User login
FDA approves new HPV assay
Manufacturer Becton Dickinson announced on Feb. 13 that it had received premarket approval from the U.S. Food and Drug Administration for a human papillomavirus (HPV) assay.
The Onclarity assay detects 14 types of high-risk HPV from specimens collected from cervical cancer screening via a SurePath liquid-based Pap test. It has previously been approved in Europe, Canada, and Japan. The bench-top molecular testing platform used for the assay has received prior FDA approval for chlamydia and gonorrhea infection testing.
The assay in particular identifies the HPV genotypes 16, 18, and 45, which are associated with more than two-thirds of cervical cancers and precancerous cervical lesions and as many as 94% of glandular cervical cancer cases.
Manufacturer Becton Dickinson announced on Feb. 13 that it had received premarket approval from the U.S. Food and Drug Administration for a human papillomavirus (HPV) assay.
The Onclarity assay detects 14 types of high-risk HPV from specimens collected from cervical cancer screening via a SurePath liquid-based Pap test. It has previously been approved in Europe, Canada, and Japan. The bench-top molecular testing platform used for the assay has received prior FDA approval for chlamydia and gonorrhea infection testing.
The assay in particular identifies the HPV genotypes 16, 18, and 45, which are associated with more than two-thirds of cervical cancers and precancerous cervical lesions and as many as 94% of glandular cervical cancer cases.
Manufacturer Becton Dickinson announced on Feb. 13 that it had received premarket approval from the U.S. Food and Drug Administration for a human papillomavirus (HPV) assay.
The Onclarity assay detects 14 types of high-risk HPV from specimens collected from cervical cancer screening via a SurePath liquid-based Pap test. It has previously been approved in Europe, Canada, and Japan. The bench-top molecular testing platform used for the assay has received prior FDA approval for chlamydia and gonorrhea infection testing.
The assay in particular identifies the HPV genotypes 16, 18, and 45, which are associated with more than two-thirds of cervical cancers and precancerous cervical lesions and as many as 94% of glandular cervical cancer cases.
Blood test approved for patients with concussions
The Food and Drug Administration approved a new blood test on Feb. 14 – the Banyan Brain Trauma Indicator – for assessing patients with mild traumatic brain injuries, also known as concussions.
Most traumatic brain injuries are classified as “mild,” and the majority of patients have negative CT scans, according to an FDA announcement. Within a matter of hours, this test can help predict which patients will have negative scans by measuring certain proteins released by brain tissue, thereby potentially eliminating unnecessary imaging – and the costs and radiation exposure that would go along with it.![]()
Read more in the FDA’s press release.
The Food and Drug Administration approved a new blood test on Feb. 14 – the Banyan Brain Trauma Indicator – for assessing patients with mild traumatic brain injuries, also known as concussions.
Most traumatic brain injuries are classified as “mild,” and the majority of patients have negative CT scans, according to an FDA announcement. Within a matter of hours, this test can help predict which patients will have negative scans by measuring certain proteins released by brain tissue, thereby potentially eliminating unnecessary imaging – and the costs and radiation exposure that would go along with it.![]()
Read more in the FDA’s press release.
The Food and Drug Administration approved a new blood test on Feb. 14 – the Banyan Brain Trauma Indicator – for assessing patients with mild traumatic brain injuries, also known as concussions.
Most traumatic brain injuries are classified as “mild,” and the majority of patients have negative CT scans, according to an FDA announcement. Within a matter of hours, this test can help predict which patients will have negative scans by measuring certain proteins released by brain tissue, thereby potentially eliminating unnecessary imaging – and the costs and radiation exposure that would go along with it.![]()
Read more in the FDA’s press release.
MMWR: Current flu vaccine does not protect elderly
, according to the Feb. 16 issue of Morbidity and Mortality Weekly Report.
The elderly are not among them. Although the vaccine was somewhat protective in children and adults up to 49 years old, “no statistically significant protection was observed in other age groups,” including people 65 years and older, reported investigators led by Brendan Flannery, PhD, of the Centers for Disease Control and Prevention influenza division.![]()
They also reported that the cumulative hospitalization rate attributed to laboratory-confirmed influenza for the week ending Feb. 3, 2018 (59.9/100,000), exceeded the rate for the same week in 2014-2015 (50.9/100,000), an A(H3N2) virus–predominant season, and is the highest rate observed for this week since the system expanded to include adults during the 2005-2006 season.
This year’s overall effectiveness rating was in contrast to the 2016-2017 seasonal effectiveness of 48% (MMWR. 2017 Feb 17;66[6];167-71).
The CDC noted that influenza is going to be active for several more weeks, so “vaccination is still recommended,” but “treatment with influenza antiviral medications, where appropriate, is especially important this season.” Meanwhile, “influenza vaccines with improved effectiveness are needed,” the CDC said.
The estimates are based on 4,562 patients 6 months to over 65 years old presenting with acute respiratory illness in 2018 from Nov. 2 to Feb. 3 at five outpatient medical clinics scattered across the United States. Nasal and oropharyngeal swabs were tested with reverse transcription polymerase chain reaction for the presence of influenza viruses; 413 subjects were 65 years or older.
Vaccine effectiveness against the less common virus A(H1N1)pdm09 was 67%, and 42% against the even rarer influenza B viruses. Estimates were adjusted for a range of confounders, including study site, age, general health, and week of illness. Vaccination rates ranged from 45% to 59% across the study sites; 38% of the subjects tested positive for influenza, most for type A viruses. The shot didn’t work too well: 43% of the influenza cases had gotten it.
The 25% effectiveness against A(H3N2) is a bit higher than recent reports of 17% from Canada and 10% from Australia, but similar to the 32% efficacy reported in the United States for the 2016-2017 season.
“These interim estimates reflect ongoing challenges with the A(H3N2) vaccine component since the 2011-12 season,” the investigators wrote. “Multiple factors might be contributing to the reported [vaccine effectiveness] against A(H3N2) viruses this season. … Genetic changes in the vaccine virus hemagglutinin protein that arise during passage in eggs might result in a vaccine immune response that is less effective against circulating viruses.”
On a related note, on Feb. 18, Senators Edward J. Markey (D-Mass.), Richard Blumenthal (D-Conn.), and Amy Klobuchar (D-Minn.) held a press conference to announce they were introducing the Flu Vaccine Bill to dedicate $1 billion over a 5-year period in order to develop a flu vaccine that could provide lifetime protection.
The investigators had no conflicts of interest.
SOURCE: Flannery B. et al. MMWR. 2018 Feb 16;67(6):180-5; Budd A. et al. MMWR. 2018 Feb 16;67(6):169-79.
, according to the Feb. 16 issue of Morbidity and Mortality Weekly Report.
The elderly are not among them. Although the vaccine was somewhat protective in children and adults up to 49 years old, “no statistically significant protection was observed in other age groups,” including people 65 years and older, reported investigators led by Brendan Flannery, PhD, of the Centers for Disease Control and Prevention influenza division.![]()
They also reported that the cumulative hospitalization rate attributed to laboratory-confirmed influenza for the week ending Feb. 3, 2018 (59.9/100,000), exceeded the rate for the same week in 2014-2015 (50.9/100,000), an A(H3N2) virus–predominant season, and is the highest rate observed for this week since the system expanded to include adults during the 2005-2006 season.
This year’s overall effectiveness rating was in contrast to the 2016-2017 seasonal effectiveness of 48% (MMWR. 2017 Feb 17;66[6];167-71).
The CDC noted that influenza is going to be active for several more weeks, so “vaccination is still recommended,” but “treatment with influenza antiviral medications, where appropriate, is especially important this season.” Meanwhile, “influenza vaccines with improved effectiveness are needed,” the CDC said.
The estimates are based on 4,562 patients 6 months to over 65 years old presenting with acute respiratory illness in 2018 from Nov. 2 to Feb. 3 at five outpatient medical clinics scattered across the United States. Nasal and oropharyngeal swabs were tested with reverse transcription polymerase chain reaction for the presence of influenza viruses; 413 subjects were 65 years or older.
Vaccine effectiveness against the less common virus A(H1N1)pdm09 was 67%, and 42% against the even rarer influenza B viruses. Estimates were adjusted for a range of confounders, including study site, age, general health, and week of illness. Vaccination rates ranged from 45% to 59% across the study sites; 38% of the subjects tested positive for influenza, most for type A viruses. The shot didn’t work too well: 43% of the influenza cases had gotten it.
The 25% effectiveness against A(H3N2) is a bit higher than recent reports of 17% from Canada and 10% from Australia, but similar to the 32% efficacy reported in the United States for the 2016-2017 season.
“These interim estimates reflect ongoing challenges with the A(H3N2) vaccine component since the 2011-12 season,” the investigators wrote. “Multiple factors might be contributing to the reported [vaccine effectiveness] against A(H3N2) viruses this season. … Genetic changes in the vaccine virus hemagglutinin protein that arise during passage in eggs might result in a vaccine immune response that is less effective against circulating viruses.”
On a related note, on Feb. 18, Senators Edward J. Markey (D-Mass.), Richard Blumenthal (D-Conn.), and Amy Klobuchar (D-Minn.) held a press conference to announce they were introducing the Flu Vaccine Bill to dedicate $1 billion over a 5-year period in order to develop a flu vaccine that could provide lifetime protection.
The investigators had no conflicts of interest.
SOURCE: Flannery B. et al. MMWR. 2018 Feb 16;67(6):180-5; Budd A. et al. MMWR. 2018 Feb 16;67(6):169-79.
, according to the Feb. 16 issue of Morbidity and Mortality Weekly Report.
The elderly are not among them. Although the vaccine was somewhat protective in children and adults up to 49 years old, “no statistically significant protection was observed in other age groups,” including people 65 years and older, reported investigators led by Brendan Flannery, PhD, of the Centers for Disease Control and Prevention influenza division.![]()
They also reported that the cumulative hospitalization rate attributed to laboratory-confirmed influenza for the week ending Feb. 3, 2018 (59.9/100,000), exceeded the rate for the same week in 2014-2015 (50.9/100,000), an A(H3N2) virus–predominant season, and is the highest rate observed for this week since the system expanded to include adults during the 2005-2006 season.
This year’s overall effectiveness rating was in contrast to the 2016-2017 seasonal effectiveness of 48% (MMWR. 2017 Feb 17;66[6];167-71).
The CDC noted that influenza is going to be active for several more weeks, so “vaccination is still recommended,” but “treatment with influenza antiviral medications, where appropriate, is especially important this season.” Meanwhile, “influenza vaccines with improved effectiveness are needed,” the CDC said.
The estimates are based on 4,562 patients 6 months to over 65 years old presenting with acute respiratory illness in 2018 from Nov. 2 to Feb. 3 at five outpatient medical clinics scattered across the United States. Nasal and oropharyngeal swabs were tested with reverse transcription polymerase chain reaction for the presence of influenza viruses; 413 subjects were 65 years or older.
Vaccine effectiveness against the less common virus A(H1N1)pdm09 was 67%, and 42% against the even rarer influenza B viruses. Estimates were adjusted for a range of confounders, including study site, age, general health, and week of illness. Vaccination rates ranged from 45% to 59% across the study sites; 38% of the subjects tested positive for influenza, most for type A viruses. The shot didn’t work too well: 43% of the influenza cases had gotten it.
The 25% effectiveness against A(H3N2) is a bit higher than recent reports of 17% from Canada and 10% from Australia, but similar to the 32% efficacy reported in the United States for the 2016-2017 season.
“These interim estimates reflect ongoing challenges with the A(H3N2) vaccine component since the 2011-12 season,” the investigators wrote. “Multiple factors might be contributing to the reported [vaccine effectiveness] against A(H3N2) viruses this season. … Genetic changes in the vaccine virus hemagglutinin protein that arise during passage in eggs might result in a vaccine immune response that is less effective against circulating viruses.”
On a related note, on Feb. 18, Senators Edward J. Markey (D-Mass.), Richard Blumenthal (D-Conn.), and Amy Klobuchar (D-Minn.) held a press conference to announce they were introducing the Flu Vaccine Bill to dedicate $1 billion over a 5-year period in order to develop a flu vaccine that could provide lifetime protection.
The investigators had no conflicts of interest.
SOURCE: Flannery B. et al. MMWR. 2018 Feb 16;67(6):180-5; Budd A. et al. MMWR. 2018 Feb 16;67(6):169-79.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
FDA approves apalutamide for castration-resistant nonmetastatic prostate cancer
The Food and Drug Administration has approved apalutamide for the treatment of patients with castration-resistant nonmetastatic prostate cancer.
Approval was based on a median metastasis-free survival for patients taking apalutamide of 40.5 months, compared with 16.2 months for patients taking a placebo in a randomized clinical trial of 1,207 patients with nonmetastatic, castration-resistant prostate cancer. All patients also received hormone therapy, either with gonadotropin-releasing hormone analogue therapy or with surgical castration.
Common side effects of apalutamide include fatigue, hypertension, rash, diarrhea, nausea, weight loss, arthralgia, falls, hot flush, decreased appetite, fractures, and peripheral edema.
Severe side effects of apalutamide include falls, fractures. and seizures, the FDA said.
Apalutamide is marketed as Erleada by Janssen Biotech.
The Food and Drug Administration has approved apalutamide for the treatment of patients with castration-resistant nonmetastatic prostate cancer.
Approval was based on a median metastasis-free survival for patients taking apalutamide of 40.5 months, compared with 16.2 months for patients taking a placebo in a randomized clinical trial of 1,207 patients with nonmetastatic, castration-resistant prostate cancer. All patients also received hormone therapy, either with gonadotropin-releasing hormone analogue therapy or with surgical castration.
Common side effects of apalutamide include fatigue, hypertension, rash, diarrhea, nausea, weight loss, arthralgia, falls, hot flush, decreased appetite, fractures, and peripheral edema.
Severe side effects of apalutamide include falls, fractures. and seizures, the FDA said.
Apalutamide is marketed as Erleada by Janssen Biotech.
The Food and Drug Administration has approved apalutamide for the treatment of patients with castration-resistant nonmetastatic prostate cancer.
Approval was based on a median metastasis-free survival for patients taking apalutamide of 40.5 months, compared with 16.2 months for patients taking a placebo in a randomized clinical trial of 1,207 patients with nonmetastatic, castration-resistant prostate cancer. All patients also received hormone therapy, either with gonadotropin-releasing hormone analogue therapy or with surgical castration.
Common side effects of apalutamide include fatigue, hypertension, rash, diarrhea, nausea, weight loss, arthralgia, falls, hot flush, decreased appetite, fractures, and peripheral edema.
Severe side effects of apalutamide include falls, fractures. and seizures, the FDA said.
Apalutamide is marketed as Erleada by Janssen Biotech.
FDA approves abiraterone acetate for metastatic high-risk CSPC
The Food and Drug Administration has approved abiraterone acetate tablets in combination with prednisone for metastatic high-risk castration-sensitive prostate cancer (CSPC).
The FDA first approved abiraterone acetate with prednisone in 2011 for patients with metastatic castration-resistant prostate cancer who had received prior chemotherapy. The indication was expanded in 2012 to patients with metastatic castration-resistant prostate cancer, the FDA said in a press statement.
The most common adverse reactions in LATITUDE for patients in the abiraterone acetate arm included hypertension, hot flush, hypokalemia, increased alanine aminotransferase or aspartate aminotransferase, headache, urinary tract infection, upper respiratory tract infection, and cough.
The recommended dose for abiraterone acetate for metastatic CSPC is 1,000 mg orally once daily with prednisone 5 mg orally once daily. Patients receiving the drug should also receive a gonadotropin-releasing hormone analogue concurrently or should have had bilateral orchiectomy, the FDA said.
Abiraterone acetate is marketed as Zytiga by Janssen Biotech.
The Food and Drug Administration has approved abiraterone acetate tablets in combination with prednisone for metastatic high-risk castration-sensitive prostate cancer (CSPC).
The FDA first approved abiraterone acetate with prednisone in 2011 for patients with metastatic castration-resistant prostate cancer who had received prior chemotherapy. The indication was expanded in 2012 to patients with metastatic castration-resistant prostate cancer, the FDA said in a press statement.
The most common adverse reactions in LATITUDE for patients in the abiraterone acetate arm included hypertension, hot flush, hypokalemia, increased alanine aminotransferase or aspartate aminotransferase, headache, urinary tract infection, upper respiratory tract infection, and cough.
The recommended dose for abiraterone acetate for metastatic CSPC is 1,000 mg orally once daily with prednisone 5 mg orally once daily. Patients receiving the drug should also receive a gonadotropin-releasing hormone analogue concurrently or should have had bilateral orchiectomy, the FDA said.
Abiraterone acetate is marketed as Zytiga by Janssen Biotech.
The Food and Drug Administration has approved abiraterone acetate tablets in combination with prednisone for metastatic high-risk castration-sensitive prostate cancer (CSPC).
The FDA first approved abiraterone acetate with prednisone in 2011 for patients with metastatic castration-resistant prostate cancer who had received prior chemotherapy. The indication was expanded in 2012 to patients with metastatic castration-resistant prostate cancer, the FDA said in a press statement.
The most common adverse reactions in LATITUDE for patients in the abiraterone acetate arm included hypertension, hot flush, hypokalemia, increased alanine aminotransferase or aspartate aminotransferase, headache, urinary tract infection, upper respiratory tract infection, and cough.
The recommended dose for abiraterone acetate for metastatic CSPC is 1,000 mg orally once daily with prednisone 5 mg orally once daily. Patients receiving the drug should also receive a gonadotropin-releasing hormone analogue concurrently or should have had bilateral orchiectomy, the FDA said.
Abiraterone acetate is marketed as Zytiga by Janssen Biotech.
FDA approves new combination drug for HIV patients
The Food and Drug Administration has approved a combination drug intended to treat HIV-1 infections – bictegravir, emtricitabine, tenofovir alafenamide – in virologically suppressed (HIV-1 RNA less than 50 copies/mL) adults who have no history of antiretroviral treatment or as a replacement for their current antiretroviral regimen.
The approval of the new combination drug (Biktarvy) was based on four active, randomized, controlled trials comprising three double-blind studies and one open label study. After 48 weeks of treatment in all trials, CD4+ cell count was evaluated to determine the efficacy of bictegravir, emtricitabine, tenofovir alafenamide, compared with other antiretroviral therapies.
In trial 1489, patients were randomized 1:1 to receive Biktarvy (n = 314) or ABC/DTG/3TC (abacavir 600 mg/dolutegravir 50 mg/lamivudine 300 mg) (n = 315). Trial 1490 was similar to trial 1489, with patients randomized 1:1 to receive either Biktarvy (n = 320) or DTG + FTC/TAF (dolutegravir + 50 mg, emtricitabine 200 mg/tenofovir alafenamide fumarate 25 mg) (n = 325). In trial 1489, the mean increase in CD4+ count after 48 weeks was 233 cells per mm3 and 229/mm3 in the Biktarvy and ABC/DTG/3TC groups, respectively. Similarly, counts in trial 1490 CD4+ after 48 weeks were 180/mm3 and 201/mm3 in the Biktarvy and DTG + FTC/TAF groups, respectively.
Trial 1844, another randomized trial, was composed of patients who switched to Biktarvy from their older treatment. Patients were randomized 1:1 to the new treatment (n = 282) or their previous antiretroviral regimen (n = 281).
Over 48 weeks, the mean change in CD4+ cell count was –31 cells per mm3 in subjects who switched to Biktarvy and 4/mm3 in subjects who stayed on ABC/DTG/3TC.
The open-label portion of the study, trial 1878, evaluated the safety and efficacy of switching from other retroviral treatments (n = 287) to Biktarvy (n = 290). The average change in CD4+ count after 48 weeks was 25 cells per mm3 who switched to Biktarvy and 0/mm3 who stayed on their previous regimens.
The new combination has received a boxed warning regarding the risk of severe acute exacerbations of hepatitis B that have been reported in patients coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF) and may occur with discontinuation of Biktarvy. Such patients should be closely monitored for hepatic function with both clinical and laboratory follow-up for at least several months in patients if they discontinue Biktarvy. If appropriate, antihepatitis B therapy may be warranted.
The approved recommended dosage for bictegravir, emtricitabine, tenofovir alafenamide combination drug is one tablet daily with or without food. More information is available on the FDA website.
The Food and Drug Administration has approved a combination drug intended to treat HIV-1 infections – bictegravir, emtricitabine, tenofovir alafenamide – in virologically suppressed (HIV-1 RNA less than 50 copies/mL) adults who have no history of antiretroviral treatment or as a replacement for their current antiretroviral regimen.
The approval of the new combination drug (Biktarvy) was based on four active, randomized, controlled trials comprising three double-blind studies and one open label study. After 48 weeks of treatment in all trials, CD4+ cell count was evaluated to determine the efficacy of bictegravir, emtricitabine, tenofovir alafenamide, compared with other antiretroviral therapies.
In trial 1489, patients were randomized 1:1 to receive Biktarvy (n = 314) or ABC/DTG/3TC (abacavir 600 mg/dolutegravir 50 mg/lamivudine 300 mg) (n = 315). Trial 1490 was similar to trial 1489, with patients randomized 1:1 to receive either Biktarvy (n = 320) or DTG + FTC/TAF (dolutegravir + 50 mg, emtricitabine 200 mg/tenofovir alafenamide fumarate 25 mg) (n = 325). In trial 1489, the mean increase in CD4+ count after 48 weeks was 233 cells per mm3 and 229/mm3 in the Biktarvy and ABC/DTG/3TC groups, respectively. Similarly, counts in trial 1490 CD4+ after 48 weeks were 180/mm3 and 201/mm3 in the Biktarvy and DTG + FTC/TAF groups, respectively.
Trial 1844, another randomized trial, was composed of patients who switched to Biktarvy from their older treatment. Patients were randomized 1:1 to the new treatment (n = 282) or their previous antiretroviral regimen (n = 281).
Over 48 weeks, the mean change in CD4+ cell count was –31 cells per mm3 in subjects who switched to Biktarvy and 4/mm3 in subjects who stayed on ABC/DTG/3TC.
The open-label portion of the study, trial 1878, evaluated the safety and efficacy of switching from other retroviral treatments (n = 287) to Biktarvy (n = 290). The average change in CD4+ count after 48 weeks was 25 cells per mm3 who switched to Biktarvy and 0/mm3 who stayed on their previous regimens.
The new combination has received a boxed warning regarding the risk of severe acute exacerbations of hepatitis B that have been reported in patients coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF) and may occur with discontinuation of Biktarvy. Such patients should be closely monitored for hepatic function with both clinical and laboratory follow-up for at least several months in patients if they discontinue Biktarvy. If appropriate, antihepatitis B therapy may be warranted.
The approved recommended dosage for bictegravir, emtricitabine, tenofovir alafenamide combination drug is one tablet daily with or without food. More information is available on the FDA website.
The Food and Drug Administration has approved a combination drug intended to treat HIV-1 infections – bictegravir, emtricitabine, tenofovir alafenamide – in virologically suppressed (HIV-1 RNA less than 50 copies/mL) adults who have no history of antiretroviral treatment or as a replacement for their current antiretroviral regimen.
The approval of the new combination drug (Biktarvy) was based on four active, randomized, controlled trials comprising three double-blind studies and one open label study. After 48 weeks of treatment in all trials, CD4+ cell count was evaluated to determine the efficacy of bictegravir, emtricitabine, tenofovir alafenamide, compared with other antiretroviral therapies.
In trial 1489, patients were randomized 1:1 to receive Biktarvy (n = 314) or ABC/DTG/3TC (abacavir 600 mg/dolutegravir 50 mg/lamivudine 300 mg) (n = 315). Trial 1490 was similar to trial 1489, with patients randomized 1:1 to receive either Biktarvy (n = 320) or DTG + FTC/TAF (dolutegravir + 50 mg, emtricitabine 200 mg/tenofovir alafenamide fumarate 25 mg) (n = 325). In trial 1489, the mean increase in CD4+ count after 48 weeks was 233 cells per mm3 and 229/mm3 in the Biktarvy and ABC/DTG/3TC groups, respectively. Similarly, counts in trial 1490 CD4+ after 48 weeks were 180/mm3 and 201/mm3 in the Biktarvy and DTG + FTC/TAF groups, respectively.
Trial 1844, another randomized trial, was composed of patients who switched to Biktarvy from their older treatment. Patients were randomized 1:1 to the new treatment (n = 282) or their previous antiretroviral regimen (n = 281).
Over 48 weeks, the mean change in CD4+ cell count was –31 cells per mm3 in subjects who switched to Biktarvy and 4/mm3 in subjects who stayed on ABC/DTG/3TC.
The open-label portion of the study, trial 1878, evaluated the safety and efficacy of switching from other retroviral treatments (n = 287) to Biktarvy (n = 290). The average change in CD4+ count after 48 weeks was 25 cells per mm3 who switched to Biktarvy and 0/mm3 who stayed on their previous regimens.
The new combination has received a boxed warning regarding the risk of severe acute exacerbations of hepatitis B that have been reported in patients coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF) and may occur with discontinuation of Biktarvy. Such patients should be closely monitored for hepatic function with both clinical and laboratory follow-up for at least several months in patients if they discontinue Biktarvy. If appropriate, antihepatitis B therapy may be warranted.
The approved recommended dosage for bictegravir, emtricitabine, tenofovir alafenamide combination drug is one tablet daily with or without food. More information is available on the FDA website.
Duodenoscope redesign prompts voluntary recall by Pentax
Pentax has issued a voluntary recall for Pentax ED-3490TK duodenoscopes because of infections associated with reprocessed duodenoscopes, and the Food and Drug Administration has cleared the 510(k) to improve the device. The new design will, it is hoped, improve cleaning and disinfection for these devices.
“Reducing infections associated with duodenoscopes remains a top priority for the FDA, and we believe the new design changes to the Pentax duodenoscope will make these devices easier to clean and high-level disinfect to help enhance their safety,” said Suzanne Schwartz, MD, associate director for science and strategic partnerships at the FDA’s Center for Devices and Radiological Health. “We will continue to encourage new innovations for these devices to protect public health while enabling patients to have continued access to minimally invasive, life-saving endoscopy procedures.”
. In one study, even after double high-level disinfection or standard high-level disinfection followed by ethylene oxide gas sterilization, duodenoscopes had similar rates of contamination. These contamination events were associated with outbreaks of carbapenem-resistant Enterobacteriaceae infections. One of the culprits behind residual contamination may be the presence of biofilms, which are notoriously difficult to clean with standard disinfection methods.
Prior to the clearance of the elevator channel sealing mechanism, the first duodenoscope with a disposable distal cap was introduced, the Pentax ED34-i10T. The use of a disposable tip for the duodenoscope is meant to decrease the risk of future infections associated with these devices. The use of a disposable tip also improves cleaning and reprocessing of the duodenoscopes.
The FDA continues to work with manufacturers to improve the safety of duodenscopes and other reusable medical devices to protect patients from bacterial infections.
Pentax has issued a voluntary recall for Pentax ED-3490TK duodenoscopes because of infections associated with reprocessed duodenoscopes, and the Food and Drug Administration has cleared the 510(k) to improve the device. The new design will, it is hoped, improve cleaning and disinfection for these devices.
“Reducing infections associated with duodenoscopes remains a top priority for the FDA, and we believe the new design changes to the Pentax duodenoscope will make these devices easier to clean and high-level disinfect to help enhance their safety,” said Suzanne Schwartz, MD, associate director for science and strategic partnerships at the FDA’s Center for Devices and Radiological Health. “We will continue to encourage new innovations for these devices to protect public health while enabling patients to have continued access to minimally invasive, life-saving endoscopy procedures.”
. In one study, even after double high-level disinfection or standard high-level disinfection followed by ethylene oxide gas sterilization, duodenoscopes had similar rates of contamination. These contamination events were associated with outbreaks of carbapenem-resistant Enterobacteriaceae infections. One of the culprits behind residual contamination may be the presence of biofilms, which are notoriously difficult to clean with standard disinfection methods.
Prior to the clearance of the elevator channel sealing mechanism, the first duodenoscope with a disposable distal cap was introduced, the Pentax ED34-i10T. The use of a disposable tip for the duodenoscope is meant to decrease the risk of future infections associated with these devices. The use of a disposable tip also improves cleaning and reprocessing of the duodenoscopes.
The FDA continues to work with manufacturers to improve the safety of duodenscopes and other reusable medical devices to protect patients from bacterial infections.
Pentax has issued a voluntary recall for Pentax ED-3490TK duodenoscopes because of infections associated with reprocessed duodenoscopes, and the Food and Drug Administration has cleared the 510(k) to improve the device. The new design will, it is hoped, improve cleaning and disinfection for these devices.
“Reducing infections associated with duodenoscopes remains a top priority for the FDA, and we believe the new design changes to the Pentax duodenoscope will make these devices easier to clean and high-level disinfect to help enhance their safety,” said Suzanne Schwartz, MD, associate director for science and strategic partnerships at the FDA’s Center for Devices and Radiological Health. “We will continue to encourage new innovations for these devices to protect public health while enabling patients to have continued access to minimally invasive, life-saving endoscopy procedures.”
. In one study, even after double high-level disinfection or standard high-level disinfection followed by ethylene oxide gas sterilization, duodenoscopes had similar rates of contamination. These contamination events were associated with outbreaks of carbapenem-resistant Enterobacteriaceae infections. One of the culprits behind residual contamination may be the presence of biofilms, which are notoriously difficult to clean with standard disinfection methods.
Prior to the clearance of the elevator channel sealing mechanism, the first duodenoscope with a disposable distal cap was introduced, the Pentax ED34-i10T. The use of a disposable tip for the duodenoscope is meant to decrease the risk of future infections associated with these devices. The use of a disposable tip also improves cleaning and reprocessing of the duodenoscopes.
The FDA continues to work with manufacturers to improve the safety of duodenscopes and other reusable medical devices to protect patients from bacterial infections.
FDA approves complete combo tablet for HIV
The Food and Drug Administration announced that it has approved Symfi Lo tablets, a fixed-dose combination product containing efavirenz (400 mg), lamivudine (300 mg), and tenofovir disoproxil fumarate (300 mg, equivalent to 245 mg of tenofovir disoproxil). The tablets are indicated as a complete regimen for treating HIV-1 in adults and in pediatric patients weighing at least 35 kg.
The recommended dose is one tablet taken daily by mouth on an empty stomach, preferably at bedtime, as that may improve the tolerability of nervous system symptoms, according to an email release by the FDA Office of Health and Constituent Affairs.
The most common adverse reactions (in more than 5% of patients taking Symfi Lo) were rash and dizziness. The warnings and precautions contained in the label are: lactic acidosis/severe hepatomegaly with steatosis; new-onset or worsening renal impairment; and serious psychiatric symptoms, such as severe depression and suicidal ideation.
The FDA approval was primarily based upon the results of two randomized trials: Study 903 and Encore-1.
SOURCE: FDA email release and full label with prescribing information.
The Food and Drug Administration announced that it has approved Symfi Lo tablets, a fixed-dose combination product containing efavirenz (400 mg), lamivudine (300 mg), and tenofovir disoproxil fumarate (300 mg, equivalent to 245 mg of tenofovir disoproxil). The tablets are indicated as a complete regimen for treating HIV-1 in adults and in pediatric patients weighing at least 35 kg.
The recommended dose is one tablet taken daily by mouth on an empty stomach, preferably at bedtime, as that may improve the tolerability of nervous system symptoms, according to an email release by the FDA Office of Health and Constituent Affairs.
The most common adverse reactions (in more than 5% of patients taking Symfi Lo) were rash and dizziness. The warnings and precautions contained in the label are: lactic acidosis/severe hepatomegaly with steatosis; new-onset or worsening renal impairment; and serious psychiatric symptoms, such as severe depression and suicidal ideation.
The FDA approval was primarily based upon the results of two randomized trials: Study 903 and Encore-1.
SOURCE: FDA email release and full label with prescribing information.
The Food and Drug Administration announced that it has approved Symfi Lo tablets, a fixed-dose combination product containing efavirenz (400 mg), lamivudine (300 mg), and tenofovir disoproxil fumarate (300 mg, equivalent to 245 mg of tenofovir disoproxil). The tablets are indicated as a complete regimen for treating HIV-1 in adults and in pediatric patients weighing at least 35 kg.
The recommended dose is one tablet taken daily by mouth on an empty stomach, preferably at bedtime, as that may improve the tolerability of nervous system symptoms, according to an email release by the FDA Office of Health and Constituent Affairs.
The most common adverse reactions (in more than 5% of patients taking Symfi Lo) were rash and dizziness. The warnings and precautions contained in the label are: lactic acidosis/severe hepatomegaly with steatosis; new-onset or worsening renal impairment; and serious psychiatric symptoms, such as severe depression and suicidal ideation.
The FDA approval was primarily based upon the results of two randomized trials: Study 903 and Encore-1.
SOURCE: FDA email release and full label with prescribing information.
nPEP for HIV: Updated CDC guidelines available for primary care physicians
In 2016, the Centers for Disease Control and Prevention provided health care providers with updated recommendations for nonoccupational postexposure prophylaxis (nPEP) with antiretroviral drugs to prevent transmission of HIV following sexual interaction, injection-drug use, or other nonoccupational exposures.1 The new recommendations include the use of more effective and more tolerable drug regimens that employ antiretroviral medications that were approved since the previous guidelines came out in 2005; they also provide updated guidance on exposure assessment, baseline and follow-up HIV testing, and longer-term prevention measures, such as pre-exposure prophylaxis (PrEP).
Screening for HIV infection has been expanding broadly in all health care settings over the past decade, so primary care physicians play an increasingly vital role in preventing HIV infection. Today, primary care physicians are also often the most likely “go-to” health care provider when patients think they may have been exposed to HIV. Clinically, this is an emergency situation, so time is of the essence: Treatment with three powerful antiretrovirals must be initiated within a few hours of – but no later than 72 hours after – an isolated exposure to blood, genital secretions, or other potentially infectious body fluids that may contain HIV.
The key issue for primary care physicians, especially those who have never prescribed PEP before, is advance planning. What you do up front, in terms of organizing materials and training staff, is worth the effort because there is so much at stake – for your patients and for society. The good news is that once you have an established nPEP protocol in place, it stays in place. When a patient asks for help, the protocol kicks in automatically.
Getting ready for nPEP
Prepare your staff:
- Educate your whole staff about the urgency of seeing potential nPEP patients immediately.
- Choose the staff person in your office who will submit requests for PEP medications to the pharmacy and/or pharmaceutical companies; your financial reimbursement staff person is likely a good candidate for this job.
- Learn about patient assistance programs (for uninsured or underinsured patients) and crime victims compensation programs (reimbursement or emergency awards for victims of violent crimes, including rape, for various out-of-pocket expenses including medical expenses).
Keep paperwork and materials on hand:
- Have information and forms for patient assistance programs for pharmaceutical companies supplying the drugs. Pharmaceutical companies are aware of the urgency for nPEP medications and are ready to respond immediately. They may mail the medication so it arrives the next day or, more likely, fax a voucher or other information for the patient to present to a local pharmacist who will fill the prescription.
- Have information on your state’s crime victims compensation program available.
- Consider keeping nPEP Starter Packs (with an initial 3-7 days’ worth of medication) readily available in your office.
Rapid evaluation of patients seeking care after potential exposure to HIV
Effective delivery of nPEP requires prompt initial evaluation of patients and assessment of HIV transmission risk. Take a methodical, step-by-step history of the exposure to address the following basic questions:
- Date and time of exposure? nPEP should be initiated as soon as possible after HIV exposure; it is unlikely to be effective if not initiated within 72 hours or less.
- Frequency of exposure? Type/route of exposure? nPEP is generally reserved for isolated or infrequent exposures that present a substantial risk for HIV acquisition (see Table 1 on HIV acquisition risk below).
- HIV status of exposure source? If the source is positive, is the source person on HIV treatment with antiretroviral therapy? If unknown, is the source person an injecting drug user or a man who has sex with men (MSM)?
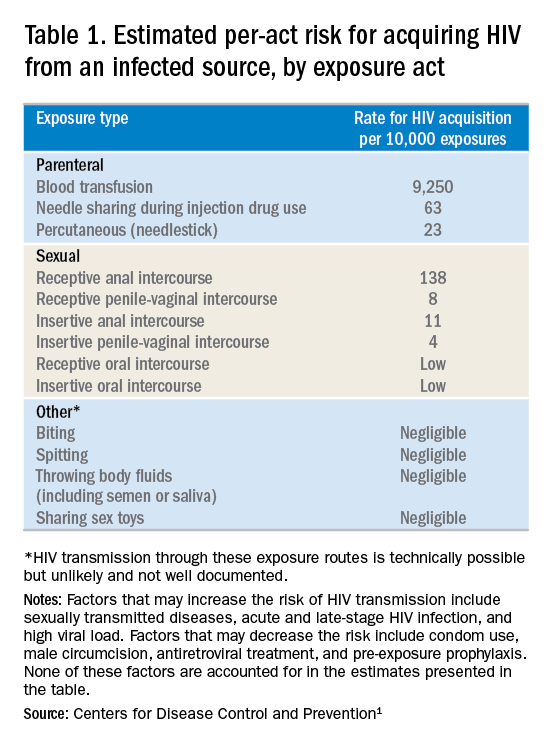
Based on the initial evaluation, is nPEP recommended?
Answers to the questions asked during the initial evaluation of the patient will determine whether nPEP is indicated. Along with its updated recommendations, the CDC provided an algorithm to help guide evaluation and treatment.
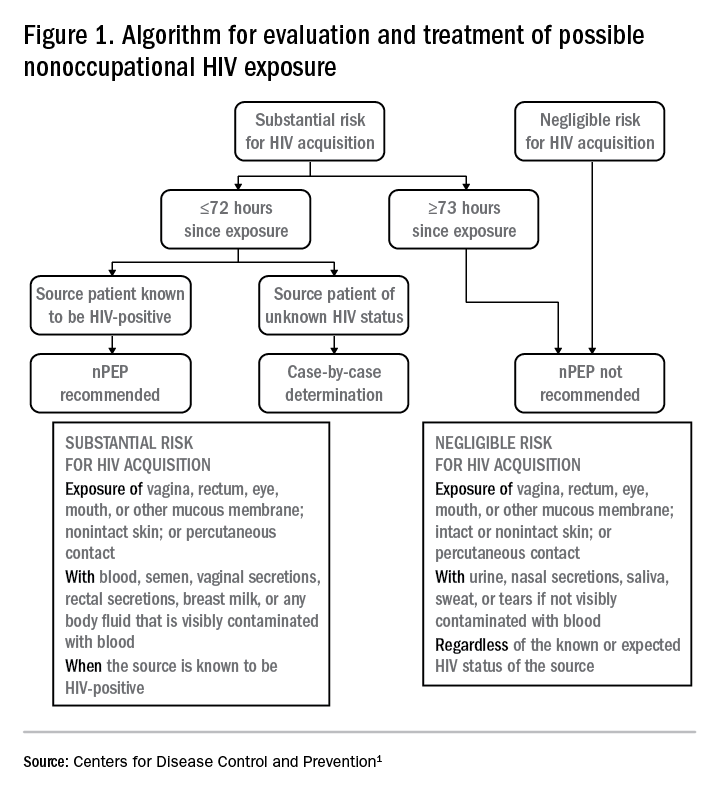
Preferred HIV test
Administer an HIV test to all patients considered for nPEP, preferably the rapid combined antigen and antibody test (Ag/Ab), or just the antibody test if the Ag/Ab test is not available. nPEP is indicated only for persons without HIV infections. However, if results are not available during the initial evaluation, assume the patient is not infected. If indicated and started, nPEP can be discontinued if tests later shown the patient already has an HIV infection.
Laboratory testing
If nPEP is indicated, conduct laboratory testing. Lab testing is required to document the patient’s HIV status (and that of the source person, when available), identify and manage other conditions potentially resulting from exposure, identify conditions that may affect the nPEP medication regimen, and monitor safety or toxicities to the prescribed regimen.
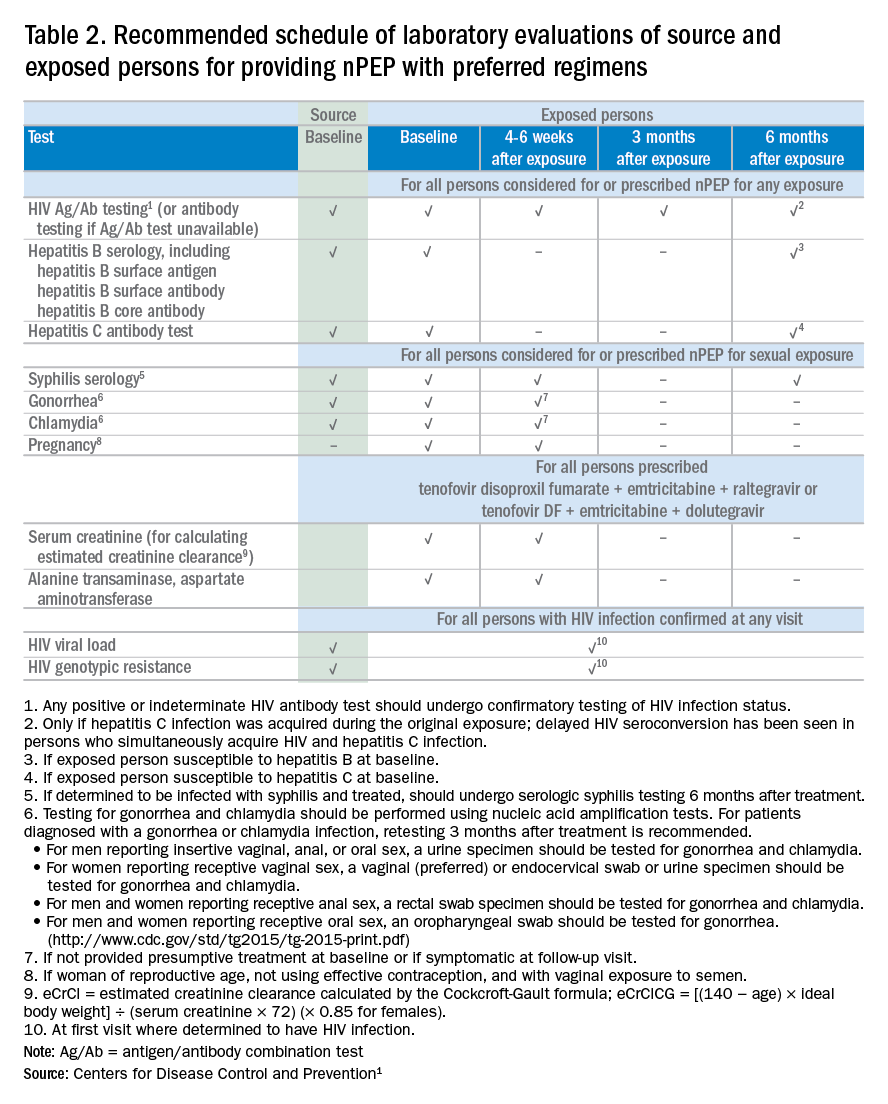
nPEP treatment regimen for otherwise healthy adults and adolescents
In the absence of randomized clinical trials, data from a case/control study demonstrating an 81% reduction of HIV transmission after use of occupational PEP among hospital workers remains the strongest evidence for the benefit of nPEP.1,2 For patients offered nPEP, recommended treatment includes prescribing either of the following regimens for 28 days:
- Preferred regimen: tenofovir disoproxil fumarate (TDF) (300 mg) with emtricitabine (FTC) (200 mg) once daily plus either raltegravir (RAL) 400 mg twice daily or dolutegravir (DTG) 50 mg daily.
- Alternative regimen: TDF (300 mg) with FTC (200 mg) once daily plus darunavir (DRV) (800 mg) and ritonavir (RTV) (100 mg) once daily.
Additional considerations and nPEP treatment regimens for children, patients with decreased renal function, and pregnant women are included in the CDC guidelines.
Crucial Information for Patients on nPEP
Emphasize the importance of proper dosing and adherence.
Review the patient information for each drug in the regimen, specifically the black boxes, warnings, and side effects, and counsel your patients accordingly.
Transitioning from nPEP to PrEP or from PrEP to nPEP
If you have a patient who engages in behavior that places them at risk for frequent, recurrent exposures to HIV, consider transitioning them to PrEP (pre-exposure prophylaxis) following their 28-day course of nPEP.3 PrEP is a two-drug regimen taken daily on an ongoing basis.
Additionally, for patients who are already on PrEP but who have not taken their medications within a week before the possible exposure, consider initiating nPEP for 28 days and then reintroducing PrEP if their HIV status is negative and the problems with adherence can be addressed moving forward.
Raising Awareness About nPEP
Many people never expect to be exposed to HIV and may not know about the availability of PEP in an emergency situation. You can help raise awareness by making educational materials available in your waiting rooms and exam rooms. Brochures and other HIV/AIDS educational materials for patients are available from the CDC Act Against AIDS campaign.
Summary

Dr. Dominguez is a Captain, U.S. Public Health Service, epidemiology branch, division of HIV/AIDS prevention, CDC.
Additional resources
- The CDC recommends that everyone between the ages of 13 and 64 get tested for HIV at least once as part of routine health care. As part of its Act Against AIDS initiative, the CDC developed the HIV Screening Standard Care program, which provides free tools and resources to help clinicians and nurses incorporate routine HIV screening into primary care settings.
- HIV guidelines and recommendations .
- Postexposure prophylaxis (PEP)
- Pre-Exposure prophylaxis (PrEP)
References
1. Centers for Disease Control and Prevention. Updated guidelines for antiretroviral postexposure prophylaxis after sexual, injection drug use, or other nonoccupational exposure to HIV. United States, 2016. Accessed March 6, 2017.
2. Cardo DM et al. New Engl J Med. 1997;337(21):1485-90.
3. Centers for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States–2014: a clinical practice guideline. Accessed March 6, 2017.
In 2016, the Centers for Disease Control and Prevention provided health care providers with updated recommendations for nonoccupational postexposure prophylaxis (nPEP) with antiretroviral drugs to prevent transmission of HIV following sexual interaction, injection-drug use, or other nonoccupational exposures.1 The new recommendations include the use of more effective and more tolerable drug regimens that employ antiretroviral medications that were approved since the previous guidelines came out in 2005; they also provide updated guidance on exposure assessment, baseline and follow-up HIV testing, and longer-term prevention measures, such as pre-exposure prophylaxis (PrEP).
Screening for HIV infection has been expanding broadly in all health care settings over the past decade, so primary care physicians play an increasingly vital role in preventing HIV infection. Today, primary care physicians are also often the most likely “go-to” health care provider when patients think they may have been exposed to HIV. Clinically, this is an emergency situation, so time is of the essence: Treatment with three powerful antiretrovirals must be initiated within a few hours of – but no later than 72 hours after – an isolated exposure to blood, genital secretions, or other potentially infectious body fluids that may contain HIV.
The key issue for primary care physicians, especially those who have never prescribed PEP before, is advance planning. What you do up front, in terms of organizing materials and training staff, is worth the effort because there is so much at stake – for your patients and for society. The good news is that once you have an established nPEP protocol in place, it stays in place. When a patient asks for help, the protocol kicks in automatically.
Getting ready for nPEP
Prepare your staff:
- Educate your whole staff about the urgency of seeing potential nPEP patients immediately.
- Choose the staff person in your office who will submit requests for PEP medications to the pharmacy and/or pharmaceutical companies; your financial reimbursement staff person is likely a good candidate for this job.
- Learn about patient assistance programs (for uninsured or underinsured patients) and crime victims compensation programs (reimbursement or emergency awards for victims of violent crimes, including rape, for various out-of-pocket expenses including medical expenses).
Keep paperwork and materials on hand:
- Have information and forms for patient assistance programs for pharmaceutical companies supplying the drugs. Pharmaceutical companies are aware of the urgency for nPEP medications and are ready to respond immediately. They may mail the medication so it arrives the next day or, more likely, fax a voucher or other information for the patient to present to a local pharmacist who will fill the prescription.
- Have information on your state’s crime victims compensation program available.
- Consider keeping nPEP Starter Packs (with an initial 3-7 days’ worth of medication) readily available in your office.
Rapid evaluation of patients seeking care after potential exposure to HIV
Effective delivery of nPEP requires prompt initial evaluation of patients and assessment of HIV transmission risk. Take a methodical, step-by-step history of the exposure to address the following basic questions:
- Date and time of exposure? nPEP should be initiated as soon as possible after HIV exposure; it is unlikely to be effective if not initiated within 72 hours or less.
- Frequency of exposure? Type/route of exposure? nPEP is generally reserved for isolated or infrequent exposures that present a substantial risk for HIV acquisition (see Table 1 on HIV acquisition risk below).
- HIV status of exposure source? If the source is positive, is the source person on HIV treatment with antiretroviral therapy? If unknown, is the source person an injecting drug user or a man who has sex with men (MSM)?
Based on the initial evaluation, is nPEP recommended?
Answers to the questions asked during the initial evaluation of the patient will determine whether nPEP is indicated. Along with its updated recommendations, the CDC provided an algorithm to help guide evaluation and treatment.
Preferred HIV test
Administer an HIV test to all patients considered for nPEP, preferably the rapid combined antigen and antibody test (Ag/Ab), or just the antibody test if the Ag/Ab test is not available. nPEP is indicated only for persons without HIV infections. However, if results are not available during the initial evaluation, assume the patient is not infected. If indicated and started, nPEP can be discontinued if tests later shown the patient already has an HIV infection.
Laboratory testing
If nPEP is indicated, conduct laboratory testing. Lab testing is required to document the patient’s HIV status (and that of the source person, when available), identify and manage other conditions potentially resulting from exposure, identify conditions that may affect the nPEP medication regimen, and monitor safety or toxicities to the prescribed regimen.
nPEP treatment regimen for otherwise healthy adults and adolescents
In the absence of randomized clinical trials, data from a case/control study demonstrating an 81% reduction of HIV transmission after use of occupational PEP among hospital workers remains the strongest evidence for the benefit of nPEP.1,2 For patients offered nPEP, recommended treatment includes prescribing either of the following regimens for 28 days:
- Preferred regimen: tenofovir disoproxil fumarate (TDF) (300 mg) with emtricitabine (FTC) (200 mg) once daily plus either raltegravir (RAL) 400 mg twice daily or dolutegravir (DTG) 50 mg daily.
- Alternative regimen: TDF (300 mg) with FTC (200 mg) once daily plus darunavir (DRV) (800 mg) and ritonavir (RTV) (100 mg) once daily.
Additional considerations and nPEP treatment regimens for children, patients with decreased renal function, and pregnant women are included in the CDC guidelines.
Crucial Information for Patients on nPEP
Emphasize the importance of proper dosing and adherence.
Review the patient information for each drug in the regimen, specifically the black boxes, warnings, and side effects, and counsel your patients accordingly.
Transitioning from nPEP to PrEP or from PrEP to nPEP
If you have a patient who engages in behavior that places them at risk for frequent, recurrent exposures to HIV, consider transitioning them to PrEP (pre-exposure prophylaxis) following their 28-day course of nPEP.3 PrEP is a two-drug regimen taken daily on an ongoing basis.
Additionally, for patients who are already on PrEP but who have not taken their medications within a week before the possible exposure, consider initiating nPEP for 28 days and then reintroducing PrEP if their HIV status is negative and the problems with adherence can be addressed moving forward.
Raising Awareness About nPEP
Many people never expect to be exposed to HIV and may not know about the availability of PEP in an emergency situation. You can help raise awareness by making educational materials available in your waiting rooms and exam rooms. Brochures and other HIV/AIDS educational materials for patients are available from the CDC Act Against AIDS campaign.
Summary
Dr. Dominguez is a Captain, U.S. Public Health Service, epidemiology branch, division of HIV/AIDS prevention, CDC.
Additional resources
- The CDC recommends that everyone between the ages of 13 and 64 get tested for HIV at least once as part of routine health care. As part of its Act Against AIDS initiative, the CDC developed the HIV Screening Standard Care program, which provides free tools and resources to help clinicians and nurses incorporate routine HIV screening into primary care settings.
- HIV guidelines and recommendations .
- Postexposure prophylaxis (PEP)
- Pre-Exposure prophylaxis (PrEP)
References
1. Centers for Disease Control and Prevention. Updated guidelines for antiretroviral postexposure prophylaxis after sexual, injection drug use, or other nonoccupational exposure to HIV. United States, 2016. Accessed March 6, 2017.
2. Cardo DM et al. New Engl J Med. 1997;337(21):1485-90.
3. Centers for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States–2014: a clinical practice guideline. Accessed March 6, 2017.
In 2016, the Centers for Disease Control and Prevention provided health care providers with updated recommendations for nonoccupational postexposure prophylaxis (nPEP) with antiretroviral drugs to prevent transmission of HIV following sexual interaction, injection-drug use, or other nonoccupational exposures.1 The new recommendations include the use of more effective and more tolerable drug regimens that employ antiretroviral medications that were approved since the previous guidelines came out in 2005; they also provide updated guidance on exposure assessment, baseline and follow-up HIV testing, and longer-term prevention measures, such as pre-exposure prophylaxis (PrEP).
Screening for HIV infection has been expanding broadly in all health care settings over the past decade, so primary care physicians play an increasingly vital role in preventing HIV infection. Today, primary care physicians are also often the most likely “go-to” health care provider when patients think they may have been exposed to HIV. Clinically, this is an emergency situation, so time is of the essence: Treatment with three powerful antiretrovirals must be initiated within a few hours of – but no later than 72 hours after – an isolated exposure to blood, genital secretions, or other potentially infectious body fluids that may contain HIV.
The key issue for primary care physicians, especially those who have never prescribed PEP before, is advance planning. What you do up front, in terms of organizing materials and training staff, is worth the effort because there is so much at stake – for your patients and for society. The good news is that once you have an established nPEP protocol in place, it stays in place. When a patient asks for help, the protocol kicks in automatically.
Getting ready for nPEP
Prepare your staff:
- Educate your whole staff about the urgency of seeing potential nPEP patients immediately.
- Choose the staff person in your office who will submit requests for PEP medications to the pharmacy and/or pharmaceutical companies; your financial reimbursement staff person is likely a good candidate for this job.
- Learn about patient assistance programs (for uninsured or underinsured patients) and crime victims compensation programs (reimbursement or emergency awards for victims of violent crimes, including rape, for various out-of-pocket expenses including medical expenses).
Keep paperwork and materials on hand:
- Have information and forms for patient assistance programs for pharmaceutical companies supplying the drugs. Pharmaceutical companies are aware of the urgency for nPEP medications and are ready to respond immediately. They may mail the medication so it arrives the next day or, more likely, fax a voucher or other information for the patient to present to a local pharmacist who will fill the prescription.
- Have information on your state’s crime victims compensation program available.
- Consider keeping nPEP Starter Packs (with an initial 3-7 days’ worth of medication) readily available in your office.
Rapid evaluation of patients seeking care after potential exposure to HIV
Effective delivery of nPEP requires prompt initial evaluation of patients and assessment of HIV transmission risk. Take a methodical, step-by-step history of the exposure to address the following basic questions:
- Date and time of exposure? nPEP should be initiated as soon as possible after HIV exposure; it is unlikely to be effective if not initiated within 72 hours or less.
- Frequency of exposure? Type/route of exposure? nPEP is generally reserved for isolated or infrequent exposures that present a substantial risk for HIV acquisition (see Table 1 on HIV acquisition risk below).
- HIV status of exposure source? If the source is positive, is the source person on HIV treatment with antiretroviral therapy? If unknown, is the source person an injecting drug user or a man who has sex with men (MSM)?
Based on the initial evaluation, is nPEP recommended?
Answers to the questions asked during the initial evaluation of the patient will determine whether nPEP is indicated. Along with its updated recommendations, the CDC provided an algorithm to help guide evaluation and treatment.
Preferred HIV test
Administer an HIV test to all patients considered for nPEP, preferably the rapid combined antigen and antibody test (Ag/Ab), or just the antibody test if the Ag/Ab test is not available. nPEP is indicated only for persons without HIV infections. However, if results are not available during the initial evaluation, assume the patient is not infected. If indicated and started, nPEP can be discontinued if tests later shown the patient already has an HIV infection.
Laboratory testing
If nPEP is indicated, conduct laboratory testing. Lab testing is required to document the patient’s HIV status (and that of the source person, when available), identify and manage other conditions potentially resulting from exposure, identify conditions that may affect the nPEP medication regimen, and monitor safety or toxicities to the prescribed regimen.
nPEP treatment regimen for otherwise healthy adults and adolescents
In the absence of randomized clinical trials, data from a case/control study demonstrating an 81% reduction of HIV transmission after use of occupational PEP among hospital workers remains the strongest evidence for the benefit of nPEP.1,2 For patients offered nPEP, recommended treatment includes prescribing either of the following regimens for 28 days:
- Preferred regimen: tenofovir disoproxil fumarate (TDF) (300 mg) with emtricitabine (FTC) (200 mg) once daily plus either raltegravir (RAL) 400 mg twice daily or dolutegravir (DTG) 50 mg daily.
- Alternative regimen: TDF (300 mg) with FTC (200 mg) once daily plus darunavir (DRV) (800 mg) and ritonavir (RTV) (100 mg) once daily.
Additional considerations and nPEP treatment regimens for children, patients with decreased renal function, and pregnant women are included in the CDC guidelines.
Crucial Information for Patients on nPEP
Emphasize the importance of proper dosing and adherence.
Review the patient information for each drug in the regimen, specifically the black boxes, warnings, and side effects, and counsel your patients accordingly.
Transitioning from nPEP to PrEP or from PrEP to nPEP
If you have a patient who engages in behavior that places them at risk for frequent, recurrent exposures to HIV, consider transitioning them to PrEP (pre-exposure prophylaxis) following their 28-day course of nPEP.3 PrEP is a two-drug regimen taken daily on an ongoing basis.
Additionally, for patients who are already on PrEP but who have not taken their medications within a week before the possible exposure, consider initiating nPEP for 28 days and then reintroducing PrEP if their HIV status is negative and the problems with adherence can be addressed moving forward.
Raising Awareness About nPEP
Many people never expect to be exposed to HIV and may not know about the availability of PEP in an emergency situation. You can help raise awareness by making educational materials available in your waiting rooms and exam rooms. Brochures and other HIV/AIDS educational materials for patients are available from the CDC Act Against AIDS campaign.
Summary
Dr. Dominguez is a Captain, U.S. Public Health Service, epidemiology branch, division of HIV/AIDS prevention, CDC.
Additional resources
- The CDC recommends that everyone between the ages of 13 and 64 get tested for HIV at least once as part of routine health care. As part of its Act Against AIDS initiative, the CDC developed the HIV Screening Standard Care program, which provides free tools and resources to help clinicians and nurses incorporate routine HIV screening into primary care settings.
- HIV guidelines and recommendations .
- Postexposure prophylaxis (PEP)
- Pre-Exposure prophylaxis (PrEP)
References
1. Centers for Disease Control and Prevention. Updated guidelines for antiretroviral postexposure prophylaxis after sexual, injection drug use, or other nonoccupational exposure to HIV. United States, 2016. Accessed March 6, 2017.
2. Cardo DM et al. New Engl J Med. 1997;337(21):1485-90.
3. Centers for Disease Control and Prevention. Preexposure prophylaxis for the prevention of HIV infection in the United States–2014: a clinical practice guideline. Accessed March 6, 2017.
Drug combo indicated for bacterial pneumonia
(Avycaz) to include hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) in adults.
Specifically, the approved indication is for infections caused by certain Gram-negative bacteria – some of which are increasingly resistant to available antibiotics – including, Klebsiella pneumoniae, Enterobacter cloacae, Escherichia coli, Serratia marcescens, Proteus mirabilis, Pseudomonas aeruginosa, and Haemophilus influenzae.
There have not been new treatment options for HABP/VABP caused by Gram-negative bacteria in more than 15 years, according to Allergan, the drug’s manufacturer.
This is the third approved indication for ceftazidime/avibactam; the other two indications are for complicated intra-abdominal infections (in combination with metronidazole) and for complicated urinary tract infections.
(Avycaz) to include hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) in adults.
Specifically, the approved indication is for infections caused by certain Gram-negative bacteria – some of which are increasingly resistant to available antibiotics – including, Klebsiella pneumoniae, Enterobacter cloacae, Escherichia coli, Serratia marcescens, Proteus mirabilis, Pseudomonas aeruginosa, and Haemophilus influenzae.
There have not been new treatment options for HABP/VABP caused by Gram-negative bacteria in more than 15 years, according to Allergan, the drug’s manufacturer.
This is the third approved indication for ceftazidime/avibactam; the other two indications are for complicated intra-abdominal infections (in combination with metronidazole) and for complicated urinary tract infections.
(Avycaz) to include hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) in adults.
Specifically, the approved indication is for infections caused by certain Gram-negative bacteria – some of which are increasingly resistant to available antibiotics – including, Klebsiella pneumoniae, Enterobacter cloacae, Escherichia coli, Serratia marcescens, Proteus mirabilis, Pseudomonas aeruginosa, and Haemophilus influenzae.
There have not been new treatment options for HABP/VABP caused by Gram-negative bacteria in more than 15 years, according to Allergan, the drug’s manufacturer.
The approval of the expanded indication was based on data from the phase 3, multinational, double-blind REPROVE trial. The study showed that ceftazidime/avibactam was noninferior to meropenem with respect to 28-day all-cause mortality.
This is the third approved indication for ceftazidime/avibactam; the other two indications are for complicated intra-abdominal infections (in combination with metronidazole) and for complicated urinary tract infections.