User login
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series
Prostate cancer is the most common noncutaneous cancer in men, accounting for 29% of all incident cancer cases.1 Typically, prostate cancer metastasizes to bone and regional lymph nodes.2 However, intrathoracic manifestation may occur. This report presents 3 cases of rare intrathoracic manifestations of metastatic prostate cancer with a review of the current literature.
CASE PRESENTATIONS
Case 1
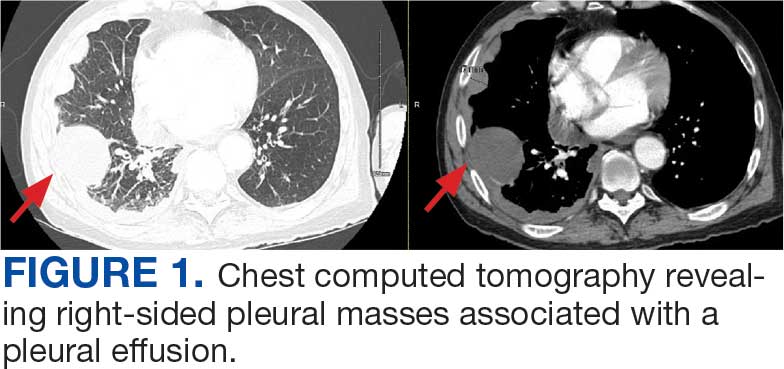
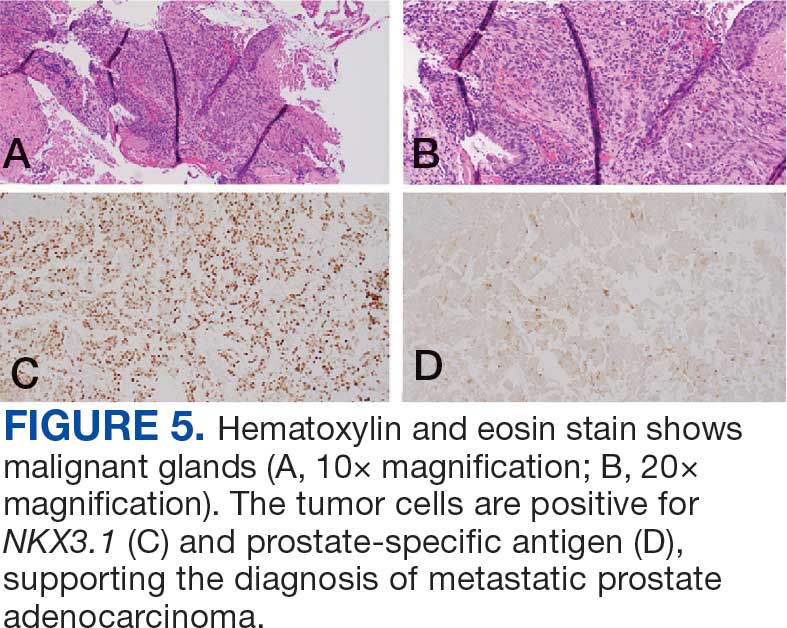
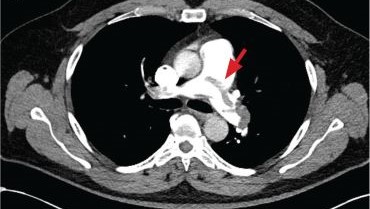
A 71-year-old male who was an active smoker and a long-standing employment as a plumber was diagnosed with rectal cancer in 2022. He completed neoadjuvant capecitabine and radiation therapy followed by a rectosigmoidectomy. Several weeks after surgery, the patient presented to the emergency department (ED) with a dry cough and worsening shortness of breath. Point-of-care ultrasound of the lungs revealed a moderate right pleural effusion with several nodular pleural masses. A chest computed tomography (CT) confirmed these findings (Figure 1). A CT of the abdomen and pelvis revealed prostatomegaly with the medial lobe of the prostate protruding into the bladder; however, no enlarged retroperitoneal, mesenteric or pelvic lymph nodes were noted. The patient underwent a right pleural fluid drainage and pleural mass biopsy. Pleural mass histomorphology as well as immunohistochemical (IHC) stains were consistent with metastatic prostate adenocarcinoma. The pleural fluid cytology also was consistent with metastatic prostate adenocarcinoma.
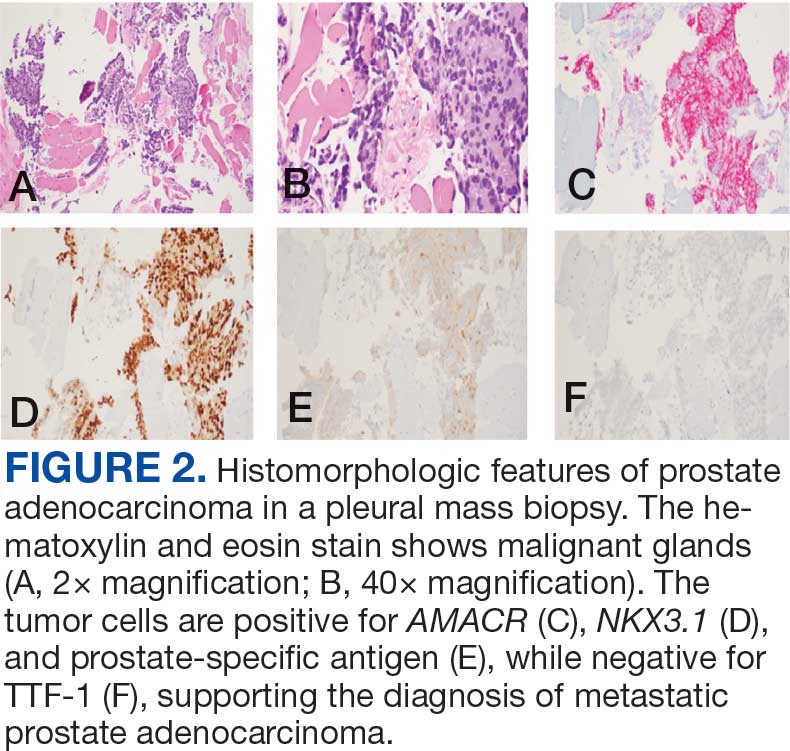
Immunohistochemistry showed weak positive staining for prostate-specific NK3 homeobox 1 gene (NKX3.1), alpha-methylacyl-CoA racemase gene (AMACR), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), keratin-20, and caudal type homeobox 2 gene (CDX2) (Figure 2) 2). The patient's prostate-specific antigen (PSA) was found to be elevated at 33.9 ng/mL (reference range, < 4 ng/mL).
Case 2
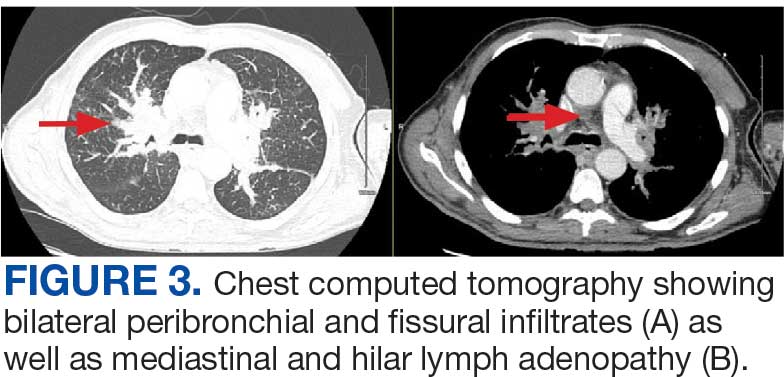
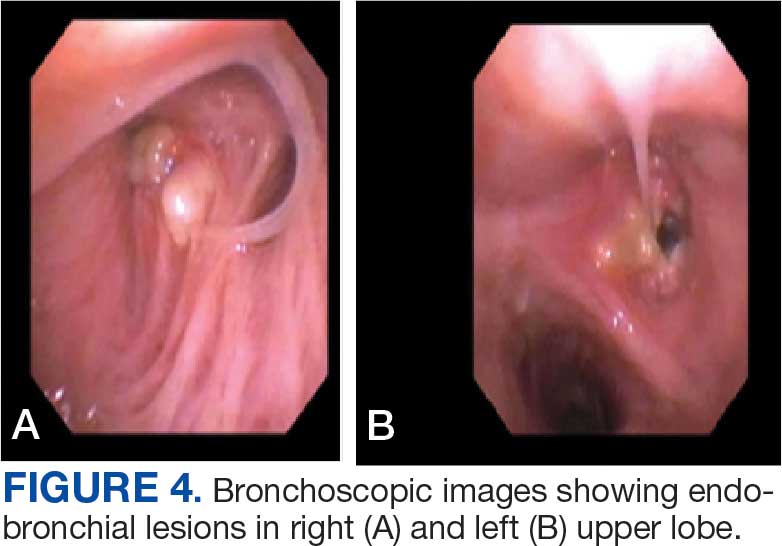
A 71-year-old male with a history of alcohol use disorder and a 30-year smoking history presented to the ED with worsening dyspnea on exertion. The patient’s baseline exercise tolerance decreased to walking for only 1 block. He reported unintentional weight loss of about 30 pounds over the prior year, no recent respiratory infections, no prior breathing problems, and no personal or family history of cancer. Chest CT revealed findings of bilateral peribronchial opacities as well as mediastinal and hilar lymphadenopathy (Figure 3). The patient developed hypoxic respiratory failure necessitating intubation, mechanical ventilation, and management in the medical intensive care unit, where he was treated for postobstructive pneumonia. Fiberoptic bronchoscopy revealed endobronchial lesions in the right and left upper lobe that were partially obstructing the airway (Figure 4).
The endobronchial masses were debulked using forceps, and samples were sent for surgical pathology evaluation. Staging was completed using linear endobronchial ultrasound, which revealed an enlarged subcarinal lymph node (S7). The surgical pathology of the endobronchial mass and the subcarinal lymph node cytology were consistent with metastatic adenocarcinoma of the prostate. The tumor cells were positive for AE1/AE3, PSA, and NKX3.1, but were negative for CK7 and TTF-1 (Figure 5). Further imaging revealed an enlarged heterogeneous prostate gland, prominent pelvic nodes, and left retroperitoneal lymphadenopathy, as well as sclerotic foci within the T10 vertebral body and right inferior pubic ramus. PSA was also found to be significantly elevated at 700 ng/mL.
Case 3
An 80-year-old male veteran with a history of prostate cancer and recently diagnosed T2N1M0 head and neck squamous cell carcinoma was referred to the Pulmonary service for evaluation of a pulmonary nodule. His medical history was notable for prostate cancer diagnosed 12 years earlier, with an unknown Gleason score. Initial treatment included prostatectomy followed by whole pelvic radiation therapy a year after, due to elevated PSA in surveillance monitoring. This treatment led to remission. After establishing remission for > 10 years, the patient was started on low-dose testosterone replacement therapy to address complications of radiation therapy, namely hypogonadism.
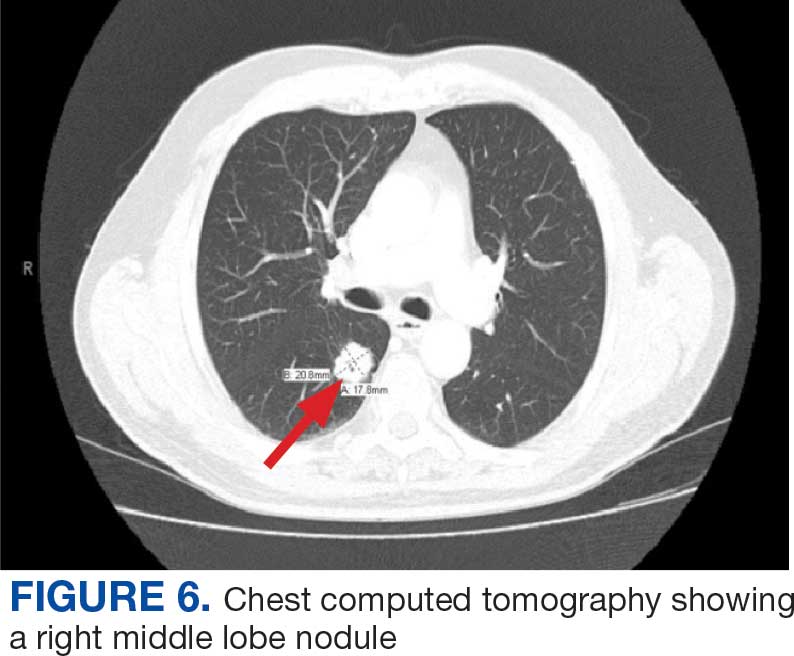
On evaluation, a chest CT was significant for a large 2-cm right middle lobe nodule (Figure 6). At that time, PSA was noted to be borderline elevated at 4.2 ng/mL, and whole-body imaging did not reveal any lesions elsewhere, specifically no bone metastasis. Biopsies of the right middle lobe lung nodule revealed adenocarcinoma consistent with metastatic prostate cancer. Testosterone therapy was promptly discontinued.
The patient initially refused androgen deprivation therapy owing to the antiandrogenic adverse effects. However, subsequent chest CTs revealed growing lung nodules, which convinced him to proceed with androgen deprivation therapy followed by palliative radiation, and chemotherapy and management of malignant pleural effusion with indwelling small bore pleural catheter for about 10 years. He died from COVID-19 during the pandemic.
DISCUSSION
These cases highlight the importance of including prostate cancer in the differential diagnoses of male patients with intrathoracic abnormalities, even in the absence of metastasis to the more common sites. In a large cohort study of 74,826 patients with metastatic prostate cancer, Gandaglia et al found that the most frequent sites of metastasis were bone (84.0%) and distant lymph nodes (10.6%).2 However, thoracic involvement was observed in 9.1% of cases, with isolated thoracic metastasis being rare. The cases described in this report exemplify exceptionally uncommon occurrences within that 9.1%.
Pleural metastases, as observed in Case 1, are a particularly rare manifestation. In a 10-year retrospective assessment, Vinjamoori et al discovered pleural nodules or masses in only 6 of 82 patients (7.3%) with atypical metastases.3 Adrenal and liver metastases accounted for 15% and 37% of cases with atypical distribution. As such, isolated pleural disease is rare even in atypical presentations.3
As seen in Case 2, endobronchial metastases producing airway obstruction are also rare, with the most common primary cancers associated with endobronchial metastasis being breast, colon, and renal cancer.4 The available literature on this presentation is confined to case reports. Hameed et al reported a case of synchronous biopsy-proven endobronchial metastasis from prostate cancer.5 These cases highlight the importance of maintaining a high level of clinical awareness when encountering endobronchial lesions in patients with prostate cancer.
Case 3 presents a unique situation of lung metastases without any involvement of the bones. It is well known—and was confirmed by Heidenreich et al—that lung metastases in prostate adenocarcinoma usually coincide with extensive osseous disease.6 This instance highlights the importance of watchful monitoring for unusual patterns of cancer recurrence.
Immunohistochemistry stains that are specific to prostate cancer include antibodies against PSA. Prostate-specific membrane antigen is another marker that is far more present in malignant than in benign prostate tissue.
The NKX3.1 gene encodes a homeobox protein, which is a transcription factor and tumor suppressor. In prostate cancer, there is loss of heterozygosity of the gene and stains for the IHC antibody to NKX3.1.7
On the other hand, lung cells stain positive for TTF-1, which is produced by surfactant-producing type 2 pneumocytes and club cells in the lung. Antibodies to TTF-1, a common IHC stain, are used to identify adenocarcinoma of lung origin and may carry a prognostic value.7
The immunohistochemistry profiles, specifically the presence of prostate-specific markers such as PSA and NKX3.1, played a vital role in making the diagnosis.
In Case 1, weak TTF-1 positivity was noted, an unusual finding in metastatic prostate adenocarcinoma. Marak et al documented a rare case of TTF-1–positive metastatic prostate cancer, illustrating the potential for diagnostic confusion with primary lung malignancies.8
The 3 cases described in this report demonstrate the importance of clinical consideration, serial follow-up of PSA levels, using more prostate-specific positron emission tomography tracers (eg, Pylarify) alongside traditional imaging, and tissue biopsy to detect unusual metastases.
CONCLUSIONS
Although thoracic metastases from prostate cancer are rare, these presentations highlight the importance of clinical awareness regarding atypical cases. Pleural disease, endobronchial lesions, and isolated pulmonary nodules might be the first clinical manifestation of metastatic prostate cancer. A high index of suspicion, appropriate imaging, and judicious use of immunohistochemistry are important to ensure accurate diagnosis and optimal patient management.
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49. doi:10.3322/caac.21820
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74(2):210-216. doi:10.1002/pros.22742
- Vinjamoori AH, Jagannathan JP, Shinagare AB, et al. Atypical metastases from prostate cancer: 10-year experience at a single institution. AJR Am J Roentgenol. 2012;199(2):367-372. doi:10.2214/AJR.11.7533
- Salud A, Porcel JM, Rovirosa A, Bellmunt J. Endobronchial metastatic disease: analysis of 32 cases. J Surg Oncol. 1996;62(4):249-252. doi:10.1002/(SICI)1096- 9098(199608)62:4<249::AID-JSO4>3.0.CO;2-6
- Hameed M, Haq IU, Yousaf M, Hussein M, Rashid U, Al-Bozom I. Endobronchial metastases secondary to prostate cancer: a case report and literature review. Respir Med Case Rep. 2020;32:101326. doi:10.1016/j.rmcr.2020.101326
- Heidenreich A, Bastian PJ, Bellmunt J, et al; for the European Association of Urology. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration- resistant prostate cancer. Eur Urol. 2014;65(2):467- 479. doi:10.1016/j.eururo.2013.11.002
- Schallenberg S, Dernbach G, Dragomir MP, et al. TTF-1 status in early-stage lung adenocarcinoma is an independent predictor of relapse and survival superior to tumor grading. Eur J Cancer. 2024;197:113474. doi:10.1016/j.ejca.2023.113474
- Marak C, Guddati AK, Ashraf A, Smith J, Kaushik P. Prostate adenocarcinoma with atypical immunohistochemistry presenting with a Cheerio sign. AIM Clinical Cases. 2023;1:e220508. doi:10.7326/aimcc.2022.0508
Prostate cancer is the most common noncutaneous cancer in men, accounting for 29% of all incident cancer cases.1 Typically, prostate cancer metastasizes to bone and regional lymph nodes.2 However, intrathoracic manifestation may occur. This report presents 3 cases of rare intrathoracic manifestations of metastatic prostate cancer with a review of the current literature.
CASE PRESENTATIONS
Case 1
A 71-year-old male who was an active smoker and a long-standing employment as a plumber was diagnosed with rectal cancer in 2022. He completed neoadjuvant capecitabine and radiation therapy followed by a rectosigmoidectomy. Several weeks after surgery, the patient presented to the emergency department (ED) with a dry cough and worsening shortness of breath. Point-of-care ultrasound of the lungs revealed a moderate right pleural effusion with several nodular pleural masses. A chest computed tomography (CT) confirmed these findings (Figure 1). A CT of the abdomen and pelvis revealed prostatomegaly with the medial lobe of the prostate protruding into the bladder; however, no enlarged retroperitoneal, mesenteric or pelvic lymph nodes were noted. The patient underwent a right pleural fluid drainage and pleural mass biopsy. Pleural mass histomorphology as well as immunohistochemical (IHC) stains were consistent with metastatic prostate adenocarcinoma. The pleural fluid cytology also was consistent with metastatic prostate adenocarcinoma.
Immunohistochemistry showed weak positive staining for prostate-specific NK3 homeobox 1 gene (NKX3.1), alpha-methylacyl-CoA racemase gene (AMACR), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), keratin-20, and caudal type homeobox 2 gene (CDX2) (Figure 2) 2). The patient's prostate-specific antigen (PSA) was found to be elevated at 33.9 ng/mL (reference range, < 4 ng/mL).
Case 2
A 71-year-old male with a history of alcohol use disorder and a 30-year smoking history presented to the ED with worsening dyspnea on exertion. The patient’s baseline exercise tolerance decreased to walking for only 1 block. He reported unintentional weight loss of about 30 pounds over the prior year, no recent respiratory infections, no prior breathing problems, and no personal or family history of cancer. Chest CT revealed findings of bilateral peribronchial opacities as well as mediastinal and hilar lymphadenopathy (Figure 3). The patient developed hypoxic respiratory failure necessitating intubation, mechanical ventilation, and management in the medical intensive care unit, where he was treated for postobstructive pneumonia. Fiberoptic bronchoscopy revealed endobronchial lesions in the right and left upper lobe that were partially obstructing the airway (Figure 4).
The endobronchial masses were debulked using forceps, and samples were sent for surgical pathology evaluation. Staging was completed using linear endobronchial ultrasound, which revealed an enlarged subcarinal lymph node (S7). The surgical pathology of the endobronchial mass and the subcarinal lymph node cytology were consistent with metastatic adenocarcinoma of the prostate. The tumor cells were positive for AE1/AE3, PSA, and NKX3.1, but were negative for CK7 and TTF-1 (Figure 5). Further imaging revealed an enlarged heterogeneous prostate gland, prominent pelvic nodes, and left retroperitoneal lymphadenopathy, as well as sclerotic foci within the T10 vertebral body and right inferior pubic ramus. PSA was also found to be significantly elevated at 700 ng/mL.
Case 3
An 80-year-old male veteran with a history of prostate cancer and recently diagnosed T2N1M0 head and neck squamous cell carcinoma was referred to the Pulmonary service for evaluation of a pulmonary nodule. His medical history was notable for prostate cancer diagnosed 12 years earlier, with an unknown Gleason score. Initial treatment included prostatectomy followed by whole pelvic radiation therapy a year after, due to elevated PSA in surveillance monitoring. This treatment led to remission. After establishing remission for > 10 years, the patient was started on low-dose testosterone replacement therapy to address complications of radiation therapy, namely hypogonadism.
On evaluation, a chest CT was significant for a large 2-cm right middle lobe nodule (Figure 6). At that time, PSA was noted to be borderline elevated at 4.2 ng/mL, and whole-body imaging did not reveal any lesions elsewhere, specifically no bone metastasis. Biopsies of the right middle lobe lung nodule revealed adenocarcinoma consistent with metastatic prostate cancer. Testosterone therapy was promptly discontinued.
The patient initially refused androgen deprivation therapy owing to the antiandrogenic adverse effects. However, subsequent chest CTs revealed growing lung nodules, which convinced him to proceed with androgen deprivation therapy followed by palliative radiation, and chemotherapy and management of malignant pleural effusion with indwelling small bore pleural catheter for about 10 years. He died from COVID-19 during the pandemic.
DISCUSSION
These cases highlight the importance of including prostate cancer in the differential diagnoses of male patients with intrathoracic abnormalities, even in the absence of metastasis to the more common sites. In a large cohort study of 74,826 patients with metastatic prostate cancer, Gandaglia et al found that the most frequent sites of metastasis were bone (84.0%) and distant lymph nodes (10.6%).2 However, thoracic involvement was observed in 9.1% of cases, with isolated thoracic metastasis being rare. The cases described in this report exemplify exceptionally uncommon occurrences within that 9.1%.
Pleural metastases, as observed in Case 1, are a particularly rare manifestation. In a 10-year retrospective assessment, Vinjamoori et al discovered pleural nodules or masses in only 6 of 82 patients (7.3%) with atypical metastases.3 Adrenal and liver metastases accounted for 15% and 37% of cases with atypical distribution. As such, isolated pleural disease is rare even in atypical presentations.3
As seen in Case 2, endobronchial metastases producing airway obstruction are also rare, with the most common primary cancers associated with endobronchial metastasis being breast, colon, and renal cancer.4 The available literature on this presentation is confined to case reports. Hameed et al reported a case of synchronous biopsy-proven endobronchial metastasis from prostate cancer.5 These cases highlight the importance of maintaining a high level of clinical awareness when encountering endobronchial lesions in patients with prostate cancer.
Case 3 presents a unique situation of lung metastases without any involvement of the bones. It is well known—and was confirmed by Heidenreich et al—that lung metastases in prostate adenocarcinoma usually coincide with extensive osseous disease.6 This instance highlights the importance of watchful monitoring for unusual patterns of cancer recurrence.
Immunohistochemistry stains that are specific to prostate cancer include antibodies against PSA. Prostate-specific membrane antigen is another marker that is far more present in malignant than in benign prostate tissue.
The NKX3.1 gene encodes a homeobox protein, which is a transcription factor and tumor suppressor. In prostate cancer, there is loss of heterozygosity of the gene and stains for the IHC antibody to NKX3.1.7
On the other hand, lung cells stain positive for TTF-1, which is produced by surfactant-producing type 2 pneumocytes and club cells in the lung. Antibodies to TTF-1, a common IHC stain, are used to identify adenocarcinoma of lung origin and may carry a prognostic value.7
The immunohistochemistry profiles, specifically the presence of prostate-specific markers such as PSA and NKX3.1, played a vital role in making the diagnosis.
In Case 1, weak TTF-1 positivity was noted, an unusual finding in metastatic prostate adenocarcinoma. Marak et al documented a rare case of TTF-1–positive metastatic prostate cancer, illustrating the potential for diagnostic confusion with primary lung malignancies.8
The 3 cases described in this report demonstrate the importance of clinical consideration, serial follow-up of PSA levels, using more prostate-specific positron emission tomography tracers (eg, Pylarify) alongside traditional imaging, and tissue biopsy to detect unusual metastases.
CONCLUSIONS
Although thoracic metastases from prostate cancer are rare, these presentations highlight the importance of clinical awareness regarding atypical cases. Pleural disease, endobronchial lesions, and isolated pulmonary nodules might be the first clinical manifestation of metastatic prostate cancer. A high index of suspicion, appropriate imaging, and judicious use of immunohistochemistry are important to ensure accurate diagnosis and optimal patient management.
Prostate cancer is the most common noncutaneous cancer in men, accounting for 29% of all incident cancer cases.1 Typically, prostate cancer metastasizes to bone and regional lymph nodes.2 However, intrathoracic manifestation may occur. This report presents 3 cases of rare intrathoracic manifestations of metastatic prostate cancer with a review of the current literature.
CASE PRESENTATIONS
Case 1
A 71-year-old male who was an active smoker and a long-standing employment as a plumber was diagnosed with rectal cancer in 2022. He completed neoadjuvant capecitabine and radiation therapy followed by a rectosigmoidectomy. Several weeks after surgery, the patient presented to the emergency department (ED) with a dry cough and worsening shortness of breath. Point-of-care ultrasound of the lungs revealed a moderate right pleural effusion with several nodular pleural masses. A chest computed tomography (CT) confirmed these findings (Figure 1). A CT of the abdomen and pelvis revealed prostatomegaly with the medial lobe of the prostate protruding into the bladder; however, no enlarged retroperitoneal, mesenteric or pelvic lymph nodes were noted. The patient underwent a right pleural fluid drainage and pleural mass biopsy. Pleural mass histomorphology as well as immunohistochemical (IHC) stains were consistent with metastatic prostate adenocarcinoma. The pleural fluid cytology also was consistent with metastatic prostate adenocarcinoma.
Immunohistochemistry showed weak positive staining for prostate-specific NK3 homeobox 1 gene (NKX3.1), alpha-methylacyl-CoA racemase gene (AMACR), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), and prosaposin, and negative transcription termination factor (TTF-1), keratin-7 (CK7), keratin-20, and caudal type homeobox 2 gene (CDX2) (Figure 2) 2). The patient's prostate-specific antigen (PSA) was found to be elevated at 33.9 ng/mL (reference range, < 4 ng/mL).
Case 2
A 71-year-old male with a history of alcohol use disorder and a 30-year smoking history presented to the ED with worsening dyspnea on exertion. The patient’s baseline exercise tolerance decreased to walking for only 1 block. He reported unintentional weight loss of about 30 pounds over the prior year, no recent respiratory infections, no prior breathing problems, and no personal or family history of cancer. Chest CT revealed findings of bilateral peribronchial opacities as well as mediastinal and hilar lymphadenopathy (Figure 3). The patient developed hypoxic respiratory failure necessitating intubation, mechanical ventilation, and management in the medical intensive care unit, where he was treated for postobstructive pneumonia. Fiberoptic bronchoscopy revealed endobronchial lesions in the right and left upper lobe that were partially obstructing the airway (Figure 4).
The endobronchial masses were debulked using forceps, and samples were sent for surgical pathology evaluation. Staging was completed using linear endobronchial ultrasound, which revealed an enlarged subcarinal lymph node (S7). The surgical pathology of the endobronchial mass and the subcarinal lymph node cytology were consistent with metastatic adenocarcinoma of the prostate. The tumor cells were positive for AE1/AE3, PSA, and NKX3.1, but were negative for CK7 and TTF-1 (Figure 5). Further imaging revealed an enlarged heterogeneous prostate gland, prominent pelvic nodes, and left retroperitoneal lymphadenopathy, as well as sclerotic foci within the T10 vertebral body and right inferior pubic ramus. PSA was also found to be significantly elevated at 700 ng/mL.
Case 3
An 80-year-old male veteran with a history of prostate cancer and recently diagnosed T2N1M0 head and neck squamous cell carcinoma was referred to the Pulmonary service for evaluation of a pulmonary nodule. His medical history was notable for prostate cancer diagnosed 12 years earlier, with an unknown Gleason score. Initial treatment included prostatectomy followed by whole pelvic radiation therapy a year after, due to elevated PSA in surveillance monitoring. This treatment led to remission. After establishing remission for > 10 years, the patient was started on low-dose testosterone replacement therapy to address complications of radiation therapy, namely hypogonadism.
On evaluation, a chest CT was significant for a large 2-cm right middle lobe nodule (Figure 6). At that time, PSA was noted to be borderline elevated at 4.2 ng/mL, and whole-body imaging did not reveal any lesions elsewhere, specifically no bone metastasis. Biopsies of the right middle lobe lung nodule revealed adenocarcinoma consistent with metastatic prostate cancer. Testosterone therapy was promptly discontinued.
The patient initially refused androgen deprivation therapy owing to the antiandrogenic adverse effects. However, subsequent chest CTs revealed growing lung nodules, which convinced him to proceed with androgen deprivation therapy followed by palliative radiation, and chemotherapy and management of malignant pleural effusion with indwelling small bore pleural catheter for about 10 years. He died from COVID-19 during the pandemic.
DISCUSSION
These cases highlight the importance of including prostate cancer in the differential diagnoses of male patients with intrathoracic abnormalities, even in the absence of metastasis to the more common sites. In a large cohort study of 74,826 patients with metastatic prostate cancer, Gandaglia et al found that the most frequent sites of metastasis were bone (84.0%) and distant lymph nodes (10.6%).2 However, thoracic involvement was observed in 9.1% of cases, with isolated thoracic metastasis being rare. The cases described in this report exemplify exceptionally uncommon occurrences within that 9.1%.
Pleural metastases, as observed in Case 1, are a particularly rare manifestation. In a 10-year retrospective assessment, Vinjamoori et al discovered pleural nodules or masses in only 6 of 82 patients (7.3%) with atypical metastases.3 Adrenal and liver metastases accounted for 15% and 37% of cases with atypical distribution. As such, isolated pleural disease is rare even in atypical presentations.3
As seen in Case 2, endobronchial metastases producing airway obstruction are also rare, with the most common primary cancers associated with endobronchial metastasis being breast, colon, and renal cancer.4 The available literature on this presentation is confined to case reports. Hameed et al reported a case of synchronous biopsy-proven endobronchial metastasis from prostate cancer.5 These cases highlight the importance of maintaining a high level of clinical awareness when encountering endobronchial lesions in patients with prostate cancer.
Case 3 presents a unique situation of lung metastases without any involvement of the bones. It is well known—and was confirmed by Heidenreich et al—that lung metastases in prostate adenocarcinoma usually coincide with extensive osseous disease.6 This instance highlights the importance of watchful monitoring for unusual patterns of cancer recurrence.
Immunohistochemistry stains that are specific to prostate cancer include antibodies against PSA. Prostate-specific membrane antigen is another marker that is far more present in malignant than in benign prostate tissue.
The NKX3.1 gene encodes a homeobox protein, which is a transcription factor and tumor suppressor. In prostate cancer, there is loss of heterozygosity of the gene and stains for the IHC antibody to NKX3.1.7
On the other hand, lung cells stain positive for TTF-1, which is produced by surfactant-producing type 2 pneumocytes and club cells in the lung. Antibodies to TTF-1, a common IHC stain, are used to identify adenocarcinoma of lung origin and may carry a prognostic value.7
The immunohistochemistry profiles, specifically the presence of prostate-specific markers such as PSA and NKX3.1, played a vital role in making the diagnosis.
In Case 1, weak TTF-1 positivity was noted, an unusual finding in metastatic prostate adenocarcinoma. Marak et al documented a rare case of TTF-1–positive metastatic prostate cancer, illustrating the potential for diagnostic confusion with primary lung malignancies.8
The 3 cases described in this report demonstrate the importance of clinical consideration, serial follow-up of PSA levels, using more prostate-specific positron emission tomography tracers (eg, Pylarify) alongside traditional imaging, and tissue biopsy to detect unusual metastases.
CONCLUSIONS
Although thoracic metastases from prostate cancer are rare, these presentations highlight the importance of clinical awareness regarding atypical cases. Pleural disease, endobronchial lesions, and isolated pulmonary nodules might be the first clinical manifestation of metastatic prostate cancer. A high index of suspicion, appropriate imaging, and judicious use of immunohistochemistry are important to ensure accurate diagnosis and optimal patient management.
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49. doi:10.3322/caac.21820
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74(2):210-216. doi:10.1002/pros.22742
- Vinjamoori AH, Jagannathan JP, Shinagare AB, et al. Atypical metastases from prostate cancer: 10-year experience at a single institution. AJR Am J Roentgenol. 2012;199(2):367-372. doi:10.2214/AJR.11.7533
- Salud A, Porcel JM, Rovirosa A, Bellmunt J. Endobronchial metastatic disease: analysis of 32 cases. J Surg Oncol. 1996;62(4):249-252. doi:10.1002/(SICI)1096- 9098(199608)62:4<249::AID-JSO4>3.0.CO;2-6
- Hameed M, Haq IU, Yousaf M, Hussein M, Rashid U, Al-Bozom I. Endobronchial metastases secondary to prostate cancer: a case report and literature review. Respir Med Case Rep. 2020;32:101326. doi:10.1016/j.rmcr.2020.101326
- Heidenreich A, Bastian PJ, Bellmunt J, et al; for the European Association of Urology. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration- resistant prostate cancer. Eur Urol. 2014;65(2):467- 479. doi:10.1016/j.eururo.2013.11.002
- Schallenberg S, Dernbach G, Dragomir MP, et al. TTF-1 status in early-stage lung adenocarcinoma is an independent predictor of relapse and survival superior to tumor grading. Eur J Cancer. 2024;197:113474. doi:10.1016/j.ejca.2023.113474
- Marak C, Guddati AK, Ashraf A, Smith J, Kaushik P. Prostate adenocarcinoma with atypical immunohistochemistry presenting with a Cheerio sign. AIM Clinical Cases. 2023;1:e220508. doi:10.7326/aimcc.2022.0508
- Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49. doi:10.3322/caac.21820
- Gandaglia G, Abdollah F, Schiffmann J, et al. Distribution of metastatic sites in patients with prostate cancer: a population-based analysis. Prostate. 2014;74(2):210-216. doi:10.1002/pros.22742
- Vinjamoori AH, Jagannathan JP, Shinagare AB, et al. Atypical metastases from prostate cancer: 10-year experience at a single institution. AJR Am J Roentgenol. 2012;199(2):367-372. doi:10.2214/AJR.11.7533
- Salud A, Porcel JM, Rovirosa A, Bellmunt J. Endobronchial metastatic disease: analysis of 32 cases. J Surg Oncol. 1996;62(4):249-252. doi:10.1002/(SICI)1096- 9098(199608)62:4<249::AID-JSO4>3.0.CO;2-6
- Hameed M, Haq IU, Yousaf M, Hussein M, Rashid U, Al-Bozom I. Endobronchial metastases secondary to prostate cancer: a case report and literature review. Respir Med Case Rep. 2020;32:101326. doi:10.1016/j.rmcr.2020.101326
- Heidenreich A, Bastian PJ, Bellmunt J, et al; for the European Association of Urology. EAU guidelines on prostate cancer. Part II: treatment of advanced, relapsing, and castration- resistant prostate cancer. Eur Urol. 2014;65(2):467- 479. doi:10.1016/j.eururo.2013.11.002
- Schallenberg S, Dernbach G, Dragomir MP, et al. TTF-1 status in early-stage lung adenocarcinoma is an independent predictor of relapse and survival superior to tumor grading. Eur J Cancer. 2024;197:113474. doi:10.1016/j.ejca.2023.113474
- Marak C, Guddati AK, Ashraf A, Smith J, Kaushik P. Prostate adenocarcinoma with atypical immunohistochemistry presenting with a Cheerio sign. AIM Clinical Cases. 2023;1:e220508. doi:10.7326/aimcc.2022.0508
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series
Atypical Intrathoracic Manifestations of Metastatic Prostate Cancer: A Case Series

The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
Pulmonary embolism (PE) is a common cause of morbidity and mortality in the general population.1 The incidence of PE has been reported to range from 39 to 115 per 100,000 persons per year and has remained stable.2 Although mortality rates have declined, they remain high.3 The clinical presentation is nonspecific, making diagnosis and management challenging. A crucial and difficult aspect in the management of patients with PE is weighing the risks vs benefits of treatment, including thrombolytic therapy and other invasive procedures, which carry inherent risks. These factors have led to the development of PE response teams (PERTs) in some hospitals to implement effective multidisciplinary protocols that facilitate prompt diagnosis, management, and follow-up.4
CASE PRESENTATIONS
Case 1
New onset seizures and cardiac arrest in the treatment of saddle PE. A 54-year-old male who worked as a draftsman and truck driver with a history of hypertension and nephrolithiasis presented to the emergency department (ED) with progressive shortness of breath for 2 weeks. On the morning of ED presentation the patient experienced an episode of severe shortness of breath, lightheadedness, and chest pressure. He reported no other symptoms such as palpitations, nausea, vomiting, abdominal discomfort, or extremity pain or swelling. He reported no recent travel, immunization, falls, or surgery. Upon evaluation, the patient was found to be in no acute distress, with stable vital signs and laboratory results except for 2 elevated results: > 20 μg/mL D-dimer (reference range, < 0.5 μg/mL) and N-terminal prohormone brain natriuretic peptide (proBNP) level, 3455 pg/mL (reference range, < 125 pg/mL for patients aged < 75 years). Electrocardiogram showed T-wave inversions in leads V2 to V4. Imaging revealed a saddle PE and left popliteal deep venous thrombosis (Figure 1). The patient received an anticoagulation loading dose and was started on heparin drip upon admission to the medical intensive care unit (MICU) for further management and monitoring. The Interventional Radiology Service recommended full anticoagulation with consideration of reperfusion therapies if deterioration developed.
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
While in the MICU, point-of-care ultrasound findings were confirmed with official echocardiogram by the cardiology service, which demonstrated a preserved ejection fraction of 60% to 65%, a D-shaped left ventricle with septal wall hypokinesis secondary to right heart strain (Figure 2), a markedly elevated right ventricular systolic pressure (RVSP) of 73 mm Hg, and a mean pulmonary artery pressure (mPAP) of 38 mm Hg. The patient’s blood pressure progressively decreased, heart rate increased, and he required increased oxygen supplementation. The case was discussed with the Pharmacy Service, and since the patient had no contraindications to thrombolytic therapy, the appropriate dosage was calculated and 100 mg intravenous (IV) tissue plasminogen activator (tPA) was administered over 2 hours.
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
About 40 minutes into tPA infusion, the patient suddenly experienced marked shortness of breath, diaphoresis, and anxiety with seizure-like involuntary movements; as a result, the infusion was stopped. He also had episodes of posturing, mental status decline, and briefly going in and out of consciousness, which lasted about 3 minutes before he lost consciousness and pulse. High-quality advanced cardiac life support was initiated, followed by endotracheal intubation. Despite a secured airway and return of spontaneous circulation, the patient remained hypotensive and continued to have seizure-like activity.
The patient was administered a total of 8 mg of lorazepam, sedated with propofol, initiated at 5 μg/kg/min, titrated to stop seizure activity at 15μg/kg/min, and later maintained at 10 μg/kg/min, for a RASS of -1, and started on norepinephrine 0.1 μg/kg/min for acute stabilization. Head computed tomography without contrast showed no acute intracranial pathology as etiology of seizures. Seizure etiology differential at this time was broad; however, hypoxemia due to PE and medication adverse effects were strongly suspected.
The patient’s condition improved, and vasopressor therapy was tapered off the next day. Four days later, the patient was weaned from mechanical ventilation and transferred to the step-down unit. Echocardiogram obtained 48 hours after tPA infusion showed essentially normal left ventricular function (60%-65%), a RVSP of 17 mm Hg and mPAP of 13 mm Hg. The patient’s ProBNP levels markedly decreased to 137 pg/mL. Postextubation, the neurologic examination was at baseline. The Neurology Service recommended temporary treatment with levetiracetam, 1000 mg every 12 hours, and the Hematology Service recommended transitioning to direct oral anticoagulation with follow-up. The patient presented significant clinical and respiratory improvement and was referred for home-based physical rehabilitation as recommended by the physical medicine and rehabilitation service before being discharged.
Case 2
Localized tPA infusion for bilateral PEs via infusion catheters. A 91-year-old male with no history of smoking and a medical history of hypertension, diabetes mellitus, prostate cancer (> 20 years postradiotherapy) and severe osteoarthritis was receiving treatment in the medical ward for medication-induced liver injury secondary to an antibiotic for a urinary tract infection. During the night the patient developed hypotension (86/46 mm Hg), shortness of breath, tachypnea, desaturation, nonradiating retrosternal chest pain, and tachycardia. The hypotension resolved after a 500-mL 0.9 normal saline bolus, and hypoxemia improved with supplemental oxygen via Venturi mask. Chest computed tomography angiography was performed immediately and revealed extensive bilateral acute PE, located most proximally in the right main pulmonary artery (PA) and on the left in the proximal lobar branches, with associated right heart strain. The patient was started on IV heparin with a bolus of 5000 units (80 u/kg) followed by a drip with a partial thromboplastin time goal of 62-103 seconds and transferred to MICU.
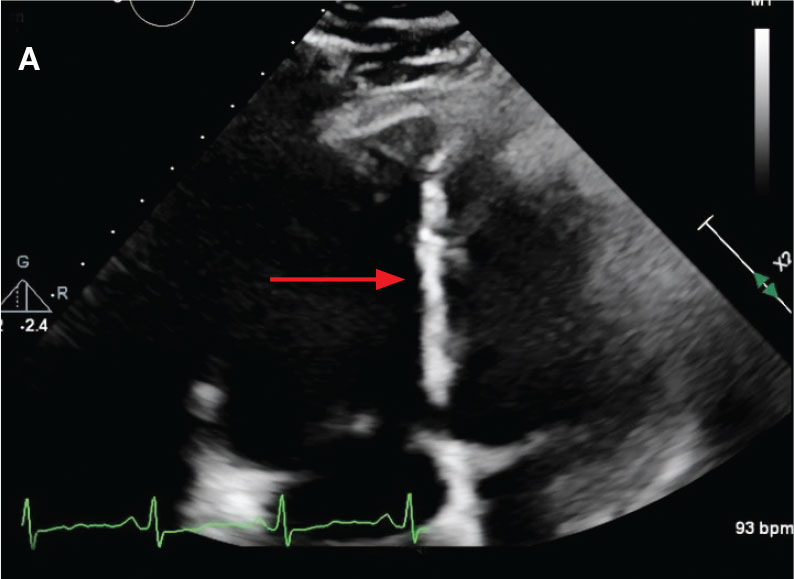
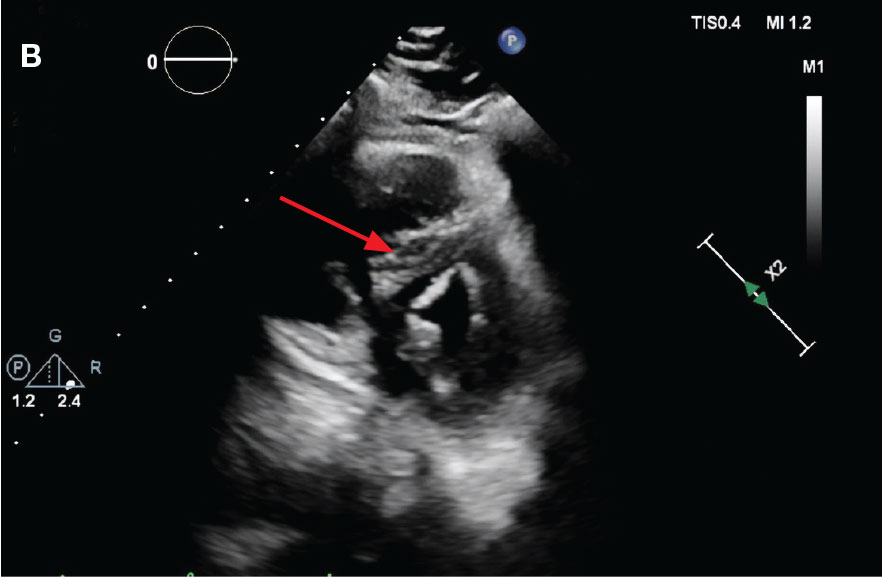
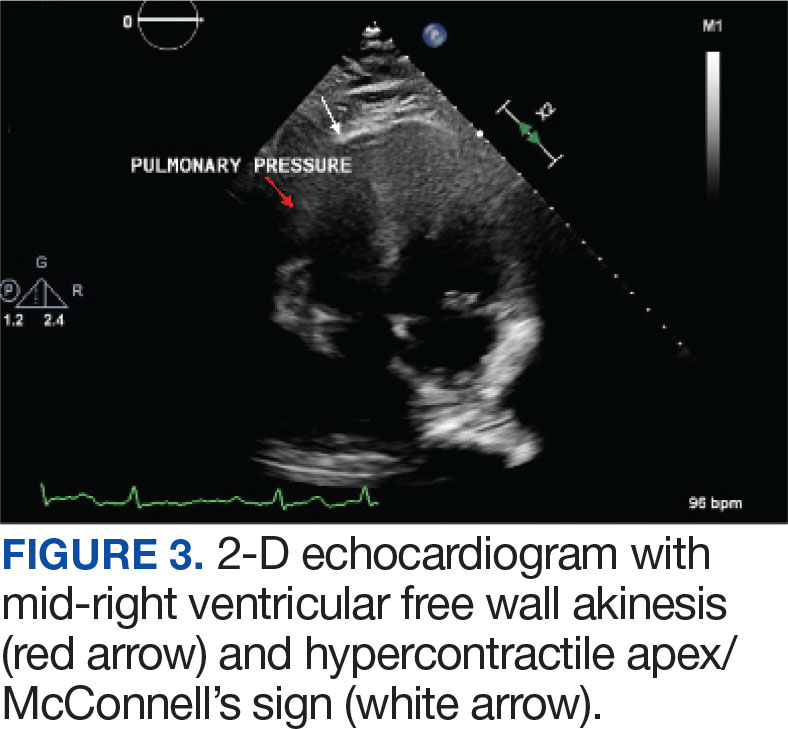
Laboratory findings were notable for proBNP that increased from 115 pg/mL to 4470 pg/mL (reference range, < 450 pg/mL for patients aged 75 years) and elevated troponin levels at 218 ng/L to 295 ng/L (reference range, < 22 ng/L), exhibiting chemical evidence of right heart strain. Initial echocardiogram showed mid-right ventricular free wall akinesis with a hypercontractile apex, suggestive of PE (McConnell’s sign) (Figure 3). Interventional Radiology Service was consulted and recommended tPA infusion given that the patient had bilateral PEs and stable blood pressure.
Pulmonary angiogram showed elevated pressures in the right PA of 64/21 mm Hg and the left PA pressures of 63/20 mm Hg. Mechanical disruption of the larger right lower PA thrombus was achieved via a pigtail catheter followed by bilateral catheter bolus infusions of 2 mg tPA (alteplase) and a continuous tPA infusion 0.5 mg/h for 24 hours, in conjunction with a heparin infusion.
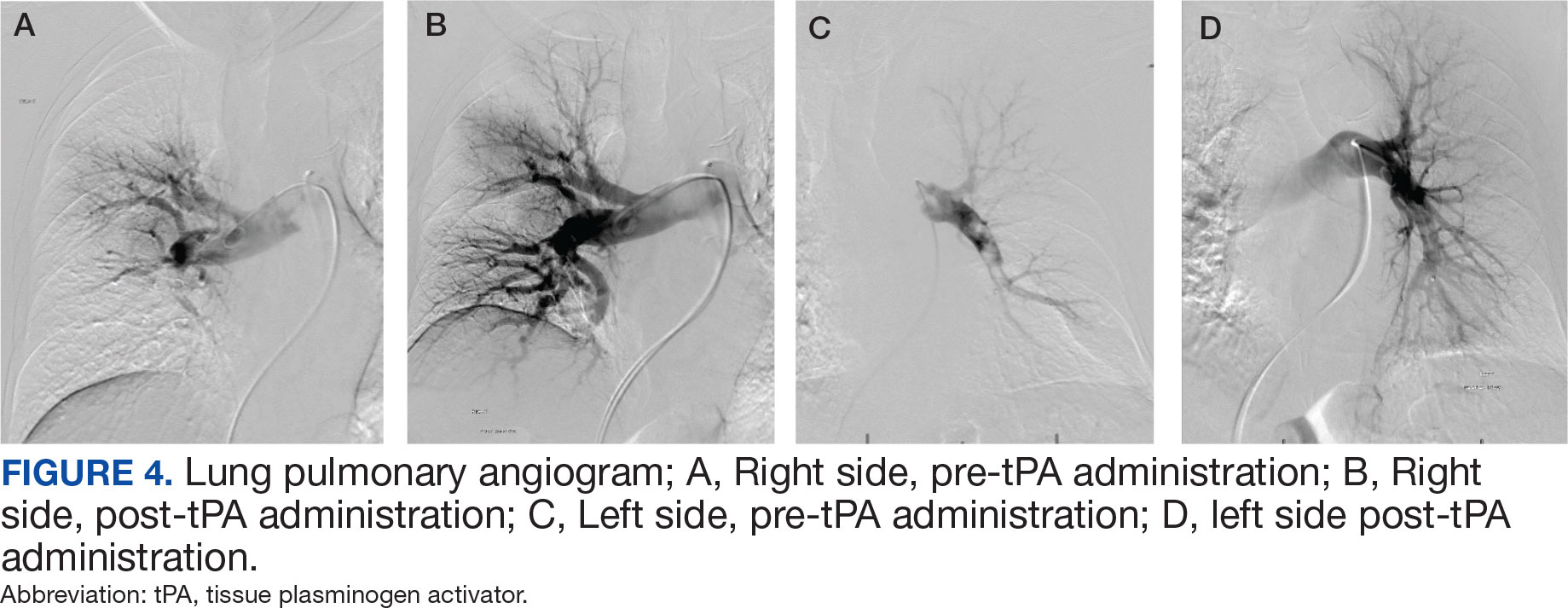
After 24 hours of tPA infusion, the catheters were removed, with posttreatment pulmonary angiography demonstrating right and left PA pressures of 42/15 mm Hg and 40/16 mm Hg, respectively. Pre- and postlocalized tPA infusion treatment images are provided for visual comparison (Figure 4). An echocardiogram performed after tPA infusion showed no signs of pulmonary hypertension. The Hematology Service provided recommendations regarding anticoagulation, and after completion of tPA infusion, the patient was transitioned to an unfractioned heparin infusion and subsequently to direct oral anticoagulation prior to transfer back to the medical ward, hemodynamically stable and asymptomatic.
DISCUSSION
PE management can be a straightforward decision when the patient meets criteria for hemodynamic instability, or with small PE burden. In contrast, management can be more challenging in intermediate-risk (submassive) PE when patients remain hemodynamically stable but show signs of cardiopulmonary stress, such as right heart strain, elevated troponins, or increased proBNP levels.2 In these situations, case-by- case evaluation is warranted. A PERT can assess the most beneficial treatment approach by considering factors such as right ventricular dysfunction, hemodynamic status, clot burden, and clinical deterioration despite appropriate anticoagulation. The evidence supporting the benefits these organized teams can provide is growing. These case reports emphasize the need for a multidisciplinary and systematic approach in these complex cases, especially in the management of intermediate-risk PE patients.
Currently, the Veterans Affairs Caribbean Healthcare System does not have an organized PERT, although a multidisciplinary approach was applied in the management of these patients. A systematic, structured team could have decreased time to interventions and alleviated the burden of physician decision-making. Having such a team would streamline the diagnostic pathway for patients presenting from a ward or emergency department with suspected PE.
We present 2 cases of patients found to have a high clot burden from PEs. The patients were initially hemodynamically stable (intermediate-risk PE), but later required systemic or localized thrombolysis due to hemodynamic deterioration despite adequate anticoagulation. Despite similar diagnoses and etiologies, these patients were successfully managed using different approaches, yielding positive outcomes. This reflects the complexity and variability in diagnosing and managing intermediate-risk PE in patients with different comorbidities and clot burden effects. In Case 1, our multidisciplinary approach was obtained via consults to selected services such as interventional radiology, cardiology, and direct involvement of pharmacy. An organized PERT conceivably would have allowed quicker discussions among these services, including hematology, to provide recommendations and collaborative support upon the patient’s arrival to the ED. Additionally, with a PERT team, a systematic approach to these patients could have allowed for an earlier official echocardiogram report for evaluation of right heart strain and develop an adequate therapeutic plan in a timely manner.
In Case 2, consultation with the Interventional Radiology Service yielded a better therapeutic plan, utilizing localized tPA infusion for this older adult patient with increased risk of bleeding with systemic tPA infusion. Having a PERT presents an opportunity to optimize PE management through early recognition, diagnosis, and treatment by institutional consensus from an interdisciplinary team.5,6 These response teams may improve outcomes and prognosis for patients with PE, especially where diagnosis and management is not clear.
The definite etiology of seizure activity in the first case pre- and postcardiac arrest, in the context of no acute intracranial process, remains unknown. Reports have emerged about postreperfusion seizures in acute ischemic stroke, as well as cases of seizures masquerading as PE as the primary presentation. 7,8 However, there were no reports of patients developing seizures post tPA infusion for the treatment of PE. This report may shed light into possible complications secondary to tPA infusion, raising awareness among physicians and encouraging further investigation into its possible etiologies.
CONCLUSIONS
Management of PE can be challenging in patients that meet criteria for intermediate risk. PERTs are a tool that allow for a multidisciplinary, standardized and systematic approach with a diagnostic and treatment algorithm that conceivably would result in a better consensus and therapeutic approach.
- Thompson BT, Kabrhel C. Epidemiology and pathogenesis of acute pulmonary embolism in adults. UpToDate. Wolters Kluwer. Updated December 4, 2023. Accessed February 26, 2025. https://www.uptodate.cn/contents/epidemiology-and-pathogenesis-of-acute-pulmonary-embolism-in-adults
- Kulka HC, Zeller A, Fornaro J, Wuillemin WA, Konstantinides S, Christ M. Acute pulmonary embolism– its diagnosis and treatment from a multidisciplinary viewpoint. Dtsch Arztebl Int. 2021;118(37):618-628. doi:10.3238/arztebl.m2021.0226
- Zghouzi M, Mwansa H, Shore S, et al. Sex, racial, and geographic disparities in pulmonary embolism-related mortality nationwide. Ann Am Thorac Soc. 2023;20(11):1571-1577. doi:10.1513/AnnalsATS.202302-091OC
- Channick RN. The pulmonary embolism response team: why and how? Semin Respir Crit Care Med. 2021;42(2):212-217. doi:10.1055/s-0041-1722963
- Rosovsky R, Zhao K, Sista A, Rivera-Lebron B, Kabrhel C. Pulmonary embolism response teams: purpose, evidence for efficacy, and future research directions. Res Pract Thromb Haemost. 2019;3(3):315-330. doi:10.1002/rth2.12216
- Glazier JJ, Patiño-Velasquez S, Oviedo C. The pulmonary embolism response team: rationale, operation, and outcomes. Int J Angiol. 2022;31(3):198-202. doi:10.1055/s-0042-1750328
- Lekoubou A, Fox J, Ssentongo P. Incidence and association of reperfusion therapies with poststroke seizures: a systematic review and meta-analysis. Stroke. 2020;51(9):2715-2723.doi:10.1161/STROKEAHA.119. 028899
- Alemany M, Nuñez A, Falip M, et al. Acute symptomatic seizures and epilepsy after mechanical thrombectomy. A prospective long-term follow-up study. Seizure. 2021;89:5-9. doi:10.1016/j.seizure.2021.04.011
Pulmonary embolism (PE) is a common cause of morbidity and mortality in the general population.1 The incidence of PE has been reported to range from 39 to 115 per 100,000 persons per year and has remained stable.2 Although mortality rates have declined, they remain high.3 The clinical presentation is nonspecific, making diagnosis and management challenging. A crucial and difficult aspect in the management of patients with PE is weighing the risks vs benefits of treatment, including thrombolytic therapy and other invasive procedures, which carry inherent risks. These factors have led to the development of PE response teams (PERTs) in some hospitals to implement effective multidisciplinary protocols that facilitate prompt diagnosis, management, and follow-up.4
CASE PRESENTATIONS
Case 1
New onset seizures and cardiac arrest in the treatment of saddle PE. A 54-year-old male who worked as a draftsman and truck driver with a history of hypertension and nephrolithiasis presented to the emergency department (ED) with progressive shortness of breath for 2 weeks. On the morning of ED presentation the patient experienced an episode of severe shortness of breath, lightheadedness, and chest pressure. He reported no other symptoms such as palpitations, nausea, vomiting, abdominal discomfort, or extremity pain or swelling. He reported no recent travel, immunization, falls, or surgery. Upon evaluation, the patient was found to be in no acute distress, with stable vital signs and laboratory results except for 2 elevated results: > 20 μg/mL D-dimer (reference range, < 0.5 μg/mL) and N-terminal prohormone brain natriuretic peptide (proBNP) level, 3455 pg/mL (reference range, < 125 pg/mL for patients aged < 75 years). Electrocardiogram showed T-wave inversions in leads V2 to V4. Imaging revealed a saddle PE and left popliteal deep venous thrombosis (Figure 1). The patient received an anticoagulation loading dose and was started on heparin drip upon admission to the medical intensive care unit (MICU) for further management and monitoring. The Interventional Radiology Service recommended full anticoagulation with consideration of reperfusion therapies if deterioration developed.
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
While in the MICU, point-of-care ultrasound findings were confirmed with official echocardiogram by the cardiology service, which demonstrated a preserved ejection fraction of 60% to 65%, a D-shaped left ventricle with septal wall hypokinesis secondary to right heart strain (Figure 2), a markedly elevated right ventricular systolic pressure (RVSP) of 73 mm Hg, and a mean pulmonary artery pressure (mPAP) of 38 mm Hg. The patient’s blood pressure progressively decreased, heart rate increased, and he required increased oxygen supplementation. The case was discussed with the Pharmacy Service, and since the patient had no contraindications to thrombolytic therapy, the appropriate dosage was calculated and 100 mg intravenous (IV) tissue plasminogen activator (tPA) was administered over 2 hours.
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
About 40 minutes into tPA infusion, the patient suddenly experienced marked shortness of breath, diaphoresis, and anxiety with seizure-like involuntary movements; as a result, the infusion was stopped. He also had episodes of posturing, mental status decline, and briefly going in and out of consciousness, which lasted about 3 minutes before he lost consciousness and pulse. High-quality advanced cardiac life support was initiated, followed by endotracheal intubation. Despite a secured airway and return of spontaneous circulation, the patient remained hypotensive and continued to have seizure-like activity.
The patient was administered a total of 8 mg of lorazepam, sedated with propofol, initiated at 5 μg/kg/min, titrated to stop seizure activity at 15μg/kg/min, and later maintained at 10 μg/kg/min, for a RASS of -1, and started on norepinephrine 0.1 μg/kg/min for acute stabilization. Head computed tomography without contrast showed no acute intracranial pathology as etiology of seizures. Seizure etiology differential at this time was broad; however, hypoxemia due to PE and medication adverse effects were strongly suspected.
The patient’s condition improved, and vasopressor therapy was tapered off the next day. Four days later, the patient was weaned from mechanical ventilation and transferred to the step-down unit. Echocardiogram obtained 48 hours after tPA infusion showed essentially normal left ventricular function (60%-65%), a RVSP of 17 mm Hg and mPAP of 13 mm Hg. The patient’s ProBNP levels markedly decreased to 137 pg/mL. Postextubation, the neurologic examination was at baseline. The Neurology Service recommended temporary treatment with levetiracetam, 1000 mg every 12 hours, and the Hematology Service recommended transitioning to direct oral anticoagulation with follow-up. The patient presented significant clinical and respiratory improvement and was referred for home-based physical rehabilitation as recommended by the physical medicine and rehabilitation service before being discharged.
Case 2
Localized tPA infusion for bilateral PEs via infusion catheters. A 91-year-old male with no history of smoking and a medical history of hypertension, diabetes mellitus, prostate cancer (> 20 years postradiotherapy) and severe osteoarthritis was receiving treatment in the medical ward for medication-induced liver injury secondary to an antibiotic for a urinary tract infection. During the night the patient developed hypotension (86/46 mm Hg), shortness of breath, tachypnea, desaturation, nonradiating retrosternal chest pain, and tachycardia. The hypotension resolved after a 500-mL 0.9 normal saline bolus, and hypoxemia improved with supplemental oxygen via Venturi mask. Chest computed tomography angiography was performed immediately and revealed extensive bilateral acute PE, located most proximally in the right main pulmonary artery (PA) and on the left in the proximal lobar branches, with associated right heart strain. The patient was started on IV heparin with a bolus of 5000 units (80 u/kg) followed by a drip with a partial thromboplastin time goal of 62-103 seconds and transferred to MICU.
Laboratory findings were notable for proBNP that increased from 115 pg/mL to 4470 pg/mL (reference range, < 450 pg/mL for patients aged 75 years) and elevated troponin levels at 218 ng/L to 295 ng/L (reference range, < 22 ng/L), exhibiting chemical evidence of right heart strain. Initial echocardiogram showed mid-right ventricular free wall akinesis with a hypercontractile apex, suggestive of PE (McConnell’s sign) (Figure 3). Interventional Radiology Service was consulted and recommended tPA infusion given that the patient had bilateral PEs and stable blood pressure.
Pulmonary angiogram showed elevated pressures in the right PA of 64/21 mm Hg and the left PA pressures of 63/20 mm Hg. Mechanical disruption of the larger right lower PA thrombus was achieved via a pigtail catheter followed by bilateral catheter bolus infusions of 2 mg tPA (alteplase) and a continuous tPA infusion 0.5 mg/h for 24 hours, in conjunction with a heparin infusion.
After 24 hours of tPA infusion, the catheters were removed, with posttreatment pulmonary angiography demonstrating right and left PA pressures of 42/15 mm Hg and 40/16 mm Hg, respectively. Pre- and postlocalized tPA infusion treatment images are provided for visual comparison (Figure 4). An echocardiogram performed after tPA infusion showed no signs of pulmonary hypertension. The Hematology Service provided recommendations regarding anticoagulation, and after completion of tPA infusion, the patient was transitioned to an unfractioned heparin infusion and subsequently to direct oral anticoagulation prior to transfer back to the medical ward, hemodynamically stable and asymptomatic.
DISCUSSION
PE management can be a straightforward decision when the patient meets criteria for hemodynamic instability, or with small PE burden. In contrast, management can be more challenging in intermediate-risk (submassive) PE when patients remain hemodynamically stable but show signs of cardiopulmonary stress, such as right heart strain, elevated troponins, or increased proBNP levels.2 In these situations, case-by- case evaluation is warranted. A PERT can assess the most beneficial treatment approach by considering factors such as right ventricular dysfunction, hemodynamic status, clot burden, and clinical deterioration despite appropriate anticoagulation. The evidence supporting the benefits these organized teams can provide is growing. These case reports emphasize the need for a multidisciplinary and systematic approach in these complex cases, especially in the management of intermediate-risk PE patients.
Currently, the Veterans Affairs Caribbean Healthcare System does not have an organized PERT, although a multidisciplinary approach was applied in the management of these patients. A systematic, structured team could have decreased time to interventions and alleviated the burden of physician decision-making. Having such a team would streamline the diagnostic pathway for patients presenting from a ward or emergency department with suspected PE.
We present 2 cases of patients found to have a high clot burden from PEs. The patients were initially hemodynamically stable (intermediate-risk PE), but later required systemic or localized thrombolysis due to hemodynamic deterioration despite adequate anticoagulation. Despite similar diagnoses and etiologies, these patients were successfully managed using different approaches, yielding positive outcomes. This reflects the complexity and variability in diagnosing and managing intermediate-risk PE in patients with different comorbidities and clot burden effects. In Case 1, our multidisciplinary approach was obtained via consults to selected services such as interventional radiology, cardiology, and direct involvement of pharmacy. An organized PERT conceivably would have allowed quicker discussions among these services, including hematology, to provide recommendations and collaborative support upon the patient’s arrival to the ED. Additionally, with a PERT team, a systematic approach to these patients could have allowed for an earlier official echocardiogram report for evaluation of right heart strain and develop an adequate therapeutic plan in a timely manner.
In Case 2, consultation with the Interventional Radiology Service yielded a better therapeutic plan, utilizing localized tPA infusion for this older adult patient with increased risk of bleeding with systemic tPA infusion. Having a PERT presents an opportunity to optimize PE management through early recognition, diagnosis, and treatment by institutional consensus from an interdisciplinary team.5,6 These response teams may improve outcomes and prognosis for patients with PE, especially where diagnosis and management is not clear.
The definite etiology of seizure activity in the first case pre- and postcardiac arrest, in the context of no acute intracranial process, remains unknown. Reports have emerged about postreperfusion seizures in acute ischemic stroke, as well as cases of seizures masquerading as PE as the primary presentation. 7,8 However, there were no reports of patients developing seizures post tPA infusion for the treatment of PE. This report may shed light into possible complications secondary to tPA infusion, raising awareness among physicians and encouraging further investigation into its possible etiologies.
CONCLUSIONS
Management of PE can be challenging in patients that meet criteria for intermediate risk. PERTs are a tool that allow for a multidisciplinary, standardized and systematic approach with a diagnostic and treatment algorithm that conceivably would result in a better consensus and therapeutic approach.
Pulmonary embolism (PE) is a common cause of morbidity and mortality in the general population.1 The incidence of PE has been reported to range from 39 to 115 per 100,000 persons per year and has remained stable.2 Although mortality rates have declined, they remain high.3 The clinical presentation is nonspecific, making diagnosis and management challenging. A crucial and difficult aspect in the management of patients with PE is weighing the risks vs benefits of treatment, including thrombolytic therapy and other invasive procedures, which carry inherent risks. These factors have led to the development of PE response teams (PERTs) in some hospitals to implement effective multidisciplinary protocols that facilitate prompt diagnosis, management, and follow-up.4
CASE PRESENTATIONS
Case 1
New onset seizures and cardiac arrest in the treatment of saddle PE. A 54-year-old male who worked as a draftsman and truck driver with a history of hypertension and nephrolithiasis presented to the emergency department (ED) with progressive shortness of breath for 2 weeks. On the morning of ED presentation the patient experienced an episode of severe shortness of breath, lightheadedness, and chest pressure. He reported no other symptoms such as palpitations, nausea, vomiting, abdominal discomfort, or extremity pain or swelling. He reported no recent travel, immunization, falls, or surgery. Upon evaluation, the patient was found to be in no acute distress, with stable vital signs and laboratory results except for 2 elevated results: > 20 μg/mL D-dimer (reference range, < 0.5 μg/mL) and N-terminal prohormone brain natriuretic peptide (proBNP) level, 3455 pg/mL (reference range, < 125 pg/mL for patients aged < 75 years). Electrocardiogram showed T-wave inversions in leads V2 to V4. Imaging revealed a saddle PE and left popliteal deep venous thrombosis (Figure 1). The patient received an anticoagulation loading dose and was started on heparin drip upon admission to the medical intensive care unit (MICU) for further management and monitoring. The Interventional Radiology Service recommended full anticoagulation with consideration of reperfusion therapies if deterioration developed.
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
indicated by arrows in the pulmonary trunk extending to the left pulmonary artery (A),
and obliterating right pulmonary artery and branches of left pulmonary artery (B).
While in the MICU, point-of-care ultrasound findings were confirmed with official echocardiogram by the cardiology service, which demonstrated a preserved ejection fraction of 60% to 65%, a D-shaped left ventricle with septal wall hypokinesis secondary to right heart strain (Figure 2), a markedly elevated right ventricular systolic pressure (RVSP) of 73 mm Hg, and a mean pulmonary artery pressure (mPAP) of 38 mm Hg. The patient’s blood pressure progressively decreased, heart rate increased, and he required increased oxygen supplementation. The case was discussed with the Pharmacy Service, and since the patient had no contraindications to thrombolytic therapy, the appropriate dosage was calculated and 100 mg intravenous (IV) tissue plasminogen activator (tPA) was administered over 2 hours.
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
flattening and deviation to left in direction (A) and septal deviation to left with
formation of D-sign (B).
About 40 minutes into tPA infusion, the patient suddenly experienced marked shortness of breath, diaphoresis, and anxiety with seizure-like involuntary movements; as a result, the infusion was stopped. He also had episodes of posturing, mental status decline, and briefly going in and out of consciousness, which lasted about 3 minutes before he lost consciousness and pulse. High-quality advanced cardiac life support was initiated, followed by endotracheal intubation. Despite a secured airway and return of spontaneous circulation, the patient remained hypotensive and continued to have seizure-like activity.
The patient was administered a total of 8 mg of lorazepam, sedated with propofol, initiated at 5 μg/kg/min, titrated to stop seizure activity at 15μg/kg/min, and later maintained at 10 μg/kg/min, for a RASS of -1, and started on norepinephrine 0.1 μg/kg/min for acute stabilization. Head computed tomography without contrast showed no acute intracranial pathology as etiology of seizures. Seizure etiology differential at this time was broad; however, hypoxemia due to PE and medication adverse effects were strongly suspected.
The patient’s condition improved, and vasopressor therapy was tapered off the next day. Four days later, the patient was weaned from mechanical ventilation and transferred to the step-down unit. Echocardiogram obtained 48 hours after tPA infusion showed essentially normal left ventricular function (60%-65%), a RVSP of 17 mm Hg and mPAP of 13 mm Hg. The patient’s ProBNP levels markedly decreased to 137 pg/mL. Postextubation, the neurologic examination was at baseline. The Neurology Service recommended temporary treatment with levetiracetam, 1000 mg every 12 hours, and the Hematology Service recommended transitioning to direct oral anticoagulation with follow-up. The patient presented significant clinical and respiratory improvement and was referred for home-based physical rehabilitation as recommended by the physical medicine and rehabilitation service before being discharged.
Case 2
Localized tPA infusion for bilateral PEs via infusion catheters. A 91-year-old male with no history of smoking and a medical history of hypertension, diabetes mellitus, prostate cancer (> 20 years postradiotherapy) and severe osteoarthritis was receiving treatment in the medical ward for medication-induced liver injury secondary to an antibiotic for a urinary tract infection. During the night the patient developed hypotension (86/46 mm Hg), shortness of breath, tachypnea, desaturation, nonradiating retrosternal chest pain, and tachycardia. The hypotension resolved after a 500-mL 0.9 normal saline bolus, and hypoxemia improved with supplemental oxygen via Venturi mask. Chest computed tomography angiography was performed immediately and revealed extensive bilateral acute PE, located most proximally in the right main pulmonary artery (PA) and on the left in the proximal lobar branches, with associated right heart strain. The patient was started on IV heparin with a bolus of 5000 units (80 u/kg) followed by a drip with a partial thromboplastin time goal of 62-103 seconds and transferred to MICU.
Laboratory findings were notable for proBNP that increased from 115 pg/mL to 4470 pg/mL (reference range, < 450 pg/mL for patients aged 75 years) and elevated troponin levels at 218 ng/L to 295 ng/L (reference range, < 22 ng/L), exhibiting chemical evidence of right heart strain. Initial echocardiogram showed mid-right ventricular free wall akinesis with a hypercontractile apex, suggestive of PE (McConnell’s sign) (Figure 3). Interventional Radiology Service was consulted and recommended tPA infusion given that the patient had bilateral PEs and stable blood pressure.
Pulmonary angiogram showed elevated pressures in the right PA of 64/21 mm Hg and the left PA pressures of 63/20 mm Hg. Mechanical disruption of the larger right lower PA thrombus was achieved via a pigtail catheter followed by bilateral catheter bolus infusions of 2 mg tPA (alteplase) and a continuous tPA infusion 0.5 mg/h for 24 hours, in conjunction with a heparin infusion.
After 24 hours of tPA infusion, the catheters were removed, with posttreatment pulmonary angiography demonstrating right and left PA pressures of 42/15 mm Hg and 40/16 mm Hg, respectively. Pre- and postlocalized tPA infusion treatment images are provided for visual comparison (Figure 4). An echocardiogram performed after tPA infusion showed no signs of pulmonary hypertension. The Hematology Service provided recommendations regarding anticoagulation, and after completion of tPA infusion, the patient was transitioned to an unfractioned heparin infusion and subsequently to direct oral anticoagulation prior to transfer back to the medical ward, hemodynamically stable and asymptomatic.
DISCUSSION
PE management can be a straightforward decision when the patient meets criteria for hemodynamic instability, or with small PE burden. In contrast, management can be more challenging in intermediate-risk (submassive) PE when patients remain hemodynamically stable but show signs of cardiopulmonary stress, such as right heart strain, elevated troponins, or increased proBNP levels.2 In these situations, case-by- case evaluation is warranted. A PERT can assess the most beneficial treatment approach by considering factors such as right ventricular dysfunction, hemodynamic status, clot burden, and clinical deterioration despite appropriate anticoagulation. The evidence supporting the benefits these organized teams can provide is growing. These case reports emphasize the need for a multidisciplinary and systematic approach in these complex cases, especially in the management of intermediate-risk PE patients.
Currently, the Veterans Affairs Caribbean Healthcare System does not have an organized PERT, although a multidisciplinary approach was applied in the management of these patients. A systematic, structured team could have decreased time to interventions and alleviated the burden of physician decision-making. Having such a team would streamline the diagnostic pathway for patients presenting from a ward or emergency department with suspected PE.
We present 2 cases of patients found to have a high clot burden from PEs. The patients were initially hemodynamically stable (intermediate-risk PE), but later required systemic or localized thrombolysis due to hemodynamic deterioration despite adequate anticoagulation. Despite similar diagnoses and etiologies, these patients were successfully managed using different approaches, yielding positive outcomes. This reflects the complexity and variability in diagnosing and managing intermediate-risk PE in patients with different comorbidities and clot burden effects. In Case 1, our multidisciplinary approach was obtained via consults to selected services such as interventional radiology, cardiology, and direct involvement of pharmacy. An organized PERT conceivably would have allowed quicker discussions among these services, including hematology, to provide recommendations and collaborative support upon the patient’s arrival to the ED. Additionally, with a PERT team, a systematic approach to these patients could have allowed for an earlier official echocardiogram report for evaluation of right heart strain and develop an adequate therapeutic plan in a timely manner.
In Case 2, consultation with the Interventional Radiology Service yielded a better therapeutic plan, utilizing localized tPA infusion for this older adult patient with increased risk of bleeding with systemic tPA infusion. Having a PERT presents an opportunity to optimize PE management through early recognition, diagnosis, and treatment by institutional consensus from an interdisciplinary team.5,6 These response teams may improve outcomes and prognosis for patients with PE, especially where diagnosis and management is not clear.
The definite etiology of seizure activity in the first case pre- and postcardiac arrest, in the context of no acute intracranial process, remains unknown. Reports have emerged about postreperfusion seizures in acute ischemic stroke, as well as cases of seizures masquerading as PE as the primary presentation. 7,8 However, there were no reports of patients developing seizures post tPA infusion for the treatment of PE. This report may shed light into possible complications secondary to tPA infusion, raising awareness among physicians and encouraging further investigation into its possible etiologies.
CONCLUSIONS
Management of PE can be challenging in patients that meet criteria for intermediate risk. PERTs are a tool that allow for a multidisciplinary, standardized and systematic approach with a diagnostic and treatment algorithm that conceivably would result in a better consensus and therapeutic approach.
- Thompson BT, Kabrhel C. Epidemiology and pathogenesis of acute pulmonary embolism in adults. UpToDate. Wolters Kluwer. Updated December 4, 2023. Accessed February 26, 2025. https://www.uptodate.cn/contents/epidemiology-and-pathogenesis-of-acute-pulmonary-embolism-in-adults
- Kulka HC, Zeller A, Fornaro J, Wuillemin WA, Konstantinides S, Christ M. Acute pulmonary embolism– its diagnosis and treatment from a multidisciplinary viewpoint. Dtsch Arztebl Int. 2021;118(37):618-628. doi:10.3238/arztebl.m2021.0226
- Zghouzi M, Mwansa H, Shore S, et al. Sex, racial, and geographic disparities in pulmonary embolism-related mortality nationwide. Ann Am Thorac Soc. 2023;20(11):1571-1577. doi:10.1513/AnnalsATS.202302-091OC
- Channick RN. The pulmonary embolism response team: why and how? Semin Respir Crit Care Med. 2021;42(2):212-217. doi:10.1055/s-0041-1722963
- Rosovsky R, Zhao K, Sista A, Rivera-Lebron B, Kabrhel C. Pulmonary embolism response teams: purpose, evidence for efficacy, and future research directions. Res Pract Thromb Haemost. 2019;3(3):315-330. doi:10.1002/rth2.12216
- Glazier JJ, Patiño-Velasquez S, Oviedo C. The pulmonary embolism response team: rationale, operation, and outcomes. Int J Angiol. 2022;31(3):198-202. doi:10.1055/s-0042-1750328
- Lekoubou A, Fox J, Ssentongo P. Incidence and association of reperfusion therapies with poststroke seizures: a systematic review and meta-analysis. Stroke. 2020;51(9):2715-2723.doi:10.1161/STROKEAHA.119. 028899
- Alemany M, Nuñez A, Falip M, et al. Acute symptomatic seizures and epilepsy after mechanical thrombectomy. A prospective long-term follow-up study. Seizure. 2021;89:5-9. doi:10.1016/j.seizure.2021.04.011
- Thompson BT, Kabrhel C. Epidemiology and pathogenesis of acute pulmonary embolism in adults. UpToDate. Wolters Kluwer. Updated December 4, 2023. Accessed February 26, 2025. https://www.uptodate.cn/contents/epidemiology-and-pathogenesis-of-acute-pulmonary-embolism-in-adults
- Kulka HC, Zeller A, Fornaro J, Wuillemin WA, Konstantinides S, Christ M. Acute pulmonary embolism– its diagnosis and treatment from a multidisciplinary viewpoint. Dtsch Arztebl Int. 2021;118(37):618-628. doi:10.3238/arztebl.m2021.0226
- Zghouzi M, Mwansa H, Shore S, et al. Sex, racial, and geographic disparities in pulmonary embolism-related mortality nationwide. Ann Am Thorac Soc. 2023;20(11):1571-1577. doi:10.1513/AnnalsATS.202302-091OC
- Channick RN. The pulmonary embolism response team: why and how? Semin Respir Crit Care Med. 2021;42(2):212-217. doi:10.1055/s-0041-1722963
- Rosovsky R, Zhao K, Sista A, Rivera-Lebron B, Kabrhel C. Pulmonary embolism response teams: purpose, evidence for efficacy, and future research directions. Res Pract Thromb Haemost. 2019;3(3):315-330. doi:10.1002/rth2.12216
- Glazier JJ, Patiño-Velasquez S, Oviedo C. The pulmonary embolism response team: rationale, operation, and outcomes. Int J Angiol. 2022;31(3):198-202. doi:10.1055/s-0042-1750328
- Lekoubou A, Fox J, Ssentongo P. Incidence and association of reperfusion therapies with poststroke seizures: a systematic review and meta-analysis. Stroke. 2020;51(9):2715-2723.doi:10.1161/STROKEAHA.119. 028899
- Alemany M, Nuñez A, Falip M, et al. Acute symptomatic seizures and epilepsy after mechanical thrombectomy. A prospective long-term follow-up study. Seizure. 2021;89:5-9. doi:10.1016/j.seizure.2021.04.011
The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
The Need for a Multidisciplinary Approach for Successful High-Risk Pulmonary Embolism Treatment
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Obesity is a growing worldwide epidemic with significant implications for individual health and public health care costs. It is also associated with several medical conditions, including diabetes, cardiovascular disease, cancer, and mental health disorders.1 Comprehensive lifestyle intervention is a first-line therapy for obesity consisting of dietary and exercise interventions. Despite initial success, long-term results and durability of weight loss with lifestyle modifications are limited. 2 Bariatric surgery, including sleeve gastrectomy and gastric bypass surgery, is a more invasive approach that is highly effective in weight loss. However, these operations are not reversible, and patients may not be eligible for or may not desire surgery. Overall, bariatric surgery is widely underutilized, with < 1% of eligible patients ultimately undergoing surgery.3,4
Endoscopic bariatric therapies are increasingly popular procedures that address the need for additional treatments for obesity among individuals who have not had success with lifestyle changes and are not surgical candidates. The most common procedure is the endoscopic sleeve gastroplasty (ESG), which applies full-thickness sutures in the stomach to reduce gastric volume, delay gastric emptying, and limit food intake while keeping the fundus intact compared with sleeve gastrectomy. This procedure is typically considered in patients with body mass index (BMI) ≥ 30, who do not qualify for or do not want traditional bariatric surgery. The literature supports robust outcomes after ESG, with studies demonstrating significant and sustained total body weight loss of up to 14% to 16% at 5 years and significant improvement in ≥ 1 metabolic comorbidities in 80% of patients.5,6 ESG adverse events (AEs) include abdominal pain, nausea, and vomiting that are typically self-limited to 1 week. Rarer but more serious AEs include bleeding, perforation, or infection, and occur in 2% of cases based on large trial data.5,7
Although the weight loss benefits of ESG are well established, to date, there are limited data on the effects of endoscopic bariatric therapies like ESG on mental health conditions. Here, we describe a case of a veteran with a history of mental health disorders that prevented him from completing bariatric surgery. The patient underwent ESG and had a successful clinical course.
CASE PRESENTATION
A 59-year-old male veteran with a medical history of class III obesity (42.4 BMI), obstructive sleep apnea, hypothyroidism, hypertension, type 2 diabetes mellitus, and a large ventral hernia was referred to the MOVE! (Management of Overweight/ Obese Veterans Everywhere!) multidisciplinary high-intensity weight loss program at the US Department of Veterans Affairs (VA) West Los Angeles VA Medical Center (WLAVAMC). His psychiatric history included generalized anxiety disorder, posttraumatic stress disorder (PTSD), and panic disorder, managed by the Psychiatry Service and treated with sertraline 25 mg daily, lorazepam 0.5 mg twice daily, and hydroxyzine 20 mg nightly. He had previously implemented lifestyle changes and attended MOVE! classes and nutrition coaching for 1 year but was unsuccessful in losing weight. He had also tried liraglutide 3 mg daily for weight loss but was unable to tolerate it and reported worsening medication-related anxiety.
The patient declined further weight loss pharmacotherapy and was referred to bariatric surgery. He was scheduled for a surgical sleeve gastrectomy. However, on the day he arrived at the hospital for surgery, he developed severe anxiety and had a panic attack, and it was canceled. Due to his mental health issues, he was no longer comfortable proceeding with surgery and was left without other options for obesity treatment. The veteran was extremely disappointed because the ventral hernia caused significant quality of life impairment, limited his ability to exercise, and caused him embarrassment in public settings. The hernia could not be surgically repaired until there was significant weight loss.
A bariatric endoscopy program within the Division of Gastroenterology was developed and implemented at the WLAVAMC in February 2023 in conjunction with MOVE! The patient was referred for consideration of an endoscopic weight loss procedure. He was determined to be a suitable candidate for ESG based on his BMI being > 40 and personal preference not to proceed with surgery to lose enough weight to qualify for hernia repair. The veteran underwent an endoscopy, which showed normal anatomy and gastric mucosa. ESG was performed in standard fashion (Figure).8 Three vertical lines were made using argon plasma coagulation from the incisura to 2 cm below the gastroesophageal junction along the anterior, posterior, and greater curvature of the stomach to mark the area for endoscopic suture placement. Starting at the incisura, 7 full-thickness sutures were placed to create a volume reduction plication, with preservation of the fundus. The patient did well postprocedure with no immediate or delayed AEs and was discharged home the same day.
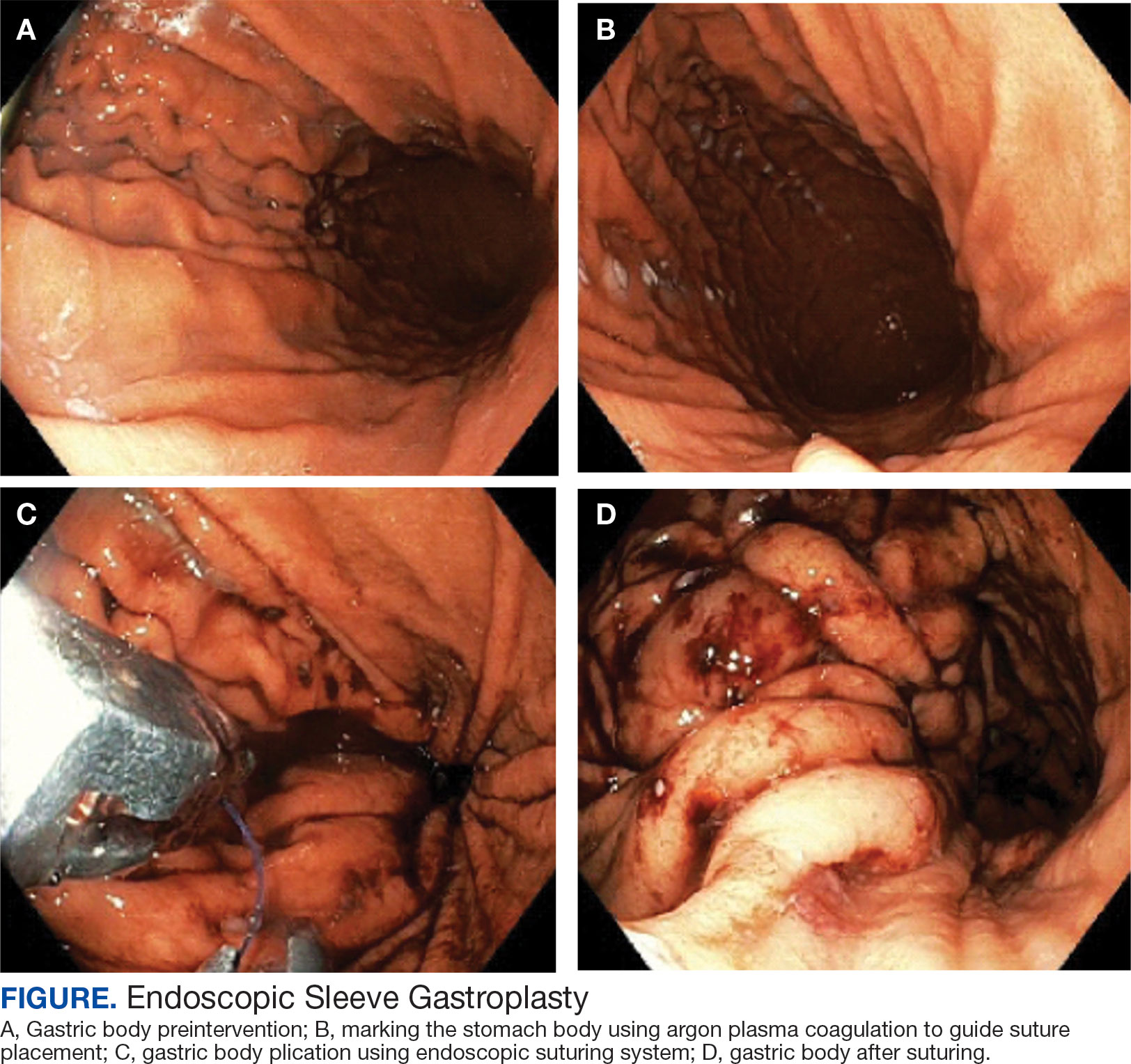
Follow-up
The veteran followed a gradual dietary advancement from a clear liquid diet to pureed and soft texture food. The patient’s weight dropped from 359 lbs preprocedure to 304 lbs 6 months postprocedure, a total body weight loss (TWBL) of 15.3%. At 12 months the veteran weighed 299 lbs (16.7% TBWL). He also had notable improvements in metabolic parameters. His systolic blood pressure decreased from ≥ 140 mm Hg to 120 to 130 mm Hg and hemoglobin A1c dropped from 7.0% to 6.3%. Remarkably, his psychiatrist noted significant improvement in his overall mental health. The veteran reported complete cessation of panic attacks since the ESG, improvements in PTSD and anxiety, and was able to discontinue lorazepam and decrease his dose of sertraline to 12.5 mg daily. He reported feeling more energetic and goal-oriented with increased clarity of thought. Perhaps the most significant outcome was that after the 55-lb weight loss at 6 months, the patient was eligible to undergo ventral hernia surgical repair, which had previously contributed to shame and social isolation. This, in turn, improved his quality of life, allowed him to start walking again, up to 8 miles daily, and to feel comfortable again going out in public settings.
DISCUSSION
Bariatric surgeries are an effective method of achieving weight loss and improving obesity-related comorbidities. However, only a small percentage of individuals with obesity are candidates for bariatric surgery. Given the dramatic increase in the prevalence of obesity, other options are needed. Specifically, within the VA, an estimated 80% of veterans are overweight or obese, but only about 500 bariatric surgeries are performed annually.9 With the need for additional weight loss therapies, VA programs are starting to offer endoscopic bariatric procedures as an alternative option. This may be a desirable choice for patients with obesity (BMI > 30), with or without associated metabolic comorbidities, who need more aggressive intervention beyond dietary and lifestyle changes and are either not interested in or not eligible for bariatric surgery or weight loss medications.
Although there is evidence that metabolic comorbidities are associated with obesity, there has been less research on obesity and mental health comorbidities such as depression and anxiety. These psychiatric conditions may even be more common among patients seeking weight loss procedures and more prominent in certain groups such as veterans, which may ultimately exclude these patients from bariatric surgery.10 Prior studies suggest that bariatric surgery can reduce the severity of depression and, to a lesser extent, anxiety symptoms at 2 years following the initial surgery; however, there is limited literature describing the impact of weight loss procedure on panic disorders.11-14 We suspect that a weight loss procedure such as ESG may have indirectly improved the veteran’s mood disorder due to the weight loss it induced, increasing the ability to exercise, quality of sleep, and participation in public settings.
This case highlights a veteran who did not tolerate weight loss medication and had severe anxiety and PTSD that prevented him from going through with bariatric surgery. He then underwent an endoscopic weight loss procedure. The ESG helped him successfully achieve significant weight loss, increase his physical activity, reduce his anxiety and panic disorder, and overall, significantly improve his quality of life. More than 1 year after the procedure, the patient has sustained improvements in his psychiatric and emotional health along with durable weight loss, maintaining > 15% of his total weight lost. Additional studies are needed to further understand the prevalence and long-term outcomes of mental health comorbidities, as well as weight loss outcomes in this group of patients who undergo endoscopic bariatric procedures.
CONCLUSIONS
We describe a case of a veteran with severe obesity and significant psychiatric comorbidities that prevented him from undergoing bariatric surgery, who underwent an ESG. This procedure led to significant weight loss, improvement of metabolic parameters, reduction in anxiety and PTSD, and enhancement of his quality of life. This case emphasizes the unique advantages of ESG and supports the expansion of endoscopic bariatric programs in the VA.
- Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr Metab Cardiovasc Dis. 2007;17(4):319-326. doi:10.1016/j.numecd.2006.07.005
- Bray GA, Kim KK, Wilding JPH; World Obesity Federation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev. 2017;18(7):715-723. doi:10.1111/obr.12551
- Imbus JR, Voils CI, Funk LM. Bariatric surgery barriers: a review using andersen’s model of health services use. Surg Obes Relat Dis. 2018;14(3):404-412. doi:10.1016/j.soard.2017.11.012
- Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: a meta-analysis. JAMA. 2016;315(2):150- 163. doi:10.1001/jama.2015.18118
- Abu Dayyeh BK, Bazerbachi F, Vargas EJ, et al.. Endoscopic sleeve gastroplasty for treatment of class 1 and 2 obesity (MERIT): a prospective, multicentre, randomised trial. Lancet. 2022;400(10350):441-451. doi:10.1016/S0140-6736(22)01280-6
- Matteo MV, Bove V, Ciasca G, et al. Success predictors of endoscopic sleeve gastroplasty. Obes Surg. 2024;34(5):1496-1504. doi:10.1007/s11695-024-07109-4
- Maselli DB, Hoff AC, Kucera A, et al. Endoscopic sleeve gastroplasty in class III obesity: efficacy, safety, and durability outcomes in 404 consecutive patients. World J Gastrointest Endosc. 2023;15(6):469-479. doi:10.4253/wjge.v15.i6.469
- Kumar N, Abu Dayyeh BK, Lopez-Nava Breviere G, et al. Endoscopic sutured gastroplasty: procedure evolution from first-in-man cases through current technique. Surg Endosc. 2018;32(4):2159-2164. doi:10.1007/s00464-017-5869-2
- Maggard-Gibbons M, Shekelle PG, Girgis MD, et al. Endoscopic Bariatric Interventions versus lifestyle interventions or surgery for weight loss in patients with obesity: a systematic review and meta-analysis. Department of Veterans Affairs (US); 2022. https://www.ncbi.nlm.nih.gov/books/NBK587943/
- Maggard Gibbons MA, Maher AM, Dawes AJ, et al. Psychological clearance for bariatric surgery: a systematic review. VA-ESP project #05-2262014.
- van Hout GC, Verschure SK, van Heck GL. Psychosocial predictors of success following bariatric surgery. Obes Surg. 2005;15(4):552-560. doi:10.1381/0960892053723484
- Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the national comorbidity survey replication. Biol Psychiatry. 2007;61(3):348-358. doi:10.1016/j.biopsych.2006.03.040
- Aylward L, Lilly C, Konsor M, et al. How soon do depression and anxiety symptoms improve after bariatric surgery?. Healthcare (Basel). 2023;11(6):862. doi:10.3390/healthcare11060862
- Law S, Dong S, Zhou F, Zheng D, Wang C, Dong Z. Bariatric surgery and mental health outcomes: an umbrella review. Front Endocrinol (Lausanne). 2023;14:1283621. doi:10.3389/fendo.2023.1283621
Obesity is a growing worldwide epidemic with significant implications for individual health and public health care costs. It is also associated with several medical conditions, including diabetes, cardiovascular disease, cancer, and mental health disorders.1 Comprehensive lifestyle intervention is a first-line therapy for obesity consisting of dietary and exercise interventions. Despite initial success, long-term results and durability of weight loss with lifestyle modifications are limited. 2 Bariatric surgery, including sleeve gastrectomy and gastric bypass surgery, is a more invasive approach that is highly effective in weight loss. However, these operations are not reversible, and patients may not be eligible for or may not desire surgery. Overall, bariatric surgery is widely underutilized, with < 1% of eligible patients ultimately undergoing surgery.3,4
Endoscopic bariatric therapies are increasingly popular procedures that address the need for additional treatments for obesity among individuals who have not had success with lifestyle changes and are not surgical candidates. The most common procedure is the endoscopic sleeve gastroplasty (ESG), which applies full-thickness sutures in the stomach to reduce gastric volume, delay gastric emptying, and limit food intake while keeping the fundus intact compared with sleeve gastrectomy. This procedure is typically considered in patients with body mass index (BMI) ≥ 30, who do not qualify for or do not want traditional bariatric surgery. The literature supports robust outcomes after ESG, with studies demonstrating significant and sustained total body weight loss of up to 14% to 16% at 5 years and significant improvement in ≥ 1 metabolic comorbidities in 80% of patients.5,6 ESG adverse events (AEs) include abdominal pain, nausea, and vomiting that are typically self-limited to 1 week. Rarer but more serious AEs include bleeding, perforation, or infection, and occur in 2% of cases based on large trial data.5,7
Although the weight loss benefits of ESG are well established, to date, there are limited data on the effects of endoscopic bariatric therapies like ESG on mental health conditions. Here, we describe a case of a veteran with a history of mental health disorders that prevented him from completing bariatric surgery. The patient underwent ESG and had a successful clinical course.
CASE PRESENTATION
A 59-year-old male veteran with a medical history of class III obesity (42.4 BMI), obstructive sleep apnea, hypothyroidism, hypertension, type 2 diabetes mellitus, and a large ventral hernia was referred to the MOVE! (Management of Overweight/ Obese Veterans Everywhere!) multidisciplinary high-intensity weight loss program at the US Department of Veterans Affairs (VA) West Los Angeles VA Medical Center (WLAVAMC). His psychiatric history included generalized anxiety disorder, posttraumatic stress disorder (PTSD), and panic disorder, managed by the Psychiatry Service and treated with sertraline 25 mg daily, lorazepam 0.5 mg twice daily, and hydroxyzine 20 mg nightly. He had previously implemented lifestyle changes and attended MOVE! classes and nutrition coaching for 1 year but was unsuccessful in losing weight. He had also tried liraglutide 3 mg daily for weight loss but was unable to tolerate it and reported worsening medication-related anxiety.
The patient declined further weight loss pharmacotherapy and was referred to bariatric surgery. He was scheduled for a surgical sleeve gastrectomy. However, on the day he arrived at the hospital for surgery, he developed severe anxiety and had a panic attack, and it was canceled. Due to his mental health issues, he was no longer comfortable proceeding with surgery and was left without other options for obesity treatment. The veteran was extremely disappointed because the ventral hernia caused significant quality of life impairment, limited his ability to exercise, and caused him embarrassment in public settings. The hernia could not be surgically repaired until there was significant weight loss.
A bariatric endoscopy program within the Division of Gastroenterology was developed and implemented at the WLAVAMC in February 2023 in conjunction with MOVE! The patient was referred for consideration of an endoscopic weight loss procedure. He was determined to be a suitable candidate for ESG based on his BMI being > 40 and personal preference not to proceed with surgery to lose enough weight to qualify for hernia repair. The veteran underwent an endoscopy, which showed normal anatomy and gastric mucosa. ESG was performed in standard fashion (Figure).8 Three vertical lines were made using argon plasma coagulation from the incisura to 2 cm below the gastroesophageal junction along the anterior, posterior, and greater curvature of the stomach to mark the area for endoscopic suture placement. Starting at the incisura, 7 full-thickness sutures were placed to create a volume reduction plication, with preservation of the fundus. The patient did well postprocedure with no immediate or delayed AEs and was discharged home the same day.
Follow-up
The veteran followed a gradual dietary advancement from a clear liquid diet to pureed and soft texture food. The patient’s weight dropped from 359 lbs preprocedure to 304 lbs 6 months postprocedure, a total body weight loss (TWBL) of 15.3%. At 12 months the veteran weighed 299 lbs (16.7% TBWL). He also had notable improvements in metabolic parameters. His systolic blood pressure decreased from ≥ 140 mm Hg to 120 to 130 mm Hg and hemoglobin A1c dropped from 7.0% to 6.3%. Remarkably, his psychiatrist noted significant improvement in his overall mental health. The veteran reported complete cessation of panic attacks since the ESG, improvements in PTSD and anxiety, and was able to discontinue lorazepam and decrease his dose of sertraline to 12.5 mg daily. He reported feeling more energetic and goal-oriented with increased clarity of thought. Perhaps the most significant outcome was that after the 55-lb weight loss at 6 months, the patient was eligible to undergo ventral hernia surgical repair, which had previously contributed to shame and social isolation. This, in turn, improved his quality of life, allowed him to start walking again, up to 8 miles daily, and to feel comfortable again going out in public settings.
DISCUSSION
Bariatric surgeries are an effective method of achieving weight loss and improving obesity-related comorbidities. However, only a small percentage of individuals with obesity are candidates for bariatric surgery. Given the dramatic increase in the prevalence of obesity, other options are needed. Specifically, within the VA, an estimated 80% of veterans are overweight or obese, but only about 500 bariatric surgeries are performed annually.9 With the need for additional weight loss therapies, VA programs are starting to offer endoscopic bariatric procedures as an alternative option. This may be a desirable choice for patients with obesity (BMI > 30), with or without associated metabolic comorbidities, who need more aggressive intervention beyond dietary and lifestyle changes and are either not interested in or not eligible for bariatric surgery or weight loss medications.
Although there is evidence that metabolic comorbidities are associated with obesity, there has been less research on obesity and mental health comorbidities such as depression and anxiety. These psychiatric conditions may even be more common among patients seeking weight loss procedures and more prominent in certain groups such as veterans, which may ultimately exclude these patients from bariatric surgery.10 Prior studies suggest that bariatric surgery can reduce the severity of depression and, to a lesser extent, anxiety symptoms at 2 years following the initial surgery; however, there is limited literature describing the impact of weight loss procedure on panic disorders.11-14 We suspect that a weight loss procedure such as ESG may have indirectly improved the veteran’s mood disorder due to the weight loss it induced, increasing the ability to exercise, quality of sleep, and participation in public settings.
This case highlights a veteran who did not tolerate weight loss medication and had severe anxiety and PTSD that prevented him from going through with bariatric surgery. He then underwent an endoscopic weight loss procedure. The ESG helped him successfully achieve significant weight loss, increase his physical activity, reduce his anxiety and panic disorder, and overall, significantly improve his quality of life. More than 1 year after the procedure, the patient has sustained improvements in his psychiatric and emotional health along with durable weight loss, maintaining > 15% of his total weight lost. Additional studies are needed to further understand the prevalence and long-term outcomes of mental health comorbidities, as well as weight loss outcomes in this group of patients who undergo endoscopic bariatric procedures.
CONCLUSIONS
We describe a case of a veteran with severe obesity and significant psychiatric comorbidities that prevented him from undergoing bariatric surgery, who underwent an ESG. This procedure led to significant weight loss, improvement of metabolic parameters, reduction in anxiety and PTSD, and enhancement of his quality of life. This case emphasizes the unique advantages of ESG and supports the expansion of endoscopic bariatric programs in the VA.
Obesity is a growing worldwide epidemic with significant implications for individual health and public health care costs. It is also associated with several medical conditions, including diabetes, cardiovascular disease, cancer, and mental health disorders.1 Comprehensive lifestyle intervention is a first-line therapy for obesity consisting of dietary and exercise interventions. Despite initial success, long-term results and durability of weight loss with lifestyle modifications are limited. 2 Bariatric surgery, including sleeve gastrectomy and gastric bypass surgery, is a more invasive approach that is highly effective in weight loss. However, these operations are not reversible, and patients may not be eligible for or may not desire surgery. Overall, bariatric surgery is widely underutilized, with < 1% of eligible patients ultimately undergoing surgery.3,4
Endoscopic bariatric therapies are increasingly popular procedures that address the need for additional treatments for obesity among individuals who have not had success with lifestyle changes and are not surgical candidates. The most common procedure is the endoscopic sleeve gastroplasty (ESG), which applies full-thickness sutures in the stomach to reduce gastric volume, delay gastric emptying, and limit food intake while keeping the fundus intact compared with sleeve gastrectomy. This procedure is typically considered in patients with body mass index (BMI) ≥ 30, who do not qualify for or do not want traditional bariatric surgery. The literature supports robust outcomes after ESG, with studies demonstrating significant and sustained total body weight loss of up to 14% to 16% at 5 years and significant improvement in ≥ 1 metabolic comorbidities in 80% of patients.5,6 ESG adverse events (AEs) include abdominal pain, nausea, and vomiting that are typically self-limited to 1 week. Rarer but more serious AEs include bleeding, perforation, or infection, and occur in 2% of cases based on large trial data.5,7
Although the weight loss benefits of ESG are well established, to date, there are limited data on the effects of endoscopic bariatric therapies like ESG on mental health conditions. Here, we describe a case of a veteran with a history of mental health disorders that prevented him from completing bariatric surgery. The patient underwent ESG and had a successful clinical course.
CASE PRESENTATION
A 59-year-old male veteran with a medical history of class III obesity (42.4 BMI), obstructive sleep apnea, hypothyroidism, hypertension, type 2 diabetes mellitus, and a large ventral hernia was referred to the MOVE! (Management of Overweight/ Obese Veterans Everywhere!) multidisciplinary high-intensity weight loss program at the US Department of Veterans Affairs (VA) West Los Angeles VA Medical Center (WLAVAMC). His psychiatric history included generalized anxiety disorder, posttraumatic stress disorder (PTSD), and panic disorder, managed by the Psychiatry Service and treated with sertraline 25 mg daily, lorazepam 0.5 mg twice daily, and hydroxyzine 20 mg nightly. He had previously implemented lifestyle changes and attended MOVE! classes and nutrition coaching for 1 year but was unsuccessful in losing weight. He had also tried liraglutide 3 mg daily for weight loss but was unable to tolerate it and reported worsening medication-related anxiety.
The patient declined further weight loss pharmacotherapy and was referred to bariatric surgery. He was scheduled for a surgical sleeve gastrectomy. However, on the day he arrived at the hospital for surgery, he developed severe anxiety and had a panic attack, and it was canceled. Due to his mental health issues, he was no longer comfortable proceeding with surgery and was left without other options for obesity treatment. The veteran was extremely disappointed because the ventral hernia caused significant quality of life impairment, limited his ability to exercise, and caused him embarrassment in public settings. The hernia could not be surgically repaired until there was significant weight loss.
A bariatric endoscopy program within the Division of Gastroenterology was developed and implemented at the WLAVAMC in February 2023 in conjunction with MOVE! The patient was referred for consideration of an endoscopic weight loss procedure. He was determined to be a suitable candidate for ESG based on his BMI being > 40 and personal preference not to proceed with surgery to lose enough weight to qualify for hernia repair. The veteran underwent an endoscopy, which showed normal anatomy and gastric mucosa. ESG was performed in standard fashion (Figure).8 Three vertical lines were made using argon plasma coagulation from the incisura to 2 cm below the gastroesophageal junction along the anterior, posterior, and greater curvature of the stomach to mark the area for endoscopic suture placement. Starting at the incisura, 7 full-thickness sutures were placed to create a volume reduction plication, with preservation of the fundus. The patient did well postprocedure with no immediate or delayed AEs and was discharged home the same day.
Follow-up
The veteran followed a gradual dietary advancement from a clear liquid diet to pureed and soft texture food. The patient’s weight dropped from 359 lbs preprocedure to 304 lbs 6 months postprocedure, a total body weight loss (TWBL) of 15.3%. At 12 months the veteran weighed 299 lbs (16.7% TBWL). He also had notable improvements in metabolic parameters. His systolic blood pressure decreased from ≥ 140 mm Hg to 120 to 130 mm Hg and hemoglobin A1c dropped from 7.0% to 6.3%. Remarkably, his psychiatrist noted significant improvement in his overall mental health. The veteran reported complete cessation of panic attacks since the ESG, improvements in PTSD and anxiety, and was able to discontinue lorazepam and decrease his dose of sertraline to 12.5 mg daily. He reported feeling more energetic and goal-oriented with increased clarity of thought. Perhaps the most significant outcome was that after the 55-lb weight loss at 6 months, the patient was eligible to undergo ventral hernia surgical repair, which had previously contributed to shame and social isolation. This, in turn, improved his quality of life, allowed him to start walking again, up to 8 miles daily, and to feel comfortable again going out in public settings.
DISCUSSION
Bariatric surgeries are an effective method of achieving weight loss and improving obesity-related comorbidities. However, only a small percentage of individuals with obesity are candidates for bariatric surgery. Given the dramatic increase in the prevalence of obesity, other options are needed. Specifically, within the VA, an estimated 80% of veterans are overweight or obese, but only about 500 bariatric surgeries are performed annually.9 With the need for additional weight loss therapies, VA programs are starting to offer endoscopic bariatric procedures as an alternative option. This may be a desirable choice for patients with obesity (BMI > 30), with or without associated metabolic comorbidities, who need more aggressive intervention beyond dietary and lifestyle changes and are either not interested in or not eligible for bariatric surgery or weight loss medications.
Although there is evidence that metabolic comorbidities are associated with obesity, there has been less research on obesity and mental health comorbidities such as depression and anxiety. These psychiatric conditions may even be more common among patients seeking weight loss procedures and more prominent in certain groups such as veterans, which may ultimately exclude these patients from bariatric surgery.10 Prior studies suggest that bariatric surgery can reduce the severity of depression and, to a lesser extent, anxiety symptoms at 2 years following the initial surgery; however, there is limited literature describing the impact of weight loss procedure on panic disorders.11-14 We suspect that a weight loss procedure such as ESG may have indirectly improved the veteran’s mood disorder due to the weight loss it induced, increasing the ability to exercise, quality of sleep, and participation in public settings.
This case highlights a veteran who did not tolerate weight loss medication and had severe anxiety and PTSD that prevented him from going through with bariatric surgery. He then underwent an endoscopic weight loss procedure. The ESG helped him successfully achieve significant weight loss, increase his physical activity, reduce his anxiety and panic disorder, and overall, significantly improve his quality of life. More than 1 year after the procedure, the patient has sustained improvements in his psychiatric and emotional health along with durable weight loss, maintaining > 15% of his total weight lost. Additional studies are needed to further understand the prevalence and long-term outcomes of mental health comorbidities, as well as weight loss outcomes in this group of patients who undergo endoscopic bariatric procedures.
CONCLUSIONS
We describe a case of a veteran with severe obesity and significant psychiatric comorbidities that prevented him from undergoing bariatric surgery, who underwent an ESG. This procedure led to significant weight loss, improvement of metabolic parameters, reduction in anxiety and PTSD, and enhancement of his quality of life. This case emphasizes the unique advantages of ESG and supports the expansion of endoscopic bariatric programs in the VA.
- Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr Metab Cardiovasc Dis. 2007;17(4):319-326. doi:10.1016/j.numecd.2006.07.005
- Bray GA, Kim KK, Wilding JPH; World Obesity Federation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev. 2017;18(7):715-723. doi:10.1111/obr.12551
- Imbus JR, Voils CI, Funk LM. Bariatric surgery barriers: a review using andersen’s model of health services use. Surg Obes Relat Dis. 2018;14(3):404-412. doi:10.1016/j.soard.2017.11.012
- Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: a meta-analysis. JAMA. 2016;315(2):150- 163. doi:10.1001/jama.2015.18118
- Abu Dayyeh BK, Bazerbachi F, Vargas EJ, et al.. Endoscopic sleeve gastroplasty for treatment of class 1 and 2 obesity (MERIT): a prospective, multicentre, randomised trial. Lancet. 2022;400(10350):441-451. doi:10.1016/S0140-6736(22)01280-6
- Matteo MV, Bove V, Ciasca G, et al. Success predictors of endoscopic sleeve gastroplasty. Obes Surg. 2024;34(5):1496-1504. doi:10.1007/s11695-024-07109-4
- Maselli DB, Hoff AC, Kucera A, et al. Endoscopic sleeve gastroplasty in class III obesity: efficacy, safety, and durability outcomes in 404 consecutive patients. World J Gastrointest Endosc. 2023;15(6):469-479. doi:10.4253/wjge.v15.i6.469
- Kumar N, Abu Dayyeh BK, Lopez-Nava Breviere G, et al. Endoscopic sutured gastroplasty: procedure evolution from first-in-man cases through current technique. Surg Endosc. 2018;32(4):2159-2164. doi:10.1007/s00464-017-5869-2
- Maggard-Gibbons M, Shekelle PG, Girgis MD, et al. Endoscopic Bariatric Interventions versus lifestyle interventions or surgery for weight loss in patients with obesity: a systematic review and meta-analysis. Department of Veterans Affairs (US); 2022. https://www.ncbi.nlm.nih.gov/books/NBK587943/
- Maggard Gibbons MA, Maher AM, Dawes AJ, et al. Psychological clearance for bariatric surgery: a systematic review. VA-ESP project #05-2262014.
- van Hout GC, Verschure SK, van Heck GL. Psychosocial predictors of success following bariatric surgery. Obes Surg. 2005;15(4):552-560. doi:10.1381/0960892053723484
- Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the national comorbidity survey replication. Biol Psychiatry. 2007;61(3):348-358. doi:10.1016/j.biopsych.2006.03.040
- Aylward L, Lilly C, Konsor M, et al. How soon do depression and anxiety symptoms improve after bariatric surgery?. Healthcare (Basel). 2023;11(6):862. doi:10.3390/healthcare11060862
- Law S, Dong S, Zhou F, Zheng D, Wang C, Dong Z. Bariatric surgery and mental health outcomes: an umbrella review. Front Endocrinol (Lausanne). 2023;14:1283621. doi:10.3389/fendo.2023.1283621
- Ritchie SA, Connell JM. The link between abdominal obesity, metabolic syndrome and cardiovascular disease. Nutr Metab Cardiovasc Dis. 2007;17(4):319-326. doi:10.1016/j.numecd.2006.07.005
- Bray GA, Kim KK, Wilding JPH; World Obesity Federation. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev. 2017;18(7):715-723. doi:10.1111/obr.12551
- Imbus JR, Voils CI, Funk LM. Bariatric surgery barriers: a review using andersen’s model of health services use. Surg Obes Relat Dis. 2018;14(3):404-412. doi:10.1016/j.soard.2017.11.012
- Dawes AJ, Maggard-Gibbons M, Maher AR, et al. Mental health conditions among patients seeking and undergoing bariatric surgery: a meta-analysis. JAMA. 2016;315(2):150- 163. doi:10.1001/jama.2015.18118
- Abu Dayyeh BK, Bazerbachi F, Vargas EJ, et al.. Endoscopic sleeve gastroplasty for treatment of class 1 and 2 obesity (MERIT): a prospective, multicentre, randomised trial. Lancet. 2022;400(10350):441-451. doi:10.1016/S0140-6736(22)01280-6
- Matteo MV, Bove V, Ciasca G, et al. Success predictors of endoscopic sleeve gastroplasty. Obes Surg. 2024;34(5):1496-1504. doi:10.1007/s11695-024-07109-4
- Maselli DB, Hoff AC, Kucera A, et al. Endoscopic sleeve gastroplasty in class III obesity: efficacy, safety, and durability outcomes in 404 consecutive patients. World J Gastrointest Endosc. 2023;15(6):469-479. doi:10.4253/wjge.v15.i6.469
- Kumar N, Abu Dayyeh BK, Lopez-Nava Breviere G, et al. Endoscopic sutured gastroplasty: procedure evolution from first-in-man cases through current technique. Surg Endosc. 2018;32(4):2159-2164. doi:10.1007/s00464-017-5869-2
- Maggard-Gibbons M, Shekelle PG, Girgis MD, et al. Endoscopic Bariatric Interventions versus lifestyle interventions or surgery for weight loss in patients with obesity: a systematic review and meta-analysis. Department of Veterans Affairs (US); 2022. https://www.ncbi.nlm.nih.gov/books/NBK587943/
- Maggard Gibbons MA, Maher AM, Dawes AJ, et al. Psychological clearance for bariatric surgery: a systematic review. VA-ESP project #05-2262014.
- van Hout GC, Verschure SK, van Heck GL. Psychosocial predictors of success following bariatric surgery. Obes Surg. 2005;15(4):552-560. doi:10.1381/0960892053723484
- Hudson JI, Hiripi E, Pope HG Jr, Kessler RC. The prevalence and correlates of eating disorders in the national comorbidity survey replication. Biol Psychiatry. 2007;61(3):348-358. doi:10.1016/j.biopsych.2006.03.040
- Aylward L, Lilly C, Konsor M, et al. How soon do depression and anxiety symptoms improve after bariatric surgery?. Healthcare (Basel). 2023;11(6):862. doi:10.3390/healthcare11060862
- Law S, Dong S, Zhou F, Zheng D, Wang C, Dong Z. Bariatric surgery and mental health outcomes: an umbrella review. Front Endocrinol (Lausanne). 2023;14:1283621. doi:10.3389/fendo.2023.1283621
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Endoscopic Sleeve Gastroplasty is an Effective Treatment for Obesity in a Veteran With Metabolic and Psychiatric Comorbidities
Anti-Tumor Necrosis Factor Treatment for Glomerulopathy: Case Report and Review of Literature
Podocytes are terminally differentiated, highly specialized cells located in juxtaposition to the basement membrane over the abluminal surfaces of endothelial cells within the glomerular tuft. This triad structure is the site of the filtration barrier, which forms highly delicate and tightly regulated architecture to carry out the ultrafiltration function of the kidney.1 The filtration barrier is characterized by foot processes that are connected by specialized junctions called slit diaphragms.
Insults to components of the filtration barrier can initiate cascading events and perpetuate structural alterations that may eventually result in sclerotic changes.2 Common causes among children include minimal change disease (MCD) with the collapse of foot processes resulting in proteinuria, Alport syndrome due to mutation of collagen fibers within the basement membrane leading to hematuria and proteinuria, immune complex mediated nephropathy following common infections or autoimmune diseases, and focal segmental glomerulosclerosis (FSGS) that can show variable histopathology toward eventual glomerular scarring.3,4 These children often clinically have minimal, if any, signs of systemic inflammation.3-5 This has been a limiting factor for the commitment to immunomodulatory treatment, except for steroids for the treatment of MCD.6 Although prolonged steroid treatment may be efficacious, adverse effects are significant in a growing child. Alternative treatments, such as tacrolimus and rituximab have been suggested as second-line steroid-sparing agents.7,8 Not uncommonly, however, these cases are managed by supportive measures only during the progression of the natural course of the disease, which may eventually lead to renal failure, requiring transplant for survival.8,9
This case report highlights a child with a variant of uncertain significance (VUS) in genes involved in Alport syndrome and FSGS who developed an abrupt onset of proteinuria and hematuria after a respiratory illness. To our knowledge, he represents the youngest case demonstrating the benefit of targeted treatment against tumor necrosis factor-α (TNF-α) for glomerulopathy using biologic response modifiers.
Case Description
This is currently a 7-year-old male patient who was born at 39 weeks gestation to gravida 3 para 3 following induced labor due to elevated maternal blood pressure. During the first 2 years of life, his growth and development were normal and his immunizations were up to date. The patient's medical history included upper respiratory tract infections (URIs), respiratory syncytial virus, as well as 3 bouts of pneumonia and multiple otitis media that resulted in 18 rounds of antibiotics. The child was also allergic to nuts and milk protein. The patient’s parents are of Northern European and Native American descent. There is no known family history of eye, ear, or kidney diseases.
Renal concerns were first noted at the age of 2 years and 6 months when he presented to an emergency department in Fall 2019 (week 0) for several weeks of intermittent dark-colored urine. His mother reported that the discoloration recently progressed in intensity to cola-colored, along with the onset of persistent vomiting without any fever or diarrhea. On physical examination, the patient had normal vitals: weight 14.8 kg (68th percentile), height 91 cm (24th percentile), and body surface area 0.6 m2. There was no edema, rash, or lymphadenopathy, but he appeared pale.
The patient’s initial laboratory results included: complete blood count with white blood cells (WBC) 10 x 103/L (reference range, 4.5-13.5 x 103/L); differential lymphocytes 69%; neutrophils 21%; hemoglobin 10 g/dL (reference range, 12-16 g/dL); hematocrit, 30%; (reference range, 37%-45%); platelets 437 103/L (reference range, 150-450 x 103/L); serum creatinine 0.46 mg/dL (reference range, 0.5-0.9 mg/dL); and albumin 3.1 g/dL (reference range, 3.5-5.2 g/dL). Serum electrolyte levels and liver enzymes were normal. A urine analysis revealed 3+ protein and 3+ blood with dysmorphic red blood cells (RBC) and RBC casts without WBC. The patient's spot urine protein-to-creatinine ratio was 4.3 and his renal ultrasound was normal. The patient was referred to Nephrology.
During the next 2 weeks, his protein-to-creatinine ratio progressed to 5.9 and serum albumin fell to 2.7 g/dL. His urine remained red colored, and a microscopic examination with RBC > 500 and WBC up to 10 on a high powered field. His workup was negative for antinuclear antibodies, antineutrophil cytoplasmic antibody, antistreptolysin-O (ASO) and anti-DNase B. Serum C3 was low at 81 mg/dL (reference range, 90-180 mg/dL), C4 was 13.3 mg/dL (reference range, 10-40 mg/dL), and immunoglobulin G was low at 452 mg/dL (reference range 719-1475 mg/dL). A baseline audiology test revealed normal hearing.
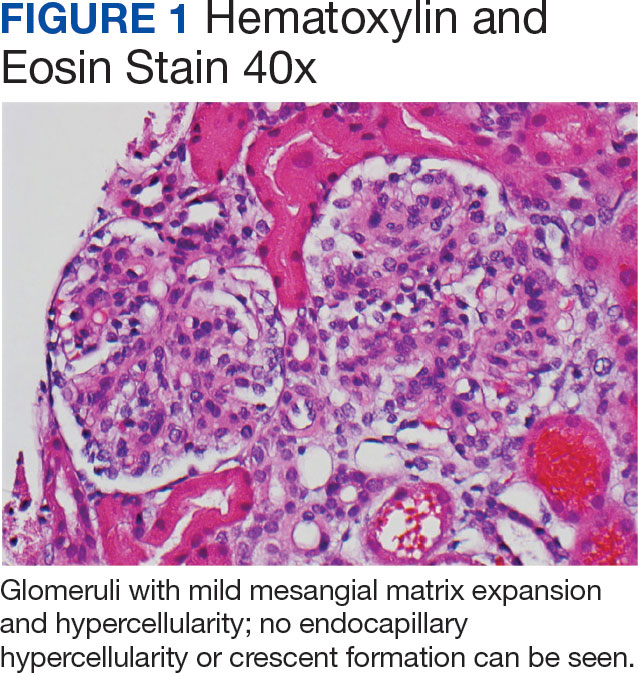
Percutaneous renal biopsy yielded about 12 glomeruli, all exhibiting mild mesangial matrix expansion and hypercellularity (Figure 1). One glomerulus had prominent parietal epithelial cells without endocapillary hypercellularity or crescent formation. There was no interstitial fibrosis or tubular atrophy. Immunofluorescence studies showed no evidence of immune complex deposition with negative staining for immunoglobulin heavy and light chains, C3 and C1q. Staining for α 2 and α 5 units of collagen was normal. Electron microscopy showed patchy areas of severe basement membrane thinning with frequent foci of mild to moderate lamina densa splitting and associated visceral epithelial cell foot process effacement (Figure 2).
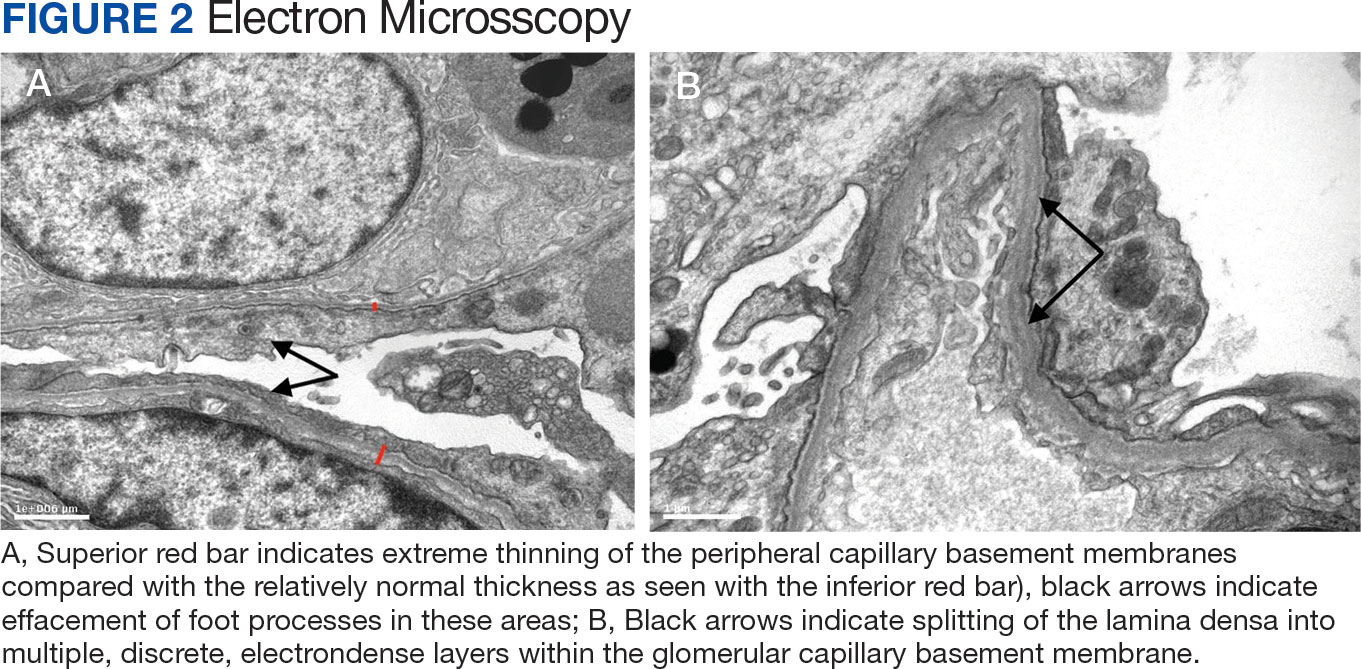
These were reported as concerning findings for possible Alport syndrome by 3 independent pathology teams. The genetic testing was submitted at a commercial laboratory to screen 17 mutations, including COL4A3, COL4A4, and COL4A5. Results showed the presence of a heterozygous VUS in the COL4A4 gene (c.1055C > T; p.Pro352Leu; dbSNP ID: rs371717486; PolyPhen-2: Probably Damaging; SIFT: Deleterious) as well as the presence of a heterozygous VUS in TRPC6 gene (c2463A>T; p.Lys821Asn; dbSNP ID: rs199948731; PolyPhen-2: Benign; SIFT: Tolerated). Further genetic investigation by whole exome sequencing on approximately 20,000 genes through MNG Laboratories showed a new heterozygous VUS in the OSGEP gene [c.328T>C; p.Cys110Arg]. Additional studies ruled out mitochondrial disease, CoQ10 deficiency, and metabolic disorders upon normal findings for mitochondrial DNA, urine amino acids, plasma acylcarnitine profile, orotic acid, ammonia, and homocysteine levels.
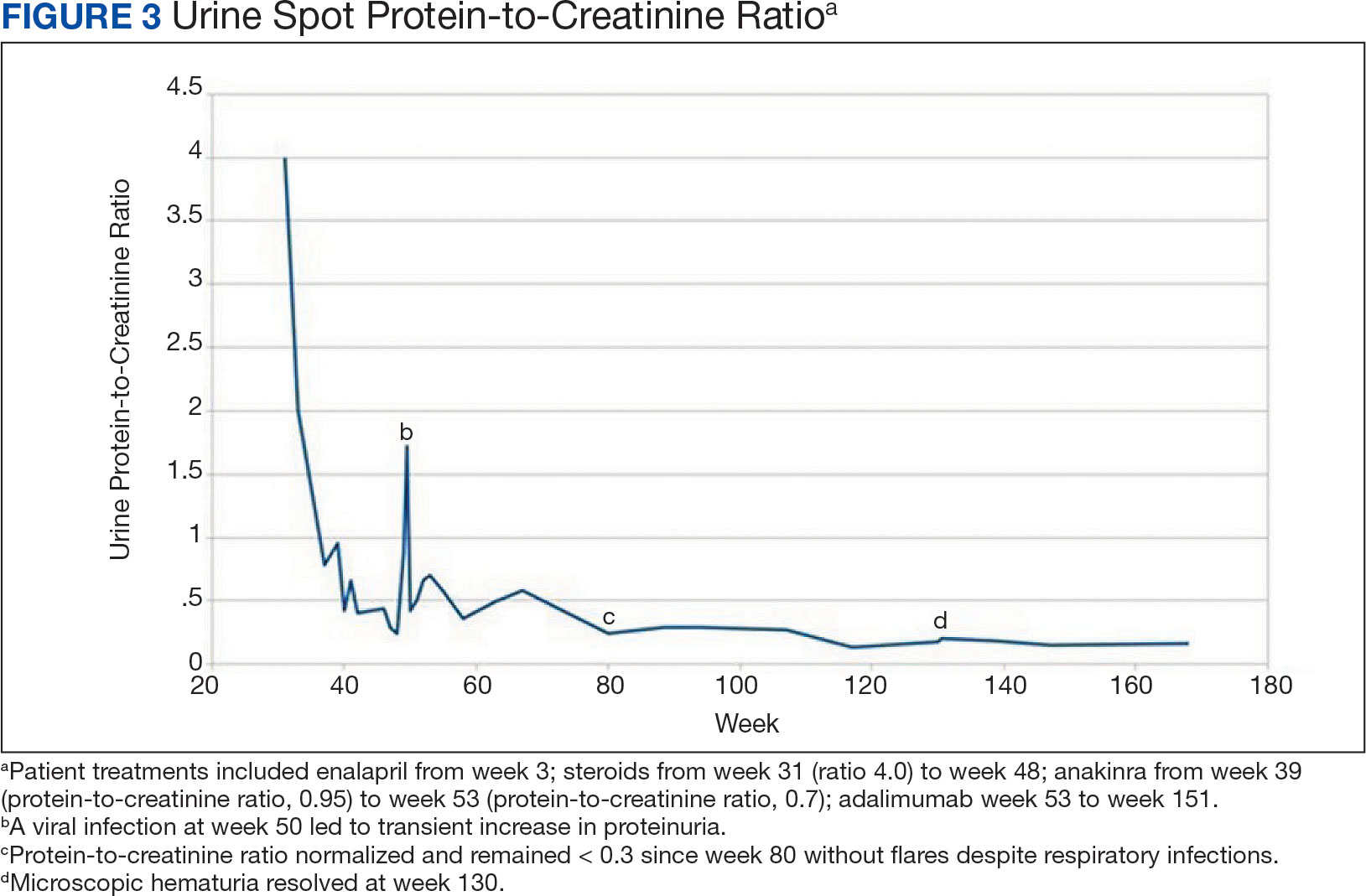
Figure 3 summarizes the patient’s treatment response during 170 weeks of follow-up (Fall 2019 to Summer 2023). The patient was started on enalapril 0.6 mg/kg daily at week 3, which continued throughout treatment. Following a rheumatology consult at week 30, the patient was started on prednisolone 3 mg/mL to assess the role of inflammation through the treatment response. An initial dose of 2 mg/kg daily (9 mL) for 1 month was followed by every other day treatment that was tapered off by week 48. To control mild but noticeably increasing proteinuria in the interim, subcutaneous anakinra 50 mg (3 mg/kg daily) was added as a steroid
DISCUSSION
This case describes a child with rapidly progressive proteinuria and hematuria following a URI who was found to have VUS mutations in 3 different genes associated with chronic kidney disease. Serology tests on the patient were negative for streptococcal antibodies and antinuclear antibodies, ruling out poststreptococcal glomerulonephritis, or systemic lupus erythematosus. His renal biopsy findings were concerning for altered podocytes, mesangial cells, and basement membrane without inflammatory infiltrate, immune complex, complements, immunoglobulin A, or vasculopathy. His blood inflammatory markers, erythrocyte sedimentation rate, C-reactive protein, and ferritin were normal when his care team initiated daily steroids.
Overall, the patient’s clinical presentation and histopathology findings were suggestive of Alport syndrome or thin basement membrane nephropathy with a high potential to progress into FSGS.10-12 Alport syndrome affects 1 in 5000 to 10,000 children annually due to S-linked inheritance of COL4A5, or autosomal recessive inheritance of COL4A3 or COL4A4 genes. It presents with hematuria and hearing loss.10 Our patient had a single copy COL4A4 gene mutation that was classified as VUS. He also had 2 additional VUS affecting the TRPC6 and OSGEP genes. TRPC6 gene mutation can be associated with FSGS through autosomal dominant inheritance. Both COL4A4 and TRPC6 gene mutations were paternally inherited. Although the patient’s father not having renal disease argues against the clinical significance of these findings, there is literature on the potential role of heterozygous COL4A4 variant mimicking thin basement membrane nephropathy that can lead to renal impairment upon copresence of superimposed conditions.13 The patient’s rapidly progressing hematuria and changes in the basement membrane were worrisome for emerging FSGS. Furthermore, VUS of TRPC6 has been reported in late onset autosomal dominant FSGS and can be associated with early onset steroid-resistant nephrotic syndrome (NS) in children.14 This concern was voiced by 3 nephrology consultants during the initial evaluation, leading to the consensus that steroid treatment for podocytopathy would not alter the patient’s long-term outcomes (ie, progression to FSGS).
Immunomodulation
Our rationale for immunomodulatory treatment was based on the abrupt onset of renal concerns following a URI, suggesting the importance of an inflammatory trigger causing altered homeostasis in a genetically susceptible host. Preclinical models show that microbial products such as lipopolysaccharides can lead to podocytopathy by several mechanisms through activation of toll-like receptor signaling. It can directly cause apoptosis by downregulation of the intracellular Akt survival pathway.15 Lipopolysaccharide can also activate the NF-αB pathway and upregulate the production of interleukin-1 (IL-1) and TNF-α in mesangial cells.16,17
Both cytokines can promote mesangial cell proliferation.18 Through autocrine and paracrine mechanisms, proinflammatory cytokines can further perpetuate somatic tissue changes and contribute to the development of podocytopathy. For instance, TNF-α can promote podocyte injury and proteinuria by downregulation of the slit diaphragm protein expression (ie, nephrin, ezrin, or podocin), and disruption of podocyte cytoskeleton.19,20 TNF-α promotes the influx and activation of macrophages and inflammatory cells. It is actively involved in chronic alterations within the glomeruli by the upregulation of matrix metalloproteases by integrins, as well as activation of myofibroblast progenitors and extracellular matrix deposition in crosstalk with transforming growth factor and other key mediators.17,21,22
For the patient described in this case report, initial improvement on steroids encouraged the pursuit of additional treatment to downregulate inflammatory pathways within the glomerular milieu. However, within the COVID-19 environment, escalating the patient’s treatment using traditional immunomodulators (ie, calcineurin inhibitors or mycophenolate mofetil) was not favored due to the risk of infection. Initially, anakinra, a recombinant IL-1 receptor antagonist, was preferred as a steroid-sparing agent for its short life and safety profile during the pandemic. At first, the patient responded well to anakinra and was allowed a steroid wean when the dose was titrated up to 6 mg/kg daily. However, anakinra did not prevent the escalation of proteinuria following a URI. After the treatment was changed to adalimumab, a fully humanized monoclonal antibody to TNF-α, the patient continued to improve and reach full remission despite experiencing a cold and the flu in the following months.
Literature Review
There is a paucity of literature on applications of biological response modifiers for idiopathic NS and FSGS.23,24 Angeletti and colleagues reported that 3 patients with severe long-standing FSGS benefited from anakinra 4 mg/kg daily to reduce proteinuria and improve kidney function. All the patients had positive C3 staining in renal biopsy and treatment response, which supported the role of C3a in inducing podocyte injury through upregulated expression of IL-1 and IL-1R.23 Trachtman and colleagues reported on the phase II FONT trial that included 14 of 21 patients aged < 18 years with advanced FSGS who were treated with adalimumab 24 mg/m2, or ≤ 40 mg every other week.24 Although, during a 6-month period, none of the 7 patients met the endpoint of reduced proteinuria by ≥ 50%, and the authors suggested that careful patient selection may improve the treatment response in future trials.24
A recent study involving transcriptomics on renal tissue samples combined with available pathology (fibrosis), urinary markers, and clinical characteristics on 285 patients with MCD or FSGS from 3 different continents identified 3 distinct clusters. Patients with evidence of activated kidney TNF pathway (n = 72, aged > 18 years) were found to have poor clinical outcomes.25 The study identified 2 urine markers associated with the TNF pathway (ie, tissue inhibitor of metalloproteinases-1 and monocyte chemoattractant protein-1), which aligns with the preclinical findings previously mentioned.25
Conclusions
The patient’s condition in this case illustrates the complex nature of biologically predetermined cascading events in the emergence of glomerular disease upon environmental triggers under the influence of genetic factors.
Chronic kidney disease affects 7.7% of veterans annually, illustrating the need for new therapeutics.26 Based on our experience and literature review, upregulation of TNF-α is a root cause of glomerulopathy; further studies are warranted to evaluate the efficacy of anti-TNF biologic response modifiers for the treatment of these patients. Long-term postmarketing safety profile and steroid-sparing properties of adalimumab should allow inclusion of pediatric cases in future trials. Results may also contribute to identifying new predictive biomarkers related to the basement membrane when combined with precision nephrology to further advance patient selection and targeted treatment.25,27
Acknowledgments
The authors thank the patient’s mother for providing consent to allow publication of this case report.
1. Arif E, Nihalani D. Glomerular filtration barrier assembly: an insight. Postdoc J. 2013;1(4):33-45.
2. Garg PA. Review of podocyte biology. Am J Nephrol. 2018;47(suppl 1):3-13. doi:10.1159/000481633SUPPL
3. Warady BA, Agarwal R, Bangalore S, et al. Alport syndrome classification and management. Kidney Med. 2020;2(5):639-649. doi:10.1016/j.xkme.2020.05.014
4. Angioi A, Pani A. FSGS: from pathogenesis to the histological lesion. J Nephrol. 2016;29(4):517-523. doi:10.1007/s40620-016-0333-2
5. Roca N, Martinez C, Jatem E, Madrid A, Lopez M, Segarra A. Activation of the acute inflammatory phase response in idiopathic nephrotic syndrome: association with clinicopathological phenotypes and with response to corticosteroids. Clin Kidney J. 2021;14(4):1207-1215. doi:10.1093/ckj/sfaa247
6. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. 2017;12(2):332-345.
7. Medjeral-Thomas NR, Lawrence C, Condon M, et al. Randomized, controlled trial of tacrolimus and prednisolone monotherapy for adults with De Novo minimal change disease: a multicenter, randomized, controlled trial. Clin J Am Soc Nephrol. 2020;15(2):209-218. doi:10.2215/CJN.06290420
8. Ye Q, Lan B, Liu H, Persson PB, Lai EY, Mao J. A critical role of the podocyte cytoskeleton in the pathogenesis of glomerular proteinuria and autoimmune podocytopathies. Acta Physiol (Oxf). 2022;235(4):e13850. doi:10.1111/apha.13850
9. Trautmann A, Schnaidt S, Lipska-Ziμtkiewicz BS, et al. Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol. 2017;28:3055-3065. doi:10.1681/ASN.2016101121
10. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):711-719. doi:10.1007/s00467-020-04819-6
11. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169-78. doi:10.1046/j.1523-1755.2003.00234.x
12. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2017; 12(3):502-517. doi:10.2215/CJN.05960616
13. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant? Kidney Int Rep. 2018;3(6):1239-1241. doi:10.1016/j.ekir.2018.08.002
14. Gigante M, Caridi G, Montemurno E, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626-1634. doi:10.2215/CJN.07830910
15. Saurus P, Kuusela S, Lehtonen E, et al. Podocyte apoptosis is prevented by blocking the toll-like receptor pathway. Cell Death Dis. 2015;6(5):e1752. doi:10.1038/cddis.2015.125
16. Baud L, Oudinet JP, Bens M, et al. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35(5):1111-1118. doi:10.1038/ki.1989.98
17. White S, Lin L, Hu K. NF-κB and tPA signaling in kidney and other diseases. Cells. 2020;9(6):1348. doi:10.3390/cells9061348
18. Tesch GH, Lan HY, Atkins RC, Nikolic-Paterson DJ. Role of interleukin-1 in mesangial cell proliferation and matrix deposition in experimental mesangioproliferative nephritis. Am J Pathol. 1997;151(1):141-150.
19. Lai KN, Leung JCK, Chan LYY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA Nephropathy. Nephrol Dial Transplant. 2009;24(1):62-72. doi:10.1093/ndt/gfn441
20. Saleem MA, Kobayashi Y. Cell biology and genetics of minimal change disease. F1000 Res. 2016;5: F1000 Faculty Rev-412. doi:10.12688/f1000research.7300.1
21. Kim KP, Williams CE, Lemmon CA. Cell-matrix interactions in renal fibrosis. Kidney Dial. 2022;2(4):607-624. doi:10.3390/kidneydial2040055
22. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8(3):210. doi:10.1186/ar1963
23. Angeletti A, Magnasco A, Trivelli A, et al. Refractory minimal change disease and focal segmental glomerular sclerosis treated with Anakinra. Kidney Int Rep. 2021;7(1):121-124. doi:10.1016/j.ekir.2021.10.018
24. Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol. 2015;16:111. doi:10.1186/s12882-015-0094-5
25. Mariani LH, Eddy S, AlAkwaa FM, et al. Precision nephrology identified tumor necrosis factor activation variability in minimal change disease and focal segmental glomerulosclerosis. Kidney Int. 2023;103(3):565-579. doi:10.1016/j.kint.2022.10.023
26. Korshak L, Washington DL, Powell J, Nylen E, Kokkinos P. Kidney Disease in Veterans. US Dept of Veterans Affairs, Office of Health Equity. Updated May 13, 2020. Accessed June 28, 2024. https://www.va.gov/HEALTHEQUITY/Kidney_Disease_In_Veterans.asp
27. Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253-1259. doi:10.1038/ki.2014.305
Podocytes are terminally differentiated, highly specialized cells located in juxtaposition to the basement membrane over the abluminal surfaces of endothelial cells within the glomerular tuft. This triad structure is the site of the filtration barrier, which forms highly delicate and tightly regulated architecture to carry out the ultrafiltration function of the kidney.1 The filtration barrier is characterized by foot processes that are connected by specialized junctions called slit diaphragms.
Insults to components of the filtration barrier can initiate cascading events and perpetuate structural alterations that may eventually result in sclerotic changes.2 Common causes among children include minimal change disease (MCD) with the collapse of foot processes resulting in proteinuria, Alport syndrome due to mutation of collagen fibers within the basement membrane leading to hematuria and proteinuria, immune complex mediated nephropathy following common infections or autoimmune diseases, and focal segmental glomerulosclerosis (FSGS) that can show variable histopathology toward eventual glomerular scarring.3,4 These children often clinically have minimal, if any, signs of systemic inflammation.3-5 This has been a limiting factor for the commitment to immunomodulatory treatment, except for steroids for the treatment of MCD.6 Although prolonged steroid treatment may be efficacious, adverse effects are significant in a growing child. Alternative treatments, such as tacrolimus and rituximab have been suggested as second-line steroid-sparing agents.7,8 Not uncommonly, however, these cases are managed by supportive measures only during the progression of the natural course of the disease, which may eventually lead to renal failure, requiring transplant for survival.8,9
This case report highlights a child with a variant of uncertain significance (VUS) in genes involved in Alport syndrome and FSGS who developed an abrupt onset of proteinuria and hematuria after a respiratory illness. To our knowledge, he represents the youngest case demonstrating the benefit of targeted treatment against tumor necrosis factor-α (TNF-α) for glomerulopathy using biologic response modifiers.
Case Description
This is currently a 7-year-old male patient who was born at 39 weeks gestation to gravida 3 para 3 following induced labor due to elevated maternal blood pressure. During the first 2 years of life, his growth and development were normal and his immunizations were up to date. The patient's medical history included upper respiratory tract infections (URIs), respiratory syncytial virus, as well as 3 bouts of pneumonia and multiple otitis media that resulted in 18 rounds of antibiotics. The child was also allergic to nuts and milk protein. The patient’s parents are of Northern European and Native American descent. There is no known family history of eye, ear, or kidney diseases.
Renal concerns were first noted at the age of 2 years and 6 months when he presented to an emergency department in Fall 2019 (week 0) for several weeks of intermittent dark-colored urine. His mother reported that the discoloration recently progressed in intensity to cola-colored, along with the onset of persistent vomiting without any fever or diarrhea. On physical examination, the patient had normal vitals: weight 14.8 kg (68th percentile), height 91 cm (24th percentile), and body surface area 0.6 m2. There was no edema, rash, or lymphadenopathy, but he appeared pale.
The patient’s initial laboratory results included: complete blood count with white blood cells (WBC) 10 x 103/L (reference range, 4.5-13.5 x 103/L); differential lymphocytes 69%; neutrophils 21%; hemoglobin 10 g/dL (reference range, 12-16 g/dL); hematocrit, 30%; (reference range, 37%-45%); platelets 437 103/L (reference range, 150-450 x 103/L); serum creatinine 0.46 mg/dL (reference range, 0.5-0.9 mg/dL); and albumin 3.1 g/dL (reference range, 3.5-5.2 g/dL). Serum electrolyte levels and liver enzymes were normal. A urine analysis revealed 3+ protein and 3+ blood with dysmorphic red blood cells (RBC) and RBC casts without WBC. The patient's spot urine protein-to-creatinine ratio was 4.3 and his renal ultrasound was normal. The patient was referred to Nephrology.
During the next 2 weeks, his protein-to-creatinine ratio progressed to 5.9 and serum albumin fell to 2.7 g/dL. His urine remained red colored, and a microscopic examination with RBC > 500 and WBC up to 10 on a high powered field. His workup was negative for antinuclear antibodies, antineutrophil cytoplasmic antibody, antistreptolysin-O (ASO) and anti-DNase B. Serum C3 was low at 81 mg/dL (reference range, 90-180 mg/dL), C4 was 13.3 mg/dL (reference range, 10-40 mg/dL), and immunoglobulin G was low at 452 mg/dL (reference range 719-1475 mg/dL). A baseline audiology test revealed normal hearing.
Percutaneous renal biopsy yielded about 12 glomeruli, all exhibiting mild mesangial matrix expansion and hypercellularity (Figure 1). One glomerulus had prominent parietal epithelial cells without endocapillary hypercellularity or crescent formation. There was no interstitial fibrosis or tubular atrophy. Immunofluorescence studies showed no evidence of immune complex deposition with negative staining for immunoglobulin heavy and light chains, C3 and C1q. Staining for α 2 and α 5 units of collagen was normal. Electron microscopy showed patchy areas of severe basement membrane thinning with frequent foci of mild to moderate lamina densa splitting and associated visceral epithelial cell foot process effacement (Figure 2).
These were reported as concerning findings for possible Alport syndrome by 3 independent pathology teams. The genetic testing was submitted at a commercial laboratory to screen 17 mutations, including COL4A3, COL4A4, and COL4A5. Results showed the presence of a heterozygous VUS in the COL4A4 gene (c.1055C > T; p.Pro352Leu; dbSNP ID: rs371717486; PolyPhen-2: Probably Damaging; SIFT: Deleterious) as well as the presence of a heterozygous VUS in TRPC6 gene (c2463A>T; p.Lys821Asn; dbSNP ID: rs199948731; PolyPhen-2: Benign; SIFT: Tolerated). Further genetic investigation by whole exome sequencing on approximately 20,000 genes through MNG Laboratories showed a new heterozygous VUS in the OSGEP gene [c.328T>C; p.Cys110Arg]. Additional studies ruled out mitochondrial disease, CoQ10 deficiency, and metabolic disorders upon normal findings for mitochondrial DNA, urine amino acids, plasma acylcarnitine profile, orotic acid, ammonia, and homocysteine levels.
Figure 3 summarizes the patient’s treatment response during 170 weeks of follow-up (Fall 2019 to Summer 2023). The patient was started on enalapril 0.6 mg/kg daily at week 3, which continued throughout treatment. Following a rheumatology consult at week 30, the patient was started on prednisolone 3 mg/mL to assess the role of inflammation through the treatment response. An initial dose of 2 mg/kg daily (9 mL) for 1 month was followed by every other day treatment that was tapered off by week 48. To control mild but noticeably increasing proteinuria in the interim, subcutaneous anakinra 50 mg (3 mg/kg daily) was added as a steroid
DISCUSSION
This case describes a child with rapidly progressive proteinuria and hematuria following a URI who was found to have VUS mutations in 3 different genes associated with chronic kidney disease. Serology tests on the patient were negative for streptococcal antibodies and antinuclear antibodies, ruling out poststreptococcal glomerulonephritis, or systemic lupus erythematosus. His renal biopsy findings were concerning for altered podocytes, mesangial cells, and basement membrane without inflammatory infiltrate, immune complex, complements, immunoglobulin A, or vasculopathy. His blood inflammatory markers, erythrocyte sedimentation rate, C-reactive protein, and ferritin were normal when his care team initiated daily steroids.
Overall, the patient’s clinical presentation and histopathology findings were suggestive of Alport syndrome or thin basement membrane nephropathy with a high potential to progress into FSGS.10-12 Alport syndrome affects 1 in 5000 to 10,000 children annually due to S-linked inheritance of COL4A5, or autosomal recessive inheritance of COL4A3 or COL4A4 genes. It presents with hematuria and hearing loss.10 Our patient had a single copy COL4A4 gene mutation that was classified as VUS. He also had 2 additional VUS affecting the TRPC6 and OSGEP genes. TRPC6 gene mutation can be associated with FSGS through autosomal dominant inheritance. Both COL4A4 and TRPC6 gene mutations were paternally inherited. Although the patient’s father not having renal disease argues against the clinical significance of these findings, there is literature on the potential role of heterozygous COL4A4 variant mimicking thin basement membrane nephropathy that can lead to renal impairment upon copresence of superimposed conditions.13 The patient’s rapidly progressing hematuria and changes in the basement membrane were worrisome for emerging FSGS. Furthermore, VUS of TRPC6 has been reported in late onset autosomal dominant FSGS and can be associated with early onset steroid-resistant nephrotic syndrome (NS) in children.14 This concern was voiced by 3 nephrology consultants during the initial evaluation, leading to the consensus that steroid treatment for podocytopathy would not alter the patient’s long-term outcomes (ie, progression to FSGS).
Immunomodulation
Our rationale for immunomodulatory treatment was based on the abrupt onset of renal concerns following a URI, suggesting the importance of an inflammatory trigger causing altered homeostasis in a genetically susceptible host. Preclinical models show that microbial products such as lipopolysaccharides can lead to podocytopathy by several mechanisms through activation of toll-like receptor signaling. It can directly cause apoptosis by downregulation of the intracellular Akt survival pathway.15 Lipopolysaccharide can also activate the NF-αB pathway and upregulate the production of interleukin-1 (IL-1) and TNF-α in mesangial cells.16,17
Both cytokines can promote mesangial cell proliferation.18 Through autocrine and paracrine mechanisms, proinflammatory cytokines can further perpetuate somatic tissue changes and contribute to the development of podocytopathy. For instance, TNF-α can promote podocyte injury and proteinuria by downregulation of the slit diaphragm protein expression (ie, nephrin, ezrin, or podocin), and disruption of podocyte cytoskeleton.19,20 TNF-α promotes the influx and activation of macrophages and inflammatory cells. It is actively involved in chronic alterations within the glomeruli by the upregulation of matrix metalloproteases by integrins, as well as activation of myofibroblast progenitors and extracellular matrix deposition in crosstalk with transforming growth factor and other key mediators.17,21,22
For the patient described in this case report, initial improvement on steroids encouraged the pursuit of additional treatment to downregulate inflammatory pathways within the glomerular milieu. However, within the COVID-19 environment, escalating the patient’s treatment using traditional immunomodulators (ie, calcineurin inhibitors or mycophenolate mofetil) was not favored due to the risk of infection. Initially, anakinra, a recombinant IL-1 receptor antagonist, was preferred as a steroid-sparing agent for its short life and safety profile during the pandemic. At first, the patient responded well to anakinra and was allowed a steroid wean when the dose was titrated up to 6 mg/kg daily. However, anakinra did not prevent the escalation of proteinuria following a URI. After the treatment was changed to adalimumab, a fully humanized monoclonal antibody to TNF-α, the patient continued to improve and reach full remission despite experiencing a cold and the flu in the following months.
Literature Review
There is a paucity of literature on applications of biological response modifiers for idiopathic NS and FSGS.23,24 Angeletti and colleagues reported that 3 patients with severe long-standing FSGS benefited from anakinra 4 mg/kg daily to reduce proteinuria and improve kidney function. All the patients had positive C3 staining in renal biopsy and treatment response, which supported the role of C3a in inducing podocyte injury through upregulated expression of IL-1 and IL-1R.23 Trachtman and colleagues reported on the phase II FONT trial that included 14 of 21 patients aged < 18 years with advanced FSGS who were treated with adalimumab 24 mg/m2, or ≤ 40 mg every other week.24 Although, during a 6-month period, none of the 7 patients met the endpoint of reduced proteinuria by ≥ 50%, and the authors suggested that careful patient selection may improve the treatment response in future trials.24
A recent study involving transcriptomics on renal tissue samples combined with available pathology (fibrosis), urinary markers, and clinical characteristics on 285 patients with MCD or FSGS from 3 different continents identified 3 distinct clusters. Patients with evidence of activated kidney TNF pathway (n = 72, aged > 18 years) were found to have poor clinical outcomes.25 The study identified 2 urine markers associated with the TNF pathway (ie, tissue inhibitor of metalloproteinases-1 and monocyte chemoattractant protein-1), which aligns with the preclinical findings previously mentioned.25
Conclusions
The patient’s condition in this case illustrates the complex nature of biologically predetermined cascading events in the emergence of glomerular disease upon environmental triggers under the influence of genetic factors.
Chronic kidney disease affects 7.7% of veterans annually, illustrating the need for new therapeutics.26 Based on our experience and literature review, upregulation of TNF-α is a root cause of glomerulopathy; further studies are warranted to evaluate the efficacy of anti-TNF biologic response modifiers for the treatment of these patients. Long-term postmarketing safety profile and steroid-sparing properties of adalimumab should allow inclusion of pediatric cases in future trials. Results may also contribute to identifying new predictive biomarkers related to the basement membrane when combined with precision nephrology to further advance patient selection and targeted treatment.25,27
Acknowledgments
The authors thank the patient’s mother for providing consent to allow publication of this case report.
Podocytes are terminally differentiated, highly specialized cells located in juxtaposition to the basement membrane over the abluminal surfaces of endothelial cells within the glomerular tuft. This triad structure is the site of the filtration barrier, which forms highly delicate and tightly regulated architecture to carry out the ultrafiltration function of the kidney.1 The filtration barrier is characterized by foot processes that are connected by specialized junctions called slit diaphragms.
Insults to components of the filtration barrier can initiate cascading events and perpetuate structural alterations that may eventually result in sclerotic changes.2 Common causes among children include minimal change disease (MCD) with the collapse of foot processes resulting in proteinuria, Alport syndrome due to mutation of collagen fibers within the basement membrane leading to hematuria and proteinuria, immune complex mediated nephropathy following common infections or autoimmune diseases, and focal segmental glomerulosclerosis (FSGS) that can show variable histopathology toward eventual glomerular scarring.3,4 These children often clinically have minimal, if any, signs of systemic inflammation.3-5 This has been a limiting factor for the commitment to immunomodulatory treatment, except for steroids for the treatment of MCD.6 Although prolonged steroid treatment may be efficacious, adverse effects are significant in a growing child. Alternative treatments, such as tacrolimus and rituximab have been suggested as second-line steroid-sparing agents.7,8 Not uncommonly, however, these cases are managed by supportive measures only during the progression of the natural course of the disease, which may eventually lead to renal failure, requiring transplant for survival.8,9
This case report highlights a child with a variant of uncertain significance (VUS) in genes involved in Alport syndrome and FSGS who developed an abrupt onset of proteinuria and hematuria after a respiratory illness. To our knowledge, he represents the youngest case demonstrating the benefit of targeted treatment against tumor necrosis factor-α (TNF-α) for glomerulopathy using biologic response modifiers.
Case Description
This is currently a 7-year-old male patient who was born at 39 weeks gestation to gravida 3 para 3 following induced labor due to elevated maternal blood pressure. During the first 2 years of life, his growth and development were normal and his immunizations were up to date. The patient's medical history included upper respiratory tract infections (URIs), respiratory syncytial virus, as well as 3 bouts of pneumonia and multiple otitis media that resulted in 18 rounds of antibiotics. The child was also allergic to nuts and milk protein. The patient’s parents are of Northern European and Native American descent. There is no known family history of eye, ear, or kidney diseases.
Renal concerns were first noted at the age of 2 years and 6 months when he presented to an emergency department in Fall 2019 (week 0) for several weeks of intermittent dark-colored urine. His mother reported that the discoloration recently progressed in intensity to cola-colored, along with the onset of persistent vomiting without any fever or diarrhea. On physical examination, the patient had normal vitals: weight 14.8 kg (68th percentile), height 91 cm (24th percentile), and body surface area 0.6 m2. There was no edema, rash, or lymphadenopathy, but he appeared pale.
The patient’s initial laboratory results included: complete blood count with white blood cells (WBC) 10 x 103/L (reference range, 4.5-13.5 x 103/L); differential lymphocytes 69%; neutrophils 21%; hemoglobin 10 g/dL (reference range, 12-16 g/dL); hematocrit, 30%; (reference range, 37%-45%); platelets 437 103/L (reference range, 150-450 x 103/L); serum creatinine 0.46 mg/dL (reference range, 0.5-0.9 mg/dL); and albumin 3.1 g/dL (reference range, 3.5-5.2 g/dL). Serum electrolyte levels and liver enzymes were normal. A urine analysis revealed 3+ protein and 3+ blood with dysmorphic red blood cells (RBC) and RBC casts without WBC. The patient's spot urine protein-to-creatinine ratio was 4.3 and his renal ultrasound was normal. The patient was referred to Nephrology.
During the next 2 weeks, his protein-to-creatinine ratio progressed to 5.9 and serum albumin fell to 2.7 g/dL. His urine remained red colored, and a microscopic examination with RBC > 500 and WBC up to 10 on a high powered field. His workup was negative for antinuclear antibodies, antineutrophil cytoplasmic antibody, antistreptolysin-O (ASO) and anti-DNase B. Serum C3 was low at 81 mg/dL (reference range, 90-180 mg/dL), C4 was 13.3 mg/dL (reference range, 10-40 mg/dL), and immunoglobulin G was low at 452 mg/dL (reference range 719-1475 mg/dL). A baseline audiology test revealed normal hearing.
Percutaneous renal biopsy yielded about 12 glomeruli, all exhibiting mild mesangial matrix expansion and hypercellularity (Figure 1). One glomerulus had prominent parietal epithelial cells without endocapillary hypercellularity or crescent formation. There was no interstitial fibrosis or tubular atrophy. Immunofluorescence studies showed no evidence of immune complex deposition with negative staining for immunoglobulin heavy and light chains, C3 and C1q. Staining for α 2 and α 5 units of collagen was normal. Electron microscopy showed patchy areas of severe basement membrane thinning with frequent foci of mild to moderate lamina densa splitting and associated visceral epithelial cell foot process effacement (Figure 2).
These were reported as concerning findings for possible Alport syndrome by 3 independent pathology teams. The genetic testing was submitted at a commercial laboratory to screen 17 mutations, including COL4A3, COL4A4, and COL4A5. Results showed the presence of a heterozygous VUS in the COL4A4 gene (c.1055C > T; p.Pro352Leu; dbSNP ID: rs371717486; PolyPhen-2: Probably Damaging; SIFT: Deleterious) as well as the presence of a heterozygous VUS in TRPC6 gene (c2463A>T; p.Lys821Asn; dbSNP ID: rs199948731; PolyPhen-2: Benign; SIFT: Tolerated). Further genetic investigation by whole exome sequencing on approximately 20,000 genes through MNG Laboratories showed a new heterozygous VUS in the OSGEP gene [c.328T>C; p.Cys110Arg]. Additional studies ruled out mitochondrial disease, CoQ10 deficiency, and metabolic disorders upon normal findings for mitochondrial DNA, urine amino acids, plasma acylcarnitine profile, orotic acid, ammonia, and homocysteine levels.
Figure 3 summarizes the patient’s treatment response during 170 weeks of follow-up (Fall 2019 to Summer 2023). The patient was started on enalapril 0.6 mg/kg daily at week 3, which continued throughout treatment. Following a rheumatology consult at week 30, the patient was started on prednisolone 3 mg/mL to assess the role of inflammation through the treatment response. An initial dose of 2 mg/kg daily (9 mL) for 1 month was followed by every other day treatment that was tapered off by week 48. To control mild but noticeably increasing proteinuria in the interim, subcutaneous anakinra 50 mg (3 mg/kg daily) was added as a steroid
DISCUSSION
This case describes a child with rapidly progressive proteinuria and hematuria following a URI who was found to have VUS mutations in 3 different genes associated with chronic kidney disease. Serology tests on the patient were negative for streptococcal antibodies and antinuclear antibodies, ruling out poststreptococcal glomerulonephritis, or systemic lupus erythematosus. His renal biopsy findings were concerning for altered podocytes, mesangial cells, and basement membrane without inflammatory infiltrate, immune complex, complements, immunoglobulin A, or vasculopathy. His blood inflammatory markers, erythrocyte sedimentation rate, C-reactive protein, and ferritin were normal when his care team initiated daily steroids.
Overall, the patient’s clinical presentation and histopathology findings were suggestive of Alport syndrome or thin basement membrane nephropathy with a high potential to progress into FSGS.10-12 Alport syndrome affects 1 in 5000 to 10,000 children annually due to S-linked inheritance of COL4A5, or autosomal recessive inheritance of COL4A3 or COL4A4 genes. It presents with hematuria and hearing loss.10 Our patient had a single copy COL4A4 gene mutation that was classified as VUS. He also had 2 additional VUS affecting the TRPC6 and OSGEP genes. TRPC6 gene mutation can be associated with FSGS through autosomal dominant inheritance. Both COL4A4 and TRPC6 gene mutations were paternally inherited. Although the patient’s father not having renal disease argues against the clinical significance of these findings, there is literature on the potential role of heterozygous COL4A4 variant mimicking thin basement membrane nephropathy that can lead to renal impairment upon copresence of superimposed conditions.13 The patient’s rapidly progressing hematuria and changes in the basement membrane were worrisome for emerging FSGS. Furthermore, VUS of TRPC6 has been reported in late onset autosomal dominant FSGS and can be associated with early onset steroid-resistant nephrotic syndrome (NS) in children.14 This concern was voiced by 3 nephrology consultants during the initial evaluation, leading to the consensus that steroid treatment for podocytopathy would not alter the patient’s long-term outcomes (ie, progression to FSGS).
Immunomodulation
Our rationale for immunomodulatory treatment was based on the abrupt onset of renal concerns following a URI, suggesting the importance of an inflammatory trigger causing altered homeostasis in a genetically susceptible host. Preclinical models show that microbial products such as lipopolysaccharides can lead to podocytopathy by several mechanisms through activation of toll-like receptor signaling. It can directly cause apoptosis by downregulation of the intracellular Akt survival pathway.15 Lipopolysaccharide can also activate the NF-αB pathway and upregulate the production of interleukin-1 (IL-1) and TNF-α in mesangial cells.16,17
Both cytokines can promote mesangial cell proliferation.18 Through autocrine and paracrine mechanisms, proinflammatory cytokines can further perpetuate somatic tissue changes and contribute to the development of podocytopathy. For instance, TNF-α can promote podocyte injury and proteinuria by downregulation of the slit diaphragm protein expression (ie, nephrin, ezrin, or podocin), and disruption of podocyte cytoskeleton.19,20 TNF-α promotes the influx and activation of macrophages and inflammatory cells. It is actively involved in chronic alterations within the glomeruli by the upregulation of matrix metalloproteases by integrins, as well as activation of myofibroblast progenitors and extracellular matrix deposition in crosstalk with transforming growth factor and other key mediators.17,21,22
For the patient described in this case report, initial improvement on steroids encouraged the pursuit of additional treatment to downregulate inflammatory pathways within the glomerular milieu. However, within the COVID-19 environment, escalating the patient’s treatment using traditional immunomodulators (ie, calcineurin inhibitors or mycophenolate mofetil) was not favored due to the risk of infection. Initially, anakinra, a recombinant IL-1 receptor antagonist, was preferred as a steroid-sparing agent for its short life and safety profile during the pandemic. At first, the patient responded well to anakinra and was allowed a steroid wean when the dose was titrated up to 6 mg/kg daily. However, anakinra did not prevent the escalation of proteinuria following a URI. After the treatment was changed to adalimumab, a fully humanized monoclonal antibody to TNF-α, the patient continued to improve and reach full remission despite experiencing a cold and the flu in the following months.
Literature Review
There is a paucity of literature on applications of biological response modifiers for idiopathic NS and FSGS.23,24 Angeletti and colleagues reported that 3 patients with severe long-standing FSGS benefited from anakinra 4 mg/kg daily to reduce proteinuria and improve kidney function. All the patients had positive C3 staining in renal biopsy and treatment response, which supported the role of C3a in inducing podocyte injury through upregulated expression of IL-1 and IL-1R.23 Trachtman and colleagues reported on the phase II FONT trial that included 14 of 21 patients aged < 18 years with advanced FSGS who were treated with adalimumab 24 mg/m2, or ≤ 40 mg every other week.24 Although, during a 6-month period, none of the 7 patients met the endpoint of reduced proteinuria by ≥ 50%, and the authors suggested that careful patient selection may improve the treatment response in future trials.24
A recent study involving transcriptomics on renal tissue samples combined with available pathology (fibrosis), urinary markers, and clinical characteristics on 285 patients with MCD or FSGS from 3 different continents identified 3 distinct clusters. Patients with evidence of activated kidney TNF pathway (n = 72, aged > 18 years) were found to have poor clinical outcomes.25 The study identified 2 urine markers associated with the TNF pathway (ie, tissue inhibitor of metalloproteinases-1 and monocyte chemoattractant protein-1), which aligns with the preclinical findings previously mentioned.25
Conclusions
The patient’s condition in this case illustrates the complex nature of biologically predetermined cascading events in the emergence of glomerular disease upon environmental triggers under the influence of genetic factors.
Chronic kidney disease affects 7.7% of veterans annually, illustrating the need for new therapeutics.26 Based on our experience and literature review, upregulation of TNF-α is a root cause of glomerulopathy; further studies are warranted to evaluate the efficacy of anti-TNF biologic response modifiers for the treatment of these patients. Long-term postmarketing safety profile and steroid-sparing properties of adalimumab should allow inclusion of pediatric cases in future trials. Results may also contribute to identifying new predictive biomarkers related to the basement membrane when combined with precision nephrology to further advance patient selection and targeted treatment.25,27
Acknowledgments
The authors thank the patient’s mother for providing consent to allow publication of this case report.
1. Arif E, Nihalani D. Glomerular filtration barrier assembly: an insight. Postdoc J. 2013;1(4):33-45.
2. Garg PA. Review of podocyte biology. Am J Nephrol. 2018;47(suppl 1):3-13. doi:10.1159/000481633SUPPL
3. Warady BA, Agarwal R, Bangalore S, et al. Alport syndrome classification and management. Kidney Med. 2020;2(5):639-649. doi:10.1016/j.xkme.2020.05.014
4. Angioi A, Pani A. FSGS: from pathogenesis to the histological lesion. J Nephrol. 2016;29(4):517-523. doi:10.1007/s40620-016-0333-2
5. Roca N, Martinez C, Jatem E, Madrid A, Lopez M, Segarra A. Activation of the acute inflammatory phase response in idiopathic nephrotic syndrome: association with clinicopathological phenotypes and with response to corticosteroids. Clin Kidney J. 2021;14(4):1207-1215. doi:10.1093/ckj/sfaa247
6. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. 2017;12(2):332-345.
7. Medjeral-Thomas NR, Lawrence C, Condon M, et al. Randomized, controlled trial of tacrolimus and prednisolone monotherapy for adults with De Novo minimal change disease: a multicenter, randomized, controlled trial. Clin J Am Soc Nephrol. 2020;15(2):209-218. doi:10.2215/CJN.06290420
8. Ye Q, Lan B, Liu H, Persson PB, Lai EY, Mao J. A critical role of the podocyte cytoskeleton in the pathogenesis of glomerular proteinuria and autoimmune podocytopathies. Acta Physiol (Oxf). 2022;235(4):e13850. doi:10.1111/apha.13850
9. Trautmann A, Schnaidt S, Lipska-Ziμtkiewicz BS, et al. Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol. 2017;28:3055-3065. doi:10.1681/ASN.2016101121
10. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):711-719. doi:10.1007/s00467-020-04819-6
11. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169-78. doi:10.1046/j.1523-1755.2003.00234.x
12. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2017; 12(3):502-517. doi:10.2215/CJN.05960616
13. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant? Kidney Int Rep. 2018;3(6):1239-1241. doi:10.1016/j.ekir.2018.08.002
14. Gigante M, Caridi G, Montemurno E, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626-1634. doi:10.2215/CJN.07830910
15. Saurus P, Kuusela S, Lehtonen E, et al. Podocyte apoptosis is prevented by blocking the toll-like receptor pathway. Cell Death Dis. 2015;6(5):e1752. doi:10.1038/cddis.2015.125
16. Baud L, Oudinet JP, Bens M, et al. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35(5):1111-1118. doi:10.1038/ki.1989.98
17. White S, Lin L, Hu K. NF-κB and tPA signaling in kidney and other diseases. Cells. 2020;9(6):1348. doi:10.3390/cells9061348
18. Tesch GH, Lan HY, Atkins RC, Nikolic-Paterson DJ. Role of interleukin-1 in mesangial cell proliferation and matrix deposition in experimental mesangioproliferative nephritis. Am J Pathol. 1997;151(1):141-150.
19. Lai KN, Leung JCK, Chan LYY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA Nephropathy. Nephrol Dial Transplant. 2009;24(1):62-72. doi:10.1093/ndt/gfn441
20. Saleem MA, Kobayashi Y. Cell biology and genetics of minimal change disease. F1000 Res. 2016;5: F1000 Faculty Rev-412. doi:10.12688/f1000research.7300.1
21. Kim KP, Williams CE, Lemmon CA. Cell-matrix interactions in renal fibrosis. Kidney Dial. 2022;2(4):607-624. doi:10.3390/kidneydial2040055
22. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8(3):210. doi:10.1186/ar1963
23. Angeletti A, Magnasco A, Trivelli A, et al. Refractory minimal change disease and focal segmental glomerular sclerosis treated with Anakinra. Kidney Int Rep. 2021;7(1):121-124. doi:10.1016/j.ekir.2021.10.018
24. Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol. 2015;16:111. doi:10.1186/s12882-015-0094-5
25. Mariani LH, Eddy S, AlAkwaa FM, et al. Precision nephrology identified tumor necrosis factor activation variability in minimal change disease and focal segmental glomerulosclerosis. Kidney Int. 2023;103(3):565-579. doi:10.1016/j.kint.2022.10.023
26. Korshak L, Washington DL, Powell J, Nylen E, Kokkinos P. Kidney Disease in Veterans. US Dept of Veterans Affairs, Office of Health Equity. Updated May 13, 2020. Accessed June 28, 2024. https://www.va.gov/HEALTHEQUITY/Kidney_Disease_In_Veterans.asp
27. Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253-1259. doi:10.1038/ki.2014.305
1. Arif E, Nihalani D. Glomerular filtration barrier assembly: an insight. Postdoc J. 2013;1(4):33-45.
2. Garg PA. Review of podocyte biology. Am J Nephrol. 2018;47(suppl 1):3-13. doi:10.1159/000481633SUPPL
3. Warady BA, Agarwal R, Bangalore S, et al. Alport syndrome classification and management. Kidney Med. 2020;2(5):639-649. doi:10.1016/j.xkme.2020.05.014
4. Angioi A, Pani A. FSGS: from pathogenesis to the histological lesion. J Nephrol. 2016;29(4):517-523. doi:10.1007/s40620-016-0333-2
5. Roca N, Martinez C, Jatem E, Madrid A, Lopez M, Segarra A. Activation of the acute inflammatory phase response in idiopathic nephrotic syndrome: association with clinicopathological phenotypes and with response to corticosteroids. Clin Kidney J. 2021;14(4):1207-1215. doi:10.1093/ckj/sfaa247
6. Vivarelli M, Massella L, Ruggiero B, Emma F. Minimal change disease. Clin J Am Soc Nephrol. 2017;12(2):332-345.
7. Medjeral-Thomas NR, Lawrence C, Condon M, et al. Randomized, controlled trial of tacrolimus and prednisolone monotherapy for adults with De Novo minimal change disease: a multicenter, randomized, controlled trial. Clin J Am Soc Nephrol. 2020;15(2):209-218. doi:10.2215/CJN.06290420
8. Ye Q, Lan B, Liu H, Persson PB, Lai EY, Mao J. A critical role of the podocyte cytoskeleton in the pathogenesis of glomerular proteinuria and autoimmune podocytopathies. Acta Physiol (Oxf). 2022;235(4):e13850. doi:10.1111/apha.13850
9. Trautmann A, Schnaidt S, Lipska-Ziμtkiewicz BS, et al. Long-term outcome of steroid-resistant nephrotic syndrome in children. J Am Soc Nephrol. 2017;28:3055-3065. doi:10.1681/ASN.2016101121
10. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. 2021;36(3):711-719. doi:10.1007/s00467-020-04819-6
11. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY. Thin basement membrane nephropathy. Kidney Int. 2003;64(4):1169-78. doi:10.1046/j.1523-1755.2003.00234.x
12. Rosenberg AZ, Kopp JB. Focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2017; 12(3):502-517. doi:10.2215/CJN.05960616
13. Savige J. Should we diagnose autosomal dominant Alport syndrome when there is a pathogenic heterozygous COL4A3 or COL4A4 variant? Kidney Int Rep. 2018;3(6):1239-1241. doi:10.1016/j.ekir.2018.08.002
14. Gigante M, Caridi G, Montemurno E, et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626-1634. doi:10.2215/CJN.07830910
15. Saurus P, Kuusela S, Lehtonen E, et al. Podocyte apoptosis is prevented by blocking the toll-like receptor pathway. Cell Death Dis. 2015;6(5):e1752. doi:10.1038/cddis.2015.125
16. Baud L, Oudinet JP, Bens M, et al. Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int. 1989;35(5):1111-1118. doi:10.1038/ki.1989.98
17. White S, Lin L, Hu K. NF-κB and tPA signaling in kidney and other diseases. Cells. 2020;9(6):1348. doi:10.3390/cells9061348
18. Tesch GH, Lan HY, Atkins RC, Nikolic-Paterson DJ. Role of interleukin-1 in mesangial cell proliferation and matrix deposition in experimental mesangioproliferative nephritis. Am J Pathol. 1997;151(1):141-150.
19. Lai KN, Leung JCK, Chan LYY, et al. Podocyte injury induced by mesangial-derived cytokines in IgA Nephropathy. Nephrol Dial Transplant. 2009;24(1):62-72. doi:10.1093/ndt/gfn441
20. Saleem MA, Kobayashi Y. Cell biology and genetics of minimal change disease. F1000 Res. 2016;5: F1000 Faculty Rev-412. doi:10.12688/f1000research.7300.1
21. Kim KP, Williams CE, Lemmon CA. Cell-matrix interactions in renal fibrosis. Kidney Dial. 2022;2(4):607-624. doi:10.3390/kidneydial2040055
22. Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8(3):210. doi:10.1186/ar1963
23. Angeletti A, Magnasco A, Trivelli A, et al. Refractory minimal change disease and focal segmental glomerular sclerosis treated with Anakinra. Kidney Int Rep. 2021;7(1):121-124. doi:10.1016/j.ekir.2021.10.018
24. Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and adalimumab in patients with resistant focal segmental glomerulosclerosis: report of the font clinical trial group. BMC Nephrol. 2015;16:111. doi:10.1186/s12882-015-0094-5
25. Mariani LH, Eddy S, AlAkwaa FM, et al. Precision nephrology identified tumor necrosis factor activation variability in minimal change disease and focal segmental glomerulosclerosis. Kidney Int. 2023;103(3):565-579. doi:10.1016/j.kint.2022.10.023
26. Korshak L, Washington DL, Powell J, Nylen E, Kokkinos P. Kidney Disease in Veterans. US Dept of Veterans Affairs, Office of Health Equity. Updated May 13, 2020. Accessed June 28, 2024. https://www.va.gov/HEALTHEQUITY/Kidney_Disease_In_Veterans.asp
27. Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86(6):1253-1259. doi:10.1038/ki.2014.305
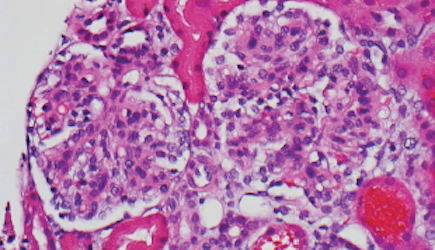
Three Anomalies and a Complication: Ruptured Noncoronary Sinus of Valsalva Aneurysm, Atrial Septal Aneurysm, and Patent Foramen Ovale
A 53 year-old white male with a past medical history of hypertension, hyperlipidemia, and former tobacco use was referred to the Dayton VAMC in Ohio for symptoms that included shortness of breath and a recent abnormal stress test. The patient reported no history of known coronary artery disease (CAD), congestive heart failure, or other cardiovascular diseases. The patient also reported no recent fever, bacterial blood infection, syphilis infection, recreational drug use, or chest trauma.
A physical examination was remarkable for grade 3/6 continuous murmur at the 5th interspace to the left of the sternum and a loud “pistol shot” sound heard over the femoral artery. The patient had jugular venous distension and 2+ leg edema bilaterally. His vital signs were normal, and laboratory blood tests showed normal hemoglobin level and kidney function.
An electrocardiogram showed nonspecific ST segment changes and a transthoracic echocardiogram (TTE) revealed a high-velocity jet in the right atrium (RA) above the tricuspid valve concerning for sinus of Valsalva aneurysm (SVA). 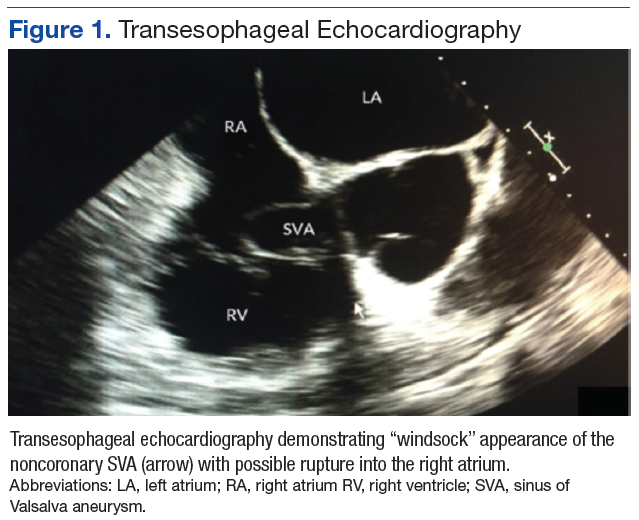
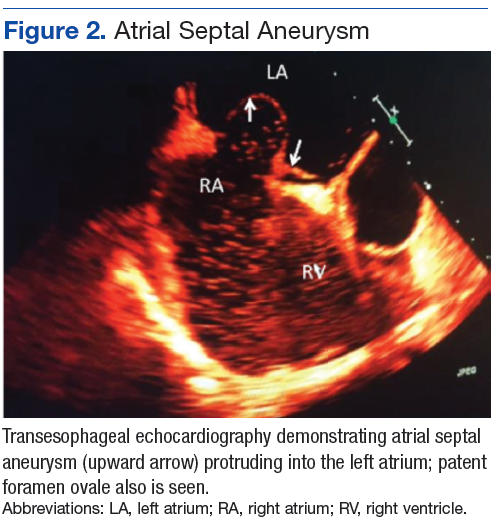
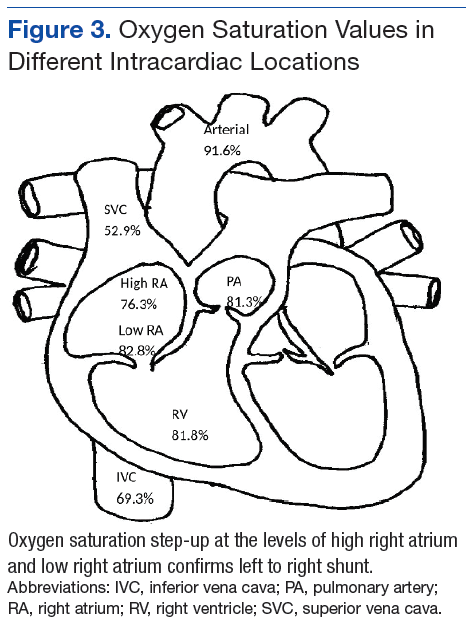
Right heart catheterization revealed elevated RA pressures with positive shunt study showing oxygen saturation step-up in the RA (Figure 3). Left heart hemodynamic measurement from an aortic approach to the distal part of the noncoronary cusp SVA revealed an RA pressure-tracing pattern consistent with rupture of the noncoronary SVA into the RA (Figure 4). 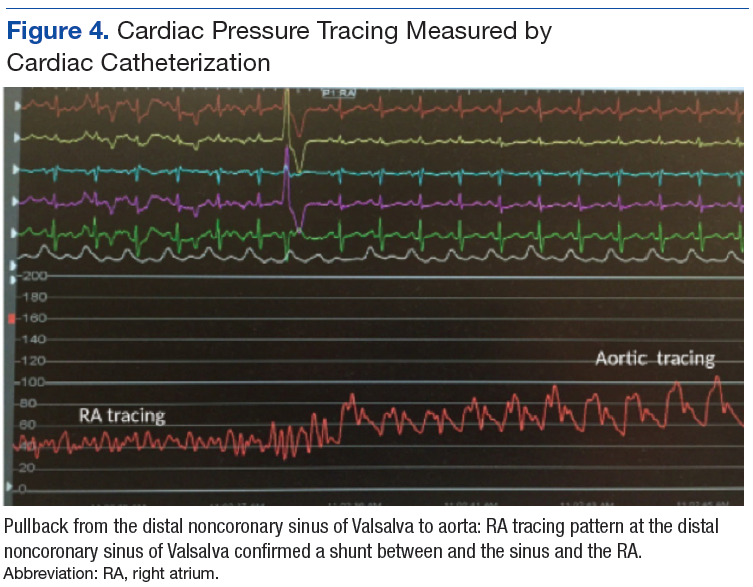
The primary diagnosis was of acute heart failure secondary to ruptured aneurysm of the noncoronary SVA into RA. The patient also received a secondary diagnosis of atrial septal aneurysm and PFO.
Treatment & Outcome
The patient was treated with aggressive diuresis and responded well to therapy. Considering the high mortality rate associated with a ruptured SVA, the patient was referred to a tertiary care center for surgical evaluation. He underwent repair of aorto-right atrial communication with a Cormatrix patch (Roswell, GA) from the aortic side and with primary closure from the right atrial side with resection of the windsock tract; coronary artery bypass graft x1 with right internal mammary artery to the right coronary artery; closure of the PFO with the Cormatrix patch.
The postoperative TEE confirmed preserved LV and RV function, no shunts, no aortic or tricuspid insufficiency. Biopsy of the tissue resected showed intimal fibroplasia. A TTE completed 1 year after surgery showed normal valvular function and without any structural abnormalities. The patient had improvement in symptoms and an uneventful year after surgical intervention followed by 24 session of cardiac rehabilitation.
Discussion
Sinus of Valsalva aneurysm is a dilation of the aortic wall between the aortic valve and the sinotubular junction that is caused by the lack of continuity between the middle layer of the aortic wall and the aortic valve.1 Cases of SVA are rare cardiac anomalies with prevalence of 1% in patients undergoing open-heart surgery.2 Between 65% and 85% of SVA cases originate from the right coronary sinus, 10% to 20% from the noncoronary sinus, and < 5% from the left coronary sinus.3
Sinus of Valsalva aneurysm is usually congenital, although cases associated with syphilis, bacterial endocarditis, trauma, Behçet disease, and aortic dissection have been reported. Structural defects associated with congenital SVAs include ventricular septal defect, bicuspid aortic valve, and aortic regurgitation. It is less commonly associated with pulmonary stenosis, coarctation of the aorta, patent ductus arteriosus, tricuspid regurgitation, and atrial septal defects.
The most common complication of the SVA is rupture into another cardiac chamber, frequently the right ventricle (60%) or RA (29%) and less frequently into left atrium (6%), left ventricle (4%), or pericardium (1%).1 Patients with ruptured SVA mainly develop dyspnea and chest pain, but cough, fatigue, peripheral edema, and continuous murmur have been reported.1
Atrial septal aneurysm is an uncommon finding in adults, with an incidence of 2.2 % in the general population, and it is often associated with atrial septal defect and PFO.1,4 Although ASA formation can be secondary to interatrial differences in pressures, it can be a primary malformation involving the region of the fossa ovalis or the entire atrial septum.4 Atrial septal aneurysm may be an isolated anomaly, but often is found in association with other structural cardiac anomalies, including SVA and PFO.4,5
Conclusion
Although coexistence of SVA and ASA has been reported previously, the case reported here, a ruptured noncoronary SVA that was associated with a large ASA and a PFO, has not been previously documented in the English literature. This patient’s anomalies are most likely congenital in origin. Progressive dyspnea and chest pain in the presence of a continuous loud murmur should raise the suspicion of ruptured sinus of Valsalva. Although no significant aortic regurgitation was noted on echocardiography, the pistol shot sound heard over the femoral artery was believed to be due to the rapid diastolic runoff into the RA through the ruptured SVA.
The significant increase in the RA pressure made the ASA and PFO more prominent. A TEE, left and right heart catheterizations with shunt study are vital for the diagnosis of SVA. If left untreated, SVA has an ominous prognosis. Surgical repair of ruptured SVA has an accepted risk and good prognosis with 10-year survival rate of 90%, whereas the mean survival of untreated ruptured SVA is about 4 years.6,7 Hence, the patient in this study was referred to a tertiary care center for surgical intervention.
1. Galicia-Tornell MM, Marín-Solís B, Mercado-Astorga O, Espinoza-Anguiano S, Martínez-Martínez M, Villalpando-Mendoza E. Sinus of Valsalva aneurysm with rupture. Case report and literature review. Cir Cir. 2009;77(6):441-445.
2. Takach TJ, Reul GJ, Duncan JM, et al. Sinus of Valsalva aneurysm or fistula: management and outcome. Ann Thorac Surg. 1999;68(5):1573-1577.
3. Meier JH, Seward JB, Miller FA Jr, Oh JK, Enriquez-Sarano M. Aneurysms in the left ventricular outflow tract: clinical presentation, causes, and echocardiographic features. J Am Soc Echocardiogr. 1998;11(7):729-745.
4. Mügge A, Daniel WG, Angermann C et al. Atrial septal aneurysm in adult patients: a multicenter study using transthoracic and transesophageal echocardiography. Circulation. 1995;91(11):2785-2792.
5. Silver MD, Dorsey JS. Aneurysms of the septum primum in adults. Arch Pathol Lab Med. 1978;102(2):62-65.
6. Wang ZJ, Zou CW, Li DC, et al. Surgical repair of sinus of Valsalva aneurysm in Asian patients. Ann Thorac Surg. 2007;84(1):156-160.
7. Yan F, Huo Q, Qiao J, Murat V, Ma SF. Surgery for sinus of valsalva aneurysm: 27-year experience with 100 patients. Asian Cardiovasc Thorac Ann. 2008;16(5):361-365.
A 53 year-old white male with a past medical history of hypertension, hyperlipidemia, and former tobacco use was referred to the Dayton VAMC in Ohio for symptoms that included shortness of breath and a recent abnormal stress test. The patient reported no history of known coronary artery disease (CAD), congestive heart failure, or other cardiovascular diseases. The patient also reported no recent fever, bacterial blood infection, syphilis infection, recreational drug use, or chest trauma.
A physical examination was remarkable for grade 3/6 continuous murmur at the 5th interspace to the left of the sternum and a loud “pistol shot” sound heard over the femoral artery. The patient had jugular venous distension and 2+ leg edema bilaterally. His vital signs were normal, and laboratory blood tests showed normal hemoglobin level and kidney function.
An electrocardiogram showed nonspecific ST segment changes and a transthoracic echocardiogram (TTE) revealed a high-velocity jet in the right atrium (RA) above the tricuspid valve concerning for sinus of Valsalva aneurysm (SVA).
Right heart catheterization revealed elevated RA pressures with positive shunt study showing oxygen saturation step-up in the RA (Figure 3). Left heart hemodynamic measurement from an aortic approach to the distal part of the noncoronary cusp SVA revealed an RA pressure-tracing pattern consistent with rupture of the noncoronary SVA into the RA (Figure 4).
The primary diagnosis was of acute heart failure secondary to ruptured aneurysm of the noncoronary SVA into RA. The patient also received a secondary diagnosis of atrial septal aneurysm and PFO.
Treatment & Outcome
The patient was treated with aggressive diuresis and responded well to therapy. Considering the high mortality rate associated with a ruptured SVA, the patient was referred to a tertiary care center for surgical evaluation. He underwent repair of aorto-right atrial communication with a Cormatrix patch (Roswell, GA) from the aortic side and with primary closure from the right atrial side with resection of the windsock tract; coronary artery bypass graft x1 with right internal mammary artery to the right coronary artery; closure of the PFO with the Cormatrix patch.
The postoperative TEE confirmed preserved LV and RV function, no shunts, no aortic or tricuspid insufficiency. Biopsy of the tissue resected showed intimal fibroplasia. A TTE completed 1 year after surgery showed normal valvular function and without any structural abnormalities. The patient had improvement in symptoms and an uneventful year after surgical intervention followed by 24 session of cardiac rehabilitation.
Discussion
Sinus of Valsalva aneurysm is a dilation of the aortic wall between the aortic valve and the sinotubular junction that is caused by the lack of continuity between the middle layer of the aortic wall and the aortic valve.1 Cases of SVA are rare cardiac anomalies with prevalence of 1% in patients undergoing open-heart surgery.2 Between 65% and 85% of SVA cases originate from the right coronary sinus, 10% to 20% from the noncoronary sinus, and < 5% from the left coronary sinus.3
Sinus of Valsalva aneurysm is usually congenital, although cases associated with syphilis, bacterial endocarditis, trauma, Behçet disease, and aortic dissection have been reported. Structural defects associated with congenital SVAs include ventricular septal defect, bicuspid aortic valve, and aortic regurgitation. It is less commonly associated with pulmonary stenosis, coarctation of the aorta, patent ductus arteriosus, tricuspid regurgitation, and atrial septal defects.
The most common complication of the SVA is rupture into another cardiac chamber, frequently the right ventricle (60%) or RA (29%) and less frequently into left atrium (6%), left ventricle (4%), or pericardium (1%).1 Patients with ruptured SVA mainly develop dyspnea and chest pain, but cough, fatigue, peripheral edema, and continuous murmur have been reported.1
Atrial septal aneurysm is an uncommon finding in adults, with an incidence of 2.2 % in the general population, and it is often associated with atrial septal defect and PFO.1,4 Although ASA formation can be secondary to interatrial differences in pressures, it can be a primary malformation involving the region of the fossa ovalis or the entire atrial septum.4 Atrial septal aneurysm may be an isolated anomaly, but often is found in association with other structural cardiac anomalies, including SVA and PFO.4,5
Conclusion
Although coexistence of SVA and ASA has been reported previously, the case reported here, a ruptured noncoronary SVA that was associated with a large ASA and a PFO, has not been previously documented in the English literature. This patient’s anomalies are most likely congenital in origin. Progressive dyspnea and chest pain in the presence of a continuous loud murmur should raise the suspicion of ruptured sinus of Valsalva. Although no significant aortic regurgitation was noted on echocardiography, the pistol shot sound heard over the femoral artery was believed to be due to the rapid diastolic runoff into the RA through the ruptured SVA.
The significant increase in the RA pressure made the ASA and PFO more prominent. A TEE, left and right heart catheterizations with shunt study are vital for the diagnosis of SVA. If left untreated, SVA has an ominous prognosis. Surgical repair of ruptured SVA has an accepted risk and good prognosis with 10-year survival rate of 90%, whereas the mean survival of untreated ruptured SVA is about 4 years.6,7 Hence, the patient in this study was referred to a tertiary care center for surgical intervention.
A 53 year-old white male with a past medical history of hypertension, hyperlipidemia, and former tobacco use was referred to the Dayton VAMC in Ohio for symptoms that included shortness of breath and a recent abnormal stress test. The patient reported no history of known coronary artery disease (CAD), congestive heart failure, or other cardiovascular diseases. The patient also reported no recent fever, bacterial blood infection, syphilis infection, recreational drug use, or chest trauma.
A physical examination was remarkable for grade 3/6 continuous murmur at the 5th interspace to the left of the sternum and a loud “pistol shot” sound heard over the femoral artery. The patient had jugular venous distension and 2+ leg edema bilaterally. His vital signs were normal, and laboratory blood tests showed normal hemoglobin level and kidney function.
An electrocardiogram showed nonspecific ST segment changes and a transthoracic echocardiogram (TTE) revealed a high-velocity jet in the right atrium (RA) above the tricuspid valve concerning for sinus of Valsalva aneurysm (SVA).
Right heart catheterization revealed elevated RA pressures with positive shunt study showing oxygen saturation step-up in the RA (Figure 3). Left heart hemodynamic measurement from an aortic approach to the distal part of the noncoronary cusp SVA revealed an RA pressure-tracing pattern consistent with rupture of the noncoronary SVA into the RA (Figure 4).
The primary diagnosis was of acute heart failure secondary to ruptured aneurysm of the noncoronary SVA into RA. The patient also received a secondary diagnosis of atrial septal aneurysm and PFO.
Treatment & Outcome
The patient was treated with aggressive diuresis and responded well to therapy. Considering the high mortality rate associated with a ruptured SVA, the patient was referred to a tertiary care center for surgical evaluation. He underwent repair of aorto-right atrial communication with a Cormatrix patch (Roswell, GA) from the aortic side and with primary closure from the right atrial side with resection of the windsock tract; coronary artery bypass graft x1 with right internal mammary artery to the right coronary artery; closure of the PFO with the Cormatrix patch.
The postoperative TEE confirmed preserved LV and RV function, no shunts, no aortic or tricuspid insufficiency. Biopsy of the tissue resected showed intimal fibroplasia. A TTE completed 1 year after surgery showed normal valvular function and without any structural abnormalities. The patient had improvement in symptoms and an uneventful year after surgical intervention followed by 24 session of cardiac rehabilitation.
Discussion
Sinus of Valsalva aneurysm is a dilation of the aortic wall between the aortic valve and the sinotubular junction that is caused by the lack of continuity between the middle layer of the aortic wall and the aortic valve.1 Cases of SVA are rare cardiac anomalies with prevalence of 1% in patients undergoing open-heart surgery.2 Between 65% and 85% of SVA cases originate from the right coronary sinus, 10% to 20% from the noncoronary sinus, and < 5% from the left coronary sinus.3
Sinus of Valsalva aneurysm is usually congenital, although cases associated with syphilis, bacterial endocarditis, trauma, Behçet disease, and aortic dissection have been reported. Structural defects associated with congenital SVAs include ventricular septal defect, bicuspid aortic valve, and aortic regurgitation. It is less commonly associated with pulmonary stenosis, coarctation of the aorta, patent ductus arteriosus, tricuspid regurgitation, and atrial septal defects.
The most common complication of the SVA is rupture into another cardiac chamber, frequently the right ventricle (60%) or RA (29%) and less frequently into left atrium (6%), left ventricle (4%), or pericardium (1%).1 Patients with ruptured SVA mainly develop dyspnea and chest pain, but cough, fatigue, peripheral edema, and continuous murmur have been reported.1
Atrial septal aneurysm is an uncommon finding in adults, with an incidence of 2.2 % in the general population, and it is often associated with atrial septal defect and PFO.1,4 Although ASA formation can be secondary to interatrial differences in pressures, it can be a primary malformation involving the region of the fossa ovalis or the entire atrial septum.4 Atrial septal aneurysm may be an isolated anomaly, but often is found in association with other structural cardiac anomalies, including SVA and PFO.4,5
Conclusion
Although coexistence of SVA and ASA has been reported previously, the case reported here, a ruptured noncoronary SVA that was associated with a large ASA and a PFO, has not been previously documented in the English literature. This patient’s anomalies are most likely congenital in origin. Progressive dyspnea and chest pain in the presence of a continuous loud murmur should raise the suspicion of ruptured sinus of Valsalva. Although no significant aortic regurgitation was noted on echocardiography, the pistol shot sound heard over the femoral artery was believed to be due to the rapid diastolic runoff into the RA through the ruptured SVA.
The significant increase in the RA pressure made the ASA and PFO more prominent. A TEE, left and right heart catheterizations with shunt study are vital for the diagnosis of SVA. If left untreated, SVA has an ominous prognosis. Surgical repair of ruptured SVA has an accepted risk and good prognosis with 10-year survival rate of 90%, whereas the mean survival of untreated ruptured SVA is about 4 years.6,7 Hence, the patient in this study was referred to a tertiary care center for surgical intervention.
1. Galicia-Tornell MM, Marín-Solís B, Mercado-Astorga O, Espinoza-Anguiano S, Martínez-Martínez M, Villalpando-Mendoza E. Sinus of Valsalva aneurysm with rupture. Case report and literature review. Cir Cir. 2009;77(6):441-445.
2. Takach TJ, Reul GJ, Duncan JM, et al. Sinus of Valsalva aneurysm or fistula: management and outcome. Ann Thorac Surg. 1999;68(5):1573-1577.
3. Meier JH, Seward JB, Miller FA Jr, Oh JK, Enriquez-Sarano M. Aneurysms in the left ventricular outflow tract: clinical presentation, causes, and echocardiographic features. J Am Soc Echocardiogr. 1998;11(7):729-745.
4. Mügge A, Daniel WG, Angermann C et al. Atrial septal aneurysm in adult patients: a multicenter study using transthoracic and transesophageal echocardiography. Circulation. 1995;91(11):2785-2792.
5. Silver MD, Dorsey JS. Aneurysms of the septum primum in adults. Arch Pathol Lab Med. 1978;102(2):62-65.
6. Wang ZJ, Zou CW, Li DC, et al. Surgical repair of sinus of Valsalva aneurysm in Asian patients. Ann Thorac Surg. 2007;84(1):156-160.
7. Yan F, Huo Q, Qiao J, Murat V, Ma SF. Surgery for sinus of valsalva aneurysm: 27-year experience with 100 patients. Asian Cardiovasc Thorac Ann. 2008;16(5):361-365.
1. Galicia-Tornell MM, Marín-Solís B, Mercado-Astorga O, Espinoza-Anguiano S, Martínez-Martínez M, Villalpando-Mendoza E. Sinus of Valsalva aneurysm with rupture. Case report and literature review. Cir Cir. 2009;77(6):441-445.
2. Takach TJ, Reul GJ, Duncan JM, et al. Sinus of Valsalva aneurysm or fistula: management and outcome. Ann Thorac Surg. 1999;68(5):1573-1577.
3. Meier JH, Seward JB, Miller FA Jr, Oh JK, Enriquez-Sarano M. Aneurysms in the left ventricular outflow tract: clinical presentation, causes, and echocardiographic features. J Am Soc Echocardiogr. 1998;11(7):729-745.
4. Mügge A, Daniel WG, Angermann C et al. Atrial septal aneurysm in adult patients: a multicenter study using transthoracic and transesophageal echocardiography. Circulation. 1995;91(11):2785-2792.
5. Silver MD, Dorsey JS. Aneurysms of the septum primum in adults. Arch Pathol Lab Med. 1978;102(2):62-65.
6. Wang ZJ, Zou CW, Li DC, et al. Surgical repair of sinus of Valsalva aneurysm in Asian patients. Ann Thorac Surg. 2007;84(1):156-160.
7. Yan F, Huo Q, Qiao J, Murat V, Ma SF. Surgery for sinus of valsalva aneurysm: 27-year experience with 100 patients. Asian Cardiovasc Thorac Ann. 2008;16(5):361-365.
Pembrolizumab-Induced Bullous Pemphigoid: Navigating Diagnostic Challenges and Treatment Resistance
Pembrolizumab-Induced Bullous Pemphigoid: Navigating Diagnostic Challenges and Treatment Resistance
Bullous pemphigoid (BP) is an autoimmune blistering disorder characterized by the development of tense subepidermal blisters and erosions primarily on the skin, commonly affecting the elderly.1 It is attributed to autoantibodies targeting 2 hemidesmosomal components within the dermoepidermal junction—transmembrane collagen XVII (BP180/BPAG2) and plakin family protein BP230 (BPAG1)—resulting in blister formation due to loss of structural integrity.2 Typically, patients present with pruritic urticarial plaques and tense bullae localized on flexural areas, but cutaneous manifestations vary and can be nonspecific. Histologically, a subepidermal blister with eosinophilic infiltration is characteristic, and detection of circulating autoantibodies against BP180 and BP230 antigens aids in diagnosis.3,4
Drug-induced BP (DIBP) is a subset triggered by medications, including immune checkpoint inhibitors (ICIs) targeting programmed cell death protein-1 (PD-1) or its ligand, programmed death ligand-1 (PD-L1).5,6 Often overexpressed in malignant tumors, PD-L1 inhibits host lymphocytic and apoptotic immune responses. Anti‒PD-1 and anti‒PD-L1 agents, designed to enhance the immune system’s ability to recognize and eliminate cancer cells,7,8 have improved oncologic outcomes for various cancers, including urothelial cancer.9-11 Before 2016, platinum-based chemotherapy was the mainstay for metastatic urothelial cancer management, but US Food and Drug Administration approval of 5 ICIs—nivolumab, pembrolizumab, avelumab, atezolizumab, and durvalumab—transformed treatment options.12Despite robust antitumor responses to ICIs, these medications are increasingly associated with immune-related adverse events (IRAEs), including DIBP, due to inhibition of negative regulators of immunity crucial for maintaining immunologic homeostasis.13,14 Up to 30% to 40% of patients treated with PD-1 inhibitors experience dermatologic complications, such as lichenoid reactions, eczema, vitiligo, and pruritus,15 and patients undergoing treatment with the PD-1 inhibitor pembrolizumab are estimated to be 2.6 times more likely to develop a rash than those receiving standard chemotherapy.16,17 The pathogenesis of DIBP involves autoreactive T-cell activation and subsequent autoantibody production against BP antigens.18 We present the case of DIBP secondary to pembrolizumab immunotherapy in a man with PD-L1–negative metastatic bladder cancer.
Case Report
An 81-year-old man with metastatic urothelial carcinoma presented to dermatology with a pruritic rash characterized by blisters of 5 months’ duration following treatment with pembrolizumab. He had a history of non–muscle invasive urothelial carcinoma and underwent intravesical bacillus Calmette-Guerin treatment. Thirty years later, after surveillance cystoscopies, the patient developed hematuria, which prompted pelvic ultrasonography and cystoscopy that revealed a tumor. Transurethral resection of the bladder tumor confirmed invasive, high-grade papillary urothelial carcinoma with vascular and muscle invasion (clinical stage T2NxMx). Due to elevated creatinine levels, neoadjuvant chemotherapy was contraindicated. Instead, the patient underwent cystoprostatectomy with ureteroileal conduit creation and pelvic lymphadenectomy one month later; final pathology revealed pT2aN0M0 disease with multifocal carcinoma in situ. At that time, there was no evidence of distant metastasis. Surveillance 5 months later identified pulmonary nodules that were confirmed as metastatic urothelial cancer by positron emission tomography/computed tomography (CT). The patient received 6 cycles of paclitaxel (175 mg/m² on day 1) and gemcitabine (1000 mg/m² on days 1 and 8 every 21 days), with progressive disease 16 months later. Despite 0% PD-L1 expression, pembrolizumab 400 mg intravenous (IV) treatment every 6 weeks was initiated 2 months later, and subsequent positron emission tomography/CT showed a positive response at 3 and 7 months after treatment initiation. After the patient’s sixth cycle of pembrolizumab, a generalized maculopapular rash involving approximately 50% of the body surface area led to discontinuation of pembrolizumab, initiation of multiple courses of prednisone and prednisolone, and a dermatology referral.
At the current presentation, the patient exhibited excoriated red patches on the abdomen, wrists, arms, upper chest, and legs (Figure 1). Tense blisters were observed on various areas, including the ear and arms. The provisional diagnosis was pembrolizumab-induced BP, supported by the clinical history, presentation, and an initial positive response to steroids. Treatment included topical triamcinolone 0.1% ointment and prednisone 40 mg daily. Biopsies revealed subepidermal blisters with underlying eosinophils on histopathology (Figure 2). Direct immunofluorescence showed strong linear basement membrane zone staining with IgG and C3, consistent with a diagnosis of BP.
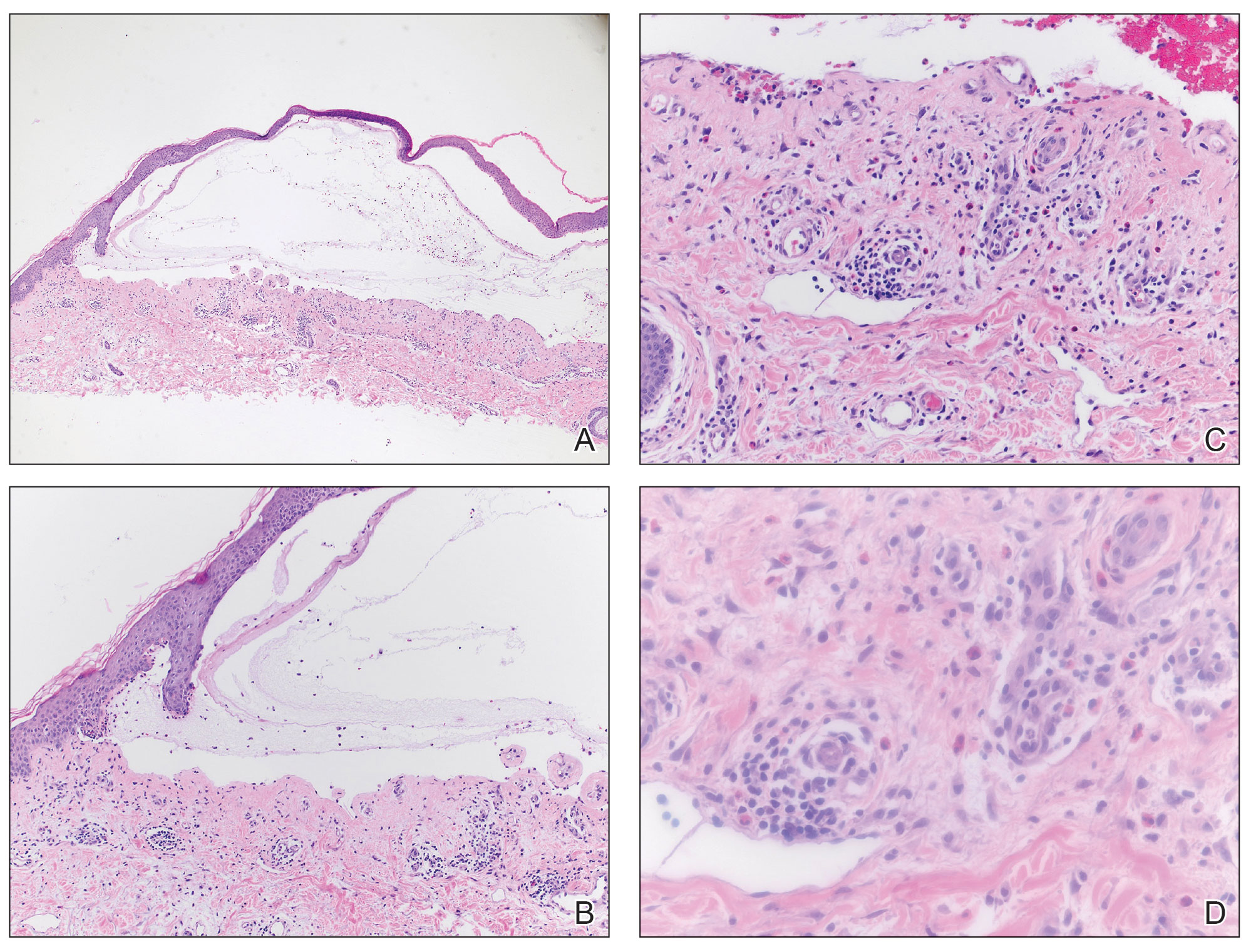
One month later, the patient was given the first of two 1-g doses of rituximab, chosen as a treatment due to metastatic cancer history and ongoing severity of the DIBP. In addition, a slow prednisone taper was initiated. Atovaquone 1500 mg daily was ordered for Pneumocystis jirovecii prophylaxis. Following the first rituximab dose, the patient became clear of DIBP but required treatment for a chronic urinary tract infection, delaying the second rituximab dose. The prednisone taper continued, however, and the patient reported re-emergence of several blisters, followed by resolution of pruritus following the second rituximab dose. Bilateral pulmonary embolisms were noted on a restaging CT, attributed to the underlying malignancy and inflammation from DIBP. Doxycycline was initiated at 100 mg twice daily, and prednisone was slowly tapered (as tolerated by the patient’s symptoms) down to 2.5 mg daily approximately 6 months after rituximab initiation. The patient remains in clinical remission at last follow-up; however, considerations for further treatments have included intravenous immunoglobulin.
Comment
This case highlights major clinical challenges in the diagnosis and management of DIBP in a patient with metastatic urothelial carcinoma receiving ICI therapy. Our patient’s clinical course offers several high-yield lessons regarding diagnostic latency, treatment resistance, and a multidisciplinary approach to management.
Pruritus as a Precursor—Since an initial report in 2015, the emergence of DIBP postpembrolizumab has been well described in the literature.19-22 Pruritus is frequently the earliest symptom, preceding bullous eruption. Similar to our case—in which DIBP developed 30 weeks after pembrolizumab initiation—the classic clinical presentation and formation of bullae often are delayed, typically occurring 28 and 39 weeks.
Beyond Corticosteroids to Manage Refractory DIBP—Our patient’s DIBP persisted despite multiple interventions, including pembrolizumab discontinuation, corticosteroid therapy, and rituximab administration. Although cases of DIBP in pembrolizumab-treated metastatic urothelial carcinoma patients have been reported, they did not exhibit similar treatment resistance.23-25 As observed in our patient, immunotherapy discontinuation has been reported in at least 40% of all ICI-mediated cases of BP.14 Subsequent management involves low-dose oral corticosteroids and potent topical corticosteroids; the duration of steroid treatment varies widely, ranging from a few weeks to longer than 12 months, with no standardized approach.26 In cases where ICI withdrawal and corticosteroids fail to produce a complete response, monoclonal antibodies such as rituximab, dupilumab, and omalizumab have been used as alternative treatments, with dupilumab recently receiving US Food and Drug Administration approval for moderate to severe BP.27-31 These biologics selectively inhibit autoantibody formation and the inflammatory cascade, and research has pointed toward them as safe and effective options for refractory BP. Although robust randomized, controlled clinical trials on rituximab for DIBP still are lacking, prospective and retrospective cohort studies have shown promising results, including complete remission rates of 67% to 90%, along with a decline in circulating BP180-specific B lymphocytes, anti-BP180 IgG, and the expression of proinflammatory IL-15 and IL-6.32
Despite receiving 2 doses of rituximab, our patient experienced recurrence of blisters when prednisone was tapered, prompting discussions about alternative tapering timelines and additional therapies such as doxycycline33 or intravenous immunoglobulin,34 which have emerged as steroid-sparing agents for BP following initial steroid therapy.
Systemic Barriers and the Need for Multidisciplinary Care—This case underscores systemic barriers within the health care system that impede prompt diagnosis and management of conditions such as DIBP. The 5-month delay between the patient’s referral to dermatology and the actual consultation, potentially due to shortages of dermatologists, highlights the need for more systematic urgent dermatologic referrals and streamlined diagnostic pathways in suspected cases of IRAEs. Diagnosis requires comprehensive evaluation, including skin biopsy for histopathologic examination and immunofluorescence studies. Ruling out alternative blistering disorders, such as epidermolysis bullosa acquisita, is crucial before confirming a BP diagnosis. Encouraging direct communication between referring physicians and consultants often can expedite the process, as a call from the referring physician can alert the consultant and speed up scheduling. Notably, the patient’s daughter, who was a patient of the dermatologist herself, played a crucial role in advocating for the dermatology referral. Although this should not be necessary, it highlights the pivotal role families can play in ensuring timely access to specialized care for challenging conditions such as BP.
Lastly, the refractory nature of the patient’s condition, coupled with concurrent chronic urinary tract infection and bilateral pulmonary embolisms, emphasizes the necessity of multidisciplinary collaboration among oncology, dermatology, and primary care in managing DIBP. Consulting experts on IRAEs and coordinating with the oncologist were essential for making informed treatment decisions and facilitating the timely exchange of clinical information.
Conclusion
This case underscores the importance of timely recognition and diagnosis of DIBP in patients undergoing ICI therapy but also highlights the need for individualized treatment approaches and multidisciplinary collaboration when managing adverse cutaneous reactions.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-332.
- Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Sci. 2014;73:179-186.
- Sárdy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69:748-753.
- Smith EP, Taylor TB, Meyer LJ, et al. Antigen identification in drug-induced bullous pemphigoid. J Am Acad Dermatol. 1993;29(5 Pt 2):879-882.
- Siegel J, Totonchy M, Damsky W, et al. Bullous disorders associated with anti-PD-1 and anti-PD-L1 therapy: a retrospective analysis evaluating the clinical and histopathologic features, frequency, and impact on cancer therapy. J Am Acad Dermatol. 2018;79:1081-1088.
- Asdourian MS, Shah N, Jacoby TV, et al. Association of bullous pemphigoid with immune checkpoint inhibitor therapy in patients with cancer: a systematic review. JAMA Dermatol. 2022;158:933-941.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264.
- Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
- Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558-562.
- Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015-1026.
- Fradet Y, Bellmunt J, Vaughn DJ, et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: results of >2 years of follow-up. Ann Oncol. 2019;30:970-976.
- Felsenstein KM, Theodorescu D. Precision medicine for urothelial bladder cancer: update on tumour genomics and immunotherapy. Nat Rev Urol. 2018;15:92-111.
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Hwang SJE, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.e1.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26:2375-2391.
- Weber JS, Yang JC, Atkins MB, et al. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33:2092-2099.
- Carlos G, Anforth R, Chou S, et al. A case of bullous pemphigoid in a patient with metastatic melanoma treated with pembrolizumab. Melanoma Res. 2015;25:265-268.
- Adachi E, Honda T, Nonoyama S, al. Severe bullous pemphigoid in a metastatic lung cancer patient treated with pembrolizumab. J Dermatol. 2019;46:E232-E233.
- Cardona AF, Ruiz-Patiño A, Zatarain-Barron ZL, et al. Refractory bullous pemphigoid in a patient with metastatic lung adenocarcinoma treated with pembrolizumab. Case Rep Oncol. 2021;14:386-390.
- Sun CW, Grossman SK, Aphale A, et al. Pembrolizumab-induced bullous pemphigoid. JAAD Case Rep. 2019;5:362-364.
- Correia C, Fernandes S, Soares-de-Almeida L, et al. Bullous pemphigoid probably associated with pembrolizumab: a case of delayed toxicity. Int J Dermatol. 2022;61:E129-E131.
- Shalata W, Weissmann S, Itzhaki Gabay S, et al. A retrospective, single-institution experience of bullous pemphigoid as an adverse effect of immune checkpoint inhibitors. Cancers. 2022;14:5451. doi:10.3390/cancers14215451
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Wang J, Hu X, Jiang W, et al. Analysis of the clinical characteristics of pembrolizumab-induced bullous pemphigoid. Front Oncol. 2023;13:1095694.
- Thomas RM, Colon A, Motaparthi K. Rituximab in autoimmune pemphigoid diseases: indications, optimized regimens, and practice gaps. Clin Dermatol. 2020;38:384-396.
- Sowerby L, Dewan AK, Granter S, et al. Rituximab treatment of nivolumab-induced bullous pemphigoid. JAMA Dermatol. 2017;153:603-605.
- Sharma P, Barnes M, Nabeel S, et al. Pembrolizumab-induced bullous pemphigoid treated with rituximab. JCO Oncol Pract. 2020;16:764-766.
- Abdat R, Waldman RA, de Bedout V, et al. Dupilumab as a novel therapy for bullous pemphigoid: a multicenter case series. J Am Acad Dermatol. 2020;83:46-52.
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621.
- Karakioulaki M, Eyerich K, Patsatsi A. Advancements in bullous pemphigoid treatment: a comprehensive pipeline update. Am J Clin Dermatol. 2024;25:195-212.
- Jin XX, Wang X, Shan Y, et al. Efficacy and safety of tetracyclines for pemphigoid: a systematic review and meta-analysis. Arch Dermatol Res. 2022;314:191-201.
- Kianfar N, Dasdar S, Daneshpazhooh M, et al. A systematic review on efficacy, safety and treatment durability of intravenous immunoglobulin in autoimmune bullous dermatoses: special focus on indication and combination therapy. Exp Dermatol. 2023;32:934-944.
Bullous pemphigoid (BP) is an autoimmune blistering disorder characterized by the development of tense subepidermal blisters and erosions primarily on the skin, commonly affecting the elderly.1 It is attributed to autoantibodies targeting 2 hemidesmosomal components within the dermoepidermal junction—transmembrane collagen XVII (BP180/BPAG2) and plakin family protein BP230 (BPAG1)—resulting in blister formation due to loss of structural integrity.2 Typically, patients present with pruritic urticarial plaques and tense bullae localized on flexural areas, but cutaneous manifestations vary and can be nonspecific. Histologically, a subepidermal blister with eosinophilic infiltration is characteristic, and detection of circulating autoantibodies against BP180 and BP230 antigens aids in diagnosis.3,4
Drug-induced BP (DIBP) is a subset triggered by medications, including immune checkpoint inhibitors (ICIs) targeting programmed cell death protein-1 (PD-1) or its ligand, programmed death ligand-1 (PD-L1).5,6 Often overexpressed in malignant tumors, PD-L1 inhibits host lymphocytic and apoptotic immune responses. Anti‒PD-1 and anti‒PD-L1 agents, designed to enhance the immune system’s ability to recognize and eliminate cancer cells,7,8 have improved oncologic outcomes for various cancers, including urothelial cancer.9-11 Before 2016, platinum-based chemotherapy was the mainstay for metastatic urothelial cancer management, but US Food and Drug Administration approval of 5 ICIs—nivolumab, pembrolizumab, avelumab, atezolizumab, and durvalumab—transformed treatment options.12Despite robust antitumor responses to ICIs, these medications are increasingly associated with immune-related adverse events (IRAEs), including DIBP, due to inhibition of negative regulators of immunity crucial for maintaining immunologic homeostasis.13,14 Up to 30% to 40% of patients treated with PD-1 inhibitors experience dermatologic complications, such as lichenoid reactions, eczema, vitiligo, and pruritus,15 and patients undergoing treatment with the PD-1 inhibitor pembrolizumab are estimated to be 2.6 times more likely to develop a rash than those receiving standard chemotherapy.16,17 The pathogenesis of DIBP involves autoreactive T-cell activation and subsequent autoantibody production against BP antigens.18 We present the case of DIBP secondary to pembrolizumab immunotherapy in a man with PD-L1–negative metastatic bladder cancer.
Case Report
An 81-year-old man with metastatic urothelial carcinoma presented to dermatology with a pruritic rash characterized by blisters of 5 months’ duration following treatment with pembrolizumab. He had a history of non–muscle invasive urothelial carcinoma and underwent intravesical bacillus Calmette-Guerin treatment. Thirty years later, after surveillance cystoscopies, the patient developed hematuria, which prompted pelvic ultrasonography and cystoscopy that revealed a tumor. Transurethral resection of the bladder tumor confirmed invasive, high-grade papillary urothelial carcinoma with vascular and muscle invasion (clinical stage T2NxMx). Due to elevated creatinine levels, neoadjuvant chemotherapy was contraindicated. Instead, the patient underwent cystoprostatectomy with ureteroileal conduit creation and pelvic lymphadenectomy one month later; final pathology revealed pT2aN0M0 disease with multifocal carcinoma in situ. At that time, there was no evidence of distant metastasis. Surveillance 5 months later identified pulmonary nodules that were confirmed as metastatic urothelial cancer by positron emission tomography/computed tomography (CT). The patient received 6 cycles of paclitaxel (175 mg/m² on day 1) and gemcitabine (1000 mg/m² on days 1 and 8 every 21 days), with progressive disease 16 months later. Despite 0% PD-L1 expression, pembrolizumab 400 mg intravenous (IV) treatment every 6 weeks was initiated 2 months later, and subsequent positron emission tomography/CT showed a positive response at 3 and 7 months after treatment initiation. After the patient’s sixth cycle of pembrolizumab, a generalized maculopapular rash involving approximately 50% of the body surface area led to discontinuation of pembrolizumab, initiation of multiple courses of prednisone and prednisolone, and a dermatology referral.
At the current presentation, the patient exhibited excoriated red patches on the abdomen, wrists, arms, upper chest, and legs (Figure 1). Tense blisters were observed on various areas, including the ear and arms. The provisional diagnosis was pembrolizumab-induced BP, supported by the clinical history, presentation, and an initial positive response to steroids. Treatment included topical triamcinolone 0.1% ointment and prednisone 40 mg daily. Biopsies revealed subepidermal blisters with underlying eosinophils on histopathology (Figure 2). Direct immunofluorescence showed strong linear basement membrane zone staining with IgG and C3, consistent with a diagnosis of BP.
One month later, the patient was given the first of two 1-g doses of rituximab, chosen as a treatment due to metastatic cancer history and ongoing severity of the DIBP. In addition, a slow prednisone taper was initiated. Atovaquone 1500 mg daily was ordered for Pneumocystis jirovecii prophylaxis. Following the first rituximab dose, the patient became clear of DIBP but required treatment for a chronic urinary tract infection, delaying the second rituximab dose. The prednisone taper continued, however, and the patient reported re-emergence of several blisters, followed by resolution of pruritus following the second rituximab dose. Bilateral pulmonary embolisms were noted on a restaging CT, attributed to the underlying malignancy and inflammation from DIBP. Doxycycline was initiated at 100 mg twice daily, and prednisone was slowly tapered (as tolerated by the patient’s symptoms) down to 2.5 mg daily approximately 6 months after rituximab initiation. The patient remains in clinical remission at last follow-up; however, considerations for further treatments have included intravenous immunoglobulin.
Comment
This case highlights major clinical challenges in the diagnosis and management of DIBP in a patient with metastatic urothelial carcinoma receiving ICI therapy. Our patient’s clinical course offers several high-yield lessons regarding diagnostic latency, treatment resistance, and a multidisciplinary approach to management.
Pruritus as a Precursor—Since an initial report in 2015, the emergence of DIBP postpembrolizumab has been well described in the literature.19-22 Pruritus is frequently the earliest symptom, preceding bullous eruption. Similar to our case—in which DIBP developed 30 weeks after pembrolizumab initiation—the classic clinical presentation and formation of bullae often are delayed, typically occurring 28 and 39 weeks.
Beyond Corticosteroids to Manage Refractory DIBP—Our patient’s DIBP persisted despite multiple interventions, including pembrolizumab discontinuation, corticosteroid therapy, and rituximab administration. Although cases of DIBP in pembrolizumab-treated metastatic urothelial carcinoma patients have been reported, they did not exhibit similar treatment resistance.23-25 As observed in our patient, immunotherapy discontinuation has been reported in at least 40% of all ICI-mediated cases of BP.14 Subsequent management involves low-dose oral corticosteroids and potent topical corticosteroids; the duration of steroid treatment varies widely, ranging from a few weeks to longer than 12 months, with no standardized approach.26 In cases where ICI withdrawal and corticosteroids fail to produce a complete response, monoclonal antibodies such as rituximab, dupilumab, and omalizumab have been used as alternative treatments, with dupilumab recently receiving US Food and Drug Administration approval for moderate to severe BP.27-31 These biologics selectively inhibit autoantibody formation and the inflammatory cascade, and research has pointed toward them as safe and effective options for refractory BP. Although robust randomized, controlled clinical trials on rituximab for DIBP still are lacking, prospective and retrospective cohort studies have shown promising results, including complete remission rates of 67% to 90%, along with a decline in circulating BP180-specific B lymphocytes, anti-BP180 IgG, and the expression of proinflammatory IL-15 and IL-6.32
Despite receiving 2 doses of rituximab, our patient experienced recurrence of blisters when prednisone was tapered, prompting discussions about alternative tapering timelines and additional therapies such as doxycycline33 or intravenous immunoglobulin,34 which have emerged as steroid-sparing agents for BP following initial steroid therapy.
Systemic Barriers and the Need for Multidisciplinary Care—This case underscores systemic barriers within the health care system that impede prompt diagnosis and management of conditions such as DIBP. The 5-month delay between the patient’s referral to dermatology and the actual consultation, potentially due to shortages of dermatologists, highlights the need for more systematic urgent dermatologic referrals and streamlined diagnostic pathways in suspected cases of IRAEs. Diagnosis requires comprehensive evaluation, including skin biopsy for histopathologic examination and immunofluorescence studies. Ruling out alternative blistering disorders, such as epidermolysis bullosa acquisita, is crucial before confirming a BP diagnosis. Encouraging direct communication between referring physicians and consultants often can expedite the process, as a call from the referring physician can alert the consultant and speed up scheduling. Notably, the patient’s daughter, who was a patient of the dermatologist herself, played a crucial role in advocating for the dermatology referral. Although this should not be necessary, it highlights the pivotal role families can play in ensuring timely access to specialized care for challenging conditions such as BP.
Lastly, the refractory nature of the patient’s condition, coupled with concurrent chronic urinary tract infection and bilateral pulmonary embolisms, emphasizes the necessity of multidisciplinary collaboration among oncology, dermatology, and primary care in managing DIBP. Consulting experts on IRAEs and coordinating with the oncologist were essential for making informed treatment decisions and facilitating the timely exchange of clinical information.
Conclusion
This case underscores the importance of timely recognition and diagnosis of DIBP in patients undergoing ICI therapy but also highlights the need for individualized treatment approaches and multidisciplinary collaboration when managing adverse cutaneous reactions.
Bullous pemphigoid (BP) is an autoimmune blistering disorder characterized by the development of tense subepidermal blisters and erosions primarily on the skin, commonly affecting the elderly.1 It is attributed to autoantibodies targeting 2 hemidesmosomal components within the dermoepidermal junction—transmembrane collagen XVII (BP180/BPAG2) and plakin family protein BP230 (BPAG1)—resulting in blister formation due to loss of structural integrity.2 Typically, patients present with pruritic urticarial plaques and tense bullae localized on flexural areas, but cutaneous manifestations vary and can be nonspecific. Histologically, a subepidermal blister with eosinophilic infiltration is characteristic, and detection of circulating autoantibodies against BP180 and BP230 antigens aids in diagnosis.3,4
Drug-induced BP (DIBP) is a subset triggered by medications, including immune checkpoint inhibitors (ICIs) targeting programmed cell death protein-1 (PD-1) or its ligand, programmed death ligand-1 (PD-L1).5,6 Often overexpressed in malignant tumors, PD-L1 inhibits host lymphocytic and apoptotic immune responses. Anti‒PD-1 and anti‒PD-L1 agents, designed to enhance the immune system’s ability to recognize and eliminate cancer cells,7,8 have improved oncologic outcomes for various cancers, including urothelial cancer.9-11 Before 2016, platinum-based chemotherapy was the mainstay for metastatic urothelial cancer management, but US Food and Drug Administration approval of 5 ICIs—nivolumab, pembrolizumab, avelumab, atezolizumab, and durvalumab—transformed treatment options.12Despite robust antitumor responses to ICIs, these medications are increasingly associated with immune-related adverse events (IRAEs), including DIBP, due to inhibition of negative regulators of immunity crucial for maintaining immunologic homeostasis.13,14 Up to 30% to 40% of patients treated with PD-1 inhibitors experience dermatologic complications, such as lichenoid reactions, eczema, vitiligo, and pruritus,15 and patients undergoing treatment with the PD-1 inhibitor pembrolizumab are estimated to be 2.6 times more likely to develop a rash than those receiving standard chemotherapy.16,17 The pathogenesis of DIBP involves autoreactive T-cell activation and subsequent autoantibody production against BP antigens.18 We present the case of DIBP secondary to pembrolizumab immunotherapy in a man with PD-L1–negative metastatic bladder cancer.
Case Report
An 81-year-old man with metastatic urothelial carcinoma presented to dermatology with a pruritic rash characterized by blisters of 5 months’ duration following treatment with pembrolizumab. He had a history of non–muscle invasive urothelial carcinoma and underwent intravesical bacillus Calmette-Guerin treatment. Thirty years later, after surveillance cystoscopies, the patient developed hematuria, which prompted pelvic ultrasonography and cystoscopy that revealed a tumor. Transurethral resection of the bladder tumor confirmed invasive, high-grade papillary urothelial carcinoma with vascular and muscle invasion (clinical stage T2NxMx). Due to elevated creatinine levels, neoadjuvant chemotherapy was contraindicated. Instead, the patient underwent cystoprostatectomy with ureteroileal conduit creation and pelvic lymphadenectomy one month later; final pathology revealed pT2aN0M0 disease with multifocal carcinoma in situ. At that time, there was no evidence of distant metastasis. Surveillance 5 months later identified pulmonary nodules that were confirmed as metastatic urothelial cancer by positron emission tomography/computed tomography (CT). The patient received 6 cycles of paclitaxel (175 mg/m² on day 1) and gemcitabine (1000 mg/m² on days 1 and 8 every 21 days), with progressive disease 16 months later. Despite 0% PD-L1 expression, pembrolizumab 400 mg intravenous (IV) treatment every 6 weeks was initiated 2 months later, and subsequent positron emission tomography/CT showed a positive response at 3 and 7 months after treatment initiation. After the patient’s sixth cycle of pembrolizumab, a generalized maculopapular rash involving approximately 50% of the body surface area led to discontinuation of pembrolizumab, initiation of multiple courses of prednisone and prednisolone, and a dermatology referral.
At the current presentation, the patient exhibited excoriated red patches on the abdomen, wrists, arms, upper chest, and legs (Figure 1). Tense blisters were observed on various areas, including the ear and arms. The provisional diagnosis was pembrolizumab-induced BP, supported by the clinical history, presentation, and an initial positive response to steroids. Treatment included topical triamcinolone 0.1% ointment and prednisone 40 mg daily. Biopsies revealed subepidermal blisters with underlying eosinophils on histopathology (Figure 2). Direct immunofluorescence showed strong linear basement membrane zone staining with IgG and C3, consistent with a diagnosis of BP.
One month later, the patient was given the first of two 1-g doses of rituximab, chosen as a treatment due to metastatic cancer history and ongoing severity of the DIBP. In addition, a slow prednisone taper was initiated. Atovaquone 1500 mg daily was ordered for Pneumocystis jirovecii prophylaxis. Following the first rituximab dose, the patient became clear of DIBP but required treatment for a chronic urinary tract infection, delaying the second rituximab dose. The prednisone taper continued, however, and the patient reported re-emergence of several blisters, followed by resolution of pruritus following the second rituximab dose. Bilateral pulmonary embolisms were noted on a restaging CT, attributed to the underlying malignancy and inflammation from DIBP. Doxycycline was initiated at 100 mg twice daily, and prednisone was slowly tapered (as tolerated by the patient’s symptoms) down to 2.5 mg daily approximately 6 months after rituximab initiation. The patient remains in clinical remission at last follow-up; however, considerations for further treatments have included intravenous immunoglobulin.
Comment
This case highlights major clinical challenges in the diagnosis and management of DIBP in a patient with metastatic urothelial carcinoma receiving ICI therapy. Our patient’s clinical course offers several high-yield lessons regarding diagnostic latency, treatment resistance, and a multidisciplinary approach to management.
Pruritus as a Precursor—Since an initial report in 2015, the emergence of DIBP postpembrolizumab has been well described in the literature.19-22 Pruritus is frequently the earliest symptom, preceding bullous eruption. Similar to our case—in which DIBP developed 30 weeks after pembrolizumab initiation—the classic clinical presentation and formation of bullae often are delayed, typically occurring 28 and 39 weeks.
Beyond Corticosteroids to Manage Refractory DIBP—Our patient’s DIBP persisted despite multiple interventions, including pembrolizumab discontinuation, corticosteroid therapy, and rituximab administration. Although cases of DIBP in pembrolizumab-treated metastatic urothelial carcinoma patients have been reported, they did not exhibit similar treatment resistance.23-25 As observed in our patient, immunotherapy discontinuation has been reported in at least 40% of all ICI-mediated cases of BP.14 Subsequent management involves low-dose oral corticosteroids and potent topical corticosteroids; the duration of steroid treatment varies widely, ranging from a few weeks to longer than 12 months, with no standardized approach.26 In cases where ICI withdrawal and corticosteroids fail to produce a complete response, monoclonal antibodies such as rituximab, dupilumab, and omalizumab have been used as alternative treatments, with dupilumab recently receiving US Food and Drug Administration approval for moderate to severe BP.27-31 These biologics selectively inhibit autoantibody formation and the inflammatory cascade, and research has pointed toward them as safe and effective options for refractory BP. Although robust randomized, controlled clinical trials on rituximab for DIBP still are lacking, prospective and retrospective cohort studies have shown promising results, including complete remission rates of 67% to 90%, along with a decline in circulating BP180-specific B lymphocytes, anti-BP180 IgG, and the expression of proinflammatory IL-15 and IL-6.32
Despite receiving 2 doses of rituximab, our patient experienced recurrence of blisters when prednisone was tapered, prompting discussions about alternative tapering timelines and additional therapies such as doxycycline33 or intravenous immunoglobulin,34 which have emerged as steroid-sparing agents for BP following initial steroid therapy.
Systemic Barriers and the Need for Multidisciplinary Care—This case underscores systemic barriers within the health care system that impede prompt diagnosis and management of conditions such as DIBP. The 5-month delay between the patient’s referral to dermatology and the actual consultation, potentially due to shortages of dermatologists, highlights the need for more systematic urgent dermatologic referrals and streamlined diagnostic pathways in suspected cases of IRAEs. Diagnosis requires comprehensive evaluation, including skin biopsy for histopathologic examination and immunofluorescence studies. Ruling out alternative blistering disorders, such as epidermolysis bullosa acquisita, is crucial before confirming a BP diagnosis. Encouraging direct communication between referring physicians and consultants often can expedite the process, as a call from the referring physician can alert the consultant and speed up scheduling. Notably, the patient’s daughter, who was a patient of the dermatologist herself, played a crucial role in advocating for the dermatology referral. Although this should not be necessary, it highlights the pivotal role families can play in ensuring timely access to specialized care for challenging conditions such as BP.
Lastly, the refractory nature of the patient’s condition, coupled with concurrent chronic urinary tract infection and bilateral pulmonary embolisms, emphasizes the necessity of multidisciplinary collaboration among oncology, dermatology, and primary care in managing DIBP. Consulting experts on IRAEs and coordinating with the oncologist were essential for making informed treatment decisions and facilitating the timely exchange of clinical information.
Conclusion
This case underscores the importance of timely recognition and diagnosis of DIBP in patients undergoing ICI therapy but also highlights the need for individualized treatment approaches and multidisciplinary collaboration when managing adverse cutaneous reactions.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-332.
- Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Sci. 2014;73:179-186.
- Sárdy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69:748-753.
- Smith EP, Taylor TB, Meyer LJ, et al. Antigen identification in drug-induced bullous pemphigoid. J Am Acad Dermatol. 1993;29(5 Pt 2):879-882.
- Siegel J, Totonchy M, Damsky W, et al. Bullous disorders associated with anti-PD-1 and anti-PD-L1 therapy: a retrospective analysis evaluating the clinical and histopathologic features, frequency, and impact on cancer therapy. J Am Acad Dermatol. 2018;79:1081-1088.
- Asdourian MS, Shah N, Jacoby TV, et al. Association of bullous pemphigoid with immune checkpoint inhibitor therapy in patients with cancer: a systematic review. JAMA Dermatol. 2022;158:933-941.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264.
- Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
- Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558-562.
- Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015-1026.
- Fradet Y, Bellmunt J, Vaughn DJ, et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: results of >2 years of follow-up. Ann Oncol. 2019;30:970-976.
- Felsenstein KM, Theodorescu D. Precision medicine for urothelial bladder cancer: update on tumour genomics and immunotherapy. Nat Rev Urol. 2018;15:92-111.
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Hwang SJE, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.e1.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26:2375-2391.
- Weber JS, Yang JC, Atkins MB, et al. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33:2092-2099.
- Carlos G, Anforth R, Chou S, et al. A case of bullous pemphigoid in a patient with metastatic melanoma treated with pembrolizumab. Melanoma Res. 2015;25:265-268.
- Adachi E, Honda T, Nonoyama S, al. Severe bullous pemphigoid in a metastatic lung cancer patient treated with pembrolizumab. J Dermatol. 2019;46:E232-E233.
- Cardona AF, Ruiz-Patiño A, Zatarain-Barron ZL, et al. Refractory bullous pemphigoid in a patient with metastatic lung adenocarcinoma treated with pembrolizumab. Case Rep Oncol. 2021;14:386-390.
- Sun CW, Grossman SK, Aphale A, et al. Pembrolizumab-induced bullous pemphigoid. JAAD Case Rep. 2019;5:362-364.
- Correia C, Fernandes S, Soares-de-Almeida L, et al. Bullous pemphigoid probably associated with pembrolizumab: a case of delayed toxicity. Int J Dermatol. 2022;61:E129-E131.
- Shalata W, Weissmann S, Itzhaki Gabay S, et al. A retrospective, single-institution experience of bullous pemphigoid as an adverse effect of immune checkpoint inhibitors. Cancers. 2022;14:5451. doi:10.3390/cancers14215451
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Wang J, Hu X, Jiang W, et al. Analysis of the clinical characteristics of pembrolizumab-induced bullous pemphigoid. Front Oncol. 2023;13:1095694.
- Thomas RM, Colon A, Motaparthi K. Rituximab in autoimmune pemphigoid diseases: indications, optimized regimens, and practice gaps. Clin Dermatol. 2020;38:384-396.
- Sowerby L, Dewan AK, Granter S, et al. Rituximab treatment of nivolumab-induced bullous pemphigoid. JAMA Dermatol. 2017;153:603-605.
- Sharma P, Barnes M, Nabeel S, et al. Pembrolizumab-induced bullous pemphigoid treated with rituximab. JCO Oncol Pract. 2020;16:764-766.
- Abdat R, Waldman RA, de Bedout V, et al. Dupilumab as a novel therapy for bullous pemphigoid: a multicenter case series. J Am Acad Dermatol. 2020;83:46-52.
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621.
- Karakioulaki M, Eyerich K, Patsatsi A. Advancements in bullous pemphigoid treatment: a comprehensive pipeline update. Am J Clin Dermatol. 2024;25:195-212.
- Jin XX, Wang X, Shan Y, et al. Efficacy and safety of tetracyclines for pemphigoid: a systematic review and meta-analysis. Arch Dermatol Res. 2022;314:191-201.
- Kianfar N, Dasdar S, Daneshpazhooh M, et al. A systematic review on efficacy, safety and treatment durability of intravenous immunoglobulin in autoimmune bullous dermatoses: special focus on indication and combination therapy. Exp Dermatol. 2023;32:934-944.
- Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320-332.
- Nishie W. Update on the pathogenesis of bullous pemphigoid: an autoantibody-mediated blistering disease targeting collagen XVII. J Dermatol Sci. 2014;73:179-186.
- Sárdy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69:748-753.
- Smith EP, Taylor TB, Meyer LJ, et al. Antigen identification in drug-induced bullous pemphigoid. J Am Acad Dermatol. 1993;29(5 Pt 2):879-882.
- Siegel J, Totonchy M, Damsky W, et al. Bullous disorders associated with anti-PD-1 and anti-PD-L1 therapy: a retrospective analysis evaluating the clinical and histopathologic features, frequency, and impact on cancer therapy. J Am Acad Dermatol. 2018;79:1081-1088.
- Asdourian MS, Shah N, Jacoby TV, et al. Association of bullous pemphigoid with immune checkpoint inhibitor therapy in patients with cancer: a systematic review. JAMA Dermatol. 2022;158:933-941.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264.
- Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991-998.
- Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558-562.
- Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015-1026.
- Fradet Y, Bellmunt J, Vaughn DJ, et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: results of >2 years of follow-up. Ann Oncol. 2019;30:970-976.
- Felsenstein KM, Theodorescu D. Precision medicine for urothelial bladder cancer: update on tumour genomics and immunotherapy. Nat Rev Urol. 2018;15:92-111.
- Sibaud V. Dermatologic reactions to immune checkpoint inhibitors: skin toxicities and immunotherapy. Am J Clin Dermatol. 2018;19:345-361.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Hwang SJE, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.e1.
- Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
- Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26:2375-2391.
- Weber JS, Yang JC, Atkins MB, et al. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33:2092-2099.
- Carlos G, Anforth R, Chou S, et al. A case of bullous pemphigoid in a patient with metastatic melanoma treated with pembrolizumab. Melanoma Res. 2015;25:265-268.
- Adachi E, Honda T, Nonoyama S, al. Severe bullous pemphigoid in a metastatic lung cancer patient treated with pembrolizumab. J Dermatol. 2019;46:E232-E233.
- Cardona AF, Ruiz-Patiño A, Zatarain-Barron ZL, et al. Refractory bullous pemphigoid in a patient with metastatic lung adenocarcinoma treated with pembrolizumab. Case Rep Oncol. 2021;14:386-390.
- Sun CW, Grossman SK, Aphale A, et al. Pembrolizumab-induced bullous pemphigoid. JAAD Case Rep. 2019;5:362-364.
- Correia C, Fernandes S, Soares-de-Almeida L, et al. Bullous pemphigoid probably associated with pembrolizumab: a case of delayed toxicity. Int J Dermatol. 2022;61:E129-E131.
- Shalata W, Weissmann S, Itzhaki Gabay S, et al. A retrospective, single-institution experience of bullous pemphigoid as an adverse effect of immune checkpoint inhibitors. Cancers. 2022;14:5451. doi:10.3390/cancers14215451
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Wang J, Hu X, Jiang W, et al. Analysis of the clinical characteristics of pembrolizumab-induced bullous pemphigoid. Front Oncol. 2023;13:1095694.
- Thomas RM, Colon A, Motaparthi K. Rituximab in autoimmune pemphigoid diseases: indications, optimized regimens, and practice gaps. Clin Dermatol. 2020;38:384-396.
- Sowerby L, Dewan AK, Granter S, et al. Rituximab treatment of nivolumab-induced bullous pemphigoid. JAMA Dermatol. 2017;153:603-605.
- Sharma P, Barnes M, Nabeel S, et al. Pembrolizumab-induced bullous pemphigoid treated with rituximab. JCO Oncol Pract. 2020;16:764-766.
- Abdat R, Waldman RA, de Bedout V, et al. Dupilumab as a novel therapy for bullous pemphigoid: a multicenter case series. J Am Acad Dermatol. 2020;83:46-52.
- Cao P, Xu W, Zhang L. Rituximab, omalizumab, and dupilumab treatment outcomes in bullous pemphigoid: a systematic review. Front Immunol. 2022;13:928621.
- Karakioulaki M, Eyerich K, Patsatsi A. Advancements in bullous pemphigoid treatment: a comprehensive pipeline update. Am J Clin Dermatol. 2024;25:195-212.
- Jin XX, Wang X, Shan Y, et al. Efficacy and safety of tetracyclines for pemphigoid: a systematic review and meta-analysis. Arch Dermatol Res. 2022;314:191-201.
- Kianfar N, Dasdar S, Daneshpazhooh M, et al. A systematic review on efficacy, safety and treatment durability of intravenous immunoglobulin in autoimmune bullous dermatoses: special focus on indication and combination therapy. Exp Dermatol. 2023;32:934-944.
Pembrolizumab-Induced Bullous Pemphigoid: Navigating Diagnostic Challenges and Treatment Resistance
Pembrolizumab-Induced Bullous Pemphigoid: Navigating Diagnostic Challenges and Treatment Resistance
Practice Points
- Suspect bullous pemphigoid (BP) in patients receiving immune checkpoint inhibitors (ICIs) with new-onset pruritus or dermatologic lesions; blisters may be delayed for months.
- Treatment-resistant cases of drug-induced BP warrant consideration of alternative therapies, including rituximab, doxycycline, or intravenous immunoglobulin.
- Multidisciplinary management with dermatology and oncology is essential, as immune-related effects may persist even after ICI discontinuation.
- Encourage patients to report new skin changes promptly to their primary care physician to allow for early intervention.
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
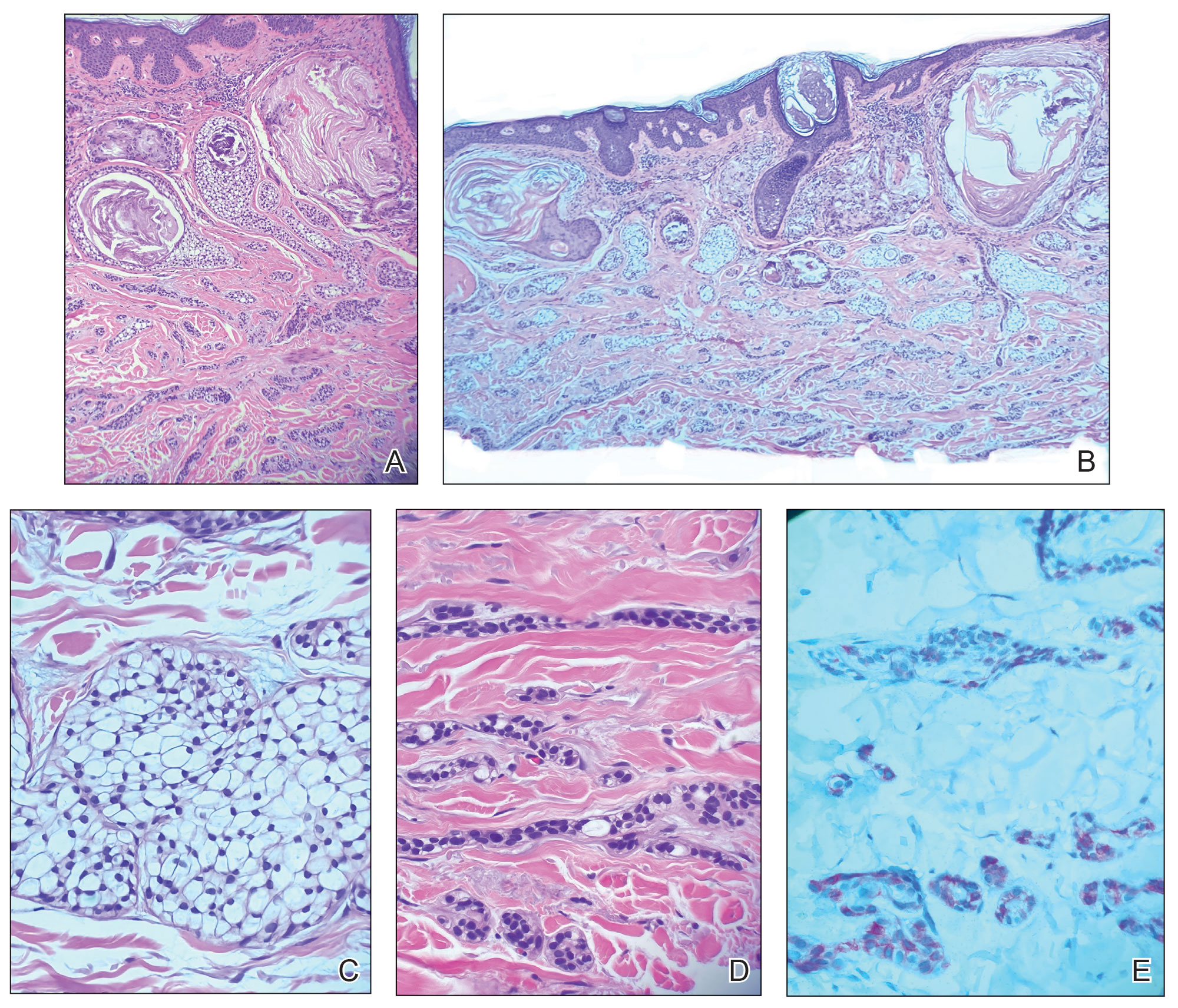
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
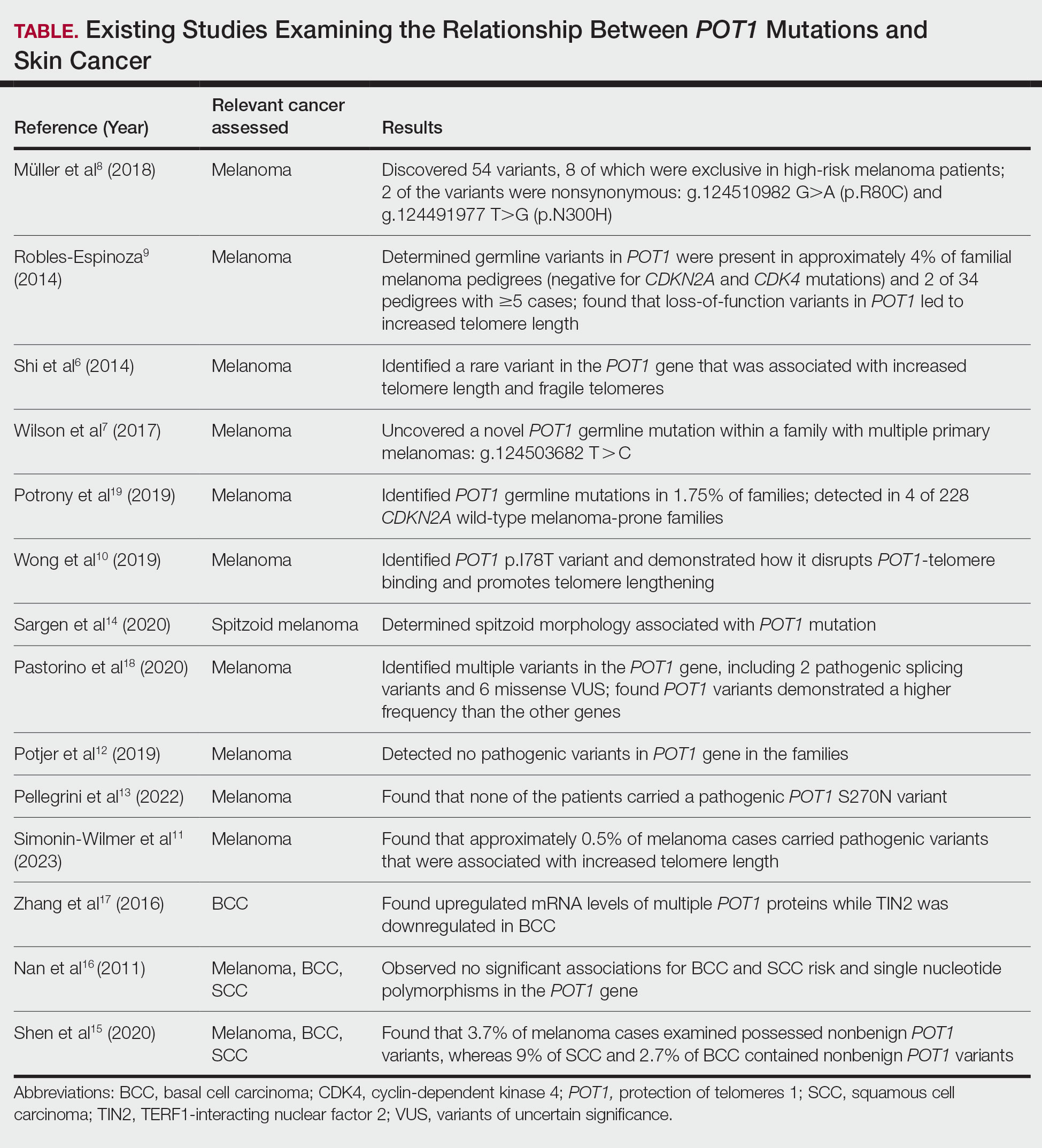
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
A 72-year-old man with a history of multiple cancers, including melanoma, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC), presented to the dermatology clinic for a regularly scheduled full-body skin examination. His family history was negative for malignancy, but due to his personal history of both primary internal cancers and skin cancers, the patient previously had been referred by dermatology to a medical geneticist for evaluation. He tested positive for a pathogenic POT1 (protection of telomeres 1) variant associated with tumor predisposition, which most often is associated with cutaneous melanoma, chronic lymphocytic leukemia (CLL), angiosarcoma, and gliomas.1
At the current presentation, physical examination revealed a small, asymmetric, pink papule on the superior thoracic spine. A biopsy of the lesion was performed (Figure 1). Pathology demonstrated cornifying cystic structures with a granulomatous response at the surface of the tumor, ductal differentiation with depth, and infiltrative strands and cords of hyperchromatic cells within a collagenous stroma at the base of the specimen (Figures 2A and 2B). One unusual finding was the presence of prominent clear-cell change within the superficial portion of the neoplasm (Figure 2C). Immunohistochemical stains revealed strong p63 and p40 positivity. Epithelial membrane antigen staining was positive in the hyperchromatic strands and cords with depth but not in the clear-cell superficial portion. Similarly, periodic acid–Schiff–positive material increased within tumor cells in proportion to depth of infiltration. Additional immunohistochemical staining showed carcinoembryonic antigen was largely negative (with rare positivity in a few ductal lumina), with negative results for S100, SOX10, CD117, BerEP4, factor XIIIa, CD34, and cytokeratin 7 (Figures 2D and 2E).
The differential diagnoses included trichilemmal carcinoma (which may manifest with CD34 expression),2 clear cell BCC, adenoid cystic carcinoma (tubular variant), sebaceous carcinoma, and eccrine carcinoma. Importantly, the patient was under continuous oncologic surveillance, with no evidence of a primary internal tumor to suggest metastasis. Despite negative carcinoembryonic antigen staining, the immunohistochemical and histopathologic findings fit best with a primary cutaneous malignant eccrine tumor, specifically microcystic adnexal carcinoma (MAC), in which p63 typically stains peripheral cells but solid variants have been described.3
Eccrine carcinoma is exceedingly rare, reported in 0.01% of diagnosed cutaneous malignancies, and demonstrates overlapping features to other malignant eccrine tumors. It possesses an inconsistent immunohistochemical staining profile, making the distinction from other malignant sweat gland tumors challenging.4 Given that the morphologic features were otherwise classic for MAC in our patient, we favored a clear-cell variant.
Sixteen years prior to the current presentation, our patient presented to urology with a history of prostatitis and increasing prostate-specific antigen levels. Biopsies were negative until prostate-specific antigen reached 13 ng/mL, confirming stage 1A prostate cancer. The patient subsequently underwent a robot-assisted radical prostatectomy. At age 63 years, dysphagia that was unresponsive to antibiotics led to a tonsillar biopsy revealing T2N2bM0 stage IVA SCC of the right tonsil with confirmed HPV type 16 with extracapsular extension. The patient underwent transoral robotic radical tonsillectomy and right neck dissection, followed by adjuvant chemoradiation consisting of intensity-modulated radiation therapy (IMRT) to a total dose of 63 Gy in 33 fractions, with concurrent weekly cisplatin. At age 67 years, dyspepsia, dysphagia, pyrosis, and gastroesophageal reflux prompted endoscopy, revealing T1aNxMx esophageal adenocarcinoma. Three months later, the patient underwent laparoscopic-assisted esophagectomy, with no recurrence. At age 68 years, an atypical intramelanocytic proliferation was found on the left cheek and was treated with Mohs micrographic surgery.
At age 71 years, acral lentiginous malignant melanoma (Breslow thickness 0.8 mm; Clark level IV; American Joint Committee on Cancer T1b) was diagnosed on the left plantar foot and treated with Mohs micrographic surgery. Sentinel lymph node biopsy was negative. Squamous cell carcinoma in situ on the frontal scalp and nodular BCC on the right upper back also were diagnosed.
While there are no guidelines for surveillance of individuals with POT1, recommendations were given in consensus from a medical genetics team,1 including comprehensive monitoring—specifically baseline imaging utilizing brain and full-body magnetic resonance imaging. Furthermore, considering the crucial role of POT1 in maintaining telomeres, it was advised to measure telomere length as part of the surveillance process. Given the patient’s susceptibility to CLL, routine complete blood count assessments were recommended. Additionally, we advised close monitoring for seizures and consideration of genetic testing in first-degree relatives.
Literature Review
Given our patient’s history of multiple skin cancers, including the most recent MAC, we sought to conduct a review of the literature to evaluate existing skin cancer associations and reports for patients with known POT1 mutations to guide recommendations for dermatologic surveillance (Table). A search of PubMed articles indexed for MEDLINE through April 2023 using the terms microcystic adnexal carcinoma, POT1, melanoma, basal cell carcinoma, squamous cell carcinoma, and skin cancer yielded no reported cases of MAC associated with POT1 mutations. POT1 is one of 6 proteins (TERF1, TERF2, RAP1, TIN2, TPP1, and POT1) belonging to the shelterin complex, which plays a crucial role in telomeric DNA remodeling and regulation of telomere length.5 Mutation in the POT1 gene disrupts the shelterin complex, causing telomeres to become elongated and unstable, resulting in chromosomal abnormalities and promoting cancer development.5
While our literature review did not reveal any associations between the shelterin complex genes and MAC, mutations in the POT1 gene have been studied in other types of skin cancer, particularly melanoma.1 One of the earliest studies was conducted in 2014 by Shi et al,6 in which whole-exome sequencing was performed on families with a history of melanoma. Multiple POT1 gene pathogenic variants associated with increased telomere length and fragility were identified in unrelated families. Subsequent studies have confirmed POT1 variants in melanoma-prone families,7 supporting an association between increased telomere length and melanoma risk8-11; however, other studies have yielded nonsignificant findings.12,13 Further investigation also has identified morphologic characteristics consistent with POT1 mutation, including spitzoid morphology.14
The association between POT1 mutations and nonmelanoma skin cancers has been relatively understudied. While a few studies have explored this link, results have shown mixed findings. Some studies have suggested a potential role for POT1 mutations in cutaneous SCC risk,15 while other studies have shown no significant associations for both BCC and SCC risk and telomere gene mutations.16 Additionally, mRNA levels of POT1 were upregulated in BCC cases compared to normal tissue in a gene expression.17
Comment
In the literature, POT1 mutations are well established as high-penetrance alterations associated with melanoma.9,18,19 However, the correlation between POT1 and other forms of skin cancer is not yet delineated. Recent insights suggest that POT1 mutations play a major role in promoting melanoma progression through telomere elongation, an established driver of melanoma progression, thereby extending the proliferative capacity of incipient cancer cells.20 This notion is supported by observations of increased telomere length in melanomaprone families with POT1 mutations. Given this association, research has focused on examining the relationship between telomere length and skin cancer.
Several studies have examined the relationship between telomere length and the risk for various types of skin cancer, including melanoma, BCC, and SCC. Prior investigations have suggested that shorter telomere length is associated with a decreased risk for melanoma and an increased risk for BCC, while no significant association has been observed for SCC.16 However, subsequent reports analyzing POT1 variants have failed to reveal any conclusive associations between BCC and SCC and telomere length.16,21
In contrast, other genetic variants associated with melanoma susceptibility have demonstrated notable associations with BCC and SCC; for instance, the CDKN2A (cyclin-dependent kinase inhibitor 2A) gene, which is the first gene linked to high-risk familial melanoma, exhibits an increased presence of mutations in individuals with BCC and SCC.22 Similarly, the MC1R (melanocortin 1 receptor) variant, a gene involved in human pigmentation and known to increase the risk for melanoma, carries a statistically significantly higher risk for BCC (summary odds ratio, 1.39; 95% CI, 1.15-1.69) and SCC (summary odds ratio, 1.61; 95% CI, 1.35-1.91) when at least one variant is present and an even greater risk with 2 or more variants.23
Considering the potential importance of POT1 mutations and their association with melanoma, as well as the inconsistencies surrounding POT1 mutations and their associations with BCC and SCC, further research may clarify the impact of POT1 mutations on the development and progression of different types of skin cancers and improve understanding of the complex interplay among telomere length, genetic variants, and skin cancer susceptibility. Given the established risk for melanoma with POT1 mutations, regular dermatology surveillance seems prudent. Dermatologists should consider referring patients with multiple skin cancers (especially melanoma) and any strong family history of internal malignancies to genetic testing for POT1. Though melanoma, CLL, angiosarcoma, and gliomas are the most commonly associated malignancies with POT1 mutations, as our case demonstrates, presentations can be heterogeneous, and the spectrum of malignancies associated with POT1 may be more expansive than previously thought.
For our patient, the current surveillance plan is fullbody skin examinations every 3 months. Given no prior family history of malignancies, presumably our patient’s case was a spontaneous mutation. Interestingly, despite his many primary cancer diagnoses and metastases, our patient has responded well to all treatments without recurrence. It is unclear if these characteristics and treatment successes are features of POT1associated cancers. Further research is needed to refine recommendations for screening and management of patients with identified POT1 mutations.
Conclusion
This case report highlights a rare occurrence of MAC in a patient with a POT1 mutation. Given the limited research conducted on investigating POT1 mutations and skin cancer, it is important to consider various forms of skin cancer, in addition to melanoma, when treating patients with a POT1 mutation.
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
- Accardo ML, Osborne J, Else T. POT1 tumor predisposition. GeneReviews®. October 29, 2020. Updated December 4, 2025. University of Washington.
- Chaichamnan K, Satayasoontorn K, Puttanupaab S, et al. Malignant proliferating trichilemmal tumors with CD34 expression. J Med Assoc Thai. 2010;93(suppl 6):S28-S34.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Kaseb H, Babiker HM. Eccrine carcinoma. StatPearls [Internet]. Updated June 26, 2023. Accessed May 11, 2026. https://www.ncbi.nlm.nih.gov/books/NBK541042
- Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649-1654. doi:10.1101/gad.1215404
- Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482-486. doi:10.1038/ng.2941
- Wilson TL, Hattangady N, Lerario AM, et al. A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer. 2017;16:561-566. doi:10.1007/s10689-017-9984-y
- Müller C, Krunic M, Wendt J, et al. Germline variants in the POT1- gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8:1475-1480. doi:10.1534/g3.117.300394
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-offunction variants predispose to familial melanoma. Nat Genet. 2014;46:478-481. doi:10.1038/ng.2947
- Wong K, Robles-Espinoza CD, Rodriguez D, et al. Association of the POT1 germline missense variant p.I78T with familial melanoma. JAMA Dermatol. 2019;155:604-609. doi:10.1001/jamadermatol.2018.3662
- Simonin-Wilmer I, Ossio R, Leddin EM, et al. Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet. 2023;60:692-696. doi:10.1136/jmg-2022-108776
- Potjer TP, Bollen S, Grimbergen AJEM, et al; Dutch Working Group for Clinical Oncogenetics. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144:2453- 2464. doi:10.1002/ijc.31984
- Pellegrini C, Raimondi S, Di Nardo L, et al; Italian Melanoma Intergroup (IMI). Melanoma in children and adolescents: analysis of susceptibility genes in 123 Italian patients. J Eur Acad Dermatol Venereol. 2022;36:213-221. doi:10.1111/jdv.17735
- Sargen MR, Calista D, Elder DE, et al. Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol. 2020;83:860-869. doi:10.1016/j.jaad.2020.03.100
- Shen E, Xiu J, Lopez GY, et al. POT1 mutation spectrum in tumour types commonly diagnosed among POT1-associated hereditary cancer syndrome families. J Med Genet. 2020;57:664-670. doi:10.1136 /jmedgenet-2019-106657
- Nan H, Qureshi AA, Prescott J, et al. Genetic variants in telomere-maintaining genes and skin cancer risk. Hum Genet. 2011;129:247-253. doi:10.1007/s00439-010-0921-5
- Zhang L, Huang X, Zhu X, et al. Differential senescence capacities in meibomian gland carcinoma and basal cell carcinoma. Int J Cancer. 2016;138:1442-1452. doi:10.1002/ijc.29882
- Pastorino L, Andreotti V, Dalmasso B, et al. Insights into genetic susceptibility to melanoma by gene panel testing: potential pathogenic variants in ACD, ATM, BAP1, and POT1. Cancers (Basel). 2020;12:1007. doi:10.3390/cancers12041007
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al. POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol. 2019;181:105-113. doi:10.1111/bjd.17443
- Kim WT, Hennick K, Johnson J, et al. Cancer-associated POT1 mutations lead to telomere elongation without induction of a DNA damage response. EMBO J. 2021;40:e107346.
- Ventura A, Pellegrini C, Cardelli L, et al. Telomeres and telomerase in cutaneous squamous cell carcinoma. Int J Mol Sci. 2019;20:1333. doi:10.3390/ijms20061333
- Helgadottir H, Höiom V, Jönsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51:545-552. doi:10.1136/jmedgenet-2014-102320
- Tagliabue E, Fargnoli MC, Gandini S, et al; M-SKIP Study Group. MC1R gene variants and non-melanoma skin cancer: a pooledanalysis from the M-SKIP project. Br J Cancer. 2015;113:354-363. doi:10.1038/bjc.2015.231
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
Microcystic Adnexal Carcinoma– like Neoplasm in a Patient With POT1 Mutation
PRACTICE POINTS
- Dermatologists should consider referring patients with both a history of skin cancer and a strong family history of internal malignancy for genetic testing for POT1 (protection of telomeres 1) mutations.
- Although melanoma, chronic lymphocytic leukemia, angiosarcoma, and gliomas are most commonly associated with POT1 mutations, this case suggests a broader and more heterogeneous malignancy spectrum than previously recognized.

Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
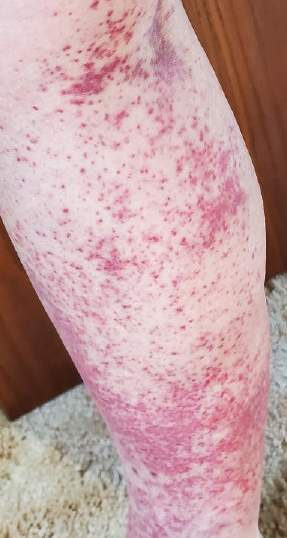
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Practice Points
- Elevation of the erythrocyte sedimentation rate (ESR) is nonspecific for inflammation and may be observed in the setting of increased immunoglobulin levels.
- Patients with elevated ESR and clinical evidence of recurrent petechiae and purpura should be screened for monoclonal and polyclonal gammopathies.

Metastatic Primary Extramammary Paget Disease: A Case Series
Metastatic Primary Extramammary Paget Disease: A Case Series
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
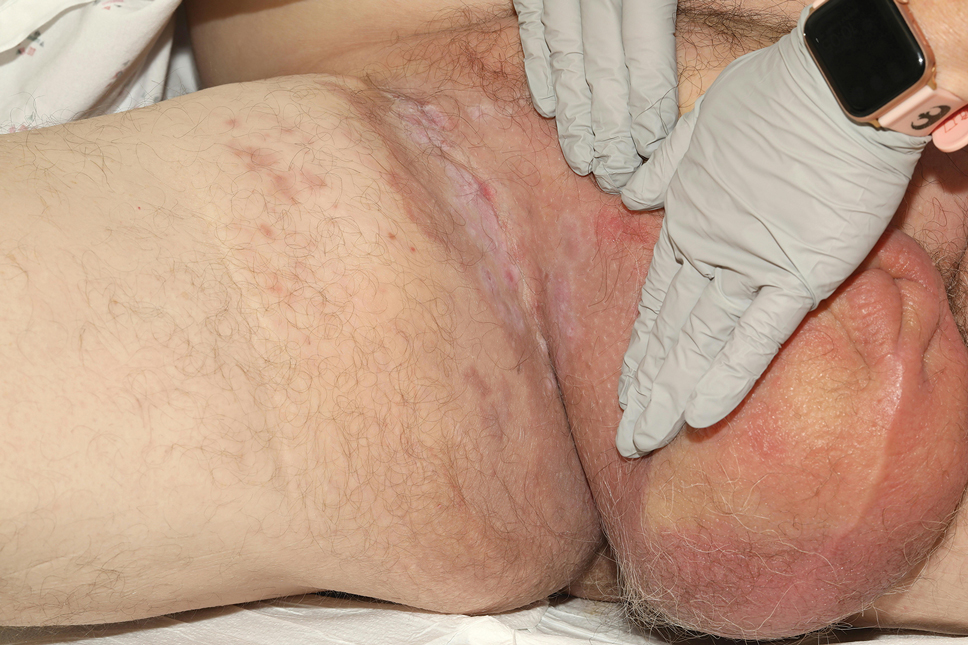
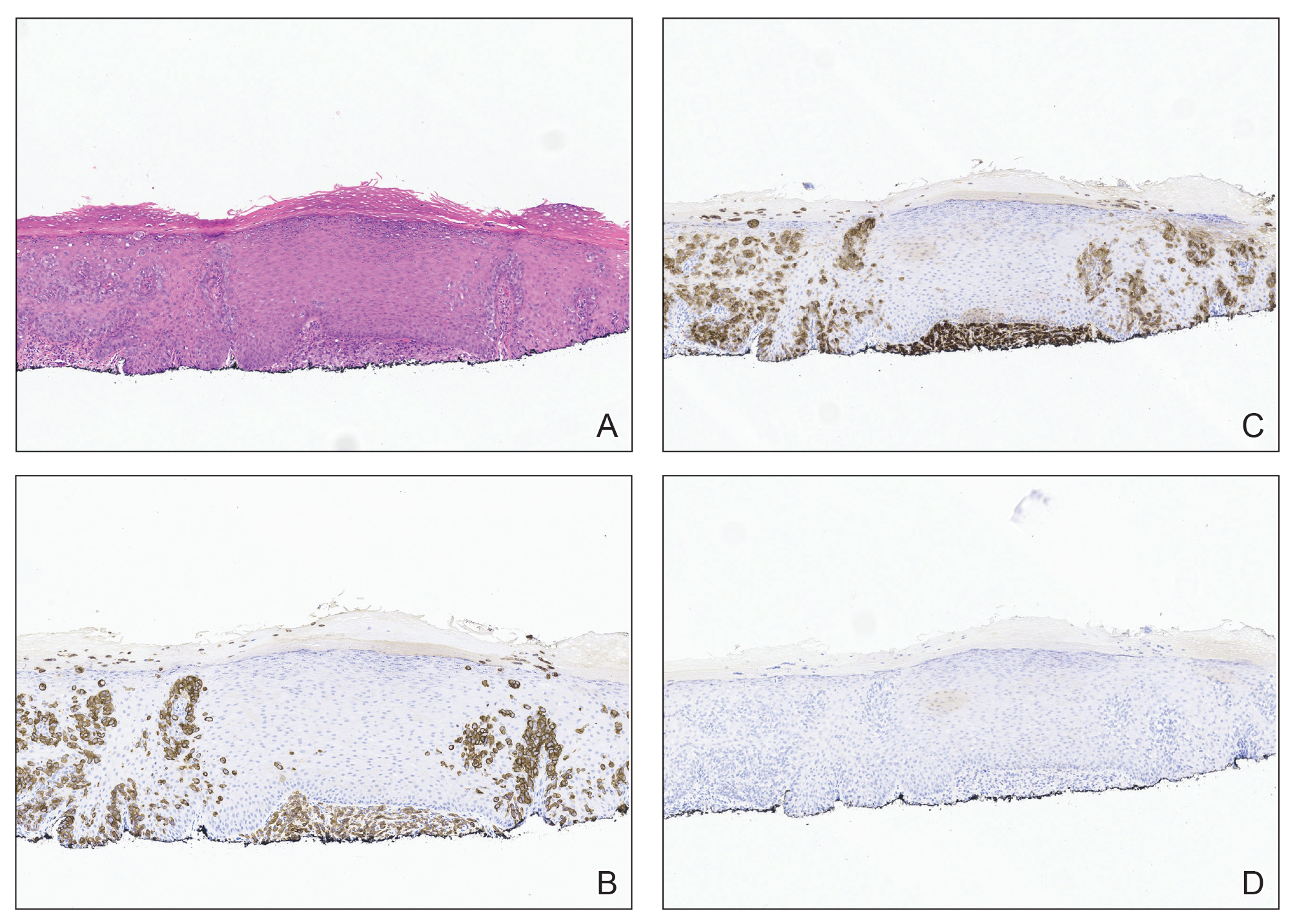
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
Metastatic Primary Extramammary Paget Disease: A Case Series
Metastatic Primary Extramammary Paget Disease: A Case Series
Practice Points
- Invasive primary extramammary Paget disease has a higher risk for lymph node metastasis.
- Consider extramammary Paget disease in patients presenting with erythematous pruritic plaques in apocrine-rich areas that fail to respond to topical steroids or antifungals.
- Prompt diagnosis can expedite comprehensive malignancy work-up and multidisciplinary management, potentially impacting patient outcomes.

Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses
Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses
Hyperkalemia involves elevated serum potassium levels (> 5.0 mEq/L) and represents an important electrolyte disturbance due to its potentially severe consequences, including cardiac effects that can lead to dysrhythmia and even asystole and death.1,2 In a US Medicare population, the prevalence of hyperkalemia has been estimated at 2.7% and is associated with substantial health care costs.3 The prevalence is even more marked in patients with preexisting conditions such as chronic kidney disease (CKD) and heart failure.4,5
Hyperkalemia can result from multiple factors, including impaired renal function, adrenal disease, adverse drug reactions of angiotensin-converting enzyme inhibitors (ACEIs) and other medications, and heritable mutations.6 Hyperkalemia poses a considerable clinical risk, associated with adverse outcomes such as myocardial infarction and increased mortality in patients with CKD.5,7,8 Electrocardiographic (ECG) changes associated with hyperkalemia play a vital role in guiding clinical decisions and treatment strategies.9 Understanding the pathophysiology, risk factors, and consequences of hyperkalemia, as well as the significance of ECG changes in its management, is essential for health care practitioners.
Case Presentation
An 81-year-old Hispanic man with a history of hypertension, hypothyroidism, gout, and CKD stage 3B presented to the emergency department with progressive weakness resulting in falls and culminating in an inability to ambulate independently. Additional symptoms included nausea, diarrhea, and myalgia. His vital signs were notable for a pulse of 41 beats/min. The physical examination was remarkable for significant weakness of the bilateral upper extremities, inability to bear his own weight, and bilateral lower extremity edema. His initial ECG upon arrival showed bradycardia with wide QRS, absent P waves, and peaked T waves (Figure 1a). These findings differed from his baseline ECG taken 1 year earlier, which showed sinus rhythm with premature atrial complexes and an old right bundle branch block (Figure 1b).
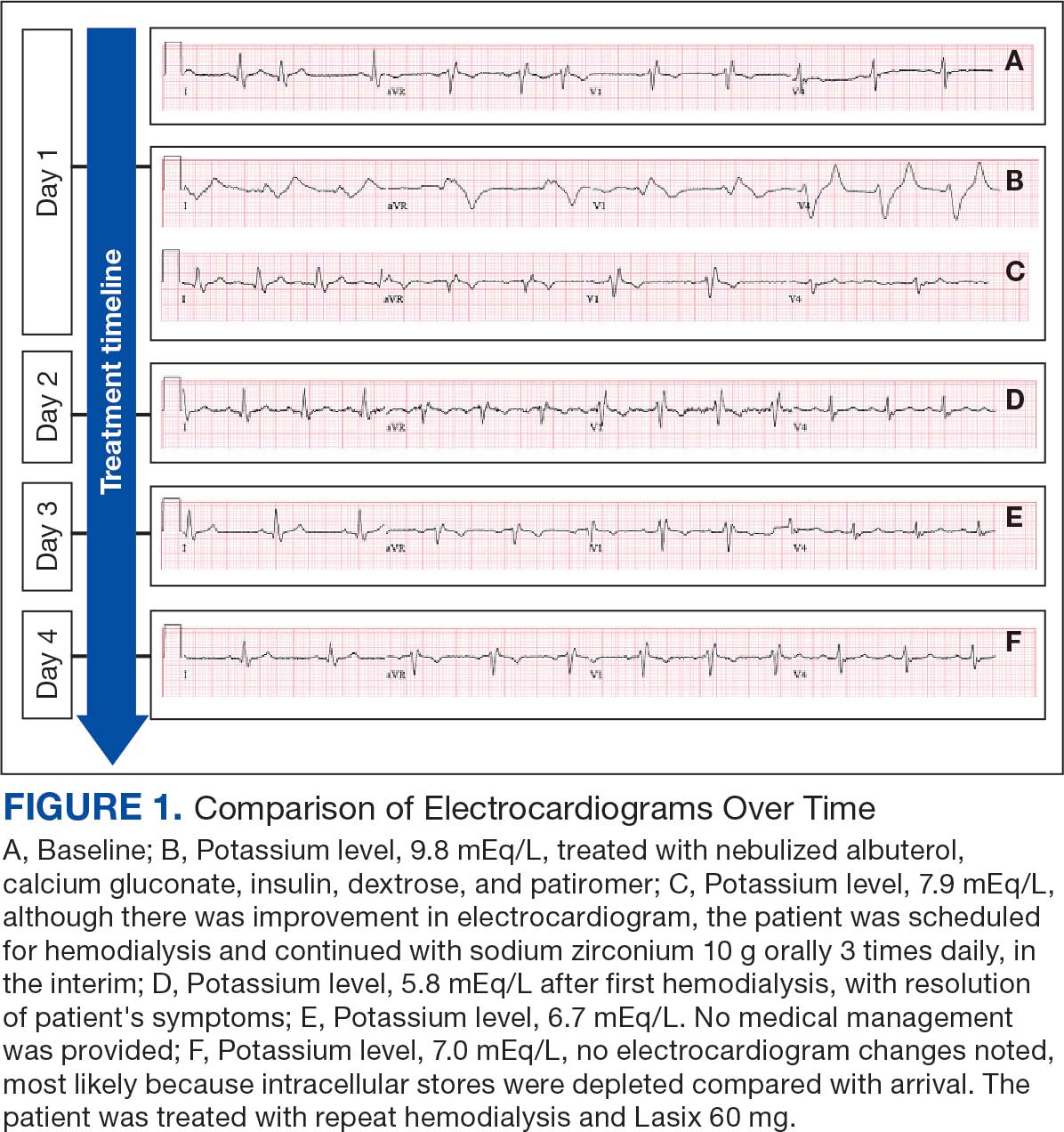
Medication review revealed that the patient was currently prescribed 100 mg allopurinol daily, 2.5 mg amlodipine daily, 10 mg atorvastatin at bedtime, 4 mg doxazosin daily, 112 mcg levothyroxine daily, 100 mg losartan daily, 25 mg metoprolol daily, and 0.4 mg tamsulosin daily. The patient had also been taking over-the-counter indomethacin for knee pain.
Based on the ECG results, he was treated with 0.083%/6 mL nebulized albuterol, 4.65 Mq/250 mL saline solution intravenous (IV) calcium gluconate, 10 units IV insulin with concomitant 50%/25 mL IV dextrose and 8.4 g of oral patiromer suspension. IV furosemide was held due to concern for renal function. The decision to proceed with hemodialysis was made. Repeat laboratory tests were performed, and an ECG obtained after treatment initiation but prior to hemodialysis demonstrated improvement of rate and T wave shortening (Figure 1c). The serum potassium level dropped from 9.8 mEq/L to 7.9 mEq/L (reference range, 3.5-5.0 mEq/L) (Table 1).
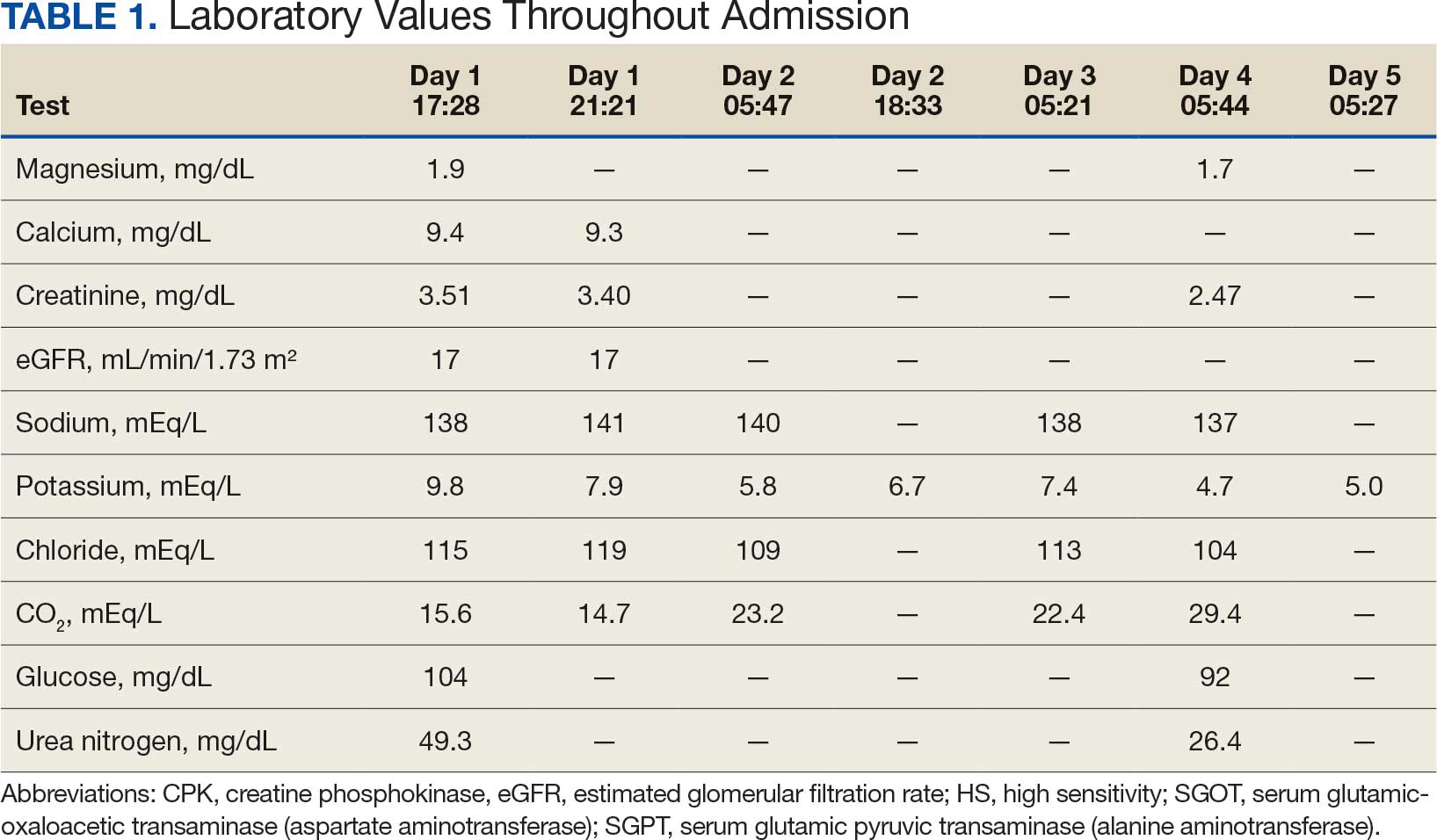
In addition to hemodialysis, sodium zirconium 10 g orally 3 times daily was added. Laboratory test results and an ECG was performed after dialysis continued to demonstrate improvement (Figure 1d). The patient’s potassium level decreased to 5.8 mEq/L, with the ECG demonstrating stability of heart rate and further improvement of the PR interval, QRS complex, and T waves.
Despite the established treatment regimen, potassium levels again rose to 6.7 mEq/L, but there were no significant changes in the ECG, and thus no medication changes were made (Figure 1e). Subsequent monitoring demonstrated a further increase in potassium to 7.4 mEq/L, with an ECG demonstrating a return to the baseline of 1 year prior. The patient underwent hemodialysis again and was given oral furosemide 60 mg every 12 hours. The potassium concentration after dialysis decreased to 4.7 mEq/L and remained stable, not going above 5.0 mEq/L on subsequent monitoring. The patient had resolution of all symptoms and was discharged.
Discussion
We have described in detail the presentation of each pathology and mechanisms of each treatment, starting with the patient’s initial condition that brought him to the emergency room—muscle weakness. Skeletal muscle weakness is a common manifestation of hyperkalemia, occurring in 20% to 40% of cases, and is more prevalent in severe elevations of potassium. Rarely, the weakness can progress to flaccid paralysis of the patient’s extremities and, in extreme cases, the diaphragm.
Muscle weakness progression occurs in a manner that resembles Guillain-Barré syndrome, starting in the lower extremities and ascending toward the upper extremities.10 This is known as secondary hyperkalemic periodic paralysis. Hyperkalemia lowers the transmembrane gradient in neurons, leading to neuronal depolarization independent of the degree of hyperkalemia. If the degree of hyperkalemia is large enough, this depolarization inactivates voltage-gated sodium channels, making neurons refractory to excitation. Electromyographical studies have shown reduction in the compounded muscle action potential.11 The transient nature of this paralysis is reflected by rapid correction of weakness and paralysis when the electrolyte disorder is corrected.
The patient in this case also presented with bradycardia. The ECG manifestations of hyperkalemia can include atrial asystole, intraventricular conduction disturbances, peaked T waves, and widened QRS complexes. However, some patients with renal insufficiency may not exhibit ECG changes despite significantly elevated serum potassium levels.12
The severity of hyperkalemia is crucial in determining the associated ECG changes, with levels > 6.0 mEq/L presenting with abnormalities.13 ECG findings alone may not always accurately reflect the severity of hyperkalemia, as up to 60% of patients with potassium levels > 6.0 mEq/L may not show ECG changes.14 Additionally, extreme hyperkalemia can lead to inconsistent ECG findings, making it challenging to rely solely on ECG for diagnosis and monitoring.8 The level of potassium that causes these effects varies widely through patient populations.
The main mechanism by which hyperkalemia affects the heart’s conduction system is through voltage differences across the conduction fibers and eventual steady-state inactivation of sodium channels. This combination of mechanisms shortens the action potential duration, allowing more cardiomyocytes to undergo synchronized depolarization. This amalgamation of cardiomyocytes repolarizing can be reflected on ECGs as peaked T waves. As the action potential decreases, there is a period during which cardiomyocytes are prone to tachyarrhythmias and ventricular fibrillation.
A reduced action potential may lead to increased rates of depolarization and thus conduction, which in some scenarios may increase heart rate. As the levels of potassium rise, intracellular accumulation impedes the entry of sodium by decreasing the cation gradient across the cell membrane. This effectively slows the sinus nodes and prolongs the QRS by slowing the overall propagation of action potentials. By this mechanism, conduction delays, blocks, or asystole are manifested. The patient in this case showed conduction delays, peaked T waves, and disappearance of P waves when he first arrived.
Hyperkalemia Treatment
Hyperkalemia develops most commonly due to acute or chronic kidney diseases, as was the case with this patient. The patient’s hyperkalemia was also augmented by the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which can directly affect renal function. A properly functioning kidney is responsible for excretion of up to 90% of ingested potassium, while the remainder is excreted through the gastrointestinal (GI) tract. Definitive treatment of hyperkalemia is mitigated primarily through these 2 organ systems. The treatment also includes transitory mechanisms of potassium reduction. The goal of each method is to preserve the action potential of cardiomyocytes and myocytes. This patient presented with acute symptomatic hyperkalemia and received various medications to acutely, transitorily, and definitively treat it.
Initial therapy included calcium gluconate, which functions to stabilize the myocardial cell membrane. Hyperkalemia decreases the resting membrane action potential of excitable cells and predisposes them to early depolarization and thus dysrhythmias. Calcium decreases the threshold potential across cells and offsets the overall gradient back to near normal levels.15 Calcium can be delivered through calcium gluconate or calcium chloride. Calcium chloride is not preferred because extravasation can cause pain, blistering and tissue ischemia. Central venous access is required, potentially delaying prompt treatment. Calcium acts rapidly after administration—within 1 to 3 minutes—but only lasts 30 to 60 minutes.16 Administration of calcium gluconate can be repeated as often as necessary, but patients must be monitored for adverse effects of calcium such as nausea, abdominal pain, polydipsia, polyuria, muscle weakness, and paresthesia. Care must be taken when patients are taking digoxin, because calcium may potentiate toxicity.17 Although calcium provides immediate benefits it does little to correct the underlying cause; other medications are required to remove potassium from the body.
Two medication classes have been proven to shift potassium intracellularly. The first are β-2 agonists, such as albuterol/levalbuterol, and the second is insulin. Both work through sodium-potassium-ATPase in a direct manner. β-2 agonists stimulate sodium-potassium-ATPase to move more potassium intracellularly, but these effects have been seen only with high doses of albuterol, typically 4× the standard dose of 0.5 mg in nebulized solutions to achieve decreases in potassium of 0.3 to 0.6 mEq/L, although some trials have reported decreases of 0.62 to 0.98 mEq/L.15,18 These potassium-lowering effects of β-2 agonist are modest, but can be seen 20 to 30 minutes after administration and persist up to 1 to 2 hours. β-2 agonists are also readily affected by β blockers, which may reduce or negate the desired effect in hyperkalemia. For these reasons, a β-2 agonist should not be given as monotherapy and should be provided as an adjuvant to more independent therapies such as insulin. Insulin binds to receptors on muscle cells and increases the quantity of sodium-potassium-ATPase and glucose transporters. With this increase in influx pumps, surrounding tissues with higher resting membrane potentials can absorb the potassium load, thereby protecting cardiomyocytes.
Potassium Removal
Three methods are currently available to remove potassium from the body: GI excretion, renal excretion, and direct removal from the bloodstream. Under normal physiologic conditions, the kidneys account for about 90% of the body’s ability to remove potassium. Loop diuretics facilitate the removal of potassium by increasing urine production and have an additional potassium-wasting effect. Although the onset of action of loop diuretics is typically 30 to 60 minutes after oral administration, their effect can last for several hours. In this patient, furosemide was introduced later in the treatment plan to manage recurring hyperkalemia by enhancing renal potassium excretion.
Potassium binders such as patiromer act in the GI tract, effectively reducing serum potassium levels although with a slower onset of action than furosemide, generally taking hours to days to exert its effect. Both medications illustrate a tailored approach to managing potassium levels, adapted to the evolving needs and renal function of the patient. The last method is using hemodialysis—by far the most rapid method to remove potassium, but also the most invasive. The different methods of treating hyperkalemia are summarized in Table 2. This patient required multiple days of hemodialysis to completely correct the electrolyte disorder. Upon discharge, the patient continued oral furosemide 40 mg daily and eventually discontinued hemodialysis due to stable renal function.
Often, after correcting an inciting event, potassium stores in the body eventually stabilize and do not require additional follow-up. Patients prone to hyperkalemia should be thoroughly educated on medications to avoid (NSAIDs, ACEIs/ARBs, trimethoprim), an adequate low potassium diet, and symptoms that may warrant medical attention.19
Conclusions
This case illustrates the importance of recognizing the spectrum of manifestations of hyperkalemia, which ranged from muscle weakness to cardiac dysrhythmias. Management strategies for the patient included stabilization of cardiac membranes, potassium shifting, and potassium removal, each tailored to the patient’s individual clinical findings.
The case further illustrates the critical role of continuous monitoring and dynamic adjustment of therapeutic strategies in response to evolving clinical and laboratory findings. The initial and subsequent ECGs, alongside laboratory tests, were instrumental in guiding the adjustments needed in the treatment regimen, ensuring both the efficacy and safety of the interventions. This proactive approach can mitigate the risk of recurrent hyperkalemia and its complications.
- Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381-401. doi:10.1146/annurev.physiol.010908.163241 2.
- Simon LV, Hashmi MF, Farrell MW. Hyperkalemia. In: StatPearls. StatPearls Publishing; September 4, 2023. Accessed October 22, 2025.
- Mu F, Betts KA, Woolley JM, et al. Prevalence and economic burden of hyperkalemia in the United States Medicare population. Curr Med Res Opin. 2020;36:1333-1341. doi:10.1080/03007995.2020.1775072
- Loutradis C, Tolika P, Skodra A, et al. Prevalence of hyperkalemia in diabetic and non-diabetic patients with chronic kidney disease: a nested case-control study. Am J Nephrol. 2015;42:351-360. doi:10.1159/000442393
- Grodzinsky A, Goyal A, Gosch K, et al. Prevalence and prognosis of hyperkalemia in patients with acute myocardial infarction. Am J Med. 2016;129:858-865. doi:10.1016/j.amjmed.2016.03.008
- Hunter RW, Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant. 2019;34(suppl 3):iii2-iii11. doi:10.1093/ndt/gfz206
- Luo J, Brunelli SM, Jensen DE, Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90-100. doi:10.2215/CJN.01730215
- Montford JR, Linas S. How dangerous is hyperkalemia? J Am Soc Nephrol. 2017;28:3155-3165. doi:10.1681/ASN.2016121344
- Mattu A, Brady WJ, Robinson DA. Electrocardiographic manifestations of hyperkalemia. Am J Emerg Med. 2000;18:721-729. doi:10.1053/ajem.2000.7344
- Kimmons LA, Usery JB. Acute ascending muscle weakness secondary to medication-induced hyperkalemia. Case Rep Med. 2014;2014:789529. doi:10.1155/2014/789529
- Naik KR, Saroja AO, Khanpet MS. Reversible electrophysiological abnormalities in acute secondary hyperkalemic paralysis. Ann Indian Acad Neurol. 2012;15:339-343. doi:10.4103/0972-2327.104354
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330. doi:10.2215/CJN.04611007
- Larivée NL, Michaud JB, More KM, Wilson JA, Tennankore KK. Hyperkalemia: prevalence, predictors and emerging treatments. Cardiol Ther. 2023;12:35-63. doi:10.1007/s40119-022-00289-z
- Shingarev R, Allon M. A physiologic-based approach to the treatment of acute hyperkalemia. Am J Kidney Dis. 2010;56:578-584. doi:10.1053/j.ajkd.2010.03.014
- Parham WA, Mehdirad AA, Biermann KM, Fredman CS. Hyperkalemia revisited. Tex Heart Inst J. 2006;33:40-47.
- Ng KE, Lee CS. Updated treatment options in the management of hyperkalemia. U.S. Pharmacist. February 16, 2017. Accessed October 1, 2025. www.uspharmacist.com/article/updated-treatment-options-in-the-management-of-hyperkalemia
- Quick G, Bastani B. Prolonged asystolic hyperkalemic cardiac arrest with no neurologic sequelae. Ann Emerg Med. 1994;24:305-311. doi:10.1016/s0196-0644(94)70144-x 18.
- Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426-429. doi:10.7326/0003-4819-110-6-42619.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024;105(4 suppl):S117-S314. doi:10.1016/j.kint.2023.10.018
Hyperkalemia involves elevated serum potassium levels (> 5.0 mEq/L) and represents an important electrolyte disturbance due to its potentially severe consequences, including cardiac effects that can lead to dysrhythmia and even asystole and death.1,2 In a US Medicare population, the prevalence of hyperkalemia has been estimated at 2.7% and is associated with substantial health care costs.3 The prevalence is even more marked in patients with preexisting conditions such as chronic kidney disease (CKD) and heart failure.4,5
Hyperkalemia can result from multiple factors, including impaired renal function, adrenal disease, adverse drug reactions of angiotensin-converting enzyme inhibitors (ACEIs) and other medications, and heritable mutations.6 Hyperkalemia poses a considerable clinical risk, associated with adverse outcomes such as myocardial infarction and increased mortality in patients with CKD.5,7,8 Electrocardiographic (ECG) changes associated with hyperkalemia play a vital role in guiding clinical decisions and treatment strategies.9 Understanding the pathophysiology, risk factors, and consequences of hyperkalemia, as well as the significance of ECG changes in its management, is essential for health care practitioners.
Case Presentation
An 81-year-old Hispanic man with a history of hypertension, hypothyroidism, gout, and CKD stage 3B presented to the emergency department with progressive weakness resulting in falls and culminating in an inability to ambulate independently. Additional symptoms included nausea, diarrhea, and myalgia. His vital signs were notable for a pulse of 41 beats/min. The physical examination was remarkable for significant weakness of the bilateral upper extremities, inability to bear his own weight, and bilateral lower extremity edema. His initial ECG upon arrival showed bradycardia with wide QRS, absent P waves, and peaked T waves (Figure 1a). These findings differed from his baseline ECG taken 1 year earlier, which showed sinus rhythm with premature atrial complexes and an old right bundle branch block (Figure 1b).
Medication review revealed that the patient was currently prescribed 100 mg allopurinol daily, 2.5 mg amlodipine daily, 10 mg atorvastatin at bedtime, 4 mg doxazosin daily, 112 mcg levothyroxine daily, 100 mg losartan daily, 25 mg metoprolol daily, and 0.4 mg tamsulosin daily. The patient had also been taking over-the-counter indomethacin for knee pain.
Based on the ECG results, he was treated with 0.083%/6 mL nebulized albuterol, 4.65 Mq/250 mL saline solution intravenous (IV) calcium gluconate, 10 units IV insulin with concomitant 50%/25 mL IV dextrose and 8.4 g of oral patiromer suspension. IV furosemide was held due to concern for renal function. The decision to proceed with hemodialysis was made. Repeat laboratory tests were performed, and an ECG obtained after treatment initiation but prior to hemodialysis demonstrated improvement of rate and T wave shortening (Figure 1c). The serum potassium level dropped from 9.8 mEq/L to 7.9 mEq/L (reference range, 3.5-5.0 mEq/L) (Table 1).
In addition to hemodialysis, sodium zirconium 10 g orally 3 times daily was added. Laboratory test results and an ECG was performed after dialysis continued to demonstrate improvement (Figure 1d). The patient’s potassium level decreased to 5.8 mEq/L, with the ECG demonstrating stability of heart rate and further improvement of the PR interval, QRS complex, and T waves.
Despite the established treatment regimen, potassium levels again rose to 6.7 mEq/L, but there were no significant changes in the ECG, and thus no medication changes were made (Figure 1e). Subsequent monitoring demonstrated a further increase in potassium to 7.4 mEq/L, with an ECG demonstrating a return to the baseline of 1 year prior. The patient underwent hemodialysis again and was given oral furosemide 60 mg every 12 hours. The potassium concentration after dialysis decreased to 4.7 mEq/L and remained stable, not going above 5.0 mEq/L on subsequent monitoring. The patient had resolution of all symptoms and was discharged.
Discussion
We have described in detail the presentation of each pathology and mechanisms of each treatment, starting with the patient’s initial condition that brought him to the emergency room—muscle weakness. Skeletal muscle weakness is a common manifestation of hyperkalemia, occurring in 20% to 40% of cases, and is more prevalent in severe elevations of potassium. Rarely, the weakness can progress to flaccid paralysis of the patient’s extremities and, in extreme cases, the diaphragm.
Muscle weakness progression occurs in a manner that resembles Guillain-Barré syndrome, starting in the lower extremities and ascending toward the upper extremities.10 This is known as secondary hyperkalemic periodic paralysis. Hyperkalemia lowers the transmembrane gradient in neurons, leading to neuronal depolarization independent of the degree of hyperkalemia. If the degree of hyperkalemia is large enough, this depolarization inactivates voltage-gated sodium channels, making neurons refractory to excitation. Electromyographical studies have shown reduction in the compounded muscle action potential.11 The transient nature of this paralysis is reflected by rapid correction of weakness and paralysis when the electrolyte disorder is corrected.
The patient in this case also presented with bradycardia. The ECG manifestations of hyperkalemia can include atrial asystole, intraventricular conduction disturbances, peaked T waves, and widened QRS complexes. However, some patients with renal insufficiency may not exhibit ECG changes despite significantly elevated serum potassium levels.12
The severity of hyperkalemia is crucial in determining the associated ECG changes, with levels > 6.0 mEq/L presenting with abnormalities.13 ECG findings alone may not always accurately reflect the severity of hyperkalemia, as up to 60% of patients with potassium levels > 6.0 mEq/L may not show ECG changes.14 Additionally, extreme hyperkalemia can lead to inconsistent ECG findings, making it challenging to rely solely on ECG for diagnosis and monitoring.8 The level of potassium that causes these effects varies widely through patient populations.
The main mechanism by which hyperkalemia affects the heart’s conduction system is through voltage differences across the conduction fibers and eventual steady-state inactivation of sodium channels. This combination of mechanisms shortens the action potential duration, allowing more cardiomyocytes to undergo synchronized depolarization. This amalgamation of cardiomyocytes repolarizing can be reflected on ECGs as peaked T waves. As the action potential decreases, there is a period during which cardiomyocytes are prone to tachyarrhythmias and ventricular fibrillation.
A reduced action potential may lead to increased rates of depolarization and thus conduction, which in some scenarios may increase heart rate. As the levels of potassium rise, intracellular accumulation impedes the entry of sodium by decreasing the cation gradient across the cell membrane. This effectively slows the sinus nodes and prolongs the QRS by slowing the overall propagation of action potentials. By this mechanism, conduction delays, blocks, or asystole are manifested. The patient in this case showed conduction delays, peaked T waves, and disappearance of P waves when he first arrived.
Hyperkalemia Treatment
Hyperkalemia develops most commonly due to acute or chronic kidney diseases, as was the case with this patient. The patient’s hyperkalemia was also augmented by the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which can directly affect renal function. A properly functioning kidney is responsible for excretion of up to 90% of ingested potassium, while the remainder is excreted through the gastrointestinal (GI) tract. Definitive treatment of hyperkalemia is mitigated primarily through these 2 organ systems. The treatment also includes transitory mechanisms of potassium reduction. The goal of each method is to preserve the action potential of cardiomyocytes and myocytes. This patient presented with acute symptomatic hyperkalemia and received various medications to acutely, transitorily, and definitively treat it.
Initial therapy included calcium gluconate, which functions to stabilize the myocardial cell membrane. Hyperkalemia decreases the resting membrane action potential of excitable cells and predisposes them to early depolarization and thus dysrhythmias. Calcium decreases the threshold potential across cells and offsets the overall gradient back to near normal levels.15 Calcium can be delivered through calcium gluconate or calcium chloride. Calcium chloride is not preferred because extravasation can cause pain, blistering and tissue ischemia. Central venous access is required, potentially delaying prompt treatment. Calcium acts rapidly after administration—within 1 to 3 minutes—but only lasts 30 to 60 minutes.16 Administration of calcium gluconate can be repeated as often as necessary, but patients must be monitored for adverse effects of calcium such as nausea, abdominal pain, polydipsia, polyuria, muscle weakness, and paresthesia. Care must be taken when patients are taking digoxin, because calcium may potentiate toxicity.17 Although calcium provides immediate benefits it does little to correct the underlying cause; other medications are required to remove potassium from the body.
Two medication classes have been proven to shift potassium intracellularly. The first are β-2 agonists, such as albuterol/levalbuterol, and the second is insulin. Both work through sodium-potassium-ATPase in a direct manner. β-2 agonists stimulate sodium-potassium-ATPase to move more potassium intracellularly, but these effects have been seen only with high doses of albuterol, typically 4× the standard dose of 0.5 mg in nebulized solutions to achieve decreases in potassium of 0.3 to 0.6 mEq/L, although some trials have reported decreases of 0.62 to 0.98 mEq/L.15,18 These potassium-lowering effects of β-2 agonist are modest, but can be seen 20 to 30 minutes after administration and persist up to 1 to 2 hours. β-2 agonists are also readily affected by β blockers, which may reduce or negate the desired effect in hyperkalemia. For these reasons, a β-2 agonist should not be given as monotherapy and should be provided as an adjuvant to more independent therapies such as insulin. Insulin binds to receptors on muscle cells and increases the quantity of sodium-potassium-ATPase and glucose transporters. With this increase in influx pumps, surrounding tissues with higher resting membrane potentials can absorb the potassium load, thereby protecting cardiomyocytes.
Potassium Removal
Three methods are currently available to remove potassium from the body: GI excretion, renal excretion, and direct removal from the bloodstream. Under normal physiologic conditions, the kidneys account for about 90% of the body’s ability to remove potassium. Loop diuretics facilitate the removal of potassium by increasing urine production and have an additional potassium-wasting effect. Although the onset of action of loop diuretics is typically 30 to 60 minutes after oral administration, their effect can last for several hours. In this patient, furosemide was introduced later in the treatment plan to manage recurring hyperkalemia by enhancing renal potassium excretion.
Potassium binders such as patiromer act in the GI tract, effectively reducing serum potassium levels although with a slower onset of action than furosemide, generally taking hours to days to exert its effect. Both medications illustrate a tailored approach to managing potassium levels, adapted to the evolving needs and renal function of the patient. The last method is using hemodialysis—by far the most rapid method to remove potassium, but also the most invasive. The different methods of treating hyperkalemia are summarized in Table 2. This patient required multiple days of hemodialysis to completely correct the electrolyte disorder. Upon discharge, the patient continued oral furosemide 40 mg daily and eventually discontinued hemodialysis due to stable renal function.
Often, after correcting an inciting event, potassium stores in the body eventually stabilize and do not require additional follow-up. Patients prone to hyperkalemia should be thoroughly educated on medications to avoid (NSAIDs, ACEIs/ARBs, trimethoprim), an adequate low potassium diet, and symptoms that may warrant medical attention.19
Conclusions
This case illustrates the importance of recognizing the spectrum of manifestations of hyperkalemia, which ranged from muscle weakness to cardiac dysrhythmias. Management strategies for the patient included stabilization of cardiac membranes, potassium shifting, and potassium removal, each tailored to the patient’s individual clinical findings.
The case further illustrates the critical role of continuous monitoring and dynamic adjustment of therapeutic strategies in response to evolving clinical and laboratory findings. The initial and subsequent ECGs, alongside laboratory tests, were instrumental in guiding the adjustments needed in the treatment regimen, ensuring both the efficacy and safety of the interventions. This proactive approach can mitigate the risk of recurrent hyperkalemia and its complications.
Hyperkalemia involves elevated serum potassium levels (> 5.0 mEq/L) and represents an important electrolyte disturbance due to its potentially severe consequences, including cardiac effects that can lead to dysrhythmia and even asystole and death.1,2 In a US Medicare population, the prevalence of hyperkalemia has been estimated at 2.7% and is associated with substantial health care costs.3 The prevalence is even more marked in patients with preexisting conditions such as chronic kidney disease (CKD) and heart failure.4,5
Hyperkalemia can result from multiple factors, including impaired renal function, adrenal disease, adverse drug reactions of angiotensin-converting enzyme inhibitors (ACEIs) and other medications, and heritable mutations.6 Hyperkalemia poses a considerable clinical risk, associated with adverse outcomes such as myocardial infarction and increased mortality in patients with CKD.5,7,8 Electrocardiographic (ECG) changes associated with hyperkalemia play a vital role in guiding clinical decisions and treatment strategies.9 Understanding the pathophysiology, risk factors, and consequences of hyperkalemia, as well as the significance of ECG changes in its management, is essential for health care practitioners.
Case Presentation
An 81-year-old Hispanic man with a history of hypertension, hypothyroidism, gout, and CKD stage 3B presented to the emergency department with progressive weakness resulting in falls and culminating in an inability to ambulate independently. Additional symptoms included nausea, diarrhea, and myalgia. His vital signs were notable for a pulse of 41 beats/min. The physical examination was remarkable for significant weakness of the bilateral upper extremities, inability to bear his own weight, and bilateral lower extremity edema. His initial ECG upon arrival showed bradycardia with wide QRS, absent P waves, and peaked T waves (Figure 1a). These findings differed from his baseline ECG taken 1 year earlier, which showed sinus rhythm with premature atrial complexes and an old right bundle branch block (Figure 1b).
Medication review revealed that the patient was currently prescribed 100 mg allopurinol daily, 2.5 mg amlodipine daily, 10 mg atorvastatin at bedtime, 4 mg doxazosin daily, 112 mcg levothyroxine daily, 100 mg losartan daily, 25 mg metoprolol daily, and 0.4 mg tamsulosin daily. The patient had also been taking over-the-counter indomethacin for knee pain.
Based on the ECG results, he was treated with 0.083%/6 mL nebulized albuterol, 4.65 Mq/250 mL saline solution intravenous (IV) calcium gluconate, 10 units IV insulin with concomitant 50%/25 mL IV dextrose and 8.4 g of oral patiromer suspension. IV furosemide was held due to concern for renal function. The decision to proceed with hemodialysis was made. Repeat laboratory tests were performed, and an ECG obtained after treatment initiation but prior to hemodialysis demonstrated improvement of rate and T wave shortening (Figure 1c). The serum potassium level dropped from 9.8 mEq/L to 7.9 mEq/L (reference range, 3.5-5.0 mEq/L) (Table 1).
In addition to hemodialysis, sodium zirconium 10 g orally 3 times daily was added. Laboratory test results and an ECG was performed after dialysis continued to demonstrate improvement (Figure 1d). The patient’s potassium level decreased to 5.8 mEq/L, with the ECG demonstrating stability of heart rate and further improvement of the PR interval, QRS complex, and T waves.
Despite the established treatment regimen, potassium levels again rose to 6.7 mEq/L, but there were no significant changes in the ECG, and thus no medication changes were made (Figure 1e). Subsequent monitoring demonstrated a further increase in potassium to 7.4 mEq/L, with an ECG demonstrating a return to the baseline of 1 year prior. The patient underwent hemodialysis again and was given oral furosemide 60 mg every 12 hours. The potassium concentration after dialysis decreased to 4.7 mEq/L and remained stable, not going above 5.0 mEq/L on subsequent monitoring. The patient had resolution of all symptoms and was discharged.
Discussion
We have described in detail the presentation of each pathology and mechanisms of each treatment, starting with the patient’s initial condition that brought him to the emergency room—muscle weakness. Skeletal muscle weakness is a common manifestation of hyperkalemia, occurring in 20% to 40% of cases, and is more prevalent in severe elevations of potassium. Rarely, the weakness can progress to flaccid paralysis of the patient’s extremities and, in extreme cases, the diaphragm.
Muscle weakness progression occurs in a manner that resembles Guillain-Barré syndrome, starting in the lower extremities and ascending toward the upper extremities.10 This is known as secondary hyperkalemic periodic paralysis. Hyperkalemia lowers the transmembrane gradient in neurons, leading to neuronal depolarization independent of the degree of hyperkalemia. If the degree of hyperkalemia is large enough, this depolarization inactivates voltage-gated sodium channels, making neurons refractory to excitation. Electromyographical studies have shown reduction in the compounded muscle action potential.11 The transient nature of this paralysis is reflected by rapid correction of weakness and paralysis when the electrolyte disorder is corrected.
The patient in this case also presented with bradycardia. The ECG manifestations of hyperkalemia can include atrial asystole, intraventricular conduction disturbances, peaked T waves, and widened QRS complexes. However, some patients with renal insufficiency may not exhibit ECG changes despite significantly elevated serum potassium levels.12
The severity of hyperkalemia is crucial in determining the associated ECG changes, with levels > 6.0 mEq/L presenting with abnormalities.13 ECG findings alone may not always accurately reflect the severity of hyperkalemia, as up to 60% of patients with potassium levels > 6.0 mEq/L may not show ECG changes.14 Additionally, extreme hyperkalemia can lead to inconsistent ECG findings, making it challenging to rely solely on ECG for diagnosis and monitoring.8 The level of potassium that causes these effects varies widely through patient populations.
The main mechanism by which hyperkalemia affects the heart’s conduction system is through voltage differences across the conduction fibers and eventual steady-state inactivation of sodium channels. This combination of mechanisms shortens the action potential duration, allowing more cardiomyocytes to undergo synchronized depolarization. This amalgamation of cardiomyocytes repolarizing can be reflected on ECGs as peaked T waves. As the action potential decreases, there is a period during which cardiomyocytes are prone to tachyarrhythmias and ventricular fibrillation.
A reduced action potential may lead to increased rates of depolarization and thus conduction, which in some scenarios may increase heart rate. As the levels of potassium rise, intracellular accumulation impedes the entry of sodium by decreasing the cation gradient across the cell membrane. This effectively slows the sinus nodes and prolongs the QRS by slowing the overall propagation of action potentials. By this mechanism, conduction delays, blocks, or asystole are manifested. The patient in this case showed conduction delays, peaked T waves, and disappearance of P waves when he first arrived.
Hyperkalemia Treatment
Hyperkalemia develops most commonly due to acute or chronic kidney diseases, as was the case with this patient. The patient’s hyperkalemia was also augmented by the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which can directly affect renal function. A properly functioning kidney is responsible for excretion of up to 90% of ingested potassium, while the remainder is excreted through the gastrointestinal (GI) tract. Definitive treatment of hyperkalemia is mitigated primarily through these 2 organ systems. The treatment also includes transitory mechanisms of potassium reduction. The goal of each method is to preserve the action potential of cardiomyocytes and myocytes. This patient presented with acute symptomatic hyperkalemia and received various medications to acutely, transitorily, and definitively treat it.
Initial therapy included calcium gluconate, which functions to stabilize the myocardial cell membrane. Hyperkalemia decreases the resting membrane action potential of excitable cells and predisposes them to early depolarization and thus dysrhythmias. Calcium decreases the threshold potential across cells and offsets the overall gradient back to near normal levels.15 Calcium can be delivered through calcium gluconate or calcium chloride. Calcium chloride is not preferred because extravasation can cause pain, blistering and tissue ischemia. Central venous access is required, potentially delaying prompt treatment. Calcium acts rapidly after administration—within 1 to 3 minutes—but only lasts 30 to 60 minutes.16 Administration of calcium gluconate can be repeated as often as necessary, but patients must be monitored for adverse effects of calcium such as nausea, abdominal pain, polydipsia, polyuria, muscle weakness, and paresthesia. Care must be taken when patients are taking digoxin, because calcium may potentiate toxicity.17 Although calcium provides immediate benefits it does little to correct the underlying cause; other medications are required to remove potassium from the body.
Two medication classes have been proven to shift potassium intracellularly. The first are β-2 agonists, such as albuterol/levalbuterol, and the second is insulin. Both work through sodium-potassium-ATPase in a direct manner. β-2 agonists stimulate sodium-potassium-ATPase to move more potassium intracellularly, but these effects have been seen only with high doses of albuterol, typically 4× the standard dose of 0.5 mg in nebulized solutions to achieve decreases in potassium of 0.3 to 0.6 mEq/L, although some trials have reported decreases of 0.62 to 0.98 mEq/L.15,18 These potassium-lowering effects of β-2 agonist are modest, but can be seen 20 to 30 minutes after administration and persist up to 1 to 2 hours. β-2 agonists are also readily affected by β blockers, which may reduce or negate the desired effect in hyperkalemia. For these reasons, a β-2 agonist should not be given as monotherapy and should be provided as an adjuvant to more independent therapies such as insulin. Insulin binds to receptors on muscle cells and increases the quantity of sodium-potassium-ATPase and glucose transporters. With this increase in influx pumps, surrounding tissues with higher resting membrane potentials can absorb the potassium load, thereby protecting cardiomyocytes.
Potassium Removal
Three methods are currently available to remove potassium from the body: GI excretion, renal excretion, and direct removal from the bloodstream. Under normal physiologic conditions, the kidneys account for about 90% of the body’s ability to remove potassium. Loop diuretics facilitate the removal of potassium by increasing urine production and have an additional potassium-wasting effect. Although the onset of action of loop diuretics is typically 30 to 60 minutes after oral administration, their effect can last for several hours. In this patient, furosemide was introduced later in the treatment plan to manage recurring hyperkalemia by enhancing renal potassium excretion.
Potassium binders such as patiromer act in the GI tract, effectively reducing serum potassium levels although with a slower onset of action than furosemide, generally taking hours to days to exert its effect. Both medications illustrate a tailored approach to managing potassium levels, adapted to the evolving needs and renal function of the patient. The last method is using hemodialysis—by far the most rapid method to remove potassium, but also the most invasive. The different methods of treating hyperkalemia are summarized in Table 2. This patient required multiple days of hemodialysis to completely correct the electrolyte disorder. Upon discharge, the patient continued oral furosemide 40 mg daily and eventually discontinued hemodialysis due to stable renal function.
Often, after correcting an inciting event, potassium stores in the body eventually stabilize and do not require additional follow-up. Patients prone to hyperkalemia should be thoroughly educated on medications to avoid (NSAIDs, ACEIs/ARBs, trimethoprim), an adequate low potassium diet, and symptoms that may warrant medical attention.19
Conclusions
This case illustrates the importance of recognizing the spectrum of manifestations of hyperkalemia, which ranged from muscle weakness to cardiac dysrhythmias. Management strategies for the patient included stabilization of cardiac membranes, potassium shifting, and potassium removal, each tailored to the patient’s individual clinical findings.
The case further illustrates the critical role of continuous monitoring and dynamic adjustment of therapeutic strategies in response to evolving clinical and laboratory findings. The initial and subsequent ECGs, alongside laboratory tests, were instrumental in guiding the adjustments needed in the treatment regimen, ensuring both the efficacy and safety of the interventions. This proactive approach can mitigate the risk of recurrent hyperkalemia and its complications.
- Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381-401. doi:10.1146/annurev.physiol.010908.163241 2.
- Simon LV, Hashmi MF, Farrell MW. Hyperkalemia. In: StatPearls. StatPearls Publishing; September 4, 2023. Accessed October 22, 2025.
- Mu F, Betts KA, Woolley JM, et al. Prevalence and economic burden of hyperkalemia in the United States Medicare population. Curr Med Res Opin. 2020;36:1333-1341. doi:10.1080/03007995.2020.1775072
- Loutradis C, Tolika P, Skodra A, et al. Prevalence of hyperkalemia in diabetic and non-diabetic patients with chronic kidney disease: a nested case-control study. Am J Nephrol. 2015;42:351-360. doi:10.1159/000442393
- Grodzinsky A, Goyal A, Gosch K, et al. Prevalence and prognosis of hyperkalemia in patients with acute myocardial infarction. Am J Med. 2016;129:858-865. doi:10.1016/j.amjmed.2016.03.008
- Hunter RW, Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant. 2019;34(suppl 3):iii2-iii11. doi:10.1093/ndt/gfz206
- Luo J, Brunelli SM, Jensen DE, Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90-100. doi:10.2215/CJN.01730215
- Montford JR, Linas S. How dangerous is hyperkalemia? J Am Soc Nephrol. 2017;28:3155-3165. doi:10.1681/ASN.2016121344
- Mattu A, Brady WJ, Robinson DA. Electrocardiographic manifestations of hyperkalemia. Am J Emerg Med. 2000;18:721-729. doi:10.1053/ajem.2000.7344
- Kimmons LA, Usery JB. Acute ascending muscle weakness secondary to medication-induced hyperkalemia. Case Rep Med. 2014;2014:789529. doi:10.1155/2014/789529
- Naik KR, Saroja AO, Khanpet MS. Reversible electrophysiological abnormalities in acute secondary hyperkalemic paralysis. Ann Indian Acad Neurol. 2012;15:339-343. doi:10.4103/0972-2327.104354
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330. doi:10.2215/CJN.04611007
- Larivée NL, Michaud JB, More KM, Wilson JA, Tennankore KK. Hyperkalemia: prevalence, predictors and emerging treatments. Cardiol Ther. 2023;12:35-63. doi:10.1007/s40119-022-00289-z
- Shingarev R, Allon M. A physiologic-based approach to the treatment of acute hyperkalemia. Am J Kidney Dis. 2010;56:578-584. doi:10.1053/j.ajkd.2010.03.014
- Parham WA, Mehdirad AA, Biermann KM, Fredman CS. Hyperkalemia revisited. Tex Heart Inst J. 2006;33:40-47.
- Ng KE, Lee CS. Updated treatment options in the management of hyperkalemia. U.S. Pharmacist. February 16, 2017. Accessed October 1, 2025. www.uspharmacist.com/article/updated-treatment-options-in-the-management-of-hyperkalemia
- Quick G, Bastani B. Prolonged asystolic hyperkalemic cardiac arrest with no neurologic sequelae. Ann Emerg Med. 1994;24:305-311. doi:10.1016/s0196-0644(94)70144-x 18.
- Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426-429. doi:10.7326/0003-4819-110-6-42619.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024;105(4 suppl):S117-S314. doi:10.1016/j.kint.2023.10.018
- Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381-401. doi:10.1146/annurev.physiol.010908.163241 2.
- Simon LV, Hashmi MF, Farrell MW. Hyperkalemia. In: StatPearls. StatPearls Publishing; September 4, 2023. Accessed October 22, 2025.
- Mu F, Betts KA, Woolley JM, et al. Prevalence and economic burden of hyperkalemia in the United States Medicare population. Curr Med Res Opin. 2020;36:1333-1341. doi:10.1080/03007995.2020.1775072
- Loutradis C, Tolika P, Skodra A, et al. Prevalence of hyperkalemia in diabetic and non-diabetic patients with chronic kidney disease: a nested case-control study. Am J Nephrol. 2015;42:351-360. doi:10.1159/000442393
- Grodzinsky A, Goyal A, Gosch K, et al. Prevalence and prognosis of hyperkalemia in patients with acute myocardial infarction. Am J Med. 2016;129:858-865. doi:10.1016/j.amjmed.2016.03.008
- Hunter RW, Bailey MA. Hyperkalemia: pathophysiology, risk factors and consequences. Nephrol Dial Transplant. 2019;34(suppl 3):iii2-iii11. doi:10.1093/ndt/gfz206
- Luo J, Brunelli SM, Jensen DE, Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90-100. doi:10.2215/CJN.01730215
- Montford JR, Linas S. How dangerous is hyperkalemia? J Am Soc Nephrol. 2017;28:3155-3165. doi:10.1681/ASN.2016121344
- Mattu A, Brady WJ, Robinson DA. Electrocardiographic manifestations of hyperkalemia. Am J Emerg Med. 2000;18:721-729. doi:10.1053/ajem.2000.7344
- Kimmons LA, Usery JB. Acute ascending muscle weakness secondary to medication-induced hyperkalemia. Case Rep Med. 2014;2014:789529. doi:10.1155/2014/789529
- Naik KR, Saroja AO, Khanpet MS. Reversible electrophysiological abnormalities in acute secondary hyperkalemic paralysis. Ann Indian Acad Neurol. 2012;15:339-343. doi:10.4103/0972-2327.104354
- Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330. doi:10.2215/CJN.04611007
- Larivée NL, Michaud JB, More KM, Wilson JA, Tennankore KK. Hyperkalemia: prevalence, predictors and emerging treatments. Cardiol Ther. 2023;12:35-63. doi:10.1007/s40119-022-00289-z
- Shingarev R, Allon M. A physiologic-based approach to the treatment of acute hyperkalemia. Am J Kidney Dis. 2010;56:578-584. doi:10.1053/j.ajkd.2010.03.014
- Parham WA, Mehdirad AA, Biermann KM, Fredman CS. Hyperkalemia revisited. Tex Heart Inst J. 2006;33:40-47.
- Ng KE, Lee CS. Updated treatment options in the management of hyperkalemia. U.S. Pharmacist. February 16, 2017. Accessed October 1, 2025. www.uspharmacist.com/article/updated-treatment-options-in-the-management-of-hyperkalemia
- Quick G, Bastani B. Prolonged asystolic hyperkalemic cardiac arrest with no neurologic sequelae. Ann Emerg Med. 1994;24:305-311. doi:10.1016/s0196-0644(94)70144-x 18.
- Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426-429. doi:10.7326/0003-4819-110-6-42619.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024;105(4 suppl):S117-S314. doi:10.1016/j.kint.2023.10.018
Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses
Following the Hyperkalemia Trail: A Case Report of ECG Changes and Treatment Responses
