latency to treatment, Mr. Lay and his coauthors said.
The median age of onset of IS was 6.6 months in the study population, 45% of whom were female.
A total of 64 patients were first seen for IS concerns by a provider who was not a neurologist. Of those, the cumulative latency to seeing the first effective provider was over 100 days, with some patients’ wait time stretching to over 200 days.
The survey asked about socioeconomic factors, including income, health insurance status, proximity to specialty care, ethnicity, and parental language preference. Of these, only a non-English parental language preference was associated with increased time to seeing an effective provider (hazard ratio, 0.37; 95% confidence interval, 0.20-0.68; P = .002).
“These results suggest that a simple lack of awareness of infantile spasms among health care providers may be responsible for potentially catastrophic delays in IS care,” wrote Mr. Lay and his coauthors. Dr. Hussain concurred: “The clear driver of treatment delay is the failure of health care providers to recognize the appearance, importance, and urgency of infantile spasms. Delay in diagnosis and treatment of infantile spasms is common, costly – to both patients and the health care system – and almost entirely preventable.”
The Elsie and Isaac Fogelman Endowment, the Hughes Family Foundation, and the UCLA Children’s Discovery and Innovation Institute funded the study. Dr. Hussain has received research support from and been on the advisory boards of several pharmaceutical companies. Mr. Lay reported no disclosures.
koakes@frontlinemedcom.com
On Twitter @karioakes
AT AES 2016
Key clinical point:
Major finding: The cumulative latency to seeing an effective provider was over 100 days for IS patients initially seen by a nonneurologist.
Data source: Survey of parent(s) of 100 children with infantile spasms.
Disclosures: The Elsie and Isaac Fogelman Endowment, the Hughes Family Foundation, and the UCLA Children’s Discovery and Innovation Institute funded the study. Dr. Hussain has received research support from and been on the advisory boards of several pharmaceutical companies. Mr. Lay reported no disclosures.

Enterovirus D68 – An emerging threat to child health
In August 2014, we first heard of increased pediatric cases of severe respiratory tract disease, many requiring management in the ICU, and of acute flaccid myelitis/paralysis (AFM) of unknown etiology from many states across the United States. Concurrently with this outbreak in the United States, similar clinical cases were reported in Canada and Europe. Subsequently, enterovirus D68 was confirmed in some, but not all, of the paralyzed children. Although new to many of us, enterovirus D68 was already known as an atypical enterovirus sharing many of its structural and chemical properties with rhinovirus. For example, it most often was reported from respiratory samples and less common from stool samples. It also had been associated with clusters of respiratory disease since 2000 and a 2008 case of fatal AFM.

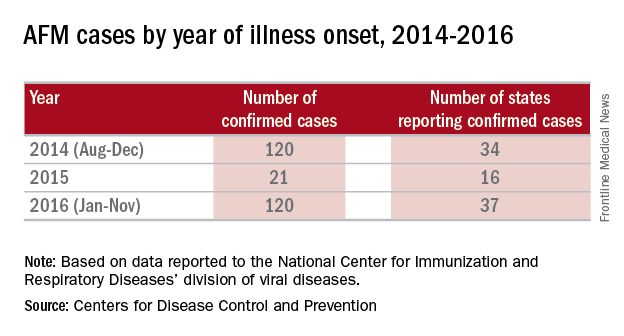
There were 120 cases of AFM, coinciding with the nationwide outbreak of enteroviral D68 disease, reported in 2014. The Centers for Disease Control and Prevention has evaluated the cerebrospinal fluid in many of these cases, and no pathogen has consistently been detected. The children were mostly school age, aged 7-11 years, presented with acute, febrile respiratory illness followed by acute onset of cranial nerve dysfunction or flaccid paralysis of one or more limbs. The CSF revealed mild pleocytosis, most often with mild elevation of protein and a normal glucose. However, the MRI was distinctly abnormal with focal lesion in the cranial nerve nuclei (in those with bulbar dysfunction) and/or in the anterior horn or spinal cord gray matter. Long-term prognosis is unknown, although most patients have persistent weakness, despite some improvement, to date.
In 2016, the CDC has reported an increase in cases after a decline in 2015 despite the absence of epidemic respiratory tract disease in the United States from enterovirus D68. In the Netherlands, an increase in respiratory disease from enterovirus D68 in children and adults also has been reported since June 2016. Respiratory disease has been observed in children as young as 3 months of age, and most of the children have underlying comorbidity, many with asthma or other pulmonary conditions. Thirteen of 17 (77%) cases in children have required ICU admission, while most of the adult cases were mild and influenzalike. One child developed bulbar dysfunction and limb weakness.
Enterovirus D68 infection should be suspected in children with moderate to severe respiratory tract infection or acute onset bulbar or flaccid paralysis of unknown etiology, especially in summer and fall. In such cases, respiratory specimens (nasopharyngeal or oral swabs or wash, tracheal secretions or bronchoalveolar lavage) should be obtained. Increasingly, hospitals and laboratories can perform multiplex polymerase chain reaction testing for enterovirus/rhinovirus. However, most do not determine the specific enterovirus. CDC and some state health departments use real-time reverse transcription polymerase chain reaction (rRT-PCR), which enables reporting of specific enterovirus species within days. CDC recommends that clinicians consider enterovirus D68 testing for children with unknown, severe respiratory illness or AFM. Details for sending specimens should be available from your state’s Department of Public Health website or the CDC.
Prevention strategies may be critical for limiting the spread of enterovirus D68 in the community. The CDC recommends:
- Wash your hands often with soap and water for 20 seconds.
- Avoid touching your eyes, nose and mouth with unwashed hands.
- Avoid close contact such as kissing, hugging, and sharing cups with people who are ill.
- Cover your coughs and sneezes with a tissue or shirt sleeve, not your hands.
- Clean and disinfect frequently touched surfaces, such as toys and doorknobs, especially if someone is sick.
- Stay home when you are ill.
In 2014, it was speculated that the epidemic might have been a one-time event. It now appears more likely that enterovirus D68 activity has been increasing since 2000, and that children and immunocompromised hosts will be at greatest risk because of a lack of neutralizing antibody. Ongoing enterovirus surveillance will be critical to understand the potential for severe respiratory disease as will the development of new and effective antivirals. A vaccine for enterovirus 71 recently demonstrated efficacy against hand, foot, and mouth disease in children and may provide insights into the development of vaccines against enterovirus D68.
References
Lancet Infect Dis. 2016 May;16(5):e64-75
Emerg Infect Dis. 2017 Jan;23(1):140-3.
J Med Virol. 2016 May;88(5):739-45
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. Dr. Pelton said he had no relevant financial disclosures. Email him at pdnews@frontlinemedcom.com.
In August 2014, we first heard of increased pediatric cases of severe respiratory tract disease, many requiring management in the ICU, and of acute flaccid myelitis/paralysis (AFM) of unknown etiology from many states across the United States. Concurrently with this outbreak in the United States, similar clinical cases were reported in Canada and Europe. Subsequently, enterovirus D68 was confirmed in some, but not all, of the paralyzed children. Although new to many of us, enterovirus D68 was already known as an atypical enterovirus sharing many of its structural and chemical properties with rhinovirus. For example, it most often was reported from respiratory samples and less common from stool samples. It also had been associated with clusters of respiratory disease since 2000 and a 2008 case of fatal AFM.
There were 120 cases of AFM, coinciding with the nationwide outbreak of enteroviral D68 disease, reported in 2014. The Centers for Disease Control and Prevention has evaluated the cerebrospinal fluid in many of these cases, and no pathogen has consistently been detected. The children were mostly school age, aged 7-11 years, presented with acute, febrile respiratory illness followed by acute onset of cranial nerve dysfunction or flaccid paralysis of one or more limbs. The CSF revealed mild pleocytosis, most often with mild elevation of protein and a normal glucose. However, the MRI was distinctly abnormal with focal lesion in the cranial nerve nuclei (in those with bulbar dysfunction) and/or in the anterior horn or spinal cord gray matter. Long-term prognosis is unknown, although most patients have persistent weakness, despite some improvement, to date.
In 2016, the CDC has reported an increase in cases after a decline in 2015 despite the absence of epidemic respiratory tract disease in the United States from enterovirus D68. In the Netherlands, an increase in respiratory disease from enterovirus D68 in children and adults also has been reported since June 2016. Respiratory disease has been observed in children as young as 3 months of age, and most of the children have underlying comorbidity, many with asthma or other pulmonary conditions. Thirteen of 17 (77%) cases in children have required ICU admission, while most of the adult cases were mild and influenzalike. One child developed bulbar dysfunction and limb weakness.
Enterovirus D68 infection should be suspected in children with moderate to severe respiratory tract infection or acute onset bulbar or flaccid paralysis of unknown etiology, especially in summer and fall. In such cases, respiratory specimens (nasopharyngeal or oral swabs or wash, tracheal secretions or bronchoalveolar lavage) should be obtained. Increasingly, hospitals and laboratories can perform multiplex polymerase chain reaction testing for enterovirus/rhinovirus. However, most do not determine the specific enterovirus. CDC and some state health departments use real-time reverse transcription polymerase chain reaction (rRT-PCR), which enables reporting of specific enterovirus species within days. CDC recommends that clinicians consider enterovirus D68 testing for children with unknown, severe respiratory illness or AFM. Details for sending specimens should be available from your state’s Department of Public Health website or the CDC.
Prevention strategies may be critical for limiting the spread of enterovirus D68 in the community. The CDC recommends:
- Wash your hands often with soap and water for 20 seconds.
- Avoid touching your eyes, nose and mouth with unwashed hands.
- Avoid close contact such as kissing, hugging, and sharing cups with people who are ill.
- Cover your coughs and sneezes with a tissue or shirt sleeve, not your hands.
- Clean and disinfect frequently touched surfaces, such as toys and doorknobs, especially if someone is sick.
- Stay home when you are ill.
In 2014, it was speculated that the epidemic might have been a one-time event. It now appears more likely that enterovirus D68 activity has been increasing since 2000, and that children and immunocompromised hosts will be at greatest risk because of a lack of neutralizing antibody. Ongoing enterovirus surveillance will be critical to understand the potential for severe respiratory disease as will the development of new and effective antivirals. A vaccine for enterovirus 71 recently demonstrated efficacy against hand, foot, and mouth disease in children and may provide insights into the development of vaccines against enterovirus D68.
References
Lancet Infect Dis. 2016 May;16(5):e64-75
Emerg Infect Dis. 2017 Jan;23(1):140-3.
J Med Virol. 2016 May;88(5):739-45
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. Dr. Pelton said he had no relevant financial disclosures. Email him at pdnews@frontlinemedcom.com.
In August 2014, we first heard of increased pediatric cases of severe respiratory tract disease, many requiring management in the ICU, and of acute flaccid myelitis/paralysis (AFM) of unknown etiology from many states across the United States. Concurrently with this outbreak in the United States, similar clinical cases were reported in Canada and Europe. Subsequently, enterovirus D68 was confirmed in some, but not all, of the paralyzed children. Although new to many of us, enterovirus D68 was already known as an atypical enterovirus sharing many of its structural and chemical properties with rhinovirus. For example, it most often was reported from respiratory samples and less common from stool samples. It also had been associated with clusters of respiratory disease since 2000 and a 2008 case of fatal AFM.
There were 120 cases of AFM, coinciding with the nationwide outbreak of enteroviral D68 disease, reported in 2014. The Centers for Disease Control and Prevention has evaluated the cerebrospinal fluid in many of these cases, and no pathogen has consistently been detected. The children were mostly school age, aged 7-11 years, presented with acute, febrile respiratory illness followed by acute onset of cranial nerve dysfunction or flaccid paralysis of one or more limbs. The CSF revealed mild pleocytosis, most often with mild elevation of protein and a normal glucose. However, the MRI was distinctly abnormal with focal lesion in the cranial nerve nuclei (in those with bulbar dysfunction) and/or in the anterior horn or spinal cord gray matter. Long-term prognosis is unknown, although most patients have persistent weakness, despite some improvement, to date.
In 2016, the CDC has reported an increase in cases after a decline in 2015 despite the absence of epidemic respiratory tract disease in the United States from enterovirus D68. In the Netherlands, an increase in respiratory disease from enterovirus D68 in children and adults also has been reported since June 2016. Respiratory disease has been observed in children as young as 3 months of age, and most of the children have underlying comorbidity, many with asthma or other pulmonary conditions. Thirteen of 17 (77%) cases in children have required ICU admission, while most of the adult cases were mild and influenzalike. One child developed bulbar dysfunction and limb weakness.
Enterovirus D68 infection should be suspected in children with moderate to severe respiratory tract infection or acute onset bulbar or flaccid paralysis of unknown etiology, especially in summer and fall. In such cases, respiratory specimens (nasopharyngeal or oral swabs or wash, tracheal secretions or bronchoalveolar lavage) should be obtained. Increasingly, hospitals and laboratories can perform multiplex polymerase chain reaction testing for enterovirus/rhinovirus. However, most do not determine the specific enterovirus. CDC and some state health departments use real-time reverse transcription polymerase chain reaction (rRT-PCR), which enables reporting of specific enterovirus species within days. CDC recommends that clinicians consider enterovirus D68 testing for children with unknown, severe respiratory illness or AFM. Details for sending specimens should be available from your state’s Department of Public Health website or the CDC.
Prevention strategies may be critical for limiting the spread of enterovirus D68 in the community. The CDC recommends:
- Wash your hands often with soap and water for 20 seconds.
- Avoid touching your eyes, nose and mouth with unwashed hands.
- Avoid close contact such as kissing, hugging, and sharing cups with people who are ill.
- Cover your coughs and sneezes with a tissue or shirt sleeve, not your hands.
- Clean and disinfect frequently touched surfaces, such as toys and doorknobs, especially if someone is sick.
- Stay home when you are ill.
In 2014, it was speculated that the epidemic might have been a one-time event. It now appears more likely that enterovirus D68 activity has been increasing since 2000, and that children and immunocompromised hosts will be at greatest risk because of a lack of neutralizing antibody. Ongoing enterovirus surveillance will be critical to understand the potential for severe respiratory disease as will the development of new and effective antivirals. A vaccine for enterovirus 71 recently demonstrated efficacy against hand, foot, and mouth disease in children and may provide insights into the development of vaccines against enterovirus D68.
References
Lancet Infect Dis. 2016 May;16(5):e64-75
Emerg Infect Dis. 2017 Jan;23(1):140-3.
J Med Virol. 2016 May;88(5):739-45
Dr. Pelton is chief of pediatric infectious disease and coordinator of the maternal-child HIV program at Boston Medical Center. Dr. Pelton said he had no relevant financial disclosures. Email him at pdnews@frontlinemedcom.com.
Infection in AML patient prompts discovery

Photo courtesy of
Janice Carr/CDC
The quest to understand a prolonged infection in an infant with acute myeloid leukemia (AML) has led to the discovery of a mutation that allows bacteria to tolerate antibiotic therapy.
Researchers described this discovery in the journal mBio.
“These findings detail a ‘perfect storm’ for development of antibiotic tolerance by bacteria that already pose a clinical challenge,” said study author Jason Rosch, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The same conditions may be present in other patients with immune systems that have been compromised by chemotherapy or disease,” added co-author Joshua Wolf, MBBS, also of St. Jude.
The “perfect storm” involved a patient who was 6 weeks old when she was diagnosed with AML. The treatment wiped out her white blood cells, and, despite infection-control measures, she developed a bloodstream infection with vancomycin-resistant Enterococcus faecium (VRE).
The infection persisted for 26 days and only resolved after her immune system recovered. She then successfully completed AML treatment.
In-depth DNA sequencing of 22 VRE samples collected during the patient’s infection helped researchers link the prolonged infection to a point mutation in the relA gene of VRE.
The mutation inappropriately activated the stringent response pathway, which bacteria use to survive under stress and to tolerate antibiotics.
The mutation resulted in elevated levels of the signaling molecule alarmone, and this likely primed the bacteria to survive exposure to multiple antibiotics, the researchers said.
The team also noted that relA-mutant VRE was susceptible to the antibiotics linezolid and daptomycin in minimum inhibitory concentration testing and during planktonic growth.
However, when growing in biofilm, relA-mutant VRE could tolerate high doses of both antibiotics.
“This mutation has particular clinical significance because the antibiotics involved, linezolid and daptomycin, are the last line of defense against VRE infection,” Dr Wolf said.
Among the compounds in development for the treatment of bacterial biofilms is the experimental antibiotic ADEP-4. In this study, ADEP-4 killed relA-mutant and non-mutant VRE growing in biofilm in the lab.
“In the future, compounds like ADEP-4 may provide a new approach to resolving persistent infections,” Dr Wolf said.
Dr Rosch noted that evidence gleaned from tracking the evolution of VRE throughout the infection suggested the patient’s immune-compromised state was essential to survival of the mutant VRE.
Gene transcription was altered significantly in relA-mutant VRE and produced biofilms that were less robust and possibly unlikely to otherwise survive.
“The case expands our understanding of the role of the stringent response in susceptibility and tolerance to a wide range of antibiotics, especially in biofilms,” Dr Rosch said. “It also demonstrates that these mutations can develop and gain a foothold during a human infection.” ![]()
Photo courtesy of
Janice Carr/CDC
The quest to understand a prolonged infection in an infant with acute myeloid leukemia (AML) has led to the discovery of a mutation that allows bacteria to tolerate antibiotic therapy.
Researchers described this discovery in the journal mBio.
“These findings detail a ‘perfect storm’ for development of antibiotic tolerance by bacteria that already pose a clinical challenge,” said study author Jason Rosch, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The same conditions may be present in other patients with immune systems that have been compromised by chemotherapy or disease,” added co-author Joshua Wolf, MBBS, also of St. Jude.
The “perfect storm” involved a patient who was 6 weeks old when she was diagnosed with AML. The treatment wiped out her white blood cells, and, despite infection-control measures, she developed a bloodstream infection with vancomycin-resistant Enterococcus faecium (VRE).
The infection persisted for 26 days and only resolved after her immune system recovered. She then successfully completed AML treatment.
In-depth DNA sequencing of 22 VRE samples collected during the patient’s infection helped researchers link the prolonged infection to a point mutation in the relA gene of VRE.
The mutation inappropriately activated the stringent response pathway, which bacteria use to survive under stress and to tolerate antibiotics.
The mutation resulted in elevated levels of the signaling molecule alarmone, and this likely primed the bacteria to survive exposure to multiple antibiotics, the researchers said.
The team also noted that relA-mutant VRE was susceptible to the antibiotics linezolid and daptomycin in minimum inhibitory concentration testing and during planktonic growth.
However, when growing in biofilm, relA-mutant VRE could tolerate high doses of both antibiotics.
“This mutation has particular clinical significance because the antibiotics involved, linezolid and daptomycin, are the last line of defense against VRE infection,” Dr Wolf said.
Among the compounds in development for the treatment of bacterial biofilms is the experimental antibiotic ADEP-4. In this study, ADEP-4 killed relA-mutant and non-mutant VRE growing in biofilm in the lab.
“In the future, compounds like ADEP-4 may provide a new approach to resolving persistent infections,” Dr Wolf said.
Dr Rosch noted that evidence gleaned from tracking the evolution of VRE throughout the infection suggested the patient’s immune-compromised state was essential to survival of the mutant VRE.
Gene transcription was altered significantly in relA-mutant VRE and produced biofilms that were less robust and possibly unlikely to otherwise survive.
“The case expands our understanding of the role of the stringent response in susceptibility and tolerance to a wide range of antibiotics, especially in biofilms,” Dr Rosch said. “It also demonstrates that these mutations can develop and gain a foothold during a human infection.” ![]()
Photo courtesy of
Janice Carr/CDC
The quest to understand a prolonged infection in an infant with acute myeloid leukemia (AML) has led to the discovery of a mutation that allows bacteria to tolerate antibiotic therapy.
Researchers described this discovery in the journal mBio.
“These findings detail a ‘perfect storm’ for development of antibiotic tolerance by bacteria that already pose a clinical challenge,” said study author Jason Rosch, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“The same conditions may be present in other patients with immune systems that have been compromised by chemotherapy or disease,” added co-author Joshua Wolf, MBBS, also of St. Jude.
The “perfect storm” involved a patient who was 6 weeks old when she was diagnosed with AML. The treatment wiped out her white blood cells, and, despite infection-control measures, she developed a bloodstream infection with vancomycin-resistant Enterococcus faecium (VRE).
The infection persisted for 26 days and only resolved after her immune system recovered. She then successfully completed AML treatment.
In-depth DNA sequencing of 22 VRE samples collected during the patient’s infection helped researchers link the prolonged infection to a point mutation in the relA gene of VRE.
The mutation inappropriately activated the stringent response pathway, which bacteria use to survive under stress and to tolerate antibiotics.
The mutation resulted in elevated levels of the signaling molecule alarmone, and this likely primed the bacteria to survive exposure to multiple antibiotics, the researchers said.
The team also noted that relA-mutant VRE was susceptible to the antibiotics linezolid and daptomycin in minimum inhibitory concentration testing and during planktonic growth.
However, when growing in biofilm, relA-mutant VRE could tolerate high doses of both antibiotics.
“This mutation has particular clinical significance because the antibiotics involved, linezolid and daptomycin, are the last line of defense against VRE infection,” Dr Wolf said.
Among the compounds in development for the treatment of bacterial biofilms is the experimental antibiotic ADEP-4. In this study, ADEP-4 killed relA-mutant and non-mutant VRE growing in biofilm in the lab.
“In the future, compounds like ADEP-4 may provide a new approach to resolving persistent infections,” Dr Wolf said.
Dr Rosch noted that evidence gleaned from tracking the evolution of VRE throughout the infection suggested the patient’s immune-compromised state was essential to survival of the mutant VRE.
Gene transcription was altered significantly in relA-mutant VRE and produced biofilms that were less robust and possibly unlikely to otherwise survive.
“The case expands our understanding of the role of the stringent response in susceptibility and tolerance to a wide range of antibiotics, especially in biofilms,” Dr Rosch said. “It also demonstrates that these mutations can develop and gain a foothold during a human infection.” ![]()
Shedding Light on Onychomadesis
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23

Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
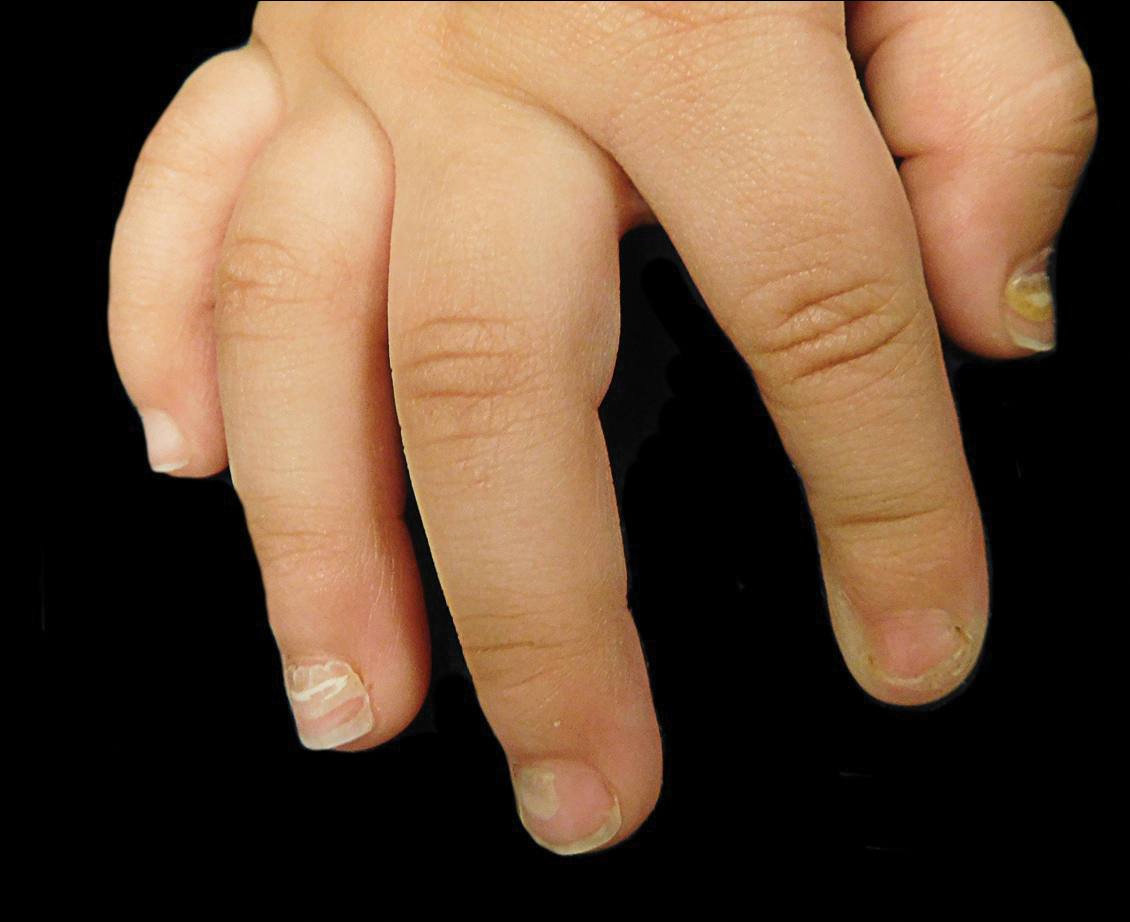
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
Onychomadesis is an acute, noninflammatory, painless, proximal separation of the nail plate from the nail matrix. It occurs due to an abrupt stoppage of nail production by matrix cells, producing temporary cessation of nail growth with or without subsequent complete shedding of nails.1-10 Onychomadesis has a wide spectrum of clinical presentations ranging from mild transverse ridges of the nail plate (Beau lines) to complete nail shedding.4,11 Onychomadesis may be related to systemic and dermatologic diseases, drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids), nail trauma, fever, or infection,5 and a connection between onychomadesis and hand-foot-and-mouth disease (HFMD) was first described by Clementz et al12 following outbreaks in Europe, Asia, and the United States.
Epidemiology
Onychomadesis has been observed in children of all ages including neonates. Neonatal onychomadesis is thought to be related to perinatal stressors and birth trauma, with possible exacerbation by superimposed candidiasis.10 Depending on the underlying cause, there may be involvement of a single nail or multiple nails. Nag et al1 noted that onychomadesis was most commonly observed in nails of the middle finger (73.7%), followed by the thumb (63.2%) and ring finger (52.6%). Fingernails are more commonly involved than toenails.1
Clementz et al12 first proposed the association between onychomadesis and HFMD in 2000. Patients with a history of HFMD were found to be 14 times more likely to develop onychomadesis (relative risk, 14; 95% confidence interval, 4.57-42.86).4 A common pathogen for HFMD is coxsackievirus A6 (CVA6),13,14 but the mechanism of onychomadesis in HFMD remains unclear.5,7,13 Outbreaks of HFMD have been reported in Spain, Finland, Japan, Thailand, the United States, Singapore, and China.15 During an outbreak of HFMD in Taiwan, the incidence of onychomadesis following CVA6 infection was 37% (48/130) compared to 5% (7/145) in cases with non-CVA6 causative strains.16 There also have been observed differences in the prevalence of onychomadesis by age: a 55% (18/33) occurrence rate was noted in the youngest age group (range, 9–23 months), 30% (8/27) in the middle age group (range, 24–32 months), and 4% (1/28) in the oldest age group (range, 33–42 months), with an average of 4 nails shed per case.17 A study in Spain also found a high occurrence of onychomadesis in a nursery setting, with 92% (11/12) of onychomadesis cases preceded by HFMD 2 months prior.18
Etiology
Local trauma to the nail bed is the most common cause of single-digit onychomadesis.4 Multiple-digit involvement suggests a systemic etiology such as fever, erythroderma, and Kawasaki disease; use of drugs (eg, chemotherapeutic agents, anticonvulsants, lithium, retinoids); and viral infections such as HFMD and varicella at the infantile age (Table).5,9,19 Most drug-related nail changes are the outcome of acute toxicity to the proliferating nail matrix epithelium. If onychomadesis affects all nails at the same level, the patient’s history of medication use and other treatments taken 2 to 3 weeks prior to the appearance of the nail findings should be evaluated. Chemotherapeutic agents produce nail changes in a high proportion of patients, which often are related to drug dosage. These effects also are reproducible with re-administration of the drug.20 Onychomadesis also has been reported as a possible side effect of anticonvulsants such as valproic acid (VPA).21 One study evaluating the link between VPA and onychomadesis indicated that nail changes may be due to a disturbance of zinc metabolism.22 However, the pathomechanism of onychomadesis associated with VPA treatment remains unclear.21 Onychomadesis also has developed after an allergic drug reaction to oral penicillin V after treatment of a sore throat in a 23-month-old child.23
Nail involvement has been reported in 10% of cases of inflammatory conditions such as lichen planus21; however, it may be more common but underrecognized and underreported. Grover et al9 indicated that lichen planus–induced severe inflammation in the matrix of the nail unit leading to a temporary growth arrest was the possible mechanism leading to nail shedding. Prompt systemic and intramatricial steroid treatment of lichen planus is required to avoid potential scarring of the nail matrix and permanent damage.9
Onychomadesis also has been reported following varicella infection (chickenpox). Podder et al19 reported the case of a 7-year-old girl who had recovered from a varicella infection 5 weeks prior and presented with onychomadesis of the right index fingernail with all other fingernails and toenails appearing normal. Kocak and Koçak5 reported onychomadesis in 2 sisters with varicella infection. There are few reported cases, so it is still unclear whether varicella infection is an inciting factor.19
One of the most studied viral infections linked to onychomadesis is HFMD, which is a common viral infection that mostly affects children younger than 10 years.1 The precise mechanism of onychomadesis for these viral infection events remains unclear.7,10,13 Several theories have been delineated, including nail matrix arrest from fever occurring during HFMD.6 However, this cause is unlikely, as fevers are typically low grade and present only for a few hours.4,6,13 Direct inflammation spreading from skin lesions of HFMD around the nails or maceration associated with finger blisters could cause onychomadesis.1,5,7 Haneke24 hypothesized that nail shedding may be the consequence of vesicles localized in the periungual tissue, but studies have shown incidence without prior lesions on the fingers and no relationship between nail matrix arrest and severity of HFMD.5,6,13 Bettoli et al25 reported that inflammation secondary to viral infection around the nail matrix might be induced directly by viruses or indirectly by virus-specific immunocomplexes and consequent distal embolism. Osterback et al14 used reverse transcription–polymerase chain reaction to detect CVA6 in fragmented nails from 2 children and 1 parent following an HFMD episode, suggesting that virus replication could damage the nail matrix, resulting in onychomadesis. Cabrerizo et al18 also suggested that virus replication directly damages the nail matrix based on the presence of CVA6 in shed nails. Because fingernails with onychomadesis are not always of the fingers affected by HFMD, an indirect effect of viral infection on the nail matrix is more plausible.8 Additional studies are needed to clarify the virus-associated mechanism of nail matrix arrest.6 Finally, frequent washing of hands15 resulting in maceration, Candida infection, and allergic contact dermatitis2 may be possible causes. It is unclear if onychomadesis following HFMD is related to viral replication, inflammation, or intensive hygienic measures, and further investigation is needed.2,15
Clinical Characteristics
The ventral floor is the site of the germinal matrix and is responsible for 90% of nail production. As a result, more of the nail plate substance is produced proximally, leading to a natural convex curvature from the proximal to distal nail.11 Beau lines are transverse ridging of the nail plates.6 Onychomadesis may be viewed as a more severe form of Beau lines, with complete separation and possible shedding of the nail plate (Figure).3,4 In both cases, an insult to the nail matrix is followed by recovery and production of the nail plate at the nail matrix.4 In Beau lines, slowing or disruption of cell growth from the proximal matrix results in a thinner nail plate, leading to transverse depressions. Onychomadesis has a similar pathophysiology but is associated with a complete halt in the nail plate production.3
Diagnosis
The diagnosis of onychomadesis is made clinically.3,10 Distinct nail changes can be detected by inspection and palpation of the nail plate,3,11 which allows for differentiation between Beau lines and complete nail shedding. Additionally, any signs of nail trauma need to be noted, as well as pain, swelling, or pruritus, as these symptoms also can guide in determining the etiology of the nail dystrophy. Ultrasonography can confirm the diagnosis, as the defect can be identified beneath the proximal nail fold.3,26 When it occurs after HFMD or varicella, onychomadesis tends to present in 28 to 40 days following infection.4,6,10 Physicians should consider underlying associations. A review of viral illnesses within 1 to 2 months prior to development of nail changes often will identify the causative disease.4 Each patient should be evaluated for recent nail trauma; medications; viral infection; and autoimmune, systemic, and inflammatory diseases.
Treatment
Onychomadesis typically is mild and self-limited.4,10 There is no specific treatment,10 but a conservative approach to management is recommended. Treatment of any underlying medical conditions or discontinuation of an offending medication may help to prevent recurrent onychomadesis.3 Supportive care along with protection of the nail bed by maintaining short nails and using adhesive bandages over the affected nails to avoid snagging the nail or ripping off the partially attached nails is recommended.4 In some cases, onychomadesis has been treated with topical application of urea cream 40% under occlusion27 or halcinonide cream 0.1% under occlusion for 5 to 6 days,28 but these treatments have not been universally effective.3 External use of basic fibroblast growth factor to stimulate new regrowth of the nail plate has been advocated.3 It is important to reassure patients that as long as the underlying causes are eliminated and the nail matrix has not been permanently scarred, the nails should grow back within 12 weeks or sooner in children. Thus, typically only reassurance and counseling of parents/guardians is required for onychomadesis in children.1,2 However, the nails may be dystrophic or fail to regrow if there is poor peripheral circulation or permanent nail matrix damage.
Conclusion
Fortunately, onychomadesis is self-limited. Physicians should look for underlying causes of onychomadesis, including a history of viral infections such as HFMD and varicella as well as systemic diseases and use of medications. As long as any underlying disorder or condition has been resolved, spontaneous regrowth of healthy nails usually but not always occurs within 12 weeks or sooner in children.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
- Nag SS, Dutta A, Mandal RK. Delayed cutaneous findings of hand, foot, and mouth disease. Indian Pediatr. 2016;53:42-44.
- Tan ZH, Koh MJ. Nail shedding following hand, foot and mouth disease. Arch Dis Child. 2013;98:665.
- Braswell MA, Daniel CR, Brodell RT. Beau lines, onychomadesis, and retronychia: a unifying hypothesis. J Am Acad Dermatol. 2015;73:849-855.
- Clark CM, Silverberg NB, Weinberg JM. What is your diagnosis? onychomadesis following hand-foot-and-mouth disease. Cutis. 2015;95:312, 319-320.
- Kocak AY, Koçak O. Onychomadesis in two sisters induced by varicella infection. Pediatr Dermatol. 2013;30:E108-E109.
- Shin JY, Cho BK, Park HJ. A clinical study of nail changes occurring secondary to hand-foot-mouth disease: onychomadesis and Beau’s lines. Ann Dermatol. 2014;26:280-283.
- Shikuma E, Endo Y, Fujisawa A, et al. Onychomadesis developed only on the nails having cutaneous lesions of severe hand-foot-mouth disease. Case Rep Dermatol Med. 2011;2011:324193.
- Kim EJ, Park HS, Yoon HS, et al. Four cases of onychomadesis after hand-foot-mouth disease. Ann Dermatol. 2014;26:777-778.
- Grover C, Vohra S. Onychomadesis with lichen planus: an under-recognized manifestation. Indian J Dermatol. 2015;60:420.
- Chu DH, Rubin AI. Diagnosis and management of nail disorders. In: Holland K, ed. The Pediatric Clinics of North America. Vol 61. Philadelphia, PA: Elsevier; 2014:301-302.
- Kowalewski C, Schwartz RA. Components, growth, and composition of the nail. In: Demis D, ed. Clinical Dermatology. Philadelphia, PA: Lippincott-Raven; 1998.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Scarfì F, Arunachalam M, Galeone M, et al. An uncommon onychomadesis in adults. Int J Dermatol. 2014;53:1392-1394.
- Osterback R, Vuorinen T, Linna M, et al. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg Infect Dis. 2009;15:1485-1488.
- Yan X, Zhang ZZ, Yang ZH, et al. Clinical and etiological characteristics of atypical hand-foot-and-mouth disease in children from Chongqing, China: a retrospective study [published online November 26, 2015]. Biomed Res Int. 2015;2015:802046.
- Wei SH, Huang YP, Liu MC, et al. An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis. 2011;11:346.
- Guimbao J, Rodrigo P, Alberto MJ, et al. Onychomadesis outbreak linked to hand, foot, and mouth disease, Spain, July 2008. Euro Surveill. 2010;15:19663.
- Cabrerizo M, De Miguel T, Armada A, et al. Onychomadesis after a hand, foot, and mouth disease outbreak in Spain, 2009. Epidemiol Infect. 2010;138:1775-1778.
- Podder I, Das A, Gharami RC. Onychomadesis following varicella infection: is it a mere co-incidence? Indian J Dermatol. 2015;60:626-627.
- Piraccini BM, Iorizzo M, Tosti A. Drug-induced nail abnormalities. Am J Clin Dermatol. 2003;4:31-37.
- Poretti A, Lips U, Belvedere M, et al. Onychomadesis: a rare side-effect of valproic acid medication? Pediatr Dermatol. 2009;26:749-750.
- Grech V, Vella C. Generalized onycholoysis associated with sodium valproate therapy. Eur Neurol. 1999;42:64-65.
- Shah RK, Uddin M, Fatunde OJ. Onychomadesis secondary to penicillin allergy in a child. J Pediatr. 2012;161:166.
- Haneke E. Onychomadesis and hand, foot and mouth disease—is there a connection? Euro Surveill. 2010;15(37).
- Bettoli V, Zauli S, Toni G, et al. Onychomadesis following hand, foot, and mouth disease: a case report from Italy and review of the literature. Int J Dermatol. 2013;52:728-730.
- Wortsman X, Wortsman J, Guerrero R, et al. Anatomical changes in retronychia and onychomadesis detected using ultrasound. Dermatol Surg. 2010;36:1615-1620.
- Fleming CJ, Hunt MJ, Barnetson RS. Mycosis fungoides with onychomadesis. Br J Dermatol. 1996;135:1012-1013.
- Mishra D, Singh G, Pandey SS. Possible carbamazepine-induced reversible onychomadesis. Int J Dermatol. 1989;28:460-461.
Practice Points
- Onychomadesis in a child may be a cutaneous sign of systemic disease.
- In childhood, onychomadesis is sometimes linked with hand-foot-and-mouth disease.
- Spontaneous nail regrowth usually occurs within 12 weeks but may occur faster in children.
Transition to adult epilepsy care done right
Has this ever happened to you? You are an adult neurologist who has been asked to take on the care of a pediatric neurology patient. The patient who comes to your clinic is a 20-year-old young woman with a history of moderate developmental delay and intractable epilepsy. She is on numerous medications including valproic acid with a previous trial of the ketogenic diet. You receive a report that she has focal epilepsy and is having frequent seizures and last had an MRI at age 2 years. Prior notes talk about her summer vacations but not much about the future plans for her epilepsy. You see the patient in clinic, and the family is not happy to be in the adult clinic. They are disappointed that you don’t spend more time with them or fill out myriad forms. You find out that they have not obtained legal guardianship for their daughter and have no plan for work placement after school. She also has various other medical comorbidities that were previously addressed by the pediatric neurologist.

There is a not much evidence on the right way to do this. In 2013, the American Epilepsy Society approved a Transition Tool that is helpful in outlining the steps for a successful transition, and in 2016, the Child Neurology Foundation put forth a consensus statement with eight principles to guide a successful transition. Transitions are an expectation of good care and they recommend that a written policy be present for all offices.
Talking about transitioning should start as early as 10-12 years of age and should be discussed every year. Thinking about prognosis and a realistic plan for each child as they enter adult life is important. Patients and families should be able to understand how the disease affects them, what their medications are and how to independently obtain them, what comorbidities are associated with their disease, how to stay healthy, how to improve their quality of life, and how to advocate for themselves. As children become teenagers they should have a concrete plan for ongoing education, work, women’s issues, and an understanding of decision-making capacity and whether legal guardianship or a power of attorney needs to be implemented.
, even if they are still seen in the pediatric setting. A transition packet should be created that includes a summary of the diagnosis, work-up, previous treatments, and considerations for future treatments and emergency care. Also included is a plan for who will continue to address any non–seizure-related diagnoses the pediatric neurologist may have been managing. The patient and family also have an opportunity to review and contribute to this. This packet enables the adult neurologist to easily understand all issues and assume care of the patient, easing this aspect of the transition.
An advance meeting of the patient and family with the adult provider should be arranged whenever possible. To address this, some centers are now creating a transition clinic staffed by both pediatric and adult neurologists and/or nurses. This ideally takes place in the adult setting and is an excellent way to smooth the transition for the patient, family, and providers. Good transition is important to help prevent gaps in care, avoid reinventing the wheel, and improve satisfaction for everyone involved (patient, family, nurses, and neurologists). The key points are that transition discussions start early, patients and families should be involved and empowered in the process, and the creation of a transition packet for the adult provider is very helpful. Care transitions are something we will be hearing a lot more about in the upcoming years. And, hopefully, next time, the patient scenario seen above will go more smoothly!
Dr. Felton is an epilepsy specialist at the University of Wisconsin, Madison, and Dr. Kelley is director of the Pediatric Epilepsy Monitoring Unit at Johns Hopkins University, Baltimore. This editorial reflects the content of a presentation given by Dr. Felton and Dr. Kelley at the annual meeting of the American Epilepsy Society in Houston. The authors report no conflict of interest.
Has this ever happened to you? You are an adult neurologist who has been asked to take on the care of a pediatric neurology patient. The patient who comes to your clinic is a 20-year-old young woman with a history of moderate developmental delay and intractable epilepsy. She is on numerous medications including valproic acid with a previous trial of the ketogenic diet. You receive a report that she has focal epilepsy and is having frequent seizures and last had an MRI at age 2 years. Prior notes talk about her summer vacations but not much about the future plans for her epilepsy. You see the patient in clinic, and the family is not happy to be in the adult clinic. They are disappointed that you don’t spend more time with them or fill out myriad forms. You find out that they have not obtained legal guardianship for their daughter and have no plan for work placement after school. She also has various other medical comorbidities that were previously addressed by the pediatric neurologist.
There is a not much evidence on the right way to do this. In 2013, the American Epilepsy Society approved a Transition Tool that is helpful in outlining the steps for a successful transition, and in 2016, the Child Neurology Foundation put forth a consensus statement with eight principles to guide a successful transition. Transitions are an expectation of good care and they recommend that a written policy be present for all offices.
Talking about transitioning should start as early as 10-12 years of age and should be discussed every year. Thinking about prognosis and a realistic plan for each child as they enter adult life is important. Patients and families should be able to understand how the disease affects them, what their medications are and how to independently obtain them, what comorbidities are associated with their disease, how to stay healthy, how to improve their quality of life, and how to advocate for themselves. As children become teenagers they should have a concrete plan for ongoing education, work, women’s issues, and an understanding of decision-making capacity and whether legal guardianship or a power of attorney needs to be implemented.
, even if they are still seen in the pediatric setting. A transition packet should be created that includes a summary of the diagnosis, work-up, previous treatments, and considerations for future treatments and emergency care. Also included is a plan for who will continue to address any non–seizure-related diagnoses the pediatric neurologist may have been managing. The patient and family also have an opportunity to review and contribute to this. This packet enables the adult neurologist to easily understand all issues and assume care of the patient, easing this aspect of the transition.
An advance meeting of the patient and family with the adult provider should be arranged whenever possible. To address this, some centers are now creating a transition clinic staffed by both pediatric and adult neurologists and/or nurses. This ideally takes place in the adult setting and is an excellent way to smooth the transition for the patient, family, and providers. Good transition is important to help prevent gaps in care, avoid reinventing the wheel, and improve satisfaction for everyone involved (patient, family, nurses, and neurologists). The key points are that transition discussions start early, patients and families should be involved and empowered in the process, and the creation of a transition packet for the adult provider is very helpful. Care transitions are something we will be hearing a lot more about in the upcoming years. And, hopefully, next time, the patient scenario seen above will go more smoothly!
Dr. Felton is an epilepsy specialist at the University of Wisconsin, Madison, and Dr. Kelley is director of the Pediatric Epilepsy Monitoring Unit at Johns Hopkins University, Baltimore. This editorial reflects the content of a presentation given by Dr. Felton and Dr. Kelley at the annual meeting of the American Epilepsy Society in Houston. The authors report no conflict of interest.
Has this ever happened to you? You are an adult neurologist who has been asked to take on the care of a pediatric neurology patient. The patient who comes to your clinic is a 20-year-old young woman with a history of moderate developmental delay and intractable epilepsy. She is on numerous medications including valproic acid with a previous trial of the ketogenic diet. You receive a report that she has focal epilepsy and is having frequent seizures and last had an MRI at age 2 years. Prior notes talk about her summer vacations but not much about the future plans for her epilepsy. You see the patient in clinic, and the family is not happy to be in the adult clinic. They are disappointed that you don’t spend more time with them or fill out myriad forms. You find out that they have not obtained legal guardianship for their daughter and have no plan for work placement after school. She also has various other medical comorbidities that were previously addressed by the pediatric neurologist.
There is a not much evidence on the right way to do this. In 2013, the American Epilepsy Society approved a Transition Tool that is helpful in outlining the steps for a successful transition, and in 2016, the Child Neurology Foundation put forth a consensus statement with eight principles to guide a successful transition. Transitions are an expectation of good care and they recommend that a written policy be present for all offices.
Talking about transitioning should start as early as 10-12 years of age and should be discussed every year. Thinking about prognosis and a realistic plan for each child as they enter adult life is important. Patients and families should be able to understand how the disease affects them, what their medications are and how to independently obtain them, what comorbidities are associated with their disease, how to stay healthy, how to improve their quality of life, and how to advocate for themselves. As children become teenagers they should have a concrete plan for ongoing education, work, women’s issues, and an understanding of decision-making capacity and whether legal guardianship or a power of attorney needs to be implemented.
, even if they are still seen in the pediatric setting. A transition packet should be created that includes a summary of the diagnosis, work-up, previous treatments, and considerations for future treatments and emergency care. Also included is a plan for who will continue to address any non–seizure-related diagnoses the pediatric neurologist may have been managing. The patient and family also have an opportunity to review and contribute to this. This packet enables the adult neurologist to easily understand all issues and assume care of the patient, easing this aspect of the transition.
An advance meeting of the patient and family with the adult provider should be arranged whenever possible. To address this, some centers are now creating a transition clinic staffed by both pediatric and adult neurologists and/or nurses. This ideally takes place in the adult setting and is an excellent way to smooth the transition for the patient, family, and providers. Good transition is important to help prevent gaps in care, avoid reinventing the wheel, and improve satisfaction for everyone involved (patient, family, nurses, and neurologists). The key points are that transition discussions start early, patients and families should be involved and empowered in the process, and the creation of a transition packet for the adult provider is very helpful. Care transitions are something we will be hearing a lot more about in the upcoming years. And, hopefully, next time, the patient scenario seen above will go more smoothly!
Dr. Felton is an epilepsy specialist at the University of Wisconsin, Madison, and Dr. Kelley is director of the Pediatric Epilepsy Monitoring Unit at Johns Hopkins University, Baltimore. This editorial reflects the content of a presentation given by Dr. Felton and Dr. Kelley at the annual meeting of the American Epilepsy Society in Houston. The authors report no conflict of interest.

Delays in Diagnosis and Treatment of Infantile Spasms Are Common
HOUSTON—Children with infantile spasms commonly endure substantial delays in diagnosis and treatment, according to research presented at the 70th Annual Meeting of the American Epilepsy Society. “A simple lack of awareness of infantile spasms among healthcare providers may be responsible for potentially catastrophic delays in diagnosis and treatment,” said Shaun Hussain, MD, Director of the University of California, Los Angeles Infantile Spasms Program, and colleagues. “There is a desperate need for effective interventions to increase basic familiarity with infantile spasms among healthcare providers.”

Dr. Hussain and his colleagues performed a study to measure delays in diagnosis and treatment of infantile spasms and identify barriers to optimal care. The researchers retrospectively identified children with video-EEG-confirmed infantile spasms in a clinical database.
When the children’s parents presented for follow-up, they were surveyed about their experiences with diagnosis and treatment. The investigators asked about medical and sociodemographic factors that could affect the care of infantile spasms. Specifically, the researchers determined the dates of infantile spasms onset, first visit with any healthcare provider, first visit with any neurologist, and first visit with an effective provider. Dr. Hussain and colleagues defined an effective provider as a healthcare provider who identified infantile spasms and prescribed a first-line treatment (ie, ACTH, corticosteroids, vigabatrin, or surgical resection). The investigators reviewed medical records to corroborate parents’ survey responses. They calculated the time to first effective provider using Cox proportional hazards regression.
The parents of 100 children with previous or ongoing infantile spasms were included in the study. Approximately 29% of patients were seen by an effective provider within one week of spasms onset. The median time from spasms onset to the first visit with an effective provider was 24.5 days. In sequential univariate analyses, parental sociodemographic attributes (eg, race, ethnicity, religion, household income, education level, type of healthcare insurance, and distance from patients’ home to the tertiary center) did not predict time to first effective provider. In open-ended discussions, numerous parents reported that their suspicions that “something was wrong” often had been discounted by pediatricians, emergency room physicians, and neurologists. In a qualitative analysis, many parents reported self-diagnosis using Internet resources and self-referral after various diagnostic difficulties and false reassurance by health care providers.
HOUSTON—Children with infantile spasms commonly endure substantial delays in diagnosis and treatment, according to research presented at the 70th Annual Meeting of the American Epilepsy Society. “A simple lack of awareness of infantile spasms among healthcare providers may be responsible for potentially catastrophic delays in diagnosis and treatment,” said Shaun Hussain, MD, Director of the University of California, Los Angeles Infantile Spasms Program, and colleagues. “There is a desperate need for effective interventions to increase basic familiarity with infantile spasms among healthcare providers.”
Dr. Hussain and his colleagues performed a study to measure delays in diagnosis and treatment of infantile spasms and identify barriers to optimal care. The researchers retrospectively identified children with video-EEG-confirmed infantile spasms in a clinical database.
When the children’s parents presented for follow-up, they were surveyed about their experiences with diagnosis and treatment. The investigators asked about medical and sociodemographic factors that could affect the care of infantile spasms. Specifically, the researchers determined the dates of infantile spasms onset, first visit with any healthcare provider, first visit with any neurologist, and first visit with an effective provider. Dr. Hussain and colleagues defined an effective provider as a healthcare provider who identified infantile spasms and prescribed a first-line treatment (ie, ACTH, corticosteroids, vigabatrin, or surgical resection). The investigators reviewed medical records to corroborate parents’ survey responses. They calculated the time to first effective provider using Cox proportional hazards regression.
The parents of 100 children with previous or ongoing infantile spasms were included in the study. Approximately 29% of patients were seen by an effective provider within one week of spasms onset. The median time from spasms onset to the first visit with an effective provider was 24.5 days. In sequential univariate analyses, parental sociodemographic attributes (eg, race, ethnicity, religion, household income, education level, type of healthcare insurance, and distance from patients’ home to the tertiary center) did not predict time to first effective provider. In open-ended discussions, numerous parents reported that their suspicions that “something was wrong” often had been discounted by pediatricians, emergency room physicians, and neurologists. In a qualitative analysis, many parents reported self-diagnosis using Internet resources and self-referral after various diagnostic difficulties and false reassurance by health care providers.
HOUSTON—Children with infantile spasms commonly endure substantial delays in diagnosis and treatment, according to research presented at the 70th Annual Meeting of the American Epilepsy Society. “A simple lack of awareness of infantile spasms among healthcare providers may be responsible for potentially catastrophic delays in diagnosis and treatment,” said Shaun Hussain, MD, Director of the University of California, Los Angeles Infantile Spasms Program, and colleagues. “There is a desperate need for effective interventions to increase basic familiarity with infantile spasms among healthcare providers.”
Dr. Hussain and his colleagues performed a study to measure delays in diagnosis and treatment of infantile spasms and identify barriers to optimal care. The researchers retrospectively identified children with video-EEG-confirmed infantile spasms in a clinical database.
When the children’s parents presented for follow-up, they were surveyed about their experiences with diagnosis and treatment. The investigators asked about medical and sociodemographic factors that could affect the care of infantile spasms. Specifically, the researchers determined the dates of infantile spasms onset, first visit with any healthcare provider, first visit with any neurologist, and first visit with an effective provider. Dr. Hussain and colleagues defined an effective provider as a healthcare provider who identified infantile spasms and prescribed a first-line treatment (ie, ACTH, corticosteroids, vigabatrin, or surgical resection). The investigators reviewed medical records to corroborate parents’ survey responses. They calculated the time to first effective provider using Cox proportional hazards regression.
The parents of 100 children with previous or ongoing infantile spasms were included in the study. Approximately 29% of patients were seen by an effective provider within one week of spasms onset. The median time from spasms onset to the first visit with an effective provider was 24.5 days. In sequential univariate analyses, parental sociodemographic attributes (eg, race, ethnicity, religion, household income, education level, type of healthcare insurance, and distance from patients’ home to the tertiary center) did not predict time to first effective provider. In open-ended discussions, numerous parents reported that their suspicions that “something was wrong” often had been discounted by pediatricians, emergency room physicians, and neurologists. In a qualitative analysis, many parents reported self-diagnosis using Internet resources and self-referral after various diagnostic difficulties and false reassurance by health care providers.

Recognizing Congenital Zika Syndrome
VANCOUVER—Infants infected with Zika virus in utero may develop a syndrome characterized by brain volume loss, intracerebral calcifications, and spasticity. They may develop dyskinesia or seizures after several months, and a subset of children has severe arthrogryposis.
And although microcephaly at birth is common, infants may have a normal head size at birth, but develop postnatal microcephaly or other neurologic symptoms at six months, according to research described at the 45th Annual Meeting of the Child Neurology Society.
“The spectrum of congenital Zika syndrome is expanding as we come to understand it better,” said William B. Dobyns, MD, Professor of Pediatrics at the University of Washington in Seattle and a faculty member at the Center for Integrative Brain Research at Seattle Children’s Research Institute. “We all need to stop calling this microcephaly. This is much more than that. This is the congenital Zika syndrome.”

Zika virus is trophic for neural stem cells, and the first reports of microcephaly associated with prenatal Zika virus infection came from Brazil in January 2016. In the US, mosquitoes that transmit Zika virus, Aedes aegypti and albopictus, are present year round in Florida and seasonally in about a quarter of the states. “It is pretty clear that it will be coming.… We need to take precautions until treatments or preventives are available,” he said. In addition, child neurologists need to be able to recognize congenital Zika syndrome. “It is entirely possible for us to do so,” Dr. Dobyns said. “You do not even need viral titers in the more classically affected children.”
A Case Series of 57 Children
Dr. Dobyns worked with André Pessoa, MD, a child neurologist at Hospital Infantil Albert Sabin in Fortaleza, Brazil, and other neurologists in the region to compile data on a series of 57 children with congenital microcephaly and presumed or proven Zika exposure of the mothers during pregnancy. Microcephaly was defined as occipitofrontal head circumference of at least two standard deviations below the mean.
About half of the children had a bony protuberance of the occipital bone, known as an occipital shelf, Dr. Dobyns said. This feature occurs when the fetal brain, instead of growing and pushing out the skull plates, is severely injured and shrinks. The frontal and parietal bones, but not the occipital bone, collapse over the injured brain.
Almost all of the children had prominent calcifications in the brain. Unlike in children infected with cytomegalovirus, periventricular calcifications are the exception in children infected with Zika virus. Researchers observed subcortical or cortical calcifications on CT in 51 of the 57 children infected with Zika virus and basal ganglia calcifications in 33 of the 57 children.
Furthermore, calcifications with Zika virus infection tend to be diffuse and bilateral, whereas calcifications with cytomegalovirus infection tend to be patchy, Dr. Dobyns said.
All patients had the same general pattern of enlarged extra-axial space, ventriculomegaly, or both, indicating brain volume loss.
About 20% of patients had severe arthrogryposis multiplex congenita, and all of these children had abnormally positioned proximal joints.
Twenty of the children underwent brain MRI. MRI showed an abnormal cortex in all 20 children. The patients appear to have a diffuse cortical malformation that is most consistent with polymicrogyria, Dr. Dobyns said.
Nearly 20% of children in the series had microcephaly between two and three standard deviations below the mean. But 81% had microcephaly of three or more standard deviations below the mean. The mean occipitofrontal head circumference was four standard deviations below the mean.
Neurologic features included spasticity in 94% of the children and severe irritability or tremor in 64% of the children. About 20% had seizures after several months. Some patients had eye abnormalities, including optic nerve pallor, macular atrophy, and strabismus.
“The exam is characteristic,” Dr. Dobyns said. “They all develop a dyskinesia later in the first year of life. They have spastic quadriparesis. They frequently have tremors at birth. They feed poorly. They tend to be irritable and scream all the time. They are starting to have seizures as they get past six months of age.”
As in other studies, data from this series suggest that children whose mothers have a symptomatic illness or are infected earlier in pregnancy may be at higher risk of congenital Zika syndrome.
Infants Without Microcephaly at Birth
Dr. Dobyns presented preliminary data from children who were exposed to Zika virus but did not have microcephaly at birth. These children had most of the same features on exam as children with microcephaly, although the features tended to be less severe. The children started to have seizures after several months. When their head size was measured at six months or older, it fell below the second percentile, meaning that these children had postnatal microcephaly. The children did not have congenital contractures, Dr. Dobyns said.
Vanessa van der Linden, MD, a pediatric neurologist at the Association for Assistance of Disabled Children in Recife, Brazil, Dr. Pessoa, Dr. Dobyns, and colleagues on November 22, 2016, published a description of 13 infants who had evidence of congenital Zika infection but did not have microcephaly at birth. Their report was published online in the CDC’s Morbidity and Mortality Weekly Report. The researchers found that head growth decelerated in all 13 of the infants by as early as age 5 months, and 11 of the infants had microcephaly. The findings suggest that infants exposed to Zika virus prenatally should receive comprehensive medical and developmental follow-up, even in the absence of microcephaly at birth, the investigators said.
That infants with prenatal Zika infection may develop postnatal microcephaly is not surprising, Dr. Dobyns said. Microcephaly, however, remains only one possible symptom of congenital Zika syndrome. “It is a pattern of features seen clinically, on CT scans, and behaviorally that will mark this syndrome,” he said.
—Jake Remaly
Suggested Reading
Moore CA, Staples JE, Dobyns WB, et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2016 Nov 3 [Epub ahead of print].
van der Linden V, Pessoa A, Dobyns W, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb Mortal Wkly Rep. 22 Nov 2016 [Epub ahead of print].
VANCOUVER—Infants infected with Zika virus in utero may develop a syndrome characterized by brain volume loss, intracerebral calcifications, and spasticity. They may develop dyskinesia or seizures after several months, and a subset of children has severe arthrogryposis.
And although microcephaly at birth is common, infants may have a normal head size at birth, but develop postnatal microcephaly or other neurologic symptoms at six months, according to research described at the 45th Annual Meeting of the Child Neurology Society.
“The spectrum of congenital Zika syndrome is expanding as we come to understand it better,” said William B. Dobyns, MD, Professor of Pediatrics at the University of Washington in Seattle and a faculty member at the Center for Integrative Brain Research at Seattle Children’s Research Institute. “We all need to stop calling this microcephaly. This is much more than that. This is the congenital Zika syndrome.”
Zika virus is trophic for neural stem cells, and the first reports of microcephaly associated with prenatal Zika virus infection came from Brazil in January 2016. In the US, mosquitoes that transmit Zika virus, Aedes aegypti and albopictus, are present year round in Florida and seasonally in about a quarter of the states. “It is pretty clear that it will be coming.… We need to take precautions until treatments or preventives are available,” he said. In addition, child neurologists need to be able to recognize congenital Zika syndrome. “It is entirely possible for us to do so,” Dr. Dobyns said. “You do not even need viral titers in the more classically affected children.”
A Case Series of 57 Children
Dr. Dobyns worked with André Pessoa, MD, a child neurologist at Hospital Infantil Albert Sabin in Fortaleza, Brazil, and other neurologists in the region to compile data on a series of 57 children with congenital microcephaly and presumed or proven Zika exposure of the mothers during pregnancy. Microcephaly was defined as occipitofrontal head circumference of at least two standard deviations below the mean.
About half of the children had a bony protuberance of the occipital bone, known as an occipital shelf, Dr. Dobyns said. This feature occurs when the fetal brain, instead of growing and pushing out the skull plates, is severely injured and shrinks. The frontal and parietal bones, but not the occipital bone, collapse over the injured brain.
Almost all of the children had prominent calcifications in the brain. Unlike in children infected with cytomegalovirus, periventricular calcifications are the exception in children infected with Zika virus. Researchers observed subcortical or cortical calcifications on CT in 51 of the 57 children infected with Zika virus and basal ganglia calcifications in 33 of the 57 children.
Furthermore, calcifications with Zika virus infection tend to be diffuse and bilateral, whereas calcifications with cytomegalovirus infection tend to be patchy, Dr. Dobyns said.
All patients had the same general pattern of enlarged extra-axial space, ventriculomegaly, or both, indicating brain volume loss.
About 20% of patients had severe arthrogryposis multiplex congenita, and all of these children had abnormally positioned proximal joints.
Twenty of the children underwent brain MRI. MRI showed an abnormal cortex in all 20 children. The patients appear to have a diffuse cortical malformation that is most consistent with polymicrogyria, Dr. Dobyns said.
Nearly 20% of children in the series had microcephaly between two and three standard deviations below the mean. But 81% had microcephaly of three or more standard deviations below the mean. The mean occipitofrontal head circumference was four standard deviations below the mean.
Neurologic features included spasticity in 94% of the children and severe irritability or tremor in 64% of the children. About 20% had seizures after several months. Some patients had eye abnormalities, including optic nerve pallor, macular atrophy, and strabismus.
“The exam is characteristic,” Dr. Dobyns said. “They all develop a dyskinesia later in the first year of life. They have spastic quadriparesis. They frequently have tremors at birth. They feed poorly. They tend to be irritable and scream all the time. They are starting to have seizures as they get past six months of age.”
As in other studies, data from this series suggest that children whose mothers have a symptomatic illness or are infected earlier in pregnancy may be at higher risk of congenital Zika syndrome.
Infants Without Microcephaly at Birth
Dr. Dobyns presented preliminary data from children who were exposed to Zika virus but did not have microcephaly at birth. These children had most of the same features on exam as children with microcephaly, although the features tended to be less severe. The children started to have seizures after several months. When their head size was measured at six months or older, it fell below the second percentile, meaning that these children had postnatal microcephaly. The children did not have congenital contractures, Dr. Dobyns said.
Vanessa van der Linden, MD, a pediatric neurologist at the Association for Assistance of Disabled Children in Recife, Brazil, Dr. Pessoa, Dr. Dobyns, and colleagues on November 22, 2016, published a description of 13 infants who had evidence of congenital Zika infection but did not have microcephaly at birth. Their report was published online in the CDC’s Morbidity and Mortality Weekly Report. The researchers found that head growth decelerated in all 13 of the infants by as early as age 5 months, and 11 of the infants had microcephaly. The findings suggest that infants exposed to Zika virus prenatally should receive comprehensive medical and developmental follow-up, even in the absence of microcephaly at birth, the investigators said.
That infants with prenatal Zika infection may develop postnatal microcephaly is not surprising, Dr. Dobyns said. Microcephaly, however, remains only one possible symptom of congenital Zika syndrome. “It is a pattern of features seen clinically, on CT scans, and behaviorally that will mark this syndrome,” he said.
—Jake Remaly
Suggested Reading
Moore CA, Staples JE, Dobyns WB, et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2016 Nov 3 [Epub ahead of print].
van der Linden V, Pessoa A, Dobyns W, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb Mortal Wkly Rep. 22 Nov 2016 [Epub ahead of print].
VANCOUVER—Infants infected with Zika virus in utero may develop a syndrome characterized by brain volume loss, intracerebral calcifications, and spasticity. They may develop dyskinesia or seizures after several months, and a subset of children has severe arthrogryposis.
And although microcephaly at birth is common, infants may have a normal head size at birth, but develop postnatal microcephaly or other neurologic symptoms at six months, according to research described at the 45th Annual Meeting of the Child Neurology Society.
“The spectrum of congenital Zika syndrome is expanding as we come to understand it better,” said William B. Dobyns, MD, Professor of Pediatrics at the University of Washington in Seattle and a faculty member at the Center for Integrative Brain Research at Seattle Children’s Research Institute. “We all need to stop calling this microcephaly. This is much more than that. This is the congenital Zika syndrome.”
Zika virus is trophic for neural stem cells, and the first reports of microcephaly associated with prenatal Zika virus infection came from Brazil in January 2016. In the US, mosquitoes that transmit Zika virus, Aedes aegypti and albopictus, are present year round in Florida and seasonally in about a quarter of the states. “It is pretty clear that it will be coming.… We need to take precautions until treatments or preventives are available,” he said. In addition, child neurologists need to be able to recognize congenital Zika syndrome. “It is entirely possible for us to do so,” Dr. Dobyns said. “You do not even need viral titers in the more classically affected children.”
A Case Series of 57 Children
Dr. Dobyns worked with André Pessoa, MD, a child neurologist at Hospital Infantil Albert Sabin in Fortaleza, Brazil, and other neurologists in the region to compile data on a series of 57 children with congenital microcephaly and presumed or proven Zika exposure of the mothers during pregnancy. Microcephaly was defined as occipitofrontal head circumference of at least two standard deviations below the mean.
About half of the children had a bony protuberance of the occipital bone, known as an occipital shelf, Dr. Dobyns said. This feature occurs when the fetal brain, instead of growing and pushing out the skull plates, is severely injured and shrinks. The frontal and parietal bones, but not the occipital bone, collapse over the injured brain.
Almost all of the children had prominent calcifications in the brain. Unlike in children infected with cytomegalovirus, periventricular calcifications are the exception in children infected with Zika virus. Researchers observed subcortical or cortical calcifications on CT in 51 of the 57 children infected with Zika virus and basal ganglia calcifications in 33 of the 57 children.
Furthermore, calcifications with Zika virus infection tend to be diffuse and bilateral, whereas calcifications with cytomegalovirus infection tend to be patchy, Dr. Dobyns said.
All patients had the same general pattern of enlarged extra-axial space, ventriculomegaly, or both, indicating brain volume loss.
About 20% of patients had severe arthrogryposis multiplex congenita, and all of these children had abnormally positioned proximal joints.
Twenty of the children underwent brain MRI. MRI showed an abnormal cortex in all 20 children. The patients appear to have a diffuse cortical malformation that is most consistent with polymicrogyria, Dr. Dobyns said.
Nearly 20% of children in the series had microcephaly between two and three standard deviations below the mean. But 81% had microcephaly of three or more standard deviations below the mean. The mean occipitofrontal head circumference was four standard deviations below the mean.
Neurologic features included spasticity in 94% of the children and severe irritability or tremor in 64% of the children. About 20% had seizures after several months. Some patients had eye abnormalities, including optic nerve pallor, macular atrophy, and strabismus.
“The exam is characteristic,” Dr. Dobyns said. “They all develop a dyskinesia later in the first year of life. They have spastic quadriparesis. They frequently have tremors at birth. They feed poorly. They tend to be irritable and scream all the time. They are starting to have seizures as they get past six months of age.”
As in other studies, data from this series suggest that children whose mothers have a symptomatic illness or are infected earlier in pregnancy may be at higher risk of congenital Zika syndrome.
Infants Without Microcephaly at Birth
Dr. Dobyns presented preliminary data from children who were exposed to Zika virus but did not have microcephaly at birth. These children had most of the same features on exam as children with microcephaly, although the features tended to be less severe. The children started to have seizures after several months. When their head size was measured at six months or older, it fell below the second percentile, meaning that these children had postnatal microcephaly. The children did not have congenital contractures, Dr. Dobyns said.
Vanessa van der Linden, MD, a pediatric neurologist at the Association for Assistance of Disabled Children in Recife, Brazil, Dr. Pessoa, Dr. Dobyns, and colleagues on November 22, 2016, published a description of 13 infants who had evidence of congenital Zika infection but did not have microcephaly at birth. Their report was published online in the CDC’s Morbidity and Mortality Weekly Report. The researchers found that head growth decelerated in all 13 of the infants by as early as age 5 months, and 11 of the infants had microcephaly. The findings suggest that infants exposed to Zika virus prenatally should receive comprehensive medical and developmental follow-up, even in the absence of microcephaly at birth, the investigators said.
That infants with prenatal Zika infection may develop postnatal microcephaly is not surprising, Dr. Dobyns said. Microcephaly, however, remains only one possible symptom of congenital Zika syndrome. “It is a pattern of features seen clinically, on CT scans, and behaviorally that will mark this syndrome,” he said.
—Jake Remaly
Suggested Reading
Moore CA, Staples JE, Dobyns WB, et al. Characterizing the pattern of anomalies in congenital Zika syndrome for pediatric clinicians. JAMA Pediatr. 2016 Nov 3 [Epub ahead of print].
van der Linden V, Pessoa A, Dobyns W, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb Mortal Wkly Rep. 22 Nov 2016 [Epub ahead of print].

Pleth Variability Index shows promise for asthma assessments
Clinical question: Does pulse variability on plethysmography, or the Pleth Variability Index (PVI), correlate with disease severity in obstructive airway disease in children?
Background: Asthma is the most common reason for hospitalization in the United S. for children 3-12 years old. Asthma accounts for a quarter of ED visits for children aged 1-9 years old.1 Although systems have been developed to assess asthma exacerbation severity and the need for hospitalization, many of these depend on reassessments over time or have been proven to be invalid in larger studies.2,3,4 Pulsus paradoxus (PP), which is defined as a drop in systolic blood pressure greater than 10 mm Hg, correlates with the severity of obstruction in asthma exacerbations, but it is not practical in the children being evaluated in the ED or hospital.5,6 PP measurement using plethysmography has been found to correlate with measurement by sphygmomanometry.7 Furthermore, PVI, which is derived from amplitude variability in the pulse oximeter waveform, has been found to correlate with fluid responsiveness in mechanically ventilated patients. To this date, no study has assessed the correlation between PVI and exacerbation severity in asthma.

Setting: A 137-bed, tertiary-care children’s hospital.
Synopsis: Over a 6-month period on weekdays, researchers enrolled patients aged 1-18 years evaluated in the ED for asthma exacerbations or reactive airway disease. ED staff diagnosed patients clinically, and other patients with conditions known to affect PP – such as dehydration, croup, and cardiac disease – were excluded. PVI was calculated by measuring the minimum perfusion index (PImin) and the maximum perfusion index (PImax) using the following formula:
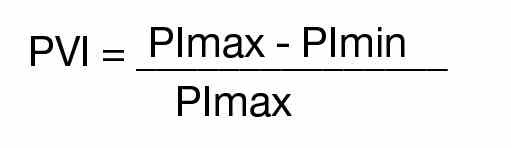
A printout of the first ED pulse oximetry reading was used to obtain the PImax and PImin as below:
Researchers followed patients after the initial evaluation to determine disposition from the ED, which included either discharge to home, admission to a general pediatrics floor, or admission to the PICU. The hospital utilized specific criteria for disposition from the ED (see Table 1).
Of the 117 patients who were analyzed after application of exclusion criteria, 48 were discharged to home, 61 were admitted to a general pediatrics floor, and eight were admitted to the PICU. The three groups were found to be demographically similar. Researchers found a significant difference between the PVI of the three groups, but pairwise analysis showed no significant difference between the PVI of patients admitted to the general pediatrics floor versus discharged to home (see Table 2).
Bottom line: PVI shows promise as a tool to rapidly assess disease severity in pediatric patients being evaluated and treated for asthma, but further studies are needed to validate this in the ED and hospital setting.
Citation: Brandwein A, Patel K, Kline M, Silver P, Gangadharan S. Using pleth variability as a triage tool for children with obstructive airway disease in a pediatric emergency department [published online ahead of print Oct. 6, 2016]. Pediatr Emerg Care. doi: 10.1097/PEC.0000000000000887.
References
1. Care of children and adolescents in U.S. hospitals. Agency for Healthcare Research and Quality website. Available at: https://archive.ahrq.gov/data/hcup/factbk4/factbk4.htm. Accessed Nov. 18, 2016.
2. Kelly AM, Kerr D, Powell C. Is severity assessment after one hour of treatment better for predicting the need for admission in acute asthma? Respir Med. 2004;98(8):777-781.
3. Keogh KA, Macarthur C, Parkin PC, et al. Predictors of hospitalization in children with acute asthma. J Pediatr. 2001;139(2):273-277.
4. Keahey L, Bulloch B, Becker AB, et al. Initial oxygen saturation as a predictor of admission in children presenting to the emergency department with acute asthma. Ann Emerg Med. 2002;40(3):300-307.
5. Guntheroth WG, Morgan BC, Mullins GL. Effect of respiration on venous return and stroke volume in cardiac tamponade. Mechanism of pulsus paradoxus. Circ Res. 1967;20(4):381-390.
6. Frey B, Freezer N. Diagnostic value and pathophysiologic basis of pulsus paradoxus in infants and children with respiratory disease. Pediatr Pulmonol. 2001;31(2):138-143.
7. Clark JA, Lieh-Lai M, Thomas R, Raghavan K, Sarnaik AP. Comparison of traditional and plethysmographic methods for measuring pulsus paradoxus. Arch Pediatr Adolesc Med. 2004;158(1):48-51.
Clinical question: Does pulse variability on plethysmography, or the Pleth Variability Index (PVI), correlate with disease severity in obstructive airway disease in children?
Background: Asthma is the most common reason for hospitalization in the United S. for children 3-12 years old. Asthma accounts for a quarter of ED visits for children aged 1-9 years old.1 Although systems have been developed to assess asthma exacerbation severity and the need for hospitalization, many of these depend on reassessments over time or have been proven to be invalid in larger studies.2,3,4 Pulsus paradoxus (PP), which is defined as a drop in systolic blood pressure greater than 10 mm Hg, correlates with the severity of obstruction in asthma exacerbations, but it is not practical in the children being evaluated in the ED or hospital.5,6 PP measurement using plethysmography has been found to correlate with measurement by sphygmomanometry.7 Furthermore, PVI, which is derived from amplitude variability in the pulse oximeter waveform, has been found to correlate with fluid responsiveness in mechanically ventilated patients. To this date, no study has assessed the correlation between PVI and exacerbation severity in asthma.
Setting: A 137-bed, tertiary-care children’s hospital.
Synopsis: Over a 6-month period on weekdays, researchers enrolled patients aged 1-18 years evaluated in the ED for asthma exacerbations or reactive airway disease. ED staff diagnosed patients clinically, and other patients with conditions known to affect PP – such as dehydration, croup, and cardiac disease – were excluded. PVI was calculated by measuring the minimum perfusion index (PImin) and the maximum perfusion index (PImax) using the following formula:
A printout of the first ED pulse oximetry reading was used to obtain the PImax and PImin as below:
Researchers followed patients after the initial evaluation to determine disposition from the ED, which included either discharge to home, admission to a general pediatrics floor, or admission to the PICU. The hospital utilized specific criteria for disposition from the ED (see Table 1).
Of the 117 patients who were analyzed after application of exclusion criteria, 48 were discharged to home, 61 were admitted to a general pediatrics floor, and eight were admitted to the PICU. The three groups were found to be demographically similar. Researchers found a significant difference between the PVI of the three groups, but pairwise analysis showed no significant difference between the PVI of patients admitted to the general pediatrics floor versus discharged to home (see Table 2).
Bottom line: PVI shows promise as a tool to rapidly assess disease severity in pediatric patients being evaluated and treated for asthma, but further studies are needed to validate this in the ED and hospital setting.
Citation: Brandwein A, Patel K, Kline M, Silver P, Gangadharan S. Using pleth variability as a triage tool for children with obstructive airway disease in a pediatric emergency department [published online ahead of print Oct. 6, 2016]. Pediatr Emerg Care. doi: 10.1097/PEC.0000000000000887.
References
1. Care of children and adolescents in U.S. hospitals. Agency for Healthcare Research and Quality website. Available at: https://archive.ahrq.gov/data/hcup/factbk4/factbk4.htm. Accessed Nov. 18, 2016.
2. Kelly AM, Kerr D, Powell C. Is severity assessment after one hour of treatment better for predicting the need for admission in acute asthma? Respir Med. 2004;98(8):777-781.
3. Keogh KA, Macarthur C, Parkin PC, et al. Predictors of hospitalization in children with acute asthma. J Pediatr. 2001;139(2):273-277.
4. Keahey L, Bulloch B, Becker AB, et al. Initial oxygen saturation as a predictor of admission in children presenting to the emergency department with acute asthma. Ann Emerg Med. 2002;40(3):300-307.
5. Guntheroth WG, Morgan BC, Mullins GL. Effect of respiration on venous return and stroke volume in cardiac tamponade. Mechanism of pulsus paradoxus. Circ Res. 1967;20(4):381-390.
6. Frey B, Freezer N. Diagnostic value and pathophysiologic basis of pulsus paradoxus in infants and children with respiratory disease. Pediatr Pulmonol. 2001;31(2):138-143.
7. Clark JA, Lieh-Lai M, Thomas R, Raghavan K, Sarnaik AP. Comparison of traditional and plethysmographic methods for measuring pulsus paradoxus. Arch Pediatr Adolesc Med. 2004;158(1):48-51.
Clinical question: Does pulse variability on plethysmography, or the Pleth Variability Index (PVI), correlate with disease severity in obstructive airway disease in children?
Background: Asthma is the most common reason for hospitalization in the United S. for children 3-12 years old. Asthma accounts for a quarter of ED visits for children aged 1-9 years old.1 Although systems have been developed to assess asthma exacerbation severity and the need for hospitalization, many of these depend on reassessments over time or have been proven to be invalid in larger studies.2,3,4 Pulsus paradoxus (PP), which is defined as a drop in systolic blood pressure greater than 10 mm Hg, correlates with the severity of obstruction in asthma exacerbations, but it is not practical in the children being evaluated in the ED or hospital.5,6 PP measurement using plethysmography has been found to correlate with measurement by sphygmomanometry.7 Furthermore, PVI, which is derived from amplitude variability in the pulse oximeter waveform, has been found to correlate with fluid responsiveness in mechanically ventilated patients. To this date, no study has assessed the correlation between PVI and exacerbation severity in asthma.
Setting: A 137-bed, tertiary-care children’s hospital.
Synopsis: Over a 6-month period on weekdays, researchers enrolled patients aged 1-18 years evaluated in the ED for asthma exacerbations or reactive airway disease. ED staff diagnosed patients clinically, and other patients with conditions known to affect PP – such as dehydration, croup, and cardiac disease – were excluded. PVI was calculated by measuring the minimum perfusion index (PImin) and the maximum perfusion index (PImax) using the following formula:
A printout of the first ED pulse oximetry reading was used to obtain the PImax and PImin as below:
Researchers followed patients after the initial evaluation to determine disposition from the ED, which included either discharge to home, admission to a general pediatrics floor, or admission to the PICU. The hospital utilized specific criteria for disposition from the ED (see Table 1).
Of the 117 patients who were analyzed after application of exclusion criteria, 48 were discharged to home, 61 were admitted to a general pediatrics floor, and eight were admitted to the PICU. The three groups were found to be demographically similar. Researchers found a significant difference between the PVI of the three groups, but pairwise analysis showed no significant difference between the PVI of patients admitted to the general pediatrics floor versus discharged to home (see Table 2).
Bottom line: PVI shows promise as a tool to rapidly assess disease severity in pediatric patients being evaluated and treated for asthma, but further studies are needed to validate this in the ED and hospital setting.
Citation: Brandwein A, Patel K, Kline M, Silver P, Gangadharan S. Using pleth variability as a triage tool for children with obstructive airway disease in a pediatric emergency department [published online ahead of print Oct. 6, 2016]. Pediatr Emerg Care. doi: 10.1097/PEC.0000000000000887.
References
1. Care of children and adolescents in U.S. hospitals. Agency for Healthcare Research and Quality website. Available at: https://archive.ahrq.gov/data/hcup/factbk4/factbk4.htm. Accessed Nov. 18, 2016.
2. Kelly AM, Kerr D, Powell C. Is severity assessment after one hour of treatment better for predicting the need for admission in acute asthma? Respir Med. 2004;98(8):777-781.
3. Keogh KA, Macarthur C, Parkin PC, et al. Predictors of hospitalization in children with acute asthma. J Pediatr. 2001;139(2):273-277.
4. Keahey L, Bulloch B, Becker AB, et al. Initial oxygen saturation as a predictor of admission in children presenting to the emergency department with acute asthma. Ann Emerg Med. 2002;40(3):300-307.
5. Guntheroth WG, Morgan BC, Mullins GL. Effect of respiration on venous return and stroke volume in cardiac tamponade. Mechanism of pulsus paradoxus. Circ Res. 1967;20(4):381-390.
6. Frey B, Freezer N. Diagnostic value and pathophysiologic basis of pulsus paradoxus in infants and children with respiratory disease. Pediatr Pulmonol. 2001;31(2):138-143.
7. Clark JA, Lieh-Lai M, Thomas R, Raghavan K, Sarnaik AP. Comparison of traditional and plethysmographic methods for measuring pulsus paradoxus. Arch Pediatr Adolesc Med. 2004;158(1):48-51.