User login
Febrile Seizures: Evaluation and Treatment
From the Nationwide Children’s Hospital, Columbus, OH (Dr. Patel) and Cook Children’s Medical Center, Fort Worth, TX (Dr. Perry).
Abstract
- Objective: To review the current understanding and management of febrile seizures.
- Methods: Review of the literature.
- Results: Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as-needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted.
- Conclusion: Providers caring for pediatric patients should be aware of the clinical considerations in managing patients with febrile seizures.
Key words: febrile seizure; Dravat syndrome; GEFS+; PCDH19; FIRES; complex febrile seizure.
A febrile seizure is defined as a seizure in association with a febrile illness in the absence of a central nervous system infection or acute electrolyte imbalance in children older than 1 month of age without prior afebrile seizures [1]. The mechanism by which fever provokes a febrile seizure is unclear [2]. Febrile seizures are the most common type of childhood seizures, affecting 2% to 5% of children [1]. The age of onset is between 6 months and 5 years [3]; peak incidence occurs at about 18 months of age. Simple febrile seizures are the most common type of febrile seizure. By definition, they are generalized, last less than 10 minutes and only occur once in a 24-hour time-period. A complex febrile seizure is one with focal onset or one that occurs more than once during a febrile illness, or lasts more than 10 minutes. Febrile status epilepticus, a subtype of complex febrile seizures, represents about 25% of all episodes of childhood status epilepticus. They account for more than two-thirds of cases during the first 2 years of life.
The risk of reoccurrence after presenting with one febrile seizure is approximately 30%, with the risk being 60% after 2 febrile seizures and 90% after 3 [4–6]. Some families have an autosomal dominant inheritance pattern with polygenic inheritance suspected for the majority of patients presenting with febrile seizures.
Multiple chromosomes have been postulated to be associated with genetic susceptibility for febrile seizures, with siblings having a 25% increased risk and high concordance noted in monozygotic twins [7]. The pathophysiology for febrile seizures has been associated with a genetic risk associated with the rate of temperature rise with animal studies suggesting temperature regulation of c-aminobutyric acid (GABA) a receptors [2]. Other studies propose a link between genetic and environmental factors resulting in an inflammatory process which influences neuronal excitement predisposing one to a febrile seizure [8].
Debate exists between the relation of febrile seizures and childhood vaccinations. Seizures are rare following administration of childhood vaccines. Most seizures following administration of vaccines are simple febrile seizures [9]. Febrile seizures associated with vaccines are more associated with underlying epilepsy. In a study of patients with vaccine-related encephalopathy and febrile status epilepticus, the majority of patients were found to have Dravet syndrome; it was determined that the vaccine may have triggered an earlier onset of the presentation for Dravet in those predestined to develop this disease but did not adversely impact ultimate outcome [10].
In this article, we review simple and complex febrile seizures with a focus on clinical management. Epilepsy syndromes associated with febrile seizures are also discussed. Cases are provided to highlight important clinical considerations.
Case 1: Simple Febrile Seizure
A 9-month-old infant and his mother present to the pediatrician. The mother notes that the infant had an event of concern. She notes the infant had stiffness in all 4 extremities followed by jerking that lasted 30 to 60 seconds. The infant was not responsive during the event. He was sleepy afterward, but returned to normal soon after the event ended. After, she noted that the infant felt warm and she checked his temperature. He had a fever of 101°F. The infant has normal development and no other medical problems.
What are management considerations for simple febrile seizure?
A simple febrile seizure is the most common type of febrile seizure. They are generalized, lasting less than 10 minutes and only occur once in a 24-hour period. There is no increased risk of developing epilepsy or developmental delay for patients after the first simple febrile seizures when compared to other children [5,6]. The diagnosis is based on history provided and a physical examination including evaluation of body temperature [11,12].
No routine laboratory tests are needed as a result of a simple febrile seizure unless obtained to assist in identifying the fever source [3,11]. Routine EEG testing is not recommended for these patients [3,11]. Routine imaging of the brain is also not needed [3,11]. Only if a patient has signs of meningitis should a lumbar puncture be performed [11]. The American Academy of Pediatrics states that a lumbar puncture is strongly considered for those younger than 12 months if they present with their first complex febrile seizure as signs of meningitis may be absent in young children. For infants 6 to 12 months of age, a lumbar puncture can be considered when immunization status is deficient or unknown [13,14]. Also, a lumbar puncture is an option for children who are pretreated with antibiotics [11]. For patients younger than 6 months, data is lacking on the percentage of patients with bacterial meningitis following a simple febrile seizure.
Daily preventative therapy with an anti-epilepsy medication is not necessary [3,11]. A review of several treatment studies shows that some anti-epileptic medications are effective in preventing recurrent simple febrile seizures. Studies have demonstrated the effectiveness of phenobarbital, primidone, and valproic acid in preventing the recurrence of simple febrile seizures; however, the side effects of each medication outweighed the benefit [3]. Carbamazepine and phenytoin have not been shown to be effective in preventing recurrent febrile seizures [3].
For anxious caregivers with children having recurrent febrile seizures, a daily medication or treating with an abortive seizure medication at the time of a febrile illness can be considered [3,5,6,15]. Treating with an abortive medication may mask signs and symptoms of meningitis making evaluation more challenging [16]. Evidence does not support that using antipyretic medications such as acetaminophen or ibuprofen will reduce the recurrence of febrile seizures. The seizure usually is the first noticed symptom due to the rise of temperature being the cause of the febrile seizure in an otherwise well child prior to the seizure [11,17]. Damage to the brain and associated structures is not found with patients presenting with simple febrile seizures [5,6]. Education on all of these principles is strongly recommended for caregiver reassurance.
Case 2: Complex Febrile Seizure
A 1-year-old child presents to the emergency department. Mother was with the child and she noticed stiffness followed by jerking of the left arm and leg, which quickly became noted in both arms and legs. The episode appeared to last for 15 minutes before EMS arrived to the house. A medication was given to the child by EMS that stopped the event. EMS noted the child had a temperature of 101.5°F. The child was previously healthy and has had normal development thus far.
What is the epidemiology of complex febrile seizure?
A complex febrile seizure is one with focal onset, or one that occurs more than once during a febrile illness or lasts more than 10 minutes. They are less common, representing only 20% to 30% of all febrile seizures [18–20]. In The National Collaborative Perinatal Project (NCPP), 1706 children with febrile seizures were identified from 54,000 and were followed from birth until 7 years of age. The initial febrile seizure was defined as complex in about 28%. For all febrile seizures, focal features were present in 4%, prolonged duration (> 10 minutes) in 7.6%, and recurrent episodes within 24 hours in 16.2% [21]. Similar observations have been reported by Berg and Shinnar [5,6]. Of 136 children who had recurrences, 41.2% had one or more complex features and the strongest correlate of having recurrent complex febrile seizure was the number of recurrent seizures. They also found that children with complex recurrences had other recurrences that were not complex; however, complex features had a tendency to recur. Further, a strong association between focal onset and prolonged duration was found [5,6]. Previous studies established a correlation between complex attacks, particularly prolonged ones and young age (age < 1 year) [5,6]. Additionally, children with seizures with a relatively low fever (< 102°F) were slightly more likely to have a complex febrile seizure as the initial episode [5,6].
Children with febrile seizures are already at 4- to 5-fold increased risk for subsequent unprovoked seizures. A history of febrile seizures has been found in 13% to 18% of children with new-onset epilepsy. In the NCPP study, the predictors identified for the development of epilepsy following febrile seizures were an abnormal neurological and developmental status of the child before the seizure, a history of afebrile seizures in a parent or prior-born sibling, or complex features [21]. Ten percent of children with 2 or more of the previously mentioned risk factors (including complex features) developed epilepsy and 13% of them had seizures without fever [20,22]. Further, intractable epilepsy and neurological impair-ment have been found to be more common in children with prior prolonged febrile seizure, with no association to any specific seizure type [18,23–25]. The association between febrile seizures and mesial temporal sclerosis (MTS) is a commonly debated topic. Retrospective studies have reported an association between prolonged or atypical febrile seizures and intractable temporal lobe epilepsy. Epidemiological studies fail to show a causal relationship between febrile seizures and temporal lobe epilepsy [26]. This suggests that febrile seizures are a marker of susceptibility to seizures and future epilepsy (in some cases) rather than a direct cause. It is clear that a minority of cases of MTS or complex partial seizures are associated with prior febrile seizures [20,22].
What is the risk of intracranial pathology in complex febrile seizure?
Patients with complex febrile seizures usually seek medical attention [27]. However, the risk of acute pathology necessitating treatment changes based on neuroimaging was found to be very low and likely not necessary in the evaluation of complex febrile seizures during the acute presentation [27]. Imaging with a high-resolution brain MRI could be considered later on a routine basis for prolonged febrile seizures due to the possible association between prolonged febrile seizures and mesial temporal sclerosis [19,28,29].
Neuroimaging has provided evidence that hippocampal injury can occasionally occur during prolonged and focal febrile seizures in infants who otherwise appear normal. It has been speculated that a pre-existing abnormality increases the propensity to focal prolonged seizures and further hippocampal damage. Hesdorffer and colleagues [30] found definite abnormalities on MRI in 14.8% of children with complex febrile seizures and 11.4 % of simple febrile seizures among 159 children with a first febrile seizure. However, MRI abnormalities were related to a specific subtype of complex seizures: focal and prolonged. The most common abnormalities observed were subcortical focal hyperintensity, an abnormal white matter signal, and focal cortical dysplasia.
What are important aspects of the clinical evaluation?
The evaluation and management of the child with complex febrile seizures is debated as well. The most important part in the history and examination is to look for the source of the fever and rule out the presence of a CNS infection, since complex febrile seizures are much more frequently associated with meningitis than simple febrile seizures [16]. The American Academy of Pediatrics recommended that a lumbar puncture be strongly considered in infants younger than 12 months after a first febrile seizure and should be considered in children between 12 and 18 months of age, since signs of meningitis may be absent in young children [13]. If the threshold for a lumbar puncture is low in infants with febrile seizures in general, it should be even lower for children with complex febrile episodes for all the factors mentioned above. The guidelines developed in 1990 by the Royal College of Physicians and the British Paediatric Association concluded that indications for performing an lumbar puncture were complex febrile seizure, signs of meningismus, or a child who is unduly drowsy and irritable or systematically ill [21].
Obtaining an EEG within 24 hours of presentation may show generalized background slowing, which could make identifying possible epileptiform abnormalities difficult [22]. Therefore, a routine sleep deprived EEG when the child is back to baseline can be more useful in identifying if epileptiform abnormalities are present. If epileptiform abnormalities are present on a routine sleep deprived EEG, this may suggest the patient is at higher risk for developing future epilepsy and the febrile illness lowered the seizure threshold; however, it is unclear whether clinical management would change as a result [31].
What treatment options are available?
Complications with prolonged and/or recurrent seizures can occur. Treatments options can be stratified into 3 possible categories: emergency rescue treatment for prolonged or a cluster of febrile seizures, intermittent treatment at the time of illness, and chronic use of medication. Treatment options for complex febrile seizures may include the use of a rescue seizure medication when the febrile seizure is prolonged. Rectal preparations of diazepam gel can be effective in stopping an ongoing seizure and can be provided for home use in patients with known recurrence of febrile status epilepticus [3]. For children and adolescents where a rectal administration is not ideal, intranasal versed can be utilized instead of rectal diazepam. In addition, the use of an intermittent benzodiazepine at the onset of febrile illness can also be considered a treatment option. Using oral diazepam at the time of a febrile illness has been demonstrated in reducing the recurrence of febrile seizures [3]. Other studies have shown similar results when using buccal midazolam [32]. No adequate studies have been performed using second- or third-generation anti-epilepsy medications in the treatment of recurrent of complex febrile seizures [3].
It is unclear whether benefit is present to using intermittent benzodiazepine doses prior or during a febrile illness for those prone for recurrent febrile seizures [33]. Physicians may consider this option in patients with frequent recurrent seizures, when caregivers can identify the fever before the seizure occurs.
Overall, parental education of efficacy and side effect profiles should be discussed in detail when considering any treatment options for complex febrile seizures [34]. It is important to remember that the long-term prognosis in terms of developing epilepsy or neurological and cognitive problems is not influenced by the use of antiepileptic medications for recurrent febrile seizures [17]. Even in the case of prolonged febrile seizures in otherwise neurodevelopmentally normal children, antiepileptics have not been shown to cause damage to the brain [19].
Febrile Status Epilepticus
Febrile status epilepticus is a subtype of complex febrile seizures and is defined as a febrile seizure lasting greater than 30 minutes. Overall, febrile status epilepticus accounts for approximately 5% of all presentations of febrile seizures [35]. It represents about 25% of all episodes of childhood status epilepticus and more than two-thirds of cases during the first 2 years of life. Literature suggests that an increased risk for focal epilepsy exists [36]. Children presenting with febrile status epilepticus are more likely to have a family history of epilepsy and a history of a previous neurological abnormality [22]. It is likely to reoccur if the first presentation was febrile status epilepticus. However, increased risk for death or developmental disability as a result of the seizure is not seen [37].
The prospective multicenter study of the consequences of prolonged febrile seizures in childhood (FEBSTAT) has been conducted. The study reported that febrile status epilepticus is usually focal (67% of episodes), occurs in very young children (median age 1.3 years), and is frequently the first febrile seizure [22]. In this study, the median duration of the seizure was about 68 minutes and 24% of children had an episode lasting more than 2 hours. In 87% of the events, seizures did not stop spontaneously and benzodiazepines were needed. Focal features observed were eye and head deviation, staring, and impaired consciousness prior to the seizure and an asymmetric convulsion or Todd’s paresis.
Case 3: Epilepsy Syndromes Associated With Febrile Seizures
A 1-year-old female presents for evaluation of seizures that began at age 8 months. Seizures are described as occurring in the setting of fever with bilateral symmetric tonic clonic activity lasting durations of less than 10 minutes on average, but at least 2 instances of seizure lasting 20 minutes or more. The family notes that seizures have occurred almost every time the child has had a febrile illness and often cluster over several days. They report at least 1 seizure that occurred in the absence of fever. Development has been normal to date and an EEG done by their primary provider was also normal.
What epilepsy syndromes are associated with febrile seizures?
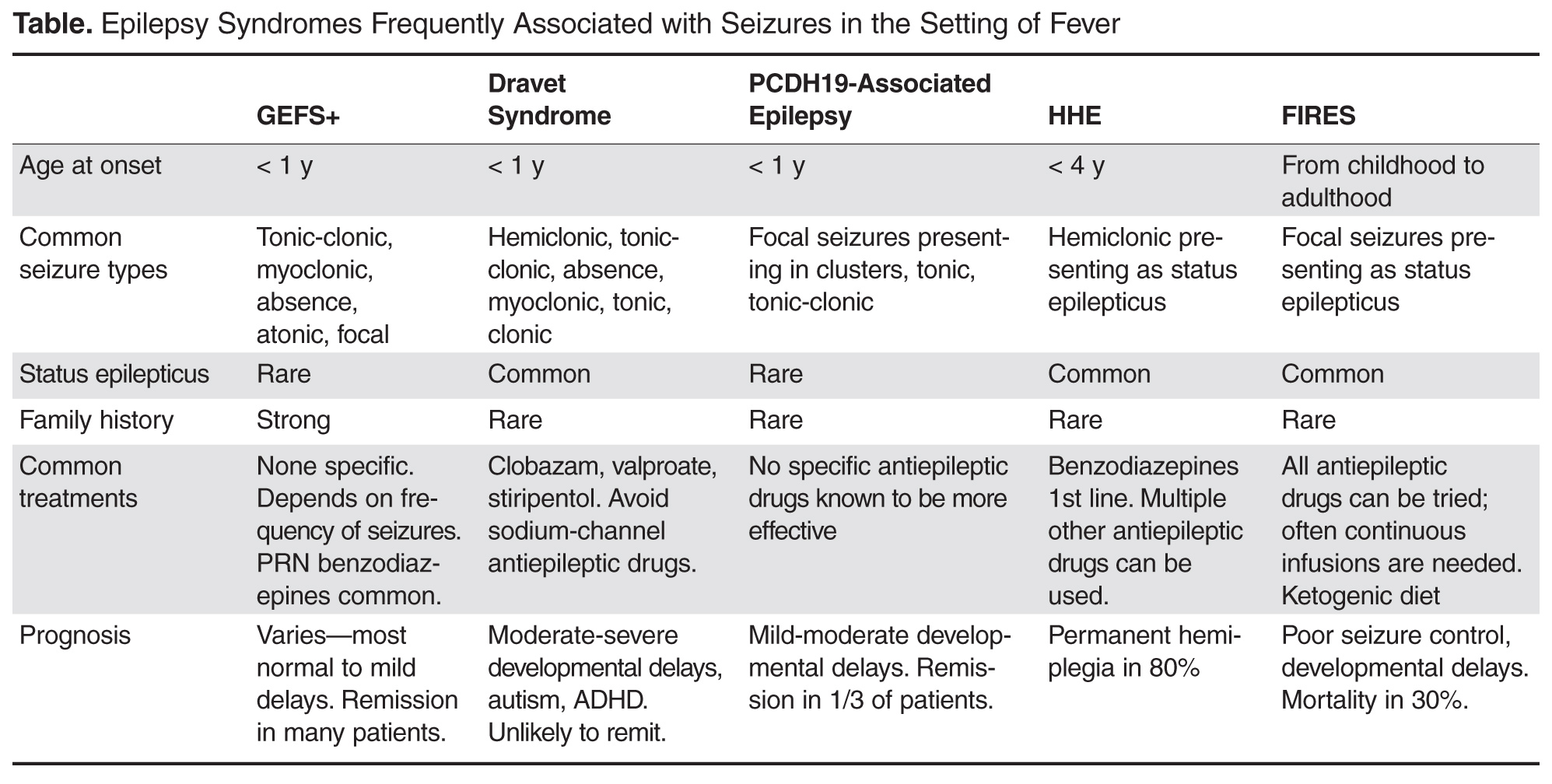
Genetic Epilepsy with Febrile Seizures Plus
GEFS+ was first described in 1997 following recognition of a pattern of febrile seizures followed later by the development of various epilepsy syndromes within the same family [38]. As such, the syndrome is defined based on the familial occurrence of febrile and afebrile seizures in at least 2 family members and can have a wide range of phenotypes. The most common presentation is of typical febrile seizures which can persist beyond the typical upper age limit of 6 years. Unprovoked generalized seizures of multiple types (ie, myoclonic, absence, atonic) occur at a later age, though focal seizures may also be present. The presence of focal onset seizures led to the naming change from “generalized” epilepsy with febrile seizures plus as it was previously referred. Seizure frequency and severity may vary between family members, as can response to treatment, making prognosis difficult to predict. As even in typical febrile seizures a family history of febrile seizure may be common, it may be difficult to diagnose the syndrome after the initial febrile seizure. However, if the family history is strong for a family member with a GEFS+ phenotype, one can appropriately counsel the family on the possibility that a similar course may evolve. While the majority of GEFS+ patients have milder phenotypes, some more severe phenotypes can have cognitive delays. Dravet syndrome falls within the spectrum of GEFS+ and is a prime example of the phenotypic continuum to more severe presentations in some patients.
The syndrome is believed to be inherited in an auto-somal dominant fashion with incomplete penetrance. Multiple genes have been implicated as a cause, though only 11.5% of families with clinical GEFS+ may have mutations [39]. SCN1A, encoding the α-subunit of the voltage-gated sodium channel is most frequently reported in GEFS+ families, yet is only found in 10% [38]. When associated with GEFS+, SCN1A mutations are more often missense type, whereas truncating and nonsense mutations are more commonly encountered in Dravet syndrome. Mutations in SCN1B encoding the β1 subunit of the voltage-gated sodium channel has also been reported [40]. Finally, the GABA(A) receptor gamma 2 subunit GABRG2 has been found in < 1% of GEFS+ families [39]. The variability in causative genes underscores the reasons for phenotype variability and it is likely that other modifier genes are responsible for the heterogeneity within GEFS+ families [41].
Dravet Syndrome
Dravet syndrome, often referred to as severe myoclonic epilepsy of infancy, was first described in 1978 and has since become one of the most recognized genetic epilepsy syndromes [42]. The clinical presentation often begins with seizures in the first year of life, frequently in the setting of febrile illness. The initial seizures are generalized or hemiclonic in the majority and are often prolonged evolving to status epilepticus. Unlike typical febrile seizures, one should suspect Dravet syndrome in children that present with repetitive bouts of complex febrile seizures or febrile status epilepticus, especially if the associated seizure semiology is of hemiclonic type. In addition, seizures in the setting of modest hyperthermia (ie, hot baths) should raise suspicion for this condition. Commonly EEG and MRI are normal in the first year of life and psychomotor development remains normal until typically the second year of life [43].
By the second year, other seizure types including myoclonic, atypical absence, clonic, and tonic seizures arise. The EEG frequently begins to show generalized spike wave and polyspike wave discharges. Seizures continue occurring frequently during early childhood, often resulting in status epilepticus. Cognitive development begins to stagnate between the ages of 1 and 4 years with emergence of autistic traits and hyperactivity [44]. Development may stabilize between the ages of 5 and 16 years, but fails to demonstrate much improvement [44]. Higher frequency of seizures may correlate with increase in cognitive impairment and behavior problems, supporting the need for rapid diagnosis and appropriate therapy [44].
Over the years, several cases of atypical or borderline Dravet syndrome have been described, most highlighting the absence of myoclonic seizures [45]. Others may present with primarily clonic or tonic-clonic type seizures only [46]. Despite these differences, all cases share a similar drug resistance and cognitive delay and are categorized as Dravet syndrome.
In 2001, Claus et al discovered the genetic alteration in SCN1A responsible for 70% of Dravet syndrome cases [47]. The disorder is inherited in an autosomal dominant fashion, though 40% to 80% of mutations resulting in Dravet syndrome are de novo [48]. Mutations can be present in other family members, as this syndrome is part of the spectrum of GEFS+, though parental phenotypes are often much less severe. Approximately 50% of mutations resulting in Dravet syndrome are truncating, while the other 50% are missense mutations involving splice site or pore forming regions leading to loss of function [49]. Finally, small and large chromosome rearrangements make up 2% to 3% of cases [50]. Other genes reported to result in Dravet syndrome include SCN1B and GABRG2 mutations. In addition, PCDH19 can produce a phenotype similar to Dravet syndrome in females and is discussed in more detail below.
With the emergence of more rapid and cheaper forms of genetic testing, molecular diagnosis can now be made earlier in life before all the typical clinical features of Dravet syndrome arise. As a result, one might hope to alter treatment strategy and gear therapy towards the most effective medications. While drug resistance is the norm for the condition, certain drugs such as benzodiazepines, valproate, and stiripentol may be most effective [43]. Topiramate and levetiracetam have been reported as effi-cacious in small series, as has the ketogenic diet [51–55]. Varieties of medications which target sodium channels are known to exacerbate seizures in Dravet syndrome and should be avoided, including lamotrigine, carbamazepine, oxcarbazepine, and phenytoin [56]. In addition to maintenance therapy, it is important to provide patients with a rescue plan for acute seizures in an effort to avoid status epilepticus. In addition, measures to avoid overheating may provide additional benefit.
Case 3 Continued
After a careful history, the physician discovers that the child also has frequent myoclonic seizures described as brief jerks of the extremities or sudden forward falls. The family notes they have seen these seizures more frequently since antiepileptic therapy was started. The physician recognize that this child may have Dravet syndrome and suspect her medication may be resulting in aggravation of seizures.
The physician decides to discontinue the medication suspected to aggravate the seizures and chooses to start the child on clobazam. The physician also begins evaluation for Dravet syndrome by sending directed SCN1A genetic testing. The testing comes back negative for mutations in the SCN1A gene.
What other investigations would be warranted now?
PCDH19
PCDH19 was first recognized as a cause of epilepsy and mental retardation limited to females (EFMR), a syndrome characterized by onset of seizures in infancy or early childhood with predominantly generalized type seizures including tonic-clonic, absence, myoclonic, tonic, and atonic [57]. Since that initial description, the phenotype associated with PCDH19 mutations has expanded to include female patients with primarily focal epilepsy, variable cognitive impairment, and commonly onset with seizures in the setting of fever [58,59]. Typically seizures begin around age 10 months presenting as a cluster of focal seizures in the setting of fever, often followed by a second cluster 6 months later [59]. Generalized seizures occur in a small proportion of patients (9%) and this feature, along with relatively fewer bouts of status epilepticus and less frequent seizures (most monthly to yearly frequency) can differentiate PCDH19 associated epilepsy from Dravet syndrome [59]. Seizures tend to improve with age and no particular antiepileptic drug has been found especially efficacious in the syndrome. Unlike Dravet syndrome, up to a third of patients with this syndrome may ultimately become seizure-free [59].
Cognitive development is normal prior to seizure onset in the majority of patients and most but not all patients will develop some cognitive impairment ranging from mild to severe [59]. It is the more severe patients that most often have overlapping characteristics of Dravet syndrome, thus PCDH19 mutations should be investigated in female patients with Dravet phenotype yet negative SCN1A testing.
PCDH19 is a calcium-dependent adhesion protein involved in neuronal circuit formation during development and in the maintenance of normal synaptic circuits in adulthood [60,61]. Disease causing mutations in PCDH19 are primarily missense (48.5%) or frameshift, nonsense, and splice-site mutations resulting in premature termination codon [59]. Ninety percent of mutations are de novo. When inherited, the disorder is X-linked and may come from an unaffected father or a mother that is similarly affected or not, suggesting variable clinical severity in females and gender-related protections in males [59].
Case 3 Continued
Given the negative SCN1A testing, the physician chooses to pursue other genetic testing that may explain the patient’s phenotype. A more extensive “epilepsy gene panel” that includes 70 different genes associated with epilepsy syndromes is ordered.
Hemiconvulsion-Hemiplegia Epilepsy Syndrome
Hemiconvulsion-hemiplegia epilepsy syndrome (HHE) is characterized by the occurrence of unilateral convulsive status epilepticus followed by transient or permanent ipsilateral hemiplegia. The syndrome occurs in otherwise healthy children often in the setting of nonspecific febrile illness before the age of 4 years, with peak occurrence in the first 2 years of life [62]. Seizures are characterized by unilateral clonic activity with EEG demonstrating rhythmic 2–3 Hz slow wave activity and spikes in the hemisphere contralateral to the body involvement. MRI frequently demonstrates diffusion changes congruent with EEG findings, often in the perisylvian region. The hemiplegia that remains following status epilepticus is permanent in up to 80% of cases [63]. As hemiplegia can occur following complex febrile seizures, it is recommended a minimum duration of hemiplegia of 1 week be used to differentiate HHE [64]. Status epilepticus is persistent in this syndrome and can last for hours if untreated. Focal onset seizures will often continue to occur in the patient even after the status has been aborted.
The etiology of HHE is variable with many cases idiopathic. Some cases are reported as symptomatic, as the syndrome can present in the setting of other underlying brain disorders such as Sturge-Weber and tuberous sclerosis complex. While some viruses have been proposed as a cause, they are not found in the cerebral spinal fluid of patients [65]. Treatment consists of rapid treatment of status epilepticus with benzodiazepines as first-line therapy, often followed by other intravenous antiepileptic drugs as necessary.
Febrile Infection–Related Epilepsy Syndrome
Febrile infection–related epilepsy syndrome (FIRES) is presented under several names in the literature including idiopathic catastrophic epileptic encephalopathy [66], devastating encephalopathy in school-age children [67], new-onset refractory status epilepticus [68], as well as fever-induced refractory epileptic encephalopathy syndrome [69] and fever-induced refractory epileptic encephalopathy in school-age children [70]. All describe rare catastrophic epilepsy presenting in otherwise healthy children during or days following a febrile illness. While febrile illness precedes the epilepsy in 96% of cases, up to 50% of patients may not have fever at the time they present [41,65]. While age of onset is typically in early childhood, presentation in adulthood also occurs. Initial seizures are often focal, presenting as forced lateral head or eye deviation, oral or manual automatisms, and clonic movements of the face and extremities. Seizures will inevitably progress to status epilepticus with ictal onset often multifocal predominating in the perisylvian regions [41]. MRI is often normal at onset or shows only subtle swelling of the mesial temporal structures. Over months, MRI often shows T2-hyperintensity and atrophy of the mesial temporal structures, though as many as 50% of MRIs may remain normal [71].
The evaluation for cause in FIRES is often unrewarding. Inflammatory markers are typically absent from both serum and CSF. CSF may show minimal pleocytosis with negative oligoclonal bands and absence of common receptor antibodies. Treatment is equally unrewarding with patients typically failing conventional antiepileptic drugs and continuous infusions titrated to burst suppression. Immunomodulatory therapies are mostly ineffective as well. The most useful therapy reported has been the keto-genic diet with efficacy in up to 50% of patients [72]. Recently, therapeutic hypothermia has also been reported to be effective in 2 cases [73]. For the majority of patients, therapy will remain ineffective and seizures will continue for weeks to months with gradual resolution, though seizures often continue intermittently following the end of status epilepticus. Prognosis is poor for seizure control and neurocognitive recovery with mortality of 30% reported [41].
Case 3 Conclusion
The epilepsy gene panel ordered returns with the result of a disease-causing mutation in the PCDH19 gene. The child is diagnosed with PCDH19-associated epilepsy and is treated with phenobarbital. For the first years of life, she presents on average once per year with a cluster of seizures in the setting of febrile illness which is often managed with short durations of scheduled benzodiazepines. Seizures slow by age 6. She has mild delays in speech and receives some accommodations through her school system. By age 10, she has been seizure-free for several years. She is able to be weaned off medications without recurrence of seizures.
Summary
Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted. Providers should have a high index of suspicion for these syndromes when they encounter children that repeatedly present with prolonged febrile seizures, clusters of febrile seizures, or febrile seizures in addition to afebrile seizure events. Early referral, diagnosis, and treatment has the potential to alter outcome in some of these syndromes, thus the importance of becoming familiar with these diagnoses.
Corresponding author: Anup D. Patel, MD, Nationwide Children's Hospital, Columbus, OH 43205, anup.patel@nationwidechildrens.org.
Financial disclosures: Dr. Patel disclosed that he has consulted for GW Pharmaceuticals and Supernus and is on the Scientific Advisory Board for UCB Pharma.
1. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002;17 Suppl 1:S44–52.
2. Kang J-Q, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci 2006;26:2590–7.
3. Baumann RJ, Duffner PK. Treatment of children with simple febrile seizures: the AAP practice parameter. American Academy of Pediatrics. Pediatr Neurol 2000;23:11–7.
4. Tarkka R, Rantala H, Uhari M, Pokka T. Risk of recurrence and outcome after the first febrile seizure. Pediatr Neurol 1998;18:218–20.
5. Berg AT, Shinnar S. Complex febrile seizures. Epilepsia 1996;37:126–33.
6. Berg AT, Shinnar S. Unprovoked seizures in children with febrile seizures: short-term outcome. Neurology 1996;47:562–8.
7. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006;67:687–90.
8. Dube CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev 2009;31:366–71.
9. Vestergaard M, Christensen J. Register-based studies on febrile seizures in Denmark. Brain Dev 2009;31:372–7.
10. Berkovic SF, Petrou S. Febrile seizures: traffic slows in the heat. Trends Molecul Med 2006;12:343–4.
11. Practice parameter: the neurodiagnostic evaluation of the child with a first simple febrile seizure. American Academy of Pediatrics. Provisional Committee on Quality Improvement, Subcommittee on Febrile Seizures. Pediatrics 1996;97:769–72; discussion 773–765.
12. Fukuyama Y, Seki T, Ohtsuka C, et al. Practical guidelines for physicians in the management of febrile seizures. Brain Dev 1996;18:479–84.
13. Neurodiagnostic evaluation of the child with a simple febrile seizure. Pediatrics 2011;127:389–94.
14. Guedj R, Chappuy H, Titomanlio L, et al. Risk of bacterial meningitis in children 6 to 11 months of age with a first simple febrile seizure: a retrospective, cross-sectional, observational study. Acad Emerg Med 2015;22:1290–7.
15. Wo SB, Lee JH, Lee YJ, et al. Risk for developing epilepsy and epileptiform discharges on EEG in patients with febrile seizures. Brain Dev 2013;35:307–11.
16. Green SM, Rothrock SG, Clem KJ, et al. Can seizures be the sole manifestation of meningitis in febrile children? Pediatrics 1993;92:527–34.
17. Knudsen FU. Febrile seizures: treatment and prognosis. Epilepsia 2000;41:2–9.
18. Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 1987;316:493–8.
19. Teng D, Dayan P, Tyler S, et al. Risk of intracranial pathologic conditions requiring emergency intervention after a first complex febrile seizure episode among children. Pediatrics 2006;117:304–8.
20. Janszky J, Schulz R, Ebner A. Clinical features and surgical outcome of medial temporal lobe epilepsy with a history of complex febrile convulsions. Epilepsy Res 2003;55:1–8.
21. Capovilla G, Mastrangelo M, Romeo A, Vigevano F. Recommendations for the management of «febrile seizures»: Ad Hoc Task Force of LICE Guidelines Commission. Epilepsia 2009;50 Suppl 1:2–6.
22. Shinnar S, Hesdorffer DC, Nordli DR Jr, et al. Phenomenology of prolonged febrile seizures: results of the FEBSTAT study. Neurology 2008;71:170–6.
23. Camfield P, Camfield C, Gordon K, Dooley J. What types of epilepsy are preceded by febrile seizures? A population-based study of children. Dev Med Child Neurol 1994;36:887–92.
24. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976;295:1029–33.
25. Hamati-Haddad A, Abou-Khalil B. Epilepsy diagnosis and localization in patients with antecedent childhood febrile convulsions. Neurology 1998;50:917–22.
26. Davies KG, Hermann BP, Dohan FC Jr, et al. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 1996;24:119–26.
27. Kimia AA, Ben-Joseph E, Prabhu S, et al. Yield of emergent neuroimaging among children presenting with a first complex febrile seizure. Pediatr Emerg Care 2012;28:316–21.
28. Abou-Khalil B, Andermann E, Andermann F, et al. Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia 1993;34:878–83.
29. Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol 2004;17:161–4.
30. Hesdorffer DC, Chan S, Tian H, et al. Are MRI-detected brain abnormalities associated with febrile seizure type? Epilepsia 2008;49:765–71.
31. Patel AD, Vidaurre J. Complex febrile seizures: a practical guide to evaluation and treatment. J Child Neurol 2013;28:762–67.
32. Pavlidou E, Tzitiridou M, Panteliadis C. Effectiveness of intermittent diazepam prophylaxis in febrile seizures: long-term prospective controlled study. J Child Neurol 2006;21:1036–40.
33. Offringa M, Newton R. Prophylactic drug management for febrile seizures in children (Review). Evid Based Child Health 2013;8:1376–485.
34. Gordon KE, Dooley JM, Camfield PR, et al. Treatment of febrile seizures: the influence of treatment efficacy and side-effect profile on value to parents. Pediatrics 2001;108:1080–8.
35. Maytal J, Shinnar S. Febrile status epilepticus. Pediatrics 1990;86:611–6.
36. Ahmad S, Marsh ED. Febrile status epilepticus: current state of clinical and basic research. Semin Pediatr Neurol 2010;17:150–4.
37. Maytal J, Shinnar S, Moshe SL, Alvarez LA. Low morbidity and mortality of status epilepticus in children. Pediatrics 1989;83:323–31.
38. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120 (Pt 3):479–90.
39. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–85.
40. Wallace RH, Scheffer IE, Parasivam G, et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 2002;58:1426–9.
41. Cross JH. Fever and fever-related epilepsies. Epilepsia 2012;53 Suppl 4:3–8.
42. Dravet C. Les epilepsies graves de l’enfant. Vie Med 1978;8:543–8.
43. Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53 Suppl 2:1–6.
44. Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 2006;47 Suppl 2:45–8.
45. Ogino T, Ohtsuka Y, Amano R, et al. An investigation on the borderland of severe myoclonic epilepsy in infancy. Jap J Psych Neurol 1988;42:554–5.
46. Kanazawa O. Refractory grand mal seizures with onset during infancy including severe myoclonic epilepsy in infancy. Brain Dev 2001;23:749–56.
47. van der Worp HB, Claus SP, Bar PR, et al. Reproducibility of measurements of cerebral infarct volume on CT scans. Stroke 2001;32:424–30.
48. Wang JW, Kurahashi H, Ishii A, et al. Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia 2008;49:1528–34.
49. Madia F, Striano P, Gennaro E, et al. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology 2006;67:1230–5.
50. Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–8.
51. Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002;49:45–8.
52. Nieto-Barrera M, Candau R, Nieto-Jimenez M, et al. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure 2000;9:590–4.
53. Striano P, Coppola A, Pezzella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007;69:250–4.
54. Caraballo RH, Cersosimo RO, Sakr D, et al. Ketogenic diet in patients with Dravet syndrome. Epilepsia 2005;46:1539–44.
55. Kang HC, Kim YJ, Kim DW, Kim HD. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia 2005;46:272–9.
56. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011;53 Suppl 2:16–8.
57. Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81.
58. Specchio N, Marini C, Terracciano A, et al. Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 2011;52:1251–7.
59. Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–9.
60. Hirano S, Yan Q, Suzuki ST. Expression of a novel protocadherin, OL-protocadherin, in a subset of functional systems of the developing mouse brain. J Neurosci 1999;19:995–1005.
61. Kim SY, Chung HS, Sun W, Kim H. Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 2007;147:996–1021.
62. Gastaut H, Poirier F, Payan H, et al. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia 1960;1:418–47.
63. Panayiotopoulos CP. The epilepsies: seizures, syndromes and management. Oxfordshire (UK): Bladon Medical Publishing; 2005.
64. Chauvel P, Dravet C. The HHE syndrome. In: Roger J, Bureau M, Dravet C, editors. Epileptic syndromes in infancy, childhood and adolescence. 4th ed. John Libbey; 2005; 247–60.
65. Nabbout R. FIRES and IHHE: Delineation of the syndromes. Epilepsia 2013;54 Suppl 6:54–6.
66. Baxter P, Clarke A, Cross H, et al. Idiopathic catastrophic epileptic encephalopathy presenting with acute onset intractable status. Seizure 2003;12:379–87.
67. Mikaeloff Y, Jambaque I, Hertz-Pannier L, et al. Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis. Epilepsy Res 2006;69:67–79.
68. Wilder-Smith EP, Lim EC, Teoh HL, et al. The NORSE (new-onset refractory status epilepticus) syndrome: defining a disease entity. Ann Acad Med Singapore 2005;34:417–20.
69. van Baalen A, Hausler M, Boor R, et al. Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood. Epilepsia 2010;51:1323–8.
70. Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011;10:99–108.
71. Howell KB, Katanyuwong K, Mackay MT, et al. Long-term follow-up of febrile infection-related epilepsy syndrome. Epilepsia 2012;53:101–10.
72. Nabbout R, Mazzuca M, Hubert P, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010;51:2033–7.
73. Lin JJ, Lin KL, Hsia SH, Wang HS. Therapeutic hypothermia for febrile infection-related epilepsy syndrome in two patients. Pediatr Neurol 2012;47:448–50.
From the Nationwide Children’s Hospital, Columbus, OH (Dr. Patel) and Cook Children’s Medical Center, Fort Worth, TX (Dr. Perry).
Abstract
- Objective: To review the current understanding and management of febrile seizures.
- Methods: Review of the literature.
- Results: Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as-needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted.
- Conclusion: Providers caring for pediatric patients should be aware of the clinical considerations in managing patients with febrile seizures.
Key words: febrile seizure; Dravat syndrome; GEFS+; PCDH19; FIRES; complex febrile seizure.
A febrile seizure is defined as a seizure in association with a febrile illness in the absence of a central nervous system infection or acute electrolyte imbalance in children older than 1 month of age without prior afebrile seizures [1]. The mechanism by which fever provokes a febrile seizure is unclear [2]. Febrile seizures are the most common type of childhood seizures, affecting 2% to 5% of children [1]. The age of onset is between 6 months and 5 years [3]; peak incidence occurs at about 18 months of age. Simple febrile seizures are the most common type of febrile seizure. By definition, they are generalized, last less than 10 minutes and only occur once in a 24-hour time-period. A complex febrile seizure is one with focal onset or one that occurs more than once during a febrile illness, or lasts more than 10 minutes. Febrile status epilepticus, a subtype of complex febrile seizures, represents about 25% of all episodes of childhood status epilepticus. They account for more than two-thirds of cases during the first 2 years of life.
The risk of reoccurrence after presenting with one febrile seizure is approximately 30%, with the risk being 60% after 2 febrile seizures and 90% after 3 [4–6]. Some families have an autosomal dominant inheritance pattern with polygenic inheritance suspected for the majority of patients presenting with febrile seizures.
Multiple chromosomes have been postulated to be associated with genetic susceptibility for febrile seizures, with siblings having a 25% increased risk and high concordance noted in monozygotic twins [7]. The pathophysiology for febrile seizures has been associated with a genetic risk associated with the rate of temperature rise with animal studies suggesting temperature regulation of c-aminobutyric acid (GABA) a receptors [2]. Other studies propose a link between genetic and environmental factors resulting in an inflammatory process which influences neuronal excitement predisposing one to a febrile seizure [8].
Debate exists between the relation of febrile seizures and childhood vaccinations. Seizures are rare following administration of childhood vaccines. Most seizures following administration of vaccines are simple febrile seizures [9]. Febrile seizures associated with vaccines are more associated with underlying epilepsy. In a study of patients with vaccine-related encephalopathy and febrile status epilepticus, the majority of patients were found to have Dravet syndrome; it was determined that the vaccine may have triggered an earlier onset of the presentation for Dravet in those predestined to develop this disease but did not adversely impact ultimate outcome [10].
In this article, we review simple and complex febrile seizures with a focus on clinical management. Epilepsy syndromes associated with febrile seizures are also discussed. Cases are provided to highlight important clinical considerations.
Case 1: Simple Febrile Seizure
A 9-month-old infant and his mother present to the pediatrician. The mother notes that the infant had an event of concern. She notes the infant had stiffness in all 4 extremities followed by jerking that lasted 30 to 60 seconds. The infant was not responsive during the event. He was sleepy afterward, but returned to normal soon after the event ended. After, she noted that the infant felt warm and she checked his temperature. He had a fever of 101°F. The infant has normal development and no other medical problems.
What are management considerations for simple febrile seizure?
A simple febrile seizure is the most common type of febrile seizure. They are generalized, lasting less than 10 minutes and only occur once in a 24-hour period. There is no increased risk of developing epilepsy or developmental delay for patients after the first simple febrile seizures when compared to other children [5,6]. The diagnosis is based on history provided and a physical examination including evaluation of body temperature [11,12].
No routine laboratory tests are needed as a result of a simple febrile seizure unless obtained to assist in identifying the fever source [3,11]. Routine EEG testing is not recommended for these patients [3,11]. Routine imaging of the brain is also not needed [3,11]. Only if a patient has signs of meningitis should a lumbar puncture be performed [11]. The American Academy of Pediatrics states that a lumbar puncture is strongly considered for those younger than 12 months if they present with their first complex febrile seizure as signs of meningitis may be absent in young children. For infants 6 to 12 months of age, a lumbar puncture can be considered when immunization status is deficient or unknown [13,14]. Also, a lumbar puncture is an option for children who are pretreated with antibiotics [11]. For patients younger than 6 months, data is lacking on the percentage of patients with bacterial meningitis following a simple febrile seizure.
Daily preventative therapy with an anti-epilepsy medication is not necessary [3,11]. A review of several treatment studies shows that some anti-epileptic medications are effective in preventing recurrent simple febrile seizures. Studies have demonstrated the effectiveness of phenobarbital, primidone, and valproic acid in preventing the recurrence of simple febrile seizures; however, the side effects of each medication outweighed the benefit [3]. Carbamazepine and phenytoin have not been shown to be effective in preventing recurrent febrile seizures [3].
For anxious caregivers with children having recurrent febrile seizures, a daily medication or treating with an abortive seizure medication at the time of a febrile illness can be considered [3,5,6,15]. Treating with an abortive medication may mask signs and symptoms of meningitis making evaluation more challenging [16]. Evidence does not support that using antipyretic medications such as acetaminophen or ibuprofen will reduce the recurrence of febrile seizures. The seizure usually is the first noticed symptom due to the rise of temperature being the cause of the febrile seizure in an otherwise well child prior to the seizure [11,17]. Damage to the brain and associated structures is not found with patients presenting with simple febrile seizures [5,6]. Education on all of these principles is strongly recommended for caregiver reassurance.
Case 2: Complex Febrile Seizure
A 1-year-old child presents to the emergency department. Mother was with the child and she noticed stiffness followed by jerking of the left arm and leg, which quickly became noted in both arms and legs. The episode appeared to last for 15 minutes before EMS arrived to the house. A medication was given to the child by EMS that stopped the event. EMS noted the child had a temperature of 101.5°F. The child was previously healthy and has had normal development thus far.
What is the epidemiology of complex febrile seizure?
A complex febrile seizure is one with focal onset, or one that occurs more than once during a febrile illness or lasts more than 10 minutes. They are less common, representing only 20% to 30% of all febrile seizures [18–20]. In The National Collaborative Perinatal Project (NCPP), 1706 children with febrile seizures were identified from 54,000 and were followed from birth until 7 years of age. The initial febrile seizure was defined as complex in about 28%. For all febrile seizures, focal features were present in 4%, prolonged duration (> 10 minutes) in 7.6%, and recurrent episodes within 24 hours in 16.2% [21]. Similar observations have been reported by Berg and Shinnar [5,6]. Of 136 children who had recurrences, 41.2% had one or more complex features and the strongest correlate of having recurrent complex febrile seizure was the number of recurrent seizures. They also found that children with complex recurrences had other recurrences that were not complex; however, complex features had a tendency to recur. Further, a strong association between focal onset and prolonged duration was found [5,6]. Previous studies established a correlation between complex attacks, particularly prolonged ones and young age (age < 1 year) [5,6]. Additionally, children with seizures with a relatively low fever (< 102°F) were slightly more likely to have a complex febrile seizure as the initial episode [5,6].
Children with febrile seizures are already at 4- to 5-fold increased risk for subsequent unprovoked seizures. A history of febrile seizures has been found in 13% to 18% of children with new-onset epilepsy. In the NCPP study, the predictors identified for the development of epilepsy following febrile seizures were an abnormal neurological and developmental status of the child before the seizure, a history of afebrile seizures in a parent or prior-born sibling, or complex features [21]. Ten percent of children with 2 or more of the previously mentioned risk factors (including complex features) developed epilepsy and 13% of them had seizures without fever [20,22]. Further, intractable epilepsy and neurological impair-ment have been found to be more common in children with prior prolonged febrile seizure, with no association to any specific seizure type [18,23–25]. The association between febrile seizures and mesial temporal sclerosis (MTS) is a commonly debated topic. Retrospective studies have reported an association between prolonged or atypical febrile seizures and intractable temporal lobe epilepsy. Epidemiological studies fail to show a causal relationship between febrile seizures and temporal lobe epilepsy [26]. This suggests that febrile seizures are a marker of susceptibility to seizures and future epilepsy (in some cases) rather than a direct cause. It is clear that a minority of cases of MTS or complex partial seizures are associated with prior febrile seizures [20,22].
What is the risk of intracranial pathology in complex febrile seizure?
Patients with complex febrile seizures usually seek medical attention [27]. However, the risk of acute pathology necessitating treatment changes based on neuroimaging was found to be very low and likely not necessary in the evaluation of complex febrile seizures during the acute presentation [27]. Imaging with a high-resolution brain MRI could be considered later on a routine basis for prolonged febrile seizures due to the possible association between prolonged febrile seizures and mesial temporal sclerosis [19,28,29].
Neuroimaging has provided evidence that hippocampal injury can occasionally occur during prolonged and focal febrile seizures in infants who otherwise appear normal. It has been speculated that a pre-existing abnormality increases the propensity to focal prolonged seizures and further hippocampal damage. Hesdorffer and colleagues [30] found definite abnormalities on MRI in 14.8% of children with complex febrile seizures and 11.4 % of simple febrile seizures among 159 children with a first febrile seizure. However, MRI abnormalities were related to a specific subtype of complex seizures: focal and prolonged. The most common abnormalities observed were subcortical focal hyperintensity, an abnormal white matter signal, and focal cortical dysplasia.
What are important aspects of the clinical evaluation?
The evaluation and management of the child with complex febrile seizures is debated as well. The most important part in the history and examination is to look for the source of the fever and rule out the presence of a CNS infection, since complex febrile seizures are much more frequently associated with meningitis than simple febrile seizures [16]. The American Academy of Pediatrics recommended that a lumbar puncture be strongly considered in infants younger than 12 months after a first febrile seizure and should be considered in children between 12 and 18 months of age, since signs of meningitis may be absent in young children [13]. If the threshold for a lumbar puncture is low in infants with febrile seizures in general, it should be even lower for children with complex febrile episodes for all the factors mentioned above. The guidelines developed in 1990 by the Royal College of Physicians and the British Paediatric Association concluded that indications for performing an lumbar puncture were complex febrile seizure, signs of meningismus, or a child who is unduly drowsy and irritable or systematically ill [21].
Obtaining an EEG within 24 hours of presentation may show generalized background slowing, which could make identifying possible epileptiform abnormalities difficult [22]. Therefore, a routine sleep deprived EEG when the child is back to baseline can be more useful in identifying if epileptiform abnormalities are present. If epileptiform abnormalities are present on a routine sleep deprived EEG, this may suggest the patient is at higher risk for developing future epilepsy and the febrile illness lowered the seizure threshold; however, it is unclear whether clinical management would change as a result [31].
What treatment options are available?
Complications with prolonged and/or recurrent seizures can occur. Treatments options can be stratified into 3 possible categories: emergency rescue treatment for prolonged or a cluster of febrile seizures, intermittent treatment at the time of illness, and chronic use of medication. Treatment options for complex febrile seizures may include the use of a rescue seizure medication when the febrile seizure is prolonged. Rectal preparations of diazepam gel can be effective in stopping an ongoing seizure and can be provided for home use in patients with known recurrence of febrile status epilepticus [3]. For children and adolescents where a rectal administration is not ideal, intranasal versed can be utilized instead of rectal diazepam. In addition, the use of an intermittent benzodiazepine at the onset of febrile illness can also be considered a treatment option. Using oral diazepam at the time of a febrile illness has been demonstrated in reducing the recurrence of febrile seizures [3]. Other studies have shown similar results when using buccal midazolam [32]. No adequate studies have been performed using second- or third-generation anti-epilepsy medications in the treatment of recurrent of complex febrile seizures [3].
It is unclear whether benefit is present to using intermittent benzodiazepine doses prior or during a febrile illness for those prone for recurrent febrile seizures [33]. Physicians may consider this option in patients with frequent recurrent seizures, when caregivers can identify the fever before the seizure occurs.
Overall, parental education of efficacy and side effect profiles should be discussed in detail when considering any treatment options for complex febrile seizures [34]. It is important to remember that the long-term prognosis in terms of developing epilepsy or neurological and cognitive problems is not influenced by the use of antiepileptic medications for recurrent febrile seizures [17]. Even in the case of prolonged febrile seizures in otherwise neurodevelopmentally normal children, antiepileptics have not been shown to cause damage to the brain [19].
Febrile Status Epilepticus
Febrile status epilepticus is a subtype of complex febrile seizures and is defined as a febrile seizure lasting greater than 30 minutes. Overall, febrile status epilepticus accounts for approximately 5% of all presentations of febrile seizures [35]. It represents about 25% of all episodes of childhood status epilepticus and more than two-thirds of cases during the first 2 years of life. Literature suggests that an increased risk for focal epilepsy exists [36]. Children presenting with febrile status epilepticus are more likely to have a family history of epilepsy and a history of a previous neurological abnormality [22]. It is likely to reoccur if the first presentation was febrile status epilepticus. However, increased risk for death or developmental disability as a result of the seizure is not seen [37].
The prospective multicenter study of the consequences of prolonged febrile seizures in childhood (FEBSTAT) has been conducted. The study reported that febrile status epilepticus is usually focal (67% of episodes), occurs in very young children (median age 1.3 years), and is frequently the first febrile seizure [22]. In this study, the median duration of the seizure was about 68 minutes and 24% of children had an episode lasting more than 2 hours. In 87% of the events, seizures did not stop spontaneously and benzodiazepines were needed. Focal features observed were eye and head deviation, staring, and impaired consciousness prior to the seizure and an asymmetric convulsion or Todd’s paresis.
Case 3: Epilepsy Syndromes Associated With Febrile Seizures
A 1-year-old female presents for evaluation of seizures that began at age 8 months. Seizures are described as occurring in the setting of fever with bilateral symmetric tonic clonic activity lasting durations of less than 10 minutes on average, but at least 2 instances of seizure lasting 20 minutes or more. The family notes that seizures have occurred almost every time the child has had a febrile illness and often cluster over several days. They report at least 1 seizure that occurred in the absence of fever. Development has been normal to date and an EEG done by their primary provider was also normal.
What epilepsy syndromes are associated with febrile seizures?
Genetic Epilepsy with Febrile Seizures Plus
GEFS+ was first described in 1997 following recognition of a pattern of febrile seizures followed later by the development of various epilepsy syndromes within the same family [38]. As such, the syndrome is defined based on the familial occurrence of febrile and afebrile seizures in at least 2 family members and can have a wide range of phenotypes. The most common presentation is of typical febrile seizures which can persist beyond the typical upper age limit of 6 years. Unprovoked generalized seizures of multiple types (ie, myoclonic, absence, atonic) occur at a later age, though focal seizures may also be present. The presence of focal onset seizures led to the naming change from “generalized” epilepsy with febrile seizures plus as it was previously referred. Seizure frequency and severity may vary between family members, as can response to treatment, making prognosis difficult to predict. As even in typical febrile seizures a family history of febrile seizure may be common, it may be difficult to diagnose the syndrome after the initial febrile seizure. However, if the family history is strong for a family member with a GEFS+ phenotype, one can appropriately counsel the family on the possibility that a similar course may evolve. While the majority of GEFS+ patients have milder phenotypes, some more severe phenotypes can have cognitive delays. Dravet syndrome falls within the spectrum of GEFS+ and is a prime example of the phenotypic continuum to more severe presentations in some patients.
The syndrome is believed to be inherited in an auto-somal dominant fashion with incomplete penetrance. Multiple genes have been implicated as a cause, though only 11.5% of families with clinical GEFS+ may have mutations [39]. SCN1A, encoding the α-subunit of the voltage-gated sodium channel is most frequently reported in GEFS+ families, yet is only found in 10% [38]. When associated with GEFS+, SCN1A mutations are more often missense type, whereas truncating and nonsense mutations are more commonly encountered in Dravet syndrome. Mutations in SCN1B encoding the β1 subunit of the voltage-gated sodium channel has also been reported [40]. Finally, the GABA(A) receptor gamma 2 subunit GABRG2 has been found in < 1% of GEFS+ families [39]. The variability in causative genes underscores the reasons for phenotype variability and it is likely that other modifier genes are responsible for the heterogeneity within GEFS+ families [41].
Dravet Syndrome
Dravet syndrome, often referred to as severe myoclonic epilepsy of infancy, was first described in 1978 and has since become one of the most recognized genetic epilepsy syndromes [42]. The clinical presentation often begins with seizures in the first year of life, frequently in the setting of febrile illness. The initial seizures are generalized or hemiclonic in the majority and are often prolonged evolving to status epilepticus. Unlike typical febrile seizures, one should suspect Dravet syndrome in children that present with repetitive bouts of complex febrile seizures or febrile status epilepticus, especially if the associated seizure semiology is of hemiclonic type. In addition, seizures in the setting of modest hyperthermia (ie, hot baths) should raise suspicion for this condition. Commonly EEG and MRI are normal in the first year of life and psychomotor development remains normal until typically the second year of life [43].
By the second year, other seizure types including myoclonic, atypical absence, clonic, and tonic seizures arise. The EEG frequently begins to show generalized spike wave and polyspike wave discharges. Seizures continue occurring frequently during early childhood, often resulting in status epilepticus. Cognitive development begins to stagnate between the ages of 1 and 4 years with emergence of autistic traits and hyperactivity [44]. Development may stabilize between the ages of 5 and 16 years, but fails to demonstrate much improvement [44]. Higher frequency of seizures may correlate with increase in cognitive impairment and behavior problems, supporting the need for rapid diagnosis and appropriate therapy [44].
Over the years, several cases of atypical or borderline Dravet syndrome have been described, most highlighting the absence of myoclonic seizures [45]. Others may present with primarily clonic or tonic-clonic type seizures only [46]. Despite these differences, all cases share a similar drug resistance and cognitive delay and are categorized as Dravet syndrome.
In 2001, Claus et al discovered the genetic alteration in SCN1A responsible for 70% of Dravet syndrome cases [47]. The disorder is inherited in an autosomal dominant fashion, though 40% to 80% of mutations resulting in Dravet syndrome are de novo [48]. Mutations can be present in other family members, as this syndrome is part of the spectrum of GEFS+, though parental phenotypes are often much less severe. Approximately 50% of mutations resulting in Dravet syndrome are truncating, while the other 50% are missense mutations involving splice site or pore forming regions leading to loss of function [49]. Finally, small and large chromosome rearrangements make up 2% to 3% of cases [50]. Other genes reported to result in Dravet syndrome include SCN1B and GABRG2 mutations. In addition, PCDH19 can produce a phenotype similar to Dravet syndrome in females and is discussed in more detail below.
With the emergence of more rapid and cheaper forms of genetic testing, molecular diagnosis can now be made earlier in life before all the typical clinical features of Dravet syndrome arise. As a result, one might hope to alter treatment strategy and gear therapy towards the most effective medications. While drug resistance is the norm for the condition, certain drugs such as benzodiazepines, valproate, and stiripentol may be most effective [43]. Topiramate and levetiracetam have been reported as effi-cacious in small series, as has the ketogenic diet [51–55]. Varieties of medications which target sodium channels are known to exacerbate seizures in Dravet syndrome and should be avoided, including lamotrigine, carbamazepine, oxcarbazepine, and phenytoin [56]. In addition to maintenance therapy, it is important to provide patients with a rescue plan for acute seizures in an effort to avoid status epilepticus. In addition, measures to avoid overheating may provide additional benefit.
Case 3 Continued
After a careful history, the physician discovers that the child also has frequent myoclonic seizures described as brief jerks of the extremities or sudden forward falls. The family notes they have seen these seizures more frequently since antiepileptic therapy was started. The physician recognize that this child may have Dravet syndrome and suspect her medication may be resulting in aggravation of seizures.
The physician decides to discontinue the medication suspected to aggravate the seizures and chooses to start the child on clobazam. The physician also begins evaluation for Dravet syndrome by sending directed SCN1A genetic testing. The testing comes back negative for mutations in the SCN1A gene.
What other investigations would be warranted now?
PCDH19
PCDH19 was first recognized as a cause of epilepsy and mental retardation limited to females (EFMR), a syndrome characterized by onset of seizures in infancy or early childhood with predominantly generalized type seizures including tonic-clonic, absence, myoclonic, tonic, and atonic [57]. Since that initial description, the phenotype associated with PCDH19 mutations has expanded to include female patients with primarily focal epilepsy, variable cognitive impairment, and commonly onset with seizures in the setting of fever [58,59]. Typically seizures begin around age 10 months presenting as a cluster of focal seizures in the setting of fever, often followed by a second cluster 6 months later [59]. Generalized seizures occur in a small proportion of patients (9%) and this feature, along with relatively fewer bouts of status epilepticus and less frequent seizures (most monthly to yearly frequency) can differentiate PCDH19 associated epilepsy from Dravet syndrome [59]. Seizures tend to improve with age and no particular antiepileptic drug has been found especially efficacious in the syndrome. Unlike Dravet syndrome, up to a third of patients with this syndrome may ultimately become seizure-free [59].
Cognitive development is normal prior to seizure onset in the majority of patients and most but not all patients will develop some cognitive impairment ranging from mild to severe [59]. It is the more severe patients that most often have overlapping characteristics of Dravet syndrome, thus PCDH19 mutations should be investigated in female patients with Dravet phenotype yet negative SCN1A testing.
PCDH19 is a calcium-dependent adhesion protein involved in neuronal circuit formation during development and in the maintenance of normal synaptic circuits in adulthood [60,61]. Disease causing mutations in PCDH19 are primarily missense (48.5%) or frameshift, nonsense, and splice-site mutations resulting in premature termination codon [59]. Ninety percent of mutations are de novo. When inherited, the disorder is X-linked and may come from an unaffected father or a mother that is similarly affected or not, suggesting variable clinical severity in females and gender-related protections in males [59].
Case 3 Continued
Given the negative SCN1A testing, the physician chooses to pursue other genetic testing that may explain the patient’s phenotype. A more extensive “epilepsy gene panel” that includes 70 different genes associated with epilepsy syndromes is ordered.
Hemiconvulsion-Hemiplegia Epilepsy Syndrome
Hemiconvulsion-hemiplegia epilepsy syndrome (HHE) is characterized by the occurrence of unilateral convulsive status epilepticus followed by transient or permanent ipsilateral hemiplegia. The syndrome occurs in otherwise healthy children often in the setting of nonspecific febrile illness before the age of 4 years, with peak occurrence in the first 2 years of life [62]. Seizures are characterized by unilateral clonic activity with EEG demonstrating rhythmic 2–3 Hz slow wave activity and spikes in the hemisphere contralateral to the body involvement. MRI frequently demonstrates diffusion changes congruent with EEG findings, often in the perisylvian region. The hemiplegia that remains following status epilepticus is permanent in up to 80% of cases [63]. As hemiplegia can occur following complex febrile seizures, it is recommended a minimum duration of hemiplegia of 1 week be used to differentiate HHE [64]. Status epilepticus is persistent in this syndrome and can last for hours if untreated. Focal onset seizures will often continue to occur in the patient even after the status has been aborted.
The etiology of HHE is variable with many cases idiopathic. Some cases are reported as symptomatic, as the syndrome can present in the setting of other underlying brain disorders such as Sturge-Weber and tuberous sclerosis complex. While some viruses have been proposed as a cause, they are not found in the cerebral spinal fluid of patients [65]. Treatment consists of rapid treatment of status epilepticus with benzodiazepines as first-line therapy, often followed by other intravenous antiepileptic drugs as necessary.
Febrile Infection–Related Epilepsy Syndrome
Febrile infection–related epilepsy syndrome (FIRES) is presented under several names in the literature including idiopathic catastrophic epileptic encephalopathy [66], devastating encephalopathy in school-age children [67], new-onset refractory status epilepticus [68], as well as fever-induced refractory epileptic encephalopathy syndrome [69] and fever-induced refractory epileptic encephalopathy in school-age children [70]. All describe rare catastrophic epilepsy presenting in otherwise healthy children during or days following a febrile illness. While febrile illness precedes the epilepsy in 96% of cases, up to 50% of patients may not have fever at the time they present [41,65]. While age of onset is typically in early childhood, presentation in adulthood also occurs. Initial seizures are often focal, presenting as forced lateral head or eye deviation, oral or manual automatisms, and clonic movements of the face and extremities. Seizures will inevitably progress to status epilepticus with ictal onset often multifocal predominating in the perisylvian regions [41]. MRI is often normal at onset or shows only subtle swelling of the mesial temporal structures. Over months, MRI often shows T2-hyperintensity and atrophy of the mesial temporal structures, though as many as 50% of MRIs may remain normal [71].
The evaluation for cause in FIRES is often unrewarding. Inflammatory markers are typically absent from both serum and CSF. CSF may show minimal pleocytosis with negative oligoclonal bands and absence of common receptor antibodies. Treatment is equally unrewarding with patients typically failing conventional antiepileptic drugs and continuous infusions titrated to burst suppression. Immunomodulatory therapies are mostly ineffective as well. The most useful therapy reported has been the keto-genic diet with efficacy in up to 50% of patients [72]. Recently, therapeutic hypothermia has also been reported to be effective in 2 cases [73]. For the majority of patients, therapy will remain ineffective and seizures will continue for weeks to months with gradual resolution, though seizures often continue intermittently following the end of status epilepticus. Prognosis is poor for seizure control and neurocognitive recovery with mortality of 30% reported [41].
Case 3 Conclusion
The epilepsy gene panel ordered returns with the result of a disease-causing mutation in the PCDH19 gene. The child is diagnosed with PCDH19-associated epilepsy and is treated with phenobarbital. For the first years of life, she presents on average once per year with a cluster of seizures in the setting of febrile illness which is often managed with short durations of scheduled benzodiazepines. Seizures slow by age 6. She has mild delays in speech and receives some accommodations through her school system. By age 10, she has been seizure-free for several years. She is able to be weaned off medications without recurrence of seizures.
Summary
Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted. Providers should have a high index of suspicion for these syndromes when they encounter children that repeatedly present with prolonged febrile seizures, clusters of febrile seizures, or febrile seizures in addition to afebrile seizure events. Early referral, diagnosis, and treatment has the potential to alter outcome in some of these syndromes, thus the importance of becoming familiar with these diagnoses.
Corresponding author: Anup D. Patel, MD, Nationwide Children's Hospital, Columbus, OH 43205, anup.patel@nationwidechildrens.org.
Financial disclosures: Dr. Patel disclosed that he has consulted for GW Pharmaceuticals and Supernus and is on the Scientific Advisory Board for UCB Pharma.
From the Nationwide Children’s Hospital, Columbus, OH (Dr. Patel) and Cook Children’s Medical Center, Fort Worth, TX (Dr. Perry).
Abstract
- Objective: To review the current understanding and management of febrile seizures.
- Methods: Review of the literature.
- Results: Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as-needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted.
- Conclusion: Providers caring for pediatric patients should be aware of the clinical considerations in managing patients with febrile seizures.
Key words: febrile seizure; Dravat syndrome; GEFS+; PCDH19; FIRES; complex febrile seizure.
A febrile seizure is defined as a seizure in association with a febrile illness in the absence of a central nervous system infection or acute electrolyte imbalance in children older than 1 month of age without prior afebrile seizures [1]. The mechanism by which fever provokes a febrile seizure is unclear [2]. Febrile seizures are the most common type of childhood seizures, affecting 2% to 5% of children [1]. The age of onset is between 6 months and 5 years [3]; peak incidence occurs at about 18 months of age. Simple febrile seizures are the most common type of febrile seizure. By definition, they are generalized, last less than 10 minutes and only occur once in a 24-hour time-period. A complex febrile seizure is one with focal onset or one that occurs more than once during a febrile illness, or lasts more than 10 minutes. Febrile status epilepticus, a subtype of complex febrile seizures, represents about 25% of all episodes of childhood status epilepticus. They account for more than two-thirds of cases during the first 2 years of life.
The risk of reoccurrence after presenting with one febrile seizure is approximately 30%, with the risk being 60% after 2 febrile seizures and 90% after 3 [4–6]. Some families have an autosomal dominant inheritance pattern with polygenic inheritance suspected for the majority of patients presenting with febrile seizures.
Multiple chromosomes have been postulated to be associated with genetic susceptibility for febrile seizures, with siblings having a 25% increased risk and high concordance noted in monozygotic twins [7]. The pathophysiology for febrile seizures has been associated with a genetic risk associated with the rate of temperature rise with animal studies suggesting temperature regulation of c-aminobutyric acid (GABA) a receptors [2]. Other studies propose a link between genetic and environmental factors resulting in an inflammatory process which influences neuronal excitement predisposing one to a febrile seizure [8].
Debate exists between the relation of febrile seizures and childhood vaccinations. Seizures are rare following administration of childhood vaccines. Most seizures following administration of vaccines are simple febrile seizures [9]. Febrile seizures associated with vaccines are more associated with underlying epilepsy. In a study of patients with vaccine-related encephalopathy and febrile status epilepticus, the majority of patients were found to have Dravet syndrome; it was determined that the vaccine may have triggered an earlier onset of the presentation for Dravet in those predestined to develop this disease but did not adversely impact ultimate outcome [10].
In this article, we review simple and complex febrile seizures with a focus on clinical management. Epilepsy syndromes associated with febrile seizures are also discussed. Cases are provided to highlight important clinical considerations.
Case 1: Simple Febrile Seizure
A 9-month-old infant and his mother present to the pediatrician. The mother notes that the infant had an event of concern. She notes the infant had stiffness in all 4 extremities followed by jerking that lasted 30 to 60 seconds. The infant was not responsive during the event. He was sleepy afterward, but returned to normal soon after the event ended. After, she noted that the infant felt warm and she checked his temperature. He had a fever of 101°F. The infant has normal development and no other medical problems.
What are management considerations for simple febrile seizure?
A simple febrile seizure is the most common type of febrile seizure. They are generalized, lasting less than 10 minutes and only occur once in a 24-hour period. There is no increased risk of developing epilepsy or developmental delay for patients after the first simple febrile seizures when compared to other children [5,6]. The diagnosis is based on history provided and a physical examination including evaluation of body temperature [11,12].
No routine laboratory tests are needed as a result of a simple febrile seizure unless obtained to assist in identifying the fever source [3,11]. Routine EEG testing is not recommended for these patients [3,11]. Routine imaging of the brain is also not needed [3,11]. Only if a patient has signs of meningitis should a lumbar puncture be performed [11]. The American Academy of Pediatrics states that a lumbar puncture is strongly considered for those younger than 12 months if they present with their first complex febrile seizure as signs of meningitis may be absent in young children. For infants 6 to 12 months of age, a lumbar puncture can be considered when immunization status is deficient or unknown [13,14]. Also, a lumbar puncture is an option for children who are pretreated with antibiotics [11]. For patients younger than 6 months, data is lacking on the percentage of patients with bacterial meningitis following a simple febrile seizure.
Daily preventative therapy with an anti-epilepsy medication is not necessary [3,11]. A review of several treatment studies shows that some anti-epileptic medications are effective in preventing recurrent simple febrile seizures. Studies have demonstrated the effectiveness of phenobarbital, primidone, and valproic acid in preventing the recurrence of simple febrile seizures; however, the side effects of each medication outweighed the benefit [3]. Carbamazepine and phenytoin have not been shown to be effective in preventing recurrent febrile seizures [3].
For anxious caregivers with children having recurrent febrile seizures, a daily medication or treating with an abortive seizure medication at the time of a febrile illness can be considered [3,5,6,15]. Treating with an abortive medication may mask signs and symptoms of meningitis making evaluation more challenging [16]. Evidence does not support that using antipyretic medications such as acetaminophen or ibuprofen will reduce the recurrence of febrile seizures. The seizure usually is the first noticed symptom due to the rise of temperature being the cause of the febrile seizure in an otherwise well child prior to the seizure [11,17]. Damage to the brain and associated structures is not found with patients presenting with simple febrile seizures [5,6]. Education on all of these principles is strongly recommended for caregiver reassurance.
Case 2: Complex Febrile Seizure
A 1-year-old child presents to the emergency department. Mother was with the child and she noticed stiffness followed by jerking of the left arm and leg, which quickly became noted in both arms and legs. The episode appeared to last for 15 minutes before EMS arrived to the house. A medication was given to the child by EMS that stopped the event. EMS noted the child had a temperature of 101.5°F. The child was previously healthy and has had normal development thus far.
What is the epidemiology of complex febrile seizure?
A complex febrile seizure is one with focal onset, or one that occurs more than once during a febrile illness or lasts more than 10 minutes. They are less common, representing only 20% to 30% of all febrile seizures [18–20]. In The National Collaborative Perinatal Project (NCPP), 1706 children with febrile seizures were identified from 54,000 and were followed from birth until 7 years of age. The initial febrile seizure was defined as complex in about 28%. For all febrile seizures, focal features were present in 4%, prolonged duration (> 10 minutes) in 7.6%, and recurrent episodes within 24 hours in 16.2% [21]. Similar observations have been reported by Berg and Shinnar [5,6]. Of 136 children who had recurrences, 41.2% had one or more complex features and the strongest correlate of having recurrent complex febrile seizure was the number of recurrent seizures. They also found that children with complex recurrences had other recurrences that were not complex; however, complex features had a tendency to recur. Further, a strong association between focal onset and prolonged duration was found [5,6]. Previous studies established a correlation between complex attacks, particularly prolonged ones and young age (age < 1 year) [5,6]. Additionally, children with seizures with a relatively low fever (< 102°F) were slightly more likely to have a complex febrile seizure as the initial episode [5,6].
Children with febrile seizures are already at 4- to 5-fold increased risk for subsequent unprovoked seizures. A history of febrile seizures has been found in 13% to 18% of children with new-onset epilepsy. In the NCPP study, the predictors identified for the development of epilepsy following febrile seizures were an abnormal neurological and developmental status of the child before the seizure, a history of afebrile seizures in a parent or prior-born sibling, or complex features [21]. Ten percent of children with 2 or more of the previously mentioned risk factors (including complex features) developed epilepsy and 13% of them had seizures without fever [20,22]. Further, intractable epilepsy and neurological impair-ment have been found to be more common in children with prior prolonged febrile seizure, with no association to any specific seizure type [18,23–25]. The association between febrile seizures and mesial temporal sclerosis (MTS) is a commonly debated topic. Retrospective studies have reported an association between prolonged or atypical febrile seizures and intractable temporal lobe epilepsy. Epidemiological studies fail to show a causal relationship between febrile seizures and temporal lobe epilepsy [26]. This suggests that febrile seizures are a marker of susceptibility to seizures and future epilepsy (in some cases) rather than a direct cause. It is clear that a minority of cases of MTS or complex partial seizures are associated with prior febrile seizures [20,22].
What is the risk of intracranial pathology in complex febrile seizure?
Patients with complex febrile seizures usually seek medical attention [27]. However, the risk of acute pathology necessitating treatment changes based on neuroimaging was found to be very low and likely not necessary in the evaluation of complex febrile seizures during the acute presentation [27]. Imaging with a high-resolution brain MRI could be considered later on a routine basis for prolonged febrile seizures due to the possible association between prolonged febrile seizures and mesial temporal sclerosis [19,28,29].
Neuroimaging has provided evidence that hippocampal injury can occasionally occur during prolonged and focal febrile seizures in infants who otherwise appear normal. It has been speculated that a pre-existing abnormality increases the propensity to focal prolonged seizures and further hippocampal damage. Hesdorffer and colleagues [30] found definite abnormalities on MRI in 14.8% of children with complex febrile seizures and 11.4 % of simple febrile seizures among 159 children with a first febrile seizure. However, MRI abnormalities were related to a specific subtype of complex seizures: focal and prolonged. The most common abnormalities observed were subcortical focal hyperintensity, an abnormal white matter signal, and focal cortical dysplasia.
What are important aspects of the clinical evaluation?
The evaluation and management of the child with complex febrile seizures is debated as well. The most important part in the history and examination is to look for the source of the fever and rule out the presence of a CNS infection, since complex febrile seizures are much more frequently associated with meningitis than simple febrile seizures [16]. The American Academy of Pediatrics recommended that a lumbar puncture be strongly considered in infants younger than 12 months after a first febrile seizure and should be considered in children between 12 and 18 months of age, since signs of meningitis may be absent in young children [13]. If the threshold for a lumbar puncture is low in infants with febrile seizures in general, it should be even lower for children with complex febrile episodes for all the factors mentioned above. The guidelines developed in 1990 by the Royal College of Physicians and the British Paediatric Association concluded that indications for performing an lumbar puncture were complex febrile seizure, signs of meningismus, or a child who is unduly drowsy and irritable or systematically ill [21].
Obtaining an EEG within 24 hours of presentation may show generalized background slowing, which could make identifying possible epileptiform abnormalities difficult [22]. Therefore, a routine sleep deprived EEG when the child is back to baseline can be more useful in identifying if epileptiform abnormalities are present. If epileptiform abnormalities are present on a routine sleep deprived EEG, this may suggest the patient is at higher risk for developing future epilepsy and the febrile illness lowered the seizure threshold; however, it is unclear whether clinical management would change as a result [31].
What treatment options are available?
Complications with prolonged and/or recurrent seizures can occur. Treatments options can be stratified into 3 possible categories: emergency rescue treatment for prolonged or a cluster of febrile seizures, intermittent treatment at the time of illness, and chronic use of medication. Treatment options for complex febrile seizures may include the use of a rescue seizure medication when the febrile seizure is prolonged. Rectal preparations of diazepam gel can be effective in stopping an ongoing seizure and can be provided for home use in patients with known recurrence of febrile status epilepticus [3]. For children and adolescents where a rectal administration is not ideal, intranasal versed can be utilized instead of rectal diazepam. In addition, the use of an intermittent benzodiazepine at the onset of febrile illness can also be considered a treatment option. Using oral diazepam at the time of a febrile illness has been demonstrated in reducing the recurrence of febrile seizures [3]. Other studies have shown similar results when using buccal midazolam [32]. No adequate studies have been performed using second- or third-generation anti-epilepsy medications in the treatment of recurrent of complex febrile seizures [3].
It is unclear whether benefit is present to using intermittent benzodiazepine doses prior or during a febrile illness for those prone for recurrent febrile seizures [33]. Physicians may consider this option in patients with frequent recurrent seizures, when caregivers can identify the fever before the seizure occurs.
Overall, parental education of efficacy and side effect profiles should be discussed in detail when considering any treatment options for complex febrile seizures [34]. It is important to remember that the long-term prognosis in terms of developing epilepsy or neurological and cognitive problems is not influenced by the use of antiepileptic medications for recurrent febrile seizures [17]. Even in the case of prolonged febrile seizures in otherwise neurodevelopmentally normal children, antiepileptics have not been shown to cause damage to the brain [19].
Febrile Status Epilepticus
Febrile status epilepticus is a subtype of complex febrile seizures and is defined as a febrile seizure lasting greater than 30 minutes. Overall, febrile status epilepticus accounts for approximately 5% of all presentations of febrile seizures [35]. It represents about 25% of all episodes of childhood status epilepticus and more than two-thirds of cases during the first 2 years of life. Literature suggests that an increased risk for focal epilepsy exists [36]. Children presenting with febrile status epilepticus are more likely to have a family history of epilepsy and a history of a previous neurological abnormality [22]. It is likely to reoccur if the first presentation was febrile status epilepticus. However, increased risk for death or developmental disability as a result of the seizure is not seen [37].
The prospective multicenter study of the consequences of prolonged febrile seizures in childhood (FEBSTAT) has been conducted. The study reported that febrile status epilepticus is usually focal (67% of episodes), occurs in very young children (median age 1.3 years), and is frequently the first febrile seizure [22]. In this study, the median duration of the seizure was about 68 minutes and 24% of children had an episode lasting more than 2 hours. In 87% of the events, seizures did not stop spontaneously and benzodiazepines were needed. Focal features observed were eye and head deviation, staring, and impaired consciousness prior to the seizure and an asymmetric convulsion or Todd’s paresis.
Case 3: Epilepsy Syndromes Associated With Febrile Seizures
A 1-year-old female presents for evaluation of seizures that began at age 8 months. Seizures are described as occurring in the setting of fever with bilateral symmetric tonic clonic activity lasting durations of less than 10 minutes on average, but at least 2 instances of seizure lasting 20 minutes or more. The family notes that seizures have occurred almost every time the child has had a febrile illness and often cluster over several days. They report at least 1 seizure that occurred in the absence of fever. Development has been normal to date and an EEG done by their primary provider was also normal.
What epilepsy syndromes are associated with febrile seizures?
Genetic Epilepsy with Febrile Seizures Plus
GEFS+ was first described in 1997 following recognition of a pattern of febrile seizures followed later by the development of various epilepsy syndromes within the same family [38]. As such, the syndrome is defined based on the familial occurrence of febrile and afebrile seizures in at least 2 family members and can have a wide range of phenotypes. The most common presentation is of typical febrile seizures which can persist beyond the typical upper age limit of 6 years. Unprovoked generalized seizures of multiple types (ie, myoclonic, absence, atonic) occur at a later age, though focal seizures may also be present. The presence of focal onset seizures led to the naming change from “generalized” epilepsy with febrile seizures plus as it was previously referred. Seizure frequency and severity may vary between family members, as can response to treatment, making prognosis difficult to predict. As even in typical febrile seizures a family history of febrile seizure may be common, it may be difficult to diagnose the syndrome after the initial febrile seizure. However, if the family history is strong for a family member with a GEFS+ phenotype, one can appropriately counsel the family on the possibility that a similar course may evolve. While the majority of GEFS+ patients have milder phenotypes, some more severe phenotypes can have cognitive delays. Dravet syndrome falls within the spectrum of GEFS+ and is a prime example of the phenotypic continuum to more severe presentations in some patients.
The syndrome is believed to be inherited in an auto-somal dominant fashion with incomplete penetrance. Multiple genes have been implicated as a cause, though only 11.5% of families with clinical GEFS+ may have mutations [39]. SCN1A, encoding the α-subunit of the voltage-gated sodium channel is most frequently reported in GEFS+ families, yet is only found in 10% [38]. When associated with GEFS+, SCN1A mutations are more often missense type, whereas truncating and nonsense mutations are more commonly encountered in Dravet syndrome. Mutations in SCN1B encoding the β1 subunit of the voltage-gated sodium channel has also been reported [40]. Finally, the GABA(A) receptor gamma 2 subunit GABRG2 has been found in < 1% of GEFS+ families [39]. The variability in causative genes underscores the reasons for phenotype variability and it is likely that other modifier genes are responsible for the heterogeneity within GEFS+ families [41].
Dravet Syndrome
Dravet syndrome, often referred to as severe myoclonic epilepsy of infancy, was first described in 1978 and has since become one of the most recognized genetic epilepsy syndromes [42]. The clinical presentation often begins with seizures in the first year of life, frequently in the setting of febrile illness. The initial seizures are generalized or hemiclonic in the majority and are often prolonged evolving to status epilepticus. Unlike typical febrile seizures, one should suspect Dravet syndrome in children that present with repetitive bouts of complex febrile seizures or febrile status epilepticus, especially if the associated seizure semiology is of hemiclonic type. In addition, seizures in the setting of modest hyperthermia (ie, hot baths) should raise suspicion for this condition. Commonly EEG and MRI are normal in the first year of life and psychomotor development remains normal until typically the second year of life [43].
By the second year, other seizure types including myoclonic, atypical absence, clonic, and tonic seizures arise. The EEG frequently begins to show generalized spike wave and polyspike wave discharges. Seizures continue occurring frequently during early childhood, often resulting in status epilepticus. Cognitive development begins to stagnate between the ages of 1 and 4 years with emergence of autistic traits and hyperactivity [44]. Development may stabilize between the ages of 5 and 16 years, but fails to demonstrate much improvement [44]. Higher frequency of seizures may correlate with increase in cognitive impairment and behavior problems, supporting the need for rapid diagnosis and appropriate therapy [44].
Over the years, several cases of atypical or borderline Dravet syndrome have been described, most highlighting the absence of myoclonic seizures [45]. Others may present with primarily clonic or tonic-clonic type seizures only [46]. Despite these differences, all cases share a similar drug resistance and cognitive delay and are categorized as Dravet syndrome.
In 2001, Claus et al discovered the genetic alteration in SCN1A responsible for 70% of Dravet syndrome cases [47]. The disorder is inherited in an autosomal dominant fashion, though 40% to 80% of mutations resulting in Dravet syndrome are de novo [48]. Mutations can be present in other family members, as this syndrome is part of the spectrum of GEFS+, though parental phenotypes are often much less severe. Approximately 50% of mutations resulting in Dravet syndrome are truncating, while the other 50% are missense mutations involving splice site or pore forming regions leading to loss of function [49]. Finally, small and large chromosome rearrangements make up 2% to 3% of cases [50]. Other genes reported to result in Dravet syndrome include SCN1B and GABRG2 mutations. In addition, PCDH19 can produce a phenotype similar to Dravet syndrome in females and is discussed in more detail below.
With the emergence of more rapid and cheaper forms of genetic testing, molecular diagnosis can now be made earlier in life before all the typical clinical features of Dravet syndrome arise. As a result, one might hope to alter treatment strategy and gear therapy towards the most effective medications. While drug resistance is the norm for the condition, certain drugs such as benzodiazepines, valproate, and stiripentol may be most effective [43]. Topiramate and levetiracetam have been reported as effi-cacious in small series, as has the ketogenic diet [51–55]. Varieties of medications which target sodium channels are known to exacerbate seizures in Dravet syndrome and should be avoided, including lamotrigine, carbamazepine, oxcarbazepine, and phenytoin [56]. In addition to maintenance therapy, it is important to provide patients with a rescue plan for acute seizures in an effort to avoid status epilepticus. In addition, measures to avoid overheating may provide additional benefit.
Case 3 Continued
After a careful history, the physician discovers that the child also has frequent myoclonic seizures described as brief jerks of the extremities or sudden forward falls. The family notes they have seen these seizures more frequently since antiepileptic therapy was started. The physician recognize that this child may have Dravet syndrome and suspect her medication may be resulting in aggravation of seizures.
The physician decides to discontinue the medication suspected to aggravate the seizures and chooses to start the child on clobazam. The physician also begins evaluation for Dravet syndrome by sending directed SCN1A genetic testing. The testing comes back negative for mutations in the SCN1A gene.
What other investigations would be warranted now?
PCDH19
PCDH19 was first recognized as a cause of epilepsy and mental retardation limited to females (EFMR), a syndrome characterized by onset of seizures in infancy or early childhood with predominantly generalized type seizures including tonic-clonic, absence, myoclonic, tonic, and atonic [57]. Since that initial description, the phenotype associated with PCDH19 mutations has expanded to include female patients with primarily focal epilepsy, variable cognitive impairment, and commonly onset with seizures in the setting of fever [58,59]. Typically seizures begin around age 10 months presenting as a cluster of focal seizures in the setting of fever, often followed by a second cluster 6 months later [59]. Generalized seizures occur in a small proportion of patients (9%) and this feature, along with relatively fewer bouts of status epilepticus and less frequent seizures (most monthly to yearly frequency) can differentiate PCDH19 associated epilepsy from Dravet syndrome [59]. Seizures tend to improve with age and no particular antiepileptic drug has been found especially efficacious in the syndrome. Unlike Dravet syndrome, up to a third of patients with this syndrome may ultimately become seizure-free [59].
Cognitive development is normal prior to seizure onset in the majority of patients and most but not all patients will develop some cognitive impairment ranging from mild to severe [59]. It is the more severe patients that most often have overlapping characteristics of Dravet syndrome, thus PCDH19 mutations should be investigated in female patients with Dravet phenotype yet negative SCN1A testing.
PCDH19 is a calcium-dependent adhesion protein involved in neuronal circuit formation during development and in the maintenance of normal synaptic circuits in adulthood [60,61]. Disease causing mutations in PCDH19 are primarily missense (48.5%) or frameshift, nonsense, and splice-site mutations resulting in premature termination codon [59]. Ninety percent of mutations are de novo. When inherited, the disorder is X-linked and may come from an unaffected father or a mother that is similarly affected or not, suggesting variable clinical severity in females and gender-related protections in males [59].
Case 3 Continued
Given the negative SCN1A testing, the physician chooses to pursue other genetic testing that may explain the patient’s phenotype. A more extensive “epilepsy gene panel” that includes 70 different genes associated with epilepsy syndromes is ordered.
Hemiconvulsion-Hemiplegia Epilepsy Syndrome
Hemiconvulsion-hemiplegia epilepsy syndrome (HHE) is characterized by the occurrence of unilateral convulsive status epilepticus followed by transient or permanent ipsilateral hemiplegia. The syndrome occurs in otherwise healthy children often in the setting of nonspecific febrile illness before the age of 4 years, with peak occurrence in the first 2 years of life [62]. Seizures are characterized by unilateral clonic activity with EEG demonstrating rhythmic 2–3 Hz slow wave activity and spikes in the hemisphere contralateral to the body involvement. MRI frequently demonstrates diffusion changes congruent with EEG findings, often in the perisylvian region. The hemiplegia that remains following status epilepticus is permanent in up to 80% of cases [63]. As hemiplegia can occur following complex febrile seizures, it is recommended a minimum duration of hemiplegia of 1 week be used to differentiate HHE [64]. Status epilepticus is persistent in this syndrome and can last for hours if untreated. Focal onset seizures will often continue to occur in the patient even after the status has been aborted.
The etiology of HHE is variable with many cases idiopathic. Some cases are reported as symptomatic, as the syndrome can present in the setting of other underlying brain disorders such as Sturge-Weber and tuberous sclerosis complex. While some viruses have been proposed as a cause, they are not found in the cerebral spinal fluid of patients [65]. Treatment consists of rapid treatment of status epilepticus with benzodiazepines as first-line therapy, often followed by other intravenous antiepileptic drugs as necessary.
Febrile Infection–Related Epilepsy Syndrome
Febrile infection–related epilepsy syndrome (FIRES) is presented under several names in the literature including idiopathic catastrophic epileptic encephalopathy [66], devastating encephalopathy in school-age children [67], new-onset refractory status epilepticus [68], as well as fever-induced refractory epileptic encephalopathy syndrome [69] and fever-induced refractory epileptic encephalopathy in school-age children [70]. All describe rare catastrophic epilepsy presenting in otherwise healthy children during or days following a febrile illness. While febrile illness precedes the epilepsy in 96% of cases, up to 50% of patients may not have fever at the time they present [41,65]. While age of onset is typically in early childhood, presentation in adulthood also occurs. Initial seizures are often focal, presenting as forced lateral head or eye deviation, oral or manual automatisms, and clonic movements of the face and extremities. Seizures will inevitably progress to status epilepticus with ictal onset often multifocal predominating in the perisylvian regions [41]. MRI is often normal at onset or shows only subtle swelling of the mesial temporal structures. Over months, MRI often shows T2-hyperintensity and atrophy of the mesial temporal structures, though as many as 50% of MRIs may remain normal [71].
The evaluation for cause in FIRES is often unrewarding. Inflammatory markers are typically absent from both serum and CSF. CSF may show minimal pleocytosis with negative oligoclonal bands and absence of common receptor antibodies. Treatment is equally unrewarding with patients typically failing conventional antiepileptic drugs and continuous infusions titrated to burst suppression. Immunomodulatory therapies are mostly ineffective as well. The most useful therapy reported has been the keto-genic diet with efficacy in up to 50% of patients [72]. Recently, therapeutic hypothermia has also been reported to be effective in 2 cases [73]. For the majority of patients, therapy will remain ineffective and seizures will continue for weeks to months with gradual resolution, though seizures often continue intermittently following the end of status epilepticus. Prognosis is poor for seizure control and neurocognitive recovery with mortality of 30% reported [41].
Case 3 Conclusion
The epilepsy gene panel ordered returns with the result of a disease-causing mutation in the PCDH19 gene. The child is diagnosed with PCDH19-associated epilepsy and is treated with phenobarbital. For the first years of life, she presents on average once per year with a cluster of seizures in the setting of febrile illness which is often managed with short durations of scheduled benzodiazepines. Seizures slow by age 6. She has mild delays in speech and receives some accommodations through her school system. By age 10, she has been seizure-free for several years. She is able to be weaned off medications without recurrence of seizures.
Summary
Febrile seizures are a common manifestation in early childhood and very often a benign occurrence. For simple febrile seizures, minimal evaluation is necessary and treatment typically not warranted beyond reassurance and education of caregivers. For complex febrile seizures, additional evaluation in rare cases may suggest an underlying seizure tendency, though most follow a typical benign course of febrile seizures. In some cases, as needed benzodiazepines used for prolonged or recurrent febrile seizures may be of value. There are well described epilepsy syndromes for which febrile seizures may be the initial manifestation and it is paramount that providers recognize the signs and symptoms of these syndromes in order to appropriately counsel families and initiate treatment or referral when warranted. Providers should have a high index of suspicion for these syndromes when they encounter children that repeatedly present with prolonged febrile seizures, clusters of febrile seizures, or febrile seizures in addition to afebrile seizure events. Early referral, diagnosis, and treatment has the potential to alter outcome in some of these syndromes, thus the importance of becoming familiar with these diagnoses.
Corresponding author: Anup D. Patel, MD, Nationwide Children's Hospital, Columbus, OH 43205, anup.patel@nationwidechildrens.org.
Financial disclosures: Dr. Patel disclosed that he has consulted for GW Pharmaceuticals and Supernus and is on the Scientific Advisory Board for UCB Pharma.
1. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002;17 Suppl 1:S44–52.
2. Kang J-Q, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci 2006;26:2590–7.
3. Baumann RJ, Duffner PK. Treatment of children with simple febrile seizures: the AAP practice parameter. American Academy of Pediatrics. Pediatr Neurol 2000;23:11–7.
4. Tarkka R, Rantala H, Uhari M, Pokka T. Risk of recurrence and outcome after the first febrile seizure. Pediatr Neurol 1998;18:218–20.
5. Berg AT, Shinnar S. Complex febrile seizures. Epilepsia 1996;37:126–33.
6. Berg AT, Shinnar S. Unprovoked seizures in children with febrile seizures: short-term outcome. Neurology 1996;47:562–8.
7. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006;67:687–90.
8. Dube CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev 2009;31:366–71.
9. Vestergaard M, Christensen J. Register-based studies on febrile seizures in Denmark. Brain Dev 2009;31:372–7.
10. Berkovic SF, Petrou S. Febrile seizures: traffic slows in the heat. Trends Molecul Med 2006;12:343–4.
11. Practice parameter: the neurodiagnostic evaluation of the child with a first simple febrile seizure. American Academy of Pediatrics. Provisional Committee on Quality Improvement, Subcommittee on Febrile Seizures. Pediatrics 1996;97:769–72; discussion 773–765.
12. Fukuyama Y, Seki T, Ohtsuka C, et al. Practical guidelines for physicians in the management of febrile seizures. Brain Dev 1996;18:479–84.
13. Neurodiagnostic evaluation of the child with a simple febrile seizure. Pediatrics 2011;127:389–94.
14. Guedj R, Chappuy H, Titomanlio L, et al. Risk of bacterial meningitis in children 6 to 11 months of age with a first simple febrile seizure: a retrospective, cross-sectional, observational study. Acad Emerg Med 2015;22:1290–7.
15. Wo SB, Lee JH, Lee YJ, et al. Risk for developing epilepsy and epileptiform discharges on EEG in patients with febrile seizures. Brain Dev 2013;35:307–11.
16. Green SM, Rothrock SG, Clem KJ, et al. Can seizures be the sole manifestation of meningitis in febrile children? Pediatrics 1993;92:527–34.
17. Knudsen FU. Febrile seizures: treatment and prognosis. Epilepsia 2000;41:2–9.
18. Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 1987;316:493–8.
19. Teng D, Dayan P, Tyler S, et al. Risk of intracranial pathologic conditions requiring emergency intervention after a first complex febrile seizure episode among children. Pediatrics 2006;117:304–8.
20. Janszky J, Schulz R, Ebner A. Clinical features and surgical outcome of medial temporal lobe epilepsy with a history of complex febrile convulsions. Epilepsy Res 2003;55:1–8.
21. Capovilla G, Mastrangelo M, Romeo A, Vigevano F. Recommendations for the management of «febrile seizures»: Ad Hoc Task Force of LICE Guidelines Commission. Epilepsia 2009;50 Suppl 1:2–6.
22. Shinnar S, Hesdorffer DC, Nordli DR Jr, et al. Phenomenology of prolonged febrile seizures: results of the FEBSTAT study. Neurology 2008;71:170–6.
23. Camfield P, Camfield C, Gordon K, Dooley J. What types of epilepsy are preceded by febrile seizures? A population-based study of children. Dev Med Child Neurol 1994;36:887–92.
24. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976;295:1029–33.
25. Hamati-Haddad A, Abou-Khalil B. Epilepsy diagnosis and localization in patients with antecedent childhood febrile convulsions. Neurology 1998;50:917–22.
26. Davies KG, Hermann BP, Dohan FC Jr, et al. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 1996;24:119–26.
27. Kimia AA, Ben-Joseph E, Prabhu S, et al. Yield of emergent neuroimaging among children presenting with a first complex febrile seizure. Pediatr Emerg Care 2012;28:316–21.
28. Abou-Khalil B, Andermann E, Andermann F, et al. Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia 1993;34:878–83.
29. Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol 2004;17:161–4.
30. Hesdorffer DC, Chan S, Tian H, et al. Are MRI-detected brain abnormalities associated with febrile seizure type? Epilepsia 2008;49:765–71.
31. Patel AD, Vidaurre J. Complex febrile seizures: a practical guide to evaluation and treatment. J Child Neurol 2013;28:762–67.
32. Pavlidou E, Tzitiridou M, Panteliadis C. Effectiveness of intermittent diazepam prophylaxis in febrile seizures: long-term prospective controlled study. J Child Neurol 2006;21:1036–40.
33. Offringa M, Newton R. Prophylactic drug management for febrile seizures in children (Review). Evid Based Child Health 2013;8:1376–485.
34. Gordon KE, Dooley JM, Camfield PR, et al. Treatment of febrile seizures: the influence of treatment efficacy and side-effect profile on value to parents. Pediatrics 2001;108:1080–8.
35. Maytal J, Shinnar S. Febrile status epilepticus. Pediatrics 1990;86:611–6.
36. Ahmad S, Marsh ED. Febrile status epilepticus: current state of clinical and basic research. Semin Pediatr Neurol 2010;17:150–4.
37. Maytal J, Shinnar S, Moshe SL, Alvarez LA. Low morbidity and mortality of status epilepticus in children. Pediatrics 1989;83:323–31.
38. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120 (Pt 3):479–90.
39. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–85.
40. Wallace RH, Scheffer IE, Parasivam G, et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 2002;58:1426–9.
41. Cross JH. Fever and fever-related epilepsies. Epilepsia 2012;53 Suppl 4:3–8.
42. Dravet C. Les epilepsies graves de l’enfant. Vie Med 1978;8:543–8.
43. Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53 Suppl 2:1–6.
44. Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 2006;47 Suppl 2:45–8.
45. Ogino T, Ohtsuka Y, Amano R, et al. An investigation on the borderland of severe myoclonic epilepsy in infancy. Jap J Psych Neurol 1988;42:554–5.
46. Kanazawa O. Refractory grand mal seizures with onset during infancy including severe myoclonic epilepsy in infancy. Brain Dev 2001;23:749–56.
47. van der Worp HB, Claus SP, Bar PR, et al. Reproducibility of measurements of cerebral infarct volume on CT scans. Stroke 2001;32:424–30.
48. Wang JW, Kurahashi H, Ishii A, et al. Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia 2008;49:1528–34.
49. Madia F, Striano P, Gennaro E, et al. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology 2006;67:1230–5.
50. Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–8.
51. Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002;49:45–8.
52. Nieto-Barrera M, Candau R, Nieto-Jimenez M, et al. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure 2000;9:590–4.
53. Striano P, Coppola A, Pezzella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007;69:250–4.
54. Caraballo RH, Cersosimo RO, Sakr D, et al. Ketogenic diet in patients with Dravet syndrome. Epilepsia 2005;46:1539–44.
55. Kang HC, Kim YJ, Kim DW, Kim HD. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia 2005;46:272–9.
56. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011;53 Suppl 2:16–8.
57. Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81.
58. Specchio N, Marini C, Terracciano A, et al. Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 2011;52:1251–7.
59. Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–9.
60. Hirano S, Yan Q, Suzuki ST. Expression of a novel protocadherin, OL-protocadherin, in a subset of functional systems of the developing mouse brain. J Neurosci 1999;19:995–1005.
61. Kim SY, Chung HS, Sun W, Kim H. Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 2007;147:996–1021.
62. Gastaut H, Poirier F, Payan H, et al. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia 1960;1:418–47.
63. Panayiotopoulos CP. The epilepsies: seizures, syndromes and management. Oxfordshire (UK): Bladon Medical Publishing; 2005.
64. Chauvel P, Dravet C. The HHE syndrome. In: Roger J, Bureau M, Dravet C, editors. Epileptic syndromes in infancy, childhood and adolescence. 4th ed. John Libbey; 2005; 247–60.
65. Nabbout R. FIRES and IHHE: Delineation of the syndromes. Epilepsia 2013;54 Suppl 6:54–6.
66. Baxter P, Clarke A, Cross H, et al. Idiopathic catastrophic epileptic encephalopathy presenting with acute onset intractable status. Seizure 2003;12:379–87.
67. Mikaeloff Y, Jambaque I, Hertz-Pannier L, et al. Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis. Epilepsy Res 2006;69:67–79.
68. Wilder-Smith EP, Lim EC, Teoh HL, et al. The NORSE (new-onset refractory status epilepticus) syndrome: defining a disease entity. Ann Acad Med Singapore 2005;34:417–20.
69. van Baalen A, Hausler M, Boor R, et al. Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood. Epilepsia 2010;51:1323–8.
70. Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011;10:99–108.
71. Howell KB, Katanyuwong K, Mackay MT, et al. Long-term follow-up of febrile infection-related epilepsy syndrome. Epilepsia 2012;53:101–10.
72. Nabbout R, Mazzuca M, Hubert P, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010;51:2033–7.
73. Lin JJ, Lin KL, Hsia SH, Wang HS. Therapeutic hypothermia for febrile infection-related epilepsy syndrome in two patients. Pediatr Neurol 2012;47:448–50.
1. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002;17 Suppl 1:S44–52.
2. Kang J-Q, Shen W, Macdonald RL. Why does fever trigger febrile seizures? GABAA receptor γ2 subunit mutations associated with idiopathic generalized epilepsies have temperature-dependent trafficking deficiencies. J Neurosci 2006;26:2590–7.
3. Baumann RJ, Duffner PK. Treatment of children with simple febrile seizures: the AAP practice parameter. American Academy of Pediatrics. Pediatr Neurol 2000;23:11–7.
4. Tarkka R, Rantala H, Uhari M, Pokka T. Risk of recurrence and outcome after the first febrile seizure. Pediatr Neurol 1998;18:218–20.
5. Berg AT, Shinnar S. Complex febrile seizures. Epilepsia 1996;37:126–33.
6. Berg AT, Shinnar S. Unprovoked seizures in children with febrile seizures: short-term outcome. Neurology 1996;47:562–8.
7. Audenaert D, Schwartz E, Claeys KG, et al. A novel GABRG2 mutation associated with febrile seizures. Neurology 2006;67:687–90.
8. Dube CM, Brewster AL, Baram TZ. Febrile seizures: mechanisms and relationship to epilepsy. Brain Dev 2009;31:366–71.
9. Vestergaard M, Christensen J. Register-based studies on febrile seizures in Denmark. Brain Dev 2009;31:372–7.
10. Berkovic SF, Petrou S. Febrile seizures: traffic slows in the heat. Trends Molecul Med 2006;12:343–4.
11. Practice parameter: the neurodiagnostic evaluation of the child with a first simple febrile seizure. American Academy of Pediatrics. Provisional Committee on Quality Improvement, Subcommittee on Febrile Seizures. Pediatrics 1996;97:769–72; discussion 773–765.
12. Fukuyama Y, Seki T, Ohtsuka C, et al. Practical guidelines for physicians in the management of febrile seizures. Brain Dev 1996;18:479–84.
13. Neurodiagnostic evaluation of the child with a simple febrile seizure. Pediatrics 2011;127:389–94.
14. Guedj R, Chappuy H, Titomanlio L, et al. Risk of bacterial meningitis in children 6 to 11 months of age with a first simple febrile seizure: a retrospective, cross-sectional, observational study. Acad Emerg Med 2015;22:1290–7.
15. Wo SB, Lee JH, Lee YJ, et al. Risk for developing epilepsy and epileptiform discharges on EEG in patients with febrile seizures. Brain Dev 2013;35:307–11.
16. Green SM, Rothrock SG, Clem KJ, et al. Can seizures be the sole manifestation of meningitis in febrile children? Pediatrics 1993;92:527–34.
17. Knudsen FU. Febrile seizures: treatment and prognosis. Epilepsia 2000;41:2–9.
18. Annegers JF, Hauser WA, Shirts SB, Kurland LT. Factors prognostic of unprovoked seizures after febrile convulsions. N Engl J Med 1987;316:493–8.
19. Teng D, Dayan P, Tyler S, et al. Risk of intracranial pathologic conditions requiring emergency intervention after a first complex febrile seizure episode among children. Pediatrics 2006;117:304–8.
20. Janszky J, Schulz R, Ebner A. Clinical features and surgical outcome of medial temporal lobe epilepsy with a history of complex febrile convulsions. Epilepsy Res 2003;55:1–8.
21. Capovilla G, Mastrangelo M, Romeo A, Vigevano F. Recommendations for the management of «febrile seizures»: Ad Hoc Task Force of LICE Guidelines Commission. Epilepsia 2009;50 Suppl 1:2–6.
22. Shinnar S, Hesdorffer DC, Nordli DR Jr, et al. Phenomenology of prolonged febrile seizures: results of the FEBSTAT study. Neurology 2008;71:170–6.
23. Camfield P, Camfield C, Gordon K, Dooley J. What types of epilepsy are preceded by febrile seizures? A population-based study of children. Dev Med Child Neurol 1994;36:887–92.
24. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures. N Engl J Med 1976;295:1029–33.
25. Hamati-Haddad A, Abou-Khalil B. Epilepsy diagnosis and localization in patients with antecedent childhood febrile convulsions. Neurology 1998;50:917–22.
26. Davies KG, Hermann BP, Dohan FC Jr, et al. Relationship of hippocampal sclerosis to duration and age of onset of epilepsy, and childhood febrile seizures in temporal lobectomy patients. Epilepsy Res 1996;24:119–26.
27. Kimia AA, Ben-Joseph E, Prabhu S, et al. Yield of emergent neuroimaging among children presenting with a first complex febrile seizure. Pediatr Emerg Care 2012;28:316–21.
28. Abou-Khalil B, Andermann E, Andermann F, et al. Temporal lobe epilepsy after prolonged febrile convulsions: excellent outcome after surgical treatment. Epilepsia 1993;34:878–83.
29. Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol 2004;17:161–4.
30. Hesdorffer DC, Chan S, Tian H, et al. Are MRI-detected brain abnormalities associated with febrile seizure type? Epilepsia 2008;49:765–71.
31. Patel AD, Vidaurre J. Complex febrile seizures: a practical guide to evaluation and treatment. J Child Neurol 2013;28:762–67.
32. Pavlidou E, Tzitiridou M, Panteliadis C. Effectiveness of intermittent diazepam prophylaxis in febrile seizures: long-term prospective controlled study. J Child Neurol 2006;21:1036–40.
33. Offringa M, Newton R. Prophylactic drug management for febrile seizures in children (Review). Evid Based Child Health 2013;8:1376–485.
34. Gordon KE, Dooley JM, Camfield PR, et al. Treatment of febrile seizures: the influence of treatment efficacy and side-effect profile on value to parents. Pediatrics 2001;108:1080–8.
35. Maytal J, Shinnar S. Febrile status epilepticus. Pediatrics 1990;86:611–6.
36. Ahmad S, Marsh ED. Febrile status epilepticus: current state of clinical and basic research. Semin Pediatr Neurol 2010;17:150–4.
37. Maytal J, Shinnar S, Moshe SL, Alvarez LA. Low morbidity and mortality of status epilepticus in children. Pediatrics 1989;83:323–31.
38. Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120 (Pt 3):479–90.
39. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–85.
40. Wallace RH, Scheffer IE, Parasivam G, et al. Generalized epilepsy with febrile seizures plus: mutation of the sodium channel subunit SCN1B. Neurology 2002;58:1426–9.
41. Cross JH. Fever and fever-related epilepsies. Epilepsia 2012;53 Suppl 4:3–8.
42. Dravet C. Les epilepsies graves de l’enfant. Vie Med 1978;8:543–8.
43. Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53 Suppl 2:1–6.
44. Wolff M, Casse-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia 2006;47 Suppl 2:45–8.
45. Ogino T, Ohtsuka Y, Amano R, et al. An investigation on the borderland of severe myoclonic epilepsy in infancy. Jap J Psych Neurol 1988;42:554–5.
46. Kanazawa O. Refractory grand mal seizures with onset during infancy including severe myoclonic epilepsy in infancy. Brain Dev 2001;23:749–56.
47. van der Worp HB, Claus SP, Bar PR, et al. Reproducibility of measurements of cerebral infarct volume on CT scans. Stroke 2001;32:424–30.
48. Wang JW, Kurahashi H, Ishii A, et al. Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia 2008;49:1528–34.
49. Madia F, Striano P, Gennaro E, et al. Cryptic chromosome deletions involving SCN1A in severe myoclonic epilepsy of infancy. Neurology 2006;67:1230–5.
50. Marini C, Scheffer IE, Nabbout R, et al. SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 2009;50:1670–8.
51. Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res 2002;49:45–8.
52. Nieto-Barrera M, Candau R, Nieto-Jimenez M, et al. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure 2000;9:590–4.
53. Striano P, Coppola A, Pezzella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007;69:250–4.
54. Caraballo RH, Cersosimo RO, Sakr D, et al. Ketogenic diet in patients with Dravet syndrome. Epilepsia 2005;46:1539–44.
55. Kang HC, Kim YJ, Kim DW, Kim HD. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia 2005;46:272–9.
56. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011;53 Suppl 2:16–8.
57. Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet 2008;40:776–81.
58. Specchio N, Marini C, Terracciano A, et al. Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 2011;52:1251–7.
59. Marini C, Darra F, Specchio N, et al. Focal seizures with affective symptoms are a major feature of PCDH19 gene-related epilepsy. Epilepsia 2012;53:2111–9.
60. Hirano S, Yan Q, Suzuki ST. Expression of a novel protocadherin, OL-protocadherin, in a subset of functional systems of the developing mouse brain. J Neurosci 1999;19:995–1005.
61. Kim SY, Chung HS, Sun W, Kim H. Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience 2007;147:996–1021.
62. Gastaut H, Poirier F, Payan H, et al. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia 1960;1:418–47.
63. Panayiotopoulos CP. The epilepsies: seizures, syndromes and management. Oxfordshire (UK): Bladon Medical Publishing; 2005.
64. Chauvel P, Dravet C. The HHE syndrome. In: Roger J, Bureau M, Dravet C, editors. Epileptic syndromes in infancy, childhood and adolescence. 4th ed. John Libbey; 2005; 247–60.
65. Nabbout R. FIRES and IHHE: Delineation of the syndromes. Epilepsia 2013;54 Suppl 6:54–6.
66. Baxter P, Clarke A, Cross H, et al. Idiopathic catastrophic epileptic encephalopathy presenting with acute onset intractable status. Seizure 2003;12:379–87.
67. Mikaeloff Y, Jambaque I, Hertz-Pannier L, et al. Devastating epileptic encephalopathy in school-aged children (DESC): a pseudo encephalitis. Epilepsy Res 2006;69:67–79.
68. Wilder-Smith EP, Lim EC, Teoh HL, et al. The NORSE (new-onset refractory status epilepticus) syndrome: defining a disease entity. Ann Acad Med Singapore 2005;34:417–20.
69. van Baalen A, Hausler M, Boor R, et al. Febrile infection-related epilepsy syndrome (FIRES): a nonencephalitic encephalopathy in childhood. Epilepsia 2010;51:1323–8.
70. Nabbout R, Vezzani A, Dulac O, Chiron C. Acute encephalopathy with inflammation-mediated status epilepticus. Lancet Neurol 2011;10:99–108.
71. Howell KB, Katanyuwong K, Mackay MT, et al. Long-term follow-up of febrile infection-related epilepsy syndrome. Epilepsia 2012;53:101–10.
72. Nabbout R, Mazzuca M, Hubert P, et al. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010;51:2033–7.
73. Lin JJ, Lin KL, Hsia SH, Wang HS. Therapeutic hypothermia for febrile infection-related epilepsy syndrome in two patients. Pediatr Neurol 2012;47:448–50.
Childhood vaccine trauma decreases adolescent HPV immunization uptake
The more vaccines children get at any one office visit between the ages of 4 and 6 years, the more fearful they are of needles later on and the less likely they are to start human papillomavirus vaccine (HPV) as adolescents, according to an investigation of 120 children.
Shot-stacking between ages 4 and 6 years is not uncommon, especially if children might not be back in the office any time soon. Although it’s convenient and often better reimbursed to give all the recommended 4- to 6-year-old shots – MMR, DTaP, varicella, IPV, and maybe flu and hepatitis B vaccines – in one visit, the investigators found a hidden cost.

“We don’t need to change the vaccine schedule,” but we do have to be careful because 4- to 6-year-olds are especially vulnerable to phobias. “I think the best thing would be to give one vaccine at age 4, two at age 5, and two at age 6,” years, said lead investigator and pediatric emergency physician Amy Baxter, MD, a clinical associate professor at the Medical College of Georgia, Augusta.
“I’ve dealt with needle phobia in the ED. These kids come in and they are already freaked out about needles. If they get diagnosed with diabetes or leukemia, parents don’t think they are going to be able to handle it,” she said in an interview.
Expanded school vaccination programs might help by making it more convenient for parents to space out shots. The development of patch, microneedle, or effective sublingual or intranasal options also would help.
Dr. Baxter and her colleagues asked 120 children aged 10 to 12 years old to rate their fear of needles on a visual analogue scale (VAS), from 0 points meaning no fear to 100. The investigators matched the scores against the children’s immunization records (Vaccine. 2017 Jun 20. doi: 10.1016/j.vaccine.2017.06.029).
There was no significant relationship between needle fear and the total number of injections. However, fear did correlate with the number of shots children received in 1 day from ages 4 to 6 years old. Six children (50%) who had four same-day injections during that period scored in the upper quartile of anxiety as adolescents (VAS greater than 83), versus 22 (27%) who had three, and 2 (10%) who had two. None of the children who had just one shot per office visit as preschoolers were very worried about needles (P = .0387).
The investigators found that the likelihood of being in the upper quartile of fear as adolescents increased with each additional same-day injection between the ages of 4 and 6 years (odds ratio, 3.108; 95% confidence interval, 1.311-7.367; P = .01).
The team checked back with the children a few years later to see who had started HPV immunizations by age 14 years. Just 27% of children in the upper quartile of needle fear had done so, versus 48% in the least anxious quartile (VAS less than 32). The study wasn’t powered to detect a statistically significant difference in HPV vaccine uptake, but decreased uptake with higher needle fear came close (OR, 2.57; 95% CI, 0.864-7.621; P = .0889).
“It’s the stacking that causes fear. There was no difference in the uptake of vaccines when they were spread out, but more shots” at one time “causes more distress, so there was a difference in fear 5 years later,” Dr. Baxter said.
The investigators also assessed parental concerns about immunizations. “What was interesting was that whether the parents were in the upper or lower quartile of anxiety didn’t impact HPV initiation at all. The children’s anxiety 2 years earlier seemed much more relevant,” Dr. Baxter said.
In a literature review of 15 studies from 1958-2016 that included over 8,000 subjects, the investigators also found that needle fear tracked neatly with the sixfold increase in scheduled vaccines since the 1970s, with more than 60% of people now endorsing some degree of injection anxiety – more than ever before. “The curve of increasing needle fear correlated strongly with increasing vaccine number,” they found (Kendall’s tau b = 0.747; 95% CI, 0.513-0.982; P = .0003).
As two-thirds of the literature review sample were adults, “these results imply that fear acquired in childhood persists,” they concluded.
The majority of children in the study were white. Their mothers were a mean of 44 years, with a mean education level of 18 years. Demographic differences didn’t affect needle fear.
Dr. Baxter is a pediatric pain researcher and the inventor of Buzzy, a bumblebee shaped vibrator to distract children and relieve pain during shots. When asked if readers could trust her study findings given her interest in selling the device, she noted that, in a previous study, “Buzzy for 15 seconds did not work well for 4 to 6 year olds” getting multiple injections at one office visit. “Buzzy is not the solution for giving four shots at once.”
The more vaccines children get at any one office visit between the ages of 4 and 6 years, the more fearful they are of needles later on and the less likely they are to start human papillomavirus vaccine (HPV) as adolescents, according to an investigation of 120 children.
Shot-stacking between ages 4 and 6 years is not uncommon, especially if children might not be back in the office any time soon. Although it’s convenient and often better reimbursed to give all the recommended 4- to 6-year-old shots – MMR, DTaP, varicella, IPV, and maybe flu and hepatitis B vaccines – in one visit, the investigators found a hidden cost.
“We don’t need to change the vaccine schedule,” but we do have to be careful because 4- to 6-year-olds are especially vulnerable to phobias. “I think the best thing would be to give one vaccine at age 4, two at age 5, and two at age 6,” years, said lead investigator and pediatric emergency physician Amy Baxter, MD, a clinical associate professor at the Medical College of Georgia, Augusta.
“I’ve dealt with needle phobia in the ED. These kids come in and they are already freaked out about needles. If they get diagnosed with diabetes or leukemia, parents don’t think they are going to be able to handle it,” she said in an interview.
Expanded school vaccination programs might help by making it more convenient for parents to space out shots. The development of patch, microneedle, or effective sublingual or intranasal options also would help.
Dr. Baxter and her colleagues asked 120 children aged 10 to 12 years old to rate their fear of needles on a visual analogue scale (VAS), from 0 points meaning no fear to 100. The investigators matched the scores against the children’s immunization records (Vaccine. 2017 Jun 20. doi: 10.1016/j.vaccine.2017.06.029).
There was no significant relationship between needle fear and the total number of injections. However, fear did correlate with the number of shots children received in 1 day from ages 4 to 6 years old. Six children (50%) who had four same-day injections during that period scored in the upper quartile of anxiety as adolescents (VAS greater than 83), versus 22 (27%) who had three, and 2 (10%) who had two. None of the children who had just one shot per office visit as preschoolers were very worried about needles (P = .0387).
The investigators found that the likelihood of being in the upper quartile of fear as adolescents increased with each additional same-day injection between the ages of 4 and 6 years (odds ratio, 3.108; 95% confidence interval, 1.311-7.367; P = .01).
The team checked back with the children a few years later to see who had started HPV immunizations by age 14 years. Just 27% of children in the upper quartile of needle fear had done so, versus 48% in the least anxious quartile (VAS less than 32). The study wasn’t powered to detect a statistically significant difference in HPV vaccine uptake, but decreased uptake with higher needle fear came close (OR, 2.57; 95% CI, 0.864-7.621; P = .0889).
“It’s the stacking that causes fear. There was no difference in the uptake of vaccines when they were spread out, but more shots” at one time “causes more distress, so there was a difference in fear 5 years later,” Dr. Baxter said.
The investigators also assessed parental concerns about immunizations. “What was interesting was that whether the parents were in the upper or lower quartile of anxiety didn’t impact HPV initiation at all. The children’s anxiety 2 years earlier seemed much more relevant,” Dr. Baxter said.
In a literature review of 15 studies from 1958-2016 that included over 8,000 subjects, the investigators also found that needle fear tracked neatly with the sixfold increase in scheduled vaccines since the 1970s, with more than 60% of people now endorsing some degree of injection anxiety – more than ever before. “The curve of increasing needle fear correlated strongly with increasing vaccine number,” they found (Kendall’s tau b = 0.747; 95% CI, 0.513-0.982; P = .0003).
As two-thirds of the literature review sample were adults, “these results imply that fear acquired in childhood persists,” they concluded.
The majority of children in the study were white. Their mothers were a mean of 44 years, with a mean education level of 18 years. Demographic differences didn’t affect needle fear.
Dr. Baxter is a pediatric pain researcher and the inventor of Buzzy, a bumblebee shaped vibrator to distract children and relieve pain during shots. When asked if readers could trust her study findings given her interest in selling the device, she noted that, in a previous study, “Buzzy for 15 seconds did not work well for 4 to 6 year olds” getting multiple injections at one office visit. “Buzzy is not the solution for giving four shots at once.”
The more vaccines children get at any one office visit between the ages of 4 and 6 years, the more fearful they are of needles later on and the less likely they are to start human papillomavirus vaccine (HPV) as adolescents, according to an investigation of 120 children.
Shot-stacking between ages 4 and 6 years is not uncommon, especially if children might not be back in the office any time soon. Although it’s convenient and often better reimbursed to give all the recommended 4- to 6-year-old shots – MMR, DTaP, varicella, IPV, and maybe flu and hepatitis B vaccines – in one visit, the investigators found a hidden cost.
“We don’t need to change the vaccine schedule,” but we do have to be careful because 4- to 6-year-olds are especially vulnerable to phobias. “I think the best thing would be to give one vaccine at age 4, two at age 5, and two at age 6,” years, said lead investigator and pediatric emergency physician Amy Baxter, MD, a clinical associate professor at the Medical College of Georgia, Augusta.
“I’ve dealt with needle phobia in the ED. These kids come in and they are already freaked out about needles. If they get diagnosed with diabetes or leukemia, parents don’t think they are going to be able to handle it,” she said in an interview.
Expanded school vaccination programs might help by making it more convenient for parents to space out shots. The development of patch, microneedle, or effective sublingual or intranasal options also would help.
Dr. Baxter and her colleagues asked 120 children aged 10 to 12 years old to rate their fear of needles on a visual analogue scale (VAS), from 0 points meaning no fear to 100. The investigators matched the scores against the children’s immunization records (Vaccine. 2017 Jun 20. doi: 10.1016/j.vaccine.2017.06.029).
There was no significant relationship between needle fear and the total number of injections. However, fear did correlate with the number of shots children received in 1 day from ages 4 to 6 years old. Six children (50%) who had four same-day injections during that period scored in the upper quartile of anxiety as adolescents (VAS greater than 83), versus 22 (27%) who had three, and 2 (10%) who had two. None of the children who had just one shot per office visit as preschoolers were very worried about needles (P = .0387).
The investigators found that the likelihood of being in the upper quartile of fear as adolescents increased with each additional same-day injection between the ages of 4 and 6 years (odds ratio, 3.108; 95% confidence interval, 1.311-7.367; P = .01).
The team checked back with the children a few years later to see who had started HPV immunizations by age 14 years. Just 27% of children in the upper quartile of needle fear had done so, versus 48% in the least anxious quartile (VAS less than 32). The study wasn’t powered to detect a statistically significant difference in HPV vaccine uptake, but decreased uptake with higher needle fear came close (OR, 2.57; 95% CI, 0.864-7.621; P = .0889).
“It’s the stacking that causes fear. There was no difference in the uptake of vaccines when they were spread out, but more shots” at one time “causes more distress, so there was a difference in fear 5 years later,” Dr. Baxter said.
The investigators also assessed parental concerns about immunizations. “What was interesting was that whether the parents were in the upper or lower quartile of anxiety didn’t impact HPV initiation at all. The children’s anxiety 2 years earlier seemed much more relevant,” Dr. Baxter said.
In a literature review of 15 studies from 1958-2016 that included over 8,000 subjects, the investigators also found that needle fear tracked neatly with the sixfold increase in scheduled vaccines since the 1970s, with more than 60% of people now endorsing some degree of injection anxiety – more than ever before. “The curve of increasing needle fear correlated strongly with increasing vaccine number,” they found (Kendall’s tau b = 0.747; 95% CI, 0.513-0.982; P = .0003).
As two-thirds of the literature review sample were adults, “these results imply that fear acquired in childhood persists,” they concluded.
The majority of children in the study were white. Their mothers were a mean of 44 years, with a mean education level of 18 years. Demographic differences didn’t affect needle fear.
Dr. Baxter is a pediatric pain researcher and the inventor of Buzzy, a bumblebee shaped vibrator to distract children and relieve pain during shots. When asked if readers could trust her study findings given her interest in selling the device, she noted that, in a previous study, “Buzzy for 15 seconds did not work well for 4 to 6 year olds” getting multiple injections at one office visit. “Buzzy is not the solution for giving four shots at once.”
FROM VACCINE
Key clinical point:
Major finding: Six children (50%) who had four same-day injections during that period scored in the upper quartile of anxiety as adolescents (VAS greater than 83), versus 22 (27%) who had three, and 2 (10%) who had two same-day injections. Just 27% of children in the upper quartile of needle fear had started HPV immunization at age 14 years, versus 48% in the least anxious quartile.
Data source: Survey and follow-up of 120 children and their parents.
Disclosures: The lead investigator is a pediatric pain researcher and the inventor of Buzzy, a bumblebee shaped vibrator to distract children and relieve pain during shots. When asked if readers could trust her findings given her interest in selling the device, she noted that, in a previous study, “Buzzy for 15 seconds did not work well for 4-6 year olds” getting multiple injections in one office visit. “Buzzy is not the solution for giving four shots at once.”
English infant quadravalent group B meningococcal vaccine pays off
MADRID – Cases of invasive meningococcal disease in English 1 year olds dropped fourfold in the first year after the multicomponent group B meningococcal (MenB) vaccine (4CMenB, Bexsero) was added to the national infant immunization list, Shamez Ladhani, MD, Ph.D, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“In 1 year olds, we have seen a significant decrease in the number of cases, compared with the number of cases predicted. I suspect the vaccine effectiveness is close to 94% against meningococcal group B,” said Dr. Ladhani of Public Health England, London.

Meningococcal meningitis and septicemia remain the leading cause of death for children under 5 in the UK. Public awareness of this threat is high, so 4CMenB has been well accepted. Of eligible infants, 96% have received the first dose and 89% the second. The first cohort became available for the 12-month booster dose in May 2016.
Between May and December of 2016, the Public Health England intensive surveillance program confirmed six cases of invasive meningococcal disease. During the same time frame in 2015, there were 24 cases, whereas, during the 4 years prior to introduction of 4CMenB, the average was 18 cases per year.
Three of the six 1-year-olds had meningitis, and three had septicemia. One was admitted to an ICU. None died. None had comorbid conditions placing them at increased risk. Five of the six children with invasive meningococcal disease had received two doses of the vaccine, and one child became ill 3 months after receiving the third dose.
The vaccine is licensed to be given as a four-dose series: three primary doses plus one booster dose. UK health officials deemed that excessive and not cost effective. Based on data from the vaccine trials, they determined that three doses are sufficient. This cut the cost of the national program by 25% while maintaining protection (Lancet. 2016 Dec 3;388(10061):2775-82).
Pediatricians from other countries were extremely curious about this innovative immunization program. What about vaccine side effects in infants? they asked.
Dr. Ladhani replied that studies in Australia, Northern Ireland, and Scotland have all shown a small uptick in mild fever and irritability in the first 3 days after the first dose of 4CMenB. It’s less of an issue with the second dose.
“One dose is not protective. You need two. Parents understand that, while these side effects are annoying, the risks associated with meningitis are far greater. They are very, very accepting of the vaccine,” according to Dr. Ladhani.
What about using 4CMenB in teenagers? physicians asked.
Dr. Ladhani predicted that the infant immunization program will have zero effect in adolescents. Teens are the main carriers of meningococcal bacteria. So, in theory, vaccinating them could not only protect the adolescents themselves but could benefit the whole population through herd immunity.
“The problem is we don’t have solid evidence that Bexsero protects against carriage. A massive carriage study is underway in Australia. It should provide data in 2 or 3 years. If we do see a carriage effect, then vaccinating teenagers becomes a very attractive option because you protect them and others around them,” he explained.
Dr. Ladhani reported having no financial conflicts regarding his study, funded by Public Health England.
MADRID – Cases of invasive meningococcal disease in English 1 year olds dropped fourfold in the first year after the multicomponent group B meningococcal (MenB) vaccine (4CMenB, Bexsero) was added to the national infant immunization list, Shamez Ladhani, MD, Ph.D, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“In 1 year olds, we have seen a significant decrease in the number of cases, compared with the number of cases predicted. I suspect the vaccine effectiveness is close to 94% against meningococcal group B,” said Dr. Ladhani of Public Health England, London.
Meningococcal meningitis and septicemia remain the leading cause of death for children under 5 in the UK. Public awareness of this threat is high, so 4CMenB has been well accepted. Of eligible infants, 96% have received the first dose and 89% the second. The first cohort became available for the 12-month booster dose in May 2016.
Between May and December of 2016, the Public Health England intensive surveillance program confirmed six cases of invasive meningococcal disease. During the same time frame in 2015, there were 24 cases, whereas, during the 4 years prior to introduction of 4CMenB, the average was 18 cases per year.
Three of the six 1-year-olds had meningitis, and three had septicemia. One was admitted to an ICU. None died. None had comorbid conditions placing them at increased risk. Five of the six children with invasive meningococcal disease had received two doses of the vaccine, and one child became ill 3 months after receiving the third dose.
The vaccine is licensed to be given as a four-dose series: three primary doses plus one booster dose. UK health officials deemed that excessive and not cost effective. Based on data from the vaccine trials, they determined that three doses are sufficient. This cut the cost of the national program by 25% while maintaining protection (Lancet. 2016 Dec 3;388(10061):2775-82).
Pediatricians from other countries were extremely curious about this innovative immunization program. What about vaccine side effects in infants? they asked.
Dr. Ladhani replied that studies in Australia, Northern Ireland, and Scotland have all shown a small uptick in mild fever and irritability in the first 3 days after the first dose of 4CMenB. It’s less of an issue with the second dose.
“One dose is not protective. You need two. Parents understand that, while these side effects are annoying, the risks associated with meningitis are far greater. They are very, very accepting of the vaccine,” according to Dr. Ladhani.
What about using 4CMenB in teenagers? physicians asked.
Dr. Ladhani predicted that the infant immunization program will have zero effect in adolescents. Teens are the main carriers of meningococcal bacteria. So, in theory, vaccinating them could not only protect the adolescents themselves but could benefit the whole population through herd immunity.
“The problem is we don’t have solid evidence that Bexsero protects against carriage. A massive carriage study is underway in Australia. It should provide data in 2 or 3 years. If we do see a carriage effect, then vaccinating teenagers becomes a very attractive option because you protect them and others around them,” he explained.
Dr. Ladhani reported having no financial conflicts regarding his study, funded by Public Health England.
MADRID – Cases of invasive meningococcal disease in English 1 year olds dropped fourfold in the first year after the multicomponent group B meningococcal (MenB) vaccine (4CMenB, Bexsero) was added to the national infant immunization list, Shamez Ladhani, MD, Ph.D, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“In 1 year olds, we have seen a significant decrease in the number of cases, compared with the number of cases predicted. I suspect the vaccine effectiveness is close to 94% against meningococcal group B,” said Dr. Ladhani of Public Health England, London.
Meningococcal meningitis and septicemia remain the leading cause of death for children under 5 in the UK. Public awareness of this threat is high, so 4CMenB has been well accepted. Of eligible infants, 96% have received the first dose and 89% the second. The first cohort became available for the 12-month booster dose in May 2016.
Between May and December of 2016, the Public Health England intensive surveillance program confirmed six cases of invasive meningococcal disease. During the same time frame in 2015, there were 24 cases, whereas, during the 4 years prior to introduction of 4CMenB, the average was 18 cases per year.
Three of the six 1-year-olds had meningitis, and three had septicemia. One was admitted to an ICU. None died. None had comorbid conditions placing them at increased risk. Five of the six children with invasive meningococcal disease had received two doses of the vaccine, and one child became ill 3 months after receiving the third dose.
The vaccine is licensed to be given as a four-dose series: three primary doses plus one booster dose. UK health officials deemed that excessive and not cost effective. Based on data from the vaccine trials, they determined that three doses are sufficient. This cut the cost of the national program by 25% while maintaining protection (Lancet. 2016 Dec 3;388(10061):2775-82).
Pediatricians from other countries were extremely curious about this innovative immunization program. What about vaccine side effects in infants? they asked.
Dr. Ladhani replied that studies in Australia, Northern Ireland, and Scotland have all shown a small uptick in mild fever and irritability in the first 3 days after the first dose of 4CMenB. It’s less of an issue with the second dose.
“One dose is not protective. You need two. Parents understand that, while these side effects are annoying, the risks associated with meningitis are far greater. They are very, very accepting of the vaccine,” according to Dr. Ladhani.
What about using 4CMenB in teenagers? physicians asked.
Dr. Ladhani predicted that the infant immunization program will have zero effect in adolescents. Teens are the main carriers of meningococcal bacteria. So, in theory, vaccinating them could not only protect the adolescents themselves but could benefit the whole population through herd immunity.
“The problem is we don’t have solid evidence that Bexsero protects against carriage. A massive carriage study is underway in Australia. It should provide data in 2 or 3 years. If we do see a carriage effect, then vaccinating teenagers becomes a very attractive option because you protect them and others around them,” he explained.
Dr. Ladhani reported having no financial conflicts regarding his study, funded by Public Health England.
AT ESPID 2017
Key clinical point:
Major finding: The number of confirmed cases of invasive meningococcal disease in English 1-year-olds dropped to six the year after introduction of the 4CMenB vaccine in the infant immunization schedule, down from 24 cases the previous year.
Data source: An enhanced surveillance study utilizing a combination of laboratory, public health, and clinical reporting to track and confirm all cases of invasive meningococcal disease in English 1-year-olds.
Disclosures: The study was funded by Public Health England. The presenter, who is employed by the agency, reported having no financial conflicts.
Transient tachypnea of newborn increases bronchiolitis risk
MADRID – A new Finnish study raises a provocative question: Is transient tachypnea of the newborn really transient?
Transient tachypnea of the newborn (TTN) has traditionally been viewed as a benign, self-limited condition involving 1-3 days of respiratory distress. But data from Finland’s comprehensive national health registries indicate that TTN in term babies is associated with significantly increased risk of subsequent bronchiolitis during infancy, Otto Helve, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“This association suggests similar pathogenic mechanisms in transient tachypnea of the newborn and bronchiolitis. We suggest that an intrinsic defect in sodium ion–driven pulmonary fluid transport may predispose to clinically significant bronchiolitis during the first year of life,” said Dr. Helve, a pediatrician at the National Institute for Health and Welfare, Helsinki, and the University of Helsinki.
Of more than 1 million term babies born in Finland during 1996-2015, 17,569 were diagnosed with TTN. During the same period, 40,338 infants were hospitalized with a diagnosis of bronchiolitis attributable to respiratory syncytial virus infection.
In a multivariate analysis adjusted for birth year, gender, delivery method, gestational age, and parity, TTN was independently associated with a 1.2-fold increased risk of bronchiolitis during the first year of life.
Dr. Helve reported having no financial conflicts of interest regarding his study.

MADRID – A new Finnish study raises a provocative question: Is transient tachypnea of the newborn really transient?
Transient tachypnea of the newborn (TTN) has traditionally been viewed as a benign, self-limited condition involving 1-3 days of respiratory distress. But data from Finland’s comprehensive national health registries indicate that TTN in term babies is associated with significantly increased risk of subsequent bronchiolitis during infancy, Otto Helve, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“This association suggests similar pathogenic mechanisms in transient tachypnea of the newborn and bronchiolitis. We suggest that an intrinsic defect in sodium ion–driven pulmonary fluid transport may predispose to clinically significant bronchiolitis during the first year of life,” said Dr. Helve, a pediatrician at the National Institute for Health and Welfare, Helsinki, and the University of Helsinki.
Of more than 1 million term babies born in Finland during 1996-2015, 17,569 were diagnosed with TTN. During the same period, 40,338 infants were hospitalized with a diagnosis of bronchiolitis attributable to respiratory syncytial virus infection.
In a multivariate analysis adjusted for birth year, gender, delivery method, gestational age, and parity, TTN was independently associated with a 1.2-fold increased risk of bronchiolitis during the first year of life.
Dr. Helve reported having no financial conflicts of interest regarding his study.
MADRID – A new Finnish study raises a provocative question: Is transient tachypnea of the newborn really transient?
Transient tachypnea of the newborn (TTN) has traditionally been viewed as a benign, self-limited condition involving 1-3 days of respiratory distress. But data from Finland’s comprehensive national health registries indicate that TTN in term babies is associated with significantly increased risk of subsequent bronchiolitis during infancy, Otto Helve, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
“This association suggests similar pathogenic mechanisms in transient tachypnea of the newborn and bronchiolitis. We suggest that an intrinsic defect in sodium ion–driven pulmonary fluid transport may predispose to clinically significant bronchiolitis during the first year of life,” said Dr. Helve, a pediatrician at the National Institute for Health and Welfare, Helsinki, and the University of Helsinki.
Of more than 1 million term babies born in Finland during 1996-2015, 17,569 were diagnosed with TTN. During the same period, 40,338 infants were hospitalized with a diagnosis of bronchiolitis attributable to respiratory syncytial virus infection.
In a multivariate analysis adjusted for birth year, gender, delivery method, gestational age, and parity, TTN was independently associated with a 1.2-fold increased risk of bronchiolitis during the first year of life.
Dr. Helve reported having no financial conflicts of interest regarding his study.
AT ESPID 2017
Key clinical point:
Major finding: Transient tachypnea of the newborn was independently associated with a 1.2-fold increased risk of bronchiolitis during the first year of life of Finnish term babies.
Data source: This population-based case-control study included 17,569 Finnish term babies diagnosed with transient tachypnea of the newborn and 40,338 diagnosed with bronchiolitis.
Disclosures: Dr. Helve reported having no financial conflicts of interest.
Adoptions increasingly involve special needs, prenatal drug exposures
SAN FRANCISCO – International adoptions today almost exclusively involve children with special needs, and domestic adoptions are far more likely to involve children exposed prenatally to marijuana, opiates, or another drug, Lisa Prock, MD, MPH, found in a study.
“Pediatric health care providers reviewing referrals for adoption should be knowledgeable about the changing demographics of this population and the long-term implications of prenatal substance exposure, given increasing opiate and marijuana use nationally,” said Dr. Prock of Boston Children’s Hospital.

During the initial flood of international adoptions in the 1990s, families sought preadoption consultations, often centering on infectious disease, at an increasing number of adoption clinics. Even with the decline of international adoptions, however, prospective parents still frequently seek information from pediatric providers about children they are considering adopting, whether internationally or domestically.
Prospective parents can receive medical record reviews of children they are considering adopting, provided by adoption agencies or attorneys. Dr. Prock retrospectively reviewed all the preadoptive charts submitted to one adoption clinic during 2 years a decade apart: 63 charts in 2006 and 91 charts in 2016.
Domestic records for both years usually included information about family history, prenatal history, maternal substance abuse history, lab results, and, for newborns, a physical exam. International records, however, generally included only lab results and a physical exam, sometimes with limited information on substance abuse as well, at both time points.
The records reveal just how dramatically referrals have flipped from an international focus to a domestic one in the past decade. International adoption referrals dropped from 84% in 2006 to 29% in 2016 as domestic ones increased from 16% to 71%.
Children with special needs also account for a larger proportion of all adoption referrals today. Just a quarter of international adoption referrals in 2006 involved children with special needs, including cleft lip and/or palate and congenital heart disease, but nearly all international referrals (96%) involved special needs in 2016.
Similarly, domestic adoption referrals in which the child is known to have prenatal exposure to alcohol or drugs doubled from 30% in 2006 to 66% in 2016, driven predominantly by maternal use of marijuana and opiates, Dr. Prock found. It’s important both for providers and for prospective parents to be aware of the higher likelihood that an internationally adopted child will have significant medical and/or developmental needs and that domestically adopted children are more likely to have experienced prenatal drug or alcohol exposures.
“Prospective adoptive parents may benefit from increased understanding of the long-term impact of prenatal substance exposure to marijuana, opiates and other substances,” Dr. Prock said. She also noted the need for prospective parents to be aware of the mental health concerns that can co-occur with substance use even though they may not be reported in preadoptive medical records.
No external funding was used for the research. Dr. Prock had no relevant financial disclosures.
SAN FRANCISCO – International adoptions today almost exclusively involve children with special needs, and domestic adoptions are far more likely to involve children exposed prenatally to marijuana, opiates, or another drug, Lisa Prock, MD, MPH, found in a study.
“Pediatric health care providers reviewing referrals for adoption should be knowledgeable about the changing demographics of this population and the long-term implications of prenatal substance exposure, given increasing opiate and marijuana use nationally,” said Dr. Prock of Boston Children’s Hospital.
During the initial flood of international adoptions in the 1990s, families sought preadoption consultations, often centering on infectious disease, at an increasing number of adoption clinics. Even with the decline of international adoptions, however, prospective parents still frequently seek information from pediatric providers about children they are considering adopting, whether internationally or domestically.
Prospective parents can receive medical record reviews of children they are considering adopting, provided by adoption agencies or attorneys. Dr. Prock retrospectively reviewed all the preadoptive charts submitted to one adoption clinic during 2 years a decade apart: 63 charts in 2006 and 91 charts in 2016.
Domestic records for both years usually included information about family history, prenatal history, maternal substance abuse history, lab results, and, for newborns, a physical exam. International records, however, generally included only lab results and a physical exam, sometimes with limited information on substance abuse as well, at both time points.
The records reveal just how dramatically referrals have flipped from an international focus to a domestic one in the past decade. International adoption referrals dropped from 84% in 2006 to 29% in 2016 as domestic ones increased from 16% to 71%.
Children with special needs also account for a larger proportion of all adoption referrals today. Just a quarter of international adoption referrals in 2006 involved children with special needs, including cleft lip and/or palate and congenital heart disease, but nearly all international referrals (96%) involved special needs in 2016.
Similarly, domestic adoption referrals in which the child is known to have prenatal exposure to alcohol or drugs doubled from 30% in 2006 to 66% in 2016, driven predominantly by maternal use of marijuana and opiates, Dr. Prock found. It’s important both for providers and for prospective parents to be aware of the higher likelihood that an internationally adopted child will have significant medical and/or developmental needs and that domestically adopted children are more likely to have experienced prenatal drug or alcohol exposures.
“Prospective adoptive parents may benefit from increased understanding of the long-term impact of prenatal substance exposure to marijuana, opiates and other substances,” Dr. Prock said. She also noted the need for prospective parents to be aware of the mental health concerns that can co-occur with substance use even though they may not be reported in preadoptive medical records.
No external funding was used for the research. Dr. Prock had no relevant financial disclosures.
SAN FRANCISCO – International adoptions today almost exclusively involve children with special needs, and domestic adoptions are far more likely to involve children exposed prenatally to marijuana, opiates, or another drug, Lisa Prock, MD, MPH, found in a study.
“Pediatric health care providers reviewing referrals for adoption should be knowledgeable about the changing demographics of this population and the long-term implications of prenatal substance exposure, given increasing opiate and marijuana use nationally,” said Dr. Prock of Boston Children’s Hospital.
During the initial flood of international adoptions in the 1990s, families sought preadoption consultations, often centering on infectious disease, at an increasing number of adoption clinics. Even with the decline of international adoptions, however, prospective parents still frequently seek information from pediatric providers about children they are considering adopting, whether internationally or domestically.
Prospective parents can receive medical record reviews of children they are considering adopting, provided by adoption agencies or attorneys. Dr. Prock retrospectively reviewed all the preadoptive charts submitted to one adoption clinic during 2 years a decade apart: 63 charts in 2006 and 91 charts in 2016.
Domestic records for both years usually included information about family history, prenatal history, maternal substance abuse history, lab results, and, for newborns, a physical exam. International records, however, generally included only lab results and a physical exam, sometimes with limited information on substance abuse as well, at both time points.
The records reveal just how dramatically referrals have flipped from an international focus to a domestic one in the past decade. International adoption referrals dropped from 84% in 2006 to 29% in 2016 as domestic ones increased from 16% to 71%.
Children with special needs also account for a larger proportion of all adoption referrals today. Just a quarter of international adoption referrals in 2006 involved children with special needs, including cleft lip and/or palate and congenital heart disease, but nearly all international referrals (96%) involved special needs in 2016.
Similarly, domestic adoption referrals in which the child is known to have prenatal exposure to alcohol or drugs doubled from 30% in 2006 to 66% in 2016, driven predominantly by maternal use of marijuana and opiates, Dr. Prock found. It’s important both for providers and for prospective parents to be aware of the higher likelihood that an internationally adopted child will have significant medical and/or developmental needs and that domestically adopted children are more likely to have experienced prenatal drug or alcohol exposures.
“Prospective adoptive parents may benefit from increased understanding of the long-term impact of prenatal substance exposure to marijuana, opiates and other substances,” Dr. Prock said. She also noted the need for prospective parents to be aware of the mental health concerns that can co-occur with substance use even though they may not be reported in preadoptive medical records.
No external funding was used for the research. Dr. Prock had no relevant financial disclosures.
AT PAS 17
Key clinical point:
Major finding: International adoption referrals with special needs increased from 25% to 96% from 2006 to 2016; domestic referrals with prenatal substance exposure increased from 30% to 66% over the same time period.
Data source: A retrospective review of all preadoptive medical records at a single adoption clinic in 2006 and 2016.
Disclosures: The researchers did not receive external funding. Dr. Prock had no relevant financial disclosures.
T-cell product improves outcomes of haplo-HSCT
MADRID—Updated results of a phase 1/2 study suggest the T-cell product BPX-501 lowers the risks associated with haploidentical hematopoietic stem cell transplant (haplo-HSCT).
In this ongoing study, researchers are testing BPX-501 in pediatric patients undergoing haplo-HSCT to treat a range of hematologic disorders.
Patients treated thus far have experienced rapid engraftment and early hospital discharge, a low rate of acute graft-versus-host disease (GHVD), no extensive chronic GVHD, and a low rate of transplant-related mortality at 180 days.
“The combination of haploidentical transplantation and BPX-501 infusion is an effective strategy for children in need of an allograft lacking a compatible donor,” said study investigator Mattia Algeri, MD, of Ospedale Pediatrico Bambino Gesù in Rome, Italy.
Dr Algeri presented these results during the Presidential Symposium at the 22nd Congress of the European Hematology Association (EHA) as abstract S146.
The research was sponsored by Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate the T cells in the event of toxicity.
Rimiducid is used to activate the CaspaCIDe safety switch, which consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway.
The goal of this therapy is to allow physicians to more safely perform haplo-HSCTs.
Patients
Dr Algeri and his colleagues have tested BPX-501 in 98 pediatric patients treated at centers in Europe and the US.
Fifty-nine patients had non-malignant conditions, including primary immune deficiency (n=26), thalassemia major (n=8), sickle cell disease (n=5), Diamond-Blackfan anemia (n=2), Swachman-Diamond syndrome (n=1), Fanconi anemia (n=9), hemophagocytic lymphohistiocytosis (n=6), aplastic anemia (n=1), and osteoporosis (n=1).
Thirty-nine patients had malignant conditions, including acute lymphoblastic leukemia (ALL, n=21), acute myeloid leukemia (AML, n=14), myelodysplastic syndromes (MDS, n=3), and non-Hodgkin lymphoma (NHL, n=1).
The patients received BPX-501 after an alpha/beta T-cell-depleted haplo-HSCT. All patients had at least 6 months of follow-up.
Overall results
Ninety-five percent of the patients engrafted (93/98), and the researchers said they observed rapid recovery of T cells, B cells, and immunoglobulins.
At 180 days, the incidence of transplant-related mortality was 5%, and there were no cases of post-transplant lymphoproliferative disorder.
The cumulative incidence of grade 2-4 acute GVHD was 14%. For patients with at least 1 year of follow-up, the cumulative incidence of chronic GVHD at 1 year was 3%.
Eleven patients received rimiducid—10 who had uncontrollable acute GVHD and 1 who developed late acute GVHD. In all of these patients, GVHD resolved and has not recurred.
There were no adverse events associated with BPX-501 or rimiducid.
European cohort
Dr Algeri presented more detailed data on the 61 patients treated at centers in Europe.
Fifteen of these patients had ALL, 10 had AML, 16 had primary immune deficiency, 7 had thalassemia major, 1 had sickle cell disease, 2 had Diamond-Blackfan anemia, 5 had Fanconi anemia, 4 had hemophagocytic lymphohistiocytosis, and 1 had osteoporosis.
Their median age was 4.8 (range, 0.25-17), and 56% were male. The patients received busulfan-based conditioning (41%), total body irradiation (36%), treosulfan-based conditioning (18%), and other conditioning (5%).
Ninety-five percent of the patients had a parent donor, and the other 5% had a sibling donor. The median donor age was 36 (range, 19-50).
The patients’ median time to neutrophil recovery was 15 days (range, 9-75), and their median time to platelet recovery was 10 days (range, 4-64). Their median time to discharge was 25 days (range, 14-122).
The cumulative incidence of acute grade 2-4 GVHD was 9.9%, and the cumulative incidence of acute grade 3-4 GVHD was 3.3%.
There were no cases of transplant-related mortality at 180 days and no cases of extensive chronic GVHD.
“These preliminary data compare favorably to previously published data on matched, unrelated donor transplantation,” Dr Algeri said. “And for this reason, an observational matched, unrelated donor study is being initiated to enable comparison of the safety and efficacy of haploidentical transplantation and BPX-501 infusion to the standard of care for patients without a matched sibling donor.” ![]()
MADRID—Updated results of a phase 1/2 study suggest the T-cell product BPX-501 lowers the risks associated with haploidentical hematopoietic stem cell transplant (haplo-HSCT).
In this ongoing study, researchers are testing BPX-501 in pediatric patients undergoing haplo-HSCT to treat a range of hematologic disorders.
Patients treated thus far have experienced rapid engraftment and early hospital discharge, a low rate of acute graft-versus-host disease (GHVD), no extensive chronic GVHD, and a low rate of transplant-related mortality at 180 days.
“The combination of haploidentical transplantation and BPX-501 infusion is an effective strategy for children in need of an allograft lacking a compatible donor,” said study investigator Mattia Algeri, MD, of Ospedale Pediatrico Bambino Gesù in Rome, Italy.
Dr Algeri presented these results during the Presidential Symposium at the 22nd Congress of the European Hematology Association (EHA) as abstract S146.
The research was sponsored by Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate the T cells in the event of toxicity.
Rimiducid is used to activate the CaspaCIDe safety switch, which consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway.
The goal of this therapy is to allow physicians to more safely perform haplo-HSCTs.
Patients
Dr Algeri and his colleagues have tested BPX-501 in 98 pediatric patients treated at centers in Europe and the US.
Fifty-nine patients had non-malignant conditions, including primary immune deficiency (n=26), thalassemia major (n=8), sickle cell disease (n=5), Diamond-Blackfan anemia (n=2), Swachman-Diamond syndrome (n=1), Fanconi anemia (n=9), hemophagocytic lymphohistiocytosis (n=6), aplastic anemia (n=1), and osteoporosis (n=1).
Thirty-nine patients had malignant conditions, including acute lymphoblastic leukemia (ALL, n=21), acute myeloid leukemia (AML, n=14), myelodysplastic syndromes (MDS, n=3), and non-Hodgkin lymphoma (NHL, n=1).
The patients received BPX-501 after an alpha/beta T-cell-depleted haplo-HSCT. All patients had at least 6 months of follow-up.
Overall results
Ninety-five percent of the patients engrafted (93/98), and the researchers said they observed rapid recovery of T cells, B cells, and immunoglobulins.
At 180 days, the incidence of transplant-related mortality was 5%, and there were no cases of post-transplant lymphoproliferative disorder.
The cumulative incidence of grade 2-4 acute GVHD was 14%. For patients with at least 1 year of follow-up, the cumulative incidence of chronic GVHD at 1 year was 3%.
Eleven patients received rimiducid—10 who had uncontrollable acute GVHD and 1 who developed late acute GVHD. In all of these patients, GVHD resolved and has not recurred.
There were no adverse events associated with BPX-501 or rimiducid.
European cohort
Dr Algeri presented more detailed data on the 61 patients treated at centers in Europe.
Fifteen of these patients had ALL, 10 had AML, 16 had primary immune deficiency, 7 had thalassemia major, 1 had sickle cell disease, 2 had Diamond-Blackfan anemia, 5 had Fanconi anemia, 4 had hemophagocytic lymphohistiocytosis, and 1 had osteoporosis.
Their median age was 4.8 (range, 0.25-17), and 56% were male. The patients received busulfan-based conditioning (41%), total body irradiation (36%), treosulfan-based conditioning (18%), and other conditioning (5%).
Ninety-five percent of the patients had a parent donor, and the other 5% had a sibling donor. The median donor age was 36 (range, 19-50).
The patients’ median time to neutrophil recovery was 15 days (range, 9-75), and their median time to platelet recovery was 10 days (range, 4-64). Their median time to discharge was 25 days (range, 14-122).
The cumulative incidence of acute grade 2-4 GVHD was 9.9%, and the cumulative incidence of acute grade 3-4 GVHD was 3.3%.
There were no cases of transplant-related mortality at 180 days and no cases of extensive chronic GVHD.
“These preliminary data compare favorably to previously published data on matched, unrelated donor transplantation,” Dr Algeri said. “And for this reason, an observational matched, unrelated donor study is being initiated to enable comparison of the safety and efficacy of haploidentical transplantation and BPX-501 infusion to the standard of care for patients without a matched sibling donor.” ![]()
MADRID—Updated results of a phase 1/2 study suggest the T-cell product BPX-501 lowers the risks associated with haploidentical hematopoietic stem cell transplant (haplo-HSCT).
In this ongoing study, researchers are testing BPX-501 in pediatric patients undergoing haplo-HSCT to treat a range of hematologic disorders.
Patients treated thus far have experienced rapid engraftment and early hospital discharge, a low rate of acute graft-versus-host disease (GHVD), no extensive chronic GVHD, and a low rate of transplant-related mortality at 180 days.
“The combination of haploidentical transplantation and BPX-501 infusion is an effective strategy for children in need of an allograft lacking a compatible donor,” said study investigator Mattia Algeri, MD, of Ospedale Pediatrico Bambino Gesù in Rome, Italy.
Dr Algeri presented these results during the Presidential Symposium at the 22nd Congress of the European Hematology Association (EHA) as abstract S146.
The research was sponsored by Bellicum Pharmaceuticals, Inc., the company developing BPX-501.
About BPX-501
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate the T cells in the event of toxicity.
Rimiducid is used to activate the CaspaCIDe safety switch, which consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway.
The goal of this therapy is to allow physicians to more safely perform haplo-HSCTs.
Patients
Dr Algeri and his colleagues have tested BPX-501 in 98 pediatric patients treated at centers in Europe and the US.
Fifty-nine patients had non-malignant conditions, including primary immune deficiency (n=26), thalassemia major (n=8), sickle cell disease (n=5), Diamond-Blackfan anemia (n=2), Swachman-Diamond syndrome (n=1), Fanconi anemia (n=9), hemophagocytic lymphohistiocytosis (n=6), aplastic anemia (n=1), and osteoporosis (n=1).
Thirty-nine patients had malignant conditions, including acute lymphoblastic leukemia (ALL, n=21), acute myeloid leukemia (AML, n=14), myelodysplastic syndromes (MDS, n=3), and non-Hodgkin lymphoma (NHL, n=1).
The patients received BPX-501 after an alpha/beta T-cell-depleted haplo-HSCT. All patients had at least 6 months of follow-up.
Overall results
Ninety-five percent of the patients engrafted (93/98), and the researchers said they observed rapid recovery of T cells, B cells, and immunoglobulins.
At 180 days, the incidence of transplant-related mortality was 5%, and there were no cases of post-transplant lymphoproliferative disorder.
The cumulative incidence of grade 2-4 acute GVHD was 14%. For patients with at least 1 year of follow-up, the cumulative incidence of chronic GVHD at 1 year was 3%.
Eleven patients received rimiducid—10 who had uncontrollable acute GVHD and 1 who developed late acute GVHD. In all of these patients, GVHD resolved and has not recurred.
There were no adverse events associated with BPX-501 or rimiducid.
European cohort
Dr Algeri presented more detailed data on the 61 patients treated at centers in Europe.
Fifteen of these patients had ALL, 10 had AML, 16 had primary immune deficiency, 7 had thalassemia major, 1 had sickle cell disease, 2 had Diamond-Blackfan anemia, 5 had Fanconi anemia, 4 had hemophagocytic lymphohistiocytosis, and 1 had osteoporosis.
Their median age was 4.8 (range, 0.25-17), and 56% were male. The patients received busulfan-based conditioning (41%), total body irradiation (36%), treosulfan-based conditioning (18%), and other conditioning (5%).
Ninety-five percent of the patients had a parent donor, and the other 5% had a sibling donor. The median donor age was 36 (range, 19-50).
The patients’ median time to neutrophil recovery was 15 days (range, 9-75), and their median time to platelet recovery was 10 days (range, 4-64). Their median time to discharge was 25 days (range, 14-122).
The cumulative incidence of acute grade 2-4 GVHD was 9.9%, and the cumulative incidence of acute grade 3-4 GVHD was 3.3%.
There were no cases of transplant-related mortality at 180 days and no cases of extensive chronic GVHD.
“These preliminary data compare favorably to previously published data on matched, unrelated donor transplantation,” Dr Algeri said. “And for this reason, an observational matched, unrelated donor study is being initiated to enable comparison of the safety and efficacy of haploidentical transplantation and BPX-501 infusion to the standard of care for patients without a matched sibling donor.” ![]()
Solving stool refusal
When parents bring in their delightful, verbal 3-year-old for refusing to poop on the potty, it may seem laughable. But with impending preschool and costs of diapers, stool refusal can be a major aggravation for families! Fortunately,
Commonly a healthy, typically developing boy stands and urinates in the toilet just fine, but sneaks off behind the sofa to poop. Parent gyrations have gone from cajoling, to punishing, to offering trips to Disney! Flaring tempers can set the stage for stool refusal to be a power play.
There are a number of reasons stool refusal may give clues to child and family tendencies and relevant intervention. We always should be alert to rare medical problems such as Hirschsprung disease or traumas (from slammed toilet lids to sexual abuse). But while learning to use the toilet for urination and defecation generally occur around the same time, there are pitfalls making pooping in the potty different. An impending stool provides stronger sensations and more advance warning than urine and tends to come at regular times, making it logical to start toilet learning with sitting on the potty after meals.
But once seated on the potty, stools can require some waiting – not a typical toddler forte! While running to sit has novelty at first and may be reinforced by celebration, this quickly becomes routine and boring. Very active or very intense children especially hate having their play interrupted by a trip to the bathroom. Oppositional children just won’t perform if they think the parent cares! And unlike for urination, everyone can inhibit defecation long enough for the urge to pass. Repeated stool retention from ignoring the urge makes stools dessicated and harder, with resulting pain when finally passed. One painful stool makes many a young child decide “Never again!” and simply refuse the toilet. A rectal fissure can both start 
During unclogging and establishing a new stool pattern, the toddler should be matter-of-factly put back in diapers (not pull ups) saying “Oh well, you are just not ready for pants yet.” Dramatically placing the treasured Superhero underwear on the top shelf increases motivation (or promised if none have been acquired). Returning to diapers without shaming the child is key, and all caregivers need to buy in. They need to be good “actors,” conveying that they don’t really care about toileting to reduce the power struggle. If controlling poop is a battle, only the child can win!
When the soft stools are occurring several times per day, I suggest “M&M treatment”: 1 for sitting, 2 for peeing, and 3 for pooping = 6 potential M&Ms per episode. The “1 for sitting” (the easiest part), is not painful and restores the habit of complying. Remember, M&Ms are no match for a game on an iPad! By charting the times of stools, the parent can remove electronics ½ hour before the expected poop and restrict the child to one room of the house with a potty nearby. Parents can interact, but should avoid making this a rewarding playtime. When the child uses the potty rather than their pants, the room restriction is removed until the next window for pooping. If they poop outside the toilet, they remain restricted (and no electronics) until the next window (even the next day).
Some parents are especially sensitive to the smell and mess of stools and pass that attitude along to their child by saying “Ugh, you stink!” or “I can’t stand this mess!” or even handing the child to another caregiver in a gesture of rejection. These messages are not missed by the child, who may then not want to deal with the mess, either. I coach parents to stay at least neutral about stools, reminding them that, “Your child is going to have to poop her whole life!”
Demanding a diaper and then getting the special intimacy of bottom cleaning can be reinforcing. If there is a younger sibling, diaper changes may be a desired opportunity for the toddler to regress and retain some “baby privileges.” Other clues to this dynamic include thumb sucking, baby talk, clinginess, or being rough on the sibling. One part of addressing this issue is to prescribe “babying” the toddler by holding in arms, rocking, talking baby talk, offering a pacifier, and feeding him during daily parent-child one-on-one Special Time. This sounds crazy to parents aiming for grown up toileting, but I promise them the child will not go backwards! It addresses the child’s deep fear that the nurturing of infancy is no longer available.
You may have noticed that boys are much more likely to refuse stools than girls. Some of this difference may be that high activity, but learning to urinate standing up also is fun, a Big Boy feat, and a source of pride to fathers. If regular sitting to poop has not been well established before the fun of standing to pee is offered, the little guys are not so interested in sitting again to poop. Plus the wiping and hand washing after poops are further aggravations delaying return to the Legos. But more! By around age 3 years, both genders make the horrifying discovery that boys have a penis and girls don’t. At this age of confusion about potential transformations, the obvious conclusion is that the girl’s penis was lost! And that turd disappearing down the toilet looks a lot like a dismembered body part! Reassurance and education is in order. I address this with my “Penis Talk”: “Boys are made with a penis and girls are made with a vagina. (For boys:) When you get big like your Dad, your penis will be big, too. No one can ever take your penis away. (For girls, a less common concern.) You have always had a vagina. You did not lose a penis.” I recommend that you practice this in front of a mirror before first use!
Another cognitive milestone concerns what sorts of things can disappear dow

Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at pdnews@frontlinemedcom.com.
When parents bring in their delightful, verbal 3-year-old for refusing to poop on the potty, it may seem laughable. But with impending preschool and costs of diapers, stool refusal can be a major aggravation for families! Fortunately,
Commonly a healthy, typically developing boy stands and urinates in the toilet just fine, but sneaks off behind the sofa to poop. Parent gyrations have gone from cajoling, to punishing, to offering trips to Disney! Flaring tempers can set the stage for stool refusal to be a power play.
There are a number of reasons stool refusal may give clues to child and family tendencies and relevant intervention. We always should be alert to rare medical problems such as Hirschsprung disease or traumas (from slammed toilet lids to sexual abuse). But while learning to use the toilet for urination and defecation generally occur around the same time, there are pitfalls making pooping in the potty different. An impending stool provides stronger sensations and more advance warning than urine and tends to come at regular times, making it logical to start toilet learning with sitting on the potty after meals.
But once seated on the potty, stools can require some waiting – not a typical toddler forte! While running to sit has novelty at first and may be reinforced by celebration, this quickly becomes routine and boring. Very active or very intense children especially hate having their play interrupted by a trip to the bathroom. Oppositional children just won’t perform if they think the parent cares! And unlike for urination, everyone can inhibit defecation long enough for the urge to pass. Repeated stool retention from ignoring the urge makes stools dessicated and harder, with resulting pain when finally passed. One painful stool makes many a young child decide “Never again!” and simply refuse the toilet. A rectal fissure can both start
During unclogging and establishing a new stool pattern, the toddler should be matter-of-factly put back in diapers (not pull ups) saying “Oh well, you are just not ready for pants yet.” Dramatically placing the treasured Superhero underwear on the top shelf increases motivation (or promised if none have been acquired). Returning to diapers without shaming the child is key, and all caregivers need to buy in. They need to be good “actors,” conveying that they don’t really care about toileting to reduce the power struggle. If controlling poop is a battle, only the child can win!
When the soft stools are occurring several times per day, I suggest “M&M treatment”: 1 for sitting, 2 for peeing, and 3 for pooping = 6 potential M&Ms per episode. The “1 for sitting” (the easiest part), is not painful and restores the habit of complying. Remember, M&Ms are no match for a game on an iPad! By charting the times of stools, the parent can remove electronics ½ hour before the expected poop and restrict the child to one room of the house with a potty nearby. Parents can interact, but should avoid making this a rewarding playtime. When the child uses the potty rather than their pants, the room restriction is removed until the next window for pooping. If they poop outside the toilet, they remain restricted (and no electronics) until the next window (even the next day).
Some parents are especially sensitive to the smell and mess of stools and pass that attitude along to their child by saying “Ugh, you stink!” or “I can’t stand this mess!” or even handing the child to another caregiver in a gesture of rejection. These messages are not missed by the child, who may then not want to deal with the mess, either. I coach parents to stay at least neutral about stools, reminding them that, “Your child is going to have to poop her whole life!”
Demanding a diaper and then getting the special intimacy of bottom cleaning can be reinforcing. If there is a younger sibling, diaper changes may be a desired opportunity for the toddler to regress and retain some “baby privileges.” Other clues to this dynamic include thumb sucking, baby talk, clinginess, or being rough on the sibling. One part of addressing this issue is to prescribe “babying” the toddler by holding in arms, rocking, talking baby talk, offering a pacifier, and feeding him during daily parent-child one-on-one Special Time. This sounds crazy to parents aiming for grown up toileting, but I promise them the child will not go backwards! It addresses the child’s deep fear that the nurturing of infancy is no longer available.
You may have noticed that boys are much more likely to refuse stools than girls. Some of this difference may be that high activity, but learning to urinate standing up also is fun, a Big Boy feat, and a source of pride to fathers. If regular sitting to poop has not been well established before the fun of standing to pee is offered, the little guys are not so interested in sitting again to poop. Plus the wiping and hand washing after poops are further aggravations delaying return to the Legos. But more! By around age 3 years, both genders make the horrifying discovery that boys have a penis and girls don’t. At this age of confusion about potential transformations, the obvious conclusion is that the girl’s penis was lost! And that turd disappearing down the toilet looks a lot like a dismembered body part! Reassurance and education is in order. I address this with my “Penis Talk”: “Boys are made with a penis and girls are made with a vagina. (For boys:) When you get big like your Dad, your penis will be big, too. No one can ever take your penis away. (For girls, a less common concern.) You have always had a vagina. You did not lose a penis.” I recommend that you practice this in front of a mirror before first use!
Another cognitive milestone concerns what sorts of things can disappear dow
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at pdnews@frontlinemedcom.com.
When parents bring in their delightful, verbal 3-year-old for refusing to poop on the potty, it may seem laughable. But with impending preschool and costs of diapers, stool refusal can be a major aggravation for families! Fortunately,
Commonly a healthy, typically developing boy stands and urinates in the toilet just fine, but sneaks off behind the sofa to poop. Parent gyrations have gone from cajoling, to punishing, to offering trips to Disney! Flaring tempers can set the stage for stool refusal to be a power play.
There are a number of reasons stool refusal may give clues to child and family tendencies and relevant intervention. We always should be alert to rare medical problems such as Hirschsprung disease or traumas (from slammed toilet lids to sexual abuse). But while learning to use the toilet for urination and defecation generally occur around the same time, there are pitfalls making pooping in the potty different. An impending stool provides stronger sensations and more advance warning than urine and tends to come at regular times, making it logical to start toilet learning with sitting on the potty after meals.
But once seated on the potty, stools can require some waiting – not a typical toddler forte! While running to sit has novelty at first and may be reinforced by celebration, this quickly becomes routine and boring. Very active or very intense children especially hate having their play interrupted by a trip to the bathroom. Oppositional children just won’t perform if they think the parent cares! And unlike for urination, everyone can inhibit defecation long enough for the urge to pass. Repeated stool retention from ignoring the urge makes stools dessicated and harder, with resulting pain when finally passed. One painful stool makes many a young child decide “Never again!” and simply refuse the toilet. A rectal fissure can both start
During unclogging and establishing a new stool pattern, the toddler should be matter-of-factly put back in diapers (not pull ups) saying “Oh well, you are just not ready for pants yet.” Dramatically placing the treasured Superhero underwear on the top shelf increases motivation (or promised if none have been acquired). Returning to diapers without shaming the child is key, and all caregivers need to buy in. They need to be good “actors,” conveying that they don’t really care about toileting to reduce the power struggle. If controlling poop is a battle, only the child can win!
When the soft stools are occurring several times per day, I suggest “M&M treatment”: 1 for sitting, 2 for peeing, and 3 for pooping = 6 potential M&Ms per episode. The “1 for sitting” (the easiest part), is not painful and restores the habit of complying. Remember, M&Ms are no match for a game on an iPad! By charting the times of stools, the parent can remove electronics ½ hour before the expected poop and restrict the child to one room of the house with a potty nearby. Parents can interact, but should avoid making this a rewarding playtime. When the child uses the potty rather than their pants, the room restriction is removed until the next window for pooping. If they poop outside the toilet, they remain restricted (and no electronics) until the next window (even the next day).
Some parents are especially sensitive to the smell and mess of stools and pass that attitude along to their child by saying “Ugh, you stink!” or “I can’t stand this mess!” or even handing the child to another caregiver in a gesture of rejection. These messages are not missed by the child, who may then not want to deal with the mess, either. I coach parents to stay at least neutral about stools, reminding them that, “Your child is going to have to poop her whole life!”
Demanding a diaper and then getting the special intimacy of bottom cleaning can be reinforcing. If there is a younger sibling, diaper changes may be a desired opportunity for the toddler to regress and retain some “baby privileges.” Other clues to this dynamic include thumb sucking, baby talk, clinginess, or being rough on the sibling. One part of addressing this issue is to prescribe “babying” the toddler by holding in arms, rocking, talking baby talk, offering a pacifier, and feeding him during daily parent-child one-on-one Special Time. This sounds crazy to parents aiming for grown up toileting, but I promise them the child will not go backwards! It addresses the child’s deep fear that the nurturing of infancy is no longer available.
You may have noticed that boys are much more likely to refuse stools than girls. Some of this difference may be that high activity, but learning to urinate standing up also is fun, a Big Boy feat, and a source of pride to fathers. If regular sitting to poop has not been well established before the fun of standing to pee is offered, the little guys are not so interested in sitting again to poop. Plus the wiping and hand washing after poops are further aggravations delaying return to the Legos. But more! By around age 3 years, both genders make the horrifying discovery that boys have a penis and girls don’t. At this age of confusion about potential transformations, the obvious conclusion is that the girl’s penis was lost! And that turd disappearing down the toilet looks a lot like a dismembered body part! Reassurance and education is in order. I address this with my “Penis Talk”: “Boys are made with a penis and girls are made with a vagina. (For boys:) When you get big like your Dad, your penis will be big, too. No one can ever take your penis away. (For girls, a less common concern.) You have always had a vagina. You did not lose a penis.” I recommend that you practice this in front of a mirror before first use!
Another cognitive milestone concerns what sorts of things can disappear dow
Dr. Howard is assistant professor of pediatrics at Johns Hopkins University, Baltimore, and creator of CHADIS (www.CHADIS.com). She had no other relevant disclosures. Dr. Howard’s contribution to this publication was as a paid expert to Frontline Medical News. Email her at pdnews@frontlinemedcom.com.
Obtaining coverage for transgender and gender-expansive youth
Transgender and gender-expansive youth face many barriers to health care. (Gender-expansive youth are defined as “youth who do not identify with traditional gender roles but are otherwise not confined to one gender narrative or experience.”) Although some of these youth may be fortunate to have a supportive family and access to health care providers proficient in transgender health care, they still face difficulties in having their insurance cover transgender-related services. This is not an impossible task, but it is a constant struggle for many clinicians.
In this column, I will provide some tips and strategies to help clinicians get insurance companies to cover these critical services. However, keep in mind that there is no one-size-fits-all approach to obtaining insurance coverage. In addition, growing uncertainty over the repeal of the Affordable Care Act (ACA) – which was critical in lifting many of the barriers to insurance coverage for transgender individuals – will make this task challenging.
Health insurance is extraordinarily complex. There are multiple private and public plans that vary in the services they cover. This variation is state dependent. And even within states, there is additional variability. Most health insurance plans are purchased by employers, and employers have a choice of what can be covered in their health plans. So even though an insurance company may state that it covers transgender-related services, the patient’s employer may pay for a plan that doesn’t cover such services. The only way to be sure whether a patient’s insurance will cover transgender-related services or not is to contact the insurance provider directly, but with extremely busy schedules and heavy patient loads, this is easier said than done. It would be helpful to have a social worker perform this task, but even having a social worker can be a luxury for some clinics.
The ACA made it easier for transgender individuals to obtain insurance coverage. Three years ago, the U.S. Department of Health and Human Services stated that Medicare’s longstanding exclusion of “transsexual surgical procedures” was no longer valid.1 Although it did not universally ban transgender exclusion policies, it did allow individual states to do so. Thirteen states have explicit policies that ban exclusions of transgender-related services in both private insurance and in Medicaid, and an additional five states have some policies that discourage such practices.2 This allowed some insurance providers and state Medicaid plans to offer coverage of transgender-related services.
Another challenge in obtaining insurance coverage for transgender and gender-expansive youth is claims denial for sex-specific procedures. For example, if a transwoman is designated as “male” in the electronic medical record and requires a breast ultrasound, the insurance company may automatically reject this claim because this procedure is covered for bodies designated as “female.” If the patient’s insurance plan covers transgender-related services, the clinic can notify the insurance company that the patient is transgender; if the patient’s plan does not, then the clinic will need to appeal to the insurance provider. Alternatively, for clinics associated with federally-funded institutions (e.g., most hospitals), the clinician can use Condition Code 45 in the billing to override the sex mismatch, although not all hospitals have implemented this code.3
1. Patient’s identifying information. Usually the patient’s name and date of birth is sufficient. Clinicians should use the patient’s preferred name in the letter, but provide the insurance or legal name of the patient so that the insurance provider can locate the patient’s records.
2. Result of a psychosocial evaluation and diagnosis (if any). Many insurance providers are looking specifically for the gender dysphoria diagnosis.
3. The duration of the referring health professional’s relationship with the patient, which includes the type of evaluation and therapy or counseling (e.g., cognitive behavior therapy or gender coaching).
4. An explanation that the criteria (usually from the World Professional Association for Transgender Health standard of care4 or the Endocrine Society Guidelines titled Endocrine Treatment of Transsexual Persons5) for hormone therapy have been met, and a brief description of the clinical rationale for supporting the client’s request for hormone therapy.
5. A statement that informed consent has been obtained from the patient (or parental permission if the patient is younger than 18 years).
6. A statement that the referring health professional is available for coordination of care.
If the clinician fails to convince the insurance provider of the necessity of covering transgender-related services, the patient still can pay out of pocket. Some hormones can be affordable to certain patients. In the state of Pennsylvania, for example, a 10-mL vial of testosterone can cost anywhere from $60 to $80, and may generally last anywhere from 10 weeks to a year, depending on dosage. Nevertheless, these costs still may be prohibitive for many transgender youth. Many are chronically unemployed or underemployed, or struggle with homelessness.6 Some transgender youth have to the face the excruciatingly difficult choice between having something to eat for the day or living another day with gender dysphoria.
Clinicians should work very hard to make sure that their transgender and gender-expansive patients obtain the care they need. The above strategies may help navigate the complex insurance system. However, insurance policies vary by state, and anti-trans discrimination creates additional barriers to health care. Therefore, clinicians who take care of transgender youth also should advocate for policies that protect these patients from discrimination, and they should advocate for policies that expand medical coverage for this vulnerable population.

Resources
• The Human Rights Campaign keeps a list of insurance plans that cover transgender-related services, but this list is far from comprehensive.
• Healthcare.gov provides some guidance on how to obtain coverage and navigate the insurance system for transgender individuals.
• UCSF Center of Excellence for Transgender Health provides some excellent resources and guidance on obtaining insurance coverage for transgender individuals.
References
1. LGBT Health 2014;1(4):256-8.
2. Map: State Health Insurance Rules: National Center for Transgender Equality, 2016 [Available from: www.transequality.org/issues/resources/map-state-health-insurance-rules].
3. Health insurance coverage issues for transgender people in the United States: University of California, San Fransisco Center of Excellence for Transgender Health, 2017 [Available from: http://transhealth.ucsf.edu/trans?page=guidelines-insurance].
4. International Journal of Transgenderism 2012;13(4):165-232.
5. J Clin Endocrinol Metab 2009;94(9):3132-54.
6. Injustice at Every Turn: A Report of the National Transgender Discrimination Survey. Washington: National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011.
Transgender and gender-expansive youth face many barriers to health care. (Gender-expansive youth are defined as “youth who do not identify with traditional gender roles but are otherwise not confined to one gender narrative or experience.”) Although some of these youth may be fortunate to have a supportive family and access to health care providers proficient in transgender health care, they still face difficulties in having their insurance cover transgender-related services. This is not an impossible task, but it is a constant struggle for many clinicians.
In this column, I will provide some tips and strategies to help clinicians get insurance companies to cover these critical services. However, keep in mind that there is no one-size-fits-all approach to obtaining insurance coverage. In addition, growing uncertainty over the repeal of the Affordable Care Act (ACA) – which was critical in lifting many of the barriers to insurance coverage for transgender individuals – will make this task challenging.
Health insurance is extraordinarily complex. There are multiple private and public plans that vary in the services they cover. This variation is state dependent. And even within states, there is additional variability. Most health insurance plans are purchased by employers, and employers have a choice of what can be covered in their health plans. So even though an insurance company may state that it covers transgender-related services, the patient’s employer may pay for a plan that doesn’t cover such services. The only way to be sure whether a patient’s insurance will cover transgender-related services or not is to contact the insurance provider directly, but with extremely busy schedules and heavy patient loads, this is easier said than done. It would be helpful to have a social worker perform this task, but even having a social worker can be a luxury for some clinics.
The ACA made it easier for transgender individuals to obtain insurance coverage. Three years ago, the U.S. Department of Health and Human Services stated that Medicare’s longstanding exclusion of “transsexual surgical procedures” was no longer valid.1 Although it did not universally ban transgender exclusion policies, it did allow individual states to do so. Thirteen states have explicit policies that ban exclusions of transgender-related services in both private insurance and in Medicaid, and an additional five states have some policies that discourage such practices.2 This allowed some insurance providers and state Medicaid plans to offer coverage of transgender-related services.
Another challenge in obtaining insurance coverage for transgender and gender-expansive youth is claims denial for sex-specific procedures. For example, if a transwoman is designated as “male” in the electronic medical record and requires a breast ultrasound, the insurance company may automatically reject this claim because this procedure is covered for bodies designated as “female.” If the patient’s insurance plan covers transgender-related services, the clinic can notify the insurance company that the patient is transgender; if the patient’s plan does not, then the clinic will need to appeal to the insurance provider. Alternatively, for clinics associated with federally-funded institutions (e.g., most hospitals), the clinician can use Condition Code 45 in the billing to override the sex mismatch, although not all hospitals have implemented this code.3
1. Patient’s identifying information. Usually the patient’s name and date of birth is sufficient. Clinicians should use the patient’s preferred name in the letter, but provide the insurance or legal name of the patient so that the insurance provider can locate the patient’s records.
2. Result of a psychosocial evaluation and diagnosis (if any). Many insurance providers are looking specifically for the gender dysphoria diagnosis.
3. The duration of the referring health professional’s relationship with the patient, which includes the type of evaluation and therapy or counseling (e.g., cognitive behavior therapy or gender coaching).
4. An explanation that the criteria (usually from the World Professional Association for Transgender Health standard of care4 or the Endocrine Society Guidelines titled Endocrine Treatment of Transsexual Persons5) for hormone therapy have been met, and a brief description of the clinical rationale for supporting the client’s request for hormone therapy.
5. A statement that informed consent has been obtained from the patient (or parental permission if the patient is younger than 18 years).
6. A statement that the referring health professional is available for coordination of care.
If the clinician fails to convince the insurance provider of the necessity of covering transgender-related services, the patient still can pay out of pocket. Some hormones can be affordable to certain patients. In the state of Pennsylvania, for example, a 10-mL vial of testosterone can cost anywhere from $60 to $80, and may generally last anywhere from 10 weeks to a year, depending on dosage. Nevertheless, these costs still may be prohibitive for many transgender youth. Many are chronically unemployed or underemployed, or struggle with homelessness.6 Some transgender youth have to the face the excruciatingly difficult choice between having something to eat for the day or living another day with gender dysphoria.
Clinicians should work very hard to make sure that their transgender and gender-expansive patients obtain the care they need. The above strategies may help navigate the complex insurance system. However, insurance policies vary by state, and anti-trans discrimination creates additional barriers to health care. Therefore, clinicians who take care of transgender youth also should advocate for policies that protect these patients from discrimination, and they should advocate for policies that expand medical coverage for this vulnerable population.
Resources
• The Human Rights Campaign keeps a list of insurance plans that cover transgender-related services, but this list is far from comprehensive.
• Healthcare.gov provides some guidance on how to obtain coverage and navigate the insurance system for transgender individuals.
• UCSF Center of Excellence for Transgender Health provides some excellent resources and guidance on obtaining insurance coverage for transgender individuals.
References
1. LGBT Health 2014;1(4):256-8.
2. Map: State Health Insurance Rules: National Center for Transgender Equality, 2016 [Available from: www.transequality.org/issues/resources/map-state-health-insurance-rules].
3. Health insurance coverage issues for transgender people in the United States: University of California, San Fransisco Center of Excellence for Transgender Health, 2017 [Available from: http://transhealth.ucsf.edu/trans?page=guidelines-insurance].
4. International Journal of Transgenderism 2012;13(4):165-232.
5. J Clin Endocrinol Metab 2009;94(9):3132-54.
6. Injustice at Every Turn: A Report of the National Transgender Discrimination Survey. Washington: National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011.
Transgender and gender-expansive youth face many barriers to health care. (Gender-expansive youth are defined as “youth who do not identify with traditional gender roles but are otherwise not confined to one gender narrative or experience.”) Although some of these youth may be fortunate to have a supportive family and access to health care providers proficient in transgender health care, they still face difficulties in having their insurance cover transgender-related services. This is not an impossible task, but it is a constant struggle for many clinicians.
In this column, I will provide some tips and strategies to help clinicians get insurance companies to cover these critical services. However, keep in mind that there is no one-size-fits-all approach to obtaining insurance coverage. In addition, growing uncertainty over the repeal of the Affordable Care Act (ACA) – which was critical in lifting many of the barriers to insurance coverage for transgender individuals – will make this task challenging.
Health insurance is extraordinarily complex. There are multiple private and public plans that vary in the services they cover. This variation is state dependent. And even within states, there is additional variability. Most health insurance plans are purchased by employers, and employers have a choice of what can be covered in their health plans. So even though an insurance company may state that it covers transgender-related services, the patient’s employer may pay for a plan that doesn’t cover such services. The only way to be sure whether a patient’s insurance will cover transgender-related services or not is to contact the insurance provider directly, but with extremely busy schedules and heavy patient loads, this is easier said than done. It would be helpful to have a social worker perform this task, but even having a social worker can be a luxury for some clinics.
The ACA made it easier for transgender individuals to obtain insurance coverage. Three years ago, the U.S. Department of Health and Human Services stated that Medicare’s longstanding exclusion of “transsexual surgical procedures” was no longer valid.1 Although it did not universally ban transgender exclusion policies, it did allow individual states to do so. Thirteen states have explicit policies that ban exclusions of transgender-related services in both private insurance and in Medicaid, and an additional five states have some policies that discourage such practices.2 This allowed some insurance providers and state Medicaid plans to offer coverage of transgender-related services.
Another challenge in obtaining insurance coverage for transgender and gender-expansive youth is claims denial for sex-specific procedures. For example, if a transwoman is designated as “male” in the electronic medical record and requires a breast ultrasound, the insurance company may automatically reject this claim because this procedure is covered for bodies designated as “female.” If the patient’s insurance plan covers transgender-related services, the clinic can notify the insurance company that the patient is transgender; if the patient’s plan does not, then the clinic will need to appeal to the insurance provider. Alternatively, for clinics associated with federally-funded institutions (e.g., most hospitals), the clinician can use Condition Code 45 in the billing to override the sex mismatch, although not all hospitals have implemented this code.3
1. Patient’s identifying information. Usually the patient’s name and date of birth is sufficient. Clinicians should use the patient’s preferred name in the letter, but provide the insurance or legal name of the patient so that the insurance provider can locate the patient’s records.
2. Result of a psychosocial evaluation and diagnosis (if any). Many insurance providers are looking specifically for the gender dysphoria diagnosis.
3. The duration of the referring health professional’s relationship with the patient, which includes the type of evaluation and therapy or counseling (e.g., cognitive behavior therapy or gender coaching).
4. An explanation that the criteria (usually from the World Professional Association for Transgender Health standard of care4 or the Endocrine Society Guidelines titled Endocrine Treatment of Transsexual Persons5) for hormone therapy have been met, and a brief description of the clinical rationale for supporting the client’s request for hormone therapy.
5. A statement that informed consent has been obtained from the patient (or parental permission if the patient is younger than 18 years).
6. A statement that the referring health professional is available for coordination of care.
If the clinician fails to convince the insurance provider of the necessity of covering transgender-related services, the patient still can pay out of pocket. Some hormones can be affordable to certain patients. In the state of Pennsylvania, for example, a 10-mL vial of testosterone can cost anywhere from $60 to $80, and may generally last anywhere from 10 weeks to a year, depending on dosage. Nevertheless, these costs still may be prohibitive for many transgender youth. Many are chronically unemployed or underemployed, or struggle with homelessness.6 Some transgender youth have to the face the excruciatingly difficult choice between having something to eat for the day or living another day with gender dysphoria.
Clinicians should work very hard to make sure that their transgender and gender-expansive patients obtain the care they need. The above strategies may help navigate the complex insurance system. However, insurance policies vary by state, and anti-trans discrimination creates additional barriers to health care. Therefore, clinicians who take care of transgender youth also should advocate for policies that protect these patients from discrimination, and they should advocate for policies that expand medical coverage for this vulnerable population.
Resources
• The Human Rights Campaign keeps a list of insurance plans that cover transgender-related services, but this list is far from comprehensive.
• Healthcare.gov provides some guidance on how to obtain coverage and navigate the insurance system for transgender individuals.
• UCSF Center of Excellence for Transgender Health provides some excellent resources and guidance on obtaining insurance coverage for transgender individuals.
References
1. LGBT Health 2014;1(4):256-8.
2. Map: State Health Insurance Rules: National Center for Transgender Equality, 2016 [Available from: www.transequality.org/issues/resources/map-state-health-insurance-rules].
3. Health insurance coverage issues for transgender people in the United States: University of California, San Fransisco Center of Excellence for Transgender Health, 2017 [Available from: http://transhealth.ucsf.edu/trans?page=guidelines-insurance].
4. International Journal of Transgenderism 2012;13(4):165-232.
5. J Clin Endocrinol Metab 2009;94(9):3132-54.
6. Injustice at Every Turn: A Report of the National Transgender Discrimination Survey. Washington: National Center for Transgender Equality and National Gay and Lesbian Task Force, 2011.
CSF p-Tau predicts neurocognitive sequelae in survivors of childhood cancer

A small retrospective study of survivors of childhood acute lymphoblastic leukemia (ALL) or non-Hodgkin lymphoma (NHL) has found that phosphorylated Tau (p-Tau) in patients’ cerebrospinal fluid (CSF) is a predictor of late neurocognitive consequences.
Investigators compared intellectual performance, memory, and executive functioning between survivors and control subjects and observed that CSF levels of p-Tau during treatment and total intrathecal methotrexate dose negatively correlated with intellectual performance.
They suggest that identifying at-risk children early “could inspire interventions to prevent or remediate chemotherapy-induced cognitive sequelae.”
The investigators enrolled 31 nonirradiated adults, 27 who had had ALL and 4 NHL. They compared the survivors to 35 age-matched controls.
All study participants were a mean age of 21.5 years (range, 16.1–29.8). The mean age of the survivors at diagnosis was 6.4 years.
"Our team collected samples of brain fluid during the cancer treatment,” Rudi D’Hooge, PhD, of KU Leuven in Belgium, said. “We analyzed the p-Tau levels to measure the damage to the brain cells."
Investigators assessed intelligence, memory, and executive function using Wechsler Adult Intelligence Scale (WAIS IV), Rey Auditory Verbal Learning Test (AVLT), and Amsterdam Neuropsychological Tasks (ANT), respectively.
Statistical analysis included two-sided, one-way analysis of covariance (ANCOVA) with survivor group vs control group as an independent factor. Parental socioeconomic status was a covariate.
Dr D’Hooge and colleagues published their findings in JNCI, the Journal of the National Cancer Institute.
Findings
Investigators found that survivors had statistically significant lower total intelligence (P=0.001), verbal intelligence (P=0.02), and performance intelligence (P=0.007) quotients than controls.
They also found a negative correlation between CSF p-Tau, but not CSF Tau, levels and total intelligence (P=0.02), verbal intelligence (P=0.001), and performance intelligence (P=0.04) quotients.
Only performance intelligence was negatively correlated with total intrathecal methotrexate (P=0.007).
And because total intrathecal methotrexate dose and CSF p-Tau were not significantly correlated (P=0.29), the investigators believe intervening factors increase CSF p-Tau independently from methotrexate dose.
Results of subset tests revealed that cognitive flexibility (set-shifting and working memory), and processing speed were affected (P<0.05).
However, long-term memory, focused and sustained attention, and inhibition appeared unaffected (P>0.05).
The investigators believe these differences in vulnerability of cognitive functions parallel patient age at time of development.
For example, long-term memory, focused and sustained attention, and inhibition develop before children reach 6 years, the mean age at which the survivor cohort was diagnosed.
But set-shifting, working memory, and processing speed mature during adolescence, after the patients were diagnosed and treated.
Limitations of the study, according to the investigators, include its retrospective and cross-sectional design, the relatively small sample size, and the lack of pretreatment neurocognitive data.
Nevertheless, they believe the study should encourage the use of the CSF biomarker, intrathecal methotrexate dose, and age at therapy initiation in neurotoxicity assessments to identify children at risk for long-term sequelae.
"If we systematically measure these p-Tau levels in the future," Iris Elens, MD, also of KU Leuven, said, "we can offer specific help to children with high values. With early coaching aimed at the most relevant functions we can prevent problems that would otherwise manifest 10 to 15 years after the treatment."
The Olivia Hendrickx Research Fund supported the study. ![]()
A small retrospective study of survivors of childhood acute lymphoblastic leukemia (ALL) or non-Hodgkin lymphoma (NHL) has found that phosphorylated Tau (p-Tau) in patients’ cerebrospinal fluid (CSF) is a predictor of late neurocognitive consequences.
Investigators compared intellectual performance, memory, and executive functioning between survivors and control subjects and observed that CSF levels of p-Tau during treatment and total intrathecal methotrexate dose negatively correlated with intellectual performance.
They suggest that identifying at-risk children early “could inspire interventions to prevent or remediate chemotherapy-induced cognitive sequelae.”
The investigators enrolled 31 nonirradiated adults, 27 who had had ALL and 4 NHL. They compared the survivors to 35 age-matched controls.
All study participants were a mean age of 21.5 years (range, 16.1–29.8). The mean age of the survivors at diagnosis was 6.4 years.
"Our team collected samples of brain fluid during the cancer treatment,” Rudi D’Hooge, PhD, of KU Leuven in Belgium, said. “We analyzed the p-Tau levels to measure the damage to the brain cells."
Investigators assessed intelligence, memory, and executive function using Wechsler Adult Intelligence Scale (WAIS IV), Rey Auditory Verbal Learning Test (AVLT), and Amsterdam Neuropsychological Tasks (ANT), respectively.
Statistical analysis included two-sided, one-way analysis of covariance (ANCOVA) with survivor group vs control group as an independent factor. Parental socioeconomic status was a covariate.
Dr D’Hooge and colleagues published their findings in JNCI, the Journal of the National Cancer Institute.
Findings
Investigators found that survivors had statistically significant lower total intelligence (P=0.001), verbal intelligence (P=0.02), and performance intelligence (P=0.007) quotients than controls.
They also found a negative correlation between CSF p-Tau, but not CSF Tau, levels and total intelligence (P=0.02), verbal intelligence (P=0.001), and performance intelligence (P=0.04) quotients.
Only performance intelligence was negatively correlated with total intrathecal methotrexate (P=0.007).
And because total intrathecal methotrexate dose and CSF p-Tau were not significantly correlated (P=0.29), the investigators believe intervening factors increase CSF p-Tau independently from methotrexate dose.
Results of subset tests revealed that cognitive flexibility (set-shifting and working memory), and processing speed were affected (P<0.05).
However, long-term memory, focused and sustained attention, and inhibition appeared unaffected (P>0.05).
The investigators believe these differences in vulnerability of cognitive functions parallel patient age at time of development.
For example, long-term memory, focused and sustained attention, and inhibition develop before children reach 6 years, the mean age at which the survivor cohort was diagnosed.
But set-shifting, working memory, and processing speed mature during adolescence, after the patients were diagnosed and treated.
Limitations of the study, according to the investigators, include its retrospective and cross-sectional design, the relatively small sample size, and the lack of pretreatment neurocognitive data.
Nevertheless, they believe the study should encourage the use of the CSF biomarker, intrathecal methotrexate dose, and age at therapy initiation in neurotoxicity assessments to identify children at risk for long-term sequelae.
"If we systematically measure these p-Tau levels in the future," Iris Elens, MD, also of KU Leuven, said, "we can offer specific help to children with high values. With early coaching aimed at the most relevant functions we can prevent problems that would otherwise manifest 10 to 15 years after the treatment."
The Olivia Hendrickx Research Fund supported the study. ![]()
A small retrospective study of survivors of childhood acute lymphoblastic leukemia (ALL) or non-Hodgkin lymphoma (NHL) has found that phosphorylated Tau (p-Tau) in patients’ cerebrospinal fluid (CSF) is a predictor of late neurocognitive consequences.
Investigators compared intellectual performance, memory, and executive functioning between survivors and control subjects and observed that CSF levels of p-Tau during treatment and total intrathecal methotrexate dose negatively correlated with intellectual performance.
They suggest that identifying at-risk children early “could inspire interventions to prevent or remediate chemotherapy-induced cognitive sequelae.”
The investigators enrolled 31 nonirradiated adults, 27 who had had ALL and 4 NHL. They compared the survivors to 35 age-matched controls.
All study participants were a mean age of 21.5 years (range, 16.1–29.8). The mean age of the survivors at diagnosis was 6.4 years.
"Our team collected samples of brain fluid during the cancer treatment,” Rudi D’Hooge, PhD, of KU Leuven in Belgium, said. “We analyzed the p-Tau levels to measure the damage to the brain cells."
Investigators assessed intelligence, memory, and executive function using Wechsler Adult Intelligence Scale (WAIS IV), Rey Auditory Verbal Learning Test (AVLT), and Amsterdam Neuropsychological Tasks (ANT), respectively.
Statistical analysis included two-sided, one-way analysis of covariance (ANCOVA) with survivor group vs control group as an independent factor. Parental socioeconomic status was a covariate.
Dr D’Hooge and colleagues published their findings in JNCI, the Journal of the National Cancer Institute.
Findings
Investigators found that survivors had statistically significant lower total intelligence (P=0.001), verbal intelligence (P=0.02), and performance intelligence (P=0.007) quotients than controls.
They also found a negative correlation between CSF p-Tau, but not CSF Tau, levels and total intelligence (P=0.02), verbal intelligence (P=0.001), and performance intelligence (P=0.04) quotients.
Only performance intelligence was negatively correlated with total intrathecal methotrexate (P=0.007).
And because total intrathecal methotrexate dose and CSF p-Tau were not significantly correlated (P=0.29), the investigators believe intervening factors increase CSF p-Tau independently from methotrexate dose.
Results of subset tests revealed that cognitive flexibility (set-shifting and working memory), and processing speed were affected (P<0.05).
However, long-term memory, focused and sustained attention, and inhibition appeared unaffected (P>0.05).
The investigators believe these differences in vulnerability of cognitive functions parallel patient age at time of development.
For example, long-term memory, focused and sustained attention, and inhibition develop before children reach 6 years, the mean age at which the survivor cohort was diagnosed.
But set-shifting, working memory, and processing speed mature during adolescence, after the patients were diagnosed and treated.
Limitations of the study, according to the investigators, include its retrospective and cross-sectional design, the relatively small sample size, and the lack of pretreatment neurocognitive data.
Nevertheless, they believe the study should encourage the use of the CSF biomarker, intrathecal methotrexate dose, and age at therapy initiation in neurotoxicity assessments to identify children at risk for long-term sequelae.
"If we systematically measure these p-Tau levels in the future," Iris Elens, MD, also of KU Leuven, said, "we can offer specific help to children with high values. With early coaching aimed at the most relevant functions we can prevent problems that would otherwise manifest 10 to 15 years after the treatment."
The Olivia Hendrickx Research Fund supported the study. ![]()
Privacy and maternal records
At the undergraduate level, classes on medical ethics tend to focus on the big ticket items like abortion, euthanasia, and social justice. Personally, I find the more interesting clinical cases involve relatively minor issues that accumulate to create problems. Privacy is one example.
A large amount of information in the mother’s prenatal records potentially impacts a newborn’s care. Ideally, the EHR is transferring data to the newborn’s chart, but not everything automatically populates in the newborn record, so there will be times when a pediatrician needs to review the mother’s chart.

- The criteria for selecting which mothers’ charts to review involve racial profiling.
- Access to mental health records involving addiction treatment requires special authorization. State laws and hospital policies will vary.
- Mom is a Hollywood celebrity and, while reviewing her chart, prurient curiosity extends the search to records of her cosmetic surgeries.
In my opinion, most of what is and isn’t permissible is determined by medical custom and not by statutes. The judiciary reserves the power to intervene, so medical custom should be informed by laws and by legal principles. But, the primary basis for these decisions should be a commitment to patient advocacy and to common sense, which in this situation means, “Would the typical reasonable person be upset if she learned I had done something without telling her?” If the answer to that question is yes, or in any way equivocal, I think ethics would dictate obtaining consent or at least assent.
Opiate addiction has quadrupled in the past 15 years. Almost all states now have prescription registries to help detect doctor shopping, multiple prescribers, and misdirection. If you are prescribing an opiate, it is ethically reasonable (and now the law) for you to make writing the prescription contingent on your patient agreeing to your consulting the registry. No consent, no prescription.
I think the facts of that case (writing a prescription) can be distinguished (a legal term) from the case of a neonatologist accessing the narcotic registry of the mother while on a fishing expedition to find evidence that might help the baby. Perhaps it is okay with the mother’s uncoerced consent, but otherwise I think that practice reeks as an unreasonable search. Ethically and legally, it has parallels to Ferguson v. City of Charleston (SCOTUS 2001).
That was a 6-3 Supreme Court decision, so, while I agree with the majority, you may find hospital lawyers who disagree. Overall, I assert that consent and privacy are best considered ethically as advocacy for the patient and not as legalistic forms that the physician must complete.
The reverse situation also occurs. Sometimes maternal health information is placed into the newborn’s chart that doesn’t need to be there. For example, common practice has been to designate mom, after delivery, as G4P2022. This contains the information that mother has had two therapeutic abortions. Does that information belong in a newborn’s chart? Especially in the era of the EHR where this information will hang around forever and will be easily obtained by the baby 16 years later when she can access all her medical information online. Will the mother be upset for her teenage daughter to learn that mom has had two abortions? Is that private information, belonging to the mother, that was given in confidence to her obstetrician? I advocate respecting privacy.
I have similar concerns about STD information being transferred from maternal charts to the newborn’s EHR. A maternal history of gonorrhea treated 8 years previously is unlikely to be relevant and should not populate the newborn’s EHR. I can make an argument that chlamydia detected and treated during the pregnancy might be useful to the baby’s pediatrician because neither treatment nor tests of cure are perfect. Perhaps, it could exist as a Snapchat-type record and disappear from the newborn’s record in a year if no respiratory symptoms occur.
I’m aware of efforts to destigmatize abortion and STDs, but, until that occurs, sensitive information should be handled delicately to preserve privacy. That is a major component of the Hippocratic Oath.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis.
At the undergraduate level, classes on medical ethics tend to focus on the big ticket items like abortion, euthanasia, and social justice. Personally, I find the more interesting clinical cases involve relatively minor issues that accumulate to create problems. Privacy is one example.
A large amount of information in the mother’s prenatal records potentially impacts a newborn’s care. Ideally, the EHR is transferring data to the newborn’s chart, but not everything automatically populates in the newborn record, so there will be times when a pediatrician needs to review the mother’s chart.
- The criteria for selecting which mothers’ charts to review involve racial profiling.
- Access to mental health records involving addiction treatment requires special authorization. State laws and hospital policies will vary.
- Mom is a Hollywood celebrity and, while reviewing her chart, prurient curiosity extends the search to records of her cosmetic surgeries.
In my opinion, most of what is and isn’t permissible is determined by medical custom and not by statutes. The judiciary reserves the power to intervene, so medical custom should be informed by laws and by legal principles. But, the primary basis for these decisions should be a commitment to patient advocacy and to common sense, which in this situation means, “Would the typical reasonable person be upset if she learned I had done something without telling her?” If the answer to that question is yes, or in any way equivocal, I think ethics would dictate obtaining consent or at least assent.
Opiate addiction has quadrupled in the past 15 years. Almost all states now have prescription registries to help detect doctor shopping, multiple prescribers, and misdirection. If you are prescribing an opiate, it is ethically reasonable (and now the law) for you to make writing the prescription contingent on your patient agreeing to your consulting the registry. No consent, no prescription.
I think the facts of that case (writing a prescription) can be distinguished (a legal term) from the case of a neonatologist accessing the narcotic registry of the mother while on a fishing expedition to find evidence that might help the baby. Perhaps it is okay with the mother’s uncoerced consent, but otherwise I think that practice reeks as an unreasonable search. Ethically and legally, it has parallels to Ferguson v. City of Charleston (SCOTUS 2001).
That was a 6-3 Supreme Court decision, so, while I agree with the majority, you may find hospital lawyers who disagree. Overall, I assert that consent and privacy are best considered ethically as advocacy for the patient and not as legalistic forms that the physician must complete.
The reverse situation also occurs. Sometimes maternal health information is placed into the newborn’s chart that doesn’t need to be there. For example, common practice has been to designate mom, after delivery, as G4P2022. This contains the information that mother has had two therapeutic abortions. Does that information belong in a newborn’s chart? Especially in the era of the EHR where this information will hang around forever and will be easily obtained by the baby 16 years later when she can access all her medical information online. Will the mother be upset for her teenage daughter to learn that mom has had two abortions? Is that private information, belonging to the mother, that was given in confidence to her obstetrician? I advocate respecting privacy.
I have similar concerns about STD information being transferred from maternal charts to the newborn’s EHR. A maternal history of gonorrhea treated 8 years previously is unlikely to be relevant and should not populate the newborn’s EHR. I can make an argument that chlamydia detected and treated during the pregnancy might be useful to the baby’s pediatrician because neither treatment nor tests of cure are perfect. Perhaps, it could exist as a Snapchat-type record and disappear from the newborn’s record in a year if no respiratory symptoms occur.
I’m aware of efforts to destigmatize abortion and STDs, but, until that occurs, sensitive information should be handled delicately to preserve privacy. That is a major component of the Hippocratic Oath.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis.
At the undergraduate level, classes on medical ethics tend to focus on the big ticket items like abortion, euthanasia, and social justice. Personally, I find the more interesting clinical cases involve relatively minor issues that accumulate to create problems. Privacy is one example.
A large amount of information in the mother’s prenatal records potentially impacts a newborn’s care. Ideally, the EHR is transferring data to the newborn’s chart, but not everything automatically populates in the newborn record, so there will be times when a pediatrician needs to review the mother’s chart.
- The criteria for selecting which mothers’ charts to review involve racial profiling.
- Access to mental health records involving addiction treatment requires special authorization. State laws and hospital policies will vary.
- Mom is a Hollywood celebrity and, while reviewing her chart, prurient curiosity extends the search to records of her cosmetic surgeries.
In my opinion, most of what is and isn’t permissible is determined by medical custom and not by statutes. The judiciary reserves the power to intervene, so medical custom should be informed by laws and by legal principles. But, the primary basis for these decisions should be a commitment to patient advocacy and to common sense, which in this situation means, “Would the typical reasonable person be upset if she learned I had done something without telling her?” If the answer to that question is yes, or in any way equivocal, I think ethics would dictate obtaining consent or at least assent.
Opiate addiction has quadrupled in the past 15 years. Almost all states now have prescription registries to help detect doctor shopping, multiple prescribers, and misdirection. If you are prescribing an opiate, it is ethically reasonable (and now the law) for you to make writing the prescription contingent on your patient agreeing to your consulting the registry. No consent, no prescription.
I think the facts of that case (writing a prescription) can be distinguished (a legal term) from the case of a neonatologist accessing the narcotic registry of the mother while on a fishing expedition to find evidence that might help the baby. Perhaps it is okay with the mother’s uncoerced consent, but otherwise I think that practice reeks as an unreasonable search. Ethically and legally, it has parallels to Ferguson v. City of Charleston (SCOTUS 2001).
That was a 6-3 Supreme Court decision, so, while I agree with the majority, you may find hospital lawyers who disagree. Overall, I assert that consent and privacy are best considered ethically as advocacy for the patient and not as legalistic forms that the physician must complete.
The reverse situation also occurs. Sometimes maternal health information is placed into the newborn’s chart that doesn’t need to be there. For example, common practice has been to designate mom, after delivery, as G4P2022. This contains the information that mother has had two therapeutic abortions. Does that information belong in a newborn’s chart? Especially in the era of the EHR where this information will hang around forever and will be easily obtained by the baby 16 years later when she can access all her medical information online. Will the mother be upset for her teenage daughter to learn that mom has had two abortions? Is that private information, belonging to the mother, that was given in confidence to her obstetrician? I advocate respecting privacy.
I have similar concerns about STD information being transferred from maternal charts to the newborn’s EHR. A maternal history of gonorrhea treated 8 years previously is unlikely to be relevant and should not populate the newborn’s EHR. I can make an argument that chlamydia detected and treated during the pregnancy might be useful to the baby’s pediatrician because neither treatment nor tests of cure are perfect. Perhaps, it could exist as a Snapchat-type record and disappear from the newborn’s record in a year if no respiratory symptoms occur.
I’m aware of efforts to destigmatize abortion and STDs, but, until that occurs, sensitive information should be handled delicately to preserve privacy. That is a major component of the Hippocratic Oath.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis.