User login
Society of Hospital Medicine’s Project BOOST Reduces Medicare Penalties and Readmissions
SHM’s Project BOOST is accepting applications for its 2014 cohort, giving hospitalists and hospital-based team members time to complete the application and receive buy-in from hospital executives to participate in the program.
And this year is the best year yet to make the case to hospital leadership for using Project BOOST to reduce hospital readmissions. More than 180 hospitals throughout the U.S. have used Project BOOST to systematically tackle readmissions.
Last year, the first peer-reviewed research on Project BOOST, published in the Journal of Hospital Medicine, showed that the program reduced 30-day readmissions to 12.7% from 14.7% among 11 hospitals participating in the study. In addition, media and government agencies taking a hard look at readmissions rates have also used Project BOOST as an example of programs that can reduce readmissions and avoid Medicare penalties.1
Accepted Project BOOST sites begin the yearlong program with an in-person training conference with other BOOST sites. After the training, participants utilize a comprehensive toolkit to begin implementing their own programs, followed by ongoing mentoring with national experts in reducing readmissions and collaboration with other hospitals tackling similar challenges.
Details, educational resources, and free on-demand webinars are available at www.hospitalmedicine.org/projectboost.
Brendon Shank is SHM’s associate vice president of communications.
Reference
SHM’s Project BOOST is accepting applications for its 2014 cohort, giving hospitalists and hospital-based team members time to complete the application and receive buy-in from hospital executives to participate in the program.
And this year is the best year yet to make the case to hospital leadership for using Project BOOST to reduce hospital readmissions. More than 180 hospitals throughout the U.S. have used Project BOOST to systematically tackle readmissions.
Last year, the first peer-reviewed research on Project BOOST, published in the Journal of Hospital Medicine, showed that the program reduced 30-day readmissions to 12.7% from 14.7% among 11 hospitals participating in the study. In addition, media and government agencies taking a hard look at readmissions rates have also used Project BOOST as an example of programs that can reduce readmissions and avoid Medicare penalties.1
Accepted Project BOOST sites begin the yearlong program with an in-person training conference with other BOOST sites. After the training, participants utilize a comprehensive toolkit to begin implementing their own programs, followed by ongoing mentoring with national experts in reducing readmissions and collaboration with other hospitals tackling similar challenges.
Details, educational resources, and free on-demand webinars are available at www.hospitalmedicine.org/projectboost.
Brendon Shank is SHM’s associate vice president of communications.
Reference
SHM’s Project BOOST is accepting applications for its 2014 cohort, giving hospitalists and hospital-based team members time to complete the application and receive buy-in from hospital executives to participate in the program.
And this year is the best year yet to make the case to hospital leadership for using Project BOOST to reduce hospital readmissions. More than 180 hospitals throughout the U.S. have used Project BOOST to systematically tackle readmissions.
Last year, the first peer-reviewed research on Project BOOST, published in the Journal of Hospital Medicine, showed that the program reduced 30-day readmissions to 12.7% from 14.7% among 11 hospitals participating in the study. In addition, media and government agencies taking a hard look at readmissions rates have also used Project BOOST as an example of programs that can reduce readmissions and avoid Medicare penalties.1
Accepted Project BOOST sites begin the yearlong program with an in-person training conference with other BOOST sites. After the training, participants utilize a comprehensive toolkit to begin implementing their own programs, followed by ongoing mentoring with national experts in reducing readmissions and collaboration with other hospitals tackling similar challenges.
Details, educational resources, and free on-demand webinars are available at www.hospitalmedicine.org/projectboost.
Brendon Shank is SHM’s associate vice president of communications.
Reference
Centers for Medicare & Medicaid Services Modify Physician Quality Reporting System

Only 27% of eligible providers participated in the Physician Quality Reporting System (PQRS) in 2011—roughly 26,500 medical practices and 266,500 medical professionals, according to the Centers for Medicare & Medicaid Services (CMS).
“A lot of physicians have walked away [from PQRS] feeling like there are not sufficient measures for them to be measured against,” says Cheryl Damberg, senior principal researcher at RAND corporation and professor at the Pardee RAND Graduate School in Santa Monica, Calif.
Encouraging more participation from hospitalists has been the goal of the Society of Hospital Medicine (SHM) for the last several years, says Gregory Seymann, MD, SFHM, clinical professor and chief in the division of hospital medicine at University of California San Diego Health Sciences and chair of SHM’s Performance Measurement and Reporting Committee (PMRC).
“The committee has tried to champion it the best we can, making sure the measures that are there and in development meet the needs of the specialty,” Dr. Seymann says.
In just one year, the SHM committee managed to increase hospitalist reportable measures in PQRS from a paltry 11—half of which were only for stroke patients—to 21, which now includes things like diabetes exams, osteoporosis management, documentation of current medications, and community-acquired pneumonia treatment.
For Comparison’s Sake
For the first couple of phases of PQRS reporting, very few measures were relevant to hospitalists, Dr. Seymann says. The committee worked to ensure that more measures were added and billing codes modified to include those used by the specialty. Hospital medicine is relatively new, not officially recognized by the American Board of Medical Specialties (ABMS), and hospitalists serve a unique role. Most hospitalists are in internal medicine, family medicine, or pediatrics, but they aren’t doing what the average primary care doctor does, like referral for breast cancer or colon cancer screening, Dr. Seymann adds. Additionally, they aren’t always the provider performing specific cardiac or neurological care.
Hospitalists’ patients usually are in the hospital because they are sick. They may have chronic disease or more complex medical needs (e.g. osteoporosis-related hip fracture) than the average population seen by a non-hospitalist PCP.
If hospitalists are compared to other PCPs, as is the plan in the Physician Value-Based Payment Modifier, it “looks like our patients are dying a lot more frequently, we’re spending a lot of money, and we’re not doing primary care,” Dr. Seymann explains.
New Brand, New Push
PQRS is not new; it is the rebranding of CMS’ Physician Quality Reporting Initiative (PQRI), launched in 2006. But changes to the program are part of a national push to improve healthcare quality and patient care while reimbursing for performance on outcome- and process-based measures instead of simply for the volume of services provided. Each year, CMS updates PQRS rules.
This year is the last one in which providers will receive a bonus for reporting through PQRS. Beginning next year, practitioners that don’t meet the reporting requirements for 2013 will incur a 1.5% penalty—with additional penalties for physicians in groups of 100 or more from the value-based payment modifier. This year also serves as the performance year for 2016, when a 2% penalty for insufficient reporting will be assessed.
In early December 2013, the Centers for Medicare & Medicaid Services (CMS) published the 2014 Physician Fee Schedule and, with it, the final rules for the PQRS. Although many physicians and specialist groups believed the measures included in PQRS in previous years were too limited, CMS has added the additional reporting methodology of qualified clinical data registries (QCDR), which can include measures outside of the PQRS—a marked shift from previous policies.
The rule change, Damberg says, should take some energy out of the discussion surrounding the program and allow more physicians to participate.
“From CMS’ perspective, they want doctors delivering the recommended care and they want doctors to be able to report it out easily,” Damberg says.
Moving Forward
In 2014, providers can submit measures through the new QCDR option, or submit PQRS-identified measures through a Medicare qualified registry, through electronic health records, through the group practice reporting option (GPRO), and through claims-based reporting (though this last option is expected to be phased out over time).
Registries themselves are not new, but they can cost millions of dollars to establish and as much as a million a year to maintain. They typically contain more clinical depth and specificity than claims data, and numerous studies show the use of registries leads to improved patient outcomes.
“We don’t know how many [existing] registries are going to qualify to become these qualified clinical data registries,” says Tom Granatir, senior vice president for health policy and external relations at ABMS. “It’s going to take some time for these registries to evolve.”
Qualified clinical data registries must be in operation for at least one year to be eligible for certification by Medicare. They must include performance data from other payers beyond Medicare. Not only must QCDRs be capable of capturing and sending data, they must also provide national benchmarks to those who submit and must report back at least four times per year.
Granatir believes the QCDR rule, which allows QCDR’s to report measures beyond those included in the PQRS program, will help increase participation and will lead to more practice-based measures, but he fears it may exclude some important nuances of day-to-day patient care.
“The whole point [of quality measure reporting] is to create more public transparency…but if you have measures that are not relevant to what is actually done in practices, then it’s not a useful dataset,” he says.
Ideally, Damberg says, PQRS and other performance measures should enable physicians to do what they do better.
“I think this is really going to raise the stakes for [hospitalists] if they want to control their destiny,” Damberg says. “I think they have to get really engaged in this game and take a pro-active role in looking at where the quality gaps are and how can they better benefit patients. That’s the ultimate goal.”
Kelly April Tyrrell is a freelance writer in Wilmington, Del.
Only 27% of eligible providers participated in the Physician Quality Reporting System (PQRS) in 2011—roughly 26,500 medical practices and 266,500 medical professionals, according to the Centers for Medicare & Medicaid Services (CMS).
“A lot of physicians have walked away [from PQRS] feeling like there are not sufficient measures for them to be measured against,” says Cheryl Damberg, senior principal researcher at RAND corporation and professor at the Pardee RAND Graduate School in Santa Monica, Calif.
Encouraging more participation from hospitalists has been the goal of the Society of Hospital Medicine (SHM) for the last several years, says Gregory Seymann, MD, SFHM, clinical professor and chief in the division of hospital medicine at University of California San Diego Health Sciences and chair of SHM’s Performance Measurement and Reporting Committee (PMRC).
“The committee has tried to champion it the best we can, making sure the measures that are there and in development meet the needs of the specialty,” Dr. Seymann says.
In just one year, the SHM committee managed to increase hospitalist reportable measures in PQRS from a paltry 11—half of which were only for stroke patients—to 21, which now includes things like diabetes exams, osteoporosis management, documentation of current medications, and community-acquired pneumonia treatment.
For Comparison’s Sake
For the first couple of phases of PQRS reporting, very few measures were relevant to hospitalists, Dr. Seymann says. The committee worked to ensure that more measures were added and billing codes modified to include those used by the specialty. Hospital medicine is relatively new, not officially recognized by the American Board of Medical Specialties (ABMS), and hospitalists serve a unique role. Most hospitalists are in internal medicine, family medicine, or pediatrics, but they aren’t doing what the average primary care doctor does, like referral for breast cancer or colon cancer screening, Dr. Seymann adds. Additionally, they aren’t always the provider performing specific cardiac or neurological care.
Hospitalists’ patients usually are in the hospital because they are sick. They may have chronic disease or more complex medical needs (e.g. osteoporosis-related hip fracture) than the average population seen by a non-hospitalist PCP.
If hospitalists are compared to other PCPs, as is the plan in the Physician Value-Based Payment Modifier, it “looks like our patients are dying a lot more frequently, we’re spending a lot of money, and we’re not doing primary care,” Dr. Seymann explains.
New Brand, New Push
PQRS is not new; it is the rebranding of CMS’ Physician Quality Reporting Initiative (PQRI), launched in 2006. But changes to the program are part of a national push to improve healthcare quality and patient care while reimbursing for performance on outcome- and process-based measures instead of simply for the volume of services provided. Each year, CMS updates PQRS rules.
This year is the last one in which providers will receive a bonus for reporting through PQRS. Beginning next year, practitioners that don’t meet the reporting requirements for 2013 will incur a 1.5% penalty—with additional penalties for physicians in groups of 100 or more from the value-based payment modifier. This year also serves as the performance year for 2016, when a 2% penalty for insufficient reporting will be assessed.
In early December 2013, the Centers for Medicare & Medicaid Services (CMS) published the 2014 Physician Fee Schedule and, with it, the final rules for the PQRS. Although many physicians and specialist groups believed the measures included in PQRS in previous years were too limited, CMS has added the additional reporting methodology of qualified clinical data registries (QCDR), which can include measures outside of the PQRS—a marked shift from previous policies.
The rule change, Damberg says, should take some energy out of the discussion surrounding the program and allow more physicians to participate.
“From CMS’ perspective, they want doctors delivering the recommended care and they want doctors to be able to report it out easily,” Damberg says.
Moving Forward
In 2014, providers can submit measures through the new QCDR option, or submit PQRS-identified measures through a Medicare qualified registry, through electronic health records, through the group practice reporting option (GPRO), and through claims-based reporting (though this last option is expected to be phased out over time).
Registries themselves are not new, but they can cost millions of dollars to establish and as much as a million a year to maintain. They typically contain more clinical depth and specificity than claims data, and numerous studies show the use of registries leads to improved patient outcomes.
“We don’t know how many [existing] registries are going to qualify to become these qualified clinical data registries,” says Tom Granatir, senior vice president for health policy and external relations at ABMS. “It’s going to take some time for these registries to evolve.”
Qualified clinical data registries must be in operation for at least one year to be eligible for certification by Medicare. They must include performance data from other payers beyond Medicare. Not only must QCDRs be capable of capturing and sending data, they must also provide national benchmarks to those who submit and must report back at least four times per year.
Granatir believes the QCDR rule, which allows QCDR’s to report measures beyond those included in the PQRS program, will help increase participation and will lead to more practice-based measures, but he fears it may exclude some important nuances of day-to-day patient care.
“The whole point [of quality measure reporting] is to create more public transparency…but if you have measures that are not relevant to what is actually done in practices, then it’s not a useful dataset,” he says.
Ideally, Damberg says, PQRS and other performance measures should enable physicians to do what they do better.
“I think this is really going to raise the stakes for [hospitalists] if they want to control their destiny,” Damberg says. “I think they have to get really engaged in this game and take a pro-active role in looking at where the quality gaps are and how can they better benefit patients. That’s the ultimate goal.”
Kelly April Tyrrell is a freelance writer in Wilmington, Del.
Only 27% of eligible providers participated in the Physician Quality Reporting System (PQRS) in 2011—roughly 26,500 medical practices and 266,500 medical professionals, according to the Centers for Medicare & Medicaid Services (CMS).
“A lot of physicians have walked away [from PQRS] feeling like there are not sufficient measures for them to be measured against,” says Cheryl Damberg, senior principal researcher at RAND corporation and professor at the Pardee RAND Graduate School in Santa Monica, Calif.
Encouraging more participation from hospitalists has been the goal of the Society of Hospital Medicine (SHM) for the last several years, says Gregory Seymann, MD, SFHM, clinical professor and chief in the division of hospital medicine at University of California San Diego Health Sciences and chair of SHM’s Performance Measurement and Reporting Committee (PMRC).
“The committee has tried to champion it the best we can, making sure the measures that are there and in development meet the needs of the specialty,” Dr. Seymann says.
In just one year, the SHM committee managed to increase hospitalist reportable measures in PQRS from a paltry 11—half of which were only for stroke patients—to 21, which now includes things like diabetes exams, osteoporosis management, documentation of current medications, and community-acquired pneumonia treatment.
For Comparison’s Sake
For the first couple of phases of PQRS reporting, very few measures were relevant to hospitalists, Dr. Seymann says. The committee worked to ensure that more measures were added and billing codes modified to include those used by the specialty. Hospital medicine is relatively new, not officially recognized by the American Board of Medical Specialties (ABMS), and hospitalists serve a unique role. Most hospitalists are in internal medicine, family medicine, or pediatrics, but they aren’t doing what the average primary care doctor does, like referral for breast cancer or colon cancer screening, Dr. Seymann adds. Additionally, they aren’t always the provider performing specific cardiac or neurological care.
Hospitalists’ patients usually are in the hospital because they are sick. They may have chronic disease or more complex medical needs (e.g. osteoporosis-related hip fracture) than the average population seen by a non-hospitalist PCP.
If hospitalists are compared to other PCPs, as is the plan in the Physician Value-Based Payment Modifier, it “looks like our patients are dying a lot more frequently, we’re spending a lot of money, and we’re not doing primary care,” Dr. Seymann explains.
New Brand, New Push
PQRS is not new; it is the rebranding of CMS’ Physician Quality Reporting Initiative (PQRI), launched in 2006. But changes to the program are part of a national push to improve healthcare quality and patient care while reimbursing for performance on outcome- and process-based measures instead of simply for the volume of services provided. Each year, CMS updates PQRS rules.
This year is the last one in which providers will receive a bonus for reporting through PQRS. Beginning next year, practitioners that don’t meet the reporting requirements for 2013 will incur a 1.5% penalty—with additional penalties for physicians in groups of 100 or more from the value-based payment modifier. This year also serves as the performance year for 2016, when a 2% penalty for insufficient reporting will be assessed.
In early December 2013, the Centers for Medicare & Medicaid Services (CMS) published the 2014 Physician Fee Schedule and, with it, the final rules for the PQRS. Although many physicians and specialist groups believed the measures included in PQRS in previous years were too limited, CMS has added the additional reporting methodology of qualified clinical data registries (QCDR), which can include measures outside of the PQRS—a marked shift from previous policies.
The rule change, Damberg says, should take some energy out of the discussion surrounding the program and allow more physicians to participate.
“From CMS’ perspective, they want doctors delivering the recommended care and they want doctors to be able to report it out easily,” Damberg says.
Moving Forward
In 2014, providers can submit measures through the new QCDR option, or submit PQRS-identified measures through a Medicare qualified registry, through electronic health records, through the group practice reporting option (GPRO), and through claims-based reporting (though this last option is expected to be phased out over time).
Registries themselves are not new, but they can cost millions of dollars to establish and as much as a million a year to maintain. They typically contain more clinical depth and specificity than claims data, and numerous studies show the use of registries leads to improved patient outcomes.
“We don’t know how many [existing] registries are going to qualify to become these qualified clinical data registries,” says Tom Granatir, senior vice president for health policy and external relations at ABMS. “It’s going to take some time for these registries to evolve.”
Qualified clinical data registries must be in operation for at least one year to be eligible for certification by Medicare. They must include performance data from other payers beyond Medicare. Not only must QCDRs be capable of capturing and sending data, they must also provide national benchmarks to those who submit and must report back at least four times per year.
Granatir believes the QCDR rule, which allows QCDR’s to report measures beyond those included in the PQRS program, will help increase participation and will lead to more practice-based measures, but he fears it may exclude some important nuances of day-to-day patient care.
“The whole point [of quality measure reporting] is to create more public transparency…but if you have measures that are not relevant to what is actually done in practices, then it’s not a useful dataset,” he says.
Ideally, Damberg says, PQRS and other performance measures should enable physicians to do what they do better.
“I think this is really going to raise the stakes for [hospitalists] if they want to control their destiny,” Damberg says. “I think they have to get really engaged in this game and take a pro-active role in looking at where the quality gaps are and how can they better benefit patients. That’s the ultimate goal.”
Kelly April Tyrrell is a freelance writer in Wilmington, Del.
Aggregate Early Warning Score Results Mixed for High-Risk Pediatric Patients

Clinical question: Do rapid response systems (RRS) reduce the rate of critical deterioration (CD) in hospitalized children?
Background: Over the past decade, a majority of pediatric inpatient units and freestanding children’s hospitals have instituted RRSs that utilize medical emergency teams (MET) to rapidly evaluate clinically deteriorating patients. Pediatric RRSs manifest variability between institutions in MET composition and RRS triggers. Prior studies of RRSs in the pediatric population have been mixed, with a lack of robust evidence that RRSs reduce hospital mortality and cardiopulmonary arrest rates. Evaluation of the effect of RRSs in pediatric units and hospitals is complicated by the heterogeneity of pediatric RRS implementation and the overall low rate of in-hospital cardiopulmonary arrest and death. In a 2012 study, the authors defined CD events as those leading to ICU transfer and subsequent mechanical ventilation (noninvasive or invasive) or vasopressor infusion within 12 hours. CD event rates, quantified as an event rate per 1,000 non-ICU patient-days, were found to be associated with a >13-fold increase in risk of in-hospital death and were believed to be a valid proximate outcome for in-hospital mortality.
Study design: Single-center interrupted time series analysis
Setting: 516-bed urban, tertiary care, freestanding children’s hospital.
Synopsis: The RRS at this institution consisted of an aggregate early warning score (EWS), which triggered the response of a MET (within 30 minutes) 24 hours a day, seven days a week. Distinct from the code-blue team, the MET comprised PICU staff, including: (1) a fellow, attending, or nurse practitioner; (2) a nurse; and (3) a respiratory therapist. Researchers compared the 32 months prior to implementation to the 27 months after implementation. An interrupted time series analysis was performed using advanced statistical modeling with adjustments for season, ward, and case-mix index.
Although there were no significant differences in rates of cardiopulmonary arrest or mortality, adjusted analysis revealed a net 62% reduction in CD event rate (IRR=0.38) after initiation of the RRS. Adjusted analysis also found a net 83% reduction in mechanical ventilation events (IRR=0.17) and a net 80% reduction in vasopressor use (IRR=0.20). Of note, these reductions were not significant in the unadjusted analyses. After transfer to the ICU, time elapsed until initiation of either mechanical ventilation or vasopressor use was longer.
Bottom line: With an aggregate EWS identifying high-risk patients requiring rapid evaluation by a MET, a pediatric RRS reduces adjusted rates of CD events but might not yield significant reductions in unadjusted rates of mortality, cardiopulmonary arrest, or CD events.
Citation: Bonafide CP, Localio AR, Roberts KE, Nadkarni VM, Weirich CM, Keren R. Impact of rapid response system imple-mentation on critical deterioration events in children. JAMA Pediatr. 2014;168(1):25-33.
Reviewed by Pediatric Editor Weijen Chang, MD, SFHM, FAAP, associate clinical professor of medicine and pediatrics at the University of California at San Diego School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital.
Clinical question: Do rapid response systems (RRS) reduce the rate of critical deterioration (CD) in hospitalized children?
Background: Over the past decade, a majority of pediatric inpatient units and freestanding children’s hospitals have instituted RRSs that utilize medical emergency teams (MET) to rapidly evaluate clinically deteriorating patients. Pediatric RRSs manifest variability between institutions in MET composition and RRS triggers. Prior studies of RRSs in the pediatric population have been mixed, with a lack of robust evidence that RRSs reduce hospital mortality and cardiopulmonary arrest rates. Evaluation of the effect of RRSs in pediatric units and hospitals is complicated by the heterogeneity of pediatric RRS implementation and the overall low rate of in-hospital cardiopulmonary arrest and death. In a 2012 study, the authors defined CD events as those leading to ICU transfer and subsequent mechanical ventilation (noninvasive or invasive) or vasopressor infusion within 12 hours. CD event rates, quantified as an event rate per 1,000 non-ICU patient-days, were found to be associated with a >13-fold increase in risk of in-hospital death and were believed to be a valid proximate outcome for in-hospital mortality.
Study design: Single-center interrupted time series analysis
Setting: 516-bed urban, tertiary care, freestanding children’s hospital.
Synopsis: The RRS at this institution consisted of an aggregate early warning score (EWS), which triggered the response of a MET (within 30 minutes) 24 hours a day, seven days a week. Distinct from the code-blue team, the MET comprised PICU staff, including: (1) a fellow, attending, or nurse practitioner; (2) a nurse; and (3) a respiratory therapist. Researchers compared the 32 months prior to implementation to the 27 months after implementation. An interrupted time series analysis was performed using advanced statistical modeling with adjustments for season, ward, and case-mix index.
Although there were no significant differences in rates of cardiopulmonary arrest or mortality, adjusted analysis revealed a net 62% reduction in CD event rate (IRR=0.38) after initiation of the RRS. Adjusted analysis also found a net 83% reduction in mechanical ventilation events (IRR=0.17) and a net 80% reduction in vasopressor use (IRR=0.20). Of note, these reductions were not significant in the unadjusted analyses. After transfer to the ICU, time elapsed until initiation of either mechanical ventilation or vasopressor use was longer.
Bottom line: With an aggregate EWS identifying high-risk patients requiring rapid evaluation by a MET, a pediatric RRS reduces adjusted rates of CD events but might not yield significant reductions in unadjusted rates of mortality, cardiopulmonary arrest, or CD events.
Citation: Bonafide CP, Localio AR, Roberts KE, Nadkarni VM, Weirich CM, Keren R. Impact of rapid response system imple-mentation on critical deterioration events in children. JAMA Pediatr. 2014;168(1):25-33.
Reviewed by Pediatric Editor Weijen Chang, MD, SFHM, FAAP, associate clinical professor of medicine and pediatrics at the University of California at San Diego School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital.
Clinical question: Do rapid response systems (RRS) reduce the rate of critical deterioration (CD) in hospitalized children?
Background: Over the past decade, a majority of pediatric inpatient units and freestanding children’s hospitals have instituted RRSs that utilize medical emergency teams (MET) to rapidly evaluate clinically deteriorating patients. Pediatric RRSs manifest variability between institutions in MET composition and RRS triggers. Prior studies of RRSs in the pediatric population have been mixed, with a lack of robust evidence that RRSs reduce hospital mortality and cardiopulmonary arrest rates. Evaluation of the effect of RRSs in pediatric units and hospitals is complicated by the heterogeneity of pediatric RRS implementation and the overall low rate of in-hospital cardiopulmonary arrest and death. In a 2012 study, the authors defined CD events as those leading to ICU transfer and subsequent mechanical ventilation (noninvasive or invasive) or vasopressor infusion within 12 hours. CD event rates, quantified as an event rate per 1,000 non-ICU patient-days, were found to be associated with a >13-fold increase in risk of in-hospital death and were believed to be a valid proximate outcome for in-hospital mortality.
Study design: Single-center interrupted time series analysis
Setting: 516-bed urban, tertiary care, freestanding children’s hospital.
Synopsis: The RRS at this institution consisted of an aggregate early warning score (EWS), which triggered the response of a MET (within 30 minutes) 24 hours a day, seven days a week. Distinct from the code-blue team, the MET comprised PICU staff, including: (1) a fellow, attending, or nurse practitioner; (2) a nurse; and (3) a respiratory therapist. Researchers compared the 32 months prior to implementation to the 27 months after implementation. An interrupted time series analysis was performed using advanced statistical modeling with adjustments for season, ward, and case-mix index.
Although there were no significant differences in rates of cardiopulmonary arrest or mortality, adjusted analysis revealed a net 62% reduction in CD event rate (IRR=0.38) after initiation of the RRS. Adjusted analysis also found a net 83% reduction in mechanical ventilation events (IRR=0.17) and a net 80% reduction in vasopressor use (IRR=0.20). Of note, these reductions were not significant in the unadjusted analyses. After transfer to the ICU, time elapsed until initiation of either mechanical ventilation or vasopressor use was longer.
Bottom line: With an aggregate EWS identifying high-risk patients requiring rapid evaluation by a MET, a pediatric RRS reduces adjusted rates of CD events but might not yield significant reductions in unadjusted rates of mortality, cardiopulmonary arrest, or CD events.
Citation: Bonafide CP, Localio AR, Roberts KE, Nadkarni VM, Weirich CM, Keren R. Impact of rapid response system imple-mentation on critical deterioration events in children. JAMA Pediatr. 2014;168(1):25-33.
Reviewed by Pediatric Editor Weijen Chang, MD, SFHM, FAAP, associate clinical professor of medicine and pediatrics at the University of California at San Diego School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital.
Benefits of Membership in Society of Hospital Medicine Plentiful
Hospital medicine has always been more than a medical specialty; it is a movement to improve healthcare delivered in the hospital and to make the hospital a vital part of the healthcare system in every community served.
SHM’s members have always been at the forefront of that movement. The benefits of SHM membership now keep more than 12,000 SHM members ahead of the curve in a rapidly growing and evolving medical specialty. Although SHM membership offers numerous benefits, many SHM members tell others that simply being part of the movement to transform healthcare and revolutionize patient care is the main reason they joined SHM and renew every year.
Exclusive member benefits include:
- Access to the Hospital Medicine Exchange, an exclusive online member community for networking and collaboration;
- Opportunities to make your voice heard on important policy and regulatory issues affecting hospitalists and their patients;
- Enhanced professional satisfaction gained through networking with hospitalists on a local and national level;
- Complimentary subscription to The Hospitalist news magazine and to the Journal of Hospital Medicine, the first peer-reviewed journal in hospital medicine;
- Member discounts on educational events, including our annual meeting, and select resources available in our online store;
- Access to SHM’s Career Center, with hundreds of positions and top-of-the-line functionality;
- Fellowship in Hospital Medicine designation opportunities (FHM, SFHM, MHM);
- Program discounts on medical liability insurance through The Doctors Company;
- Member discounts on Wiley educational products; and
- NEW: Free and discounted coding services from The Coding Network.
To become a member, visit www.hospitalmedicine.org/membership.
Hospital medicine has always been more than a medical specialty; it is a movement to improve healthcare delivered in the hospital and to make the hospital a vital part of the healthcare system in every community served.
SHM’s members have always been at the forefront of that movement. The benefits of SHM membership now keep more than 12,000 SHM members ahead of the curve in a rapidly growing and evolving medical specialty. Although SHM membership offers numerous benefits, many SHM members tell others that simply being part of the movement to transform healthcare and revolutionize patient care is the main reason they joined SHM and renew every year.
Exclusive member benefits include:
- Access to the Hospital Medicine Exchange, an exclusive online member community for networking and collaboration;
- Opportunities to make your voice heard on important policy and regulatory issues affecting hospitalists and their patients;
- Enhanced professional satisfaction gained through networking with hospitalists on a local and national level;
- Complimentary subscription to The Hospitalist news magazine and to the Journal of Hospital Medicine, the first peer-reviewed journal in hospital medicine;
- Member discounts on educational events, including our annual meeting, and select resources available in our online store;
- Access to SHM’s Career Center, with hundreds of positions and top-of-the-line functionality;
- Fellowship in Hospital Medicine designation opportunities (FHM, SFHM, MHM);
- Program discounts on medical liability insurance through The Doctors Company;
- Member discounts on Wiley educational products; and
- NEW: Free and discounted coding services from The Coding Network.
To become a member, visit www.hospitalmedicine.org/membership.
Hospital medicine has always been more than a medical specialty; it is a movement to improve healthcare delivered in the hospital and to make the hospital a vital part of the healthcare system in every community served.
SHM’s members have always been at the forefront of that movement. The benefits of SHM membership now keep more than 12,000 SHM members ahead of the curve in a rapidly growing and evolving medical specialty. Although SHM membership offers numerous benefits, many SHM members tell others that simply being part of the movement to transform healthcare and revolutionize patient care is the main reason they joined SHM and renew every year.
Exclusive member benefits include:
- Access to the Hospital Medicine Exchange, an exclusive online member community for networking and collaboration;
- Opportunities to make your voice heard on important policy and regulatory issues affecting hospitalists and their patients;
- Enhanced professional satisfaction gained through networking with hospitalists on a local and national level;
- Complimentary subscription to The Hospitalist news magazine and to the Journal of Hospital Medicine, the first peer-reviewed journal in hospital medicine;
- Member discounts on educational events, including our annual meeting, and select resources available in our online store;
- Access to SHM’s Career Center, with hundreds of positions and top-of-the-line functionality;
- Fellowship in Hospital Medicine designation opportunities (FHM, SFHM, MHM);
- Program discounts on medical liability insurance through The Doctors Company;
- Member discounts on Wiley educational products; and
- NEW: Free and discounted coding services from The Coding Network.
To become a member, visit www.hospitalmedicine.org/membership.
Society of Hospital Medicine Uses Social Media to Connect Members
Now more than ever, social media and SHM are connecting hospitalists with peers and experts in healthcare. Last year, SHM launched a new blog, The Hospital Leader (http://blogs.hospitalmedicine.org/Blog), and introduced hospitalists to eight prominent voices in the HM movement. In January, SHM used Twitter (http://www.twitter.com/shmlive) to educate hospitalists and others on CMS’ changes to the “two-midnight rule” through a live tweet of a free webinar.
Since its inception in October 2012, nearly 3,000 hospitalists have logged into HMX more than 72,000 times to ask questions, share experiences, and learn from other hospitalists across the country.
Want to connect with the other hospitalists and experts in healthcare? Here’s how:
- Ask questions and get real-world answers at Hospital Medicine Exchange (HMX): http://connect.hospitalmedicine.org/home.
- Join SHM’s 2,200-plus followers on Twitter for quick news and information about the hospital medicine movement: https://twitter.com/shmlive.
- For new research in hospital medicine, follow the Journal of Hospital Medicine on Twitter: https://twitter.com/JHospMedicine.
- Get regular insight from leaders and experts in the movement at SHM’s blog, The Hospital Leader: http://blogs.hospitalmedicine.org/Blog.
- “Like” and “share” away with SHM on Facebook: www.facebook.com/Hospitalists.
Tweeting at HM14? Use #HospMed14
Last year, tweets about SHM’s annual meeting reached thousands, creating more than two million impressions. This year will be even bigger. Join the conversation with leaders in the specialty, using #hospmed14 whenever you do.
Now more than ever, social media and SHM are connecting hospitalists with peers and experts in healthcare. Last year, SHM launched a new blog, The Hospital Leader (http://blogs.hospitalmedicine.org/Blog), and introduced hospitalists to eight prominent voices in the HM movement. In January, SHM used Twitter (http://www.twitter.com/shmlive) to educate hospitalists and others on CMS’ changes to the “two-midnight rule” through a live tweet of a free webinar.
Since its inception in October 2012, nearly 3,000 hospitalists have logged into HMX more than 72,000 times to ask questions, share experiences, and learn from other hospitalists across the country.
Want to connect with the other hospitalists and experts in healthcare? Here’s how:
- Ask questions and get real-world answers at Hospital Medicine Exchange (HMX): http://connect.hospitalmedicine.org/home.
- Join SHM’s 2,200-plus followers on Twitter for quick news and information about the hospital medicine movement: https://twitter.com/shmlive.
- For new research in hospital medicine, follow the Journal of Hospital Medicine on Twitter: https://twitter.com/JHospMedicine.
- Get regular insight from leaders and experts in the movement at SHM’s blog, The Hospital Leader: http://blogs.hospitalmedicine.org/Blog.
- “Like” and “share” away with SHM on Facebook: www.facebook.com/Hospitalists.
Tweeting at HM14? Use #HospMed14
Last year, tweets about SHM’s annual meeting reached thousands, creating more than two million impressions. This year will be even bigger. Join the conversation with leaders in the specialty, using #hospmed14 whenever you do.
Now more than ever, social media and SHM are connecting hospitalists with peers and experts in healthcare. Last year, SHM launched a new blog, The Hospital Leader (http://blogs.hospitalmedicine.org/Blog), and introduced hospitalists to eight prominent voices in the HM movement. In January, SHM used Twitter (http://www.twitter.com/shmlive) to educate hospitalists and others on CMS’ changes to the “two-midnight rule” through a live tweet of a free webinar.
Since its inception in October 2012, nearly 3,000 hospitalists have logged into HMX more than 72,000 times to ask questions, share experiences, and learn from other hospitalists across the country.
Want to connect with the other hospitalists and experts in healthcare? Here’s how:
- Ask questions and get real-world answers at Hospital Medicine Exchange (HMX): http://connect.hospitalmedicine.org/home.
- Join SHM’s 2,200-plus followers on Twitter for quick news and information about the hospital medicine movement: https://twitter.com/shmlive.
- For new research in hospital medicine, follow the Journal of Hospital Medicine on Twitter: https://twitter.com/JHospMedicine.
- Get regular insight from leaders and experts in the movement at SHM’s blog, The Hospital Leader: http://blogs.hospitalmedicine.org/Blog.
- “Like” and “share” away with SHM on Facebook: www.facebook.com/Hospitalists.
Tweeting at HM14? Use #HospMed14
Last year, tweets about SHM’s annual meeting reached thousands, creating more than two million impressions. This year will be even bigger. Join the conversation with leaders in the specialty, using #hospmed14 whenever you do.
The Costs of Quality Care in Pediatric Hospital Medicine

“Dr. Chang? Oh my, it’s Dr. Chang! And his little son!” I called them “mall moments.” I would be at the local shopping mall with my father, picking up new clothes for the upcoming school year, when suddenly an elderly woman would approach. My father, despite his inability to remember my own birthday, would warmly grasp the woman’s hands, gaze into her eyes, ask about her family, then reminisce about her late husband and his last days in the hospital. After a few minutes, she would say something like, “Well, your father is the best doctor in Bakersfield, and you’ll be lucky to grow up to be just like him.”
And this would be fine, except the same scene would replay at the supermarket, the dry cleaners, and the local Chinese restaurant (the only place my father would eat out until he discovered the exotic pleasures of sushi). I wondered how my father ever got any errands done, with all his patients chatting with him along the way. Looking back on these “moments,” it is clear to me that this was my father’s measure of quality—his patients loved him. Other doctors loved him. The nurses—well, maybe not so much. He was a doctor’s doctor.
Quality measures? After working in his office, I only knew of two: The waiting room must be empty before the doors are closed and locked, and no patient ever gets turned away, for any reason. By seven o’clock in the evening, these measures got pretty old. But simple credos made him one of the most beloved physicians in Kern County, Calif.
Quality, in whatever form it takes, has a cost, however. My father divorced twice. My own “quality” time with him was spent making weekend rounds at the seemingly innumerable nursing homes around Bakersfield, Calif., although this was great olfactory training for my future career as a hospitalist. Many a parent’s day was spent with only my mother present, and I would be lying if I said I didn’t envy the other children with both parents doting over their science projects.
As we in pediatric hospital medicine (PHM) embark on a journey to define and promote quality in our care of children, we are well aware that adhering to our defined standards of quality will have a cost. What has been discussed less, but is perhaps even more elementary, is the cost of simply endeavoring to define and measure quality itself. This has not slowed down the onslaught of newly defined quality measures in PHM. Quality measures from the adult HM world, such as readmission rates, adherence to national guidelines, and communication with primary care providers, have been extracted and repurposed.
Attempts to extrapolate these measures to PHM have been less than successful. Alverson and O’Callaghan recently made a compelling case debunking readmission rates as a valid quality measure in PHM.1 Compliance with Children’s Asthma Care (CAC) measures was not found to decrease asthma-related readmissions or subsequent ED visits in a 2011 study, although a study published in 2012 showed an association between compliance with asthma action plans at discharge and lower readmission rates.2,3 Documentation of primary care follow-up for patients discharged from a free-standing children’s hospital actually increased the readmission rate (if that is believed to be a quality measure).4
Yet quality measures continue to be created, espoused, and studied. Payments to accountable care organizations (ACO), hospitals, and individual providers are being tied to performance on quality measures. Medicare is considering quality measures that can be applied to PHM, which might affect future payments to children’s hospitals. Paciorkowski and colleagues recently described the development of 87 performance indicators specific to PHM that could be used to track quality of care on a division level, 79 of which were provider specific.5 A committee of pediatric hospitalists led by Paul Hain, MD, recently proposed a “dashboard” of metrics pertaining to descriptive, quality, productivity, and other data that could be used to compare PHM groups across the country.6 Many hospitalist groups already have instituted financial incentives tied to provider or group-specific quality measures.7 Pay-for-performance has arrived in adult HM and is now pulling out of the station: next stop, PHM.
Source: From Crosby P. B. Quality Is Free: The Art of Making Quality Certain. New York: McGraw-Hill; 1979.
The Rest of the Cost Story
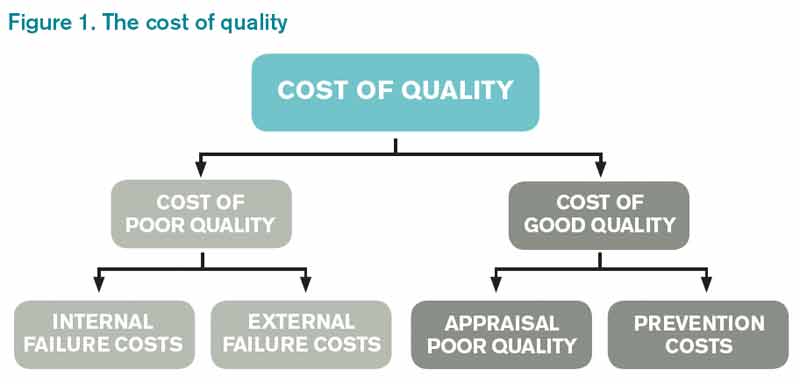
Like any labor-intensive process in medicine, defining, measuring, and improving quality has a cost. A 2007 survey of four urban teaching hospitals found that core QI activities required 1%-2% of the total operating revenue.8 The QI activity costs fall into the category of the “cost of good quality,” as defined by Philip Crosby in his book, Quality is Free (see Figure 1).9 Although hospital operations with better process “sigma” will have lower prevention and appraisal costs, these can never be fully eliminated.
Despite our attempts at controlling costs, most ongoing QI efforts focused on improving clinical quality alone are doomed to fail with regard to providing bottom-line cost reductions.10 QI efforts that focus on decreasing variability in the use of best practices, such as the National Surgical Quality Improvement Program (NSQIP), have brought improvements in both outcomes and reduced costs of complications.11 Not only do these QI efforts lower the “cost of poor quality,” but they may provide less measurable benefits, such as reduced opportunity costs. Whether these efforts can compensate by reducing the cost of poor quality can be speculative. Some HM authorities, such as Duke University Health CMO Thomas Owens, have made the case, especially to hospital administrators, for espousing a more formulaic return on investment (ROI) calculation for HM QI efforts, taking into account reduced opportunity costs.12
But measured costs tell only part of the story. For every new quality measure that is defined, there are also unmeasured costs to measuring and collecting evidence of quality. Being constantly measured and assessed often leads to a perceived loss of autonomy, and this can lead to burnout; more than 40% of respondents from local hospitalist groups in the most recent SHM Career Satisfaction Survey indicated that optimal autonomy was among the four most important factors for job satisfaction.13 The same survey found that hospitalists were least satisfied with organizational climate, autonomy, and availability of personal time.14
As many a hospitalist can relate, although involvement in QI processes is considered a cornerstone of hospitalist practice, increased time spent in a given QI activity rarely translates to increased compensation. Fourteen percent of hospitalists in a recent SHM Focused Survey reported not even having dedicated time for or being compensated for QI.
Which is not to say, of course, that defining and measuring quality is not a worthy pursuit. On the contrary, QI is a pillar of hospital medicine practice. A recent survey showed that 84% of pediatric hospitalists participated in QI initiatives, and 72% considered the variety of pursuits inherent in a PHM career as a factor influencing career choice.15 But just as we are now focused on choosing wisely in diagnosing and treating our patients, we should also be choosing wisely in diagnosing and treating our systems. What is true for our patients is true for our system of care—simply ordering the test can lead to a cascade of interventions that can be not only costly but also potentially dangerous for the patient.
Physician-defined quality measures in adult HM have now been adopted as yardsticks with which to measure all hospitals—and with which to punish those who do not measure up. In 1984, Dr. Earl Steinberg, then a professor of medicine at Johns Hopkins, published a seminal article in the New England Journal of Medicine describing potential cost savings to the Medicare program from reductions in hospital readmissions.16 This was the match that lit the fuse to what is now the Affordable Care Act Hospital Readmissions Reduction Program. Yet, this quality measure might not even be a quality measure of…quality. A 2013 JAMA study showed that readmission rates for acute myocardial infarction and pneumonia were not correlated with mortality, the time-tested gold standard for quality in medicine.17 That has not stopped Medicare from levying $227 million in fines on 2,225 hospitals across the country beginning Oct. 1, 2013 for excess readmissions in Year 2 of the Hospital Readmissions Reduction Program.18 It seems that we have built it, and they have come, and now they won’t leave.
In Sum
What is the lesson for PHM? Assessing and improving quality of care remains a necessary cornerstone of PHM, but choosing meaningful quality measures is difficult and can have long-term consequences. The choices we make with regard to the direction of QI will, however, define the future of pediatric healthcare for decades to come. As such, we cannot waste both financial and human resources on defining and assessing quality measures that may sound superficially important but, in the end, are not reflective of the real quality of care provided to our patients.
My father, in his adherence to his own ideal of quality medical care, reaped the unintended consequences of his pursuit of quality medical care. Sometimes, though just sometimes, there are unintended consequences to the unintended consequences. I learned, and was perhaps inspired, just by watching him interact with patients and their families. Somehow I don’t think my own children will learn much by watching me interact with my computer.
Dr. Chang is pediatric editor of The Hospitalist. He is associate clinical professor of medicine and pediatrics at the University of California at San Diego (UCSD) School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital. Send comments and questions to wwch@ucsd.edu.
“Dr. Chang? Oh my, it’s Dr. Chang! And his little son!” I called them “mall moments.” I would be at the local shopping mall with my father, picking up new clothes for the upcoming school year, when suddenly an elderly woman would approach. My father, despite his inability to remember my own birthday, would warmly grasp the woman’s hands, gaze into her eyes, ask about her family, then reminisce about her late husband and his last days in the hospital. After a few minutes, she would say something like, “Well, your father is the best doctor in Bakersfield, and you’ll be lucky to grow up to be just like him.”
And this would be fine, except the same scene would replay at the supermarket, the dry cleaners, and the local Chinese restaurant (the only place my father would eat out until he discovered the exotic pleasures of sushi). I wondered how my father ever got any errands done, with all his patients chatting with him along the way. Looking back on these “moments,” it is clear to me that this was my father’s measure of quality—his patients loved him. Other doctors loved him. The nurses—well, maybe not so much. He was a doctor’s doctor.
Quality measures? After working in his office, I only knew of two: The waiting room must be empty before the doors are closed and locked, and no patient ever gets turned away, for any reason. By seven o’clock in the evening, these measures got pretty old. But simple credos made him one of the most beloved physicians in Kern County, Calif.
Quality, in whatever form it takes, has a cost, however. My father divorced twice. My own “quality” time with him was spent making weekend rounds at the seemingly innumerable nursing homes around Bakersfield, Calif., although this was great olfactory training for my future career as a hospitalist. Many a parent’s day was spent with only my mother present, and I would be lying if I said I didn’t envy the other children with both parents doting over their science projects.
As we in pediatric hospital medicine (PHM) embark on a journey to define and promote quality in our care of children, we are well aware that adhering to our defined standards of quality will have a cost. What has been discussed less, but is perhaps even more elementary, is the cost of simply endeavoring to define and measure quality itself. This has not slowed down the onslaught of newly defined quality measures in PHM. Quality measures from the adult HM world, such as readmission rates, adherence to national guidelines, and communication with primary care providers, have been extracted and repurposed.
Attempts to extrapolate these measures to PHM have been less than successful. Alverson and O’Callaghan recently made a compelling case debunking readmission rates as a valid quality measure in PHM.1 Compliance with Children’s Asthma Care (CAC) measures was not found to decrease asthma-related readmissions or subsequent ED visits in a 2011 study, although a study published in 2012 showed an association between compliance with asthma action plans at discharge and lower readmission rates.2,3 Documentation of primary care follow-up for patients discharged from a free-standing children’s hospital actually increased the readmission rate (if that is believed to be a quality measure).4
Yet quality measures continue to be created, espoused, and studied. Payments to accountable care organizations (ACO), hospitals, and individual providers are being tied to performance on quality measures. Medicare is considering quality measures that can be applied to PHM, which might affect future payments to children’s hospitals. Paciorkowski and colleagues recently described the development of 87 performance indicators specific to PHM that could be used to track quality of care on a division level, 79 of which were provider specific.5 A committee of pediatric hospitalists led by Paul Hain, MD, recently proposed a “dashboard” of metrics pertaining to descriptive, quality, productivity, and other data that could be used to compare PHM groups across the country.6 Many hospitalist groups already have instituted financial incentives tied to provider or group-specific quality measures.7 Pay-for-performance has arrived in adult HM and is now pulling out of the station: next stop, PHM.
Source: From Crosby P. B. Quality Is Free: The Art of Making Quality Certain. New York: McGraw-Hill; 1979.
The Rest of the Cost Story
Like any labor-intensive process in medicine, defining, measuring, and improving quality has a cost. A 2007 survey of four urban teaching hospitals found that core QI activities required 1%-2% of the total operating revenue.8 The QI activity costs fall into the category of the “cost of good quality,” as defined by Philip Crosby in his book, Quality is Free (see Figure 1).9 Although hospital operations with better process “sigma” will have lower prevention and appraisal costs, these can never be fully eliminated.
Despite our attempts at controlling costs, most ongoing QI efforts focused on improving clinical quality alone are doomed to fail with regard to providing bottom-line cost reductions.10 QI efforts that focus on decreasing variability in the use of best practices, such as the National Surgical Quality Improvement Program (NSQIP), have brought improvements in both outcomes and reduced costs of complications.11 Not only do these QI efforts lower the “cost of poor quality,” but they may provide less measurable benefits, such as reduced opportunity costs. Whether these efforts can compensate by reducing the cost of poor quality can be speculative. Some HM authorities, such as Duke University Health CMO Thomas Owens, have made the case, especially to hospital administrators, for espousing a more formulaic return on investment (ROI) calculation for HM QI efforts, taking into account reduced opportunity costs.12
But measured costs tell only part of the story. For every new quality measure that is defined, there are also unmeasured costs to measuring and collecting evidence of quality. Being constantly measured and assessed often leads to a perceived loss of autonomy, and this can lead to burnout; more than 40% of respondents from local hospitalist groups in the most recent SHM Career Satisfaction Survey indicated that optimal autonomy was among the four most important factors for job satisfaction.13 The same survey found that hospitalists were least satisfied with organizational climate, autonomy, and availability of personal time.14
As many a hospitalist can relate, although involvement in QI processes is considered a cornerstone of hospitalist practice, increased time spent in a given QI activity rarely translates to increased compensation. Fourteen percent of hospitalists in a recent SHM Focused Survey reported not even having dedicated time for or being compensated for QI.
Which is not to say, of course, that defining and measuring quality is not a worthy pursuit. On the contrary, QI is a pillar of hospital medicine practice. A recent survey showed that 84% of pediatric hospitalists participated in QI initiatives, and 72% considered the variety of pursuits inherent in a PHM career as a factor influencing career choice.15 But just as we are now focused on choosing wisely in diagnosing and treating our patients, we should also be choosing wisely in diagnosing and treating our systems. What is true for our patients is true for our system of care—simply ordering the test can lead to a cascade of interventions that can be not only costly but also potentially dangerous for the patient.
Physician-defined quality measures in adult HM have now been adopted as yardsticks with which to measure all hospitals—and with which to punish those who do not measure up. In 1984, Dr. Earl Steinberg, then a professor of medicine at Johns Hopkins, published a seminal article in the New England Journal of Medicine describing potential cost savings to the Medicare program from reductions in hospital readmissions.16 This was the match that lit the fuse to what is now the Affordable Care Act Hospital Readmissions Reduction Program. Yet, this quality measure might not even be a quality measure of…quality. A 2013 JAMA study showed that readmission rates for acute myocardial infarction and pneumonia were not correlated with mortality, the time-tested gold standard for quality in medicine.17 That has not stopped Medicare from levying $227 million in fines on 2,225 hospitals across the country beginning Oct. 1, 2013 for excess readmissions in Year 2 of the Hospital Readmissions Reduction Program.18 It seems that we have built it, and they have come, and now they won’t leave.
In Sum
What is the lesson for PHM? Assessing and improving quality of care remains a necessary cornerstone of PHM, but choosing meaningful quality measures is difficult and can have long-term consequences. The choices we make with regard to the direction of QI will, however, define the future of pediatric healthcare for decades to come. As such, we cannot waste both financial and human resources on defining and assessing quality measures that may sound superficially important but, in the end, are not reflective of the real quality of care provided to our patients.
My father, in his adherence to his own ideal of quality medical care, reaped the unintended consequences of his pursuit of quality medical care. Sometimes, though just sometimes, there are unintended consequences to the unintended consequences. I learned, and was perhaps inspired, just by watching him interact with patients and their families. Somehow I don’t think my own children will learn much by watching me interact with my computer.
Dr. Chang is pediatric editor of The Hospitalist. He is associate clinical professor of medicine and pediatrics at the University of California at San Diego (UCSD) School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital. Send comments and questions to wwch@ucsd.edu.
“Dr. Chang? Oh my, it’s Dr. Chang! And his little son!” I called them “mall moments.” I would be at the local shopping mall with my father, picking up new clothes for the upcoming school year, when suddenly an elderly woman would approach. My father, despite his inability to remember my own birthday, would warmly grasp the woman’s hands, gaze into her eyes, ask about her family, then reminisce about her late husband and his last days in the hospital. After a few minutes, she would say something like, “Well, your father is the best doctor in Bakersfield, and you’ll be lucky to grow up to be just like him.”
And this would be fine, except the same scene would replay at the supermarket, the dry cleaners, and the local Chinese restaurant (the only place my father would eat out until he discovered the exotic pleasures of sushi). I wondered how my father ever got any errands done, with all his patients chatting with him along the way. Looking back on these “moments,” it is clear to me that this was my father’s measure of quality—his patients loved him. Other doctors loved him. The nurses—well, maybe not so much. He was a doctor’s doctor.
Quality measures? After working in his office, I only knew of two: The waiting room must be empty before the doors are closed and locked, and no patient ever gets turned away, for any reason. By seven o’clock in the evening, these measures got pretty old. But simple credos made him one of the most beloved physicians in Kern County, Calif.
Quality, in whatever form it takes, has a cost, however. My father divorced twice. My own “quality” time with him was spent making weekend rounds at the seemingly innumerable nursing homes around Bakersfield, Calif., although this was great olfactory training for my future career as a hospitalist. Many a parent’s day was spent with only my mother present, and I would be lying if I said I didn’t envy the other children with both parents doting over their science projects.
As we in pediatric hospital medicine (PHM) embark on a journey to define and promote quality in our care of children, we are well aware that adhering to our defined standards of quality will have a cost. What has been discussed less, but is perhaps even more elementary, is the cost of simply endeavoring to define and measure quality itself. This has not slowed down the onslaught of newly defined quality measures in PHM. Quality measures from the adult HM world, such as readmission rates, adherence to national guidelines, and communication with primary care providers, have been extracted and repurposed.
Attempts to extrapolate these measures to PHM have been less than successful. Alverson and O’Callaghan recently made a compelling case debunking readmission rates as a valid quality measure in PHM.1 Compliance with Children’s Asthma Care (CAC) measures was not found to decrease asthma-related readmissions or subsequent ED visits in a 2011 study, although a study published in 2012 showed an association between compliance with asthma action plans at discharge and lower readmission rates.2,3 Documentation of primary care follow-up for patients discharged from a free-standing children’s hospital actually increased the readmission rate (if that is believed to be a quality measure).4
Yet quality measures continue to be created, espoused, and studied. Payments to accountable care organizations (ACO), hospitals, and individual providers are being tied to performance on quality measures. Medicare is considering quality measures that can be applied to PHM, which might affect future payments to children’s hospitals. Paciorkowski and colleagues recently described the development of 87 performance indicators specific to PHM that could be used to track quality of care on a division level, 79 of which were provider specific.5 A committee of pediatric hospitalists led by Paul Hain, MD, recently proposed a “dashboard” of metrics pertaining to descriptive, quality, productivity, and other data that could be used to compare PHM groups across the country.6 Many hospitalist groups already have instituted financial incentives tied to provider or group-specific quality measures.7 Pay-for-performance has arrived in adult HM and is now pulling out of the station: next stop, PHM.
Source: From Crosby P. B. Quality Is Free: The Art of Making Quality Certain. New York: McGraw-Hill; 1979.
The Rest of the Cost Story
Like any labor-intensive process in medicine, defining, measuring, and improving quality has a cost. A 2007 survey of four urban teaching hospitals found that core QI activities required 1%-2% of the total operating revenue.8 The QI activity costs fall into the category of the “cost of good quality,” as defined by Philip Crosby in his book, Quality is Free (see Figure 1).9 Although hospital operations with better process “sigma” will have lower prevention and appraisal costs, these can never be fully eliminated.
Despite our attempts at controlling costs, most ongoing QI efforts focused on improving clinical quality alone are doomed to fail with regard to providing bottom-line cost reductions.10 QI efforts that focus on decreasing variability in the use of best practices, such as the National Surgical Quality Improvement Program (NSQIP), have brought improvements in both outcomes and reduced costs of complications.11 Not only do these QI efforts lower the “cost of poor quality,” but they may provide less measurable benefits, such as reduced opportunity costs. Whether these efforts can compensate by reducing the cost of poor quality can be speculative. Some HM authorities, such as Duke University Health CMO Thomas Owens, have made the case, especially to hospital administrators, for espousing a more formulaic return on investment (ROI) calculation for HM QI efforts, taking into account reduced opportunity costs.12
But measured costs tell only part of the story. For every new quality measure that is defined, there are also unmeasured costs to measuring and collecting evidence of quality. Being constantly measured and assessed often leads to a perceived loss of autonomy, and this can lead to burnout; more than 40% of respondents from local hospitalist groups in the most recent SHM Career Satisfaction Survey indicated that optimal autonomy was among the four most important factors for job satisfaction.13 The same survey found that hospitalists were least satisfied with organizational climate, autonomy, and availability of personal time.14
As many a hospitalist can relate, although involvement in QI processes is considered a cornerstone of hospitalist practice, increased time spent in a given QI activity rarely translates to increased compensation. Fourteen percent of hospitalists in a recent SHM Focused Survey reported not even having dedicated time for or being compensated for QI.
Which is not to say, of course, that defining and measuring quality is not a worthy pursuit. On the contrary, QI is a pillar of hospital medicine practice. A recent survey showed that 84% of pediatric hospitalists participated in QI initiatives, and 72% considered the variety of pursuits inherent in a PHM career as a factor influencing career choice.15 But just as we are now focused on choosing wisely in diagnosing and treating our patients, we should also be choosing wisely in diagnosing and treating our systems. What is true for our patients is true for our system of care—simply ordering the test can lead to a cascade of interventions that can be not only costly but also potentially dangerous for the patient.
Physician-defined quality measures in adult HM have now been adopted as yardsticks with which to measure all hospitals—and with which to punish those who do not measure up. In 1984, Dr. Earl Steinberg, then a professor of medicine at Johns Hopkins, published a seminal article in the New England Journal of Medicine describing potential cost savings to the Medicare program from reductions in hospital readmissions.16 This was the match that lit the fuse to what is now the Affordable Care Act Hospital Readmissions Reduction Program. Yet, this quality measure might not even be a quality measure of…quality. A 2013 JAMA study showed that readmission rates for acute myocardial infarction and pneumonia were not correlated with mortality, the time-tested gold standard for quality in medicine.17 That has not stopped Medicare from levying $227 million in fines on 2,225 hospitals across the country beginning Oct. 1, 2013 for excess readmissions in Year 2 of the Hospital Readmissions Reduction Program.18 It seems that we have built it, and they have come, and now they won’t leave.
In Sum
What is the lesson for PHM? Assessing and improving quality of care remains a necessary cornerstone of PHM, but choosing meaningful quality measures is difficult and can have long-term consequences. The choices we make with regard to the direction of QI will, however, define the future of pediatric healthcare for decades to come. As such, we cannot waste both financial and human resources on defining and assessing quality measures that may sound superficially important but, in the end, are not reflective of the real quality of care provided to our patients.
My father, in his adherence to his own ideal of quality medical care, reaped the unintended consequences of his pursuit of quality medical care. Sometimes, though just sometimes, there are unintended consequences to the unintended consequences. I learned, and was perhaps inspired, just by watching him interact with patients and their families. Somehow I don’t think my own children will learn much by watching me interact with my computer.
Dr. Chang is pediatric editor of The Hospitalist. He is associate clinical professor of medicine and pediatrics at the University of California at San Diego (UCSD) School of Medicine, and a hospitalist at both UCSD Medical Center and Rady Children’s Hospital. Send comments and questions to wwch@ucsd.edu.
Hospitalist Pay Shifts from Volume to Value with Global Payment System

The move to paying hospitals and physicians based on value instead of volume is well underway. As programs ultimately designed to offer a global payment for a population (ACOs) or an episode of care (bundled payment) expand, we are left with this paradox: How do we reward physicians for working harder and seeing more patients under a global payment system that encourages physicians and hospitals to do less?
It appears that the existing fee-for-service payment system will need to form the scaffolding of any new, value-based system. Physicians must document the services they provide, leaving a “footprint” that can be recognized and rewarded. Without a record of the volume of services, physicians will have no incentive to see more patients during times of increased demand. This is what we often experience with straight-salary arrangements—physicians question why they should work harder for no additional compensation.
Through the ACO lens, Bruce Landon, professor of health care policy at Harvard Medical School, states the challenge in a different way: “The fundamental questions become how ACOs will divide their global budgets and how their physicians and service providers will be reimbursed. Thus, this system for determining who has earned what portion of payments—keeping score—is likely to be crucially important to the success of these new models of care.”1
In another article addressing value-based payment for physicians, Eric Stecker, MD, MPH, and Steve Schroeder, MD, argue that, due to their longevity and resilience, relative value units (RVUs), instead of physician-level capitation, straight salary, or salary with pay for performance incentives, should be the preferred mechanism to reimburse physicians based on value.2

I’d like to further develop the idea of an RVU-centric approach to value-based physician reimbursement, specifically discussing the case of hospitalists.
In Table 1, I provide examples of “value-based elements” to be added to an RVU reimbursement system. I chose measures related to three hospital-based quality programs: readmission reduction, hospital-acquired conditions, and value-based purchasing; however, one could choose hospitalist-relevant quality measures from other programs, such as ACOs, meaningful use, outpatient quality reporting (for observation patients), bundled payments, or a broad range of other domains. I selected only process measures, because outcome measures such as mortality or readmission rates suffer from sample size that is too small and risk adjustment too inadequate to be applied to individual physician payment.
Drs. Stecker and Schroeder offer an observation that is especially important to hospitalists: “Although RVUs are traditionally used for episodes of care provided by individual clinicians for individual patients, activities linked to RVUs could be more broadly defined to include team-based and supervisory clinical activities as well.”2 In the table, I include “multidisciplinary discharge planning rounds” as a potential measure. One can envision other team-based or supervisory activities involving hospitalists collaborating with nurses, pharmacists, or case managers working on a catheter-UTI bundle, high-risk medication counseling, or readmission risk assessment—with each activity linked to RVUs.
The implementation of an RVU system incorporating quality measures would be aided by documentation templates in the electronic medical record, similar to templates emerging for care bundles like central line blood stream infection. Value-based RVUs would have challenges, such as the need to change the measures over time and the system gaming inherent in any incentive design. Details of implementing the program would need to be worked out, such as attributing measures to individual physicians/providers or limiting to one the number of times certain measures are fulfilled per hospitalization.
Once established, a value-based RVU system could replace the complex and variable physician compensation landscape that exists today. As has always been the case, an RVU system could form the basis of a production incentive. Such a system could be implemented on existing billing software systems, would not require additional resources to administer, and is likely to find acceptance among hospitalists, because it is something most are already accustomed to.
Current efforts to pay physicians based on value are facing substantial headwinds. The Value-Based Payment Modifier has been criticized for being too complex, while the Physician Quality Reporting System, in place since 2007, has been plagued by a “dismal” adoption rate by physicians and has been noted to “reflect a vanishingly small part of professional activities in most clinical specialties.”3 The time may be right to rethink physician value-based payment and integrate it into the existing, time-honored RVU payment system.
Dr. Whitcomb is Chief Medical Officer of Remedy Partners. He is co-founder and past president of SHM. Email him at wfwhit@comcast.net.
References
- Landon BE. Keeping score under a global payment system. N Engl J Med. 2012;366(5):393-395.
- Stecker EC, Schroeder SA. Adding value to relative-value units. N Engl J Med. 2013;369(23):2176-2179.
- Berenson RA, Kaye DR. Grading a physician’s value — the misapplication of performance measurement. N Engl J Med. 2013;369(22):2079-2078.
The move to paying hospitals and physicians based on value instead of volume is well underway. As programs ultimately designed to offer a global payment for a population (ACOs) or an episode of care (bundled payment) expand, we are left with this paradox: How do we reward physicians for working harder and seeing more patients under a global payment system that encourages physicians and hospitals to do less?
It appears that the existing fee-for-service payment system will need to form the scaffolding of any new, value-based system. Physicians must document the services they provide, leaving a “footprint” that can be recognized and rewarded. Without a record of the volume of services, physicians will have no incentive to see more patients during times of increased demand. This is what we often experience with straight-salary arrangements—physicians question why they should work harder for no additional compensation.
Through the ACO lens, Bruce Landon, professor of health care policy at Harvard Medical School, states the challenge in a different way: “The fundamental questions become how ACOs will divide their global budgets and how their physicians and service providers will be reimbursed. Thus, this system for determining who has earned what portion of payments—keeping score—is likely to be crucially important to the success of these new models of care.”1
In another article addressing value-based payment for physicians, Eric Stecker, MD, MPH, and Steve Schroeder, MD, argue that, due to their longevity and resilience, relative value units (RVUs), instead of physician-level capitation, straight salary, or salary with pay for performance incentives, should be the preferred mechanism to reimburse physicians based on value.2
I’d like to further develop the idea of an RVU-centric approach to value-based physician reimbursement, specifically discussing the case of hospitalists.
In Table 1, I provide examples of “value-based elements” to be added to an RVU reimbursement system. I chose measures related to three hospital-based quality programs: readmission reduction, hospital-acquired conditions, and value-based purchasing; however, one could choose hospitalist-relevant quality measures from other programs, such as ACOs, meaningful use, outpatient quality reporting (for observation patients), bundled payments, or a broad range of other domains. I selected only process measures, because outcome measures such as mortality or readmission rates suffer from sample size that is too small and risk adjustment too inadequate to be applied to individual physician payment.
Drs. Stecker and Schroeder offer an observation that is especially important to hospitalists: “Although RVUs are traditionally used for episodes of care provided by individual clinicians for individual patients, activities linked to RVUs could be more broadly defined to include team-based and supervisory clinical activities as well.”2 In the table, I include “multidisciplinary discharge planning rounds” as a potential measure. One can envision other team-based or supervisory activities involving hospitalists collaborating with nurses, pharmacists, or case managers working on a catheter-UTI bundle, high-risk medication counseling, or readmission risk assessment—with each activity linked to RVUs.
The implementation of an RVU system incorporating quality measures would be aided by documentation templates in the electronic medical record, similar to templates emerging for care bundles like central line blood stream infection. Value-based RVUs would have challenges, such as the need to change the measures over time and the system gaming inherent in any incentive design. Details of implementing the program would need to be worked out, such as attributing measures to individual physicians/providers or limiting to one the number of times certain measures are fulfilled per hospitalization.
Once established, a value-based RVU system could replace the complex and variable physician compensation landscape that exists today. As has always been the case, an RVU system could form the basis of a production incentive. Such a system could be implemented on existing billing software systems, would not require additional resources to administer, and is likely to find acceptance among hospitalists, because it is something most are already accustomed to.
Current efforts to pay physicians based on value are facing substantial headwinds. The Value-Based Payment Modifier has been criticized for being too complex, while the Physician Quality Reporting System, in place since 2007, has been plagued by a “dismal” adoption rate by physicians and has been noted to “reflect a vanishingly small part of professional activities in most clinical specialties.”3 The time may be right to rethink physician value-based payment and integrate it into the existing, time-honored RVU payment system.
Dr. Whitcomb is Chief Medical Officer of Remedy Partners. He is co-founder and past president of SHM. Email him at wfwhit@comcast.net.
References
- Landon BE. Keeping score under a global payment system. N Engl J Med. 2012;366(5):393-395.
- Stecker EC, Schroeder SA. Adding value to relative-value units. N Engl J Med. 2013;369(23):2176-2179.
- Berenson RA, Kaye DR. Grading a physician’s value — the misapplication of performance measurement. N Engl J Med. 2013;369(22):2079-2078.
The move to paying hospitals and physicians based on value instead of volume is well underway. As programs ultimately designed to offer a global payment for a population (ACOs) or an episode of care (bundled payment) expand, we are left with this paradox: How do we reward physicians for working harder and seeing more patients under a global payment system that encourages physicians and hospitals to do less?
It appears that the existing fee-for-service payment system will need to form the scaffolding of any new, value-based system. Physicians must document the services they provide, leaving a “footprint” that can be recognized and rewarded. Without a record of the volume of services, physicians will have no incentive to see more patients during times of increased demand. This is what we often experience with straight-salary arrangements—physicians question why they should work harder for no additional compensation.
Through the ACO lens, Bruce Landon, professor of health care policy at Harvard Medical School, states the challenge in a different way: “The fundamental questions become how ACOs will divide their global budgets and how their physicians and service providers will be reimbursed. Thus, this system for determining who has earned what portion of payments—keeping score—is likely to be crucially important to the success of these new models of care.”1
In another article addressing value-based payment for physicians, Eric Stecker, MD, MPH, and Steve Schroeder, MD, argue that, due to their longevity and resilience, relative value units (RVUs), instead of physician-level capitation, straight salary, or salary with pay for performance incentives, should be the preferred mechanism to reimburse physicians based on value.2
I’d like to further develop the idea of an RVU-centric approach to value-based physician reimbursement, specifically discussing the case of hospitalists.
In Table 1, I provide examples of “value-based elements” to be added to an RVU reimbursement system. I chose measures related to three hospital-based quality programs: readmission reduction, hospital-acquired conditions, and value-based purchasing; however, one could choose hospitalist-relevant quality measures from other programs, such as ACOs, meaningful use, outpatient quality reporting (for observation patients), bundled payments, or a broad range of other domains. I selected only process measures, because outcome measures such as mortality or readmission rates suffer from sample size that is too small and risk adjustment too inadequate to be applied to individual physician payment.
Drs. Stecker and Schroeder offer an observation that is especially important to hospitalists: “Although RVUs are traditionally used for episodes of care provided by individual clinicians for individual patients, activities linked to RVUs could be more broadly defined to include team-based and supervisory clinical activities as well.”2 In the table, I include “multidisciplinary discharge planning rounds” as a potential measure. One can envision other team-based or supervisory activities involving hospitalists collaborating with nurses, pharmacists, or case managers working on a catheter-UTI bundle, high-risk medication counseling, or readmission risk assessment—with each activity linked to RVUs.
The implementation of an RVU system incorporating quality measures would be aided by documentation templates in the electronic medical record, similar to templates emerging for care bundles like central line blood stream infection. Value-based RVUs would have challenges, such as the need to change the measures over time and the system gaming inherent in any incentive design. Details of implementing the program would need to be worked out, such as attributing measures to individual physicians/providers or limiting to one the number of times certain measures are fulfilled per hospitalization.
Once established, a value-based RVU system could replace the complex and variable physician compensation landscape that exists today. As has always been the case, an RVU system could form the basis of a production incentive. Such a system could be implemented on existing billing software systems, would not require additional resources to administer, and is likely to find acceptance among hospitalists, because it is something most are already accustomed to.
Current efforts to pay physicians based on value are facing substantial headwinds. The Value-Based Payment Modifier has been criticized for being too complex, while the Physician Quality Reporting System, in place since 2007, has been plagued by a “dismal” adoption rate by physicians and has been noted to “reflect a vanishingly small part of professional activities in most clinical specialties.”3 The time may be right to rethink physician value-based payment and integrate it into the existing, time-honored RVU payment system.
Dr. Whitcomb is Chief Medical Officer of Remedy Partners. He is co-founder and past president of SHM. Email him at wfwhit@comcast.net.
References
- Landon BE. Keeping score under a global payment system. N Engl J Med. 2012;366(5):393-395.
- Stecker EC, Schroeder SA. Adding value to relative-value units. N Engl J Med. 2013;369(23):2176-2179.
- Berenson RA, Kaye DR. Grading a physician’s value — the misapplication of performance measurement. N Engl J Med. 2013;369(22):2079-2078.
The confused binge drinker
CASE Paranoid and confused
Mr. P, age 46, presents to the emergency department (ED) with a chief complaint of feeling “very weird.” Although he has seen a number of psychiatrists in the past, he does not recall being given a specific diagnosis. He describes his feelings as “1 minute I am fine and the next minute I am confused.” He endorses feeling paranoid for the past 6 to 12 months and reports a history of passive suicidal ideations. On the day he presents to the ED, however, he has a specific plan to shoot himself. He does not report audiovisual hallucinations, but has noticed that he talks to himself often.
Mr. P reports feeling worthless at times. He has a history of manic symptoms, including decreased need for sleep and hypersexuality. He describes verbal and sexual abuse by his foster parents. Mr. P reports using Cannabis and opioids occasionally and to drinking every “now and then” but not every day. He denies using benzodiazepines. When he is evaluated, he is not taking any medication and has no significant medical problems. Mr. P reports a history of several hospitalizations, but he could not describe the reasons or timing of past admissions.
Mr. P has a 10th-grade education. He lives with his fiancée, who reports that he has been behaving oddly for some time. She noticed that he has memory problems and describes violent behavior, such as shaking his fist at her, breaking the television, and attempting to cut his throat once when he was “intoxicated.” She says she does not feel safe around him because of his labile mood and history of
aggression. She confirms that Mr. P does not drink daily but binge-drinks at times.
Initial mental status examination of evaluation reveals hyperverbal, rapid speech. Mr. P is circumstantial and tangential in his thought process. He has poor judgment and insight and exhibits suicidal ideations with a plan. Toxicology screening reveals a blood alcohol level of 50 mg/dL and is positive for Cannabis and opiates.
Which condition most likely accounts for Mr. P’s presentation?
a) bipolar disorder, currently manic
b) substance-induced mood disorder
c) cognitive disorder
d) delirium
TREATMENT Rapid improvement
From the ED, Mr. P was admitted to an inpatient psychiatric unit, where he was found initially to be disoriented to time, place, and person. His thought process remained disorganized and irrational, with significant memory difficulties. He is noted to have an unsteady gait. Nursing staff observes that Mr. P has significant difficulties with activities of daily living and requires assistance. He talks in circles
and uses nonsensical words.
His serum vitamin B12 level, folate level, rapid plasma reagin, magnesium level, and thiamine level are within normal limits; CT scan of the brain is unremarkable. Neuropsychological testing reveals significant and diffuse cognitive deficits suggestive of frontal lobe dysfunction. He is deemed to not have decision-making capacity; because he has no family, his fiancée is appointed as his temporary health care proxy.
Thiamine and lorazepam are prescribed as needed because of Mr. P’s history of alcohol abuse. However, it’s determined that he does not need lorazepam because his vital signs are stable and there is no evidence of alcohol withdrawal symptoms.
During the course of his 10-day hospitalization, Mr. P’s cognitive difficulties resolved. He regains orientation to time, place, and person. He gains skill in all his activities of daily living, to the point of independence, and is discharged with minimal supervision. Vitamin B supplementation is prescribed, with close follow up in an outpatient day program. MRI/SPECT scan is considered to rule out frontotemporal dementia as recommended by the results of his neurocognitive testing profile.
Which condition likely account for Mr. P’s presentation during inpatient hospitalization?
a) Wernicke’s encephalopathy
b) Korsakoff’s syndrome
c) malingering
d) frontotemporal dementia
e) a neurodegenerative disease
The author's observations
Mr. P’s fluctuating mental status, gait instability, and confabulation create high suspicion for Wernicke’s encephalopathy; his dramatic improvement with IV thiamine supports that diagnosis. Mr. P attends the outpatient day program once after his discharge, and is then lost to follow-up.
During inpatient stay, Mr. P eventually admits to binge drinking several times a week, and drinking early in the morning, which would continue throughout the day. His significant cognitive deficits revealed by neuropsychological testing suggests consideration of a differential diagnosis of multifactorial cognitive dysfunction because of:
• long-term substance use
• Korsakoff’s syndrome
• frontotemporal dementia
• a neurodegenerative disease
• malingering (Table 1).
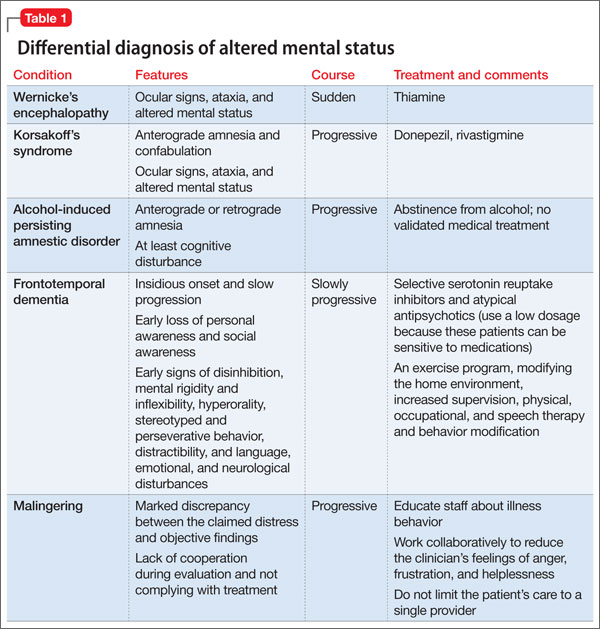
Wernicke’s encephalopathy
Wernicke’s encephalopathy is a life-threatening neurologic disorder caused by thiamine deficiency. The disease is rare, catastrophic in onset, and clinically complex1; as in Mr. P’s case, diagnosis often is delayed. In autopsy studies, the reported prevalence of Wernicke’s encephalopathy is 0.4% to 2.8%.1 Wernicke’s encephalopathy was suspected before death in 33% of alcohol-dependent patientsand 6% of nonalcoholics.1 Other causes of Wernicke’s encephalopathy include cancer, gastrointestinal surgery, hyperemesis gravidarum, a starvation or malnutrition state, GI tract disease, AIDS, parenteral nutrition, repetitive vomiting, and infection.1
Diagnosis. Making the correct diagnosis is challenging because the clinical presentation can be variable. No lab or imaging studies confirm the diagnosis. The triad of signs considered to support the diagnosis include ocular signs such as nystagmus, cerebellar signs, and confusion. These signs occur in only 8% to 10% of patients in whom the diagnosis likely.1,2
Attempts to increase the likelihood of making an accurate lifetime diagnosis of
Wernicke’s encephalopathy include expanding the focus to 8 clinical domains:
• dietary deficiency
• eye signs
• cerebellar signs
• seizures
• frontal lobe dysfunction
• amnesia
• mild memory impairment
• altered mental status.1
The sensitivity of making a correct diagnosis increases to 85% if at least 2 of 4 features—namely dietary deficiency, eye signs, cerebellar signs, memory impairment, and altered mental status—are present. These criteria can be applied to alcoholic and nonalcoholic patients.1Table 23 lists common and uncommon symptoms of Wernicke’s encephalopathy.
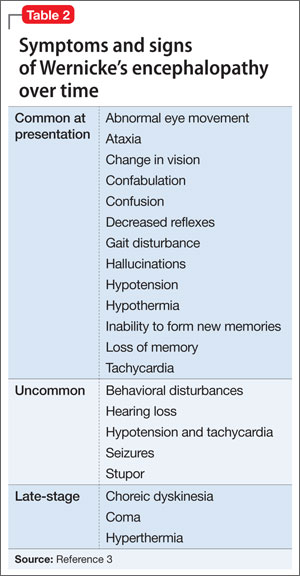
Although CT scan of the brain is not a reliable test for the disorder, MRI can be powerful tool that could support a diagnosis of acute Wernicke’s encephalopathy.1 We did not consider MRI in Mr. P’s case because the consulting neurologist thought this was unnecessary because of the quick improvement in his cognitive status with IV thiamine—although MRI might have helped to detect the disease earlier. In some studies, brain MRI revealed lesions in two-thirds of Wernicke’s encephalopathy patients.1 Typically, lesions are symmetrical and seen in the thalamus, mammillary body, and periaqueductal areas.1,4 Atypical lesions commonly are seen in the cerebellum, dentate nuclei, caudate nucleus, and cerebral cortex.1
Treatment. Evidence supports use of IV thiamine, 200 mg 3 times a day, when the disease is suspected or established.1,2 Thiamine has been associated with sporadic anaphylactic reactions, and should be administered when resuscitation facilities are available. Do not delay treatment because resuscitation measures are unavailable because you risk causing irreversible brain damage.1
In Mr. P’s case, prompt recognition of the need for thiamine likely led to a better outcome. Thiamine supplementation can prevent Wernicke’s encephalopathy in some patients. Prophylactic parenteral administration of thiamine before administration of glucose in the ED is recommended, as well as vitamin B supplementation with thiamine included upon discharge.1,2 Studies support several treatment regimens for patients with Wernicke’s encephalopathy and those at risk of it.1,3,5
Neither the optimal dosage of thiamine nor the appropriate duration of treatment have been determined by randomized, double-blind, controlled studies; empirical clinical practice and recommendations by Royal College of Physicians, London, suggest that a more prolonged course of thiamine—administered as long as improvement continues—might be beneficial.6
Left untreated, Wernicke’s encephalopathy can lead to irreversible brain damage.2
Mortality has been reported as 17% to 20%; 82% of patients develop Korsakoff’s syndrome, a chronic condition characterized by short-term memory loss. One-quarter of patients who develop Korsakoff’s syndrome require long-term residential care because of permanent brain damage.2
Making a diagnosis of Wernicke’s encephalopathy is a challenge because no specific symptom or diagnostic test can be relied upon to confirm the diagnosis. Also, patients might deny that they have an alcohol problem or give an inaccurate history of their alcohol use,2 as Mr. P did. The disorder is substantially underdiagnosed; as a consequence, patients are at risk of brain damage.2
Bottom Line
Not all patients who present with aggressive behavior, mania, and psychiatric
symptoms have a primary psychiatric diagnosis. It is important to consider
nutritional deficiencies caused by chronic alcohol abuse in patients presenting
with acute onset of confusion or altered mental status. Wernicke’s encephalopathy
might be the result of alcohol abuse and can be treated with IV thiamine.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Galvin R, Bråthen G, Ivashynka A, et al; EFNS. Guidelines for diagnosis, therapy and prevention of Wernicke’s encephalopathy. Eur J Neurol. 2010;17(12):
1408-1418.
2. Robinson K. Wernicke’s encephalopathy. Emerg Nurse. 2003;11(5):30-33.
3. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442-455.
4. Celik Y, Kaya M. Brain SPECT findings in Wernicke’s encephalopathy. Neurol Sci. 2004;25(1):23-26.
5. Thomson AD, Guerrini I, Marshall JE. Wernicke’s encephalopathy: role of thiamine. Practical Gastroenterology. 2009;33(6):21-30.
6. Thomson AD, Cook CCH, Guerrini I, et al. Wernicke’s encephalopathy: ‘plus ca change, plus c’est la meme chose’. Alcohol Alcohol. 2008;43:180-186.
CASE Paranoid and confused
Mr. P, age 46, presents to the emergency department (ED) with a chief complaint of feeling “very weird.” Although he has seen a number of psychiatrists in the past, he does not recall being given a specific diagnosis. He describes his feelings as “1 minute I am fine and the next minute I am confused.” He endorses feeling paranoid for the past 6 to 12 months and reports a history of passive suicidal ideations. On the day he presents to the ED, however, he has a specific plan to shoot himself. He does not report audiovisual hallucinations, but has noticed that he talks to himself often.
Mr. P reports feeling worthless at times. He has a history of manic symptoms, including decreased need for sleep and hypersexuality. He describes verbal and sexual abuse by his foster parents. Mr. P reports using Cannabis and opioids occasionally and to drinking every “now and then” but not every day. He denies using benzodiazepines. When he is evaluated, he is not taking any medication and has no significant medical problems. Mr. P reports a history of several hospitalizations, but he could not describe the reasons or timing of past admissions.
Mr. P has a 10th-grade education. He lives with his fiancée, who reports that he has been behaving oddly for some time. She noticed that he has memory problems and describes violent behavior, such as shaking his fist at her, breaking the television, and attempting to cut his throat once when he was “intoxicated.” She says she does not feel safe around him because of his labile mood and history of
aggression. She confirms that Mr. P does not drink daily but binge-drinks at times.
Initial mental status examination of evaluation reveals hyperverbal, rapid speech. Mr. P is circumstantial and tangential in his thought process. He has poor judgment and insight and exhibits suicidal ideations with a plan. Toxicology screening reveals a blood alcohol level of 50 mg/dL and is positive for Cannabis and opiates.
Which condition most likely accounts for Mr. P’s presentation?
a) bipolar disorder, currently manic
b) substance-induced mood disorder
c) cognitive disorder
d) delirium
TREATMENT Rapid improvement
From the ED, Mr. P was admitted to an inpatient psychiatric unit, where he was found initially to be disoriented to time, place, and person. His thought process remained disorganized and irrational, with significant memory difficulties. He is noted to have an unsteady gait. Nursing staff observes that Mr. P has significant difficulties with activities of daily living and requires assistance. He talks in circles
and uses nonsensical words.
His serum vitamin B12 level, folate level, rapid plasma reagin, magnesium level, and thiamine level are within normal limits; CT scan of the brain is unremarkable. Neuropsychological testing reveals significant and diffuse cognitive deficits suggestive of frontal lobe dysfunction. He is deemed to not have decision-making capacity; because he has no family, his fiancée is appointed as his temporary health care proxy.
Thiamine and lorazepam are prescribed as needed because of Mr. P’s history of alcohol abuse. However, it’s determined that he does not need lorazepam because his vital signs are stable and there is no evidence of alcohol withdrawal symptoms.
During the course of his 10-day hospitalization, Mr. P’s cognitive difficulties resolved. He regains orientation to time, place, and person. He gains skill in all his activities of daily living, to the point of independence, and is discharged with minimal supervision. Vitamin B supplementation is prescribed, with close follow up in an outpatient day program. MRI/SPECT scan is considered to rule out frontotemporal dementia as recommended by the results of his neurocognitive testing profile.
Which condition likely account for Mr. P’s presentation during inpatient hospitalization?
a) Wernicke’s encephalopathy
b) Korsakoff’s syndrome
c) malingering
d) frontotemporal dementia
e) a neurodegenerative disease
The author's observations
Mr. P’s fluctuating mental status, gait instability, and confabulation create high suspicion for Wernicke’s encephalopathy; his dramatic improvement with IV thiamine supports that diagnosis. Mr. P attends the outpatient day program once after his discharge, and is then lost to follow-up.
During inpatient stay, Mr. P eventually admits to binge drinking several times a week, and drinking early in the morning, which would continue throughout the day. His significant cognitive deficits revealed by neuropsychological testing suggests consideration of a differential diagnosis of multifactorial cognitive dysfunction because of:
• long-term substance use
• Korsakoff’s syndrome
• frontotemporal dementia
• a neurodegenerative disease
• malingering (Table 1).
Wernicke’s encephalopathy
Wernicke’s encephalopathy is a life-threatening neurologic disorder caused by thiamine deficiency. The disease is rare, catastrophic in onset, and clinically complex1; as in Mr. P’s case, diagnosis often is delayed. In autopsy studies, the reported prevalence of Wernicke’s encephalopathy is 0.4% to 2.8%.1 Wernicke’s encephalopathy was suspected before death in 33% of alcohol-dependent patientsand 6% of nonalcoholics.1 Other causes of Wernicke’s encephalopathy include cancer, gastrointestinal surgery, hyperemesis gravidarum, a starvation or malnutrition state, GI tract disease, AIDS, parenteral nutrition, repetitive vomiting, and infection.1
Diagnosis. Making the correct diagnosis is challenging because the clinical presentation can be variable. No lab or imaging studies confirm the diagnosis. The triad of signs considered to support the diagnosis include ocular signs such as nystagmus, cerebellar signs, and confusion. These signs occur in only 8% to 10% of patients in whom the diagnosis likely.1,2
Attempts to increase the likelihood of making an accurate lifetime diagnosis of
Wernicke’s encephalopathy include expanding the focus to 8 clinical domains:
• dietary deficiency
• eye signs
• cerebellar signs
• seizures
• frontal lobe dysfunction
• amnesia
• mild memory impairment
• altered mental status.1
The sensitivity of making a correct diagnosis increases to 85% if at least 2 of 4 features—namely dietary deficiency, eye signs, cerebellar signs, memory impairment, and altered mental status—are present. These criteria can be applied to alcoholic and nonalcoholic patients.1Table 23 lists common and uncommon symptoms of Wernicke’s encephalopathy.
Although CT scan of the brain is not a reliable test for the disorder, MRI can be powerful tool that could support a diagnosis of acute Wernicke’s encephalopathy.1 We did not consider MRI in Mr. P’s case because the consulting neurologist thought this was unnecessary because of the quick improvement in his cognitive status with IV thiamine—although MRI might have helped to detect the disease earlier. In some studies, brain MRI revealed lesions in two-thirds of Wernicke’s encephalopathy patients.1 Typically, lesions are symmetrical and seen in the thalamus, mammillary body, and periaqueductal areas.1,4 Atypical lesions commonly are seen in the cerebellum, dentate nuclei, caudate nucleus, and cerebral cortex.1
Treatment. Evidence supports use of IV thiamine, 200 mg 3 times a day, when the disease is suspected or established.1,2 Thiamine has been associated with sporadic anaphylactic reactions, and should be administered when resuscitation facilities are available. Do not delay treatment because resuscitation measures are unavailable because you risk causing irreversible brain damage.1
In Mr. P’s case, prompt recognition of the need for thiamine likely led to a better outcome. Thiamine supplementation can prevent Wernicke’s encephalopathy in some patients. Prophylactic parenteral administration of thiamine before administration of glucose in the ED is recommended, as well as vitamin B supplementation with thiamine included upon discharge.1,2 Studies support several treatment regimens for patients with Wernicke’s encephalopathy and those at risk of it.1,3,5
Neither the optimal dosage of thiamine nor the appropriate duration of treatment have been determined by randomized, double-blind, controlled studies; empirical clinical practice and recommendations by Royal College of Physicians, London, suggest that a more prolonged course of thiamine—administered as long as improvement continues—might be beneficial.6
Left untreated, Wernicke’s encephalopathy can lead to irreversible brain damage.2
Mortality has been reported as 17% to 20%; 82% of patients develop Korsakoff’s syndrome, a chronic condition characterized by short-term memory loss. One-quarter of patients who develop Korsakoff’s syndrome require long-term residential care because of permanent brain damage.2
Making a diagnosis of Wernicke’s encephalopathy is a challenge because no specific symptom or diagnostic test can be relied upon to confirm the diagnosis. Also, patients might deny that they have an alcohol problem or give an inaccurate history of their alcohol use,2 as Mr. P did. The disorder is substantially underdiagnosed; as a consequence, patients are at risk of brain damage.2
Bottom Line
Not all patients who present with aggressive behavior, mania, and psychiatric
symptoms have a primary psychiatric diagnosis. It is important to consider
nutritional deficiencies caused by chronic alcohol abuse in patients presenting
with acute onset of confusion or altered mental status. Wernicke’s encephalopathy
might be the result of alcohol abuse and can be treated with IV thiamine.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE Paranoid and confused
Mr. P, age 46, presents to the emergency department (ED) with a chief complaint of feeling “very weird.” Although he has seen a number of psychiatrists in the past, he does not recall being given a specific diagnosis. He describes his feelings as “1 minute I am fine and the next minute I am confused.” He endorses feeling paranoid for the past 6 to 12 months and reports a history of passive suicidal ideations. On the day he presents to the ED, however, he has a specific plan to shoot himself. He does not report audiovisual hallucinations, but has noticed that he talks to himself often.
Mr. P reports feeling worthless at times. He has a history of manic symptoms, including decreased need for sleep and hypersexuality. He describes verbal and sexual abuse by his foster parents. Mr. P reports using Cannabis and opioids occasionally and to drinking every “now and then” but not every day. He denies using benzodiazepines. When he is evaluated, he is not taking any medication and has no significant medical problems. Mr. P reports a history of several hospitalizations, but he could not describe the reasons or timing of past admissions.
Mr. P has a 10th-grade education. He lives with his fiancée, who reports that he has been behaving oddly for some time. She noticed that he has memory problems and describes violent behavior, such as shaking his fist at her, breaking the television, and attempting to cut his throat once when he was “intoxicated.” She says she does not feel safe around him because of his labile mood and history of
aggression. She confirms that Mr. P does not drink daily but binge-drinks at times.
Initial mental status examination of evaluation reveals hyperverbal, rapid speech. Mr. P is circumstantial and tangential in his thought process. He has poor judgment and insight and exhibits suicidal ideations with a plan. Toxicology screening reveals a blood alcohol level of 50 mg/dL and is positive for Cannabis and opiates.
Which condition most likely accounts for Mr. P’s presentation?
a) bipolar disorder, currently manic
b) substance-induced mood disorder
c) cognitive disorder
d) delirium
TREATMENT Rapid improvement
From the ED, Mr. P was admitted to an inpatient psychiatric unit, where he was found initially to be disoriented to time, place, and person. His thought process remained disorganized and irrational, with significant memory difficulties. He is noted to have an unsteady gait. Nursing staff observes that Mr. P has significant difficulties with activities of daily living and requires assistance. He talks in circles
and uses nonsensical words.
His serum vitamin B12 level, folate level, rapid plasma reagin, magnesium level, and thiamine level are within normal limits; CT scan of the brain is unremarkable. Neuropsychological testing reveals significant and diffuse cognitive deficits suggestive of frontal lobe dysfunction. He is deemed to not have decision-making capacity; because he has no family, his fiancée is appointed as his temporary health care proxy.
Thiamine and lorazepam are prescribed as needed because of Mr. P’s history of alcohol abuse. However, it’s determined that he does not need lorazepam because his vital signs are stable and there is no evidence of alcohol withdrawal symptoms.
During the course of his 10-day hospitalization, Mr. P’s cognitive difficulties resolved. He regains orientation to time, place, and person. He gains skill in all his activities of daily living, to the point of independence, and is discharged with minimal supervision. Vitamin B supplementation is prescribed, with close follow up in an outpatient day program. MRI/SPECT scan is considered to rule out frontotemporal dementia as recommended by the results of his neurocognitive testing profile.
Which condition likely account for Mr. P’s presentation during inpatient hospitalization?
a) Wernicke’s encephalopathy
b) Korsakoff’s syndrome
c) malingering
d) frontotemporal dementia
e) a neurodegenerative disease
The author's observations
Mr. P’s fluctuating mental status, gait instability, and confabulation create high suspicion for Wernicke’s encephalopathy; his dramatic improvement with IV thiamine supports that diagnosis. Mr. P attends the outpatient day program once after his discharge, and is then lost to follow-up.
During inpatient stay, Mr. P eventually admits to binge drinking several times a week, and drinking early in the morning, which would continue throughout the day. His significant cognitive deficits revealed by neuropsychological testing suggests consideration of a differential diagnosis of multifactorial cognitive dysfunction because of:
• long-term substance use
• Korsakoff’s syndrome
• frontotemporal dementia
• a neurodegenerative disease
• malingering (Table 1).
Wernicke’s encephalopathy
Wernicke’s encephalopathy is a life-threatening neurologic disorder caused by thiamine deficiency. The disease is rare, catastrophic in onset, and clinically complex1; as in Mr. P’s case, diagnosis often is delayed. In autopsy studies, the reported prevalence of Wernicke’s encephalopathy is 0.4% to 2.8%.1 Wernicke’s encephalopathy was suspected before death in 33% of alcohol-dependent patientsand 6% of nonalcoholics.1 Other causes of Wernicke’s encephalopathy include cancer, gastrointestinal surgery, hyperemesis gravidarum, a starvation or malnutrition state, GI tract disease, AIDS, parenteral nutrition, repetitive vomiting, and infection.1
Diagnosis. Making the correct diagnosis is challenging because the clinical presentation can be variable. No lab or imaging studies confirm the diagnosis. The triad of signs considered to support the diagnosis include ocular signs such as nystagmus, cerebellar signs, and confusion. These signs occur in only 8% to 10% of patients in whom the diagnosis likely.1,2
Attempts to increase the likelihood of making an accurate lifetime diagnosis of
Wernicke’s encephalopathy include expanding the focus to 8 clinical domains:
• dietary deficiency
• eye signs
• cerebellar signs
• seizures
• frontal lobe dysfunction
• amnesia
• mild memory impairment
• altered mental status.1
The sensitivity of making a correct diagnosis increases to 85% if at least 2 of 4 features—namely dietary deficiency, eye signs, cerebellar signs, memory impairment, and altered mental status—are present. These criteria can be applied to alcoholic and nonalcoholic patients.1Table 23 lists common and uncommon symptoms of Wernicke’s encephalopathy.
Although CT scan of the brain is not a reliable test for the disorder, MRI can be powerful tool that could support a diagnosis of acute Wernicke’s encephalopathy.1 We did not consider MRI in Mr. P’s case because the consulting neurologist thought this was unnecessary because of the quick improvement in his cognitive status with IV thiamine—although MRI might have helped to detect the disease earlier. In some studies, brain MRI revealed lesions in two-thirds of Wernicke’s encephalopathy patients.1 Typically, lesions are symmetrical and seen in the thalamus, mammillary body, and periaqueductal areas.1,4 Atypical lesions commonly are seen in the cerebellum, dentate nuclei, caudate nucleus, and cerebral cortex.1
Treatment. Evidence supports use of IV thiamine, 200 mg 3 times a day, when the disease is suspected or established.1,2 Thiamine has been associated with sporadic anaphylactic reactions, and should be administered when resuscitation facilities are available. Do not delay treatment because resuscitation measures are unavailable because you risk causing irreversible brain damage.1
In Mr. P’s case, prompt recognition of the need for thiamine likely led to a better outcome. Thiamine supplementation can prevent Wernicke’s encephalopathy in some patients. Prophylactic parenteral administration of thiamine before administration of glucose in the ED is recommended, as well as vitamin B supplementation with thiamine included upon discharge.1,2 Studies support several treatment regimens for patients with Wernicke’s encephalopathy and those at risk of it.1,3,5
Neither the optimal dosage of thiamine nor the appropriate duration of treatment have been determined by randomized, double-blind, controlled studies; empirical clinical practice and recommendations by Royal College of Physicians, London, suggest that a more prolonged course of thiamine—administered as long as improvement continues—might be beneficial.6
Left untreated, Wernicke’s encephalopathy can lead to irreversible brain damage.2
Mortality has been reported as 17% to 20%; 82% of patients develop Korsakoff’s syndrome, a chronic condition characterized by short-term memory loss. One-quarter of patients who develop Korsakoff’s syndrome require long-term residential care because of permanent brain damage.2
Making a diagnosis of Wernicke’s encephalopathy is a challenge because no specific symptom or diagnostic test can be relied upon to confirm the diagnosis. Also, patients might deny that they have an alcohol problem or give an inaccurate history of their alcohol use,2 as Mr. P did. The disorder is substantially underdiagnosed; as a consequence, patients are at risk of brain damage.2
Bottom Line
Not all patients who present with aggressive behavior, mania, and psychiatric
symptoms have a primary psychiatric diagnosis. It is important to consider
nutritional deficiencies caused by chronic alcohol abuse in patients presenting
with acute onset of confusion or altered mental status. Wernicke’s encephalopathy
might be the result of alcohol abuse and can be treated with IV thiamine.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Galvin R, Bråthen G, Ivashynka A, et al; EFNS. Guidelines for diagnosis, therapy and prevention of Wernicke’s encephalopathy. Eur J Neurol. 2010;17(12):
1408-1418.
2. Robinson K. Wernicke’s encephalopathy. Emerg Nurse. 2003;11(5):30-33.
3. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442-455.
4. Celik Y, Kaya M. Brain SPECT findings in Wernicke’s encephalopathy. Neurol Sci. 2004;25(1):23-26.
5. Thomson AD, Guerrini I, Marshall JE. Wernicke’s encephalopathy: role of thiamine. Practical Gastroenterology. 2009;33(6):21-30.
6. Thomson AD, Cook CCH, Guerrini I, et al. Wernicke’s encephalopathy: ‘plus ca change, plus c’est la meme chose’. Alcohol Alcohol. 2008;43:180-186.
1. Galvin R, Bråthen G, Ivashynka A, et al; EFNS. Guidelines for diagnosis, therapy and prevention of Wernicke’s encephalopathy. Eur J Neurol. 2010;17(12):
1408-1418.
2. Robinson K. Wernicke’s encephalopathy. Emerg Nurse. 2003;11(5):30-33.
3. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442-455.
4. Celik Y, Kaya M. Brain SPECT findings in Wernicke’s encephalopathy. Neurol Sci. 2004;25(1):23-26.
5. Thomson AD, Guerrini I, Marshall JE. Wernicke’s encephalopathy: role of thiamine. Practical Gastroenterology. 2009;33(6):21-30.
6. Thomson AD, Cook CCH, Guerrini I, et al. Wernicke’s encephalopathy: ‘plus ca change, plus c’est la meme chose’. Alcohol Alcohol. 2008;43:180-186.
Are the people we serve ‘patients’ or ‘customers’?
Dear Dr. Mossman,
At the multispecialty hospital where I work, administrators refer to patients as “customers” and tell us that, by improving “the customer experience,” we can reduce complaints and avoid malpractice suits. This business lingo offends me. Doesn’t providing good care do more to prevent malpractice claims than calling sick patients “customers”?
Submitted by “Dr. H”
“All words are pegs to hang ideas on.” As was true when Reverend Henry Ward Beecher uttered this phrase in the 19th century,1 names affect how we relate to and feel about people. Many doctors don’t think of themselves as “selling” services, and they find calling patients “customers” distasteful.
But for at least 4 decades, mental health professionals themselves have used a “customer approach” to think about certain aspects of psychiatrist–patient encounters.2 More pertinent to Dr. H’s questions, many attorneys who advise physicians are convinced that giving patients a satisfying “customer experience” is a sound strategy for reducing the risk of malpractice litigation.3
If the attorneys are right, taking a customer service perspective can lower the likelihood that psychiatrists will be sued. To understand why, this article looks at:
• terms for referring to health care recipients
• the feelings those terms generate
• how the “customer service” perspective has become a malpractice prevention
strategy.
Off-putting connotations
All the currently used ways of referring to persons served by doctors have off-putting features.
The word “patient” dates back to the 14th century and comes from Latin present
participles of pati, “to suffer.” Although Alpha Omega Alpha’s motto—“be worthy
to serve the suffering”4—expresses doctors’ commitment to help others, “patient”
carries emotional baggage. A “patient” is “a sick individual” who seeks treatment
from a physician,5 a circumstance that most people (including doctors) find unpleasant and hope is only temporary. The adjective “patient” means “bearing pains or trials calmly or without complaint” and “manifesting forbearance under provocation or strain,”5 phrases associated with passivity, deference, and a long wait to see the doctor.
Because “patient” evokes notions of helplessness and need for direction, non-medical psychotherapists often use “client” to designate care recipients. “Client” has the same Latin root as “to lean” and refers to someone “under the protection of another.” More pertinent to discussions of mental health care, a “client” also is “a person who pays a professional person or organization for services” or “a customer.”5 The latter definition explains what makes “client” feel wrong to medical practitioners, who regard those they treat as deserving more compassion and sacrifice than someone who simply purchases professional services.
“Consumer,” a word of French origin derived from the Latin consumere (“to take
up”), refers to “a person who buys goods and services.”5 If “consumers” are buyers, then those from whom they make purchases are merchants or sellers. Western marketplace concepts often regard consumers as sovereign judges of their needs, and the role of commodity producers is to try to satisfy those needs.6
The problem with viewing health care recipients this way is that sellers don’t caution customers about buying things when only principles of supply-and-demand govern exchange relationships.7 Quite the contrary: producers sometimes promote their products through “advertising [that] distorts reality and creates artificial needs to make profit for a firm.”8 If physicians behave this way, however, they get criticism and deserve it.
A “customer” in 15th-century Middle English was a tax collector, but in modern
usage, a customer is someone who, like a consumer, “purchases some commodity or service.”5 By the early 20th century, “customer” became associated with notions of empowerment embodied in the merchants’ credo, “The customer is always right.”9 Chronic illnesses often require self-management and collaboration with those labeled the “givers” and “recipients” of medical care. Research shows that “patients are more trusting of, and committed to, physicians who adopt an empowering communication style with them,” which suggests “that empowering
patients presents a means to improve the patient–physician relationship.”10
Feelings about names
People have strong feelings about what they are called. In opposing calling patients “consumers,” Nobel Prize-winning economist Paul Krugman explains: “Medical care is an area in which crucial decisions—life and death decisions—must be made; yet making those decisions intelligently requires a vast amount of specialized knowledge; and often those decisions must also be made under conditions in which the patient …needs action immediately, with no time for discussion, let alone comparison shopping. …That’s why doctors have traditionally…been expected to behave according to higher standards than the average professional…The idea that all this can be reduced to money—that doctors are just people selling services to consumers of health care—is, well, sickening.”11
Less famous recipients of nonpsychiatric medical services also prefer being called
“patients” over “clients” or “consumers.”12-14 Recipients of mental health services have a different view, however. In some surveys, “patient” gets a plurality or majority of service recipients’ votes,15,16 but in others, recipients prefer to be called “clients” or other terms.17,18 Of note, people who prefer being called “patients” tend to strongly dislike being called “clients.”19 On the professional
side, psychiatrists—along with other physicians—prefer to speak of treating “patients” and to criticize letting economic phrases infect medical discourse.20-22
Names: A practical difference?
Does what psychiatrists call those they serve make any practical difference? Perhaps not, but evidence suggests that the attitudes that doctors take toward patients affects economic success and malpractice risk.
When they have choices about where they can seek health care, medical patients value physicians’ competence, but they also consider nonclinical factors such as family members’ opinions and convenience.23 Knowing this, the federal government’s Centers for Medicare & Medicaid Services publishes results from its Hospital Consumer Assessment of Healthcare Providers and Systems to “create incentives for hospitals to improve their quality of care.”24
Nonclinical factors play a big part in patients’ decisions about suing their doctors, too. Many malpractice claims turn out to be groundless in the sense that they do not involve medical errors,25 and most errors that result in injury do not lead to malpractice suits.26
What explains this disparity? Often when a lawsuit is filed, whatever injury may have occurred is coupled with an aggravating factor, such as a communication gaffe,27 a physician’s domineering tone of voice,28 or failure to acknowledge error.29 The lower a physician’s patient satisfaction ratings, the higher the physician’s likelihood of receiving complaints and getting sued for malpractice.30,31
These kinds of considerations probably lie behind the recommendation of one hospital manager to doctors: “Continue to call them patients but treat them like
customers.”32 More insights into this view come from responses solicited from Yale
students, staff members, and alumni about whether it seems preferable to be a “patient” or a “customer” (Box).33

Bottom Line
When patients get injured during medical care, evidence suggests that how they feel about their doctors makes a big difference in whether they decide to file suit. If you’re like most psychiatrists, you prefer to call persons whom you treat “patients.” But watching and improving the things that affect your patients’ “customer experience” may help you avoid malpractice litigation.
Related Resource
• Goldhill D. To fix healthcare, turn patients into customers. Bloomberg Personal Finance. www.bloomberg.com/news/2013-01-03/to-fix-health-care-turn-patients-intocustomers.html.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Beecher HW, Drysdale W. Proverbs from Plymouth pulpit. New York, NY: D. Appleton & Co.;1887.
2. Lazare A, Eisenthal S, Wasserman L. The customer approach to patienthood: attending to patient requests in a walk-in clinic. Arch Gen Psychiatry. 1975;32:553-558.
3. Schleiter KE. Difficult patient-physician relationships and the risk of medical malpractice litigation. Virtual Mentor. 2009;11:242-246.
4. Alpha Omega Alpha Honor Medical Society. Alpha Omega Alpha constitution. http://www.alphaomegaalpha.org/constitution.html. Accessed December 13, 2013. Accessed December 13, 2013.
5. Merriam-Webster. Dictionary. http://www.merriamwebster.com. Accessed December 9, 2013.
6. Kotler P, Burton S, Deans K, et al. Marketing, 9th ed. Frenchs Forest, Australia: Pearson Education Australia; 2013.
7. Deber RB. Getting what we pay for: myths and realities about financing Canada’s health care system. Health Law Can. 2000;21(2):9-56.
8. Takala T, Uusitalo O. An alternative view of relationship marketing: a framework for ethical analysis. Eur J Mark. 1996;30:45-60.
9. Van Vuren FS. The Yankee who taught Britishers that ‘the customer is always right.’ Milwaukee Journal. http://www.wisconsinhistory.org/wlhba/articleView.
asp?pg=1&id=11176. Published September 9, 1932. Accessed December 20, 2013.
10. Ouschan T, Sweeney J, Johnson L. Customer empowerment and relationship outcomes in healthcare consultations. Eur J Mark. 2006;40:1068-1086.
11. Krugman P. Patients are not consumers. The New York Times. http://krugman.blogs.nytimes.com/2011/04/20/patients-are-not-consumers. Published April 20, 2011. Accessed December 13, 2013.
12. Nair BR. Patient, client or customer? Med J Aust. 1998;169:593.
13. Wing PC. Patient or client? If in doubt, ask. CMAJ. 1997;157:287-289.
14. Deber RB, Kraetschmer N, Urowitz S, et al. Patient, consumer, client, or customer: what do people want to be called? Health Expect. 2005;8(4):345-351.
15. Sharma V, Whitney D, Kazarian SS, et al. Preferred terms for users of mental health services among service providers and recipients. Psychiatr Serv. 2000;51(2): 203-209.
16. Simmons P, Hawley CJ, Gale TM, et al. Service user, patient, client, user or survivor: describing recipients of mental health services. Psychiatrist. 2010;34:20-23.
17. Lloyd C, King R, Bassett H, et al. Patient, client or consumer? A survey of preferred terms. Australas Psychiatry. 2001; 9(4):321-324.
18. Covell NH, McCorkle BH, Weissman EM, et al. What’s in a name? Terms preferred by service recipients. Adm Policy Ment Health. 2007;34(5):443-447.
19. Ritchie CW, Hayes D, Ames DJ. Patient or client? The opinions of people attending a psychiatric clinic. Psychiatrist. 2000;24(12):447-450.
20. Andreasen NC. Clients, consumers, providers, and products: where will it all end? Am J Psychiatry. 1995;152:1107-1109.
21. Editorial. What’s in a name? Lancet. 2000;356(9248):2111.
22. Torrey EF. Patients, clients, consumers, survivors et al: what’s in a name? Schizophr Bull. 2011;37(3):466-468.
23. Wilson CT, Woloshin S, Schwartz L. Choosing where to have major surgery: who makes the decision? Arch Surg. 2007;142(3):242-246.
24. Centers for Medicare & Medicaid Services. Hospital consumer assessment of healthcare providers and systems. http://www.hcahpsonline.org. Accessed
January 26, 2014.
25. Studdert DM, Mello MM, Gawande AA, et al. Claims, errors, and compensation payments in medical malpractice litigation. N Engl J Med. 2006;354:2024-2033.
26. Localio AR, Lawthers AG, Brennan TA, et al. Relation between malpractice claims and adverse events due to negligence—results of the Harvard Medical Practice Study III. N Engl J Med. 1991;325:245-251.
27. Huntington B, Kuhn N. Communication gaffes: a root cause of malpractice claims. Bayl Univ Med Cent. 2003;16(2):157-161.
28. Ambady N, Laplante D, Nguyen T, et al. Surgeons’ tone of voice: a clue to malpractice history. Surgery. 2002;132(1):5-9.
29. Witman AB, Park DM, Hardin SB. How do patients want physicians to handle mistakes? A survey of internal medicine patients in an academic setting. Arch Intern Med. 1996;156(22):2565-2569.
30. Stelfox HT, Gandhi TK, Orav EJ, et al. The relation of patient satisfaction with complaints against physicians and malpractice lawsuits. Am J Med. 2005;118(10):
1126-1133.
31. Hickson GB, Federspiel CF, Pichert JW, et al. Patient complaints and malpractice risk. JAMA. 2002;287(22):2951-2957.
32. Bain W. Do we need a new word for patients? Continue to call them patients but treat them like customers. BMJ. 1999;319(7222):1436.
33. Johnson R, Moskowitz E, Thomas J, et al. Would you rather be treated as a patient or a customer? Yale Insights. http://insights.som.yale.edu/insights/would-you-rather-betreated-patient-or-customer. Accessed December 13, 2013.
Dear Dr. Mossman,
At the multispecialty hospital where I work, administrators refer to patients as “customers” and tell us that, by improving “the customer experience,” we can reduce complaints and avoid malpractice suits. This business lingo offends me. Doesn’t providing good care do more to prevent malpractice claims than calling sick patients “customers”?
Submitted by “Dr. H”
“All words are pegs to hang ideas on.” As was true when Reverend Henry Ward Beecher uttered this phrase in the 19th century,1 names affect how we relate to and feel about people. Many doctors don’t think of themselves as “selling” services, and they find calling patients “customers” distasteful.
But for at least 4 decades, mental health professionals themselves have used a “customer approach” to think about certain aspects of psychiatrist–patient encounters.2 More pertinent to Dr. H’s questions, many attorneys who advise physicians are convinced that giving patients a satisfying “customer experience” is a sound strategy for reducing the risk of malpractice litigation.3
If the attorneys are right, taking a customer service perspective can lower the likelihood that psychiatrists will be sued. To understand why, this article looks at:
• terms for referring to health care recipients
• the feelings those terms generate
• how the “customer service” perspective has become a malpractice prevention
strategy.
Off-putting connotations
All the currently used ways of referring to persons served by doctors have off-putting features.
The word “patient” dates back to the 14th century and comes from Latin present
participles of pati, “to suffer.” Although Alpha Omega Alpha’s motto—“be worthy
to serve the suffering”4—expresses doctors’ commitment to help others, “patient”
carries emotional baggage. A “patient” is “a sick individual” who seeks treatment
from a physician,5 a circumstance that most people (including doctors) find unpleasant and hope is only temporary. The adjective “patient” means “bearing pains or trials calmly or without complaint” and “manifesting forbearance under provocation or strain,”5 phrases associated with passivity, deference, and a long wait to see the doctor.
Because “patient” evokes notions of helplessness and need for direction, non-medical psychotherapists often use “client” to designate care recipients. “Client” has the same Latin root as “to lean” and refers to someone “under the protection of another.” More pertinent to discussions of mental health care, a “client” also is “a person who pays a professional person or organization for services” or “a customer.”5 The latter definition explains what makes “client” feel wrong to medical practitioners, who regard those they treat as deserving more compassion and sacrifice than someone who simply purchases professional services.
“Consumer,” a word of French origin derived from the Latin consumere (“to take
up”), refers to “a person who buys goods and services.”5 If “consumers” are buyers, then those from whom they make purchases are merchants or sellers. Western marketplace concepts often regard consumers as sovereign judges of their needs, and the role of commodity producers is to try to satisfy those needs.6
The problem with viewing health care recipients this way is that sellers don’t caution customers about buying things when only principles of supply-and-demand govern exchange relationships.7 Quite the contrary: producers sometimes promote their products through “advertising [that] distorts reality and creates artificial needs to make profit for a firm.”8 If physicians behave this way, however, they get criticism and deserve it.
A “customer” in 15th-century Middle English was a tax collector, but in modern
usage, a customer is someone who, like a consumer, “purchases some commodity or service.”5 By the early 20th century, “customer” became associated with notions of empowerment embodied in the merchants’ credo, “The customer is always right.”9 Chronic illnesses often require self-management and collaboration with those labeled the “givers” and “recipients” of medical care. Research shows that “patients are more trusting of, and committed to, physicians who adopt an empowering communication style with them,” which suggests “that empowering
patients presents a means to improve the patient–physician relationship.”10
Feelings about names
People have strong feelings about what they are called. In opposing calling patients “consumers,” Nobel Prize-winning economist Paul Krugman explains: “Medical care is an area in which crucial decisions—life and death decisions—must be made; yet making those decisions intelligently requires a vast amount of specialized knowledge; and often those decisions must also be made under conditions in which the patient …needs action immediately, with no time for discussion, let alone comparison shopping. …That’s why doctors have traditionally…been expected to behave according to higher standards than the average professional…The idea that all this can be reduced to money—that doctors are just people selling services to consumers of health care—is, well, sickening.”11
Less famous recipients of nonpsychiatric medical services also prefer being called
“patients” over “clients” or “consumers.”12-14 Recipients of mental health services have a different view, however. In some surveys, “patient” gets a plurality or majority of service recipients’ votes,15,16 but in others, recipients prefer to be called “clients” or other terms.17,18 Of note, people who prefer being called “patients” tend to strongly dislike being called “clients.”19 On the professional
side, psychiatrists—along with other physicians—prefer to speak of treating “patients” and to criticize letting economic phrases infect medical discourse.20-22
Names: A practical difference?
Does what psychiatrists call those they serve make any practical difference? Perhaps not, but evidence suggests that the attitudes that doctors take toward patients affects economic success and malpractice risk.
When they have choices about where they can seek health care, medical patients value physicians’ competence, but they also consider nonclinical factors such as family members’ opinions and convenience.23 Knowing this, the federal government’s Centers for Medicare & Medicaid Services publishes results from its Hospital Consumer Assessment of Healthcare Providers and Systems to “create incentives for hospitals to improve their quality of care.”24
Nonclinical factors play a big part in patients’ decisions about suing their doctors, too. Many malpractice claims turn out to be groundless in the sense that they do not involve medical errors,25 and most errors that result in injury do not lead to malpractice suits.26
What explains this disparity? Often when a lawsuit is filed, whatever injury may have occurred is coupled with an aggravating factor, such as a communication gaffe,27 a physician’s domineering tone of voice,28 or failure to acknowledge error.29 The lower a physician’s patient satisfaction ratings, the higher the physician’s likelihood of receiving complaints and getting sued for malpractice.30,31
These kinds of considerations probably lie behind the recommendation of one hospital manager to doctors: “Continue to call them patients but treat them like
customers.”32 More insights into this view come from responses solicited from Yale
students, staff members, and alumni about whether it seems preferable to be a “patient” or a “customer” (Box).33
Bottom Line
When patients get injured during medical care, evidence suggests that how they feel about their doctors makes a big difference in whether they decide to file suit. If you’re like most psychiatrists, you prefer to call persons whom you treat “patients.” But watching and improving the things that affect your patients’ “customer experience” may help you avoid malpractice litigation.
Related Resource
• Goldhill D. To fix healthcare, turn patients into customers. Bloomberg Personal Finance. www.bloomberg.com/news/2013-01-03/to-fix-health-care-turn-patients-intocustomers.html.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dear Dr. Mossman,
At the multispecialty hospital where I work, administrators refer to patients as “customers” and tell us that, by improving “the customer experience,” we can reduce complaints and avoid malpractice suits. This business lingo offends me. Doesn’t providing good care do more to prevent malpractice claims than calling sick patients “customers”?
Submitted by “Dr. H”
“All words are pegs to hang ideas on.” As was true when Reverend Henry Ward Beecher uttered this phrase in the 19th century,1 names affect how we relate to and feel about people. Many doctors don’t think of themselves as “selling” services, and they find calling patients “customers” distasteful.
But for at least 4 decades, mental health professionals themselves have used a “customer approach” to think about certain aspects of psychiatrist–patient encounters.2 More pertinent to Dr. H’s questions, many attorneys who advise physicians are convinced that giving patients a satisfying “customer experience” is a sound strategy for reducing the risk of malpractice litigation.3
If the attorneys are right, taking a customer service perspective can lower the likelihood that psychiatrists will be sued. To understand why, this article looks at:
• terms for referring to health care recipients
• the feelings those terms generate
• how the “customer service” perspective has become a malpractice prevention
strategy.
Off-putting connotations
All the currently used ways of referring to persons served by doctors have off-putting features.
The word “patient” dates back to the 14th century and comes from Latin present
participles of pati, “to suffer.” Although Alpha Omega Alpha’s motto—“be worthy
to serve the suffering”4—expresses doctors’ commitment to help others, “patient”
carries emotional baggage. A “patient” is “a sick individual” who seeks treatment
from a physician,5 a circumstance that most people (including doctors) find unpleasant and hope is only temporary. The adjective “patient” means “bearing pains or trials calmly or without complaint” and “manifesting forbearance under provocation or strain,”5 phrases associated with passivity, deference, and a long wait to see the doctor.
Because “patient” evokes notions of helplessness and need for direction, non-medical psychotherapists often use “client” to designate care recipients. “Client” has the same Latin root as “to lean” and refers to someone “under the protection of another.” More pertinent to discussions of mental health care, a “client” also is “a person who pays a professional person or organization for services” or “a customer.”5 The latter definition explains what makes “client” feel wrong to medical practitioners, who regard those they treat as deserving more compassion and sacrifice than someone who simply purchases professional services.
“Consumer,” a word of French origin derived from the Latin consumere (“to take
up”), refers to “a person who buys goods and services.”5 If “consumers” are buyers, then those from whom they make purchases are merchants or sellers. Western marketplace concepts often regard consumers as sovereign judges of their needs, and the role of commodity producers is to try to satisfy those needs.6
The problem with viewing health care recipients this way is that sellers don’t caution customers about buying things when only principles of supply-and-demand govern exchange relationships.7 Quite the contrary: producers sometimes promote their products through “advertising [that] distorts reality and creates artificial needs to make profit for a firm.”8 If physicians behave this way, however, they get criticism and deserve it.
A “customer” in 15th-century Middle English was a tax collector, but in modern
usage, a customer is someone who, like a consumer, “purchases some commodity or service.”5 By the early 20th century, “customer” became associated with notions of empowerment embodied in the merchants’ credo, “The customer is always right.”9 Chronic illnesses often require self-management and collaboration with those labeled the “givers” and “recipients” of medical care. Research shows that “patients are more trusting of, and committed to, physicians who adopt an empowering communication style with them,” which suggests “that empowering
patients presents a means to improve the patient–physician relationship.”10
Feelings about names
People have strong feelings about what they are called. In opposing calling patients “consumers,” Nobel Prize-winning economist Paul Krugman explains: “Medical care is an area in which crucial decisions—life and death decisions—must be made; yet making those decisions intelligently requires a vast amount of specialized knowledge; and often those decisions must also be made under conditions in which the patient …needs action immediately, with no time for discussion, let alone comparison shopping. …That’s why doctors have traditionally…been expected to behave according to higher standards than the average professional…The idea that all this can be reduced to money—that doctors are just people selling services to consumers of health care—is, well, sickening.”11
Less famous recipients of nonpsychiatric medical services also prefer being called
“patients” over “clients” or “consumers.”12-14 Recipients of mental health services have a different view, however. In some surveys, “patient” gets a plurality or majority of service recipients’ votes,15,16 but in others, recipients prefer to be called “clients” or other terms.17,18 Of note, people who prefer being called “patients” tend to strongly dislike being called “clients.”19 On the professional
side, psychiatrists—along with other physicians—prefer to speak of treating “patients” and to criticize letting economic phrases infect medical discourse.20-22
Names: A practical difference?
Does what psychiatrists call those they serve make any practical difference? Perhaps not, but evidence suggests that the attitudes that doctors take toward patients affects economic success and malpractice risk.
When they have choices about where they can seek health care, medical patients value physicians’ competence, but they also consider nonclinical factors such as family members’ opinions and convenience.23 Knowing this, the federal government’s Centers for Medicare & Medicaid Services publishes results from its Hospital Consumer Assessment of Healthcare Providers and Systems to “create incentives for hospitals to improve their quality of care.”24
Nonclinical factors play a big part in patients’ decisions about suing their doctors, too. Many malpractice claims turn out to be groundless in the sense that they do not involve medical errors,25 and most errors that result in injury do not lead to malpractice suits.26
What explains this disparity? Often when a lawsuit is filed, whatever injury may have occurred is coupled with an aggravating factor, such as a communication gaffe,27 a physician’s domineering tone of voice,28 or failure to acknowledge error.29 The lower a physician’s patient satisfaction ratings, the higher the physician’s likelihood of receiving complaints and getting sued for malpractice.30,31
These kinds of considerations probably lie behind the recommendation of one hospital manager to doctors: “Continue to call them patients but treat them like
customers.”32 More insights into this view come from responses solicited from Yale
students, staff members, and alumni about whether it seems preferable to be a “patient” or a “customer” (Box).33
Bottom Line
When patients get injured during medical care, evidence suggests that how they feel about their doctors makes a big difference in whether they decide to file suit. If you’re like most psychiatrists, you prefer to call persons whom you treat “patients.” But watching and improving the things that affect your patients’ “customer experience” may help you avoid malpractice litigation.
Related Resource
• Goldhill D. To fix healthcare, turn patients into customers. Bloomberg Personal Finance. www.bloomberg.com/news/2013-01-03/to-fix-health-care-turn-patients-intocustomers.html.
Disclosure
Dr. Mossman reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Beecher HW, Drysdale W. Proverbs from Plymouth pulpit. New York, NY: D. Appleton & Co.;1887.
2. Lazare A, Eisenthal S, Wasserman L. The customer approach to patienthood: attending to patient requests in a walk-in clinic. Arch Gen Psychiatry. 1975;32:553-558.
3. Schleiter KE. Difficult patient-physician relationships and the risk of medical malpractice litigation. Virtual Mentor. 2009;11:242-246.
4. Alpha Omega Alpha Honor Medical Society. Alpha Omega Alpha constitution. http://www.alphaomegaalpha.org/constitution.html. Accessed December 13, 2013. Accessed December 13, 2013.
5. Merriam-Webster. Dictionary. http://www.merriamwebster.com. Accessed December 9, 2013.
6. Kotler P, Burton S, Deans K, et al. Marketing, 9th ed. Frenchs Forest, Australia: Pearson Education Australia; 2013.
7. Deber RB. Getting what we pay for: myths and realities about financing Canada’s health care system. Health Law Can. 2000;21(2):9-56.
8. Takala T, Uusitalo O. An alternative view of relationship marketing: a framework for ethical analysis. Eur J Mark. 1996;30:45-60.
9. Van Vuren FS. The Yankee who taught Britishers that ‘the customer is always right.’ Milwaukee Journal. http://www.wisconsinhistory.org/wlhba/articleView.
asp?pg=1&id=11176. Published September 9, 1932. Accessed December 20, 2013.
10. Ouschan T, Sweeney J, Johnson L. Customer empowerment and relationship outcomes in healthcare consultations. Eur J Mark. 2006;40:1068-1086.
11. Krugman P. Patients are not consumers. The New York Times. http://krugman.blogs.nytimes.com/2011/04/20/patients-are-not-consumers. Published April 20, 2011. Accessed December 13, 2013.
12. Nair BR. Patient, client or customer? Med J Aust. 1998;169:593.
13. Wing PC. Patient or client? If in doubt, ask. CMAJ. 1997;157:287-289.
14. Deber RB, Kraetschmer N, Urowitz S, et al. Patient, consumer, client, or customer: what do people want to be called? Health Expect. 2005;8(4):345-351.
15. Sharma V, Whitney D, Kazarian SS, et al. Preferred terms for users of mental health services among service providers and recipients. Psychiatr Serv. 2000;51(2): 203-209.
16. Simmons P, Hawley CJ, Gale TM, et al. Service user, patient, client, user or survivor: describing recipients of mental health services. Psychiatrist. 2010;34:20-23.
17. Lloyd C, King R, Bassett H, et al. Patient, client or consumer? A survey of preferred terms. Australas Psychiatry. 2001; 9(4):321-324.
18. Covell NH, McCorkle BH, Weissman EM, et al. What’s in a name? Terms preferred by service recipients. Adm Policy Ment Health. 2007;34(5):443-447.
19. Ritchie CW, Hayes D, Ames DJ. Patient or client? The opinions of people attending a psychiatric clinic. Psychiatrist. 2000;24(12):447-450.
20. Andreasen NC. Clients, consumers, providers, and products: where will it all end? Am J Psychiatry. 1995;152:1107-1109.
21. Editorial. What’s in a name? Lancet. 2000;356(9248):2111.
22. Torrey EF. Patients, clients, consumers, survivors et al: what’s in a name? Schizophr Bull. 2011;37(3):466-468.
23. Wilson CT, Woloshin S, Schwartz L. Choosing where to have major surgery: who makes the decision? Arch Surg. 2007;142(3):242-246.
24. Centers for Medicare & Medicaid Services. Hospital consumer assessment of healthcare providers and systems. http://www.hcahpsonline.org. Accessed
January 26, 2014.
25. Studdert DM, Mello MM, Gawande AA, et al. Claims, errors, and compensation payments in medical malpractice litigation. N Engl J Med. 2006;354:2024-2033.
26. Localio AR, Lawthers AG, Brennan TA, et al. Relation between malpractice claims and adverse events due to negligence—results of the Harvard Medical Practice Study III. N Engl J Med. 1991;325:245-251.
27. Huntington B, Kuhn N. Communication gaffes: a root cause of malpractice claims. Bayl Univ Med Cent. 2003;16(2):157-161.
28. Ambady N, Laplante D, Nguyen T, et al. Surgeons’ tone of voice: a clue to malpractice history. Surgery. 2002;132(1):5-9.
29. Witman AB, Park DM, Hardin SB. How do patients want physicians to handle mistakes? A survey of internal medicine patients in an academic setting. Arch Intern Med. 1996;156(22):2565-2569.
30. Stelfox HT, Gandhi TK, Orav EJ, et al. The relation of patient satisfaction with complaints against physicians and malpractice lawsuits. Am J Med. 2005;118(10):
1126-1133.
31. Hickson GB, Federspiel CF, Pichert JW, et al. Patient complaints and malpractice risk. JAMA. 2002;287(22):2951-2957.
32. Bain W. Do we need a new word for patients? Continue to call them patients but treat them like customers. BMJ. 1999;319(7222):1436.
33. Johnson R, Moskowitz E, Thomas J, et al. Would you rather be treated as a patient or a customer? Yale Insights. http://insights.som.yale.edu/insights/would-you-rather-betreated-patient-or-customer. Accessed December 13, 2013.
1. Beecher HW, Drysdale W. Proverbs from Plymouth pulpit. New York, NY: D. Appleton & Co.;1887.
2. Lazare A, Eisenthal S, Wasserman L. The customer approach to patienthood: attending to patient requests in a walk-in clinic. Arch Gen Psychiatry. 1975;32:553-558.
3. Schleiter KE. Difficult patient-physician relationships and the risk of medical malpractice litigation. Virtual Mentor. 2009;11:242-246.
4. Alpha Omega Alpha Honor Medical Society. Alpha Omega Alpha constitution. http://www.alphaomegaalpha.org/constitution.html. Accessed December 13, 2013. Accessed December 13, 2013.
5. Merriam-Webster. Dictionary. http://www.merriamwebster.com. Accessed December 9, 2013.
6. Kotler P, Burton S, Deans K, et al. Marketing, 9th ed. Frenchs Forest, Australia: Pearson Education Australia; 2013.
7. Deber RB. Getting what we pay for: myths and realities about financing Canada’s health care system. Health Law Can. 2000;21(2):9-56.
8. Takala T, Uusitalo O. An alternative view of relationship marketing: a framework for ethical analysis. Eur J Mark. 1996;30:45-60.
9. Van Vuren FS. The Yankee who taught Britishers that ‘the customer is always right.’ Milwaukee Journal. http://www.wisconsinhistory.org/wlhba/articleView.
asp?pg=1&id=11176. Published September 9, 1932. Accessed December 20, 2013.
10. Ouschan T, Sweeney J, Johnson L. Customer empowerment and relationship outcomes in healthcare consultations. Eur J Mark. 2006;40:1068-1086.
11. Krugman P. Patients are not consumers. The New York Times. http://krugman.blogs.nytimes.com/2011/04/20/patients-are-not-consumers. Published April 20, 2011. Accessed December 13, 2013.
12. Nair BR. Patient, client or customer? Med J Aust. 1998;169:593.
13. Wing PC. Patient or client? If in doubt, ask. CMAJ. 1997;157:287-289.
14. Deber RB, Kraetschmer N, Urowitz S, et al. Patient, consumer, client, or customer: what do people want to be called? Health Expect. 2005;8(4):345-351.
15. Sharma V, Whitney D, Kazarian SS, et al. Preferred terms for users of mental health services among service providers and recipients. Psychiatr Serv. 2000;51(2): 203-209.
16. Simmons P, Hawley CJ, Gale TM, et al. Service user, patient, client, user or survivor: describing recipients of mental health services. Psychiatrist. 2010;34:20-23.
17. Lloyd C, King R, Bassett H, et al. Patient, client or consumer? A survey of preferred terms. Australas Psychiatry. 2001; 9(4):321-324.
18. Covell NH, McCorkle BH, Weissman EM, et al. What’s in a name? Terms preferred by service recipients. Adm Policy Ment Health. 2007;34(5):443-447.
19. Ritchie CW, Hayes D, Ames DJ. Patient or client? The opinions of people attending a psychiatric clinic. Psychiatrist. 2000;24(12):447-450.
20. Andreasen NC. Clients, consumers, providers, and products: where will it all end? Am J Psychiatry. 1995;152:1107-1109.
21. Editorial. What’s in a name? Lancet. 2000;356(9248):2111.
22. Torrey EF. Patients, clients, consumers, survivors et al: what’s in a name? Schizophr Bull. 2011;37(3):466-468.
23. Wilson CT, Woloshin S, Schwartz L. Choosing where to have major surgery: who makes the decision? Arch Surg. 2007;142(3):242-246.
24. Centers for Medicare & Medicaid Services. Hospital consumer assessment of healthcare providers and systems. http://www.hcahpsonline.org. Accessed
January 26, 2014.
25. Studdert DM, Mello MM, Gawande AA, et al. Claims, errors, and compensation payments in medical malpractice litigation. N Engl J Med. 2006;354:2024-2033.
26. Localio AR, Lawthers AG, Brennan TA, et al. Relation between malpractice claims and adverse events due to negligence—results of the Harvard Medical Practice Study III. N Engl J Med. 1991;325:245-251.
27. Huntington B, Kuhn N. Communication gaffes: a root cause of malpractice claims. Bayl Univ Med Cent. 2003;16(2):157-161.
28. Ambady N, Laplante D, Nguyen T, et al. Surgeons’ tone of voice: a clue to malpractice history. Surgery. 2002;132(1):5-9.
29. Witman AB, Park DM, Hardin SB. How do patients want physicians to handle mistakes? A survey of internal medicine patients in an academic setting. Arch Intern Med. 1996;156(22):2565-2569.
30. Stelfox HT, Gandhi TK, Orav EJ, et al. The relation of patient satisfaction with complaints against physicians and malpractice lawsuits. Am J Med. 2005;118(10):
1126-1133.
31. Hickson GB, Federspiel CF, Pichert JW, et al. Patient complaints and malpractice risk. JAMA. 2002;287(22):2951-2957.
32. Bain W. Do we need a new word for patients? Continue to call them patients but treat them like customers. BMJ. 1999;319(7222):1436.
33. Johnson R, Moskowitz E, Thomas J, et al. Would you rather be treated as a patient or a customer? Yale Insights. http://insights.som.yale.edu/insights/would-you-rather-betreated-patient-or-customer. Accessed December 13, 2013.
Answers to 7 questions about using neuropsychological testing in your practice
Neuropsychological evaluation, consisting of a thorough examination of cognitive and behavioral functioning, can make an invaluable contribution to the care of psychiatric patients. Through the vehicle of standardized measures of abilities, patients’ cognitive strengths and weaknesses can be elucidated—revealing potential areas for further interventions or to explain impediments to treatment. A licensed clinical psychologist provides this service.
You, as a consumer of reported findings, can use the results to inform your diagnosis and treatment plan. Recommendations from the neuropsychologist often address dispositional planning, cognitive intervention, psychiatric intervention, and work and school accommodations.
Probing the brain−behavior relationship
Neuropsychology is a subspecialty of clinical psychology that is focused on understanding the brain–behavior relationship. Drawing information from multiple disciplines, including psychiatry and neurology, neuropsychology seeks to uncover the cognitive, behavioral, and emotional difficulties that can result from known or suspected brain dysfunction. Increasingly, to protect the public and referral sources, clinical psychologists who perform neuropsychological testing demonstrate their competence through board certification (eg, the American Board of Clinical Neuropsychology).
How is testing conducted? Evaluations comprise measures that are standardized, scored objectively, and have established psychometric properties. Testing can performed on an outpatient or inpatient basis; the duration of testing depends on the question for which the referring practitioner seeks an answer.
Measures typically are administered by paper and pencil, although computer-based
assessments are increasingly being employed. Because of the influence of demographic variables (age, sex, years of education, race), scores are compared with normative samples that resemble those of the patient’s background as closely as possible.
A thorough clinical interview with the patient, a collateral interview with caregivers
and family, and review of relevant medical records are crucial parts of the assessment. Multiple areas of cognition are assessed:
• intelligence
• academic functioning
• attention
• working memory
• speed of processing
• learning and memory
• visual spatial skills
• fine motor skills
• executive functioning.
Essentially, the evaluation speaks to a patient’s neurocognitive functioning and cerebral integrity.
How are results scored? Interpretation of test scores is contingent on expectations of how a patient should perform in the absence of neurologic or psychiatric illness (ie, based on normative data and performancebased estimates of premorbid functioning).1 The overall pattern of intact scores and deficit scores can be used to form specific impressions about a diagnosis, cognitive strengths and weaknesses, and strategies for intervention.
Personality testing. In addition to the cognitive aspect of the evaluation, personality measures are incorporated when relevant to the referral question or presenting concern.
Personality tests can be broadly divided into objective and projective measures.
Objective personality measures, such as the Minnesota Multiphasic Personality
Inventory-Second Edition, require the examinee to respond to a set of items as
true or false or on a Likert-type scale from strongly agree to disagree. Responses are then scored in standardized fashion, making comparisons to normative data, which are then analyzed to determine the extent to which the examinee experiences psychiatric symptoms.
As part of testing, patients’ responses to ambiguous or unstructured standard
stimuli—such as a series of drawings, abstract patterns, or incomplete sentences—
are analyzed to determine underlying personality traits, feelings, and attitudes.
Classic examples of these measures include the Rorschach Test and the Thematic
Apperception Test.
Personality measures and psychiatric testing are designed to answer questions
related to patients’ emotional status. These measures assess psychiatric symptoms and diagnoses, whereas neuropsychological measures provide an understanding of patients’ cognitive assets and limitations.
7 Common questions about neuropsychological testing
1 Will my patient’s insurance cover these assessments? The question is common from practitioners who are considering requesting an assessment for a patient. The short answer is “Yes.”
Most payers follow Medicare guidelines for reimbursement of neuropsychological
testing; if testing is determined to be medically necessary, insurance companies often cover the assessment. Medicaid also pays for psychometric testing services. Neuropsychologists who have a hospital-based practice typically include patients
with all types of insurance coverage. For example, 40% of patients seen in a hospital are covered by Medicare or Medicaid.2
A caveat: Local intermediaries interpret policies and procedures in different ways,
so there is variability in coverage by geographic region. That is why it is crucial
for neuropsychologists to obtain preauthorization, as would be the case with other medical procedures and services sought by referral.
Last, insurance companies do not pay for assessment of a learning disability. The
rationale typically offered for this lack of coverage? The assessment is for academic, not medical, purposes. In such a situation, patients and their families are offered a private-pay option.
2 What are the indications for neuropsychological assessment? Psychiatric practitioners are one of the top medical specialties that refer their patients for neuropsychological testing.3 This is because many patients with a psychiatric or
neurologic disorder experience changes in cognition, mood, and personality. Such
changes can range in severity from subtle to dramatic, and might reflect an underlying disease state or a side effect of medication or other treatment. Whatever the nature of a patient’s problem, careful assessment might help elucidate specific areas with which he (she) is struggling—so that you can better target your interventions. Table 1 lists common reasons for referring a patient for neuropsychological evaluation. Throughout this discussion, we describe examples of clinical situations in which neuropsychological testing is useful for establishing a differential diagnosis and dispositional planning.

3 How does neuropsychological testing help with the differential diagnosis? As an example, one area in which cognitive testing can be beneficial is in geriatric psychiatry.Dementia. Aging often is accompanied by a normal decline in memory and other cognitive functions. But because subtle changes in memory and cognition also canbe the sign of a progressive cognitive disorder, differentiating normal aging from early dementia is essential. Table 2 summarizes typical changes in cognition with aging.
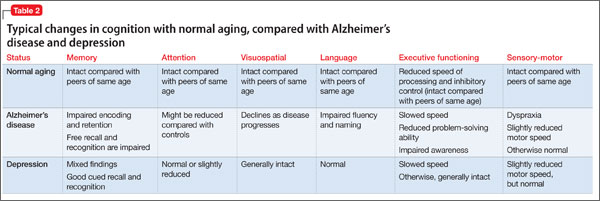
Neuropsychologists, through knowledge of psychometric testing and the brain−behavior relationship, can help you detect dementia and plan treatment early. To determine if cognitive changes are progressive, patients might undergo re-evaluation—typically, every 6 to 12 months—to ascertain if changes have occurred. Mood disorders. Neuropsychological evaluation can be useful in building a differential diagnosis when determining whether cognitive symptoms are attributable to a mood disorder or a medical illness. Cognitive deficits associated with an affective disturbance generally include impairments in attention, memory, and executive functioning.4 The severity of deficits has been linked to severity of illness. When patients with a mood disorder demonstrate localizing impairments or those of greater severity than expected, suspicion arises that another cause likely better explains those deficits, and further medical testing then is often recommended. Medical procedures. Increasingly, neuropsychological assessment is used to assist in determining the appropriateness of medical procedures. For example, neurosurgical patients being considered for deep brain stimulation, brain tumor resectioning, and epilepsy surgery often are referred for preoperative and postoperative testing. Treating clinicians need an understanding of current cognitive status, localization of functioning, and psychological status to make appropriate decisions about a patient’s candidacy for one of these procedures,and to understand associated risk.
4 How is neuropsychological testing used for dispositional planning? The
results of cognitive and psychological testing have implications for dispositional
planning for patients who are receiving psychiatric care. The primary issue often is
to determine the patient’s level of independence and ability to make decisions about his affairs.5
Neuropsychological testing can help determine if cognitive deficits limit aspects of functional independence—for example, can the patient live alone, or must he live with family or in a residential care facility? Generally, the greater the cognitive impairment, the more supervision and assistance are required. This relationship between cognitive ability and independence in activities of daily living has been demonstrated in many groups of psychiatric patients, including older adults with dementia,6 patients with schizophrenia,7 and those with bipolar disorder.8
Specific recommendations can be made regarding management of finances, administering medications, and driving. To formulate an appropriate dispositional plan, the referring psychiatrist might integrate recommendations from the neuropsychological assessment with findings of other evaluations and with information that has been collected about the patient.
5 Can neuropsychological testing be used to refer a patient for neurological and cognitive rehabilitation? Yes. The neuropsychologist is singularly qualified to make recommendations about a range of interventions for cognitive deficits that have been identified on formal testing.
Typically, recommendations for addressing cognitive deficits involve rehabilitation
focused on development and use of compensatory strategies and modification to promote brain health.9,10 Rehabilitation therapy typically is aimed at increasing functioning independence and reducing physical and cognitive deficits associated
with illness (eg, traumatic brain injury [TBI], stroke, orthopedic injury, debility).
Patients who have a TBI or who have had a stroke often have comorbid psychiatric problems, including mood and anxiety disorders, that can exacerbate deficits and impede engagement in rehabilitation. The neuropsychological evaluation can determine if this is the case and if psychiatric consultation is warranted to assist with managing symptoms.
Premorbid psychiatric illness can affect rehabilitation. Formal neuropsychological testing can assist with parsing out deficits associated with new-onset illness compared with premorbid psychiatric problems. The evaluation of a patient before he begins rehabilitation also can be compared with evaluations made during treatment and after discharge to 1) assess for changes and 2) update recommendations about management.
Recommendations about cognitive interventions might include specific compensatory strategies to address areas of weakness and capitalize on strengths. Such strategies can include using internal mnemonics, such as visual imagery (ie, using a visual image to help encode verbal information) or semantic elaboration (using semantic cues to aid in encoding and recall of information). Methods can help train patients to capitalize on areas of stronger cognitive functioning in compensating for their weaknesses; an example is the spaced-retrieval technique, which relies on repetition of information that is to be learned over time.11
Perhaps the most practical strategies for addressing areas of weakness are nonmnemonic-based external memory aids, such as diaries, notebooks, calendars, alarms, and lists.12 For example, for a patient with a TBI who has impaired memory, recommendations might include using written notes or a calendar system; using a pillbox for medication management; and using labels to promote structure and consistency in the home. These strategies are meant to promote increased independence and to minimize the effect of cognitive deficits on daily functioning.
Recommended strategies can include lifestyle changes to promote improved cognitive functioning and overall health, such as:
• sleep hygiene, to reduce the effects of fatigue
• encouraging the patient to adhere to a diet, take prescribed medications, and follow up with his health care providers
• developing cognitive and physical exercise routines.
In addition, a patient who has had a stroke or who have a TBI might benefit from psychotherapy or referral to a group program or community resources to help cope with the effects of illness.13
6 How does neuropsychological testing help determine the appropriate psychiatric intervention for a patient? Results of neuropsychological testing can help determine appropriate interventions for a psychiatric condition that might be the principal factor affecting the patient’s functioning.
Concerning psychoactive medications, consider the following:
Mood and anxiety disorders. Neuropsychological measures can help substantiate the need for pharmacotherapy in a comorbid mood or anxiety disorder in a patient who has a neurologic illness, such as stroke or TBI.
ADHD. In a patient who has attentiondeficit/hyperactivity disorder (ADHD), results of cognitive testing might help determine if attention issues undermine daily functioning. Testing provides information beyond rating scale scores to justify diagnosis and psychopharmacotherapy.14
Dementia. Geriatric patients who have dementia often have coexisting behavioral and mood changes that, once evaluated, might improve with pharmacotherapy.
Other areas. Cognitive side effects of medications can be monitored by conducting testing before and after medication is started. The evaluation can address the patient’s ability to engage in psychotherapeutic interventions. Patients who have severe cognitive deficits might have greater difficulty engaging in psychotherapy, compared with patients who have less severe, or no, cognitive
impairment.15
7 Does neuropsychological testing help patients make return-to-work
and return-to-school decisions? Yes. Cognitive and psychiatric functioning have
implications for decisions about occupational and academic pursuits.
Patients who have severe cognitive or psychiatric symptoms might be or might not be able to maintain gainful employment or participate in school. Testing can help 1) document and justify disability and 2) establish recommendations about disability status. Those whose cognitive impairments or psychiatric symptoms are less severe might benefit from neuropsychological testing so that recommendations can be made regarding accommodations at work or in school, such as:
• reduced work or school schedule
• reduced level of occupational or academic demand
• change in supervision or evaluation procedures by employer or school.
Cognitive strengths and weaknesses can be used to help a patient devise and implement compensatory strategies at work or school, such as:
• note-taking
• audio recording of meetings and lectures
• using a calendar.
Patients sometimes benefit from formal vocational rehabilitation services to facilitate finding appropriate employment, returning to employment, and implementing workplace accommodations.
In conclusion
Neuropsychological evaluation, typically covered by health insurance, provides the
referring clinician with objective information about patients’ cognitive assets and limitations. In turn, this information can help you make a diagnosis and plan
treatment.
Unlike psychological testing, in which the patient is assessed for psychiatric
symptoms and conditions, neuropsychological measures offer insight into such
abilities as attention, memory, and reasoning. Neuropsychological evaluations also
can add insight to your determination of the cause of symptoms, thereby influencing decisions about medical therapy.
Last, these evaluations can aid with decision-making about dispositional planning
and whether adjunctive services, such as rehabilitation, would be of benefit.
Bottom Line
Neuropsychological assessments are a useful consultation to consider for patients
in a psychiatric setting. These evaluations can aid you in building and narrowing the differential diagnosis; identifying patients’ strengths and weakness; and making informed recommendations about functional independence.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Donders J. A survey of report writing by neuropsychologists, I: general characteristics and content. Clin Neuropsychol. 2001;15(2):137-149.
2. Lamberty GT, Courtney JC, Heilbronner RC. The practice of clinical neuropsychology: a survey of practices and settings. New York, NY: Taylor & Francis; 2005.
3. Sweet JJ, Meyer DG, Nelson NW, et al. The TCN/AACN 2010 “salary survey”: professional practices, beliefs, and incomes of U.S. neuropsychologists. Clin Neuropsychol. 2011;25(1):12-61.
4. Marvel CL, Paradiso S. Cognitive and neurologic impairment in mood disorders. Psychiatr Clin North Am. 2004;27(1):19-36,vii-viii.
5. Moberg PJ, Rick JH. Decision-making capacity and competency in the elderly: a clinical and neuropsychological perspective. NeuroRehabilitation. 2008;23(5):
403-413.
6. Bradshaw LE, Goldberg SE, Lewis SA, et al. Six-month outcomes following an emergency hospital admission for older adults with co-morbid mental health problems indicate complexity of care needs. Age Ageing. 2013; 42(5):582-588.
7. Medalia A, Lim RW. Self-awareness of cognitive functioning in schizophrenia. Schizophr Res. 2004;71(2-3):331-338.
8. Andreou C, Bozikas VP. The predictive significance of neurocognitive factors for functional outcome in bipolar disorder. Curr Opin Psychiatry. 2013;26(1):54-59.
9. Stuss DT, Winocur G, Robertson IH, eds. Cognitive neurorehabilitation: evidence and application. 2nd ed. New York, NY: Cambridge University Press; 2008.
10. Raskin SA, ed. Neuroplasticity and rehabilitation. New York, NY: The Guilford Press; 2011.
11. Glisky EL, Glisky ML. Memory rehabilitation in older adults. In: Stuss DT, Winocur G, Robertson IH. Cognitive neurorehabilitation. 1st ed. New York, NY: Cambridge University Press; 2008.
12. Kapur N, Glisky EL, Wilson BA. External memory aids and computers in memory rehabilitation. In: Baddeley AD, Kopelman MD, Wilson BA. Handbook of memory disorders. Chichester, United Kingdom: Wiley; 2002:757-784.
13. Stalder-Luthy F, Messerli-Burgy N, Hofer H, et al. Effect of psychological interventions on depressive symptoms in long-term rehabilitation after an acquired brain injury: a systematic review and meta-analysis. Arch Phys Med Rehabil.
2013;94(7):1386-1397.
14. Hale JB, Reddy LA, Semrud-Clikeman M, et al. Executive impairment determines ADHD medication response: implications for academic achievement. J Learn Disabil. 2011;44(2):196-212.
15. Medalia A, Lim R. Treatment of cognitive dysfunction in psychiatric disorders. J Psychiatr Pract. 2004;10(1):17-25.
Neuropsychological evaluation, consisting of a thorough examination of cognitive and behavioral functioning, can make an invaluable contribution to the care of psychiatric patients. Through the vehicle of standardized measures of abilities, patients’ cognitive strengths and weaknesses can be elucidated—revealing potential areas for further interventions or to explain impediments to treatment. A licensed clinical psychologist provides this service.
You, as a consumer of reported findings, can use the results to inform your diagnosis and treatment plan. Recommendations from the neuropsychologist often address dispositional planning, cognitive intervention, psychiatric intervention, and work and school accommodations.
Probing the brain−behavior relationship
Neuropsychology is a subspecialty of clinical psychology that is focused on understanding the brain–behavior relationship. Drawing information from multiple disciplines, including psychiatry and neurology, neuropsychology seeks to uncover the cognitive, behavioral, and emotional difficulties that can result from known or suspected brain dysfunction. Increasingly, to protect the public and referral sources, clinical psychologists who perform neuropsychological testing demonstrate their competence through board certification (eg, the American Board of Clinical Neuropsychology).
How is testing conducted? Evaluations comprise measures that are standardized, scored objectively, and have established psychometric properties. Testing can performed on an outpatient or inpatient basis; the duration of testing depends on the question for which the referring practitioner seeks an answer.
Measures typically are administered by paper and pencil, although computer-based
assessments are increasingly being employed. Because of the influence of demographic variables (age, sex, years of education, race), scores are compared with normative samples that resemble those of the patient’s background as closely as possible.
A thorough clinical interview with the patient, a collateral interview with caregivers
and family, and review of relevant medical records are crucial parts of the assessment. Multiple areas of cognition are assessed:
• intelligence
• academic functioning
• attention
• working memory
• speed of processing
• learning and memory
• visual spatial skills
• fine motor skills
• executive functioning.
Essentially, the evaluation speaks to a patient’s neurocognitive functioning and cerebral integrity.
How are results scored? Interpretation of test scores is contingent on expectations of how a patient should perform in the absence of neurologic or psychiatric illness (ie, based on normative data and performancebased estimates of premorbid functioning).1 The overall pattern of intact scores and deficit scores can be used to form specific impressions about a diagnosis, cognitive strengths and weaknesses, and strategies for intervention.
Personality testing. In addition to the cognitive aspect of the evaluation, personality measures are incorporated when relevant to the referral question or presenting concern.
Personality tests can be broadly divided into objective and projective measures.
Objective personality measures, such as the Minnesota Multiphasic Personality
Inventory-Second Edition, require the examinee to respond to a set of items as
true or false or on a Likert-type scale from strongly agree to disagree. Responses are then scored in standardized fashion, making comparisons to normative data, which are then analyzed to determine the extent to which the examinee experiences psychiatric symptoms.
As part of testing, patients’ responses to ambiguous or unstructured standard
stimuli—such as a series of drawings, abstract patterns, or incomplete sentences—
are analyzed to determine underlying personality traits, feelings, and attitudes.
Classic examples of these measures include the Rorschach Test and the Thematic
Apperception Test.
Personality measures and psychiatric testing are designed to answer questions
related to patients’ emotional status. These measures assess psychiatric symptoms and diagnoses, whereas neuropsychological measures provide an understanding of patients’ cognitive assets and limitations.
7 Common questions about neuropsychological testing
1 Will my patient’s insurance cover these assessments? The question is common from practitioners who are considering requesting an assessment for a patient. The short answer is “Yes.”
Most payers follow Medicare guidelines for reimbursement of neuropsychological
testing; if testing is determined to be medically necessary, insurance companies often cover the assessment. Medicaid also pays for psychometric testing services. Neuropsychologists who have a hospital-based practice typically include patients
with all types of insurance coverage. For example, 40% of patients seen in a hospital are covered by Medicare or Medicaid.2
A caveat: Local intermediaries interpret policies and procedures in different ways,
so there is variability in coverage by geographic region. That is why it is crucial
for neuropsychologists to obtain preauthorization, as would be the case with other medical procedures and services sought by referral.
Last, insurance companies do not pay for assessment of a learning disability. The
rationale typically offered for this lack of coverage? The assessment is for academic, not medical, purposes. In such a situation, patients and their families are offered a private-pay option.
2 What are the indications for neuropsychological assessment? Psychiatric practitioners are one of the top medical specialties that refer their patients for neuropsychological testing.3 This is because many patients with a psychiatric or
neurologic disorder experience changes in cognition, mood, and personality. Such
changes can range in severity from subtle to dramatic, and might reflect an underlying disease state or a side effect of medication or other treatment. Whatever the nature of a patient’s problem, careful assessment might help elucidate specific areas with which he (she) is struggling—so that you can better target your interventions. Table 1 lists common reasons for referring a patient for neuropsychological evaluation. Throughout this discussion, we describe examples of clinical situations in which neuropsychological testing is useful for establishing a differential diagnosis and dispositional planning.
3 How does neuropsychological testing help with the differential diagnosis? As an example, one area in which cognitive testing can be beneficial is in geriatric psychiatry.Dementia. Aging often is accompanied by a normal decline in memory and other cognitive functions. But because subtle changes in memory and cognition also canbe the sign of a progressive cognitive disorder, differentiating normal aging from early dementia is essential. Table 2 summarizes typical changes in cognition with aging.
Neuropsychologists, through knowledge of psychometric testing and the brain−behavior relationship, can help you detect dementia and plan treatment early. To determine if cognitive changes are progressive, patients might undergo re-evaluation—typically, every 6 to 12 months—to ascertain if changes have occurred. Mood disorders. Neuropsychological evaluation can be useful in building a differential diagnosis when determining whether cognitive symptoms are attributable to a mood disorder or a medical illness. Cognitive deficits associated with an affective disturbance generally include impairments in attention, memory, and executive functioning.4 The severity of deficits has been linked to severity of illness. When patients with a mood disorder demonstrate localizing impairments or those of greater severity than expected, suspicion arises that another cause likely better explains those deficits, and further medical testing then is often recommended. Medical procedures. Increasingly, neuropsychological assessment is used to assist in determining the appropriateness of medical procedures. For example, neurosurgical patients being considered for deep brain stimulation, brain tumor resectioning, and epilepsy surgery often are referred for preoperative and postoperative testing. Treating clinicians need an understanding of current cognitive status, localization of functioning, and psychological status to make appropriate decisions about a patient’s candidacy for one of these procedures,and to understand associated risk.
4 How is neuropsychological testing used for dispositional planning? The
results of cognitive and psychological testing have implications for dispositional
planning for patients who are receiving psychiatric care. The primary issue often is
to determine the patient’s level of independence and ability to make decisions about his affairs.5
Neuropsychological testing can help determine if cognitive deficits limit aspects of functional independence—for example, can the patient live alone, or must he live with family or in a residential care facility? Generally, the greater the cognitive impairment, the more supervision and assistance are required. This relationship between cognitive ability and independence in activities of daily living has been demonstrated in many groups of psychiatric patients, including older adults with dementia,6 patients with schizophrenia,7 and those with bipolar disorder.8
Specific recommendations can be made regarding management of finances, administering medications, and driving. To formulate an appropriate dispositional plan, the referring psychiatrist might integrate recommendations from the neuropsychological assessment with findings of other evaluations and with information that has been collected about the patient.
5 Can neuropsychological testing be used to refer a patient for neurological and cognitive rehabilitation? Yes. The neuropsychologist is singularly qualified to make recommendations about a range of interventions for cognitive deficits that have been identified on formal testing.
Typically, recommendations for addressing cognitive deficits involve rehabilitation
focused on development and use of compensatory strategies and modification to promote brain health.9,10 Rehabilitation therapy typically is aimed at increasing functioning independence and reducing physical and cognitive deficits associated
with illness (eg, traumatic brain injury [TBI], stroke, orthopedic injury, debility).
Patients who have a TBI or who have had a stroke often have comorbid psychiatric problems, including mood and anxiety disorders, that can exacerbate deficits and impede engagement in rehabilitation. The neuropsychological evaluation can determine if this is the case and if psychiatric consultation is warranted to assist with managing symptoms.
Premorbid psychiatric illness can affect rehabilitation. Formal neuropsychological testing can assist with parsing out deficits associated with new-onset illness compared with premorbid psychiatric problems. The evaluation of a patient before he begins rehabilitation also can be compared with evaluations made during treatment and after discharge to 1) assess for changes and 2) update recommendations about management.
Recommendations about cognitive interventions might include specific compensatory strategies to address areas of weakness and capitalize on strengths. Such strategies can include using internal mnemonics, such as visual imagery (ie, using a visual image to help encode verbal information) or semantic elaboration (using semantic cues to aid in encoding and recall of information). Methods can help train patients to capitalize on areas of stronger cognitive functioning in compensating for their weaknesses; an example is the spaced-retrieval technique, which relies on repetition of information that is to be learned over time.11
Perhaps the most practical strategies for addressing areas of weakness are nonmnemonic-based external memory aids, such as diaries, notebooks, calendars, alarms, and lists.12 For example, for a patient with a TBI who has impaired memory, recommendations might include using written notes or a calendar system; using a pillbox for medication management; and using labels to promote structure and consistency in the home. These strategies are meant to promote increased independence and to minimize the effect of cognitive deficits on daily functioning.
Recommended strategies can include lifestyle changes to promote improved cognitive functioning and overall health, such as:
• sleep hygiene, to reduce the effects of fatigue
• encouraging the patient to adhere to a diet, take prescribed medications, and follow up with his health care providers
• developing cognitive and physical exercise routines.
In addition, a patient who has had a stroke or who have a TBI might benefit from psychotherapy or referral to a group program or community resources to help cope with the effects of illness.13
6 How does neuropsychological testing help determine the appropriate psychiatric intervention for a patient? Results of neuropsychological testing can help determine appropriate interventions for a psychiatric condition that might be the principal factor affecting the patient’s functioning.
Concerning psychoactive medications, consider the following:
Mood and anxiety disorders. Neuropsychological measures can help substantiate the need for pharmacotherapy in a comorbid mood or anxiety disorder in a patient who has a neurologic illness, such as stroke or TBI.
ADHD. In a patient who has attentiondeficit/hyperactivity disorder (ADHD), results of cognitive testing might help determine if attention issues undermine daily functioning. Testing provides information beyond rating scale scores to justify diagnosis and psychopharmacotherapy.14
Dementia. Geriatric patients who have dementia often have coexisting behavioral and mood changes that, once evaluated, might improve with pharmacotherapy.
Other areas. Cognitive side effects of medications can be monitored by conducting testing before and after medication is started. The evaluation can address the patient’s ability to engage in psychotherapeutic interventions. Patients who have severe cognitive deficits might have greater difficulty engaging in psychotherapy, compared with patients who have less severe, or no, cognitive
impairment.15
7 Does neuropsychological testing help patients make return-to-work
and return-to-school decisions? Yes. Cognitive and psychiatric functioning have
implications for decisions about occupational and academic pursuits.
Patients who have severe cognitive or psychiatric symptoms might be or might not be able to maintain gainful employment or participate in school. Testing can help 1) document and justify disability and 2) establish recommendations about disability status. Those whose cognitive impairments or psychiatric symptoms are less severe might benefit from neuropsychological testing so that recommendations can be made regarding accommodations at work or in school, such as:
• reduced work or school schedule
• reduced level of occupational or academic demand
• change in supervision or evaluation procedures by employer or school.
Cognitive strengths and weaknesses can be used to help a patient devise and implement compensatory strategies at work or school, such as:
• note-taking
• audio recording of meetings and lectures
• using a calendar.
Patients sometimes benefit from formal vocational rehabilitation services to facilitate finding appropriate employment, returning to employment, and implementing workplace accommodations.
In conclusion
Neuropsychological evaluation, typically covered by health insurance, provides the
referring clinician with objective information about patients’ cognitive assets and limitations. In turn, this information can help you make a diagnosis and plan
treatment.
Unlike psychological testing, in which the patient is assessed for psychiatric
symptoms and conditions, neuropsychological measures offer insight into such
abilities as attention, memory, and reasoning. Neuropsychological evaluations also
can add insight to your determination of the cause of symptoms, thereby influencing decisions about medical therapy.
Last, these evaluations can aid with decision-making about dispositional planning
and whether adjunctive services, such as rehabilitation, would be of benefit.
Bottom Line
Neuropsychological assessments are a useful consultation to consider for patients
in a psychiatric setting. These evaluations can aid you in building and narrowing the differential diagnosis; identifying patients’ strengths and weakness; and making informed recommendations about functional independence.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Neuropsychological evaluation, consisting of a thorough examination of cognitive and behavioral functioning, can make an invaluable contribution to the care of psychiatric patients. Through the vehicle of standardized measures of abilities, patients’ cognitive strengths and weaknesses can be elucidated—revealing potential areas for further interventions or to explain impediments to treatment. A licensed clinical psychologist provides this service.
You, as a consumer of reported findings, can use the results to inform your diagnosis and treatment plan. Recommendations from the neuropsychologist often address dispositional planning, cognitive intervention, psychiatric intervention, and work and school accommodations.
Probing the brain−behavior relationship
Neuropsychology is a subspecialty of clinical psychology that is focused on understanding the brain–behavior relationship. Drawing information from multiple disciplines, including psychiatry and neurology, neuropsychology seeks to uncover the cognitive, behavioral, and emotional difficulties that can result from known or suspected brain dysfunction. Increasingly, to protect the public and referral sources, clinical psychologists who perform neuropsychological testing demonstrate their competence through board certification (eg, the American Board of Clinical Neuropsychology).
How is testing conducted? Evaluations comprise measures that are standardized, scored objectively, and have established psychometric properties. Testing can performed on an outpatient or inpatient basis; the duration of testing depends on the question for which the referring practitioner seeks an answer.
Measures typically are administered by paper and pencil, although computer-based
assessments are increasingly being employed. Because of the influence of demographic variables (age, sex, years of education, race), scores are compared with normative samples that resemble those of the patient’s background as closely as possible.
A thorough clinical interview with the patient, a collateral interview with caregivers
and family, and review of relevant medical records are crucial parts of the assessment. Multiple areas of cognition are assessed:
• intelligence
• academic functioning
• attention
• working memory
• speed of processing
• learning and memory
• visual spatial skills
• fine motor skills
• executive functioning.
Essentially, the evaluation speaks to a patient’s neurocognitive functioning and cerebral integrity.
How are results scored? Interpretation of test scores is contingent on expectations of how a patient should perform in the absence of neurologic or psychiatric illness (ie, based on normative data and performancebased estimates of premorbid functioning).1 The overall pattern of intact scores and deficit scores can be used to form specific impressions about a diagnosis, cognitive strengths and weaknesses, and strategies for intervention.
Personality testing. In addition to the cognitive aspect of the evaluation, personality measures are incorporated when relevant to the referral question or presenting concern.
Personality tests can be broadly divided into objective and projective measures.
Objective personality measures, such as the Minnesota Multiphasic Personality
Inventory-Second Edition, require the examinee to respond to a set of items as
true or false or on a Likert-type scale from strongly agree to disagree. Responses are then scored in standardized fashion, making comparisons to normative data, which are then analyzed to determine the extent to which the examinee experiences psychiatric symptoms.
As part of testing, patients’ responses to ambiguous or unstructured standard
stimuli—such as a series of drawings, abstract patterns, or incomplete sentences—
are analyzed to determine underlying personality traits, feelings, and attitudes.
Classic examples of these measures include the Rorschach Test and the Thematic
Apperception Test.
Personality measures and psychiatric testing are designed to answer questions
related to patients’ emotional status. These measures assess psychiatric symptoms and diagnoses, whereas neuropsychological measures provide an understanding of patients’ cognitive assets and limitations.
7 Common questions about neuropsychological testing
1 Will my patient’s insurance cover these assessments? The question is common from practitioners who are considering requesting an assessment for a patient. The short answer is “Yes.”
Most payers follow Medicare guidelines for reimbursement of neuropsychological
testing; if testing is determined to be medically necessary, insurance companies often cover the assessment. Medicaid also pays for psychometric testing services. Neuropsychologists who have a hospital-based practice typically include patients
with all types of insurance coverage. For example, 40% of patients seen in a hospital are covered by Medicare or Medicaid.2
A caveat: Local intermediaries interpret policies and procedures in different ways,
so there is variability in coverage by geographic region. That is why it is crucial
for neuropsychologists to obtain preauthorization, as would be the case with other medical procedures and services sought by referral.
Last, insurance companies do not pay for assessment of a learning disability. The
rationale typically offered for this lack of coverage? The assessment is for academic, not medical, purposes. In such a situation, patients and their families are offered a private-pay option.
2 What are the indications for neuropsychological assessment? Psychiatric practitioners are one of the top medical specialties that refer their patients for neuropsychological testing.3 This is because many patients with a psychiatric or
neurologic disorder experience changes in cognition, mood, and personality. Such
changes can range in severity from subtle to dramatic, and might reflect an underlying disease state or a side effect of medication or other treatment. Whatever the nature of a patient’s problem, careful assessment might help elucidate specific areas with which he (she) is struggling—so that you can better target your interventions. Table 1 lists common reasons for referring a patient for neuropsychological evaluation. Throughout this discussion, we describe examples of clinical situations in which neuropsychological testing is useful for establishing a differential diagnosis and dispositional planning.
3 How does neuropsychological testing help with the differential diagnosis? As an example, one area in which cognitive testing can be beneficial is in geriatric psychiatry.Dementia. Aging often is accompanied by a normal decline in memory and other cognitive functions. But because subtle changes in memory and cognition also canbe the sign of a progressive cognitive disorder, differentiating normal aging from early dementia is essential. Table 2 summarizes typical changes in cognition with aging.
Neuropsychologists, through knowledge of psychometric testing and the brain−behavior relationship, can help you detect dementia and plan treatment early. To determine if cognitive changes are progressive, patients might undergo re-evaluation—typically, every 6 to 12 months—to ascertain if changes have occurred. Mood disorders. Neuropsychological evaluation can be useful in building a differential diagnosis when determining whether cognitive symptoms are attributable to a mood disorder or a medical illness. Cognitive deficits associated with an affective disturbance generally include impairments in attention, memory, and executive functioning.4 The severity of deficits has been linked to severity of illness. When patients with a mood disorder demonstrate localizing impairments or those of greater severity than expected, suspicion arises that another cause likely better explains those deficits, and further medical testing then is often recommended. Medical procedures. Increasingly, neuropsychological assessment is used to assist in determining the appropriateness of medical procedures. For example, neurosurgical patients being considered for deep brain stimulation, brain tumor resectioning, and epilepsy surgery often are referred for preoperative and postoperative testing. Treating clinicians need an understanding of current cognitive status, localization of functioning, and psychological status to make appropriate decisions about a patient’s candidacy for one of these procedures,and to understand associated risk.
4 How is neuropsychological testing used for dispositional planning? The
results of cognitive and psychological testing have implications for dispositional
planning for patients who are receiving psychiatric care. The primary issue often is
to determine the patient’s level of independence and ability to make decisions about his affairs.5
Neuropsychological testing can help determine if cognitive deficits limit aspects of functional independence—for example, can the patient live alone, or must he live with family or in a residential care facility? Generally, the greater the cognitive impairment, the more supervision and assistance are required. This relationship between cognitive ability and independence in activities of daily living has been demonstrated in many groups of psychiatric patients, including older adults with dementia,6 patients with schizophrenia,7 and those with bipolar disorder.8
Specific recommendations can be made regarding management of finances, administering medications, and driving. To formulate an appropriate dispositional plan, the referring psychiatrist might integrate recommendations from the neuropsychological assessment with findings of other evaluations and with information that has been collected about the patient.
5 Can neuropsychological testing be used to refer a patient for neurological and cognitive rehabilitation? Yes. The neuropsychologist is singularly qualified to make recommendations about a range of interventions for cognitive deficits that have been identified on formal testing.
Typically, recommendations for addressing cognitive deficits involve rehabilitation
focused on development and use of compensatory strategies and modification to promote brain health.9,10 Rehabilitation therapy typically is aimed at increasing functioning independence and reducing physical and cognitive deficits associated
with illness (eg, traumatic brain injury [TBI], stroke, orthopedic injury, debility).
Patients who have a TBI or who have had a stroke often have comorbid psychiatric problems, including mood and anxiety disorders, that can exacerbate deficits and impede engagement in rehabilitation. The neuropsychological evaluation can determine if this is the case and if psychiatric consultation is warranted to assist with managing symptoms.
Premorbid psychiatric illness can affect rehabilitation. Formal neuropsychological testing can assist with parsing out deficits associated with new-onset illness compared with premorbid psychiatric problems. The evaluation of a patient before he begins rehabilitation also can be compared with evaluations made during treatment and after discharge to 1) assess for changes and 2) update recommendations about management.
Recommendations about cognitive interventions might include specific compensatory strategies to address areas of weakness and capitalize on strengths. Such strategies can include using internal mnemonics, such as visual imagery (ie, using a visual image to help encode verbal information) or semantic elaboration (using semantic cues to aid in encoding and recall of information). Methods can help train patients to capitalize on areas of stronger cognitive functioning in compensating for their weaknesses; an example is the spaced-retrieval technique, which relies on repetition of information that is to be learned over time.11
Perhaps the most practical strategies for addressing areas of weakness are nonmnemonic-based external memory aids, such as diaries, notebooks, calendars, alarms, and lists.12 For example, for a patient with a TBI who has impaired memory, recommendations might include using written notes or a calendar system; using a pillbox for medication management; and using labels to promote structure and consistency in the home. These strategies are meant to promote increased independence and to minimize the effect of cognitive deficits on daily functioning.
Recommended strategies can include lifestyle changes to promote improved cognitive functioning and overall health, such as:
• sleep hygiene, to reduce the effects of fatigue
• encouraging the patient to adhere to a diet, take prescribed medications, and follow up with his health care providers
• developing cognitive and physical exercise routines.
In addition, a patient who has had a stroke or who have a TBI might benefit from psychotherapy or referral to a group program or community resources to help cope with the effects of illness.13
6 How does neuropsychological testing help determine the appropriate psychiatric intervention for a patient? Results of neuropsychological testing can help determine appropriate interventions for a psychiatric condition that might be the principal factor affecting the patient’s functioning.
Concerning psychoactive medications, consider the following:
Mood and anxiety disorders. Neuropsychological measures can help substantiate the need for pharmacotherapy in a comorbid mood or anxiety disorder in a patient who has a neurologic illness, such as stroke or TBI.
ADHD. In a patient who has attentiondeficit/hyperactivity disorder (ADHD), results of cognitive testing might help determine if attention issues undermine daily functioning. Testing provides information beyond rating scale scores to justify diagnosis and psychopharmacotherapy.14
Dementia. Geriatric patients who have dementia often have coexisting behavioral and mood changes that, once evaluated, might improve with pharmacotherapy.
Other areas. Cognitive side effects of medications can be monitored by conducting testing before and after medication is started. The evaluation can address the patient’s ability to engage in psychotherapeutic interventions. Patients who have severe cognitive deficits might have greater difficulty engaging in psychotherapy, compared with patients who have less severe, or no, cognitive
impairment.15
7 Does neuropsychological testing help patients make return-to-work
and return-to-school decisions? Yes. Cognitive and psychiatric functioning have
implications for decisions about occupational and academic pursuits.
Patients who have severe cognitive or psychiatric symptoms might be or might not be able to maintain gainful employment or participate in school. Testing can help 1) document and justify disability and 2) establish recommendations about disability status. Those whose cognitive impairments or psychiatric symptoms are less severe might benefit from neuropsychological testing so that recommendations can be made regarding accommodations at work or in school, such as:
• reduced work or school schedule
• reduced level of occupational or academic demand
• change in supervision or evaluation procedures by employer or school.
Cognitive strengths and weaknesses can be used to help a patient devise and implement compensatory strategies at work or school, such as:
• note-taking
• audio recording of meetings and lectures
• using a calendar.
Patients sometimes benefit from formal vocational rehabilitation services to facilitate finding appropriate employment, returning to employment, and implementing workplace accommodations.
In conclusion
Neuropsychological evaluation, typically covered by health insurance, provides the
referring clinician with objective information about patients’ cognitive assets and limitations. In turn, this information can help you make a diagnosis and plan
treatment.
Unlike psychological testing, in which the patient is assessed for psychiatric
symptoms and conditions, neuropsychological measures offer insight into such
abilities as attention, memory, and reasoning. Neuropsychological evaluations also
can add insight to your determination of the cause of symptoms, thereby influencing decisions about medical therapy.
Last, these evaluations can aid with decision-making about dispositional planning
and whether adjunctive services, such as rehabilitation, would be of benefit.
Bottom Line
Neuropsychological assessments are a useful consultation to consider for patients
in a psychiatric setting. These evaluations can aid you in building and narrowing the differential diagnosis; identifying patients’ strengths and weakness; and making informed recommendations about functional independence.
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Donders J. A survey of report writing by neuropsychologists, I: general characteristics and content. Clin Neuropsychol. 2001;15(2):137-149.
2. Lamberty GT, Courtney JC, Heilbronner RC. The practice of clinical neuropsychology: a survey of practices and settings. New York, NY: Taylor & Francis; 2005.
3. Sweet JJ, Meyer DG, Nelson NW, et al. The TCN/AACN 2010 “salary survey”: professional practices, beliefs, and incomes of U.S. neuropsychologists. Clin Neuropsychol. 2011;25(1):12-61.
4. Marvel CL, Paradiso S. Cognitive and neurologic impairment in mood disorders. Psychiatr Clin North Am. 2004;27(1):19-36,vii-viii.
5. Moberg PJ, Rick JH. Decision-making capacity and competency in the elderly: a clinical and neuropsychological perspective. NeuroRehabilitation. 2008;23(5):
403-413.
6. Bradshaw LE, Goldberg SE, Lewis SA, et al. Six-month outcomes following an emergency hospital admission for older adults with co-morbid mental health problems indicate complexity of care needs. Age Ageing. 2013; 42(5):582-588.
7. Medalia A, Lim RW. Self-awareness of cognitive functioning in schizophrenia. Schizophr Res. 2004;71(2-3):331-338.
8. Andreou C, Bozikas VP. The predictive significance of neurocognitive factors for functional outcome in bipolar disorder. Curr Opin Psychiatry. 2013;26(1):54-59.
9. Stuss DT, Winocur G, Robertson IH, eds. Cognitive neurorehabilitation: evidence and application. 2nd ed. New York, NY: Cambridge University Press; 2008.
10. Raskin SA, ed. Neuroplasticity and rehabilitation. New York, NY: The Guilford Press; 2011.
11. Glisky EL, Glisky ML. Memory rehabilitation in older adults. In: Stuss DT, Winocur G, Robertson IH. Cognitive neurorehabilitation. 1st ed. New York, NY: Cambridge University Press; 2008.
12. Kapur N, Glisky EL, Wilson BA. External memory aids and computers in memory rehabilitation. In: Baddeley AD, Kopelman MD, Wilson BA. Handbook of memory disorders. Chichester, United Kingdom: Wiley; 2002:757-784.
13. Stalder-Luthy F, Messerli-Burgy N, Hofer H, et al. Effect of psychological interventions on depressive symptoms in long-term rehabilitation after an acquired brain injury: a systematic review and meta-analysis. Arch Phys Med Rehabil.
2013;94(7):1386-1397.
14. Hale JB, Reddy LA, Semrud-Clikeman M, et al. Executive impairment determines ADHD medication response: implications for academic achievement. J Learn Disabil. 2011;44(2):196-212.
15. Medalia A, Lim R. Treatment of cognitive dysfunction in psychiatric disorders. J Psychiatr Pract. 2004;10(1):17-25.
1. Donders J. A survey of report writing by neuropsychologists, I: general characteristics and content. Clin Neuropsychol. 2001;15(2):137-149.
2. Lamberty GT, Courtney JC, Heilbronner RC. The practice of clinical neuropsychology: a survey of practices and settings. New York, NY: Taylor & Francis; 2005.
3. Sweet JJ, Meyer DG, Nelson NW, et al. The TCN/AACN 2010 “salary survey”: professional practices, beliefs, and incomes of U.S. neuropsychologists. Clin Neuropsychol. 2011;25(1):12-61.
4. Marvel CL, Paradiso S. Cognitive and neurologic impairment in mood disorders. Psychiatr Clin North Am. 2004;27(1):19-36,vii-viii.
5. Moberg PJ, Rick JH. Decision-making capacity and competency in the elderly: a clinical and neuropsychological perspective. NeuroRehabilitation. 2008;23(5):
403-413.
6. Bradshaw LE, Goldberg SE, Lewis SA, et al. Six-month outcomes following an emergency hospital admission for older adults with co-morbid mental health problems indicate complexity of care needs. Age Ageing. 2013; 42(5):582-588.
7. Medalia A, Lim RW. Self-awareness of cognitive functioning in schizophrenia. Schizophr Res. 2004;71(2-3):331-338.
8. Andreou C, Bozikas VP. The predictive significance of neurocognitive factors for functional outcome in bipolar disorder. Curr Opin Psychiatry. 2013;26(1):54-59.
9. Stuss DT, Winocur G, Robertson IH, eds. Cognitive neurorehabilitation: evidence and application. 2nd ed. New York, NY: Cambridge University Press; 2008.
10. Raskin SA, ed. Neuroplasticity and rehabilitation. New York, NY: The Guilford Press; 2011.
11. Glisky EL, Glisky ML. Memory rehabilitation in older adults. In: Stuss DT, Winocur G, Robertson IH. Cognitive neurorehabilitation. 1st ed. New York, NY: Cambridge University Press; 2008.
12. Kapur N, Glisky EL, Wilson BA. External memory aids and computers in memory rehabilitation. In: Baddeley AD, Kopelman MD, Wilson BA. Handbook of memory disorders. Chichester, United Kingdom: Wiley; 2002:757-784.
13. Stalder-Luthy F, Messerli-Burgy N, Hofer H, et al. Effect of psychological interventions on depressive symptoms in long-term rehabilitation after an acquired brain injury: a systematic review and meta-analysis. Arch Phys Med Rehabil.
2013;94(7):1386-1397.
14. Hale JB, Reddy LA, Semrud-Clikeman M, et al. Executive impairment determines ADHD medication response: implications for academic achievement. J Learn Disabil. 2011;44(2):196-212.
15. Medalia A, Lim R. Treatment of cognitive dysfunction in psychiatric disorders. J Psychiatr Pract. 2004;10(1):17-25.
