User login
Specialist offers pain medication pro tips for rheumatologists
LAS VEGAS – Be prepared to prescribe pain medications off label. Understand that some of these drugs are double- or even triple-action, although the three-in-ones aren’t necessarily the best option. And beware of prescribing both opioid painkillers and benzodiazepines over the long term.
Xavier Jimenez, MD, a psychiatrist and chronic pain specialist at the Cleveland Clinic, offered these held by Global Academy for Medical Education.
Here’s a closer look at his tips:
• “Avoid chronic opioid therapy for the most part because of the preponderance of negative effects,” Dr. Jimenez said. “You should also try as best as possible to avoid chronic benzodiazepine therapy, which hasn’t gotten as much media attention but has been linked to all sorts of negative outcomes.”
As he described it, a 2010 study (Pain Med. 2010 Jun;11[6]:805-14) revealed that “benzodiazepines predict opioid use better than pain itself. One thing you can do [to help patients] is start to remove the benzodiazepines.”
Are benzodiazepines even useful for rheumatic conditions? A 2012 Cochrane review of the use of diazepam and triazolam found that they “do not appear to be beneficial in improving pain over 24 hours or 1 week” (Cochrane Database Syst Rev. 2012 Jan 18;1:CD008922. doi: 10.1002/14651858.CD008922.pub2).
• When it comes to choosing medications for pain, “there isn’t really a good textbook algorithm. You’ll have to be creative,” he said. “There are a lot of off-label uses going on here. It’s the norm, not the exception.”
• Some painkillers can have antidepressant, anxiolytic (anti-anxiety) or analgesic effects, and a few like duloxetine (Cymbalta) have “double” or “triple action,” Dr. Jimenez said. Some doctors may be tempted to immediately go for something like duloxetine, but he questioned whether “it’s really that simple.”
“That’s done and is effective for a large proportion of these patients, but not everyone. You will have a lot of patients who’ve tried these and not benefited.”
Is Cymbalta appropriate for rheumatic disease? It may be, at least for chronic knee pain due to osteoarthritis: A 2013 review found that three studies supported its pain-relieving properties relative to placebo at about 4 weeks (Ther Adv Musculoskelet Dis. 2013 Dec;5[6]:291-304).
• Consider treating unresolved anxiety, which Dr. Jimenez said he sees more often in moderate and refractory chronic pain. “No one’s asking you to become psychiatrists,” he said, “but I imagine you are asked to act as psychiatrists.”
Researchers continue to explore connections between anxiety and rheumatic disease. A 2016 study of 56 patients with rheumatoid arthritis (RA) over 1 year found that “inflated [28-joint Disease Activity Score] despite well controlled inflammatory disease markers may indicate significant psychological morbidity and related non-inflammatory pain, rather than true disease activity” (BMC Musculoskelet Disord. 2016;17:155).
• Among the many painkiller options are: Gabapentin (“widely used, relatively safe”), pregabalin (keep an eye on kidney values) and topiramate/Topamax (“a pretty decent medication if you’ve got someone who’s not too depressed,” but watch out for the brain-dulling effect that’s spawned a nickname for the drug: “Dopamax”).
• Cannabinoids aren’t ready for prime time as a pain treatment. “Generally, the message here is ‘not yet,’ ” Dr. Jimenez said. “We don’t have enough compelling evidence to suggest cannabinoids are effective, uniformly or across rheumatic diseases. We know much more about the deficits than the benefits.”
What about rheumatic disease and cannabinoids specifically? A 2018 review of research said “preliminary clinical trials have explored the effects of cannabis on rheumatoid arthritis, osteoarthritis, and fibromyalgia; preliminary evidence has also found an association between the cannabinoid system and other rheumatic conditions, including systemic sclerosis and juvenile idiopathic arthritis.”
However, the report says, evidence so far is insufficient to recommend cannabis in rheumatic disease (Nat Rev Rheumatol. 2018;14:488-98).
For now, Dr. Jimenez advised: “Sit tight and wait.”
Dr. Jimenez reported no relevant disclosures. Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – Be prepared to prescribe pain medications off label. Understand that some of these drugs are double- or even triple-action, although the three-in-ones aren’t necessarily the best option. And beware of prescribing both opioid painkillers and benzodiazepines over the long term.
Xavier Jimenez, MD, a psychiatrist and chronic pain specialist at the Cleveland Clinic, offered these held by Global Academy for Medical Education.
Here’s a closer look at his tips:
• “Avoid chronic opioid therapy for the most part because of the preponderance of negative effects,” Dr. Jimenez said. “You should also try as best as possible to avoid chronic benzodiazepine therapy, which hasn’t gotten as much media attention but has been linked to all sorts of negative outcomes.”
As he described it, a 2010 study (Pain Med. 2010 Jun;11[6]:805-14) revealed that “benzodiazepines predict opioid use better than pain itself. One thing you can do [to help patients] is start to remove the benzodiazepines.”
Are benzodiazepines even useful for rheumatic conditions? A 2012 Cochrane review of the use of diazepam and triazolam found that they “do not appear to be beneficial in improving pain over 24 hours or 1 week” (Cochrane Database Syst Rev. 2012 Jan 18;1:CD008922. doi: 10.1002/14651858.CD008922.pub2).
• When it comes to choosing medications for pain, “there isn’t really a good textbook algorithm. You’ll have to be creative,” he said. “There are a lot of off-label uses going on here. It’s the norm, not the exception.”
• Some painkillers can have antidepressant, anxiolytic (anti-anxiety) or analgesic effects, and a few like duloxetine (Cymbalta) have “double” or “triple action,” Dr. Jimenez said. Some doctors may be tempted to immediately go for something like duloxetine, but he questioned whether “it’s really that simple.”
“That’s done and is effective for a large proportion of these patients, but not everyone. You will have a lot of patients who’ve tried these and not benefited.”
Is Cymbalta appropriate for rheumatic disease? It may be, at least for chronic knee pain due to osteoarthritis: A 2013 review found that three studies supported its pain-relieving properties relative to placebo at about 4 weeks (Ther Adv Musculoskelet Dis. 2013 Dec;5[6]:291-304).
• Consider treating unresolved anxiety, which Dr. Jimenez said he sees more often in moderate and refractory chronic pain. “No one’s asking you to become psychiatrists,” he said, “but I imagine you are asked to act as psychiatrists.”
Researchers continue to explore connections between anxiety and rheumatic disease. A 2016 study of 56 patients with rheumatoid arthritis (RA) over 1 year found that “inflated [28-joint Disease Activity Score] despite well controlled inflammatory disease markers may indicate significant psychological morbidity and related non-inflammatory pain, rather than true disease activity” (BMC Musculoskelet Disord. 2016;17:155).
• Among the many painkiller options are: Gabapentin (“widely used, relatively safe”), pregabalin (keep an eye on kidney values) and topiramate/Topamax (“a pretty decent medication if you’ve got someone who’s not too depressed,” but watch out for the brain-dulling effect that’s spawned a nickname for the drug: “Dopamax”).
• Cannabinoids aren’t ready for prime time as a pain treatment. “Generally, the message here is ‘not yet,’ ” Dr. Jimenez said. “We don’t have enough compelling evidence to suggest cannabinoids are effective, uniformly or across rheumatic diseases. We know much more about the deficits than the benefits.”
What about rheumatic disease and cannabinoids specifically? A 2018 review of research said “preliminary clinical trials have explored the effects of cannabis on rheumatoid arthritis, osteoarthritis, and fibromyalgia; preliminary evidence has also found an association between the cannabinoid system and other rheumatic conditions, including systemic sclerosis and juvenile idiopathic arthritis.”
However, the report says, evidence so far is insufficient to recommend cannabis in rheumatic disease (Nat Rev Rheumatol. 2018;14:488-98).
For now, Dr. Jimenez advised: “Sit tight and wait.”
Dr. Jimenez reported no relevant disclosures. Global Academy for Medical Education and this news organization are owned by the same parent company.
LAS VEGAS – Be prepared to prescribe pain medications off label. Understand that some of these drugs are double- or even triple-action, although the three-in-ones aren’t necessarily the best option. And beware of prescribing both opioid painkillers and benzodiazepines over the long term.
Xavier Jimenez, MD, a psychiatrist and chronic pain specialist at the Cleveland Clinic, offered these held by Global Academy for Medical Education.
Here’s a closer look at his tips:
• “Avoid chronic opioid therapy for the most part because of the preponderance of negative effects,” Dr. Jimenez said. “You should also try as best as possible to avoid chronic benzodiazepine therapy, which hasn’t gotten as much media attention but has been linked to all sorts of negative outcomes.”
As he described it, a 2010 study (Pain Med. 2010 Jun;11[6]:805-14) revealed that “benzodiazepines predict opioid use better than pain itself. One thing you can do [to help patients] is start to remove the benzodiazepines.”
Are benzodiazepines even useful for rheumatic conditions? A 2012 Cochrane review of the use of diazepam and triazolam found that they “do not appear to be beneficial in improving pain over 24 hours or 1 week” (Cochrane Database Syst Rev. 2012 Jan 18;1:CD008922. doi: 10.1002/14651858.CD008922.pub2).
• When it comes to choosing medications for pain, “there isn’t really a good textbook algorithm. You’ll have to be creative,” he said. “There are a lot of off-label uses going on here. It’s the norm, not the exception.”
• Some painkillers can have antidepressant, anxiolytic (anti-anxiety) or analgesic effects, and a few like duloxetine (Cymbalta) have “double” or “triple action,” Dr. Jimenez said. Some doctors may be tempted to immediately go for something like duloxetine, but he questioned whether “it’s really that simple.”
“That’s done and is effective for a large proportion of these patients, but not everyone. You will have a lot of patients who’ve tried these and not benefited.”
Is Cymbalta appropriate for rheumatic disease? It may be, at least for chronic knee pain due to osteoarthritis: A 2013 review found that three studies supported its pain-relieving properties relative to placebo at about 4 weeks (Ther Adv Musculoskelet Dis. 2013 Dec;5[6]:291-304).
• Consider treating unresolved anxiety, which Dr. Jimenez said he sees more often in moderate and refractory chronic pain. “No one’s asking you to become psychiatrists,” he said, “but I imagine you are asked to act as psychiatrists.”
Researchers continue to explore connections between anxiety and rheumatic disease. A 2016 study of 56 patients with rheumatoid arthritis (RA) over 1 year found that “inflated [28-joint Disease Activity Score] despite well controlled inflammatory disease markers may indicate significant psychological morbidity and related non-inflammatory pain, rather than true disease activity” (BMC Musculoskelet Disord. 2016;17:155).
• Among the many painkiller options are: Gabapentin (“widely used, relatively safe”), pregabalin (keep an eye on kidney values) and topiramate/Topamax (“a pretty decent medication if you’ve got someone who’s not too depressed,” but watch out for the brain-dulling effect that’s spawned a nickname for the drug: “Dopamax”).
• Cannabinoids aren’t ready for prime time as a pain treatment. “Generally, the message here is ‘not yet,’ ” Dr. Jimenez said. “We don’t have enough compelling evidence to suggest cannabinoids are effective, uniformly or across rheumatic diseases. We know much more about the deficits than the benefits.”
What about rheumatic disease and cannabinoids specifically? A 2018 review of research said “preliminary clinical trials have explored the effects of cannabis on rheumatoid arthritis, osteoarthritis, and fibromyalgia; preliminary evidence has also found an association between the cannabinoid system and other rheumatic conditions, including systemic sclerosis and juvenile idiopathic arthritis.”
However, the report says, evidence so far is insufficient to recommend cannabis in rheumatic disease (Nat Rev Rheumatol. 2018;14:488-98).
For now, Dr. Jimenez advised: “Sit tight and wait.”
Dr. Jimenez reported no relevant disclosures. Global Academy for Medical Education and this news organization are owned by the same parent company.
REPORTING FROM THE ANNUAL PERSPECTIVES IN RHEUMATIC DISEASES
AYA cancer: Bridging the divide
and older adult patients, a trend that has been going on for decades. But clinicians and researchers are getting serious about an important question: Why?
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist, and director of pediatric bone marrow transplantation at Cleveland Clinic Children’s Hospital, Ohio, said in an interview.
He is referring to the cancers that affect adolescents and young adults (AYAs), who are broadly defined as patients aged 15-39 years.
“A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults; biology may be different in the adolescent and young adult patients, which may lead to different outcomes,” Dr. Hanna said.
In addition, the psychosocial needs in this age group differ vastly from those of other groups, he said.
“Many of these patients are in college or have just started their families, so we have to pay attention more to financial toxicities and fertility, for example,” he said.

Another factor that likely contributes to the disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs. That’s a point on which Dr. Hanna and many other experts agree.
A recent series of articles published in Blood addressed these and other issues, including whether AYAs with ALL or aggressive B-cell non-Hodgkin lymphomas (NHLs) should be treated as children or adults, treatment strategies for those with acute myeloid leukemias, management of Hodgkin lymphoma, and psychosocial challenges and health-related quality of life (QOL) of AYAs with hematologic malignancies.
“Hematological malignancies occurring in AYAs represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population,” Jorge Cortes, MD, an assistant editor for Blood, wrote in an introduction to the series.

“Unfortunately, not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs frequently centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults. Clinical trials specifically designed for AYAs are scanty,” noted Dr. Cortes, who directs the chronic myeloid leukemia (CML) and acute myeloid leukemia programs (AML) at the University of Texas MD Anderson Cancer Center, Houston.
Treatment approach and setting
In the Blood article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15-20 years is supported by numerous studies, and that in young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” as they have been shown to dramatically improve outcomes, with long-term survival rates nearing 70% (2018;132:351-61).
Patients in these age groups require specific programs that take into account factors such as care access and trial access, increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
Kristen O’Dwyer, MD, and her colleagues, in their article on AML treatment in AYAs, argue that based on “the distinguishing characteristics of AYAs with AML,” neither the pediatric nor adult approaches are ideally suited for them.
Rather, AYA-specific approaches merit consideration, they concluded (Blood 2018;132:362-68).
Similarly, Kieron Dunleavy, MD, and Thomas G. Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that a “remarkable divide” in the treatment of patients under age 18 years with lymphoma versus their young adult counterparts underscores the need for collaboration in developing consensus regarding treatment of AYAs (Blood 2018;132:369-75).
But recent findings from a study by Paul C. Nathan, MD, and his colleagues focuses more on where that treatment should take place (J Natl Cancer Inst. 2018 Jul 19. doi: 10.1093/jnci/djy119).
The study provides new insights into the understanding of treatment differences for adolescents seen in pediatric vs. adult cancer facilities. And the findings suggest that the trade-off for improved outcomes among those treated in the pediatric setting – as emerging literature demonstrates – is higher resource use and cost, Helen M. Parsons, PhD, and her colleagues wrote in an accompanying editorial (J Natl Cancer Inst. 2018 Jul 19. doi: 10.1093/jnci/djy123).
Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the cost of care was higher when treatment took place in a pediatric setting vs. an adult institution. This was driven in part by higher hospitalization rates and longer hospital stays, the investigators found.
“Additionally, adolescents treated in the pediatric setting tended to seek more [emergency department] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship,” Dr. Parsons and her colleagues wrote.
This was true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
The findings of higher inpatient days in the pediatric setting is not surprising given that induction therapies for pediatric ALL are generally more complex and intensive than therapies commonly used in adults with ALL, and given that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase ... more work on this topic is needed to more fully understand these patterns,” they wrote, adding that “the finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.”
Disease and developmental biology
As Dr. Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr. Dwyer and her colleagues said, explaining that a recent European study showed that in 5,564 patients with de novo AML, the frequency of favorable cytogenetics was low in infants, increased in children and young adults, and decreased again in middle age and older age (Cancer. 2016 Dec 15;122[24]:3821-30).
“Normal karyotype increases in prevalence from 13.7% in infants to approximately 25% in children, 44% in AYAs, and 50% in adults. Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr. Dwyer and her colleagues wrote, noting that it also is becoming more apparent that age influences the presence of AML-related molecular abnormalities.
The authors argue that recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr. Boissel and Dr. Baruchel also note that the “black hole” of understanding of ALL biology in AYAs that characterized the last 15 years has been “nearly brought to light and revealed a continuum between childhood and adult ALL.”
One example of this involves data from the NOPHO-ALL-2008 trial, showing that the proportion of patients with intrachromosomal amplification of the long arm of chromosome 21 (iAMP21), which is a rare event occurring in about 2% of children with ALL, is more frequent in older children and adolescents and is associated with higher relapse risk that is only partially diminished by intensified treatment.
In NOPHO-2008, iAMP21 occurred in 1.5% of patients aged 1-9 years, 5.8% of those aged 10-17 years, and 12% of those aged 17-45 years. The authors provided numerous other examples of such age-related differences in disease biology and concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
The “financial toxicity” mentioned by Dr. Hanna – the high cost of care, lost work time, and delays in reaching educational and career goals, for example – is a major factor that must be addressed in this population, but there are also many others.
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John M. Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, N.C., said in an interview.
These patients not only have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, but in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added.

“Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... These are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so,” he said.
In a 2014 study, he and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life. The latter finding was surprising, and highlights the “critical importance” of the social dimension in AYAs’ lives.
“Patient after patient will say ‘I found out who my real friends are,’ ” Dr. Salsman said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr. Salsman and his colleagues are using those findings to develop interventions that can maximize self care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves.
A randomized controlled pilot study incorporating social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions, for example, is underway.
Another intervention targets emotional well-being via web-based tools to increase positive affect. A proof-of-concept study showed that the approach is feasible and well received, and efforts are underway to plan a larger-scale randomized controlled trial, he said.
Dr. Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital.
PRISM was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis, and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr. Cortes noted a paucity of clinical trials specifically designed for this population.
“At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, adding that “enrollment of AYAs in clinical trials in cancer in general has been suboptimal at best.”
The dismal numbers with respect to enrollment of AYAs with cancer in clinical trials may be related in part to treatment setting, Dr. Salsman said.
Data suggest that the majority of AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers, where the bulk of research is being done, he explained.
The bottom line is that more research involving AYAs is needed, as is greater understanding of why enrollment is so much lower among AYA patients, Dr. Hanna said, noting that in 2017, The American Society of Clinical Oncology (ASCO) and Friends of Cancer Research (FOCR) released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.
Individuals aged 12 years and older should routinely be included in such trials as their drug metabolism is similar to that of adults, and inclusion of younger patients may also be appropriate if they are part of the population impacted by the disease, depending on specific disease biology, action of the drug, and available safety information, the organizations said.
Officials at the Food and Drug Administration are considering that possibility, Dr. Hanna said.
Attention to the disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups, has definitely increased in recent years, Dr. Salsman added, noting that in addition to ASCO and FOCR, several other organizations are working to address the problem.
About 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population; the Institute of Medicine held a forum on the care of AYAs with cancer; and the National Cancer Institute (NCI) held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology, he noted.
An article in Cancer provides a summary of the progress toward the priorities identified during the NCI meeting, which convened five working groups to address various topics, including clinical trial enrollment (Cancer. 2016 Apr 1;122[7]:988-99).
Dr. Hanna added that groups such as the Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now.
“One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically coordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr. Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.
What’s next?
Among the recommendations of the authors of the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams with involvement of multidisciplinary teams.
Many centers are already working on models for collaborative care, Dr. Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in “getting stakeholders on the same page, helping them have a shared vision, and working to maximize improvements in outcomes.”
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr. Hanna said. In the case of the community-powered advocacy organization Critical Mass, they succeeded in getting lawmakers to introduce a bill in the U.S. House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
and older adult patients, a trend that has been going on for decades. But clinicians and researchers are getting serious about an important question: Why?
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist, and director of pediatric bone marrow transplantation at Cleveland Clinic Children’s Hospital, Ohio, said in an interview.
He is referring to the cancers that affect adolescents and young adults (AYAs), who are broadly defined as patients aged 15-39 years.
“A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults; biology may be different in the adolescent and young adult patients, which may lead to different outcomes,” Dr. Hanna said.
In addition, the psychosocial needs in this age group differ vastly from those of other groups, he said.
“Many of these patients are in college or have just started their families, so we have to pay attention more to financial toxicities and fertility, for example,” he said.
Another factor that likely contributes to the disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs. That’s a point on which Dr. Hanna and many other experts agree.
A recent series of articles published in Blood addressed these and other issues, including whether AYAs with ALL or aggressive B-cell non-Hodgkin lymphomas (NHLs) should be treated as children or adults, treatment strategies for those with acute myeloid leukemias, management of Hodgkin lymphoma, and psychosocial challenges and health-related quality of life (QOL) of AYAs with hematologic malignancies.
“Hematological malignancies occurring in AYAs represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population,” Jorge Cortes, MD, an assistant editor for Blood, wrote in an introduction to the series.
“Unfortunately, not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs frequently centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults. Clinical trials specifically designed for AYAs are scanty,” noted Dr. Cortes, who directs the chronic myeloid leukemia (CML) and acute myeloid leukemia programs (AML) at the University of Texas MD Anderson Cancer Center, Houston.
Treatment approach and setting
In the Blood article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15-20 years is supported by numerous studies, and that in young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” as they have been shown to dramatically improve outcomes, with long-term survival rates nearing 70% (2018;132:351-61).
Patients in these age groups require specific programs that take into account factors such as care access and trial access, increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
Kristen O’Dwyer, MD, and her colleagues, in their article on AML treatment in AYAs, argue that based on “the distinguishing characteristics of AYAs with AML,” neither the pediatric nor adult approaches are ideally suited for them.
Rather, AYA-specific approaches merit consideration, they concluded (Blood 2018;132:362-68).
Similarly, Kieron Dunleavy, MD, and Thomas G. Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that a “remarkable divide” in the treatment of patients under age 18 years with lymphoma versus their young adult counterparts underscores the need for collaboration in developing consensus regarding treatment of AYAs (Blood 2018;132:369-75).
But recent findings from a study by Paul C. Nathan, MD, and his colleagues focuses more on where that treatment should take place (J Natl Cancer Inst. 2018 Jul 19. doi: 10.1093/jnci/djy119).
The study provides new insights into the understanding of treatment differences for adolescents seen in pediatric vs. adult cancer facilities. And the findings suggest that the trade-off for improved outcomes among those treated in the pediatric setting – as emerging literature demonstrates – is higher resource use and cost, Helen M. Parsons, PhD, and her colleagues wrote in an accompanying editorial (J Natl Cancer Inst. 2018 Jul 19. doi: 10.1093/jnci/djy123).
Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the cost of care was higher when treatment took place in a pediatric setting vs. an adult institution. This was driven in part by higher hospitalization rates and longer hospital stays, the investigators found.
“Additionally, adolescents treated in the pediatric setting tended to seek more [emergency department] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship,” Dr. Parsons and her colleagues wrote.
This was true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
The findings of higher inpatient days in the pediatric setting is not surprising given that induction therapies for pediatric ALL are generally more complex and intensive than therapies commonly used in adults with ALL, and given that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase ... more work on this topic is needed to more fully understand these patterns,” they wrote, adding that “the finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.”
Disease and developmental biology
As Dr. Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr. Dwyer and her colleagues said, explaining that a recent European study showed that in 5,564 patients with de novo AML, the frequency of favorable cytogenetics was low in infants, increased in children and young adults, and decreased again in middle age and older age (Cancer. 2016 Dec 15;122[24]:3821-30).
“Normal karyotype increases in prevalence from 13.7% in infants to approximately 25% in children, 44% in AYAs, and 50% in adults. Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr. Dwyer and her colleagues wrote, noting that it also is becoming more apparent that age influences the presence of AML-related molecular abnormalities.
The authors argue that recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr. Boissel and Dr. Baruchel also note that the “black hole” of understanding of ALL biology in AYAs that characterized the last 15 years has been “nearly brought to light and revealed a continuum between childhood and adult ALL.”
One example of this involves data from the NOPHO-ALL-2008 trial, showing that the proportion of patients with intrachromosomal amplification of the long arm of chromosome 21 (iAMP21), which is a rare event occurring in about 2% of children with ALL, is more frequent in older children and adolescents and is associated with higher relapse risk that is only partially diminished by intensified treatment.
In NOPHO-2008, iAMP21 occurred in 1.5% of patients aged 1-9 years, 5.8% of those aged 10-17 years, and 12% of those aged 17-45 years. The authors provided numerous other examples of such age-related differences in disease biology and concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
The “financial toxicity” mentioned by Dr. Hanna – the high cost of care, lost work time, and delays in reaching educational and career goals, for example – is a major factor that must be addressed in this population, but there are also many others.
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John M. Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, N.C., said in an interview.
These patients not only have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, but in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added.
“Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... These are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so,” he said.
In a 2014 study, he and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life. The latter finding was surprising, and highlights the “critical importance” of the social dimension in AYAs’ lives.
“Patient after patient will say ‘I found out who my real friends are,’ ” Dr. Salsman said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr. Salsman and his colleagues are using those findings to develop interventions that can maximize self care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves.
A randomized controlled pilot study incorporating social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions, for example, is underway.
Another intervention targets emotional well-being via web-based tools to increase positive affect. A proof-of-concept study showed that the approach is feasible and well received, and efforts are underway to plan a larger-scale randomized controlled trial, he said.
Dr. Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital.
PRISM was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis, and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr. Cortes noted a paucity of clinical trials specifically designed for this population.
“At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, adding that “enrollment of AYAs in clinical trials in cancer in general has been suboptimal at best.”
The dismal numbers with respect to enrollment of AYAs with cancer in clinical trials may be related in part to treatment setting, Dr. Salsman said.
Data suggest that the majority of AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers, where the bulk of research is being done, he explained.
The bottom line is that more research involving AYAs is needed, as is greater understanding of why enrollment is so much lower among AYA patients, Dr. Hanna said, noting that in 2017, The American Society of Clinical Oncology (ASCO) and Friends of Cancer Research (FOCR) released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.
Individuals aged 12 years and older should routinely be included in such trials as their drug metabolism is similar to that of adults, and inclusion of younger patients may also be appropriate if they are part of the population impacted by the disease, depending on specific disease biology, action of the drug, and available safety information, the organizations said.
Officials at the Food and Drug Administration are considering that possibility, Dr. Hanna said.
Attention to the disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups, has definitely increased in recent years, Dr. Salsman added, noting that in addition to ASCO and FOCR, several other organizations are working to address the problem.
About 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population; the Institute of Medicine held a forum on the care of AYAs with cancer; and the National Cancer Institute (NCI) held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology, he noted.
An article in Cancer provides a summary of the progress toward the priorities identified during the NCI meeting, which convened five working groups to address various topics, including clinical trial enrollment (Cancer. 2016 Apr 1;122[7]:988-99).
Dr. Hanna added that groups such as the Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now.
“One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically coordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr. Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.
What’s next?
Among the recommendations of the authors of the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams with involvement of multidisciplinary teams.
Many centers are already working on models for collaborative care, Dr. Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in “getting stakeholders on the same page, helping them have a shared vision, and working to maximize improvements in outcomes.”
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr. Hanna said. In the case of the community-powered advocacy organization Critical Mass, they succeeded in getting lawmakers to introduce a bill in the U.S. House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
and older adult patients, a trend that has been going on for decades. But clinicians and researchers are getting serious about an important question: Why?
“This is a very heterogeneous group of disorders,” Rabi Hanna, MD, a pediatric hematologist and oncologist, and director of pediatric bone marrow transplantation at Cleveland Clinic Children’s Hospital, Ohio, said in an interview.
He is referring to the cancers that affect adolescents and young adults (AYAs), who are broadly defined as patients aged 15-39 years.
“A few cancers, such as [acute lymphoblastic leukemia], are more common in children, and others, such as breast cancer, are more common in adults; biology may be different in the adolescent and young adult patients, which may lead to different outcomes,” Dr. Hanna said.
In addition, the psychosocial needs in this age group differ vastly from those of other groups, he said.
“Many of these patients are in college or have just started their families, so we have to pay attention more to financial toxicities and fertility, for example,” he said.
Another factor that likely contributes to the disparities between AYAs and other populations with cancer is the relative lack of clinical trial involvement among AYAs. That’s a point on which Dr. Hanna and many other experts agree.
A recent series of articles published in Blood addressed these and other issues, including whether AYAs with ALL or aggressive B-cell non-Hodgkin lymphomas (NHLs) should be treated as children or adults, treatment strategies for those with acute myeloid leukemias, management of Hodgkin lymphoma, and psychosocial challenges and health-related quality of life (QOL) of AYAs with hematologic malignancies.
“Hematological malignancies occurring in AYAs represent a unique challenge because of their special biological features and distinctive therapeutic requirements, as well as the unique medical, social, and psychological characteristics of this patient population,” Jorge Cortes, MD, an assistant editor for Blood, wrote in an introduction to the series.
“Unfortunately, not much has been done to explore unique molecular and biological features of AYA hematologic malignancies. The discussion on the management of AYAs frequently centers on whether these patients should be treated in a pediatric setting or an adult setting, or with regimens designed for children or for adults. Clinical trials specifically designed for AYAs are scanty,” noted Dr. Cortes, who directs the chronic myeloid leukemia (CML) and acute myeloid leukemia programs (AML) at the University of Texas MD Anderson Cancer Center, Houston.
Treatment approach and setting
In the Blood article on ALL in AYAs, Nicolas Boissel, MD, and André Baruchel, MD, note that the use of “fully pediatric protocols” in patients aged 15-20 years is supported by numerous studies, and that in young adults, evidence increasingly supports “pediatric-inspired or even fully pediatric approaches” as they have been shown to dramatically improve outcomes, with long-term survival rates nearing 70% (2018;132:351-61).
Patients in these age groups require specific programs that take into account factors such as care access and trial access, increased risk of acute toxicities, and treatment adherence, which can be particularly problematic in AYAs, they concluded.
Kristen O’Dwyer, MD, and her colleagues, in their article on AML treatment in AYAs, argue that based on “the distinguishing characteristics of AYAs with AML,” neither the pediatric nor adult approaches are ideally suited for them.
Rather, AYA-specific approaches merit consideration, they concluded (Blood 2018;132:362-68).
Similarly, Kieron Dunleavy, MD, and Thomas G. Gross, MD, note in an article on managing aggressive B-cell NHLs in AYAs that a “remarkable divide” in the treatment of patients under age 18 years with lymphoma versus their young adult counterparts underscores the need for collaboration in developing consensus regarding treatment of AYAs (Blood 2018;132:369-75).
But recent findings from a study by Paul C. Nathan, MD, and his colleagues focuses more on where that treatment should take place (J Natl Cancer Inst. 2018 Jul 19. doi: 10.1093/jnci/djy119).
The study provides new insights into the understanding of treatment differences for adolescents seen in pediatric vs. adult cancer facilities. And the findings suggest that the trade-off for improved outcomes among those treated in the pediatric setting – as emerging literature demonstrates – is higher resource use and cost, Helen M. Parsons, PhD, and her colleagues wrote in an accompanying editorial (J Natl Cancer Inst. 2018 Jul 19. doi: 10.1093/jnci/djy123).
Among 1,356 patients aged 15-17 years who were diagnosed with cancer between 1996 and 2010, the cost of care was higher when treatment took place in a pediatric setting vs. an adult institution. This was driven in part by higher hospitalization rates and longer hospital stays, the investigators found.
“Additionally, adolescents treated in the pediatric setting tended to seek more [emergency department] care immediately before diagnosis and during the initial treatment phase; these adolescents also used more home care services during initial treatment and survivorship,” Dr. Parsons and her colleagues wrote.
This was true across different diagnoses, including leukemias, lymphomas, sarcomas, and germ cell tumors, but only during the initial treatment phase.
The findings of higher inpatient days in the pediatric setting is not surprising given that induction therapies for pediatric ALL are generally more complex and intensive than therapies commonly used in adults with ALL, and given that pediatric cancer hospitals tend to have a wider array of services, including psychosocial and family support services.
“What is less clear is why individuals seen in pediatric settings have higher rates of ED care directly before diagnosis and during the initial treatment phase ... more work on this topic is needed to more fully understand these patterns,” they wrote, adding that “the finding that adolescents treated in pediatric institutions had higher resource use across diagnostic groups demonstrates that resource utilization may be driven just as much by care setting as diagnosis.”
Disease and developmental biology
As Dr. Hanna noted, biological differences and changes over time suggest that different age groups need varying approaches to treatment and may have different outcomes with the same treatments.
For example, the biology of AML is known to change with age, Dr. Dwyer and her colleagues said, explaining that a recent European study showed that in 5,564 patients with de novo AML, the frequency of favorable cytogenetics was low in infants, increased in children and young adults, and decreased again in middle age and older age (Cancer. 2016 Dec 15;122[24]:3821-30).
“Normal karyotype increases in prevalence from 13.7% in infants to approximately 25% in children, 44% in AYAs, and 50% in adults. Most unfavorable cytogenetic abnormalities are rare across all age groups, though complex cytogenetics are relatively more frequent in infants, decrease in frequency in AYAs, and then increase in frequency beyond AYA,” Dr. Dwyer and her colleagues wrote, noting that it also is becoming more apparent that age influences the presence of AML-related molecular abnormalities.
The authors argue that recognition of age-related differences in disease biology “will provide the best opportunity to improve the clinical outcomes that have been static for decades.”
Dr. Boissel and Dr. Baruchel also note that the “black hole” of understanding of ALL biology in AYAs that characterized the last 15 years has been “nearly brought to light and revealed a continuum between childhood and adult ALL.”
One example of this involves data from the NOPHO-ALL-2008 trial, showing that the proportion of patients with intrachromosomal amplification of the long arm of chromosome 21 (iAMP21), which is a rare event occurring in about 2% of children with ALL, is more frequent in older children and adolescents and is associated with higher relapse risk that is only partially diminished by intensified treatment.
In NOPHO-2008, iAMP21 occurred in 1.5% of patients aged 1-9 years, 5.8% of those aged 10-17 years, and 12% of those aged 17-45 years. The authors provided numerous other examples of such age-related differences in disease biology and concluded that “risk stratification based on recent biology findings and sequential [minimum residual disease] evaluations should now be implemented, as well as new therapeutic options including immunotherapy and targeted therapies, at best within the setting of integrated pediatric and AYA protocols.”
Psychosocial factors
The “financial toxicity” mentioned by Dr. Hanna – the high cost of care, lost work time, and delays in reaching educational and career goals, for example – is a major factor that must be addressed in this population, but there are also many others.
“Cancer is a non-normative event for AYAs. It is extremely disruptive to them physically, psychologically, and vocationally ... and this poses significant challenges,” John M. Salsman, PhD, director of clinical research in AYA oncology at Wake Forest University, Winston-Salem, N.C., said in an interview.
These patients not only have 5-year survival rates that haven’t improved in tandem with those in pediatric and adult populations over the last 3 decades, but in addition to the financial toxicity and strain, they also have higher rates of depression and anxiety, including fear of recurrence, he added.
“Quality of life is incredibly important, and these things need to be addressed because of the developmental changes AYAs are navigating; there are issues of positive body image, family and career decisions ... These are challenging for anyone, and when you throw a cancer diagnosis into the mix they become disproportionately so,” he said.
In a 2014 study, he and his colleagues found that AYAs with cancer had poorer physical and emotional quality of life when compared with matched controls, but better social quality of life. The latter finding was surprising, and highlights the “critical importance” of the social dimension in AYAs’ lives.
“Patient after patient will say ‘I found out who my real friends are,’ ” Dr. Salsman said. “There’s this refinement and deepening of the social network among some posttreatment survivors.”
Dr. Salsman and his colleagues are using those findings to develop interventions that can maximize self care in posttreatment survivorship – a time when AYAs may feel they have a new lease on life and may be more motivated to adhere to recommendations and take care of themselves.
A randomized controlled pilot study incorporating social media apps and other technologies to build on the positive social components of their lives in promoting physical activity interventions, for example, is underway.
Another intervention targets emotional well-being via web-based tools to increase positive affect. A proof-of-concept study showed that the approach is feasible and well received, and efforts are underway to plan a larger-scale randomized controlled trial, he said.
Dr. Salsman also praised the PRISM (Promoting Resilience in Stress Management) tool developed by researchers at Seattle Children’s Hospital.
PRISM was created to help AYAs with cancer and other illnesses learn coping skills to manage stress after their diagnosis, and to boost quality of life beyond treatment. A digital app has also been developed to be used in conjunction with the program.
Trial enrollment
In his editorial introducing the Blood series on AYAs and cancer, Dr. Cortes noted a paucity of clinical trials specifically designed for this population.
“At the time of this writing, I could identify four therapeutic trials registered at www.clinicaltrials.gov that appeared to be somewhat specifically designed for AYAs (some included children also),” he wrote, adding that “enrollment of AYAs in clinical trials in cancer in general has been suboptimal at best.”
The dismal numbers with respect to enrollment of AYAs with cancer in clinical trials may be related in part to treatment setting, Dr. Salsman said.
Data suggest that the majority of AYAs with cancer are treated in community-based practices rather than comprehensive cancer centers, where the bulk of research is being done, he explained.
The bottom line is that more research involving AYAs is needed, as is greater understanding of why enrollment is so much lower among AYA patients, Dr. Hanna said, noting that in 2017, The American Society of Clinical Oncology (ASCO) and Friends of Cancer Research (FOCR) released a statement recommending that pediatric patients be considered for enrollment in later-phase trials for cancer types that span both adults and children.
Individuals aged 12 years and older should routinely be included in such trials as their drug metabolism is similar to that of adults, and inclusion of younger patients may also be appropriate if they are part of the population impacted by the disease, depending on specific disease biology, action of the drug, and available safety information, the organizations said.
Officials at the Food and Drug Administration are considering that possibility, Dr. Hanna said.
Attention to the disparities in survival improvements and trial involvement among AYAs with cancer, compared with other age groups, has definitely increased in recent years, Dr. Salsman added, noting that in addition to ASCO and FOCR, several other organizations are working to address the problem.
About 5 years ago, the National Clinical Trials Network formed a working group that developed a number of specific objectives for incorporating more AYAs into cancer trials and finding better ways to study this population; the Institute of Medicine held a forum on the care of AYAs with cancer; and the National Cancer Institute (NCI) held a state-of-the-science meeting that focused on identifying strategic priorities for AYA oncology, he noted.
An article in Cancer provides a summary of the progress toward the priorities identified during the NCI meeting, which convened five working groups to address various topics, including clinical trial enrollment (Cancer. 2016 Apr 1;122[7]:988-99).
Dr. Hanna added that groups such as the Southwest Oncology Group (SWOG) and Children’s Oncology Group (COG) also have AYA committees now.
“One of the success stories of working together between SWOG and COG was the intergroup study C10403 for patients with ALL. And now there are efforts for an intergroup AYA-AML task force to include representatives from each of the cooperative groups that historically coordinated myeloid disease clinical trials – COG, SWOG, Alliance, and ECOG-ACRIN,” he said.
In fact, all of the National Clinical Trials Network groups have some initiative in place to address AYA concerns, said Dr. Salsman, who chairs the ECOG-ACRIN AYA oncology subcommittee.
Despite these efforts, and many others, long-term survival improvements among AYAs with cancer still fall short, compared with those of other age groups.
What’s next?
Among the recommendations of the authors of the AYA series in Blood is a call for assessing AYA-specific therapy in future clinical trials, as well as improved collaboration between adult and pediatric teams with involvement of multidisciplinary teams.
Many centers are already working on models for collaborative care, Dr. Salsman said, citing the Fort Worth AYA Oncology Coalition led by medical director Karen Albritton, MD, as an example of a program that has been successful in “getting stakeholders on the same page, helping them have a shared vision, and working to maximize improvements in outcomes.”
Patients are also taking the lead in demanding better care and attention to their psychosocial needs, Dr. Hanna said. In the case of the community-powered advocacy organization Critical Mass, they succeeded in getting lawmakers to introduce a bill in the U.S. House of Representatives that would allow college students to defer loan payments while undergoing cancer treatment.
Bacteremic sepsis in ALL linked to later cognitive issues
Bacteremic sepsis during acute lymphoblastic leukemia (ALL) treatment may contribute to neurocognitive dysfunction later in life, results of a cohort study suggest.

Pediatric ALL survivors who had sepsis while on treatment performed worse on measures of intelligence, attention, executive function, and processing speed than survivors with no sepsis history, according to study results.
Links between sepsis and impaired neurocognitive function found in this study have “practice-changing implications” for cancer survivors, investigators reported in JAMA Pediatrics.
“Prevention of infection, early recognition and appropriate management of sepsis, and preemptive neurocognitive interventions should be prioritized, because these might prevent or ameliorate neurologic damage,” said Joshua Wolf, MBBS, of St. Jude Children’s Research Hospital, Memphis, and the coauthors of the report.
The study included 212 children who, at a median age of 5 years, had received risk-adapted chemotherapy for ALL with no hematopoietic cell transplant or cranial irradiation.
Sixteen of the patients (7.5%) had a history of bacteremic sepsis during ALL therapy, according to retrospectively obtained data.
As a part of the study, all participants participated in neurocognitive testing, which was done at a median of 7.7 years after diagnosis.
Patients with a history of bacteremic sepsis performed poorly on multiple measures of neurocognitive function, as compared with all other participants, according to results of analyses that were adjusted for multiple potentially confounding factors, such as age, race, and leukemia risk category.
Although not all neurocognitive measures were significantly different between groups, survivors with a sepsis history performed worse on evaluations of spatial planning (difference, 0.78; 95% confidence interval, 0.57-1.00), verbal fluency (0.38; 95% CI, 0.14-0.62), and attention (0.63; 95% CI, 0.30-0.95), among other measures, investigators said.
This is believed to be the first published study looking at potential links between sepsis during ALL treatment and long-term neurocognitive dysfunction, investigators said. However, similar observations have been made in other patient populations, they added.
Exactly how sepsis might lead to neurocognitive deficits remains unclear. “In the population of children with cancer, these mechanisms might be augmented by increased blood-brain barrier permeability to neurotoxic chemotherapy drugs,” they said in their report.
Further study is needed to look at potential brain injury mechanisms, and to validate the current findings in other ALL patient cohorts, they concluded.
The study was supported by the National Institute of Mental Health, the National Cancer Institute, and the American Lebanese Syrian Associated Charities. The researchers reported having no conflicts of interest.
SOURCE: Cheung YT et al. JAMA Pediatr. 2018 Sep 24. doi:10.1001/jamapediatrics.2018.2500.
Bacteremic sepsis during acute lymphoblastic leukemia (ALL) treatment may contribute to neurocognitive dysfunction later in life, results of a cohort study suggest.
Pediatric ALL survivors who had sepsis while on treatment performed worse on measures of intelligence, attention, executive function, and processing speed than survivors with no sepsis history, according to study results.
Links between sepsis and impaired neurocognitive function found in this study have “practice-changing implications” for cancer survivors, investigators reported in JAMA Pediatrics.
“Prevention of infection, early recognition and appropriate management of sepsis, and preemptive neurocognitive interventions should be prioritized, because these might prevent or ameliorate neurologic damage,” said Joshua Wolf, MBBS, of St. Jude Children’s Research Hospital, Memphis, and the coauthors of the report.
The study included 212 children who, at a median age of 5 years, had received risk-adapted chemotherapy for ALL with no hematopoietic cell transplant or cranial irradiation.
Sixteen of the patients (7.5%) had a history of bacteremic sepsis during ALL therapy, according to retrospectively obtained data.
As a part of the study, all participants participated in neurocognitive testing, which was done at a median of 7.7 years after diagnosis.
Patients with a history of bacteremic sepsis performed poorly on multiple measures of neurocognitive function, as compared with all other participants, according to results of analyses that were adjusted for multiple potentially confounding factors, such as age, race, and leukemia risk category.
Although not all neurocognitive measures were significantly different between groups, survivors with a sepsis history performed worse on evaluations of spatial planning (difference, 0.78; 95% confidence interval, 0.57-1.00), verbal fluency (0.38; 95% CI, 0.14-0.62), and attention (0.63; 95% CI, 0.30-0.95), among other measures, investigators said.
This is believed to be the first published study looking at potential links between sepsis during ALL treatment and long-term neurocognitive dysfunction, investigators said. However, similar observations have been made in other patient populations, they added.
Exactly how sepsis might lead to neurocognitive deficits remains unclear. “In the population of children with cancer, these mechanisms might be augmented by increased blood-brain barrier permeability to neurotoxic chemotherapy drugs,” they said in their report.
Further study is needed to look at potential brain injury mechanisms, and to validate the current findings in other ALL patient cohorts, they concluded.
The study was supported by the National Institute of Mental Health, the National Cancer Institute, and the American Lebanese Syrian Associated Charities. The researchers reported having no conflicts of interest.
SOURCE: Cheung YT et al. JAMA Pediatr. 2018 Sep 24. doi:10.1001/jamapediatrics.2018.2500.
Bacteremic sepsis during acute lymphoblastic leukemia (ALL) treatment may contribute to neurocognitive dysfunction later in life, results of a cohort study suggest.
Pediatric ALL survivors who had sepsis while on treatment performed worse on measures of intelligence, attention, executive function, and processing speed than survivors with no sepsis history, according to study results.
Links between sepsis and impaired neurocognitive function found in this study have “practice-changing implications” for cancer survivors, investigators reported in JAMA Pediatrics.
“Prevention of infection, early recognition and appropriate management of sepsis, and preemptive neurocognitive interventions should be prioritized, because these might prevent or ameliorate neurologic damage,” said Joshua Wolf, MBBS, of St. Jude Children’s Research Hospital, Memphis, and the coauthors of the report.
The study included 212 children who, at a median age of 5 years, had received risk-adapted chemotherapy for ALL with no hematopoietic cell transplant or cranial irradiation.
Sixteen of the patients (7.5%) had a history of bacteremic sepsis during ALL therapy, according to retrospectively obtained data.
As a part of the study, all participants participated in neurocognitive testing, which was done at a median of 7.7 years after diagnosis.
Patients with a history of bacteremic sepsis performed poorly on multiple measures of neurocognitive function, as compared with all other participants, according to results of analyses that were adjusted for multiple potentially confounding factors, such as age, race, and leukemia risk category.
Although not all neurocognitive measures were significantly different between groups, survivors with a sepsis history performed worse on evaluations of spatial planning (difference, 0.78; 95% confidence interval, 0.57-1.00), verbal fluency (0.38; 95% CI, 0.14-0.62), and attention (0.63; 95% CI, 0.30-0.95), among other measures, investigators said.
This is believed to be the first published study looking at potential links between sepsis during ALL treatment and long-term neurocognitive dysfunction, investigators said. However, similar observations have been made in other patient populations, they added.
Exactly how sepsis might lead to neurocognitive deficits remains unclear. “In the population of children with cancer, these mechanisms might be augmented by increased blood-brain barrier permeability to neurotoxic chemotherapy drugs,” they said in their report.
Further study is needed to look at potential brain injury mechanisms, and to validate the current findings in other ALL patient cohorts, they concluded.
The study was supported by the National Institute of Mental Health, the National Cancer Institute, and the American Lebanese Syrian Associated Charities. The researchers reported having no conflicts of interest.
SOURCE: Cheung YT et al. JAMA Pediatr. 2018 Sep 24. doi:10.1001/jamapediatrics.2018.2500.
FROM JAMA PEDIATRICS
Key clinical point:
Major finding: ALL survivors with a sepsis history performed worse than did those with no sepsis history on evaluations of spatial planning (difference, 0.78), verbal fluency (0.38), and attention (0.63).
Study details: Prospective cohort study of 212 ALL survivors who underwent neurocognitive testing at a median of nearly 8 years after diagnosis.
Disclosures: The study was supported by the National Institute of Mental Health, the National Cancer Institute, and the American Lebanese Syrian Associated Charities. The researchers reported having no conflicts of interest.
Source: Cheung YT et al. JAMA Pediatr. 2018 Sep 24. doi:10.1001/jamapediatrics.2018.2500.
Ketamine infusions may be helpful in central sensitization pain syndromes
LAS VEGAS – Ketamine infusions are a reasonable option for patients with central sensitization pain syndromes, Jay Joshi, MD, said at the annual PAINWeek.
This disparate group of disorders includes pain experienced by both the body and mind: anxiety and depression, complex regional pain syndrome, opioid-induced hyperalgesia, phantom limb pain, fibromyalgia, and PTSD. In trained hands, ketamine infusions can benefit all of them, often providing the first relief for patients frustrated by years of seeking help for a medical disorder that has no obvious physical cause, said Dr. Joshi, CEO and medical director of the National Pain Centers in Vernon Hills, Ill.
Central sensitization is a CNS response to pain, often chronic, that results in increased neural activity or an increased response to stimuli that wouldn’t normally be interpreted as pain. The root causes can be peripheral injury, persistent inflammation, or neural injury.
“Central sensitization is produced by increases in excitability and reduction in inhibitory transmission, which may produce a persistent enhancement of pain sensitivity,” Dr. Joshi said. These changes include increased glutaminergic signaling – the target of ketamine’s action as an N-methyl-D-aspartate (NMDA) receptor blocker.
By blocking glutamate reuptake and increasing it in the synapse, ketamine “resets the hyperalgesia hyperexcitatory pathway that’s been stuck in this ‘on’ position,” Dr. Joshi said in an interview. “As a selective NMDA receptor antagonist, ketamine seems to be binding to a subreceptor that’s responsible for the symptoms that patients with these syndromes experience. Other NMDA receptor antagonists don’t give the same results. By turning off the signal, we’re giving the nervous system a chance to reset” and return to a more normally functioning state.
“Even though these people have had an injury and aren’t functioning normally, they still have normal neural pathways that can perceive sensation correctly,” he added.
Ketamine is only approved as an injectable anesthetic, but has been gaining popularity as a treatment for depression and other psychiatric disorders, as well as pain. Reports have been so positive that the Food and Drug Administration is considering approval of a ketamine-based nasal spray – esketamine – that’s being developed by Johnson & Johnson. The company reported positive phase 3 data during the May meeting of the American Society of Clinical Psychopharmacology.
When paired with an oral antidepressant, and compared with a placebo spray, esketamine significantly increased the number of responders and remitters and decreased relapses. Based on these results, and three other positive phase 3 studies, Johnson & Johnson submitted for FDA approval in September 2018.
Anecdotal reports of significant relief of chronic pain associated with ketamine have made pain another attractive off-label use, despite a paucity of high-quality data. In July 2018, a consortium of the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists published evidence-based guidelines for the drug’s use in chronic pain (Reg Anesth Pain Med. 2018 Jul;43[5]:521-46). Overall, the panel found weak evidence supporting its use for most conditions, except for moderate evidence for complex regional pain syndrome.
- Spinal cord injury pain: Weak evidence for short-term benefit at doses of 0.42-0.4 mg/kg per hour ranging from 17 minutes to 5 hours for 7 consecutive days.
- Complex regional pain syndrome: Moderate evidence for pain improvement up to 12 weeks at doses of 22 mg/hour for 4 days or 0.35 mg/kg per hour over 4 hours daily for 10 days.
- Mixed neuropathic pain, phantom limb pain, postherpetic neuralgia, fibromyalgia, cancer pain, ischemic pain, migraine headache, and low-back pain: weak to no evidence.
Nevertheless, Dr. Joshi is a firm believer in ketamine’s benefit for pain patients, when it’s administered at appropriate doses by clinicians trained in anesthesia. “Our main clinic is in a surgical center and we administer ketamine under a surgical protocol. This is a powerful anesthetic and should be treated as such,” he said. Patients are risk-stratified with the Anesthesiology Society of America physical status classification system and constantly monitored during the infusions.
These kinds of precautions are not generally taken in the dozens of unregulated “ketamine clinics” continue to open across the country, Dr. Joshi said. “They’re typically not staffed by anesthesiologists or nurse anesthetists, but by other providers without adequate training who may have only taken a weekend or online course in how to administer the drug.”
Dr. Joshi reported no disclosures relevant to his presentation.
LAS VEGAS – Ketamine infusions are a reasonable option for patients with central sensitization pain syndromes, Jay Joshi, MD, said at the annual PAINWeek.
This disparate group of disorders includes pain experienced by both the body and mind: anxiety and depression, complex regional pain syndrome, opioid-induced hyperalgesia, phantom limb pain, fibromyalgia, and PTSD. In trained hands, ketamine infusions can benefit all of them, often providing the first relief for patients frustrated by years of seeking help for a medical disorder that has no obvious physical cause, said Dr. Joshi, CEO and medical director of the National Pain Centers in Vernon Hills, Ill.
Central sensitization is a CNS response to pain, often chronic, that results in increased neural activity or an increased response to stimuli that wouldn’t normally be interpreted as pain. The root causes can be peripheral injury, persistent inflammation, or neural injury.
“Central sensitization is produced by increases in excitability and reduction in inhibitory transmission, which may produce a persistent enhancement of pain sensitivity,” Dr. Joshi said. These changes include increased glutaminergic signaling – the target of ketamine’s action as an N-methyl-D-aspartate (NMDA) receptor blocker.
By blocking glutamate reuptake and increasing it in the synapse, ketamine “resets the hyperalgesia hyperexcitatory pathway that’s been stuck in this ‘on’ position,” Dr. Joshi said in an interview. “As a selective NMDA receptor antagonist, ketamine seems to be binding to a subreceptor that’s responsible for the symptoms that patients with these syndromes experience. Other NMDA receptor antagonists don’t give the same results. By turning off the signal, we’re giving the nervous system a chance to reset” and return to a more normally functioning state.
“Even though these people have had an injury and aren’t functioning normally, they still have normal neural pathways that can perceive sensation correctly,” he added.
Ketamine is only approved as an injectable anesthetic, but has been gaining popularity as a treatment for depression and other psychiatric disorders, as well as pain. Reports have been so positive that the Food and Drug Administration is considering approval of a ketamine-based nasal spray – esketamine – that’s being developed by Johnson & Johnson. The company reported positive phase 3 data during the May meeting of the American Society of Clinical Psychopharmacology.
When paired with an oral antidepressant, and compared with a placebo spray, esketamine significantly increased the number of responders and remitters and decreased relapses. Based on these results, and three other positive phase 3 studies, Johnson & Johnson submitted for FDA approval in September 2018.
Anecdotal reports of significant relief of chronic pain associated with ketamine have made pain another attractive off-label use, despite a paucity of high-quality data. In July 2018, a consortium of the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists published evidence-based guidelines for the drug’s use in chronic pain (Reg Anesth Pain Med. 2018 Jul;43[5]:521-46). Overall, the panel found weak evidence supporting its use for most conditions, except for moderate evidence for complex regional pain syndrome.
- Spinal cord injury pain: Weak evidence for short-term benefit at doses of 0.42-0.4 mg/kg per hour ranging from 17 minutes to 5 hours for 7 consecutive days.
- Complex regional pain syndrome: Moderate evidence for pain improvement up to 12 weeks at doses of 22 mg/hour for 4 days or 0.35 mg/kg per hour over 4 hours daily for 10 days.
- Mixed neuropathic pain, phantom limb pain, postherpetic neuralgia, fibromyalgia, cancer pain, ischemic pain, migraine headache, and low-back pain: weak to no evidence.
Nevertheless, Dr. Joshi is a firm believer in ketamine’s benefit for pain patients, when it’s administered at appropriate doses by clinicians trained in anesthesia. “Our main clinic is in a surgical center and we administer ketamine under a surgical protocol. This is a powerful anesthetic and should be treated as such,” he said. Patients are risk-stratified with the Anesthesiology Society of America physical status classification system and constantly monitored during the infusions.
These kinds of precautions are not generally taken in the dozens of unregulated “ketamine clinics” continue to open across the country, Dr. Joshi said. “They’re typically not staffed by anesthesiologists or nurse anesthetists, but by other providers without adequate training who may have only taken a weekend or online course in how to administer the drug.”
Dr. Joshi reported no disclosures relevant to his presentation.
LAS VEGAS – Ketamine infusions are a reasonable option for patients with central sensitization pain syndromes, Jay Joshi, MD, said at the annual PAINWeek.
This disparate group of disorders includes pain experienced by both the body and mind: anxiety and depression, complex regional pain syndrome, opioid-induced hyperalgesia, phantom limb pain, fibromyalgia, and PTSD. In trained hands, ketamine infusions can benefit all of them, often providing the first relief for patients frustrated by years of seeking help for a medical disorder that has no obvious physical cause, said Dr. Joshi, CEO and medical director of the National Pain Centers in Vernon Hills, Ill.
Central sensitization is a CNS response to pain, often chronic, that results in increased neural activity or an increased response to stimuli that wouldn’t normally be interpreted as pain. The root causes can be peripheral injury, persistent inflammation, or neural injury.
“Central sensitization is produced by increases in excitability and reduction in inhibitory transmission, which may produce a persistent enhancement of pain sensitivity,” Dr. Joshi said. These changes include increased glutaminergic signaling – the target of ketamine’s action as an N-methyl-D-aspartate (NMDA) receptor blocker.
By blocking glutamate reuptake and increasing it in the synapse, ketamine “resets the hyperalgesia hyperexcitatory pathway that’s been stuck in this ‘on’ position,” Dr. Joshi said in an interview. “As a selective NMDA receptor antagonist, ketamine seems to be binding to a subreceptor that’s responsible for the symptoms that patients with these syndromes experience. Other NMDA receptor antagonists don’t give the same results. By turning off the signal, we’re giving the nervous system a chance to reset” and return to a more normally functioning state.
“Even though these people have had an injury and aren’t functioning normally, they still have normal neural pathways that can perceive sensation correctly,” he added.
Ketamine is only approved as an injectable anesthetic, but has been gaining popularity as a treatment for depression and other psychiatric disorders, as well as pain. Reports have been so positive that the Food and Drug Administration is considering approval of a ketamine-based nasal spray – esketamine – that’s being developed by Johnson & Johnson. The company reported positive phase 3 data during the May meeting of the American Society of Clinical Psychopharmacology.
When paired with an oral antidepressant, and compared with a placebo spray, esketamine significantly increased the number of responders and remitters and decreased relapses. Based on these results, and three other positive phase 3 studies, Johnson & Johnson submitted for FDA approval in September 2018.
Anecdotal reports of significant relief of chronic pain associated with ketamine have made pain another attractive off-label use, despite a paucity of high-quality data. In July 2018, a consortium of the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists published evidence-based guidelines for the drug’s use in chronic pain (Reg Anesth Pain Med. 2018 Jul;43[5]:521-46). Overall, the panel found weak evidence supporting its use for most conditions, except for moderate evidence for complex regional pain syndrome.
- Spinal cord injury pain: Weak evidence for short-term benefit at doses of 0.42-0.4 mg/kg per hour ranging from 17 minutes to 5 hours for 7 consecutive days.
- Complex regional pain syndrome: Moderate evidence for pain improvement up to 12 weeks at doses of 22 mg/hour for 4 days or 0.35 mg/kg per hour over 4 hours daily for 10 days.
- Mixed neuropathic pain, phantom limb pain, postherpetic neuralgia, fibromyalgia, cancer pain, ischemic pain, migraine headache, and low-back pain: weak to no evidence.
Nevertheless, Dr. Joshi is a firm believer in ketamine’s benefit for pain patients, when it’s administered at appropriate doses by clinicians trained in anesthesia. “Our main clinic is in a surgical center and we administer ketamine under a surgical protocol. This is a powerful anesthetic and should be treated as such,” he said. Patients are risk-stratified with the Anesthesiology Society of America physical status classification system and constantly monitored during the infusions.
These kinds of precautions are not generally taken in the dozens of unregulated “ketamine clinics” continue to open across the country, Dr. Joshi said. “They’re typically not staffed by anesthesiologists or nurse anesthetists, but by other providers without adequate training who may have only taken a weekend or online course in how to administer the drug.”
Dr. Joshi reported no disclosures relevant to his presentation.
EXPERT ANALYSIS FROM PAINWEEK 2018
Register for October Coding Course
Learn to use the correct codes, use all the codes necessary and submit everything properly the first time at the SVS Coding and Reimbursement Workshop, Oct. 19 and 20 at the Renaissance Chicago Downtown Hotel in Chicago. These skills learned can help vascular surgeons avoid an audit, as correct coding lessens the risk for one. And an audit costs staff time and money no matter what the outcome. Book hotel rooms by Sept. 27 to receive the special room rate for participants.
Learn to use the correct codes, use all the codes necessary and submit everything properly the first time at the SVS Coding and Reimbursement Workshop, Oct. 19 and 20 at the Renaissance Chicago Downtown Hotel in Chicago. These skills learned can help vascular surgeons avoid an audit, as correct coding lessens the risk for one. And an audit costs staff time and money no matter what the outcome. Book hotel rooms by Sept. 27 to receive the special room rate for participants.
Learn to use the correct codes, use all the codes necessary and submit everything properly the first time at the SVS Coding and Reimbursement Workshop, Oct. 19 and 20 at the Renaissance Chicago Downtown Hotel in Chicago. These skills learned can help vascular surgeons avoid an audit, as correct coding lessens the risk for one. And an audit costs staff time and money no matter what the outcome. Book hotel rooms by Sept. 27 to receive the special room rate for participants.
Bridge Grant Applications Due Oct. 1
Applications are due Oct. 1 for the SVS Foundation's new Bridge Grant, which provides funding from one grant to another. The grant targets those investigators with previous national funding for basic research, such as an NIH R01 grant, and who applied for another R01 grant but did not receive a high enough priority score to be funded again. Apply today.
Applications are due Oct. 1 for the SVS Foundation's new Bridge Grant, which provides funding from one grant to another. The grant targets those investigators with previous national funding for basic research, such as an NIH R01 grant, and who applied for another R01 grant but did not receive a high enough priority score to be funded again. Apply today.
Applications are due Oct. 1 for the SVS Foundation's new Bridge Grant, which provides funding from one grant to another. The grant targets those investigators with previous national funding for basic research, such as an NIH R01 grant, and who applied for another R01 grant but did not receive a high enough priority score to be funded again. Apply today.
Outcomes of Palliative Care Consults With Hospitalized Veterans
Families and patients receive emotional support and better care planning after palliative care consultations.
Inpatient palliative care (IPC) consultation services have been widely adopted in US hospitals. Outcomes research has demonstrated improved quality of life (QOL) for palliative inpatients for symptom control and satisfaction with care.1-5 Families benefit from emotional support, care planning, and transitions of care.4,6-8 Outcomes, including hospital length of stay, hospital costs, and discharge dispositionalso seem to improve.9-17 The Department of Veterans Affairs (VA) provides palliative care (PC) consultation teams at its hospitals nationwide; however, few studies exist to show how a PC service is used at a VA hospital. The following study of a PC consult team at an urban VA facility provides a unique picture of how a PC team is used.
Methods
The John Cochran Division of the VA St. Louis Health Care System (VASLHCS) in Missouri is a 509-bed adult acute care hospital with medical and surgical specialties and subspecialties available for veterans, including an intensive care unit (ICU). The PC team is one of the subspecialty teams following patients after consultation and consists of a PC physician, nurse practitioner, chaplain, social worker, and psychologist.
Data Collection
This study was exempt from internal review board approval. The attending physician kept track of each IPC encounter between September 2014 and April 2016. Data were retrieved from the Computerized Patient Record System by identifying charts that included family meeting notes during the specified time. All 130 patients included in this study were followed by the PC team. Patient charts were reviewed, and information was uploaded to spreadsheets, which became the database for this study. The data included age, patient location, diagnosis, number of days between admission and PC consultation, and number of days between admission and family meeting. Other data included code status changes and discharge dispositions. Only consultations that resulted in direct patient contact were included.
The VASLHCS requires therapeutic support level (TSL), or code status, documentation by the attending physician regarding the discussion with a competent patient or valid representative if the patient is incapacitated. Levels of support are TSL I ‘‘no limitation on care,’’ TSL II ‘‘partial code,’’ that is, usually no cardiopulmonary resuscitation or do not intubate with selected medical measures to continue, and TSL III ‘‘comfort measures only.’’ If a patient’s code level changed after IPC consultation, the change is recorded.
Data Analysis
The files were purged of all unique personal health history. Because there was no control group, multivariable analyses of association were not warranted. Analysis was confined to descriptive measures.
Results
A total of 130 patients with IPC consultations were included in this retrospective study conducted from September 2014 to April 2016 (Table 1). 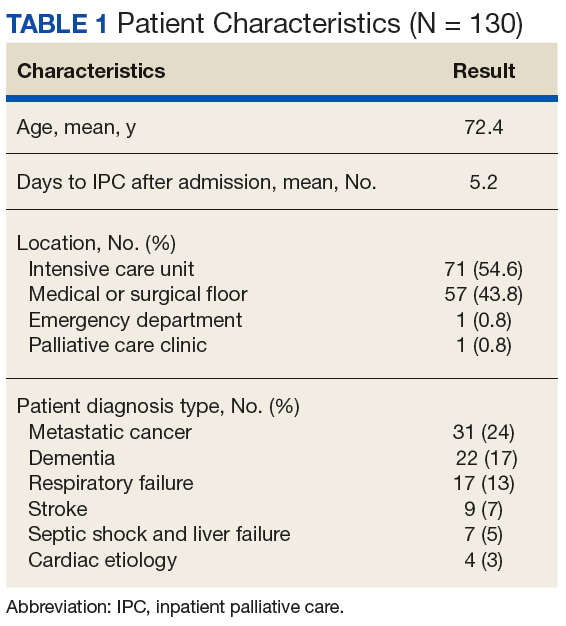
The scope of IPC consultations usually include medical recommendations about symptom management, discharge planning, discussion about goals of care (GOC), code status and prognosis, managing expected in-hospital expirations (deaths), and determination of hospice eligibility. Of the IPC cohort, 74% were aged > 65 years; 26.1% were aged < 65 years (Table 2). 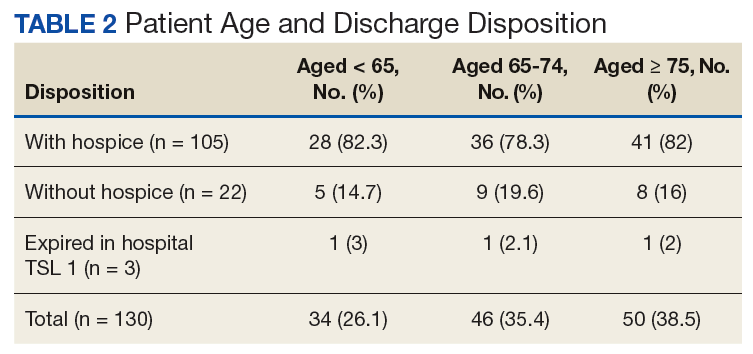
The mean days for an initial IPC consultation following admission was 3 on the medical/surgical floors and 7 days for ICU (P = .003; 95% CI, -6.37 to 1.36). 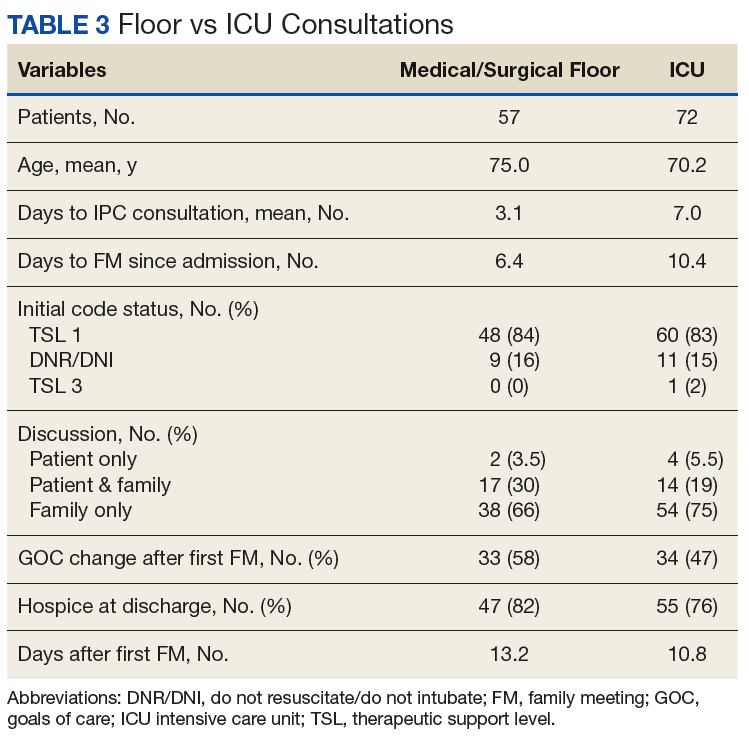
Discussion
Although small, the proportion of patients with serious illness or multiple chronic conditions account for a disproportionately large portion of health care spending.18 Despite the high cost, evidence demonstrates that these patients receive health care of inadequate quality characterized by fragmentation, overuse, medical errors, and poor QOL. Multiple studies show that IPC consultation provides improved patient outcomes and decreased hospital costs.9-17
From a purely outcomes-based interpretation, IPC consultation was associated with 83% of patients receiving a change in code status from full code/TSL 1. The study team drew 2 main conclusions from the data: (1) The IPC consultation is an effective way to broach GOC discussion and adjust code status; and (2) These data suggest room for earlier PC involvement. Remarkably, only 3 patients (2%) expired while inpatient with full code status.
The data also provide a unique comparison of timing of PC referrals. Pantilat and colleagues published characteristics of PC consultation services in California hospitals, and on average, patients were in the hospital 5.9 days (median 5.5; SD 3.3) prior to referral.19 This study’s average number of days for initial IPC consultation following admission was 3 days on the medical/surgical floors and 7 days in the ICU. Both time frames seem reasonable but again indicate some potential improvement for earlier IPC utilization.
Although the time frame of the intervention limited the number of patients in this study, early PC consultations in the acute care setting are a helpful intervention for veterans and families to better understand the complexity of their medical condition and prognosis and allow for a frank and open discussion about realistic goals. The importance of these discussions also were reflected in the high percentage of patients transitioning to hospice level of care (80%) and the low number of patients who remained full code (3 of 130). Other studies have shown conflicting results when interventions have been exclusively for cancer patients. In this study, 45% of patients were admitted with diagnoses other than cancer compared with 24% of patients with related diagnoses in a study by Gonsalves and colleagues.20
In this study, the majority (71.6%) of family meetings were held only with family (no patient involvement), resulting in missed opportunities for earlier patient and PC involvement especially for those patients with serious medical illnesses.
A systematic review published by Wendler and colleagues found that surrogate decision makers often find that role troubling and traumatizing even with advance directive documents.21 Earlier identification and PC consultations could initiate discussions between patients and their loved ones to decide “when enough is enough,” and about whether or not to prolong the dying process, when compatible with the patient’s wishes.
Early PC consultations also could highlight a potential highly vulnerable population of medically unbefriended patients (elder orphans). These patients may have no one in their lives to act as surrogate decision makers. This situation calls for further interventions regarding early identification of these patients and better processes to assist in their decision making. Many physicians believe it is not appropriate to begin advance directive planning on an outpatient basis. However, multiple studies have shown that patients want their doctors to discuss advance care planning with them before they become ill.22 Many other doctors have shown a positive response from patients when advance directive discussions are held during outpatient visits.23
The goals of this study were to evaluate the effectiveness of IPC consultation on goals of care and to address code status with patients and their families. Along with these conversations, the study team provided comprehensive PC evaluation. The PC team focused on providing excellent symptom management. The team of PC physicians, pain specialists, pain pharmacists, a chaplain, psychologists, and social workers addressed all the bio-psycho-social needs of patients/families and provided comprehensive recommendations. This multidimensional approach has gained significant acceptance.24
At VASLHCS, the program has grown to about 600 new consults per year, with a dedicated inpatient hospice unit, daily outpatient clinic, and myriad learning opportunities for trainees; the center has become a main site of rotation for hospice and palliative care fellows from training programs in St. Louis.
Utilization of PC consultation to help meeting the veterans’ needs at the bio-psycho-social level will also provide a benefit for the facility as it will decrease observed/expected standardized mortality ratio (SMR) data. This reduction of SMR data will be a result of successful patient transitions to hospice level of care at least 12 months prior to their passing or if their level of care is changed to inpatient hospice after they are admitted, the patients won’t be included as acute care mortality. However, with this initial small group of patients it was not possible to retrospectively calculate the impact on SMR or SAIL (Strategic Analytics for Improvement and Learning) indicators. The long-term expectation is to have a positive impact on those indicators represented by decreased inpatient mortality and improved SAIL.
Limitations
This study was a single-institution study, but every institution has its own internal culture. The team did not have a concurrent or historic control for comparison or use a questionnaire for patients and families rating their satisfaction.
Conclusion
This study provides multiple future directions of research as the authors now have baseline data about how the service is used. Future areas of interest would be to study the effectiveness of early palliative care interventions, such as a provider education series, implementation of consultation criteria, and prospective measurement of the impact of palliative care consultations on the SMR and SAIL indicators. This research could help identify which early interventions show the best efficacy, an area where research is notably lacking.25
1. El-Jawahri A, Greer JA, Temel JS. Does palliative care improve outcomes for patients with incurable illness? A review of the evidence. J Support Oncol. 2011;9(3):87-94.
2. Higginson IJ, Finlay I, Goodwin DM, et al. Do hospital-based palliative teams improve care for patients or families at the end of life? J Pain Symptom Manage. 2002;23(2):96-106.
3. Gade G, Venohr I, Conner D, et al. Impact of an inpatient palliative care team: a randomized control trial. J Palliat Med. 2008;11(2):180-190.
4. Benzar E, Hansen L, Kneitel AW, Fromme EK. Discharge planning for palliative care patients: a qualitative analysis. J Palliat Med. 2011;14(1):65-69.
5. Enguidanos S, Housen P, Penido M, Mejia B, Miller JA. Family members’ perceptions of inpatient palliative care consult services: a qualitative study. Palliat Med. 2014;28(1):42-48.
6. Granda-Cameron C, Viola SR, Lynch MP, Polomano RC. Measuring patient-oriented outcomes in palliative care: functionality and quality of life. Clin J Oncol Nurs. 2008;12(1):65-77.
7. Chand P, Gabriel T, Wallace CL, Nelson CM. Inpatient palliative care consultation: describing patient satisfaction. Perm J. 2013;17(1):53-55.
8. Tangeman JC, Rudra CB, Kerr CW, Grant PC. A hospice-hospital partnership: reducing hospitalization costs and 30-day readmissions among seriously ill adults. J Palliat Med. 2014;17(9):1005-1010.
9. Fromme EK, Bascom PB, Smith MD, et al. Survival, mortality, and location of death for patients seen by a hospital-based palliative care team. J Palliat Med. 2006;9(4):903-911.
10. Penrod JD, Deb P, Dellenbaugh C, et al. Hospital-based palliative care consultation: effects on hospital cost. J Palliat Med. 2010;13(8):973-979.
11. Ranganathan A, Dougherty M, Waite D, Casarett D. Can palliative home care reduce 30-day readmissions? Results of a propensity score matched cohort study. J Palliat Med. 2013;16(10):1290-1293.
12. Starks H, Wang S, Farber S, Owens DA, Curtis JR. Cost savings vary by length of stay for inpatients receiving palliative care consultation services. J Palliat Med. 2013;16(10):1215-1220.
13. Goldenheim A, Oates D, Parker V, Russell M, Winter M, Silliman RA. Rehospitalization of older adults discharged to home hospice care. J Palliat Med. 2014;17(7):841-844.
14. May P, Normand C, Morrison RS. Economic impact of hospital inpatient palliative care consultation: review of current evidence and directions for future research. J Palliat Med. 2014;17(9):1054-1063.
15. Granda-Cameron C, Behta M, Hovinga M, Rundio A, Mintzer D. Risk factors associated with unplanned hospital readmissions in adults with cancer. Oncol Nurs Forum. 2015;42(3):e257-e268.
16. Brody AA, Ciemins E, Newman J, Harrington C. The effects of an inpatient palliative care team on discharge disposition. J Palliat Med. 2010;13(5):541-548.
17. Penrod JD, Deb P, Luhrs C, et al. Cost and utilization outcomes of patients receiving hospital-based palliative care consultation. J Palliat Med. 2006;9(4):855-860.
18. Aldridge MD, Kelley AS. Appendix E, Epidemiology of serious illness and high utilization of health care. In: Institute of Medicine, Committee on Approaching Death: Addressing Key End of Life Issues. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. Washington, DC: National Academies Press; 2015.
19. Pantilat SZ, Kerr KM, Billings JA, Bruno KA, O’Riordan DL. Characteristics of palliative care consultation services in California hospitals. J Palliat Med. 2012;15(5):555-560.
20. Gonsalves WI, Tashi T, Krishnamurthy J, et al. Effect of palliative care services on the aggressiveness of end-of-life care in the Veterans Affairs cancer population. J Palliat Med. 2011;14(11):1231-1235.
21. Wendler D, Rid A. Systematic review: the effect on surrogates of making treatment decisions for others. Ann Intern Med. 2011;154(5):336-346.
22. American Bar Association Commission on Law and Aging. Myths and facts about health care advance directives. https://www.americanbar.org/content/dam/aba/publications/bifocal/BIFOCALSept-Oct2015.authcheckdam.pdf. Accessed July 10, 2018.
23. Tierney WM, Dexter PR, Gramelspacher GP, Perkins AJ, Zhou X-H, Wolinsky FD. The effect of discussions about advance directives on patients’ satisfaction with primary care. J Gen Intern Med. 2001;16(1):32-40.
24. Bailey FA, Williams BR, Woodby LL, et al. Intervention to improve care at life’s end in inpatient settings: the BEACON trial. J Gen Intern Med. 2014;29(6):836-843.
25. Dalgaard K, Bergenholtz H, Nielsen M, Timm H. Early integration of palliative care in hospitals: a systematic review on methods, barriers, and outcome. Palliat Support Care. 2014;12(6):495-513.
Families and patients receive emotional support and better care planning after palliative care consultations.
Families and patients receive emotional support and better care planning after palliative care consultations.
Inpatient palliative care (IPC) consultation services have been widely adopted in US hospitals. Outcomes research has demonstrated improved quality of life (QOL) for palliative inpatients for symptom control and satisfaction with care.1-5 Families benefit from emotional support, care planning, and transitions of care.4,6-8 Outcomes, including hospital length of stay, hospital costs, and discharge dispositionalso seem to improve.9-17 The Department of Veterans Affairs (VA) provides palliative care (PC) consultation teams at its hospitals nationwide; however, few studies exist to show how a PC service is used at a VA hospital. The following study of a PC consult team at an urban VA facility provides a unique picture of how a PC team is used.
Methods
The John Cochran Division of the VA St. Louis Health Care System (VASLHCS) in Missouri is a 509-bed adult acute care hospital with medical and surgical specialties and subspecialties available for veterans, including an intensive care unit (ICU). The PC team is one of the subspecialty teams following patients after consultation and consists of a PC physician, nurse practitioner, chaplain, social worker, and psychologist.
Data Collection
This study was exempt from internal review board approval. The attending physician kept track of each IPC encounter between September 2014 and April 2016. Data were retrieved from the Computerized Patient Record System by identifying charts that included family meeting notes during the specified time. All 130 patients included in this study were followed by the PC team. Patient charts were reviewed, and information was uploaded to spreadsheets, which became the database for this study. The data included age, patient location, diagnosis, number of days between admission and PC consultation, and number of days between admission and family meeting. Other data included code status changes and discharge dispositions. Only consultations that resulted in direct patient contact were included.
The VASLHCS requires therapeutic support level (TSL), or code status, documentation by the attending physician regarding the discussion with a competent patient or valid representative if the patient is incapacitated. Levels of support are TSL I ‘‘no limitation on care,’’ TSL II ‘‘partial code,’’ that is, usually no cardiopulmonary resuscitation or do not intubate with selected medical measures to continue, and TSL III ‘‘comfort measures only.’’ If a patient’s code level changed after IPC consultation, the change is recorded.
Data Analysis
The files were purged of all unique personal health history. Because there was no control group, multivariable analyses of association were not warranted. Analysis was confined to descriptive measures.
Results
A total of 130 patients with IPC consultations were included in this retrospective study conducted from September 2014 to April 2016 (Table 1).
The scope of IPC consultations usually include medical recommendations about symptom management, discharge planning, discussion about goals of care (GOC), code status and prognosis, managing expected in-hospital expirations (deaths), and determination of hospice eligibility. Of the IPC cohort, 74% were aged > 65 years; 26.1% were aged < 65 years (Table 2).
The mean days for an initial IPC consultation following admission was 3 on the medical/surgical floors and 7 days for ICU (P = .003; 95% CI, -6.37 to 1.36).
Discussion
Although small, the proportion of patients with serious illness or multiple chronic conditions account for a disproportionately large portion of health care spending.18 Despite the high cost, evidence demonstrates that these patients receive health care of inadequate quality characterized by fragmentation, overuse, medical errors, and poor QOL. Multiple studies show that IPC consultation provides improved patient outcomes and decreased hospital costs.9-17
From a purely outcomes-based interpretation, IPC consultation was associated with 83% of patients receiving a change in code status from full code/TSL 1. The study team drew 2 main conclusions from the data: (1) The IPC consultation is an effective way to broach GOC discussion and adjust code status; and (2) These data suggest room for earlier PC involvement. Remarkably, only 3 patients (2%) expired while inpatient with full code status.
The data also provide a unique comparison of timing of PC referrals. Pantilat and colleagues published characteristics of PC consultation services in California hospitals, and on average, patients were in the hospital 5.9 days (median 5.5; SD 3.3) prior to referral.19 This study’s average number of days for initial IPC consultation following admission was 3 days on the medical/surgical floors and 7 days in the ICU. Both time frames seem reasonable but again indicate some potential improvement for earlier IPC utilization.
Although the time frame of the intervention limited the number of patients in this study, early PC consultations in the acute care setting are a helpful intervention for veterans and families to better understand the complexity of their medical condition and prognosis and allow for a frank and open discussion about realistic goals. The importance of these discussions also were reflected in the high percentage of patients transitioning to hospice level of care (80%) and the low number of patients who remained full code (3 of 130). Other studies have shown conflicting results when interventions have been exclusively for cancer patients. In this study, 45% of patients were admitted with diagnoses other than cancer compared with 24% of patients with related diagnoses in a study by Gonsalves and colleagues.20
In this study, the majority (71.6%) of family meetings were held only with family (no patient involvement), resulting in missed opportunities for earlier patient and PC involvement especially for those patients with serious medical illnesses.
A systematic review published by Wendler and colleagues found that surrogate decision makers often find that role troubling and traumatizing even with advance directive documents.21 Earlier identification and PC consultations could initiate discussions between patients and their loved ones to decide “when enough is enough,” and about whether or not to prolong the dying process, when compatible with the patient’s wishes.
Early PC consultations also could highlight a potential highly vulnerable population of medically unbefriended patients (elder orphans). These patients may have no one in their lives to act as surrogate decision makers. This situation calls for further interventions regarding early identification of these patients and better processes to assist in their decision making. Many physicians believe it is not appropriate to begin advance directive planning on an outpatient basis. However, multiple studies have shown that patients want their doctors to discuss advance care planning with them before they become ill.22 Many other doctors have shown a positive response from patients when advance directive discussions are held during outpatient visits.23
The goals of this study were to evaluate the effectiveness of IPC consultation on goals of care and to address code status with patients and their families. Along with these conversations, the study team provided comprehensive PC evaluation. The PC team focused on providing excellent symptom management. The team of PC physicians, pain specialists, pain pharmacists, a chaplain, psychologists, and social workers addressed all the bio-psycho-social needs of patients/families and provided comprehensive recommendations. This multidimensional approach has gained significant acceptance.24
At VASLHCS, the program has grown to about 600 new consults per year, with a dedicated inpatient hospice unit, daily outpatient clinic, and myriad learning opportunities for trainees; the center has become a main site of rotation for hospice and palliative care fellows from training programs in St. Louis.
Utilization of PC consultation to help meeting the veterans’ needs at the bio-psycho-social level will also provide a benefit for the facility as it will decrease observed/expected standardized mortality ratio (SMR) data. This reduction of SMR data will be a result of successful patient transitions to hospice level of care at least 12 months prior to their passing or if their level of care is changed to inpatient hospice after they are admitted, the patients won’t be included as acute care mortality. However, with this initial small group of patients it was not possible to retrospectively calculate the impact on SMR or SAIL (Strategic Analytics for Improvement and Learning) indicators. The long-term expectation is to have a positive impact on those indicators represented by decreased inpatient mortality and improved SAIL.
Limitations
This study was a single-institution study, but every institution has its own internal culture. The team did not have a concurrent or historic control for comparison or use a questionnaire for patients and families rating their satisfaction.
Conclusion
This study provides multiple future directions of research as the authors now have baseline data about how the service is used. Future areas of interest would be to study the effectiveness of early palliative care interventions, such as a provider education series, implementation of consultation criteria, and prospective measurement of the impact of palliative care consultations on the SMR and SAIL indicators. This research could help identify which early interventions show the best efficacy, an area where research is notably lacking.25
Inpatient palliative care (IPC) consultation services have been widely adopted in US hospitals. Outcomes research has demonstrated improved quality of life (QOL) for palliative inpatients for symptom control and satisfaction with care.1-5 Families benefit from emotional support, care planning, and transitions of care.4,6-8 Outcomes, including hospital length of stay, hospital costs, and discharge dispositionalso seem to improve.9-17 The Department of Veterans Affairs (VA) provides palliative care (PC) consultation teams at its hospitals nationwide; however, few studies exist to show how a PC service is used at a VA hospital. The following study of a PC consult team at an urban VA facility provides a unique picture of how a PC team is used.
Methods
The John Cochran Division of the VA St. Louis Health Care System (VASLHCS) in Missouri is a 509-bed adult acute care hospital with medical and surgical specialties and subspecialties available for veterans, including an intensive care unit (ICU). The PC team is one of the subspecialty teams following patients after consultation and consists of a PC physician, nurse practitioner, chaplain, social worker, and psychologist.
Data Collection
This study was exempt from internal review board approval. The attending physician kept track of each IPC encounter between September 2014 and April 2016. Data were retrieved from the Computerized Patient Record System by identifying charts that included family meeting notes during the specified time. All 130 patients included in this study were followed by the PC team. Patient charts were reviewed, and information was uploaded to spreadsheets, which became the database for this study. The data included age, patient location, diagnosis, number of days between admission and PC consultation, and number of days between admission and family meeting. Other data included code status changes and discharge dispositions. Only consultations that resulted in direct patient contact were included.
The VASLHCS requires therapeutic support level (TSL), or code status, documentation by the attending physician regarding the discussion with a competent patient or valid representative if the patient is incapacitated. Levels of support are TSL I ‘‘no limitation on care,’’ TSL II ‘‘partial code,’’ that is, usually no cardiopulmonary resuscitation or do not intubate with selected medical measures to continue, and TSL III ‘‘comfort measures only.’’ If a patient’s code level changed after IPC consultation, the change is recorded.
Data Analysis
The files were purged of all unique personal health history. Because there was no control group, multivariable analyses of association were not warranted. Analysis was confined to descriptive measures.
Results
A total of 130 patients with IPC consultations were included in this retrospective study conducted from September 2014 to April 2016 (Table 1).
The scope of IPC consultations usually include medical recommendations about symptom management, discharge planning, discussion about goals of care (GOC), code status and prognosis, managing expected in-hospital expirations (deaths), and determination of hospice eligibility. Of the IPC cohort, 74% were aged > 65 years; 26.1% were aged < 65 years (Table 2).
The mean days for an initial IPC consultation following admission was 3 on the medical/surgical floors and 7 days for ICU (P = .003; 95% CI, -6.37 to 1.36).
Discussion
Although small, the proportion of patients with serious illness or multiple chronic conditions account for a disproportionately large portion of health care spending.18 Despite the high cost, evidence demonstrates that these patients receive health care of inadequate quality characterized by fragmentation, overuse, medical errors, and poor QOL. Multiple studies show that IPC consultation provides improved patient outcomes and decreased hospital costs.9-17
From a purely outcomes-based interpretation, IPC consultation was associated with 83% of patients receiving a change in code status from full code/TSL 1. The study team drew 2 main conclusions from the data: (1) The IPC consultation is an effective way to broach GOC discussion and adjust code status; and (2) These data suggest room for earlier PC involvement. Remarkably, only 3 patients (2%) expired while inpatient with full code status.
The data also provide a unique comparison of timing of PC referrals. Pantilat and colleagues published characteristics of PC consultation services in California hospitals, and on average, patients were in the hospital 5.9 days (median 5.5; SD 3.3) prior to referral.19 This study’s average number of days for initial IPC consultation following admission was 3 days on the medical/surgical floors and 7 days in the ICU. Both time frames seem reasonable but again indicate some potential improvement for earlier IPC utilization.
Although the time frame of the intervention limited the number of patients in this study, early PC consultations in the acute care setting are a helpful intervention for veterans and families to better understand the complexity of their medical condition and prognosis and allow for a frank and open discussion about realistic goals. The importance of these discussions also were reflected in the high percentage of patients transitioning to hospice level of care (80%) and the low number of patients who remained full code (3 of 130). Other studies have shown conflicting results when interventions have been exclusively for cancer patients. In this study, 45% of patients were admitted with diagnoses other than cancer compared with 24% of patients with related diagnoses in a study by Gonsalves and colleagues.20
In this study, the majority (71.6%) of family meetings were held only with family (no patient involvement), resulting in missed opportunities for earlier patient and PC involvement especially for those patients with serious medical illnesses.
A systematic review published by Wendler and colleagues found that surrogate decision makers often find that role troubling and traumatizing even with advance directive documents.21 Earlier identification and PC consultations could initiate discussions between patients and their loved ones to decide “when enough is enough,” and about whether or not to prolong the dying process, when compatible with the patient’s wishes.
Early PC consultations also could highlight a potential highly vulnerable population of medically unbefriended patients (elder orphans). These patients may have no one in their lives to act as surrogate decision makers. This situation calls for further interventions regarding early identification of these patients and better processes to assist in their decision making. Many physicians believe it is not appropriate to begin advance directive planning on an outpatient basis. However, multiple studies have shown that patients want their doctors to discuss advance care planning with them before they become ill.22 Many other doctors have shown a positive response from patients when advance directive discussions are held during outpatient visits.23
The goals of this study were to evaluate the effectiveness of IPC consultation on goals of care and to address code status with patients and their families. Along with these conversations, the study team provided comprehensive PC evaluation. The PC team focused on providing excellent symptom management. The team of PC physicians, pain specialists, pain pharmacists, a chaplain, psychologists, and social workers addressed all the bio-psycho-social needs of patients/families and provided comprehensive recommendations. This multidimensional approach has gained significant acceptance.24
At VASLHCS, the program has grown to about 600 new consults per year, with a dedicated inpatient hospice unit, daily outpatient clinic, and myriad learning opportunities for trainees; the center has become a main site of rotation for hospice and palliative care fellows from training programs in St. Louis.
Utilization of PC consultation to help meeting the veterans’ needs at the bio-psycho-social level will also provide a benefit for the facility as it will decrease observed/expected standardized mortality ratio (SMR) data. This reduction of SMR data will be a result of successful patient transitions to hospice level of care at least 12 months prior to their passing or if their level of care is changed to inpatient hospice after they are admitted, the patients won’t be included as acute care mortality. However, with this initial small group of patients it was not possible to retrospectively calculate the impact on SMR or SAIL (Strategic Analytics for Improvement and Learning) indicators. The long-term expectation is to have a positive impact on those indicators represented by decreased inpatient mortality and improved SAIL.
Limitations
This study was a single-institution study, but every institution has its own internal culture. The team did not have a concurrent or historic control for comparison or use a questionnaire for patients and families rating their satisfaction.
Conclusion
This study provides multiple future directions of research as the authors now have baseline data about how the service is used. Future areas of interest would be to study the effectiveness of early palliative care interventions, such as a provider education series, implementation of consultation criteria, and prospective measurement of the impact of palliative care consultations on the SMR and SAIL indicators. This research could help identify which early interventions show the best efficacy, an area where research is notably lacking.25
1. El-Jawahri A, Greer JA, Temel JS. Does palliative care improve outcomes for patients with incurable illness? A review of the evidence. J Support Oncol. 2011;9(3):87-94.
2. Higginson IJ, Finlay I, Goodwin DM, et al. Do hospital-based palliative teams improve care for patients or families at the end of life? J Pain Symptom Manage. 2002;23(2):96-106.
3. Gade G, Venohr I, Conner D, et al. Impact of an inpatient palliative care team: a randomized control trial. J Palliat Med. 2008;11(2):180-190.
4. Benzar E, Hansen L, Kneitel AW, Fromme EK. Discharge planning for palliative care patients: a qualitative analysis. J Palliat Med. 2011;14(1):65-69.
5. Enguidanos S, Housen P, Penido M, Mejia B, Miller JA. Family members’ perceptions of inpatient palliative care consult services: a qualitative study. Palliat Med. 2014;28(1):42-48.
6. Granda-Cameron C, Viola SR, Lynch MP, Polomano RC. Measuring patient-oriented outcomes in palliative care: functionality and quality of life. Clin J Oncol Nurs. 2008;12(1):65-77.
7. Chand P, Gabriel T, Wallace CL, Nelson CM. Inpatient palliative care consultation: describing patient satisfaction. Perm J. 2013;17(1):53-55.
8. Tangeman JC, Rudra CB, Kerr CW, Grant PC. A hospice-hospital partnership: reducing hospitalization costs and 30-day readmissions among seriously ill adults. J Palliat Med. 2014;17(9):1005-1010.
9. Fromme EK, Bascom PB, Smith MD, et al. Survival, mortality, and location of death for patients seen by a hospital-based palliative care team. J Palliat Med. 2006;9(4):903-911.
10. Penrod JD, Deb P, Dellenbaugh C, et al. Hospital-based palliative care consultation: effects on hospital cost. J Palliat Med. 2010;13(8):973-979.
11. Ranganathan A, Dougherty M, Waite D, Casarett D. Can palliative home care reduce 30-day readmissions? Results of a propensity score matched cohort study. J Palliat Med. 2013;16(10):1290-1293.
12. Starks H, Wang S, Farber S, Owens DA, Curtis JR. Cost savings vary by length of stay for inpatients receiving palliative care consultation services. J Palliat Med. 2013;16(10):1215-1220.
13. Goldenheim A, Oates D, Parker V, Russell M, Winter M, Silliman RA. Rehospitalization of older adults discharged to home hospice care. J Palliat Med. 2014;17(7):841-844.
14. May P, Normand C, Morrison RS. Economic impact of hospital inpatient palliative care consultation: review of current evidence and directions for future research. J Palliat Med. 2014;17(9):1054-1063.
15. Granda-Cameron C, Behta M, Hovinga M, Rundio A, Mintzer D. Risk factors associated with unplanned hospital readmissions in adults with cancer. Oncol Nurs Forum. 2015;42(3):e257-e268.
16. Brody AA, Ciemins E, Newman J, Harrington C. The effects of an inpatient palliative care team on discharge disposition. J Palliat Med. 2010;13(5):541-548.
17. Penrod JD, Deb P, Luhrs C, et al. Cost and utilization outcomes of patients receiving hospital-based palliative care consultation. J Palliat Med. 2006;9(4):855-860.
18. Aldridge MD, Kelley AS. Appendix E, Epidemiology of serious illness and high utilization of health care. In: Institute of Medicine, Committee on Approaching Death: Addressing Key End of Life Issues. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. Washington, DC: National Academies Press; 2015.
19. Pantilat SZ, Kerr KM, Billings JA, Bruno KA, O’Riordan DL. Characteristics of palliative care consultation services in California hospitals. J Palliat Med. 2012;15(5):555-560.
20. Gonsalves WI, Tashi T, Krishnamurthy J, et al. Effect of palliative care services on the aggressiveness of end-of-life care in the Veterans Affairs cancer population. J Palliat Med. 2011;14(11):1231-1235.
21. Wendler D, Rid A. Systematic review: the effect on surrogates of making treatment decisions for others. Ann Intern Med. 2011;154(5):336-346.
22. American Bar Association Commission on Law and Aging. Myths and facts about health care advance directives. https://www.americanbar.org/content/dam/aba/publications/bifocal/BIFOCALSept-Oct2015.authcheckdam.pdf. Accessed July 10, 2018.
23. Tierney WM, Dexter PR, Gramelspacher GP, Perkins AJ, Zhou X-H, Wolinsky FD. The effect of discussions about advance directives on patients’ satisfaction with primary care. J Gen Intern Med. 2001;16(1):32-40.
24. Bailey FA, Williams BR, Woodby LL, et al. Intervention to improve care at life’s end in inpatient settings: the BEACON trial. J Gen Intern Med. 2014;29(6):836-843.
25. Dalgaard K, Bergenholtz H, Nielsen M, Timm H. Early integration of palliative care in hospitals: a systematic review on methods, barriers, and outcome. Palliat Support Care. 2014;12(6):495-513.
1. El-Jawahri A, Greer JA, Temel JS. Does palliative care improve outcomes for patients with incurable illness? A review of the evidence. J Support Oncol. 2011;9(3):87-94.
2. Higginson IJ, Finlay I, Goodwin DM, et al. Do hospital-based palliative teams improve care for patients or families at the end of life? J Pain Symptom Manage. 2002;23(2):96-106.
3. Gade G, Venohr I, Conner D, et al. Impact of an inpatient palliative care team: a randomized control trial. J Palliat Med. 2008;11(2):180-190.
4. Benzar E, Hansen L, Kneitel AW, Fromme EK. Discharge planning for palliative care patients: a qualitative analysis. J Palliat Med. 2011;14(1):65-69.
5. Enguidanos S, Housen P, Penido M, Mejia B, Miller JA. Family members’ perceptions of inpatient palliative care consult services: a qualitative study. Palliat Med. 2014;28(1):42-48.
6. Granda-Cameron C, Viola SR, Lynch MP, Polomano RC. Measuring patient-oriented outcomes in palliative care: functionality and quality of life. Clin J Oncol Nurs. 2008;12(1):65-77.
7. Chand P, Gabriel T, Wallace CL, Nelson CM. Inpatient palliative care consultation: describing patient satisfaction. Perm J. 2013;17(1):53-55.
8. Tangeman JC, Rudra CB, Kerr CW, Grant PC. A hospice-hospital partnership: reducing hospitalization costs and 30-day readmissions among seriously ill adults. J Palliat Med. 2014;17(9):1005-1010.
9. Fromme EK, Bascom PB, Smith MD, et al. Survival, mortality, and location of death for patients seen by a hospital-based palliative care team. J Palliat Med. 2006;9(4):903-911.
10. Penrod JD, Deb P, Dellenbaugh C, et al. Hospital-based palliative care consultation: effects on hospital cost. J Palliat Med. 2010;13(8):973-979.
11. Ranganathan A, Dougherty M, Waite D, Casarett D. Can palliative home care reduce 30-day readmissions? Results of a propensity score matched cohort study. J Palliat Med. 2013;16(10):1290-1293.
12. Starks H, Wang S, Farber S, Owens DA, Curtis JR. Cost savings vary by length of stay for inpatients receiving palliative care consultation services. J Palliat Med. 2013;16(10):1215-1220.
13. Goldenheim A, Oates D, Parker V, Russell M, Winter M, Silliman RA. Rehospitalization of older adults discharged to home hospice care. J Palliat Med. 2014;17(7):841-844.
14. May P, Normand C, Morrison RS. Economic impact of hospital inpatient palliative care consultation: review of current evidence and directions for future research. J Palliat Med. 2014;17(9):1054-1063.
15. Granda-Cameron C, Behta M, Hovinga M, Rundio A, Mintzer D. Risk factors associated with unplanned hospital readmissions in adults with cancer. Oncol Nurs Forum. 2015;42(3):e257-e268.
16. Brody AA, Ciemins E, Newman J, Harrington C. The effects of an inpatient palliative care team on discharge disposition. J Palliat Med. 2010;13(5):541-548.
17. Penrod JD, Deb P, Luhrs C, et al. Cost and utilization outcomes of patients receiving hospital-based palliative care consultation. J Palliat Med. 2006;9(4):855-860.
18. Aldridge MD, Kelley AS. Appendix E, Epidemiology of serious illness and high utilization of health care. In: Institute of Medicine, Committee on Approaching Death: Addressing Key End of Life Issues. Dying in America: Improving Quality and Honoring Individual Preferences Near the End of Life. Washington, DC: National Academies Press; 2015.
19. Pantilat SZ, Kerr KM, Billings JA, Bruno KA, O’Riordan DL. Characteristics of palliative care consultation services in California hospitals. J Palliat Med. 2012;15(5):555-560.
20. Gonsalves WI, Tashi T, Krishnamurthy J, et al. Effect of palliative care services on the aggressiveness of end-of-life care in the Veterans Affairs cancer population. J Palliat Med. 2011;14(11):1231-1235.
21. Wendler D, Rid A. Systematic review: the effect on surrogates of making treatment decisions for others. Ann Intern Med. 2011;154(5):336-346.
22. American Bar Association Commission on Law and Aging. Myths and facts about health care advance directives. https://www.americanbar.org/content/dam/aba/publications/bifocal/BIFOCALSept-Oct2015.authcheckdam.pdf. Accessed July 10, 2018.
23. Tierney WM, Dexter PR, Gramelspacher GP, Perkins AJ, Zhou X-H, Wolinsky FD. The effect of discussions about advance directives on patients’ satisfaction with primary care. J Gen Intern Med. 2001;16(1):32-40.
24. Bailey FA, Williams BR, Woodby LL, et al. Intervention to improve care at life’s end in inpatient settings: the BEACON trial. J Gen Intern Med. 2014;29(6):836-843.
25. Dalgaard K, Bergenholtz H, Nielsen M, Timm H. Early integration of palliative care in hospitals: a systematic review on methods, barriers, and outcome. Palliat Support Care. 2014;12(6):495-513.
CHMP reconsiders new indication for blinatumomab

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) said it will re-examine a recent opinion on blinatumomab (Blincyto).
In July, the CHMP recommended against approving blinatumomab to treat patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) who have minimal residual disease (MRD).
However, the CHMP has agreed to re-examine its position and issue a final recommendation.
Blinatumomab is currently approved by the European Commission (EC) as monotherapy for adults with Philadelphia chromosome-negative, CD19-positive, relapsed or refractory BCP-ALL.
Blinatumomab is also approved as monotherapy for pediatric patients age 1 year or older who have relapsed/refractory, Philadelphia chromosome-negative, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
Amgen is seeking an extension of the marketing authorization for blinatumomab to include BCP-ALL patients with MRD.
The CHMP previously recommended against approving blinatumomab for these patients based on data from the BLAST study. Results from this phase 2 trial were published in Blood in April.
The CHMP noted that, although blinatumomab helped clear away residual cells in many patients in the BLAST trial, there is no strong evidence that this leads to improved survival.
Given the uncertainty, the CHMP was of the opinion that the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen request a re-examination of the CHMP’s opinion, and the CHMP has complied.
The CHMP’s recommendations are reviewed by the EC, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein. The EC usually makes a decision within 67 days of CHMP recommendations.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) said it will re-examine a recent opinion on blinatumomab (Blincyto).
In July, the CHMP recommended against approving blinatumomab to treat patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) who have minimal residual disease (MRD).
However, the CHMP has agreed to re-examine its position and issue a final recommendation.
Blinatumomab is currently approved by the European Commission (EC) as monotherapy for adults with Philadelphia chromosome-negative, CD19-positive, relapsed or refractory BCP-ALL.
Blinatumomab is also approved as monotherapy for pediatric patients age 1 year or older who have relapsed/refractory, Philadelphia chromosome-negative, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
Amgen is seeking an extension of the marketing authorization for blinatumomab to include BCP-ALL patients with MRD.
The CHMP previously recommended against approving blinatumomab for these patients based on data from the BLAST study. Results from this phase 2 trial were published in Blood in April.
The CHMP noted that, although blinatumomab helped clear away residual cells in many patients in the BLAST trial, there is no strong evidence that this leads to improved survival.
Given the uncertainty, the CHMP was of the opinion that the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen request a re-examination of the CHMP’s opinion, and the CHMP has complied.
The CHMP’s recommendations are reviewed by the EC, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein. The EC usually makes a decision within 67 days of CHMP recommendations.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) said it will re-examine a recent opinion on blinatumomab (Blincyto).
In July, the CHMP recommended against approving blinatumomab to treat patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL) who have minimal residual disease (MRD).
However, the CHMP has agreed to re-examine its position and issue a final recommendation.
Blinatumomab is currently approved by the European Commission (EC) as monotherapy for adults with Philadelphia chromosome-negative, CD19-positive, relapsed or refractory BCP-ALL.
Blinatumomab is also approved as monotherapy for pediatric patients age 1 year or older who have relapsed/refractory, Philadelphia chromosome-negative, CD19-positive BCP-ALL and have received at least two prior therapies or relapsed after allogeneic hematopoietic stem cell transplant.
Amgen is seeking an extension of the marketing authorization for blinatumomab to include BCP-ALL patients with MRD.
The CHMP previously recommended against approving blinatumomab for these patients based on data from the BLAST study. Results from this phase 2 trial were published in Blood in April.
The CHMP noted that, although blinatumomab helped clear away residual cells in many patients in the BLAST trial, there is no strong evidence that this leads to improved survival.
Given the uncertainty, the CHMP was of the opinion that the benefits of blinatumomab do not outweigh its risks in MRD-positive BCP-ALL patients.
However, Amgen request a re-examination of the CHMP’s opinion, and the CHMP has complied.
The CHMP’s recommendations are reviewed by the EC, which has the authority to approve medicines for use in the European Union, Norway, Iceland, and Liechtenstein. The EC usually makes a decision within 67 days of CHMP recommendations.
CHMP backs proposed biosimilars of pegfilgrastim
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for three proposed biosimilars of pegfilgrastim—Ziextenzo, Pelmeg, and Fulphila.
If approved by the European Commission (EC), these products would be used for the same indication as the reference medicine, Neulasta (pegfilgrastim).
Neulasta has been EC-approved since 2002 to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults who receive cytotoxic chemotherapy to treat malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
According to the CHMP, data suggest that Ziextenzo, Pelmeg, and Fulphila all have quality, efficacy, and safety profiles comparable to Neulasta.
The EC is expected to make a decision on the approval of Ziextenzo, Pelmeg, and Fulphila within 67 days of the CHMP’s opinion.
The EC’s decision will apply to the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions based on the EC’s judgement.
Ziextenzo is being developed by Sandoz GmbH, Pelmeg is being developed by Cinfa Biotech S.L., and Fulphila is being developed by MYLAN S.A.S.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for three proposed biosimilars of pegfilgrastim—Ziextenzo, Pelmeg, and Fulphila.
If approved by the European Commission (EC), these products would be used for the same indication as the reference medicine, Neulasta (pegfilgrastim).
Neulasta has been EC-approved since 2002 to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults who receive cytotoxic chemotherapy to treat malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
According to the CHMP, data suggest that Ziextenzo, Pelmeg, and Fulphila all have quality, efficacy, and safety profiles comparable to Neulasta.
The EC is expected to make a decision on the approval of Ziextenzo, Pelmeg, and Fulphila within 67 days of the CHMP’s opinion.
The EC’s decision will apply to the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions based on the EC’s judgement.
Ziextenzo is being developed by Sandoz GmbH, Pelmeg is being developed by Cinfa Biotech S.L., and Fulphila is being developed by MYLAN S.A.S.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for three proposed biosimilars of pegfilgrastim—Ziextenzo, Pelmeg, and Fulphila.
If approved by the European Commission (EC), these products would be used for the same indication as the reference medicine, Neulasta (pegfilgrastim).
Neulasta has been EC-approved since 2002 to reduce the duration of neutropenia and the incidence of febrile neutropenia in adults who receive cytotoxic chemotherapy to treat malignancies except chronic myeloid leukemia and myelodysplastic syndromes.
According to the CHMP, data suggest that Ziextenzo, Pelmeg, and Fulphila all have quality, efficacy, and safety profiles comparable to Neulasta.
The EC is expected to make a decision on the approval of Ziextenzo, Pelmeg, and Fulphila within 67 days of the CHMP’s opinion.
The EC’s decision will apply to the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions based on the EC’s judgement.
Ziextenzo is being developed by Sandoz GmbH, Pelmeg is being developed by Cinfa Biotech S.L., and Fulphila is being developed by MYLAN S.A.S.
CHMP recommends factor VIII therapy for hemophilia A
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for damoctocog alfa pegol, a recombinant human factor VIII therapy.
Bayer is seeking European marketing authorization for damoctocog alfa pegol (formerly BAY94-9027) for the treatment and prophylaxis of bleeding in previously treated patients age 12 and older with hemophilia A.
The CHMP’s recommendation for damoctocog alfa pegol, which is approved in the U.S. under the name Jivi, will be reviewed by the European Commission (EC).
The EC typically makes a decision about marketing authorization within 67 days of the CHMP’s opinion. The EC’s decision will apply to the European Union, Norway, Iceland, and Liechtenstein.
The CHMP’s recommendation for damoctocog alfa pegol is supported by the phase 2/3 PROTECT VIII trial. Some results from this trial were published in the Journal of Thrombosis and Haemostasis in 2016. Additional results are available in the U.S. prescribing information for Jivi.
PROTECT VIII enrolled previously treated adults and adolescents (ages 12 to 65) with severe hemophilia A.
In part A, researchers evaluated different dosing regimens for damoctocog alfa pegol used as prophylaxis and on-demand treatment. An optional extension study was available to patients who completed part A.
In part B, researchers evaluated damoctocog alfa pegol for perioperative management.
Efficacy
In part A, there were 132 patients in the intent‐to‐treat population—112 in the prophylaxis group and 20 in the on-demand group.
Patients received damoctocog alfa pegol for 36 weeks. For the first 10 weeks, patients in the prophylaxis group received twice-weekly dosing at 25 IU/kg.
Patients with more than one bleed during this time went on to receive 30–40 IU/kg twice weekly. Patients with one or fewer bleeds were eligible for randomization to dosing every 5 days (45–60 IU/kg) or every 7 days (60 IU/kg).
The median annualized bleeding rate (ABR) was 4.1 for the patients who were treated twice weekly and were not eligible for randomization (n=13) and 1.9 for patients who were eligible for randomization but continued on twice-weekly treatment (n=11).
The median ABR was 1.9 for patients who were randomized to treatment every 5 days (n=43) and 0.96 for patients who completed prophylaxis with dosing every 7 days (32/43).
The median ABR for patients treated on demand was 24.1.
There were 388 treated bleeds in the on-demand group and 317 treated bleeds in the prophylaxis group. Overall, 73.3% of responses to treatment were considered “excellent” or “good,” 23.3% were considered “moderate,” and 3.3% were considered “poor.”
There were 17 patients who underwent 20 major surgeries in part B or the extension study and 10 patients who underwent minor surgeries in part A. Damoctocog alfa pegol provided “good” or “excellent” hemostatic control during all surgeries.
Safety
Safety data are available for 148 patients age 12 and older.
Adverse events in these patients included abdominal pain (3%), nausea (5%), vomiting (3%), injection site reactions (1%), pyrexia (5%), hypersensitivity (2%), dizziness (2%), headache (14%), insomnia (3%), cough (7%), erythema (1%), pruritus (1%), rash (2%), and flushing (1%).
A factor VIII inhibitor was reported in one adult patient, but repeat testing did not confirm the report.
One adult with asthma had a clinical hypersensitivity reaction and a transient increase of IgM anti-PEG antibody titer, which was negative upon retesting.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for damoctocog alfa pegol, a recombinant human factor VIII therapy.
Bayer is seeking European marketing authorization for damoctocog alfa pegol (formerly BAY94-9027) for the treatment and prophylaxis of bleeding in previously treated patients age 12 and older with hemophilia A.
The CHMP’s recommendation for damoctocog alfa pegol, which is approved in the U.S. under the name Jivi, will be reviewed by the European Commission (EC).
The EC typically makes a decision about marketing authorization within 67 days of the CHMP’s opinion. The EC’s decision will apply to the European Union, Norway, Iceland, and Liechtenstein.
The CHMP’s recommendation for damoctocog alfa pegol is supported by the phase 2/3 PROTECT VIII trial. Some results from this trial were published in the Journal of Thrombosis and Haemostasis in 2016. Additional results are available in the U.S. prescribing information for Jivi.
PROTECT VIII enrolled previously treated adults and adolescents (ages 12 to 65) with severe hemophilia A.
In part A, researchers evaluated different dosing regimens for damoctocog alfa pegol used as prophylaxis and on-demand treatment. An optional extension study was available to patients who completed part A.
In part B, researchers evaluated damoctocog alfa pegol for perioperative management.
Efficacy
In part A, there were 132 patients in the intent‐to‐treat population—112 in the prophylaxis group and 20 in the on-demand group.
Patients received damoctocog alfa pegol for 36 weeks. For the first 10 weeks, patients in the prophylaxis group received twice-weekly dosing at 25 IU/kg.
Patients with more than one bleed during this time went on to receive 30–40 IU/kg twice weekly. Patients with one or fewer bleeds were eligible for randomization to dosing every 5 days (45–60 IU/kg) or every 7 days (60 IU/kg).
The median annualized bleeding rate (ABR) was 4.1 for the patients who were treated twice weekly and were not eligible for randomization (n=13) and 1.9 for patients who were eligible for randomization but continued on twice-weekly treatment (n=11).
The median ABR was 1.9 for patients who were randomized to treatment every 5 days (n=43) and 0.96 for patients who completed prophylaxis with dosing every 7 days (32/43).
The median ABR for patients treated on demand was 24.1.
There were 388 treated bleeds in the on-demand group and 317 treated bleeds in the prophylaxis group. Overall, 73.3% of responses to treatment were considered “excellent” or “good,” 23.3% were considered “moderate,” and 3.3% were considered “poor.”
There were 17 patients who underwent 20 major surgeries in part B or the extension study and 10 patients who underwent minor surgeries in part A. Damoctocog alfa pegol provided “good” or “excellent” hemostatic control during all surgeries.
Safety
Safety data are available for 148 patients age 12 and older.
Adverse events in these patients included abdominal pain (3%), nausea (5%), vomiting (3%), injection site reactions (1%), pyrexia (5%), hypersensitivity (2%), dizziness (2%), headache (14%), insomnia (3%), cough (7%), erythema (1%), pruritus (1%), rash (2%), and flushing (1%).
A factor VIII inhibitor was reported in one adult patient, but repeat testing did not confirm the report.
One adult with asthma had a clinical hypersensitivity reaction and a transient increase of IgM anti-PEG antibody titer, which was negative upon retesting.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval for damoctocog alfa pegol, a recombinant human factor VIII therapy.
Bayer is seeking European marketing authorization for damoctocog alfa pegol (formerly BAY94-9027) for the treatment and prophylaxis of bleeding in previously treated patients age 12 and older with hemophilia A.
The CHMP’s recommendation for damoctocog alfa pegol, which is approved in the U.S. under the name Jivi, will be reviewed by the European Commission (EC).
The EC typically makes a decision about marketing authorization within 67 days of the CHMP’s opinion. The EC’s decision will apply to the European Union, Norway, Iceland, and Liechtenstein.
The CHMP’s recommendation for damoctocog alfa pegol is supported by the phase 2/3 PROTECT VIII trial. Some results from this trial were published in the Journal of Thrombosis and Haemostasis in 2016. Additional results are available in the U.S. prescribing information for Jivi.
PROTECT VIII enrolled previously treated adults and adolescents (ages 12 to 65) with severe hemophilia A.
In part A, researchers evaluated different dosing regimens for damoctocog alfa pegol used as prophylaxis and on-demand treatment. An optional extension study was available to patients who completed part A.
In part B, researchers evaluated damoctocog alfa pegol for perioperative management.
Efficacy
In part A, there were 132 patients in the intent‐to‐treat population—112 in the prophylaxis group and 20 in the on-demand group.
Patients received damoctocog alfa pegol for 36 weeks. For the first 10 weeks, patients in the prophylaxis group received twice-weekly dosing at 25 IU/kg.
Patients with more than one bleed during this time went on to receive 30–40 IU/kg twice weekly. Patients with one or fewer bleeds were eligible for randomization to dosing every 5 days (45–60 IU/kg) or every 7 days (60 IU/kg).
The median annualized bleeding rate (ABR) was 4.1 for the patients who were treated twice weekly and were not eligible for randomization (n=13) and 1.9 for patients who were eligible for randomization but continued on twice-weekly treatment (n=11).
The median ABR was 1.9 for patients who were randomized to treatment every 5 days (n=43) and 0.96 for patients who completed prophylaxis with dosing every 7 days (32/43).
The median ABR for patients treated on demand was 24.1.
There were 388 treated bleeds in the on-demand group and 317 treated bleeds in the prophylaxis group. Overall, 73.3% of responses to treatment were considered “excellent” or “good,” 23.3% were considered “moderate,” and 3.3% were considered “poor.”
There were 17 patients who underwent 20 major surgeries in part B or the extension study and 10 patients who underwent minor surgeries in part A. Damoctocog alfa pegol provided “good” or “excellent” hemostatic control during all surgeries.
Safety
Safety data are available for 148 patients age 12 and older.
Adverse events in these patients included abdominal pain (3%), nausea (5%), vomiting (3%), injection site reactions (1%), pyrexia (5%), hypersensitivity (2%), dizziness (2%), headache (14%), insomnia (3%), cough (7%), erythema (1%), pruritus (1%), rash (2%), and flushing (1%).
A factor VIII inhibitor was reported in one adult patient, but repeat testing did not confirm the report.
One adult with asthma had a clinical hypersensitivity reaction and a transient increase of IgM anti-PEG antibody titer, which was negative upon retesting.