User login
FDA wants data on role of flavored tobacco products in youth initiation
The Food and Drug Administration is seeking data on the role that flavors, including menthol, in tobacco products play in the initiation, use, and cessation of tobacco products, with an emphasis on how flavoring impacts young people.
“In the spirit of our commitment to preventing kids from using tobacco, we are taking a closer look at flavors in tobacco products to better understand their level of impact on youth initiation,” FDA Commissioner Scott Gottlieb, MD, said in statement. It is important “that we also explore how flavors, under a properly regulated framework that protects youth, may also be helping some currently addicted adult cigarette smokers switch to certain noncombustible forms of tobacco products.”
“Youth consistently report product flavoring as a leading reason for using tobacco products,” Dr. Gottlieb noted. “In fact, there is evidence indicating that youth tobacco users who reported their first tobacco was flavored had a higher prevalence of current tobacco product use, compared to youth whose product was not flavored.”
The advance notice calls for information across a number of areas, including the role of flavors other than tobacco in tobacco products; flavors and initiation and patterns of tobacco product use, particularly among youths and young adults; and flavors and cessation, dual-use, and relapse among current and former tobacco product users.
It also is seeking comment on whether standards should be set on tobacco flavoring, including whether there should a prohibition or restriction on flavors and to which types of products these standards should apply. The notice specifically asks about menthol and its role in cigarette initiation and whether limitations on menthol could lead to use of other tobacco products.
“Because almost 90% of adult smokers started smoking by the age of 18, it’s imperative we look at new ways we can ensure that kids don’t progress from experimentation to regular use,” Commissioner Gottlieb said.
The American Heart Association called the action “long overdue.”
“We encourage the FDA to quickly move beyond information gathering and develop a strong flavoring product standard,” CEO Nancy Brown said in a statement. “There is already clear evidence that flavored tobacco products, including menthol, harm the public health. To make it worse, fruit- and candy-flavored e-cigarettes, cigars, and other tobacco products are highly attractive to kids and make it more likely that they will take up this addiction.”
The action comes less than a week after FDA published an advance notice seeking information comments on reducing nicotine levels in cigarettes to help combat nicotine addiction.
The advance notice will be published March 21 in the Federal Register; comments will be accepted at www.regulations.gov for 90 days.
The Food and Drug Administration is seeking data on the role that flavors, including menthol, in tobacco products play in the initiation, use, and cessation of tobacco products, with an emphasis on how flavoring impacts young people.
“In the spirit of our commitment to preventing kids from using tobacco, we are taking a closer look at flavors in tobacco products to better understand their level of impact on youth initiation,” FDA Commissioner Scott Gottlieb, MD, said in statement. It is important “that we also explore how flavors, under a properly regulated framework that protects youth, may also be helping some currently addicted adult cigarette smokers switch to certain noncombustible forms of tobacco products.”
“Youth consistently report product flavoring as a leading reason for using tobacco products,” Dr. Gottlieb noted. “In fact, there is evidence indicating that youth tobacco users who reported their first tobacco was flavored had a higher prevalence of current tobacco product use, compared to youth whose product was not flavored.”
The advance notice calls for information across a number of areas, including the role of flavors other than tobacco in tobacco products; flavors and initiation and patterns of tobacco product use, particularly among youths and young adults; and flavors and cessation, dual-use, and relapse among current and former tobacco product users.
It also is seeking comment on whether standards should be set on tobacco flavoring, including whether there should a prohibition or restriction on flavors and to which types of products these standards should apply. The notice specifically asks about menthol and its role in cigarette initiation and whether limitations on menthol could lead to use of other tobacco products.
“Because almost 90% of adult smokers started smoking by the age of 18, it’s imperative we look at new ways we can ensure that kids don’t progress from experimentation to regular use,” Commissioner Gottlieb said.
The American Heart Association called the action “long overdue.”
“We encourage the FDA to quickly move beyond information gathering and develop a strong flavoring product standard,” CEO Nancy Brown said in a statement. “There is already clear evidence that flavored tobacco products, including menthol, harm the public health. To make it worse, fruit- and candy-flavored e-cigarettes, cigars, and other tobacco products are highly attractive to kids and make it more likely that they will take up this addiction.”
The action comes less than a week after FDA published an advance notice seeking information comments on reducing nicotine levels in cigarettes to help combat nicotine addiction.
The advance notice will be published March 21 in the Federal Register; comments will be accepted at www.regulations.gov for 90 days.
The Food and Drug Administration is seeking data on the role that flavors, including menthol, in tobacco products play in the initiation, use, and cessation of tobacco products, with an emphasis on how flavoring impacts young people.
“In the spirit of our commitment to preventing kids from using tobacco, we are taking a closer look at flavors in tobacco products to better understand their level of impact on youth initiation,” FDA Commissioner Scott Gottlieb, MD, said in statement. It is important “that we also explore how flavors, under a properly regulated framework that protects youth, may also be helping some currently addicted adult cigarette smokers switch to certain noncombustible forms of tobacco products.”
“Youth consistently report product flavoring as a leading reason for using tobacco products,” Dr. Gottlieb noted. “In fact, there is evidence indicating that youth tobacco users who reported their first tobacco was flavored had a higher prevalence of current tobacco product use, compared to youth whose product was not flavored.”
The advance notice calls for information across a number of areas, including the role of flavors other than tobacco in tobacco products; flavors and initiation and patterns of tobacco product use, particularly among youths and young adults; and flavors and cessation, dual-use, and relapse among current and former tobacco product users.
It also is seeking comment on whether standards should be set on tobacco flavoring, including whether there should a prohibition or restriction on flavors and to which types of products these standards should apply. The notice specifically asks about menthol and its role in cigarette initiation and whether limitations on menthol could lead to use of other tobacco products.
“Because almost 90% of adult smokers started smoking by the age of 18, it’s imperative we look at new ways we can ensure that kids don’t progress from experimentation to regular use,” Commissioner Gottlieb said.
The American Heart Association called the action “long overdue.”
“We encourage the FDA to quickly move beyond information gathering and develop a strong flavoring product standard,” CEO Nancy Brown said in a statement. “There is already clear evidence that flavored tobacco products, including menthol, harm the public health. To make it worse, fruit- and candy-flavored e-cigarettes, cigars, and other tobacco products are highly attractive to kids and make it more likely that they will take up this addiction.”
The action comes less than a week after FDA published an advance notice seeking information comments on reducing nicotine levels in cigarettes to help combat nicotine addiction.
The advance notice will be published March 21 in the Federal Register; comments will be accepted at www.regulations.gov for 90 days.
CDC: Beware Brazil yellow fever outbreak
The Centers for Disease Control and Prevention recommends that travelers who haven’t been vaccinated against yellow fever should avoid travel to Brazil, according to a media teleconference by CDC officials.
“The most important new recommendation ... is that travelers should not go to these yellow fever hot spots in Brazil, unless they are vaccinated,” stated Martin Cetron, MD, director of the Division of Global Migration and Quarantine at the CDC. “Health officials in Brazil recently confirmed more than 920 cases of yellow fever, including more than 300 deaths, during this outbreak” he added.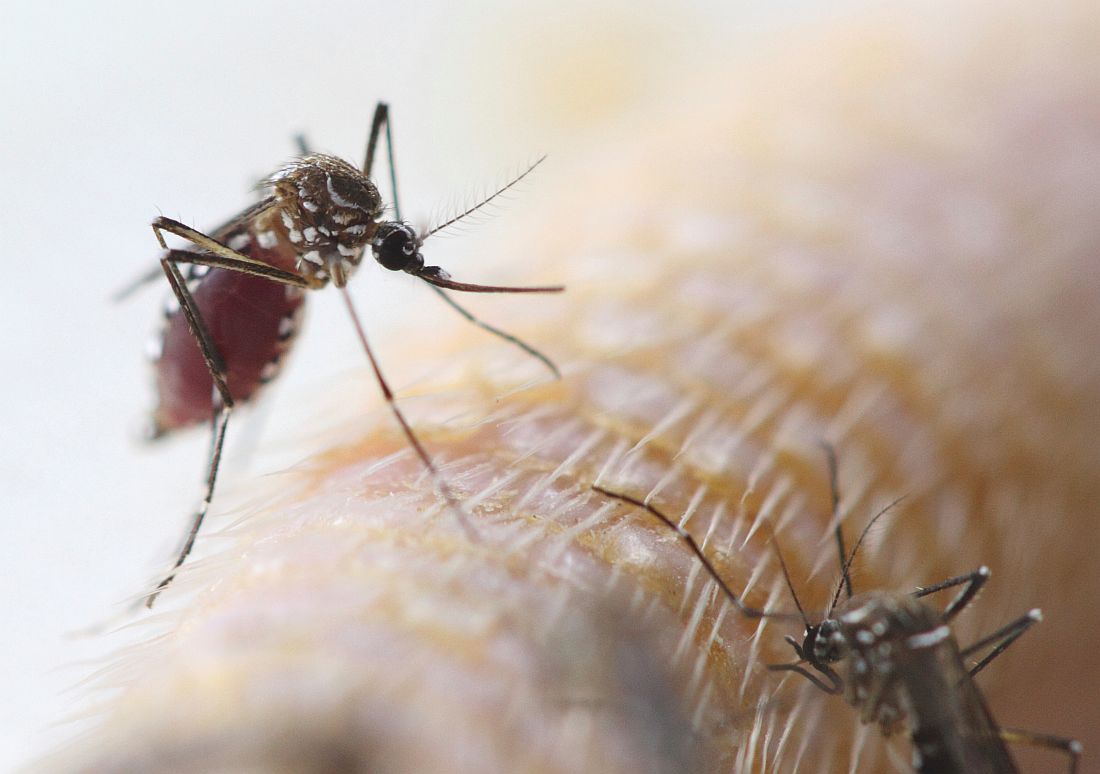
Since the beginning of 2018, 10 travel-related cases of yellow fever have been reported among international travelers returning from Brazil. Four of these travelers died. All 10 travelers had not received the yellow fever vaccine. Of these 10 travelers, 8 acquired the disease on Ilha Grande, an island off the coast of Rio De Janeiro that is popular among tourists.
The CDC is urging travelers to get vaccinated because of the potentially fatal effects of yellow fever. Vaccinations are recommended for any eligible person 9 months of age or older traveling to Brazil, specifically Espírito Santo, São Paulo, and Rio de Janeiro states, and certain cities in Bahia state, as well as Ilha Grande in particular.
Individuals heading to Brazil should receive the vaccination at least 10 days before travel. If a traveler is unvaccinated and cannot get the vaccination in the appropriate amount of time, areas where vaccination is recommended should be avoided.
The Food and Drug Administration–approved yellow fever vaccine, YF-VAX, is not currently available because of manufacturing issues. Stamaril, another yellow fever vaccine, is available at a limited number of yellow fever vaccination clinics in the United States.
In light of these supply issues, the CDC has provided resources to locate yellow fever vaccination clinics.
Dr. Cetron reemphasized that unvaccinated individuals planning to vacation in Brazil may want to reconsider their travel plans.
“People who have never been vaccinated against yellow fever should not travel to the areas in Brazil affected by the outbreak. Particularly the hot spot of Ilha Grande.”
Information for clinicians and travelers is available on the travel notice portion of the CDC site. The travel notice for Brazil includes a map of the yellow fever–affected areas in Brazil, as well as other informational resources.
The related CDC Morbidity and Mortality Weekly Report is available online.
The Centers for Disease Control and Prevention recommends that travelers who haven’t been vaccinated against yellow fever should avoid travel to Brazil, according to a media teleconference by CDC officials.
“The most important new recommendation ... is that travelers should not go to these yellow fever hot spots in Brazil, unless they are vaccinated,” stated Martin Cetron, MD, director of the Division of Global Migration and Quarantine at the CDC. “Health officials in Brazil recently confirmed more than 920 cases of yellow fever, including more than 300 deaths, during this outbreak” he added.
Since the beginning of 2018, 10 travel-related cases of yellow fever have been reported among international travelers returning from Brazil. Four of these travelers died. All 10 travelers had not received the yellow fever vaccine. Of these 10 travelers, 8 acquired the disease on Ilha Grande, an island off the coast of Rio De Janeiro that is popular among tourists.
The CDC is urging travelers to get vaccinated because of the potentially fatal effects of yellow fever. Vaccinations are recommended for any eligible person 9 months of age or older traveling to Brazil, specifically Espírito Santo, São Paulo, and Rio de Janeiro states, and certain cities in Bahia state, as well as Ilha Grande in particular.
Individuals heading to Brazil should receive the vaccination at least 10 days before travel. If a traveler is unvaccinated and cannot get the vaccination in the appropriate amount of time, areas where vaccination is recommended should be avoided.
The Food and Drug Administration–approved yellow fever vaccine, YF-VAX, is not currently available because of manufacturing issues. Stamaril, another yellow fever vaccine, is available at a limited number of yellow fever vaccination clinics in the United States.
In light of these supply issues, the CDC has provided resources to locate yellow fever vaccination clinics.
Dr. Cetron reemphasized that unvaccinated individuals planning to vacation in Brazil may want to reconsider their travel plans.
“People who have never been vaccinated against yellow fever should not travel to the areas in Brazil affected by the outbreak. Particularly the hot spot of Ilha Grande.”
Information for clinicians and travelers is available on the travel notice portion of the CDC site. The travel notice for Brazil includes a map of the yellow fever–affected areas in Brazil, as well as other informational resources.
The related CDC Morbidity and Mortality Weekly Report is available online.
The Centers for Disease Control and Prevention recommends that travelers who haven’t been vaccinated against yellow fever should avoid travel to Brazil, according to a media teleconference by CDC officials.
“The most important new recommendation ... is that travelers should not go to these yellow fever hot spots in Brazil, unless they are vaccinated,” stated Martin Cetron, MD, director of the Division of Global Migration and Quarantine at the CDC. “Health officials in Brazil recently confirmed more than 920 cases of yellow fever, including more than 300 deaths, during this outbreak” he added.
Since the beginning of 2018, 10 travel-related cases of yellow fever have been reported among international travelers returning from Brazil. Four of these travelers died. All 10 travelers had not received the yellow fever vaccine. Of these 10 travelers, 8 acquired the disease on Ilha Grande, an island off the coast of Rio De Janeiro that is popular among tourists.
The CDC is urging travelers to get vaccinated because of the potentially fatal effects of yellow fever. Vaccinations are recommended for any eligible person 9 months of age or older traveling to Brazil, specifically Espírito Santo, São Paulo, and Rio de Janeiro states, and certain cities in Bahia state, as well as Ilha Grande in particular.
Individuals heading to Brazil should receive the vaccination at least 10 days before travel. If a traveler is unvaccinated and cannot get the vaccination in the appropriate amount of time, areas where vaccination is recommended should be avoided.
The Food and Drug Administration–approved yellow fever vaccine, YF-VAX, is not currently available because of manufacturing issues. Stamaril, another yellow fever vaccine, is available at a limited number of yellow fever vaccination clinics in the United States.
In light of these supply issues, the CDC has provided resources to locate yellow fever vaccination clinics.
Dr. Cetron reemphasized that unvaccinated individuals planning to vacation in Brazil may want to reconsider their travel plans.
“People who have never been vaccinated against yellow fever should not travel to the areas in Brazil affected by the outbreak. Particularly the hot spot of Ilha Grande.”
Information for clinicians and travelers is available on the travel notice portion of the CDC site. The travel notice for Brazil includes a map of the yellow fever–affected areas in Brazil, as well as other informational resources.
The related CDC Morbidity and Mortality Weekly Report is available online.
FDA proposes lower nicotine levels in cigarettes
Nicotine levels in cigarettes could see a significant reduction under regulatory options being considered by the Food and Drug Administration.
Cigarettes “are the only legal consumer product that, when used as intended, will kill half all long-term users,” FDA Commissioner Scott Gottlieb, MD, said in a statement announcing the effort.
The agency is seeking comment on a proposed regulation regarding “a potential maximum nicotine level that would be appropriate for the protection of public health, in light of scientific evidence about the addictive properties of nicotine in cigarettes.” An advance notice of proposed rule making was posted online March 15 and is scheduled for publication in the Federal Register on March 16.
The FDA also is seeking comments on a number of other areas to help inform potential regulatory action down the road, including whether a new standard for lower nicotine levels should be implemented at once or whether a phased-in approach should be taken; whether FDA should specify a method for manufacturers to use in order to detect nicotine levels in their products; and whether the proposed lower level is technically achievable.
The agency also is seeking comment on potential unintended effects of lowering the amount of nicotine in cigarettes, such as turning to other combustible tobacco products such as cigars in conjunction with or as a replacement for cigarette use; increasing the number of cigarettes smoked, or seeking comparable nicotine from noncombustible tobacco sources.
At this time, FDA is not suggesting what the target might be on a specific nicotine level. While the advanced notice asks specifically about the “merits of nicotine levels like 0.3, 0.4, and 0.5 mg nicotine/g of tobacco filler,” it is not suggesting that this is the range being considered.
[polldaddy:9960560]
“Not to prejudge any possible proposed rule that we would do or any possible level, that is the purpose of an advanced proposed rule making, but we share all the science that we are aware of, and we characterize the studies that have been done to date in trying to find out what that right level is,” Mitch Zeller, director of the FDA Center for Tobacco Products, said during a March 15 press call.
He said that the FDA aiming to make sure the level is low enough that it cannot be compensated for by smoking more or inhaling deeper and holding the breath in longer, much like how smokers compensated when they smoked “light” cigarettes in the unregulated market.
Mr. Zeller said that seeking comments on those levels is based on the scientific evidence that is laid out in the advanced notice, but it is not necessarily foreshadowing where the standard will be set.
Drastically reducing the amount of nicotine in cigarettes is expected to significantly lower not only the number of people addicted to cigarettes but also as the negative health effects of nicotine addiction, FDA experts wrote in a perspective piece published March 15 in the New England Journal of Medicine (doi: 10.1065/NEJMsr1714617).
“Our findings show that reducing the nicotine level in cigarettes has the potential to substantially reduce the enormous burden of smoking-related death and disease,” Benjamin J. Apelberg, PhD, director of the Division of Population Health Science, Office of Science, within the FDA Center for Tobacco Products, and his colleagues, wrote in the report.
Modeling for the implementation of a lower nicotine level policy suggests that smoking prevalence will decline from a median of 12.8% in baseline scenario to a median of 10.8% within a year of implementation, with the increase related to smoking cessation.
“We estimate that approximately 5 million additional smokers would quit smoking within a year after implementation of the hypothetical policy,” Dr. Apelberg and his colleagues wrote. “By 2060, smoking prevalence drops from 7.9% in the baseline scenario to 1.4% in the policy scenario.”
Their analysis is based on a nicotine level that is “so low that there would not be enough nicotine available in cigarette tobacco for smokers to sustain addiction,” they noted.
The FDA plans to release two more advanced notices of proposed rule making related to using regulatory levers to reduce cigarette smoking, including one that addresses flavoring in tobacco and one related to the regulation of premium cigars. Other than the 90-day comment period related to the advanced notices, the agency has not set any deadlines on when it will act.
“Legally, we are not going to prejudge how long this will take or what will happen,” Mr. Zeller said. “This is an early step in what could be a potential rule-making process using the product standard authority. We will take a long and hard look at all the comments that come in over the next 90 days and based upon our review of all the information, all the comments that come in, all the feedback that we have, we will then make a decision about taking the next step in the rule-making process.”
Nicotine levels in cigarettes could see a significant reduction under regulatory options being considered by the Food and Drug Administration.
Cigarettes “are the only legal consumer product that, when used as intended, will kill half all long-term users,” FDA Commissioner Scott Gottlieb, MD, said in a statement announcing the effort.
The agency is seeking comment on a proposed regulation regarding “a potential maximum nicotine level that would be appropriate for the protection of public health, in light of scientific evidence about the addictive properties of nicotine in cigarettes.” An advance notice of proposed rule making was posted online March 15 and is scheduled for publication in the Federal Register on March 16.
The FDA also is seeking comments on a number of other areas to help inform potential regulatory action down the road, including whether a new standard for lower nicotine levels should be implemented at once or whether a phased-in approach should be taken; whether FDA should specify a method for manufacturers to use in order to detect nicotine levels in their products; and whether the proposed lower level is technically achievable.
The agency also is seeking comment on potential unintended effects of lowering the amount of nicotine in cigarettes, such as turning to other combustible tobacco products such as cigars in conjunction with or as a replacement for cigarette use; increasing the number of cigarettes smoked, or seeking comparable nicotine from noncombustible tobacco sources.
At this time, FDA is not suggesting what the target might be on a specific nicotine level. While the advanced notice asks specifically about the “merits of nicotine levels like 0.3, 0.4, and 0.5 mg nicotine/g of tobacco filler,” it is not suggesting that this is the range being considered.
[polldaddy:9960560]
“Not to prejudge any possible proposed rule that we would do or any possible level, that is the purpose of an advanced proposed rule making, but we share all the science that we are aware of, and we characterize the studies that have been done to date in trying to find out what that right level is,” Mitch Zeller, director of the FDA Center for Tobacco Products, said during a March 15 press call.
He said that the FDA aiming to make sure the level is low enough that it cannot be compensated for by smoking more or inhaling deeper and holding the breath in longer, much like how smokers compensated when they smoked “light” cigarettes in the unregulated market.
Mr. Zeller said that seeking comments on those levels is based on the scientific evidence that is laid out in the advanced notice, but it is not necessarily foreshadowing where the standard will be set.
Drastically reducing the amount of nicotine in cigarettes is expected to significantly lower not only the number of people addicted to cigarettes but also as the negative health effects of nicotine addiction, FDA experts wrote in a perspective piece published March 15 in the New England Journal of Medicine (doi: 10.1065/NEJMsr1714617).
“Our findings show that reducing the nicotine level in cigarettes has the potential to substantially reduce the enormous burden of smoking-related death and disease,” Benjamin J. Apelberg, PhD, director of the Division of Population Health Science, Office of Science, within the FDA Center for Tobacco Products, and his colleagues, wrote in the report.
Modeling for the implementation of a lower nicotine level policy suggests that smoking prevalence will decline from a median of 12.8% in baseline scenario to a median of 10.8% within a year of implementation, with the increase related to smoking cessation.
“We estimate that approximately 5 million additional smokers would quit smoking within a year after implementation of the hypothetical policy,” Dr. Apelberg and his colleagues wrote. “By 2060, smoking prevalence drops from 7.9% in the baseline scenario to 1.4% in the policy scenario.”
Their analysis is based on a nicotine level that is “so low that there would not be enough nicotine available in cigarette tobacco for smokers to sustain addiction,” they noted.
The FDA plans to release two more advanced notices of proposed rule making related to using regulatory levers to reduce cigarette smoking, including one that addresses flavoring in tobacco and one related to the regulation of premium cigars. Other than the 90-day comment period related to the advanced notices, the agency has not set any deadlines on when it will act.
“Legally, we are not going to prejudge how long this will take or what will happen,” Mr. Zeller said. “This is an early step in what could be a potential rule-making process using the product standard authority. We will take a long and hard look at all the comments that come in over the next 90 days and based upon our review of all the information, all the comments that come in, all the feedback that we have, we will then make a decision about taking the next step in the rule-making process.”
Nicotine levels in cigarettes could see a significant reduction under regulatory options being considered by the Food and Drug Administration.
Cigarettes “are the only legal consumer product that, when used as intended, will kill half all long-term users,” FDA Commissioner Scott Gottlieb, MD, said in a statement announcing the effort.
The agency is seeking comment on a proposed regulation regarding “a potential maximum nicotine level that would be appropriate for the protection of public health, in light of scientific evidence about the addictive properties of nicotine in cigarettes.” An advance notice of proposed rule making was posted online March 15 and is scheduled for publication in the Federal Register on March 16.
The FDA also is seeking comments on a number of other areas to help inform potential regulatory action down the road, including whether a new standard for lower nicotine levels should be implemented at once or whether a phased-in approach should be taken; whether FDA should specify a method for manufacturers to use in order to detect nicotine levels in their products; and whether the proposed lower level is technically achievable.
The agency also is seeking comment on potential unintended effects of lowering the amount of nicotine in cigarettes, such as turning to other combustible tobacco products such as cigars in conjunction with or as a replacement for cigarette use; increasing the number of cigarettes smoked, or seeking comparable nicotine from noncombustible tobacco sources.
At this time, FDA is not suggesting what the target might be on a specific nicotine level. While the advanced notice asks specifically about the “merits of nicotine levels like 0.3, 0.4, and 0.5 mg nicotine/g of tobacco filler,” it is not suggesting that this is the range being considered.
[polldaddy:9960560]
“Not to prejudge any possible proposed rule that we would do or any possible level, that is the purpose of an advanced proposed rule making, but we share all the science that we are aware of, and we characterize the studies that have been done to date in trying to find out what that right level is,” Mitch Zeller, director of the FDA Center for Tobacco Products, said during a March 15 press call.
He said that the FDA aiming to make sure the level is low enough that it cannot be compensated for by smoking more or inhaling deeper and holding the breath in longer, much like how smokers compensated when they smoked “light” cigarettes in the unregulated market.
Mr. Zeller said that seeking comments on those levels is based on the scientific evidence that is laid out in the advanced notice, but it is not necessarily foreshadowing where the standard will be set.
Drastically reducing the amount of nicotine in cigarettes is expected to significantly lower not only the number of people addicted to cigarettes but also as the negative health effects of nicotine addiction, FDA experts wrote in a perspective piece published March 15 in the New England Journal of Medicine (doi: 10.1065/NEJMsr1714617).
“Our findings show that reducing the nicotine level in cigarettes has the potential to substantially reduce the enormous burden of smoking-related death and disease,” Benjamin J. Apelberg, PhD, director of the Division of Population Health Science, Office of Science, within the FDA Center for Tobacco Products, and his colleagues, wrote in the report.
Modeling for the implementation of a lower nicotine level policy suggests that smoking prevalence will decline from a median of 12.8% in baseline scenario to a median of 10.8% within a year of implementation, with the increase related to smoking cessation.
“We estimate that approximately 5 million additional smokers would quit smoking within a year after implementation of the hypothetical policy,” Dr. Apelberg and his colleagues wrote. “By 2060, smoking prevalence drops from 7.9% in the baseline scenario to 1.4% in the policy scenario.”
Their analysis is based on a nicotine level that is “so low that there would not be enough nicotine available in cigarette tobacco for smokers to sustain addiction,” they noted.
The FDA plans to release two more advanced notices of proposed rule making related to using regulatory levers to reduce cigarette smoking, including one that addresses flavoring in tobacco and one related to the regulation of premium cigars. Other than the 90-day comment period related to the advanced notices, the agency has not set any deadlines on when it will act.
“Legally, we are not going to prejudge how long this will take or what will happen,” Mr. Zeller said. “This is an early step in what could be a potential rule-making process using the product standard authority. We will take a long and hard look at all the comments that come in over the next 90 days and based upon our review of all the information, all the comments that come in, all the feedback that we have, we will then make a decision about taking the next step in the rule-making process.”
FDA approves continuous glucose monitor with AI assistant
between the ages of 14 and 75 years old.
The first CGM to use artificial intelligence for this purpose, the Guardian Connect system is intended to help people with diabetes who use multiple daily injections of insulin to manage their diabetes by helping them prevent hyperglycemia and hypoglycemia, according to a statement from Medtronic.
Guardian Connect utilizes a predictive algorithm that alerts patients of significant swings in blood glucose levels up to 60 minutes prior to the event. When combined with the Guardian Sensor 3, which is placed on the abdomen to monitor blood glucose levels, the Guardian Connect system was accurate and was able to alert patients about 98.5% of hypoglycemic events, according to the results of a clinical trial. This information can also be shared and monitored with care takers and family member in real time or via text message.
Perhaps the most intriguing aspect of using the Guardian Connect system is exclusive access to the Sugar.IQ smart diabetes assistant. Utilizing artificial intelligence technology from IBM Watson Health, the Sugar.IQ assistant continually analyzes how patient’s blood glucose levels respond to factors like food intake, insulin dosages, and daily routines. The combination of continuous monitoring and real-time analysis may be able to identify patterns and lead to personalized insights that will help patients with diabetes keep their blood glucose levels under control.
“Despite proven benefits and advances in technology, only a minority of insulin-using people with diabetes currently use continuous glucose monitors,” Timothy Bailey, MD, the director of the AMCR Institute and a clinical associate professor at University of California, San Diego, said in a statement. “Newer sensors paired with intelligent algorithms that help to both predict and understand glucose excursions, particularly hypoglycemia, will make diabetes safer and more comprehensible for people who inject insulin. Greater utilization of smarter CGM systems promises to allow our patients to achieve more glycemic time-in-range and to further reduce the risk of hypoglycemia,” he said.
Similar products have been approved in the last 6 months, but they have not utilized artificial intelligence and continuous analysis.
No serious adverse events were reported in the clinical trial of Guardian Connect, but less serious adverse events were observed, including gastroenteritis, upper respiratory infection, worsening of benign prostatic hyperplasia, rash, and blisters.
Guardian Connect should be available commercially sometime this summer.
between the ages of 14 and 75 years old.
The first CGM to use artificial intelligence for this purpose, the Guardian Connect system is intended to help people with diabetes who use multiple daily injections of insulin to manage their diabetes by helping them prevent hyperglycemia and hypoglycemia, according to a statement from Medtronic.
Guardian Connect utilizes a predictive algorithm that alerts patients of significant swings in blood glucose levels up to 60 minutes prior to the event. When combined with the Guardian Sensor 3, which is placed on the abdomen to monitor blood glucose levels, the Guardian Connect system was accurate and was able to alert patients about 98.5% of hypoglycemic events, according to the results of a clinical trial. This information can also be shared and monitored with care takers and family member in real time or via text message.
Perhaps the most intriguing aspect of using the Guardian Connect system is exclusive access to the Sugar.IQ smart diabetes assistant. Utilizing artificial intelligence technology from IBM Watson Health, the Sugar.IQ assistant continually analyzes how patient’s blood glucose levels respond to factors like food intake, insulin dosages, and daily routines. The combination of continuous monitoring and real-time analysis may be able to identify patterns and lead to personalized insights that will help patients with diabetes keep their blood glucose levels under control.
“Despite proven benefits and advances in technology, only a minority of insulin-using people with diabetes currently use continuous glucose monitors,” Timothy Bailey, MD, the director of the AMCR Institute and a clinical associate professor at University of California, San Diego, said in a statement. “Newer sensors paired with intelligent algorithms that help to both predict and understand glucose excursions, particularly hypoglycemia, will make diabetes safer and more comprehensible for people who inject insulin. Greater utilization of smarter CGM systems promises to allow our patients to achieve more glycemic time-in-range and to further reduce the risk of hypoglycemia,” he said.
Similar products have been approved in the last 6 months, but they have not utilized artificial intelligence and continuous analysis.
No serious adverse events were reported in the clinical trial of Guardian Connect, but less serious adverse events were observed, including gastroenteritis, upper respiratory infection, worsening of benign prostatic hyperplasia, rash, and blisters.
Guardian Connect should be available commercially sometime this summer.
between the ages of 14 and 75 years old.
The first CGM to use artificial intelligence for this purpose, the Guardian Connect system is intended to help people with diabetes who use multiple daily injections of insulin to manage their diabetes by helping them prevent hyperglycemia and hypoglycemia, according to a statement from Medtronic.
Guardian Connect utilizes a predictive algorithm that alerts patients of significant swings in blood glucose levels up to 60 minutes prior to the event. When combined with the Guardian Sensor 3, which is placed on the abdomen to monitor blood glucose levels, the Guardian Connect system was accurate and was able to alert patients about 98.5% of hypoglycemic events, according to the results of a clinical trial. This information can also be shared and monitored with care takers and family member in real time or via text message.
Perhaps the most intriguing aspect of using the Guardian Connect system is exclusive access to the Sugar.IQ smart diabetes assistant. Utilizing artificial intelligence technology from IBM Watson Health, the Sugar.IQ assistant continually analyzes how patient’s blood glucose levels respond to factors like food intake, insulin dosages, and daily routines. The combination of continuous monitoring and real-time analysis may be able to identify patterns and lead to personalized insights that will help patients with diabetes keep their blood glucose levels under control.
“Despite proven benefits and advances in technology, only a minority of insulin-using people with diabetes currently use continuous glucose monitors,” Timothy Bailey, MD, the director of the AMCR Institute and a clinical associate professor at University of California, San Diego, said in a statement. “Newer sensors paired with intelligent algorithms that help to both predict and understand glucose excursions, particularly hypoglycemia, will make diabetes safer and more comprehensible for people who inject insulin. Greater utilization of smarter CGM systems promises to allow our patients to achieve more glycemic time-in-range and to further reduce the risk of hypoglycemia,” he said.
Similar products have been approved in the last 6 months, but they have not utilized artificial intelligence and continuous analysis.
No serious adverse events were reported in the clinical trial of Guardian Connect, but less serious adverse events were observed, including gastroenteritis, upper respiratory infection, worsening of benign prostatic hyperplasia, rash, and blisters.
Guardian Connect should be available commercially sometime this summer.
FDA issues warning to all duodenoscope manufacturers
The Food and Drug Administration on March 9 issued warning letters to all three duodenoscope manufacturers for failing to comply with the requirements of federal law under which they were ordered to conduct postmarket surveillance studies to assess the effectiveness of reprocessing the devices.
The warning is part of an ongoing effort to prevent patient infections associated with the transmission of bacteria from contaminated duodenoscopes. The three manufacturers – Olympus, Fujifilm, and Pentax – are required to conduct studies to sample and culture reprocessed duodenoscopes that are in clinical use to learn more about issues that contribute to contamination, and to study human factors to determine how hospital staff who have had training are following the reprocessing instructions. In 2015, the FDA ordered the companies to conduct a postmarket surveillance study to determine whether health care facilities were able to properly clean and disinfect the devices.
Currently, the Olympus manufacturer has failed to start data collection, while both Pentax and Fujifilm have failed to provide sufficient data required for their respective studies to sample and culture reprocessed duodenoscopes that are in clinical use. In addition, Olympus and Pentax have not complied with requirements to assess how well staff members have followed the reprocessing instructions after the human factors studies and Fujifilm has been meeting its requirements for its human factors study only.
“The FDA has taken important steps to improve the reprocessing of duodenoscopes, and we’ve seen a reduction in reports of patient infections, but we need the required postmarket studies to determine whether these measures are being properly implemented in real-world clinical settings and whether we need to take additional action to further improve the safety of these devices,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in a press release. “We expect these device manufacturers to meet their study obligations to ensure patient safety.”
The companies have until March 24 to submit a plan that outlines how study milestones will be achieved. If the companies fail to respond to the warning letter, the FDA states that they may take additional action, such as seizure, injunction, and civil monetary penalties.
Read the full press release on the FDA’s website.
The Food and Drug Administration on March 9 issued warning letters to all three duodenoscope manufacturers for failing to comply with the requirements of federal law under which they were ordered to conduct postmarket surveillance studies to assess the effectiveness of reprocessing the devices.
The warning is part of an ongoing effort to prevent patient infections associated with the transmission of bacteria from contaminated duodenoscopes. The three manufacturers – Olympus, Fujifilm, and Pentax – are required to conduct studies to sample and culture reprocessed duodenoscopes that are in clinical use to learn more about issues that contribute to contamination, and to study human factors to determine how hospital staff who have had training are following the reprocessing instructions. In 2015, the FDA ordered the companies to conduct a postmarket surveillance study to determine whether health care facilities were able to properly clean and disinfect the devices.
Currently, the Olympus manufacturer has failed to start data collection, while both Pentax and Fujifilm have failed to provide sufficient data required for their respective studies to sample and culture reprocessed duodenoscopes that are in clinical use. In addition, Olympus and Pentax have not complied with requirements to assess how well staff members have followed the reprocessing instructions after the human factors studies and Fujifilm has been meeting its requirements for its human factors study only.
“The FDA has taken important steps to improve the reprocessing of duodenoscopes, and we’ve seen a reduction in reports of patient infections, but we need the required postmarket studies to determine whether these measures are being properly implemented in real-world clinical settings and whether we need to take additional action to further improve the safety of these devices,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in a press release. “We expect these device manufacturers to meet their study obligations to ensure patient safety.”
The companies have until March 24 to submit a plan that outlines how study milestones will be achieved. If the companies fail to respond to the warning letter, the FDA states that they may take additional action, such as seizure, injunction, and civil monetary penalties.
Read the full press release on the FDA’s website.
The Food and Drug Administration on March 9 issued warning letters to all three duodenoscope manufacturers for failing to comply with the requirements of federal law under which they were ordered to conduct postmarket surveillance studies to assess the effectiveness of reprocessing the devices.
The warning is part of an ongoing effort to prevent patient infections associated with the transmission of bacteria from contaminated duodenoscopes. The three manufacturers – Olympus, Fujifilm, and Pentax – are required to conduct studies to sample and culture reprocessed duodenoscopes that are in clinical use to learn more about issues that contribute to contamination, and to study human factors to determine how hospital staff who have had training are following the reprocessing instructions. In 2015, the FDA ordered the companies to conduct a postmarket surveillance study to determine whether health care facilities were able to properly clean and disinfect the devices.
Currently, the Olympus manufacturer has failed to start data collection, while both Pentax and Fujifilm have failed to provide sufficient data required for their respective studies to sample and culture reprocessed duodenoscopes that are in clinical use. In addition, Olympus and Pentax have not complied with requirements to assess how well staff members have followed the reprocessing instructions after the human factors studies and Fujifilm has been meeting its requirements for its human factors study only.
“The FDA has taken important steps to improve the reprocessing of duodenoscopes, and we’ve seen a reduction in reports of patient infections, but we need the required postmarket studies to determine whether these measures are being properly implemented in real-world clinical settings and whether we need to take additional action to further improve the safety of these devices,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health in a press release. “We expect these device manufacturers to meet their study obligations to ensure patient safety.”
The companies have until March 24 to submit a plan that outlines how study milestones will be achieved. If the companies fail to respond to the warning letter, the FDA states that they may take additional action, such as seizure, injunction, and civil monetary penalties.
Read the full press release on the FDA’s website.
Xeljanz: FDA panel recommends ulcerative colitis indication
SILVER SPRING, MD. – Federal advisors to the Food and Drug Administration on March 8 voted unanimously to recommend approval of an additional indication for tofacitinib (Xeljanz), this time for ulcerative colitis.
Members of the Gastrointestinal Drugs Advisory Committee unanimously voted to recommend two different dosing regimens: 10 mg twice daily for 16 weeks in patients who have not experienced a therapeutic benefit after 8 weeks of treatment, as well as 10 mg twice daily for patients who have an inadequate or loss of response to TNF-blocker therapy, based on the results of several phase 3 clinical trials.
The recommended ulcerative colitis (UC) indication was based on the OCTAVE trials (N Engl J Med 2017;376:1723-36), including a phase 2 study, two identical phase 3 induction trials (OCTAVE Induction 1 and OCTAVE Induction 2), a 53-week, phase 3 maintenance trial (OCTAVE Sustain), and an open-label extension study.
The induction trials enrolled a total of 1,139 patients with moderate to severe UC. Patients in both studies were administered tofacitinib 10 mg twice daily or placebo and were assessed after 8 weeks to judge clinical response. Patients in both studies displayed notable remission rates (18.5% and 16.6%), compared with placebo, according to Eric Maller, MD, executive director of the UC development program at Pfizer.*
Patients who did not achieve remission, but showed some clinical response (decrease in Mayo score of at least 3 points), were then enrolled in the 53-week OCTAVE Sustain, where they were randomized to receive tofacitinib 10 mg twice daily, 5 mg twice daily, or placebo.
During maintenance treatment, both 5 mg and 10 mg doses demonstrated substantial treatment benefits, with 32.4% and 41.0% of patients achieving remission, an increase of 22.0% and 30.7%, compared with placebo, respectively.
As part of the maintenance study, Pfizer analyzed patients with or without prior TNF-blocker failure. This analysis revealed that patients who had previously failed TNF-blocker therapy experienced a greater treatment benefit than those who had not. While the benefit was noticeable in both dosage groups, patients taking the 10-mg dose experienced the greatest benefit, with 70% increase in remission rates, 39% increase in mucosal healing, and 75% increase in steroid-free remission among baseline remitters, compared with patients in the 5-mg group, Dr. Maller said.
Researchers also looked at a subgroup of 295 patients as part of an open-label extension study who had no clinical response to tofacitinib 10 mg twice daily after 8 weeks and subsequently treated them for an additional 8 weeks. After the additional 8 weeks of treatment, over half (51.2%) displayed clinical responses and 8.6% were in remission.
“This is a desperate patient population. These are impressive results,” stated Darrell Pardi, MD, vice chair of the advisory committee and professor of medicine at the Mayo Clinic, Rochester, Minn.
Serious adverse events were seen in 4% of tofacitinib-treated patients in the induction trials, compared with 6% of placebo-treated, according to Lesley Hanes, MD, medical officer with the FDA Center for Drug Evaluation and Research.
Adverse events appeared to be dose dependent, with risk of deaths and malignancies (excluding nonmelanoma skin cancer), opportunistic infections, herpes zoster infection (HZ), “possible” drug-induced liver injury, and cardiovascular and thromboembolic events more common with the 10-mg dose, Dr. Hanes said.
“I think the safety concerns, though, they are dose dependent, the difference between the 5 [mg] and 10 [mg] were not large,” according to Dr. Pardi. “Several of these are mitigatable by dermatologic exam or, hopefully, a vaccine.”
Several of the advisory committee members submitted conflict of interest waivers. Chair and vice chair Jean-Pierre Raufman, MD, and Darrell Pardi, MD, disclosed funding from competing pharmaceutical manufacturers.
*This article was updated on March 12, 2018.
SILVER SPRING, MD. – Federal advisors to the Food and Drug Administration on March 8 voted unanimously to recommend approval of an additional indication for tofacitinib (Xeljanz), this time for ulcerative colitis.
Members of the Gastrointestinal Drugs Advisory Committee unanimously voted to recommend two different dosing regimens: 10 mg twice daily for 16 weeks in patients who have not experienced a therapeutic benefit after 8 weeks of treatment, as well as 10 mg twice daily for patients who have an inadequate or loss of response to TNF-blocker therapy, based on the results of several phase 3 clinical trials.
The recommended ulcerative colitis (UC) indication was based on the OCTAVE trials (N Engl J Med 2017;376:1723-36), including a phase 2 study, two identical phase 3 induction trials (OCTAVE Induction 1 and OCTAVE Induction 2), a 53-week, phase 3 maintenance trial (OCTAVE Sustain), and an open-label extension study.
The induction trials enrolled a total of 1,139 patients with moderate to severe UC. Patients in both studies were administered tofacitinib 10 mg twice daily or placebo and were assessed after 8 weeks to judge clinical response. Patients in both studies displayed notable remission rates (18.5% and 16.6%), compared with placebo, according to Eric Maller, MD, executive director of the UC development program at Pfizer.*
Patients who did not achieve remission, but showed some clinical response (decrease in Mayo score of at least 3 points), were then enrolled in the 53-week OCTAVE Sustain, where they were randomized to receive tofacitinib 10 mg twice daily, 5 mg twice daily, or placebo.
During maintenance treatment, both 5 mg and 10 mg doses demonstrated substantial treatment benefits, with 32.4% and 41.0% of patients achieving remission, an increase of 22.0% and 30.7%, compared with placebo, respectively.
As part of the maintenance study, Pfizer analyzed patients with or without prior TNF-blocker failure. This analysis revealed that patients who had previously failed TNF-blocker therapy experienced a greater treatment benefit than those who had not. While the benefit was noticeable in both dosage groups, patients taking the 10-mg dose experienced the greatest benefit, with 70% increase in remission rates, 39% increase in mucosal healing, and 75% increase in steroid-free remission among baseline remitters, compared with patients in the 5-mg group, Dr. Maller said.
Researchers also looked at a subgroup of 295 patients as part of an open-label extension study who had no clinical response to tofacitinib 10 mg twice daily after 8 weeks and subsequently treated them for an additional 8 weeks. After the additional 8 weeks of treatment, over half (51.2%) displayed clinical responses and 8.6% were in remission.
“This is a desperate patient population. These are impressive results,” stated Darrell Pardi, MD, vice chair of the advisory committee and professor of medicine at the Mayo Clinic, Rochester, Minn.
Serious adverse events were seen in 4% of tofacitinib-treated patients in the induction trials, compared with 6% of placebo-treated, according to Lesley Hanes, MD, medical officer with the FDA Center for Drug Evaluation and Research.
Adverse events appeared to be dose dependent, with risk of deaths and malignancies (excluding nonmelanoma skin cancer), opportunistic infections, herpes zoster infection (HZ), “possible” drug-induced liver injury, and cardiovascular and thromboembolic events more common with the 10-mg dose, Dr. Hanes said.
“I think the safety concerns, though, they are dose dependent, the difference between the 5 [mg] and 10 [mg] were not large,” according to Dr. Pardi. “Several of these are mitigatable by dermatologic exam or, hopefully, a vaccine.”
Several of the advisory committee members submitted conflict of interest waivers. Chair and vice chair Jean-Pierre Raufman, MD, and Darrell Pardi, MD, disclosed funding from competing pharmaceutical manufacturers.
*This article was updated on March 12, 2018.
SILVER SPRING, MD. – Federal advisors to the Food and Drug Administration on March 8 voted unanimously to recommend approval of an additional indication for tofacitinib (Xeljanz), this time for ulcerative colitis.
Members of the Gastrointestinal Drugs Advisory Committee unanimously voted to recommend two different dosing regimens: 10 mg twice daily for 16 weeks in patients who have not experienced a therapeutic benefit after 8 weeks of treatment, as well as 10 mg twice daily for patients who have an inadequate or loss of response to TNF-blocker therapy, based on the results of several phase 3 clinical trials.
The recommended ulcerative colitis (UC) indication was based on the OCTAVE trials (N Engl J Med 2017;376:1723-36), including a phase 2 study, two identical phase 3 induction trials (OCTAVE Induction 1 and OCTAVE Induction 2), a 53-week, phase 3 maintenance trial (OCTAVE Sustain), and an open-label extension study.
The induction trials enrolled a total of 1,139 patients with moderate to severe UC. Patients in both studies were administered tofacitinib 10 mg twice daily or placebo and were assessed after 8 weeks to judge clinical response. Patients in both studies displayed notable remission rates (18.5% and 16.6%), compared with placebo, according to Eric Maller, MD, executive director of the UC development program at Pfizer.*
Patients who did not achieve remission, but showed some clinical response (decrease in Mayo score of at least 3 points), were then enrolled in the 53-week OCTAVE Sustain, where they were randomized to receive tofacitinib 10 mg twice daily, 5 mg twice daily, or placebo.
During maintenance treatment, both 5 mg and 10 mg doses demonstrated substantial treatment benefits, with 32.4% and 41.0% of patients achieving remission, an increase of 22.0% and 30.7%, compared with placebo, respectively.
As part of the maintenance study, Pfizer analyzed patients with or without prior TNF-blocker failure. This analysis revealed that patients who had previously failed TNF-blocker therapy experienced a greater treatment benefit than those who had not. While the benefit was noticeable in both dosage groups, patients taking the 10-mg dose experienced the greatest benefit, with 70% increase in remission rates, 39% increase in mucosal healing, and 75% increase in steroid-free remission among baseline remitters, compared with patients in the 5-mg group, Dr. Maller said.
Researchers also looked at a subgroup of 295 patients as part of an open-label extension study who had no clinical response to tofacitinib 10 mg twice daily after 8 weeks and subsequently treated them for an additional 8 weeks. After the additional 8 weeks of treatment, over half (51.2%) displayed clinical responses and 8.6% were in remission.
“This is a desperate patient population. These are impressive results,” stated Darrell Pardi, MD, vice chair of the advisory committee and professor of medicine at the Mayo Clinic, Rochester, Minn.
Serious adverse events were seen in 4% of tofacitinib-treated patients in the induction trials, compared with 6% of placebo-treated, according to Lesley Hanes, MD, medical officer with the FDA Center for Drug Evaluation and Research.
Adverse events appeared to be dose dependent, with risk of deaths and malignancies (excluding nonmelanoma skin cancer), opportunistic infections, herpes zoster infection (HZ), “possible” drug-induced liver injury, and cardiovascular and thromboembolic events more common with the 10-mg dose, Dr. Hanes said.
“I think the safety concerns, though, they are dose dependent, the difference between the 5 [mg] and 10 [mg] were not large,” according to Dr. Pardi. “Several of these are mitigatable by dermatologic exam or, hopefully, a vaccine.”
Several of the advisory committee members submitted conflict of interest waivers. Chair and vice chair Jean-Pierre Raufman, MD, and Darrell Pardi, MD, disclosed funding from competing pharmaceutical manufacturers.
*This article was updated on March 12, 2018.
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING
FDA approves tests to screen for tickborne parasite in blood supply
The Food and Drug Administration has approved two new tests to screen whole blood and plasma supplies for the tickborne parasite Babesia microti.
The Imugen Babesia microti Arrayed Fluorescent Immunoassay (AFIA) detects antibodies to B. microti in human plasma samples and the Imugen Babesia microti Nucleic Acid Test (NAT) detects B. microti DNA in human whole blood samples. The tests can be used as donor screening tests on individual samples from donors of whole blood and blood components, as well as living organ and tissue donors, according to the FDA. The tests are not intended as a way to diagnose babesiosis infections.
The new screening tests are the first approvals for B. microti detection. The tests have been available under investigational use since 2012. The tests are manufactured by Oxford Immunotec and are currently only available as in-house tests at the company’s Norwood, Mass., facility.
There is no FDA guidance for testing blood and plasma samples for B. microti, but the agency is planning to issue draft guidance later in 2018.
The Food and Drug Administration has approved two new tests to screen whole blood and plasma supplies for the tickborne parasite Babesia microti.
The Imugen Babesia microti Arrayed Fluorescent Immunoassay (AFIA) detects antibodies to B. microti in human plasma samples and the Imugen Babesia microti Nucleic Acid Test (NAT) detects B. microti DNA in human whole blood samples. The tests can be used as donor screening tests on individual samples from donors of whole blood and blood components, as well as living organ and tissue donors, according to the FDA. The tests are not intended as a way to diagnose babesiosis infections.
The new screening tests are the first approvals for B. microti detection. The tests have been available under investigational use since 2012. The tests are manufactured by Oxford Immunotec and are currently only available as in-house tests at the company’s Norwood, Mass., facility.
There is no FDA guidance for testing blood and plasma samples for B. microti, but the agency is planning to issue draft guidance later in 2018.
The Food and Drug Administration has approved two new tests to screen whole blood and plasma supplies for the tickborne parasite Babesia microti.
The Imugen Babesia microti Arrayed Fluorescent Immunoassay (AFIA) detects antibodies to B. microti in human plasma samples and the Imugen Babesia microti Nucleic Acid Test (NAT) detects B. microti DNA in human whole blood samples. The tests can be used as donor screening tests on individual samples from donors of whole blood and blood components, as well as living organ and tissue donors, according to the FDA. The tests are not intended as a way to diagnose babesiosis infections.
The new screening tests are the first approvals for B. microti detection. The tests have been available under investigational use since 2012. The tests are manufactured by Oxford Immunotec and are currently only available as in-house tests at the company’s Norwood, Mass., facility.
There is no FDA guidance for testing blood and plasma samples for B. microti, but the agency is planning to issue draft guidance later in 2018.
Lurasidone approved for bipolar I depression for children aged 10-17
The Food and Drug Administration has approved lurasidone HCI (Latuda) for treating bipolar I depression in children and adolescents, according to a March 6 statement from the drug’s manufacturer.
“We know that children who have been diagnosed with bipolar depression can be at risk for poor school performance and impairments in social functioning,” said Robert L. Findling, MD, professor of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore, in the statement.
Approval of the atypical antipsychotic is based on results of a 6-week, randomized placebo-controlled phase 3 study of 347 children and adolescents diagnosed with bipolar I depression. Patients received either 20-80 mg/day of lurasidone or placebo.
Patients who received lurasidone reportedly experienced improved bipolar depression symptoms, compared with placebo, based on “the primary efficacy endpoint of change from baseline to week 6 on the Children’s Depression Rating Scale–Revised total score (–21.0 vs. –15.3; effect size = 0.45; P less than .0001),” the statement said. Clinically relevant changes also were found among patients who took the medication on other measures, including the Clinical Global Impressions-Bipolar Scale.
The most common adverse effects were nausea (16% vs. 5.8%), weight gain (6.9% vs. 1.7%), and insomnia (5.1% vs. 2.3%).
Lurasidone also has been approved for treating schizophrenia and bipolar I depression in adults. Last year, the drug was approved for treating schizophrenia in adolescents.
The Food and Drug Administration has approved lurasidone HCI (Latuda) for treating bipolar I depression in children and adolescents, according to a March 6 statement from the drug’s manufacturer.
“We know that children who have been diagnosed with bipolar depression can be at risk for poor school performance and impairments in social functioning,” said Robert L. Findling, MD, professor of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore, in the statement.
Approval of the atypical antipsychotic is based on results of a 6-week, randomized placebo-controlled phase 3 study of 347 children and adolescents diagnosed with bipolar I depression. Patients received either 20-80 mg/day of lurasidone or placebo.
Patients who received lurasidone reportedly experienced improved bipolar depression symptoms, compared with placebo, based on “the primary efficacy endpoint of change from baseline to week 6 on the Children’s Depression Rating Scale–Revised total score (–21.0 vs. –15.3; effect size = 0.45; P less than .0001),” the statement said. Clinically relevant changes also were found among patients who took the medication on other measures, including the Clinical Global Impressions-Bipolar Scale.
The most common adverse effects were nausea (16% vs. 5.8%), weight gain (6.9% vs. 1.7%), and insomnia (5.1% vs. 2.3%).
Lurasidone also has been approved for treating schizophrenia and bipolar I depression in adults. Last year, the drug was approved for treating schizophrenia in adolescents.
The Food and Drug Administration has approved lurasidone HCI (Latuda) for treating bipolar I depression in children and adolescents, according to a March 6 statement from the drug’s manufacturer.
“We know that children who have been diagnosed with bipolar depression can be at risk for poor school performance and impairments in social functioning,” said Robert L. Findling, MD, professor of psychiatry and behavioral sciences at Johns Hopkins University, Baltimore, in the statement.
Approval of the atypical antipsychotic is based on results of a 6-week, randomized placebo-controlled phase 3 study of 347 children and adolescents diagnosed with bipolar I depression. Patients received either 20-80 mg/day of lurasidone or placebo.
Patients who received lurasidone reportedly experienced improved bipolar depression symptoms, compared with placebo, based on “the primary efficacy endpoint of change from baseline to week 6 on the Children’s Depression Rating Scale–Revised total score (–21.0 vs. –15.3; effect size = 0.45; P less than .0001),” the statement said. Clinically relevant changes also were found among patients who took the medication on other measures, including the Clinical Global Impressions-Bipolar Scale.
The most common adverse effects were nausea (16% vs. 5.8%), weight gain (6.9% vs. 1.7%), and insomnia (5.1% vs. 2.3%).
Lurasidone also has been approved for treating schizophrenia and bipolar I depression in adults. Last year, the drug was approved for treating schizophrenia in adolescents.
FDA approves new treatment for multidrug-resistant HIV
The Food and Drug Administration Feb. 6 approved ibalizumab-uiyk (Trogarzo) to treat multidrug-resistant HIV (MDR HIV) in adults, the agency announced.
“While most patients living with HIV can be successfully treated using a combination of two or more antiretroviral drugs, a small percentage of patients who have taken many HIV drugs in the past have multidrug-resistant HIV, limiting their treatment options and putting them at a high risk of HIV-related complications and progression to death,” Jeff Murray, MD, deputy director of the FDA Division of Antiviral Products, said in a statement. “Trogarzo is the first drug in a new class of antiretroviral medications that can provide significant benefit to patients who have run out of HIV treatment options. New treatment options may be able to improve their outcomes.”

Ibalizumab-uiyk, an HIV-1 inhibitor, was approved based on the results of a phase 3, single-arm study of 40 patients with MDR-HIV-1 who had high virus levels in their blood despite antiretroviral treatment. To be included in the study, all patients had to have received highly active antiretroviral therapy for at least 6 months prior. Over 24 weeks, patients were monitored to compare previous, infective treatments with ibalizumab-uiyk in conjunction with an optimized background regimen of antiretroviral drugs.
The majority of patients showed a significant decrease in their HIV-RNA levels 1 week after ibalizumab-uiyk was added to their previous drug regimens. After 24 weeks of treatment with ibalizumab-uiyk, in conjunction with other antiretroviral drugs, 43% of patients achieved HIV RNA suppression.
Ibalizumab-uiyk was granted Fast Track, Priority Review, and Breakthrough Therapy designations from the FDA. In addition, it was granted Orphan Drug designation, a program that encourages the development of drugs to treat rare diseases.
Ibalizumab-uiyk is administered once every 14 days in conjunction with other retroviral medications.
The most common adverse reactions to ibalizumab-uiyk were diarrhea, dizziness, nausea, and rash. Less common, and more severe, reactions were changes in the immune system.
Ibalizumab-uiyk will be marketed by Taimed Biologics USA.
The Food and Drug Administration Feb. 6 approved ibalizumab-uiyk (Trogarzo) to treat multidrug-resistant HIV (MDR HIV) in adults, the agency announced.
“While most patients living with HIV can be successfully treated using a combination of two or more antiretroviral drugs, a small percentage of patients who have taken many HIV drugs in the past have multidrug-resistant HIV, limiting their treatment options and putting them at a high risk of HIV-related complications and progression to death,” Jeff Murray, MD, deputy director of the FDA Division of Antiviral Products, said in a statement. “Trogarzo is the first drug in a new class of antiretroviral medications that can provide significant benefit to patients who have run out of HIV treatment options. New treatment options may be able to improve their outcomes.”
Ibalizumab-uiyk, an HIV-1 inhibitor, was approved based on the results of a phase 3, single-arm study of 40 patients with MDR-HIV-1 who had high virus levels in their blood despite antiretroviral treatment. To be included in the study, all patients had to have received highly active antiretroviral therapy for at least 6 months prior. Over 24 weeks, patients were monitored to compare previous, infective treatments with ibalizumab-uiyk in conjunction with an optimized background regimen of antiretroviral drugs.
The majority of patients showed a significant decrease in their HIV-RNA levels 1 week after ibalizumab-uiyk was added to their previous drug regimens. After 24 weeks of treatment with ibalizumab-uiyk, in conjunction with other antiretroviral drugs, 43% of patients achieved HIV RNA suppression.
Ibalizumab-uiyk was granted Fast Track, Priority Review, and Breakthrough Therapy designations from the FDA. In addition, it was granted Orphan Drug designation, a program that encourages the development of drugs to treat rare diseases.
Ibalizumab-uiyk is administered once every 14 days in conjunction with other retroviral medications.
The most common adverse reactions to ibalizumab-uiyk were diarrhea, dizziness, nausea, and rash. Less common, and more severe, reactions were changes in the immune system.
Ibalizumab-uiyk will be marketed by Taimed Biologics USA.
The Food and Drug Administration Feb. 6 approved ibalizumab-uiyk (Trogarzo) to treat multidrug-resistant HIV (MDR HIV) in adults, the agency announced.
“While most patients living with HIV can be successfully treated using a combination of two or more antiretroviral drugs, a small percentage of patients who have taken many HIV drugs in the past have multidrug-resistant HIV, limiting their treatment options and putting them at a high risk of HIV-related complications and progression to death,” Jeff Murray, MD, deputy director of the FDA Division of Antiviral Products, said in a statement. “Trogarzo is the first drug in a new class of antiretroviral medications that can provide significant benefit to patients who have run out of HIV treatment options. New treatment options may be able to improve their outcomes.”
Ibalizumab-uiyk, an HIV-1 inhibitor, was approved based on the results of a phase 3, single-arm study of 40 patients with MDR-HIV-1 who had high virus levels in their blood despite antiretroviral treatment. To be included in the study, all patients had to have received highly active antiretroviral therapy for at least 6 months prior. Over 24 weeks, patients were monitored to compare previous, infective treatments with ibalizumab-uiyk in conjunction with an optimized background regimen of antiretroviral drugs.
The majority of patients showed a significant decrease in their HIV-RNA levels 1 week after ibalizumab-uiyk was added to their previous drug regimens. After 24 weeks of treatment with ibalizumab-uiyk, in conjunction with other antiretroviral drugs, 43% of patients achieved HIV RNA suppression.
Ibalizumab-uiyk was granted Fast Track, Priority Review, and Breakthrough Therapy designations from the FDA. In addition, it was granted Orphan Drug designation, a program that encourages the development of drugs to treat rare diseases.
Ibalizumab-uiyk is administered once every 14 days in conjunction with other retroviral medications.
The most common adverse reactions to ibalizumab-uiyk were diarrhea, dizziness, nausea, and rash. Less common, and more severe, reactions were changes in the immune system.
Ibalizumab-uiyk will be marketed by Taimed Biologics USA.
Opioid deaths in the ED increase nationally
Opioid-related deaths in emergency departments increased by approximately 30% across all regions of the United States between 2016 and 2017, according to the Centers for Disease Control and Prevention.
Analysis of 91 million ED visits from the CDC’s National Syndromic Surveillance Program and Enhanced State Opioid Overdose Surveillance database found significant increases in opioid overdose deaths in 16 states, reaching as high as 109% in Wisconsin and 106% in Delaware, CDC officials said during a press briefing.

“We are currently seeing the highest drug overdose death rate ever recorded in the United States, driven by prescription opioids and by illicit opioids such as heroin and illicitly manufactured fentanyl,” said Anne Schuchat, MD, acting CDC director. “In 2016, there were more than 63,000 drug overdose deaths, and more than 42,000 of those deaths involved an opioid.”
Of the 91 million visits, a total of 261,755 were suspected of opioid overdoses across both databases.
The greatest increase was seen in the Midwest region (69.7%), followed by the West (40.3%), Northeast (21.3%), Southwest (20.2%), and Southeast (14%).
Death rates rose across all demographics, regardless of sex or age.
While Delaware recorded some of the highest increases in deaths, Massachusetts, New Hampshire, and Rhode Island decreased, although not within statistical significance.
“These decreases may possibly be related to implementation of interventions, including expansion of access to medication-assisted treatment,” said Dr. Schuchat. “The decrease in Kentucky during this period of time may reflect some fluctuations in drug supply.”
In a comparison of urban and rural areas, large and medium metropolitan communities had the sharpest increase, at 45%.
To combat the rise in deaths, the CDC is encouraging an increase in naloxone distribution and training for first responders and community members.
The agency also recommends that local health departments begin using ED data to alert local communities when opioid-related deaths rise.
“This is a very difficult and fast-moving epidemic, and there are no easy solutions,” Dr. Schuchat said. [These data send] “a wake-up call about the need to improve what happens when patients leave the emergency department; all of us working together, government, public health, the medical community, law enforcement, and community members themselves can help fight this epidemic and save lives.”
ezimmerman@frontlinemedcom.com
SOURCE: Vivolo-Kantor AM et al. MMWR Morb Mortal Wkly Rep. 6 Mar 2018. doi: 10.15585/mmwr.mm6709e1.
Opioid-related deaths in emergency departments increased by approximately 30% across all regions of the United States between 2016 and 2017, according to the Centers for Disease Control and Prevention.
Analysis of 91 million ED visits from the CDC’s National Syndromic Surveillance Program and Enhanced State Opioid Overdose Surveillance database found significant increases in opioid overdose deaths in 16 states, reaching as high as 109% in Wisconsin and 106% in Delaware, CDC officials said during a press briefing.
“We are currently seeing the highest drug overdose death rate ever recorded in the United States, driven by prescription opioids and by illicit opioids such as heroin and illicitly manufactured fentanyl,” said Anne Schuchat, MD, acting CDC director. “In 2016, there were more than 63,000 drug overdose deaths, and more than 42,000 of those deaths involved an opioid.”
Of the 91 million visits, a total of 261,755 were suspected of opioid overdoses across both databases.
The greatest increase was seen in the Midwest region (69.7%), followed by the West (40.3%), Northeast (21.3%), Southwest (20.2%), and Southeast (14%).
Death rates rose across all demographics, regardless of sex or age.
While Delaware recorded some of the highest increases in deaths, Massachusetts, New Hampshire, and Rhode Island decreased, although not within statistical significance.
“These decreases may possibly be related to implementation of interventions, including expansion of access to medication-assisted treatment,” said Dr. Schuchat. “The decrease in Kentucky during this period of time may reflect some fluctuations in drug supply.”
In a comparison of urban and rural areas, large and medium metropolitan communities had the sharpest increase, at 45%.
To combat the rise in deaths, the CDC is encouraging an increase in naloxone distribution and training for first responders and community members.
The agency also recommends that local health departments begin using ED data to alert local communities when opioid-related deaths rise.
“This is a very difficult and fast-moving epidemic, and there are no easy solutions,” Dr. Schuchat said. [These data send] “a wake-up call about the need to improve what happens when patients leave the emergency department; all of us working together, government, public health, the medical community, law enforcement, and community members themselves can help fight this epidemic and save lives.”
ezimmerman@frontlinemedcom.com
SOURCE: Vivolo-Kantor AM et al. MMWR Morb Mortal Wkly Rep. 6 Mar 2018. doi: 10.15585/mmwr.mm6709e1.
Opioid-related deaths in emergency departments increased by approximately 30% across all regions of the United States between 2016 and 2017, according to the Centers for Disease Control and Prevention.
Analysis of 91 million ED visits from the CDC’s National Syndromic Surveillance Program and Enhanced State Opioid Overdose Surveillance database found significant increases in opioid overdose deaths in 16 states, reaching as high as 109% in Wisconsin and 106% in Delaware, CDC officials said during a press briefing.
“We are currently seeing the highest drug overdose death rate ever recorded in the United States, driven by prescription opioids and by illicit opioids such as heroin and illicitly manufactured fentanyl,” said Anne Schuchat, MD, acting CDC director. “In 2016, there were more than 63,000 drug overdose deaths, and more than 42,000 of those deaths involved an opioid.”
Of the 91 million visits, a total of 261,755 were suspected of opioid overdoses across both databases.
The greatest increase was seen in the Midwest region (69.7%), followed by the West (40.3%), Northeast (21.3%), Southwest (20.2%), and Southeast (14%).
Death rates rose across all demographics, regardless of sex or age.
While Delaware recorded some of the highest increases in deaths, Massachusetts, New Hampshire, and Rhode Island decreased, although not within statistical significance.
“These decreases may possibly be related to implementation of interventions, including expansion of access to medication-assisted treatment,” said Dr. Schuchat. “The decrease in Kentucky during this period of time may reflect some fluctuations in drug supply.”
In a comparison of urban and rural areas, large and medium metropolitan communities had the sharpest increase, at 45%.
To combat the rise in deaths, the CDC is encouraging an increase in naloxone distribution and training for first responders and community members.
The agency also recommends that local health departments begin using ED data to alert local communities when opioid-related deaths rise.
“This is a very difficult and fast-moving epidemic, and there are no easy solutions,” Dr. Schuchat said. [These data send] “a wake-up call about the need to improve what happens when patients leave the emergency department; all of us working together, government, public health, the medical community, law enforcement, and community members themselves can help fight this epidemic and save lives.”
ezimmerman@frontlinemedcom.com
SOURCE: Vivolo-Kantor AM et al. MMWR Morb Mortal Wkly Rep. 6 Mar 2018. doi: 10.15585/mmwr.mm6709e1.
FROM MMWR