User login
VIDEO: Intestinal remodeling contributes to HbA1c drop after Roux-en-Y gastric bypass
CHICAGO – Of medical and surgical tactics to tackle long-term weight loss, , and gene expression in the Roux limb may hold the key to the surgery’s efficacy, according to an ongoing study.
“We know that Roux-en-Y gastric bypass surgery is highly effective as not only a weight-loss therapy, but more and more we’re appreciating its role as a diabetes therapy as well,” said Margaret Stefater, MD, PhD, speaking in an interview at the annual meeting of the Endocrine Society.
The study, she said, was designed to learn more about the intestine’s contribution to the salubrious effect that Roux-en-Y surgery has on diabetes.
“We used microarray in order to characterize gene expression in the intestine” to gain a broad understanding of the processes that are altered after surgery, said Dr. Stefater, a pediatric endocrinology fellow at Boston Children’s Hospital. More specifically, though, the study looked at an individual’s changes in gene expression over time and correlated those changes with that patient’s clinical picture.
The data reported by Dr. Stefater and shared in a press conference, represent part of an ongoing longitudinal prospective study of 32 patients.
“The study aims to characterize gene expression for the first postoperative year,” and findings from the first 6 postoperative months of 19 patients were shared at the meeting, said Dr. Stefater. “This is the first look at our cohort.”
So far, she and her colleagues have compared gene expression using microarray at 1 month and 6 months post-surgery, comparing change across time and change from baseline data.
From hundreds of candidate genes, Dr. Stefater and her colleagues have developed a smaller gene list that, even in the first postoperative month, is predictive of changes in hemoglobin A1c levels over time. “Remarkably, the changes in a select list of genes out to 1 month is actually able to predict hemoglobin A1c levels out to 1 year,” she said. “This speaks to the fact that biological reprogramming in the intestine is somehow related to glycemic response in patients.
“We hope that by understanding these processes, we can home in on those processes that are most likely to be mechanistically responsible for these changes, and then to reverse-engineer this surgery to identify processes or targets which may be good places to start when we think about creating better, or nonsurgical, therapies for people who have obesity and diabetes,” said Dr. Stefater.
Dr. Stefater reported no relevant financial disclosures.
SOURCE: Stefater MA et al. ENDO 2018, Abstract OR 12-6.
CHICAGO – Of medical and surgical tactics to tackle long-term weight loss, , and gene expression in the Roux limb may hold the key to the surgery’s efficacy, according to an ongoing study.
“We know that Roux-en-Y gastric bypass surgery is highly effective as not only a weight-loss therapy, but more and more we’re appreciating its role as a diabetes therapy as well,” said Margaret Stefater, MD, PhD, speaking in an interview at the annual meeting of the Endocrine Society.
The study, she said, was designed to learn more about the intestine’s contribution to the salubrious effect that Roux-en-Y surgery has on diabetes.
“We used microarray in order to characterize gene expression in the intestine” to gain a broad understanding of the processes that are altered after surgery, said Dr. Stefater, a pediatric endocrinology fellow at Boston Children’s Hospital. More specifically, though, the study looked at an individual’s changes in gene expression over time and correlated those changes with that patient’s clinical picture.
The data reported by Dr. Stefater and shared in a press conference, represent part of an ongoing longitudinal prospective study of 32 patients.
“The study aims to characterize gene expression for the first postoperative year,” and findings from the first 6 postoperative months of 19 patients were shared at the meeting, said Dr. Stefater. “This is the first look at our cohort.”
So far, she and her colleagues have compared gene expression using microarray at 1 month and 6 months post-surgery, comparing change across time and change from baseline data.
From hundreds of candidate genes, Dr. Stefater and her colleagues have developed a smaller gene list that, even in the first postoperative month, is predictive of changes in hemoglobin A1c levels over time. “Remarkably, the changes in a select list of genes out to 1 month is actually able to predict hemoglobin A1c levels out to 1 year,” she said. “This speaks to the fact that biological reprogramming in the intestine is somehow related to glycemic response in patients.
“We hope that by understanding these processes, we can home in on those processes that are most likely to be mechanistically responsible for these changes, and then to reverse-engineer this surgery to identify processes or targets which may be good places to start when we think about creating better, or nonsurgical, therapies for people who have obesity and diabetes,” said Dr. Stefater.
Dr. Stefater reported no relevant financial disclosures.
SOURCE: Stefater MA et al. ENDO 2018, Abstract OR 12-6.
CHICAGO – Of medical and surgical tactics to tackle long-term weight loss, , and gene expression in the Roux limb may hold the key to the surgery’s efficacy, according to an ongoing study.
“We know that Roux-en-Y gastric bypass surgery is highly effective as not only a weight-loss therapy, but more and more we’re appreciating its role as a diabetes therapy as well,” said Margaret Stefater, MD, PhD, speaking in an interview at the annual meeting of the Endocrine Society.
The study, she said, was designed to learn more about the intestine’s contribution to the salubrious effect that Roux-en-Y surgery has on diabetes.
“We used microarray in order to characterize gene expression in the intestine” to gain a broad understanding of the processes that are altered after surgery, said Dr. Stefater, a pediatric endocrinology fellow at Boston Children’s Hospital. More specifically, though, the study looked at an individual’s changes in gene expression over time and correlated those changes with that patient’s clinical picture.
The data reported by Dr. Stefater and shared in a press conference, represent part of an ongoing longitudinal prospective study of 32 patients.
“The study aims to characterize gene expression for the first postoperative year,” and findings from the first 6 postoperative months of 19 patients were shared at the meeting, said Dr. Stefater. “This is the first look at our cohort.”
So far, she and her colleagues have compared gene expression using microarray at 1 month and 6 months post-surgery, comparing change across time and change from baseline data.
From hundreds of candidate genes, Dr. Stefater and her colleagues have developed a smaller gene list that, even in the first postoperative month, is predictive of changes in hemoglobin A1c levels over time. “Remarkably, the changes in a select list of genes out to 1 month is actually able to predict hemoglobin A1c levels out to 1 year,” she said. “This speaks to the fact that biological reprogramming in the intestine is somehow related to glycemic response in patients.
“We hope that by understanding these processes, we can home in on those processes that are most likely to be mechanistically responsible for these changes, and then to reverse-engineer this surgery to identify processes or targets which may be good places to start when we think about creating better, or nonsurgical, therapies for people who have obesity and diabetes,” said Dr. Stefater.
Dr. Stefater reported no relevant financial disclosures.
SOURCE: Stefater MA et al. ENDO 2018, Abstract OR 12-6.
REPORTING FROM ENDO 2018
FDA expands indication for blinatumomab in treating ALL
The Food and Drug Administration has granted accelerated who are in remission but still have minimal residual disease (MRD).
This is the first FDA-approved treatment for those with MRD, the FDA said in a statement.
The current approval was based on a single-arm clinical trial of 86 patients in first or second complete remission who had detectable MRD in at least 1 out of 1,000 cells in their bone marrow. Undetectable MRD was achieved by 70 patients after one cycle of blinatumomab treatment. More than half of the patients remained alive and in remission for at least 22.3 months, the FDA said.
Common side effects include bacterial and pathogen-unspecified infections, pyrexia, headache, infusion-related reactions, neutropenia, anemia, febrile neutropenia, and thrombocytopenia. The drug carries a boxed warning about cytokine release syndrome at the start of the first treatment. The FDA also warns that children weighing less than 22 kg should receive the drug prepared with preservative-free saline because of the risk of serious adverse reactions in pediatric patients from a benzyl alcohol preservative.
Blinatumomab is marketed as Blincyto by Amgen.
The Food and Drug Administration has granted accelerated who are in remission but still have minimal residual disease (MRD).
This is the first FDA-approved treatment for those with MRD, the FDA said in a statement.
The current approval was based on a single-arm clinical trial of 86 patients in first or second complete remission who had detectable MRD in at least 1 out of 1,000 cells in their bone marrow. Undetectable MRD was achieved by 70 patients after one cycle of blinatumomab treatment. More than half of the patients remained alive and in remission for at least 22.3 months, the FDA said.
Common side effects include bacterial and pathogen-unspecified infections, pyrexia, headache, infusion-related reactions, neutropenia, anemia, febrile neutropenia, and thrombocytopenia. The drug carries a boxed warning about cytokine release syndrome at the start of the first treatment. The FDA also warns that children weighing less than 22 kg should receive the drug prepared with preservative-free saline because of the risk of serious adverse reactions in pediatric patients from a benzyl alcohol preservative.
Blinatumomab is marketed as Blincyto by Amgen.
The Food and Drug Administration has granted accelerated who are in remission but still have minimal residual disease (MRD).
This is the first FDA-approved treatment for those with MRD, the FDA said in a statement.
The current approval was based on a single-arm clinical trial of 86 patients in first or second complete remission who had detectable MRD in at least 1 out of 1,000 cells in their bone marrow. Undetectable MRD was achieved by 70 patients after one cycle of blinatumomab treatment. More than half of the patients remained alive and in remission for at least 22.3 months, the FDA said.
Common side effects include bacterial and pathogen-unspecified infections, pyrexia, headache, infusion-related reactions, neutropenia, anemia, febrile neutropenia, and thrombocytopenia. The drug carries a boxed warning about cytokine release syndrome at the start of the first treatment. The FDA also warns that children weighing less than 22 kg should receive the drug prepared with preservative-free saline because of the risk of serious adverse reactions in pediatric patients from a benzyl alcohol preservative.
Blinatumomab is marketed as Blincyto by Amgen.
FDA approves highest capacity insulin pen
This will be the highest capacity long-acting insulin pen to be available commercially, according to a company press release.
The Max SoloStar pen holds more than 900 units of insulin glargine and can provide up to 160 units/mL in a single injection, which may reduce the number of injections needed to deliver the necessary dosage to adults. Another benefit of the higher capacity is that the device will require fewer refills and the associated copays, which could potentially lower costs for patients, depending on their insurance coverage.
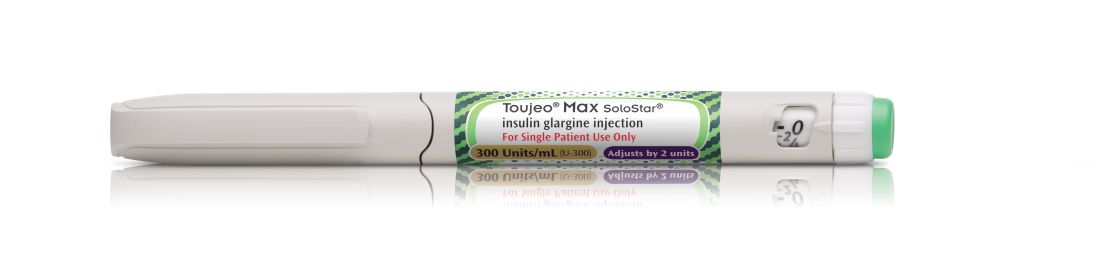
The dosing instructions for insulin glargine vary depending on whether patients use the older SoloStar or the high capacity Max SoloStar. The SoloStar holds 450 units (1.5mL) of insulin glargine and delivers doses in 1 unit increments, delivering a maximum dose of 80 units per injection. The MaxSoloStar has twice the capacity of the original – 900 units (3 mL) of insulin glargine – and delivers doses in 2 unit increments up to a maximum 160 unit injection. This is recommended for patients who require at least 20 units per day. All insulin glargine injections should be administered subcutaneously in the abdomen, thigh, or deltoid at the same time each day.
The most common side effect of insulin products, such as insulin glargine, is hypoglycemia.
The Max SoloStar will be available in retail pharmacies throughout the United States in the third quarter of 2018.
This will be the highest capacity long-acting insulin pen to be available commercially, according to a company press release.
The Max SoloStar pen holds more than 900 units of insulin glargine and can provide up to 160 units/mL in a single injection, which may reduce the number of injections needed to deliver the necessary dosage to adults. Another benefit of the higher capacity is that the device will require fewer refills and the associated copays, which could potentially lower costs for patients, depending on their insurance coverage.
The dosing instructions for insulin glargine vary depending on whether patients use the older SoloStar or the high capacity Max SoloStar. The SoloStar holds 450 units (1.5mL) of insulin glargine and delivers doses in 1 unit increments, delivering a maximum dose of 80 units per injection. The MaxSoloStar has twice the capacity of the original – 900 units (3 mL) of insulin glargine – and delivers doses in 2 unit increments up to a maximum 160 unit injection. This is recommended for patients who require at least 20 units per day. All insulin glargine injections should be administered subcutaneously in the abdomen, thigh, or deltoid at the same time each day.
The most common side effect of insulin products, such as insulin glargine, is hypoglycemia.
The Max SoloStar will be available in retail pharmacies throughout the United States in the third quarter of 2018.
This will be the highest capacity long-acting insulin pen to be available commercially, according to a company press release.
The Max SoloStar pen holds more than 900 units of insulin glargine and can provide up to 160 units/mL in a single injection, which may reduce the number of injections needed to deliver the necessary dosage to adults. Another benefit of the higher capacity is that the device will require fewer refills and the associated copays, which could potentially lower costs for patients, depending on their insurance coverage.
The dosing instructions for insulin glargine vary depending on whether patients use the older SoloStar or the high capacity Max SoloStar. The SoloStar holds 450 units (1.5mL) of insulin glargine and delivers doses in 1 unit increments, delivering a maximum dose of 80 units per injection. The MaxSoloStar has twice the capacity of the original – 900 units (3 mL) of insulin glargine – and delivers doses in 2 unit increments up to a maximum 160 unit injection. This is recommended for patients who require at least 20 units per day. All insulin glargine injections should be administered subcutaneously in the abdomen, thigh, or deltoid at the same time each day.
The most common side effect of insulin products, such as insulin glargine, is hypoglycemia.
The Max SoloStar will be available in retail pharmacies throughout the United States in the third quarter of 2018.
Dexcom G6 gets FDA nod
from the Food and Drug Administration.
The Dexcom G6 is about 28% smaller than its predecessor, the G5, can be worn for up to 10 days – 43% longer than the G5 – and doesn’t require any finger-stick calibrations or treatment decisions. It’s the first FDA-approved integrated continuous glucose monitoring (iCGM) system that can link electronically to other compatible devices, including automated insulin dosing systems, insulin pumps, blood glucose meters, and other electronic devices used for diabetes management, the FDA said in a press statement. Its revamped sensor doesn’t interact with acetaminophen – another distinct advantage over the G5.
The device will be commercially available sometime this year, the Dexcom website noted.
The device also set a new premarketing review standard for CGM’s, which can now utilize the less-burdensome 510(k) clearance pathway. Until now, they have been treated as the highest-risk Class III medical devices.
According to the FDA statement, the agency “…recognized this as an opportunity to reduce the regulatory burden for this type of device by establishing criteria that would classify these as ‘moderate risk,’ class II medical devices with special controls.”
G6 was approved through this new pathway, dedicated to novel, low-to-moderate-risk devices that are not “substantially equivalent” to an already legally marketed device, the press statement said.

The FDA evaluated data from two clinical studies of the Dexcom G6, which included 324 adults and children aged 2 years and older with diabetes. Both studies included multiple clinical visits within a 10-day period where system readings were compared to a laboratory test method that measures blood glucose values. No serious adverse events were reported during the studies.
from the Food and Drug Administration.
The Dexcom G6 is about 28% smaller than its predecessor, the G5, can be worn for up to 10 days – 43% longer than the G5 – and doesn’t require any finger-stick calibrations or treatment decisions. It’s the first FDA-approved integrated continuous glucose monitoring (iCGM) system that can link electronically to other compatible devices, including automated insulin dosing systems, insulin pumps, blood glucose meters, and other electronic devices used for diabetes management, the FDA said in a press statement. Its revamped sensor doesn’t interact with acetaminophen – another distinct advantage over the G5.
The device will be commercially available sometime this year, the Dexcom website noted.
The device also set a new premarketing review standard for CGM’s, which can now utilize the less-burdensome 510(k) clearance pathway. Until now, they have been treated as the highest-risk Class III medical devices.
According to the FDA statement, the agency “…recognized this as an opportunity to reduce the regulatory burden for this type of device by establishing criteria that would classify these as ‘moderate risk,’ class II medical devices with special controls.”
G6 was approved through this new pathway, dedicated to novel, low-to-moderate-risk devices that are not “substantially equivalent” to an already legally marketed device, the press statement said.
The FDA evaluated data from two clinical studies of the Dexcom G6, which included 324 adults and children aged 2 years and older with diabetes. Both studies included multiple clinical visits within a 10-day period where system readings were compared to a laboratory test method that measures blood glucose values. No serious adverse events were reported during the studies.
from the Food and Drug Administration.
The Dexcom G6 is about 28% smaller than its predecessor, the G5, can be worn for up to 10 days – 43% longer than the G5 – and doesn’t require any finger-stick calibrations or treatment decisions. It’s the first FDA-approved integrated continuous glucose monitoring (iCGM) system that can link electronically to other compatible devices, including automated insulin dosing systems, insulin pumps, blood glucose meters, and other electronic devices used for diabetes management, the FDA said in a press statement. Its revamped sensor doesn’t interact with acetaminophen – another distinct advantage over the G5.
The device will be commercially available sometime this year, the Dexcom website noted.
The device also set a new premarketing review standard for CGM’s, which can now utilize the less-burdensome 510(k) clearance pathway. Until now, they have been treated as the highest-risk Class III medical devices.
According to the FDA statement, the agency “…recognized this as an opportunity to reduce the regulatory burden for this type of device by establishing criteria that would classify these as ‘moderate risk,’ class II medical devices with special controls.”
G6 was approved through this new pathway, dedicated to novel, low-to-moderate-risk devices that are not “substantially equivalent” to an already legally marketed device, the press statement said.
The FDA evaluated data from two clinical studies of the Dexcom G6, which included 324 adults and children aged 2 years and older with diabetes. Both studies included multiple clinical visits within a 10-day period where system readings were compared to a laboratory test method that measures blood glucose values. No serious adverse events were reported during the studies.
FDA advisors recommend lofexidine for opioid withdrawal
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.

Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.
Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
SILVER SPRING, MD. – Members of the Food and Drug Administration Psychopharmacologic Drugs Advisory Committee voted 11 to 1 to recommend approval of lofexidine as the first nonopioid treatment option for the symptomatic treatment of opioid withdrawal.
Opioid withdrawal symptoms are the largest barrier to discontinuing opioid use, according to Louis Baxter, MD, executive medical director of the Professional Assistance Program in Princeton, N.J., who presented on behalf of U.S. WorldMeds, which plans to market lofexidine as Lucemyra.
Lofexidine, a selective alpha2-adrenergic receptor agonist that regulates norepinephrine release has been approved for management of opioid withdrawal in the United Kingdom since 1992.
The advisory committee voted to recommend lofexidine on the strength of the results of two randomized, double-blind, and placebo controlled phase 3 studies on the safety and efficacy of lofexidine for symptomatic treatment of opioid withdrawal between days 1 through 7. One study randomized 264 patients to lofexidine (134) or placebo (130), with patients in the treatment arm received 3.2 mg of lofexidine on days 1-5, then placebo until day 7. The second study randomized 603 patients to three groups, comparing high dose (3.2 mg/day) and low dose (2.4 mg/day) regimens of lofexidine to placebo; patients in the treatment arms took four smaller doses of lofexidine throughout the day to achieve the cumulative dose.
Researchers enrolled heavy users of short-acting opioids; heroin was the predominant agent. Both studies were conducted in the scenario of abrupt withdrawal, or the most intense withdrawal situation.
Symptomatic benefit was measured using the Short Opiate Withdrawal Scale of Gossop (SOWS-Gossop), a patient reported outcome. Patients were asked to rank their symptoms as none, mild, moderate or severe across measures like feeling sick, stomach cramps, and heart pounding among other symptoms.
Lofexidine increased completion of withdrawal treatment compared to placebo. Patients in the first study had a 5-day completion rate of 53%, compared to 35% for the placebo group. Researchers observed similar results in the 7-day completion rates the second study, with low and high dose completion rates of 42% and 40%, respectively, both of which were much higher than placebo (28%).
Lofexidine also reduced patient withdrawal symptoms, according to SOWS-Gossop scores during peak withdrawal. In the first study, SOWS-Gossop scores were 2-4 points lower in the lofexidine group compared to placebo. Similarly, the scores were significantly better in both lofexidine groups in the second study, compared to placebo, particularly on days 1 to 4. Decreasing withdrawal symptoms during this period is particularly important because this is the most vulnerable window for patient dropout, briefing documents from US WorldMeds.
Several notable adverse events occurred during the study, particularly at higher doses of lofexidine. The risk of bradycardia and hypotension are prominent in patients taking lofexidine, but these are risks associated with this class of drug, according to Mark Pirner, MD, senior medical director at US WorldMeds, who noted that “the lower dose, if that’s what ultimately gets approved [by the FDA], is safe and effective too.”
Development of lofexidine was conducted in collaboration with the National Institute on Drug Abuse and the FDA, according to briefing documents from US WorldMeds.
The Prescription Drug User Fee Act (PDUFA) for lofexidine is May 26; FDA actions on new drug applications often occur at near the PDUFA date.
The FDA is not obligated to follow the recommendations of its advisory committees.
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING
FDA approves nilotinib for children with CML
Nilotinib is now approved for use by children aged 1 year and older with Philadelphia chromosome–positive chronic myeloid leukemia (Ph+ CML) in the chronic phase.
The Food and Drug Administration expanded the drug’s indication to include use as first- and second-line treatment in children.
Adverse events in the pediatric studies were similar to those observed in adults. However, children experienced hyperbilirubinemia (grade 3/4: 13%) and transaminase elevation (AST grade 3/4: 1%; ALT grade 3/4: 9%). Additionally, one previously treated pediatric patient progressed with advance phase/blast crisis after about 10 months of treatment.
Nilotinib (Tasigna) was already approved in adults with newly diagnosed Ph+ CML in the chronic phase and adults with chronic phase and accelerated phase Ph+ CML resistant or intolerant to prior therapy.
Nilotinib is now approved for use by children aged 1 year and older with Philadelphia chromosome–positive chronic myeloid leukemia (Ph+ CML) in the chronic phase.
The Food and Drug Administration expanded the drug’s indication to include use as first- and second-line treatment in children.
Adverse events in the pediatric studies were similar to those observed in adults. However, children experienced hyperbilirubinemia (grade 3/4: 13%) and transaminase elevation (AST grade 3/4: 1%; ALT grade 3/4: 9%). Additionally, one previously treated pediatric patient progressed with advance phase/blast crisis after about 10 months of treatment.
Nilotinib (Tasigna) was already approved in adults with newly diagnosed Ph+ CML in the chronic phase and adults with chronic phase and accelerated phase Ph+ CML resistant or intolerant to prior therapy.
Nilotinib is now approved for use by children aged 1 year and older with Philadelphia chromosome–positive chronic myeloid leukemia (Ph+ CML) in the chronic phase.
The Food and Drug Administration expanded the drug’s indication to include use as first- and second-line treatment in children.
Adverse events in the pediatric studies were similar to those observed in adults. However, children experienced hyperbilirubinemia (grade 3/4: 13%) and transaminase elevation (AST grade 3/4: 1%; ALT grade 3/4: 9%). Additionally, one previously treated pediatric patient progressed with advance phase/blast crisis after about 10 months of treatment.
Nilotinib (Tasigna) was already approved in adults with newly diagnosed Ph+ CML in the chronic phase and adults with chronic phase and accelerated phase Ph+ CML resistant or intolerant to prior therapy.
FDA approves certolizumab label update for pregnancy, breastfeeding
The manufacturer of certolizumab pegol, UCB, announced March 22 that the Food and Drug Administration approved a label update to the biologic that includes pharmacokinetic data showing negligible to low transfer of the biologic through the placenta and minimal mother-to-infant transfer from breast milk.

In the CRIB study, certolizumab levels were below the lower limit of quantification (defined as 0.032 mcg/mL) in 13 out of 15 infant blood samples at birth and in all samples at weeks 4 and 8. No anticertolizumab antibodies were detected in mothers, umbilical cords, or infants.
In the CRADLE study, 56% of 137 breast milk samples from 17 mothers had no measurable certolizumab, and the remaining samples showed minimal levels of the biologic. No serious adverse reactions were noted in the 17 infants in the study.
“It is well recognized that women with chronic inflammatory disease face uncertainty during motherhood given the lack of information on treatment during pregnancy and breastfeeding. Many women with chronic inflammatory disease discontinue their biologic treatment during pregnancy, often when they need disease control the most,” said CRADLE lead study author Megan E. B. Clowse, MD, of Duke University, Durham, N.C., in a press release issued by UCB. “These data for Cimzia provide important information to empower women and healthcare providers making decisions about treatment during pregnancy and breastfeeding.”
UCB said that limited data from an ongoing pregnancy registry regarding the use of certolizumab in pregnant women are not sufficient to inform a risk of major birth defects or other adverse pregnancy outcomes.
The manufacturer of certolizumab pegol, UCB, announced March 22 that the Food and Drug Administration approved a label update to the biologic that includes pharmacokinetic data showing negligible to low transfer of the biologic through the placenta and minimal mother-to-infant transfer from breast milk.
In the CRIB study, certolizumab levels were below the lower limit of quantification (defined as 0.032 mcg/mL) in 13 out of 15 infant blood samples at birth and in all samples at weeks 4 and 8. No anticertolizumab antibodies were detected in mothers, umbilical cords, or infants.
In the CRADLE study, 56% of 137 breast milk samples from 17 mothers had no measurable certolizumab, and the remaining samples showed minimal levels of the biologic. No serious adverse reactions were noted in the 17 infants in the study.
“It is well recognized that women with chronic inflammatory disease face uncertainty during motherhood given the lack of information on treatment during pregnancy and breastfeeding. Many women with chronic inflammatory disease discontinue their biologic treatment during pregnancy, often when they need disease control the most,” said CRADLE lead study author Megan E. B. Clowse, MD, of Duke University, Durham, N.C., in a press release issued by UCB. “These data for Cimzia provide important information to empower women and healthcare providers making decisions about treatment during pregnancy and breastfeeding.”
UCB said that limited data from an ongoing pregnancy registry regarding the use of certolizumab in pregnant women are not sufficient to inform a risk of major birth defects or other adverse pregnancy outcomes.
The manufacturer of certolizumab pegol, UCB, announced March 22 that the Food and Drug Administration approved a label update to the biologic that includes pharmacokinetic data showing negligible to low transfer of the biologic through the placenta and minimal mother-to-infant transfer from breast milk.
In the CRIB study, certolizumab levels were below the lower limit of quantification (defined as 0.032 mcg/mL) in 13 out of 15 infant blood samples at birth and in all samples at weeks 4 and 8. No anticertolizumab antibodies were detected in mothers, umbilical cords, or infants.
In the CRADLE study, 56% of 137 breast milk samples from 17 mothers had no measurable certolizumab, and the remaining samples showed minimal levels of the biologic. No serious adverse reactions were noted in the 17 infants in the study.
“It is well recognized that women with chronic inflammatory disease face uncertainty during motherhood given the lack of information on treatment during pregnancy and breastfeeding. Many women with chronic inflammatory disease discontinue their biologic treatment during pregnancy, often when they need disease control the most,” said CRADLE lead study author Megan E. B. Clowse, MD, of Duke University, Durham, N.C., in a press release issued by UCB. “These data for Cimzia provide important information to empower women and healthcare providers making decisions about treatment during pregnancy and breastfeeding.”
UCB said that limited data from an ongoing pregnancy registry regarding the use of certolizumab in pregnant women are not sufficient to inform a risk of major birth defects or other adverse pregnancy outcomes.
FDA approves IL-23 antagonist for plaque psoriasis
in adults who are eligible for systemic therapy or phototherapy, according to a statement from Sun Pharma.
Tildrakizumab is administered at a dose of 100 mg, subcutaneously, at weeks 0 and 4, then every 12 weeks. Approval is based on data from two phase 3, identically designed clinical trials, reSURFACE1 and reSURFACE2. Both studies were multicenter, randomized, double-blind, and placebo controlled. In the studies, 926 patients received tildrakizumab (616 patients) or placebo (310 patients).
The effectiveness of tildrakizumab extended beyond 12 weeks, with 74% of patients achieving a PASI 75 at 28 weeks after three doses. This percentage grew to 84% at week 64 in patients who continued treatment. Similar results were observed with PGA scores, with 69% of patients who had a PGA score of 0 or 1 at 12 weeks maintaining that score at week 28.
Tildrakizumab has been associated with serious side effects, including serious allergic reactions including skin rash, swelling of the face and mouth, trouble breathing, and chest tightness. It may also increase patient susceptibility to infection. It is approved with a Medication Guide for patients, explaining the potential risks associated with treatment.
Tildrakizumab will be marketed as Ilumya.
Sun Pharma is working with the FDA on postapproval commitments, and once that has been completed, they will have a better idea of when it will become available, according to a spokesperson for the manufacturer. The cost is not yet available.
in adults who are eligible for systemic therapy or phototherapy, according to a statement from Sun Pharma.
Tildrakizumab is administered at a dose of 100 mg, subcutaneously, at weeks 0 and 4, then every 12 weeks. Approval is based on data from two phase 3, identically designed clinical trials, reSURFACE1 and reSURFACE2. Both studies were multicenter, randomized, double-blind, and placebo controlled. In the studies, 926 patients received tildrakizumab (616 patients) or placebo (310 patients).
The effectiveness of tildrakizumab extended beyond 12 weeks, with 74% of patients achieving a PASI 75 at 28 weeks after three doses. This percentage grew to 84% at week 64 in patients who continued treatment. Similar results were observed with PGA scores, with 69% of patients who had a PGA score of 0 or 1 at 12 weeks maintaining that score at week 28.
Tildrakizumab has been associated with serious side effects, including serious allergic reactions including skin rash, swelling of the face and mouth, trouble breathing, and chest tightness. It may also increase patient susceptibility to infection. It is approved with a Medication Guide for patients, explaining the potential risks associated with treatment.
Tildrakizumab will be marketed as Ilumya.
Sun Pharma is working with the FDA on postapproval commitments, and once that has been completed, they will have a better idea of when it will become available, according to a spokesperson for the manufacturer. The cost is not yet available.
in adults who are eligible for systemic therapy or phototherapy, according to a statement from Sun Pharma.
Tildrakizumab is administered at a dose of 100 mg, subcutaneously, at weeks 0 and 4, then every 12 weeks. Approval is based on data from two phase 3, identically designed clinical trials, reSURFACE1 and reSURFACE2. Both studies were multicenter, randomized, double-blind, and placebo controlled. In the studies, 926 patients received tildrakizumab (616 patients) or placebo (310 patients).
The effectiveness of tildrakizumab extended beyond 12 weeks, with 74% of patients achieving a PASI 75 at 28 weeks after three doses. This percentage grew to 84% at week 64 in patients who continued treatment. Similar results were observed with PGA scores, with 69% of patients who had a PGA score of 0 or 1 at 12 weeks maintaining that score at week 28.
Tildrakizumab has been associated with serious side effects, including serious allergic reactions including skin rash, swelling of the face and mouth, trouble breathing, and chest tightness. It may also increase patient susceptibility to infection. It is approved with a Medication Guide for patients, explaining the potential risks associated with treatment.
Tildrakizumab will be marketed as Ilumya.
Sun Pharma is working with the FDA on postapproval commitments, and once that has been completed, they will have a better idea of when it will become available, according to a spokesperson for the manufacturer. The cost is not yet available.
FDA updates breast implant–associated lymphoma cases, risk
(BIA-ALCL), including nine deaths.
This figure includes all medical device reports received by the agency between 2011 and September 2017. The FDA recently provided an update on ALCL linked to breast implants and an estimate of lifetime risk of developing ALCL.
Based on available medical literature, the lifetime risk of developing BIA-ALCL for patients with textured breast implants ranges from 1 in 3,817 to 1 in 30,000, according to the update.
Of the 272 reports with data on surface type, 242 were textured implants and 30 were smooth implants. In addition, 413 reports include information on the implant fill type: 234 used silicone gel and 179 were saline filled.
“The FDA has been closely tracking the relationship between breast implants and a rare type of non-Hodgkin’s lymphoma since we first identified this possible association. We’ve been working to gather additional information to better characterize and quantify the risk so that patients and providers can have more informed discussions about breast implants,” said Binita Ashar, MD, director of the division of surgical devices in the FDA’s Center for Devices and Radiological Health. “As part of that effort, we are working to update and enhance the information we have on this association, including updating the total number of known cases of BIA-ALCL and the lifetime risk of developing BIA-ALCL as reported in medical literature.”
The possible association between breast implants and the development of anaplastic large cell lymphoma (ALCL) was first identified in 2011. At that time, there were not enough cases of to determine what factors increased a patient’s risk of developing the disease. As more information became available, the World Health Organization designated BIA-ALCL as a T-cell lymphoma that can develop following breast implants.
(BIA-ALCL), including nine deaths.
This figure includes all medical device reports received by the agency between 2011 and September 2017. The FDA recently provided an update on ALCL linked to breast implants and an estimate of lifetime risk of developing ALCL.
Based on available medical literature, the lifetime risk of developing BIA-ALCL for patients with textured breast implants ranges from 1 in 3,817 to 1 in 30,000, according to the update.
Of the 272 reports with data on surface type, 242 were textured implants and 30 were smooth implants. In addition, 413 reports include information on the implant fill type: 234 used silicone gel and 179 were saline filled.
“The FDA has been closely tracking the relationship between breast implants and a rare type of non-Hodgkin’s lymphoma since we first identified this possible association. We’ve been working to gather additional information to better characterize and quantify the risk so that patients and providers can have more informed discussions about breast implants,” said Binita Ashar, MD, director of the division of surgical devices in the FDA’s Center for Devices and Radiological Health. “As part of that effort, we are working to update and enhance the information we have on this association, including updating the total number of known cases of BIA-ALCL and the lifetime risk of developing BIA-ALCL as reported in medical literature.”
The possible association between breast implants and the development of anaplastic large cell lymphoma (ALCL) was first identified in 2011. At that time, there were not enough cases of to determine what factors increased a patient’s risk of developing the disease. As more information became available, the World Health Organization designated BIA-ALCL as a T-cell lymphoma that can develop following breast implants.
(BIA-ALCL), including nine deaths.
This figure includes all medical device reports received by the agency between 2011 and September 2017. The FDA recently provided an update on ALCL linked to breast implants and an estimate of lifetime risk of developing ALCL.
Based on available medical literature, the lifetime risk of developing BIA-ALCL for patients with textured breast implants ranges from 1 in 3,817 to 1 in 30,000, according to the update.
Of the 272 reports with data on surface type, 242 were textured implants and 30 were smooth implants. In addition, 413 reports include information on the implant fill type: 234 used silicone gel and 179 were saline filled.
“The FDA has been closely tracking the relationship between breast implants and a rare type of non-Hodgkin’s lymphoma since we first identified this possible association. We’ve been working to gather additional information to better characterize and quantify the risk so that patients and providers can have more informed discussions about breast implants,” said Binita Ashar, MD, director of the division of surgical devices in the FDA’s Center for Devices and Radiological Health. “As part of that effort, we are working to update and enhance the information we have on this association, including updating the total number of known cases of BIA-ALCL and the lifetime risk of developing BIA-ALCL as reported in medical literature.”
The possible association between breast implants and the development of anaplastic large cell lymphoma (ALCL) was first identified in 2011. At that time, there were not enough cases of to determine what factors increased a patient’s risk of developing the disease. As more information became available, the World Health Organization designated BIA-ALCL as a T-cell lymphoma that can develop following breast implants.
FDA approves new option in Hodgkin lymphoma treatment
The Food and Drug Administration has approved brentuximab vedotin, in combination with chemotherapy, for previously untreated adults with stage III or IV classical Hodgkin lymphoma.
The drug, which is marketed by Seattle Genetics as Adcetris, is already approved in classical Hodgkin lymphoma after relapse and after stem cell transplant when the patient is at risk of relapse or progression. The drug is also approved to treat both systemic anaplastic large cell lymphoma (ALCL) and primary cutaneous ALCL after failure on other treatments.
The modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD versus 77.2% for ABVD (P = .03), a 23% relative risk reduction (N Engl J Med. 2018;378:331-44).
Common side effects of brentuximab vedotin include neutropenia, anemia, peripheral neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia. The drug carries a boxed warning highlighting the risk of John Cunningham virus infection resulting in progressive multifocal leukoencephalopathy.
The Food and Drug Administration has approved brentuximab vedotin, in combination with chemotherapy, for previously untreated adults with stage III or IV classical Hodgkin lymphoma.
The drug, which is marketed by Seattle Genetics as Adcetris, is already approved in classical Hodgkin lymphoma after relapse and after stem cell transplant when the patient is at risk of relapse or progression. The drug is also approved to treat both systemic anaplastic large cell lymphoma (ALCL) and primary cutaneous ALCL after failure on other treatments.
The modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD versus 77.2% for ABVD (P = .03), a 23% relative risk reduction (N Engl J Med. 2018;378:331-44).
Common side effects of brentuximab vedotin include neutropenia, anemia, peripheral neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia. The drug carries a boxed warning highlighting the risk of John Cunningham virus infection resulting in progressive multifocal leukoencephalopathy.
The Food and Drug Administration has approved brentuximab vedotin, in combination with chemotherapy, for previously untreated adults with stage III or IV classical Hodgkin lymphoma.
The drug, which is marketed by Seattle Genetics as Adcetris, is already approved in classical Hodgkin lymphoma after relapse and after stem cell transplant when the patient is at risk of relapse or progression. The drug is also approved to treat both systemic anaplastic large cell lymphoma (ALCL) and primary cutaneous ALCL after failure on other treatments.
The modified 2-year progression-free survival in the trial was 82.1% for patients receiving brentuximab plus AVD versus 77.2% for ABVD (P = .03), a 23% relative risk reduction (N Engl J Med. 2018;378:331-44).
Common side effects of brentuximab vedotin include neutropenia, anemia, peripheral neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia. The drug carries a boxed warning highlighting the risk of John Cunningham virus infection resulting in progressive multifocal leukoencephalopathy.