User login
FDA authorizes first direct-to-consumer BRCA1/2 test
The Food and Drug Administration has authorized the first direct-to-consumer (DTC) test to report on three specific BRCA1/BRCA2 breast cancer gene mutations.
Personal Genome Service Genetic Health Risk (GHR) Report for BRCA1/BRCA2 (Selected Variants) does not identify the most common BRCA1/2 mutations but rather the three most common in people of Ashkenazi (Eastern European) Jewish descent, the FDA said in a press statement.

The test, marketed by 23andMe, analyzes DNA from a self-collected saliva sample.
The three mutations identified by the test are present in about 2% of Ashkenazi Jewish women, but rarely in other ethnic populations. Any individual who takes the test may have other mutations in BRCA1 or BRCA2 genes, or other cancer-related gene mutations that are not detected by this test.
“This test provides information to certain individuals who may be at increased breast, ovarian, or prostate cancer risk and who might not otherwise get genetic screening and is a step forward in the availability of DTC genetic tests. But it has a lot of caveats,” Donald St. Pierre, acting director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the press statement. “While the detection of a BRCA mutation on this test does indicate an increased risk, only a small percentage of Americans carry one of these three mutations and most BRCA mutations that increase an individual’s risk are not detected by this test. The test should not be used as a substitute for seeing your doctor for cancer screenings or counseling on genetic and lifestyle factors that can increase or decrease cancer risk.”
The authorization was based on data provided by the company to indicate the test correctly identifies the three genetic variants in saliva samples and is reproducible. In addition, the company submitted data to demonstrate that the instructions are comprehensible and easy to follow.
The FDA cautions that consumers and health care professionals “should not use the test results to determine any treatments, including antihormone therapies and prophylactic removal of the breasts or ovaries.” Decisions should be made only after confirmatory testing and genetic counseling, they said.
The Food and Drug Administration has authorized the first direct-to-consumer (DTC) test to report on three specific BRCA1/BRCA2 breast cancer gene mutations.
Personal Genome Service Genetic Health Risk (GHR) Report for BRCA1/BRCA2 (Selected Variants) does not identify the most common BRCA1/2 mutations but rather the three most common in people of Ashkenazi (Eastern European) Jewish descent, the FDA said in a press statement.
The test, marketed by 23andMe, analyzes DNA from a self-collected saliva sample.
The three mutations identified by the test are present in about 2% of Ashkenazi Jewish women, but rarely in other ethnic populations. Any individual who takes the test may have other mutations in BRCA1 or BRCA2 genes, or other cancer-related gene mutations that are not detected by this test.
“This test provides information to certain individuals who may be at increased breast, ovarian, or prostate cancer risk and who might not otherwise get genetic screening and is a step forward in the availability of DTC genetic tests. But it has a lot of caveats,” Donald St. Pierre, acting director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the press statement. “While the detection of a BRCA mutation on this test does indicate an increased risk, only a small percentage of Americans carry one of these three mutations and most BRCA mutations that increase an individual’s risk are not detected by this test. The test should not be used as a substitute for seeing your doctor for cancer screenings or counseling on genetic and lifestyle factors that can increase or decrease cancer risk.”
The authorization was based on data provided by the company to indicate the test correctly identifies the three genetic variants in saliva samples and is reproducible. In addition, the company submitted data to demonstrate that the instructions are comprehensible and easy to follow.
The FDA cautions that consumers and health care professionals “should not use the test results to determine any treatments, including antihormone therapies and prophylactic removal of the breasts or ovaries.” Decisions should be made only after confirmatory testing and genetic counseling, they said.
The Food and Drug Administration has authorized the first direct-to-consumer (DTC) test to report on three specific BRCA1/BRCA2 breast cancer gene mutations.
Personal Genome Service Genetic Health Risk (GHR) Report for BRCA1/BRCA2 (Selected Variants) does not identify the most common BRCA1/2 mutations but rather the three most common in people of Ashkenazi (Eastern European) Jewish descent, the FDA said in a press statement.
The test, marketed by 23andMe, analyzes DNA from a self-collected saliva sample.
The three mutations identified by the test are present in about 2% of Ashkenazi Jewish women, but rarely in other ethnic populations. Any individual who takes the test may have other mutations in BRCA1 or BRCA2 genes, or other cancer-related gene mutations that are not detected by this test.
“This test provides information to certain individuals who may be at increased breast, ovarian, or prostate cancer risk and who might not otherwise get genetic screening and is a step forward in the availability of DTC genetic tests. But it has a lot of caveats,” Donald St. Pierre, acting director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health, said in the press statement. “While the detection of a BRCA mutation on this test does indicate an increased risk, only a small percentage of Americans carry one of these three mutations and most BRCA mutations that increase an individual’s risk are not detected by this test. The test should not be used as a substitute for seeing your doctor for cancer screenings or counseling on genetic and lifestyle factors that can increase or decrease cancer risk.”
The authorization was based on data provided by the company to indicate the test correctly identifies the three genetic variants in saliva samples and is reproducible. In addition, the company submitted data to demonstrate that the instructions are comprehensible and easy to follow.
The FDA cautions that consumers and health care professionals “should not use the test results to determine any treatments, including antihormone therapies and prophylactic removal of the breasts or ovaries.” Decisions should be made only after confirmatory testing and genetic counseling, they said.
FDA alerts doctors to stop administering compounded products
The Food and Drug Administration today alerted physicians to stop administering or providing drugs manufactured by Cantrell Drug, including opioid products and other drugs intended for sterile injection, because the company has failed to ensure quality and sterility in its compounding operations, which could seriously compromise patient safety.
Health care providers should check medical supplies and separate anything produced by Cantrell Drug, which can be identified by finding the company’s name on the label. The FDA also recommended that physicians seek alternative arrangements for medicines and that any patients who have any products made by Cantrell contact their physicians.

So far, the FDA is not aware of any illness or injuries caused by potentially contaminated products from Cantrell, but any that do arise should be reported to the FDA’s MedWatch program.
Read more about this alert in the FDA website.
The Food and Drug Administration today alerted physicians to stop administering or providing drugs manufactured by Cantrell Drug, including opioid products and other drugs intended for sterile injection, because the company has failed to ensure quality and sterility in its compounding operations, which could seriously compromise patient safety.
Health care providers should check medical supplies and separate anything produced by Cantrell Drug, which can be identified by finding the company’s name on the label. The FDA also recommended that physicians seek alternative arrangements for medicines and that any patients who have any products made by Cantrell contact their physicians.
So far, the FDA is not aware of any illness or injuries caused by potentially contaminated products from Cantrell, but any that do arise should be reported to the FDA’s MedWatch program.
Read more about this alert in the FDA website.
The Food and Drug Administration today alerted physicians to stop administering or providing drugs manufactured by Cantrell Drug, including opioid products and other drugs intended for sterile injection, because the company has failed to ensure quality and sterility in its compounding operations, which could seriously compromise patient safety.
Health care providers should check medical supplies and separate anything produced by Cantrell Drug, which can be identified by finding the company’s name on the label. The FDA also recommended that physicians seek alternative arrangements for medicines and that any patients who have any products made by Cantrell contact their physicians.
So far, the FDA is not aware of any illness or injuries caused by potentially contaminated products from Cantrell, but any that do arise should be reported to the FDA’s MedWatch program.
Read more about this alert in the FDA website.
2 new influenza strains recommended for next season
SILVER SPRING, MD. – In an effort to better match the vaccine to the virus, federal advisors have recommended two new strains be swapped into the 2018-2019 quadrivalent influenza vaccine.
Singapore A(H3N2) and the B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage) are recommended be added to A/Michigan/45/2015 (H1N1)pdm09-like virus and B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage) for the upcoming season, according to a near-unanimous vote at a meeting of the Food and Drug Administration Vaccines and Related Biological Products Advisory Committee.
Trivalent vaccines should include the same strains, with the exception of B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage), the committee recommended.
The panel voted separately on the strains, and all votes were unanimous, except for the vote on the B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage) in the trivalent vaccine, which was supported with 11 positive votes with 1 abstention.
The advisory committee’s recommendation is identical to the recommendations recently made by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. The WHO recommended that trivalent vaccines contain A/Michigan/45/2015 (H1N1)pdm09-like virus, A/Singapore/INFIMH-16-0019/2016 (H3N2)-like virus, and B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage). WHO also recommended that quadrivalent vaccines contain all of the above strains and B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage) as the second influenza B strain.
Most of the influenza activity in the United States this season is due to influenza A (H3N2) viruses (67%), according to Lisa Grohskopf, MD, associate chief for policy & liaison in the Influenza Division at the Centers for Disease Control and Prevention. Fortunately, the majority of circulating strains are similar to those contained in the 2017-2018 vaccine. Only strains with B/Victoria lineage displayed antigenic drift, but represented less than 1% of all circulating viruses.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups, and have increased since last season. As of Feb. 17, the preliminary estimate of hospitalizations in this age group was 322.7 cases per 100,000 people, compared with about 290.5 per 100,000 during the 2016-2017 season. There have been 97 pediatric deaths associated with influenza, compared with 110 reported during the 2016-2017 season, 93 during 2015-2016, and 148 during 2014-2015.
These data are not final because the flu season is still ongoing, but a full analysis will be provided at the end of the season, Dr. Grohskopf pointed out.With H3N2 strains of influenza A predominating, questions on the effectiveness of the newly recommended Singapore A(H3N2) were raised by the committee. Jacqueline Katz, PhD, director of the WHO Collaborating Center for Surveillance, Epidemiology, and Control of Influenza, reassured the committee.
“Yes, in fact, it does cover them very well. The majority of the viruses that we’ve tested at the CDC were that emerging 3C2a2 [clade of H3N2] group, and the Singapore virus covered those very well. In general, that’s why we went with Singapore,” she said.
Dr. Katz added that one of the reasons Singapore is so effective is because of its position in the lineage of these flu strains. “It’s at the base of the [phylogenetic] tree; it’s not on the tip of the tree where things are changing, so it’s a more conservative selection.”
The CDC estimate of current vaccine effectiveness (VE) against influenza A (H3N2) viruses is 25%, as of Feb. 3. Effectiveness is even higher for all influenza viruses, with an estimated VE of 36%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 36% (MMWR. 2018;67:180-5).
While the FDA usually follows the recommendations of its panel members, it is not obligated to do so. None of the committee members disclosed relevant financial conflicts of interest.
SILVER SPRING, MD. – In an effort to better match the vaccine to the virus, federal advisors have recommended two new strains be swapped into the 2018-2019 quadrivalent influenza vaccine.
Singapore A(H3N2) and the B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage) are recommended be added to A/Michigan/45/2015 (H1N1)pdm09-like virus and B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage) for the upcoming season, according to a near-unanimous vote at a meeting of the Food and Drug Administration Vaccines and Related Biological Products Advisory Committee.
Trivalent vaccines should include the same strains, with the exception of B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage), the committee recommended.
The panel voted separately on the strains, and all votes were unanimous, except for the vote on the B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage) in the trivalent vaccine, which was supported with 11 positive votes with 1 abstention.
The advisory committee’s recommendation is identical to the recommendations recently made by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. The WHO recommended that trivalent vaccines contain A/Michigan/45/2015 (H1N1)pdm09-like virus, A/Singapore/INFIMH-16-0019/2016 (H3N2)-like virus, and B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage). WHO also recommended that quadrivalent vaccines contain all of the above strains and B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage) as the second influenza B strain.
Most of the influenza activity in the United States this season is due to influenza A (H3N2) viruses (67%), according to Lisa Grohskopf, MD, associate chief for policy & liaison in the Influenza Division at the Centers for Disease Control and Prevention. Fortunately, the majority of circulating strains are similar to those contained in the 2017-2018 vaccine. Only strains with B/Victoria lineage displayed antigenic drift, but represented less than 1% of all circulating viruses.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups, and have increased since last season. As of Feb. 17, the preliminary estimate of hospitalizations in this age group was 322.7 cases per 100,000 people, compared with about 290.5 per 100,000 during the 2016-2017 season. There have been 97 pediatric deaths associated with influenza, compared with 110 reported during the 2016-2017 season, 93 during 2015-2016, and 148 during 2014-2015.
These data are not final because the flu season is still ongoing, but a full analysis will be provided at the end of the season, Dr. Grohskopf pointed out.With H3N2 strains of influenza A predominating, questions on the effectiveness of the newly recommended Singapore A(H3N2) were raised by the committee. Jacqueline Katz, PhD, director of the WHO Collaborating Center for Surveillance, Epidemiology, and Control of Influenza, reassured the committee.
“Yes, in fact, it does cover them very well. The majority of the viruses that we’ve tested at the CDC were that emerging 3C2a2 [clade of H3N2] group, and the Singapore virus covered those very well. In general, that’s why we went with Singapore,” she said.
Dr. Katz added that one of the reasons Singapore is so effective is because of its position in the lineage of these flu strains. “It’s at the base of the [phylogenetic] tree; it’s not on the tip of the tree where things are changing, so it’s a more conservative selection.”
The CDC estimate of current vaccine effectiveness (VE) against influenza A (H3N2) viruses is 25%, as of Feb. 3. Effectiveness is even higher for all influenza viruses, with an estimated VE of 36%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 36% (MMWR. 2018;67:180-5).
While the FDA usually follows the recommendations of its panel members, it is not obligated to do so. None of the committee members disclosed relevant financial conflicts of interest.
SILVER SPRING, MD. – In an effort to better match the vaccine to the virus, federal advisors have recommended two new strains be swapped into the 2018-2019 quadrivalent influenza vaccine.
Singapore A(H3N2) and the B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage) are recommended be added to A/Michigan/45/2015 (H1N1)pdm09-like virus and B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage) for the upcoming season, according to a near-unanimous vote at a meeting of the Food and Drug Administration Vaccines and Related Biological Products Advisory Committee.
Trivalent vaccines should include the same strains, with the exception of B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage), the committee recommended.
The panel voted separately on the strains, and all votes were unanimous, except for the vote on the B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage) in the trivalent vaccine, which was supported with 11 positive votes with 1 abstention.
The advisory committee’s recommendation is identical to the recommendations recently made by the World Health Organization for next season’s influenza vaccines in the Northern Hemisphere. The WHO recommended that trivalent vaccines contain A/Michigan/45/2015 (H1N1)pdm09-like virus, A/Singapore/INFIMH-16-0019/2016 (H3N2)-like virus, and B/Colorado/06/2017-like virus (B/Victoria/2/87 lineage). WHO also recommended that quadrivalent vaccines contain all of the above strains and B/Phuket/3073/2013-like virus (B/Yamagata/16/88 lineage) as the second influenza B strain.
Most of the influenza activity in the United States this season is due to influenza A (H3N2) viruses (67%), according to Lisa Grohskopf, MD, associate chief for policy & liaison in the Influenza Division at the Centers for Disease Control and Prevention. Fortunately, the majority of circulating strains are similar to those contained in the 2017-2018 vaccine. Only strains with B/Victoria lineage displayed antigenic drift, but represented less than 1% of all circulating viruses.
Hospitalization rates for laboratory-confirmed influenza this season have been markedly higher among people aged 65 years and older, compared with younger age groups, and have increased since last season. As of Feb. 17, the preliminary estimate of hospitalizations in this age group was 322.7 cases per 100,000 people, compared with about 290.5 per 100,000 during the 2016-2017 season. There have been 97 pediatric deaths associated with influenza, compared with 110 reported during the 2016-2017 season, 93 during 2015-2016, and 148 during 2014-2015.
These data are not final because the flu season is still ongoing, but a full analysis will be provided at the end of the season, Dr. Grohskopf pointed out.With H3N2 strains of influenza A predominating, questions on the effectiveness of the newly recommended Singapore A(H3N2) were raised by the committee. Jacqueline Katz, PhD, director of the WHO Collaborating Center for Surveillance, Epidemiology, and Control of Influenza, reassured the committee.
“Yes, in fact, it does cover them very well. The majority of the viruses that we’ve tested at the CDC were that emerging 3C2a2 [clade of H3N2] group, and the Singapore virus covered those very well. In general, that’s why we went with Singapore,” she said.
Dr. Katz added that one of the reasons Singapore is so effective is because of its position in the lineage of these flu strains. “It’s at the base of the [phylogenetic] tree; it’s not on the tip of the tree where things are changing, so it’s a more conservative selection.”
The CDC estimate of current vaccine effectiveness (VE) against influenza A (H3N2) viruses is 25%, as of Feb. 3. Effectiveness is even higher for all influenza viruses, with an estimated VE of 36%, indicating that the flu vaccine reduced a person’s risk of having to seek medical care at a doctor’s office for flu illness by 36% (MMWR. 2018;67:180-5).
While the FDA usually follows the recommendations of its panel members, it is not obligated to do so. None of the committee members disclosed relevant financial conflicts of interest.
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING
Arm-placement indication for CGM approved by FDA
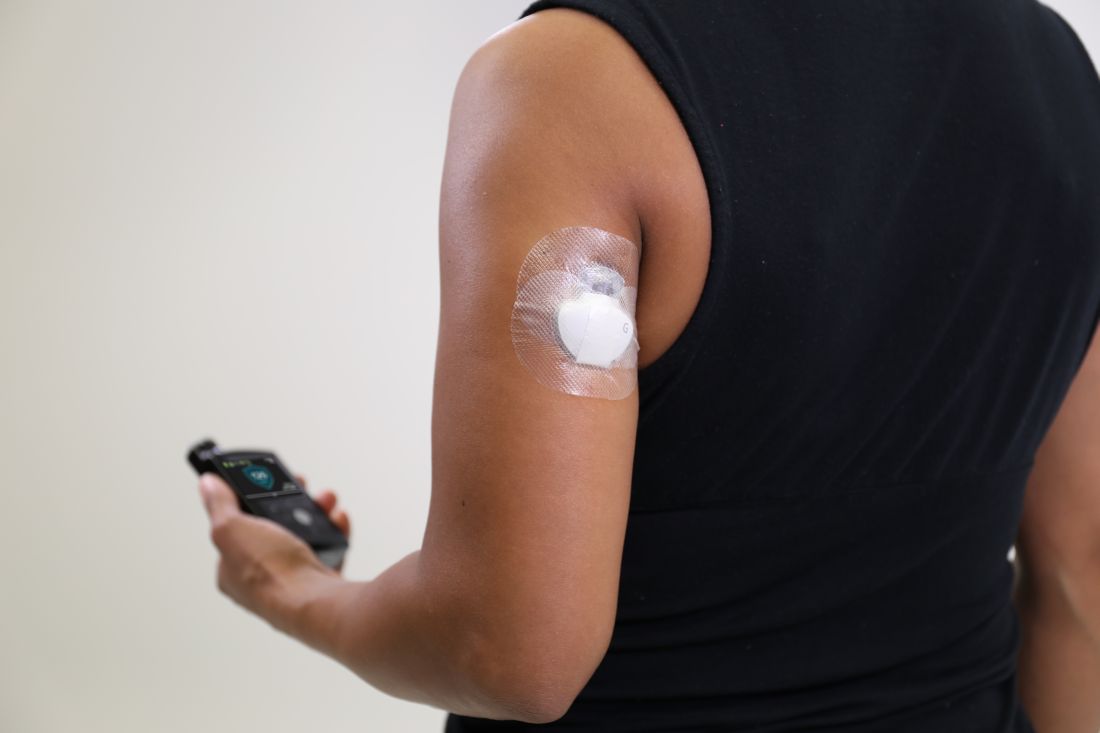
The Food and Drug Administration has approved a new indication for the Guardian Sensor 3 continuous glucose monitor (CGM), allowing patients to position it on their arms, according to an announcement by Medtronic, the sensor’s manufacturer.
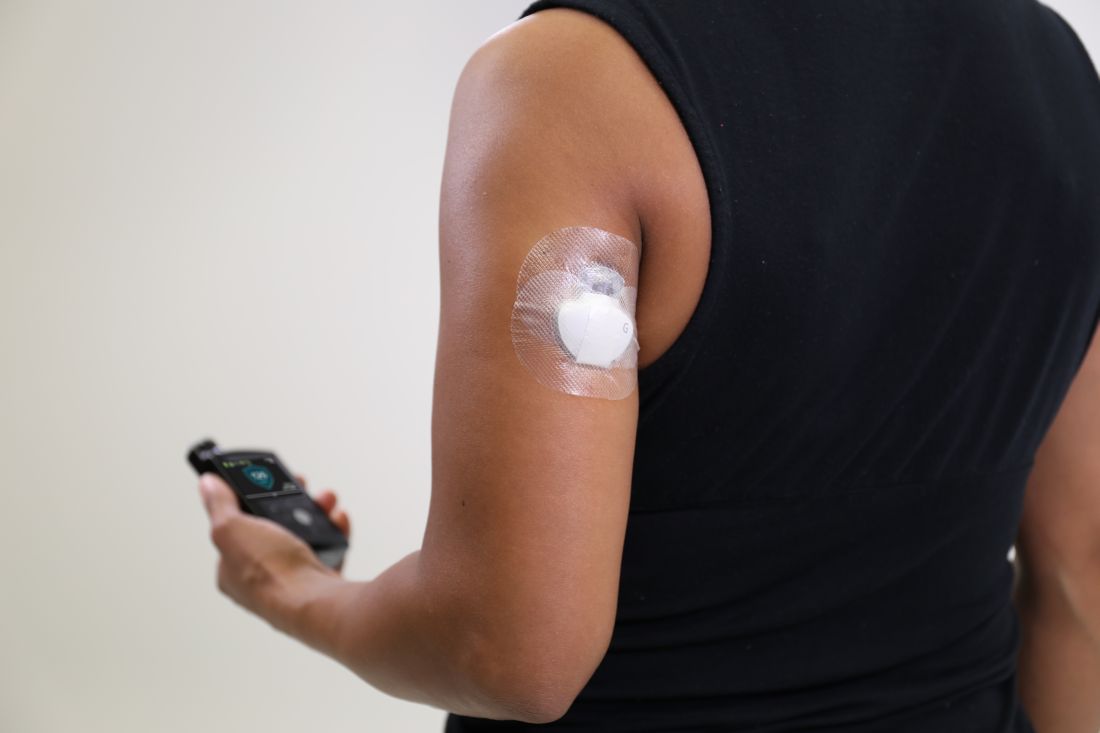
Although previously only the abdomen had FDA approval as a CGM site, findings from at least one study have shown that most patients prefer to position their sensors elsewhere. This new indication gives these patients more flexibility in where they can place their CGMs. The site of CGM placement did not affect the failure rate or noncompliance.
The sensor works in tandem with an automated insulin delivery system known as the MiniMed 670G system, also manufactured by Medtronic. This system is approved for people with type 1 diabetes aged 14 years and older and adjusts basal insulin levels every 5 minutes – “based on real-time data delivered by the Guardian Sensor 3,” according to Medtronic.
Read more in Medtronic’s press release.
Correction, 3/21/18: A previous version of this article contained a photo of a device that was not the Guardian 3 Sensor.
The Food and Drug Administration has approved a new indication for the Guardian Sensor 3 continuous glucose monitor (CGM), allowing patients to position it on their arms, according to an announcement by Medtronic, the sensor’s manufacturer.
Although previously only the abdomen had FDA approval as a CGM site, findings from at least one study have shown that most patients prefer to position their sensors elsewhere. This new indication gives these patients more flexibility in where they can place their CGMs. The site of CGM placement did not affect the failure rate or noncompliance.
The sensor works in tandem with an automated insulin delivery system known as the MiniMed 670G system, also manufactured by Medtronic. This system is approved for people with type 1 diabetes aged 14 years and older and adjusts basal insulin levels every 5 minutes – “based on real-time data delivered by the Guardian Sensor 3,” according to Medtronic.
Read more in Medtronic’s press release.
Correction, 3/21/18: A previous version of this article contained a photo of a device that was not the Guardian 3 Sensor.
The Food and Drug Administration has approved a new indication for the Guardian Sensor 3 continuous glucose monitor (CGM), allowing patients to position it on their arms, according to an announcement by Medtronic, the sensor’s manufacturer.
Although previously only the abdomen had FDA approval as a CGM site, findings from at least one study have shown that most patients prefer to position their sensors elsewhere. This new indication gives these patients more flexibility in where they can place their CGMs. The site of CGM placement did not affect the failure rate or noncompliance.
The sensor works in tandem with an automated insulin delivery system known as the MiniMed 670G system, also manufactured by Medtronic. This system is approved for people with type 1 diabetes aged 14 years and older and adjusts basal insulin levels every 5 minutes – “based on real-time data delivered by the Guardian Sensor 3,” according to Medtronic.
Read more in Medtronic’s press release.
Correction, 3/21/18: A previous version of this article contained a photo of a device that was not the Guardian 3 Sensor.
FDA approves abemaciclib plus aromatase inhibitor as initial therapy
Abemaciclib (Verzenio) in combination with an aromatase inhibitor has been approved as initial endocrine-based therapy for postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer, the US Food and Drug Administration announced in a press release.
Approval was based on the results of the MONARCH 3 study, a randomized, double-blind, placebo-controlled, multicenter clinical trial in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer. A total of 493 patients were randomized to receive either abemaciclib 150 mg or placebo orally twice daily, plus the treating physician’s choice of letrozole or anastrozole. The estimated median progression-free survival (PFS) (RECIST 1.1) was 28.2 months (95% CI: 23.5, not reached) for patients receiving abemaciclib and 14.8 months (95% CI: 11.2, 19.2) for those receiving placebo (HR 0.540; 95% CI: 0.418, 0.698; p<0.0001).
The most common adverse reactions that were seen in at least 20% of patients receiving abemaciclib in MONARCH 3 and were reported at a rate more than 2% higher than the rates seen in the placebo arm were diarrhea, neutropenia, fatigue, infections, nausea, abdominal pain, anemia, vomiting, alopecia, decreased appetite, and leukopenia.
The recommended starting dose of abemaciclib in combination with an aromatase inhibitor is 150 mg twice daily orally with or without food.
Abemaciclib (Verzenio) is manufactured by Eli Lilly.
Full prescribing information is available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208855s000lbl.pdf.
Abemaciclib (Verzenio) in combination with an aromatase inhibitor has been approved as initial endocrine-based therapy for postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer, the US Food and Drug Administration announced in a press release.
Approval was based on the results of the MONARCH 3 study, a randomized, double-blind, placebo-controlled, multicenter clinical trial in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer. A total of 493 patients were randomized to receive either abemaciclib 150 mg or placebo orally twice daily, plus the treating physician’s choice of letrozole or anastrozole. The estimated median progression-free survival (PFS) (RECIST 1.1) was 28.2 months (95% CI: 23.5, not reached) for patients receiving abemaciclib and 14.8 months (95% CI: 11.2, 19.2) for those receiving placebo (HR 0.540; 95% CI: 0.418, 0.698; p<0.0001).
The most common adverse reactions that were seen in at least 20% of patients receiving abemaciclib in MONARCH 3 and were reported at a rate more than 2% higher than the rates seen in the placebo arm were diarrhea, neutropenia, fatigue, infections, nausea, abdominal pain, anemia, vomiting, alopecia, decreased appetite, and leukopenia.
The recommended starting dose of abemaciclib in combination with an aromatase inhibitor is 150 mg twice daily orally with or without food.
Abemaciclib (Verzenio) is manufactured by Eli Lilly.
Full prescribing information is available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208855s000lbl.pdf.
Abemaciclib (Verzenio) in combination with an aromatase inhibitor has been approved as initial endocrine-based therapy for postmenopausal women with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer, the US Food and Drug Administration announced in a press release.
Approval was based on the results of the MONARCH 3 study, a randomized, double-blind, placebo-controlled, multicenter clinical trial in postmenopausal women with HR-positive, HER2-negative advanced or metastatic breast cancer. A total of 493 patients were randomized to receive either abemaciclib 150 mg or placebo orally twice daily, plus the treating physician’s choice of letrozole or anastrozole. The estimated median progression-free survival (PFS) (RECIST 1.1) was 28.2 months (95% CI: 23.5, not reached) for patients receiving abemaciclib and 14.8 months (95% CI: 11.2, 19.2) for those receiving placebo (HR 0.540; 95% CI: 0.418, 0.698; p<0.0001).
The most common adverse reactions that were seen in at least 20% of patients receiving abemaciclib in MONARCH 3 and were reported at a rate more than 2% higher than the rates seen in the placebo arm were diarrhea, neutropenia, fatigue, infections, nausea, abdominal pain, anemia, vomiting, alopecia, decreased appetite, and leukopenia.
The recommended starting dose of abemaciclib in combination with an aromatase inhibitor is 150 mg twice daily orally with or without food.
Abemaciclib (Verzenio) is manufactured by Eli Lilly.
Full prescribing information is available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208855s000lbl.pdf.
FDA warns against clarithromycin use in patients with heart disease
The Food and Drug Administration has added a new warning for an increased risk of death in patients with heart disease who have used clarithromycin (Biaxin), on the basis of results of a 10-year follow-up from the CLARICOR trial.
The CLARICOR trial followed 4,372 randomized patients for at least 2 years after undergoing 14 days of treatment with daily doses of 500 mg clarithromycin. Among these patients, researchers observed an unexpected increase in deaths in patients with coronary heart disease. (The Feb. 22 FDA statement announcing the alert did not provide data from CLARICOR.) As of yet, there is no clear explanation of how clarithromycin would lead to more deaths, compared with a placebo, the agency said.
Regardless, two of the six observational studies published found a link between clarithromycin use and long-term risks; four did not. The CLARICOR trial provides the strongest evidence of increased health risks, the statement said.
The FDA is recommending that health care professionals be aware of the risks associated with clarithromycin use and consider the benefits and risks of use in patients with heart disease. If at all possible, the use of other antibiotics may be a better option. Doctors should advise patients to be aware of signs and symptoms associated with cardiovascular issues.
Patients are also an important piece of the puzzle and should communicate with their health care providers about heart disease, particularly when taking antibiotics to treat for an infection.
The FDA has added the results of the CLARICOR trial to the clarithromycin drug labels. The agency will continue to monitor the safety reports in patients using clarithromycin.
Serious adverse events associated with clarithromycin should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
The Food and Drug Administration has added a new warning for an increased risk of death in patients with heart disease who have used clarithromycin (Biaxin), on the basis of results of a 10-year follow-up from the CLARICOR trial.
The CLARICOR trial followed 4,372 randomized patients for at least 2 years after undergoing 14 days of treatment with daily doses of 500 mg clarithromycin. Among these patients, researchers observed an unexpected increase in deaths in patients with coronary heart disease. (The Feb. 22 FDA statement announcing the alert did not provide data from CLARICOR.) As of yet, there is no clear explanation of how clarithromycin would lead to more deaths, compared with a placebo, the agency said.
Regardless, two of the six observational studies published found a link between clarithromycin use and long-term risks; four did not. The CLARICOR trial provides the strongest evidence of increased health risks, the statement said.
The FDA is recommending that health care professionals be aware of the risks associated with clarithromycin use and consider the benefits and risks of use in patients with heart disease. If at all possible, the use of other antibiotics may be a better option. Doctors should advise patients to be aware of signs and symptoms associated with cardiovascular issues.
Patients are also an important piece of the puzzle and should communicate with their health care providers about heart disease, particularly when taking antibiotics to treat for an infection.
The FDA has added the results of the CLARICOR trial to the clarithromycin drug labels. The agency will continue to monitor the safety reports in patients using clarithromycin.
Serious adverse events associated with clarithromycin should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
The Food and Drug Administration has added a new warning for an increased risk of death in patients with heart disease who have used clarithromycin (Biaxin), on the basis of results of a 10-year follow-up from the CLARICOR trial.
The CLARICOR trial followed 4,372 randomized patients for at least 2 years after undergoing 14 days of treatment with daily doses of 500 mg clarithromycin. Among these patients, researchers observed an unexpected increase in deaths in patients with coronary heart disease. (The Feb. 22 FDA statement announcing the alert did not provide data from CLARICOR.) As of yet, there is no clear explanation of how clarithromycin would lead to more deaths, compared with a placebo, the agency said.
Regardless, two of the six observational studies published found a link between clarithromycin use and long-term risks; four did not. The CLARICOR trial provides the strongest evidence of increased health risks, the statement said.
The FDA is recommending that health care professionals be aware of the risks associated with clarithromycin use and consider the benefits and risks of use in patients with heart disease. If at all possible, the use of other antibiotics may be a better option. Doctors should advise patients to be aware of signs and symptoms associated with cardiovascular issues.
Patients are also an important piece of the puzzle and should communicate with their health care providers about heart disease, particularly when taking antibiotics to treat for an infection.
The FDA has added the results of the CLARICOR trial to the clarithromycin drug labels. The agency will continue to monitor the safety reports in patients using clarithromycin.
Serious adverse events associated with clarithromycin should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch/.
ACIP unanimously recommends HEPLISAV-B
At a meeting of the Center for Disease Control and Prevention’s Advisory Committee on Immunization Practices, members unanimously voted to include HEPLISAV-B on the ACIP list of recommended products to vaccinate adults against hepatitis B.
“I think this is a huge advance, and a step forward “ said David S. Stephens, MD, of Emory University, Atlanta, who is a voting member of ACIP.![]()
According to Sarah Schillie, MD, of ACIP’s Hepatitis Work Group, the reduction from three doses to two also will improve vaccine series completion rates, providing more effective protection. This could be very important for health care professionals, with only about 60% of treated individuals fulfilling the three doses necessary for complete HBV protection.
Although fewer doses are needed with HEPLISAV-B, it displays similar immunogenicity to similar vaccines (90.0%-100% vs. 70.5%-90.2%). It is also more effective, compared with similar vaccines in those with type II diabetes (90.0% vs. 65.1%) and chronic kidney disease (89.9% vs. 81.1%).
HEPLISAV-B uses an adjuvant, but it appears to be safe and well tolerated. The rate of mild (45.6% vs. 45.7%) and serious (5.4% vs. 6.3%) reactions were similar among HEPLISAV-B and comparable vaccines, although cardiovascular events were more common with this vaccine than with others (0.27% vs. 0.14%).
Dr. Stephens, who supported the recommendation of HEPLISAV-B, voiced reservations about these findings. “I am concerned about the myocardial infarction signal and the use of this new adjuvant and certainly urge us to look at the postmarketing data carefully.”
While the reported incidence rate of acute HBV cases consistently fell during 2000-2015, it began to rise again at the end of 2015. This is linked with concomitant rise in injection drug use, which also is a risk factor in transmitting HBV. Over the same period, the incidence of HBV was highest in people aged 30-49 years. With these factors to consider, the vote by ACIP for HEPLISAV-B as a recommended vaccine is timely and may help reduce HBV infections in adults.
At a meeting of the Center for Disease Control and Prevention’s Advisory Committee on Immunization Practices, members unanimously voted to include HEPLISAV-B on the ACIP list of recommended products to vaccinate adults against hepatitis B.
“I think this is a huge advance, and a step forward “ said David S. Stephens, MD, of Emory University, Atlanta, who is a voting member of ACIP.![]()
According to Sarah Schillie, MD, of ACIP’s Hepatitis Work Group, the reduction from three doses to two also will improve vaccine series completion rates, providing more effective protection. This could be very important for health care professionals, with only about 60% of treated individuals fulfilling the three doses necessary for complete HBV protection.
Although fewer doses are needed with HEPLISAV-B, it displays similar immunogenicity to similar vaccines (90.0%-100% vs. 70.5%-90.2%). It is also more effective, compared with similar vaccines in those with type II diabetes (90.0% vs. 65.1%) and chronic kidney disease (89.9% vs. 81.1%).
HEPLISAV-B uses an adjuvant, but it appears to be safe and well tolerated. The rate of mild (45.6% vs. 45.7%) and serious (5.4% vs. 6.3%) reactions were similar among HEPLISAV-B and comparable vaccines, although cardiovascular events were more common with this vaccine than with others (0.27% vs. 0.14%).
Dr. Stephens, who supported the recommendation of HEPLISAV-B, voiced reservations about these findings. “I am concerned about the myocardial infarction signal and the use of this new adjuvant and certainly urge us to look at the postmarketing data carefully.”
While the reported incidence rate of acute HBV cases consistently fell during 2000-2015, it began to rise again at the end of 2015. This is linked with concomitant rise in injection drug use, which also is a risk factor in transmitting HBV. Over the same period, the incidence of HBV was highest in people aged 30-49 years. With these factors to consider, the vote by ACIP for HEPLISAV-B as a recommended vaccine is timely and may help reduce HBV infections in adults.
At a meeting of the Center for Disease Control and Prevention’s Advisory Committee on Immunization Practices, members unanimously voted to include HEPLISAV-B on the ACIP list of recommended products to vaccinate adults against hepatitis B.
“I think this is a huge advance, and a step forward “ said David S. Stephens, MD, of Emory University, Atlanta, who is a voting member of ACIP.![]()
According to Sarah Schillie, MD, of ACIP’s Hepatitis Work Group, the reduction from three doses to two also will improve vaccine series completion rates, providing more effective protection. This could be very important for health care professionals, with only about 60% of treated individuals fulfilling the three doses necessary for complete HBV protection.
Although fewer doses are needed with HEPLISAV-B, it displays similar immunogenicity to similar vaccines (90.0%-100% vs. 70.5%-90.2%). It is also more effective, compared with similar vaccines in those with type II diabetes (90.0% vs. 65.1%) and chronic kidney disease (89.9% vs. 81.1%).
HEPLISAV-B uses an adjuvant, but it appears to be safe and well tolerated. The rate of mild (45.6% vs. 45.7%) and serious (5.4% vs. 6.3%) reactions were similar among HEPLISAV-B and comparable vaccines, although cardiovascular events were more common with this vaccine than with others (0.27% vs. 0.14%).
Dr. Stephens, who supported the recommendation of HEPLISAV-B, voiced reservations about these findings. “I am concerned about the myocardial infarction signal and the use of this new adjuvant and certainly urge us to look at the postmarketing data carefully.”
While the reported incidence rate of acute HBV cases consistently fell during 2000-2015, it began to rise again at the end of 2015. This is linked with concomitant rise in injection drug use, which also is a risk factor in transmitting HBV. Over the same period, the incidence of HBV was highest in people aged 30-49 years. With these factors to consider, the vote by ACIP for HEPLISAV-B as a recommended vaccine is timely and may help reduce HBV infections in adults.
REPORTING FROM AN ACIP MEETING
Gene therapy for hemophilia A gets fast-track review
The investigational gene therapy SPK-8011 for the treatment of hemophilia A is getting an accelerated review at the Food and Drug Administration, according to Spark Therapeutics.
The FDA granted breakthrough therapy designation to the gene therapy product in February and orphan drug status in January.![]()
The investigational gene therapy SPK-8011 for the treatment of hemophilia A is getting an accelerated review at the Food and Drug Administration, according to Spark Therapeutics.
The FDA granted breakthrough therapy designation to the gene therapy product in February and orphan drug status in January.![]()
The investigational gene therapy SPK-8011 for the treatment of hemophilia A is getting an accelerated review at the Food and Drug Administration, according to Spark Therapeutics.
The FDA granted breakthrough therapy designation to the gene therapy product in February and orphan drug status in January.![]()
Imfinzi approved for stage III unresectable NSCLC
Durvalumab (Imfinzi) has been approved as the first treatment for patients with stage III unresectable non-small cell lung cancer (NSCLC) that has not progressed after chemotherapy and radiation, the Food and Drug Administration announced on Feb. 16.
Chemoradiation had been the only option for such patients. “Although a small number of patients may be cured with the chemoradiation, the cancer may eventually progress. Patients now have an approved therapy that has been shown to keep the cancer from progressing for a longer time after chemoradiation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Durvalumab, which targets the PD-1/PD-L1 pathway, was previously granted accelerated approval in 2017 for the treatment of certain patients with locally advanced or metastatic bladder cancer.
The approval for the treatment of stage III, unresectable NSCLC was based on the results of the randomized PACIFIC trial of 713 patients whose cancer had not progressed after completing chemotherapy and radiation. The trial measured progression-free survival with durvalumab or a placebo, and found a median progression-free survival of 16.8 months for patients taking durvalumab and 5.6 months for patients receiving a placebo. The drug's sponsor, AstraZeneca, has agreed to provide data on overall survival in the study.
Common side effects of durvalumab in patients with stage III unresectable NSCLC include cough, fatigue, inflammation in the lungs (pneumonitis/radiation pneumonitis), upper respiratory tract infections, difficulty breathing (dyspnea) and rash.
Serious risks of durvalumab include pneumonitis, hepatitis, colitis, endocrinopathies, and nephritis. Other serious side effects include infection and infusion-related reactions. Durvalumab can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
Durvalumab (Imfinzi) has been approved as the first treatment for patients with stage III unresectable non-small cell lung cancer (NSCLC) that has not progressed after chemotherapy and radiation, the Food and Drug Administration announced on Feb. 16.
Chemoradiation had been the only option for such patients. “Although a small number of patients may be cured with the chemoradiation, the cancer may eventually progress. Patients now have an approved therapy that has been shown to keep the cancer from progressing for a longer time after chemoradiation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Durvalumab, which targets the PD-1/PD-L1 pathway, was previously granted accelerated approval in 2017 for the treatment of certain patients with locally advanced or metastatic bladder cancer.
The approval for the treatment of stage III, unresectable NSCLC was based on the results of the randomized PACIFIC trial of 713 patients whose cancer had not progressed after completing chemotherapy and radiation. The trial measured progression-free survival with durvalumab or a placebo, and found a median progression-free survival of 16.8 months for patients taking durvalumab and 5.6 months for patients receiving a placebo. The drug's sponsor, AstraZeneca, has agreed to provide data on overall survival in the study.
Common side effects of durvalumab in patients with stage III unresectable NSCLC include cough, fatigue, inflammation in the lungs (pneumonitis/radiation pneumonitis), upper respiratory tract infections, difficulty breathing (dyspnea) and rash.
Serious risks of durvalumab include pneumonitis, hepatitis, colitis, endocrinopathies, and nephritis. Other serious side effects include infection and infusion-related reactions. Durvalumab can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
Durvalumab (Imfinzi) has been approved as the first treatment for patients with stage III unresectable non-small cell lung cancer (NSCLC) that has not progressed after chemotherapy and radiation, the Food and Drug Administration announced on Feb. 16.
Chemoradiation had been the only option for such patients. “Although a small number of patients may be cured with the chemoradiation, the cancer may eventually progress. Patients now have an approved therapy that has been shown to keep the cancer from progressing for a longer time after chemoradiation,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Durvalumab, which targets the PD-1/PD-L1 pathway, was previously granted accelerated approval in 2017 for the treatment of certain patients with locally advanced or metastatic bladder cancer.
The approval for the treatment of stage III, unresectable NSCLC was based on the results of the randomized PACIFIC trial of 713 patients whose cancer had not progressed after completing chemotherapy and radiation. The trial measured progression-free survival with durvalumab or a placebo, and found a median progression-free survival of 16.8 months for patients taking durvalumab and 5.6 months for patients receiving a placebo. The drug's sponsor, AstraZeneca, has agreed to provide data on overall survival in the study.
Common side effects of durvalumab in patients with stage III unresectable NSCLC include cough, fatigue, inflammation in the lungs (pneumonitis/radiation pneumonitis), upper respiratory tract infections, difficulty breathing (dyspnea) and rash.
Serious risks of durvalumab include pneumonitis, hepatitis, colitis, endocrinopathies, and nephritis. Other serious side effects include infection and infusion-related reactions. Durvalumab can cause harm to a developing fetus; women should be advised of the potential risk to the fetus and to use effective contraception.
FDA approves new injection product to reduce preterm birth risk
Makena, a progestin injection for the prevention of preterm birth that has been approved for use since 2011, has received U.S. Food and Drug Administration approval for a subcutaneous auto-injection product to supplant its intramuscular injection formulation, announced its manufacturer, AMAG Pharmaceuticals, on Feb. 14.
The intramuscular injection’s 7-year orphan drug exclusivity expired earlier in February. AMAG plans to offer both products priced at parity, according to the company’s press release. The newer product has a smaller, thinner needle. Both products are intended for use in a woman who has a singleton pregnancy of less than 37 weeks’ gestation and who has had a prior spontaneous preterm singleton delivery. The intramuscular injection is available in both single-dose and multidose vials.
Makena revenues in 2017 were nearly $400 million. The drug was the subject of a price controversy in 2011 under its maker at the time, KV Pharmaceutical, which subsequently cut the price by more than half.
The injection has several contraindications, including blood clots and hormone-sensitive cancers. Its efficacy is based on an improved number of women who used it and were delivered at more than 37 weeks’ gestation rather than on a directly demonstrated clinical benefit in neonatal morbidity or mortality.
Read more in AMAG’s press release.
Makena, a progestin injection for the prevention of preterm birth that has been approved for use since 2011, has received U.S. Food and Drug Administration approval for a subcutaneous auto-injection product to supplant its intramuscular injection formulation, announced its manufacturer, AMAG Pharmaceuticals, on Feb. 14.
The intramuscular injection’s 7-year orphan drug exclusivity expired earlier in February. AMAG plans to offer both products priced at parity, according to the company’s press release. The newer product has a smaller, thinner needle. Both products are intended for use in a woman who has a singleton pregnancy of less than 37 weeks’ gestation and who has had a prior spontaneous preterm singleton delivery. The intramuscular injection is available in both single-dose and multidose vials.
Makena revenues in 2017 were nearly $400 million. The drug was the subject of a price controversy in 2011 under its maker at the time, KV Pharmaceutical, which subsequently cut the price by more than half.
The injection has several contraindications, including blood clots and hormone-sensitive cancers. Its efficacy is based on an improved number of women who used it and were delivered at more than 37 weeks’ gestation rather than on a directly demonstrated clinical benefit in neonatal morbidity or mortality.
Read more in AMAG’s press release.
Makena, a progestin injection for the prevention of preterm birth that has been approved for use since 2011, has received U.S. Food and Drug Administration approval for a subcutaneous auto-injection product to supplant its intramuscular injection formulation, announced its manufacturer, AMAG Pharmaceuticals, on Feb. 14.
The intramuscular injection’s 7-year orphan drug exclusivity expired earlier in February. AMAG plans to offer both products priced at parity, according to the company’s press release. The newer product has a smaller, thinner needle. Both products are intended for use in a woman who has a singleton pregnancy of less than 37 weeks’ gestation and who has had a prior spontaneous preterm singleton delivery. The intramuscular injection is available in both single-dose and multidose vials.
Makena revenues in 2017 were nearly $400 million. The drug was the subject of a price controversy in 2011 under its maker at the time, KV Pharmaceutical, which subsequently cut the price by more than half.
The injection has several contraindications, including blood clots and hormone-sensitive cancers. Its efficacy is based on an improved number of women who used it and were delivered at more than 37 weeks’ gestation rather than on a directly demonstrated clinical benefit in neonatal morbidity or mortality.
Read more in AMAG’s press release.