User login
FDA approves single-dose, oral bacterial vaginosis treatment
, thanks to its approval by the Food and Drug Administration.
Symbiomix Therapeutics announced Sept. 18 that the FDA had granted approval to secnidazole (Solosec) 2 g oral granules for the treatment of bacterial vaginosis in adult women. Clinical trials of the 5-nitroimidazole antibiotic have shown it to be effective in a single dose, offering the potential for greater patient adherence to treatment over the common regimen of twice-a-day dosing for 7 days.
In a phase 2, randomized, double-blind, dose-ranging, placebo-controlled study of 215 women with bacterial vaginosis, the clinical cure rate was 65.3% for the 2-g secnidazole group, 49.3% for the 1-g secnidazole group, and 19.4% for the placebo group (Obstet Gynecol. 2017 Aug;130[2]:379-86).
Similarly, in a phase 3 double-blind, placebo-controlled study with 189 women, clinical cure rates based on the 2016 FDA guidance were 64.0% for single-dose secnidazole 2 g versus 26.4% for placebo (Am J Obstet Gynecol. 2017 Sep 1. doi: 10.1016/j.ajog.2017.08.017).
The most common adverse events in the trials were vulvovaginal candidiasis (9.6%), headache (3.6%), nausea (3.6%), dysgeusia (3.4%), vomiting (2.5%), diarrhea (2.5%), abdominal pain (2.0%), and vulvovaginal pruritus (2.0%).
The FDA designated the drug as a qualified infectious disease product and granted it fast-track designation, making it eligible for priority review and at least 10 years of market exclusivity.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
, thanks to its approval by the Food and Drug Administration.
Symbiomix Therapeutics announced Sept. 18 that the FDA had granted approval to secnidazole (Solosec) 2 g oral granules for the treatment of bacterial vaginosis in adult women. Clinical trials of the 5-nitroimidazole antibiotic have shown it to be effective in a single dose, offering the potential for greater patient adherence to treatment over the common regimen of twice-a-day dosing for 7 days.
In a phase 2, randomized, double-blind, dose-ranging, placebo-controlled study of 215 women with bacterial vaginosis, the clinical cure rate was 65.3% for the 2-g secnidazole group, 49.3% for the 1-g secnidazole group, and 19.4% for the placebo group (Obstet Gynecol. 2017 Aug;130[2]:379-86).
Similarly, in a phase 3 double-blind, placebo-controlled study with 189 women, clinical cure rates based on the 2016 FDA guidance were 64.0% for single-dose secnidazole 2 g versus 26.4% for placebo (Am J Obstet Gynecol. 2017 Sep 1. doi: 10.1016/j.ajog.2017.08.017).
The most common adverse events in the trials were vulvovaginal candidiasis (9.6%), headache (3.6%), nausea (3.6%), dysgeusia (3.4%), vomiting (2.5%), diarrhea (2.5%), abdominal pain (2.0%), and vulvovaginal pruritus (2.0%).
The FDA designated the drug as a qualified infectious disease product and granted it fast-track designation, making it eligible for priority review and at least 10 years of market exclusivity.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
, thanks to its approval by the Food and Drug Administration.
Symbiomix Therapeutics announced Sept. 18 that the FDA had granted approval to secnidazole (Solosec) 2 g oral granules for the treatment of bacterial vaginosis in adult women. Clinical trials of the 5-nitroimidazole antibiotic have shown it to be effective in a single dose, offering the potential for greater patient adherence to treatment over the common regimen of twice-a-day dosing for 7 days.
In a phase 2, randomized, double-blind, dose-ranging, placebo-controlled study of 215 women with bacterial vaginosis, the clinical cure rate was 65.3% for the 2-g secnidazole group, 49.3% for the 1-g secnidazole group, and 19.4% for the placebo group (Obstet Gynecol. 2017 Aug;130[2]:379-86).
Similarly, in a phase 3 double-blind, placebo-controlled study with 189 women, clinical cure rates based on the 2016 FDA guidance were 64.0% for single-dose secnidazole 2 g versus 26.4% for placebo (Am J Obstet Gynecol. 2017 Sep 1. doi: 10.1016/j.ajog.2017.08.017).
The most common adverse events in the trials were vulvovaginal candidiasis (9.6%), headache (3.6%), nausea (3.6%), dysgeusia (3.4%), vomiting (2.5%), diarrhea (2.5%), abdominal pain (2.0%), and vulvovaginal pruritus (2.0%).
The FDA designated the drug as a qualified infectious disease product and granted it fast-track designation, making it eligible for priority review and at least 10 years of market exclusivity.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
FDA gives nod to first mobile app for substance use disorders
The Food and Drug Administration has permitted marketing of the first mobile medical application aimed at helping to treat substance use disorders (SUDs) in adults, the agency announced Sept. 14.
“This is an example of how innovative digital technologies can help provide patients access to additional tools during their treatment,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in a statement.
“More therapy tools means a greater potential to help improve outcomes ... for patients with substance use disorder,” Dr. Peña added.
The agency’s permission is based on data reviewed from a multisite, unblinded 12-week clinical trial sponsored by the National Institute on Drug Abuse of 399 patients who received either standard treatment or standard treatment with the addition of a desktop-based version of reSET. According to the data, adherence to abstinence for patients with alcohol, cocaine, marijuana, and stimulant SUD who used reSET increased 40.3%, compared with an increase of 17.6% in adherence to abstinence for patients who did not use the system.
No side effects are associated with the reSET application. Adverse events reported in clinical trials were in line with those found in patients with SUD, including cardiovascular disease, gastrointestinal events, depression, mania, suicidal behavior and ideation, and suicide attempts.
Edward V. Nunes, MD, said in a statement issued by the company that developed reSET, Pear Therapeutics, that the clinical outcomes found in the pivotal study were remarkable. “Clinically validated digital therapeutics may become a cornerstone of future treatment,” said Dr. Nunes, lead investigator in the study submitted to the FDA and a professor of psychiatry at Columbia University in New York.
For more information about the reSET medical application system, click here.
acruz@frontlinemedcom.com
On Twitter @abbbbeeeyyy
The Food and Drug Administration has permitted marketing of the first mobile medical application aimed at helping to treat substance use disorders (SUDs) in adults, the agency announced Sept. 14.
“This is an example of how innovative digital technologies can help provide patients access to additional tools during their treatment,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in a statement.
“More therapy tools means a greater potential to help improve outcomes ... for patients with substance use disorder,” Dr. Peña added.
The agency’s permission is based on data reviewed from a multisite, unblinded 12-week clinical trial sponsored by the National Institute on Drug Abuse of 399 patients who received either standard treatment or standard treatment with the addition of a desktop-based version of reSET. According to the data, adherence to abstinence for patients with alcohol, cocaine, marijuana, and stimulant SUD who used reSET increased 40.3%, compared with an increase of 17.6% in adherence to abstinence for patients who did not use the system.
No side effects are associated with the reSET application. Adverse events reported in clinical trials were in line with those found in patients with SUD, including cardiovascular disease, gastrointestinal events, depression, mania, suicidal behavior and ideation, and suicide attempts.
Edward V. Nunes, MD, said in a statement issued by the company that developed reSET, Pear Therapeutics, that the clinical outcomes found in the pivotal study were remarkable. “Clinically validated digital therapeutics may become a cornerstone of future treatment,” said Dr. Nunes, lead investigator in the study submitted to the FDA and a professor of psychiatry at Columbia University in New York.
For more information about the reSET medical application system, click here.
acruz@frontlinemedcom.com
On Twitter @abbbbeeeyyy
The Food and Drug Administration has permitted marketing of the first mobile medical application aimed at helping to treat substance use disorders (SUDs) in adults, the agency announced Sept. 14.
“This is an example of how innovative digital technologies can help provide patients access to additional tools during their treatment,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in a statement.
“More therapy tools means a greater potential to help improve outcomes ... for patients with substance use disorder,” Dr. Peña added.
The agency’s permission is based on data reviewed from a multisite, unblinded 12-week clinical trial sponsored by the National Institute on Drug Abuse of 399 patients who received either standard treatment or standard treatment with the addition of a desktop-based version of reSET. According to the data, adherence to abstinence for patients with alcohol, cocaine, marijuana, and stimulant SUD who used reSET increased 40.3%, compared with an increase of 17.6% in adherence to abstinence for patients who did not use the system.
No side effects are associated with the reSET application. Adverse events reported in clinical trials were in line with those found in patients with SUD, including cardiovascular disease, gastrointestinal events, depression, mania, suicidal behavior and ideation, and suicide attempts.
Edward V. Nunes, MD, said in a statement issued by the company that developed reSET, Pear Therapeutics, that the clinical outcomes found in the pivotal study were remarkable. “Clinically validated digital therapeutics may become a cornerstone of future treatment,” said Dr. Nunes, lead investigator in the study submitted to the FDA and a professor of psychiatry at Columbia University in New York.
For more information about the reSET medical application system, click here.
acruz@frontlinemedcom.com
On Twitter @abbbbeeeyyy
Briviact gets monotherapy approval for partial-onset seizures
A supplemental new drug application for Briviact (brivaracetam) CV as a monotherapy treatment for partial-onset seizures in patients aged 16 years and older with epilepsy received approval from the Food and Drug Administration on Sept. 15, according to an announcement from its manufacturer, UCB.
Brivaracetam is already approved in the United States as an adjunctive treatment for partial-onset seizures in patients in this age group. As a result, adults and adolescents aged 16 years and older with partial-onset seizures in the United States can now be initiated on brivaracetam as monotherapy or adjunctive therapy.
Brivaracetam is the newest antiepileptic drug (AED) in the ‘racetam’ class of medicines and demonstrates a high and selective affinity for synaptic vesicle protein 2A (SV2A) in the brain, which may contribute to its anticonvulsant effects. Gradual dose escalation is not required when initiating treatment with brivaracetam for monotherapy or adjunctive therapy. According to the prescribing information, it is available in three formulations: film-coated tablets (10-mg, 25-mg, 50-mg, 75-mg, and 100-mg strengths), oral solution (10 mg/mL), and injection (50 mg in a 5-mL single-dose vial).
Common adverse reactions reported in at least 5% of brivaracetam users and at least 2% more frequently than placebo are somnolence and sedation, dizziness, fatigue, and nausea and vomiting.
“This new monotherapy indication builds on an already strong and compelling clinical profile for Briviact, providing doctors the flexibility to tailor their choice of AED to match individual patient needs and circumstances,” explained Pavel Klein, MD, director of the Mid-Atlantic Epilepsy and Sleep Center, Bethesda, Md., in the UCB announcement. “In helping to progress their journey towards seizure freedom by providing a choice of treatment which can be initiated as monotherapy, at a therapeutic dose, from day 1, Briviact provides an additional treatment choice for neurologists and their patients.”
A supplemental new drug application for Briviact (brivaracetam) CV as a monotherapy treatment for partial-onset seizures in patients aged 16 years and older with epilepsy received approval from the Food and Drug Administration on Sept. 15, according to an announcement from its manufacturer, UCB.
Brivaracetam is already approved in the United States as an adjunctive treatment for partial-onset seizures in patients in this age group. As a result, adults and adolescents aged 16 years and older with partial-onset seizures in the United States can now be initiated on brivaracetam as monotherapy or adjunctive therapy.
Brivaracetam is the newest antiepileptic drug (AED) in the ‘racetam’ class of medicines and demonstrates a high and selective affinity for synaptic vesicle protein 2A (SV2A) in the brain, which may contribute to its anticonvulsant effects. Gradual dose escalation is not required when initiating treatment with brivaracetam for monotherapy or adjunctive therapy. According to the prescribing information, it is available in three formulations: film-coated tablets (10-mg, 25-mg, 50-mg, 75-mg, and 100-mg strengths), oral solution (10 mg/mL), and injection (50 mg in a 5-mL single-dose vial).
Common adverse reactions reported in at least 5% of brivaracetam users and at least 2% more frequently than placebo are somnolence and sedation, dizziness, fatigue, and nausea and vomiting.
“This new monotherapy indication builds on an already strong and compelling clinical profile for Briviact, providing doctors the flexibility to tailor their choice of AED to match individual patient needs and circumstances,” explained Pavel Klein, MD, director of the Mid-Atlantic Epilepsy and Sleep Center, Bethesda, Md., in the UCB announcement. “In helping to progress their journey towards seizure freedom by providing a choice of treatment which can be initiated as monotherapy, at a therapeutic dose, from day 1, Briviact provides an additional treatment choice for neurologists and their patients.”
A supplemental new drug application for Briviact (brivaracetam) CV as a monotherapy treatment for partial-onset seizures in patients aged 16 years and older with epilepsy received approval from the Food and Drug Administration on Sept. 15, according to an announcement from its manufacturer, UCB.
Brivaracetam is already approved in the United States as an adjunctive treatment for partial-onset seizures in patients in this age group. As a result, adults and adolescents aged 16 years and older with partial-onset seizures in the United States can now be initiated on brivaracetam as monotherapy or adjunctive therapy.
Brivaracetam is the newest antiepileptic drug (AED) in the ‘racetam’ class of medicines and demonstrates a high and selective affinity for synaptic vesicle protein 2A (SV2A) in the brain, which may contribute to its anticonvulsant effects. Gradual dose escalation is not required when initiating treatment with brivaracetam for monotherapy or adjunctive therapy. According to the prescribing information, it is available in three formulations: film-coated tablets (10-mg, 25-mg, 50-mg, 75-mg, and 100-mg strengths), oral solution (10 mg/mL), and injection (50 mg in a 5-mL single-dose vial).
Common adverse reactions reported in at least 5% of brivaracetam users and at least 2% more frequently than placebo are somnolence and sedation, dizziness, fatigue, and nausea and vomiting.
“This new monotherapy indication builds on an already strong and compelling clinical profile for Briviact, providing doctors the flexibility to tailor their choice of AED to match individual patient needs and circumstances,” explained Pavel Klein, MD, director of the Mid-Atlantic Epilepsy and Sleep Center, Bethesda, Md., in the UCB announcement. “In helping to progress their journey towards seizure freedom by providing a choice of treatment which can be initiated as monotherapy, at a therapeutic dose, from day 1, Briviact provides an additional treatment choice for neurologists and their patients.”
Aptiom approved for pediatric partial-onset seizures
The Food and Drug Administration has approved Aptiom (eslicarbazepine acetate) for the treatment of partial-onset seizures in children aged 4-17 years, according to an announcement from Sunovion Pharmaceuticals.
The approval was based on results of three clinical trials where eslicarbazepine was shown to be safe and well tolerated in pediatric populations. The efficacy of eslicarbazepine has been illustrated in clinical trials in adult populations, and data were extrapolated to support usage in pediatric patients. Eslicarbazepine has previously been approved to treat partial-onset seizures in adults.
Pediatric dosing of eslicarbazepine is based on weight, and the tablets, available in 200-mg, 400-mg, 600-mg, and 800-mg strengths, can be taken whole or crushed, with or without food, according to the prescribing information.
“The unpredictable nature of seizures can be disruptive in the lives of these young people and their families, friends, and community. It is important that physicians have additional treatment options that address patient needs,” Steven Wolf, MD, director of pediatric epilepsy at Mount Sinai Health System, said in the announcement.
The Food and Drug Administration has approved Aptiom (eslicarbazepine acetate) for the treatment of partial-onset seizures in children aged 4-17 years, according to an announcement from Sunovion Pharmaceuticals.
The approval was based on results of three clinical trials where eslicarbazepine was shown to be safe and well tolerated in pediatric populations. The efficacy of eslicarbazepine has been illustrated in clinical trials in adult populations, and data were extrapolated to support usage in pediatric patients. Eslicarbazepine has previously been approved to treat partial-onset seizures in adults.
Pediatric dosing of eslicarbazepine is based on weight, and the tablets, available in 200-mg, 400-mg, 600-mg, and 800-mg strengths, can be taken whole or crushed, with or without food, according to the prescribing information.
“The unpredictable nature of seizures can be disruptive in the lives of these young people and their families, friends, and community. It is important that physicians have additional treatment options that address patient needs,” Steven Wolf, MD, director of pediatric epilepsy at Mount Sinai Health System, said in the announcement.
The Food and Drug Administration has approved Aptiom (eslicarbazepine acetate) for the treatment of partial-onset seizures in children aged 4-17 years, according to an announcement from Sunovion Pharmaceuticals.
The approval was based on results of three clinical trials where eslicarbazepine was shown to be safe and well tolerated in pediatric populations. The efficacy of eslicarbazepine has been illustrated in clinical trials in adult populations, and data were extrapolated to support usage in pediatric patients. Eslicarbazepine has previously been approved to treat partial-onset seizures in adults.
Pediatric dosing of eslicarbazepine is based on weight, and the tablets, available in 200-mg, 400-mg, 600-mg, and 800-mg strengths, can be taken whole or crushed, with or without food, according to the prescribing information.
“The unpredictable nature of seizures can be disruptive in the lives of these young people and their families, friends, and community. It is important that physicians have additional treatment options that address patient needs,” Steven Wolf, MD, director of pediatric epilepsy at Mount Sinai Health System, said in the announcement.
FDA approves biosimilar to bevacizumab
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
The Food and Drug Administration has approved a biosimilar to bevacizumab (Avastin) for the treatment of certain colorectal, lung, brain, kidney, and cervical cancers.
Bevacizumab-awwb is the first biosimilar approved in the United States for the treatment of cancer, the FDA said in a press release.
Approval is based on structural and functional characterization, animal study data, human pharmacokinetic and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrate bevacizumab-awwb is biosimilar to bevacizumab, the FDA said.
• Metastatic colorectal cancer, in combination with intravenous 5-fluorouracil-based chemotherapy for first- or second-line treatment.
• Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy for the second-line treatment of patients who have progressed on a first-line bevacizumab product–containing regimen.
• Non-squamous non–small cell lung cancer, in combination with carboplatin and paclitaxel for first line treatment of unresectable, locally advanced, recurrent, or metastatic disease.
• Glioblastoma with progressive disease following prior therapy, based on improvement in objective response rate.
• Metastatic renal cell carcinoma, in combination with interferon alfa.
• Cervical cancer that is persistent, recurrent, or metastatic, in combination with paclitaxel and cisplatin or paclitaxel and topotecan.
Common expected side effects of the biosimilar include epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, hemorrhage, lacrimation disorder, back pain, and exfoliative dermatitis.
Serious expected side effects include perforation or fistula, arterial and venous thromboembolic events, hypertension, posterior reversible encephalopathy syndrome, proteinuria, infusion-related reactions, and ovarian failure. Women who are pregnant should not take bevacizumab-awwb.
The biosimilar to bevacizumab carries a similar boxed warning regarding the increased risk of gastrointestinal perforations; surgery and wound healing complications; and severe or fatal pulmonary, gastrointestinal, central nervous system, and vaginal hemorrhage.
The biosimilar approval was granted to Amgen, which will market the drug under the trade name Mvasi.
Adjuvant-boosted shingles vaccine earns FDA panel’s unanimous nod
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
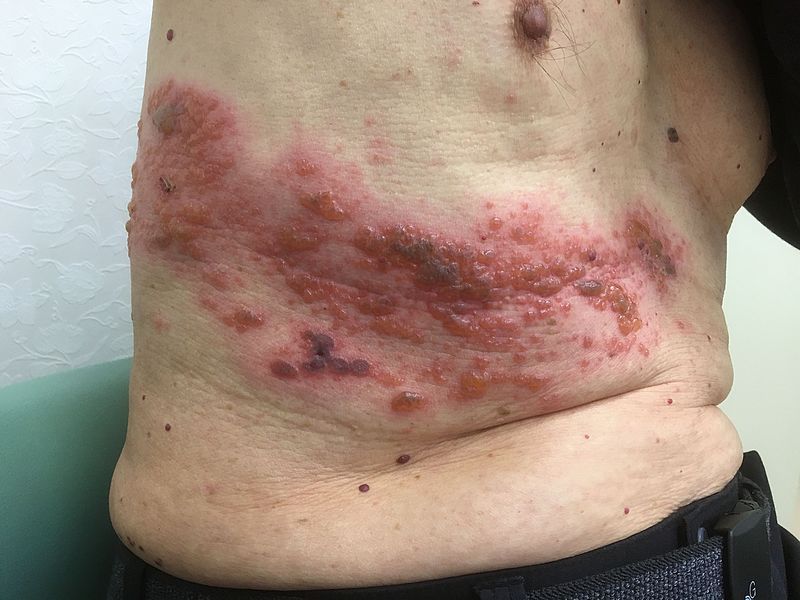
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
A new vaccine for herpes zoster is both safe and effective in preventing herpes zoster, and in reducing the incidence of postherpetic neuralgia in older adults, according to a Food and Drug Administration advisory committee, which voted unanimously to recommend the vaccine.
The FDA generally follows the recommendations of its advisory committees.
The recombinant vaccine, dubbed HZ/su during the trial phase, showed efficacy of 97.2% against herpes zoster infection in adults aged 50 years and older, and 91.3% in adults aged 70 years and older. The effect persisted for up to the 4 years of study follow-up.
HZ/su had a generally favorable safety profile, though early constitutional symptoms and local site reactions were common, according to data presented by GlaxoSmithKline. HZ/su uses an adjuvant not found in any other U.S.-approved vaccine.
The incidence of postherpetic neuralgia, a common, persistent, and costly complication of herpes zoster, was 0.1 per 1,000 person-years in those receiving vaccine, compared with 0.9-1.2 per 1,000 person-years for those receiving placebo in the pivotal clinical trials for a median follow-up of 4 years.
In the vaccine’s pivotal clinical trials, efficacy was significantly higher than the levels seen for the only currently approved zoster live vaccine, Zostavax, especially for older populations. Zostavax’s efficacy for those aged 50-59 years is 69.8%, dropping to 18% for those aged 80 years or older.
The results of the two pivotal clinical trials were presented and analyzed by the sponsor and by FDA staff during a meeting of the Vaccines and Related Biological Products Advisory Committee of the FDA’s Center for Biologics Evaluation and Research (CBER).
During pre-vote discussions, committee members were unanimous in noting with favor the high and sustained efficacy seen for HZ/su in the trial data, especially for older populations. However, some participants wondered about the generalizability of both safety and efficacy data to all populations, given the very low trial enrollment numbers for Africans, African Americans, and individuals of Hispanic origin.
The two studies, Zoster-006 and Zoster-022, were similar in design and were conducted in parallel across 18 countries; data were able to be pooled for key efficacy and safety outcomes. Study Zoster-006 enrolled patients aged 50 years and older, while study Zoster-022 began enrollment at age 70. Patients were randomized to receive vaccine or placebo, and were followed for a median of 3.1 years for efficacy in Zoster-006 and a median of 3.9 years for Zoster-022. Safety data were obtained for a median 4.4 years for both studies.
The primary outcome measure for both studies in pooled analysis was the vaccine’s effectiveness against herpes zoster and postherpetic neuropathy in adults aged 70 and over. Safety was also assessed using pooled data.
The United States was represented by 3,934 of more than 29,000 patients enrolled globally. The remainder were primarily in Western Europe, with some sites in Australia and eastern Asia, Canada, and Latin America.
The vaccine consists of a recombinant, lyophilized truncated form of the varicella zoster virus (VZV) glycoprotein E (gE) antigen protein that, at the time of administration, is reconstituted with a novel adjuvant suspension. The antigen selection was based on the fact that gE is expressed on the surface of infected cells and is the target of both humoral and cellular immune responses in the host, said GSK’s Arnaud Didierlaurent, PhD, director and head of the adjuvant platform for GSK Vaccine’s Belgium research and development division.
The adjuvant, termed ASO1B, is not currently in use for any U.S.-approved vaccine, though it was developed more than 20 years ago, said Dr. Didierlaurent. Its combination with recombinant VZV gE was found to significantly boost the antigen’s immunogenicity during GSK’s vaccine development program. The adjuvant enhances a transient innate response in the first 3 days after administration that later helps maintain durably high levels of gE-specific antibodies and strengthens gE-specific cell-mediated immunity.
Mechanistically, the robust initial innate response is responsible for the constitutional symptoms and local site reactions seen in pooled data from the two pivotal clinical trials: 70%-85% of participants receiving HZ/su reported injection site pain, 38% of participants receiving HZ/su reported redness, and about a quarter reported swelling. By comparison, 9%-13% of those receiving placebo reported injection site pain, and about 1% reported redness and swelling.
Fatigue, headache, mild fever, myalgia, and shivering were all more common in those receiving HZ/su; both local and generalized symptoms were more common in younger recipients.
“I think this is a very good case for the first licensure of this adjuvant in the United States, because the efficacy seems pretty compelling, the disease is morbid, and there are a lot of people whose lives would be changed,” said committee member Sarah Long, MD, professor of pediatrics at Drexel University, Philadelphia.
Both the GSK and FDA presentations were in agreement that serious adverse events were in the range to be expected for an older population, and balanced across study arms. However, particular attention will be given to certain potential complications during the proposed pharmacovigilance plan.
“An imbalance toward vaccine versus placebo was observed” for gout, optic ischemic neuropathy, amyotrophic lateral sclerosis, osteonecrosis, convulsion-type reactions, and supraventricular tachycardias. “All are an adverse event of interest and will be included in planned targeted safety study,” said Dr. Didierlaurent.
Several committee members remarked on the difficulty of evaluating vaccine safety in an older population, where analysis takes place against the backdrop of more comorbidities and acute illnesses than in the younger population.
“There has been a thoughtful job both by the sponsor and by CBER in looking at complicated data,” said Melinda Wharton, MD, the director of the immunization services division of the National Center for Immunization and Respiratory Diseases at the Centers for Disease Control and Prevention, Atlanta.
The committee’s chair, Kathryn Edwards, MD, agreed. “I applaud the comprehensive analysis of all these safety signals. Both the sponsor and the FDA have done a wonderful job of drilling down and answering these questions,” she said. Dr. Edwards is the Sarah H. Sell and Cornelius Vanderbilt chair in pediatrics at Vanderbilt University, Nashville, Tenn.
Herpes zoster, a reactivation of the varicella virus that lies dormant in dorsal root or cranial nerve ganglia from earlier infection, is seen in about 1 million cases per year in the United States, with about 100,000 to 200,000 cases of postherpetic neuralgia occurring, said Jeffrey Cohen, MD, chief of the laboratory of infectious diseases at the National Institute of Allergy and Infectious Diseases, Bethesda, Md. The rates of herpes zoster are increasing in the United States for unknown reasons, and direct medical costs may currently exceed $1 billion annually, he said.
Each 0.5 mL dose of the HZ/su vaccine contains 50 mcg each of the recombinant VZV gE antigen and each of the two component parts of the ASO1B adjuvant. Two doses of the vaccine are administered intramuscularly 2-6 months apart. Dose-ranging studies were conducted before the pivotal clinical trials to ascertain the optimal dose of all of the vaccine components, the need for two doses, and the optimal spacing between doses.
All committee participants submitted conflict of interest statements to the FDA, and any potential conflicts were resolved before the hearing.
koakes@frontlinemedcom.com
On Twitter @karioakes
FDA advisory committee rejects opioids in children’s cough syrup
ROCKVILLE, MD – The majority of
The voting was broken into multiple votes based on age range of patients and the specific opioid present in the cough syrup. Unlike other advisory committee meetings, this meeting did not focus on a the treatment of a disease state, but rather on the treatment of a symptom.
On Sept. 11, 2017, the FDA’s Pediatric Advisory Committee voted 21 no, 2 yes, with one abstention, that the benefit versus the risk of opioid cough suppressants for pediatric patients was not favorable.
This vote was preceded by two previous votes specifically questioning the use of codeine and hydrocodone in medications for pediatric patients. For codeine, the committee voted unanimously with 24 against that the benefit versus risk was not favorable in pediatric patients aged 12 years to less than 18 years. 
For hydrocodone, the committee asked two questions: 1) Was the benefit versus risk favorable for pediatric patients aged 6 years to less than 12 years? and 2) Was the benefit versus risk favorable for pediatric patients aged 12 years to less than 18 years? On the vote for patients aged 6 years to less than 12 years, the committee voted 23 no, 1 yes with no abstention. For the patients aged 12 years to less than 18, the committee voted 23 no, 1 yes, with no abstention.
According to Sharon Levy, MD, MPH, adolescents are the most at-risk population for opioid misuse. This susceptibility is due to the developmental neurobiology of adolescent brains. A region of the brain associated with the reward pathway, nucleus accumbens, is developing in adolescents and plays a role in salience. Salience, or the differentiation between important vs. unimportant rewards, varies widely by age group. Young children show little salience with rewards, and treat rewards equivocally. Adults have a proportional response to rewards with accurate salience. Adolescents, on the other hand, are unhappy with small rewards, but receive a massive return with large rewards. This type of neurobiological feedback makes adolescents “vulnerable to develop substance use disorders.”
Dr. Levy also noted a correlation between prescribed opioid use and alcohol, marijuana, and tobacco use as contributing factors to opioid misuse. When opioids are prescribed for pain management, there is an adjusted odds ratio (AOR) of 1.33, indicating a high likelihood of misuse. Similar AORs are seen in adolescents who have used marijuana, cigarettes, and alcohol: 2.44, 1.25, and 1.23, respectively.
Sovereign pharmaceuticals representative Leonard Lawrence presented the findings of a pharmacokinetic study for hydrocodone and guaifenesin in 25-35 pediatric patients evenly divided into groups aged 6 years to less than 12 years, and 12 years to less than 18 years. According to Mr. Lawrence, codeine appears “to be a greater risk in children younger than 12 years, and should not be used” because of difficulty breathing. Mr Lawrence went on to say that these effects were exacerbated in obese children with lung disease or obstructive sleep apnea.
Victor S. Sloan, MD, of UCB in Brussels, presented an internal review of Tussionex, a combination cough medicine (hydrocodone/chlorpheniramine). This review took into account modern pharmacovigilance methods, changes in clinical practice, and a literature review. “Upon annual review, UCB determined that benefit risk balance for use of Tussionex for cough in children was no longer favorable,” said Dr. Sloan. Based on the results of the review, UCB has filed a label supplement to limit use of Tussionex to patients aged 18 years or older.
“Codeine, in particular, is an antiquated drug,” said Kathleen Neville, MD, pediatrics and clinical pharmacology section chief of Arkansas Children’s Hospital, Little Rock. Many of the committee members echoed Dr. Neville’s opinion.
The committee members had no relevant financial disclosures.
ilacy@frontlinemedcom.com
On Twitter @ilacy_19
ROCKVILLE, MD – The majority of
The voting was broken into multiple votes based on age range of patients and the specific opioid present in the cough syrup. Unlike other advisory committee meetings, this meeting did not focus on a the treatment of a disease state, but rather on the treatment of a symptom.
On Sept. 11, 2017, the FDA’s Pediatric Advisory Committee voted 21 no, 2 yes, with one abstention, that the benefit versus the risk of opioid cough suppressants for pediatric patients was not favorable.
This vote was preceded by two previous votes specifically questioning the use of codeine and hydrocodone in medications for pediatric patients. For codeine, the committee voted unanimously with 24 against that the benefit versus risk was not favorable in pediatric patients aged 12 years to less than 18 years.
For hydrocodone, the committee asked two questions: 1) Was the benefit versus risk favorable for pediatric patients aged 6 years to less than 12 years? and 2) Was the benefit versus risk favorable for pediatric patients aged 12 years to less than 18 years? On the vote for patients aged 6 years to less than 12 years, the committee voted 23 no, 1 yes with no abstention. For the patients aged 12 years to less than 18, the committee voted 23 no, 1 yes, with no abstention.
According to Sharon Levy, MD, MPH, adolescents are the most at-risk population for opioid misuse. This susceptibility is due to the developmental neurobiology of adolescent brains. A region of the brain associated with the reward pathway, nucleus accumbens, is developing in adolescents and plays a role in salience. Salience, or the differentiation between important vs. unimportant rewards, varies widely by age group. Young children show little salience with rewards, and treat rewards equivocally. Adults have a proportional response to rewards with accurate salience. Adolescents, on the other hand, are unhappy with small rewards, but receive a massive return with large rewards. This type of neurobiological feedback makes adolescents “vulnerable to develop substance use disorders.”
Dr. Levy also noted a correlation between prescribed opioid use and alcohol, marijuana, and tobacco use as contributing factors to opioid misuse. When opioids are prescribed for pain management, there is an adjusted odds ratio (AOR) of 1.33, indicating a high likelihood of misuse. Similar AORs are seen in adolescents who have used marijuana, cigarettes, and alcohol: 2.44, 1.25, and 1.23, respectively.
Sovereign pharmaceuticals representative Leonard Lawrence presented the findings of a pharmacokinetic study for hydrocodone and guaifenesin in 25-35 pediatric patients evenly divided into groups aged 6 years to less than 12 years, and 12 years to less than 18 years. According to Mr. Lawrence, codeine appears “to be a greater risk in children younger than 12 years, and should not be used” because of difficulty breathing. Mr Lawrence went on to say that these effects were exacerbated in obese children with lung disease or obstructive sleep apnea.
Victor S. Sloan, MD, of UCB in Brussels, presented an internal review of Tussionex, a combination cough medicine (hydrocodone/chlorpheniramine). This review took into account modern pharmacovigilance methods, changes in clinical practice, and a literature review. “Upon annual review, UCB determined that benefit risk balance for use of Tussionex for cough in children was no longer favorable,” said Dr. Sloan. Based on the results of the review, UCB has filed a label supplement to limit use of Tussionex to patients aged 18 years or older.
“Codeine, in particular, is an antiquated drug,” said Kathleen Neville, MD, pediatrics and clinical pharmacology section chief of Arkansas Children’s Hospital, Little Rock. Many of the committee members echoed Dr. Neville’s opinion.
The committee members had no relevant financial disclosures.
ilacy@frontlinemedcom.com
On Twitter @ilacy_19
ROCKVILLE, MD – The majority of
The voting was broken into multiple votes based on age range of patients and the specific opioid present in the cough syrup. Unlike other advisory committee meetings, this meeting did not focus on a the treatment of a disease state, but rather on the treatment of a symptom.
On Sept. 11, 2017, the FDA’s Pediatric Advisory Committee voted 21 no, 2 yes, with one abstention, that the benefit versus the risk of opioid cough suppressants for pediatric patients was not favorable.
This vote was preceded by two previous votes specifically questioning the use of codeine and hydrocodone in medications for pediatric patients. For codeine, the committee voted unanimously with 24 against that the benefit versus risk was not favorable in pediatric patients aged 12 years to less than 18 years.
For hydrocodone, the committee asked two questions: 1) Was the benefit versus risk favorable for pediatric patients aged 6 years to less than 12 years? and 2) Was the benefit versus risk favorable for pediatric patients aged 12 years to less than 18 years? On the vote for patients aged 6 years to less than 12 years, the committee voted 23 no, 1 yes with no abstention. For the patients aged 12 years to less than 18, the committee voted 23 no, 1 yes, with no abstention.
According to Sharon Levy, MD, MPH, adolescents are the most at-risk population for opioid misuse. This susceptibility is due to the developmental neurobiology of adolescent brains. A region of the brain associated with the reward pathway, nucleus accumbens, is developing in adolescents and plays a role in salience. Salience, or the differentiation between important vs. unimportant rewards, varies widely by age group. Young children show little salience with rewards, and treat rewards equivocally. Adults have a proportional response to rewards with accurate salience. Adolescents, on the other hand, are unhappy with small rewards, but receive a massive return with large rewards. This type of neurobiological feedback makes adolescents “vulnerable to develop substance use disorders.”
Dr. Levy also noted a correlation between prescribed opioid use and alcohol, marijuana, and tobacco use as contributing factors to opioid misuse. When opioids are prescribed for pain management, there is an adjusted odds ratio (AOR) of 1.33, indicating a high likelihood of misuse. Similar AORs are seen in adolescents who have used marijuana, cigarettes, and alcohol: 2.44, 1.25, and 1.23, respectively.
Sovereign pharmaceuticals representative Leonard Lawrence presented the findings of a pharmacokinetic study for hydrocodone and guaifenesin in 25-35 pediatric patients evenly divided into groups aged 6 years to less than 12 years, and 12 years to less than 18 years. According to Mr. Lawrence, codeine appears “to be a greater risk in children younger than 12 years, and should not be used” because of difficulty breathing. Mr Lawrence went on to say that these effects were exacerbated in obese children with lung disease or obstructive sleep apnea.
Victor S. Sloan, MD, of UCB in Brussels, presented an internal review of Tussionex, a combination cough medicine (hydrocodone/chlorpheniramine). This review took into account modern pharmacovigilance methods, changes in clinical practice, and a literature review. “Upon annual review, UCB determined that benefit risk balance for use of Tussionex for cough in children was no longer favorable,” said Dr. Sloan. Based on the results of the review, UCB has filed a label supplement to limit use of Tussionex to patients aged 18 years or older.
“Codeine, in particular, is an antiquated drug,” said Kathleen Neville, MD, pediatrics and clinical pharmacology section chief of Arkansas Children’s Hospital, Little Rock. Many of the committee members echoed Dr. Neville’s opinion.
The committee members had no relevant financial disclosures.
ilacy@frontlinemedcom.com
On Twitter @ilacy_19
AT AN FDA PEDIATRIC ADVISORY COMMITTEE MEETING
FDA clears first 2D mammography device with patient-controlled compression
The Food and Drug Administration has granted premarket clearance to the first 2D digital mammography system that allows patients to control the amount of compression applied to their own breasts before the mammogram x-ray is taken.
Senographe Pristina with Self-Compression uses a handheld wireless remote control that allows women to adjust the compression force after breast positioning and during a mammography exam. The technologist positions the patient and initiates compression, then guides the patient to gradually increase compression using the remote control until adequate compression is reached. The technologist checks the applied compression and breast positioning and makes the final decision on whether the compression is adequate or needs to be adjusted, according to the FDA’s Sept. 1 announcement.
“Regular mammograms are an important tool in detecting breast cancer,” Alberto Gutierrez, PhD, director of the FDA’s Office of In Vitro Diagnostics and Radiological Health, said in a statement. “However, some patients may experience anxiety or stress about the discomfort from the compression during the mammogram. This device allows patients some control over the amount of compression for their exam.”
Read more details of the clearance on the FDA’s website.
The Food and Drug Administration has granted premarket clearance to the first 2D digital mammography system that allows patients to control the amount of compression applied to their own breasts before the mammogram x-ray is taken.
Senographe Pristina with Self-Compression uses a handheld wireless remote control that allows women to adjust the compression force after breast positioning and during a mammography exam. The technologist positions the patient and initiates compression, then guides the patient to gradually increase compression using the remote control until adequate compression is reached. The technologist checks the applied compression and breast positioning and makes the final decision on whether the compression is adequate or needs to be adjusted, according to the FDA’s Sept. 1 announcement.
“Regular mammograms are an important tool in detecting breast cancer,” Alberto Gutierrez, PhD, director of the FDA’s Office of In Vitro Diagnostics and Radiological Health, said in a statement. “However, some patients may experience anxiety or stress about the discomfort from the compression during the mammogram. This device allows patients some control over the amount of compression for their exam.”
Read more details of the clearance on the FDA’s website.
The Food and Drug Administration has granted premarket clearance to the first 2D digital mammography system that allows patients to control the amount of compression applied to their own breasts before the mammogram x-ray is taken.
Senographe Pristina with Self-Compression uses a handheld wireless remote control that allows women to adjust the compression force after breast positioning and during a mammography exam. The technologist positions the patient and initiates compression, then guides the patient to gradually increase compression using the remote control until adequate compression is reached. The technologist checks the applied compression and breast positioning and makes the final decision on whether the compression is adequate or needs to be adjusted, according to the FDA’s Sept. 1 announcement.
“Regular mammograms are an important tool in detecting breast cancer,” Alberto Gutierrez, PhD, director of the FDA’s Office of In Vitro Diagnostics and Radiological Health, said in a statement. “However, some patients may experience anxiety or stress about the discomfort from the compression during the mammogram. This device allows patients some control over the amount of compression for their exam.”
Read more details of the clearance on the FDA’s website.
Q&A: CDC director Brenda Fitzgerald stresses ‘science and service’
Brenda Fitzgerald, MD, an ob.gyn. and most recently the public health commissioner for Georgia, was named the 17th director of the Centers for Disease Control and Prevention in July.
Dr. Fitzgerald comes to the CDC after 6 years as commissioner and state health officer for the Georgia Department of Public Health. She also has been a health care policy adviser to former House Speaker Newt Gingrich (R-Ga.) and ran for Congress herself in the 1990s. In addition to practicing medicine for 3 decades, she has served on the board and as president of the Georgia Obstetrical and Gynecological Society.

Question: What are you looking forward to as the new CDC director, and what are some of your top goals during your tenure?
Answer: The federal government holds responsibility to provide for the common defense. And CDC provides the common defense of the country against health threats. That’s why CDC’s mission of saving lives and protecting people means so much to me. As a doctor and a mother, I want to do everything I can to protect our future generations and keep them safe, whether it be from infectious or chronic diseases, natural or man-made disasters.
We are going to continue a legacy of protecting Americans from the things that we’ve been fighting for a long time, like heart disease and cancer, and from newer things like opioid overdose and the Zika virus. And we’ll continue to prepare for the next health threat, whatever it may be.
Q: How does your experience as an ob.gyn. shape your approach to this job?
A: There are two things that are important at CDC: science and service. CDC researchers are at the forefront of scientific research – and also provide a service by using scientific discoveries to improve health. That is the same approach I used as a practicing ob.gyn.: I brought the clinical lens that looked at the science, and then my work with my patients emphasized the service part.
As a practicing ob.gyn., it was my responsibility to address the questions and concerns of my patients. At CDC, I am just as committed to making science-based decisions and working with our experts to make sure doctors and patients have the state-of-the-art health information they need. I said when I went from being a clinician to being the state health official for Georgia that I went from treating one patient at a time to treating 10 million at a time. Now that’s changed to over 300 million.
Even though I am now CDC director and protecting the health of millions of people, my experiences as an ob.gyn. for 3 decades will forever shape how I work to improve the health of individual people and patients.
Q: What are the next steps for the agency in terms of its response to Zika virus?
A: Zika is the first infectious disease in 50 years that has been linked to birth defects. We know that the virus attacks neural tissue. We know that about 10% of babies are born with severe birth defects – and some babies born apparently normal later turned out to have neurologic problems, like blindness or deafness. And we suspect that there may be other problems we don’t know about yet.
I’m particularly concerned about developmental delays, as we know that early brain development is key to health and achievement for a child’s future. We are still learning more about Zika every day. It’s crucial that we – the health care and public health communities – work together and remain vigilant to ensure these babies receive the care they need. What Zika has taught us is that we need to establish a pregnancy and infant registry that can flag patterns of concerning health issues so that we can work together with health officials and clinicians to get ahead of emerging health threats.
Q: How can clinicians stay up to date on the rapidly changing guidance during an outbreak like Zika?
A: During outbreaks like Zika, public health officials are continually updating guidance so clinicians can get the latest information. You can check the latest CDC guidance about testing and follow-up care for pregnant women at www.cdc.gov/zika/hc-providers. Health care providers can also contact their state, local, or territorial health department to ensure the appropriate tests are ordered and interpreted correctly. CDC also maintains a 24/7 Zika consultation service for health officials and healthcare providers caring for pregnant women with possible Zika exposure.
Q: What steps will you take to decrease maternal mortality and to understand why this number is rising?
A: As an obstetrician, survival of mothers and babies is paramount to me. It’s so basic: mothers shouldn’t die. Sadly, about 700 women die each year in the U.S. as a result of pregnancy or delivery complications.
We know the leading causes of maternal mortality: cardiovascular disease, infections or sepsis, hemorrhage, and cardiomyopathy. We also know that an increasing number of pregnant women in the U.S. have chronic health conditions such as high blood pressure, diabetes, or heart disease that puts them at risk of pregnancy complications or death.
CDC is committed to preventing pregnancy-related deaths and ensuring the best possible birth outcomes. We do this by conducting surveillance, supporting states in developing recommendations, and working with partners to promote evidence-based recommendations and best practices. State-based initiatives are important allies in bringing together OBs and the public health sector to show the importance of statewide efforts to reduce maternal mortality. There has been some exciting work done by the California Maternal Quality Care Collaborative, which has created a series of toolkits designed to help health care providers with pregnancy or childbirth complications. CMQCC’s work is inspiring other states to follow suit.
The best coalitions are between ob.gyns., public health, and hospital associations. For example, AWHONN’s [Association of Women’s Health, Obstetric and Neonatal Nurses] Postpartum Hemorrhage initiative in Georgia, New Jersey, and the District of Columbia aimed to improve the treatment of obstetric hemorrhage. Even though 54% to 93% of maternal hemorrhage-related deaths are preventable with improved clinical response, not one single hospital in Georgia or New Jersey had implemented all the recommended preparedness elements for postpartum hemorrhage events. We can save mothers’ lives – we know what to do. It just takes all of us – ob.gyns., public health and hospital associations – leading the way to make sure it happens.
Q: How can we increase HPV vaccination?
A: The latest news is that more girls and boys are getting the HPV vaccine. New CDC data showed that 60% of teens aged 13-17 had received one or more doses of HPV vaccine in 2016 – an increase of 4% from 2015. Plus, the latest statistics show that HPV vaccination has led to significant drops in HPV infections: HPV-related cancers and genital warts dropped by 71% among teen girls and 61% among young women. Still, too many children aren’t completing the HPV vaccine series, leaving them vulnerable to cancers caused by HPV infection.
Providers can help increase HPV vaccination rates by using every patient visit to review vaccination histories, providing strong clinical recommendations and education to parents for HPV and other recommended vaccines, and implementing systems to minimize missed opportunities.
agallegos@frontlinemedcom.com
On Twitter @legal_med
Brenda Fitzgerald, MD, an ob.gyn. and most recently the public health commissioner for Georgia, was named the 17th director of the Centers for Disease Control and Prevention in July.
Dr. Fitzgerald comes to the CDC after 6 years as commissioner and state health officer for the Georgia Department of Public Health. She also has been a health care policy adviser to former House Speaker Newt Gingrich (R-Ga.) and ran for Congress herself in the 1990s. In addition to practicing medicine for 3 decades, she has served on the board and as president of the Georgia Obstetrical and Gynecological Society.
Question: What are you looking forward to as the new CDC director, and what are some of your top goals during your tenure?
Answer: The federal government holds responsibility to provide for the common defense. And CDC provides the common defense of the country against health threats. That’s why CDC’s mission of saving lives and protecting people means so much to me. As a doctor and a mother, I want to do everything I can to protect our future generations and keep them safe, whether it be from infectious or chronic diseases, natural or man-made disasters.
We are going to continue a legacy of protecting Americans from the things that we’ve been fighting for a long time, like heart disease and cancer, and from newer things like opioid overdose and the Zika virus. And we’ll continue to prepare for the next health threat, whatever it may be.
Q: How does your experience as an ob.gyn. shape your approach to this job?
A: There are two things that are important at CDC: science and service. CDC researchers are at the forefront of scientific research – and also provide a service by using scientific discoveries to improve health. That is the same approach I used as a practicing ob.gyn.: I brought the clinical lens that looked at the science, and then my work with my patients emphasized the service part.
As a practicing ob.gyn., it was my responsibility to address the questions and concerns of my patients. At CDC, I am just as committed to making science-based decisions and working with our experts to make sure doctors and patients have the state-of-the-art health information they need. I said when I went from being a clinician to being the state health official for Georgia that I went from treating one patient at a time to treating 10 million at a time. Now that’s changed to over 300 million.
Even though I am now CDC director and protecting the health of millions of people, my experiences as an ob.gyn. for 3 decades will forever shape how I work to improve the health of individual people and patients.
Q: What are the next steps for the agency in terms of its response to Zika virus?
A: Zika is the first infectious disease in 50 years that has been linked to birth defects. We know that the virus attacks neural tissue. We know that about 10% of babies are born with severe birth defects – and some babies born apparently normal later turned out to have neurologic problems, like blindness or deafness. And we suspect that there may be other problems we don’t know about yet.
I’m particularly concerned about developmental delays, as we know that early brain development is key to health and achievement for a child’s future. We are still learning more about Zika every day. It’s crucial that we – the health care and public health communities – work together and remain vigilant to ensure these babies receive the care they need. What Zika has taught us is that we need to establish a pregnancy and infant registry that can flag patterns of concerning health issues so that we can work together with health officials and clinicians to get ahead of emerging health threats.
Q: How can clinicians stay up to date on the rapidly changing guidance during an outbreak like Zika?
A: During outbreaks like Zika, public health officials are continually updating guidance so clinicians can get the latest information. You can check the latest CDC guidance about testing and follow-up care for pregnant women at www.cdc.gov/zika/hc-providers. Health care providers can also contact their state, local, or territorial health department to ensure the appropriate tests are ordered and interpreted correctly. CDC also maintains a 24/7 Zika consultation service for health officials and healthcare providers caring for pregnant women with possible Zika exposure.
Q: What steps will you take to decrease maternal mortality and to understand why this number is rising?
A: As an obstetrician, survival of mothers and babies is paramount to me. It’s so basic: mothers shouldn’t die. Sadly, about 700 women die each year in the U.S. as a result of pregnancy or delivery complications.
We know the leading causes of maternal mortality: cardiovascular disease, infections or sepsis, hemorrhage, and cardiomyopathy. We also know that an increasing number of pregnant women in the U.S. have chronic health conditions such as high blood pressure, diabetes, or heart disease that puts them at risk of pregnancy complications or death.
CDC is committed to preventing pregnancy-related deaths and ensuring the best possible birth outcomes. We do this by conducting surveillance, supporting states in developing recommendations, and working with partners to promote evidence-based recommendations and best practices. State-based initiatives are important allies in bringing together OBs and the public health sector to show the importance of statewide efforts to reduce maternal mortality. There has been some exciting work done by the California Maternal Quality Care Collaborative, which has created a series of toolkits designed to help health care providers with pregnancy or childbirth complications. CMQCC’s work is inspiring other states to follow suit.
The best coalitions are between ob.gyns., public health, and hospital associations. For example, AWHONN’s [Association of Women’s Health, Obstetric and Neonatal Nurses] Postpartum Hemorrhage initiative in Georgia, New Jersey, and the District of Columbia aimed to improve the treatment of obstetric hemorrhage. Even though 54% to 93% of maternal hemorrhage-related deaths are preventable with improved clinical response, not one single hospital in Georgia or New Jersey had implemented all the recommended preparedness elements for postpartum hemorrhage events. We can save mothers’ lives – we know what to do. It just takes all of us – ob.gyns., public health and hospital associations – leading the way to make sure it happens.
Q: How can we increase HPV vaccination?
A: The latest news is that more girls and boys are getting the HPV vaccine. New CDC data showed that 60% of teens aged 13-17 had received one or more doses of HPV vaccine in 2016 – an increase of 4% from 2015. Plus, the latest statistics show that HPV vaccination has led to significant drops in HPV infections: HPV-related cancers and genital warts dropped by 71% among teen girls and 61% among young women. Still, too many children aren’t completing the HPV vaccine series, leaving them vulnerable to cancers caused by HPV infection.
Providers can help increase HPV vaccination rates by using every patient visit to review vaccination histories, providing strong clinical recommendations and education to parents for HPV and other recommended vaccines, and implementing systems to minimize missed opportunities.
agallegos@frontlinemedcom.com
On Twitter @legal_med
Brenda Fitzgerald, MD, an ob.gyn. and most recently the public health commissioner for Georgia, was named the 17th director of the Centers for Disease Control and Prevention in July.
Dr. Fitzgerald comes to the CDC after 6 years as commissioner and state health officer for the Georgia Department of Public Health. She also has been a health care policy adviser to former House Speaker Newt Gingrich (R-Ga.) and ran for Congress herself in the 1990s. In addition to practicing medicine for 3 decades, she has served on the board and as president of the Georgia Obstetrical and Gynecological Society.
Question: What are you looking forward to as the new CDC director, and what are some of your top goals during your tenure?
Answer: The federal government holds responsibility to provide for the common defense. And CDC provides the common defense of the country against health threats. That’s why CDC’s mission of saving lives and protecting people means so much to me. As a doctor and a mother, I want to do everything I can to protect our future generations and keep them safe, whether it be from infectious or chronic diseases, natural or man-made disasters.
We are going to continue a legacy of protecting Americans from the things that we’ve been fighting for a long time, like heart disease and cancer, and from newer things like opioid overdose and the Zika virus. And we’ll continue to prepare for the next health threat, whatever it may be.
Q: How does your experience as an ob.gyn. shape your approach to this job?
A: There are two things that are important at CDC: science and service. CDC researchers are at the forefront of scientific research – and also provide a service by using scientific discoveries to improve health. That is the same approach I used as a practicing ob.gyn.: I brought the clinical lens that looked at the science, and then my work with my patients emphasized the service part.
As a practicing ob.gyn., it was my responsibility to address the questions and concerns of my patients. At CDC, I am just as committed to making science-based decisions and working with our experts to make sure doctors and patients have the state-of-the-art health information they need. I said when I went from being a clinician to being the state health official for Georgia that I went from treating one patient at a time to treating 10 million at a time. Now that’s changed to over 300 million.
Even though I am now CDC director and protecting the health of millions of people, my experiences as an ob.gyn. for 3 decades will forever shape how I work to improve the health of individual people and patients.
Q: What are the next steps for the agency in terms of its response to Zika virus?
A: Zika is the first infectious disease in 50 years that has been linked to birth defects. We know that the virus attacks neural tissue. We know that about 10% of babies are born with severe birth defects – and some babies born apparently normal later turned out to have neurologic problems, like blindness or deafness. And we suspect that there may be other problems we don’t know about yet.
I’m particularly concerned about developmental delays, as we know that early brain development is key to health and achievement for a child’s future. We are still learning more about Zika every day. It’s crucial that we – the health care and public health communities – work together and remain vigilant to ensure these babies receive the care they need. What Zika has taught us is that we need to establish a pregnancy and infant registry that can flag patterns of concerning health issues so that we can work together with health officials and clinicians to get ahead of emerging health threats.
Q: How can clinicians stay up to date on the rapidly changing guidance during an outbreak like Zika?
A: During outbreaks like Zika, public health officials are continually updating guidance so clinicians can get the latest information. You can check the latest CDC guidance about testing and follow-up care for pregnant women at www.cdc.gov/zika/hc-providers. Health care providers can also contact their state, local, or territorial health department to ensure the appropriate tests are ordered and interpreted correctly. CDC also maintains a 24/7 Zika consultation service for health officials and healthcare providers caring for pregnant women with possible Zika exposure.
Q: What steps will you take to decrease maternal mortality and to understand why this number is rising?
A: As an obstetrician, survival of mothers and babies is paramount to me. It’s so basic: mothers shouldn’t die. Sadly, about 700 women die each year in the U.S. as a result of pregnancy or delivery complications.
We know the leading causes of maternal mortality: cardiovascular disease, infections or sepsis, hemorrhage, and cardiomyopathy. We also know that an increasing number of pregnant women in the U.S. have chronic health conditions such as high blood pressure, diabetes, or heart disease that puts them at risk of pregnancy complications or death.
CDC is committed to preventing pregnancy-related deaths and ensuring the best possible birth outcomes. We do this by conducting surveillance, supporting states in developing recommendations, and working with partners to promote evidence-based recommendations and best practices. State-based initiatives are important allies in bringing together OBs and the public health sector to show the importance of statewide efforts to reduce maternal mortality. There has been some exciting work done by the California Maternal Quality Care Collaborative, which has created a series of toolkits designed to help health care providers with pregnancy or childbirth complications. CMQCC’s work is inspiring other states to follow suit.
The best coalitions are between ob.gyns., public health, and hospital associations. For example, AWHONN’s [Association of Women’s Health, Obstetric and Neonatal Nurses] Postpartum Hemorrhage initiative in Georgia, New Jersey, and the District of Columbia aimed to improve the treatment of obstetric hemorrhage. Even though 54% to 93% of maternal hemorrhage-related deaths are preventable with improved clinical response, not one single hospital in Georgia or New Jersey had implemented all the recommended preparedness elements for postpartum hemorrhage events. We can save mothers’ lives – we know what to do. It just takes all of us – ob.gyns., public health and hospital associations – leading the way to make sure it happens.
Q: How can we increase HPV vaccination?
A: The latest news is that more girls and boys are getting the HPV vaccine. New CDC data showed that 60% of teens aged 13-17 had received one or more doses of HPV vaccine in 2016 – an increase of 4% from 2015. Plus, the latest statistics show that HPV vaccination has led to significant drops in HPV infections: HPV-related cancers and genital warts dropped by 71% among teen girls and 61% among young women. Still, too many children aren’t completing the HPV vaccine series, leaving them vulnerable to cancers caused by HPV infection.
Providers can help increase HPV vaccination rates by using every patient visit to review vaccination histories, providing strong clinical recommendations and education to parents for HPV and other recommended vaccines, and implementing systems to minimize missed opportunities.
agallegos@frontlinemedcom.com
On Twitter @legal_med
FDA reapproves gemtuzumab ozogamicin for CD33-positive AML treatment
The Food and Drug Administration has approved gemtuzumab ozogamicin (Mylotarg) for the treatment of newly diagnosed CD33-positive acute myeloid leukemia in adults, according to a press release.
Approval was based on results from three clinical trials. In the first, newly diagnosed AML patients who received gemtuzumab ozogamicin plus chemotherapy had significantly longer event-free survival than did patients who received chemotherapy alone. In a second trial, patients who received gemtuzumab ozogamicin alone had better overall survival compared to those who received only best supportive care. In the third clinical trial, 26% of patients who had experienced a relapse and received gemtuzumab ozogamicin experienced a remission.![]()
Common side effects of gemtuzumab ozogamicin include fever, nausea, infection, vomiting, bleeding, thrombocytopenia, stomatitis, constipation, rash, headache, elevated liver function tests, and neutropenia; it is not recommended for women who are pregnant or breastfeeding.
Gemtuzumab ozogamicin was also approved to treat patients older than 2 years old who have experienced a relapse or have not responded to initial treatment.
“Mylotarg’s history underscores the importance of examining alternative dosing, scheduling, and administration of therapies for patients with cancer, especially in those who may be most vulnerable to the side effects of treatment,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in the press release.
The Food and Drug Administration has approved gemtuzumab ozogamicin (Mylotarg) for the treatment of newly diagnosed CD33-positive acute myeloid leukemia in adults, according to a press release.
Approval was based on results from three clinical trials. In the first, newly diagnosed AML patients who received gemtuzumab ozogamicin plus chemotherapy had significantly longer event-free survival than did patients who received chemotherapy alone. In a second trial, patients who received gemtuzumab ozogamicin alone had better overall survival compared to those who received only best supportive care. In the third clinical trial, 26% of patients who had experienced a relapse and received gemtuzumab ozogamicin experienced a remission.![]()
Common side effects of gemtuzumab ozogamicin include fever, nausea, infection, vomiting, bleeding, thrombocytopenia, stomatitis, constipation, rash, headache, elevated liver function tests, and neutropenia; it is not recommended for women who are pregnant or breastfeeding.
Gemtuzumab ozogamicin was also approved to treat patients older than 2 years old who have experienced a relapse or have not responded to initial treatment.
“Mylotarg’s history underscores the importance of examining alternative dosing, scheduling, and administration of therapies for patients with cancer, especially in those who may be most vulnerable to the side effects of treatment,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in the press release.
The Food and Drug Administration has approved gemtuzumab ozogamicin (Mylotarg) for the treatment of newly diagnosed CD33-positive acute myeloid leukemia in adults, according to a press release.
Approval was based on results from three clinical trials. In the first, newly diagnosed AML patients who received gemtuzumab ozogamicin plus chemotherapy had significantly longer event-free survival than did patients who received chemotherapy alone. In a second trial, patients who received gemtuzumab ozogamicin alone had better overall survival compared to those who received only best supportive care. In the third clinical trial, 26% of patients who had experienced a relapse and received gemtuzumab ozogamicin experienced a remission.![]()
Common side effects of gemtuzumab ozogamicin include fever, nausea, infection, vomiting, bleeding, thrombocytopenia, stomatitis, constipation, rash, headache, elevated liver function tests, and neutropenia; it is not recommended for women who are pregnant or breastfeeding.
Gemtuzumab ozogamicin was also approved to treat patients older than 2 years old who have experienced a relapse or have not responded to initial treatment.
“Mylotarg’s history underscores the importance of examining alternative dosing, scheduling, and administration of therapies for patients with cancer, especially in those who may be most vulnerable to the side effects of treatment,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in the press release.