User login
Benefits and costs of accepting credit cards in your practice
Are you tired of waiting for checks in the mail? Do patients leave without paying their balance? Streamlining revenue collection by taking credit cards is a tantalizing antidote to these ills, but it has downsides. Weighing the value for you and your patients is necessary before you decide on this important practice management policy.
Clinical and practical advantages
Many patients prefer that their health care practitioners take credit cards, because it simplifies their busy lives—and who carries a checkbook anymore? Patients can put the whole session to good use without sacrificing time taking care of payment. They also can receive credit card rewards for their payment, or use health savings accounts, health reimbursement accounts, or flexible spending debit cards, making treatment more affordable.
Benefits of credit cards
Accepting credit cards has many benefits:
- Allows more time in a session to focus on clinical matters because you do not have to allocate time to collect payment, which might include dealing with a forgotten checkbook or a request for a change in your payment policies.
- Easier to collect payment for no-shows. This could result in a reduced no-show rate, because a patient might feel more accountable to show up knowing that his (her) credit card is on file.
- Saves time recording and depositing checks.
- Avoids bounced checks and collection agencies.
Money doesn’t grow on trees
Although there are advantages to accepting credit cards, several costs should be considered. Some practitioners feel that accepting credit cards makes their practice seem like a commercial business. There also is an expense of accepting credit cards, and understanding these costs can be confusing because there are different processing systems of rates. Whether the rate is flat, tiered, or wholesale, you always will pay a percentage of the transaction, plus a transaction fee.
Here are some general guidelines on rates:
- Debit cards are the least expensive to process but often have low spending limits.
- Rewards cards, such as frequent flyer cards, are the most expensive to process. Have you ever wondered who foots the bill for those frequent flyer miles? It’s not the airline; it’s the merchant (you).
- For tiered rates, swiping cards is typically cheaper than typing in the credit card info. Tiered rates often have low rates, known as “teaser” rates, because they are applicable in far fewer cases.
- For flat or wholesale rates, securely saving credit card numbers is not any more expensive than swiping a card, and saves time in the long run and potential awkwardness at the end of a session.
- A higher volume of processed credit cards might allow you to negotiate your rates.
- Check if your bank offers a less expensive option. Some banks offer preferred rates for their customers.
Also consider the time and possible expense of ensuring that you are Payment Card Industry Data Security Standard compliant (information security standards that aim to keep cardholder data secure).
Different methods of processing transactions have varying levels of requirements:
- A swiping reader with a terminal connected to a telephone line is more secure than through the Internet and carries fewer compliance burdens. Use a reader that can handle chip-cards, because you could be liable for fraudulent transactions.
- Do not save or store credit card numbers you typed yourself. Compliance is less burdensome if patients input credit card data into a secure portal.
- Store credit card data securely via your credit card processing partner, although the partner is still at risk of a data breach. Practitioners should weigh the value of convenience vs security.
- If there is a data breach and you are found negligent you could be fined $5,000 to $100,000 per month, depending on whether you are a large company or solo practice.
Bottom dollar
Credit card processing has significant advantages from both a practice management and clinical standpoint. Because prices for services vary, shop around to find the best rates and educate yourself about security requirements. Taking the time to research these matters can pay off in the long term.
Are you tired of waiting for checks in the mail? Do patients leave without paying their balance? Streamlining revenue collection by taking credit cards is a tantalizing antidote to these ills, but it has downsides. Weighing the value for you and your patients is necessary before you decide on this important practice management policy.
Clinical and practical advantages
Many patients prefer that their health care practitioners take credit cards, because it simplifies their busy lives—and who carries a checkbook anymore? Patients can put the whole session to good use without sacrificing time taking care of payment. They also can receive credit card rewards for their payment, or use health savings accounts, health reimbursement accounts, or flexible spending debit cards, making treatment more affordable.
Benefits of credit cards
Accepting credit cards has many benefits:
- Allows more time in a session to focus on clinical matters because you do not have to allocate time to collect payment, which might include dealing with a forgotten checkbook or a request for a change in your payment policies.
- Easier to collect payment for no-shows. This could result in a reduced no-show rate, because a patient might feel more accountable to show up knowing that his (her) credit card is on file.
- Saves time recording and depositing checks.
- Avoids bounced checks and collection agencies.
Money doesn’t grow on trees
Although there are advantages to accepting credit cards, several costs should be considered. Some practitioners feel that accepting credit cards makes their practice seem like a commercial business. There also is an expense of accepting credit cards, and understanding these costs can be confusing because there are different processing systems of rates. Whether the rate is flat, tiered, or wholesale, you always will pay a percentage of the transaction, plus a transaction fee.
Here are some general guidelines on rates:
- Debit cards are the least expensive to process but often have low spending limits.
- Rewards cards, such as frequent flyer cards, are the most expensive to process. Have you ever wondered who foots the bill for those frequent flyer miles? It’s not the airline; it’s the merchant (you).
- For tiered rates, swiping cards is typically cheaper than typing in the credit card info. Tiered rates often have low rates, known as “teaser” rates, because they are applicable in far fewer cases.
- For flat or wholesale rates, securely saving credit card numbers is not any more expensive than swiping a card, and saves time in the long run and potential awkwardness at the end of a session.
- A higher volume of processed credit cards might allow you to negotiate your rates.
- Check if your bank offers a less expensive option. Some banks offer preferred rates for their customers.
Also consider the time and possible expense of ensuring that you are Payment Card Industry Data Security Standard compliant (information security standards that aim to keep cardholder data secure).
Different methods of processing transactions have varying levels of requirements:
- A swiping reader with a terminal connected to a telephone line is more secure than through the Internet and carries fewer compliance burdens. Use a reader that can handle chip-cards, because you could be liable for fraudulent transactions.
- Do not save or store credit card numbers you typed yourself. Compliance is less burdensome if patients input credit card data into a secure portal.
- Store credit card data securely via your credit card processing partner, although the partner is still at risk of a data breach. Practitioners should weigh the value of convenience vs security.
- If there is a data breach and you are found negligent you could be fined $5,000 to $100,000 per month, depending on whether you are a large company or solo practice.
Bottom dollar
Credit card processing has significant advantages from both a practice management and clinical standpoint. Because prices for services vary, shop around to find the best rates and educate yourself about security requirements. Taking the time to research these matters can pay off in the long term.
Are you tired of waiting for checks in the mail? Do patients leave without paying their balance? Streamlining revenue collection by taking credit cards is a tantalizing antidote to these ills, but it has downsides. Weighing the value for you and your patients is necessary before you decide on this important practice management policy.
Clinical and practical advantages
Many patients prefer that their health care practitioners take credit cards, because it simplifies their busy lives—and who carries a checkbook anymore? Patients can put the whole session to good use without sacrificing time taking care of payment. They also can receive credit card rewards for their payment, or use health savings accounts, health reimbursement accounts, or flexible spending debit cards, making treatment more affordable.
Benefits of credit cards
Accepting credit cards has many benefits:
- Allows more time in a session to focus on clinical matters because you do not have to allocate time to collect payment, which might include dealing with a forgotten checkbook or a request for a change in your payment policies.
- Easier to collect payment for no-shows. This could result in a reduced no-show rate, because a patient might feel more accountable to show up knowing that his (her) credit card is on file.
- Saves time recording and depositing checks.
- Avoids bounced checks and collection agencies.
Money doesn’t grow on trees
Although there are advantages to accepting credit cards, several costs should be considered. Some practitioners feel that accepting credit cards makes their practice seem like a commercial business. There also is an expense of accepting credit cards, and understanding these costs can be confusing because there are different processing systems of rates. Whether the rate is flat, tiered, or wholesale, you always will pay a percentage of the transaction, plus a transaction fee.
Here are some general guidelines on rates:
- Debit cards are the least expensive to process but often have low spending limits.
- Rewards cards, such as frequent flyer cards, are the most expensive to process. Have you ever wondered who foots the bill for those frequent flyer miles? It’s not the airline; it’s the merchant (you).
- For tiered rates, swiping cards is typically cheaper than typing in the credit card info. Tiered rates often have low rates, known as “teaser” rates, because they are applicable in far fewer cases.
- For flat or wholesale rates, securely saving credit card numbers is not any more expensive than swiping a card, and saves time in the long run and potential awkwardness at the end of a session.
- A higher volume of processed credit cards might allow you to negotiate your rates.
- Check if your bank offers a less expensive option. Some banks offer preferred rates for their customers.
Also consider the time and possible expense of ensuring that you are Payment Card Industry Data Security Standard compliant (information security standards that aim to keep cardholder data secure).
Different methods of processing transactions have varying levels of requirements:
- A swiping reader with a terminal connected to a telephone line is more secure than through the Internet and carries fewer compliance burdens. Use a reader that can handle chip-cards, because you could be liable for fraudulent transactions.
- Do not save or store credit card numbers you typed yourself. Compliance is less burdensome if patients input credit card data into a secure portal.
- Store credit card data securely via your credit card processing partner, although the partner is still at risk of a data breach. Practitioners should weigh the value of convenience vs security.
- If there is a data breach and you are found negligent you could be fined $5,000 to $100,000 per month, depending on whether you are a large company or solo practice.
Bottom dollar
Credit card processing has significant advantages from both a practice management and clinical standpoint. Because prices for services vary, shop around to find the best rates and educate yourself about security requirements. Taking the time to research these matters can pay off in the long term.

How you can simplify your patient’s medication regimen to enhance adherence
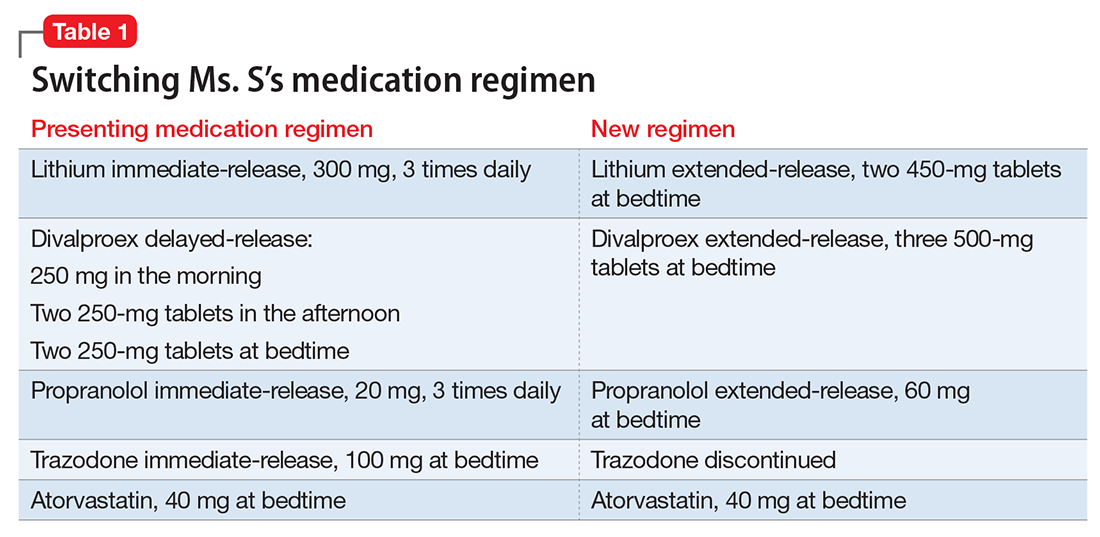
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2


Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
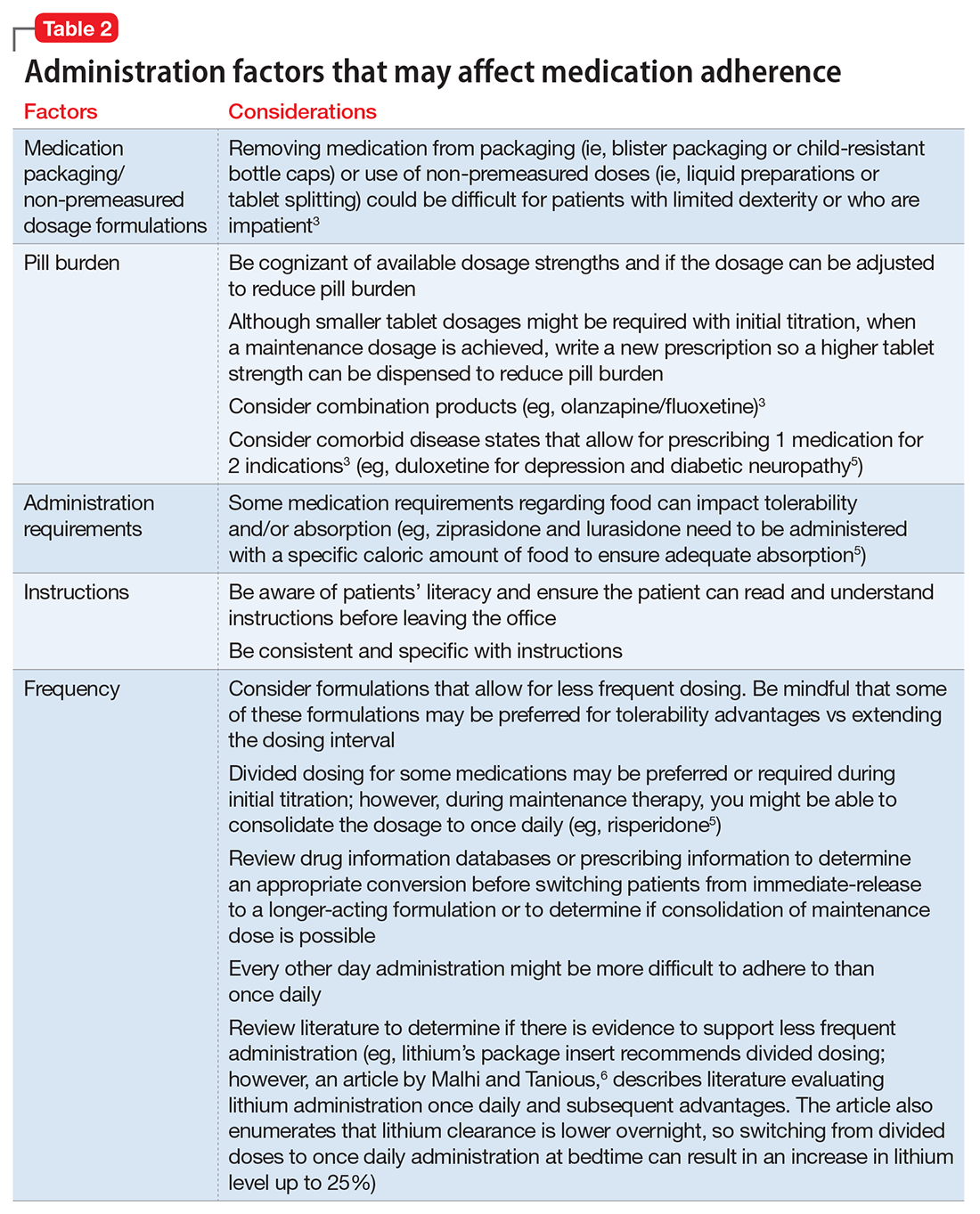
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2
Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2
Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
Valbenazine for tardive dyskinesia
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.

In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
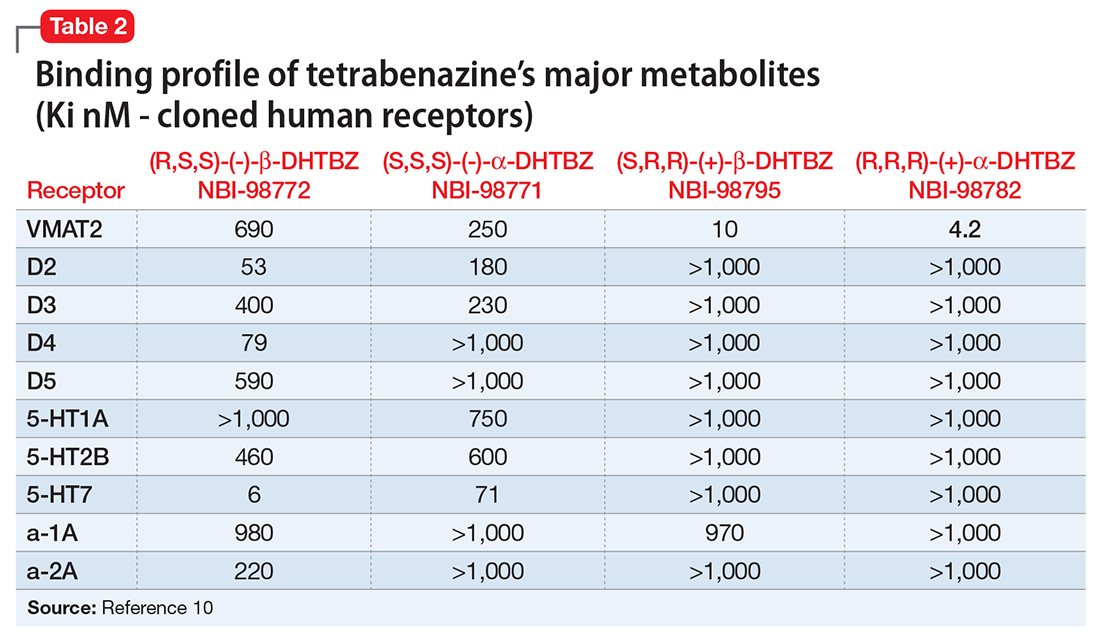
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.

Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).

Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.
In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.
Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).
Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
Despite improvements in the tolerability of antipsychotic medications, the development of tardive dyskinesia (TD) still is a significant area of concern; however, clinicians have had few treatment options. Valbenazine, a vesicular monoamine transport type 2 (VMAT2) inhibitor, is the only FDA-approved medication for TD (Table 1).1 By modulating dopamine transport into presynaptic vesicles, synaptic dopamine release is decreased, thereby reducing the postsynaptic stimulation of D2 receptors and the severity of dyskinetic movements.
In the pivotal 6-week clinical trial, valbenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) ratings.2 Study completion rates were high (87.6%), with only 2 dropouts because of adverse events in each of the placebo (n = 78) and 40-mg (n = 76) arms, and 3 in the 80-mg group (n = 80).
Before the development of valbenazine, tetrabenazine was the only effective option for treating TD. Despite tetrabenazine’s known efficacy for TD, it was not available in the United States until 2008 with the sole indication for movements related to Huntington’s disease. U.S. patients often were subjected to a litany of ineffective medications for TD, often at great expense. Moreover, tetrabenazine involved multiple daily dosing, required cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, had significant tolerability issues, and a monthly cost of $8,000 to $10,000. The availability of an agent that is effective for TD and does not have tetrabenazine’s kinetic limitations, adverse effect profile, or CYP2D6 monitoring requirements represents an enormous advance in the treatment of TD.
Clinical implications
Tardive dyskinesia remains a significant public health concern because of the increasing use of antipsychotics for disorders beyond the core indication for schizophrenia. Although exposure to dopamine D2 antagonism could result in postsynaptic receptor upregulation and supersensitivity, this process best explains what underlies withdrawal dyskinesia.3 The persistence of TD symptoms in 66% to 80% of patients after discontinuing offending agents has led to hypotheses that the underlying pathophysiology of TD might best be conceptualized as a problem with neuroplasticity. As with many disorders, environmental contributions (eg, oxidative stress) and genetic predisposition might play a role beyond that related to exposure to D2 antagonism.3
There have been trials of numerous agents, but no medication has been FDA-approved for treating TD, and limited data support the efficacy of a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]),4 albeit with small effect sizes. A medical food, consisting of branched-chain amino acids, received FDA approval for the dietary management of TD in males, but is no longer commercially available except from compounding pharmacies.5
Tetrabenazine, a molecule developed in the mid-1950s to improve on the tolerability of reserpine, was associated with significant adverse effects such as orthostasis.6 Like reserpine, tetrabenazine subsequently was found to be effective for TD7 but without the peripheral adverse effects of reserpine. However, the kinetics of tetrabenazine necessitated multiple daily doses, and required CYP2D6 genotyping for doses >50 mg/d.8
Receptor blocking. The mechanism that differentiated reserpine’s and tetrabenazine’s clinical properties became clearer in the 1980s when researchers discovered that transporters were necessary to package neurotransmitters into the synaptic vesicles of presynaptic neurons.9 The vesicular monoamine transporter (VMAT) exists in 2 isoforms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the central nervous system.10
Tetrabenazine’s improved tolerability profile was related to the fact that it is a specific and reversible VMAT2 inhibitor, while reserpine is an irreversible and nonselective antagonist of both VMAT isoforms. Investigation of tetrabenazine’s metabolism revealed that it is rapidly and extensively converted into 2 isomers, α-dihydrotetrabenazine (DH-TBZ) and β-DH-TBZ. The isomeric forms of DH-TBZ have multiple chiral centers, and therefore numerous forms of which only 2 are significantly active at VMAT2.3 The α–DH-TBZ isomer is metabolized via CYP2D6 and 3A4 into inactive metabolites, while β-DH-TBZ is metabolized solely via 2D6.3 Because of the short half-life of DH-TBZ when generated from oral tetrabenazine, the existence of 2D6 polymorphisms, and the predominant activity deriving from only 2 isomers, a molecule was synthesized (valbenazine), that when metabolized would slowly be converted into the most active isomer of α–DH-TBZ designated as NBI-98782 (Table 2). This slower conversion to NBI-98782 from valbenazine (compared with its formation from oral tetrabenazine) yielded improved kinetics and permitted once-daily dosing; moreover, because the metabolism of NBI-98782 is not solely dependent on CYP2D6, the need for genotyping was removed. Neither of the 2 metabolites from valbenazine NBI-98782 and NB-136110 have significant affinity for targets other than VMAT2.11
Use in tardive dyskinesia. Recommended starting dosage is 40 mg once daily with or without food, increased to 80 mg after 1 week, based on the design and results from the phase-III clinical trial.12 The FDA granted breakthrough therapy designation for this compound, and only 1 phase-III trial was performed. Valbenazine produced significant improvement on the AIMS, with a mean 30% reduction in AIMS scores at the Week 6 endpoint from baseline of 10.4 ± 3.6.2 The effect size was large (Cohen’s d = 0.90) for the 80-mg dosage. Continuation of 40 mg/d may be considered for some patients based on tolerability, including those who are known CYP2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors. Patients taking strong 3A4 inhibitors should not exceed 40 mg/d. The maximum daily dose is 40 mg for those who have moderate or severe hepatic impairment (Child-Pugh score, 7 to 15). Dosage adjustment is not required for mild to moderate renal impairment (creatinine clearance, 30 to 90 mL/min).
Pharmacologic profile, adverse reactions
Valbenazine and its 2 metabolites lack affinity for receptors other than VMAT2, leading to an absence of orthostasis in clinical trials.1,2 In the phase-II trial, 76% of participants receiving valbenazine (n = 51) were titrated to the maximum dosage of 75 mg/d. Common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were headache (9.8% vs 4.1% placebo), fatigue (9.8% vs 4.1% placebo), and somnolence (5.9% vs 2% placebo).1 In the phase-III trial, participants were randomized 1:1:1 to valbenazine, 40 mg (n = 72), valbenazine, 80 mg (n = 79), or placebo (n = 76). In the clinical studies the most common diagnosis was schizophrenia or schizoaffective disorder, and 40% and 85% of participants in the phase-II and phase-III studies, respectively, remained on antipsychotics.1,2 There were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III trial.2
When data from all placebo-controlled studies were pooled, only 1 adverse effect occurred with an incidence ≥5% and twice that of placebo, somnolence with a rate of 10.9% for valbenazine vs 4.2% for placebo. The incidence of akathisia in the pooled analysis was 2.7% for valbenazine vs 0.5% for placebo. Importantly, in neither study was there a safety signal related to depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms.
The mean QT prolongation for valbenazine in healthy participants was 6.7 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 8.4 milliseconds. For those taking strong 2D6 or 3A4 inhibitors, or known 2D6 poor metabolizers, the mean QT prolongation was 11.7 milliseconds (14.7 milliseconds upper bound of double-sided 90% CI). In the controlled trials, there was a dose-related increase in prolactin, alkaline phosphatase, and bilirubin. Overall, 3% of valbenazine-treated patients and 2% of placebo-treated patients discontinued because of adverse reactions.
As noted above, there were no adverse effects with an incidence ≥5% and at least twice the rate of placebo in the phase-III valbenazine trial. Aggregate data across all placebo-controlled studies found that somnolence was the only adverse effect that occurred with an incidence ≥5% and twice that of placebo (10.9% for valbenazine vs 4.2% for placebo).2 As a comparsion, rates of sedation and akathisia for tetrabenazine were higher in the pivotal Huntington’s disease trial: sedation/somnolence 31% vs 3% for placebo, and akathisia 19% vs 0% for placebo.8
How it works
Tetrabenazine, a selective VMAT2 inhibitor, is the only agent that has demonstrated significant efficacy and tolerability for TD management; however, its complex metabolism generates numerous isomers of the metabolites α-DH-TBZ and β-DH-TBZ, of which only 2 are significantly active (Table 3). By choosing an active isomer (NBI-98782) as the metabolite of interest because of its selective and potent activity at VMAT2 and having a metabolism not solely dependent on CYP2D6, a compound was generated (valbenazine) that when metabolized slowly converts into NBI-98782.
Pharmacokinetics
Valbenazine demonstrates dose-proportional pharmacokinetics after single oral dosages from 40 to 300 mg with no impact of food or fasting status on levels of the active metabolite. Valbenazine has a Tmax of 0.5 to 1.0 hours, with 49% oral bioavailability. The plasma half-life for valbenazine and for NBI-98782 ranges from 15 to 22 hours. The Tmax for NBI-98782 when formed from valbenazine occurs between 4 and 8 hours, with a Cmax of approximately 30 ng/mL. It should be noted that when NBI-98782 is generated from oral tetrabenazine, the mean half-life and Tmax are considerably shorter (6 hours and 1.5 hours, respectively), while the Cmax is much higher (approximately 77 ng/mL) (Table 4).
Valbenazine is metabolized through endogenous esterases to NBI-98782 and NBI-136110. NBI-98782, the active metabolite, is further metabolized through multiple CYP pathways, predominantly 3A4 and 2D6. Neither valbenazine nor its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that valbenazine and its active metabolite are unlikely to inhibit most major drug transporters at clinically relevant concentrations. However, valbenazine increased digoxin levels because of inhibition of intestinal P-glycoprotein; therefore plasma digoxin level monitoring is recommended when these 2 are co-administered.
Efficacy
Efficacy was established in a 6-week, fixed-dosage, double-blind, placebo-controlled trial of adult patients with TD. Eligible participants had:
- DSM-IV diagnosis of antipsychotic-induced TD for ≥3 months before screening and moderate or severe TD, as indicated by AIMS item 8 (severity of abnormal movement), which was rated by a blinded, external reviewer using a video of the participant’s AIMS assessment at screening
- a DSM-IV diagnosis of schizophrenia or schizoaffective disorder or mood disorder (and stable per investigator)
- Brief Psychiatric Rating Scale score <50 at screening.
Exclusion criteria included clinically significant and unstable medical conditions within 1 month before screening; comorbid movement disorder (eg, parkinsonism, akathisia, truncal dystonia) that was more prominent than TD; and significant risk for active suicidal ideation, suicidal behavior, or violent behavior.2 Participants had a mean age of 56, 52% were male, and 65.7% of participants in the valbenazine 40-mg group had a schizophrenia spectrum disorder diagnosis, as did 65.8% in both the placebo and valbenazine 80-mg arms.
Antipsychotic treatments were permitted during the trial and >85% of participants continued taking these medications during the study. Participants (N = 234) were randomly allocated in a 1:1:1 manner to valbenazine 40 mg, 80 mg, or matched placebo. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. Baseline AIMS scores were 9.9 ± 4.3 in the placebo group, and 9.8 ± 4.1 and 10.4 ± 3.6 in the valbenazine 40-mg and 80-mg arms, respectively.2
Outcome. A fixed-sequence testing procedure to control for family-wise error rate and multiplicity was employed, and the primary endpoint was change from baseline to Week 6 in AIMS total score (items 1 to 7) for valbenazine 80 mg vs placebo. Valbenazine, 40 mg, was associated with a 1.9 point decrease in AIMS score, while valbenazine, 80 mg, was associated with a 3.2 point decrease in AIMS score, compared with 0.1 point decrease for placebo (P < .05 for valbenazine, 40 mg, P < .001 for valbenazine, 80 mg). This difference for the 40-mg dosage did not meet the prespecified analysis endpoints; however, for the 80-mg valbenazine dosage, the effect size for this difference (Cohen’s d) was large 0.90. There also were statistically significant differences between 40 mg and 80 mg at weeks 2, 4, and 6 in the intent-to-treat population. Of the 79 participants, 43 taking the 80-mg dosage completed a 48-week extension. Efficacy was sustained in this group; however, when valbenazine was discontinued at Week 48, AIMS scores returned to baseline after 4 weeks.
Tolerability
Of the 234 randomized patients, 205 (87.6%) completed the 6-week trial. Discontinuations due to adverse events were low across all treatment groups: 2.6% and 2.8% in the placebo and valbenazine 40-mg arms, respectively, and 3.8% in valbenazine 80-mg cohort. There was no safety signal based on changes in depression, suicidality, parkinsonism rating, or changes in schizophrenia symptoms. Because valbenazine can cause somnolence, patients should not perform activities requiring mental alertness (eg, operating a vehicle or hazardous machinery) until they know how they will be affected by valbenazine.
Valbenazine should be avoided in patients with congenital long QT syndrome or with arrhythmias associated with a prolonged QT interval. For patients at increased risk of a prolonged QT interval, assess the QT interval before increasing the dosage.
Clinical considerations
Unique properties. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist (NBI-98782) in a manner that permits once daily dosing, removes the need for CYP2D6 genotyping, and provides significant efficacy.
Why Rx? The reasons to prescribe valbenazine for TD patients include:
- currently the only agent with FDA approval for TD
- fewer tolerability issues seen with the only other effective agent, tetrabenazine
- no signal for effects on mood parameters or rates of parkinsonism
- lack of multiple daily dosing and possible need for 2D6 genotyping involved with TBZ prescribing.
Dosing
The recommended dosage of valbenazine is 80 mg/d administered as a single dose with or without food, starting at 40 mg once daily for 1 week. There is no dosage adjustment required in those with mild to moderate renal impairment; however, valbenazine is not recommended in those with severe renal impairment. The maximum dose is 40 mg/d for those who with moderate or severe hepatic impairment (Child-Pugh score, 7 to 15) however, valbenazine is not recommended for patients with severe renal impairment (creatinine clearance <30 mL/min) because the exposure to the active metabolite is reduced by approximately 75%. The combined efficacy and tolerability of dosages >80 mg/d has not been evaluated. Adverse effects seen with tetrabenazine at higher dosages include akathisia, anxiety, insomnia, parkinsonism, fatigue, and depression.
A daily dose of 40 mg may be considered for some patients based on tolerability, including those who are known CYP 2D6 poor metabolizers, and those taking strong CYP2D6 inhibitors.2 For those taking strong 3A4 inhibitors, the maximum daily dose is 40 mg. Concomitant use of valbenazine with strong 3A4 inducers is not recommended as the exposure to the active metabolite is reduced by approximately 75%.2 Lastly, because VMAT2 inhibition may alter synaptic levels of other monoamines, it is recommended that valbenazine not be administered with monoamine oxidase inhibitors, such as isocarboxazid, phenelzine, or selegiline.
Contraindications
There are no reported contraindications for valbenazine. As with most medications, there is limited available data on valbenazine use in pregnant women; however, administration of valbenazine to pregnant rats during organogenesis through lactation produced an increase in the number of stillborn pups and postnatal pup mortalities at doses under the maximum recommended human dose (MRHD) using body surface area based dosing (mg/m2). Pregnant women should be advised of the potential risk to a fetus. Valbenazine and its metabolites have been detected in rat milk at concentrations higher than in plasma after oral administration of valbenazine at doses 0.1 to 1.2 times the MRHD (based on mg/m2). Based on animal findings of increased perinatal mortality in exposed fetuses and pups, woman are advised not to breastfeed during valbenazine treatment and for 5 days after the final dose. No dosage adjustment is required for geriatric patients.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
1. O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687.
2. Ingrezza [package insert]. San Diego, CA: Neurocrine Biosciences Inc.; 2017.
3. Marder S, Knesevich MA, Hauser RA, et al. KINECT 3: A randomized, double-blind, placebo-controlled phase 3 trial of valbenazine (NBI-98854) for tardive dyskinesia. Poster presented at the American Psychiatric Association Annual Meeting; May 14-18, 2016; Atlanta, GA.
4. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
5. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124.
6. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
7. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al; American Academy of Neurology. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
10. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
11. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
12. Grigoriadis DE, Smith E, Madan A, et al. Pharmacologic characteristics of valbenazine (NBI-98854) and its metabolites. Poster presented at the U.S. Psychiatric & Mental Health Congress, October 21-24, 2016; San Antonio, TX.
Neuroscience-based Nomenclature: Classifying psychotropics by mechanism of action rather than indication
An important new initiative to reclassify psychiatric medications is underway. Currently, psychotropic drugs are named primarily for their clinical use, usually as a member of 1 of 6 classes: antipsychotic, anti‑depressant, mood stabilizer, stimulant, anxiolytic, and hypnotic.1,2
This naming system creates confusion because so-called antidepressants commonly are used as anxiolytics, antipsychotics increasingly are used as antidepressants, and so on.1,2
Vocabulary based on clinical indications also leads to difficulty in classifying new agents, especially those with novel mechanisms of action or clinical uses. Therefore, there is a need to make the names of psychotropic drugs more rational and scientifically based, rather than indication-based. A task force of experts from major psychopharmacology societies around the world is developing an alternative naming system that is increasingly being accepted by the major experts and journals throughout the world, called Neuroscience-based Nomenclature (NbN).3-5
So, what is NbN?

First and foremost, NbN renames the >100 known psychotropic drugs by 1 of the 11 principle pharmacological domains that include well-known terms such as serotonin dopamine, acetylcholine, and GABA (Table 1). Also included in NbN are 9 familiar modes of action, such as agonist, antagonist, reuptake inhibitor, and enzyme inhibitors (Table 2).3-5
NbN has 4 additional dimensions or layers3-5:
- The first layer enumerates the official indications as recognized by the regulatory agencies (ie, the FDA and other government organizations).
- The second layer states efficacy based on randomized controlled trials or substantial, evidence-based clinical data, as well as side effects (not the exhaustive list provided in manufacturers’ package inserts, but only the most common ones).
- The third layer is comprised of practical notes, highlighting potentially important drug interactions, metabolic issues, and specific warnings.
- The fourth section summarizes the neurobiological effects in laboratory animals and humans.
Specific dosages and titration regimens are not provided because they can vary among different countries, and NbN is intended for nomenclature and classification, not as a prescribing guide.
How does it work in practice?
Major journals in the field have begun adapting NbN for their published papers and
What is the current status?
Two international organizations endorse NbN, and the chief editors of nearly 3 dozen scientific journals, including
Clinicians should start adopting the NbN for the psychotropic drugs they prescribe every day. It is more scientific and consistent with the mechanism of action than with a specific disorder because many psychotropic medications have been found to be useful in >1 psychiatric disorder.
1. Nutt DJ. Beyond psychoanaleptics - can we improve antidepressant drug nomenclature? J Psychopharmacol. 2009;23(4):343-345.
2. Stahl SM. Classifying psychotropic drugs by mode of action not by target disorders. CNS Spectr. 2013;18(3):113-117.
3. Zohar J, Stahl S, Moller HJ, et al. A review of the current nomenclature for psychotropic agents and an introduction to the Neuroscience-based Nomenclature. Eur Neuropsychopharmacol. 2015; 25(12):2318-2325.
4. Zohar J, Stahl S, Moller HJ, et al. Neuroscience based nomenclature. Cambridge, United Kingdom: Cambridge University Press; 2014:254.
5. Neuroscience-based nomenclature. http://nbnomenclature.org. Accessed April 12, 2017.
An important new initiative to reclassify psychiatric medications is underway. Currently, psychotropic drugs are named primarily for their clinical use, usually as a member of 1 of 6 classes: antipsychotic, anti‑depressant, mood stabilizer, stimulant, anxiolytic, and hypnotic.1,2
This naming system creates confusion because so-called antidepressants commonly are used as anxiolytics, antipsychotics increasingly are used as antidepressants, and so on.1,2
Vocabulary based on clinical indications also leads to difficulty in classifying new agents, especially those with novel mechanisms of action or clinical uses. Therefore, there is a need to make the names of psychotropic drugs more rational and scientifically based, rather than indication-based. A task force of experts from major psychopharmacology societies around the world is developing an alternative naming system that is increasingly being accepted by the major experts and journals throughout the world, called Neuroscience-based Nomenclature (NbN).3-5
So, what is NbN?
First and foremost, NbN renames the >100 known psychotropic drugs by 1 of the 11 principle pharmacological domains that include well-known terms such as serotonin dopamine, acetylcholine, and GABA (Table 1). Also included in NbN are 9 familiar modes of action, such as agonist, antagonist, reuptake inhibitor, and enzyme inhibitors (Table 2).3-5
NbN has 4 additional dimensions or layers3-5:
- The first layer enumerates the official indications as recognized by the regulatory agencies (ie, the FDA and other government organizations).
- The second layer states efficacy based on randomized controlled trials or substantial, evidence-based clinical data, as well as side effects (not the exhaustive list provided in manufacturers’ package inserts, but only the most common ones).
- The third layer is comprised of practical notes, highlighting potentially important drug interactions, metabolic issues, and specific warnings.
- The fourth section summarizes the neurobiological effects in laboratory animals and humans.
Specific dosages and titration regimens are not provided because they can vary among different countries, and NbN is intended for nomenclature and classification, not as a prescribing guide.
How does it work in practice?
Major journals in the field have begun adapting NbN for their published papers and
What is the current status?
Two international organizations endorse NbN, and the chief editors of nearly 3 dozen scientific journals, including
Clinicians should start adopting the NbN for the psychotropic drugs they prescribe every day. It is more scientific and consistent with the mechanism of action than with a specific disorder because many psychotropic medications have been found to be useful in >1 psychiatric disorder.
An important new initiative to reclassify psychiatric medications is underway. Currently, psychotropic drugs are named primarily for their clinical use, usually as a member of 1 of 6 classes: antipsychotic, anti‑depressant, mood stabilizer, stimulant, anxiolytic, and hypnotic.1,2
This naming system creates confusion because so-called antidepressants commonly are used as anxiolytics, antipsychotics increasingly are used as antidepressants, and so on.1,2
Vocabulary based on clinical indications also leads to difficulty in classifying new agents, especially those with novel mechanisms of action or clinical uses. Therefore, there is a need to make the names of psychotropic drugs more rational and scientifically based, rather than indication-based. A task force of experts from major psychopharmacology societies around the world is developing an alternative naming system that is increasingly being accepted by the major experts and journals throughout the world, called Neuroscience-based Nomenclature (NbN).3-5
So, what is NbN?
First and foremost, NbN renames the >100 known psychotropic drugs by 1 of the 11 principle pharmacological domains that include well-known terms such as serotonin dopamine, acetylcholine, and GABA (Table 1). Also included in NbN are 9 familiar modes of action, such as agonist, antagonist, reuptake inhibitor, and enzyme inhibitors (Table 2).3-5
NbN has 4 additional dimensions or layers3-5:
- The first layer enumerates the official indications as recognized by the regulatory agencies (ie, the FDA and other government organizations).
- The second layer states efficacy based on randomized controlled trials or substantial, evidence-based clinical data, as well as side effects (not the exhaustive list provided in manufacturers’ package inserts, but only the most common ones).
- The third layer is comprised of practical notes, highlighting potentially important drug interactions, metabolic issues, and specific warnings.
- The fourth section summarizes the neurobiological effects in laboratory animals and humans.
Specific dosages and titration regimens are not provided because they can vary among different countries, and NbN is intended for nomenclature and classification, not as a prescribing guide.
How does it work in practice?
Major journals in the field have begun adapting NbN for their published papers and
What is the current status?
Two international organizations endorse NbN, and the chief editors of nearly 3 dozen scientific journals, including
Clinicians should start adopting the NbN for the psychotropic drugs they prescribe every day. It is more scientific and consistent with the mechanism of action than with a specific disorder because many psychotropic medications have been found to be useful in >1 psychiatric disorder.
1. Nutt DJ. Beyond psychoanaleptics - can we improve antidepressant drug nomenclature? J Psychopharmacol. 2009;23(4):343-345.
2. Stahl SM. Classifying psychotropic drugs by mode of action not by target disorders. CNS Spectr. 2013;18(3):113-117.
3. Zohar J, Stahl S, Moller HJ, et al. A review of the current nomenclature for psychotropic agents and an introduction to the Neuroscience-based Nomenclature. Eur Neuropsychopharmacol. 2015; 25(12):2318-2325.
4. Zohar J, Stahl S, Moller HJ, et al. Neuroscience based nomenclature. Cambridge, United Kingdom: Cambridge University Press; 2014:254.
5. Neuroscience-based nomenclature. http://nbnomenclature.org. Accessed April 12, 2017.
1. Nutt DJ. Beyond psychoanaleptics - can we improve antidepressant drug nomenclature? J Psychopharmacol. 2009;23(4):343-345.
2. Stahl SM. Classifying psychotropic drugs by mode of action not by target disorders. CNS Spectr. 2013;18(3):113-117.
3. Zohar J, Stahl S, Moller HJ, et al. A review of the current nomenclature for psychotropic agents and an introduction to the Neuroscience-based Nomenclature. Eur Neuropsychopharmacol. 2015; 25(12):2318-2325.
4. Zohar J, Stahl S, Moller HJ, et al. Neuroscience based nomenclature. Cambridge, United Kingdom: Cambridge University Press; 2014:254.
5. Neuroscience-based nomenclature. http://nbnomenclature.org. Accessed April 12, 2017.

Suicide by cop: What motivates those who choose this method?
CASE Unresponsive and suicidal
Mr. Z, age 25, an unemployed immigrant from Eastern Europe, is found unresponsive at a subway station. Workup in the emergency room reveals a positive urine toxicology for benzodiazepines and a blood alcohol level of 101.6 mg/dL. When Mr. Z regains consciousness the next day, he says that he is suicidal. He recently broke up with his girlfriend and feels worthless, hopeless, and depressed. As a suicide attempt, he took quetiapine and diazepam chased with vodka.
Mr. Z reports a history of suicide attempts. He says he has been suffering from depression most of his life and has been diagnosed with bipolar I disorder and borderline personality disorder. His medication regimen consists of quetiapine, 200 mg/d, and duloxetine, 20 mg/d.
Before immigrating to the United States 5 years ago, he attempted to overdose on his mother’s prescribed diazepam and was in a coma for 2 days. Recently, he stole a bicycle with the intent of provoking the police to kill him. When caught, he deliberately disobeyed the officer’s order and advanced toward the officer in an aggressive manner. However, the officer stopped Mr. Z using a stun gun. Mr. Z reports that he still feels angry that his suicide attempt failed. He is an Orthodox Christian and says he is “very religious.”
[polldaddy:9731423]
The authors’ observations
The means of suicide differ among individuals. Some attempt suicide by themselves; others through the involuntary participation of others, such as the police. This is known as SBC. Other terms include “suicide by means of victim-precipitated homicide,”1 “hetero-suicide,”2 “suicide by proxy,”3 “copicide,”4 and “law enforcement-forced-assisted suicide.”5,6 SBC accounts for 10%7 to 36%6 of police shootings and can cause serious stress for the officers involved and creates a strain between the police and the community.8
SBC was first mentioned as “suicide by means of victim-precipitated homicide.” Wolfgang5 reported 588 cases of police officer-involved shooting in Philadelphia between January 1948 and December 31, 1952, and, concluded that 150 of these cases (26%) fit criteria for what the author termed “victim-precipitated homicide” because the victims involved were the direct precipitants of the situation leading to their death. Wolfgang stated:
Instead of a murderer performing the act of suicide by killing another person who represents the murder’s unconscious, and instead of a suicide representing the desire to kill turned on [the] self, the victim in these victim-precipitated homicide cases is considered to be a suicide prone [individual] who manifests his desire to destroy [him]self by engaging another person to perform the act.
The term “SBC” was coined in 1983 by Karl Harris, a Los Angeles County medical examiner.8 The social repercussions of this modality attracts media attention because of its negative social consequences.
Characteristics of SBC
SBC has characteristics similar to other means of suicide; it is more prevalent among men with psychiatric disorders, including major depression, bipolar disorders, schizophrenia, substance use disorders,9 poor stress response skills, recent stressors, and adverse life events,10 and history of suicide attempts.
Psychosocial characteristics include:
- mean age 31.8 years1
- male sex (98%)
- white (52%)
- approximately 40% involve some form of relationship conflict.6
In psychological autopsy studies, an estimated 70.5% of those involved in a SBC incident had ≥1 stressful life events,1 including terminal illness, loss of a job, a lawsuit, or domestic issues. However, the reason is unknown for the remaining 28% cases.2 Thirty-five percent of those involved in SBC incidents were married, 13.5% divorced, and 46.7% single.1 Seventy-seven percent had low socioeconomic status,11 with 49.3% unemployed at the time of the SBC incident.1
Pathological characteristics of SBC and other suicide means are similar. Among SBC cases, 39% had previously attempted suicide6; 56% have a psychiatric or chronic medical comorbidity. Alcohol and drug abuse were reported among 56% of individuals, and 66% had a criminal history.6 Additionally, comorbid psychiatric disorders, especially those of the impulsive and emotionally unstable types, such as borderline and antisocial personality disorder, have been found to play a major role in SBC incidents.12
Individual suicide vs SBC
Religious beliefs. The term “religiosity” is used to define an individual’s idiosyncratic religious belief or personal religious philosophy reconciling the concept of death by suicide and the afterlife. Although there are no studies that specifically reference the relationship between SBC and religiosity, religious belief and affiliation appear to be strong motivating factors. SBC victims might have an idiosyncratic view of religion related death by suicide. Whether suicide is performed while under delusional belief about God, the devil, or being possessed by demons,13 or to avoid the moral prohibition of most religious faiths in regard to suicide,6 the degree of religiosity in SBC is an important area for future research.
Mr. Z stated that his strong religious faith as an Orthodox Christian motivated the attempted SBC. He tried to provoke the officer to kill him, because as a devout Orthodox Christian, it is against his religious beliefs to kill himself. He reasoned that, because his beliefs preclude him from performing the suicidal act on his own,6,14 having an officer pull the trigger would relieve him from committing what he perceived as a sin.6
Lethal vs danger. Another difference is the level of urgency that individuals create around them when attempting SBC. Homant and Kennedy15 see this in terms of 2 ideas: lethal and danger. Lethal refers to the degree of harm posed toward the suicidal individual. Danger is the degree of harm posed by the suicidal individual toward others (ie, police officers, bystanders, hostages, family members, a spouse, etc.). SBC often is more dangerous and more lethal than other methods of suicide. SBC individuals might threaten the lives of others to provoke the police into using deadly force, such as aiming or brandishing a gun or weapon at police officers or bystanders, increasing the lethality and dangerousness of the situation. Individuals engaging in SBC might shoot or kill others to create a confrontation with the police in order to be killed in the process (Table16).

Instrumental vs expressive goals
Mohandie and Meloy6 identified 2 primary goals of those involved in SBC events: instrumental and expressive. Individuals in the instrumental category are:
- attempting to escape or avoid the consequences of criminal or shameful actions
- using the forced confrontation with police to reconcile a failed relationship
- hoping to avoid the exclusion clauses of life insurance policies
- rationalizing that while it may be morally wrong to commit suicide, being killed resolves the spiritual problem of suicide
- seeking what they believe to be a very effective and lethal means of accomplishing death.
An expressive goal is more personal and includes individuals who use the confrontation with the police to communicate:
- hopelessness, depression, and desperation
- a statement about their ultimate identification as victims
- their need to “save face” by dying or being forcibly overwhelmed rather than surrendering
- their intense power needs, rage, and revenge
- their need to draw attention to an important personal issue.
Mr. Z chose what he believed to be an efficiently lethal way of dying in accord with his religious faith, knowing that a confrontation with the police could have a fatal ending. This case represents an instrumental motivation to die by SBC that was religiously motivated.
[polldaddy:9731428]
The authors’ observations
SBC presents a specific and serious challenge for law enforcement personnel, and should be approached in a manner different than other crisis situations. Because many individuals engaging in SBC have a history of mental illness, officers with training in handling individuals with psychiatric disorders—known as Crisis Intervention Team (CIT) in many areas—should be deployed as first responders. CITs have been shown to:
- reduce arrest rates of individuals with psychiatric disorders
- increase referral rates to appropriate treatment
- decrease police injuries when responding to calls
- decrease the need for escalation with specialized tactical response teams, such as Special Weapons And Tactics.17
Identification of SBC behavior is crucial during police response. Indicators of a SBC include:
- refusal to comply with police order
- refusal to surrender
- lack of interest in getting out of a barricade or hostage situation alive.18
In approaching a SBC incident, responding officers should be non-confrontational and try to talk to the suicidal individual.8 If force is needed to resolve the crisis, non-lethal measures should be used first.8 Law enforcement and mental health professionals should suspect a SBC situation in individuals who have had prior police contact and are exhibiting behaviors outlined in the Table.16
Once suicidality is identified, it should be treated promptly. Patients who are at imminent risk to themselves or others should be hospitalized to maintain their safety. Similar to other suicide modalities, the primary risk factor for SBC is untreated or inadequately treated psychiatric illness. Therefore, the crux of managing SBC involves identifying and treating the underlying mental disorder.
Pharmacological treatment should be guided by the patient’s symptoms and psychiatric diagnosis. For suicidal behavior associated with bipolar depression and other affective disorders, lithium has evidence of reducing suicidality. Studies have shown a 5.5-fold reduction in suicide risk and a >13-fold reduction in completed suicides with lithium treatment.19 In patients with schizophrenia, clozapine has been shown to reduce suicide risk and is the only FDA-approved agent for this indication.19 Although antidepressants can effectively treat depression, there are no studies that show that 1 antidepressant is more effective than others in reducing suicidality. This might be because of the long latency period between treatment initiation and symptom relief. Ketamine, an N-methyl-
OUTCOME Medication adjustment
After Mr. Z is medically stable, he is voluntarily transferred to the inpatient psychiatric unit where he is stabilized on quetiapine, 200 mg/d, and duloxetine, 60 mg/d, and attends daily group activity, milieu, and individual therapy. Because of Mr. Z’s chronic affective instability and suicidality, we consider lithium for its anti-suicide effects, but decide against it because of lithium’s high lethality in an overdose and Mr. Z’s history of poor compliance and alcohol use.
Because of Mr. Z’s socioeconomic challenges, it is necessary to contact his extended family and social support system to be part of treatment and safety planning. After a week on the psychiatric unit, his mood symptoms stabilize and he is discharged to his family and friends in the area, with a short supply of quetiapine and duloxetine, and free follow-up care within 3 days of discharge. His mood is euthymic; his affect is broad range; his thought process is coherent and logical; he denies suicidal ideation; and can verbalize a logical and concrete safety plan. His support system assures us that Mr. Z will follow up with his appointments.
His DSM-522 discharge diagnoses are borderline personality disorder, bipolar I disorder, and suicidal behavior disorder, current.
The authors’ observations
SBC increases friction and mistrust between the police and the public, traumatizes officers who are forced to use deadly measures, and results in the death of the suicidal individual. As mental health professionals, we need to be aware of this form of suicide in our screening assessment. Training police to differentiate violent offenders from psychiatric patients could reduce the number of SBCs.9 As shown by the CIT model, educating officers on behaviors indicating a mental illness could lead to more psychiatric admissions rather than incarceration17 or death. We advocate for continuous collaborative work and cross training between the police and mental health professionals and for more research on the link between religiosity and the motivation to die by SBC, because there appears to be a not-yet quantified but strong link between them.
1. Hutson HR, Anglin D, Yarbrough J, et al. Suicide by cop. Ann Emerg Med. 1998;32(6):665-669.
2. Foote WE. Victim-precipitated homicide. In: Hall HV, ed. Lethal violence: a sourcebook on fatal domestic, acquaintance and stranger violence. London, United Kingdom: CRC Press; 1999:175-199.
3. Keram EA, Farrell BJ. Suicide by cop: issues in outcome and analysis. In: Sheehan DC, Warren JI, eds. Suicide and law enforcement. Quantico, VA: FBI Academy; 2001:587-597.
4. Violanti JM, Drylie JJ. Copicide: concepts, cases, and controversies of suicide by cop. Springfield, IL: Charles C Thomas Publisher, LTD; 2008.
5. Wolfgang ME. Suicide by means of victim-precipitated homicide. J Clin Exp Psychopathol Q Rev Psychiatry Neurol. 1959;20:335-349.
6. Mohandie K, Meloy JR. Clinical and forensic indicators of “suicide by cop.” J Forensic Sci. 2000;45(2):384-389.
7. Wright RK, Davis JH. Studies in the epidemiology of murder a proposed classification system. J Forensic Sci. 1977;22(2):464-470.
8. Miller L. Suicide by cop: causes, reactions, and practical intervention strategies. Int J Emerg Ment Health. 2006;8(3):165-174.
9. Dewey L, Allwood M, Fava J, et al. Suicide by cop: clinical risks and subtypes. Arch Suicide Res. 2013;17(4):448-461.
10. Foster T, Gillespie K, McClelland R, et al. Risk factors for suicide independent of DSM-III-R Axis I disorder. Case-control psychological autopsy study in Northern Ireland. Br J Psychiatry. 1999;175:175-179.
11. Lindsay M, Lester D. Criteria for suicide-by-cop incidents. Psychol Rep. 2008;102(2):603-605.
12. Cheng AT, Mann AH, Chan KA. Personality disorder and suicide. A case-control study. Br J Psychiatry. 1997;170:441-446.
13. Mohandie K, Meloy JR, Collins PI. Suicide by cop among officer‐involved shooting cases. J Forensic Sci. 2009;54(2):456-462.
14. Falk J, Riepert T, Rothschild MA. A case of suicide-by-cop. Leg Med (Tokyo). 2004;6(3):194-196.
15. Homant RJ, Kennedy DB. Suicide by police: a proposed typology of law enforcement officer-assisted suicide. Policing: An International Journal of Police Strategies & Management. 2000;23(3):339-355.
16. Lester D. Suicide as a staged performance. Comprehensive Psychology. 2015:4(1):1-6.
17. SpringerBriefs in psychology. Best practices for those with psychiatric disorder in the criminal justice system. In: Walker LE, Pann JM, Shapiro DL, et al. Best practices in law enforcement crisis Interventions with those with psychiatric disorder. 2015;11-18.
18. Homant RJ, Kennedy DB, Hupp R. Real and perceived danger in police officer assisted suicide. J Crim Justice. 2000;28(1):43-52.
19. Ernst CL, Goldberg JF. Antisuicide properties of psychotropic drugs: a critical review. Harv Review Psychiatry. 2004;12(1):14-41.
20. Al Jurdi RK, Swann A, Mathew SJ. Psychopharmacological agents and suicide risk reduction: ketamine and other approaches. Curr Psychiatry Rep. 2015;17(10):81.
21. Fink M, Kellner CH, McCall WV. The role of ECT in suicide prevention. Journal ECT. 2014;30(1):5-9.
22. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
CASE Unresponsive and suicidal
Mr. Z, age 25, an unemployed immigrant from Eastern Europe, is found unresponsive at a subway station. Workup in the emergency room reveals a positive urine toxicology for benzodiazepines and a blood alcohol level of 101.6 mg/dL. When Mr. Z regains consciousness the next day, he says that he is suicidal. He recently broke up with his girlfriend and feels worthless, hopeless, and depressed. As a suicide attempt, he took quetiapine and diazepam chased with vodka.
Mr. Z reports a history of suicide attempts. He says he has been suffering from depression most of his life and has been diagnosed with bipolar I disorder and borderline personality disorder. His medication regimen consists of quetiapine, 200 mg/d, and duloxetine, 20 mg/d.
Before immigrating to the United States 5 years ago, he attempted to overdose on his mother’s prescribed diazepam and was in a coma for 2 days. Recently, he stole a bicycle with the intent of provoking the police to kill him. When caught, he deliberately disobeyed the officer’s order and advanced toward the officer in an aggressive manner. However, the officer stopped Mr. Z using a stun gun. Mr. Z reports that he still feels angry that his suicide attempt failed. He is an Orthodox Christian and says he is “very religious.”
[polldaddy:9731423]
The authors’ observations
The means of suicide differ among individuals. Some attempt suicide by themselves; others through the involuntary participation of others, such as the police. This is known as SBC. Other terms include “suicide by means of victim-precipitated homicide,”1 “hetero-suicide,”2 “suicide by proxy,”3 “copicide,”4 and “law enforcement-forced-assisted suicide.”5,6 SBC accounts for 10%7 to 36%6 of police shootings and can cause serious stress for the officers involved and creates a strain between the police and the community.8
SBC was first mentioned as “suicide by means of victim-precipitated homicide.” Wolfgang5 reported 588 cases of police officer-involved shooting in Philadelphia between January 1948 and December 31, 1952, and, concluded that 150 of these cases (26%) fit criteria for what the author termed “victim-precipitated homicide” because the victims involved were the direct precipitants of the situation leading to their death. Wolfgang stated:
Instead of a murderer performing the act of suicide by killing another person who represents the murder’s unconscious, and instead of a suicide representing the desire to kill turned on [the] self, the victim in these victim-precipitated homicide cases is considered to be a suicide prone [individual] who manifests his desire to destroy [him]self by engaging another person to perform the act.
The term “SBC” was coined in 1983 by Karl Harris, a Los Angeles County medical examiner.8 The social repercussions of this modality attracts media attention because of its negative social consequences.
Characteristics of SBC
SBC has characteristics similar to other means of suicide; it is more prevalent among men with psychiatric disorders, including major depression, bipolar disorders, schizophrenia, substance use disorders,9 poor stress response skills, recent stressors, and adverse life events,10 and history of suicide attempts.
Psychosocial characteristics include:
- mean age 31.8 years1
- male sex (98%)
- white (52%)
- approximately 40% involve some form of relationship conflict.6
In psychological autopsy studies, an estimated 70.5% of those involved in a SBC incident had ≥1 stressful life events,1 including terminal illness, loss of a job, a lawsuit, or domestic issues. However, the reason is unknown for the remaining 28% cases.2 Thirty-five percent of those involved in SBC incidents were married, 13.5% divorced, and 46.7% single.1 Seventy-seven percent had low socioeconomic status,11 with 49.3% unemployed at the time of the SBC incident.1
Pathological characteristics of SBC and other suicide means are similar. Among SBC cases, 39% had previously attempted suicide6; 56% have a psychiatric or chronic medical comorbidity. Alcohol and drug abuse were reported among 56% of individuals, and 66% had a criminal history.6 Additionally, comorbid psychiatric disorders, especially those of the impulsive and emotionally unstable types, such as borderline and antisocial personality disorder, have been found to play a major role in SBC incidents.12
Individual suicide vs SBC
Religious beliefs. The term “religiosity” is used to define an individual’s idiosyncratic religious belief or personal religious philosophy reconciling the concept of death by suicide and the afterlife. Although there are no studies that specifically reference the relationship between SBC and religiosity, religious belief and affiliation appear to be strong motivating factors. SBC victims might have an idiosyncratic view of religion related death by suicide. Whether suicide is performed while under delusional belief about God, the devil, or being possessed by demons,13 or to avoid the moral prohibition of most religious faiths in regard to suicide,6 the degree of religiosity in SBC is an important area for future research.
Mr. Z stated that his strong religious faith as an Orthodox Christian motivated the attempted SBC. He tried to provoke the officer to kill him, because as a devout Orthodox Christian, it is against his religious beliefs to kill himself. He reasoned that, because his beliefs preclude him from performing the suicidal act on his own,6,14 having an officer pull the trigger would relieve him from committing what he perceived as a sin.6
Lethal vs danger. Another difference is the level of urgency that individuals create around them when attempting SBC. Homant and Kennedy15 see this in terms of 2 ideas: lethal and danger. Lethal refers to the degree of harm posed toward the suicidal individual. Danger is the degree of harm posed by the suicidal individual toward others (ie, police officers, bystanders, hostages, family members, a spouse, etc.). SBC often is more dangerous and more lethal than other methods of suicide. SBC individuals might threaten the lives of others to provoke the police into using deadly force, such as aiming or brandishing a gun or weapon at police officers or bystanders, increasing the lethality and dangerousness of the situation. Individuals engaging in SBC might shoot or kill others to create a confrontation with the police in order to be killed in the process (Table16).
Instrumental vs expressive goals
Mohandie and Meloy6 identified 2 primary goals of those involved in SBC events: instrumental and expressive. Individuals in the instrumental category are:
- attempting to escape or avoid the consequences of criminal or shameful actions
- using the forced confrontation with police to reconcile a failed relationship
- hoping to avoid the exclusion clauses of life insurance policies
- rationalizing that while it may be morally wrong to commit suicide, being killed resolves the spiritual problem of suicide
- seeking what they believe to be a very effective and lethal means of accomplishing death.
An expressive goal is more personal and includes individuals who use the confrontation with the police to communicate:
- hopelessness, depression, and desperation
- a statement about their ultimate identification as victims
- their need to “save face” by dying or being forcibly overwhelmed rather than surrendering
- their intense power needs, rage, and revenge
- their need to draw attention to an important personal issue.
Mr. Z chose what he believed to be an efficiently lethal way of dying in accord with his religious faith, knowing that a confrontation with the police could have a fatal ending. This case represents an instrumental motivation to die by SBC that was religiously motivated.
[polldaddy:9731428]
The authors’ observations
SBC presents a specific and serious challenge for law enforcement personnel, and should be approached in a manner different than other crisis situations. Because many individuals engaging in SBC have a history of mental illness, officers with training in handling individuals with psychiatric disorders—known as Crisis Intervention Team (CIT) in many areas—should be deployed as first responders. CITs have been shown to:
- reduce arrest rates of individuals with psychiatric disorders
- increase referral rates to appropriate treatment
- decrease police injuries when responding to calls
- decrease the need for escalation with specialized tactical response teams, such as Special Weapons And Tactics.17
Identification of SBC behavior is crucial during police response. Indicators of a SBC include:
- refusal to comply with police order
- refusal to surrender
- lack of interest in getting out of a barricade or hostage situation alive.18
In approaching a SBC incident, responding officers should be non-confrontational and try to talk to the suicidal individual.8 If force is needed to resolve the crisis, non-lethal measures should be used first.8 Law enforcement and mental health professionals should suspect a SBC situation in individuals who have had prior police contact and are exhibiting behaviors outlined in the Table.16
Once suicidality is identified, it should be treated promptly. Patients who are at imminent risk to themselves or others should be hospitalized to maintain their safety. Similar to other suicide modalities, the primary risk factor for SBC is untreated or inadequately treated psychiatric illness. Therefore, the crux of managing SBC involves identifying and treating the underlying mental disorder.
Pharmacological treatment should be guided by the patient’s symptoms and psychiatric diagnosis. For suicidal behavior associated with bipolar depression and other affective disorders, lithium has evidence of reducing suicidality. Studies have shown a 5.5-fold reduction in suicide risk and a >13-fold reduction in completed suicides with lithium treatment.19 In patients with schizophrenia, clozapine has been shown to reduce suicide risk and is the only FDA-approved agent for this indication.19 Although antidepressants can effectively treat depression, there are no studies that show that 1 antidepressant is more effective than others in reducing suicidality. This might be because of the long latency period between treatment initiation and symptom relief. Ketamine, an N-methyl-
OUTCOME Medication adjustment
After Mr. Z is medically stable, he is voluntarily transferred to the inpatient psychiatric unit where he is stabilized on quetiapine, 200 mg/d, and duloxetine, 60 mg/d, and attends daily group activity, milieu, and individual therapy. Because of Mr. Z’s chronic affective instability and suicidality, we consider lithium for its anti-suicide effects, but decide against it because of lithium’s high lethality in an overdose and Mr. Z’s history of poor compliance and alcohol use.
Because of Mr. Z’s socioeconomic challenges, it is necessary to contact his extended family and social support system to be part of treatment and safety planning. After a week on the psychiatric unit, his mood symptoms stabilize and he is discharged to his family and friends in the area, with a short supply of quetiapine and duloxetine, and free follow-up care within 3 days of discharge. His mood is euthymic; his affect is broad range; his thought process is coherent and logical; he denies suicidal ideation; and can verbalize a logical and concrete safety plan. His support system assures us that Mr. Z will follow up with his appointments.
His DSM-522 discharge diagnoses are borderline personality disorder, bipolar I disorder, and suicidal behavior disorder, current.
The authors’ observations
SBC increases friction and mistrust between the police and the public, traumatizes officers who are forced to use deadly measures, and results in the death of the suicidal individual. As mental health professionals, we need to be aware of this form of suicide in our screening assessment. Training police to differentiate violent offenders from psychiatric patients could reduce the number of SBCs.9 As shown by the CIT model, educating officers on behaviors indicating a mental illness could lead to more psychiatric admissions rather than incarceration17 or death. We advocate for continuous collaborative work and cross training between the police and mental health professionals and for more research on the link between religiosity and the motivation to die by SBC, because there appears to be a not-yet quantified but strong link between them.
CASE Unresponsive and suicidal
Mr. Z, age 25, an unemployed immigrant from Eastern Europe, is found unresponsive at a subway station. Workup in the emergency room reveals a positive urine toxicology for benzodiazepines and a blood alcohol level of 101.6 mg/dL. When Mr. Z regains consciousness the next day, he says that he is suicidal. He recently broke up with his girlfriend and feels worthless, hopeless, and depressed. As a suicide attempt, he took quetiapine and diazepam chased with vodka.
Mr. Z reports a history of suicide attempts. He says he has been suffering from depression most of his life and has been diagnosed with bipolar I disorder and borderline personality disorder. His medication regimen consists of quetiapine, 200 mg/d, and duloxetine, 20 mg/d.
Before immigrating to the United States 5 years ago, he attempted to overdose on his mother’s prescribed diazepam and was in a coma for 2 days. Recently, he stole a bicycle with the intent of provoking the police to kill him. When caught, he deliberately disobeyed the officer’s order and advanced toward the officer in an aggressive manner. However, the officer stopped Mr. Z using a stun gun. Mr. Z reports that he still feels angry that his suicide attempt failed. He is an Orthodox Christian and says he is “very religious.”
[polldaddy:9731423]
The authors’ observations
The means of suicide differ among individuals. Some attempt suicide by themselves; others through the involuntary participation of others, such as the police. This is known as SBC. Other terms include “suicide by means of victim-precipitated homicide,”1 “hetero-suicide,”2 “suicide by proxy,”3 “copicide,”4 and “law enforcement-forced-assisted suicide.”5,6 SBC accounts for 10%7 to 36%6 of police shootings and can cause serious stress for the officers involved and creates a strain between the police and the community.8
SBC was first mentioned as “suicide by means of victim-precipitated homicide.” Wolfgang5 reported 588 cases of police officer-involved shooting in Philadelphia between January 1948 and December 31, 1952, and, concluded that 150 of these cases (26%) fit criteria for what the author termed “victim-precipitated homicide” because the victims involved were the direct precipitants of the situation leading to their death. Wolfgang stated:
Instead of a murderer performing the act of suicide by killing another person who represents the murder’s unconscious, and instead of a suicide representing the desire to kill turned on [the] self, the victim in these victim-precipitated homicide cases is considered to be a suicide prone [individual] who manifests his desire to destroy [him]self by engaging another person to perform the act.
The term “SBC” was coined in 1983 by Karl Harris, a Los Angeles County medical examiner.8 The social repercussions of this modality attracts media attention because of its negative social consequences.
Characteristics of SBC
SBC has characteristics similar to other means of suicide; it is more prevalent among men with psychiatric disorders, including major depression, bipolar disorders, schizophrenia, substance use disorders,9 poor stress response skills, recent stressors, and adverse life events,10 and history of suicide attempts.
Psychosocial characteristics include:
- mean age 31.8 years1
- male sex (98%)
- white (52%)
- approximately 40% involve some form of relationship conflict.6
In psychological autopsy studies, an estimated 70.5% of those involved in a SBC incident had ≥1 stressful life events,1 including terminal illness, loss of a job, a lawsuit, or domestic issues. However, the reason is unknown for the remaining 28% cases.2 Thirty-five percent of those involved in SBC incidents were married, 13.5% divorced, and 46.7% single.1 Seventy-seven percent had low socioeconomic status,11 with 49.3% unemployed at the time of the SBC incident.1
Pathological characteristics of SBC and other suicide means are similar. Among SBC cases, 39% had previously attempted suicide6; 56% have a psychiatric or chronic medical comorbidity. Alcohol and drug abuse were reported among 56% of individuals, and 66% had a criminal history.6 Additionally, comorbid psychiatric disorders, especially those of the impulsive and emotionally unstable types, such as borderline and antisocial personality disorder, have been found to play a major role in SBC incidents.12
Individual suicide vs SBC
Religious beliefs. The term “religiosity” is used to define an individual’s idiosyncratic religious belief or personal religious philosophy reconciling the concept of death by suicide and the afterlife. Although there are no studies that specifically reference the relationship between SBC and religiosity, religious belief and affiliation appear to be strong motivating factors. SBC victims might have an idiosyncratic view of religion related death by suicide. Whether suicide is performed while under delusional belief about God, the devil, or being possessed by demons,13 or to avoid the moral prohibition of most religious faiths in regard to suicide,6 the degree of religiosity in SBC is an important area for future research.
Mr. Z stated that his strong religious faith as an Orthodox Christian motivated the attempted SBC. He tried to provoke the officer to kill him, because as a devout Orthodox Christian, it is against his religious beliefs to kill himself. He reasoned that, because his beliefs preclude him from performing the suicidal act on his own,6,14 having an officer pull the trigger would relieve him from committing what he perceived as a sin.6
Lethal vs danger. Another difference is the level of urgency that individuals create around them when attempting SBC. Homant and Kennedy15 see this in terms of 2 ideas: lethal and danger. Lethal refers to the degree of harm posed toward the suicidal individual. Danger is the degree of harm posed by the suicidal individual toward others (ie, police officers, bystanders, hostages, family members, a spouse, etc.). SBC often is more dangerous and more lethal than other methods of suicide. SBC individuals might threaten the lives of others to provoke the police into using deadly force, such as aiming or brandishing a gun or weapon at police officers or bystanders, increasing the lethality and dangerousness of the situation. Individuals engaging in SBC might shoot or kill others to create a confrontation with the police in order to be killed in the process (Table16).
Instrumental vs expressive goals
Mohandie and Meloy6 identified 2 primary goals of those involved in SBC events: instrumental and expressive. Individuals in the instrumental category are:
- attempting to escape or avoid the consequences of criminal or shameful actions
- using the forced confrontation with police to reconcile a failed relationship
- hoping to avoid the exclusion clauses of life insurance policies
- rationalizing that while it may be morally wrong to commit suicide, being killed resolves the spiritual problem of suicide
- seeking what they believe to be a very effective and lethal means of accomplishing death.
An expressive goal is more personal and includes individuals who use the confrontation with the police to communicate:
- hopelessness, depression, and desperation
- a statement about their ultimate identification as victims
- their need to “save face” by dying or being forcibly overwhelmed rather than surrendering
- their intense power needs, rage, and revenge
- their need to draw attention to an important personal issue.
Mr. Z chose what he believed to be an efficiently lethal way of dying in accord with his religious faith, knowing that a confrontation with the police could have a fatal ending. This case represents an instrumental motivation to die by SBC that was religiously motivated.
[polldaddy:9731428]
The authors’ observations
SBC presents a specific and serious challenge for law enforcement personnel, and should be approached in a manner different than other crisis situations. Because many individuals engaging in SBC have a history of mental illness, officers with training in handling individuals with psychiatric disorders—known as Crisis Intervention Team (CIT) in many areas—should be deployed as first responders. CITs have been shown to:
- reduce arrest rates of individuals with psychiatric disorders
- increase referral rates to appropriate treatment
- decrease police injuries when responding to calls
- decrease the need for escalation with specialized tactical response teams, such as Special Weapons And Tactics.17
Identification of SBC behavior is crucial during police response. Indicators of a SBC include:
- refusal to comply with police order
- refusal to surrender
- lack of interest in getting out of a barricade or hostage situation alive.18
In approaching a SBC incident, responding officers should be non-confrontational and try to talk to the suicidal individual.8 If force is needed to resolve the crisis, non-lethal measures should be used first.8 Law enforcement and mental health professionals should suspect a SBC situation in individuals who have had prior police contact and are exhibiting behaviors outlined in the Table.16
Once suicidality is identified, it should be treated promptly. Patients who are at imminent risk to themselves or others should be hospitalized to maintain their safety. Similar to other suicide modalities, the primary risk factor for SBC is untreated or inadequately treated psychiatric illness. Therefore, the crux of managing SBC involves identifying and treating the underlying mental disorder.
Pharmacological treatment should be guided by the patient’s symptoms and psychiatric diagnosis. For suicidal behavior associated with bipolar depression and other affective disorders, lithium has evidence of reducing suicidality. Studies have shown a 5.5-fold reduction in suicide risk and a >13-fold reduction in completed suicides with lithium treatment.19 In patients with schizophrenia, clozapine has been shown to reduce suicide risk and is the only FDA-approved agent for this indication.19 Although antidepressants can effectively treat depression, there are no studies that show that 1 antidepressant is more effective than others in reducing suicidality. This might be because of the long latency period between treatment initiation and symptom relief. Ketamine, an N-methyl-
OUTCOME Medication adjustment
After Mr. Z is medically stable, he is voluntarily transferred to the inpatient psychiatric unit where he is stabilized on quetiapine, 200 mg/d, and duloxetine, 60 mg/d, and attends daily group activity, milieu, and individual therapy. Because of Mr. Z’s chronic affective instability and suicidality, we consider lithium for its anti-suicide effects, but decide against it because of lithium’s high lethality in an overdose and Mr. Z’s history of poor compliance and alcohol use.
Because of Mr. Z’s socioeconomic challenges, it is necessary to contact his extended family and social support system to be part of treatment and safety planning. After a week on the psychiatric unit, his mood symptoms stabilize and he is discharged to his family and friends in the area, with a short supply of quetiapine and duloxetine, and free follow-up care within 3 days of discharge. His mood is euthymic; his affect is broad range; his thought process is coherent and logical; he denies suicidal ideation; and can verbalize a logical and concrete safety plan. His support system assures us that Mr. Z will follow up with his appointments.
His DSM-522 discharge diagnoses are borderline personality disorder, bipolar I disorder, and suicidal behavior disorder, current.
The authors’ observations
SBC increases friction and mistrust between the police and the public, traumatizes officers who are forced to use deadly measures, and results in the death of the suicidal individual. As mental health professionals, we need to be aware of this form of suicide in our screening assessment. Training police to differentiate violent offenders from psychiatric patients could reduce the number of SBCs.9 As shown by the CIT model, educating officers on behaviors indicating a mental illness could lead to more psychiatric admissions rather than incarceration17 or death. We advocate for continuous collaborative work and cross training between the police and mental health professionals and for more research on the link between religiosity and the motivation to die by SBC, because there appears to be a not-yet quantified but strong link between them.
1. Hutson HR, Anglin D, Yarbrough J, et al. Suicide by cop. Ann Emerg Med. 1998;32(6):665-669.
2. Foote WE. Victim-precipitated homicide. In: Hall HV, ed. Lethal violence: a sourcebook on fatal domestic, acquaintance and stranger violence. London, United Kingdom: CRC Press; 1999:175-199.
3. Keram EA, Farrell BJ. Suicide by cop: issues in outcome and analysis. In: Sheehan DC, Warren JI, eds. Suicide and law enforcement. Quantico, VA: FBI Academy; 2001:587-597.
4. Violanti JM, Drylie JJ. Copicide: concepts, cases, and controversies of suicide by cop. Springfield, IL: Charles C Thomas Publisher, LTD; 2008.
5. Wolfgang ME. Suicide by means of victim-precipitated homicide. J Clin Exp Psychopathol Q Rev Psychiatry Neurol. 1959;20:335-349.
6. Mohandie K, Meloy JR. Clinical and forensic indicators of “suicide by cop.” J Forensic Sci. 2000;45(2):384-389.
7. Wright RK, Davis JH. Studies in the epidemiology of murder a proposed classification system. J Forensic Sci. 1977;22(2):464-470.
8. Miller L. Suicide by cop: causes, reactions, and practical intervention strategies. Int J Emerg Ment Health. 2006;8(3):165-174.
9. Dewey L, Allwood M, Fava J, et al. Suicide by cop: clinical risks and subtypes. Arch Suicide Res. 2013;17(4):448-461.
10. Foster T, Gillespie K, McClelland R, et al. Risk factors for suicide independent of DSM-III-R Axis I disorder. Case-control psychological autopsy study in Northern Ireland. Br J Psychiatry. 1999;175:175-179.
11. Lindsay M, Lester D. Criteria for suicide-by-cop incidents. Psychol Rep. 2008;102(2):603-605.
12. Cheng AT, Mann AH, Chan KA. Personality disorder and suicide. A case-control study. Br J Psychiatry. 1997;170:441-446.
13. Mohandie K, Meloy JR, Collins PI. Suicide by cop among officer‐involved shooting cases. J Forensic Sci. 2009;54(2):456-462.
14. Falk J, Riepert T, Rothschild MA. A case of suicide-by-cop. Leg Med (Tokyo). 2004;6(3):194-196.
15. Homant RJ, Kennedy DB. Suicide by police: a proposed typology of law enforcement officer-assisted suicide. Policing: An International Journal of Police Strategies & Management. 2000;23(3):339-355.
16. Lester D. Suicide as a staged performance. Comprehensive Psychology. 2015:4(1):1-6.
17. SpringerBriefs in psychology. Best practices for those with psychiatric disorder in the criminal justice system. In: Walker LE, Pann JM, Shapiro DL, et al. Best practices in law enforcement crisis Interventions with those with psychiatric disorder. 2015;11-18.
18. Homant RJ, Kennedy DB, Hupp R. Real and perceived danger in police officer assisted suicide. J Crim Justice. 2000;28(1):43-52.
19. Ernst CL, Goldberg JF. Antisuicide properties of psychotropic drugs: a critical review. Harv Review Psychiatry. 2004;12(1):14-41.
20. Al Jurdi RK, Swann A, Mathew SJ. Psychopharmacological agents and suicide risk reduction: ketamine and other approaches. Curr Psychiatry Rep. 2015;17(10):81.
21. Fink M, Kellner CH, McCall WV. The role of ECT in suicide prevention. Journal ECT. 2014;30(1):5-9.
22. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
1. Hutson HR, Anglin D, Yarbrough J, et al. Suicide by cop. Ann Emerg Med. 1998;32(6):665-669.
2. Foote WE. Victim-precipitated homicide. In: Hall HV, ed. Lethal violence: a sourcebook on fatal domestic, acquaintance and stranger violence. London, United Kingdom: CRC Press; 1999:175-199.
3. Keram EA, Farrell BJ. Suicide by cop: issues in outcome and analysis. In: Sheehan DC, Warren JI, eds. Suicide and law enforcement. Quantico, VA: FBI Academy; 2001:587-597.
4. Violanti JM, Drylie JJ. Copicide: concepts, cases, and controversies of suicide by cop. Springfield, IL: Charles C Thomas Publisher, LTD; 2008.
5. Wolfgang ME. Suicide by means of victim-precipitated homicide. J Clin Exp Psychopathol Q Rev Psychiatry Neurol. 1959;20:335-349.
6. Mohandie K, Meloy JR. Clinical and forensic indicators of “suicide by cop.” J Forensic Sci. 2000;45(2):384-389.
7. Wright RK, Davis JH. Studies in the epidemiology of murder a proposed classification system. J Forensic Sci. 1977;22(2):464-470.
8. Miller L. Suicide by cop: causes, reactions, and practical intervention strategies. Int J Emerg Ment Health. 2006;8(3):165-174.
9. Dewey L, Allwood M, Fava J, et al. Suicide by cop: clinical risks and subtypes. Arch Suicide Res. 2013;17(4):448-461.
10. Foster T, Gillespie K, McClelland R, et al. Risk factors for suicide independent of DSM-III-R Axis I disorder. Case-control psychological autopsy study in Northern Ireland. Br J Psychiatry. 1999;175:175-179.
11. Lindsay M, Lester D. Criteria for suicide-by-cop incidents. Psychol Rep. 2008;102(2):603-605.
12. Cheng AT, Mann AH, Chan KA. Personality disorder and suicide. A case-control study. Br J Psychiatry. 1997;170:441-446.
13. Mohandie K, Meloy JR, Collins PI. Suicide by cop among officer‐involved shooting cases. J Forensic Sci. 2009;54(2):456-462.
14. Falk J, Riepert T, Rothschild MA. A case of suicide-by-cop. Leg Med (Tokyo). 2004;6(3):194-196.
15. Homant RJ, Kennedy DB. Suicide by police: a proposed typology of law enforcement officer-assisted suicide. Policing: An International Journal of Police Strategies & Management. 2000;23(3):339-355.
16. Lester D. Suicide as a staged performance. Comprehensive Psychology. 2015:4(1):1-6.
17. SpringerBriefs in psychology. Best practices for those with psychiatric disorder in the criminal justice system. In: Walker LE, Pann JM, Shapiro DL, et al. Best practices in law enforcement crisis Interventions with those with psychiatric disorder. 2015;11-18.
18. Homant RJ, Kennedy DB, Hupp R. Real and perceived danger in police officer assisted suicide. J Crim Justice. 2000;28(1):43-52.
19. Ernst CL, Goldberg JF. Antisuicide properties of psychotropic drugs: a critical review. Harv Review Psychiatry. 2004;12(1):14-41.
20. Al Jurdi RK, Swann A, Mathew SJ. Psychopharmacological agents and suicide risk reduction: ketamine and other approaches. Curr Psychiatry Rep. 2015;17(10):81.
21. Fink M, Kellner CH, McCall WV. The role of ECT in suicide prevention. Journal ECT. 2014;30(1):5-9.
22. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
Opioid abuse
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Reducing CV risk in diabetes: An ADA update
More than 29 million Americans have diabetes, and each year another 1.7 million are given the diagnosis.1 Prediabetes is even more common; over one-third of US adults ages 20 years and older, and more than half of those who are ages 65 and older, have attained this precursor status, representing another 86 million Americans.1
Because the evidence base for the management of diabetes is rapidly expanding, the American Diabetes Association’s (ADA) Professional Practice Committee updates its Standards of Medical Care in Diabetes annually to incorporate new evidence into its recommendations. The 2017 Standards of Care are available at: professional.diabetes.org/jfp.2
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of morbidity and mortality for people with diabetes, and is the largest contributor to the direct and indirect costs of the disease.2 As a result, all patients with diabetes should have cardiovascular (CV) risk factors, including dyslipidemia, hypertension, smoking, a family history of premature coronary disease, and the presence of albuminuria, assessed at least annually.2 Numerous studies have demonstrated the efficacy of controlling individual CV risk factors in preventing or slowing ASCVD in people with diabetes. Even larger benefits, including reduced ASCVD morbidity and mortality, can be achieved when multiple risk factors are addressed simultaneously.3
To hone your management of CV risks in patients with diabetes, we’ve put together this Q&A pointing out the elements of the ADA’s 2017 Standards of Care that are most relevant to the management of patients at risk for, or with established, ASCVD.

Screening
Since ASCVD so commonly co-occurs with diabetes, should I routinely screen asymptomatic patients with diabetes for heart disease?
No. The current evidence suggests that outcomes are NOT improved by screening people before they develop symptoms of ASCVD,4 and widespread ASCVD screening has not been shown to be cost-effective. Cardiac testing should be reserved for those with typical or atypical symptoms or those with an abnormal resting electrocardiogram (EKG).
Lifestyle modification
What are the benefits of lifestyle interventions?
The benefits include not only lost pounds, but improved mobility, physical and sexual functioning, and health-related quality of life. Recommend that all overweight patients with diabetes take advantage of intensive lifestyle interventions focusing on weight loss through decreased caloric intake and increased physical activity as per the Look AHEAD (Action for Health in Diabetes) trial.5 Although the intensive lifestyle intervention in the Look AHEAD trial did not decrease CV outcomes over 10 years of follow-up, it did improve control of CV risk factors and led to people in the intervention group taking fewer glucose-, blood pressure (BP)-, and lipid-lowering medications than those in the standard care group.
There is no one diet that is recommended for all people with diabetes. Weight reduction often requires intensive intervention. In order for weight loss diets to be sustainable, they must include patient preferences.
People with diabetes should be encouraged to receive individualized medical nutrition therapy (MNT), preferably from a registered dietitian who is well versed in nutritional management for diabetes. Such MNT is associated with a 0.5% to 2% decrease in A1c levels for people with type 2 diabetes.6-9 Specific healthy diets include the Mediterranean, Dietary Approaches to Stop Hypertension (DASH), and plant-based diets.
A new lifestyle recommendation in this year’s ADA Standards is that periods of prolonged sitting should be interrupted every 30 minutes with a period of physical activity. This appears to have glycemic benefits.2
Hypertension/BP management
When should I initiate hypertension treatment in patients with diabetes?
Nonpharmacologic therapy is reasonable in people with diabetes and mildly elevated BP (>120/80 mm Hg). If systolic blood pressure (SBP) is confirmed to be >140 mm Hg and/or diastolic blood pressure (DBP) is confirmed to be >90 mm Hg, the ADA recommends initiating pharmacologic therapy along with nonpharmacologic strategies. For patients with confirmed office-based BP >160/100 mm Hg, the ADA advises initiating lifestyle modifications as well as 2 pharmacologic medications (or a single pill combination of agents).2
What is the recommended BP target for patients with diabetes and hypertension?
These patients should be treated with a combination of measures, including lifestyle modification and pharmacologic therapy, to a target BP of <140/90 mm Hg. Randomized controlled trials (RCTs) have shown benefits with this target in terms of a reduction in the incidence of coronary heart disease (CHD) events, stroke, and diabetic kidney disease.10,11
A 2012 meta-analysis of randomized trials involving adults with type 2 diabetes mellitus (T2DM) and comparing intensive BP targets (≤130 mm Hg SBP and ≤80 mm Hg DBP) with standard targets (≤140-160 mm Hg SBP and ≤85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MIs associated with more intense BP control. There was a statistically significant 35% relative risk (RR) reduction in stroke with intensive targets, but lower BP was also associated with an increased risk of hypotension and syncope.12
The 2010 Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,13 which randomized 5518 patients with T2DM at high risk for ASCVD to either a target SBP of <120 mm Hg or 130 to 140 mm Hg, found that the patients with the lower SBP target did not benefit in the primary end point (a composite of nonfatal MI, nonfatal stroke, and CV death), but did benefit from nominally significant lower rates of total stroke and nonfatal stroke.
Based on these data, the ADA Standards of Care suggest that, “more intensive BP control may be reasonable in certain motivated, ACCORD-like patients (40-79 years of age with prior evidence of CVD or multiple CV risk factors) who have been educated about the added treatment burden, side effects, and costs of more intensive BP control and for patients who prefer to lower their risk of stroke beyond what can be achieved with usual care.”
Another major study, the 2015 Systolic Blood Pressure Intervention Trial (SPRINT) trial,14 demonstrated that treating patients with hypertension to a target SBP <120 mm Hg compared to the usual target of <140 mm Hg resulted in a 25% lower RR of the primary outcome (a composite of MI, other acute coronary syndromes, stroke, heart failure, or death from CV causes) and about a 25% reduction in all-cause mortality; however, people with diabetes were not included in the trial, so the applicability of the results to decisions about BP management in patients with diabetes is not known.
A 2015 systematic review and meta-analysis of over 100,000 participants looked at SBP lowering in adults with T2DM and found that each 10-mm Hg reduction in SBP was associated with a significantly lower risk of morbidity, CV events, CHD, stroke, albuminuria, and retinopathy.10 When trials were stratified by mean baseline SBP (<140 mm Hg or ≥140 mm Hg), RRs for outcomes other than stroke, retinopathy, and renal failure were lower in studies with greater baseline SBP.
The latest ADA Standards of Care recommend that a lower BP target of 130/80 mm Hg may be appropriate for patients at high risk of CVD if this target can be achieved without undue treatment burden. A DBP of <80 mm Hg may also be appropriate in certain patients including those with a long life expectancy, CKD, elevated urinary albumin excretion, and those with evidence of CVD or associated risk factors.15 Of note, treating older adults with diabetes to an SBP target of <130 mm Hg has not been shown to improve cardiovascular outcomes,16 and treating to a diastolic target of <70 mm Hg has been associated with a greater risk of mortality.17
What are the current recommended treatment options?
Treatment for hypertension in adults with diabetes without albuminuria should include any of the classes of medications demonstrated to reduce CV events in patients with diabetes, such as:
- angiotensin-converting enzyme (ACE) inhibitors,
- angiotensin receptor blockers (ARBs),
- thiazide-like diuretics, and
- dihydropyridine calcium channel blockers.
These recommendations are based on evidence suggesting the lack of superiority of ACE inhibitors and ARBs over other classes of antihypertensive agents for the prevention of CV outcomes in all patients with diabetes.18 However, in people with diabetes at high risk for ASCVD and/or with albuminuria, ACE inhibitors and ARBs do reduce ASCVD outcomes and the progression of kidney disease.19-24 Thus, ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and urine albumin/creatinine ratios ≥30 mg/g, as these medications are associated with a reduction in the rate of kidney disease progression.
The use of both an ACE inhibitor and an ARB in combination is not recommended.25,26 For patients treated with ACE inhibitors, ARBs, or diuretics, serum creatinine/estimated glomerular filtration rate (eGFR) and serum potassium levels should be monitored.
What are the recommended lifestyle modifications for patients with diabetes and hypertension?
Regular exercise and healthy eating are recommended for all people with diabetes to optimize glycemic control and lose weight (if they are overweight or obese). For patients with hypertension, the DASH diet (available at: https://www.nhlbi.nih.gov/health/health-topics/topics/dash/) is effective at lowering BP. The DASH diet emphasizes reducing sodium intake, increasing potassium intake, limiting alcohol intake, and increasing physical activity. Specifically, sodium intake should be restricted to <2300 mg/d and patients should consume approximately 8 to 10 servings of fruits and vegetables per day and 2 to 3 servings of low-fat dairy per day. Alcohol should be limited to 2 drinks per day for men and one drink per day for women.
Most adults with diabetes should perform 150 minutes per week of moderate to vigorous exercise, spread over at least 3 days/week. In addition, it is recommended that resistance exercises be performed at least 2 to 3 days/week. Prolonged inactivity is detrimental to health and should be interrupted with activity every 30 minutes.27
Finally, as a part of lifestyle management for all patients with diabetes, smoking cessation is important, as is attention to stress, depression, and anxiety.
Is there an advantage to nighttime dosing of antihypertensive medications?
Yes. Growing evidence suggests that there is an ASCVD benefit to avoiding nocturnal BP dipping. A 2011 RCT of 448 participants with T2DM and hypertension showed a decrease in CV events and mortality during 5.4 years of follow-up if at least one antihypertensive medication was taken at bedtime.28 As a result of this and other evidence,29 consider administering one or more antihypertensive medications at bedtime, although this is not a formal recommendation in the ADA Standards of Care.
Are there any additional issues to be aware of when treating patients with diabetes and hypertension?
Yes. Sometimes patients who have had diabetes for many years have significant orthostatic hypotension secondary to autonomic neuropathy. Postural changes in BP and pulse may require adjustment of BP targets. Home BP self-monitoring and 24-hour ambulatory BP monitoring may indicate white-coat or masked hypertension.
Lipid management
What is the current evidence for lipid treatment in diabetes?
Lipid abnormalities are common in people with diabetes and contribute to the overall high risk of ASCVD in these patients. Subgroup analyses of patients in large trials with diabetes30 and trials involving patients with diabetes31 have shown significant improvements in primary and secondary prevention of ASCVD with statin use. A 2008 meta-analysis of 18,686 people with diabetes showed a 9% reduction in all-cause mortality and a 13% reduction in vascular mortality for each 39-mg/dL reduction in low-density lipoprotein (LDL) cholesterol.32 Absolute reductions in mortality are greatest in those with highest risk, but the benefits of statin therapy are clear for low- and moderate-risk individuals with diabetes, too.33,34 As a result, statins are the medications of choice for lipid lowering and CV risk reduction and should be used in addition to lifestyle management.
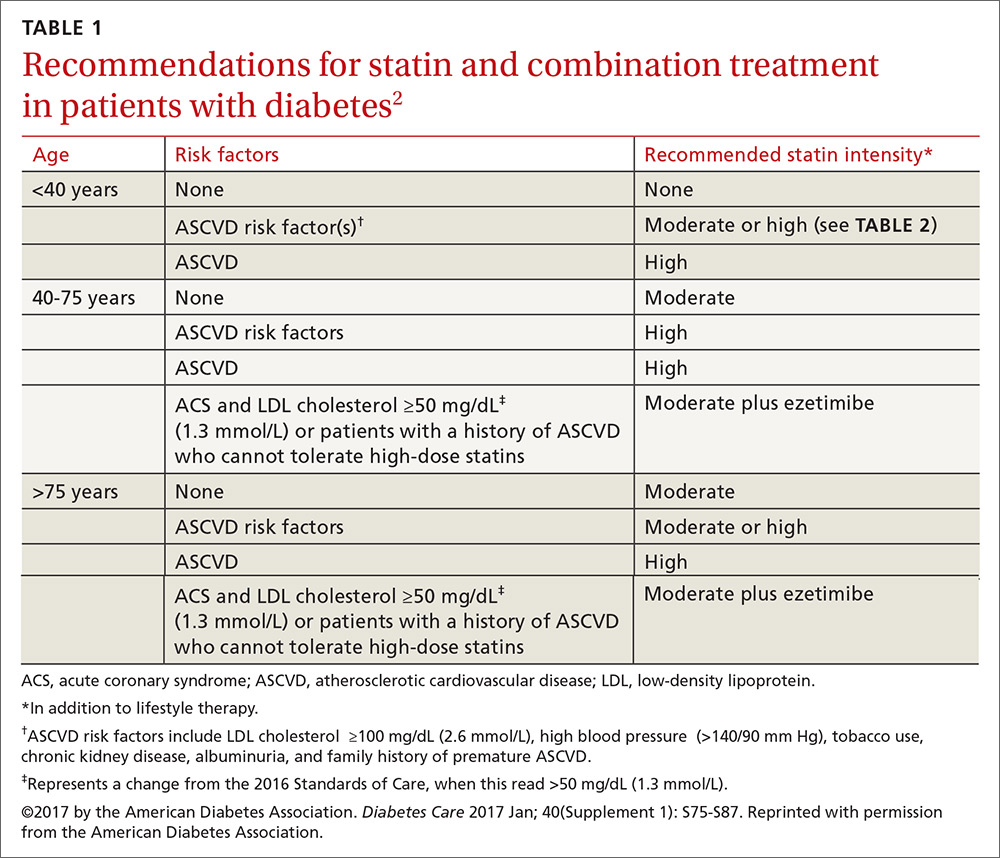
Who should get a statin, and how do I choose the optimum dosage?
Patients ages 40 to 75 years with diabetes but without additional ASCVD risk factors should receive a moderate-intensity statin, according to the ADA (see TABLES 12 and 22). For those with additional CV risk factors, a high-intensity statin should be considered. The American College of Cardiology/American Heart Association ASCVD risk calculator (available at: http://www.cvriskcalculator.com/) may be useful for some patients, but generally, risk is already known to be high for most patients with diabetes. For patients of all ages with diabetes and established ASCVD, high-intensity statin therapy should be added to lifestyle modifications.35-37
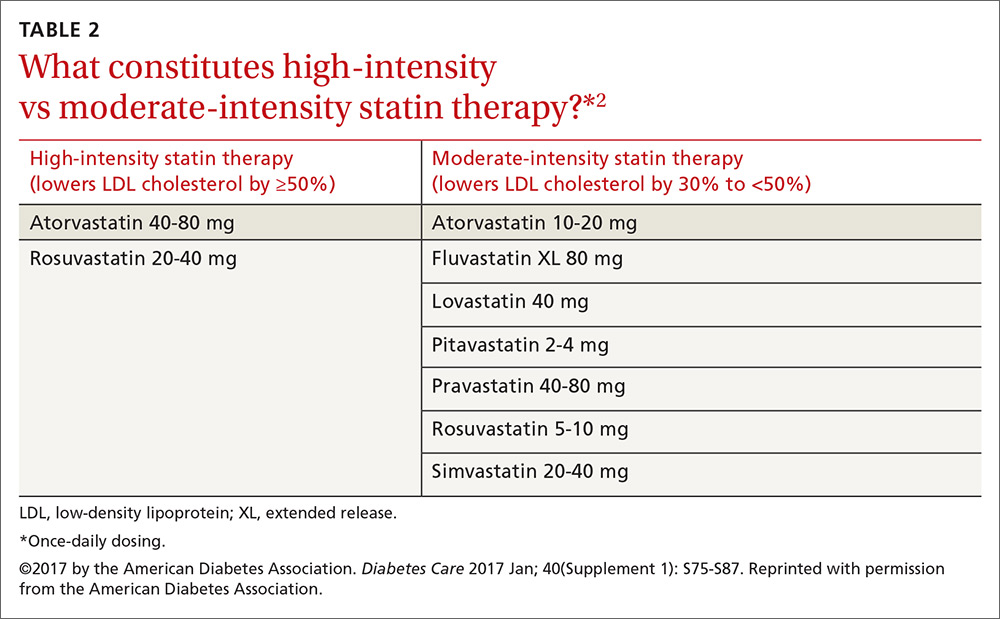
For patients with diabetes who are <40 years with additional ASCVD risk factors, few clinical trial data exist; nevertheless, consider a moderate- or high-intensity statin and lifestyle therapy. Similarly, for patients >75 years who have diabetes and no additional ASCVD risk factors, consider a moderate-intensity statin and lifestyle modifications. For older adults with additional ASCVD risk factors, consider high-intensity statin therapy.35-37
Statins and cognition. It should be noted that published data have not demonstrated an adverse effect of statins on cognition.38 Statins, however, have been linked to an increased risk of developing diabetes,39,40 although the absolute increase in risk is small, and much smaller than the benefit derived from preventing the development of coronary disease.
Should total cholesterol and LDL levels be used as targets with statin treatment?
No. Statin doses have primarily been tested against placebo in clinical trials, rather than testing to specific target LDL levels, suggesting that the initiation and intensification of statin therapy be based on a patient’s risk profile.35 When maximally tolerated doses of statins do not lower LDL cholesterol by more than 30% from the patient’s baseline, there is currently no good evidence that combination therapy would be helpful, so regular monitoring of lipid levels has limited value. A lipid profile that includes levels of total cholesterol, LDL cholesterol, high-density lipoprotein (HDL) cholesterol, and triglycerides should be obtained at initial medical evaluation, at diagnosis of diabetes, and every 5 years thereafter or before the initiation of statin therapy. Ongoing testing may be appropriate in individual circumstances and to monitor for adherence to, or efficacy of, therapy.
What should I do for my patients who can’t tolerate statins?
Try a lower dose or a different statin before eliminating the class. Research has shown that even small doses (eg, rosuvastatin 5 mg) have some benefit.41
How do combination treatments figure into the current treatment of lipids in patients with diabetes?
It depends on the agent and the patient’s profile.
Fenofibrate. The ADA does not recommend automatically adding fenofibrate to statin therapy because the combination is associated with increased risks for abnormal transaminase levels, myositis, and rhabdomyolysis. In the ACCORD trial, the combination of fenofibrate and simvastatin did not reduce the rate of fatal CV events, nonfatal MIs, or nonfatal strokes compared with simvastatin alone.42
That said, a subgroup analysis suggested a benefit for men with both a triglyceride level ≥204 mg/dL (2.3 mmol/L) and an HDL cholesterol level ≤34 mg/dL (0.9 mmol/L).42 For this reason, the combination of a statin and fenofibrate may be considered for men who meet these laboratory parameters. In addition, consider medical therapy for triglyceride levels ≥500 mg/dL to reduce the risk of pancreatitis.
Ezetimibe. Recommendations regarding ezetimibe are based on the IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), a 2015 RCT including over 18,000 patients that compared treatment with ezetimibe and simvastatin to simvastatin alone.43 Individuals in the trial were ≥50 years of age and had experienced an ACS within the preceding 10 days. In those with diabetes, the combination of moderate-intensity simvastatin (40 mg) and ezetimibe (10 mg) significantly reduced major adverse CV events with an absolute risk reduction of 5% (40% vs 45%) and an RR reduction of 14% over moderate-intensity simvastatin (40 mg) alone.
Based on these results, patients with diabetes and a recent ACS should be considered for combination therapy with ezetimibe and a moderate-intensity statin. The combination should also be considered in patients with diabetes and a history of ASCVD who cannot tolerate high-intensity statins.43
Niacin. The ADA currently does not recommend niacin in combination with a statin because of lack of efficacy on major ASCVD outcomes, possible increased risk of ischemic stroke, and adverse effects.44
What are the recommendations for the use of PCSK-9 inhibitors?
Proprotein convertase subtilisin/kexin type 9 (PCSK-9) inhibitors (ie, evolucumab and alirocumab) may be considered as adjunctive therapy to statins for patients with diabetes at high risk for ASCVD events who require additional lowering of LDL cholesterol. They may also be considered for those in whom high-intensity statin therapy is indicated, but not tolerated.
Antiplatelet agents
Who should take aspirin for primary prevention of CVD?
Both women and men ages ≥50 years who have diabetes and at least one additional CV risk factor (family history of premature ASCVD, hypertension, tobacco use, dyslipidemia, or albuminuria) should consider taking daily aspirin therapy (75-162 mg/d) if they do not have an excessive bleeding risk.45,46 The most common dose in the United States is 81 mg. This recommendation is supported by a 2010 consensus statement of the American Diabetes Association, American Heart Association, and the American College of Cardiology.47
Should patients with diabetes and heart disease receive antiplatelet therapy?
Yes. The evidence is clear that people with known diabetes and ASCVD benefit from aspirin therapy, according to the 2017 Standards of Care. Clopidogrel 75 mg/d is an appropriate alternative for patients who are allergic to aspirin. Dual antiplatelet therapy (a P2Y12 receptor antagonist and aspirin) should be used for as long as one year after an ACS and may have benefits beyond this period.48
Established heart disease
Are there specific recommendations for patients with diabetes and CHD?
According to the ADA Standards, there is good evidence that both aspirin and statin therapy are beneficial for patients with known ASCVD, and that high-intensity statin therapy should be used. In addition, consider ACE inhibitors to reduce the future risk of CV events. In patients with a prior MI, continue beta-blocker therapy for at least 2 years post event.49
Which medications should I avoid, or approach with caution, in patients with congestive heart failure (CHF)?
Thiazolidinediones, dipeptidyl peptidase 4 (DPP-4) inhibitors, and metformin all require careful attention. This is especially important to know when you consider that almost half of all patients with T2DM will develop heart failure.50
Thiazolidinediones. The 2017 Standards of Care state that patients with diabetes and symptomatic congestive heart failure should not receive thiazolidinediones, as they can worsen heart failure status via fluid retention. As such, they are contraindicated in patients with class III and IV heart failure.51
DPP-4 inhibitors. The studies on DPP-4 inhibitors and heart failure have had mixed results. The 2013 SAVOR-TIMI (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus–Thrombolysis in Myocardial Infarction) 53 trial52 showed that patients treated with saxagliptin were more likely to be hospitalized for heart failure than those taking placebo (3.5% vs 2.8%, respectively). However, the 2015 EXAMINE (Examination of Cardiovascular Outcomes with Alogliptin vs Standard of Care)53 trial and the 2015 TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin)54 trial evaluated heart failure and mortality outcomes in patients with alogliptin and sitagliptin, respectively, compared to placebo, and did not show a relationship to heart failure.
Metformin may be used in people who have T2DM and stable CHF if their eGFR remains >30 mL/min; it should be withheld from patients with unstable heart failure and those who are hospitalized with CHF.
Are there antihyperglycemic medications that reduce CV morbidity and mortality in those with established ASCVD?
Yes. This year’s ADA Standards indicate that certain glucose-lowering medications—specifically empagliflozin (a sodium–glucose cotransporter [SGLT]-2 inhibitor) and liraglutide (a glucagon-like peptide [GLP]-1 receptor agonist)—have been shown to be beneficial for those with established CVD. According to the 2017 Standards of Care, “In patients with longstanding suboptimally controlled T2DM and established ASCVD, empagliflozin or liraglutide should be considered, as they have been shown to reduce CV and all-cause mortality when added to standard care.”2 The studies that provide support for their use are summarized below. Ongoing studies are investigating the CV effects of other agents in these drug classes.
Empagliflozin. The 2015 EMPA-REG OUTCOME (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) study55 was a randomized double-blind study of empagliflozin vs placebo and usual care in patients with diabetes and established CVD. Over a median follow-up of 3.1 years, treatment with empagliflozin reduced the aggregate outcome of MI, stroke, and CV death by 14%, reduced CV deaths by 38%, and decreased deaths from any cause by 32%. In December 2016, the FDA announced a new indication for empagliflozin: to reduce the risk of CV death in adult patients with T2DM and CVD.56
Liraglutide. The LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results: A Long Term Evaluation) trial57 was a double-blind randomized trial of liraglutide vs placebo added to usual care in patients with T2DM at high risk for CVD or with existing CVD. More than 80% of the participants had existing CVD including a history of prior MI, cerebrovascular disease, or peripheral vascular disease. After a median follow-up of 3.8 years, the group taking liraglutide demonstrated a 13% reduction in the composite outcome of MI, stroke, or CV death, a 22% reduction in CV death, and a 15% reduction in death from any cause, compared with placebo.57
CORRESPONDENCE
Neil Skolnik, MD, Abington-Jefferson Health, 500 Old York Rd, Ste 108, Jenkintown, PA 19046; nskolnik@comcast.net.
The authors thank Sarah Bradley, director, professional engagement & collaboration at the American Diabetes Association, for her editorial and organizational assistance in the preparation of this manuscript.
1. Centers for Disease Control and Prevention. National Center for Chronic Disease Prevention and Health Promotion. National diabetes statistics report, 2014. Estimates of diabetes and its burden in the United States. Available at: http://templatelab.com/national-diabetes-report-2014/. Accessed April 7, 2017.
2. American Diabetes Association. Standards of Medical Care in Diabetes—2017. Available at: http://professional.diabetes.org/sites/professional.diabetes.org/files/media/dc_40_s1_final.pdf. Accessed April 7, 2017.
3. Gaede P, Lund-Andersen H, Parving HH, et al. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580-591.
4. Bax JJ, Young LH, Frye RL, et al; American Diabetes Association. Screening for coronary artery disease in patients with diabetes. Diabetes Care. 2007;30:2729-2736.
5. The Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. 2013;369:145-154.
6. UK Prospective Diabetes Study (UKDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKDS 34). Lancet. 1998;352:854-865.
7. Ziemer DC, Berkowitz KJ, Panayioto RM, et al. A simple meal plan emphasizing healthy food choices is as effective as an exchange-based meal plan for urban African Americans with type 2 diabetes. Diabetes Care. 2003;26:1719-1724.
8. Wolf AM, Conaway RM, Crowther JQ, et al; Improving Control with Activity and Nutrition (ICAN) Study. Translating lifestyle intervention to practice in obese patients with type 2 diabetes: Improving Control with Activity and Nutrition (ICAN) study. Diabetes Care. 2004;27:1570-1576.
9. Coppell KJ, Kataoka M, Williams SM, et al. Nutritional intervention in patients with type 2 diabetes who are hyperglycaemic despite optimised drug treatment-Lifestyle Over and Above Drugs in Diabetes (LOADD) study: randomised controlled trial. BMJ. 2010;341:c3337.
10. Emdin CA, Rahimi K, Neal B, et al. Blood pressure lowering in type 2 diabetes: a systematic review and meta-analysis. JAMA. 2015;313:603-615.
11. Arguedas JA, Leiva V, Wright JM. Blood pressure targets for hypertension in people with diabetes mellitus. Cochrane Database Syst Rev. 2013;10:CD008277.
12. McBrien K, Rabi DM, Campbell N, et al. Intensive and standard blood pressure targets in patients with type 2 diabetes mellitus: systematic review and meta-analysis. Arch Intern Med. 2012;172:1296-1303.
13. ACCORD Study Group, Cushman WC, Evans GW, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med. 2010;362:1575-1585.
14. SPRINT Research Group, Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
15. Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet. 1998;351:1755-1762.
16. Kirkman MS, Briscoe VJ, Clark N, et al. Diabetes in older adults. Diabetes Care. 2012;35:2650-2664.
17. Anderson RJ, Bahn GD, Moritz TE, et al; VADT Study Group. Blood pressure and cardiovascular disease risk in the Veterans Affairs Diabetes Trial. Diabetes Care. 2011;34:34-38.
18. Bangalore S, Fakheri R, Toklu B, et al. Diabetes mellitus as a compelling indication for use of renin angiotensin system blockers: systematic review and meta-analysis of randomized trials. BMJ. 2016;352:i438.
19. Heart Outcomes Prevention Evaluation Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet. 2000;355:253-259.
20. Granger CB, McMurray JJ, Yusuf S, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772-776.
21. McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362:767-771.
22. Pfeffer MA, Swedberg K, Granger CB, et al; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759-766.
23. Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861-869.
24. Palmer SC, Mavridis D, Navarese E, et al. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet. 2015;385:2047-2056.
25. The ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
26. Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. 2013;369:1892-1903.
27. Colberg SR, Sigal RJ, Yardley JE, et al. Physical activity/exercise and diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39:2065-2079.
28. Hermida RC, Ayala DE, Mojón A, et al. Influence of time of day of blood pressure-lowering treatment on cardiovascular risk in hypertensive patients with type 2 diabetes. Diabetes Care. 2011;34:1270-1276.
29. Zhao P, Xu P, Wan C, et al. Evening versus morning dosing regimen drug therapy for hypertension. Cochrane Database Syst Rev. 2011;10:CD004184.
30. Py̆orälä K, Pedersen TR, Kjekshus J, et al. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care. 1997;20:614-620.
31. Knopp RH, d’Emden M, Smilde JG, et al. Efficacy and safety of atorvastatin in the prevention of cardiovascular end points in subjects with type 2 diabetes: the Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in Non-Insulin-Dependent Diabetes Mellitus (ASPEN). Diabetes Care. 2006;29:1478-1485.
32. Cholesterol Treatment Trialists’ (CTT) Collaborators, Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117-125.
33. Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013:CD004816.
34. Carter AA, Gomes T, Camacho X, et al. Risk of incident diabetes among patients treated with statins: population based study. BMJ. 2013;346:f2610.
35. Hayward RA, Hofer TP, Vijan S. Narrative review: lack of evidence for recommended low-density lipoprotein treatment targets: a solvable problem. Ann Intern Med. 2006;145:520-530.
36. Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495-1504.
37. de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292:1307-1316.
38. Richardson K, Schoen M, French B, et al. Statins and cognitive function: a systematic review. Ann Intern Med. 2013;159:688-697.
39. Rajpathak SN, Kumbhani DJ, Crandall J, et al. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care. 2009;32:1924-1929.
40. Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735-742.
41. Meek C, Wierzbicki AS, Jewkes C, et al. Daily and intermittent rosuvastatin 5 mg therapy in statin intolerant patients: an observational study. Curr Med Res Opin. 2012;28:371-378.
42. ACCORD Study Group, Ginsberg HN, Bam MB, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563-1574.
43. Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387-2397.
44. AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255-2267.
45. Antithrombotic Trialists’ (ATT) Collaboration, Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849-1860.
46. Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J. 2012;33:1635-1701.
47. Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes. A position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Diabetes Care. 2010;33:1395-1402.
48. Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(suppl):e637S-e668S.
49. Kezerashvilli A, Marzo K, De Leon J. Beta blocker use after acute myocardial infarction in the patient with normal systolic function: when is it “ok” to discontinue? Curr Cardiol Rev. 2012;8:77-84.
50. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29-34.
51. Pioglitazone Package Insert. Available at: http://medlibrary.org/lib/rx/meds/pioglitazone-3/. Accessed April 10, 2017.
52. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
53. Zannad F, Cannon CP, Cushman WC, et al; EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
54. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
55. Zinman B, Wanner C, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
56. FDA approves Jardiance to reduce cardiovascular death in adults with type 2 diabetes. FDA News Release, December 2, 2016. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm531517.htm. Accessed February 9, 2017.
57. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
More than 29 million Americans have diabetes, and each year another 1.7 million are given the diagnosis.1 Prediabetes is even more common; over one-third of US adults ages 20 years and older, and more than half of those who are ages 65 and older, have attained this precursor status, representing another 86 million Americans.1
Because the evidence base for the management of diabetes is rapidly expanding, the American Diabetes Association’s (ADA) Professional Practice Committee updates its Standards of Medical Care in Diabetes annually to incorporate new evidence into its recommendations. The 2017 Standards of Care are available at: professional.diabetes.org/jfp.2
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of morbidity and mortality for people with diabetes, and is the largest contributor to the direct and indirect costs of the disease.2 As a result, all patients with diabetes should have cardiovascular (CV) risk factors, including dyslipidemia, hypertension, smoking, a family history of premature coronary disease, and the presence of albuminuria, assessed at least annually.2 Numerous studies have demonstrated the efficacy of controlling individual CV risk factors in preventing or slowing ASCVD in people with diabetes. Even larger benefits, including reduced ASCVD morbidity and mortality, can be achieved when multiple risk factors are addressed simultaneously.3
To hone your management of CV risks in patients with diabetes, we’ve put together this Q&A pointing out the elements of the ADA’s 2017 Standards of Care that are most relevant to the management of patients at risk for, or with established, ASCVD.
Screening
Since ASCVD so commonly co-occurs with diabetes, should I routinely screen asymptomatic patients with diabetes for heart disease?
No. The current evidence suggests that outcomes are NOT improved by screening people before they develop symptoms of ASCVD,4 and widespread ASCVD screening has not been shown to be cost-effective. Cardiac testing should be reserved for those with typical or atypical symptoms or those with an abnormal resting electrocardiogram (EKG).
Lifestyle modification
What are the benefits of lifestyle interventions?
The benefits include not only lost pounds, but improved mobility, physical and sexual functioning, and health-related quality of life. Recommend that all overweight patients with diabetes take advantage of intensive lifestyle interventions focusing on weight loss through decreased caloric intake and increased physical activity as per the Look AHEAD (Action for Health in Diabetes) trial.5 Although the intensive lifestyle intervention in the Look AHEAD trial did not decrease CV outcomes over 10 years of follow-up, it did improve control of CV risk factors and led to people in the intervention group taking fewer glucose-, blood pressure (BP)-, and lipid-lowering medications than those in the standard care group.
There is no one diet that is recommended for all people with diabetes. Weight reduction often requires intensive intervention. In order for weight loss diets to be sustainable, they must include patient preferences.
People with diabetes should be encouraged to receive individualized medical nutrition therapy (MNT), preferably from a registered dietitian who is well versed in nutritional management for diabetes. Such MNT is associated with a 0.5% to 2% decrease in A1c levels for people with type 2 diabetes.6-9 Specific healthy diets include the Mediterranean, Dietary Approaches to Stop Hypertension (DASH), and plant-based diets.
A new lifestyle recommendation in this year’s ADA Standards is that periods of prolonged sitting should be interrupted every 30 minutes with a period of physical activity. This appears to have glycemic benefits.2
Hypertension/BP management
When should I initiate hypertension treatment in patients with diabetes?
Nonpharmacologic therapy is reasonable in people with diabetes and mildly elevated BP (>120/80 mm Hg). If systolic blood pressure (SBP) is confirmed to be >140 mm Hg and/or diastolic blood pressure (DBP) is confirmed to be >90 mm Hg, the ADA recommends initiating pharmacologic therapy along with nonpharmacologic strategies. For patients with confirmed office-based BP >160/100 mm Hg, the ADA advises initiating lifestyle modifications as well as 2 pharmacologic medications (or a single pill combination of agents).2
What is the recommended BP target for patients with diabetes and hypertension?
These patients should be treated with a combination of measures, including lifestyle modification and pharmacologic therapy, to a target BP of <140/90 mm Hg. Randomized controlled trials (RCTs) have shown benefits with this target in terms of a reduction in the incidence of coronary heart disease (CHD) events, stroke, and diabetic kidney disease.10,11
A 2012 meta-analysis of randomized trials involving adults with type 2 diabetes mellitus (T2DM) and comparing intensive BP targets (≤130 mm Hg SBP and ≤80 mm Hg DBP) with standard targets (≤140-160 mm Hg SBP and ≤85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MIs associated with more intense BP control. There was a statistically significant 35% relative risk (RR) reduction in stroke with intensive targets, but lower BP was also associated with an increased risk of hypotension and syncope.12
The 2010 Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,13 which randomized 5518 patients with T2DM at high risk for ASCVD to either a target SBP of <120 mm Hg or 130 to 140 mm Hg, found that the patients with the lower SBP target did not benefit in the primary end point (a composite of nonfatal MI, nonfatal stroke, and CV death), but did benefit from nominally significant lower rates of total stroke and nonfatal stroke.
Based on these data, the ADA Standards of Care suggest that, “more intensive BP control may be reasonable in certain motivated, ACCORD-like patients (40-79 years of age with prior evidence of CVD or multiple CV risk factors) who have been educated about the added treatment burden, side effects, and costs of more intensive BP control and for patients who prefer to lower their risk of stroke beyond what can be achieved with usual care.”
Another major study, the 2015 Systolic Blood Pressure Intervention Trial (SPRINT) trial,14 demonstrated that treating patients with hypertension to a target SBP <120 mm Hg compared to the usual target of <140 mm Hg resulted in a 25% lower RR of the primary outcome (a composite of MI, other acute coronary syndromes, stroke, heart failure, or death from CV causes) and about a 25% reduction in all-cause mortality; however, people with diabetes were not included in the trial, so the applicability of the results to decisions about BP management in patients with diabetes is not known.
A 2015 systematic review and meta-analysis of over 100,000 participants looked at SBP lowering in adults with T2DM and found that each 10-mm Hg reduction in SBP was associated with a significantly lower risk of morbidity, CV events, CHD, stroke, albuminuria, and retinopathy.10 When trials were stratified by mean baseline SBP (<140 mm Hg or ≥140 mm Hg), RRs for outcomes other than stroke, retinopathy, and renal failure were lower in studies with greater baseline SBP.
The latest ADA Standards of Care recommend that a lower BP target of 130/80 mm Hg may be appropriate for patients at high risk of CVD if this target can be achieved without undue treatment burden. A DBP of <80 mm Hg may also be appropriate in certain patients including those with a long life expectancy, CKD, elevated urinary albumin excretion, and those with evidence of CVD or associated risk factors.15 Of note, treating older adults with diabetes to an SBP target of <130 mm Hg has not been shown to improve cardiovascular outcomes,16 and treating to a diastolic target of <70 mm Hg has been associated with a greater risk of mortality.17
What are the current recommended treatment options?
Treatment for hypertension in adults with diabetes without albuminuria should include any of the classes of medications demonstrated to reduce CV events in patients with diabetes, such as:
- angiotensin-converting enzyme (ACE) inhibitors,
- angiotensin receptor blockers (ARBs),
- thiazide-like diuretics, and
- dihydropyridine calcium channel blockers.
These recommendations are based on evidence suggesting the lack of superiority of ACE inhibitors and ARBs over other classes of antihypertensive agents for the prevention of CV outcomes in all patients with diabetes.18 However, in people with diabetes at high risk for ASCVD and/or with albuminuria, ACE inhibitors and ARBs do reduce ASCVD outcomes and the progression of kidney disease.19-24 Thus, ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and urine albumin/creatinine ratios ≥30 mg/g, as these medications are associated with a reduction in the rate of kidney disease progression.
The use of both an ACE inhibitor and an ARB in combination is not recommended.25,26 For patients treated with ACE inhibitors, ARBs, or diuretics, serum creatinine/estimated glomerular filtration rate (eGFR) and serum potassium levels should be monitored.
What are the recommended lifestyle modifications for patients with diabetes and hypertension?
Regular exercise and healthy eating are recommended for all people with diabetes to optimize glycemic control and lose weight (if they are overweight or obese). For patients with hypertension, the DASH diet (available at: https://www.nhlbi.nih.gov/health/health-topics/topics/dash/) is effective at lowering BP. The DASH diet emphasizes reducing sodium intake, increasing potassium intake, limiting alcohol intake, and increasing physical activity. Specifically, sodium intake should be restricted to <2300 mg/d and patients should consume approximately 8 to 10 servings of fruits and vegetables per day and 2 to 3 servings of low-fat dairy per day. Alcohol should be limited to 2 drinks per day for men and one drink per day for women.
Most adults with diabetes should perform 150 minutes per week of moderate to vigorous exercise, spread over at least 3 days/week. In addition, it is recommended that resistance exercises be performed at least 2 to 3 days/week. Prolonged inactivity is detrimental to health and should be interrupted with activity every 30 minutes.27
Finally, as a part of lifestyle management for all patients with diabetes, smoking cessation is important, as is attention to stress, depression, and anxiety.
Is there an advantage to nighttime dosing of antihypertensive medications?
Yes. Growing evidence suggests that there is an ASCVD benefit to avoiding nocturnal BP dipping. A 2011 RCT of 448 participants with T2DM and hypertension showed a decrease in CV events and mortality during 5.4 years of follow-up if at least one antihypertensive medication was taken at bedtime.28 As a result of this and other evidence,29 consider administering one or more antihypertensive medications at bedtime, although this is not a formal recommendation in the ADA Standards of Care.
Are there any additional issues to be aware of when treating patients with diabetes and hypertension?
Yes. Sometimes patients who have had diabetes for many years have significant orthostatic hypotension secondary to autonomic neuropathy. Postural changes in BP and pulse may require adjustment of BP targets. Home BP self-monitoring and 24-hour ambulatory BP monitoring may indicate white-coat or masked hypertension.
Lipid management
What is the current evidence for lipid treatment in diabetes?
Lipid abnormalities are common in people with diabetes and contribute to the overall high risk of ASCVD in these patients. Subgroup analyses of patients in large trials with diabetes30 and trials involving patients with diabetes31 have shown significant improvements in primary and secondary prevention of ASCVD with statin use. A 2008 meta-analysis of 18,686 people with diabetes showed a 9% reduction in all-cause mortality and a 13% reduction in vascular mortality for each 39-mg/dL reduction in low-density lipoprotein (LDL) cholesterol.32 Absolute reductions in mortality are greatest in those with highest risk, but the benefits of statin therapy are clear for low- and moderate-risk individuals with diabetes, too.33,34 As a result, statins are the medications of choice for lipid lowering and CV risk reduction and should be used in addition to lifestyle management.
Who should get a statin, and how do I choose the optimum dosage?
Patients ages 40 to 75 years with diabetes but without additional ASCVD risk factors should receive a moderate-intensity statin, according to the ADA (see TABLES 12 and 22). For those with additional CV risk factors, a high-intensity statin should be considered. The American College of Cardiology/American Heart Association ASCVD risk calculator (available at: http://www.cvriskcalculator.com/) may be useful for some patients, but generally, risk is already known to be high for most patients with diabetes. For patients of all ages with diabetes and established ASCVD, high-intensity statin therapy should be added to lifestyle modifications.35-37
For patients with diabetes who are <40 years with additional ASCVD risk factors, few clinical trial data exist; nevertheless, consider a moderate- or high-intensity statin and lifestyle therapy. Similarly, for patients >75 years who have diabetes and no additional ASCVD risk factors, consider a moderate-intensity statin and lifestyle modifications. For older adults with additional ASCVD risk factors, consider high-intensity statin therapy.35-37
Statins and cognition. It should be noted that published data have not demonstrated an adverse effect of statins on cognition.38 Statins, however, have been linked to an increased risk of developing diabetes,39,40 although the absolute increase in risk is small, and much smaller than the benefit derived from preventing the development of coronary disease.
Should total cholesterol and LDL levels be used as targets with statin treatment?
No. Statin doses have primarily been tested against placebo in clinical trials, rather than testing to specific target LDL levels, suggesting that the initiation and intensification of statin therapy be based on a patient’s risk profile.35 When maximally tolerated doses of statins do not lower LDL cholesterol by more than 30% from the patient’s baseline, there is currently no good evidence that combination therapy would be helpful, so regular monitoring of lipid levels has limited value. A lipid profile that includes levels of total cholesterol, LDL cholesterol, high-density lipoprotein (HDL) cholesterol, and triglycerides should be obtained at initial medical evaluation, at diagnosis of diabetes, and every 5 years thereafter or before the initiation of statin therapy. Ongoing testing may be appropriate in individual circumstances and to monitor for adherence to, or efficacy of, therapy.
What should I do for my patients who can’t tolerate statins?
Try a lower dose or a different statin before eliminating the class. Research has shown that even small doses (eg, rosuvastatin 5 mg) have some benefit.41
How do combination treatments figure into the current treatment of lipids in patients with diabetes?
It depends on the agent and the patient’s profile.
Fenofibrate. The ADA does not recommend automatically adding fenofibrate to statin therapy because the combination is associated with increased risks for abnormal transaminase levels, myositis, and rhabdomyolysis. In the ACCORD trial, the combination of fenofibrate and simvastatin did not reduce the rate of fatal CV events, nonfatal MIs, or nonfatal strokes compared with simvastatin alone.42
That said, a subgroup analysis suggested a benefit for men with both a triglyceride level ≥204 mg/dL (2.3 mmol/L) and an HDL cholesterol level ≤34 mg/dL (0.9 mmol/L).42 For this reason, the combination of a statin and fenofibrate may be considered for men who meet these laboratory parameters. In addition, consider medical therapy for triglyceride levels ≥500 mg/dL to reduce the risk of pancreatitis.
Ezetimibe. Recommendations regarding ezetimibe are based on the IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), a 2015 RCT including over 18,000 patients that compared treatment with ezetimibe and simvastatin to simvastatin alone.43 Individuals in the trial were ≥50 years of age and had experienced an ACS within the preceding 10 days. In those with diabetes, the combination of moderate-intensity simvastatin (40 mg) and ezetimibe (10 mg) significantly reduced major adverse CV events with an absolute risk reduction of 5% (40% vs 45%) and an RR reduction of 14% over moderate-intensity simvastatin (40 mg) alone.
Based on these results, patients with diabetes and a recent ACS should be considered for combination therapy with ezetimibe and a moderate-intensity statin. The combination should also be considered in patients with diabetes and a history of ASCVD who cannot tolerate high-intensity statins.43
Niacin. The ADA currently does not recommend niacin in combination with a statin because of lack of efficacy on major ASCVD outcomes, possible increased risk of ischemic stroke, and adverse effects.44
What are the recommendations for the use of PCSK-9 inhibitors?
Proprotein convertase subtilisin/kexin type 9 (PCSK-9) inhibitors (ie, evolucumab and alirocumab) may be considered as adjunctive therapy to statins for patients with diabetes at high risk for ASCVD events who require additional lowering of LDL cholesterol. They may also be considered for those in whom high-intensity statin therapy is indicated, but not tolerated.
Antiplatelet agents
Who should take aspirin for primary prevention of CVD?
Both women and men ages ≥50 years who have diabetes and at least one additional CV risk factor (family history of premature ASCVD, hypertension, tobacco use, dyslipidemia, or albuminuria) should consider taking daily aspirin therapy (75-162 mg/d) if they do not have an excessive bleeding risk.45,46 The most common dose in the United States is 81 mg. This recommendation is supported by a 2010 consensus statement of the American Diabetes Association, American Heart Association, and the American College of Cardiology.47
Should patients with diabetes and heart disease receive antiplatelet therapy?
Yes. The evidence is clear that people with known diabetes and ASCVD benefit from aspirin therapy, according to the 2017 Standards of Care. Clopidogrel 75 mg/d is an appropriate alternative for patients who are allergic to aspirin. Dual antiplatelet therapy (a P2Y12 receptor antagonist and aspirin) should be used for as long as one year after an ACS and may have benefits beyond this period.48
Established heart disease
Are there specific recommendations for patients with diabetes and CHD?
According to the ADA Standards, there is good evidence that both aspirin and statin therapy are beneficial for patients with known ASCVD, and that high-intensity statin therapy should be used. In addition, consider ACE inhibitors to reduce the future risk of CV events. In patients with a prior MI, continue beta-blocker therapy for at least 2 years post event.49
Which medications should I avoid, or approach with caution, in patients with congestive heart failure (CHF)?
Thiazolidinediones, dipeptidyl peptidase 4 (DPP-4) inhibitors, and metformin all require careful attention. This is especially important to know when you consider that almost half of all patients with T2DM will develop heart failure.50
Thiazolidinediones. The 2017 Standards of Care state that patients with diabetes and symptomatic congestive heart failure should not receive thiazolidinediones, as they can worsen heart failure status via fluid retention. As such, they are contraindicated in patients with class III and IV heart failure.51
DPP-4 inhibitors. The studies on DPP-4 inhibitors and heart failure have had mixed results. The 2013 SAVOR-TIMI (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus–Thrombolysis in Myocardial Infarction) 53 trial52 showed that patients treated with saxagliptin were more likely to be hospitalized for heart failure than those taking placebo (3.5% vs 2.8%, respectively). However, the 2015 EXAMINE (Examination of Cardiovascular Outcomes with Alogliptin vs Standard of Care)53 trial and the 2015 TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin)54 trial evaluated heart failure and mortality outcomes in patients with alogliptin and sitagliptin, respectively, compared to placebo, and did not show a relationship to heart failure.
Metformin may be used in people who have T2DM and stable CHF if their eGFR remains >30 mL/min; it should be withheld from patients with unstable heart failure and those who are hospitalized with CHF.
Are there antihyperglycemic medications that reduce CV morbidity and mortality in those with established ASCVD?
Yes. This year’s ADA Standards indicate that certain glucose-lowering medications—specifically empagliflozin (a sodium–glucose cotransporter [SGLT]-2 inhibitor) and liraglutide (a glucagon-like peptide [GLP]-1 receptor agonist)—have been shown to be beneficial for those with established CVD. According to the 2017 Standards of Care, “In patients with longstanding suboptimally controlled T2DM and established ASCVD, empagliflozin or liraglutide should be considered, as they have been shown to reduce CV and all-cause mortality when added to standard care.”2 The studies that provide support for their use are summarized below. Ongoing studies are investigating the CV effects of other agents in these drug classes.
Empagliflozin. The 2015 EMPA-REG OUTCOME (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) study55 was a randomized double-blind study of empagliflozin vs placebo and usual care in patients with diabetes and established CVD. Over a median follow-up of 3.1 years, treatment with empagliflozin reduced the aggregate outcome of MI, stroke, and CV death by 14%, reduced CV deaths by 38%, and decreased deaths from any cause by 32%. In December 2016, the FDA announced a new indication for empagliflozin: to reduce the risk of CV death in adult patients with T2DM and CVD.56
Liraglutide. The LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results: A Long Term Evaluation) trial57 was a double-blind randomized trial of liraglutide vs placebo added to usual care in patients with T2DM at high risk for CVD or with existing CVD. More than 80% of the participants had existing CVD including a history of prior MI, cerebrovascular disease, or peripheral vascular disease. After a median follow-up of 3.8 years, the group taking liraglutide demonstrated a 13% reduction in the composite outcome of MI, stroke, or CV death, a 22% reduction in CV death, and a 15% reduction in death from any cause, compared with placebo.57
CORRESPONDENCE
Neil Skolnik, MD, Abington-Jefferson Health, 500 Old York Rd, Ste 108, Jenkintown, PA 19046; nskolnik@comcast.net.
The authors thank Sarah Bradley, director, professional engagement & collaboration at the American Diabetes Association, for her editorial and organizational assistance in the preparation of this manuscript.
More than 29 million Americans have diabetes, and each year another 1.7 million are given the diagnosis.1 Prediabetes is even more common; over one-third of US adults ages 20 years and older, and more than half of those who are ages 65 and older, have attained this precursor status, representing another 86 million Americans.1
Because the evidence base for the management of diabetes is rapidly expanding, the American Diabetes Association’s (ADA) Professional Practice Committee updates its Standards of Medical Care in Diabetes annually to incorporate new evidence into its recommendations. The 2017 Standards of Care are available at: professional.diabetes.org/jfp.2
Atherosclerotic cardiovascular disease (ASCVD) is the leading cause of morbidity and mortality for people with diabetes, and is the largest contributor to the direct and indirect costs of the disease.2 As a result, all patients with diabetes should have cardiovascular (CV) risk factors, including dyslipidemia, hypertension, smoking, a family history of premature coronary disease, and the presence of albuminuria, assessed at least annually.2 Numerous studies have demonstrated the efficacy of controlling individual CV risk factors in preventing or slowing ASCVD in people with diabetes. Even larger benefits, including reduced ASCVD morbidity and mortality, can be achieved when multiple risk factors are addressed simultaneously.3
To hone your management of CV risks in patients with diabetes, we’ve put together this Q&A pointing out the elements of the ADA’s 2017 Standards of Care that are most relevant to the management of patients at risk for, or with established, ASCVD.
Screening
Since ASCVD so commonly co-occurs with diabetes, should I routinely screen asymptomatic patients with diabetes for heart disease?
No. The current evidence suggests that outcomes are NOT improved by screening people before they develop symptoms of ASCVD,4 and widespread ASCVD screening has not been shown to be cost-effective. Cardiac testing should be reserved for those with typical or atypical symptoms or those with an abnormal resting electrocardiogram (EKG).
Lifestyle modification
What are the benefits of lifestyle interventions?
The benefits include not only lost pounds, but improved mobility, physical and sexual functioning, and health-related quality of life. Recommend that all overweight patients with diabetes take advantage of intensive lifestyle interventions focusing on weight loss through decreased caloric intake and increased physical activity as per the Look AHEAD (Action for Health in Diabetes) trial.5 Although the intensive lifestyle intervention in the Look AHEAD trial did not decrease CV outcomes over 10 years of follow-up, it did improve control of CV risk factors and led to people in the intervention group taking fewer glucose-, blood pressure (BP)-, and lipid-lowering medications than those in the standard care group.
There is no one diet that is recommended for all people with diabetes. Weight reduction often requires intensive intervention. In order for weight loss diets to be sustainable, they must include patient preferences.
People with diabetes should be encouraged to receive individualized medical nutrition therapy (MNT), preferably from a registered dietitian who is well versed in nutritional management for diabetes. Such MNT is associated with a 0.5% to 2% decrease in A1c levels for people with type 2 diabetes.6-9 Specific healthy diets include the Mediterranean, Dietary Approaches to Stop Hypertension (DASH), and plant-based diets.
A new lifestyle recommendation in this year’s ADA Standards is that periods of prolonged sitting should be interrupted every 30 minutes with a period of physical activity. This appears to have glycemic benefits.2
Hypertension/BP management
When should I initiate hypertension treatment in patients with diabetes?
Nonpharmacologic therapy is reasonable in people with diabetes and mildly elevated BP (>120/80 mm Hg). If systolic blood pressure (SBP) is confirmed to be >140 mm Hg and/or diastolic blood pressure (DBP) is confirmed to be >90 mm Hg, the ADA recommends initiating pharmacologic therapy along with nonpharmacologic strategies. For patients with confirmed office-based BP >160/100 mm Hg, the ADA advises initiating lifestyle modifications as well as 2 pharmacologic medications (or a single pill combination of agents).2
What is the recommended BP target for patients with diabetes and hypertension?
These patients should be treated with a combination of measures, including lifestyle modification and pharmacologic therapy, to a target BP of <140/90 mm Hg. Randomized controlled trials (RCTs) have shown benefits with this target in terms of a reduction in the incidence of coronary heart disease (CHD) events, stroke, and diabetic kidney disease.10,11
A 2012 meta-analysis of randomized trials involving adults with type 2 diabetes mellitus (T2DM) and comparing intensive BP targets (≤130 mm Hg SBP and ≤80 mm Hg DBP) with standard targets (≤140-160 mm Hg SBP and ≤85-100 mm Hg DBP) found no significant reduction in mortality or nonfatal MIs associated with more intense BP control. There was a statistically significant 35% relative risk (RR) reduction in stroke with intensive targets, but lower BP was also associated with an increased risk of hypotension and syncope.12
The 2010 Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial,13 which randomized 5518 patients with T2DM at high risk for ASCVD to either a target SBP of <120 mm Hg or 130 to 140 mm Hg, found that the patients with the lower SBP target did not benefit in the primary end point (a composite of nonfatal MI, nonfatal stroke, and CV death), but did benefit from nominally significant lower rates of total stroke and nonfatal stroke.
Based on these data, the ADA Standards of Care suggest that, “more intensive BP control may be reasonable in certain motivated, ACCORD-like patients (40-79 years of age with prior evidence of CVD or multiple CV risk factors) who have been educated about the added treatment burden, side effects, and costs of more intensive BP control and for patients who prefer to lower their risk of stroke beyond what can be achieved with usual care.”
Another major study, the 2015 Systolic Blood Pressure Intervention Trial (SPRINT) trial,14 demonstrated that treating patients with hypertension to a target SBP <120 mm Hg compared to the usual target of <140 mm Hg resulted in a 25% lower RR of the primary outcome (a composite of MI, other acute coronary syndromes, stroke, heart failure, or death from CV causes) and about a 25% reduction in all-cause mortality; however, people with diabetes were not included in the trial, so the applicability of the results to decisions about BP management in patients with diabetes is not known.
A 2015 systematic review and meta-analysis of over 100,000 participants looked at SBP lowering in adults with T2DM and found that each 10-mm Hg reduction in SBP was associated with a significantly lower risk of morbidity, CV events, CHD, stroke, albuminuria, and retinopathy.10 When trials were stratified by mean baseline SBP (<140 mm Hg or ≥140 mm Hg), RRs for outcomes other than stroke, retinopathy, and renal failure were lower in studies with greater baseline SBP.
The latest ADA Standards of Care recommend that a lower BP target of 130/80 mm Hg may be appropriate for patients at high risk of CVD if this target can be achieved without undue treatment burden. A DBP of <80 mm Hg may also be appropriate in certain patients including those with a long life expectancy, CKD, elevated urinary albumin excretion, and those with evidence of CVD or associated risk factors.15 Of note, treating older adults with diabetes to an SBP target of <130 mm Hg has not been shown to improve cardiovascular outcomes,16 and treating to a diastolic target of <70 mm Hg has been associated with a greater risk of mortality.17
What are the current recommended treatment options?
Treatment for hypertension in adults with diabetes without albuminuria should include any of the classes of medications demonstrated to reduce CV events in patients with diabetes, such as:
- angiotensin-converting enzyme (ACE) inhibitors,
- angiotensin receptor blockers (ARBs),
- thiazide-like diuretics, and
- dihydropyridine calcium channel blockers.
These recommendations are based on evidence suggesting the lack of superiority of ACE inhibitors and ARBs over other classes of antihypertensive agents for the prevention of CV outcomes in all patients with diabetes.18 However, in people with diabetes at high risk for ASCVD and/or with albuminuria, ACE inhibitors and ARBs do reduce ASCVD outcomes and the progression of kidney disease.19-24 Thus, ACE inhibitors and ARBs continue to be recommended as first-line medications for the treatment of hypertension in patients with diabetes and urine albumin/creatinine ratios ≥30 mg/g, as these medications are associated with a reduction in the rate of kidney disease progression.
The use of both an ACE inhibitor and an ARB in combination is not recommended.25,26 For patients treated with ACE inhibitors, ARBs, or diuretics, serum creatinine/estimated glomerular filtration rate (eGFR) and serum potassium levels should be monitored.
What are the recommended lifestyle modifications for patients with diabetes and hypertension?
Regular exercise and healthy eating are recommended for all people with diabetes to optimize glycemic control and lose weight (if they are overweight or obese). For patients with hypertension, the DASH diet (available at: https://www.nhlbi.nih.gov/health/health-topics/topics/dash/) is effective at lowering BP. The DASH diet emphasizes reducing sodium intake, increasing potassium intake, limiting alcohol intake, and increasing physical activity. Specifically, sodium intake should be restricted to <2300 mg/d and patients should consume approximately 8 to 10 servings of fruits and vegetables per day and 2 to 3 servings of low-fat dairy per day. Alcohol should be limited to 2 drinks per day for men and one drink per day for women.
Most adults with diabetes should perform 150 minutes per week of moderate to vigorous exercise, spread over at least 3 days/week. In addition, it is recommended that resistance exercises be performed at least 2 to 3 days/week. Prolonged inactivity is detrimental to health and should be interrupted with activity every 30 minutes.27
Finally, as a part of lifestyle management for all patients with diabetes, smoking cessation is important, as is attention to stress, depression, and anxiety.
Is there an advantage to nighttime dosing of antihypertensive medications?
Yes. Growing evidence suggests that there is an ASCVD benefit to avoiding nocturnal BP dipping. A 2011 RCT of 448 participants with T2DM and hypertension showed a decrease in CV events and mortality during 5.4 years of follow-up if at least one antihypertensive medication was taken at bedtime.28 As a result of this and other evidence,29 consider administering one or more antihypertensive medications at bedtime, although this is not a formal recommendation in the ADA Standards of Care.
Are there any additional issues to be aware of when treating patients with diabetes and hypertension?
Yes. Sometimes patients who have had diabetes for many years have significant orthostatic hypotension secondary to autonomic neuropathy. Postural changes in BP and pulse may require adjustment of BP targets. Home BP self-monitoring and 24-hour ambulatory BP monitoring may indicate white-coat or masked hypertension.
Lipid management
What is the current evidence for lipid treatment in diabetes?
Lipid abnormalities are common in people with diabetes and contribute to the overall high risk of ASCVD in these patients. Subgroup analyses of patients in large trials with diabetes30 and trials involving patients with diabetes31 have shown significant improvements in primary and secondary prevention of ASCVD with statin use. A 2008 meta-analysis of 18,686 people with diabetes showed a 9% reduction in all-cause mortality and a 13% reduction in vascular mortality for each 39-mg/dL reduction in low-density lipoprotein (LDL) cholesterol.32 Absolute reductions in mortality are greatest in those with highest risk, but the benefits of statin therapy are clear for low- and moderate-risk individuals with diabetes, too.33,34 As a result, statins are the medications of choice for lipid lowering and CV risk reduction and should be used in addition to lifestyle management.
Who should get a statin, and how do I choose the optimum dosage?
Patients ages 40 to 75 years with diabetes but without additional ASCVD risk factors should receive a moderate-intensity statin, according to the ADA (see TABLES 12 and 22). For those with additional CV risk factors, a high-intensity statin should be considered. The American College of Cardiology/American Heart Association ASCVD risk calculator (available at: http://www.cvriskcalculator.com/) may be useful for some patients, but generally, risk is already known to be high for most patients with diabetes. For patients of all ages with diabetes and established ASCVD, high-intensity statin therapy should be added to lifestyle modifications.35-37
For patients with diabetes who are <40 years with additional ASCVD risk factors, few clinical trial data exist; nevertheless, consider a moderate- or high-intensity statin and lifestyle therapy. Similarly, for patients >75 years who have diabetes and no additional ASCVD risk factors, consider a moderate-intensity statin and lifestyle modifications. For older adults with additional ASCVD risk factors, consider high-intensity statin therapy.35-37
Statins and cognition. It should be noted that published data have not demonstrated an adverse effect of statins on cognition.38 Statins, however, have been linked to an increased risk of developing diabetes,39,40 although the absolute increase in risk is small, and much smaller than the benefit derived from preventing the development of coronary disease.
Should total cholesterol and LDL levels be used as targets with statin treatment?
No. Statin doses have primarily been tested against placebo in clinical trials, rather than testing to specific target LDL levels, suggesting that the initiation and intensification of statin therapy be based on a patient’s risk profile.35 When maximally tolerated doses of statins do not lower LDL cholesterol by more than 30% from the patient’s baseline, there is currently no good evidence that combination therapy would be helpful, so regular monitoring of lipid levels has limited value. A lipid profile that includes levels of total cholesterol, LDL cholesterol, high-density lipoprotein (HDL) cholesterol, and triglycerides should be obtained at initial medical evaluation, at diagnosis of diabetes, and every 5 years thereafter or before the initiation of statin therapy. Ongoing testing may be appropriate in individual circumstances and to monitor for adherence to, or efficacy of, therapy.
What should I do for my patients who can’t tolerate statins?
Try a lower dose or a different statin before eliminating the class. Research has shown that even small doses (eg, rosuvastatin 5 mg) have some benefit.41
How do combination treatments figure into the current treatment of lipids in patients with diabetes?
It depends on the agent and the patient’s profile.
Fenofibrate. The ADA does not recommend automatically adding fenofibrate to statin therapy because the combination is associated with increased risks for abnormal transaminase levels, myositis, and rhabdomyolysis. In the ACCORD trial, the combination of fenofibrate and simvastatin did not reduce the rate of fatal CV events, nonfatal MIs, or nonfatal strokes compared with simvastatin alone.42
That said, a subgroup analysis suggested a benefit for men with both a triglyceride level ≥204 mg/dL (2.3 mmol/L) and an HDL cholesterol level ≤34 mg/dL (0.9 mmol/L).42 For this reason, the combination of a statin and fenofibrate may be considered for men who meet these laboratory parameters. In addition, consider medical therapy for triglyceride levels ≥500 mg/dL to reduce the risk of pancreatitis.
Ezetimibe. Recommendations regarding ezetimibe are based on the IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), a 2015 RCT including over 18,000 patients that compared treatment with ezetimibe and simvastatin to simvastatin alone.43 Individuals in the trial were ≥50 years of age and had experienced an ACS within the preceding 10 days. In those with diabetes, the combination of moderate-intensity simvastatin (40 mg) and ezetimibe (10 mg) significantly reduced major adverse CV events with an absolute risk reduction of 5% (40% vs 45%) and an RR reduction of 14% over moderate-intensity simvastatin (40 mg) alone.
Based on these results, patients with diabetes and a recent ACS should be considered for combination therapy with ezetimibe and a moderate-intensity statin. The combination should also be considered in patients with diabetes and a history of ASCVD who cannot tolerate high-intensity statins.43
Niacin. The ADA currently does not recommend niacin in combination with a statin because of lack of efficacy on major ASCVD outcomes, possible increased risk of ischemic stroke, and adverse effects.44
What are the recommendations for the use of PCSK-9 inhibitors?
Proprotein convertase subtilisin/kexin type 9 (PCSK-9) inhibitors (ie, evolucumab and alirocumab) may be considered as adjunctive therapy to statins for patients with diabetes at high risk for ASCVD events who require additional lowering of LDL cholesterol. They may also be considered for those in whom high-intensity statin therapy is indicated, but not tolerated.
Antiplatelet agents
Who should take aspirin for primary prevention of CVD?
Both women and men ages ≥50 years who have diabetes and at least one additional CV risk factor (family history of premature ASCVD, hypertension, tobacco use, dyslipidemia, or albuminuria) should consider taking daily aspirin therapy (75-162 mg/d) if they do not have an excessive bleeding risk.45,46 The most common dose in the United States is 81 mg. This recommendation is supported by a 2010 consensus statement of the American Diabetes Association, American Heart Association, and the American College of Cardiology.47
Should patients with diabetes and heart disease receive antiplatelet therapy?
Yes. The evidence is clear that people with known diabetes and ASCVD benefit from aspirin therapy, according to the 2017 Standards of Care. Clopidogrel 75 mg/d is an appropriate alternative for patients who are allergic to aspirin. Dual antiplatelet therapy (a P2Y12 receptor antagonist and aspirin) should be used for as long as one year after an ACS and may have benefits beyond this period.48
Established heart disease
Are there specific recommendations for patients with diabetes and CHD?
According to the ADA Standards, there is good evidence that both aspirin and statin therapy are beneficial for patients with known ASCVD, and that high-intensity statin therapy should be used. In addition, consider ACE inhibitors to reduce the future risk of CV events. In patients with a prior MI, continue beta-blocker therapy for at least 2 years post event.49
Which medications should I avoid, or approach with caution, in patients with congestive heart failure (CHF)?
Thiazolidinediones, dipeptidyl peptidase 4 (DPP-4) inhibitors, and metformin all require careful attention. This is especially important to know when you consider that almost half of all patients with T2DM will develop heart failure.50
Thiazolidinediones. The 2017 Standards of Care state that patients with diabetes and symptomatic congestive heart failure should not receive thiazolidinediones, as they can worsen heart failure status via fluid retention. As such, they are contraindicated in patients with class III and IV heart failure.51
DPP-4 inhibitors. The studies on DPP-4 inhibitors and heart failure have had mixed results. The 2013 SAVOR-TIMI (Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus–Thrombolysis in Myocardial Infarction) 53 trial52 showed that patients treated with saxagliptin were more likely to be hospitalized for heart failure than those taking placebo (3.5% vs 2.8%, respectively). However, the 2015 EXAMINE (Examination of Cardiovascular Outcomes with Alogliptin vs Standard of Care)53 trial and the 2015 TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin)54 trial evaluated heart failure and mortality outcomes in patients with alogliptin and sitagliptin, respectively, compared to placebo, and did not show a relationship to heart failure.
Metformin may be used in people who have T2DM and stable CHF if their eGFR remains >30 mL/min; it should be withheld from patients with unstable heart failure and those who are hospitalized with CHF.
Are there antihyperglycemic medications that reduce CV morbidity and mortality in those with established ASCVD?
Yes. This year’s ADA Standards indicate that certain glucose-lowering medications—specifically empagliflozin (a sodium–glucose cotransporter [SGLT]-2 inhibitor) and liraglutide (a glucagon-like peptide [GLP]-1 receptor agonist)—have been shown to be beneficial for those with established CVD. According to the 2017 Standards of Care, “In patients with longstanding suboptimally controlled T2DM and established ASCVD, empagliflozin or liraglutide should be considered, as they have been shown to reduce CV and all-cause mortality when added to standard care.”2 The studies that provide support for their use are summarized below. Ongoing studies are investigating the CV effects of other agents in these drug classes.
Empagliflozin. The 2015 EMPA-REG OUTCOME (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) study55 was a randomized double-blind study of empagliflozin vs placebo and usual care in patients with diabetes and established CVD. Over a median follow-up of 3.1 years, treatment with empagliflozin reduced the aggregate outcome of MI, stroke, and CV death by 14%, reduced CV deaths by 38%, and decreased deaths from any cause by 32%. In December 2016, the FDA announced a new indication for empagliflozin: to reduce the risk of CV death in adult patients with T2DM and CVD.56
Liraglutide. The LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcome Results: A Long Term Evaluation) trial57 was a double-blind randomized trial of liraglutide vs placebo added to usual care in patients with T2DM at high risk for CVD or with existing CVD. More than 80% of the participants had existing CVD including a history of prior MI, cerebrovascular disease, or peripheral vascular disease. After a median follow-up of 3.8 years, the group taking liraglutide demonstrated a 13% reduction in the composite outcome of MI, stroke, or CV death, a 22% reduction in CV death, and a 15% reduction in death from any cause, compared with placebo.57
CORRESPONDENCE
Neil Skolnik, MD, Abington-Jefferson Health, 500 Old York Rd, Ste 108, Jenkintown, PA 19046; nskolnik@comcast.net.
The authors thank Sarah Bradley, director, professional engagement & collaboration at the American Diabetes Association, for her editorial and organizational assistance in the preparation of this manuscript.
1. Centers for Disease Control and Prevention. National Center for Chronic Disease Prevention and Health Promotion. National diabetes statistics report, 2014. Estimates of diabetes and its burden in the United States. Available at: http://templatelab.com/national-diabetes-report-2014/. Accessed April 7, 2017.
2. American Diabetes Association. Standards of Medical Care in Diabetes—2017. Available at: http://professional.diabetes.org/sites/professional.diabetes.org/files/media/dc_40_s1_final.pdf. Accessed April 7, 2017.
3. Gaede P, Lund-Andersen H, Parving HH, et al. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580-591.
4. Bax JJ, Young LH, Frye RL, et al; American Diabetes Association. Screening for coronary artery disease in patients with diabetes. Diabetes Care. 2007;30:2729-2736.
5. The Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. 2013;369:145-154.
6. UK Prospective Diabetes Study (UKDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKDS 34). Lancet. 1998;352:854-865.
7. Ziemer DC, Berkowitz KJ, Panayioto RM, et al. A simple meal plan emphasizing healthy food choices is as effective as an exchange-based meal plan for urban African Americans with type 2 diabetes. Diabetes Care. 2003;26:1719-1724.
8. Wolf AM, Conaway RM, Crowther JQ, et al; Improving Control with Activity and Nutrition (ICAN) Study. Translating lifestyle intervention to practice in obese patients with type 2 diabetes: Improving Control with Activity and Nutrition (ICAN) study. Diabetes Care. 2004;27:1570-1576.
9. Coppell KJ, Kataoka M, Williams SM, et al. Nutritional intervention in patients with type 2 diabetes who are hyperglycaemic despite optimised drug treatment-Lifestyle Over and Above Drugs in Diabetes (LOADD) study: randomised controlled trial. BMJ. 2010;341:c3337.
10. Emdin CA, Rahimi K, Neal B, et al. Blood pressure lowering in type 2 diabetes: a systematic review and meta-analysis. JAMA. 2015;313:603-615.
11. Arguedas JA, Leiva V, Wright JM. Blood pressure targets for hypertension in people with diabetes mellitus. Cochrane Database Syst Rev. 2013;10:CD008277.
12. McBrien K, Rabi DM, Campbell N, et al. Intensive and standard blood pressure targets in patients with type 2 diabetes mellitus: systematic review and meta-analysis. Arch Intern Med. 2012;172:1296-1303.
13. ACCORD Study Group, Cushman WC, Evans GW, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med. 2010;362:1575-1585.
14. SPRINT Research Group, Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
15. Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet. 1998;351:1755-1762.
16. Kirkman MS, Briscoe VJ, Clark N, et al. Diabetes in older adults. Diabetes Care. 2012;35:2650-2664.
17. Anderson RJ, Bahn GD, Moritz TE, et al; VADT Study Group. Blood pressure and cardiovascular disease risk in the Veterans Affairs Diabetes Trial. Diabetes Care. 2011;34:34-38.
18. Bangalore S, Fakheri R, Toklu B, et al. Diabetes mellitus as a compelling indication for use of renin angiotensin system blockers: systematic review and meta-analysis of randomized trials. BMJ. 2016;352:i438.
19. Heart Outcomes Prevention Evaluation Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet. 2000;355:253-259.
20. Granger CB, McMurray JJ, Yusuf S, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772-776.
21. McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362:767-771.
22. Pfeffer MA, Swedberg K, Granger CB, et al; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759-766.
23. Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861-869.
24. Palmer SC, Mavridis D, Navarese E, et al. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet. 2015;385:2047-2056.
25. The ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
26. Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. 2013;369:1892-1903.
27. Colberg SR, Sigal RJ, Yardley JE, et al. Physical activity/exercise and diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39:2065-2079.
28. Hermida RC, Ayala DE, Mojón A, et al. Influence of time of day of blood pressure-lowering treatment on cardiovascular risk in hypertensive patients with type 2 diabetes. Diabetes Care. 2011;34:1270-1276.
29. Zhao P, Xu P, Wan C, et al. Evening versus morning dosing regimen drug therapy for hypertension. Cochrane Database Syst Rev. 2011;10:CD004184.
30. Py̆orälä K, Pedersen TR, Kjekshus J, et al. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care. 1997;20:614-620.
31. Knopp RH, d’Emden M, Smilde JG, et al. Efficacy and safety of atorvastatin in the prevention of cardiovascular end points in subjects with type 2 diabetes: the Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in Non-Insulin-Dependent Diabetes Mellitus (ASPEN). Diabetes Care. 2006;29:1478-1485.
32. Cholesterol Treatment Trialists’ (CTT) Collaborators, Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117-125.
33. Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013:CD004816.
34. Carter AA, Gomes T, Camacho X, et al. Risk of incident diabetes among patients treated with statins: population based study. BMJ. 2013;346:f2610.
35. Hayward RA, Hofer TP, Vijan S. Narrative review: lack of evidence for recommended low-density lipoprotein treatment targets: a solvable problem. Ann Intern Med. 2006;145:520-530.
36. Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495-1504.
37. de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292:1307-1316.
38. Richardson K, Schoen M, French B, et al. Statins and cognitive function: a systematic review. Ann Intern Med. 2013;159:688-697.
39. Rajpathak SN, Kumbhani DJ, Crandall J, et al. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care. 2009;32:1924-1929.
40. Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735-742.
41. Meek C, Wierzbicki AS, Jewkes C, et al. Daily and intermittent rosuvastatin 5 mg therapy in statin intolerant patients: an observational study. Curr Med Res Opin. 2012;28:371-378.
42. ACCORD Study Group, Ginsberg HN, Bam MB, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563-1574.
43. Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387-2397.
44. AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255-2267.
45. Antithrombotic Trialists’ (ATT) Collaboration, Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849-1860.
46. Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J. 2012;33:1635-1701.
47. Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes. A position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Diabetes Care. 2010;33:1395-1402.
48. Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(suppl):e637S-e668S.
49. Kezerashvilli A, Marzo K, De Leon J. Beta blocker use after acute myocardial infarction in the patient with normal systolic function: when is it “ok” to discontinue? Curr Cardiol Rev. 2012;8:77-84.
50. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29-34.
51. Pioglitazone Package Insert. Available at: http://medlibrary.org/lib/rx/meds/pioglitazone-3/. Accessed April 10, 2017.
52. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
53. Zannad F, Cannon CP, Cushman WC, et al; EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
54. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
55. Zinman B, Wanner C, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
56. FDA approves Jardiance to reduce cardiovascular death in adults with type 2 diabetes. FDA News Release, December 2, 2016. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm531517.htm. Accessed February 9, 2017.
57. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
1. Centers for Disease Control and Prevention. National Center for Chronic Disease Prevention and Health Promotion. National diabetes statistics report, 2014. Estimates of diabetes and its burden in the United States. Available at: http://templatelab.com/national-diabetes-report-2014/. Accessed April 7, 2017.
2. American Diabetes Association. Standards of Medical Care in Diabetes—2017. Available at: http://professional.diabetes.org/sites/professional.diabetes.org/files/media/dc_40_s1_final.pdf. Accessed April 7, 2017.
3. Gaede P, Lund-Andersen H, Parving HH, et al. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580-591.
4. Bax JJ, Young LH, Frye RL, et al; American Diabetes Association. Screening for coronary artery disease in patients with diabetes. Diabetes Care. 2007;30:2729-2736.
5. The Look AHEAD Research Group. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. 2013;369:145-154.
6. UK Prospective Diabetes Study (UKDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKDS 34). Lancet. 1998;352:854-865.
7. Ziemer DC, Berkowitz KJ, Panayioto RM, et al. A simple meal plan emphasizing healthy food choices is as effective as an exchange-based meal plan for urban African Americans with type 2 diabetes. Diabetes Care. 2003;26:1719-1724.
8. Wolf AM, Conaway RM, Crowther JQ, et al; Improving Control with Activity and Nutrition (ICAN) Study. Translating lifestyle intervention to practice in obese patients with type 2 diabetes: Improving Control with Activity and Nutrition (ICAN) study. Diabetes Care. 2004;27:1570-1576.
9. Coppell KJ, Kataoka M, Williams SM, et al. Nutritional intervention in patients with type 2 diabetes who are hyperglycaemic despite optimised drug treatment-Lifestyle Over and Above Drugs in Diabetes (LOADD) study: randomised controlled trial. BMJ. 2010;341:c3337.
10. Emdin CA, Rahimi K, Neal B, et al. Blood pressure lowering in type 2 diabetes: a systematic review and meta-analysis. JAMA. 2015;313:603-615.
11. Arguedas JA, Leiva V, Wright JM. Blood pressure targets for hypertension in people with diabetes mellitus. Cochrane Database Syst Rev. 2013;10:CD008277.
12. McBrien K, Rabi DM, Campbell N, et al. Intensive and standard blood pressure targets in patients with type 2 diabetes mellitus: systematic review and meta-analysis. Arch Intern Med. 2012;172:1296-1303.
13. ACCORD Study Group, Cushman WC, Evans GW, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med. 2010;362:1575-1585.
14. SPRINT Research Group, Wright JT Jr, Williamson JD, Whelton PK, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103-2116.
15. Hansson L, Zanchetti A, Carruthers SG, et al. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. HOT Study Group. Lancet. 1998;351:1755-1762.
16. Kirkman MS, Briscoe VJ, Clark N, et al. Diabetes in older adults. Diabetes Care. 2012;35:2650-2664.
17. Anderson RJ, Bahn GD, Moritz TE, et al; VADT Study Group. Blood pressure and cardiovascular disease risk in the Veterans Affairs Diabetes Trial. Diabetes Care. 2011;34:34-38.
18. Bangalore S, Fakheri R, Toklu B, et al. Diabetes mellitus as a compelling indication for use of renin angiotensin system blockers: systematic review and meta-analysis of randomized trials. BMJ. 2016;352:i438.
19. Heart Outcomes Prevention Evaluation Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet. 2000;355:253-259.
20. Granger CB, McMurray JJ, Yusuf S, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772-776.
21. McMurray JJ, Ostergren J, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet. 2003;362:767-771.
22. Pfeffer MA, Swedberg K, Granger CB, et al; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759-766.
23. Brenner BM, Cooper ME, de Zeeuw D, et al; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861-869.
24. Palmer SC, Mavridis D, Navarese E, et al. Comparative efficacy and safety of blood pressure-lowering agents in adults with diabetes and kidney disease: a network meta-analysis. Lancet. 2015;385:2047-2056.
25. The ONTARGET Investigators. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547-1559.
26. Fried LF, Emanuele N, Zhang JH, et al; VA NEPHRON-D Investigators. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med. 2013;369:1892-1903.
27. Colberg SR, Sigal RJ, Yardley JE, et al. Physical activity/exercise and diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2016;39:2065-2079.
28. Hermida RC, Ayala DE, Mojón A, et al. Influence of time of day of blood pressure-lowering treatment on cardiovascular risk in hypertensive patients with type 2 diabetes. Diabetes Care. 2011;34:1270-1276.
29. Zhao P, Xu P, Wan C, et al. Evening versus morning dosing regimen drug therapy for hypertension. Cochrane Database Syst Rev. 2011;10:CD004184.
30. Py̆orälä K, Pedersen TR, Kjekshus J, et al. Cholesterol lowering with simvastatin improves prognosis of diabetic patients with coronary heart disease. A subgroup analysis of the Scandinavian Simvastatin Survival Study (4S). Diabetes Care. 1997;20:614-620.
31. Knopp RH, d’Emden M, Smilde JG, et al. Efficacy and safety of atorvastatin in the prevention of cardiovascular end points in subjects with type 2 diabetes: the Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in Non-Insulin-Dependent Diabetes Mellitus (ASPEN). Diabetes Care. 2006;29:1478-1485.
32. Cholesterol Treatment Trialists’ (CTT) Collaborators, Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117-125.
33. Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013:CD004816.
34. Carter AA, Gomes T, Camacho X, et al. Risk of incident diabetes among patients treated with statins: population based study. BMJ. 2013;346:f2610.
35. Hayward RA, Hofer TP, Vijan S. Narrative review: lack of evidence for recommended low-density lipoprotein treatment targets: a solvable problem. Ann Intern Med. 2006;145:520-530.
36. Cannon CP, Braunwald E, McCabe CH, et al; Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495-1504.
37. de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004;292:1307-1316.
38. Richardson K, Schoen M, French B, et al. Statins and cognitive function: a systematic review. Ann Intern Med. 2013;159:688-697.
39. Rajpathak SN, Kumbhani DJ, Crandall J, et al. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care. 2009;32:1924-1929.
40. Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735-742.
41. Meek C, Wierzbicki AS, Jewkes C, et al. Daily and intermittent rosuvastatin 5 mg therapy in statin intolerant patients: an observational study. Curr Med Res Opin. 2012;28:371-378.
42. ACCORD Study Group, Ginsberg HN, Bam MB, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563-1574.
43. Cannon CP, Blazing MA, Giugliano RP, et al; IMPROVE-IT Investigators. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387-2397.
44. AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255-2267.
45. Antithrombotic Trialists’ (ATT) Collaboration, Baigent C, Blackwell L, Collins R, et al. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849-1860.
46. Perk J, De Backer G, Gohlke H, et al; European Association for Cardiovascular Prevention & Rehabilitation (EACPR); ESC Committee for Practice Guidelines (CPG). European Guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur Heart J. 2012;33:1635-1701.
47. Pignone M, Alberts MJ, Colwell JA, et al. Aspirin for primary prevention of cardiovascular events in people with diabetes. A position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation. Diabetes Care. 2010;33:1395-1402.
48. Vandvik PO, Lincoff AM, Gore JM, et al; American College of Chest Physicians. Primary and secondary prevention of cardiovascular disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(suppl):e637S-e668S.
49. Kezerashvilli A, Marzo K, De Leon J. Beta blocker use after acute myocardial infarction in the patient with normal systolic function: when is it “ok” to discontinue? Curr Cardiol Rev. 2012;8:77-84.
50. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29-34.
51. Pioglitazone Package Insert. Available at: http://medlibrary.org/lib/rx/meds/pioglitazone-3/. Accessed April 10, 2017.
52. Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317-1326.
53. Zannad F, Cannon CP, Cushman WC, et al; EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385:2067-2076.
54. Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232-242.
55. Zinman B, Wanner C, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
56. FDA approves Jardiance to reduce cardiovascular death in adults with type 2 diabetes. FDA News Release, December 2, 2016. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm531517.htm. Accessed February 9, 2017.
57. Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.

FDA approves midostaurin to treat FLT3+ AML, advanced SM

The US Food and Drug Administration (FDA) has granted approval for the oral, multi-targeted kinase inhibitor midostaurin (Rydapt).
The drug is now approved for the treatment of adults with advanced systemic mastocytosis (SM), including aggressive SM (ASM), SM with associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL).
Midostaurin is also approved for use in combination with standard cytarabine and daunorubicin induction, followed by cytarabine consolidation, in adults with newly diagnosed acute myeloid leukemia (AML) who are FLT3 mutation-positive, as detected by an FDA-approved test.
The FDA approved a companion diagnostic, the LeukoStrat CDx FLT3 Mutation Assay, for use with midostaurin to test AML patients for the FLT3 mutation.
Midostaurin is a product of Novartis. The companion diagnostic was developed by Novartis and Invivoscribe Technologies, Inc.
Midostaurin in AML
The FDA’s approval of midostaurin in AML is based on results from the phase 3 RATIFY trial, which were presented at the 2015 ASH Annual Meeting.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.016).
The median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infections.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis. Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm.
Midostaurin in advanced SM
The FDA’s approval of midostaurin in advanced SM was based on results from a pair of phase 2, single-arm studies, hereafter referred to as Study 2 and Study 3.
Data from Study 2 were published in NEJM in June 2016, and data from Study 3 were presented at the 2010 ASH Annual Meeting.
Study 2 included 116 patients, 115 of whom were evaluable for response.
The overall response rate (ORR) was 17% in the entire cohort, 31% among patients with ASM, 11% among patients with SM-AHN, and 19% among patients with MCL. The complete response rates were 2%, 6%, 0%, and 5%, respectively.
Study 3 included 26 patients with advanced SM. In 3 of the patients, the subtype of SM was unconfirmed.
Among the 17 patients with SM-AHN, there were 10 response (ORR=59%), including 1 partial response and 9 major responses. In the 6 patients with MCL, there were 2 responses (ORR=33%), which included 1 partial response and 1 major response.
In both studies combined, there were 142 adults with ASM, SM-AHN, or MCL.
The most frequent AEs (excluding laboratory abnormalities) that occurred in at least 20% of these patients were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea.
The most frequent grade 3 or higher AEs (excluding laboratory abnormalities) that occurred in at least 5% of patients were fatigue, sepsis, gastrointestinal hemorrhage, pneumonia, diarrhea, febrile neutropenia, edema, dyspnea, nausea, vomiting, abdominal pain, and renal insufficiency.
Serious AEs occurred in 68% of patients, most commonly infections and gastrointestinal disorders. Twenty-one percent of patients discontinued treatment due to AEs, the most frequent of which were infection, nausea or vomiting, QT prolongation, and gastrointestinal hemorrhage. ![]()
The US Food and Drug Administration (FDA) has granted approval for the oral, multi-targeted kinase inhibitor midostaurin (Rydapt).
The drug is now approved for the treatment of adults with advanced systemic mastocytosis (SM), including aggressive SM (ASM), SM with associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL).
Midostaurin is also approved for use in combination with standard cytarabine and daunorubicin induction, followed by cytarabine consolidation, in adults with newly diagnosed acute myeloid leukemia (AML) who are FLT3 mutation-positive, as detected by an FDA-approved test.
The FDA approved a companion diagnostic, the LeukoStrat CDx FLT3 Mutation Assay, for use with midostaurin to test AML patients for the FLT3 mutation.
Midostaurin is a product of Novartis. The companion diagnostic was developed by Novartis and Invivoscribe Technologies, Inc.
Midostaurin in AML
The FDA’s approval of midostaurin in AML is based on results from the phase 3 RATIFY trial, which were presented at the 2015 ASH Annual Meeting.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.016).
The median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infections.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis. Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm.
Midostaurin in advanced SM
The FDA’s approval of midostaurin in advanced SM was based on results from a pair of phase 2, single-arm studies, hereafter referred to as Study 2 and Study 3.
Data from Study 2 were published in NEJM in June 2016, and data from Study 3 were presented at the 2010 ASH Annual Meeting.
Study 2 included 116 patients, 115 of whom were evaluable for response.
The overall response rate (ORR) was 17% in the entire cohort, 31% among patients with ASM, 11% among patients with SM-AHN, and 19% among patients with MCL. The complete response rates were 2%, 6%, 0%, and 5%, respectively.
Study 3 included 26 patients with advanced SM. In 3 of the patients, the subtype of SM was unconfirmed.
Among the 17 patients with SM-AHN, there were 10 response (ORR=59%), including 1 partial response and 9 major responses. In the 6 patients with MCL, there were 2 responses (ORR=33%), which included 1 partial response and 1 major response.
In both studies combined, there were 142 adults with ASM, SM-AHN, or MCL.
The most frequent AEs (excluding laboratory abnormalities) that occurred in at least 20% of these patients were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea.
The most frequent grade 3 or higher AEs (excluding laboratory abnormalities) that occurred in at least 5% of patients were fatigue, sepsis, gastrointestinal hemorrhage, pneumonia, diarrhea, febrile neutropenia, edema, dyspnea, nausea, vomiting, abdominal pain, and renal insufficiency.
Serious AEs occurred in 68% of patients, most commonly infections and gastrointestinal disorders. Twenty-one percent of patients discontinued treatment due to AEs, the most frequent of which were infection, nausea or vomiting, QT prolongation, and gastrointestinal hemorrhage. ![]()
The US Food and Drug Administration (FDA) has granted approval for the oral, multi-targeted kinase inhibitor midostaurin (Rydapt).
The drug is now approved for the treatment of adults with advanced systemic mastocytosis (SM), including aggressive SM (ASM), SM with associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL).
Midostaurin is also approved for use in combination with standard cytarabine and daunorubicin induction, followed by cytarabine consolidation, in adults with newly diagnosed acute myeloid leukemia (AML) who are FLT3 mutation-positive, as detected by an FDA-approved test.
The FDA approved a companion diagnostic, the LeukoStrat CDx FLT3 Mutation Assay, for use with midostaurin to test AML patients for the FLT3 mutation.
Midostaurin is a product of Novartis. The companion diagnostic was developed by Novartis and Invivoscribe Technologies, Inc.
Midostaurin in AML
The FDA’s approval of midostaurin in AML is based on results from the phase 3 RATIFY trial, which were presented at the 2015 ASH Annual Meeting.
In RATIFY, researchers compared midostaurin plus standard chemotherapy to placebo plus standard chemotherapy in 717 adults younger than age 60 who had FLT3-mutated AML.
Patients in the midostaurin arm experienced a statistically significant improvement in overall survival, with a 23% reduction in risk of death compared to the placebo arm (hazard ratio=0.77, P=0.016).
The median event-free survival was significantly longer in the midostaurin arm than the placebo arm—8.2 months and 3.0 months, respectively (hazard ratio=0.78, P=0.004).
The most frequent adverse events (AEs) in the midostaurin arm (occurring in at least 20% of patients) were febrile neutropenia, nausea, vomiting, mucositis, headache, musculoskeletal pain, petechiae, device-related infection, epistaxis, hyperglycemia, and upper respiratory tract infections.
The most frequent grade 3/4 AEs (occurring in at least 10% of patients) were febrile neutropenia, device-related infection, and mucositis. Nine percent of patients in the midostaurin arm stopped treatment due to AEs, as did 6% in the placebo arm.
Midostaurin in advanced SM
The FDA’s approval of midostaurin in advanced SM was based on results from a pair of phase 2, single-arm studies, hereafter referred to as Study 2 and Study 3.
Data from Study 2 were published in NEJM in June 2016, and data from Study 3 were presented at the 2010 ASH Annual Meeting.
Study 2 included 116 patients, 115 of whom were evaluable for response.
The overall response rate (ORR) was 17% in the entire cohort, 31% among patients with ASM, 11% among patients with SM-AHN, and 19% among patients with MCL. The complete response rates were 2%, 6%, 0%, and 5%, respectively.
Study 3 included 26 patients with advanced SM. In 3 of the patients, the subtype of SM was unconfirmed.
Among the 17 patients with SM-AHN, there were 10 response (ORR=59%), including 1 partial response and 9 major responses. In the 6 patients with MCL, there were 2 responses (ORR=33%), which included 1 partial response and 1 major response.
In both studies combined, there were 142 adults with ASM, SM-AHN, or MCL.
The most frequent AEs (excluding laboratory abnormalities) that occurred in at least 20% of these patients were nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, constipation, pyrexia, headache, and dyspnea.
The most frequent grade 3 or higher AEs (excluding laboratory abnormalities) that occurred in at least 5% of patients were fatigue, sepsis, gastrointestinal hemorrhage, pneumonia, diarrhea, febrile neutropenia, edema, dyspnea, nausea, vomiting, abdominal pain, and renal insufficiency.
Serious AEs occurred in 68% of patients, most commonly infections and gastrointestinal disorders. Twenty-one percent of patients discontinued treatment due to AEs, the most frequent of which were infection, nausea or vomiting, QT prolongation, and gastrointestinal hemorrhage. ![]()
FDA approves test to detect FLT3 mutations

The US Food and Drug Administration (FDA) has approved use of the LeukoStrat® CDx FLT3 Mutation Assay as a companion diagnostic test.
The LeukoStrat® CDx FLT3 Mutation Assay is a signal ratio assay that identifies both internal tandem duplication and tyrosine kinase domain mutations.
The assay is the first FDA-approved companion diagnostic for acute myeloid leukemia (AML).
It is approved for use in patients newly diagnosed with AML to determine if they have FLT3 mutations and are therefore eligible to receive treatment with midostaurin (Rydapt).
The FDA granted approval for the LeukoStrat® CDx FLT3 Mutation Assay and midostaurin simultaneously.
The assay was developed by Invivoscribe Technologies, Inc. and Novartis. Midostaurin is a product of Novartis.
Under the current labeling, FLT3 mutation testing with the LeukoStrat® CDx FLT3 Mutation Assay is exclusively performed by The Laboratory for Personalized Molecular Medicine, a subsidiary of Invivoscribe Technologies, Inc.
Under terms of a previously announced agreement with Thermo Fisher, Invivoscribe will also seek FDA approval of the LeukoStrat® CDx FLT3 Mutation Assay that will allow the sale of kits to other laboratories. ![]()
The US Food and Drug Administration (FDA) has approved use of the LeukoStrat® CDx FLT3 Mutation Assay as a companion diagnostic test.
The LeukoStrat® CDx FLT3 Mutation Assay is a signal ratio assay that identifies both internal tandem duplication and tyrosine kinase domain mutations.
The assay is the first FDA-approved companion diagnostic for acute myeloid leukemia (AML).
It is approved for use in patients newly diagnosed with AML to determine if they have FLT3 mutations and are therefore eligible to receive treatment with midostaurin (Rydapt).
The FDA granted approval for the LeukoStrat® CDx FLT3 Mutation Assay and midostaurin simultaneously.
The assay was developed by Invivoscribe Technologies, Inc. and Novartis. Midostaurin is a product of Novartis.
Under the current labeling, FLT3 mutation testing with the LeukoStrat® CDx FLT3 Mutation Assay is exclusively performed by The Laboratory for Personalized Molecular Medicine, a subsidiary of Invivoscribe Technologies, Inc.
Under terms of a previously announced agreement with Thermo Fisher, Invivoscribe will also seek FDA approval of the LeukoStrat® CDx FLT3 Mutation Assay that will allow the sale of kits to other laboratories. ![]()
The US Food and Drug Administration (FDA) has approved use of the LeukoStrat® CDx FLT3 Mutation Assay as a companion diagnostic test.
The LeukoStrat® CDx FLT3 Mutation Assay is a signal ratio assay that identifies both internal tandem duplication and tyrosine kinase domain mutations.
The assay is the first FDA-approved companion diagnostic for acute myeloid leukemia (AML).
It is approved for use in patients newly diagnosed with AML to determine if they have FLT3 mutations and are therefore eligible to receive treatment with midostaurin (Rydapt).
The FDA granted approval for the LeukoStrat® CDx FLT3 Mutation Assay and midostaurin simultaneously.
The assay was developed by Invivoscribe Technologies, Inc. and Novartis. Midostaurin is a product of Novartis.
Under the current labeling, FLT3 mutation testing with the LeukoStrat® CDx FLT3 Mutation Assay is exclusively performed by The Laboratory for Personalized Molecular Medicine, a subsidiary of Invivoscribe Technologies, Inc.
Under terms of a previously announced agreement with Thermo Fisher, Invivoscribe will also seek FDA approval of the LeukoStrat® CDx FLT3 Mutation Assay that will allow the sale of kits to other laboratories. ![]()
Consider melatonin for migraine prevention
ILLUSTRATIVE CASE
A 32-year-old woman comes to your office for help with her recurrent migraines, which she’s had since her early 20s. She is otherwise healthy and active. She is frustrated over the frequency of her migraines and the debilitation they cause. She has tried prophylactic medications in the past, but stopped taking them because of the adverse effects. What do you recommend for treatment?
Daily preventive medication can be helpful for chronic migraine sufferers whose headaches have a significant impact on their lives and who have a goal of reducing headache frequency or severity, disability, and/or avoiding acute headache medication escalation.2 An estimated 38% of patients with migraines are appropriate candidates for prophylactic therapy, but only 3% to 13% are taking preventive medications.3
Evidence-based guidelines from the American Academy of Neurology and the American Headache Society state that antiepileptic drugs (divalproex sodium, sodium valproate, topiramate) and many beta-blockers (metoprolol, propranolol, timolol) are effective and should be recommended for migraine prevention (level A recommendation; based on ≥2 class I trials).2 Medications such as antidepressants (amitriptyline, venlafaxine) and other beta-blockers (atenolol, nadolol) are probably effective and can be considered (level B recommendation; based on one class I trial or 2 class II trials).2 However, adverse effects, such as somnolence, are listed as frequent with amitriptyline and occasional to frequent with topiramate.4
Researchers have investigated melatonin before. But a 2010 double-blind, crossover, randomized controlled trial (RCT) of 46 patients with 2 to 7 migraine attacks per month found no significant difference in reduction of headache frequency with extended-release melatonin 2 mg taken one hour before bed compared to placebo over an 8-week period.5
[polldaddy:9724288]
STUDY SUMMARY
Melatonin tops amitriptyline in >50% improvement in headache frequency
This RCT conducted in Brazil compared the effectiveness of melatonin to amitriptyline and placebo for migraine prevention in 196 adults (ages 18-65 years) with chronic migraines.1 Eligible patients had a history of at least 3 migraine attacks or 4 migraine headache days per month. Patients were randomized to take identically-appearing melatonin 3 mg, amitriptyline 25 mg, or placebo nightly. The investigators appear to have concealed allocation adequately, and used double-blinding.
The primary outcome was the number of headache days per month, comparing baseline with the 4 weeks of treatment. Secondary endpoints included reduction in migraine intensity, duration, number of analgesics used, and percentage of patients with more than 50% reduction in migraine headache days.
Compared to placebo, headache days per month were reduced in both the melatonin group (6.2 days vs 4.6 days, respectively; mean difference [MD], -1.6; 95% confidence interval [CI], -2.4 to -0.9) and the amitriptyline group (6.2 days vs 5 days, respectively; MD, -1.1; 95% CI, -1.5 to -0.7) at 12 weeks, based on intention-to-treat analysis. Mean headache intensity (0-10 pain scale) was also lower at 12 weeks in the melatonin group (4.8 vs 3.6; MD, -1.2; 95% CI, -1.6 to -0.8) and in the amitriptyline group (4.8 vs 3.5; MD, -1.3; 95% CI, -1.7 to -0.9), when compared to placebo.
Headache duration (hours/month) at 12 weeks was reduced in both groups (amitriptyline MD, -4.4 hours; 95% CI, -5.1 to -3.9; melatonin MD, -4.8 hours; 95% CI, -5.7 to -3.9), as was the number of analgesics used (amitriptyline MD, -1; 95% CI, -1.5 to -0.5; melatonin MD, -1; 95% CI, -1.4 to -0.6) when compared to placebo. There was no significant difference between the melatonin and amitriptyline groups for these outcomes.
Patients taking melatonin were more likely to have a >50% improvement in headache frequency compared to amitriptyline (54% vs 39%; number needed to treat [NNT]=7; P<.05); melatonin worked much better than placebo (54% vs 20%; NNT=3; P<.01).
Adverse events were reported more often in the amitriptyline group than in the melatonin group (46 vs 16; P<.03) with daytime sleepiness being the most frequent complaint (41% of patients in the amitriptyline group vs 18% of the melatonin group; number needed to harm [NNH]=5). There was no significant difference in adverse events between melatonin and placebo (16 vs 17; P=not significant). Melatonin resulted in weight loss (mean, -0.14 kg), whereas those taking amitriptyline gained weight (+0.97 kg; P<.01).
WHAT’S NEW
An effective migraine prevention alternative with minimal adverse effects
Melatonin is an accessible and affordable option for preventing migraine headaches in chronic sufferers. The 3-mg dosing reduces headache frequency—both in terms of the number of migraine headache days per month and in terms of the percentage of patients with a >50% reduction in headache events—as well as headache intensity, with minimal adverse effects.
CAVEATS
Product consistency, missing study data
This trial used 3-mg dosing, so it is not clear if other doses are also effective. In addition, because melatonin is available over-the-counter, the quality/actual doses may be less well regulated, and thus, there may be a lack of consistency between brands. Unlike clinical practice, neither the amitriptyline nor the melatonin dose was titrated according to patient response or adverse effects. As a result, we are not sure of the actual lowest effective dose, or if greater effect (with continued minimal adverse effects) could be achieved with higher doses.
Lastly, 69% to 75% of patients in the treatment groups completed the 16-week trial, but the authors of the study reported using 3 different analytic techniques to estimate missing data. The primary outcome included 178 of 196 randomized patients (90.8%). For the primary endpoint, the authors treated all missing data as non-headache days. It is unclear how these missing data would affect the outcome, although an analysis like this would tend towards a null effect.
CHALLENGES TO IMPLEMENTATION
Challenges are negligible
There are really no challenges to implementing this practice changer; melatonin is readily available over-the-counter and it is affordable.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Gonçalves AL, Martini Ferreira A, Ribeiro RT, et al. Randomised clinical trial comparing melatonin 3 mg, amitriptyline 25 mg and placebo for migraine prevention. J Neurol Neurosurg Psychiatry. 2016;87:1127-1132.
2. Silberstein SD, Holland S, Freitag F, et al. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1337-1345.
3. Lipton RB, Bigal ME, Diamond M, et al; The American Migraine Prevalence and Prevention Advisory Group. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343-349.
4. Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:754-762.
5. Alstadhaug KB, Odeh F, Salvesen R, et al. Prophylaxis of migraine with melatonin: a randomized controlled trial. Neurology. 2010;75:1527-1532.
ILLUSTRATIVE CASE
A 32-year-old woman comes to your office for help with her recurrent migraines, which she’s had since her early 20s. She is otherwise healthy and active. She is frustrated over the frequency of her migraines and the debilitation they cause. She has tried prophylactic medications in the past, but stopped taking them because of the adverse effects. What do you recommend for treatment?
Daily preventive medication can be helpful for chronic migraine sufferers whose headaches have a significant impact on their lives and who have a goal of reducing headache frequency or severity, disability, and/or avoiding acute headache medication escalation.2 An estimated 38% of patients with migraines are appropriate candidates for prophylactic therapy, but only 3% to 13% are taking preventive medications.3
Evidence-based guidelines from the American Academy of Neurology and the American Headache Society state that antiepileptic drugs (divalproex sodium, sodium valproate, topiramate) and many beta-blockers (metoprolol, propranolol, timolol) are effective and should be recommended for migraine prevention (level A recommendation; based on ≥2 class I trials).2 Medications such as antidepressants (amitriptyline, venlafaxine) and other beta-blockers (atenolol, nadolol) are probably effective and can be considered (level B recommendation; based on one class I trial or 2 class II trials).2 However, adverse effects, such as somnolence, are listed as frequent with amitriptyline and occasional to frequent with topiramate.4
Researchers have investigated melatonin before. But a 2010 double-blind, crossover, randomized controlled trial (RCT) of 46 patients with 2 to 7 migraine attacks per month found no significant difference in reduction of headache frequency with extended-release melatonin 2 mg taken one hour before bed compared to placebo over an 8-week period.5
[polldaddy:9724288]
STUDY SUMMARY
Melatonin tops amitriptyline in >50% improvement in headache frequency
This RCT conducted in Brazil compared the effectiveness of melatonin to amitriptyline and placebo for migraine prevention in 196 adults (ages 18-65 years) with chronic migraines.1 Eligible patients had a history of at least 3 migraine attacks or 4 migraine headache days per month. Patients were randomized to take identically-appearing melatonin 3 mg, amitriptyline 25 mg, or placebo nightly. The investigators appear to have concealed allocation adequately, and used double-blinding.
The primary outcome was the number of headache days per month, comparing baseline with the 4 weeks of treatment. Secondary endpoints included reduction in migraine intensity, duration, number of analgesics used, and percentage of patients with more than 50% reduction in migraine headache days.
Compared to placebo, headache days per month were reduced in both the melatonin group (6.2 days vs 4.6 days, respectively; mean difference [MD], -1.6; 95% confidence interval [CI], -2.4 to -0.9) and the amitriptyline group (6.2 days vs 5 days, respectively; MD, -1.1; 95% CI, -1.5 to -0.7) at 12 weeks, based on intention-to-treat analysis. Mean headache intensity (0-10 pain scale) was also lower at 12 weeks in the melatonin group (4.8 vs 3.6; MD, -1.2; 95% CI, -1.6 to -0.8) and in the amitriptyline group (4.8 vs 3.5; MD, -1.3; 95% CI, -1.7 to -0.9), when compared to placebo.
Headache duration (hours/month) at 12 weeks was reduced in both groups (amitriptyline MD, -4.4 hours; 95% CI, -5.1 to -3.9; melatonin MD, -4.8 hours; 95% CI, -5.7 to -3.9), as was the number of analgesics used (amitriptyline MD, -1; 95% CI, -1.5 to -0.5; melatonin MD, -1; 95% CI, -1.4 to -0.6) when compared to placebo. There was no significant difference between the melatonin and amitriptyline groups for these outcomes.
Patients taking melatonin were more likely to have a >50% improvement in headache frequency compared to amitriptyline (54% vs 39%; number needed to treat [NNT]=7; P<.05); melatonin worked much better than placebo (54% vs 20%; NNT=3; P<.01).
Adverse events were reported more often in the amitriptyline group than in the melatonin group (46 vs 16; P<.03) with daytime sleepiness being the most frequent complaint (41% of patients in the amitriptyline group vs 18% of the melatonin group; number needed to harm [NNH]=5). There was no significant difference in adverse events between melatonin and placebo (16 vs 17; P=not significant). Melatonin resulted in weight loss (mean, -0.14 kg), whereas those taking amitriptyline gained weight (+0.97 kg; P<.01).
WHAT’S NEW
An effective migraine prevention alternative with minimal adverse effects
Melatonin is an accessible and affordable option for preventing migraine headaches in chronic sufferers. The 3-mg dosing reduces headache frequency—both in terms of the number of migraine headache days per month and in terms of the percentage of patients with a >50% reduction in headache events—as well as headache intensity, with minimal adverse effects.
CAVEATS
Product consistency, missing study data
This trial used 3-mg dosing, so it is not clear if other doses are also effective. In addition, because melatonin is available over-the-counter, the quality/actual doses may be less well regulated, and thus, there may be a lack of consistency between brands. Unlike clinical practice, neither the amitriptyline nor the melatonin dose was titrated according to patient response or adverse effects. As a result, we are not sure of the actual lowest effective dose, or if greater effect (with continued minimal adverse effects) could be achieved with higher doses.
Lastly, 69% to 75% of patients in the treatment groups completed the 16-week trial, but the authors of the study reported using 3 different analytic techniques to estimate missing data. The primary outcome included 178 of 196 randomized patients (90.8%). For the primary endpoint, the authors treated all missing data as non-headache days. It is unclear how these missing data would affect the outcome, although an analysis like this would tend towards a null effect.
CHALLENGES TO IMPLEMENTATION
Challenges are negligible
There are really no challenges to implementing this practice changer; melatonin is readily available over-the-counter and it is affordable.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 32-year-old woman comes to your office for help with her recurrent migraines, which she’s had since her early 20s. She is otherwise healthy and active. She is frustrated over the frequency of her migraines and the debilitation they cause. She has tried prophylactic medications in the past, but stopped taking them because of the adverse effects. What do you recommend for treatment?
Daily preventive medication can be helpful for chronic migraine sufferers whose headaches have a significant impact on their lives and who have a goal of reducing headache frequency or severity, disability, and/or avoiding acute headache medication escalation.2 An estimated 38% of patients with migraines are appropriate candidates for prophylactic therapy, but only 3% to 13% are taking preventive medications.3
Evidence-based guidelines from the American Academy of Neurology and the American Headache Society state that antiepileptic drugs (divalproex sodium, sodium valproate, topiramate) and many beta-blockers (metoprolol, propranolol, timolol) are effective and should be recommended for migraine prevention (level A recommendation; based on ≥2 class I trials).2 Medications such as antidepressants (amitriptyline, venlafaxine) and other beta-blockers (atenolol, nadolol) are probably effective and can be considered (level B recommendation; based on one class I trial or 2 class II trials).2 However, adverse effects, such as somnolence, are listed as frequent with amitriptyline and occasional to frequent with topiramate.4
Researchers have investigated melatonin before. But a 2010 double-blind, crossover, randomized controlled trial (RCT) of 46 patients with 2 to 7 migraine attacks per month found no significant difference in reduction of headache frequency with extended-release melatonin 2 mg taken one hour before bed compared to placebo over an 8-week period.5
[polldaddy:9724288]
STUDY SUMMARY
Melatonin tops amitriptyline in >50% improvement in headache frequency
This RCT conducted in Brazil compared the effectiveness of melatonin to amitriptyline and placebo for migraine prevention in 196 adults (ages 18-65 years) with chronic migraines.1 Eligible patients had a history of at least 3 migraine attacks or 4 migraine headache days per month. Patients were randomized to take identically-appearing melatonin 3 mg, amitriptyline 25 mg, or placebo nightly. The investigators appear to have concealed allocation adequately, and used double-blinding.
The primary outcome was the number of headache days per month, comparing baseline with the 4 weeks of treatment. Secondary endpoints included reduction in migraine intensity, duration, number of analgesics used, and percentage of patients with more than 50% reduction in migraine headache days.
Compared to placebo, headache days per month were reduced in both the melatonin group (6.2 days vs 4.6 days, respectively; mean difference [MD], -1.6; 95% confidence interval [CI], -2.4 to -0.9) and the amitriptyline group (6.2 days vs 5 days, respectively; MD, -1.1; 95% CI, -1.5 to -0.7) at 12 weeks, based on intention-to-treat analysis. Mean headache intensity (0-10 pain scale) was also lower at 12 weeks in the melatonin group (4.8 vs 3.6; MD, -1.2; 95% CI, -1.6 to -0.8) and in the amitriptyline group (4.8 vs 3.5; MD, -1.3; 95% CI, -1.7 to -0.9), when compared to placebo.
Headache duration (hours/month) at 12 weeks was reduced in both groups (amitriptyline MD, -4.4 hours; 95% CI, -5.1 to -3.9; melatonin MD, -4.8 hours; 95% CI, -5.7 to -3.9), as was the number of analgesics used (amitriptyline MD, -1; 95% CI, -1.5 to -0.5; melatonin MD, -1; 95% CI, -1.4 to -0.6) when compared to placebo. There was no significant difference between the melatonin and amitriptyline groups for these outcomes.
Patients taking melatonin were more likely to have a >50% improvement in headache frequency compared to amitriptyline (54% vs 39%; number needed to treat [NNT]=7; P<.05); melatonin worked much better than placebo (54% vs 20%; NNT=3; P<.01).
Adverse events were reported more often in the amitriptyline group than in the melatonin group (46 vs 16; P<.03) with daytime sleepiness being the most frequent complaint (41% of patients in the amitriptyline group vs 18% of the melatonin group; number needed to harm [NNH]=5). There was no significant difference in adverse events between melatonin and placebo (16 vs 17; P=not significant). Melatonin resulted in weight loss (mean, -0.14 kg), whereas those taking amitriptyline gained weight (+0.97 kg; P<.01).
WHAT’S NEW
An effective migraine prevention alternative with minimal adverse effects
Melatonin is an accessible and affordable option for preventing migraine headaches in chronic sufferers. The 3-mg dosing reduces headache frequency—both in terms of the number of migraine headache days per month and in terms of the percentage of patients with a >50% reduction in headache events—as well as headache intensity, with minimal adverse effects.
CAVEATS
Product consistency, missing study data
This trial used 3-mg dosing, so it is not clear if other doses are also effective. In addition, because melatonin is available over-the-counter, the quality/actual doses may be less well regulated, and thus, there may be a lack of consistency between brands. Unlike clinical practice, neither the amitriptyline nor the melatonin dose was titrated according to patient response or adverse effects. As a result, we are not sure of the actual lowest effective dose, or if greater effect (with continued minimal adverse effects) could be achieved with higher doses.
Lastly, 69% to 75% of patients in the treatment groups completed the 16-week trial, but the authors of the study reported using 3 different analytic techniques to estimate missing data. The primary outcome included 178 of 196 randomized patients (90.8%). For the primary endpoint, the authors treated all missing data as non-headache days. It is unclear how these missing data would affect the outcome, although an analysis like this would tend towards a null effect.
CHALLENGES TO IMPLEMENTATION
Challenges are negligible
There are really no challenges to implementing this practice changer; melatonin is readily available over-the-counter and it is affordable.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Gonçalves AL, Martini Ferreira A, Ribeiro RT, et al. Randomised clinical trial comparing melatonin 3 mg, amitriptyline 25 mg and placebo for migraine prevention. J Neurol Neurosurg Psychiatry. 2016;87:1127-1132.
2. Silberstein SD, Holland S, Freitag F, et al. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1337-1345.
3. Lipton RB, Bigal ME, Diamond M, et al; The American Migraine Prevalence and Prevention Advisory Group. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343-349.
4. Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:754-762.
5. Alstadhaug KB, Odeh F, Salvesen R, et al. Prophylaxis of migraine with melatonin: a randomized controlled trial. Neurology. 2010;75:1527-1532.
1. Gonçalves AL, Martini Ferreira A, Ribeiro RT, et al. Randomised clinical trial comparing melatonin 3 mg, amitriptyline 25 mg and placebo for migraine prevention. J Neurol Neurosurg Psychiatry. 2016;87:1127-1132.
2. Silberstein SD, Holland S, Freitag F, et al. Evidence-based guideline update: pharmacologic treatment for episodic migraine prevention in adults: report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology. 2012;78:1337-1345.
3. Lipton RB, Bigal ME, Diamond M, et al; The American Migraine Prevalence and Prevention Advisory Group. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343-349.
4. Silberstein SD. Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:754-762.
5. Alstadhaug KB, Odeh F, Salvesen R, et al. Prophylaxis of migraine with melatonin: a randomized controlled trial. Neurology. 2010;75:1527-1532.
Copyright © 2017. The Family Physicians Inquiries Network. All rights reserved.
PRACTICE CHANGER
Recommend nightly melatonin 3 mg to your patients with chronic migraines, as it appears to be as effective as amitriptyline in reducing headaches and causes fewer adverse effects.
STRENGTH OF RECOMMENDATION
B: Based on a single, good quality randomized controlled trial.
Gonçalves AL, Martini Ferreira A, Ribeiro RT, et al. Randomised clinical trial comparing melatonin 3 mg, amitriptyline 25 mg and placebo for migraine prevention. J Neurol Neurosurg Psychiatry. 2016;87:1127-1132.1
