User login
The Diagnosis: Lymphomatoid Papulosis
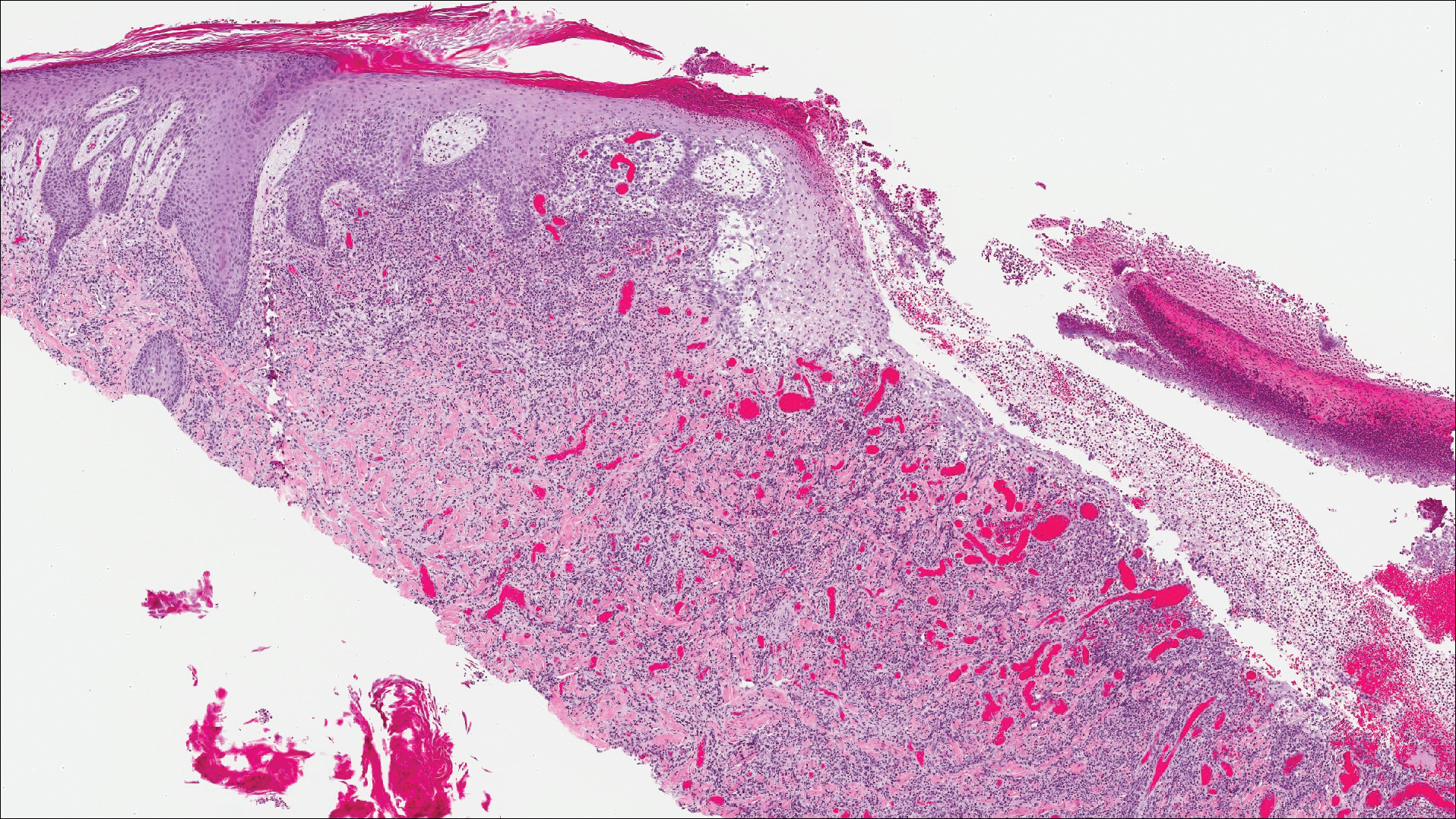
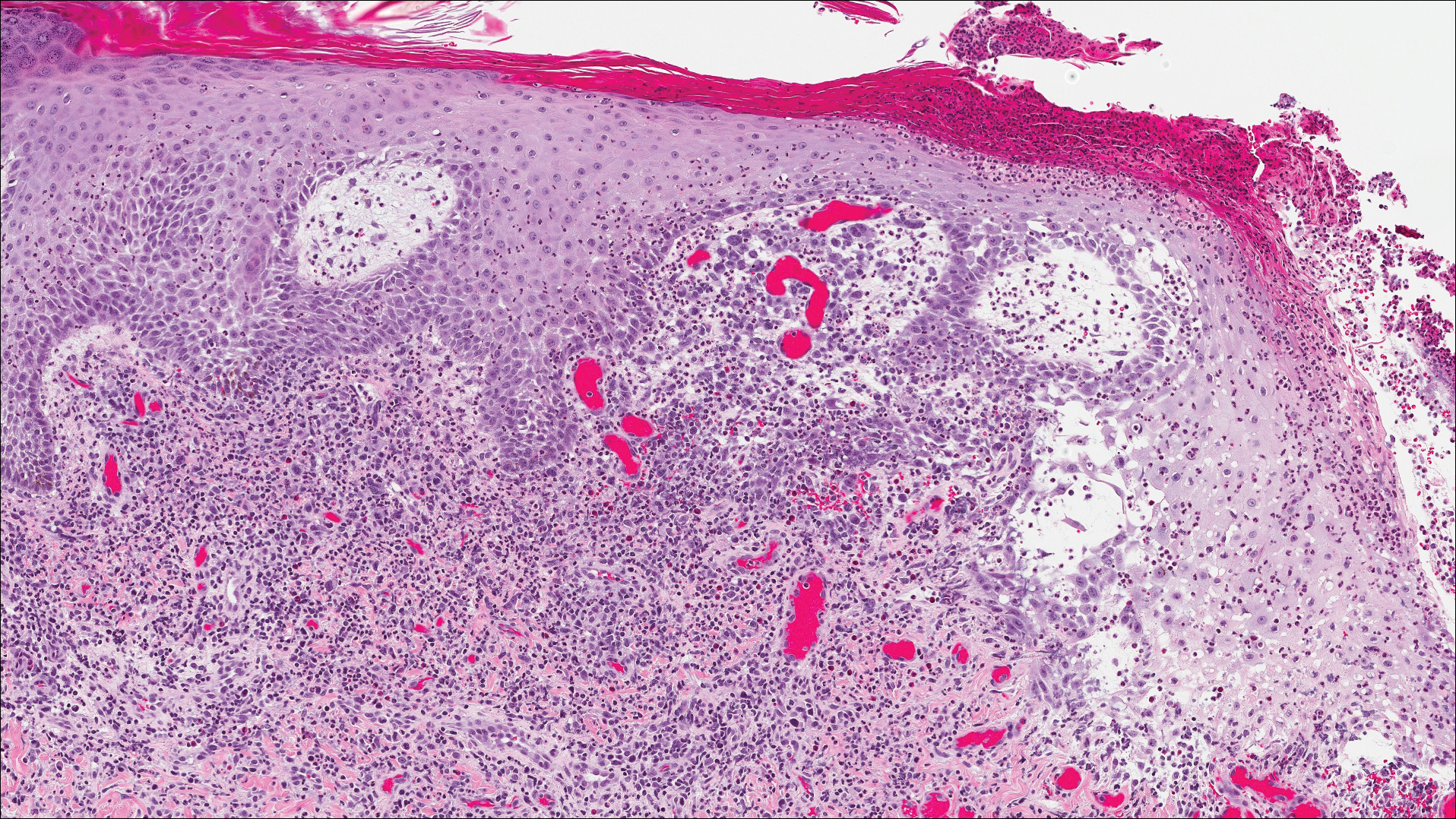
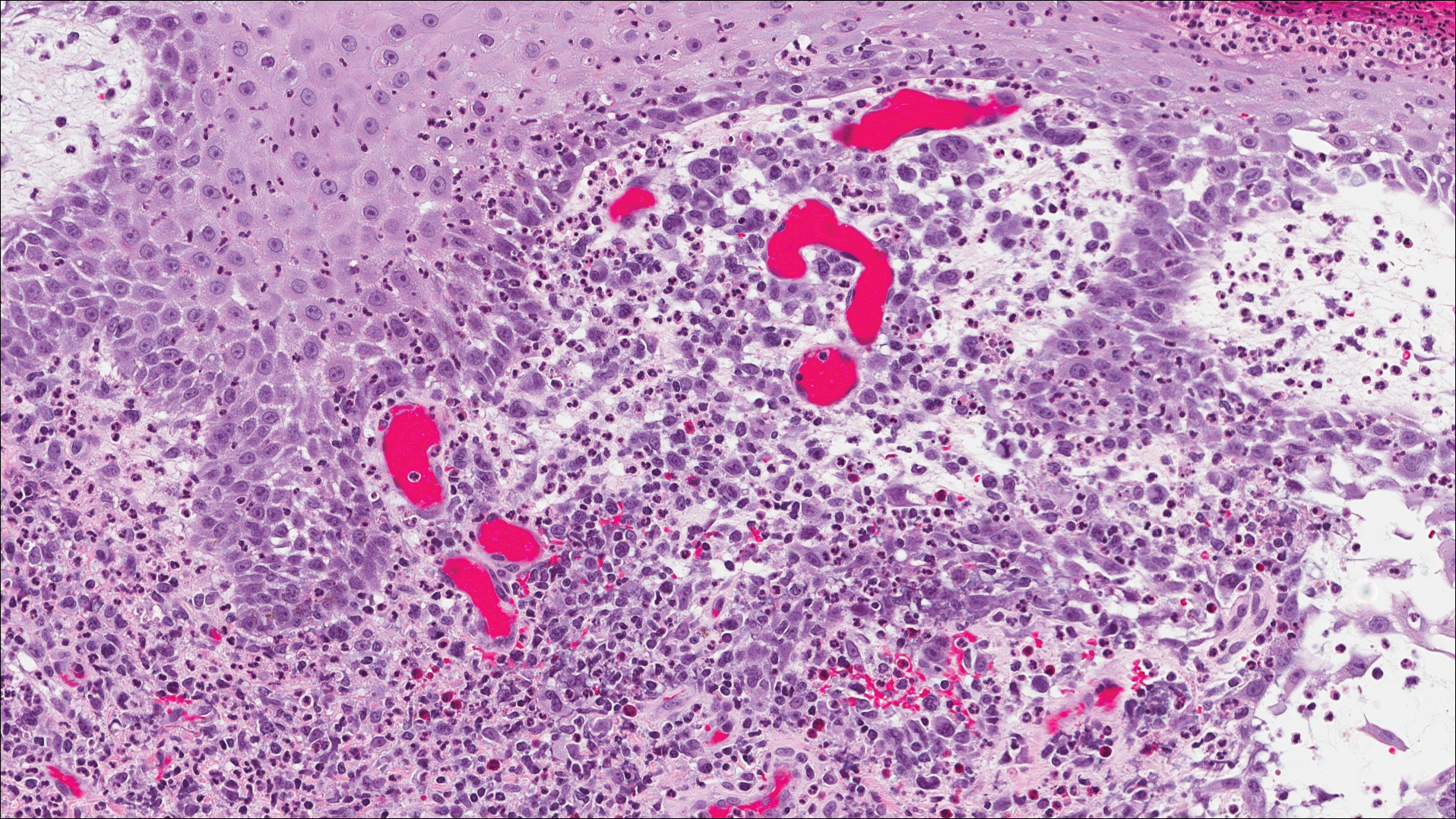
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).

Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
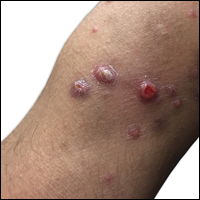
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
The Diagnosis: Lymphomatoid Papulosis
A shave biopsy of an established lesion on the volar aspect of the left wrist was performed (Figure 1). The biopsy showed an ulcerated nodular lesion characterized by a dense mixed inflammatory cell infiltrate in the dermis composed of lymphocytes, histiocytes, scattered neutrophils, and numerous eosinophils (Figure 2). Notably there was a minor population of large atypical cells with immunoblastic and anaplastic morphology present individually and in small clusters most prominently within the upper dermis (Figures 3 and 4). Immunohistochemistry of the anaplastic cells revealed a CD30+, CD3−, CD4+, CD5−, CD8−, CD2−, CD7−, CD56−, ALK1− (anaplastic lymphoma kinase-1), PAX5− (paired box protein-5), CD20−, and CD15− phenotype. These morphologic and immunohistochemical features suggested a CD30+ cutaneous lymphoproliferative disorder. The clinical history of recurrent self-healing papulonodules in an otherwise-healthy patient established the diagnosis of lymphomatoid papulosis (LyP).
Lymphomatoid papulosis is a lymphoproliferative disorder characterized by recurrent crops of self-resolving eruptive papulonodular skin lesions that may show a variety of histologic features including a CD30+ malignant T-cell lymphoma.1 Lymphomatoid papulosis was first described in 19681 but debate continues whether the condition should be considered malignant or benign.2 Although the prognosis is excellent, LyP is characterized by a protracted course, often lasting many years. Additionally, these patients have a lifelong increased risk for development of a second cutaneous or systemic lymphoma such as mycosis fungoides (MF), cutaneous or nodal anaplastic large cell lymphoma (ALCL), or Hodgkin lymphoma, among others.
Lymphomatoid papulosis is a rare disease occurring in all ethnic groups and at any age, though most commonly presenting in the fifth decade of life. Finding large atypical T cells expressing CD30 in recurring skin lesions is highly suggestive of LyP; however, large CD30+ cells also can be seen in numerous benign reactive processes such as arthropod assault, drug eruption, viral skin infections, and other dermatoses, thus clinical correlation is always paramount. The cause of LyP is largely unknown; however, spontaneous regression may be explained by CD30-CD30 ligand interaction3 as well as an increased proapoptotic milieu.4 Specific translocations such as interferon regulatory factor-4 have been hypothesized as a risk factor for malignant progression.5-7 Additionally, an inactivating gene mutation resulting in loss of transforming growth factor β1 receptor expression and subsequent unresponsiveness to the growth inhibitory effect of transforming growth factor β may play a role in progression of LyP to ALCL.8
Clinically, LyP consists of red-brown papules and nodules generally smaller than 2 cm, often with central hemorrhage, necrosis, and crusting. Lesions are at different stages of eruption and resolution. They are often grouped but may be disseminated. Spontaneous regression typically occurs within 3 to 8 weeks. Pruritus or mild tenderness may occur as well as residual hyperpigmentation or scarring. Systemic symptoms are notably absent.
The histologic features of LyP vary according to the age of the lesion and subtype.2 Early lesions may only show a few inflammatory cells, but as lesions evolve, larger immunoblastlike CD30+ atypical cells accumulate that may resemble the Reed-Sternberg cells of Hodgkin lymphoma. Of the 5 subtypes, the most common is type A. It is characterized by a wedge-shaped infiltrate with a mixed population of scattered or clustered, large, atypical CD30+ cells, lymphocytes, neutrophils, eosinophils, and histiocytes.9 Frequent mitoses often are seen. Type B appears similar to MF due to a predominantly epidermotropic infiltrate of CD3+ and often CD30− atypical cells. Spontaneously regressing papules favor LyP, whereas persistent patches or plaques favor MF. Type C appears identical to ALCL with diffuse sheets of large atypical CD30+ cells and relatively few inflammatory cells, but spontaneously regressing lesions again favor LyP, whereas persistent tumors favor ALCL. Type D appears similar to primary cutaneous aggressive epidermotropic CD8+ cytotoxic T cell lymphoma due to a markedly epidermotropic infiltrate of small atypical CD8+ and CD30+ lymphocytes, often TIA-1+ (T-cell intracytoplasmic antigen-1) or granzyme B+, but CD30 positivity and self-resolving lesions favor LyP. Type E mimics extranodal natural killer/T cell lymphoma (nasal type) due to angioinvasive CD30+ and beta F1+ T lymphocytes, often CD8+ and/or TIA-1+, but self-resolving lesions again favor LyP, as well as absence of Epstein-Barr virus and CD56−.9
The most common therapeutic approaches to LyP include topical steroids, phototherapy, and low-dose methotrexate.10 However, treatment does not change overall disease course or reduce the future risk for developing an associated lymphoma. Accordingly, abstaining from active therapeutic intervention is reasonable, especially in patients with only a few asymptomatic lesions.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153:874-880.
- Mori M, Manuelli C, Pimpinelli N, et al. CD30-CD30 ligand interaction in primary cutaneous CD30(+) T-cell lymphomas: a clue to the pathophysiology of clinical regression. Blood. 1999;94:3077-3083.
- Greisser J, Doebbeling U, Roos M, et al. Apoptosis in CD30-positive lymphoproliferative disorders of the skin. Exp Dermatol. 2005;14:380-385.
- Kiran T, Demirkesen C, Eker C, et al. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: a study of 53 cases. Leuk Res. 2013;37:396-400.
- Wada DA, Law ME, Hsi ED, et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: a multicenter study of 204 skin biopsies. Mod Pathol. 2011;24:596-605.
- Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, et al. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: a study of 54 cases. J Invest Dermatol. 2010;130:816-825.
- Schiemann WP, Pfeifer WM, Levi E, et al. A deletion in the gene for transforming growth factor β type I receptor abolishes growth regulation by transforming growth factor β in a cutaneous T-cell lymphoma. Blood. 1999;94:2854-2861.
- Kempf W, Kazakov DV, Schärer L, et al. Angioinvasive lymphomatoid papulosis: a new variant simulating aggressive lymphomas. Am J Surg Pathol. 2013;37:1-13.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
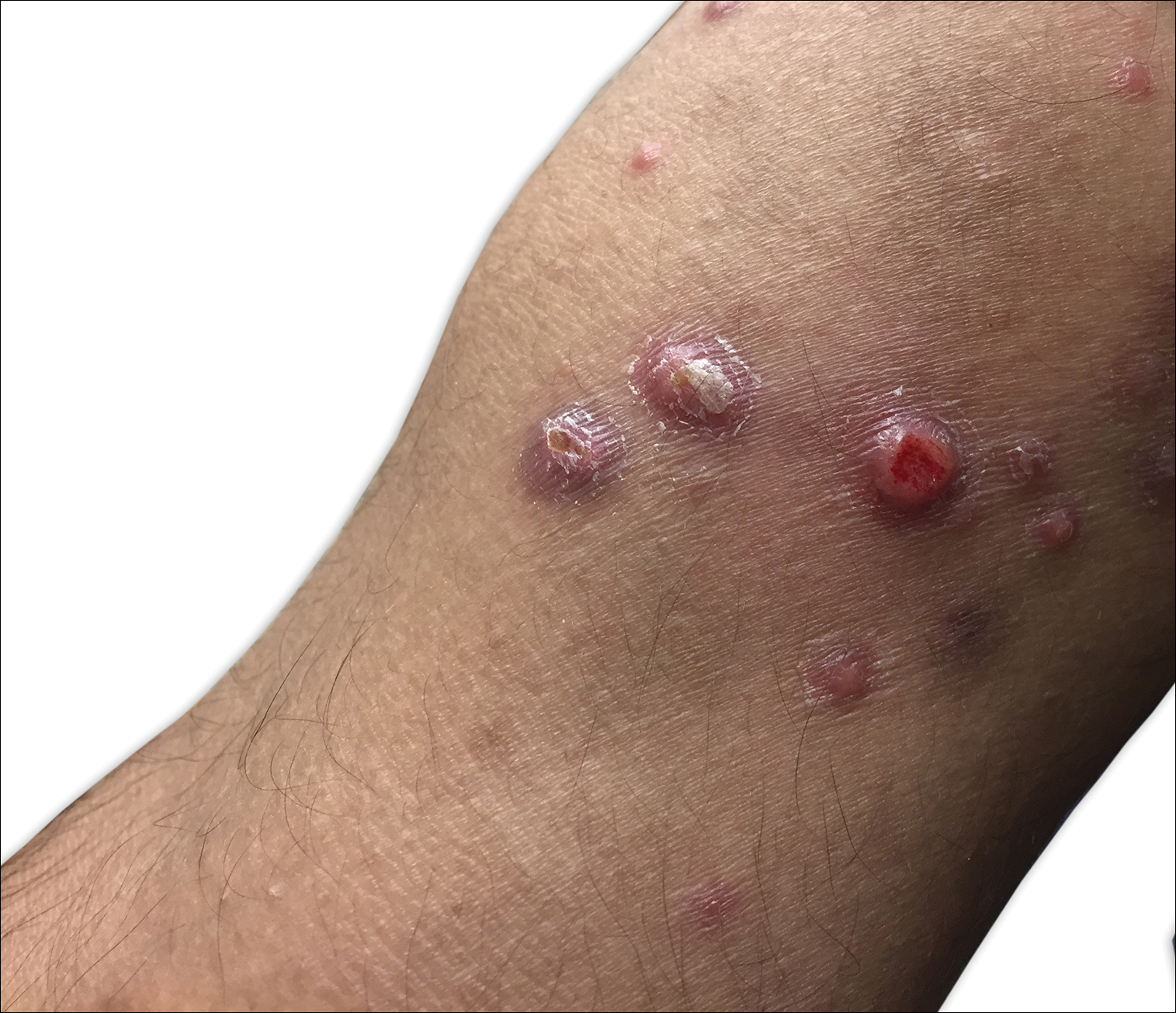
A 29-year-old man from Saudi Arabia presented with slightly tender skin lesions occurring in crops every few months over the last 7 years. The lesions typically would occur on the inguinal area, lower abdomen, buttocks, thighs, or arms, resolving within a few weeks despite no treatment. The patient denied having systemic symptoms such as fevers, chills, sweats, chest pain, shortness of breath, or unexpected weight loss. Physical examination revealed multiple erythematous papulonodules, some ulcerated with a superficial crust, grouped predominantly on the medial aspect of the right upper arm and left lower inguinal region. Isolated lesions also were present on the forearms, dorsal aspects of the hands, abdomen, and thighs. The grouped papulonodules were intermixed with faint hyperpigmented macules indicative of prior lesions. No oral lesions were noted, and there was no marked axillary or inguinal lymphadenopathy.