User login
The price of protection
It’s very likely that you have at least one or two female patients who play lacrosse. The sport has been reported to be the fastest-growing high school sport in the United States. (“Lacrosse is Actually America’s Fastest-Growing Sport,” by John Templon, BuzzFeed News, June 30, 2014). When I played in college, most of my teammates were products of prep schools in the Northeast or one of the few local hotbeds in Baltimore, Long Island, or the Finger Lakes Region of New York. But pickings were slim, and there was room for walk-ons like me looking to learn a new sport and stay in shape for football. Now hundreds of high schools in all parts of the country offer the sport for both boys and girls.

With growing awareness of the long-term effects of repeated head trauma, there has been a call from some parents and organizers of women’s lacrosse to require helmets on all players (“As Concussion Worries Rise, Girls’ Lacrosse Turns to Headgear,” by Bill Pennington, The New York Times, Nov 23, 2017). To those of us who have committed our professional lives to the health of children, the inclusion of helmets to the standard equipment for a female lacrosse player sounds like a good idea.
However, the proposed mandate has its critics, including several college coaches. Karen Corbett, women’s lacrosse coach at the University of Pennsylvania, has said that, players “will start to lead with their head because they feel protected, and that causes more injuries. We’ll become a more physical sport and a very different sport than we are today.”

Although I’m afraid that there are few data to support the validity of Dr. Hanley’s prediction, any observer of college hockey over the last 3 or 4 decades will tell you that he was unfortunately correct. There have been certainly fewer lacerations and eye injuries since face masks were introduced, but the game has become far more violent, and head, neck, and spine injuries have become more frequent. I think part of the problem is that game officials have been duped by the same false assumption as the players that more protection would make the game safer, and enforcement of the rules has not kept up with the technological changes.
There will always be injuries in any sport, but before we as physicians lend our support to a proposed change in protective equipment, we should step back and look at the broader picture. While the loss of an eye for an individual player is a tragedy, did we put several dozen more players at greater risk for spinal injury in college hockey with more protective gear? If adding headgear protects female lacrosse players from concussions, what might be the result if play becomes more physical? Protection can come with a price.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at pdnews@frontlinemedcom.com.
It’s very likely that you have at least one or two female patients who play lacrosse. The sport has been reported to be the fastest-growing high school sport in the United States. (“Lacrosse is Actually America’s Fastest-Growing Sport,” by John Templon, BuzzFeed News, June 30, 2014). When I played in college, most of my teammates were products of prep schools in the Northeast or one of the few local hotbeds in Baltimore, Long Island, or the Finger Lakes Region of New York. But pickings were slim, and there was room for walk-ons like me looking to learn a new sport and stay in shape for football. Now hundreds of high schools in all parts of the country offer the sport for both boys and girls.
With growing awareness of the long-term effects of repeated head trauma, there has been a call from some parents and organizers of women’s lacrosse to require helmets on all players (“As Concussion Worries Rise, Girls’ Lacrosse Turns to Headgear,” by Bill Pennington, The New York Times, Nov 23, 2017). To those of us who have committed our professional lives to the health of children, the inclusion of helmets to the standard equipment for a female lacrosse player sounds like a good idea.
However, the proposed mandate has its critics, including several college coaches. Karen Corbett, women’s lacrosse coach at the University of Pennsylvania, has said that, players “will start to lead with their head because they feel protected, and that causes more injuries. We’ll become a more physical sport and a very different sport than we are today.”
Although I’m afraid that there are few data to support the validity of Dr. Hanley’s prediction, any observer of college hockey over the last 3 or 4 decades will tell you that he was unfortunately correct. There have been certainly fewer lacerations and eye injuries since face masks were introduced, but the game has become far more violent, and head, neck, and spine injuries have become more frequent. I think part of the problem is that game officials have been duped by the same false assumption as the players that more protection would make the game safer, and enforcement of the rules has not kept up with the technological changes.
There will always be injuries in any sport, but before we as physicians lend our support to a proposed change in protective equipment, we should step back and look at the broader picture. While the loss of an eye for an individual player is a tragedy, did we put several dozen more players at greater risk for spinal injury in college hockey with more protective gear? If adding headgear protects female lacrosse players from concussions, what might be the result if play becomes more physical? Protection can come with a price.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at pdnews@frontlinemedcom.com.
It’s very likely that you have at least one or two female patients who play lacrosse. The sport has been reported to be the fastest-growing high school sport in the United States. (“Lacrosse is Actually America’s Fastest-Growing Sport,” by John Templon, BuzzFeed News, June 30, 2014). When I played in college, most of my teammates were products of prep schools in the Northeast or one of the few local hotbeds in Baltimore, Long Island, or the Finger Lakes Region of New York. But pickings were slim, and there was room for walk-ons like me looking to learn a new sport and stay in shape for football. Now hundreds of high schools in all parts of the country offer the sport for both boys and girls.
With growing awareness of the long-term effects of repeated head trauma, there has been a call from some parents and organizers of women’s lacrosse to require helmets on all players (“As Concussion Worries Rise, Girls’ Lacrosse Turns to Headgear,” by Bill Pennington, The New York Times, Nov 23, 2017). To those of us who have committed our professional lives to the health of children, the inclusion of helmets to the standard equipment for a female lacrosse player sounds like a good idea.
However, the proposed mandate has its critics, including several college coaches. Karen Corbett, women’s lacrosse coach at the University of Pennsylvania, has said that, players “will start to lead with their head because they feel protected, and that causes more injuries. We’ll become a more physical sport and a very different sport than we are today.”
Although I’m afraid that there are few data to support the validity of Dr. Hanley’s prediction, any observer of college hockey over the last 3 or 4 decades will tell you that he was unfortunately correct. There have been certainly fewer lacerations and eye injuries since face masks were introduced, but the game has become far more violent, and head, neck, and spine injuries have become more frequent. I think part of the problem is that game officials have been duped by the same false assumption as the players that more protection would make the game safer, and enforcement of the rules has not kept up with the technological changes.
There will always be injuries in any sport, but before we as physicians lend our support to a proposed change in protective equipment, we should step back and look at the broader picture. While the loss of an eye for an individual player is a tragedy, did we put several dozen more players at greater risk for spinal injury in college hockey with more protective gear? If adding headgear protects female lacrosse players from concussions, what might be the result if play becomes more physical? Protection can come with a price.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at pdnews@frontlinemedcom.com.
Teens with PID underscreened for HIV, syphilis
CHICAGO – Adolescents with pelvic inflammatory disease (PID) were unlikely to be screened for HIV or syphilis, and many didn’t receive an appropriate antibiotic regimen, according to a recent study reported at the annual meeting of the American Academy of Pediatrics.
Patients who were sent home rather than admitted were especially likely to miss screening, as were Hispanic patients and those with private insurance.

The Centers for Disease Control and Prevention strongly recommends that all women diagnosed with PID be tested for HIV, and that high-risk individuals also be tested for syphilis, wrote Amanda Jichlinski, MD, and her coauthors at Children’s National Health System, Washington.
The study, presented during a poster session, used data from the national Pediatric Health Information System database from 2010 to 2015. A total of 10,698 records with a diagnostic code for PID were included; patients were females aged 12-21 years seen in a pediatric emergency department.
In addition to the primary outcome of syphilis and HIV testing, the authors also looked at whether antibiotic administration for PID was in line with CDC recommendations – and it wasn’t. “Fewer than half of patients in the ED received antibiotic regimens adherent to CDC guidelines,” wrote Dr. Jichlinski and her coauthors.
Forty-six percent of patients received ceftriaxone and doxycycline, 21% received ceftriaxone and azithromycin, and 6% received ceftriaxone and metronidazole. Ceftriaxone monotherapy was given to 15% of patients. One in 10 patients with a PID diagnosis received no antibiotic at all; 2% of patients received some other regimen.
The researchers used multivariable analysis to examine separately which patient and hospital characteristics were associated with an increased likelihood of testing for both HIV and syphilis. With white, non-Hispanic adolescents used as the referent, Hispanic females with PID were less likely to receive screening for either HIV or syphilis (adjusted odds ratio, 0.8 for both; 95% confidence interval, 0.7-1.0 for both).
In contrast, black non-Hispanic females were screened more often; the aOR for HIV screening was 1.4 (95% CI, 1.2-1.6), and the aOR for syphilis screening was 1.8 (95% CI, 1.6-2.0) for this group of adolescents.
Patients were dichotomized into older (17-21 years of age; n = 4,737, 44%) and younger (12-16 years of age; n = 5,961, 56%) age groups; younger patients were slightly more likely to receive HIV (aOR, 1.2) and syphilis (aOR, 1.1) screening.
Just under a third of patients in the study were seen in a hospital with fewer than 300 beds, and these facilities were more likely to screen for HIV (aOR, 1.4) and syphilis (aOR, 1.1) than the larger hospitals.
By far the largest predictor of whether HIV and syphilis screening was done, though, was a hospital admission. Patients who were admitted (n = 4,043, 38%) were 7 times more likely to be screened for HIV and 4.6 times more likely to be screened for syphilis than those who were sent home from the emergency department.
Although the large, nationally representative study had many strengths, Dr. Jichlinski and her coauthors acknowledged that the data they were provided couldn’t account for medication that was prescribed, rather than administered in the emergency department. Also, the results may not be generalizable to adolescents treated in nonpediatric emergency departments or other facilities, such as urgent care centers.
“Adolescents with PID are underscreened for HIV and syphilis,” wrote Dr. Jichlinski and her coauthors. They called for pediatricians to receive more education about management of PID in adolescents. From a practical perspective, the investigators also suggested incorporating order sets for sexually transmitted infection testing and antibiotic administration into electronic medical records; in this way, a PID diagnosis code would trigger simplified testing and treatment choices.
Dr. Jichlinski reported no conflicts of interest. Dr. Monika Goyal, MD, senior author on the study, reported funding support by the National Institute of Child Health and Human Development. Dr. Goyal also holds an appointment at the George Washington University, Washington.
SOURCE: Jichlinski A et al. AAP 2017 Abstract 5, AAP Section on Emergency Medicine.
CHICAGO – Adolescents with pelvic inflammatory disease (PID) were unlikely to be screened for HIV or syphilis, and many didn’t receive an appropriate antibiotic regimen, according to a recent study reported at the annual meeting of the American Academy of Pediatrics.
Patients who were sent home rather than admitted were especially likely to miss screening, as were Hispanic patients and those with private insurance.
The Centers for Disease Control and Prevention strongly recommends that all women diagnosed with PID be tested for HIV, and that high-risk individuals also be tested for syphilis, wrote Amanda Jichlinski, MD, and her coauthors at Children’s National Health System, Washington.
The study, presented during a poster session, used data from the national Pediatric Health Information System database from 2010 to 2015. A total of 10,698 records with a diagnostic code for PID were included; patients were females aged 12-21 years seen in a pediatric emergency department.
In addition to the primary outcome of syphilis and HIV testing, the authors also looked at whether antibiotic administration for PID was in line with CDC recommendations – and it wasn’t. “Fewer than half of patients in the ED received antibiotic regimens adherent to CDC guidelines,” wrote Dr. Jichlinski and her coauthors.
Forty-six percent of patients received ceftriaxone and doxycycline, 21% received ceftriaxone and azithromycin, and 6% received ceftriaxone and metronidazole. Ceftriaxone monotherapy was given to 15% of patients. One in 10 patients with a PID diagnosis received no antibiotic at all; 2% of patients received some other regimen.
The researchers used multivariable analysis to examine separately which patient and hospital characteristics were associated with an increased likelihood of testing for both HIV and syphilis. With white, non-Hispanic adolescents used as the referent, Hispanic females with PID were less likely to receive screening for either HIV or syphilis (adjusted odds ratio, 0.8 for both; 95% confidence interval, 0.7-1.0 for both).
In contrast, black non-Hispanic females were screened more often; the aOR for HIV screening was 1.4 (95% CI, 1.2-1.6), and the aOR for syphilis screening was 1.8 (95% CI, 1.6-2.0) for this group of adolescents.
Patients were dichotomized into older (17-21 years of age; n = 4,737, 44%) and younger (12-16 years of age; n = 5,961, 56%) age groups; younger patients were slightly more likely to receive HIV (aOR, 1.2) and syphilis (aOR, 1.1) screening.
Just under a third of patients in the study were seen in a hospital with fewer than 300 beds, and these facilities were more likely to screen for HIV (aOR, 1.4) and syphilis (aOR, 1.1) than the larger hospitals.
By far the largest predictor of whether HIV and syphilis screening was done, though, was a hospital admission. Patients who were admitted (n = 4,043, 38%) were 7 times more likely to be screened for HIV and 4.6 times more likely to be screened for syphilis than those who were sent home from the emergency department.
Although the large, nationally representative study had many strengths, Dr. Jichlinski and her coauthors acknowledged that the data they were provided couldn’t account for medication that was prescribed, rather than administered in the emergency department. Also, the results may not be generalizable to adolescents treated in nonpediatric emergency departments or other facilities, such as urgent care centers.
“Adolescents with PID are underscreened for HIV and syphilis,” wrote Dr. Jichlinski and her coauthors. They called for pediatricians to receive more education about management of PID in adolescents. From a practical perspective, the investigators also suggested incorporating order sets for sexually transmitted infection testing and antibiotic administration into electronic medical records; in this way, a PID diagnosis code would trigger simplified testing and treatment choices.
Dr. Jichlinski reported no conflicts of interest. Dr. Monika Goyal, MD, senior author on the study, reported funding support by the National Institute of Child Health and Human Development. Dr. Goyal also holds an appointment at the George Washington University, Washington.
SOURCE: Jichlinski A et al. AAP 2017 Abstract 5, AAP Section on Emergency Medicine.
CHICAGO – Adolescents with pelvic inflammatory disease (PID) were unlikely to be screened for HIV or syphilis, and many didn’t receive an appropriate antibiotic regimen, according to a recent study reported at the annual meeting of the American Academy of Pediatrics.
Patients who were sent home rather than admitted were especially likely to miss screening, as were Hispanic patients and those with private insurance.
The Centers for Disease Control and Prevention strongly recommends that all women diagnosed with PID be tested for HIV, and that high-risk individuals also be tested for syphilis, wrote Amanda Jichlinski, MD, and her coauthors at Children’s National Health System, Washington.
The study, presented during a poster session, used data from the national Pediatric Health Information System database from 2010 to 2015. A total of 10,698 records with a diagnostic code for PID were included; patients were females aged 12-21 years seen in a pediatric emergency department.
In addition to the primary outcome of syphilis and HIV testing, the authors also looked at whether antibiotic administration for PID was in line with CDC recommendations – and it wasn’t. “Fewer than half of patients in the ED received antibiotic regimens adherent to CDC guidelines,” wrote Dr. Jichlinski and her coauthors.
Forty-six percent of patients received ceftriaxone and doxycycline, 21% received ceftriaxone and azithromycin, and 6% received ceftriaxone and metronidazole. Ceftriaxone monotherapy was given to 15% of patients. One in 10 patients with a PID diagnosis received no antibiotic at all; 2% of patients received some other regimen.
The researchers used multivariable analysis to examine separately which patient and hospital characteristics were associated with an increased likelihood of testing for both HIV and syphilis. With white, non-Hispanic adolescents used as the referent, Hispanic females with PID were less likely to receive screening for either HIV or syphilis (adjusted odds ratio, 0.8 for both; 95% confidence interval, 0.7-1.0 for both).
In contrast, black non-Hispanic females were screened more often; the aOR for HIV screening was 1.4 (95% CI, 1.2-1.6), and the aOR for syphilis screening was 1.8 (95% CI, 1.6-2.0) for this group of adolescents.
Patients were dichotomized into older (17-21 years of age; n = 4,737, 44%) and younger (12-16 years of age; n = 5,961, 56%) age groups; younger patients were slightly more likely to receive HIV (aOR, 1.2) and syphilis (aOR, 1.1) screening.
Just under a third of patients in the study were seen in a hospital with fewer than 300 beds, and these facilities were more likely to screen for HIV (aOR, 1.4) and syphilis (aOR, 1.1) than the larger hospitals.
By far the largest predictor of whether HIV and syphilis screening was done, though, was a hospital admission. Patients who were admitted (n = 4,043, 38%) were 7 times more likely to be screened for HIV and 4.6 times more likely to be screened for syphilis than those who were sent home from the emergency department.
Although the large, nationally representative study had many strengths, Dr. Jichlinski and her coauthors acknowledged that the data they were provided couldn’t account for medication that was prescribed, rather than administered in the emergency department. Also, the results may not be generalizable to adolescents treated in nonpediatric emergency departments or other facilities, such as urgent care centers.
“Adolescents with PID are underscreened for HIV and syphilis,” wrote Dr. Jichlinski and her coauthors. They called for pediatricians to receive more education about management of PID in adolescents. From a practical perspective, the investigators also suggested incorporating order sets for sexually transmitted infection testing and antibiotic administration into electronic medical records; in this way, a PID diagnosis code would trigger simplified testing and treatment choices.
Dr. Jichlinski reported no conflicts of interest. Dr. Monika Goyal, MD, senior author on the study, reported funding support by the National Institute of Child Health and Human Development. Dr. Goyal also holds an appointment at the George Washington University, Washington.
SOURCE: Jichlinski A et al. AAP 2017 Abstract 5, AAP Section on Emergency Medicine.
REPORTING FROM AAP 2017
Key clinical point:
Major finding: Hispanic females were least likely to be screened (adjusted OR, 0.8), compared with non-Hispanic white females.
Study details: Retrospective study of 10,698 adolescent patients with PID from a national database.
Disclosures: The study was funded in part by the National Institute of Child Health and Development. The authors had no relevant financial disclosures.
Source: Jichlinski A et al. AAP 2017 Abstract 5, AAP Section on Emergency Medicine
Nondrug Treatments May Benefit Patients With Epilepsy
WASHINGTON, DC—Many patients with pharmacoresistant epilepsy may benefit from nondrug treatments, including vagus nerve stimulation (VNS), the ketogenic diet, and corpus callosotomy, according to a study presented at the 71st Annual Meeting of the American Epilepsy Society. The treatments may reduce generalized and focal seizures, and most parents whose children underwent these procedures would opt for the same treatment under similar circumstances, the researchers said.
About 20% to 30% of patients have pharmacoresistant epilepsy. The ketogenic diet, corpus callosotomy, and VNS have been studied as alternatives to antiepileptic drugs (AEDs) for these patients, but few studies have compared the modalities.
Dave F. Clarke, MD, MBBS, Professor of Pediatric Neurology at the Baylor College of Medicine and Clinical Director of Epilepsy at Texas Children’s Hospital in Houston, and colleagues compared seizure control, cognitive and behavioral factors, quality of life, and parent satisfaction among patients who received VNS, underwent corpus callosotomy, or initiated the ketogenic diet. They identified 336 patients who had received one of these treatments at Dell Children’s Medical Center of Central Texas in Austin between January 2010 and November 2015. Parents of 210 of the patients completed a nine-item telephone survey.

Of the 210 patients whose parents completed the survey, 98 (33.6%) had initiated the ketogenic diet, 150 (51.4%) had received VNS, and 44 (15.1%) had undergone corpus callosotomy. Patients were between the ages of 8 months and 20 years. Patients who had initiated the ketogenic diet had a mean age of about 7, and patients who received VNS or underwent corpus callosotomy had a mean age of about 10. Patients had failed more than three AEDs on average (range, two to 13).
Parents reported a 50% or greater reduction in generalized seizures in 63% of patients who went on the ketogenic diet, 54% of patients who underwent corpus callosotomy, and 52% of patients who received VNS. Parents reported a 50% or greater reduction in focal seizures in 56% of children who went on the ketogenic diet, 56% of patients who had corpus callosotomy, and 53% of patients who received VNS.
In addition, parents reported improved quality of life in 48% of patients on the ketogenic diet, 63% of patients who had corpus callosotomy, and 44% of patients who received VNS. Overall, 80% of parents whose children were on the ketogenic diet or received VNS and 75% of parents whose children underwent corpus callosotomy reported that they were satisfied with the treatment that their child had received.
“Higher health-related quality of life after intervention was predicted by improved behavior, increased engagement, diminished frequency of atonic or generalized tonic-clonic seizures, and reduction in epilepsy-related injuries,” the researchers concluded. Parents were more likely to say that they would repeat the procedure if, after the treatment, “their child was more engaged, had diminished frequency of atonic or generalized tonic-clonic seizures, and had a reduction in epilepsy-related injuries.”
“Unfortunately, many doctors keep trying medications without considering alternatives,” said Dr. Clarke. “Based on the parents’ feedback, I would suggest doctors introduce the concept of alternatives after two AEDs fail to control seizures.” If surgery to ablate or remove the area of the brain where seizures originate is not an option, neurologists should talk to parents about the ketogenic diet, VNS, or corpus callosotomy. “If parents think the diet can be tolerated, trying it first may not be a bad option,” he said.
—Jake Remaly
WASHINGTON, DC—Many patients with pharmacoresistant epilepsy may benefit from nondrug treatments, including vagus nerve stimulation (VNS), the ketogenic diet, and corpus callosotomy, according to a study presented at the 71st Annual Meeting of the American Epilepsy Society. The treatments may reduce generalized and focal seizures, and most parents whose children underwent these procedures would opt for the same treatment under similar circumstances, the researchers said.
About 20% to 30% of patients have pharmacoresistant epilepsy. The ketogenic diet, corpus callosotomy, and VNS have been studied as alternatives to antiepileptic drugs (AEDs) for these patients, but few studies have compared the modalities.
Dave F. Clarke, MD, MBBS, Professor of Pediatric Neurology at the Baylor College of Medicine and Clinical Director of Epilepsy at Texas Children’s Hospital in Houston, and colleagues compared seizure control, cognitive and behavioral factors, quality of life, and parent satisfaction among patients who received VNS, underwent corpus callosotomy, or initiated the ketogenic diet. They identified 336 patients who had received one of these treatments at Dell Children’s Medical Center of Central Texas in Austin between January 2010 and November 2015. Parents of 210 of the patients completed a nine-item telephone survey.
Of the 210 patients whose parents completed the survey, 98 (33.6%) had initiated the ketogenic diet, 150 (51.4%) had received VNS, and 44 (15.1%) had undergone corpus callosotomy. Patients were between the ages of 8 months and 20 years. Patients who had initiated the ketogenic diet had a mean age of about 7, and patients who received VNS or underwent corpus callosotomy had a mean age of about 10. Patients had failed more than three AEDs on average (range, two to 13).
Parents reported a 50% or greater reduction in generalized seizures in 63% of patients who went on the ketogenic diet, 54% of patients who underwent corpus callosotomy, and 52% of patients who received VNS. Parents reported a 50% or greater reduction in focal seizures in 56% of children who went on the ketogenic diet, 56% of patients who had corpus callosotomy, and 53% of patients who received VNS.
In addition, parents reported improved quality of life in 48% of patients on the ketogenic diet, 63% of patients who had corpus callosotomy, and 44% of patients who received VNS. Overall, 80% of parents whose children were on the ketogenic diet or received VNS and 75% of parents whose children underwent corpus callosotomy reported that they were satisfied with the treatment that their child had received.
“Higher health-related quality of life after intervention was predicted by improved behavior, increased engagement, diminished frequency of atonic or generalized tonic-clonic seizures, and reduction in epilepsy-related injuries,” the researchers concluded. Parents were more likely to say that they would repeat the procedure if, after the treatment, “their child was more engaged, had diminished frequency of atonic or generalized tonic-clonic seizures, and had a reduction in epilepsy-related injuries.”
“Unfortunately, many doctors keep trying medications without considering alternatives,” said Dr. Clarke. “Based on the parents’ feedback, I would suggest doctors introduce the concept of alternatives after two AEDs fail to control seizures.” If surgery to ablate or remove the area of the brain where seizures originate is not an option, neurologists should talk to parents about the ketogenic diet, VNS, or corpus callosotomy. “If parents think the diet can be tolerated, trying it first may not be a bad option,” he said.
—Jake Remaly
WASHINGTON, DC—Many patients with pharmacoresistant epilepsy may benefit from nondrug treatments, including vagus nerve stimulation (VNS), the ketogenic diet, and corpus callosotomy, according to a study presented at the 71st Annual Meeting of the American Epilepsy Society. The treatments may reduce generalized and focal seizures, and most parents whose children underwent these procedures would opt for the same treatment under similar circumstances, the researchers said.
About 20% to 30% of patients have pharmacoresistant epilepsy. The ketogenic diet, corpus callosotomy, and VNS have been studied as alternatives to antiepileptic drugs (AEDs) for these patients, but few studies have compared the modalities.
Dave F. Clarke, MD, MBBS, Professor of Pediatric Neurology at the Baylor College of Medicine and Clinical Director of Epilepsy at Texas Children’s Hospital in Houston, and colleagues compared seizure control, cognitive and behavioral factors, quality of life, and parent satisfaction among patients who received VNS, underwent corpus callosotomy, or initiated the ketogenic diet. They identified 336 patients who had received one of these treatments at Dell Children’s Medical Center of Central Texas in Austin between January 2010 and November 2015. Parents of 210 of the patients completed a nine-item telephone survey.
Of the 210 patients whose parents completed the survey, 98 (33.6%) had initiated the ketogenic diet, 150 (51.4%) had received VNS, and 44 (15.1%) had undergone corpus callosotomy. Patients were between the ages of 8 months and 20 years. Patients who had initiated the ketogenic diet had a mean age of about 7, and patients who received VNS or underwent corpus callosotomy had a mean age of about 10. Patients had failed more than three AEDs on average (range, two to 13).
Parents reported a 50% or greater reduction in generalized seizures in 63% of patients who went on the ketogenic diet, 54% of patients who underwent corpus callosotomy, and 52% of patients who received VNS. Parents reported a 50% or greater reduction in focal seizures in 56% of children who went on the ketogenic diet, 56% of patients who had corpus callosotomy, and 53% of patients who received VNS.
In addition, parents reported improved quality of life in 48% of patients on the ketogenic diet, 63% of patients who had corpus callosotomy, and 44% of patients who received VNS. Overall, 80% of parents whose children were on the ketogenic diet or received VNS and 75% of parents whose children underwent corpus callosotomy reported that they were satisfied with the treatment that their child had received.
“Higher health-related quality of life after intervention was predicted by improved behavior, increased engagement, diminished frequency of atonic or generalized tonic-clonic seizures, and reduction in epilepsy-related injuries,” the researchers concluded. Parents were more likely to say that they would repeat the procedure if, after the treatment, “their child was more engaged, had diminished frequency of atonic or generalized tonic-clonic seizures, and had a reduction in epilepsy-related injuries.”
“Unfortunately, many doctors keep trying medications without considering alternatives,” said Dr. Clarke. “Based on the parents’ feedback, I would suggest doctors introduce the concept of alternatives after two AEDs fail to control seizures.” If surgery to ablate or remove the area of the brain where seizures originate is not an option, neurologists should talk to parents about the ketogenic diet, VNS, or corpus callosotomy. “If parents think the diet can be tolerated, trying it first may not be a bad option,” he said.
—Jake Remaly
Folic acid and multivitamin supplements associated with reduced autism risk
Taking folic acid and/or multivitamin supplements preceding and during pregnancy is associated with a lower risk of offspring developing autism spectrum disorder (ASD), an observational epidemiologic study published Jan. 3 showed.
The findings could have important public health implications, reported Stephen Z. Levine, PhD, and his associates.
The investigators found that 572 children, or 1.3%, received an ASD diagnosis. Dr. Levine and his associates found that children whose mothers took folic acid and multivitamin supplements during pregnancy had a lower risk of developing ASD (relative risk, 0.27; 95% confidence interval, 0.22-0.33; P less than .001), compared with those whose mothers took no supplements. Similarly, there was reduced risk among those whose mothers took only folic acid during pregnancy (RR, 0.32; CI, 0.26-0.41; P less than .001) or only multivitamins (RR, 0.35; CI, 0.28-0.44; P less than .001). Likewise, lower risks were seen among offspring whose mothers took supplements before pregnancy: Compared with no supplements, the RR was 0.39 for folic acid and/or multivitamins (CI, 0.30-0.50; P less than .001), 0.56 for just folic acid (95%CI, 0.42-0.74; P = .001), and 0.36 for just multivitamins (95%CI, 0.24-0.52; P less than .001). Similar associations were found among male and female offspring.
“This finding may reflect noncompliance, higher rates of vitamin deficiency, or poor diet among persons with psychiatric conditions,” wrote Dr. Levine, of the department of community mental health at the University of Haifa, Israel, and his associates in JAMA Psychiatry.
Another important finding is that maternal exposure to folic acid and multivitamin supplements 2 years before pregnancy is tied to a lower ASD risk.
The investigators acknowledged that the study was limited by their inability to determine possible confounding factors, such as the vehicle of vitamin dispensations, use of over-the-counter supplements, false-positive classifications from noncompliance, and absence of information on gestational age. In addition, they said, “causality cannot be inferred from observational studies such as this one.” In light of those limitations, investigators said, additional studies replicating these findings are needed.
The study was funded by several entities, including the National Institutes of Health, the Fredrik and Ingrid Thuring Foundation, and the Swedish Society of Medicine. Dr. Levine reported receiving support from Shire Pharmaceuticals, and coauthor Arad Kodesh, MD, is an employee of Meuhedet Health Services. No other relevant financial disclosures were reported.
ezimmerman@frontlinemedcom.com
SOURCE: Levine SZ et al. JAMA Psychiatry. 2018 Jan 3. doi: 10.1001/jamapsychiatry.2017.4050.
Taking folic acid and/or multivitamin supplements preceding and during pregnancy is associated with a lower risk of offspring developing autism spectrum disorder (ASD), an observational epidemiologic study published Jan. 3 showed.
The findings could have important public health implications, reported Stephen Z. Levine, PhD, and his associates.
The investigators found that 572 children, or 1.3%, received an ASD diagnosis. Dr. Levine and his associates found that children whose mothers took folic acid and multivitamin supplements during pregnancy had a lower risk of developing ASD (relative risk, 0.27; 95% confidence interval, 0.22-0.33; P less than .001), compared with those whose mothers took no supplements. Similarly, there was reduced risk among those whose mothers took only folic acid during pregnancy (RR, 0.32; CI, 0.26-0.41; P less than .001) or only multivitamins (RR, 0.35; CI, 0.28-0.44; P less than .001). Likewise, lower risks were seen among offspring whose mothers took supplements before pregnancy: Compared with no supplements, the RR was 0.39 for folic acid and/or multivitamins (CI, 0.30-0.50; P less than .001), 0.56 for just folic acid (95%CI, 0.42-0.74; P = .001), and 0.36 for just multivitamins (95%CI, 0.24-0.52; P less than .001). Similar associations were found among male and female offspring.
“This finding may reflect noncompliance, higher rates of vitamin deficiency, or poor diet among persons with psychiatric conditions,” wrote Dr. Levine, of the department of community mental health at the University of Haifa, Israel, and his associates in JAMA Psychiatry.
Another important finding is that maternal exposure to folic acid and multivitamin supplements 2 years before pregnancy is tied to a lower ASD risk.
The investigators acknowledged that the study was limited by their inability to determine possible confounding factors, such as the vehicle of vitamin dispensations, use of over-the-counter supplements, false-positive classifications from noncompliance, and absence of information on gestational age. In addition, they said, “causality cannot be inferred from observational studies such as this one.” In light of those limitations, investigators said, additional studies replicating these findings are needed.
The study was funded by several entities, including the National Institutes of Health, the Fredrik and Ingrid Thuring Foundation, and the Swedish Society of Medicine. Dr. Levine reported receiving support from Shire Pharmaceuticals, and coauthor Arad Kodesh, MD, is an employee of Meuhedet Health Services. No other relevant financial disclosures were reported.
ezimmerman@frontlinemedcom.com
SOURCE: Levine SZ et al. JAMA Psychiatry. 2018 Jan 3. doi: 10.1001/jamapsychiatry.2017.4050.
Taking folic acid and/or multivitamin supplements preceding and during pregnancy is associated with a lower risk of offspring developing autism spectrum disorder (ASD), an observational epidemiologic study published Jan. 3 showed.
The findings could have important public health implications, reported Stephen Z. Levine, PhD, and his associates.
The investigators found that 572 children, or 1.3%, received an ASD diagnosis. Dr. Levine and his associates found that children whose mothers took folic acid and multivitamin supplements during pregnancy had a lower risk of developing ASD (relative risk, 0.27; 95% confidence interval, 0.22-0.33; P less than .001), compared with those whose mothers took no supplements. Similarly, there was reduced risk among those whose mothers took only folic acid during pregnancy (RR, 0.32; CI, 0.26-0.41; P less than .001) or only multivitamins (RR, 0.35; CI, 0.28-0.44; P less than .001). Likewise, lower risks were seen among offspring whose mothers took supplements before pregnancy: Compared with no supplements, the RR was 0.39 for folic acid and/or multivitamins (CI, 0.30-0.50; P less than .001), 0.56 for just folic acid (95%CI, 0.42-0.74; P = .001), and 0.36 for just multivitamins (95%CI, 0.24-0.52; P less than .001). Similar associations were found among male and female offspring.
“This finding may reflect noncompliance, higher rates of vitamin deficiency, or poor diet among persons with psychiatric conditions,” wrote Dr. Levine, of the department of community mental health at the University of Haifa, Israel, and his associates in JAMA Psychiatry.
Another important finding is that maternal exposure to folic acid and multivitamin supplements 2 years before pregnancy is tied to a lower ASD risk.
The investigators acknowledged that the study was limited by their inability to determine possible confounding factors, such as the vehicle of vitamin dispensations, use of over-the-counter supplements, false-positive classifications from noncompliance, and absence of information on gestational age. In addition, they said, “causality cannot be inferred from observational studies such as this one.” In light of those limitations, investigators said, additional studies replicating these findings are needed.
The study was funded by several entities, including the National Institutes of Health, the Fredrik and Ingrid Thuring Foundation, and the Swedish Society of Medicine. Dr. Levine reported receiving support from Shire Pharmaceuticals, and coauthor Arad Kodesh, MD, is an employee of Meuhedet Health Services. No other relevant financial disclosures were reported.
ezimmerman@frontlinemedcom.com
SOURCE: Levine SZ et al. JAMA Psychiatry. 2018 Jan 3. doi: 10.1001/jamapsychiatry.2017.4050.
Key clinical point: Taking folic acid and multivitamin supplements before and during pregnancy can reduce risk of autism in children.
Major finding: Children whose mothers took folic acid and/or multivitamin supplements during pregnancy had a decreased risk of developing ASD, compared with those whose mothers did not (relative risk, 0.27; 95% confidence interval, 0.22-0.33; P less than .001).
Study details: Observational epidemiologic study of 45,300 Israeli children born between January 2003 and December 2007 and followed until January 2015.
Disclosures: The study was funded by several entities, including the National Institutes of Health, the Fredrik and Ingrid Thuring Foundation, and the Swedish Society of Medicine. Dr. Levine reported receiving support from Shire Pharmaceuticals, and coauthor Arad Kodesh, MD, is an employee of Meuhedet Health Services. No other relevant financial disclosures were reported.
Source: Levine SZ et al. JAMA Psychiatry. 2018 Jan 3. doi: 10.1001/jamapsychiatry.2017.4050.
FDA grants breakthrough therapy designation for severe aplastic anemia drug
The Food and Drug Administration has granted breakthrough therapy designation to eltrombopag (Promacta) for use in combination with standard immunosuppressive therapy as a first-line treatment for patients with severe aplastic anemia (SAA).

Eltrombopag already is approved as a second-line therapy in patients with refractory SAA and is approved for adults and children with refractory chronic immune thrombocytopenia (ITP).
“Promacta is a promising medicine that, if approved for first-line use in severe aplastic anemia, may redefine the standard of care for patients with this rare and serious bone marrow condition,” Samit Hirawat, MD, head of Novartis Oncology Global Drug Development, said in a statement.
The Food and Drug Administration has granted breakthrough therapy designation to eltrombopag (Promacta) for use in combination with standard immunosuppressive therapy as a first-line treatment for patients with severe aplastic anemia (SAA).
Eltrombopag already is approved as a second-line therapy in patients with refractory SAA and is approved for adults and children with refractory chronic immune thrombocytopenia (ITP).
“Promacta is a promising medicine that, if approved for first-line use in severe aplastic anemia, may redefine the standard of care for patients with this rare and serious bone marrow condition,” Samit Hirawat, MD, head of Novartis Oncology Global Drug Development, said in a statement.
The Food and Drug Administration has granted breakthrough therapy designation to eltrombopag (Promacta) for use in combination with standard immunosuppressive therapy as a first-line treatment for patients with severe aplastic anemia (SAA).
Eltrombopag already is approved as a second-line therapy in patients with refractory SAA and is approved for adults and children with refractory chronic immune thrombocytopenia (ITP).
“Promacta is a promising medicine that, if approved for first-line use in severe aplastic anemia, may redefine the standard of care for patients with this rare and serious bone marrow condition,” Samit Hirawat, MD, head of Novartis Oncology Global Drug Development, said in a statement.
See for yourself
About 800 million radiology exams were performed in this country in the past year, and they generated approximately 60 billion images, according to an article published in 2017 in the Wall Street Journal (“No need for radiologists to be negative on AI,” by Greg Ip, Nov. 24, 2017). How many of those millions of radiology studies did you order? And how many of the scores of images you requested did you see with your own two eyes? In fact, how many of the radiologists’ reports that were sent to you did you read in their entirety? How often did you just skip over the radiologist’s CYA disclaimers and simply read the final summary, “exam negative”?

I enjoy the challenge of interpreting x-ray images. In fact, I toyed with becoming a pediatric radiologist, but that career path would have meant settling in or near a large city, a compromise my wife and I were unwilling to make. I hoped to continue my habit of looking at all my patients’ x-rays, but because my practice was not in or near the hospital, I reluctantly had to bend my rules and admit I didn’t see every image I had ordered. But, I did read every report in its entirety. In one case, an offhand comment buried in the middle of the radiologist’s report referring to the “residual barium from a previous study” caught my eye, because I knew the patient hadn’t had a previous contrast study. Unfortunately, the neuroblastoma that the radiologist had missed initially, and I had seen the next day, never responded to treatment.
Toward the end of my career, digital imagery allowed me to view my patients’ x-rays without having to leave my desk, which got me closer to my goal of seeing all my patients’ studies. However, the advent of computerized axial tomography and magnetic resonance imaging meant that an increasing number of studies pushed my anatomic knowledge beyond its limits.
I suspect that many of you benefited from the if-you-order-it-look-at-it mantra during your training. How many of you have continued to follow the dictum? With the advent of digitized imagery, there is really little excuse for not taking a minute or 2 to pull up your patients’ images on your desktop. One could argue that looking inside your patient is part of a complete exam. Forcing yourself to take that extra step and look at the study may nudge you into thinking twice about whether you really needed the information the imaging study might add to the diagnostic process. Was your click to order the study just a reflex subliminally related to the fear of a lawsuit? Was it important enough to deserve a firsthand look?
At the very least, being able to say, “I’ve looked at your x-rays myself, and they look fine” may be more comforting to your patient than a third-hand relay of a “negative reading” performed by someone whom they have likely never met.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
About 800 million radiology exams were performed in this country in the past year, and they generated approximately 60 billion images, according to an article published in 2017 in the Wall Street Journal (“No need for radiologists to be negative on AI,” by Greg Ip, Nov. 24, 2017). How many of those millions of radiology studies did you order? And how many of the scores of images you requested did you see with your own two eyes? In fact, how many of the radiologists’ reports that were sent to you did you read in their entirety? How often did you just skip over the radiologist’s CYA disclaimers and simply read the final summary, “exam negative”?
I enjoy the challenge of interpreting x-ray images. In fact, I toyed with becoming a pediatric radiologist, but that career path would have meant settling in or near a large city, a compromise my wife and I were unwilling to make. I hoped to continue my habit of looking at all my patients’ x-rays, but because my practice was not in or near the hospital, I reluctantly had to bend my rules and admit I didn’t see every image I had ordered. But, I did read every report in its entirety. In one case, an offhand comment buried in the middle of the radiologist’s report referring to the “residual barium from a previous study” caught my eye, because I knew the patient hadn’t had a previous contrast study. Unfortunately, the neuroblastoma that the radiologist had missed initially, and I had seen the next day, never responded to treatment.
Toward the end of my career, digital imagery allowed me to view my patients’ x-rays without having to leave my desk, which got me closer to my goal of seeing all my patients’ studies. However, the advent of computerized axial tomography and magnetic resonance imaging meant that an increasing number of studies pushed my anatomic knowledge beyond its limits.
I suspect that many of you benefited from the if-you-order-it-look-at-it mantra during your training. How many of you have continued to follow the dictum? With the advent of digitized imagery, there is really little excuse for not taking a minute or 2 to pull up your patients’ images on your desktop. One could argue that looking inside your patient is part of a complete exam. Forcing yourself to take that extra step and look at the study may nudge you into thinking twice about whether you really needed the information the imaging study might add to the diagnostic process. Was your click to order the study just a reflex subliminally related to the fear of a lawsuit? Was it important enough to deserve a firsthand look?
At the very least, being able to say, “I’ve looked at your x-rays myself, and they look fine” may be more comforting to your patient than a third-hand relay of a “negative reading” performed by someone whom they have likely never met.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
About 800 million radiology exams were performed in this country in the past year, and they generated approximately 60 billion images, according to an article published in 2017 in the Wall Street Journal (“No need for radiologists to be negative on AI,” by Greg Ip, Nov. 24, 2017). How many of those millions of radiology studies did you order? And how many of the scores of images you requested did you see with your own two eyes? In fact, how many of the radiologists’ reports that were sent to you did you read in their entirety? How often did you just skip over the radiologist’s CYA disclaimers and simply read the final summary, “exam negative”?
I enjoy the challenge of interpreting x-ray images. In fact, I toyed with becoming a pediatric radiologist, but that career path would have meant settling in or near a large city, a compromise my wife and I were unwilling to make. I hoped to continue my habit of looking at all my patients’ x-rays, but because my practice was not in or near the hospital, I reluctantly had to bend my rules and admit I didn’t see every image I had ordered. But, I did read every report in its entirety. In one case, an offhand comment buried in the middle of the radiologist’s report referring to the “residual barium from a previous study” caught my eye, because I knew the patient hadn’t had a previous contrast study. Unfortunately, the neuroblastoma that the radiologist had missed initially, and I had seen the next day, never responded to treatment.
Toward the end of my career, digital imagery allowed me to view my patients’ x-rays without having to leave my desk, which got me closer to my goal of seeing all my patients’ studies. However, the advent of computerized axial tomography and magnetic resonance imaging meant that an increasing number of studies pushed my anatomic knowledge beyond its limits.
I suspect that many of you benefited from the if-you-order-it-look-at-it mantra during your training. How many of you have continued to follow the dictum? With the advent of digitized imagery, there is really little excuse for not taking a minute or 2 to pull up your patients’ images on your desktop. One could argue that looking inside your patient is part of a complete exam. Forcing yourself to take that extra step and look at the study may nudge you into thinking twice about whether you really needed the information the imaging study might add to the diagnostic process. Was your click to order the study just a reflex subliminally related to the fear of a lawsuit? Was it important enough to deserve a firsthand look?
At the very least, being able to say, “I’ve looked at your x-rays myself, and they look fine” may be more comforting to your patient than a third-hand relay of a “negative reading” performed by someone whom they have likely never met.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Richner-Hanhart Syndrome (Tyrosinemia Type II)
To the Editor:
Richner-Hanhart syndrome, also known as tyrosinemia type II or oculocutaneous tyrosinemia, is a rare autosomal-recessive, childhood-onset, metabolic hereditary disease.1 A deficiency of tyrosine aminotransferase leads to an accumulation of tyrosine amino acid. It is characterized by the association of palmoplantar hyperkeratosis, bilateral keratitis, and neurological disorders.
An 18-month-old girl with recurrent warts of 6 months' duration was admitted to the dermatology department. She had been treated repeatedly with acyclovir for recurrent bilateral herpetic keratitis with major photophobia since 9 months of age with no response. Clinical presentation included punctate hyperkeratosis of the fingers and toes (Figure, A), severe photophobia with decreased visual acuity, and speech delay.
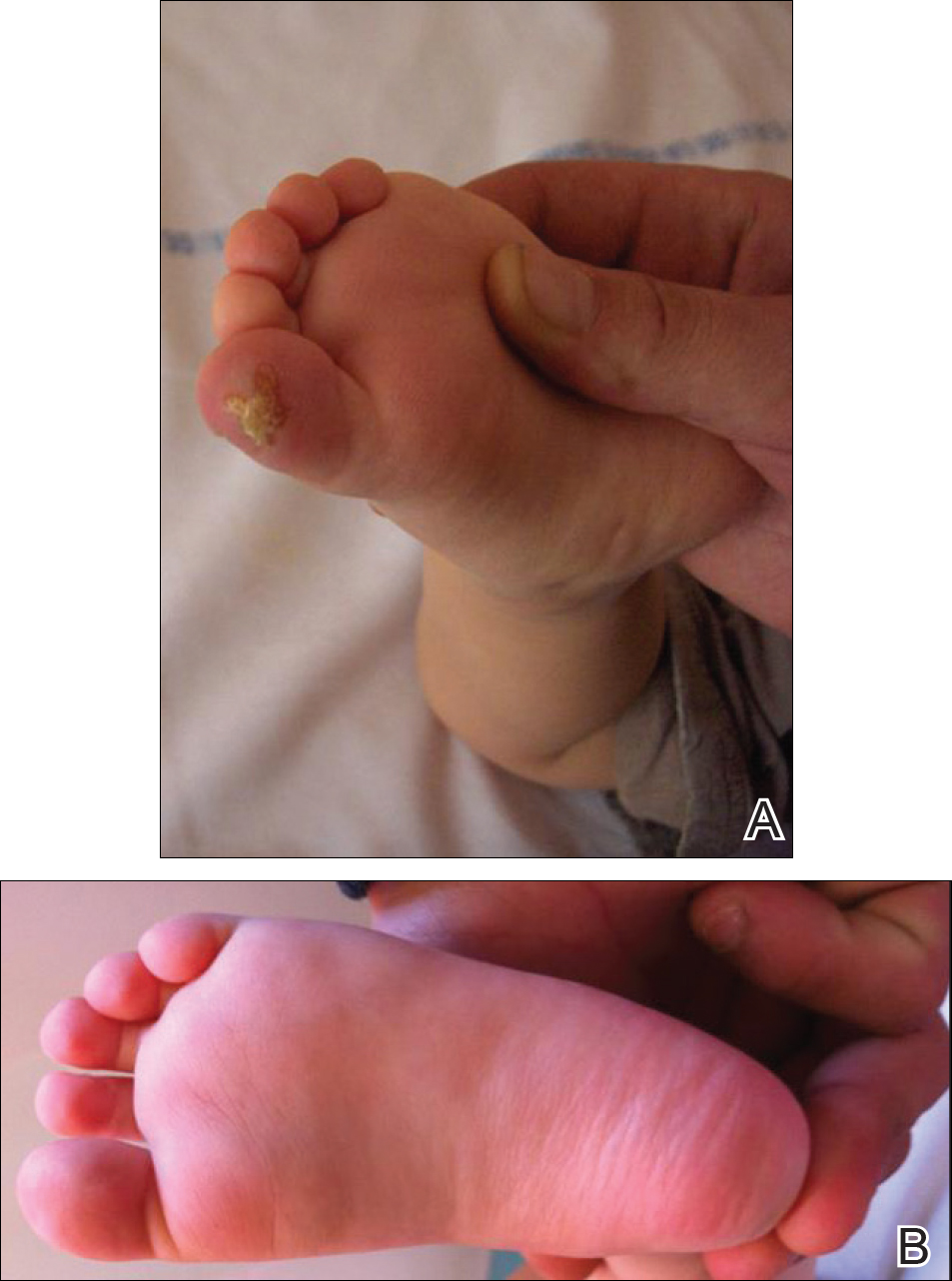
Her medical record showed a break of the growth curve with a weight of 9.25 kg (3rd percentile), a height of 80 cm (50th percentile), and a head circumference of 45 cm (50th percentile). Her parents were nonconsanguineous. The association of bilateral dendritic keratitis with punctate palmoplantar keratosis suggested a diagnosis of Richner-Hanhart syndrome. Diagnosis was confirmed by an elevated plasma level of tyrosine (1580 µmol/L; reference range, 40-80 µmol/L).
A low tyrosine and low phenylalanine diet (no animal proteins) was immediately introduced, with supplementation of amino acids, vitamins, and trace elements. After 8 days, the plasma level of tyrosinemia decreased by a factor of 4 (392 µmol/L). After 1 month, the cutaneous and ocular lesions completely resolved (Figure, B). Discrete psychomotor slowing still persisted for 1 year and then reached complete normalization. Genetic analysis showed a composite heterozygous mutation of the tyrosine aminotransferase gene, TAT, on chromosome 16. The mutation detected in the patient's mother was an A to V substitution at codon 147 (A147V). The second mutation was detected in the father; it was an 8 nucleotides duplication and then a substitution leading to a premature stop codon at codon 37 (R37X).
Richner-Hanhart syndrome is a rare autosomal-recessive disorder that is more common in Italy and in areas where inbreeding is prevalent1,2; however, no data are available on disease prevalence. It is caused by a homozygous mutation in the TAT gene located on chromosome 16q22.3 Tyrosine aminotransferase is an important enzyme involved in the tyrosine and phenylalanine metabolic degradation pathway located in the hepatic cytosol. Symptoms are due to the accumulation of tyrosine and its metabolite. Diagnosis is confirmed by an elevated plasma level of tyrosine (>500 µmol/L). This oculocutaneous syndrome is characterized by bilateral pseudodendritic keratitis, palmoplantar hyperkeratosis, and a variable degree of mental retardation.1 In contrast to tyrosinemia type II, types I and III do not affect the skin.
Intrafamilial and interfamilial phenotypic variability is reported. A large spectrum of mutations within the TAT gene have been reported.4-7 These mutations lead to a reduction or an absence in the activity of hepatic tyrosine aminotransferase. The degradation pathway of tyrosine involving TAT occurs mainly in the liver. This process also is present in the mitochondria where the enzyme is called aspartate aminotransferase.1,2 The mechanism by which Richner-Hanhart syndrome causes painful palmoplantar keratosis and keratitis remains unknown. It has been suggested that intracellular L-tyrosine crystals initiate an inflammation process resulting in the typical skin lesions and keratitis.8 There is some evidence that patients with higher values of tyrosine in early life are more likely to develop neurological problems.1 In addition, phenotype variability has been observed, even among individuals sharing the same pathogenic mutation.4
Tyrosinemia type II typically demonstrates ocular symptoms (75% of cases) that usually occur in the first year of life.8 They are characterized by photophobia, redness, and increase of lacrimation. Examination reveals a superficial and bilateral punctate keratosis with corneal dystrophy, often misdiagnosed as herpetic keratosis, as in our case, which may delay the diagnosis.9,10 Bilateral ocular lesions are suggestive, even if they are asymmetric.8,11 Furthermore, negative fluorescein staining, negative culture, and resistance to antiviral treatment exclude the diagnosis of herpetic keratosis.9,10
Skin lesions (85% of cases) typically appear in the first year of life. They are characterized by painful, irregular, limited, punctate hyperkeratosis on the palms and soles.1 They are more frequent in weight-bearing areas and tend to improve during summer, possibly due to a seasonal change in dietary behavior.4,12 Hyperkeratotic papules in a linear pattern also have been described on the flexor aspects of the fingers or toes.13 In our case, the lesions were misdiagnosed as warts for 6 months.
Retarded development affects 60% of patients with tyrosinemia type II. Expression of neurological symptoms is variable and could include mental retardation, nystagmus, tremors, ataxia, and convulsion.4 Lifetime follow-up of these patients is recommended.
Early initiation of a tyrosine-phenylalanine-restricted diet in infancy is the most effective therapy for Richner-Hanhart syndrome.13 The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into amino acid tyrosine. Thus, dietary treatment of Richner-Hanhart syndrome requires restricting or eliminating foods high in phenylalanine and tyrosine with protein "medical food" substitute. The dietary treatment allows resolution of both eye and skin symptoms after a few days or weeks and also may prevent mental retardation. It is effective in lowering the plasma level to less than 400 µmol/L. The diet must be introduced as soon as Richner-Hanhart syndrome is suspected. Supplementation with essential amino acids, vitamins, and trace elements is needed. Early screening of siblings in families with Richner-Hanhart syndrome history is recommended, even in the absence of clinical findings. Careful dietary control of maternal plasma tyrosine level must be considered during future pregnancy for women.4,14,15
Richner-Hanhart syndrome should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
- Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
- Meissner T, Betz RC, Pasternack SM, et al. Richner-Hanhart syndrome detected by expanded newborn screening. Pediatr Dermatol. 2008;25:378-380.
- Natt E, Kida K, Odievre M, et al. Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II. Proc Natl Acad Sci USA. 1992;89:9297-9301.
- Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
- Legarda M, Wlodarczyk K, Lage S, et al. A large TAT deletion in a tyrosinaemia type II patient. Mol Genet Metab. 2011;104:407-409.
- Culic V, Betz RC, Refke M, et al. Tyrosinemia type II (Richner-Hanhart syndrome): a new mutation in the TAT gene. Eur J Med Genet. 2011;54:205-208.
- Pasternack SM, Betz RC, Brandrup F, et al. Identification of two new mutations in the TAT gene in a Danish family with tyrosinaemia type II. Br J Dermatol. 2009;160:704-706.
- Macsai MS, Schwartz TL, Hinkle D, et al. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522-527.
- Kymionis GD, Kankariya VP, Kontadakis GA, et al. Isolated corneal pseudodendrites as the initial manifestation of tyrosinemia type II in monozygotic twins. J Pediatr Ophthalmol Strabismus.2012;49:E33-E36.
- Iskeleli G, Bilgeç MD, Arici C, et al. Richner-Hanhart syndrome (tyrosinemia type II): a case report of delayed diagnosis with pseudodendritic corneal lesion. Turk J Pediatr. 2011;53:692-694.
- Rehák A, Selim MM, Yadav G. Richner-Hanhart syndrome (tyrosinaemia-II)(report of four cases without ocular involvement). Br J Dermatol. 1981;104:469-475.
- Viglizzo GM, Occella C, Bleidl D, et al. Richner-Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings. Pediatr Dermatol. 2006;23:259-261.
- Machino H, Miki Y, Kawatsu T, et al. Successful dietary control of tyrosinemia II. J Am Acad Dermatol. 1983;9:533-539.
- el-Badramany MH, Fawzy AR, Farag TI. Familial Richner-Hanhart syndrome in Kuwait: twelve-year clinical reassessment by a multidisciplinary approach. Am J Med Genet. 1995;60:353-355.
- Cerone R, Fantasia AR, Castellano E, et al. Pregnancy and tyrosinaemia type II. J Inherit Metab Dis. 2002;25:317-318.
To the Editor:
Richner-Hanhart syndrome, also known as tyrosinemia type II or oculocutaneous tyrosinemia, is a rare autosomal-recessive, childhood-onset, metabolic hereditary disease.1 A deficiency of tyrosine aminotransferase leads to an accumulation of tyrosine amino acid. It is characterized by the association of palmoplantar hyperkeratosis, bilateral keratitis, and neurological disorders.
An 18-month-old girl with recurrent warts of 6 months' duration was admitted to the dermatology department. She had been treated repeatedly with acyclovir for recurrent bilateral herpetic keratitis with major photophobia since 9 months of age with no response. Clinical presentation included punctate hyperkeratosis of the fingers and toes (Figure, A), severe photophobia with decreased visual acuity, and speech delay.
Her medical record showed a break of the growth curve with a weight of 9.25 kg (3rd percentile), a height of 80 cm (50th percentile), and a head circumference of 45 cm (50th percentile). Her parents were nonconsanguineous. The association of bilateral dendritic keratitis with punctate palmoplantar keratosis suggested a diagnosis of Richner-Hanhart syndrome. Diagnosis was confirmed by an elevated plasma level of tyrosine (1580 µmol/L; reference range, 40-80 µmol/L).
A low tyrosine and low phenylalanine diet (no animal proteins) was immediately introduced, with supplementation of amino acids, vitamins, and trace elements. After 8 days, the plasma level of tyrosinemia decreased by a factor of 4 (392 µmol/L). After 1 month, the cutaneous and ocular lesions completely resolved (Figure, B). Discrete psychomotor slowing still persisted for 1 year and then reached complete normalization. Genetic analysis showed a composite heterozygous mutation of the tyrosine aminotransferase gene, TAT, on chromosome 16. The mutation detected in the patient's mother was an A to V substitution at codon 147 (A147V). The second mutation was detected in the father; it was an 8 nucleotides duplication and then a substitution leading to a premature stop codon at codon 37 (R37X).
Richner-Hanhart syndrome is a rare autosomal-recessive disorder that is more common in Italy and in areas where inbreeding is prevalent1,2; however, no data are available on disease prevalence. It is caused by a homozygous mutation in the TAT gene located on chromosome 16q22.3 Tyrosine aminotransferase is an important enzyme involved in the tyrosine and phenylalanine metabolic degradation pathway located in the hepatic cytosol. Symptoms are due to the accumulation of tyrosine and its metabolite. Diagnosis is confirmed by an elevated plasma level of tyrosine (>500 µmol/L). This oculocutaneous syndrome is characterized by bilateral pseudodendritic keratitis, palmoplantar hyperkeratosis, and a variable degree of mental retardation.1 In contrast to tyrosinemia type II, types I and III do not affect the skin.
Intrafamilial and interfamilial phenotypic variability is reported. A large spectrum of mutations within the TAT gene have been reported.4-7 These mutations lead to a reduction or an absence in the activity of hepatic tyrosine aminotransferase. The degradation pathway of tyrosine involving TAT occurs mainly in the liver. This process also is present in the mitochondria where the enzyme is called aspartate aminotransferase.1,2 The mechanism by which Richner-Hanhart syndrome causes painful palmoplantar keratosis and keratitis remains unknown. It has been suggested that intracellular L-tyrosine crystals initiate an inflammation process resulting in the typical skin lesions and keratitis.8 There is some evidence that patients with higher values of tyrosine in early life are more likely to develop neurological problems.1 In addition, phenotype variability has been observed, even among individuals sharing the same pathogenic mutation.4
Tyrosinemia type II typically demonstrates ocular symptoms (75% of cases) that usually occur in the first year of life.8 They are characterized by photophobia, redness, and increase of lacrimation. Examination reveals a superficial and bilateral punctate keratosis with corneal dystrophy, often misdiagnosed as herpetic keratosis, as in our case, which may delay the diagnosis.9,10 Bilateral ocular lesions are suggestive, even if they are asymmetric.8,11 Furthermore, negative fluorescein staining, negative culture, and resistance to antiviral treatment exclude the diagnosis of herpetic keratosis.9,10
Skin lesions (85% of cases) typically appear in the first year of life. They are characterized by painful, irregular, limited, punctate hyperkeratosis on the palms and soles.1 They are more frequent in weight-bearing areas and tend to improve during summer, possibly due to a seasonal change in dietary behavior.4,12 Hyperkeratotic papules in a linear pattern also have been described on the flexor aspects of the fingers or toes.13 In our case, the lesions were misdiagnosed as warts for 6 months.
Retarded development affects 60% of patients with tyrosinemia type II. Expression of neurological symptoms is variable and could include mental retardation, nystagmus, tremors, ataxia, and convulsion.4 Lifetime follow-up of these patients is recommended.
Early initiation of a tyrosine-phenylalanine-restricted diet in infancy is the most effective therapy for Richner-Hanhart syndrome.13 The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into amino acid tyrosine. Thus, dietary treatment of Richner-Hanhart syndrome requires restricting or eliminating foods high in phenylalanine and tyrosine with protein "medical food" substitute. The dietary treatment allows resolution of both eye and skin symptoms after a few days or weeks and also may prevent mental retardation. It is effective in lowering the plasma level to less than 400 µmol/L. The diet must be introduced as soon as Richner-Hanhart syndrome is suspected. Supplementation with essential amino acids, vitamins, and trace elements is needed. Early screening of siblings in families with Richner-Hanhart syndrome history is recommended, even in the absence of clinical findings. Careful dietary control of maternal plasma tyrosine level must be considered during future pregnancy for women.4,14,15
Richner-Hanhart syndrome should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
To the Editor:
Richner-Hanhart syndrome, also known as tyrosinemia type II or oculocutaneous tyrosinemia, is a rare autosomal-recessive, childhood-onset, metabolic hereditary disease.1 A deficiency of tyrosine aminotransferase leads to an accumulation of tyrosine amino acid. It is characterized by the association of palmoplantar hyperkeratosis, bilateral keratitis, and neurological disorders.
An 18-month-old girl with recurrent warts of 6 months' duration was admitted to the dermatology department. She had been treated repeatedly with acyclovir for recurrent bilateral herpetic keratitis with major photophobia since 9 months of age with no response. Clinical presentation included punctate hyperkeratosis of the fingers and toes (Figure, A), severe photophobia with decreased visual acuity, and speech delay.
Her medical record showed a break of the growth curve with a weight of 9.25 kg (3rd percentile), a height of 80 cm (50th percentile), and a head circumference of 45 cm (50th percentile). Her parents were nonconsanguineous. The association of bilateral dendritic keratitis with punctate palmoplantar keratosis suggested a diagnosis of Richner-Hanhart syndrome. Diagnosis was confirmed by an elevated plasma level of tyrosine (1580 µmol/L; reference range, 40-80 µmol/L).
A low tyrosine and low phenylalanine diet (no animal proteins) was immediately introduced, with supplementation of amino acids, vitamins, and trace elements. After 8 days, the plasma level of tyrosinemia decreased by a factor of 4 (392 µmol/L). After 1 month, the cutaneous and ocular lesions completely resolved (Figure, B). Discrete psychomotor slowing still persisted for 1 year and then reached complete normalization. Genetic analysis showed a composite heterozygous mutation of the tyrosine aminotransferase gene, TAT, on chromosome 16. The mutation detected in the patient's mother was an A to V substitution at codon 147 (A147V). The second mutation was detected in the father; it was an 8 nucleotides duplication and then a substitution leading to a premature stop codon at codon 37 (R37X).
Richner-Hanhart syndrome is a rare autosomal-recessive disorder that is more common in Italy and in areas where inbreeding is prevalent1,2; however, no data are available on disease prevalence. It is caused by a homozygous mutation in the TAT gene located on chromosome 16q22.3 Tyrosine aminotransferase is an important enzyme involved in the tyrosine and phenylalanine metabolic degradation pathway located in the hepatic cytosol. Symptoms are due to the accumulation of tyrosine and its metabolite. Diagnosis is confirmed by an elevated plasma level of tyrosine (>500 µmol/L). This oculocutaneous syndrome is characterized by bilateral pseudodendritic keratitis, palmoplantar hyperkeratosis, and a variable degree of mental retardation.1 In contrast to tyrosinemia type II, types I and III do not affect the skin.
Intrafamilial and interfamilial phenotypic variability is reported. A large spectrum of mutations within the TAT gene have been reported.4-7 These mutations lead to a reduction or an absence in the activity of hepatic tyrosine aminotransferase. The degradation pathway of tyrosine involving TAT occurs mainly in the liver. This process also is present in the mitochondria where the enzyme is called aspartate aminotransferase.1,2 The mechanism by which Richner-Hanhart syndrome causes painful palmoplantar keratosis and keratitis remains unknown. It has been suggested that intracellular L-tyrosine crystals initiate an inflammation process resulting in the typical skin lesions and keratitis.8 There is some evidence that patients with higher values of tyrosine in early life are more likely to develop neurological problems.1 In addition, phenotype variability has been observed, even among individuals sharing the same pathogenic mutation.4
Tyrosinemia type II typically demonstrates ocular symptoms (75% of cases) that usually occur in the first year of life.8 They are characterized by photophobia, redness, and increase of lacrimation. Examination reveals a superficial and bilateral punctate keratosis with corneal dystrophy, often misdiagnosed as herpetic keratosis, as in our case, which may delay the diagnosis.9,10 Bilateral ocular lesions are suggestive, even if they are asymmetric.8,11 Furthermore, negative fluorescein staining, negative culture, and resistance to antiviral treatment exclude the diagnosis of herpetic keratosis.9,10
Skin lesions (85% of cases) typically appear in the first year of life. They are characterized by painful, irregular, limited, punctate hyperkeratosis on the palms and soles.1 They are more frequent in weight-bearing areas and tend to improve during summer, possibly due to a seasonal change in dietary behavior.4,12 Hyperkeratotic papules in a linear pattern also have been described on the flexor aspects of the fingers or toes.13 In our case, the lesions were misdiagnosed as warts for 6 months.
Retarded development affects 60% of patients with tyrosinemia type II. Expression of neurological symptoms is variable and could include mental retardation, nystagmus, tremors, ataxia, and convulsion.4 Lifetime follow-up of these patients is recommended.
Early initiation of a tyrosine-phenylalanine-restricted diet in infancy is the most effective therapy for Richner-Hanhart syndrome.13 The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into amino acid tyrosine. Thus, dietary treatment of Richner-Hanhart syndrome requires restricting or eliminating foods high in phenylalanine and tyrosine with protein "medical food" substitute. The dietary treatment allows resolution of both eye and skin symptoms after a few days or weeks and also may prevent mental retardation. It is effective in lowering the plasma level to less than 400 µmol/L. The diet must be introduced as soon as Richner-Hanhart syndrome is suspected. Supplementation with essential amino acids, vitamins, and trace elements is needed. Early screening of siblings in families with Richner-Hanhart syndrome history is recommended, even in the absence of clinical findings. Careful dietary control of maternal plasma tyrosine level must be considered during future pregnancy for women.4,14,15
Richner-Hanhart syndrome should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
- Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
- Meissner T, Betz RC, Pasternack SM, et al. Richner-Hanhart syndrome detected by expanded newborn screening. Pediatr Dermatol. 2008;25:378-380.
- Natt E, Kida K, Odievre M, et al. Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II. Proc Natl Acad Sci USA. 1992;89:9297-9301.
- Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
- Legarda M, Wlodarczyk K, Lage S, et al. A large TAT deletion in a tyrosinaemia type II patient. Mol Genet Metab. 2011;104:407-409.
- Culic V, Betz RC, Refke M, et al. Tyrosinemia type II (Richner-Hanhart syndrome): a new mutation in the TAT gene. Eur J Med Genet. 2011;54:205-208.
- Pasternack SM, Betz RC, Brandrup F, et al. Identification of two new mutations in the TAT gene in a Danish family with tyrosinaemia type II. Br J Dermatol. 2009;160:704-706.
- Macsai MS, Schwartz TL, Hinkle D, et al. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522-527.
- Kymionis GD, Kankariya VP, Kontadakis GA, et al. Isolated corneal pseudodendrites as the initial manifestation of tyrosinemia type II in monozygotic twins. J Pediatr Ophthalmol Strabismus.2012;49:E33-E36.
- Iskeleli G, Bilgeç MD, Arici C, et al. Richner-Hanhart syndrome (tyrosinemia type II): a case report of delayed diagnosis with pseudodendritic corneal lesion. Turk J Pediatr. 2011;53:692-694.
- Rehák A, Selim MM, Yadav G. Richner-Hanhart syndrome (tyrosinaemia-II)(report of four cases without ocular involvement). Br J Dermatol. 1981;104:469-475.
- Viglizzo GM, Occella C, Bleidl D, et al. Richner-Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings. Pediatr Dermatol. 2006;23:259-261.
- Machino H, Miki Y, Kawatsu T, et al. Successful dietary control of tyrosinemia II. J Am Acad Dermatol. 1983;9:533-539.
- el-Badramany MH, Fawzy AR, Farag TI. Familial Richner-Hanhart syndrome in Kuwait: twelve-year clinical reassessment by a multidisciplinary approach. Am J Med Genet. 1995;60:353-355.
- Cerone R, Fantasia AR, Castellano E, et al. Pregnancy and tyrosinaemia type II. J Inherit Metab Dis. 2002;25:317-318.
- Scott CR. The genetic tyrosinemias. Am J Med Genet C Semin Med Genet. 2006;142C:121-126.
- Meissner T, Betz RC, Pasternack SM, et al. Richner-Hanhart syndrome detected by expanded newborn screening. Pediatr Dermatol. 2008;25:378-380.
- Natt E, Kida K, Odievre M, et al. Point mutations in the tyrosine aminotransferase gene in tyrosinemia type II. Proc Natl Acad Sci USA. 1992;89:9297-9301.
- Charfeddine C, Monastiri K, Mokni M, et al. Clinical and mutational investigations of tyrosinemia type II in Northern Tunisia: identification and structural characterization of two novel TAT mutations. Mol Genet Metab. 2006;88:184-191.
- Legarda M, Wlodarczyk K, Lage S, et al. A large TAT deletion in a tyrosinaemia type II patient. Mol Genet Metab. 2011;104:407-409.
- Culic V, Betz RC, Refke M, et al. Tyrosinemia type II (Richner-Hanhart syndrome): a new mutation in the TAT gene. Eur J Med Genet. 2011;54:205-208.
- Pasternack SM, Betz RC, Brandrup F, et al. Identification of two new mutations in the TAT gene in a Danish family with tyrosinaemia type II. Br J Dermatol. 2009;160:704-706.
- Macsai MS, Schwartz TL, Hinkle D, et al. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522-527.
- Kymionis GD, Kankariya VP, Kontadakis GA, et al. Isolated corneal pseudodendrites as the initial manifestation of tyrosinemia type II in monozygotic twins. J Pediatr Ophthalmol Strabismus.2012;49:E33-E36.
- Iskeleli G, Bilgeç MD, Arici C, et al. Richner-Hanhart syndrome (tyrosinemia type II): a case report of delayed diagnosis with pseudodendritic corneal lesion. Turk J Pediatr. 2011;53:692-694.
- Rehák A, Selim MM, Yadav G. Richner-Hanhart syndrome (tyrosinaemia-II)(report of four cases without ocular involvement). Br J Dermatol. 1981;104:469-475.
- Viglizzo GM, Occella C, Bleidl D, et al. Richner-Hanhart syndrome (tyrosinemia II): early diagnosis of an incomplete presentation with unusual findings. Pediatr Dermatol. 2006;23:259-261.
- Machino H, Miki Y, Kawatsu T, et al. Successful dietary control of tyrosinemia II. J Am Acad Dermatol. 1983;9:533-539.
- el-Badramany MH, Fawzy AR, Farag TI. Familial Richner-Hanhart syndrome in Kuwait: twelve-year clinical reassessment by a multidisciplinary approach. Am J Med Genet. 1995;60:353-355.
- Cerone R, Fantasia AR, Castellano E, et al. Pregnancy and tyrosinaemia type II. J Inherit Metab Dis. 2002;25:317-318.
Practice Points
- Richner-Hanhart syndrome (tyrosinemia type II) should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
- Early diagnosis and initiation of a tyrosinephenylalanine–restricted diet in infancy is the most effective therapy to prevent mental retardation.
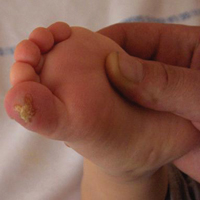
ISSOP’s Budapest Declaration: A call to action for children on the move
In late October 2017, pediatricians from over 25 countries gathered at the annual meeting of the International Society of Social Pediatrics and Child Health in Budapest to develop strategies to address the health and well-being of children on the move. With the staggering global increase in the displacement of children – due to conflict, climate change, natural disasters, and economic deprivation – there is a critical need for action.
An international slate of experts, including pediatricians who are responding to the needs of these children, provided an evidence base for intervention and action. These discussions informed the development of a consensus statement, the Budapest Declaration, that establishes a framework for a global response to the exigencies confronting these children and families.

The Budapest Declaration is grounded in children’s rights as guaranteed in the United Nation’s Convention on the Rights of the Child (CRC). All countries in the world have signed onto the CRC, with a notable exception being the United States. But leading American pediatric groups, including the American Academy of Pediatrics, have endorsed the CRC, making it a basis of organizational advocacy. The CRC entitles all children to optimal survival and development (Article 6) and optimal health and health care (Article 24).
As prescribed by these accepted legal rights for children, children and youth on the move are entitled to the same services, including health care, as resident populations of children, regardless of their legal status. Countries can be held accountable to ensure they receive the full rights due them as articulated in the CRC. Furthermore, efforts to do so should be assessed in national periodic reports to the Committee on the Rights of the Child to ensure accountability.
The relevance of the Budapest Declaration to the United States cannot be overstated. National and regional public policy throughout our country, and in particular along our Southern border, is having a devastating effect on the physical and mental health of children and youth on the move – and will continue to do so throughout their life course.
The involvement of pediatricians and other child health providers is essential to the planning and implementation of clinical and public health programs for children on the move. Child health professionals, in conjunction with their professional organizations, must be engaged in all aspects of local, national, and global responses. Leadership and contributions by pediatricians and pediatric societies and academic institutions are integral to the success of key partners, such as UNICEF, the World Health Organization, the International Organization for Migration, and the UN High Commissioner for Refugees.

Systems of care should be provided that address the physical, mental, and social health care needs of these children without bias and prejudice. Pregnant mothers on the move also should receive services to ensure the delivery of healthy newborns. Every nation should develop approaches and commitments to advance equity in the health and well-being for these children and families.

For much of the world, working within the realm of children’s rights provides a strategy and moral and legal basis for our efforts. Pediatricians need to work with colleagues in a transdisciplinary approach to ensure all children live in nurturing, rights-respecting environments. Our ongoing efforts should include encouraging academic institutions to assist with professional education, research, and evaluation in this regard. We need evaluations that contribute to continuous quality improvement in our efforts and integrate the metrics of child rights, social justice, and health equity into our care of children on the move.
We call on other national and international public, private, and academic sector organizations to advance the health and well-being of children and youth on the move. These children and families are depending on us to do so.
For the complete text of the Budapest Declaration, see www.issop.org/2017/11/10/budapest-declaration-rights-health-well-children-youth-move/.
Dr. Rushton is medical director of the South Carolina Quality Through Innovation in Pediatrics (SCQTIP) network. Dr. Goldhagen is president-elect, International Society for Social Pediatrics and Child Health, and professor of pediatrics, University of Florida, Jacksonville. Email them at pdnews@frontlinemedcom.com.
In late October 2017, pediatricians from over 25 countries gathered at the annual meeting of the International Society of Social Pediatrics and Child Health in Budapest to develop strategies to address the health and well-being of children on the move. With the staggering global increase in the displacement of children – due to conflict, climate change, natural disasters, and economic deprivation – there is a critical need for action.
An international slate of experts, including pediatricians who are responding to the needs of these children, provided an evidence base for intervention and action. These discussions informed the development of a consensus statement, the Budapest Declaration, that establishes a framework for a global response to the exigencies confronting these children and families.
The Budapest Declaration is grounded in children’s rights as guaranteed in the United Nation’s Convention on the Rights of the Child (CRC). All countries in the world have signed onto the CRC, with a notable exception being the United States. But leading American pediatric groups, including the American Academy of Pediatrics, have endorsed the CRC, making it a basis of organizational advocacy. The CRC entitles all children to optimal survival and development (Article 6) and optimal health and health care (Article 24).
As prescribed by these accepted legal rights for children, children and youth on the move are entitled to the same services, including health care, as resident populations of children, regardless of their legal status. Countries can be held accountable to ensure they receive the full rights due them as articulated in the CRC. Furthermore, efforts to do so should be assessed in national periodic reports to the Committee on the Rights of the Child to ensure accountability.
The relevance of the Budapest Declaration to the United States cannot be overstated. National and regional public policy throughout our country, and in particular along our Southern border, is having a devastating effect on the physical and mental health of children and youth on the move – and will continue to do so throughout their life course.
The involvement of pediatricians and other child health providers is essential to the planning and implementation of clinical and public health programs for children on the move. Child health professionals, in conjunction with their professional organizations, must be engaged in all aspects of local, national, and global responses. Leadership and contributions by pediatricians and pediatric societies and academic institutions are integral to the success of key partners, such as UNICEF, the World Health Organization, the International Organization for Migration, and the UN High Commissioner for Refugees.
Systems of care should be provided that address the physical, mental, and social health care needs of these children without bias and prejudice. Pregnant mothers on the move also should receive services to ensure the delivery of healthy newborns. Every nation should develop approaches and commitments to advance equity in the health and well-being for these children and families.
For much of the world, working within the realm of children’s rights provides a strategy and moral and legal basis for our efforts. Pediatricians need to work with colleagues in a transdisciplinary approach to ensure all children live in nurturing, rights-respecting environments. Our ongoing efforts should include encouraging academic institutions to assist with professional education, research, and evaluation in this regard. We need evaluations that contribute to continuous quality improvement in our efforts and integrate the metrics of child rights, social justice, and health equity into our care of children on the move.
We call on other national and international public, private, and academic sector organizations to advance the health and well-being of children and youth on the move. These children and families are depending on us to do so.
For the complete text of the Budapest Declaration, see www.issop.org/2017/11/10/budapest-declaration-rights-health-well-children-youth-move/.
Dr. Rushton is medical director of the South Carolina Quality Through Innovation in Pediatrics (SCQTIP) network. Dr. Goldhagen is president-elect, International Society for Social Pediatrics and Child Health, and professor of pediatrics, University of Florida, Jacksonville. Email them at pdnews@frontlinemedcom.com.
In late October 2017, pediatricians from over 25 countries gathered at the annual meeting of the International Society of Social Pediatrics and Child Health in Budapest to develop strategies to address the health and well-being of children on the move. With the staggering global increase in the displacement of children – due to conflict, climate change, natural disasters, and economic deprivation – there is a critical need for action.
An international slate of experts, including pediatricians who are responding to the needs of these children, provided an evidence base for intervention and action. These discussions informed the development of a consensus statement, the Budapest Declaration, that establishes a framework for a global response to the exigencies confronting these children and families.
The Budapest Declaration is grounded in children’s rights as guaranteed in the United Nation’s Convention on the Rights of the Child (CRC). All countries in the world have signed onto the CRC, with a notable exception being the United States. But leading American pediatric groups, including the American Academy of Pediatrics, have endorsed the CRC, making it a basis of organizational advocacy. The CRC entitles all children to optimal survival and development (Article 6) and optimal health and health care (Article 24).
As prescribed by these accepted legal rights for children, children and youth on the move are entitled to the same services, including health care, as resident populations of children, regardless of their legal status. Countries can be held accountable to ensure they receive the full rights due them as articulated in the CRC. Furthermore, efforts to do so should be assessed in national periodic reports to the Committee on the Rights of the Child to ensure accountability.
The relevance of the Budapest Declaration to the United States cannot be overstated. National and regional public policy throughout our country, and in particular along our Southern border, is having a devastating effect on the physical and mental health of children and youth on the move – and will continue to do so throughout their life course.
The involvement of pediatricians and other child health providers is essential to the planning and implementation of clinical and public health programs for children on the move. Child health professionals, in conjunction with their professional organizations, must be engaged in all aspects of local, national, and global responses. Leadership and contributions by pediatricians and pediatric societies and academic institutions are integral to the success of key partners, such as UNICEF, the World Health Organization, the International Organization for Migration, and the UN High Commissioner for Refugees.
Systems of care should be provided that address the physical, mental, and social health care needs of these children without bias and prejudice. Pregnant mothers on the move also should receive services to ensure the delivery of healthy newborns. Every nation should develop approaches and commitments to advance equity in the health and well-being for these children and families.
For much of the world, working within the realm of children’s rights provides a strategy and moral and legal basis for our efforts. Pediatricians need to work with colleagues in a transdisciplinary approach to ensure all children live in nurturing, rights-respecting environments. Our ongoing efforts should include encouraging academic institutions to assist with professional education, research, and evaluation in this regard. We need evaluations that contribute to continuous quality improvement in our efforts and integrate the metrics of child rights, social justice, and health equity into our care of children on the move.
We call on other national and international public, private, and academic sector organizations to advance the health and well-being of children and youth on the move. These children and families are depending on us to do so.
For the complete text of the Budapest Declaration, see www.issop.org/2017/11/10/budapest-declaration-rights-health-well-children-youth-move/.
Dr. Rushton is medical director of the South Carolina Quality Through Innovation in Pediatrics (SCQTIP) network. Dr. Goldhagen is president-elect, International Society for Social Pediatrics and Child Health, and professor of pediatrics, University of Florida, Jacksonville. Email them at pdnews@frontlinemedcom.com.
Don’t give up on influenza vaccine
I suspect most health care providers have heard the complaint, “The vaccine doesn’t work. One year I got the vaccine, and I still came down with the flu.”
Over the years, I’ve polished my responses to vaccine naysayers.
Influenza vaccine doesn’t protect you against every virus that can cause cold and flu symptoms. It only prevents influenza. It’s possible you had a different virus, such as adenovirus, coronavirus, parainfluenza virus, or respiratory syncytial virus.

Some years, the vaccine works better than others because there is a mismatch between the viruses chosen for the vaccine, and the viruses that end up circulating. Even when it doesn’t prevent flu, the vaccine can potentially reduce the severity of illness.
The discussion became a little more complicated in 2016 when the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices withdrew its support for the live attenuated influenza virus vaccine (LAIV4) because of concerns about effectiveness. During the 2015-2016 influenza season, LAIV4 demonstrated no statistically significant effectiveness in children 2-17 years of age against H1N1pdm09, the predominant influenza strain. Fortunately, inactivated injectable vaccine did offer protection. An estimated 41.8 million children aged 6 months to 17 years ultimately received this vaccine during the 2016-2017 influenza season.
Now with the 2017-2018 influenza season in full swing, some media reports are proclaiming the influenza vaccine is only 10% effective this year. This claim is based on an interim analysis of data from the most recent flu season in Australia and the effectiveness of the vaccine against the circulating H3N2 virus strain. News from the U.S. CDC is more encouraging. The H3N2 virus contained in this year’s vaccine is the same as that used last year, and so far, circulating H3N2 viruses in the United States are similar to the vaccine virus. Public health officials suggest that we can hope that the vaccine works as well as it did last year, when overall vaccine effectiveness against all circulating flu viruses was 39%, and effectiveness against the H3N2 virus specifically was 32%.
I’m upping my game when talking to parents about flu vaccine. I mention one study conducted between 2010 and 2012 in which influenza immunization reduced a child’s risk of being admitted to an intensive care unit with flu by 74% (J Infect Dis. 2014 Sep 1;210[5]:674-83). I emphasize that flu vaccine reduces the chance that a child will die from flu. According to a study published in 2017, influenza vaccine reduced the risk of death from flu by 65% in healthy children and 51% in children with high-risk medical conditions (Pediatrics. 2017 May. doi: 10.1542/peds.2016-4244).
When I’m talking to trainees, I no longer just focus on the match between circulating strains of flu and vaccine strains. I mention that viruses used to produce most seasonal flu vaccines are grown in eggs, a process that can result in minor antigenic changes in the hemagglutinin protein, especially in H3N2 viruses. These “egg-adapted changes” may result in a vaccine that stimulates a less effective immune response, even with a good match between circulating strains and vaccine strains. For example, Zost et al. found that the H3N2 virus that emerged during the 2014-2015 season possessed a new hemagglutinin-associated glycosylation site (Proc Natl Acad Sci U S A. 2017 Nov 21;114[47]:12578-83). Although this virus was represented in the 2016-2017 influenza vaccine, the egg-adapted version lost the glycosylation site, resulting in decreased vaccine immunogenicity and less protection against H3N2 viruses circulating in the community.
The real take-home message here is that we need better flu vaccines. In the short term, cell-based flu vaccines that use virus grown in animal cells are a potential alternative to egg-based vaccines. In the long term, we need a universal flu vaccine. The National Institute of Allergy and Infectious Diseases is prioritizing work on a vaccine that could provide long-lasting protection against multiple subtypes of the virus. According to a report on the National Institutes of Health website, such a vaccine could “eliminate the need to update and administer the seasonal flu vaccine each year and could provide protection against newly emerging flu strains,” including those with the potential to cause a pandemic. The NIH researchers acknowledge, however, that achieving this goal will require “a broad range of expertise and substantial resources.”
Until new vaccines are available, we need to do a better job of using available, albeit imperfect, flu vaccines. During the 2016-2017 season, only 59% of children 6 months to 17 years were immunized, and there were 110 influenza-associated deaths in children, according to the CDC. It’s likely that some of these were preventable.
The total magnitude of suffering associated with flu is more difficult to quantify, but anecdotes can be illuminating. A friend recently diagnosed with influenza shared her experience via Facebook. “Rough night. I’m seconds away from a meltdown. My body aches so bad that I can’t get comfortable on the couch or my bed. Can’t breathe, and I cough until I vomit. My head is about to burst along with my ears. Just took a hot bath hoping that would help. I don’t know what else to do. The flu really sucks.”
Indeed. Even a 1 in 10 chance of preventing the flu is better than no chance at all.
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital in Louisville. She said she had no relevant financial disclosures. Email her at pdnews@frontlinemedcom.com.
I suspect most health care providers have heard the complaint, “The vaccine doesn’t work. One year I got the vaccine, and I still came down with the flu.”
Over the years, I’ve polished my responses to vaccine naysayers.
Influenza vaccine doesn’t protect you against every virus that can cause cold and flu symptoms. It only prevents influenza. It’s possible you had a different virus, such as adenovirus, coronavirus, parainfluenza virus, or respiratory syncytial virus.
Some years, the vaccine works better than others because there is a mismatch between the viruses chosen for the vaccine, and the viruses that end up circulating. Even when it doesn’t prevent flu, the vaccine can potentially reduce the severity of illness.
The discussion became a little more complicated in 2016 when the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices withdrew its support for the live attenuated influenza virus vaccine (LAIV4) because of concerns about effectiveness. During the 2015-2016 influenza season, LAIV4 demonstrated no statistically significant effectiveness in children 2-17 years of age against H1N1pdm09, the predominant influenza strain. Fortunately, inactivated injectable vaccine did offer protection. An estimated 41.8 million children aged 6 months to 17 years ultimately received this vaccine during the 2016-2017 influenza season.
Now with the 2017-2018 influenza season in full swing, some media reports are proclaiming the influenza vaccine is only 10% effective this year. This claim is based on an interim analysis of data from the most recent flu season in Australia and the effectiveness of the vaccine against the circulating H3N2 virus strain. News from the U.S. CDC is more encouraging. The H3N2 virus contained in this year’s vaccine is the same as that used last year, and so far, circulating H3N2 viruses in the United States are similar to the vaccine virus. Public health officials suggest that we can hope that the vaccine works as well as it did last year, when overall vaccine effectiveness against all circulating flu viruses was 39%, and effectiveness against the H3N2 virus specifically was 32%.
I’m upping my game when talking to parents about flu vaccine. I mention one study conducted between 2010 and 2012 in which influenza immunization reduced a child’s risk of being admitted to an intensive care unit with flu by 74% (J Infect Dis. 2014 Sep 1;210[5]:674-83). I emphasize that flu vaccine reduces the chance that a child will die from flu. According to a study published in 2017, influenza vaccine reduced the risk of death from flu by 65% in healthy children and 51% in children with high-risk medical conditions (Pediatrics. 2017 May. doi: 10.1542/peds.2016-4244).
When I’m talking to trainees, I no longer just focus on the match between circulating strains of flu and vaccine strains. I mention that viruses used to produce most seasonal flu vaccines are grown in eggs, a process that can result in minor antigenic changes in the hemagglutinin protein, especially in H3N2 viruses. These “egg-adapted changes” may result in a vaccine that stimulates a less effective immune response, even with a good match between circulating strains and vaccine strains. For example, Zost et al. found that the H3N2 virus that emerged during the 2014-2015 season possessed a new hemagglutinin-associated glycosylation site (Proc Natl Acad Sci U S A. 2017 Nov 21;114[47]:12578-83). Although this virus was represented in the 2016-2017 influenza vaccine, the egg-adapted version lost the glycosylation site, resulting in decreased vaccine immunogenicity and less protection against H3N2 viruses circulating in the community.
The real take-home message here is that we need better flu vaccines. In the short term, cell-based flu vaccines that use virus grown in animal cells are a potential alternative to egg-based vaccines. In the long term, we need a universal flu vaccine. The National Institute of Allergy and Infectious Diseases is prioritizing work on a vaccine that could provide long-lasting protection against multiple subtypes of the virus. According to a report on the National Institutes of Health website, such a vaccine could “eliminate the need to update and administer the seasonal flu vaccine each year and could provide protection against newly emerging flu strains,” including those with the potential to cause a pandemic. The NIH researchers acknowledge, however, that achieving this goal will require “a broad range of expertise and substantial resources.”
Until new vaccines are available, we need to do a better job of using available, albeit imperfect, flu vaccines. During the 2016-2017 season, only 59% of children 6 months to 17 years were immunized, and there were 110 influenza-associated deaths in children, according to the CDC. It’s likely that some of these were preventable.
The total magnitude of suffering associated with flu is more difficult to quantify, but anecdotes can be illuminating. A friend recently diagnosed with influenza shared her experience via Facebook. “Rough night. I’m seconds away from a meltdown. My body aches so bad that I can’t get comfortable on the couch or my bed. Can’t breathe, and I cough until I vomit. My head is about to burst along with my ears. Just took a hot bath hoping that would help. I don’t know what else to do. The flu really sucks.”
Indeed. Even a 1 in 10 chance of preventing the flu is better than no chance at all.
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital in Louisville. She said she had no relevant financial disclosures. Email her at pdnews@frontlinemedcom.com.
I suspect most health care providers have heard the complaint, “The vaccine doesn’t work. One year I got the vaccine, and I still came down with the flu.”
Over the years, I’ve polished my responses to vaccine naysayers.
Influenza vaccine doesn’t protect you against every virus that can cause cold and flu symptoms. It only prevents influenza. It’s possible you had a different virus, such as adenovirus, coronavirus, parainfluenza virus, or respiratory syncytial virus.
Some years, the vaccine works better than others because there is a mismatch between the viruses chosen for the vaccine, and the viruses that end up circulating. Even when it doesn’t prevent flu, the vaccine can potentially reduce the severity of illness.
The discussion became a little more complicated in 2016 when the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices withdrew its support for the live attenuated influenza virus vaccine (LAIV4) because of concerns about effectiveness. During the 2015-2016 influenza season, LAIV4 demonstrated no statistically significant effectiveness in children 2-17 years of age against H1N1pdm09, the predominant influenza strain. Fortunately, inactivated injectable vaccine did offer protection. An estimated 41.8 million children aged 6 months to 17 years ultimately received this vaccine during the 2016-2017 influenza season.
Now with the 2017-2018 influenza season in full swing, some media reports are proclaiming the influenza vaccine is only 10% effective this year. This claim is based on an interim analysis of data from the most recent flu season in Australia and the effectiveness of the vaccine against the circulating H3N2 virus strain. News from the U.S. CDC is more encouraging. The H3N2 virus contained in this year’s vaccine is the same as that used last year, and so far, circulating H3N2 viruses in the United States are similar to the vaccine virus. Public health officials suggest that we can hope that the vaccine works as well as it did last year, when overall vaccine effectiveness against all circulating flu viruses was 39%, and effectiveness against the H3N2 virus specifically was 32%.
I’m upping my game when talking to parents about flu vaccine. I mention one study conducted between 2010 and 2012 in which influenza immunization reduced a child’s risk of being admitted to an intensive care unit with flu by 74% (J Infect Dis. 2014 Sep 1;210[5]:674-83). I emphasize that flu vaccine reduces the chance that a child will die from flu. According to a study published in 2017, influenza vaccine reduced the risk of death from flu by 65% in healthy children and 51% in children with high-risk medical conditions (Pediatrics. 2017 May. doi: 10.1542/peds.2016-4244).
When I’m talking to trainees, I no longer just focus on the match between circulating strains of flu and vaccine strains. I mention that viruses used to produce most seasonal flu vaccines are grown in eggs, a process that can result in minor antigenic changes in the hemagglutinin protein, especially in H3N2 viruses. These “egg-adapted changes” may result in a vaccine that stimulates a less effective immune response, even with a good match between circulating strains and vaccine strains. For example, Zost et al. found that the H3N2 virus that emerged during the 2014-2015 season possessed a new hemagglutinin-associated glycosylation site (Proc Natl Acad Sci U S A. 2017 Nov 21;114[47]:12578-83). Although this virus was represented in the 2016-2017 influenza vaccine, the egg-adapted version lost the glycosylation site, resulting in decreased vaccine immunogenicity and less protection against H3N2 viruses circulating in the community.
The real take-home message here is that we need better flu vaccines. In the short term, cell-based flu vaccines that use virus grown in animal cells are a potential alternative to egg-based vaccines. In the long term, we need a universal flu vaccine. The National Institute of Allergy and Infectious Diseases is prioritizing work on a vaccine that could provide long-lasting protection against multiple subtypes of the virus. According to a report on the National Institutes of Health website, such a vaccine could “eliminate the need to update and administer the seasonal flu vaccine each year and could provide protection against newly emerging flu strains,” including those with the potential to cause a pandemic. The NIH researchers acknowledge, however, that achieving this goal will require “a broad range of expertise and substantial resources.”
Until new vaccines are available, we need to do a better job of using available, albeit imperfect, flu vaccines. During the 2016-2017 season, only 59% of children 6 months to 17 years were immunized, and there were 110 influenza-associated deaths in children, according to the CDC. It’s likely that some of these were preventable.
The total magnitude of suffering associated with flu is more difficult to quantify, but anecdotes can be illuminating. A friend recently diagnosed with influenza shared her experience via Facebook. “Rough night. I’m seconds away from a meltdown. My body aches so bad that I can’t get comfortable on the couch or my bed. Can’t breathe, and I cough until I vomit. My head is about to burst along with my ears. Just took a hot bath hoping that would help. I don’t know what else to do. The flu really sucks.”
Indeed. Even a 1 in 10 chance of preventing the flu is better than no chance at all.
Dr. Bryant is a pediatrician specializing in infectious diseases at the University of Louisville (Ky.) and Norton Children’s Hospital in Louisville. She said she had no relevant financial disclosures. Email her at pdnews@frontlinemedcom.com.
Gene Replacement Improves Survival in Spinal Muscular Atrophy
A single IV infusion to replace the gene encoding survival motor neuron 1 (SMN1) increases survival among infants with spinal muscular atrophy type 1 (SMA1), according to research published in the November 2, 2017, issue of the New England Journal of Medicine. The treatment also improves motor function, and its effects are maintained for two years, the researchers said.
The loss or dysfunction of SMN1 causes SMA, a progressive disease characterized by the degeneration and loss of lower motor neurons. Onset of SMA1 typically occurs at one month of age. Children with the disease usually are weak, fail to achieve motor milestones, and have declines in respiration and swallowing. At a median age of 10.5 months, patients die or need permanent ventilatory assistance.
In December 2016, the FDA approved nusinersen for the treatment of SMA. A phase III study found that patients treated with nusinersen were more likely than controls to have improved motor function and event-free survival. The trial was stopped early because of the treatment’s efficacy.
A Small, Open-Label Trial
A mouse study indicated that IV administration of an adenoassociated viral vector (ie, AAV9) containing SMN1 reduced the effects of SMA and extended survival. Jerry R. Mendell, MD, Principal Investigator at Nationwide Children’s Hospital’s Center for Gene Therapy in Columbus, Ohio, and colleagues studied this therapeutic technique in humans.

They enrolled 15 patients with a genetically confirmed diagnosis of SMA1 into two cohorts. The first cohort received a low dose (6.7×1013 vg/kg) of treatment, and the second cohort received a high dose (2.0×1014 vg/kg). Because the first patient in cohort one had serum aminotransferase elevations, the investigators gave 1 mg/kg/day of oral prednisolone to all subsequent patients for 30 days, starting 24 hours before gene therapy.
The study’s primary outcome was treatment-related adverse events of grade 3 or higher. The secondary outcome was time until death or the need for permanent ventilatory assistance, which was defined as at least 16 hours/day of continuous respiratory assistance for at least 14 days. The achievement of motor milestones and Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scores were exploratory outcomes.
Motor Scores Improved
Three patients entered the low-dose cohort, and 12 were enrolled in the high-dose cohort. Patients’ mean age at treatment was 6.3 months in cohort 1 and 3.4 months in cohort 2. At the last follow-up, all patients had reached age 20 months, and none required permanent mechanical ventilation. Approximately 8% of patients in a historical cohort met these criteria.
All patients had increases from baseline in CHOP INTEND score and maintained these increases throughout the study. Patients in cohort 2 had mean increases of 9.8 points at one month and 15.4 points at three months. Eleven patients achieved and sustained scores greater than 40 points, which is considered clinically meaningful in SMA.
Of the patients in cohort 2, 11 sat unassisted, nine rolled over, 11 fed orally and could speak, and two walked independently. No patients in the historical cohorts achieved any of these milestones, and they rarely became able to speak.
Dr. Mendell and colleagues observed two treatment-related grade 4 adverse events. Both were elevations in serum aminotransferase levels that were attenuated after treatment with prednisolone. The researchers also noted three treatment-related nonserious adverse events (ie, asymptomatic elevations in serum aminotransferase levels that were resolved without additional prednisolone treatment).
The study results were consistent with those of the preclinical mouse study. During a follow-up period of as long as two years, Dr. Mendell and colleagues did not observe any decrease in treatment effect or regression in motor function among the study participants. The presence of antibodies to AAV9 could be a potential limitation of the therapy, however. Further research to assess the treatment’s safety and the durability of its effect are needed, according to the authors.
Comparing Two Treatments
It is difficult to compare the results of Dr. Mendell and colleagues with those of the phase III nusinersen study, because of the two trials’ different designs, said Ans T. van der Ploeg, MD, PhD, Chair of the Center for Lysosomal and Metabolic Diseases at the Erasmus MC University in Rotterdam, the Netherlands, in an accompanying editorial. One potential advantage of AAV9 gene therapy is that it might require a single IV infusion. Nusinersen, on the other hand, may require lifelong intrathecal treatment.
“The durability of the effects is uncertain for both treatments,” said Dr. van der Ploeg. “If the expression of the scAAV9 gene therapy declines over time, the same treatment may not be able to be repeated, because antibodies against AAV capsid proteins are anticipated to form.”
In addition, neither of the two therapies cures SMA type 1. Earlier treatment could be beneficial, as could a combination of both treatments, said Dr. van der Ploeg. But the high expected cost of nusinersen is “an important constraint,” she concluded.
—Erik Greb
Suggested Reading
Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-1722.
van der Ploeg AT. The dilemma of two innovative therapies for spinal muscular atrophy. N Engl J Med. 2017;377(18):1786-1787.
A single IV infusion to replace the gene encoding survival motor neuron 1 (SMN1) increases survival among infants with spinal muscular atrophy type 1 (SMA1), according to research published in the November 2, 2017, issue of the New England Journal of Medicine. The treatment also improves motor function, and its effects are maintained for two years, the researchers said.
The loss or dysfunction of SMN1 causes SMA, a progressive disease characterized by the degeneration and loss of lower motor neurons. Onset of SMA1 typically occurs at one month of age. Children with the disease usually are weak, fail to achieve motor milestones, and have declines in respiration and swallowing. At a median age of 10.5 months, patients die or need permanent ventilatory assistance.
In December 2016, the FDA approved nusinersen for the treatment of SMA. A phase III study found that patients treated with nusinersen were more likely than controls to have improved motor function and event-free survival. The trial was stopped early because of the treatment’s efficacy.
A Small, Open-Label Trial
A mouse study indicated that IV administration of an adenoassociated viral vector (ie, AAV9) containing SMN1 reduced the effects of SMA and extended survival. Jerry R. Mendell, MD, Principal Investigator at Nationwide Children’s Hospital’s Center for Gene Therapy in Columbus, Ohio, and colleagues studied this therapeutic technique in humans.
They enrolled 15 patients with a genetically confirmed diagnosis of SMA1 into two cohorts. The first cohort received a low dose (6.7×1013 vg/kg) of treatment, and the second cohort received a high dose (2.0×1014 vg/kg). Because the first patient in cohort one had serum aminotransferase elevations, the investigators gave 1 mg/kg/day of oral prednisolone to all subsequent patients for 30 days, starting 24 hours before gene therapy.
The study’s primary outcome was treatment-related adverse events of grade 3 or higher. The secondary outcome was time until death or the need for permanent ventilatory assistance, which was defined as at least 16 hours/day of continuous respiratory assistance for at least 14 days. The achievement of motor milestones and Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scores were exploratory outcomes.
Motor Scores Improved
Three patients entered the low-dose cohort, and 12 were enrolled in the high-dose cohort. Patients’ mean age at treatment was 6.3 months in cohort 1 and 3.4 months in cohort 2. At the last follow-up, all patients had reached age 20 months, and none required permanent mechanical ventilation. Approximately 8% of patients in a historical cohort met these criteria.
All patients had increases from baseline in CHOP INTEND score and maintained these increases throughout the study. Patients in cohort 2 had mean increases of 9.8 points at one month and 15.4 points at three months. Eleven patients achieved and sustained scores greater than 40 points, which is considered clinically meaningful in SMA.
Of the patients in cohort 2, 11 sat unassisted, nine rolled over, 11 fed orally and could speak, and two walked independently. No patients in the historical cohorts achieved any of these milestones, and they rarely became able to speak.
Dr. Mendell and colleagues observed two treatment-related grade 4 adverse events. Both were elevations in serum aminotransferase levels that were attenuated after treatment with prednisolone. The researchers also noted three treatment-related nonserious adverse events (ie, asymptomatic elevations in serum aminotransferase levels that were resolved without additional prednisolone treatment).
The study results were consistent with those of the preclinical mouse study. During a follow-up period of as long as two years, Dr. Mendell and colleagues did not observe any decrease in treatment effect or regression in motor function among the study participants. The presence of antibodies to AAV9 could be a potential limitation of the therapy, however. Further research to assess the treatment’s safety and the durability of its effect are needed, according to the authors.
Comparing Two Treatments
It is difficult to compare the results of Dr. Mendell and colleagues with those of the phase III nusinersen study, because of the two trials’ different designs, said Ans T. van der Ploeg, MD, PhD, Chair of the Center for Lysosomal and Metabolic Diseases at the Erasmus MC University in Rotterdam, the Netherlands, in an accompanying editorial. One potential advantage of AAV9 gene therapy is that it might require a single IV infusion. Nusinersen, on the other hand, may require lifelong intrathecal treatment.
“The durability of the effects is uncertain for both treatments,” said Dr. van der Ploeg. “If the expression of the scAAV9 gene therapy declines over time, the same treatment may not be able to be repeated, because antibodies against AAV capsid proteins are anticipated to form.”
In addition, neither of the two therapies cures SMA type 1. Earlier treatment could be beneficial, as could a combination of both treatments, said Dr. van der Ploeg. But the high expected cost of nusinersen is “an important constraint,” she concluded.
—Erik Greb
Suggested Reading
Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-1722.
van der Ploeg AT. The dilemma of two innovative therapies for spinal muscular atrophy. N Engl J Med. 2017;377(18):1786-1787.
A single IV infusion to replace the gene encoding survival motor neuron 1 (SMN1) increases survival among infants with spinal muscular atrophy type 1 (SMA1), according to research published in the November 2, 2017, issue of the New England Journal of Medicine. The treatment also improves motor function, and its effects are maintained for two years, the researchers said.
The loss or dysfunction of SMN1 causes SMA, a progressive disease characterized by the degeneration and loss of lower motor neurons. Onset of SMA1 typically occurs at one month of age. Children with the disease usually are weak, fail to achieve motor milestones, and have declines in respiration and swallowing. At a median age of 10.5 months, patients die or need permanent ventilatory assistance.
In December 2016, the FDA approved nusinersen for the treatment of SMA. A phase III study found that patients treated with nusinersen were more likely than controls to have improved motor function and event-free survival. The trial was stopped early because of the treatment’s efficacy.
A Small, Open-Label Trial
A mouse study indicated that IV administration of an adenoassociated viral vector (ie, AAV9) containing SMN1 reduced the effects of SMA and extended survival. Jerry R. Mendell, MD, Principal Investigator at Nationwide Children’s Hospital’s Center for Gene Therapy in Columbus, Ohio, and colleagues studied this therapeutic technique in humans.
They enrolled 15 patients with a genetically confirmed diagnosis of SMA1 into two cohorts. The first cohort received a low dose (6.7×1013 vg/kg) of treatment, and the second cohort received a high dose (2.0×1014 vg/kg). Because the first patient in cohort one had serum aminotransferase elevations, the investigators gave 1 mg/kg/day of oral prednisolone to all subsequent patients for 30 days, starting 24 hours before gene therapy.
The study’s primary outcome was treatment-related adverse events of grade 3 or higher. The secondary outcome was time until death or the need for permanent ventilatory assistance, which was defined as at least 16 hours/day of continuous respiratory assistance for at least 14 days. The achievement of motor milestones and Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) scores were exploratory outcomes.
Motor Scores Improved
Three patients entered the low-dose cohort, and 12 were enrolled in the high-dose cohort. Patients’ mean age at treatment was 6.3 months in cohort 1 and 3.4 months in cohort 2. At the last follow-up, all patients had reached age 20 months, and none required permanent mechanical ventilation. Approximately 8% of patients in a historical cohort met these criteria.
All patients had increases from baseline in CHOP INTEND score and maintained these increases throughout the study. Patients in cohort 2 had mean increases of 9.8 points at one month and 15.4 points at three months. Eleven patients achieved and sustained scores greater than 40 points, which is considered clinically meaningful in SMA.
Of the patients in cohort 2, 11 sat unassisted, nine rolled over, 11 fed orally and could speak, and two walked independently. No patients in the historical cohorts achieved any of these milestones, and they rarely became able to speak.
Dr. Mendell and colleagues observed two treatment-related grade 4 adverse events. Both were elevations in serum aminotransferase levels that were attenuated after treatment with prednisolone. The researchers also noted three treatment-related nonserious adverse events (ie, asymptomatic elevations in serum aminotransferase levels that were resolved without additional prednisolone treatment).
The study results were consistent with those of the preclinical mouse study. During a follow-up period of as long as two years, Dr. Mendell and colleagues did not observe any decrease in treatment effect or regression in motor function among the study participants. The presence of antibodies to AAV9 could be a potential limitation of the therapy, however. Further research to assess the treatment’s safety and the durability of its effect are needed, according to the authors.
Comparing Two Treatments
It is difficult to compare the results of Dr. Mendell and colleagues with those of the phase III nusinersen study, because of the two trials’ different designs, said Ans T. van der Ploeg, MD, PhD, Chair of the Center for Lysosomal and Metabolic Diseases at the Erasmus MC University in Rotterdam, the Netherlands, in an accompanying editorial. One potential advantage of AAV9 gene therapy is that it might require a single IV infusion. Nusinersen, on the other hand, may require lifelong intrathecal treatment.
“The durability of the effects is uncertain for both treatments,” said Dr. van der Ploeg. “If the expression of the scAAV9 gene therapy declines over time, the same treatment may not be able to be repeated, because antibodies against AAV capsid proteins are anticipated to form.”
In addition, neither of the two therapies cures SMA type 1. Earlier treatment could be beneficial, as could a combination of both treatments, said Dr. van der Ploeg. But the high expected cost of nusinersen is “an important constraint,” she concluded.
—Erik Greb
Suggested Reading
Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-1722.
van der Ploeg AT. The dilemma of two innovative therapies for spinal muscular atrophy. N Engl J Med. 2017;377(18):1786-1787.