User login
Primary Mucinous Carcinoma of the Eyelid Treated With Mohs Micrographic Surgery
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
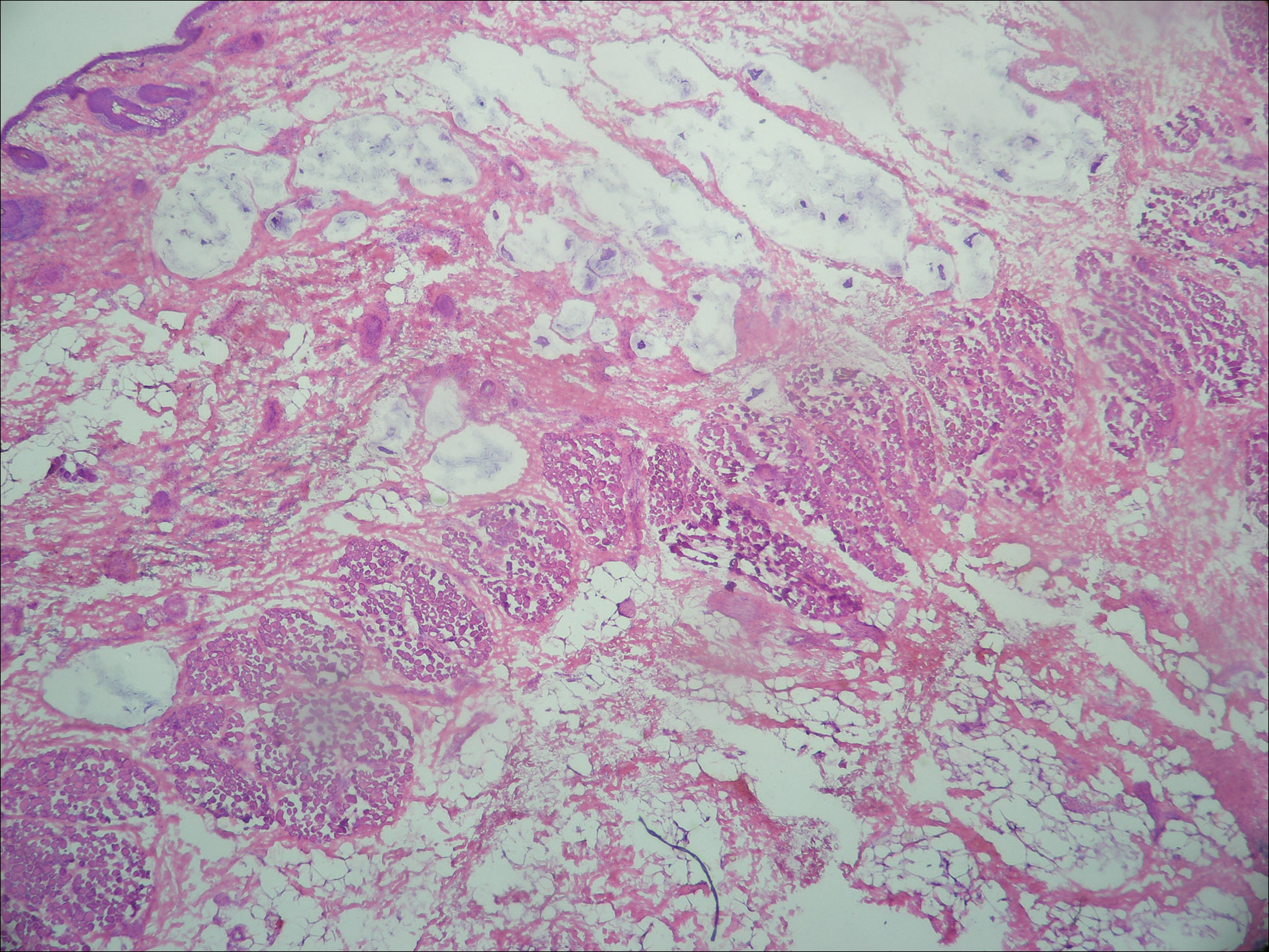
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
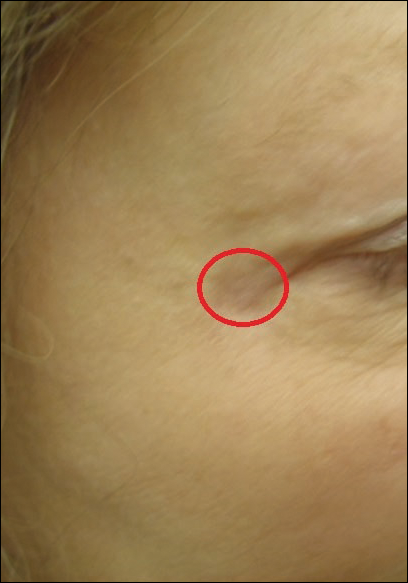
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
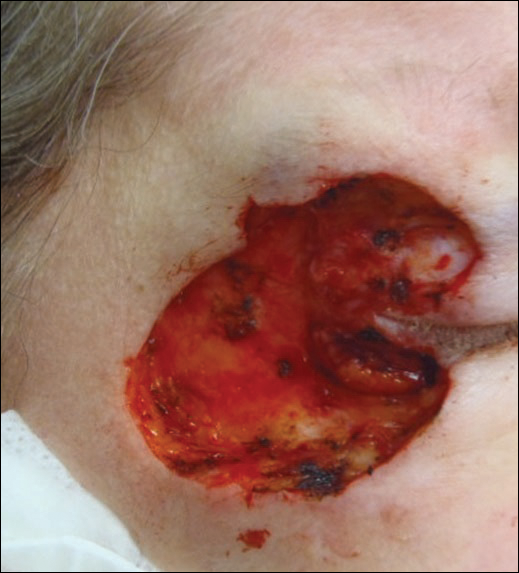
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
Practice Points
- Primary mucinous carcinoma (PMC) of the skin is a rare adnexal tumor.
- Prior to treatment, the diagnostic importance lies in distinguishing PMC from metastatic mucinous malignancies, which portend a poorer prognosis.
- Treatment primarily is surgical, with Mohs micrographic surgery offering improved tissue conservation and reduced recurrence rates.
Ustekinumab may reduce risk of nonmelanoma skin cancer
GENEVA – Ustekinumab therapy appears to protect psoriasis patients against nonmelanoma skin cancer (NMSC), according to a new analysis from the PSOLAR registry.
Compared with psoriasis patients on methotrexate, the risk of developing on-treatment NMSC was lower among patients on the interleukin-12-/23 inhibitor ustekinumab (Stelara) and those on the three tumor necrosis factor (TNF) inhibitors included in the PSOLAR registry – infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira). The lower risk was statistically significant only for ustekinumab, although there was a favorable trend with the TNF inhibitors showing a 19% relative risk reduction, Bhaskar Srivastava, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.

PSOLAR (Psoriasis Longitudinal Assessment and Registry) is an ongoing international prospective observational study evaluating long-term safety and clinical outcomes in psoriasis patients eligible for systemic therapies. The study is now fully enrolled, with 12,090 psoriasis patients and 48,870 patient-years of follow-up and climbing, noted Dr. Srivastava, an employee of Janssen Scientific Affairs, Spring House, Pa.
This analysis focused on 6,782 PSOLAR participants with a mean 18-year history of psoriasis and no history of NMSC at enrollment: 2,623 patients on ustekinumab with 7,900 patient-years of prospective follow-up, 3,727 on a TNF inhibitor with 10,580 patient-years of follow-up, and 432 controls on methotrexate with 781 patient-years of follow-up.
Patients on a biologic were significantly younger, with a mean age of 46.7 years, versus 53.6 years for those on methotrexate. Rates of past or current smoking were similar, in the 55%-60% range, regardless of which systemic agent patients were using.
The crude unadjusted incidence rate for NMSC among all patients on a biologic was 0.33 cancers/100 patient-years, compared with 1.41/100 patient-years for psoriasis patients on methotrexate.
Patients on ustekinumab had an NMSC incidence rate of 0.19/100 patient-years, with a basal cell carcinoma rate of 0.13/100 patient-years and a squamous cell carcinoma rate of 0.06/100 patient-years. Psoriasis patients on a TNF inhibitor had an NMSC incidence rate of 0.43/100 patient-years, with a basal cell carcinoma rate of 0.26/100 patient-years and a squamous cell carcinoma rate of 0.17/100 patient-years.
In a multivariate analysis adjusted for age, sex, race, location, duration of psoriasis, smoking, prior malignancy, skin type, and history of treatment with cyclosporine, methotrexate, other systemic agents, or phototherapy, patients taking ustekinumab had a statistically significant 65% reduction in the risk of NMSC compared with patients on methotrexate and a 74% relative risk reduction for basal cell carcinoma; however, the squamous cell carcinoma risk in the two patient groups was similar.
Dr. Srivastava said the PSOLAR data shouldn’t be taken as the final word regarding NMSC risk and the use of biologics. He noted that psoriasis itself is associated with an increased risk of NMSC. And methotrexate, which was used as the reference standard in this analysis, may alter the risk of NMSC.
“Overall, these results require further validation in psoriasis populations with larger numbers of exposed patients,” he said.
The PSOLAR registry is funded by Janssen, where Dr. Srivastava is employed.
GENEVA – Ustekinumab therapy appears to protect psoriasis patients against nonmelanoma skin cancer (NMSC), according to a new analysis from the PSOLAR registry.
Compared with psoriasis patients on methotrexate, the risk of developing on-treatment NMSC was lower among patients on the interleukin-12-/23 inhibitor ustekinumab (Stelara) and those on the three tumor necrosis factor (TNF) inhibitors included in the PSOLAR registry – infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira). The lower risk was statistically significant only for ustekinumab, although there was a favorable trend with the TNF inhibitors showing a 19% relative risk reduction, Bhaskar Srivastava, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
PSOLAR (Psoriasis Longitudinal Assessment and Registry) is an ongoing international prospective observational study evaluating long-term safety and clinical outcomes in psoriasis patients eligible for systemic therapies. The study is now fully enrolled, with 12,090 psoriasis patients and 48,870 patient-years of follow-up and climbing, noted Dr. Srivastava, an employee of Janssen Scientific Affairs, Spring House, Pa.
This analysis focused on 6,782 PSOLAR participants with a mean 18-year history of psoriasis and no history of NMSC at enrollment: 2,623 patients on ustekinumab with 7,900 patient-years of prospective follow-up, 3,727 on a TNF inhibitor with 10,580 patient-years of follow-up, and 432 controls on methotrexate with 781 patient-years of follow-up.
Patients on a biologic were significantly younger, with a mean age of 46.7 years, versus 53.6 years for those on methotrexate. Rates of past or current smoking were similar, in the 55%-60% range, regardless of which systemic agent patients were using.
The crude unadjusted incidence rate for NMSC among all patients on a biologic was 0.33 cancers/100 patient-years, compared with 1.41/100 patient-years for psoriasis patients on methotrexate.
Patients on ustekinumab had an NMSC incidence rate of 0.19/100 patient-years, with a basal cell carcinoma rate of 0.13/100 patient-years and a squamous cell carcinoma rate of 0.06/100 patient-years. Psoriasis patients on a TNF inhibitor had an NMSC incidence rate of 0.43/100 patient-years, with a basal cell carcinoma rate of 0.26/100 patient-years and a squamous cell carcinoma rate of 0.17/100 patient-years.
In a multivariate analysis adjusted for age, sex, race, location, duration of psoriasis, smoking, prior malignancy, skin type, and history of treatment with cyclosporine, methotrexate, other systemic agents, or phototherapy, patients taking ustekinumab had a statistically significant 65% reduction in the risk of NMSC compared with patients on methotrexate and a 74% relative risk reduction for basal cell carcinoma; however, the squamous cell carcinoma risk in the two patient groups was similar.
Dr. Srivastava said the PSOLAR data shouldn’t be taken as the final word regarding NMSC risk and the use of biologics. He noted that psoriasis itself is associated with an increased risk of NMSC. And methotrexate, which was used as the reference standard in this analysis, may alter the risk of NMSC.
“Overall, these results require further validation in psoriasis populations with larger numbers of exposed patients,” he said.
The PSOLAR registry is funded by Janssen, where Dr. Srivastava is employed.
GENEVA – Ustekinumab therapy appears to protect psoriasis patients against nonmelanoma skin cancer (NMSC), according to a new analysis from the PSOLAR registry.
Compared with psoriasis patients on methotrexate, the risk of developing on-treatment NMSC was lower among patients on the interleukin-12-/23 inhibitor ustekinumab (Stelara) and those on the three tumor necrosis factor (TNF) inhibitors included in the PSOLAR registry – infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira). The lower risk was statistically significant only for ustekinumab, although there was a favorable trend with the TNF inhibitors showing a 19% relative risk reduction, Bhaskar Srivastava, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
PSOLAR (Psoriasis Longitudinal Assessment and Registry) is an ongoing international prospective observational study evaluating long-term safety and clinical outcomes in psoriasis patients eligible for systemic therapies. The study is now fully enrolled, with 12,090 psoriasis patients and 48,870 patient-years of follow-up and climbing, noted Dr. Srivastava, an employee of Janssen Scientific Affairs, Spring House, Pa.
This analysis focused on 6,782 PSOLAR participants with a mean 18-year history of psoriasis and no history of NMSC at enrollment: 2,623 patients on ustekinumab with 7,900 patient-years of prospective follow-up, 3,727 on a TNF inhibitor with 10,580 patient-years of follow-up, and 432 controls on methotrexate with 781 patient-years of follow-up.
Patients on a biologic were significantly younger, with a mean age of 46.7 years, versus 53.6 years for those on methotrexate. Rates of past or current smoking were similar, in the 55%-60% range, regardless of which systemic agent patients were using.
The crude unadjusted incidence rate for NMSC among all patients on a biologic was 0.33 cancers/100 patient-years, compared with 1.41/100 patient-years for psoriasis patients on methotrexate.
Patients on ustekinumab had an NMSC incidence rate of 0.19/100 patient-years, with a basal cell carcinoma rate of 0.13/100 patient-years and a squamous cell carcinoma rate of 0.06/100 patient-years. Psoriasis patients on a TNF inhibitor had an NMSC incidence rate of 0.43/100 patient-years, with a basal cell carcinoma rate of 0.26/100 patient-years and a squamous cell carcinoma rate of 0.17/100 patient-years.
In a multivariate analysis adjusted for age, sex, race, location, duration of psoriasis, smoking, prior malignancy, skin type, and history of treatment with cyclosporine, methotrexate, other systemic agents, or phototherapy, patients taking ustekinumab had a statistically significant 65% reduction in the risk of NMSC compared with patients on methotrexate and a 74% relative risk reduction for basal cell carcinoma; however, the squamous cell carcinoma risk in the two patient groups was similar.
Dr. Srivastava said the PSOLAR data shouldn’t be taken as the final word regarding NMSC risk and the use of biologics. He noted that psoriasis itself is associated with an increased risk of NMSC. And methotrexate, which was used as the reference standard in this analysis, may alter the risk of NMSC.
“Overall, these results require further validation in psoriasis populations with larger numbers of exposed patients,” he said.
The PSOLAR registry is funded by Janssen, where Dr. Srivastava is employed.
AT THE EADV CONGRESS
Key clinical point:
Major finding: Psoriasis patients on ustekinumab had an adjusted 65% reduction in the risk of developing nonmelanoma skin cancer compared with patients on methotrexate.
Data source: An analysis of 6,782 psoriasis patients participating in an international prospective observational registry evaluating the long-term safety and clinical outcomes of systemic therapies.
Disclosures: The PSOLAR registry is funded by ustekinumab manufacturer Janssen; the study presenter is a company employee.
Black Eschars on the Face and Body
The Diagnosis: Lymphomatoid Papulosis
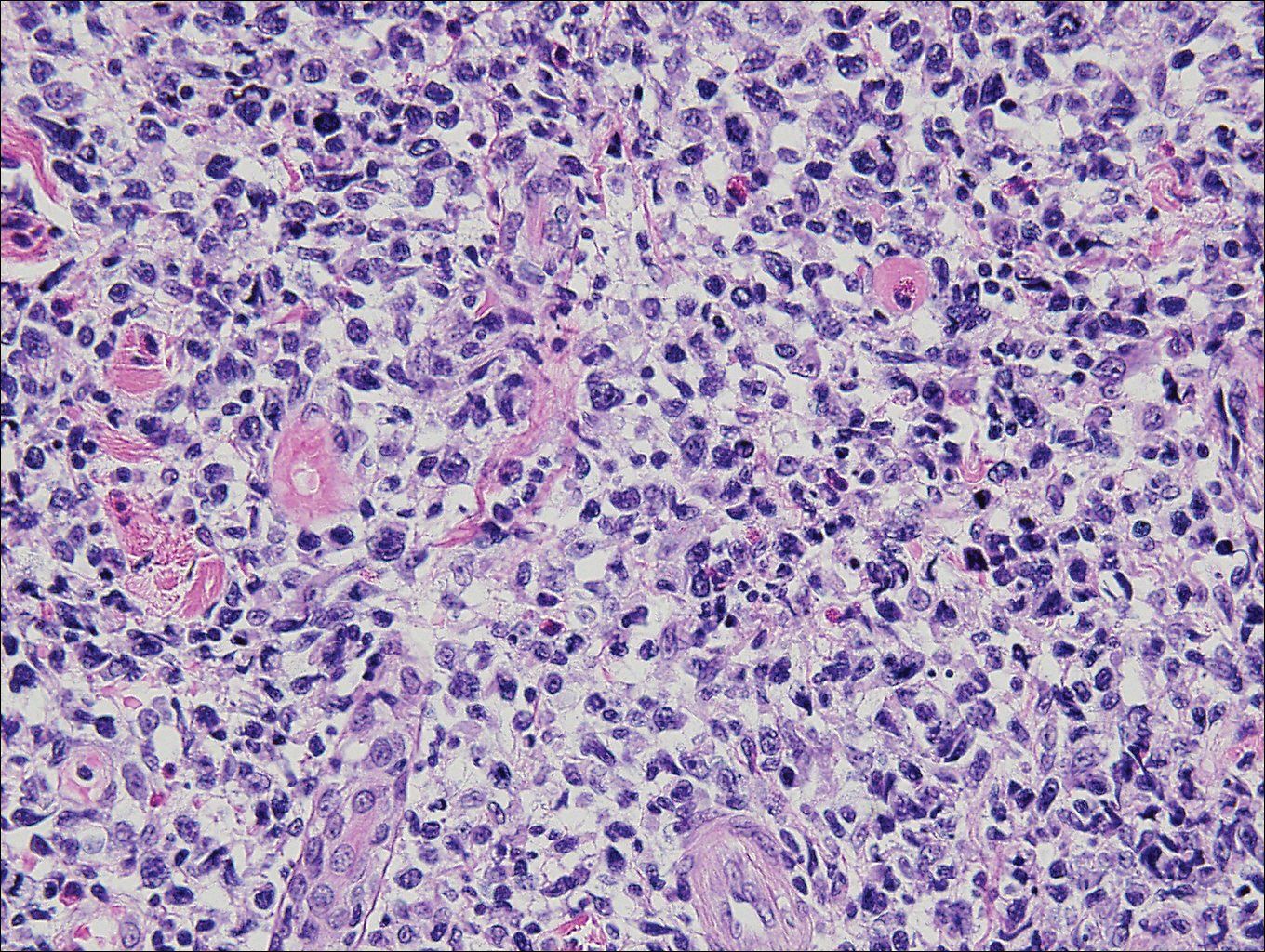
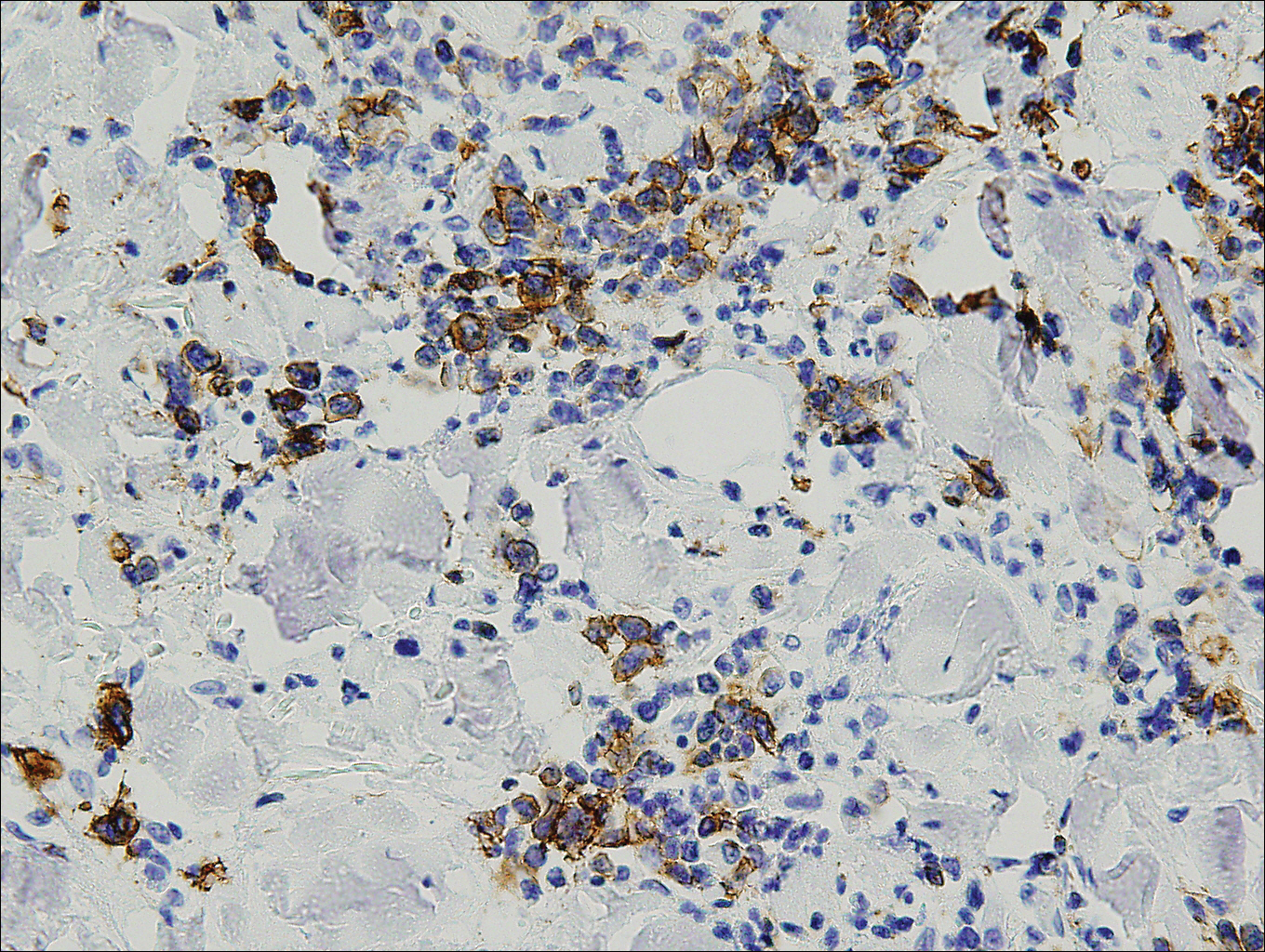
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
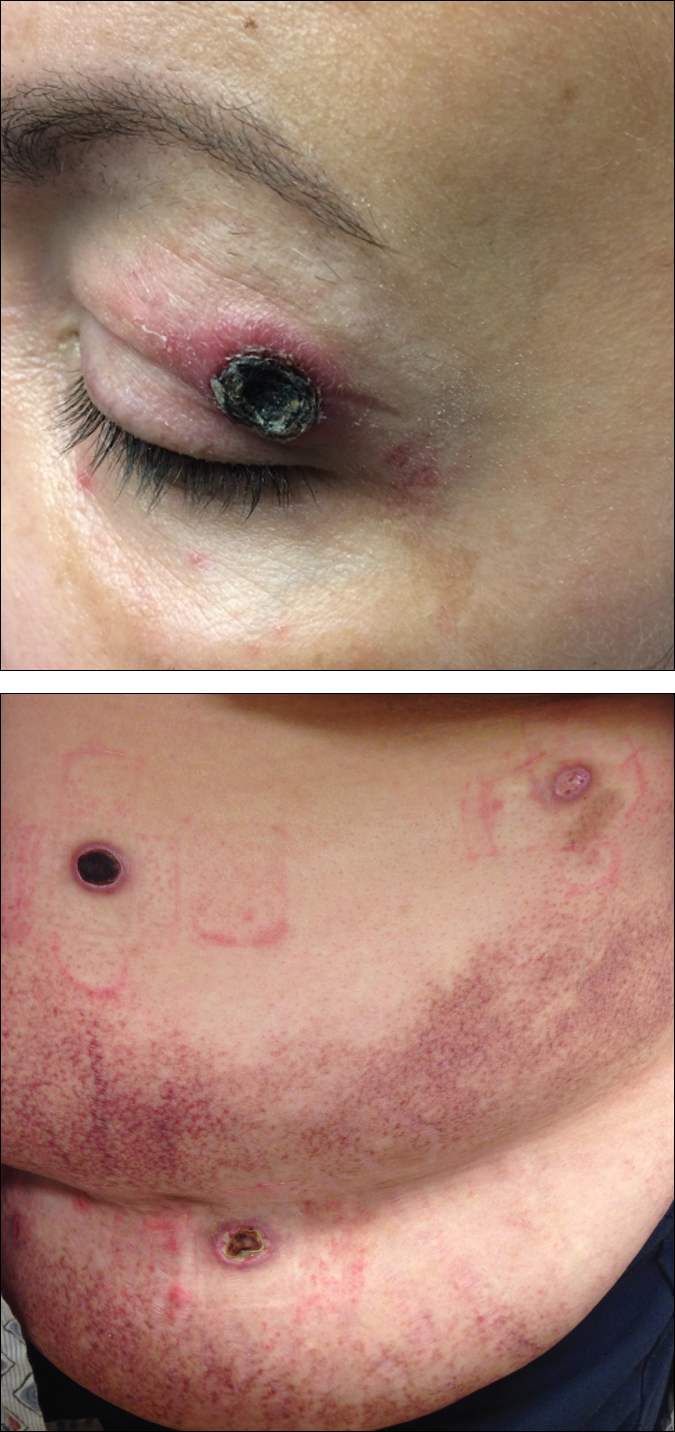
A 50-year-old woman presented for evaluation of black eschars on the face and body. Over the preceding 8 weeks she had developed several asymptomatic papules that gradually enlarged, ulcerated, and formed a black eschar, prior to gradually self-resolving over the course of several weeks. During this time, new lesions were forming. The resulting skin revealed dyspigmentation and scar formation. Prior to presentation, antimicrobial therapy had been initiated for a presumed infectious etiology; however, the eruption continued to progress. The patient denied sick contacts, livestock exposure, or recent travel. A complete review of systems, including fever, chills, or lymphadenopathy, was negative. Physical examination revealed 6 circular necrotic ulcers with an overlying black eschar on the face (top), trunk (bottom), hands, and thighs, all in various stages of healing. In addition, large, reticulated, poikilodermatous patches were incidentally noted in areas free of ulcers and eschars on the trunk (bottom) and bilateral arms and legs. Upon questioning, the patient said these patches had been present for more than 30 years. A punch biopsy from an ulcer on the chest was obtained and sent for histopathologic and immunohistochemical examination.
Debunking Actinic Keratosis Myths: Are Patients With Darker Skin At Risk for Actinic Keratoses?
Myth: Actinic keratoses are only seen in patients with lighter skin
Actinic keratoses (AKs) are precancerous lesions that may turn into squamous cell carcinoma if left untreated. UV rays cause AKs, either from outdoor sun exposure or tanning beds. According to the American Academy of Dermatology, AKs are more likely to develop in patients 40 years or older with fair skin; hair color that is naturally blonde or red; eye color that is naturally blue, green, or hazel; skin that freckles or burns when in the sun; a weakened immune system; and occupations involving substances that contain polycyclic aromatic hydrocarbons such as coal or tar.
A 2007 study compared the most common diagnoses among patients of different racial and ethnic groups in New York City. Alexis et al found that AK was in the top 10 diagnoses in white patients but not for black patients. They postulated that photoprotective factors in darkly pigmented skin such as larger and more numerous melanosomes that contain more melanin and are more dispersed throughout the epidermis result in a lower incidence of skin cancers in the skin of color (SOC) population.
RELATED ARTICLE: Common Dermatologic Disorders in Skin of Color: A Comparative Practice Survey
However, a recent skin cancer awareness study in Cutis reported that even though SOC populations have lower incidences of skin cancer such as melanoma, basal cell carcinoma, and squamous cell carcinoma, they exhibit higher death rates. Furthermore, black individuals are more likely to present with advanced-stage melanoma and acral lentiginous melanomas compared to white individuals. Kailas et al stated, “Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.” They evaluated several knowledge-based interventions for increasing skin cancer awareness, knowledge, and protective behaviors in SOC populations, including the use of visuals such as photographs to allow SOC patients to visualize different skin tones, educational interventions in another language, and pamphlets.
Dermatologists should be aware that education of SOC patients is important to eradicate the common misconception that these patients do not have to worry about AKs and other skin cancers. Remind these patients that they need to protect their skin from the sun, just as patients with fair skin do. Further research in the dermatology community should focus on educational interventions that will help increase knowledge regarding skin cancer in SOC populations.
Expert Commentary
Although more common in patients with lighter skin, actinic keratosis and skin cancer can be seen in patients of all skin types. Many patients are unaware of this risk and do not use sunscreen and other sun-protective measures. We, as a specialty, have to educate our patients and the public of the risk for actinic keratosis and skin cancer in all skin types.
—Gary Goldenberg, MD (New York, New York)
Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
American Academy of Dermatology. Actinic keratosis. https://www.aad.org/public/diseases/scaly-skin/actinic-keratosis. Accessed October 17, 2017.
Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
Myth: Actinic keratoses are only seen in patients with lighter skin
Actinic keratoses (AKs) are precancerous lesions that may turn into squamous cell carcinoma if left untreated. UV rays cause AKs, either from outdoor sun exposure or tanning beds. According to the American Academy of Dermatology, AKs are more likely to develop in patients 40 years or older with fair skin; hair color that is naturally blonde or red; eye color that is naturally blue, green, or hazel; skin that freckles or burns when in the sun; a weakened immune system; and occupations involving substances that contain polycyclic aromatic hydrocarbons such as coal or tar.
A 2007 study compared the most common diagnoses among patients of different racial and ethnic groups in New York City. Alexis et al found that AK was in the top 10 diagnoses in white patients but not for black patients. They postulated that photoprotective factors in darkly pigmented skin such as larger and more numerous melanosomes that contain more melanin and are more dispersed throughout the epidermis result in a lower incidence of skin cancers in the skin of color (SOC) population.
RELATED ARTICLE: Common Dermatologic Disorders in Skin of Color: A Comparative Practice Survey
However, a recent skin cancer awareness study in Cutis reported that even though SOC populations have lower incidences of skin cancer such as melanoma, basal cell carcinoma, and squamous cell carcinoma, they exhibit higher death rates. Furthermore, black individuals are more likely to present with advanced-stage melanoma and acral lentiginous melanomas compared to white individuals. Kailas et al stated, “Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.” They evaluated several knowledge-based interventions for increasing skin cancer awareness, knowledge, and protective behaviors in SOC populations, including the use of visuals such as photographs to allow SOC patients to visualize different skin tones, educational interventions in another language, and pamphlets.
Dermatologists should be aware that education of SOC patients is important to eradicate the common misconception that these patients do not have to worry about AKs and other skin cancers. Remind these patients that they need to protect their skin from the sun, just as patients with fair skin do. Further research in the dermatology community should focus on educational interventions that will help increase knowledge regarding skin cancer in SOC populations.
Expert Commentary
Although more common in patients with lighter skin, actinic keratosis and skin cancer can be seen in patients of all skin types. Many patients are unaware of this risk and do not use sunscreen and other sun-protective measures. We, as a specialty, have to educate our patients and the public of the risk for actinic keratosis and skin cancer in all skin types.
—Gary Goldenberg, MD (New York, New York)
Myth: Actinic keratoses are only seen in patients with lighter skin
Actinic keratoses (AKs) are precancerous lesions that may turn into squamous cell carcinoma if left untreated. UV rays cause AKs, either from outdoor sun exposure or tanning beds. According to the American Academy of Dermatology, AKs are more likely to develop in patients 40 years or older with fair skin; hair color that is naturally blonde or red; eye color that is naturally blue, green, or hazel; skin that freckles or burns when in the sun; a weakened immune system; and occupations involving substances that contain polycyclic aromatic hydrocarbons such as coal or tar.
A 2007 study compared the most common diagnoses among patients of different racial and ethnic groups in New York City. Alexis et al found that AK was in the top 10 diagnoses in white patients but not for black patients. They postulated that photoprotective factors in darkly pigmented skin such as larger and more numerous melanosomes that contain more melanin and are more dispersed throughout the epidermis result in a lower incidence of skin cancers in the skin of color (SOC) population.
RELATED ARTICLE: Common Dermatologic Disorders in Skin of Color: A Comparative Practice Survey
However, a recent skin cancer awareness study in Cutis reported that even though SOC populations have lower incidences of skin cancer such as melanoma, basal cell carcinoma, and squamous cell carcinoma, they exhibit higher death rates. Furthermore, black individuals are more likely to present with advanced-stage melanoma and acral lentiginous melanomas compared to white individuals. Kailas et al stated, “Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.” They evaluated several knowledge-based interventions for increasing skin cancer awareness, knowledge, and protective behaviors in SOC populations, including the use of visuals such as photographs to allow SOC patients to visualize different skin tones, educational interventions in another language, and pamphlets.
Dermatologists should be aware that education of SOC patients is important to eradicate the common misconception that these patients do not have to worry about AKs and other skin cancers. Remind these patients that they need to protect their skin from the sun, just as patients with fair skin do. Further research in the dermatology community should focus on educational interventions that will help increase knowledge regarding skin cancer in SOC populations.
Expert Commentary
Although more common in patients with lighter skin, actinic keratosis and skin cancer can be seen in patients of all skin types. Many patients are unaware of this risk and do not use sunscreen and other sun-protective measures. We, as a specialty, have to educate our patients and the public of the risk for actinic keratosis and skin cancer in all skin types.
—Gary Goldenberg, MD (New York, New York)
Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
American Academy of Dermatology. Actinic keratosis. https://www.aad.org/public/diseases/scaly-skin/actinic-keratosis. Accessed October 17, 2017.
Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
Alexis AF, Sergay AB, Taylor SC. Common dermatologic disorders in skin of color: a comparative practice survey. Cutis. 2007;80:387-394.
American Academy of Dermatology. Actinic keratosis. https://www.aad.org/public/diseases/scaly-skin/actinic-keratosis. Accessed October 17, 2017.
Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.

Painless Telangiectatic Lesion on the Wrist
The Diagnosis: Merkel Cell Carcinoma
A partial biopsy was performed during the dermatology examination. Histopathology demonstrated a dense dermal infiltrate of small, dark blue, pleomorphic cells (Figure 1). On high power, the individual cells were noted to have vesicular nuclei with finely granular and dusty chromatin (Figure 2). Numerous mitotic figures were present. Immunohistochemical stains were performed and revealed positive staining for cytokeratin 20 (with a perinuclear dot pattern), synaptophysin, and chromogranin.
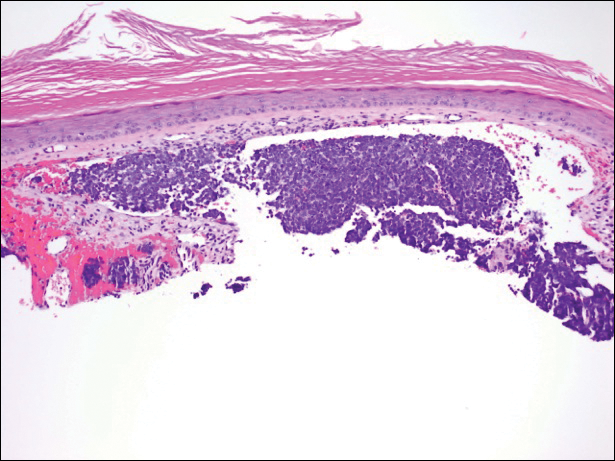
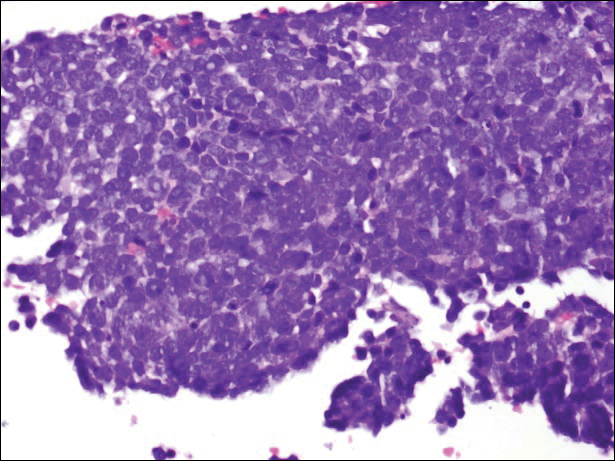
Merkel cell carcinoma (MCC) is an uncommon carcinoma of the epidermal neuroendocrine cells with approximately 1500 cases a year in the United States.1 Merkel cell carcinoma has a poor prognosis with approximately one-third of cases resulting in death within 5 years and with a survival rate strongly dependent on the stage of disease at presentation.2 A complete surgical excision with histologically verified clear margins is the main form of treatment of the primary cancer.3 Although the effectiveness of adjuvant therapy for MCC has been debated,4 retrospective analysis has shown that the high local recurrence rate of the primary tumor can be reduced by combining surgical excision with a form of radiation therapy.5
A systematic cohort study of 195 patients diagnosed with MCC summarized its most clinical factors with the acronym AEIOU: asymptomatic, expanding rapidly, immunosuppression, older than 50 years of age, and UV-exposed site on a fair-skinned individual.6 The role of immune function in MCC was highlighted by a 16-fold overrepresentation of immunosuppressed patients in the studied cohort as compared to the general US population. The immunosuppressed patients included individuals with human immunodeficiency virus, chronic lymphocytic leukemia, and iatrogenic suppression secondary to solid organ transplantation.6
In 2008, Merkel cell polyomavirus (MCPyV) was found in 80% (8/10) of MCC tumors tested.7 Since then, many different studies have suggested that MCPyV is an etiologic agent of MCC.8-10 A natural component of skin flora, MCPyV only becomes tumorigenic after integration into the host DNA and with mutations to the viral genome.11 Although there currently is no difference in treatment of MCPyV-positive and MCPyV-negative MCC,12 research is being done to determine how the discovery of the MCPyV could impact the treatment of MCC.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2009;37:20-27.
- Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300-2309.
- Eng TY, Boersma MG, Fuller CD, et al. A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol. 2007;30:624-636.
- Beenken SW, Urist MM. Treatment options for Merkel cell carcinoma. J Natl Compr Canc Netw. 2004;2:89-92.
- Decker RH, Wilson LD. Role of radiotherapy in the management of Merkel cell carcinoma of the skin. J Natl Compr Canc Netw. 2006;4:713-718.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the "AEIOU" features. J Am Acad Dermatol. 2008;58:375-381.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Sastre-Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48-56.
- Varga E, Kiss M, Szabó K, et al. Detection of Merkel cell polyomavirus DNA in Merkel cell carcinomas. Br J Dermatol. 2009;161:930-932.
- Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell carcinoma. Curr Opin Virol. 2015;11:38-43.
- Duprat JP, Landman G, Salvajoli JV, et al. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics (Sao Paulo). 2011;66:1817-1823.
The Diagnosis: Merkel Cell Carcinoma
A partial biopsy was performed during the dermatology examination. Histopathology demonstrated a dense dermal infiltrate of small, dark blue, pleomorphic cells (Figure 1). On high power, the individual cells were noted to have vesicular nuclei with finely granular and dusty chromatin (Figure 2). Numerous mitotic figures were present. Immunohistochemical stains were performed and revealed positive staining for cytokeratin 20 (with a perinuclear dot pattern), synaptophysin, and chromogranin.
Merkel cell carcinoma (MCC) is an uncommon carcinoma of the epidermal neuroendocrine cells with approximately 1500 cases a year in the United States.1 Merkel cell carcinoma has a poor prognosis with approximately one-third of cases resulting in death within 5 years and with a survival rate strongly dependent on the stage of disease at presentation.2 A complete surgical excision with histologically verified clear margins is the main form of treatment of the primary cancer.3 Although the effectiveness of adjuvant therapy for MCC has been debated,4 retrospective analysis has shown that the high local recurrence rate of the primary tumor can be reduced by combining surgical excision with a form of radiation therapy.5
A systematic cohort study of 195 patients diagnosed with MCC summarized its most clinical factors with the acronym AEIOU: asymptomatic, expanding rapidly, immunosuppression, older than 50 years of age, and UV-exposed site on a fair-skinned individual.6 The role of immune function in MCC was highlighted by a 16-fold overrepresentation of immunosuppressed patients in the studied cohort as compared to the general US population. The immunosuppressed patients included individuals with human immunodeficiency virus, chronic lymphocytic leukemia, and iatrogenic suppression secondary to solid organ transplantation.6
In 2008, Merkel cell polyomavirus (MCPyV) was found in 80% (8/10) of MCC tumors tested.7 Since then, many different studies have suggested that MCPyV is an etiologic agent of MCC.8-10 A natural component of skin flora, MCPyV only becomes tumorigenic after integration into the host DNA and with mutations to the viral genome.11 Although there currently is no difference in treatment of MCPyV-positive and MCPyV-negative MCC,12 research is being done to determine how the discovery of the MCPyV could impact the treatment of MCC.
The Diagnosis: Merkel Cell Carcinoma
A partial biopsy was performed during the dermatology examination. Histopathology demonstrated a dense dermal infiltrate of small, dark blue, pleomorphic cells (Figure 1). On high power, the individual cells were noted to have vesicular nuclei with finely granular and dusty chromatin (Figure 2). Numerous mitotic figures were present. Immunohistochemical stains were performed and revealed positive staining for cytokeratin 20 (with a perinuclear dot pattern), synaptophysin, and chromogranin.
Merkel cell carcinoma (MCC) is an uncommon carcinoma of the epidermal neuroendocrine cells with approximately 1500 cases a year in the United States.1 Merkel cell carcinoma has a poor prognosis with approximately one-third of cases resulting in death within 5 years and with a survival rate strongly dependent on the stage of disease at presentation.2 A complete surgical excision with histologically verified clear margins is the main form of treatment of the primary cancer.3 Although the effectiveness of adjuvant therapy for MCC has been debated,4 retrospective analysis has shown that the high local recurrence rate of the primary tumor can be reduced by combining surgical excision with a form of radiation therapy.5
A systematic cohort study of 195 patients diagnosed with MCC summarized its most clinical factors with the acronym AEIOU: asymptomatic, expanding rapidly, immunosuppression, older than 50 years of age, and UV-exposed site on a fair-skinned individual.6 The role of immune function in MCC was highlighted by a 16-fold overrepresentation of immunosuppressed patients in the studied cohort as compared to the general US population. The immunosuppressed patients included individuals with human immunodeficiency virus, chronic lymphocytic leukemia, and iatrogenic suppression secondary to solid organ transplantation.6
In 2008, Merkel cell polyomavirus (MCPyV) was found in 80% (8/10) of MCC tumors tested.7 Since then, many different studies have suggested that MCPyV is an etiologic agent of MCC.8-10 A natural component of skin flora, MCPyV only becomes tumorigenic after integration into the host DNA and with mutations to the viral genome.11 Although there currently is no difference in treatment of MCPyV-positive and MCPyV-negative MCC,12 research is being done to determine how the discovery of the MCPyV could impact the treatment of MCC.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2009;37:20-27.
- Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300-2309.
- Eng TY, Boersma MG, Fuller CD, et al. A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol. 2007;30:624-636.
- Beenken SW, Urist MM. Treatment options for Merkel cell carcinoma. J Natl Compr Canc Netw. 2004;2:89-92.
- Decker RH, Wilson LD. Role of radiotherapy in the management of Merkel cell carcinoma of the skin. J Natl Compr Canc Netw. 2006;4:713-718.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the "AEIOU" features. J Am Acad Dermatol. 2008;58:375-381.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Sastre-Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48-56.
- Varga E, Kiss M, Szabó K, et al. Detection of Merkel cell polyomavirus DNA in Merkel cell carcinomas. Br J Dermatol. 2009;161:930-932.
- Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell carcinoma. Curr Opin Virol. 2015;11:38-43.
- Duprat JP, Landman G, Salvajoli JV, et al. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics (Sao Paulo). 2011;66:1817-1823.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2009;37:20-27.
- Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300-2309.
- Eng TY, Boersma MG, Fuller CD, et al. A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol. 2007;30:624-636.
- Beenken SW, Urist MM. Treatment options for Merkel cell carcinoma. J Natl Compr Canc Netw. 2004;2:89-92.
- Decker RH, Wilson LD. Role of radiotherapy in the management of Merkel cell carcinoma of the skin. J Natl Compr Canc Netw. 2006;4:713-718.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the "AEIOU" features. J Am Acad Dermatol. 2008;58:375-381.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Sastre-Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48-56.
- Varga E, Kiss M, Szabó K, et al. Detection of Merkel cell polyomavirus DNA in Merkel cell carcinomas. Br J Dermatol. 2009;161:930-932.
- Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell carcinoma. Curr Opin Virol. 2015;11:38-43.
- Duprat JP, Landman G, Salvajoli JV, et al. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics (Sao Paulo). 2011;66:1817-1823.
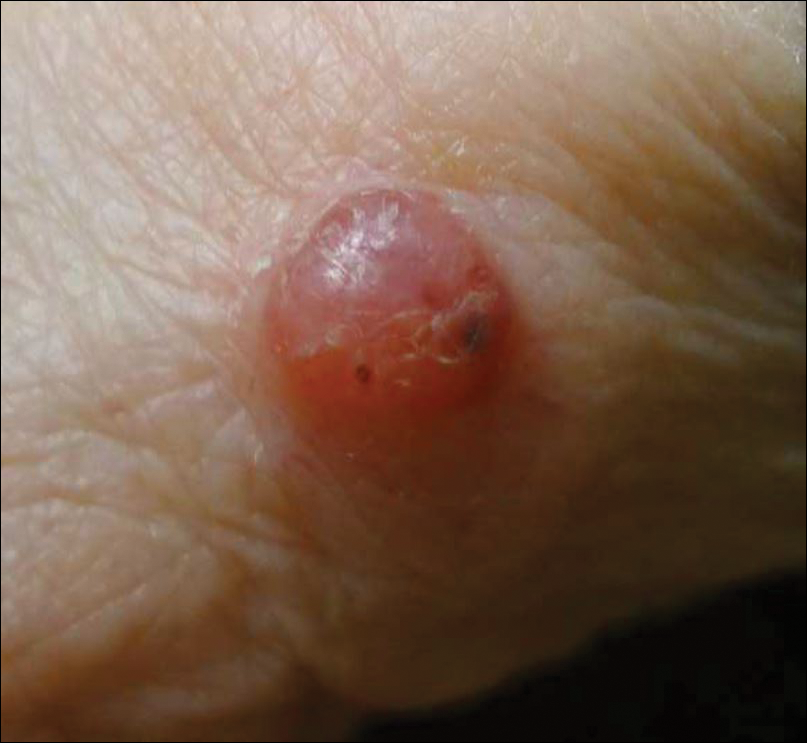
A 91-year-old white man with a history of atrial fibrillation, benign prostatic hyperplasia, dysphagia, gastroesophageal reflux disease, hypertension, hypothyroidism, osteoarthritis, and laryngeal cancer presented with an 8-mm firm, painless, pink lesion with telangiectasia on the left wrist. The lesion had been present for an unknown period of time and was asymptomatic at presentation.

Counsel fair-skinned patients on cancer prevention, says task force
The U.S. Preventive Services Task Force has recommended that clinicians counsel fair-skinned young adults, adolescents, children, and parents of young children about minimizing exposure to ultraviolet radiation to reduce their risk of skin cancer, in a draft recommendation statement that is available online.
The grade B recommendation applies to asymptomatic individuals who have fair skin, are aged 6 months to 24 years, and have no history of skin cancer; the recommendation is being issued because members of this population are at increased risk and are the subject of most of the existing research on skin cancer counseling, according to the USPSTF. The task force found a moderate net benefit when clinicians in a primary care setting offered behavioral counseling on skin cancer prevention to members of this population.
The draft recommendation is open for public comment until 8:00 p.m. Eastern Standard Time on Nov. 6, 2017.
The draft recommendation can be viewed and comments can be submitted online at the USPSTF site.
The U.S. Preventive Services Task Force has recommended that clinicians counsel fair-skinned young adults, adolescents, children, and parents of young children about minimizing exposure to ultraviolet radiation to reduce their risk of skin cancer, in a draft recommendation statement that is available online.
The grade B recommendation applies to asymptomatic individuals who have fair skin, are aged 6 months to 24 years, and have no history of skin cancer; the recommendation is being issued because members of this population are at increased risk and are the subject of most of the existing research on skin cancer counseling, according to the USPSTF. The task force found a moderate net benefit when clinicians in a primary care setting offered behavioral counseling on skin cancer prevention to members of this population.
The draft recommendation is open for public comment until 8:00 p.m. Eastern Standard Time on Nov. 6, 2017.
The draft recommendation can be viewed and comments can be submitted online at the USPSTF site.
The U.S. Preventive Services Task Force has recommended that clinicians counsel fair-skinned young adults, adolescents, children, and parents of young children about minimizing exposure to ultraviolet radiation to reduce their risk of skin cancer, in a draft recommendation statement that is available online.
The grade B recommendation applies to asymptomatic individuals who have fair skin, are aged 6 months to 24 years, and have no history of skin cancer; the recommendation is being issued because members of this population are at increased risk and are the subject of most of the existing research on skin cancer counseling, according to the USPSTF. The task force found a moderate net benefit when clinicians in a primary care setting offered behavioral counseling on skin cancer prevention to members of this population.
The draft recommendation is open for public comment until 8:00 p.m. Eastern Standard Time on Nov. 6, 2017.
The draft recommendation can be viewed and comments can be submitted online at the USPSTF site.
FROM USPSTF
Assessing the Effectiveness of Knowledge-Based Interventions in Increasing Skin Cancer Awareness, Knowledge, and Protective Behaviors in Skin of Color Populations
Malignant melanoma, basal cell carcinoma, and squamous cell carcinoma account for approximately 40% of all neoplasms among the white population in the United States. Skin cancer is the most common malignancy in the United States.1 However, despite this occurrence, there are limited data regarding skin cancer in individuals with skin of color (SOC). The 5-year survival rates for melanoma are 58.2% for black individuals, 69.7% for Hispanics, and 70.9% for Asians compared to 79.8% for white individuals in the United States.2 Even though SOC populations have lower incidences of skin cancer—melanoma, basal cell carcinoma, and squamous cell carcinoma—they exhibit higher death rates.3-7 Nonetheless, no specific guidelines exist to address sun exposure and safety habits in SOC populations.6,8 Furthermore, current demographics suggest that by the year 2050, approximately half of the US population will be nonwhite.4 Paradoxically, despite having increased sun protection from greater amounts of melanin in their skin, black individuals are more likely to present with advanced-stage melanoma (eg, stage III/IV) compared to white individuals.8-12 Furthermore, those of nonwhite populations are more likely to present with more advanced stages of acral lentiginous melanomas than white individuals.13,14 Hispanics also face an increasing incidence of more invasive acral lentiginous melanomas.15 Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.1
Although skin cancer is largely a preventable condition, the literature suggests that lack of awareness of melanoma among ethnic minorities is one of the main reasons for their poor skin cancer prognosis.16 This lack of awareness decreases the likelihood that an SOC patient would be alert to early detection of cancerous changes.17 Because educating at-risk SOC populations is key to decreasing skin cancer risk, this study focused on determining the efficacy of major knowledge-based interventions conducted to date.1 Overall, we sought to answer the question, do knowledge-based interventions increase skin cancer awareness, knowledge, and protective behavior among people of color?
Methods
For this review, the Cochrane method of analysis was used to conduct a thorough search of PubMed articles indexed for MEDLINE (1994-2016), as well as a search of CINAHL (1997-2016), PsycINFO (1999-2016), and Web of Science (1965-2016), using a combination of more than 100 search terms including but not limited to skin cancer, skin of color, intervention, and ethnic skin. The search yielded a total of 52 articles (Figure). Following review, only 8 articles met inclusion criteria, which were as follows: (1) study was related to skin cancer in SOC patients, which included an intervention to increase skin cancer awareness and knowledge; (2) study included adult participants or adolescents aged 12 to 18 years; (3) study was written in English; and (4) study was published in a peer-reviewed journal. Of the remaining 8 articles, 4 were excluded due to the following criteria: (1) study failed to provide both preintervention and postintervention data, (2) study failed to provide quantitative data, and (3) study included participants who worked as health care professionals or ancillary staff. As a result, a total of 4 articles were analyzed and discussed in this review (Table).
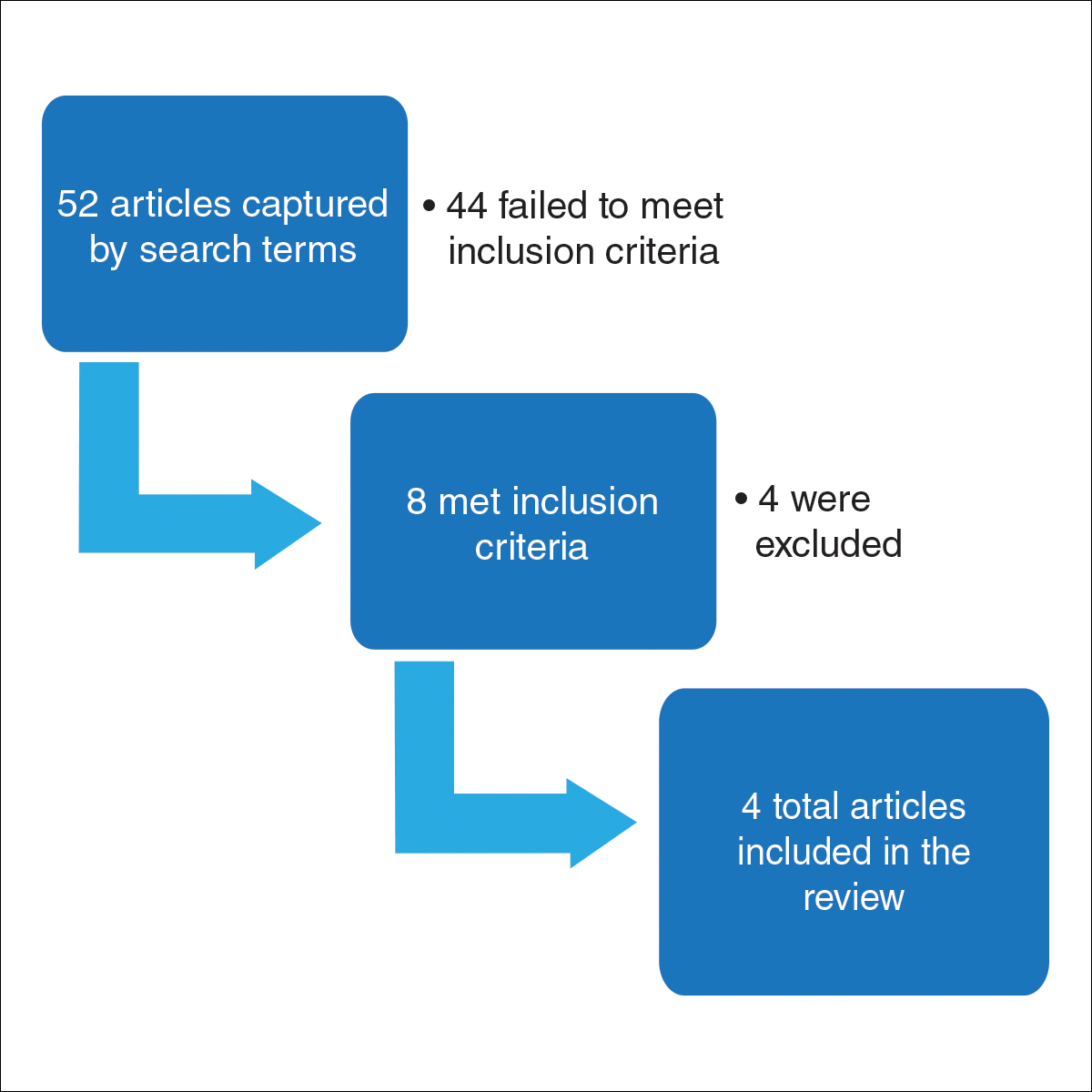
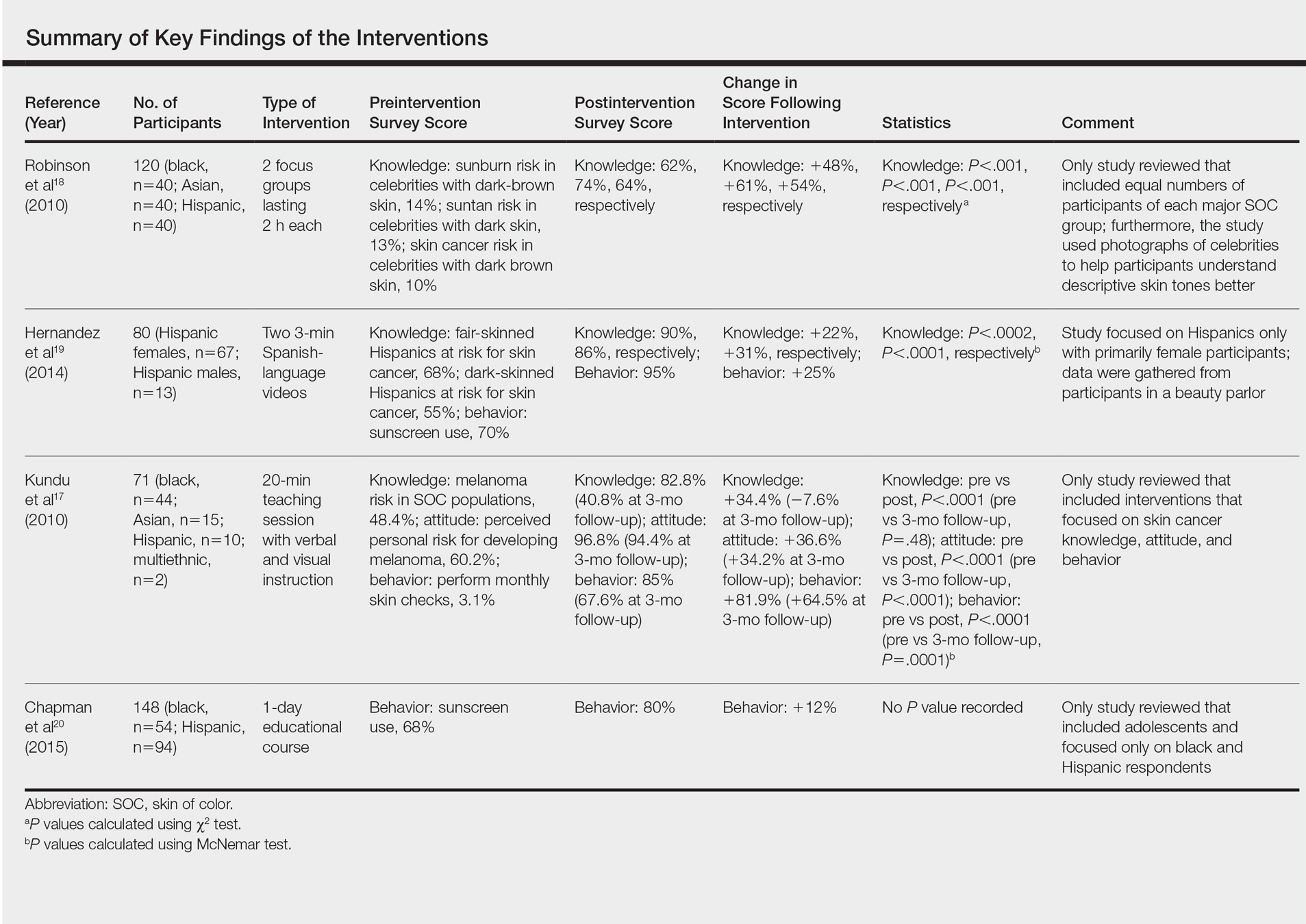
Results
Robinson et al18 conducted 12 focus groups with 120 total participants (40 black, 40 Asian, and 40 Hispanic patients). Participants engaged in a 2-hour tape-recorded focus group with a moderator guide on melanoma and skin cancer. Furthermore, they also were asked to assess skin cancer risk in 5 celebrities with different skin tones. The statistically significant preintervention results of the study (χ2=4.6, P<.001) were as follows: only 2%, 4%, and 14% correctly reported that celebrities with a very fair skin type, a fair skin type, and very dark skin type, respectively, could get sunburn, compared to 75%, 76%, and 62% post-intervention. Additionally, prior to intervention, 14% of the study population believed that dark brown skin type could get sunburn compared to 62% of the same group postintervention. This study demonstrated that the intervention helped SOC patients better identify their ability to get sunburn and identify their skin cancer risk.18
Hernandez et al19 used a video-based intervention in a Hispanic community, which was in contrast to the multiracial focus group intervention conducted by Robinson et al.18 Eighty Hispanic individuals were recruited from beauty salons to participate in the study. Participants watched two 3-minute videos in Spanish and completed a preintervention and postintervention survey. The first video emphasized the photoaging benefits of sun protection, while the second focused on skin cancer prevention. Preintervention surveys indicated that only 54 (68%) participants believed that fair-skinned Hispanics were at risk for skin cancer, which improved to 72 (90%) participants postintervention. Furthermore, initially only 44 (55%) participants thought those with darker skin types could develop skin cancer, but this number increased to 69 (86%) postintervention. For both questions regarding fair and dark skin, the agreement proportion was significantly different between the preeducation and posteducation videos (P<.0002 for the fair skin question and P<.0001 for the dark skin question). This study greatly increased awareness of skin cancer risk among Hispanics,19 similar to the Robinson et al18 study.
In contrast to 2-hour focus groups or 3-minute video–based interventions, a study by Kundu et al17 employed a 20-minute educational class-based intervention with both verbal and visual instruction. This study assessed the efficacy of an educational tutorial on improving awareness and early detection of melanoma in SOC individuals. Photographs were used to help participants recognize the ABCDEs of melanoma and to show examples of acral lentiginous melanomas in white individuals. A total of 71 participants completed a preintervention questionnaire, participated in a 20-minute class, and completed a postintervention questionnaire immediately after and 3 months following the class. The study population included 44 black, 15 Asian, 10 Hispanic, and 2 multiethnic participants. Knowledge that melanoma is a skin cancer increased from 83.9% to 100% immediately postintervention (P=.0001) and 97.2% at 3 months postintervention (P=.0075). Additionally, knowledge that people of color are at risk for melanoma increased from 48.4% preintervention to 82.8% immediately postintervention (P<.0001). However, only 40.8% of participants retained this knowledge at 3 months postintervention. Because only 1 participant reported a family history of skin cancer, the authors hypothesized that the reason for this loss of knowledge was that most participants were not personally affected by friends or family members with melanoma. A future study with an appropriate control group would be needed to support this claim. This study shed light on the potential of class-based interventions to increase both awareness and knowledge of skin cancer in SOC populations.17
A study by Chapman et al20 examined the effects of a sun protection educational program on increasing awareness of skin cancer in Hispanic and black middle school students in southern Los Angeles, California. It was the only study we reviewed that focused primarily on adolescents. Furthermore, it included the largest sample size (N=148) analyzed here. Students were given a preintervention questionnaire to evaluate their awareness of skin cancer and current sun-protection practices. Based on these results, the investigators devised a set of learning goals and incorporated them into an educational pamphlet. The intervention, called “Skin Teaching Day,” was a 1-day program discussing skin cancer and the importance of sun protection. Prior to the intervention, 68% of participants reported that they used sunscreen. Three months after completing the program, 80% of participants reported sunscreen use, an increase of 12% prior to the intervention. The results of this study demonstrated the unique effectiveness and potential of pamphlets in increasing sunscreen use.20
Comment
Overall, various methods of interventions such as focus groups, videos, pamphlets, and lectures improved knowledge of skin cancer risk and sun-protection behaviors in SOC populations. Furthermore, the unique differences of each study provided important insights into the successful design of an intervention.
An important characteristic of the Robinson et al18 study was the addition of photographs, which allowed participants not only to visualize different skin tones but also provided them with the opportunity to relate themselves to the photographs; by doing so, participants could effectively pick out the skin tone that best suited them. Written SOC scales are limited to mere descriptions and thus make it more difficult for participants to accurately identify the tone that best fits them. Kundu et al17 used photographs to teach skin self-examination and ABCDEs for detection of melanoma. Additionally, both studies used photographs to demonstrate examples of skin cancer.17,18 Recent evidence suggests the use of visuals can be efficacious for improving skin cancer knowledge and awareness; a study in 16 SOC kidney transplant recipients found that the addition of photographs of squamous cell carcinoma in various skin tones to a sun-protection educational pamphlet was more effective than the original pamphlet without photographs.21
In contrast to the Robinson et al18 study and Hernandez et al19 study, the Kundu et al17 study showed photographs of acral lentiginous melanomas in white patients rather than SOC patients. However, SOC populations may be less likely to relate to or identify skin changes in skin types that are different from their own. This technique was still beneficial, as acral lentiginous melanoma is the most common type of melanoma in SOC populations. Another benefit of the study was that it was the only study reviewed that included a follow-up postintervention questionnaire. Such data is useful, as it demonstrates how muchinformation is retained by participants and may be more likely to predict compliance with skin cancer protective behaviors.17
The Hernandez et al19 study is unique in that it was the only one to include an educational intervention entirely in Spanish, which is important to consider, as language may be a hindrance to participants’ understanding in the other studies, particularly Hispanics, possibly leading to a lack of information retention regarding sun-protective behaviors. Furthermore, it also was the only study to utilize videos as a method for interventions. The 3-minute videos demonstrated that interventions could be efficient as compared to the 2-hour in-class intervention used by Robinson et al18 and the 20-minute intervention used by Kundu et al.17 Additionally, videos also could be more cost-effective, as incentives for large focus groups would no longer be needed. Furthermore, in the Hernandez et al19 study, there was minimal to no disruption in the participants’ daily routine, as the participants were getting cosmetic services while watching the videos, perhaps allowing them to be more attentive. In contrast, both the Robinson et al18 and Kundu et al17 studies required time out from the participants’ daily schedules. In addition, these studies were notably longer than the Hernandez et al19 study. The 8-hour intervention in the Chapman et al20 study also may not be feasible for the general population because of its excessive length. However, the intervention was successful among the adolescent participants, which suggested that shorter durations are effective in the adult population and longer interventions may be more appropriate for adolescents because they benefit from peer activity.
Despite the success of the educational interventions as outlined in the 4 studies described here, a major epidemiologic flaw is that these interventions included only a small percentage of the target population. The largest total number of adults surveyed and undergoing an intervention in any of the populations was only 120.17 By failing to reach a substantial proportion of the population at risk, the number of preventable deaths likely will not decrease. The authors believe a larger-scale intervention would provide meaningful change. Australia’s SunSmart campaign to increase skin cancer awareness in the Australian population is an example of one such large-scale national intervention. The campaign focused on massive television advertisements in the summer to educate participants about the dangers of skin cancer and the importance of protective behaviors. Telephone surveys conducted from 1987 to 2011 demonstrated that more exposure to the advertisements in the SunSmart campaign meant that individuals were more likely to use sunscreen and avoid sun exposure.22 In the United States, a similar intervention would be of great benefit in educating SOC populations regarding skin cancer risk. Additionally, dermatology residents need to be adequately trained to educate patients of color about the risk for skin cancer, as survey data indicated more than 80% of Australian dermatologists desired more SOC teaching during their training and 50% indicated that they would have time to learn it during their training if offered.23 Furthermore, one study suggested that future interventions must include primary-, secondary-, and tertiary-prevention methods to effectively reduce skin cancer risk among patients of color.24 Primary prevention involves sun avoidance, secondary prevention involves detecting cancerous lesions, and tertiary prevention involves undergoing treatment of skin malignancies. However, increased knowledge does not necessarily mean increased preventative action will be employed (eg, sunscreen use, wearing sun-protective clothing and sunglasses, avoiding tanning beds and excessive sun exposure). Additional studies that demonstrate a notable increase in sun-protective behaviors related to increased knowledge are needed.
Because retention of skin cancer knowledge decreased in several postintervention surveys, there also is a dire need for continuing skin cancer education in patients of color, which may be accomplished through a combination effort of television advertisement campaigns, pamphlets, social media, community health departments, or even community members. For example, a pilot program found that Hispanic lay health workers who are educated about skin cancer may serve as a bridge between medical providers and the Hispanic community by encouraging individuals in this population to get regular skin examinations from a physician.25 Overall, there are currently gaps in the understanding and treatment of skin cancer in people of color.26 Identifying the advantages and disadvantages of all relevant skin cancer interventions conducted in the SOC population will hopefully guide future studies to help close these gaps by allowing others to design the best possible intervention. By doing so, researchers can generate an intervention that is precise, well-informed, and effective in decreasing mortality rates from skin cancer among SOC populations.
Conclusion
All of the studies reviewed demonstrated that instructional and educational interventions are promising methods for improving either knowledge, awareness, or safe skin practices and sun-protective behaviors in SOC populations to differing degrees (Table). Although each of the 4 interventions employed their own methods, they all increased 1 or more of the 3 aforementioned concepts—knowledge, awareness, or safe skin practices and sun-protective behaviors—when comparing postsurvey to presurvey data. However, the critically important message derived from this research is that there is a tremendous need for a substantial large-scale educational intervention to increase knowledge regarding skin cancer in SOC populations.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Gloster HM Jr, Neal K. Skin cancer in skin of color. J Am Acad Dermatol. 2006;55:741-760.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
- Hu S, Parmet Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites, Hispanics, and blacks in Florida. Arch Dermatol. 2009;145:1369-1374.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5, suppl 1):S26-S37.
- Byrd-Miles K, Toombs EL, Peck GL. Skin cancer in individuals of African, Asian, Latin-American, and American-Indian descent: differences in incidence, clinical presentation, and survival compared to Caucasians. J Drugs Dermatol. 2007;6:10-16.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Hu S, Parker DF, Thomas AG, et al. Advanced presentation of melanoma in African Americans: the Miami-Dade County experience. J Am Acad Dermatol. 2004;5:1031-1032.
- Bellows CF, Belafsky P, Fortgang IS, et al. Melanoma in African-Americans: trends in biological behavior and clinical characteristics over two decades. J Surg Oncol. 2001;78:10-16.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- Shin S, Palis BE, Phillips JL, et al. Cutaneous melanoma in Asian-Americans. J Surg Oncol. 2009;99:114-118.
- Stubblefield J, Kelly B. Melanoma in non-caucasian populations. Surg Clin North Am. 2014;94:1115-1126.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Pichon LC, Corral I, Landrine H, et al. Perceived skin cancer risk and sunscreen use among African American adults. J Health Psychol. 2010;15:1181-1189.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Robinson JK, Joshi KM, Ortiz S, et al. Melanoma knowledge, perception, and awareness in ethnic minorities in Chicago: recommendations regarding education. Psychooncology. 2010;20:313-320.
- Hernandez C, Wang S, Abraham I, et al. Evaluation of educational videos to increase skin cancer risk awareness and sun safe behaviors among adult Hispanics. J Cancer Educ. 2014;29:563-569.
- Chapman LW, Ochoa A, Tenconi F, et al. Dermatologic health literacy in underserved communities: a case report of south Los Angeles middle schools. Dermatol Online J. 2015;21. pii:13030/qt8671p40n.
- Yanina G, Gaber R, Clayman ML, et al. Sun protection education for diverse audiences: need for skin cancer pictures. J Cancer Educ. 2015;30:187-189.
- Dobbinson SJ, Volkov A, Wakefield MA. Continued impact of sunsmart advertising on youth and adults’ behaviors. Am J Prev Med. 2015;49:20-28.
- Rodrigues MA, Ross AL, Gilmore S, et al. Australian dermatologists’ perspective on skin of colour: results of a national survey [published online December 9, 2016]. Australas J Dermatol. doi:10.1111/ajd.12556.
- Jacobsen A, Galvan A, Lachapelle CC, et al. Defining the need for skin cancer prevention education in uninsured, minority, and immigrant communities. JAMA Dermatol. 2016;152:1342-1347.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Kailas A, Solomon JA, Mostow EN, et al. Gaps in the understanding and treatment of skin cancer in people of color. J Am Acad Dermatol. 2016;74:144-149.
Malignant melanoma, basal cell carcinoma, and squamous cell carcinoma account for approximately 40% of all neoplasms among the white population in the United States. Skin cancer is the most common malignancy in the United States.1 However, despite this occurrence, there are limited data regarding skin cancer in individuals with skin of color (SOC). The 5-year survival rates for melanoma are 58.2% for black individuals, 69.7% for Hispanics, and 70.9% for Asians compared to 79.8% for white individuals in the United States.2 Even though SOC populations have lower incidences of skin cancer—melanoma, basal cell carcinoma, and squamous cell carcinoma—they exhibit higher death rates.3-7 Nonetheless, no specific guidelines exist to address sun exposure and safety habits in SOC populations.6,8 Furthermore, current demographics suggest that by the year 2050, approximately half of the US population will be nonwhite.4 Paradoxically, despite having increased sun protection from greater amounts of melanin in their skin, black individuals are more likely to present with advanced-stage melanoma (eg, stage III/IV) compared to white individuals.8-12 Furthermore, those of nonwhite populations are more likely to present with more advanced stages of acral lentiginous melanomas than white individuals.13,14 Hispanics also face an increasing incidence of more invasive acral lentiginous melanomas.15 Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.1
Although skin cancer is largely a preventable condition, the literature suggests that lack of awareness of melanoma among ethnic minorities is one of the main reasons for their poor skin cancer prognosis.16 This lack of awareness decreases the likelihood that an SOC patient would be alert to early detection of cancerous changes.17 Because educating at-risk SOC populations is key to decreasing skin cancer risk, this study focused on determining the efficacy of major knowledge-based interventions conducted to date.1 Overall, we sought to answer the question, do knowledge-based interventions increase skin cancer awareness, knowledge, and protective behavior among people of color?
Methods
For this review, the Cochrane method of analysis was used to conduct a thorough search of PubMed articles indexed for MEDLINE (1994-2016), as well as a search of CINAHL (1997-2016), PsycINFO (1999-2016), and Web of Science (1965-2016), using a combination of more than 100 search terms including but not limited to skin cancer, skin of color, intervention, and ethnic skin. The search yielded a total of 52 articles (Figure). Following review, only 8 articles met inclusion criteria, which were as follows: (1) study was related to skin cancer in SOC patients, which included an intervention to increase skin cancer awareness and knowledge; (2) study included adult participants or adolescents aged 12 to 18 years; (3) study was written in English; and (4) study was published in a peer-reviewed journal. Of the remaining 8 articles, 4 were excluded due to the following criteria: (1) study failed to provide both preintervention and postintervention data, (2) study failed to provide quantitative data, and (3) study included participants who worked as health care professionals or ancillary staff. As a result, a total of 4 articles were analyzed and discussed in this review (Table).
Results
Robinson et al18 conducted 12 focus groups with 120 total participants (40 black, 40 Asian, and 40 Hispanic patients). Participants engaged in a 2-hour tape-recorded focus group with a moderator guide on melanoma and skin cancer. Furthermore, they also were asked to assess skin cancer risk in 5 celebrities with different skin tones. The statistically significant preintervention results of the study (χ2=4.6, P<.001) were as follows: only 2%, 4%, and 14% correctly reported that celebrities with a very fair skin type, a fair skin type, and very dark skin type, respectively, could get sunburn, compared to 75%, 76%, and 62% post-intervention. Additionally, prior to intervention, 14% of the study population believed that dark brown skin type could get sunburn compared to 62% of the same group postintervention. This study demonstrated that the intervention helped SOC patients better identify their ability to get sunburn and identify their skin cancer risk.18
Hernandez et al19 used a video-based intervention in a Hispanic community, which was in contrast to the multiracial focus group intervention conducted by Robinson et al.18 Eighty Hispanic individuals were recruited from beauty salons to participate in the study. Participants watched two 3-minute videos in Spanish and completed a preintervention and postintervention survey. The first video emphasized the photoaging benefits of sun protection, while the second focused on skin cancer prevention. Preintervention surveys indicated that only 54 (68%) participants believed that fair-skinned Hispanics were at risk for skin cancer, which improved to 72 (90%) participants postintervention. Furthermore, initially only 44 (55%) participants thought those with darker skin types could develop skin cancer, but this number increased to 69 (86%) postintervention. For both questions regarding fair and dark skin, the agreement proportion was significantly different between the preeducation and posteducation videos (P<.0002 for the fair skin question and P<.0001 for the dark skin question). This study greatly increased awareness of skin cancer risk among Hispanics,19 similar to the Robinson et al18 study.
In contrast to 2-hour focus groups or 3-minute video–based interventions, a study by Kundu et al17 employed a 20-minute educational class-based intervention with both verbal and visual instruction. This study assessed the efficacy of an educational tutorial on improving awareness and early detection of melanoma in SOC individuals. Photographs were used to help participants recognize the ABCDEs of melanoma and to show examples of acral lentiginous melanomas in white individuals. A total of 71 participants completed a preintervention questionnaire, participated in a 20-minute class, and completed a postintervention questionnaire immediately after and 3 months following the class. The study population included 44 black, 15 Asian, 10 Hispanic, and 2 multiethnic participants. Knowledge that melanoma is a skin cancer increased from 83.9% to 100% immediately postintervention (P=.0001) and 97.2% at 3 months postintervention (P=.0075). Additionally, knowledge that people of color are at risk for melanoma increased from 48.4% preintervention to 82.8% immediately postintervention (P<.0001). However, only 40.8% of participants retained this knowledge at 3 months postintervention. Because only 1 participant reported a family history of skin cancer, the authors hypothesized that the reason for this loss of knowledge was that most participants were not personally affected by friends or family members with melanoma. A future study with an appropriate control group would be needed to support this claim. This study shed light on the potential of class-based interventions to increase both awareness and knowledge of skin cancer in SOC populations.17
A study by Chapman et al20 examined the effects of a sun protection educational program on increasing awareness of skin cancer in Hispanic and black middle school students in southern Los Angeles, California. It was the only study we reviewed that focused primarily on adolescents. Furthermore, it included the largest sample size (N=148) analyzed here. Students were given a preintervention questionnaire to evaluate their awareness of skin cancer and current sun-protection practices. Based on these results, the investigators devised a set of learning goals and incorporated them into an educational pamphlet. The intervention, called “Skin Teaching Day,” was a 1-day program discussing skin cancer and the importance of sun protection. Prior to the intervention, 68% of participants reported that they used sunscreen. Three months after completing the program, 80% of participants reported sunscreen use, an increase of 12% prior to the intervention. The results of this study demonstrated the unique effectiveness and potential of pamphlets in increasing sunscreen use.20
Comment
Overall, various methods of interventions such as focus groups, videos, pamphlets, and lectures improved knowledge of skin cancer risk and sun-protection behaviors in SOC populations. Furthermore, the unique differences of each study provided important insights into the successful design of an intervention.
An important characteristic of the Robinson et al18 study was the addition of photographs, which allowed participants not only to visualize different skin tones but also provided them with the opportunity to relate themselves to the photographs; by doing so, participants could effectively pick out the skin tone that best suited them. Written SOC scales are limited to mere descriptions and thus make it more difficult for participants to accurately identify the tone that best fits them. Kundu et al17 used photographs to teach skin self-examination and ABCDEs for detection of melanoma. Additionally, both studies used photographs to demonstrate examples of skin cancer.17,18 Recent evidence suggests the use of visuals can be efficacious for improving skin cancer knowledge and awareness; a study in 16 SOC kidney transplant recipients found that the addition of photographs of squamous cell carcinoma in various skin tones to a sun-protection educational pamphlet was more effective than the original pamphlet without photographs.21
In contrast to the Robinson et al18 study and Hernandez et al19 study, the Kundu et al17 study showed photographs of acral lentiginous melanomas in white patients rather than SOC patients. However, SOC populations may be less likely to relate to or identify skin changes in skin types that are different from their own. This technique was still beneficial, as acral lentiginous melanoma is the most common type of melanoma in SOC populations. Another benefit of the study was that it was the only study reviewed that included a follow-up postintervention questionnaire. Such data is useful, as it demonstrates how muchinformation is retained by participants and may be more likely to predict compliance with skin cancer protective behaviors.17
The Hernandez et al19 study is unique in that it was the only one to include an educational intervention entirely in Spanish, which is important to consider, as language may be a hindrance to participants’ understanding in the other studies, particularly Hispanics, possibly leading to a lack of information retention regarding sun-protective behaviors. Furthermore, it also was the only study to utilize videos as a method for interventions. The 3-minute videos demonstrated that interventions could be efficient as compared to the 2-hour in-class intervention used by Robinson et al18 and the 20-minute intervention used by Kundu et al.17 Additionally, videos also could be more cost-effective, as incentives for large focus groups would no longer be needed. Furthermore, in the Hernandez et al19 study, there was minimal to no disruption in the participants’ daily routine, as the participants were getting cosmetic services while watching the videos, perhaps allowing them to be more attentive. In contrast, both the Robinson et al18 and Kundu et al17 studies required time out from the participants’ daily schedules. In addition, these studies were notably longer than the Hernandez et al19 study. The 8-hour intervention in the Chapman et al20 study also may not be feasible for the general population because of its excessive length. However, the intervention was successful among the adolescent participants, which suggested that shorter durations are effective in the adult population and longer interventions may be more appropriate for adolescents because they benefit from peer activity.
Despite the success of the educational interventions as outlined in the 4 studies described here, a major epidemiologic flaw is that these interventions included only a small percentage of the target population. The largest total number of adults surveyed and undergoing an intervention in any of the populations was only 120.17 By failing to reach a substantial proportion of the population at risk, the number of preventable deaths likely will not decrease. The authors believe a larger-scale intervention would provide meaningful change. Australia’s SunSmart campaign to increase skin cancer awareness in the Australian population is an example of one such large-scale national intervention. The campaign focused on massive television advertisements in the summer to educate participants about the dangers of skin cancer and the importance of protective behaviors. Telephone surveys conducted from 1987 to 2011 demonstrated that more exposure to the advertisements in the SunSmart campaign meant that individuals were more likely to use sunscreen and avoid sun exposure.22 In the United States, a similar intervention would be of great benefit in educating SOC populations regarding skin cancer risk. Additionally, dermatology residents need to be adequately trained to educate patients of color about the risk for skin cancer, as survey data indicated more than 80% of Australian dermatologists desired more SOC teaching during their training and 50% indicated that they would have time to learn it during their training if offered.23 Furthermore, one study suggested that future interventions must include primary-, secondary-, and tertiary-prevention methods to effectively reduce skin cancer risk among patients of color.24 Primary prevention involves sun avoidance, secondary prevention involves detecting cancerous lesions, and tertiary prevention involves undergoing treatment of skin malignancies. However, increased knowledge does not necessarily mean increased preventative action will be employed (eg, sunscreen use, wearing sun-protective clothing and sunglasses, avoiding tanning beds and excessive sun exposure). Additional studies that demonstrate a notable increase in sun-protective behaviors related to increased knowledge are needed.
Because retention of skin cancer knowledge decreased in several postintervention surveys, there also is a dire need for continuing skin cancer education in patients of color, which may be accomplished through a combination effort of television advertisement campaigns, pamphlets, social media, community health departments, or even community members. For example, a pilot program found that Hispanic lay health workers who are educated about skin cancer may serve as a bridge between medical providers and the Hispanic community by encouraging individuals in this population to get regular skin examinations from a physician.25 Overall, there are currently gaps in the understanding and treatment of skin cancer in people of color.26 Identifying the advantages and disadvantages of all relevant skin cancer interventions conducted in the SOC population will hopefully guide future studies to help close these gaps by allowing others to design the best possible intervention. By doing so, researchers can generate an intervention that is precise, well-informed, and effective in decreasing mortality rates from skin cancer among SOC populations.
Conclusion
All of the studies reviewed demonstrated that instructional and educational interventions are promising methods for improving either knowledge, awareness, or safe skin practices and sun-protective behaviors in SOC populations to differing degrees (Table). Although each of the 4 interventions employed their own methods, they all increased 1 or more of the 3 aforementioned concepts—knowledge, awareness, or safe skin practices and sun-protective behaviors—when comparing postsurvey to presurvey data. However, the critically important message derived from this research is that there is a tremendous need for a substantial large-scale educational intervention to increase knowledge regarding skin cancer in SOC populations.
Malignant melanoma, basal cell carcinoma, and squamous cell carcinoma account for approximately 40% of all neoplasms among the white population in the United States. Skin cancer is the most common malignancy in the United States.1 However, despite this occurrence, there are limited data regarding skin cancer in individuals with skin of color (SOC). The 5-year survival rates for melanoma are 58.2% for black individuals, 69.7% for Hispanics, and 70.9% for Asians compared to 79.8% for white individuals in the United States.2 Even though SOC populations have lower incidences of skin cancer—melanoma, basal cell carcinoma, and squamous cell carcinoma—they exhibit higher death rates.3-7 Nonetheless, no specific guidelines exist to address sun exposure and safety habits in SOC populations.6,8 Furthermore, current demographics suggest that by the year 2050, approximately half of the US population will be nonwhite.4 Paradoxically, despite having increased sun protection from greater amounts of melanin in their skin, black individuals are more likely to present with advanced-stage melanoma (eg, stage III/IV) compared to white individuals.8-12 Furthermore, those of nonwhite populations are more likely to present with more advanced stages of acral lentiginous melanomas than white individuals.13,14 Hispanics also face an increasing incidence of more invasive acral lentiginous melanomas.15 Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.1
Although skin cancer is largely a preventable condition, the literature suggests that lack of awareness of melanoma among ethnic minorities is one of the main reasons for their poor skin cancer prognosis.16 This lack of awareness decreases the likelihood that an SOC patient would be alert to early detection of cancerous changes.17 Because educating at-risk SOC populations is key to decreasing skin cancer risk, this study focused on determining the efficacy of major knowledge-based interventions conducted to date.1 Overall, we sought to answer the question, do knowledge-based interventions increase skin cancer awareness, knowledge, and protective behavior among people of color?
Methods
For this review, the Cochrane method of analysis was used to conduct a thorough search of PubMed articles indexed for MEDLINE (1994-2016), as well as a search of CINAHL (1997-2016), PsycINFO (1999-2016), and Web of Science (1965-2016), using a combination of more than 100 search terms including but not limited to skin cancer, skin of color, intervention, and ethnic skin. The search yielded a total of 52 articles (Figure). Following review, only 8 articles met inclusion criteria, which were as follows: (1) study was related to skin cancer in SOC patients, which included an intervention to increase skin cancer awareness and knowledge; (2) study included adult participants or adolescents aged 12 to 18 years; (3) study was written in English; and (4) study was published in a peer-reviewed journal. Of the remaining 8 articles, 4 were excluded due to the following criteria: (1) study failed to provide both preintervention and postintervention data, (2) study failed to provide quantitative data, and (3) study included participants who worked as health care professionals or ancillary staff. As a result, a total of 4 articles were analyzed and discussed in this review (Table).
Results
Robinson et al18 conducted 12 focus groups with 120 total participants (40 black, 40 Asian, and 40 Hispanic patients). Participants engaged in a 2-hour tape-recorded focus group with a moderator guide on melanoma and skin cancer. Furthermore, they also were asked to assess skin cancer risk in 5 celebrities with different skin tones. The statistically significant preintervention results of the study (χ2=4.6, P<.001) were as follows: only 2%, 4%, and 14% correctly reported that celebrities with a very fair skin type, a fair skin type, and very dark skin type, respectively, could get sunburn, compared to 75%, 76%, and 62% post-intervention. Additionally, prior to intervention, 14% of the study population believed that dark brown skin type could get sunburn compared to 62% of the same group postintervention. This study demonstrated that the intervention helped SOC patients better identify their ability to get sunburn and identify their skin cancer risk.18
Hernandez et al19 used a video-based intervention in a Hispanic community, which was in contrast to the multiracial focus group intervention conducted by Robinson et al.18 Eighty Hispanic individuals were recruited from beauty salons to participate in the study. Participants watched two 3-minute videos in Spanish and completed a preintervention and postintervention survey. The first video emphasized the photoaging benefits of sun protection, while the second focused on skin cancer prevention. Preintervention surveys indicated that only 54 (68%) participants believed that fair-skinned Hispanics were at risk for skin cancer, which improved to 72 (90%) participants postintervention. Furthermore, initially only 44 (55%) participants thought those with darker skin types could develop skin cancer, but this number increased to 69 (86%) postintervention. For both questions regarding fair and dark skin, the agreement proportion was significantly different between the preeducation and posteducation videos (P<.0002 for the fair skin question and P<.0001 for the dark skin question). This study greatly increased awareness of skin cancer risk among Hispanics,19 similar to the Robinson et al18 study.
In contrast to 2-hour focus groups or 3-minute video–based interventions, a study by Kundu et al17 employed a 20-minute educational class-based intervention with both verbal and visual instruction. This study assessed the efficacy of an educational tutorial on improving awareness and early detection of melanoma in SOC individuals. Photographs were used to help participants recognize the ABCDEs of melanoma and to show examples of acral lentiginous melanomas in white individuals. A total of 71 participants completed a preintervention questionnaire, participated in a 20-minute class, and completed a postintervention questionnaire immediately after and 3 months following the class. The study population included 44 black, 15 Asian, 10 Hispanic, and 2 multiethnic participants. Knowledge that melanoma is a skin cancer increased from 83.9% to 100% immediately postintervention (P=.0001) and 97.2% at 3 months postintervention (P=.0075). Additionally, knowledge that people of color are at risk for melanoma increased from 48.4% preintervention to 82.8% immediately postintervention (P<.0001). However, only 40.8% of participants retained this knowledge at 3 months postintervention. Because only 1 participant reported a family history of skin cancer, the authors hypothesized that the reason for this loss of knowledge was that most participants were not personally affected by friends or family members with melanoma. A future study with an appropriate control group would be needed to support this claim. This study shed light on the potential of class-based interventions to increase both awareness and knowledge of skin cancer in SOC populations.17
A study by Chapman et al20 examined the effects of a sun protection educational program on increasing awareness of skin cancer in Hispanic and black middle school students in southern Los Angeles, California. It was the only study we reviewed that focused primarily on adolescents. Furthermore, it included the largest sample size (N=148) analyzed here. Students were given a preintervention questionnaire to evaluate their awareness of skin cancer and current sun-protection practices. Based on these results, the investigators devised a set of learning goals and incorporated them into an educational pamphlet. The intervention, called “Skin Teaching Day,” was a 1-day program discussing skin cancer and the importance of sun protection. Prior to the intervention, 68% of participants reported that they used sunscreen. Three months after completing the program, 80% of participants reported sunscreen use, an increase of 12% prior to the intervention. The results of this study demonstrated the unique effectiveness and potential of pamphlets in increasing sunscreen use.20
Comment
Overall, various methods of interventions such as focus groups, videos, pamphlets, and lectures improved knowledge of skin cancer risk and sun-protection behaviors in SOC populations. Furthermore, the unique differences of each study provided important insights into the successful design of an intervention.
An important characteristic of the Robinson et al18 study was the addition of photographs, which allowed participants not only to visualize different skin tones but also provided them with the opportunity to relate themselves to the photographs; by doing so, participants could effectively pick out the skin tone that best suited them. Written SOC scales are limited to mere descriptions and thus make it more difficult for participants to accurately identify the tone that best fits them. Kundu et al17 used photographs to teach skin self-examination and ABCDEs for detection of melanoma. Additionally, both studies used photographs to demonstrate examples of skin cancer.17,18 Recent evidence suggests the use of visuals can be efficacious for improving skin cancer knowledge and awareness; a study in 16 SOC kidney transplant recipients found that the addition of photographs of squamous cell carcinoma in various skin tones to a sun-protection educational pamphlet was more effective than the original pamphlet without photographs.21
In contrast to the Robinson et al18 study and Hernandez et al19 study, the Kundu et al17 study showed photographs of acral lentiginous melanomas in white patients rather than SOC patients. However, SOC populations may be less likely to relate to or identify skin changes in skin types that are different from their own. This technique was still beneficial, as acral lentiginous melanoma is the most common type of melanoma in SOC populations. Another benefit of the study was that it was the only study reviewed that included a follow-up postintervention questionnaire. Such data is useful, as it demonstrates how muchinformation is retained by participants and may be more likely to predict compliance with skin cancer protective behaviors.17
The Hernandez et al19 study is unique in that it was the only one to include an educational intervention entirely in Spanish, which is important to consider, as language may be a hindrance to participants’ understanding in the other studies, particularly Hispanics, possibly leading to a lack of information retention regarding sun-protective behaviors. Furthermore, it also was the only study to utilize videos as a method for interventions. The 3-minute videos demonstrated that interventions could be efficient as compared to the 2-hour in-class intervention used by Robinson et al18 and the 20-minute intervention used by Kundu et al.17 Additionally, videos also could be more cost-effective, as incentives for large focus groups would no longer be needed. Furthermore, in the Hernandez et al19 study, there was minimal to no disruption in the participants’ daily routine, as the participants were getting cosmetic services while watching the videos, perhaps allowing them to be more attentive. In contrast, both the Robinson et al18 and Kundu et al17 studies required time out from the participants’ daily schedules. In addition, these studies were notably longer than the Hernandez et al19 study. The 8-hour intervention in the Chapman et al20 study also may not be feasible for the general population because of its excessive length. However, the intervention was successful among the adolescent participants, which suggested that shorter durations are effective in the adult population and longer interventions may be more appropriate for adolescents because they benefit from peer activity.
Despite the success of the educational interventions as outlined in the 4 studies described here, a major epidemiologic flaw is that these interventions included only a small percentage of the target population. The largest total number of adults surveyed and undergoing an intervention in any of the populations was only 120.17 By failing to reach a substantial proportion of the population at risk, the number of preventable deaths likely will not decrease. The authors believe a larger-scale intervention would provide meaningful change. Australia’s SunSmart campaign to increase skin cancer awareness in the Australian population is an example of one such large-scale national intervention. The campaign focused on massive television advertisements in the summer to educate participants about the dangers of skin cancer and the importance of protective behaviors. Telephone surveys conducted from 1987 to 2011 demonstrated that more exposure to the advertisements in the SunSmart campaign meant that individuals were more likely to use sunscreen and avoid sun exposure.22 In the United States, a similar intervention would be of great benefit in educating SOC populations regarding skin cancer risk. Additionally, dermatology residents need to be adequately trained to educate patients of color about the risk for skin cancer, as survey data indicated more than 80% of Australian dermatologists desired more SOC teaching during their training and 50% indicated that they would have time to learn it during their training if offered.23 Furthermore, one study suggested that future interventions must include primary-, secondary-, and tertiary-prevention methods to effectively reduce skin cancer risk among patients of color.24 Primary prevention involves sun avoidance, secondary prevention involves detecting cancerous lesions, and tertiary prevention involves undergoing treatment of skin malignancies. However, increased knowledge does not necessarily mean increased preventative action will be employed (eg, sunscreen use, wearing sun-protective clothing and sunglasses, avoiding tanning beds and excessive sun exposure). Additional studies that demonstrate a notable increase in sun-protective behaviors related to increased knowledge are needed.
Because retention of skin cancer knowledge decreased in several postintervention surveys, there also is a dire need for continuing skin cancer education in patients of color, which may be accomplished through a combination effort of television advertisement campaigns, pamphlets, social media, community health departments, or even community members. For example, a pilot program found that Hispanic lay health workers who are educated about skin cancer may serve as a bridge between medical providers and the Hispanic community by encouraging individuals in this population to get regular skin examinations from a physician.25 Overall, there are currently gaps in the understanding and treatment of skin cancer in people of color.26 Identifying the advantages and disadvantages of all relevant skin cancer interventions conducted in the SOC population will hopefully guide future studies to help close these gaps by allowing others to design the best possible intervention. By doing so, researchers can generate an intervention that is precise, well-informed, and effective in decreasing mortality rates from skin cancer among SOC populations.
Conclusion
All of the studies reviewed demonstrated that instructional and educational interventions are promising methods for improving either knowledge, awareness, or safe skin practices and sun-protective behaviors in SOC populations to differing degrees (Table). Although each of the 4 interventions employed their own methods, they all increased 1 or more of the 3 aforementioned concepts—knowledge, awareness, or safe skin practices and sun-protective behaviors—when comparing postsurvey to presurvey data. However, the critically important message derived from this research is that there is a tremendous need for a substantial large-scale educational intervention to increase knowledge regarding skin cancer in SOC populations.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Gloster HM Jr, Neal K. Skin cancer in skin of color. J Am Acad Dermatol. 2006;55:741-760.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
- Hu S, Parmet Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites, Hispanics, and blacks in Florida. Arch Dermatol. 2009;145:1369-1374.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5, suppl 1):S26-S37.
- Byrd-Miles K, Toombs EL, Peck GL. Skin cancer in individuals of African, Asian, Latin-American, and American-Indian descent: differences in incidence, clinical presentation, and survival compared to Caucasians. J Drugs Dermatol. 2007;6:10-16.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Hu S, Parker DF, Thomas AG, et al. Advanced presentation of melanoma in African Americans: the Miami-Dade County experience. J Am Acad Dermatol. 2004;5:1031-1032.
- Bellows CF, Belafsky P, Fortgang IS, et al. Melanoma in African-Americans: trends in biological behavior and clinical characteristics over two decades. J Surg Oncol. 2001;78:10-16.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- Shin S, Palis BE, Phillips JL, et al. Cutaneous melanoma in Asian-Americans. J Surg Oncol. 2009;99:114-118.
- Stubblefield J, Kelly B. Melanoma in non-caucasian populations. Surg Clin North Am. 2014;94:1115-1126.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Pichon LC, Corral I, Landrine H, et al. Perceived skin cancer risk and sunscreen use among African American adults. J Health Psychol. 2010;15:1181-1189.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Robinson JK, Joshi KM, Ortiz S, et al. Melanoma knowledge, perception, and awareness in ethnic minorities in Chicago: recommendations regarding education. Psychooncology. 2010;20:313-320.
- Hernandez C, Wang S, Abraham I, et al. Evaluation of educational videos to increase skin cancer risk awareness and sun safe behaviors among adult Hispanics. J Cancer Educ. 2014;29:563-569.
- Chapman LW, Ochoa A, Tenconi F, et al. Dermatologic health literacy in underserved communities: a case report of south Los Angeles middle schools. Dermatol Online J. 2015;21. pii:13030/qt8671p40n.
- Yanina G, Gaber R, Clayman ML, et al. Sun protection education for diverse audiences: need for skin cancer pictures. J Cancer Educ. 2015;30:187-189.
- Dobbinson SJ, Volkov A, Wakefield MA. Continued impact of sunsmart advertising on youth and adults’ behaviors. Am J Prev Med. 2015;49:20-28.
- Rodrigues MA, Ross AL, Gilmore S, et al. Australian dermatologists’ perspective on skin of colour: results of a national survey [published online December 9, 2016]. Australas J Dermatol. doi:10.1111/ajd.12556.
- Jacobsen A, Galvan A, Lachapelle CC, et al. Defining the need for skin cancer prevention education in uninsured, minority, and immigrant communities. JAMA Dermatol. 2016;152:1342-1347.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Kailas A, Solomon JA, Mostow EN, et al. Gaps in the understanding and treatment of skin cancer in people of color. J Am Acad Dermatol. 2016;74:144-149.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Gloster HM Jr, Neal K. Skin cancer in skin of color. J Am Acad Dermatol. 2006;55:741-760.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
- Hu S, Parmet Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites, Hispanics, and blacks in Florida. Arch Dermatol. 2009;145:1369-1374.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5, suppl 1):S26-S37.
- Byrd-Miles K, Toombs EL, Peck GL. Skin cancer in individuals of African, Asian, Latin-American, and American-Indian descent: differences in incidence, clinical presentation, and survival compared to Caucasians. J Drugs Dermatol. 2007;6:10-16.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Hu S, Parker DF, Thomas AG, et al. Advanced presentation of melanoma in African Americans: the Miami-Dade County experience. J Am Acad Dermatol. 2004;5:1031-1032.
- Bellows CF, Belafsky P, Fortgang IS, et al. Melanoma in African-Americans: trends in biological behavior and clinical characteristics over two decades. J Surg Oncol. 2001;78:10-16.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- Shin S, Palis BE, Phillips JL, et al. Cutaneous melanoma in Asian-Americans. J Surg Oncol. 2009;99:114-118.
- Stubblefield J, Kelly B. Melanoma in non-caucasian populations. Surg Clin North Am. 2014;94:1115-1126.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Pichon LC, Corral I, Landrine H, et al. Perceived skin cancer risk and sunscreen use among African American adults. J Health Psychol. 2010;15:1181-1189.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Robinson JK, Joshi KM, Ortiz S, et al. Melanoma knowledge, perception, and awareness in ethnic minorities in Chicago: recommendations regarding education. Psychooncology. 2010;20:313-320.
- Hernandez C, Wang S, Abraham I, et al. Evaluation of educational videos to increase skin cancer risk awareness and sun safe behaviors among adult Hispanics. J Cancer Educ. 2014;29:563-569.
- Chapman LW, Ochoa A, Tenconi F, et al. Dermatologic health literacy in underserved communities: a case report of south Los Angeles middle schools. Dermatol Online J. 2015;21. pii:13030/qt8671p40n.
- Yanina G, Gaber R, Clayman ML, et al. Sun protection education for diverse audiences: need for skin cancer pictures. J Cancer Educ. 2015;30:187-189.
- Dobbinson SJ, Volkov A, Wakefield MA. Continued impact of sunsmart advertising on youth and adults’ behaviors. Am J Prev Med. 2015;49:20-28.
- Rodrigues MA, Ross AL, Gilmore S, et al. Australian dermatologists’ perspective on skin of colour: results of a national survey [published online December 9, 2016]. Australas J Dermatol. doi:10.1111/ajd.12556.
- Jacobsen A, Galvan A, Lachapelle CC, et al. Defining the need for skin cancer prevention education in uninsured, minority, and immigrant communities. JAMA Dermatol. 2016;152:1342-1347.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Kailas A, Solomon JA, Mostow EN, et al. Gaps in the understanding and treatment of skin cancer in people of color. J Am Acad Dermatol. 2016;74:144-149.
Practice Points
- Patients of color should be informed that they are at risk for skin cancer including melanoma.
- Patients of color should be taught to identify suspicious skin lesions including the ABCDEs of melanoma.
- Patients of color should be instructed to perform self-body skin examinations, especially of the palms and soles, for any evolving skin lesions. Patients should be instructed on the importance of visiting a physician for an evolving or suspicious mole or lesion.
Do Infants Fed Rice and Rice Products Have an Increased Risk for Skin Cancer?
To the Editor:
Rice and rice products, such as rice cereal and rice snacks, contain inorganic arsenic. Exposure to arsenicin utero and during early life may be associated with adverse fetal growth, adverse infant and child immune response, and adverse neurodevelopmental outcomes. Therefore, the World Health Organization, the Food and Agriculture Organization of the United Nations, the European Union, and the US Food and Drug Administration have suggested maximum arsenic ingestion recommendations for infants: 100 ng/g for inorganic arsenic in products geared toward infants. However, infants consuming only a few servings of rice products may exceed the weekly tolerable intake of arsenic.
Karagas et al1 obtained dietary data on 759 infants who were enrolled in the New Hampshire Birth Cohort Study from 2011 to 2014. They noted that 80% of the infants had been introduced to rice cereal during the first year. Additional data on diet and total urinary arsenic at 12 months was available for 129 infants: 32.6% of these infants were fed rice snacks. In addition, the total urinary arsenic concentration was higher among infants who ate rice cereal or rice snacks as compared to infants who did not eat rice or rice products.
Chronic arsenic exposure can result in patchy dark brown hyperpigmentation with scattered pale spots referred to as “raindrops on a dusty road.” The axilla, eyelids, groin, neck, nipples, and temples often are affected. However, the hyperpigmentation can extend across the chest, abdomen, and back in severe cases.
Horizontal white lines across the nails (Mees lines) may develop. Keratoses, often on the palms (arsenic keratoses), may appear; they persist and may progress to skin cancers. In addition, patients with arsenic exposure are more susceptible to developing nonmelanoma skin cancers.2
It is unknown if exposure to inorganic arsenic in infancy predisposes these individuals to skin cancer when they become adults. Long-term longitudinal follow-up of the participants in this study may provide additional insight. Perhaps infants should not receive rice cereals and rice snacks or their parents should more carefully monitor the amount of rice and rice products that they ingest.
- Karagas MR, Punshon T, Sayarath V, et al. Association of rice and rice-product consumption with arsenic exposure early in life. JAMA Pediatr. 2016;170:609-616.
- Mayer JE, Goldman RH. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. Int J Dermatol. 2016;55;e585-e591.
To the Editor:
Rice and rice products, such as rice cereal and rice snacks, contain inorganic arsenic. Exposure to arsenicin utero and during early life may be associated with adverse fetal growth, adverse infant and child immune response, and adverse neurodevelopmental outcomes. Therefore, the World Health Organization, the Food and Agriculture Organization of the United Nations, the European Union, and the US Food and Drug Administration have suggested maximum arsenic ingestion recommendations for infants: 100 ng/g for inorganic arsenic in products geared toward infants. However, infants consuming only a few servings of rice products may exceed the weekly tolerable intake of arsenic.
Karagas et al1 obtained dietary data on 759 infants who were enrolled in the New Hampshire Birth Cohort Study from 2011 to 2014. They noted that 80% of the infants had been introduced to rice cereal during the first year. Additional data on diet and total urinary arsenic at 12 months was available for 129 infants: 32.6% of these infants were fed rice snacks. In addition, the total urinary arsenic concentration was higher among infants who ate rice cereal or rice snacks as compared to infants who did not eat rice or rice products.
Chronic arsenic exposure can result in patchy dark brown hyperpigmentation with scattered pale spots referred to as “raindrops on a dusty road.” The axilla, eyelids, groin, neck, nipples, and temples often are affected. However, the hyperpigmentation can extend across the chest, abdomen, and back in severe cases.
Horizontal white lines across the nails (Mees lines) may develop. Keratoses, often on the palms (arsenic keratoses), may appear; they persist and may progress to skin cancers. In addition, patients with arsenic exposure are more susceptible to developing nonmelanoma skin cancers.2
It is unknown if exposure to inorganic arsenic in infancy predisposes these individuals to skin cancer when they become adults. Long-term longitudinal follow-up of the participants in this study may provide additional insight. Perhaps infants should not receive rice cereals and rice snacks or their parents should more carefully monitor the amount of rice and rice products that they ingest.
To the Editor:
Rice and rice products, such as rice cereal and rice snacks, contain inorganic arsenic. Exposure to arsenicin utero and during early life may be associated with adverse fetal growth, adverse infant and child immune response, and adverse neurodevelopmental outcomes. Therefore, the World Health Organization, the Food and Agriculture Organization of the United Nations, the European Union, and the US Food and Drug Administration have suggested maximum arsenic ingestion recommendations for infants: 100 ng/g for inorganic arsenic in products geared toward infants. However, infants consuming only a few servings of rice products may exceed the weekly tolerable intake of arsenic.
Karagas et al1 obtained dietary data on 759 infants who were enrolled in the New Hampshire Birth Cohort Study from 2011 to 2014. They noted that 80% of the infants had been introduced to rice cereal during the first year. Additional data on diet and total urinary arsenic at 12 months was available for 129 infants: 32.6% of these infants were fed rice snacks. In addition, the total urinary arsenic concentration was higher among infants who ate rice cereal or rice snacks as compared to infants who did not eat rice or rice products.
Chronic arsenic exposure can result in patchy dark brown hyperpigmentation with scattered pale spots referred to as “raindrops on a dusty road.” The axilla, eyelids, groin, neck, nipples, and temples often are affected. However, the hyperpigmentation can extend across the chest, abdomen, and back in severe cases.
Horizontal white lines across the nails (Mees lines) may develop. Keratoses, often on the palms (arsenic keratoses), may appear; they persist and may progress to skin cancers. In addition, patients with arsenic exposure are more susceptible to developing nonmelanoma skin cancers.2
It is unknown if exposure to inorganic arsenic in infancy predisposes these individuals to skin cancer when they become adults. Long-term longitudinal follow-up of the participants in this study may provide additional insight. Perhaps infants should not receive rice cereals and rice snacks or their parents should more carefully monitor the amount of rice and rice products that they ingest.
- Karagas MR, Punshon T, Sayarath V, et al. Association of rice and rice-product consumption with arsenic exposure early in life. JAMA Pediatr. 2016;170:609-616.
- Mayer JE, Goldman RH. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. Int J Dermatol. 2016;55;e585-e591.
- Karagas MR, Punshon T, Sayarath V, et al. Association of rice and rice-product consumption with arsenic exposure early in life. JAMA Pediatr. 2016;170:609-616.
- Mayer JE, Goldman RH. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. Int J Dermatol. 2016;55;e585-e591.
