User login
Fluoroquinolones can cause fatal hypoglycemia, FDA warns
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.

The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
FDA recommends pooled Zika testing of blood donations
The moving away from individual testing of donations and toward pooled testing.
“This [practice of pooled testing] is usually more cost effective and less burdensome for blood establishments,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, in the statement. “However, the FDA will continue to monitor the situation closely and, as appropriate, reconsider what measures are needed to maintain the safety of the blood supply.”
The new testing recommendations reflect the decreasing number of cases of Zika virus infection in the U.S. and its territories, as well as advice from the agency’s Blood Products Advisory Committee.
The guidance makes an exception to its pooled testing recommendations: Donations from areas where a positive donation has been detected or in which the risk of mosquito-borne transmission of Zika virus is high should be tested individually. The guidance also allows the use of an FDA-approved pathogen-reduction device for plasma and certain platelet products.
The moving away from individual testing of donations and toward pooled testing.
“This [practice of pooled testing] is usually more cost effective and less burdensome for blood establishments,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, in the statement. “However, the FDA will continue to monitor the situation closely and, as appropriate, reconsider what measures are needed to maintain the safety of the blood supply.”
The new testing recommendations reflect the decreasing number of cases of Zika virus infection in the U.S. and its territories, as well as advice from the agency’s Blood Products Advisory Committee.
The guidance makes an exception to its pooled testing recommendations: Donations from areas where a positive donation has been detected or in which the risk of mosquito-borne transmission of Zika virus is high should be tested individually. The guidance also allows the use of an FDA-approved pathogen-reduction device for plasma and certain platelet products.
The moving away from individual testing of donations and toward pooled testing.
“This [practice of pooled testing] is usually more cost effective and less burdensome for blood establishments,” said Peter Marks, MD, PhD, director of the FDA’s Center for Biologics Evaluation and Research, in the statement. “However, the FDA will continue to monitor the situation closely and, as appropriate, reconsider what measures are needed to maintain the safety of the blood supply.”
The new testing recommendations reflect the decreasing number of cases of Zika virus infection in the U.S. and its territories, as well as advice from the agency’s Blood Products Advisory Committee.
The guidance makes an exception to its pooled testing recommendations: Donations from areas where a positive donation has been detected or in which the risk of mosquito-borne transmission of Zika virus is high should be tested individually. The guidance also allows the use of an FDA-approved pathogen-reduction device for plasma and certain platelet products.
FDA approves topical anticholinergic for primary axillary hyperhidrosis
The in adults and children aged 9 years and older.
Glycopyrronium, which is formulated in a cloth wipe, will be marketed as Qbrexza and is expected to be available in October, according to the approval announcement June 29, which was made by Dermira, the manufacturer. The instructions for use section in the prescribing information states that patients are advised to use one cloth to apply the medication to both axillae, wiping the cloth across each underarm. Each cloth, intended for single use, is pre-moistened with 2.4% glycopyrronium solution.
Glycopyrronium blocks sweat production “by inhibiting the interaction between acetylcholine and the cholinergic receptors responsible for sweat gland activation,” the company said in a February press release.
The approval was based on the results of two phase 3 clinical studies, ATMOS-1 and ATMOS-2, which were multicenter, randomized, double-blind, vehicle-controlled, 4-week studies in patients 9 years of age or older with primary axillary hyperhidrosis for 6 months or longer. Study subjects had production of at least 50 mg of underarm sweat over 5 minutes, and scores of four or higher on the 11-point Axillary Sweating Daily Diary (ASDD) or the Children’s ASDD (ASDD-C) – an instrument developed by the company in consultation with the FDA – and scores of three or four on the four-grade Hyperhidrosis Disease Severity Scale (HDSS). In total, 463 patients were randomized to receive glycopyrronium and 234 to vehicle. Forty four of these patients were aged 9-16 years, with 25 receiving glycopyrronium and 19 receiving vehicle.
According to the February release, the ASDD/ASDD-C severity scale responder rates were about 60% in the pediatric and adult groups treated with glycopyrronium, compared with 13.0% and 28.8% for children and adults, respectively, in the vehicle group. At week 4, the median absolute change in sweat production was a reduction of 64.2 mg in children and a reduction of 80.6 mg among adults treated with glycopyrronium, compared with reductions of 53.7 mg and 62 mg among those in the vehicle group, respectively.
In the glycopyrronium-treated group, almost 80% of the pediatric patients and 74.3% of the adults experienced at least a 50% reduction in sweat production at week 4, compared with almost 55% and 53%, respectively, in the vehicle group.
Among pediatric patients, the mean decrease from baseline in the Children’s Dermatology Quality of Life Index was 8.1 in glycopyrronium-treated patients, compared with 1.9 in the vehicle group. In adults, scores on the Dermatology Life Quality Index measure were reduced by 8.4 and 4.7 in glycopyrronium- and vehicle-treated adult patients, respectively.
Nearly 57% of adults and 44% of pediatric patients treated with glycopyrronium experienced treatment-emergent adverse events, compared with 34.3% of adults and 10.5% of the pediatric patients in the vehicle group. The majority were related to anticholinergic activity and were mild, and rarely led to drug discontinuation, according to the company.
The press release announcing the approval stated that the most common adverse effects observed after application of glycopyrronium were dry mouth, mydriasis, sore throat, headache, urinary hesitation, blurred vision, dry nose, dry throat, dry eye, dry skin and constipation. Erythema, burning/stinging, and pruritus were the most common skin reactions.
The product is contraindicated in patients with glaucoma, paralytic ileus, and other medical conditions that can be exacerbated by its anticholinergic effects, according to the prescribing information. Patients should be advised that they should wash their hands thoroughly after application, and that it can cause temporary dilation of the pupils and blurred vision if glycopyrronium comes into contact with their eyes.
The in adults and children aged 9 years and older.
Glycopyrronium, which is formulated in a cloth wipe, will be marketed as Qbrexza and is expected to be available in October, according to the approval announcement June 29, which was made by Dermira, the manufacturer. The instructions for use section in the prescribing information states that patients are advised to use one cloth to apply the medication to both axillae, wiping the cloth across each underarm. Each cloth, intended for single use, is pre-moistened with 2.4% glycopyrronium solution.
Glycopyrronium blocks sweat production “by inhibiting the interaction between acetylcholine and the cholinergic receptors responsible for sweat gland activation,” the company said in a February press release.
The approval was based on the results of two phase 3 clinical studies, ATMOS-1 and ATMOS-2, which were multicenter, randomized, double-blind, vehicle-controlled, 4-week studies in patients 9 years of age or older with primary axillary hyperhidrosis for 6 months or longer. Study subjects had production of at least 50 mg of underarm sweat over 5 minutes, and scores of four or higher on the 11-point Axillary Sweating Daily Diary (ASDD) or the Children’s ASDD (ASDD-C) – an instrument developed by the company in consultation with the FDA – and scores of three or four on the four-grade Hyperhidrosis Disease Severity Scale (HDSS). In total, 463 patients were randomized to receive glycopyrronium and 234 to vehicle. Forty four of these patients were aged 9-16 years, with 25 receiving glycopyrronium and 19 receiving vehicle.
According to the February release, the ASDD/ASDD-C severity scale responder rates were about 60% in the pediatric and adult groups treated with glycopyrronium, compared with 13.0% and 28.8% for children and adults, respectively, in the vehicle group. At week 4, the median absolute change in sweat production was a reduction of 64.2 mg in children and a reduction of 80.6 mg among adults treated with glycopyrronium, compared with reductions of 53.7 mg and 62 mg among those in the vehicle group, respectively.
In the glycopyrronium-treated group, almost 80% of the pediatric patients and 74.3% of the adults experienced at least a 50% reduction in sweat production at week 4, compared with almost 55% and 53%, respectively, in the vehicle group.
Among pediatric patients, the mean decrease from baseline in the Children’s Dermatology Quality of Life Index was 8.1 in glycopyrronium-treated patients, compared with 1.9 in the vehicle group. In adults, scores on the Dermatology Life Quality Index measure were reduced by 8.4 and 4.7 in glycopyrronium- and vehicle-treated adult patients, respectively.
Nearly 57% of adults and 44% of pediatric patients treated with glycopyrronium experienced treatment-emergent adverse events, compared with 34.3% of adults and 10.5% of the pediatric patients in the vehicle group. The majority were related to anticholinergic activity and were mild, and rarely led to drug discontinuation, according to the company.
The press release announcing the approval stated that the most common adverse effects observed after application of glycopyrronium were dry mouth, mydriasis, sore throat, headache, urinary hesitation, blurred vision, dry nose, dry throat, dry eye, dry skin and constipation. Erythema, burning/stinging, and pruritus were the most common skin reactions.
The product is contraindicated in patients with glaucoma, paralytic ileus, and other medical conditions that can be exacerbated by its anticholinergic effects, according to the prescribing information. Patients should be advised that they should wash their hands thoroughly after application, and that it can cause temporary dilation of the pupils and blurred vision if glycopyrronium comes into contact with their eyes.
The in adults and children aged 9 years and older.
Glycopyrronium, which is formulated in a cloth wipe, will be marketed as Qbrexza and is expected to be available in October, according to the approval announcement June 29, which was made by Dermira, the manufacturer. The instructions for use section in the prescribing information states that patients are advised to use one cloth to apply the medication to both axillae, wiping the cloth across each underarm. Each cloth, intended for single use, is pre-moistened with 2.4% glycopyrronium solution.
Glycopyrronium blocks sweat production “by inhibiting the interaction between acetylcholine and the cholinergic receptors responsible for sweat gland activation,” the company said in a February press release.
The approval was based on the results of two phase 3 clinical studies, ATMOS-1 and ATMOS-2, which were multicenter, randomized, double-blind, vehicle-controlled, 4-week studies in patients 9 years of age or older with primary axillary hyperhidrosis for 6 months or longer. Study subjects had production of at least 50 mg of underarm sweat over 5 minutes, and scores of four or higher on the 11-point Axillary Sweating Daily Diary (ASDD) or the Children’s ASDD (ASDD-C) – an instrument developed by the company in consultation with the FDA – and scores of three or four on the four-grade Hyperhidrosis Disease Severity Scale (HDSS). In total, 463 patients were randomized to receive glycopyrronium and 234 to vehicle. Forty four of these patients were aged 9-16 years, with 25 receiving glycopyrronium and 19 receiving vehicle.
According to the February release, the ASDD/ASDD-C severity scale responder rates were about 60% in the pediatric and adult groups treated with glycopyrronium, compared with 13.0% and 28.8% for children and adults, respectively, in the vehicle group. At week 4, the median absolute change in sweat production was a reduction of 64.2 mg in children and a reduction of 80.6 mg among adults treated with glycopyrronium, compared with reductions of 53.7 mg and 62 mg among those in the vehicle group, respectively.
In the glycopyrronium-treated group, almost 80% of the pediatric patients and 74.3% of the adults experienced at least a 50% reduction in sweat production at week 4, compared with almost 55% and 53%, respectively, in the vehicle group.
Among pediatric patients, the mean decrease from baseline in the Children’s Dermatology Quality of Life Index was 8.1 in glycopyrronium-treated patients, compared with 1.9 in the vehicle group. In adults, scores on the Dermatology Life Quality Index measure were reduced by 8.4 and 4.7 in glycopyrronium- and vehicle-treated adult patients, respectively.
Nearly 57% of adults and 44% of pediatric patients treated with glycopyrronium experienced treatment-emergent adverse events, compared with 34.3% of adults and 10.5% of the pediatric patients in the vehicle group. The majority were related to anticholinergic activity and were mild, and rarely led to drug discontinuation, according to the company.
The press release announcing the approval stated that the most common adverse effects observed after application of glycopyrronium were dry mouth, mydriasis, sore throat, headache, urinary hesitation, blurred vision, dry nose, dry throat, dry eye, dry skin and constipation. Erythema, burning/stinging, and pruritus were the most common skin reactions.
The product is contraindicated in patients with glaucoma, paralytic ileus, and other medical conditions that can be exacerbated by its anticholinergic effects, according to the prescribing information. Patients should be advised that they should wash their hands thoroughly after application, and that it can cause temporary dilation of the pupils and blurred vision if glycopyrronium comes into contact with their eyes.
Cost led to missed care for 4.5% of Americans in 2017
The percentage of Americans who went without medical care due to cost rose to 4.5% in 2017, reversing a 6-year trend, the National Center for Health Statistics reported.
The rate was 4.4% in 2016, which represented a slowdown in what had been steady decline over the previous 5 years, according to data from the National Health Interview Survey. Declining rates corresponded with the implementation of early provisions of the Affordable Care Act in 2010.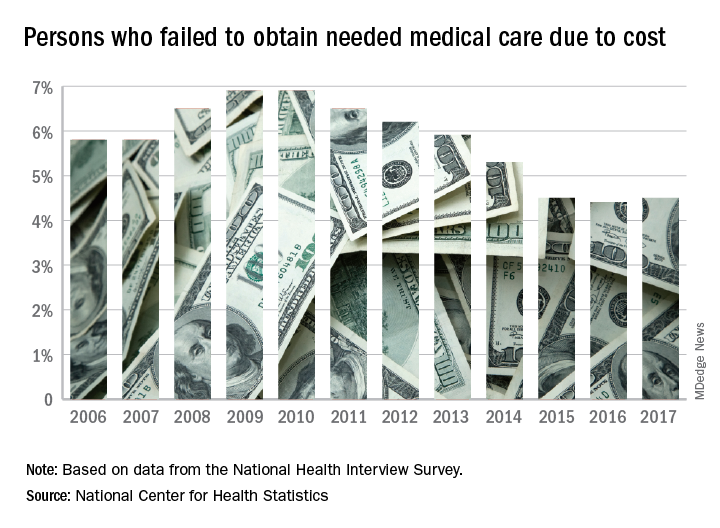
The 2017 rate varied considerably by age group. Not surprisingly, more working-age people – those aged 18-64 years – reported that they did not seek medical care at some point in the previous 12 months due to cost (6.1%). The rate was 1.2% for those under 18 years and 2.7% for the Medicare eligible – those aged 65 years and older.
For 2016, the rates were 6.2% for those aged 18-64 years, 1.2% for the under-18 group, and 2.1% for the 65+ group, the data show.
Among females of all ages in 2017, 4.8% failed to get needed care at some point in the previous year, compared with 4.1% of men. Those numbers were unchanged from 2016 but down from 4.9% for females in 2015 and up from 4.0% for males that year, the NCHS said.
In 2017, the rate also varied by race/ethnicity – 4.1% for whites, 5.3% for Hispanics, 6.1% for blacks – and by location – 4.1% for large metropolitan areas, 4.9% for small metro areas, and 5.5% for rural locales, according to the early release of survey data.
The percentage of Americans who went without medical care due to cost rose to 4.5% in 2017, reversing a 6-year trend, the National Center for Health Statistics reported.
The rate was 4.4% in 2016, which represented a slowdown in what had been steady decline over the previous 5 years, according to data from the National Health Interview Survey. Declining rates corresponded with the implementation of early provisions of the Affordable Care Act in 2010.
The 2017 rate varied considerably by age group. Not surprisingly, more working-age people – those aged 18-64 years – reported that they did not seek medical care at some point in the previous 12 months due to cost (6.1%). The rate was 1.2% for those under 18 years and 2.7% for the Medicare eligible – those aged 65 years and older.
For 2016, the rates were 6.2% for those aged 18-64 years, 1.2% for the under-18 group, and 2.1% for the 65+ group, the data show.
Among females of all ages in 2017, 4.8% failed to get needed care at some point in the previous year, compared with 4.1% of men. Those numbers were unchanged from 2016 but down from 4.9% for females in 2015 and up from 4.0% for males that year, the NCHS said.
In 2017, the rate also varied by race/ethnicity – 4.1% for whites, 5.3% for Hispanics, 6.1% for blacks – and by location – 4.1% for large metropolitan areas, 4.9% for small metro areas, and 5.5% for rural locales, according to the early release of survey data.
The percentage of Americans who went without medical care due to cost rose to 4.5% in 2017, reversing a 6-year trend, the National Center for Health Statistics reported.
The rate was 4.4% in 2016, which represented a slowdown in what had been steady decline over the previous 5 years, according to data from the National Health Interview Survey. Declining rates corresponded with the implementation of early provisions of the Affordable Care Act in 2010.
The 2017 rate varied considerably by age group. Not surprisingly, more working-age people – those aged 18-64 years – reported that they did not seek medical care at some point in the previous 12 months due to cost (6.1%). The rate was 1.2% for those under 18 years and 2.7% for the Medicare eligible – those aged 65 years and older.
For 2016, the rates were 6.2% for those aged 18-64 years, 1.2% for the under-18 group, and 2.1% for the 65+ group, the data show.
Among females of all ages in 2017, 4.8% failed to get needed care at some point in the previous year, compared with 4.1% of men. Those numbers were unchanged from 2016 but down from 4.9% for females in 2015 and up from 4.0% for males that year, the NCHS said.
In 2017, the rate also varied by race/ethnicity – 4.1% for whites, 5.3% for Hispanics, 6.1% for blacks – and by location – 4.1% for large metropolitan areas, 4.9% for small metro areas, and 5.5% for rural locales, according to the early release of survey data.
FDA approves encorafenib/binimetinib for advanced melanoma with BRAF mutations
The Food and Drug Administration has approved combination therapy of encorafenib (Braftovi) and binimetinib (Mektovi) for the treatment of unresectable or metastatic melanoma with BRAF V600E or BRAF V600K mutations; the FDA also has approved the THxID BRAF Kit as a companion diagnostic for this combination therapy.

The approval was based on results from the randomized, active-controlled, open-label, multicenter COLUMBUS trial, which included 517 patients. Progression-free survival, according to RECIST 1.1 criteria, was the major efficacy measure; the median progression-free survival was 14.9 months in the encorafenib/binimetinib combination arm versus 7.3 months in the vemurafenib (Zelboraf) monotherapy arm (hazard ratio, 0.54; 95% confidence interval, 0.41-0.71; P less than .0001).
Fatigue, nausea, diarrhea, vomiting, abdominal pain, and arthralgia were the most common adverse reactions. Discontinuation of therapy from adverse reactions occurred in 5% of patients receiving the combination, the FDA said in a press statement.
The full prescribing information for encorafenib and binimetinib can be found on the FDA website.
The Food and Drug Administration has approved combination therapy of encorafenib (Braftovi) and binimetinib (Mektovi) for the treatment of unresectable or metastatic melanoma with BRAF V600E or BRAF V600K mutations; the FDA also has approved the THxID BRAF Kit as a companion diagnostic for this combination therapy.
The approval was based on results from the randomized, active-controlled, open-label, multicenter COLUMBUS trial, which included 517 patients. Progression-free survival, according to RECIST 1.1 criteria, was the major efficacy measure; the median progression-free survival was 14.9 months in the encorafenib/binimetinib combination arm versus 7.3 months in the vemurafenib (Zelboraf) monotherapy arm (hazard ratio, 0.54; 95% confidence interval, 0.41-0.71; P less than .0001).
Fatigue, nausea, diarrhea, vomiting, abdominal pain, and arthralgia were the most common adverse reactions. Discontinuation of therapy from adverse reactions occurred in 5% of patients receiving the combination, the FDA said in a press statement.
The full prescribing information for encorafenib and binimetinib can be found on the FDA website.
The Food and Drug Administration has approved combination therapy of encorafenib (Braftovi) and binimetinib (Mektovi) for the treatment of unresectable or metastatic melanoma with BRAF V600E or BRAF V600K mutations; the FDA also has approved the THxID BRAF Kit as a companion diagnostic for this combination therapy.
The approval was based on results from the randomized, active-controlled, open-label, multicenter COLUMBUS trial, which included 517 patients. Progression-free survival, according to RECIST 1.1 criteria, was the major efficacy measure; the median progression-free survival was 14.9 months in the encorafenib/binimetinib combination arm versus 7.3 months in the vemurafenib (Zelboraf) monotherapy arm (hazard ratio, 0.54; 95% confidence interval, 0.41-0.71; P less than .0001).
Fatigue, nausea, diarrhea, vomiting, abdominal pain, and arthralgia were the most common adverse reactions. Discontinuation of therapy from adverse reactions occurred in 5% of patients receiving the combination, the FDA said in a press statement.
The full prescribing information for encorafenib and binimetinib can be found on the FDA website.
Combo treatment under review for Waldenstrom macroglobulinemia
by the Food and Drug Administration.
Ibrutinib, a Bruton’s tyrosine kinase inhibitor, is already approved as a single agent for WM. The addition of rituximab to the indication is based on positive results from the phase 3 INNOVATE study. In particular, the trial showed a superior progression-free survival rate at 30 months for the ibrutinib-rituximab combination at 82%, compared with placebo plus rituximab at 28% (N Engl J Med. 2018;378:2399-410).
The study’s lead investigator, Meletios A. Dimopoulos, MD, called the combination a “new standard of care” for WM at the recent annual meeting of the American Society of Clinical Oncology.
Ibrutinib, marketed as Imbruvica, is jointly developed and commercialized by Pharmacyclics and Janssen Biotech.
by the Food and Drug Administration.
Ibrutinib, a Bruton’s tyrosine kinase inhibitor, is already approved as a single agent for WM. The addition of rituximab to the indication is based on positive results from the phase 3 INNOVATE study. In particular, the trial showed a superior progression-free survival rate at 30 months for the ibrutinib-rituximab combination at 82%, compared with placebo plus rituximab at 28% (N Engl J Med. 2018;378:2399-410).
The study’s lead investigator, Meletios A. Dimopoulos, MD, called the combination a “new standard of care” for WM at the recent annual meeting of the American Society of Clinical Oncology.
Ibrutinib, marketed as Imbruvica, is jointly developed and commercialized by Pharmacyclics and Janssen Biotech.
by the Food and Drug Administration.
Ibrutinib, a Bruton’s tyrosine kinase inhibitor, is already approved as a single agent for WM. The addition of rituximab to the indication is based on positive results from the phase 3 INNOVATE study. In particular, the trial showed a superior progression-free survival rate at 30 months for the ibrutinib-rituximab combination at 82%, compared with placebo plus rituximab at 28% (N Engl J Med. 2018;378:2399-410).
The study’s lead investigator, Meletios A. Dimopoulos, MD, called the combination a “new standard of care” for WM at the recent annual meeting of the American Society of Clinical Oncology.
Ibrutinib, marketed as Imbruvica, is jointly developed and commercialized by Pharmacyclics and Janssen Biotech.
FDA approves Epidiolex for Lennox-Gastaut syndrome and Dravet syndrome
The Food and Drug Administration has approved cannabidiol oral solution (Epidiolex, GW Pharmaceuticals) for the treatment of two rare pediatric seizure disorders.
“This product approval demonstrates that advancing sound scientific research to investigate ingredients derived from marijuana can lead to important therapies. This new treatment provides new options for patients,” said FDA Commissioner Scott Gottlieb, MD, in a statement.
However, he cautioned, “This is an important medical advance. But it’s also important to note that this is not an approval of marijuana or all of its components. This is the approval of one specific CBD medication for a specific use. And it was based on well-controlled clinical trials evaluating the use of this compound in the treatment of a specific condition.”
The FDA Peripheral and Central Nervous System Drugs Advisory Committee’s earlier positive recommendation was based on three randomized, double-blind, placebo-controlled clinical trials. These trials showed a 50% reduction of drop seizure frequency in 40%-44% of patients with Lennox-Gastaut syndrome, and a 39% decrease in convulsive seizure frequency for trial participants with Dravet Syndrome. A total of 516 patients with one of the two seizure disorders participated in the clinical trials.
“In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition,” said Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, in a statement.
After reviewing information provided by the drug’s sponsor and the FDA, the advisory committee judged that CBD-OS, derived from a non-psychoactive chemical found in marijuana, was very unlikely to have potential for abuse.
Sedation, sleepiness, and lethargy were among the most frequently reported adverse events for the patients taking CBD-OS. In data pooled from the clinical trials, 16.3% of patients taking CBD-OS at the higher dose of 20 mg/kg/day had liver transaminase elevations above three times the upper limit of normal; this level of transaminase elevation was seen in 0.9% of patients taking placebo.
A patient medication guide detailing risks and how the drug should be used will accompany CBD-OS when it is dispensed, according to the FDA approval.
In his statement, Dr. Gottlieb put the approval in the context of the FDA’s broader efforts to encourage a strong clinical development program for marijuana-derived drugs that does not compromise standards for ensuring safety and efficacy of drugs approved by the agency. He also noted that ongoing efforts to support high quality research into marijuana-based therapies involve other federal agencies, including the National Institute on Drug Abuse and the Drug Enforcement Administration.
The FDA’s actions against companies distributing unapproved products that contain cannabidiol and making unproven marketing claims will continue, said Dr. Gottlieb. Still, “Today’s approval demonstrates our commitment to the scientific process and working with product developers to bring marijuana-based products to market,” he said.
The Food and Drug Administration has approved cannabidiol oral solution (Epidiolex, GW Pharmaceuticals) for the treatment of two rare pediatric seizure disorders.
“This product approval demonstrates that advancing sound scientific research to investigate ingredients derived from marijuana can lead to important therapies. This new treatment provides new options for patients,” said FDA Commissioner Scott Gottlieb, MD, in a statement.
However, he cautioned, “This is an important medical advance. But it’s also important to note that this is not an approval of marijuana or all of its components. This is the approval of one specific CBD medication for a specific use. And it was based on well-controlled clinical trials evaluating the use of this compound in the treatment of a specific condition.”
The FDA Peripheral and Central Nervous System Drugs Advisory Committee’s earlier positive recommendation was based on three randomized, double-blind, placebo-controlled clinical trials. These trials showed a 50% reduction of drop seizure frequency in 40%-44% of patients with Lennox-Gastaut syndrome, and a 39% decrease in convulsive seizure frequency for trial participants with Dravet Syndrome. A total of 516 patients with one of the two seizure disorders participated in the clinical trials.
“In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition,” said Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, in a statement.
After reviewing information provided by the drug’s sponsor and the FDA, the advisory committee judged that CBD-OS, derived from a non-psychoactive chemical found in marijuana, was very unlikely to have potential for abuse.
Sedation, sleepiness, and lethargy were among the most frequently reported adverse events for the patients taking CBD-OS. In data pooled from the clinical trials, 16.3% of patients taking CBD-OS at the higher dose of 20 mg/kg/day had liver transaminase elevations above three times the upper limit of normal; this level of transaminase elevation was seen in 0.9% of patients taking placebo.
A patient medication guide detailing risks and how the drug should be used will accompany CBD-OS when it is dispensed, according to the FDA approval.
In his statement, Dr. Gottlieb put the approval in the context of the FDA’s broader efforts to encourage a strong clinical development program for marijuana-derived drugs that does not compromise standards for ensuring safety and efficacy of drugs approved by the agency. He also noted that ongoing efforts to support high quality research into marijuana-based therapies involve other federal agencies, including the National Institute on Drug Abuse and the Drug Enforcement Administration.
The FDA’s actions against companies distributing unapproved products that contain cannabidiol and making unproven marketing claims will continue, said Dr. Gottlieb. Still, “Today’s approval demonstrates our commitment to the scientific process and working with product developers to bring marijuana-based products to market,” he said.
The Food and Drug Administration has approved cannabidiol oral solution (Epidiolex, GW Pharmaceuticals) for the treatment of two rare pediatric seizure disorders.
“This product approval demonstrates that advancing sound scientific research to investigate ingredients derived from marijuana can lead to important therapies. This new treatment provides new options for patients,” said FDA Commissioner Scott Gottlieb, MD, in a statement.
However, he cautioned, “This is an important medical advance. But it’s also important to note that this is not an approval of marijuana or all of its components. This is the approval of one specific CBD medication for a specific use. And it was based on well-controlled clinical trials evaluating the use of this compound in the treatment of a specific condition.”
The FDA Peripheral and Central Nervous System Drugs Advisory Committee’s earlier positive recommendation was based on three randomized, double-blind, placebo-controlled clinical trials. These trials showed a 50% reduction of drop seizure frequency in 40%-44% of patients with Lennox-Gastaut syndrome, and a 39% decrease in convulsive seizure frequency for trial participants with Dravet Syndrome. A total of 516 patients with one of the two seizure disorders participated in the clinical trials.
“In addition to another important treatment option for Lennox-Gastaut patients, this first-ever approval of a drug specifically for Dravet patients will provide a significant and needed improvement in the therapeutic approach to caring for people with this condition,” said Billy Dunn, MD, director of the Division of Neurology Products in the FDA Center for Drug Evaluation and Research, in a statement.
After reviewing information provided by the drug’s sponsor and the FDA, the advisory committee judged that CBD-OS, derived from a non-psychoactive chemical found in marijuana, was very unlikely to have potential for abuse.
Sedation, sleepiness, and lethargy were among the most frequently reported adverse events for the patients taking CBD-OS. In data pooled from the clinical trials, 16.3% of patients taking CBD-OS at the higher dose of 20 mg/kg/day had liver transaminase elevations above three times the upper limit of normal; this level of transaminase elevation was seen in 0.9% of patients taking placebo.
A patient medication guide detailing risks and how the drug should be used will accompany CBD-OS when it is dispensed, according to the FDA approval.
In his statement, Dr. Gottlieb put the approval in the context of the FDA’s broader efforts to encourage a strong clinical development program for marijuana-derived drugs that does not compromise standards for ensuring safety and efficacy of drugs approved by the agency. He also noted that ongoing efforts to support high quality research into marijuana-based therapies involve other federal agencies, including the National Institute on Drug Abuse and the Drug Enforcement Administration.
The FDA’s actions against companies distributing unapproved products that contain cannabidiol and making unproven marketing claims will continue, said Dr. Gottlieb. Still, “Today’s approval demonstrates our commitment to the scientific process and working with product developers to bring marijuana-based products to market,” he said.
Obesity didn’t just happen overnight
Is it possible to get more exercise and still gain weight? In America it is.
The steady increase in obesity prevalence among adults in the United States has been exceeded over the last decade by the percentage of adults who are getting the recommended amount of exercise, according to the National Center for Health Statistics.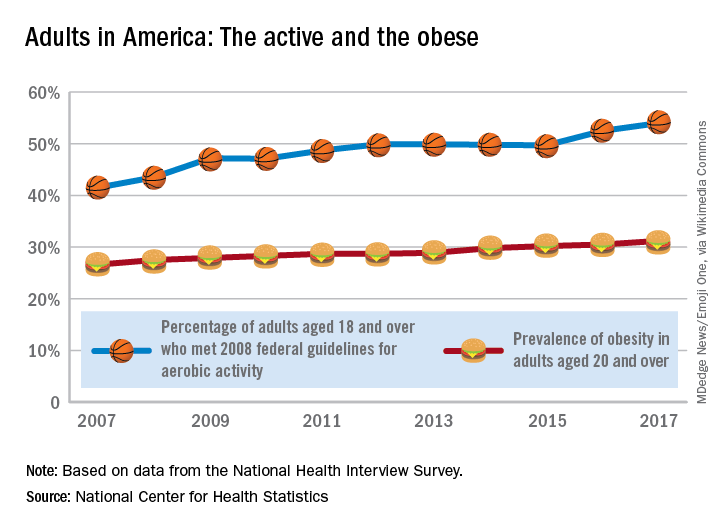
The 2008 guideline, “Physical Activity Guidelines for Americans” recommends that “adults perform at least 150 minutes a week of moderate-intensity aerobic physical activity, 75 minutes a week of vigorous-intensity aerobic physical activity, or an equivalent combination of moderate- and vigorous-intensity aerobic activity, performed in episodes of at least 10 minutes and preferably should be spread throughout the week,” the NCHS noted.
Is it possible to get more exercise and still gain weight? In America it is.
The steady increase in obesity prevalence among adults in the United States has been exceeded over the last decade by the percentage of adults who are getting the recommended amount of exercise, according to the National Center for Health Statistics.
The 2008 guideline, “Physical Activity Guidelines for Americans” recommends that “adults perform at least 150 minutes a week of moderate-intensity aerobic physical activity, 75 minutes a week of vigorous-intensity aerobic physical activity, or an equivalent combination of moderate- and vigorous-intensity aerobic activity, performed in episodes of at least 10 minutes and preferably should be spread throughout the week,” the NCHS noted.
Is it possible to get more exercise and still gain weight? In America it is.
The steady increase in obesity prevalence among adults in the United States has been exceeded over the last decade by the percentage of adults who are getting the recommended amount of exercise, according to the National Center for Health Statistics.
The 2008 guideline, “Physical Activity Guidelines for Americans” recommends that “adults perform at least 150 minutes a week of moderate-intensity aerobic physical activity, 75 minutes a week of vigorous-intensity aerobic physical activity, or an equivalent combination of moderate- and vigorous-intensity aerobic activity, performed in episodes of at least 10 minutes and preferably should be spread throughout the week,” the NCHS noted.
FDA: MiniMed 670G now available for younger diabetes patients
The MiniMed 670G hybrid closed loop system has been approved to help manage basal insulin levels in patients aged 7-13 years who have type 1 diabetes, according to a Food and Drug Administration announcement.
The system, manufactured by Medtronic, automatically measures insulin levels every 5 minutes using an included sensor and then delivers insulin as needed through its insulin pump and attached infusion patch.
As part of this approval for children aged 7-13 years, the FDA is requiring the product developer to perform a postmarket study to evaluate how the device performs in this age group in real-world settings.
“Caregivers and families of young patients with diabetes face unique challenges in managing this disease, in particular the round-the-clock glucose monitoring that can be disruptive to people’s lives,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The device was approved in September 2017 for use in patients aged 14 years and older.
Read more about this approval in the full FDA announcement.
The MiniMed 670G hybrid closed loop system has been approved to help manage basal insulin levels in patients aged 7-13 years who have type 1 diabetes, according to a Food and Drug Administration announcement.
The system, manufactured by Medtronic, automatically measures insulin levels every 5 minutes using an included sensor and then delivers insulin as needed through its insulin pump and attached infusion patch.
As part of this approval for children aged 7-13 years, the FDA is requiring the product developer to perform a postmarket study to evaluate how the device performs in this age group in real-world settings.
“Caregivers and families of young patients with diabetes face unique challenges in managing this disease, in particular the round-the-clock glucose monitoring that can be disruptive to people’s lives,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The device was approved in September 2017 for use in patients aged 14 years and older.
Read more about this approval in the full FDA announcement.
The MiniMed 670G hybrid closed loop system has been approved to help manage basal insulin levels in patients aged 7-13 years who have type 1 diabetes, according to a Food and Drug Administration announcement.
The system, manufactured by Medtronic, automatically measures insulin levels every 5 minutes using an included sensor and then delivers insulin as needed through its insulin pump and attached infusion patch.
As part of this approval for children aged 7-13 years, the FDA is requiring the product developer to perform a postmarket study to evaluate how the device performs in this age group in real-world settings.
“Caregivers and families of young patients with diabetes face unique challenges in managing this disease, in particular the round-the-clock glucose monitoring that can be disruptive to people’s lives,” FDA Commissioner Scott Gottlieb, MD, said in a statement.
The device was approved in September 2017 for use in patients aged 14 years and older.
Read more about this approval in the full FDA announcement.
FDA okays fully implantable continuous glucose monitor/mobile app combo for diabetes
to transmit continuous information about blood glucose levels for people with diabetes.
The sensor-mobile app combo, called the Eversense Continuous Glucose Monitoring (CGM) system, is designed to supplant the need for frequent blood sampling to monitor blood glucose levels.
“The FDA is committed to advancing novel products that leverage digital technology to improve patient care,” said FDA commissioner Scott Gottlieb, MD, in the agency’s press release announcing the approval. The sensor, which is roughly 1.5 cm long, is coated with a material that fluoresces when exposed to glucose; the sensor uses the amount of light emitted to calculate blood glucose levels. Patients use an adhesive patch, changed daily, to attach a “smart” transmitter that overlies the area where the sensor is implanted. This rechargeable transmitter sends blood glucose levels to the mobile app every 5 minutes, and also powers the sensor.
The FDA’s approval was based on data from 125 patients with type 1 and type 2 diabetes who used the CGM system. The bulk of clinical data was acquired from PRECISE II, which enrolled 90 patients with type 1 and type 2 diabetes. When compared with levels returned from concurrently performed conventional home glucose monitoring, the CGM system achieved a mean absolute relative difference (MARD) of 8.8% (95% confidence interval, 8.1%-9.3%). This was less than the prespecified accuracy goal of 20% MARD (P less than .0001).
During the nonrandomized, blinded, prospective PRECISE II trial, 91% of the implanted sensors were functioning through the end of 90 days. A variation of the Eversense CGM, the Eversense CGM XL, has been approved for use up to 180 days in Europe.
The overall rate of serious adverse events among patients participating in the Eversense CGM trials was less than 1%. “The safety of this novel system will also be evaluated in a post-approval study,” wrote FDA officials in the press release.
In addition to adverse effects related to the outpatient procedure in which the glucose sensor is implanted subcutaneously, the FDA said that allergic reactions, ongoing pain, discomfort, scarring, and skin changes are possible with use of the CGM. Though the system sends frequent blood glucose measurements to the accompanying mobile app, missed alerts might still result in hypo- or hyperglycemia.
The Eversense CGM is marketed by Senseonics, which funded the studies underpinning approval.
to transmit continuous information about blood glucose levels for people with diabetes.
The sensor-mobile app combo, called the Eversense Continuous Glucose Monitoring (CGM) system, is designed to supplant the need for frequent blood sampling to monitor blood glucose levels.
“The FDA is committed to advancing novel products that leverage digital technology to improve patient care,” said FDA commissioner Scott Gottlieb, MD, in the agency’s press release announcing the approval. The sensor, which is roughly 1.5 cm long, is coated with a material that fluoresces when exposed to glucose; the sensor uses the amount of light emitted to calculate blood glucose levels. Patients use an adhesive patch, changed daily, to attach a “smart” transmitter that overlies the area where the sensor is implanted. This rechargeable transmitter sends blood glucose levels to the mobile app every 5 minutes, and also powers the sensor.
The FDA’s approval was based on data from 125 patients with type 1 and type 2 diabetes who used the CGM system. The bulk of clinical data was acquired from PRECISE II, which enrolled 90 patients with type 1 and type 2 diabetes. When compared with levels returned from concurrently performed conventional home glucose monitoring, the CGM system achieved a mean absolute relative difference (MARD) of 8.8% (95% confidence interval, 8.1%-9.3%). This was less than the prespecified accuracy goal of 20% MARD (P less than .0001).
During the nonrandomized, blinded, prospective PRECISE II trial, 91% of the implanted sensors were functioning through the end of 90 days. A variation of the Eversense CGM, the Eversense CGM XL, has been approved for use up to 180 days in Europe.
The overall rate of serious adverse events among patients participating in the Eversense CGM trials was less than 1%. “The safety of this novel system will also be evaluated in a post-approval study,” wrote FDA officials in the press release.
In addition to adverse effects related to the outpatient procedure in which the glucose sensor is implanted subcutaneously, the FDA said that allergic reactions, ongoing pain, discomfort, scarring, and skin changes are possible with use of the CGM. Though the system sends frequent blood glucose measurements to the accompanying mobile app, missed alerts might still result in hypo- or hyperglycemia.
The Eversense CGM is marketed by Senseonics, which funded the studies underpinning approval.
to transmit continuous information about blood glucose levels for people with diabetes.
The sensor-mobile app combo, called the Eversense Continuous Glucose Monitoring (CGM) system, is designed to supplant the need for frequent blood sampling to monitor blood glucose levels.
“The FDA is committed to advancing novel products that leverage digital technology to improve patient care,” said FDA commissioner Scott Gottlieb, MD, in the agency’s press release announcing the approval. The sensor, which is roughly 1.5 cm long, is coated with a material that fluoresces when exposed to glucose; the sensor uses the amount of light emitted to calculate blood glucose levels. Patients use an adhesive patch, changed daily, to attach a “smart” transmitter that overlies the area where the sensor is implanted. This rechargeable transmitter sends blood glucose levels to the mobile app every 5 minutes, and also powers the sensor.
The FDA’s approval was based on data from 125 patients with type 1 and type 2 diabetes who used the CGM system. The bulk of clinical data was acquired from PRECISE II, which enrolled 90 patients with type 1 and type 2 diabetes. When compared with levels returned from concurrently performed conventional home glucose monitoring, the CGM system achieved a mean absolute relative difference (MARD) of 8.8% (95% confidence interval, 8.1%-9.3%). This was less than the prespecified accuracy goal of 20% MARD (P less than .0001).
During the nonrandomized, blinded, prospective PRECISE II trial, 91% of the implanted sensors were functioning through the end of 90 days. A variation of the Eversense CGM, the Eversense CGM XL, has been approved for use up to 180 days in Europe.
The overall rate of serious adverse events among patients participating in the Eversense CGM trials was less than 1%. “The safety of this novel system will also be evaluated in a post-approval study,” wrote FDA officials in the press release.
In addition to adverse effects related to the outpatient procedure in which the glucose sensor is implanted subcutaneously, the FDA said that allergic reactions, ongoing pain, discomfort, scarring, and skin changes are possible with use of the CGM. Though the system sends frequent blood glucose measurements to the accompanying mobile app, missed alerts might still result in hypo- or hyperglycemia.
The Eversense CGM is marketed by Senseonics, which funded the studies underpinning approval.