User login
Sicker COPD patients may be more likely to initiate arformoterol
according to study to identify predictors of its use. In addition to being sicker, being treated by a pulmonologist rather than a primary care physician and being white were factors that increased a patient’s likelihood of receiving nebulized arformoterol.

Patients less likely to receive the nebulized version of this long-acting beta2 adrenoreceptor agonist (LABA) were African Americans, patients with psychiatric comorbidities, and patients eligible for both Medicare and Medicaid.
“Studies have shown that 40% to 71% of Medicare beneficiaries receive no maintenance treatment for COPD. Although a recent longitudinal study on Medicare populations reported that use of maintenance medications has been improving, in general, it is recognized that Medicare beneficiaries with COPD remain undertreated,” Todd P. Gilmer, PhD, from the department of family medicine and public health at the University of California, San Diego, and colleagues wrote.
The investigators identified patients with COPD using Medicare administrative data; of these patients, 11,887 were arformoterol users, and 450,178 were control patients who did not use arformoterol. Patients were included in the study if they had at least one claim for COPD medication and were continuously enrolled in Medicare Parts A, B, and D. The cohort consisted of mostly white women aged 70 years or older, and 47% were dual-eligible to receive both Medicare and Medicaid benefits. A subgroup of 1,778 arformoterol users were also identified for analysis who were hospitalized and discharged within 30 days of using arformoterol, as well as a subgroup of 21,910 control patients with hospitalizations.
The researchers found COPD-related hospitalization (odds ratio, 1.31; 95% confidence interval, 1.24-1.39; P less than .001), exacerbation (OR, 1.33; 95% CI, 1.26-1.41; P less than .001), use of a systemic corticosteroid (OR, 1.50; 95% CI, 1.43-1.57; P less than .001) or methylxanthine (OR, 1.37; 95% CI, 1.28-1.47; P less than .001), use of oxygen therapy (OR, 2.01; 95% CI, 1.93-2.09; P less than .001), pulmonologist care (OR, 1.40; 95% CI, 1.34-1.46; P less than .001), and respiratory therapist care (OR, 1.23; 95% CI, 1.11-1.36; P less than .001) strongly predicted arformoterol use, while racial/ethnic minority status, psychiatric comorbidity (OR, 0.65; 95% CI, 0.56-0.76; P less than .001), acquired immune deficiency syndrome (OR, 0.69; 95% CI, 0.52-0.94; P less than .01), and dual-eligibility for Medicare and Medicaid (OR, 0.73; 95% CI, 0.70-0.77; P less than .001) lowered the odds of arformoterol use (P less than .001). In the subgroup of patients with hospitalizations, COPD-related admission (OR, 1.83; 95% CI, 1.55-2.14; P less than .001), exacerbation (OR, 2.62; 95% CI, 1.88-3.63; P less than .001)m and inpatient care from a pulmonologist (OR, 1.78; 95% CI, 1.58-2.01; P less than .001) predicted arformoterol use.
“Given the results of this study, increasing access to nebulized maintenance therapy is warranted for select populations with COPD including racial/ethnic minorities, the dual-eligible, and those with certain comorbidities, such as psychiatric disorders,” Dr. Gilmer and colleagues wrote in their study. “Future studies are needed to explore the optimal time to initiate nebulized maintenance therapy, and the potential differential impact of early versus late initiation on patient outcomes.”
Researchers noted that, although their results may seem initially counterintuitive given that COPD has a higher prevalence in men, 56% of the beneficiaries in their Medicare data were women who were 65 years or older, and the results are consistent with other studies that show similar gender distribution findings for maintenance treatment patterns among COPD patients receiving Medicare.
“Since most Medicare beneficiaries with COPD are older than 70 years of age, the higher percentage of women than men in our two cohorts can be explained by the age distributions that ensued as a result of applying our various inclusion and exclusion criteria,” they said.
This study was funded by Sunovian. Dr. Gilmer and one coauthor are paid employees of University of California San Diego, which receives research funding from Advance Health Solutions. Another coauthor is an advisory board member for Advance Health Solutions and a consultant for GlaxoSmithKline, Boehringer-Ingelheim, Astra Zeneca, Novartis, and Pulmonix. Two other coauthors are paid employees of Advance Health Solutions, and another is a paid employee of Sunovion.
SOURCE: Gilmer TP et al. COPD. 2019 Jun 19. doi: 10.1080/15412555.2019.1618256.
according to study to identify predictors of its use. In addition to being sicker, being treated by a pulmonologist rather than a primary care physician and being white were factors that increased a patient’s likelihood of receiving nebulized arformoterol.
Patients less likely to receive the nebulized version of this long-acting beta2 adrenoreceptor agonist (LABA) were African Americans, patients with psychiatric comorbidities, and patients eligible for both Medicare and Medicaid.
“Studies have shown that 40% to 71% of Medicare beneficiaries receive no maintenance treatment for COPD. Although a recent longitudinal study on Medicare populations reported that use of maintenance medications has been improving, in general, it is recognized that Medicare beneficiaries with COPD remain undertreated,” Todd P. Gilmer, PhD, from the department of family medicine and public health at the University of California, San Diego, and colleagues wrote.
The investigators identified patients with COPD using Medicare administrative data; of these patients, 11,887 were arformoterol users, and 450,178 were control patients who did not use arformoterol. Patients were included in the study if they had at least one claim for COPD medication and were continuously enrolled in Medicare Parts A, B, and D. The cohort consisted of mostly white women aged 70 years or older, and 47% were dual-eligible to receive both Medicare and Medicaid benefits. A subgroup of 1,778 arformoterol users were also identified for analysis who were hospitalized and discharged within 30 days of using arformoterol, as well as a subgroup of 21,910 control patients with hospitalizations.
The researchers found COPD-related hospitalization (odds ratio, 1.31; 95% confidence interval, 1.24-1.39; P less than .001), exacerbation (OR, 1.33; 95% CI, 1.26-1.41; P less than .001), use of a systemic corticosteroid (OR, 1.50; 95% CI, 1.43-1.57; P less than .001) or methylxanthine (OR, 1.37; 95% CI, 1.28-1.47; P less than .001), use of oxygen therapy (OR, 2.01; 95% CI, 1.93-2.09; P less than .001), pulmonologist care (OR, 1.40; 95% CI, 1.34-1.46; P less than .001), and respiratory therapist care (OR, 1.23; 95% CI, 1.11-1.36; P less than .001) strongly predicted arformoterol use, while racial/ethnic minority status, psychiatric comorbidity (OR, 0.65; 95% CI, 0.56-0.76; P less than .001), acquired immune deficiency syndrome (OR, 0.69; 95% CI, 0.52-0.94; P less than .01), and dual-eligibility for Medicare and Medicaid (OR, 0.73; 95% CI, 0.70-0.77; P less than .001) lowered the odds of arformoterol use (P less than .001). In the subgroup of patients with hospitalizations, COPD-related admission (OR, 1.83; 95% CI, 1.55-2.14; P less than .001), exacerbation (OR, 2.62; 95% CI, 1.88-3.63; P less than .001)m and inpatient care from a pulmonologist (OR, 1.78; 95% CI, 1.58-2.01; P less than .001) predicted arformoterol use.
“Given the results of this study, increasing access to nebulized maintenance therapy is warranted for select populations with COPD including racial/ethnic minorities, the dual-eligible, and those with certain comorbidities, such as psychiatric disorders,” Dr. Gilmer and colleagues wrote in their study. “Future studies are needed to explore the optimal time to initiate nebulized maintenance therapy, and the potential differential impact of early versus late initiation on patient outcomes.”
Researchers noted that, although their results may seem initially counterintuitive given that COPD has a higher prevalence in men, 56% of the beneficiaries in their Medicare data were women who were 65 years or older, and the results are consistent with other studies that show similar gender distribution findings for maintenance treatment patterns among COPD patients receiving Medicare.
“Since most Medicare beneficiaries with COPD are older than 70 years of age, the higher percentage of women than men in our two cohorts can be explained by the age distributions that ensued as a result of applying our various inclusion and exclusion criteria,” they said.
This study was funded by Sunovian. Dr. Gilmer and one coauthor are paid employees of University of California San Diego, which receives research funding from Advance Health Solutions. Another coauthor is an advisory board member for Advance Health Solutions and a consultant for GlaxoSmithKline, Boehringer-Ingelheim, Astra Zeneca, Novartis, and Pulmonix. Two other coauthors are paid employees of Advance Health Solutions, and another is a paid employee of Sunovion.
SOURCE: Gilmer TP et al. COPD. 2019 Jun 19. doi: 10.1080/15412555.2019.1618256.
according to study to identify predictors of its use. In addition to being sicker, being treated by a pulmonologist rather than a primary care physician and being white were factors that increased a patient’s likelihood of receiving nebulized arformoterol.
Patients less likely to receive the nebulized version of this long-acting beta2 adrenoreceptor agonist (LABA) were African Americans, patients with psychiatric comorbidities, and patients eligible for both Medicare and Medicaid.
“Studies have shown that 40% to 71% of Medicare beneficiaries receive no maintenance treatment for COPD. Although a recent longitudinal study on Medicare populations reported that use of maintenance medications has been improving, in general, it is recognized that Medicare beneficiaries with COPD remain undertreated,” Todd P. Gilmer, PhD, from the department of family medicine and public health at the University of California, San Diego, and colleagues wrote.
The investigators identified patients with COPD using Medicare administrative data; of these patients, 11,887 were arformoterol users, and 450,178 were control patients who did not use arformoterol. Patients were included in the study if they had at least one claim for COPD medication and were continuously enrolled in Medicare Parts A, B, and D. The cohort consisted of mostly white women aged 70 years or older, and 47% were dual-eligible to receive both Medicare and Medicaid benefits. A subgroup of 1,778 arformoterol users were also identified for analysis who were hospitalized and discharged within 30 days of using arformoterol, as well as a subgroup of 21,910 control patients with hospitalizations.
The researchers found COPD-related hospitalization (odds ratio, 1.31; 95% confidence interval, 1.24-1.39; P less than .001), exacerbation (OR, 1.33; 95% CI, 1.26-1.41; P less than .001), use of a systemic corticosteroid (OR, 1.50; 95% CI, 1.43-1.57; P less than .001) or methylxanthine (OR, 1.37; 95% CI, 1.28-1.47; P less than .001), use of oxygen therapy (OR, 2.01; 95% CI, 1.93-2.09; P less than .001), pulmonologist care (OR, 1.40; 95% CI, 1.34-1.46; P less than .001), and respiratory therapist care (OR, 1.23; 95% CI, 1.11-1.36; P less than .001) strongly predicted arformoterol use, while racial/ethnic minority status, psychiatric comorbidity (OR, 0.65; 95% CI, 0.56-0.76; P less than .001), acquired immune deficiency syndrome (OR, 0.69; 95% CI, 0.52-0.94; P less than .01), and dual-eligibility for Medicare and Medicaid (OR, 0.73; 95% CI, 0.70-0.77; P less than .001) lowered the odds of arformoterol use (P less than .001). In the subgroup of patients with hospitalizations, COPD-related admission (OR, 1.83; 95% CI, 1.55-2.14; P less than .001), exacerbation (OR, 2.62; 95% CI, 1.88-3.63; P less than .001)m and inpatient care from a pulmonologist (OR, 1.78; 95% CI, 1.58-2.01; P less than .001) predicted arformoterol use.
“Given the results of this study, increasing access to nebulized maintenance therapy is warranted for select populations with COPD including racial/ethnic minorities, the dual-eligible, and those with certain comorbidities, such as psychiatric disorders,” Dr. Gilmer and colleagues wrote in their study. “Future studies are needed to explore the optimal time to initiate nebulized maintenance therapy, and the potential differential impact of early versus late initiation on patient outcomes.”
Researchers noted that, although their results may seem initially counterintuitive given that COPD has a higher prevalence in men, 56% of the beneficiaries in their Medicare data were women who were 65 years or older, and the results are consistent with other studies that show similar gender distribution findings for maintenance treatment patterns among COPD patients receiving Medicare.
“Since most Medicare beneficiaries with COPD are older than 70 years of age, the higher percentage of women than men in our two cohorts can be explained by the age distributions that ensued as a result of applying our various inclusion and exclusion criteria,” they said.
This study was funded by Sunovian. Dr. Gilmer and one coauthor are paid employees of University of California San Diego, which receives research funding from Advance Health Solutions. Another coauthor is an advisory board member for Advance Health Solutions and a consultant for GlaxoSmithKline, Boehringer-Ingelheim, Astra Zeneca, Novartis, and Pulmonix. Two other coauthors are paid employees of Advance Health Solutions, and another is a paid employee of Sunovion.
SOURCE: Gilmer TP et al. COPD. 2019 Jun 19. doi: 10.1080/15412555.2019.1618256.
FROM COPD: JOURNAL OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
CNS-directed therapy appears more effective for synDLBCL
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
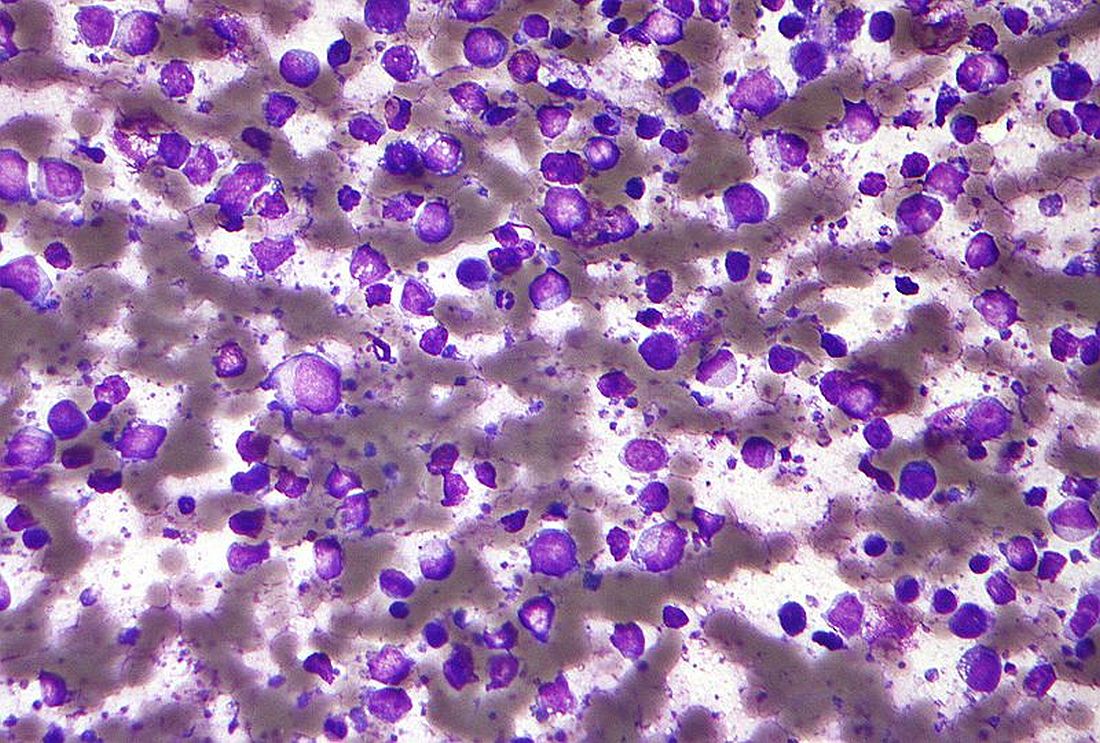
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
Controlling CNS disease is “paramount” in treating diffuse large B-cell lymphoma with synchronous CNS and systemic disease (synDLBCL), according to researchers.
In a retrospective study, the CNS was the most common site of relapse in patients with synDLBCL, and patients had better outcomes when they received CNS-directed therapy.
The 2-year progression-free survival rate was 50% in patients who received CNS-intensive therapy and 31% in those who received CNS-conservative therapy. The 2-year overall survival rate was 54% and 44%, respectively.
Dr. Joel C. Wight, of Austin Health in Heidelberg, Australia, and colleagues conducted this study and recounted their findings in the British Journal of Haematology.
The researchers retrospectively analyzed 80 patients with synDLBCL treated at 10 centers in Australia and the United Kingdom. Patients had DLBCL not otherwise specified (n = 67); high-grade B-cell lymphoma, including double-hit lymphoma (n = 12); or T-cell histiocyte-rich DLBCL (n = 1).
At baseline, all patients were treatment-naive, they had a median age of 64 years (range, 18-87 years), and 68% were male. Seventy percent of patients had high-risk disease according to the CNS International Prognostic Index (IPI), and 96% had non-CNS extranodal disease. The median number of extranodal sites outside the CNS was 2 (range, 0 to more than 10).
Patients were divided into those who received CNS-intensive therapy (n = 38) and those given CNS-conservative therapy (n = 42). The CNS-conservative group was significantly older (P less than .001), significantly more likely to have high-risk disease according to the National Comprehensive Cancer Network IPI (P = .009) or CNS IPI (P = .01) and significantly more likely to have leptomeningeal disease only (P less than .001).
Treatment
CNS-intensive therapy was defined as any established multiagent IV chemotherapy regimen with two or more CNS-penetrating drugs and cytarabine, with or without intrathecal chemotherapy and/or radiotherapy.
CNS-conservative therapy was defined as one or fewer IV CNS-penetrating chemotherapy agents in induction, with or without intrathecal chemotherapy and/or radiotherapy.
Systemic induction in the CNS-intensive group consisted of R-HyperCVAD (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with IV methotrexate and cytarabine) in 66% of patients and R-CODOX-M/IVAC (rituximab, hyperfractionated cyclophosphamide, vincristine, doxorubicin, methotrexate/ifosfamide, etoposide, cytarabine) in 24% of patients.
The most common systemic induction regimens in the CNS-conservative group were R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) or CHOP-like regimens, given to 83% of patients.
CNS-directed IV therapy was given to 100% of the CNS-intensive group and 60% of the CNS-conservative group. This consisted of IV methotrexate plus cytarabine (97%) or MATRix (methotrexate, cytarabine, and thiotepa; 3%) in the CNS-intensive group and high-dose methotrexate in the conservative group.
Intrathecal chemotherapy was given to 97% of the CNS-intensive group and 60% of the CNS-conservative group. CNS-directed radiation was given to 32% and 19%, respectively.
Thirteen patients in the CNS-intensive group and one in the CNS-conservative group underwent autologous transplant as consolidation.
Outcomes
Dose reductions were more frequent in the CNS-conservative group than in the CNS-intensive group, at 48% and 18% (P = .009), as was early cessation of chemotherapy, at 52% and 18% (P = .002). Rates of treatment-related mortality were similar, at 13% in the CNS-intensive group and 12% in the CNS-conservative group.
At the end of induction, the complete response rate was 69% in the CNS-intensive group and 51% in the CNS-conservative group (P = .16). Primary refractory disease was observed in 19% and 38% of patients, respectively (P = .07).
The CNS was the most common site of relapse or progression (n = 28). CNS progression or relapse occurred in 25% of the CNS-intensive group and 49% of the CNS-conservative group (P = .03).
The 2-year progression-free survival rate was 50% for the CNS-intensive group and 31% for the CNS-conservative group (P = .006). The 2-year overall survival rate was 54% and 44%, respectively (P = .037).
When patients were matched for induction outcomes, consolidative transplant did not improve survival.
“The most significant factor affecting survival was the ability to control the CNS disease, which was improved by the addition of IV cytarabine to [high-dose methotrexate],” the researchers wrote.
“Whilst the younger age and more intensive systemic treatment of the CNS-intensive group may have contributed to the improved survival, it is clear that CNS disease control was substantially improved by the addition of cytarabine with lower rates of CNS relapse/progression observed.”
The researchers noted that “adequate control of the CNS disease is paramount and is best achieved by intensive CNS-directed induction.”
There was no formal funding for this study, and the researchers did not provide financial disclosures.
SOURCE: Wight JC et al. Br J Haematol. 2019 Jun 24. doi: 10.1111/bjh.16064.
FROM BRITISH JOURNAL OF HAEMATOLOGY
Misguided fear is keeping benzodiazepines from elderly
SAN FRANCISCO – Used appropriately, the benefits of benzodiazepines far outweigh the risks in elderly people, according to Carl Salzman, MD, a psychiatry professor at Harvard Medical School, Boston.

Appropriate use means very low doses – 0.5 mg or less every day or b.i.d. – of short-acting benzodiazepines, either lorazepam, oxazepam, or temazepam. There’s no worry of dose escalation or addiction in the elderly, and since the drugs are not metabolized by the cytochrome P450 system, the risk of drug interactions is very small, except for a compounding effect with alcohol and other sedative hypnotics, such as zolpidem (Ambien). The fall risk is lower than it is with antidepressants and antipsychotics (Psychiatr Serv. 2003 Jul;54[7]:1006-1); (Arch Intern Med. 2009 Nov 23;169[21]:1952-60).
In short, the drugs are “wonderful” for geriatric anxiety and anxiety-related insomnia, Dr. Salzman said at the American Psychiatric Association annual meeting.
Even so, it’s “very hard to get doctors and residents to prescribe them.” It’s like the benzodiazepine scare in the 1980s, about valium. “Newspapers were filled with stories about addicts. I’m having a little bit of déjà vu all over again,” he said.
This time around, the problem is a concern that benzodiazepines cause Alzheimer’s disease, plus collateral damage from the opioid crisis. People with addiction to opioids like benzodiazepines, because they boost the high, so they have significant street value, and drug seekers demand them in the clinic. Some clinicians would rather not deal with the drugs at all.
The Alzheimer’s worry stems largely from a widely reported review that found an association between Alzheimer’s disease and previous benzodiazepine use. The finding was based on public health insurance data from Quebec; no patients were seen (BMJ. 2014 Sep 9;349:g5205).
Among many “very large questions” about the study’s validity, people “may have been on benzos because they already had memory impairment and were anxious about it,” a common occurrence. In that case, “it’s not that benzos caused dementia; it was the other way around.” Also, there was no control for substance and alcohol use, Dr. Salzman said (J Clin Psychopharmacol. 2015 Feb;35[1]:1-3).
A more robust study followed patients 65 years and older for a mean of 7.3 years, comparing benzodiazepine users to nonusers. The team found a slightly higher risk of dementia in people with minimal exposure to benzodiazepines but not with the highest level of exposure, and concluded that the finding did “not support a causal association between benzodiazepine use and dementia” (BMJ. 2016 Feb 2;352:i90).
Meanwhile, a recent review of more than a million patients found either no or a minor increased risk of mortality, another concern with benzodiazepines in the elderly. “If a detrimental effect exists, it is likely to be much smaller than previously stated and to have uncertain clinical relevance. Residual confounding likely explains at least part of” it, the investigators concluded (BMJ. 2017 Jul 6;358:j294).
in an upscale nursing home in Boston. A “dramatic” rebound was reported in short-term recall 2 weeks after volunteers tapered off benzodiazepines, mostly lorazepam, compared with those who stayed on them.
“I sat down to have lunch with the discontinuers, and I said to them, ‘Aren’t you glad that you are not taking these horrible drugs anymore, and your memory is so much better? They said, ‘No, what’s to remember? It was true that when we were taking those drugs, we might not have remembered what we watched on television the night before, but if you give a choice between feeling calm in the days, sleeping at night, and remembering what we watch on television, we’ll take the calm and the sleep every time,’ ” Dr. Salzman said.
He had no disclosures.
SAN FRANCISCO – Used appropriately, the benefits of benzodiazepines far outweigh the risks in elderly people, according to Carl Salzman, MD, a psychiatry professor at Harvard Medical School, Boston.
Appropriate use means very low doses – 0.5 mg or less every day or b.i.d. – of short-acting benzodiazepines, either lorazepam, oxazepam, or temazepam. There’s no worry of dose escalation or addiction in the elderly, and since the drugs are not metabolized by the cytochrome P450 system, the risk of drug interactions is very small, except for a compounding effect with alcohol and other sedative hypnotics, such as zolpidem (Ambien). The fall risk is lower than it is with antidepressants and antipsychotics (Psychiatr Serv. 2003 Jul;54[7]:1006-1); (Arch Intern Med. 2009 Nov 23;169[21]:1952-60).
In short, the drugs are “wonderful” for geriatric anxiety and anxiety-related insomnia, Dr. Salzman said at the American Psychiatric Association annual meeting.
Even so, it’s “very hard to get doctors and residents to prescribe them.” It’s like the benzodiazepine scare in the 1980s, about valium. “Newspapers were filled with stories about addicts. I’m having a little bit of déjà vu all over again,” he said.
This time around, the problem is a concern that benzodiazepines cause Alzheimer’s disease, plus collateral damage from the opioid crisis. People with addiction to opioids like benzodiazepines, because they boost the high, so they have significant street value, and drug seekers demand them in the clinic. Some clinicians would rather not deal with the drugs at all.
The Alzheimer’s worry stems largely from a widely reported review that found an association between Alzheimer’s disease and previous benzodiazepine use. The finding was based on public health insurance data from Quebec; no patients were seen (BMJ. 2014 Sep 9;349:g5205).
Among many “very large questions” about the study’s validity, people “may have been on benzos because they already had memory impairment and were anxious about it,” a common occurrence. In that case, “it’s not that benzos caused dementia; it was the other way around.” Also, there was no control for substance and alcohol use, Dr. Salzman said (J Clin Psychopharmacol. 2015 Feb;35[1]:1-3).
A more robust study followed patients 65 years and older for a mean of 7.3 years, comparing benzodiazepine users to nonusers. The team found a slightly higher risk of dementia in people with minimal exposure to benzodiazepines but not with the highest level of exposure, and concluded that the finding did “not support a causal association between benzodiazepine use and dementia” (BMJ. 2016 Feb 2;352:i90).
Meanwhile, a recent review of more than a million patients found either no or a minor increased risk of mortality, another concern with benzodiazepines in the elderly. “If a detrimental effect exists, it is likely to be much smaller than previously stated and to have uncertain clinical relevance. Residual confounding likely explains at least part of” it, the investigators concluded (BMJ. 2017 Jul 6;358:j294).
in an upscale nursing home in Boston. A “dramatic” rebound was reported in short-term recall 2 weeks after volunteers tapered off benzodiazepines, mostly lorazepam, compared with those who stayed on them.
“I sat down to have lunch with the discontinuers, and I said to them, ‘Aren’t you glad that you are not taking these horrible drugs anymore, and your memory is so much better? They said, ‘No, what’s to remember? It was true that when we were taking those drugs, we might not have remembered what we watched on television the night before, but if you give a choice between feeling calm in the days, sleeping at night, and remembering what we watch on television, we’ll take the calm and the sleep every time,’ ” Dr. Salzman said.
He had no disclosures.
SAN FRANCISCO – Used appropriately, the benefits of benzodiazepines far outweigh the risks in elderly people, according to Carl Salzman, MD, a psychiatry professor at Harvard Medical School, Boston.
Appropriate use means very low doses – 0.5 mg or less every day or b.i.d. – of short-acting benzodiazepines, either lorazepam, oxazepam, or temazepam. There’s no worry of dose escalation or addiction in the elderly, and since the drugs are not metabolized by the cytochrome P450 system, the risk of drug interactions is very small, except for a compounding effect with alcohol and other sedative hypnotics, such as zolpidem (Ambien). The fall risk is lower than it is with antidepressants and antipsychotics (Psychiatr Serv. 2003 Jul;54[7]:1006-1); (Arch Intern Med. 2009 Nov 23;169[21]:1952-60).
In short, the drugs are “wonderful” for geriatric anxiety and anxiety-related insomnia, Dr. Salzman said at the American Psychiatric Association annual meeting.
Even so, it’s “very hard to get doctors and residents to prescribe them.” It’s like the benzodiazepine scare in the 1980s, about valium. “Newspapers were filled with stories about addicts. I’m having a little bit of déjà vu all over again,” he said.
This time around, the problem is a concern that benzodiazepines cause Alzheimer’s disease, plus collateral damage from the opioid crisis. People with addiction to opioids like benzodiazepines, because they boost the high, so they have significant street value, and drug seekers demand them in the clinic. Some clinicians would rather not deal with the drugs at all.
The Alzheimer’s worry stems largely from a widely reported review that found an association between Alzheimer’s disease and previous benzodiazepine use. The finding was based on public health insurance data from Quebec; no patients were seen (BMJ. 2014 Sep 9;349:g5205).
Among many “very large questions” about the study’s validity, people “may have been on benzos because they already had memory impairment and were anxious about it,” a common occurrence. In that case, “it’s not that benzos caused dementia; it was the other way around.” Also, there was no control for substance and alcohol use, Dr. Salzman said (J Clin Psychopharmacol. 2015 Feb;35[1]:1-3).
A more robust study followed patients 65 years and older for a mean of 7.3 years, comparing benzodiazepine users to nonusers. The team found a slightly higher risk of dementia in people with minimal exposure to benzodiazepines but not with the highest level of exposure, and concluded that the finding did “not support a causal association between benzodiazepine use and dementia” (BMJ. 2016 Feb 2;352:i90).
Meanwhile, a recent review of more than a million patients found either no or a minor increased risk of mortality, another concern with benzodiazepines in the elderly. “If a detrimental effect exists, it is likely to be much smaller than previously stated and to have uncertain clinical relevance. Residual confounding likely explains at least part of” it, the investigators concluded (BMJ. 2017 Jul 6;358:j294).
in an upscale nursing home in Boston. A “dramatic” rebound was reported in short-term recall 2 weeks after volunteers tapered off benzodiazepines, mostly lorazepam, compared with those who stayed on them.
“I sat down to have lunch with the discontinuers, and I said to them, ‘Aren’t you glad that you are not taking these horrible drugs anymore, and your memory is so much better? They said, ‘No, what’s to remember? It was true that when we were taking those drugs, we might not have remembered what we watched on television the night before, but if you give a choice between feeling calm in the days, sleeping at night, and remembering what we watch on television, we’ll take the calm and the sleep every time,’ ” Dr. Salzman said.
He had no disclosures.
REPORTING FROM APA 2019
Recombinant vaccine cut herpes zoster rate in immunocompromised patients
Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT), results of a randomized, placebo-controlled trial indicate.
The incidence of herpes zoster was 30 per 1,000 person-years for patients who received the adjuvanted recombinant zoster vaccine (Shingrix) versus 94 per 1,000 person-years for those who received placebo, according to study results.
Recombinant zoster vaccine induced humoral and cellular responses that were strong and occurring at a rate higher than what was seen in the placebo group, said senior author Keith M. Sullivan, MD, of Duke University Medical Center, Durham, N.C., and coauthors, who reported findings on behalf of the Zoster Efficacy Study in Patients Undergoing HSCT (ZOE-HSCT) Study Group.
“The vaccinations were generally well tolerated, and most symptoms were mild and transient and did not substantially deter participants from receiving their second dose,” Dr. Sullivan and colleagues wrote in JAMA.
The risk of herpes zoster is increased for 2-3 years after autologous HSCT because of diminished T-cell immunity, according to the authors.
“Antiviral prophylaxis is commonly administered to patients after HSCT to prevent such complications, but the efficacy depends on adherence to treatment,” they said.
While vaccines could provide long-term protection, immunocompromised individuals receiving live attenuated vaccine would be at increased risk of varicella caused by spread of the vaccine strain, they added.
There have been a few encouraging recent studies of non-live vaccines in this setting, including one large phase 3 trial of a heat-inactivated varicella-zoster virus vaccine that showed patients undergoing autologous HSCT had a 63.8% estimated efficacy in preventing herpes zoster, investigators from that study said in The Lancet (2018 May 26;391[10135]:2116-27).
A phase 1/2a study of the adjuvanted recombinant zoster vaccine in patients undergoing HSCT demonstrated strong humoral and cell-mediated immunity responses, which provided the rationale for studying the vaccine further in the randomized ZOE-HSCT study, according to Dr. Sullivan and coauthors.
Their study included a total of 1,846 adults who had undergone autologous HSCT. They were randomized to receive two doses of the recombinant zoster vaccine, the first at 50-70 days after the procedure and the second 1-2 months later.
Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, which resulted in overall incidences of 30 and 94 per 1,000 person-years.
The incidence rate ratio of a first episode of herpes zoster was 0.36 for individuals receiving at least one dose, which authors said was equivalent to a vaccine efficacy of 63.7%.
That efficacy rate is “very similar” to the estimated efficacy reported for the heat-inactivated varicella-zoster virus vaccine reported in The Lancet, said Dr. Sullivan and coauthors.
However, the heat-inactivated vaccine achieved that level of protection with a four-dose schedule, including one dose given prior to autologous HSCT.
“An advantage of the short 2-dose posttransplantation schedule is that more patients might complete the vaccination program,” they said in a discussion of the results, noting that 94.7% of the recombinant zoster vaccine recipients completed two doses, compared with 81.9% of recipients who received the heat-inactivated herpes zoster vaccine in the previous report.
The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Dr. Sullivan reported disclosures related to GlaxoSmithKline (GSK), Kiadis Pharmaceutical, Roche Genentech, and the National Institute of Allergy and Infectious Diseases. Coauthors provided disclosures related to GSK, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
SOURCE: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT), results of a randomized, placebo-controlled trial indicate.
The incidence of herpes zoster was 30 per 1,000 person-years for patients who received the adjuvanted recombinant zoster vaccine (Shingrix) versus 94 per 1,000 person-years for those who received placebo, according to study results.
Recombinant zoster vaccine induced humoral and cellular responses that were strong and occurring at a rate higher than what was seen in the placebo group, said senior author Keith M. Sullivan, MD, of Duke University Medical Center, Durham, N.C., and coauthors, who reported findings on behalf of the Zoster Efficacy Study in Patients Undergoing HSCT (ZOE-HSCT) Study Group.
“The vaccinations were generally well tolerated, and most symptoms were mild and transient and did not substantially deter participants from receiving their second dose,” Dr. Sullivan and colleagues wrote in JAMA.
The risk of herpes zoster is increased for 2-3 years after autologous HSCT because of diminished T-cell immunity, according to the authors.
“Antiviral prophylaxis is commonly administered to patients after HSCT to prevent such complications, but the efficacy depends on adherence to treatment,” they said.
While vaccines could provide long-term protection, immunocompromised individuals receiving live attenuated vaccine would be at increased risk of varicella caused by spread of the vaccine strain, they added.
There have been a few encouraging recent studies of non-live vaccines in this setting, including one large phase 3 trial of a heat-inactivated varicella-zoster virus vaccine that showed patients undergoing autologous HSCT had a 63.8% estimated efficacy in preventing herpes zoster, investigators from that study said in The Lancet (2018 May 26;391[10135]:2116-27).
A phase 1/2a study of the adjuvanted recombinant zoster vaccine in patients undergoing HSCT demonstrated strong humoral and cell-mediated immunity responses, which provided the rationale for studying the vaccine further in the randomized ZOE-HSCT study, according to Dr. Sullivan and coauthors.
Their study included a total of 1,846 adults who had undergone autologous HSCT. They were randomized to receive two doses of the recombinant zoster vaccine, the first at 50-70 days after the procedure and the second 1-2 months later.
Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, which resulted in overall incidences of 30 and 94 per 1,000 person-years.
The incidence rate ratio of a first episode of herpes zoster was 0.36 for individuals receiving at least one dose, which authors said was equivalent to a vaccine efficacy of 63.7%.
That efficacy rate is “very similar” to the estimated efficacy reported for the heat-inactivated varicella-zoster virus vaccine reported in The Lancet, said Dr. Sullivan and coauthors.
However, the heat-inactivated vaccine achieved that level of protection with a four-dose schedule, including one dose given prior to autologous HSCT.
“An advantage of the short 2-dose posttransplantation schedule is that more patients might complete the vaccination program,” they said in a discussion of the results, noting that 94.7% of the recombinant zoster vaccine recipients completed two doses, compared with 81.9% of recipients who received the heat-inactivated herpes zoster vaccine in the previous report.
The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Dr. Sullivan reported disclosures related to GlaxoSmithKline (GSK), Kiadis Pharmaceutical, Roche Genentech, and the National Institute of Allergy and Infectious Diseases. Coauthors provided disclosures related to GSK, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
SOURCE: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT), results of a randomized, placebo-controlled trial indicate.
The incidence of herpes zoster was 30 per 1,000 person-years for patients who received the adjuvanted recombinant zoster vaccine (Shingrix) versus 94 per 1,000 person-years for those who received placebo, according to study results.
Recombinant zoster vaccine induced humoral and cellular responses that were strong and occurring at a rate higher than what was seen in the placebo group, said senior author Keith M. Sullivan, MD, of Duke University Medical Center, Durham, N.C., and coauthors, who reported findings on behalf of the Zoster Efficacy Study in Patients Undergoing HSCT (ZOE-HSCT) Study Group.
“The vaccinations were generally well tolerated, and most symptoms were mild and transient and did not substantially deter participants from receiving their second dose,” Dr. Sullivan and colleagues wrote in JAMA.
The risk of herpes zoster is increased for 2-3 years after autologous HSCT because of diminished T-cell immunity, according to the authors.
“Antiviral prophylaxis is commonly administered to patients after HSCT to prevent such complications, but the efficacy depends on adherence to treatment,” they said.
While vaccines could provide long-term protection, immunocompromised individuals receiving live attenuated vaccine would be at increased risk of varicella caused by spread of the vaccine strain, they added.
There have been a few encouraging recent studies of non-live vaccines in this setting, including one large phase 3 trial of a heat-inactivated varicella-zoster virus vaccine that showed patients undergoing autologous HSCT had a 63.8% estimated efficacy in preventing herpes zoster, investigators from that study said in The Lancet (2018 May 26;391[10135]:2116-27).
A phase 1/2a study of the adjuvanted recombinant zoster vaccine in patients undergoing HSCT demonstrated strong humoral and cell-mediated immunity responses, which provided the rationale for studying the vaccine further in the randomized ZOE-HSCT study, according to Dr. Sullivan and coauthors.
Their study included a total of 1,846 adults who had undergone autologous HSCT. They were randomized to receive two doses of the recombinant zoster vaccine, the first at 50-70 days after the procedure and the second 1-2 months later.
Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, which resulted in overall incidences of 30 and 94 per 1,000 person-years.
The incidence rate ratio of a first episode of herpes zoster was 0.36 for individuals receiving at least one dose, which authors said was equivalent to a vaccine efficacy of 63.7%.
That efficacy rate is “very similar” to the estimated efficacy reported for the heat-inactivated varicella-zoster virus vaccine reported in The Lancet, said Dr. Sullivan and coauthors.
However, the heat-inactivated vaccine achieved that level of protection with a four-dose schedule, including one dose given prior to autologous HSCT.
“An advantage of the short 2-dose posttransplantation schedule is that more patients might complete the vaccination program,” they said in a discussion of the results, noting that 94.7% of the recombinant zoster vaccine recipients completed two doses, compared with 81.9% of recipients who received the heat-inactivated herpes zoster vaccine in the previous report.
The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Dr. Sullivan reported disclosures related to GlaxoSmithKline (GSK), Kiadis Pharmaceutical, Roche Genentech, and the National Institute of Allergy and Infectious Diseases. Coauthors provided disclosures related to GSK, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
SOURCE: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
FROM JAMA
Key clinical point: Two doses of recombinant zoster vaccine significantly reduced incidence of herpes zoster versus placebo in adults who had undergone autologous hematopoietic stem cell transplantation (HSCT).
Major finding: Herpes zoster cases were seen in 49 and 136 individuals in the vaccine and placebo groups, respectively, resulting in overall incidences of 30 and 94 per 1,000 person-years.
Study details: A randomized clinical trial (ZOE-HSCT) including 1,846 adults who had undergone autologous HSCT.
Disclosures: The study was funded and sponsored by GlaxoSmithKline Biologicals SA. Study authors reported disclosures related to GlaxoSmithKline, Kiadis Pharmaceutical, Roche Genentech, AbbVie, Roche, Gilead, Janssen, Pharmacyclics, Morphosys, Helsinn, Celgene, and others.
Source: Bastidas A et al. JAMA. 2019 July 9. doi: 10.1001/jama.2019.9053.
Metformin linked to lower dementia risk in black patients
Black individuals who develop type 2 diabetes are more likely than their white counterparts to develop dementia. Now, findings from a new study point to a possible preventive strategy: Putting older patients on metformin when they are diagnosed could reduce their risk for dementia by as much as 40%, whereas sulfonylureas do not seem to have such an effect.
The researchers did not examine cause and effect, so their findings are not conclusive, and very few women were included in the study. Still, the authors said that their data showing a 29% lower risk of dementia associated with metformin use in black patients aged 65-74 years, and a 40% lower risk in those aged 50-64 years, suggested that “this inexpensive, widely available treatment could be broadly prescribed to substantially reduce the risk of dementia in younger [black] patients with [type 2 diabetes]” (Ann Fam Med. 2019;17:352-62).
Previous findings have suggested that black patients with type 2 diabetes face a 10%-18% higher risk of dementia, compared with white patients (Diabetes Care. 2014; 37[4]:1009-15). Another study linked type 2 diabetes in middle-aged black patients to a 41% decrease in cognition per test results over 14 years. There was no such decrease in white patients (Neuroepidemiology. 2014;43[3-4]: 220-7).
For the new study, researchers led by Jeffrey F. Scherrer, PhD, of Saint Louis University tracked 73,761 patients aged 50 years or older from 2000-2001 (when they were free of dementia and not taking diabetes) to 2015. Among the patients, 86% were white and 14% were black. In the white and black groups, 97% and 95% were men, respectively, and 61% and 55% were obese, respectively.
All participants began metformin (76%) or sulfonylurea (24%) monotherapy after the baseline period. Guidelines recommend metformin as a first-line treatment for type 2 diabetes, whereas sulfonylureas are considered second-line drugs that should be added to metformin.
After adjustment for confounders such as socioeconomic status and other medical conditions, the researchers found a significantly lower risk of dementia in black patients who took metformin, compared with those taking a sulfonylurea (hazard ratio, 0.73; 95% confidence interval, 0.6-0.89). There was no difference between the drugs among white patients (HR, 0.96; 95% CI, 0.9-1.03, both P = .008)
The results were not statistically significant among age groups, but there were trends. In black patients, the dementia-lowering benefit was largest among those aged 50-64 years (HR, 0.6; 95% CI, 0.45-0.81), followed by those aged 65-74 years (HR, 0.71; 95% CI, 0.53-0.94), and there was no benefit among those aged at least 75 (HR, 1.17; 95% CI, 0.73-1.85) all P = .055. There was a slight benefit among white patients in one of the age groups – 65-74 years (HR, 0.9; 95% CI, 0.82-0.99; P = .315).
The authors suggested that the findings could have been the result of an effect of metformin to reduce vascular disease and chronic inflammation in black patients.
They also noted that further research is needed to identify the demographic and clinical subgroups in which metformin is most strongly associated with a reduction in the risk of dementia. In addition, they emphasized that clinical trials are needed to confirm the study findings.
The National Institutes of Health funded the study. The authors report no relevant disclosures.
SOURCE: Scherrer JF et al. Ann Fam Med. 2019;17:352-62.
Black individuals who develop type 2 diabetes are more likely than their white counterparts to develop dementia. Now, findings from a new study point to a possible preventive strategy: Putting older patients on metformin when they are diagnosed could reduce their risk for dementia by as much as 40%, whereas sulfonylureas do not seem to have such an effect.
The researchers did not examine cause and effect, so their findings are not conclusive, and very few women were included in the study. Still, the authors said that their data showing a 29% lower risk of dementia associated with metformin use in black patients aged 65-74 years, and a 40% lower risk in those aged 50-64 years, suggested that “this inexpensive, widely available treatment could be broadly prescribed to substantially reduce the risk of dementia in younger [black] patients with [type 2 diabetes]” (Ann Fam Med. 2019;17:352-62).
Previous findings have suggested that black patients with type 2 diabetes face a 10%-18% higher risk of dementia, compared with white patients (Diabetes Care. 2014; 37[4]:1009-15). Another study linked type 2 diabetes in middle-aged black patients to a 41% decrease in cognition per test results over 14 years. There was no such decrease in white patients (Neuroepidemiology. 2014;43[3-4]: 220-7).
For the new study, researchers led by Jeffrey F. Scherrer, PhD, of Saint Louis University tracked 73,761 patients aged 50 years or older from 2000-2001 (when they were free of dementia and not taking diabetes) to 2015. Among the patients, 86% were white and 14% were black. In the white and black groups, 97% and 95% were men, respectively, and 61% and 55% were obese, respectively.
All participants began metformin (76%) or sulfonylurea (24%) monotherapy after the baseline period. Guidelines recommend metformin as a first-line treatment for type 2 diabetes, whereas sulfonylureas are considered second-line drugs that should be added to metformin.
After adjustment for confounders such as socioeconomic status and other medical conditions, the researchers found a significantly lower risk of dementia in black patients who took metformin, compared with those taking a sulfonylurea (hazard ratio, 0.73; 95% confidence interval, 0.6-0.89). There was no difference between the drugs among white patients (HR, 0.96; 95% CI, 0.9-1.03, both P = .008)
The results were not statistically significant among age groups, but there were trends. In black patients, the dementia-lowering benefit was largest among those aged 50-64 years (HR, 0.6; 95% CI, 0.45-0.81), followed by those aged 65-74 years (HR, 0.71; 95% CI, 0.53-0.94), and there was no benefit among those aged at least 75 (HR, 1.17; 95% CI, 0.73-1.85) all P = .055. There was a slight benefit among white patients in one of the age groups – 65-74 years (HR, 0.9; 95% CI, 0.82-0.99; P = .315).
The authors suggested that the findings could have been the result of an effect of metformin to reduce vascular disease and chronic inflammation in black patients.
They also noted that further research is needed to identify the demographic and clinical subgroups in which metformin is most strongly associated with a reduction in the risk of dementia. In addition, they emphasized that clinical trials are needed to confirm the study findings.
The National Institutes of Health funded the study. The authors report no relevant disclosures.
SOURCE: Scherrer JF et al. Ann Fam Med. 2019;17:352-62.
Black individuals who develop type 2 diabetes are more likely than their white counterparts to develop dementia. Now, findings from a new study point to a possible preventive strategy: Putting older patients on metformin when they are diagnosed could reduce their risk for dementia by as much as 40%, whereas sulfonylureas do not seem to have such an effect.
The researchers did not examine cause and effect, so their findings are not conclusive, and very few women were included in the study. Still, the authors said that their data showing a 29% lower risk of dementia associated with metformin use in black patients aged 65-74 years, and a 40% lower risk in those aged 50-64 years, suggested that “this inexpensive, widely available treatment could be broadly prescribed to substantially reduce the risk of dementia in younger [black] patients with [type 2 diabetes]” (Ann Fam Med. 2019;17:352-62).
Previous findings have suggested that black patients with type 2 diabetes face a 10%-18% higher risk of dementia, compared with white patients (Diabetes Care. 2014; 37[4]:1009-15). Another study linked type 2 diabetes in middle-aged black patients to a 41% decrease in cognition per test results over 14 years. There was no such decrease in white patients (Neuroepidemiology. 2014;43[3-4]: 220-7).
For the new study, researchers led by Jeffrey F. Scherrer, PhD, of Saint Louis University tracked 73,761 patients aged 50 years or older from 2000-2001 (when they were free of dementia and not taking diabetes) to 2015. Among the patients, 86% were white and 14% were black. In the white and black groups, 97% and 95% were men, respectively, and 61% and 55% were obese, respectively.
All participants began metformin (76%) or sulfonylurea (24%) monotherapy after the baseline period. Guidelines recommend metformin as a first-line treatment for type 2 diabetes, whereas sulfonylureas are considered second-line drugs that should be added to metformin.
After adjustment for confounders such as socioeconomic status and other medical conditions, the researchers found a significantly lower risk of dementia in black patients who took metformin, compared with those taking a sulfonylurea (hazard ratio, 0.73; 95% confidence interval, 0.6-0.89). There was no difference between the drugs among white patients (HR, 0.96; 95% CI, 0.9-1.03, both P = .008)
The results were not statistically significant among age groups, but there were trends. In black patients, the dementia-lowering benefit was largest among those aged 50-64 years (HR, 0.6; 95% CI, 0.45-0.81), followed by those aged 65-74 years (HR, 0.71; 95% CI, 0.53-0.94), and there was no benefit among those aged at least 75 (HR, 1.17; 95% CI, 0.73-1.85) all P = .055. There was a slight benefit among white patients in one of the age groups – 65-74 years (HR, 0.9; 95% CI, 0.82-0.99; P = .315).
The authors suggested that the findings could have been the result of an effect of metformin to reduce vascular disease and chronic inflammation in black patients.
They also noted that further research is needed to identify the demographic and clinical subgroups in which metformin is most strongly associated with a reduction in the risk of dementia. In addition, they emphasized that clinical trials are needed to confirm the study findings.
The National Institutes of Health funded the study. The authors report no relevant disclosures.
SOURCE: Scherrer JF et al. Ann Fam Med. 2019;17:352-62.
FROM ANNALS OF FAMILY MEDICINE
Key clinical point:
Major finding: Metformin monotherapy, compared with sulfonylurea monotherapy, was linked to a significantly lower risk for dementia in black patients (HR, 0.73; 95% CI, 0.6-0.89), but not in white patients (HR, 0.96; 95% CI, 0.9-1.03; P = .008).
Study details: Retrospective analysis of 73,761 patients aged 50 years or older in the Veterans Health Administration system who were tracked from 2000-2001 to 2015 and began metformin or sulfonylurea monotherapy after baseline.
Disclosures: The National Institutes of Health funded the study. The authors report no relevant disclosures.
Source: Scherrer JF et al. Ann Fam Med. 2019;17:352-62.
Treatment in prison systems might lead to drop in overdose deaths
Incarceration versus treatment takes center stage in a new analysis of U.S. data from researchers in the United Kingdom.

The researchers performed an observational study looking at rates of incarceration, income, and drug-related deaths from 1983 to 2014 in the United States. They found a strong association between incarceration rates and drug-related deaths. Also, a very strong association was found between lower household income and drug-related deaths. Strikingly, in the counties with the highest incarceration rates, there was a 50% higher rate of drug deaths, reported Elias Nosrati, PhD, and associates (Lancet Public Health. 2019 Jul 3;4:e326-33). It is clearer every day that our opioid epidemic was in part wrought by a zealous push to change protocols on treating chronic pain. The epidemic also appears tied to well-meaning but overprescribing doctors and allegedly unscrupulous pharmaceutical companies and distributors. What we are learning through this most recent study is that another factor tied to the opioid and overdose epidemic could be incarceration.
According to the study, an increase in crime rates combined with sentencing reforms led the number of people incarcerated in state and federal prisons to soar from less than 200,000 in 1970 to almost 1 million in 1995. Furthermore, Dr. Nosrati and associates wrote, “Incarceration is directly associated with stigma, discrimination, poor mental health, and chronic economic hardship, all of which are linked to drug use disorders.”
Treatment for drug addiction in prison systems is rare, as is adequate mental health treatment. However, treatment for this population would likely help reduce drug overdose deaths and improve the quality of life for people who are incarcerated and their families. In the Philadelphia prison system, for example, treatment for inmates is available for opioid addiction, both with methadone and now more recently with buprenorphine (Suboxone). The Philadelphia Department of Prisons also provides cognitive-behavioral therapy. In Florida, Chapter 397 of the Florida statutes – known as the Marchman Act – provides for the involuntary (and voluntary) treatment of individuals with substance abuse problems.
The court systems in South Florida have a robust drug-diversion program, aimed at directing people facing incarceration for drug offenses into treatment instead. North Carolina has studied this issue specifically and found through a model simulation that diverting 10% of drug-abusing offenders out of incarceration into treatment would save $4.8 billion in legal costs for North Carolina counties and state legal systems. Diverting 40% of individuals would close to triple that savings.
There are striking data from programs treating individuals who are leveraged into treatment in order to maintain professional licenses. These such individuals, many of whom are physicians, airline pilots, and nurses, have a rate of sobriety of 90% or greater after 5 years. This data show that
In addition to the potential reduction in morbidity and mortality as well as the financial savings, why is treatment important? Because of societal costs. When parents or family members are put in jail for a drug charge or other charge, they leave behind a community, family, and very often children who are affected economically, emotionally, and socially. Those children in particular have higher risks of depression and PTSD. Diverting an offender into treatment or treating an incarcerated person for drug and mental health problems can change the life of a child or family member, and ultimately can change society.
Dr. Jorandby is chief medical officer of Lakeview Health in Jacksonville, Fla. She trained in addiction psychiatry at Yale University, New Haven, Conn.
Incarceration versus treatment takes center stage in a new analysis of U.S. data from researchers in the United Kingdom.
The researchers performed an observational study looking at rates of incarceration, income, and drug-related deaths from 1983 to 2014 in the United States. They found a strong association between incarceration rates and drug-related deaths. Also, a very strong association was found between lower household income and drug-related deaths. Strikingly, in the counties with the highest incarceration rates, there was a 50% higher rate of drug deaths, reported Elias Nosrati, PhD, and associates (Lancet Public Health. 2019 Jul 3;4:e326-33). It is clearer every day that our opioid epidemic was in part wrought by a zealous push to change protocols on treating chronic pain. The epidemic also appears tied to well-meaning but overprescribing doctors and allegedly unscrupulous pharmaceutical companies and distributors. What we are learning through this most recent study is that another factor tied to the opioid and overdose epidemic could be incarceration.
According to the study, an increase in crime rates combined with sentencing reforms led the number of people incarcerated in state and federal prisons to soar from less than 200,000 in 1970 to almost 1 million in 1995. Furthermore, Dr. Nosrati and associates wrote, “Incarceration is directly associated with stigma, discrimination, poor mental health, and chronic economic hardship, all of which are linked to drug use disorders.”
Treatment for drug addiction in prison systems is rare, as is adequate mental health treatment. However, treatment for this population would likely help reduce drug overdose deaths and improve the quality of life for people who are incarcerated and their families. In the Philadelphia prison system, for example, treatment for inmates is available for opioid addiction, both with methadone and now more recently with buprenorphine (Suboxone). The Philadelphia Department of Prisons also provides cognitive-behavioral therapy. In Florida, Chapter 397 of the Florida statutes – known as the Marchman Act – provides for the involuntary (and voluntary) treatment of individuals with substance abuse problems.
The court systems in South Florida have a robust drug-diversion program, aimed at directing people facing incarceration for drug offenses into treatment instead. North Carolina has studied this issue specifically and found through a model simulation that diverting 10% of drug-abusing offenders out of incarceration into treatment would save $4.8 billion in legal costs for North Carolina counties and state legal systems. Diverting 40% of individuals would close to triple that savings.
There are striking data from programs treating individuals who are leveraged into treatment in order to maintain professional licenses. These such individuals, many of whom are physicians, airline pilots, and nurses, have a rate of sobriety of 90% or greater after 5 years. This data show that
In addition to the potential reduction in morbidity and mortality as well as the financial savings, why is treatment important? Because of societal costs. When parents or family members are put in jail for a drug charge or other charge, they leave behind a community, family, and very often children who are affected economically, emotionally, and socially. Those children in particular have higher risks of depression and PTSD. Diverting an offender into treatment or treating an incarcerated person for drug and mental health problems can change the life of a child or family member, and ultimately can change society.
Dr. Jorandby is chief medical officer of Lakeview Health in Jacksonville, Fla. She trained in addiction psychiatry at Yale University, New Haven, Conn.
Incarceration versus treatment takes center stage in a new analysis of U.S. data from researchers in the United Kingdom.
The researchers performed an observational study looking at rates of incarceration, income, and drug-related deaths from 1983 to 2014 in the United States. They found a strong association between incarceration rates and drug-related deaths. Also, a very strong association was found between lower household income and drug-related deaths. Strikingly, in the counties with the highest incarceration rates, there was a 50% higher rate of drug deaths, reported Elias Nosrati, PhD, and associates (Lancet Public Health. 2019 Jul 3;4:e326-33). It is clearer every day that our opioid epidemic was in part wrought by a zealous push to change protocols on treating chronic pain. The epidemic also appears tied to well-meaning but overprescribing doctors and allegedly unscrupulous pharmaceutical companies and distributors. What we are learning through this most recent study is that another factor tied to the opioid and overdose epidemic could be incarceration.
According to the study, an increase in crime rates combined with sentencing reforms led the number of people incarcerated in state and federal prisons to soar from less than 200,000 in 1970 to almost 1 million in 1995. Furthermore, Dr. Nosrati and associates wrote, “Incarceration is directly associated with stigma, discrimination, poor mental health, and chronic economic hardship, all of which are linked to drug use disorders.”
Treatment for drug addiction in prison systems is rare, as is adequate mental health treatment. However, treatment for this population would likely help reduce drug overdose deaths and improve the quality of life for people who are incarcerated and their families. In the Philadelphia prison system, for example, treatment for inmates is available for opioid addiction, both with methadone and now more recently with buprenorphine (Suboxone). The Philadelphia Department of Prisons also provides cognitive-behavioral therapy. In Florida, Chapter 397 of the Florida statutes – known as the Marchman Act – provides for the involuntary (and voluntary) treatment of individuals with substance abuse problems.
The court systems in South Florida have a robust drug-diversion program, aimed at directing people facing incarceration for drug offenses into treatment instead. North Carolina has studied this issue specifically and found through a model simulation that diverting 10% of drug-abusing offenders out of incarceration into treatment would save $4.8 billion in legal costs for North Carolina counties and state legal systems. Diverting 40% of individuals would close to triple that savings.
There are striking data from programs treating individuals who are leveraged into treatment in order to maintain professional licenses. These such individuals, many of whom are physicians, airline pilots, and nurses, have a rate of sobriety of 90% or greater after 5 years. This data show that
In addition to the potential reduction in morbidity and mortality as well as the financial savings, why is treatment important? Because of societal costs. When parents or family members are put in jail for a drug charge or other charge, they leave behind a community, family, and very often children who are affected economically, emotionally, and socially. Those children in particular have higher risks of depression and PTSD. Diverting an offender into treatment or treating an incarcerated person for drug and mental health problems can change the life of a child or family member, and ultimately can change society.
Dr. Jorandby is chief medical officer of Lakeview Health in Jacksonville, Fla. She trained in addiction psychiatry at Yale University, New Haven, Conn.
FDA approves Xpovio for relapsed/refractory multiple myeloma

The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
The oral therapy was approved for patients who have received at least four prior therapies and whose disease is resistant to several other forms of treatment, including at least two proteasome inhibitors, at least two immunomodulatory agents, and an anti-CD38 monoclonal antibody, according to the FDA.
The approval provides a “treatment option for patients with multiple myeloma with no (other) available therapy,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA Center for Drug Evaluation and Research.
The approval was based on a study that included 83 patients with RRMM who had an overall response rate of 25.3% to Xpovio in combination with dexamethasone.
“The median time to first response was 4 weeks, with a range of 1-10 weeks. The median duration of response was 3.8 months. The efficacy evaluation was supported by additional information from an ongoing, randomized trial in patients with multiple myeloma,” according to the statement.
Common side effects seen in patients taking Xpovio in combination with dexamethasone include leukopenia, neutropenia, thrombocytopenia, and anemia. Patients also reported vomiting, nausea, fatigue, diarrhea, fever, decreased appetite and weight, constipation, upper respiratory tract infections, and hyponatremia.
Patients taking Xpovio should be monitored for low blood counts, platelets, and sodium levels, and should avoid other medications that may cause dizziness or confusion. Patients’ hydration status, blood counts, and other medications should be optimized to avoid dizziness or confusion. Females of reproductive age and males with a female partner of reproductive potential must use effective contraception during treatment with Xpovio. Women who are pregnant or breastfeeding should not take Xpovio.
Xpovio must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Xpovio in combination with dexamethasone was granted accelerated approval, and further clinical trials are required to verify and describe the drug’s clinical benefit.
The FDA granted the approval of Xpovio to Karyopharm Therapeutics.
Anticholinergic drugs linked to dementia in older populations
Exposures to various types of anticholinergic medications were associated with a significantly increased risk of dementia in people aged 55 years or older in a large pharmacoepidemiologic study.

“This study was designed to assess the association between cumulative anticholinergic drug use and risk of dementia in a large, representative British population,” wrote Carol A. C. Coupland, PhD, of the division of primary care at the University of Nottingham (England), and colleagues. The findings were published in JAMA Internal Medicine.
The researchers conducted a large nested case-control study that included 58,769 patients with dementia and 225,574 matched controls from the QResearch database in England. Each study participant was matched to five controls based on various characteristics, including sex, age, and calendar time, among others.
Prescription data related to 56 different drugs with strong anticholinergic properties, including antipsychotics, bladder antimuscarinics, antiepileptics, antiparkinson agents, and antidepressants were used to measure drug exposure. The study data were analyzed from 2016 to 2018.
“The primary exposure was the total standardized daily doses (TSDDs) of anticholinergic drugs prescribed in the 1 to 11 years prior to the date of diagnosis of dementia or equivalent date in matched controls,” Dr. Coupland and colleagues wrote.
After analysis, the researchers found that exposure to antipsychotics (adjusted odds ratio, 1.70), bladder antimuscarinics (aOR, 1.65), antiepileptics (aOR, 1.39), antiparkinson agents (aOR, 1.52), and anticholinergic antidepressants (aOR, 1.29) was associated with an increased risk of dementia after adjustment for confounding factors.
“Associations were stronger in [dementia] cases diagnosed before the age of 80 years,” the researchers noted.
However, antihistamine, antivertigo/antiemetic, skeletal muscle relaxant, gastrointestinal antispasmodic, antiarrhythmic, and antimuscarinic bronchodilator anticholinergic agents were not associated with any increased risk of dementia.
One key limitation of the study was the absence of medication compliance assessment, which could result in exposure misclassification. Dr. Coupland and colleagues acknowledged this could underestimate some associations with medication exposure.
The stronger risk of dementia found among people who had dementia before age 80 “indicates that anticholinergic drugs should be prescribed with caution in middle-aged and older people,” they concluded.
One question that remains from the current study is whether anticholinergic drugs are a definite modifiable risk factor for Alzheimer’s disease and related dementias, Noll L. Campbell, PharmD, of Purdue University, West Lafayette, Ind., and colleagues wrote in an editorial accompanying the study by Dr. Coupland and associates (JAMA Intern Med. 2019 Jun 24. doi: 10.1001/jamainternmed.2019.0676).
While a pharmacologic basis for this association has been proposed, causation has yet to be established by means of prospective randomized studies. The current supposition is that deprescribing anticholinergic medications has the potential to positively effect cholinergic neurotransmission in certain regions of the brain, which could lead to improved cognitive functioning, and lower the likelihood of developing Alzheimer’s disease and related dementias, they wrote in the editorial.
However, the discontinuation of some anticholinergic agents may pose other risks, such as worsening pain or depressive symptoms, in addition to increasing the utilization of acute care facilities. As a result, high-quality, well-designed, randomized trials are needed to better understand the long-term effects of deprescribing anticholinergic medications. These trials would help inform clinicians, patients, and policymakers about the risks and benefits of deprescribing interventions, Dr. Campbell and coauthors said.
The study was supported by the National Institute for Health Research and the University of Nottingham. The authors reported financial affiliations with ClinRisk Ltd. The authors of the editorial reported receiving support from the National Institute on Aging and the Agency for Healthcare Research and Quality. Dr. Campbell reported receiving personal fees from Astellas Pharma US.
SOURCE: Coupland C et al. JAMA Intern Med. 2019 Jun 24. doi: 10.1001/jamainternmed.2019.0677
Exposures to various types of anticholinergic medications were associated with a significantly increased risk of dementia in people aged 55 years or older in a large pharmacoepidemiologic study.
“This study was designed to assess the association between cumulative anticholinergic drug use and risk of dementia in a large, representative British population,” wrote Carol A. C. Coupland, PhD, of the division of primary care at the University of Nottingham (England), and colleagues. The findings were published in JAMA Internal Medicine.
The researchers conducted a large nested case-control study that included 58,769 patients with dementia and 225,574 matched controls from the QResearch database in England. Each study participant was matched to five controls based on various characteristics, including sex, age, and calendar time, among others.
Prescription data related to 56 different drugs with strong anticholinergic properties, including antipsychotics, bladder antimuscarinics, antiepileptics, antiparkinson agents, and antidepressants were used to measure drug exposure. The study data were analyzed from 2016 to 2018.
“The primary exposure was the total standardized daily doses (TSDDs) of anticholinergic drugs prescribed in the 1 to 11 years prior to the date of diagnosis of dementia or equivalent date in matched controls,” Dr. Coupland and colleagues wrote.
After analysis, the researchers found that exposure to antipsychotics (adjusted odds ratio, 1.70), bladder antimuscarinics (aOR, 1.65), antiepileptics (aOR, 1.39), antiparkinson agents (aOR, 1.52), and anticholinergic antidepressants (aOR, 1.29) was associated with an increased risk of dementia after adjustment for confounding factors.
“Associations were stronger in [dementia] cases diagnosed before the age of 80 years,” the researchers noted.
However, antihistamine, antivertigo/antiemetic, skeletal muscle relaxant, gastrointestinal antispasmodic, antiarrhythmic, and antimuscarinic bronchodilator anticholinergic agents were not associated with any increased risk of dementia.
One key limitation of the study was the absence of medication compliance assessment, which could result in exposure misclassification. Dr. Coupland and colleagues acknowledged this could underestimate some associations with medication exposure.
The stronger risk of dementia found among people who had dementia before age 80 “indicates that anticholinergic drugs should be prescribed with caution in middle-aged and older people,” they concluded.
One question that remains from the current study is whether anticholinergic drugs are a definite modifiable risk factor for Alzheimer’s disease and related dementias, Noll L. Campbell, PharmD, of Purdue University, West Lafayette, Ind., and colleagues wrote in an editorial accompanying the study by Dr. Coupland and associates (JAMA Intern Med. 2019 Jun 24. doi: 10.1001/jamainternmed.2019.0676).
While a pharmacologic basis for this association has been proposed, causation has yet to be established by means of prospective randomized studies. The current supposition is that deprescribing anticholinergic medications has the potential to positively effect cholinergic neurotransmission in certain regions of the brain, which could lead to improved cognitive functioning, and lower the likelihood of developing Alzheimer’s disease and related dementias, they wrote in the editorial.
However, the discontinuation of some anticholinergic agents may pose other risks, such as worsening pain or depressive symptoms, in addition to increasing the utilization of acute care facilities. As a result, high-quality, well-designed, randomized trials are needed to better understand the long-term effects of deprescribing anticholinergic medications. These trials would help inform clinicians, patients, and policymakers about the risks and benefits of deprescribing interventions, Dr. Campbell and coauthors said.
The study was supported by the National Institute for Health Research and the University of Nottingham. The authors reported financial affiliations with ClinRisk Ltd. The authors of the editorial reported receiving support from the National Institute on Aging and the Agency for Healthcare Research and Quality. Dr. Campbell reported receiving personal fees from Astellas Pharma US.
SOURCE: Coupland C et al. JAMA Intern Med. 2019 Jun 24. doi: 10.1001/jamainternmed.2019.0677
Exposures to various types of anticholinergic medications were associated with a significantly increased risk of dementia in people aged 55 years or older in a large pharmacoepidemiologic study.
“This study was designed to assess the association between cumulative anticholinergic drug use and risk of dementia in a large, representative British population,” wrote Carol A. C. Coupland, PhD, of the division of primary care at the University of Nottingham (England), and colleagues. The findings were published in JAMA Internal Medicine.
The researchers conducted a large nested case-control study that included 58,769 patients with dementia and 225,574 matched controls from the QResearch database in England. Each study participant was matched to five controls based on various characteristics, including sex, age, and calendar time, among others.
Prescription data related to 56 different drugs with strong anticholinergic properties, including antipsychotics, bladder antimuscarinics, antiepileptics, antiparkinson agents, and antidepressants were used to measure drug exposure. The study data were analyzed from 2016 to 2018.
“The primary exposure was the total standardized daily doses (TSDDs) of anticholinergic drugs prescribed in the 1 to 11 years prior to the date of diagnosis of dementia or equivalent date in matched controls,” Dr. Coupland and colleagues wrote.
After analysis, the researchers found that exposure to antipsychotics (adjusted odds ratio, 1.70), bladder antimuscarinics (aOR, 1.65), antiepileptics (aOR, 1.39), antiparkinson agents (aOR, 1.52), and anticholinergic antidepressants (aOR, 1.29) was associated with an increased risk of dementia after adjustment for confounding factors.
“Associations were stronger in [dementia] cases diagnosed before the age of 80 years,” the researchers noted.
However, antihistamine, antivertigo/antiemetic, skeletal muscle relaxant, gastrointestinal antispasmodic, antiarrhythmic, and antimuscarinic bronchodilator anticholinergic agents were not associated with any increased risk of dementia.
One key limitation of the study was the absence of medication compliance assessment, which could result in exposure misclassification. Dr. Coupland and colleagues acknowledged this could underestimate some associations with medication exposure.
The stronger risk of dementia found among people who had dementia before age 80 “indicates that anticholinergic drugs should be prescribed with caution in middle-aged and older people,” they concluded.
One question that remains from the current study is whether anticholinergic drugs are a definite modifiable risk factor for Alzheimer’s disease and related dementias, Noll L. Campbell, PharmD, of Purdue University, West Lafayette, Ind., and colleagues wrote in an editorial accompanying the study by Dr. Coupland and associates (JAMA Intern Med. 2019 Jun 24. doi: 10.1001/jamainternmed.2019.0676).
While a pharmacologic basis for this association has been proposed, causation has yet to be established by means of prospective randomized studies. The current supposition is that deprescribing anticholinergic medications has the potential to positively effect cholinergic neurotransmission in certain regions of the brain, which could lead to improved cognitive functioning, and lower the likelihood of developing Alzheimer’s disease and related dementias, they wrote in the editorial.
However, the discontinuation of some anticholinergic agents may pose other risks, such as worsening pain or depressive symptoms, in addition to increasing the utilization of acute care facilities. As a result, high-quality, well-designed, randomized trials are needed to better understand the long-term effects of deprescribing anticholinergic medications. These trials would help inform clinicians, patients, and policymakers about the risks and benefits of deprescribing interventions, Dr. Campbell and coauthors said.
The study was supported by the National Institute for Health Research and the University of Nottingham. The authors reported financial affiliations with ClinRisk Ltd. The authors of the editorial reported receiving support from the National Institute on Aging and the Agency for Healthcare Research and Quality. Dr. Campbell reported receiving personal fees from Astellas Pharma US.
SOURCE: Coupland C et al. JAMA Intern Med. 2019 Jun 24. doi: 10.1001/jamainternmed.2019.0677
FROM JAMA INTERNAL MEDICINE
FDA approves drug to treat low sexual desire in women
The .
![]()
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment,” Hylton V. Joffe, MD, director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive, and Urologic Products, stated in a press release. “Today’s approval provides women with another treatment option for this condition.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not caused by a medical or psychiatric condition. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire, and generalized HSDD is a lack of desire that occurs regardless of the type of sexual activity, situation, or partner.
Vyleesi was studied in two 24-week, randomized, double-blind, placebo-controlled trials in 1,247 premenopausal women with acquired, generalized HSDD. The women used Vyleesi two or three times per month and no more than once a week. About one-quarter of patients treated with Vyleesi had an increase of 1.2 or more in their sexual desire score (scored on a range of 1.2 to 6.0, with higher scores indicating greater sexual desire), compared with about 17% of those who took placebo. About 35% of the patients treated with Vyleesi had a decrease of one or more in their distress score (scored on a range of 0-4, with higher scores indicating greater distress from low sexual desire) compared with about 31% of those who took placebo.
The drug is injected under the skin of the abdomen or thigh at least 45 minutes before anticipated sexual activity. Patients may decide the optimal time to use Vyleesi based on the duration of benefit and any side effects, such as nausea. Patients should not take more than one dose of Vyleesi within 24 hours, or more than eight doses per month. Patients should discontinue treatment after 8 weeks if they do not report an improvement in sexual desire and associated distress.
Vyleesi works by activating melanocortin receptors but the exact mechanism for improving sexual desire is unknown. Some side effects were reported. “The most common side effects of Vyleesi are nausea and vomiting, flushing, injection site reactions, and headache. About 40% of patients in the clinical trials experienced nausea, most commonly with the first Vyleesi injection, and 13% needed medications for the treatment of nausea. About 1% of patients treated with Vyleesi in the clinical trials reported darkening of the gums and parts of the skin, including the face and breasts, which did not go away in about half the patients after stopping treatment. Patients with dark skin were more likely to develop this side effect,” according to the press release.
A temporary increase in blood pressure in patients after dosing with Vyleesi was observed during the clinical trials and therefore the drug is not recommended in patients at high risk for cardiovascular disease. In addition, patients who take a naltrexone-containing medication by mouth to treat alcohol or opioid dependence should not use Vyleesi because it may significantly decrease the levels of naltrexone in the blood and could lead to naltrexone treatment failure.
The full press release can be found on the FDA website.
The .
![]()
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment,” Hylton V. Joffe, MD, director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive, and Urologic Products, stated in a press release. “Today’s approval provides women with another treatment option for this condition.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not caused by a medical or psychiatric condition. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire, and generalized HSDD is a lack of desire that occurs regardless of the type of sexual activity, situation, or partner.
Vyleesi was studied in two 24-week, randomized, double-blind, placebo-controlled trials in 1,247 premenopausal women with acquired, generalized HSDD. The women used Vyleesi two or three times per month and no more than once a week. About one-quarter of patients treated with Vyleesi had an increase of 1.2 or more in their sexual desire score (scored on a range of 1.2 to 6.0, with higher scores indicating greater sexual desire), compared with about 17% of those who took placebo. About 35% of the patients treated with Vyleesi had a decrease of one or more in their distress score (scored on a range of 0-4, with higher scores indicating greater distress from low sexual desire) compared with about 31% of those who took placebo.
The drug is injected under the skin of the abdomen or thigh at least 45 minutes before anticipated sexual activity. Patients may decide the optimal time to use Vyleesi based on the duration of benefit and any side effects, such as nausea. Patients should not take more than one dose of Vyleesi within 24 hours, or more than eight doses per month. Patients should discontinue treatment after 8 weeks if they do not report an improvement in sexual desire and associated distress.
Vyleesi works by activating melanocortin receptors but the exact mechanism for improving sexual desire is unknown. Some side effects were reported. “The most common side effects of Vyleesi are nausea and vomiting, flushing, injection site reactions, and headache. About 40% of patients in the clinical trials experienced nausea, most commonly with the first Vyleesi injection, and 13% needed medications for the treatment of nausea. About 1% of patients treated with Vyleesi in the clinical trials reported darkening of the gums and parts of the skin, including the face and breasts, which did not go away in about half the patients after stopping treatment. Patients with dark skin were more likely to develop this side effect,” according to the press release.
A temporary increase in blood pressure in patients after dosing with Vyleesi was observed during the clinical trials and therefore the drug is not recommended in patients at high risk for cardiovascular disease. In addition, patients who take a naltrexone-containing medication by mouth to treat alcohol or opioid dependence should not use Vyleesi because it may significantly decrease the levels of naltrexone in the blood and could lead to naltrexone treatment failure.
The full press release can be found on the FDA website.
The .
![]()
“There are women who, for no known reason, have reduced sexual desire that causes marked distress, and who can benefit from safe and effective pharmacologic treatment,” Hylton V. Joffe, MD, director of the Center for Drug Evaluation and Research’s Division of Bone, Reproductive, and Urologic Products, stated in a press release. “Today’s approval provides women with another treatment option for this condition.”
HSDD is characterized by low sexual desire that causes marked distress or interpersonal difficulty and is not caused by a medical or psychiatric condition. Acquired HSDD develops in a patient who previously experienced no problems with sexual desire, and generalized HSDD is a lack of desire that occurs regardless of the type of sexual activity, situation, or partner.
Vyleesi was studied in two 24-week, randomized, double-blind, placebo-controlled trials in 1,247 premenopausal women with acquired, generalized HSDD. The women used Vyleesi two or three times per month and no more than once a week. About one-quarter of patients treated with Vyleesi had an increase of 1.2 or more in their sexual desire score (scored on a range of 1.2 to 6.0, with higher scores indicating greater sexual desire), compared with about 17% of those who took placebo. About 35% of the patients treated with Vyleesi had a decrease of one or more in their distress score (scored on a range of 0-4, with higher scores indicating greater distress from low sexual desire) compared with about 31% of those who took placebo.
The drug is injected under the skin of the abdomen or thigh at least 45 minutes before anticipated sexual activity. Patients may decide the optimal time to use Vyleesi based on the duration of benefit and any side effects, such as nausea. Patients should not take more than one dose of Vyleesi within 24 hours, or more than eight doses per month. Patients should discontinue treatment after 8 weeks if they do not report an improvement in sexual desire and associated distress.
Vyleesi works by activating melanocortin receptors but the exact mechanism for improving sexual desire is unknown. Some side effects were reported. “The most common side effects of Vyleesi are nausea and vomiting, flushing, injection site reactions, and headache. About 40% of patients in the clinical trials experienced nausea, most commonly with the first Vyleesi injection, and 13% needed medications for the treatment of nausea. About 1% of patients treated with Vyleesi in the clinical trials reported darkening of the gums and parts of the skin, including the face and breasts, which did not go away in about half the patients after stopping treatment. Patients with dark skin were more likely to develop this side effect,” according to the press release.
A temporary increase in blood pressure in patients after dosing with Vyleesi was observed during the clinical trials and therefore the drug is not recommended in patients at high risk for cardiovascular disease. In addition, patients who take a naltrexone-containing medication by mouth to treat alcohol or opioid dependence should not use Vyleesi because it may significantly decrease the levels of naltrexone in the blood and could lead to naltrexone treatment failure.
The full press release can be found on the FDA website.
Medical cannabis laws appear no longer tied to drop in opioid overdose mortality
Correlations do not hold when analysis is expanded to 2017
Contrary to previous research indicating that medical cannabis laws reduced opioid overdose mortality, the association between these two has reversed, with opioid overdose mortality increased in states with comprehensive medical cannabis laws, according to Chelsea L. Shover, PhD, and associates.

The original research by Marcus A. Bachhuber, MD, and associates showed that the introduction of state medical cannabis laws was associated with a 24.8% reduction in opioid overdose deaths per 100,000 population between 1999 and 2010. In contrast, the new research – which looked at a longer time period than the original research did – found that the association between state medical cannabis laws and opioid overdose mortality reversed direction, from –21% to +23%.
“We find it unlikely that medical cannabis – used by about 2.5% of the U.S. population – has exerted large conflicting effects on opioid overdose mortality,” wrote Dr. Shover, of the department of psychiatry and behavioral sciences at Stanford (Calif.) University, and associates. “A more plausible interpretation is that this association is spurious.” Their study was published in the Proceedings of the National Academy of Sciences.
To conduct their analysis, Dr. Shover and associates extended the timeline reviewed by Dr. Bachhuber and associates to 2017. During 2010-2017, 32 states enacted medical cannabis laws, including 17 allowing only medical cannabis with low levels of tetrahydrocannabinol (THC), and 8 legalized recreational marijuana. In the expanded timeline during 1999-2017, states possessing a comprehensive medical marijuana law saw an increase in opioid overdose mortality of 28.2%. Meanwhile, states with recreational marijuana laws saw a decrease of 14.7% in opioid overdose mortality, and states with low-THC medical cannabis laws saw a decrease of 7.1%. However, the investigators noted that those values had wide confidence intervals, which indicates “compatibility with large range of true associations.”
Corporate actors with deep pockets have substantial ability to promote congenial results, and suffering people are desperate for effective solutions. Cannabinoids have demonstrated therapeutic benefits, but reducing population-level opioid overdose mortality does not appear to be among them,” Dr. Shover and associates noted.
Dr. Shover reported receiving support from National Institute on Drug Abuse and the Wu Tsai Neurosciences Institute. Another coauthor received support from the Veterans Health Administration, Wu Tsai Neurosciences Institute, and the Esther Ting Memorial Professorship at Stanford.
SOURCE: Shover CL et al. Proc Natl Acad Sci U S A. 2019 Jun 10. doi: 10.1073/pnas.1903434116.
Correlations do not hold when analysis is expanded to 2017
Correlations do not hold when analysis is expanded to 2017
Contrary to previous research indicating that medical cannabis laws reduced opioid overdose mortality, the association between these two has reversed, with opioid overdose mortality increased in states with comprehensive medical cannabis laws, according to Chelsea L. Shover, PhD, and associates.
The original research by Marcus A. Bachhuber, MD, and associates showed that the introduction of state medical cannabis laws was associated with a 24.8% reduction in opioid overdose deaths per 100,000 population between 1999 and 2010. In contrast, the new research – which looked at a longer time period than the original research did – found that the association between state medical cannabis laws and opioid overdose mortality reversed direction, from –21% to +23%.
“We find it unlikely that medical cannabis – used by about 2.5% of the U.S. population – has exerted large conflicting effects on opioid overdose mortality,” wrote Dr. Shover, of the department of psychiatry and behavioral sciences at Stanford (Calif.) University, and associates. “A more plausible interpretation is that this association is spurious.” Their study was published in the Proceedings of the National Academy of Sciences.
To conduct their analysis, Dr. Shover and associates extended the timeline reviewed by Dr. Bachhuber and associates to 2017. During 2010-2017, 32 states enacted medical cannabis laws, including 17 allowing only medical cannabis with low levels of tetrahydrocannabinol (THC), and 8 legalized recreational marijuana. In the expanded timeline during 1999-2017, states possessing a comprehensive medical marijuana law saw an increase in opioid overdose mortality of 28.2%. Meanwhile, states with recreational marijuana laws saw a decrease of 14.7% in opioid overdose mortality, and states with low-THC medical cannabis laws saw a decrease of 7.1%. However, the investigators noted that those values had wide confidence intervals, which indicates “compatibility with large range of true associations.”
Corporate actors with deep pockets have substantial ability to promote congenial results, and suffering people are desperate for effective solutions. Cannabinoids have demonstrated therapeutic benefits, but reducing population-level opioid overdose mortality does not appear to be among them,” Dr. Shover and associates noted.
Dr. Shover reported receiving support from National Institute on Drug Abuse and the Wu Tsai Neurosciences Institute. Another coauthor received support from the Veterans Health Administration, Wu Tsai Neurosciences Institute, and the Esther Ting Memorial Professorship at Stanford.
SOURCE: Shover CL et al. Proc Natl Acad Sci U S A. 2019 Jun 10. doi: 10.1073/pnas.1903434116.
Contrary to previous research indicating that medical cannabis laws reduced opioid overdose mortality, the association between these two has reversed, with opioid overdose mortality increased in states with comprehensive medical cannabis laws, according to Chelsea L. Shover, PhD, and associates.
The original research by Marcus A. Bachhuber, MD, and associates showed that the introduction of state medical cannabis laws was associated with a 24.8% reduction in opioid overdose deaths per 100,000 population between 1999 and 2010. In contrast, the new research – which looked at a longer time period than the original research did – found that the association between state medical cannabis laws and opioid overdose mortality reversed direction, from –21% to +23%.
“We find it unlikely that medical cannabis – used by about 2.5% of the U.S. population – has exerted large conflicting effects on opioid overdose mortality,” wrote Dr. Shover, of the department of psychiatry and behavioral sciences at Stanford (Calif.) University, and associates. “A more plausible interpretation is that this association is spurious.” Their study was published in the Proceedings of the National Academy of Sciences.
To conduct their analysis, Dr. Shover and associates extended the timeline reviewed by Dr. Bachhuber and associates to 2017. During 2010-2017, 32 states enacted medical cannabis laws, including 17 allowing only medical cannabis with low levels of tetrahydrocannabinol (THC), and 8 legalized recreational marijuana. In the expanded timeline during 1999-2017, states possessing a comprehensive medical marijuana law saw an increase in opioid overdose mortality of 28.2%. Meanwhile, states with recreational marijuana laws saw a decrease of 14.7% in opioid overdose mortality, and states with low-THC medical cannabis laws saw a decrease of 7.1%. However, the investigators noted that those values had wide confidence intervals, which indicates “compatibility with large range of true associations.”
Corporate actors with deep pockets have substantial ability to promote congenial results, and suffering people are desperate for effective solutions. Cannabinoids have demonstrated therapeutic benefits, but reducing population-level opioid overdose mortality does not appear to be among them,” Dr. Shover and associates noted.
Dr. Shover reported receiving support from National Institute on Drug Abuse and the Wu Tsai Neurosciences Institute. Another coauthor received support from the Veterans Health Administration, Wu Tsai Neurosciences Institute, and the Esther Ting Memorial Professorship at Stanford.
SOURCE: Shover CL et al. Proc Natl Acad Sci U S A. 2019 Jun 10. doi: 10.1073/pnas.1903434116.
FROM PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES