User login
Hidradenitis suppurativa experts reach consensus on treatment outcome measures
TOPLINE:
METHODOLOGY:
- Participants in the study were 55 HS experts from the HiSTORIC group (dermatologists, internists, surgeons, and nurses) and 24 patient research partners.
- The group identified clinician- and patient-reported HS outcome measures in the literature, then participated in an online item reduction survey, followed by an electronic Delphi survey to reach consensus on which measures should be used in clinical practice. Consensus was defined as at least 67% of participants agreeing/strongly agreeing or disagreeing/strongly disagreeing about the use of a measure in clinical practice.
- The initial literature search yielded 11 HS studies with clinician-reported outcome measures and 12 with patient-reported outcomes; of these, eight and five, respectively, were included in the final reduction survey.
TAKEAWAY:
- The group reached consensus on two HS outcome measures for use in clinical practice: the HS Investigator Global Assessment (HS-IGA) score, a clinician-reported outcome measure selected by the HS experts, and the HS Quality of Life (HiSQOL) score, a patient-reported outcome measure selected by the patient research partners.
- The HS-IGA score uses a number between 0 and 5 based on the sum of abscesses, inflammatory and noninflammatory nodules, and tunnels in regions of the upper or lower body.
- The HiSQOL, a disease-specific quality-of-life measure for adults with HS, is designed to capture unique features of HS, including symptoms (such as pain, itch, odor, and drainage) and psychosocial outcomes and activities that may be affected by the disease.
IN PRACTICE:
“The intent of these recommendations is to provide an objective framework with both clinician and patient input that can facilitate bidirectional discussion, trust building, and decision making on the current treatment strategy and the need to adjust or escalate treatment in an appropriate time frame,” the authors wrote.
SOURCE:
The study was published online in JAMA Dermatology. The lead author was Nicole Mastacouris, MS, and the corresponding author was Amit Garg, MD, both of Northwell Health, New Hyde Park, N.Y.
LIMITATIONS:
The consensus results may have been affected by variations in HS management by region. Neither measure has been studied in clinical practice, and practice variability may limit their implementation.
DISCLOSURES:
The study was supported by grants from UCB and AbbVie. Ms. Mastacouris had no financial disclosures. Dr. Garg disclosed grant support from AbbVie and UCB during the conduct of the study, as well as personal fees from AbbVie, UCB, Aclaris Therapeutics, Anaptys Bio, Aristea Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Incyte, Insmed, Janssen, Novartis, Pfizer, Sonoma Biotherapeutics, Union Therapeutics, Ventyx Biosciences, and Viela Biosciences during the conduct of the study; Dr. Garg also holds patents for HS-IGA and HiSQOL. Many other coauthors disclosed relationships with multiple companies, including AbbVie and UCB, and some also disclosed patents, including patents for HiSQOL and HS Area and Severity Index.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Participants in the study were 55 HS experts from the HiSTORIC group (dermatologists, internists, surgeons, and nurses) and 24 patient research partners.
- The group identified clinician- and patient-reported HS outcome measures in the literature, then participated in an online item reduction survey, followed by an electronic Delphi survey to reach consensus on which measures should be used in clinical practice. Consensus was defined as at least 67% of participants agreeing/strongly agreeing or disagreeing/strongly disagreeing about the use of a measure in clinical practice.
- The initial literature search yielded 11 HS studies with clinician-reported outcome measures and 12 with patient-reported outcomes; of these, eight and five, respectively, were included in the final reduction survey.
TAKEAWAY:
- The group reached consensus on two HS outcome measures for use in clinical practice: the HS Investigator Global Assessment (HS-IGA) score, a clinician-reported outcome measure selected by the HS experts, and the HS Quality of Life (HiSQOL) score, a patient-reported outcome measure selected by the patient research partners.
- The HS-IGA score uses a number between 0 and 5 based on the sum of abscesses, inflammatory and noninflammatory nodules, and tunnels in regions of the upper or lower body.
- The HiSQOL, a disease-specific quality-of-life measure for adults with HS, is designed to capture unique features of HS, including symptoms (such as pain, itch, odor, and drainage) and psychosocial outcomes and activities that may be affected by the disease.
IN PRACTICE:
“The intent of these recommendations is to provide an objective framework with both clinician and patient input that can facilitate bidirectional discussion, trust building, and decision making on the current treatment strategy and the need to adjust or escalate treatment in an appropriate time frame,” the authors wrote.
SOURCE:
The study was published online in JAMA Dermatology. The lead author was Nicole Mastacouris, MS, and the corresponding author was Amit Garg, MD, both of Northwell Health, New Hyde Park, N.Y.
LIMITATIONS:
The consensus results may have been affected by variations in HS management by region. Neither measure has been studied in clinical practice, and practice variability may limit their implementation.
DISCLOSURES:
The study was supported by grants from UCB and AbbVie. Ms. Mastacouris had no financial disclosures. Dr. Garg disclosed grant support from AbbVie and UCB during the conduct of the study, as well as personal fees from AbbVie, UCB, Aclaris Therapeutics, Anaptys Bio, Aristea Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Incyte, Insmed, Janssen, Novartis, Pfizer, Sonoma Biotherapeutics, Union Therapeutics, Ventyx Biosciences, and Viela Biosciences during the conduct of the study; Dr. Garg also holds patents for HS-IGA and HiSQOL. Many other coauthors disclosed relationships with multiple companies, including AbbVie and UCB, and some also disclosed patents, including patents for HiSQOL and HS Area and Severity Index.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Participants in the study were 55 HS experts from the HiSTORIC group (dermatologists, internists, surgeons, and nurses) and 24 patient research partners.
- The group identified clinician- and patient-reported HS outcome measures in the literature, then participated in an online item reduction survey, followed by an electronic Delphi survey to reach consensus on which measures should be used in clinical practice. Consensus was defined as at least 67% of participants agreeing/strongly agreeing or disagreeing/strongly disagreeing about the use of a measure in clinical practice.
- The initial literature search yielded 11 HS studies with clinician-reported outcome measures and 12 with patient-reported outcomes; of these, eight and five, respectively, were included in the final reduction survey.
TAKEAWAY:
- The group reached consensus on two HS outcome measures for use in clinical practice: the HS Investigator Global Assessment (HS-IGA) score, a clinician-reported outcome measure selected by the HS experts, and the HS Quality of Life (HiSQOL) score, a patient-reported outcome measure selected by the patient research partners.
- The HS-IGA score uses a number between 0 and 5 based on the sum of abscesses, inflammatory and noninflammatory nodules, and tunnels in regions of the upper or lower body.
- The HiSQOL, a disease-specific quality-of-life measure for adults with HS, is designed to capture unique features of HS, including symptoms (such as pain, itch, odor, and drainage) and psychosocial outcomes and activities that may be affected by the disease.
IN PRACTICE:
“The intent of these recommendations is to provide an objective framework with both clinician and patient input that can facilitate bidirectional discussion, trust building, and decision making on the current treatment strategy and the need to adjust or escalate treatment in an appropriate time frame,” the authors wrote.
SOURCE:
The study was published online in JAMA Dermatology. The lead author was Nicole Mastacouris, MS, and the corresponding author was Amit Garg, MD, both of Northwell Health, New Hyde Park, N.Y.
LIMITATIONS:
The consensus results may have been affected by variations in HS management by region. Neither measure has been studied in clinical practice, and practice variability may limit their implementation.
DISCLOSURES:
The study was supported by grants from UCB and AbbVie. Ms. Mastacouris had no financial disclosures. Dr. Garg disclosed grant support from AbbVie and UCB during the conduct of the study, as well as personal fees from AbbVie, UCB, Aclaris Therapeutics, Anaptys Bio, Aristea Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Incyte, Insmed, Janssen, Novartis, Pfizer, Sonoma Biotherapeutics, Union Therapeutics, Ventyx Biosciences, and Viela Biosciences during the conduct of the study; Dr. Garg also holds patents for HS-IGA and HiSQOL. Many other coauthors disclosed relationships with multiple companies, including AbbVie and UCB, and some also disclosed patents, including patents for HiSQOL and HS Area and Severity Index.
A version of this article first appeared on Medscape.com.
FROM JAMA DERMATOLOGY
Hidradenitis suppurativa experts reach consensus on treatment outcome measures
TOPLINE:
METHODOLOGY:
- Participants in the study were 55 HS experts from the HiSTORIC group (dermatologists, internists, surgeons, and nurses) and 24 patient research partners.
- The group identified clinician- and patient-reported HS outcome measures in the literature, then participated in an online item reduction survey, followed by an electronic Delphi survey to reach consensus on which measures should be used in clinical practice. Consensus was defined as at least 67% of participants agreeing/strongly agreeing or disagreeing/strongly disagreeing about the use of a measure in clinical practice.
- The initial literature search yielded 11 HS studies with clinician-reported outcome measures and 12 with patient-reported outcomes; of these, eight and five, respectively, were included in the final reduction survey.
TAKEAWAY:
- The group reached consensus on two HS outcome measures for use in clinical practice: the HS Investigator Global Assessment (HS-IGA) score, a clinician-reported outcome measure selected by the HS experts, and the HS Quality of Life (HiSQOL) score, a patient-reported outcome measure selected by the patient research partners.
- The HS-IGA score uses a number between 0 and 5 based on the sum of abscesses, inflammatory and noninflammatory nodules, and tunnels in regions of the upper or lower body.
- The HiSQOL, a disease-specific quality-of-life measure for adults with HS, is designed to capture unique features of HS, including symptoms (such as pain, itch, odor, and drainage) and psychosocial outcomes and activities that may be affected by the disease.
IN PRACTICE:
“The intent of these recommendations is to provide an objective framework with both clinician and patient input that can facilitate bidirectional discussion, trust building, and decision making on the current treatment strategy and the need to adjust or escalate treatment in an appropriate time frame,” the authors wrote.
SOURCE:
The study was published online in JAMA Dermatology. The lead author was Nicole Mastacouris, MS, and the corresponding author was Amit Garg, MD, both of Northwell Health, New Hyde Park, N.Y.
LIMITATIONS:
The consensus results may have been affected by variations in HS management by region. Neither measure has been studied in clinical practice, and practice variability may limit their implementation.
DISCLOSURES:
The study was supported by grants from UCB and AbbVie. Ms. Mastacouris had no financial disclosures. Dr. Garg disclosed grant support from AbbVie and UCB during the conduct of the study, as well as personal fees from AbbVie, UCB, Aclaris Therapeutics, Anaptys Bio, Aristea Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Incyte, Insmed, Janssen, Novartis, Pfizer, Sonoma Biotherapeutics, Union Therapeutics, Ventyx Biosciences, and Viela Biosciences during the conduct of the study; Dr. Garg also holds patents for HS-IGA and HiSQOL. Many other coauthors disclosed relationships with multiple companies, including AbbVie and UCB, and some also disclosed patents, including patents for HiSQOL and HS Area and Severity Index.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Participants in the study were 55 HS experts from the HiSTORIC group (dermatologists, internists, surgeons, and nurses) and 24 patient research partners.
- The group identified clinician- and patient-reported HS outcome measures in the literature, then participated in an online item reduction survey, followed by an electronic Delphi survey to reach consensus on which measures should be used in clinical practice. Consensus was defined as at least 67% of participants agreeing/strongly agreeing or disagreeing/strongly disagreeing about the use of a measure in clinical practice.
- The initial literature search yielded 11 HS studies with clinician-reported outcome measures and 12 with patient-reported outcomes; of these, eight and five, respectively, were included in the final reduction survey.
TAKEAWAY:
- The group reached consensus on two HS outcome measures for use in clinical practice: the HS Investigator Global Assessment (HS-IGA) score, a clinician-reported outcome measure selected by the HS experts, and the HS Quality of Life (HiSQOL) score, a patient-reported outcome measure selected by the patient research partners.
- The HS-IGA score uses a number between 0 and 5 based on the sum of abscesses, inflammatory and noninflammatory nodules, and tunnels in regions of the upper or lower body.
- The HiSQOL, a disease-specific quality-of-life measure for adults with HS, is designed to capture unique features of HS, including symptoms (such as pain, itch, odor, and drainage) and psychosocial outcomes and activities that may be affected by the disease.
IN PRACTICE:
“The intent of these recommendations is to provide an objective framework with both clinician and patient input that can facilitate bidirectional discussion, trust building, and decision making on the current treatment strategy and the need to adjust or escalate treatment in an appropriate time frame,” the authors wrote.
SOURCE:
The study was published online in JAMA Dermatology. The lead author was Nicole Mastacouris, MS, and the corresponding author was Amit Garg, MD, both of Northwell Health, New Hyde Park, N.Y.
LIMITATIONS:
The consensus results may have been affected by variations in HS management by region. Neither measure has been studied in clinical practice, and practice variability may limit their implementation.
DISCLOSURES:
The study was supported by grants from UCB and AbbVie. Ms. Mastacouris had no financial disclosures. Dr. Garg disclosed grant support from AbbVie and UCB during the conduct of the study, as well as personal fees from AbbVie, UCB, Aclaris Therapeutics, Anaptys Bio, Aristea Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Incyte, Insmed, Janssen, Novartis, Pfizer, Sonoma Biotherapeutics, Union Therapeutics, Ventyx Biosciences, and Viela Biosciences during the conduct of the study; Dr. Garg also holds patents for HS-IGA and HiSQOL. Many other coauthors disclosed relationships with multiple companies, including AbbVie and UCB, and some also disclosed patents, including patents for HiSQOL and HS Area and Severity Index.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- Participants in the study were 55 HS experts from the HiSTORIC group (dermatologists, internists, surgeons, and nurses) and 24 patient research partners.
- The group identified clinician- and patient-reported HS outcome measures in the literature, then participated in an online item reduction survey, followed by an electronic Delphi survey to reach consensus on which measures should be used in clinical practice. Consensus was defined as at least 67% of participants agreeing/strongly agreeing or disagreeing/strongly disagreeing about the use of a measure in clinical practice.
- The initial literature search yielded 11 HS studies with clinician-reported outcome measures and 12 with patient-reported outcomes; of these, eight and five, respectively, were included in the final reduction survey.
TAKEAWAY:
- The group reached consensus on two HS outcome measures for use in clinical practice: the HS Investigator Global Assessment (HS-IGA) score, a clinician-reported outcome measure selected by the HS experts, and the HS Quality of Life (HiSQOL) score, a patient-reported outcome measure selected by the patient research partners.
- The HS-IGA score uses a number between 0 and 5 based on the sum of abscesses, inflammatory and noninflammatory nodules, and tunnels in regions of the upper or lower body.
- The HiSQOL, a disease-specific quality-of-life measure for adults with HS, is designed to capture unique features of HS, including symptoms (such as pain, itch, odor, and drainage) and psychosocial outcomes and activities that may be affected by the disease.
IN PRACTICE:
“The intent of these recommendations is to provide an objective framework with both clinician and patient input that can facilitate bidirectional discussion, trust building, and decision making on the current treatment strategy and the need to adjust or escalate treatment in an appropriate time frame,” the authors wrote.
SOURCE:
The study was published online in JAMA Dermatology. The lead author was Nicole Mastacouris, MS, and the corresponding author was Amit Garg, MD, both of Northwell Health, New Hyde Park, N.Y.
LIMITATIONS:
The consensus results may have been affected by variations in HS management by region. Neither measure has been studied in clinical practice, and practice variability may limit their implementation.
DISCLOSURES:
The study was supported by grants from UCB and AbbVie. Ms. Mastacouris had no financial disclosures. Dr. Garg disclosed grant support from AbbVie and UCB during the conduct of the study, as well as personal fees from AbbVie, UCB, Aclaris Therapeutics, Anaptys Bio, Aristea Therapeutics, Boehringer Ingelheim, Bristol-Myers Squibb, Incyte, Insmed, Janssen, Novartis, Pfizer, Sonoma Biotherapeutics, Union Therapeutics, Ventyx Biosciences, and Viela Biosciences during the conduct of the study; Dr. Garg also holds patents for HS-IGA and HiSQOL. Many other coauthors disclosed relationships with multiple companies, including AbbVie and UCB, and some also disclosed patents, including patents for HiSQOL and HS Area and Severity Index.
A version of this article first appeared on Medscape.com.
FROM JAMA DERMATOLOGY
Hyperbaric oxygen therapy beneficial for calciphylaxis?
, report Daniela Kroshinsky, MD, MPH, of the department of dermatology at Massachusetts General Hospital, Boston, and colleagues.
Although intravenous sodium thiosulfate (IV STS) is considered standard care in the treatment of calciphylaxis, HBOT has been reported to have beneficial effects, they noted.
In their study, the researchers retrospectively reviewed records of 93 patients newly diagnosed with calciphylaxis, seen at Massachusetts General Hospital, between January 2006 and December 2021. They compared mortality and wound healing outcomes for 57 patients treated with IV STS only (control group) with those of 36 patients treated with HBOT plus IV STS (treatment group). Traditional survival analyses and Cox proportional hazard modeling were used to examine mortality data, and mixed effects modeling was used to analyze longitudinal wound outcomes. The study was published in the Journal of the American Academy of Dermatology.
Univariate survival analyses showed that HBOT plus IV STS was associated with significantly longer survival time than IV STS alone (P = .016), particularly for those with nonnephrogenic calciphylaxis (P < .0001), they report. An increased number of HBOT sessions conferred improved mortality outcomes, with 1, 5, 10, and 20 sessions yielding decreasing hazard ratios.
There was also a significant positive association between an increasing number of HBOT sessions and increased wound score (P = .042). Increases were seen with each session.
Anxiety/claustrophobia was the most common side effect reported among those in the HBOT group (22%).
“Given the proposed benefits and seemingly low side effect profile, it is the authors’ recommendation that HBOT be offered as an additional intervention to patients with calciphylaxis, especially if they have open wounds, to improve outcomes and expedite wound healing,” the researchers concluded.
Limitations, they noted, included the small sample size, retrospective design, and the potential for not adequately capturing patients who received external care. They were also unable to match patients by disease or wound severity. Large prospective trials would help clarify the role of HBOT for calciphylaxis, they added.
The researchers reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
, report Daniela Kroshinsky, MD, MPH, of the department of dermatology at Massachusetts General Hospital, Boston, and colleagues.
Although intravenous sodium thiosulfate (IV STS) is considered standard care in the treatment of calciphylaxis, HBOT has been reported to have beneficial effects, they noted.
In their study, the researchers retrospectively reviewed records of 93 patients newly diagnosed with calciphylaxis, seen at Massachusetts General Hospital, between January 2006 and December 2021. They compared mortality and wound healing outcomes for 57 patients treated with IV STS only (control group) with those of 36 patients treated with HBOT plus IV STS (treatment group). Traditional survival analyses and Cox proportional hazard modeling were used to examine mortality data, and mixed effects modeling was used to analyze longitudinal wound outcomes. The study was published in the Journal of the American Academy of Dermatology.
Univariate survival analyses showed that HBOT plus IV STS was associated with significantly longer survival time than IV STS alone (P = .016), particularly for those with nonnephrogenic calciphylaxis (P < .0001), they report. An increased number of HBOT sessions conferred improved mortality outcomes, with 1, 5, 10, and 20 sessions yielding decreasing hazard ratios.
There was also a significant positive association between an increasing number of HBOT sessions and increased wound score (P = .042). Increases were seen with each session.
Anxiety/claustrophobia was the most common side effect reported among those in the HBOT group (22%).
“Given the proposed benefits and seemingly low side effect profile, it is the authors’ recommendation that HBOT be offered as an additional intervention to patients with calciphylaxis, especially if they have open wounds, to improve outcomes and expedite wound healing,” the researchers concluded.
Limitations, they noted, included the small sample size, retrospective design, and the potential for not adequately capturing patients who received external care. They were also unable to match patients by disease or wound severity. Large prospective trials would help clarify the role of HBOT for calciphylaxis, they added.
The researchers reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
, report Daniela Kroshinsky, MD, MPH, of the department of dermatology at Massachusetts General Hospital, Boston, and colleagues.
Although intravenous sodium thiosulfate (IV STS) is considered standard care in the treatment of calciphylaxis, HBOT has been reported to have beneficial effects, they noted.
In their study, the researchers retrospectively reviewed records of 93 patients newly diagnosed with calciphylaxis, seen at Massachusetts General Hospital, between January 2006 and December 2021. They compared mortality and wound healing outcomes for 57 patients treated with IV STS only (control group) with those of 36 patients treated with HBOT plus IV STS (treatment group). Traditional survival analyses and Cox proportional hazard modeling were used to examine mortality data, and mixed effects modeling was used to analyze longitudinal wound outcomes. The study was published in the Journal of the American Academy of Dermatology.
Univariate survival analyses showed that HBOT plus IV STS was associated with significantly longer survival time than IV STS alone (P = .016), particularly for those with nonnephrogenic calciphylaxis (P < .0001), they report. An increased number of HBOT sessions conferred improved mortality outcomes, with 1, 5, 10, and 20 sessions yielding decreasing hazard ratios.
There was also a significant positive association between an increasing number of HBOT sessions and increased wound score (P = .042). Increases were seen with each session.
Anxiety/claustrophobia was the most common side effect reported among those in the HBOT group (22%).
“Given the proposed benefits and seemingly low side effect profile, it is the authors’ recommendation that HBOT be offered as an additional intervention to patients with calciphylaxis, especially if they have open wounds, to improve outcomes and expedite wound healing,” the researchers concluded.
Limitations, they noted, included the small sample size, retrospective design, and the potential for not adequately capturing patients who received external care. They were also unable to match patients by disease or wound severity. Large prospective trials would help clarify the role of HBOT for calciphylaxis, they added.
The researchers reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
New evidence suggests genetic risk factors in hidradenitis suppurativa
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with lesions that include deep-seated nodules and abscesses, draining tracts, and fibrotic scars, and has been reported to be highly heritable.
A genomewide association study (GWAS) that involved a meta-analysis of data from three large biobanks (UK Biobank, FinnGen, and BioVU) identified one significant locus and two suggestive loci. The researchers were able to replicate the association with HS for two loci near the SOX9 and KLF5 genes in BioVU.
In addition, they also looked at genetic correlations between HS and autoimmune and inflammatory diseases, and results suggested a positive association with inflammatory bowel disease, psoriasis, and type 2 diabetes.
However, while plausible, the variant associations near candidate genes do not prove a causal effect of these variants or genes on disease risk, and further study is needed.

“It’s possible that these genes aren’t the ones affected by the variations we describe, but both are very strong candidates,” said study author Christopher J. Sayed, MD, associate professor of dermatology, University of North Carolina at Chapel Hill. “This improves our understanding of HS as a disease that is likely related to genetic changes that result in dysregulated hair follicle maintenance, inflammation, and wound healing.”
In turn, this will allow clinicians to educate patients about the underlying genetic influences on HS, he explained, as opposed to stigmatizing misconceptions focusing only on weight, smoking, and hygiene.
“Larger studies are underway and will be needed to find variants that may predict things like disease severity and response to treatment, but this is a big first step in the right direction,” Dr. Sayed added.
The study was published online in JAMA Dermatology.
Loci identified
GWASs have been limited in HS, and previous research has not identified significant risk loci. In the current study, Dr. Sayed and colleagues sought to identify underlying genes and genetic mechanisms that may underlie pathogenesis in HS.
They performed a GWAS in a cohort of 720 patients who were part of the Hidradenitis Suppurativa Program for Research and Care Excellence (HS ProCARE) at the UNC department of dermatology, and controls from the National Longitudinal Study of Adolescent to Adult Health (Add Health) study, a U.S.-based study following adolescents through adulthood.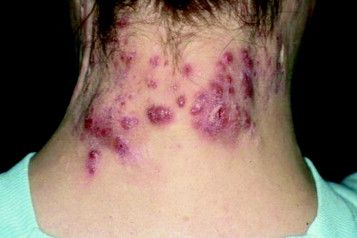
The UK Biobank (UKB) is a prospective biobank with about 500,000 individuals aged 40-69 years, and FinnGen collects and analyzes genome and health data from Finnish biobank participants. To increase power to detect associations, a GWAS was performed using participants from the UKB (247 HS cases, 453,048 controls). The HS ProCARE GWAS results were meta-analyzed with UKB, along with data from FinnGen (673 HS cases, 297,544 controls). This three-way meta-analysis revealed one genomewide significant locus and two suggestive loci.
The authors found that the most strongly associated variant, rs10512572 located on chromosome 17, showed the strongest association in FinnGen; at the second locus, the most strongly associated variant was rs17090189 located on chromosome 13 and also showed the strongest association in FinnGen; and at the third locus, the most strongly associated variant, rs5792315, located on chromosome 11, showed the strongest association in HS ProCARE.
Next, they tested the 10 most strongly associated variants at the three loci in the BioVU biobank, which has 290 HS cases, including 189 individuals of European ancestry and 101 individuals of African ancestry, with 64,234 and 12,105 controls, respectively. The locus on chromosome 17 was replicated in BioVU in the same direction of effect, while one variant at the chromosome 13 locus showed nominal evidence of association in the same direction.
In a four-way meta-analysis of BioVU combined with the other three studies, the chromosome locus became more significant and the chromosome 13 locus exceeded the genomewide significance threshold. In contrast, the chromosome 11 locus was not replicated in BioVU (P = .27).
The authors pointed out that variants at these loci are located in keratinocyte regulatory elements near the genes SOX9 and KLF5, which play a role in skin and follicular inflammation, but have not previously been associated with HS pathogenesis.
Finally, they looked to see if there were shared genetic components between HS and autoimmune and inflammatory diseases. A nominally significant genetic correlation was observed between HS and inflammatory bowel disease (P = .04), psoriasis (P = .03), and type 2 diabetes (P = .04), although none reached significance.
Different manifestation with genetic variant
In a related study, researchers assessed the prevalence of the NCSTN:c.671_682del variant among individuals with HS in Malta, the island country in the Mediterranean, which has a high prevalence of HS and its associated risk factors, particularly obesity.
Led by Dillon Mintoff, MD, of the department of pathology at the University of Malta, Msida, they conducted a cross-sectional study of 113 adults with HS. In this group, 14 (12.39%) were found to be heterozygous for the NCSTN:c.671_682del variant. Individuals who had this variant were more likely to develop symptoms earlier and to manifest them in atypical skin sites, including the scalp, neck, torso, and antecubital fossae. Additionally, even though their symptoms weren’t more severe than those without the variant, patients with the variant were more likely to require treatment with biologic agents.
Studies move genetics in HS forward
Writing in an accompanying editorial, Atlas Khan, PhD, and Lynn Petukhova, PhD, from Columbia University, New York, noted that both of these HS genetic studies “set a solid foundation for future studies aimed at understanding the biological and clinical relevance of new HS genetic evidence.”
“Each study suggests a series of experiments that will allow us to gain new knowledge about HS,” they wrote, including coming closer to providing patients with a genetic diagnosis.
In addition, evidence from the GWAS paper suggests that with “larger HS GWASs we will be able to better prioritize drug-repurposing studies,” concluded the editorialists. “Both of these goals will help to improve health outcomes for patients with HS and their family members.”
Dr. Sayed reported grants and/or personal fees from AbbVie, Novartis, UCB, Incyte, InflaRx, Alumis, and ChemoCentryx outside the submitted work; and serving as a volunteer member of the board of the HS Foundation and member of the European HS Foundation. The study in Malta was funded by the Government of Malta’s Tertiary Education Scholarship Scheme and Institutional Funds from the University of Malta. Dr. Mintoff reported grants from the Government of Malta Ministry for Education, Sport, Youth, Research and Innovation during the conduct of the study. Dr. Khan reported receiving grants from the National Institute of Diabetes and Digestive Kidney Diseases. Dr. Petukhova reported receiving grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Columbia University’s Precision Medicine Initiative, Herbert Irving Comprehensive Cancer Center, and Data Science Institute.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with lesions that include deep-seated nodules and abscesses, draining tracts, and fibrotic scars, and has been reported to be highly heritable.
A genomewide association study (GWAS) that involved a meta-analysis of data from three large biobanks (UK Biobank, FinnGen, and BioVU) identified one significant locus and two suggestive loci. The researchers were able to replicate the association with HS for two loci near the SOX9 and KLF5 genes in BioVU.
In addition, they also looked at genetic correlations between HS and autoimmune and inflammatory diseases, and results suggested a positive association with inflammatory bowel disease, psoriasis, and type 2 diabetes.
However, while plausible, the variant associations near candidate genes do not prove a causal effect of these variants or genes on disease risk, and further study is needed.
“It’s possible that these genes aren’t the ones affected by the variations we describe, but both are very strong candidates,” said study author Christopher J. Sayed, MD, associate professor of dermatology, University of North Carolina at Chapel Hill. “This improves our understanding of HS as a disease that is likely related to genetic changes that result in dysregulated hair follicle maintenance, inflammation, and wound healing.”
In turn, this will allow clinicians to educate patients about the underlying genetic influences on HS, he explained, as opposed to stigmatizing misconceptions focusing only on weight, smoking, and hygiene.
“Larger studies are underway and will be needed to find variants that may predict things like disease severity and response to treatment, but this is a big first step in the right direction,” Dr. Sayed added.
The study was published online in JAMA Dermatology.
Loci identified
GWASs have been limited in HS, and previous research has not identified significant risk loci. In the current study, Dr. Sayed and colleagues sought to identify underlying genes and genetic mechanisms that may underlie pathogenesis in HS.
They performed a GWAS in a cohort of 720 patients who were part of the Hidradenitis Suppurativa Program for Research and Care Excellence (HS ProCARE) at the UNC department of dermatology, and controls from the National Longitudinal Study of Adolescent to Adult Health (Add Health) study, a U.S.-based study following adolescents through adulthood.
The UK Biobank (UKB) is a prospective biobank with about 500,000 individuals aged 40-69 years, and FinnGen collects and analyzes genome and health data from Finnish biobank participants. To increase power to detect associations, a GWAS was performed using participants from the UKB (247 HS cases, 453,048 controls). The HS ProCARE GWAS results were meta-analyzed with UKB, along with data from FinnGen (673 HS cases, 297,544 controls). This three-way meta-analysis revealed one genomewide significant locus and two suggestive loci.
The authors found that the most strongly associated variant, rs10512572 located on chromosome 17, showed the strongest association in FinnGen; at the second locus, the most strongly associated variant was rs17090189 located on chromosome 13 and also showed the strongest association in FinnGen; and at the third locus, the most strongly associated variant, rs5792315, located on chromosome 11, showed the strongest association in HS ProCARE.
Next, they tested the 10 most strongly associated variants at the three loci in the BioVU biobank, which has 290 HS cases, including 189 individuals of European ancestry and 101 individuals of African ancestry, with 64,234 and 12,105 controls, respectively. The locus on chromosome 17 was replicated in BioVU in the same direction of effect, while one variant at the chromosome 13 locus showed nominal evidence of association in the same direction.
In a four-way meta-analysis of BioVU combined with the other three studies, the chromosome locus became more significant and the chromosome 13 locus exceeded the genomewide significance threshold. In contrast, the chromosome 11 locus was not replicated in BioVU (P = .27).
The authors pointed out that variants at these loci are located in keratinocyte regulatory elements near the genes SOX9 and KLF5, which play a role in skin and follicular inflammation, but have not previously been associated with HS pathogenesis.
Finally, they looked to see if there were shared genetic components between HS and autoimmune and inflammatory diseases. A nominally significant genetic correlation was observed between HS and inflammatory bowel disease (P = .04), psoriasis (P = .03), and type 2 diabetes (P = .04), although none reached significance.
Different manifestation with genetic variant
In a related study, researchers assessed the prevalence of the NCSTN:c.671_682del variant among individuals with HS in Malta, the island country in the Mediterranean, which has a high prevalence of HS and its associated risk factors, particularly obesity.
Led by Dillon Mintoff, MD, of the department of pathology at the University of Malta, Msida, they conducted a cross-sectional study of 113 adults with HS. In this group, 14 (12.39%) were found to be heterozygous for the NCSTN:c.671_682del variant. Individuals who had this variant were more likely to develop symptoms earlier and to manifest them in atypical skin sites, including the scalp, neck, torso, and antecubital fossae. Additionally, even though their symptoms weren’t more severe than those without the variant, patients with the variant were more likely to require treatment with biologic agents.
Studies move genetics in HS forward
Writing in an accompanying editorial, Atlas Khan, PhD, and Lynn Petukhova, PhD, from Columbia University, New York, noted that both of these HS genetic studies “set a solid foundation for future studies aimed at understanding the biological and clinical relevance of new HS genetic evidence.”
“Each study suggests a series of experiments that will allow us to gain new knowledge about HS,” they wrote, including coming closer to providing patients with a genetic diagnosis.
In addition, evidence from the GWAS paper suggests that with “larger HS GWASs we will be able to better prioritize drug-repurposing studies,” concluded the editorialists. “Both of these goals will help to improve health outcomes for patients with HS and their family members.”
Dr. Sayed reported grants and/or personal fees from AbbVie, Novartis, UCB, Incyte, InflaRx, Alumis, and ChemoCentryx outside the submitted work; and serving as a volunteer member of the board of the HS Foundation and member of the European HS Foundation. The study in Malta was funded by the Government of Malta’s Tertiary Education Scholarship Scheme and Institutional Funds from the University of Malta. Dr. Mintoff reported grants from the Government of Malta Ministry for Education, Sport, Youth, Research and Innovation during the conduct of the study. Dr. Khan reported receiving grants from the National Institute of Diabetes and Digestive Kidney Diseases. Dr. Petukhova reported receiving grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Columbia University’s Precision Medicine Initiative, Herbert Irving Comprehensive Cancer Center, and Data Science Institute.
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with lesions that include deep-seated nodules and abscesses, draining tracts, and fibrotic scars, and has been reported to be highly heritable.
A genomewide association study (GWAS) that involved a meta-analysis of data from three large biobanks (UK Biobank, FinnGen, and BioVU) identified one significant locus and two suggestive loci. The researchers were able to replicate the association with HS for two loci near the SOX9 and KLF5 genes in BioVU.
In addition, they also looked at genetic correlations between HS and autoimmune and inflammatory diseases, and results suggested a positive association with inflammatory bowel disease, psoriasis, and type 2 diabetes.
However, while plausible, the variant associations near candidate genes do not prove a causal effect of these variants or genes on disease risk, and further study is needed.
“It’s possible that these genes aren’t the ones affected by the variations we describe, but both are very strong candidates,” said study author Christopher J. Sayed, MD, associate professor of dermatology, University of North Carolina at Chapel Hill. “This improves our understanding of HS as a disease that is likely related to genetic changes that result in dysregulated hair follicle maintenance, inflammation, and wound healing.”
In turn, this will allow clinicians to educate patients about the underlying genetic influences on HS, he explained, as opposed to stigmatizing misconceptions focusing only on weight, smoking, and hygiene.
“Larger studies are underway and will be needed to find variants that may predict things like disease severity and response to treatment, but this is a big first step in the right direction,” Dr. Sayed added.
The study was published online in JAMA Dermatology.
Loci identified
GWASs have been limited in HS, and previous research has not identified significant risk loci. In the current study, Dr. Sayed and colleagues sought to identify underlying genes and genetic mechanisms that may underlie pathogenesis in HS.
They performed a GWAS in a cohort of 720 patients who were part of the Hidradenitis Suppurativa Program for Research and Care Excellence (HS ProCARE) at the UNC department of dermatology, and controls from the National Longitudinal Study of Adolescent to Adult Health (Add Health) study, a U.S.-based study following adolescents through adulthood.
The UK Biobank (UKB) is a prospective biobank with about 500,000 individuals aged 40-69 years, and FinnGen collects and analyzes genome and health data from Finnish biobank participants. To increase power to detect associations, a GWAS was performed using participants from the UKB (247 HS cases, 453,048 controls). The HS ProCARE GWAS results were meta-analyzed with UKB, along with data from FinnGen (673 HS cases, 297,544 controls). This three-way meta-analysis revealed one genomewide significant locus and two suggestive loci.
The authors found that the most strongly associated variant, rs10512572 located on chromosome 17, showed the strongest association in FinnGen; at the second locus, the most strongly associated variant was rs17090189 located on chromosome 13 and also showed the strongest association in FinnGen; and at the third locus, the most strongly associated variant, rs5792315, located on chromosome 11, showed the strongest association in HS ProCARE.
Next, they tested the 10 most strongly associated variants at the three loci in the BioVU biobank, which has 290 HS cases, including 189 individuals of European ancestry and 101 individuals of African ancestry, with 64,234 and 12,105 controls, respectively. The locus on chromosome 17 was replicated in BioVU in the same direction of effect, while one variant at the chromosome 13 locus showed nominal evidence of association in the same direction.
In a four-way meta-analysis of BioVU combined with the other three studies, the chromosome locus became more significant and the chromosome 13 locus exceeded the genomewide significance threshold. In contrast, the chromosome 11 locus was not replicated in BioVU (P = .27).
The authors pointed out that variants at these loci are located in keratinocyte regulatory elements near the genes SOX9 and KLF5, which play a role in skin and follicular inflammation, but have not previously been associated with HS pathogenesis.
Finally, they looked to see if there were shared genetic components between HS and autoimmune and inflammatory diseases. A nominally significant genetic correlation was observed between HS and inflammatory bowel disease (P = .04), psoriasis (P = .03), and type 2 diabetes (P = .04), although none reached significance.
Different manifestation with genetic variant
In a related study, researchers assessed the prevalence of the NCSTN:c.671_682del variant among individuals with HS in Malta, the island country in the Mediterranean, which has a high prevalence of HS and its associated risk factors, particularly obesity.
Led by Dillon Mintoff, MD, of the department of pathology at the University of Malta, Msida, they conducted a cross-sectional study of 113 adults with HS. In this group, 14 (12.39%) were found to be heterozygous for the NCSTN:c.671_682del variant. Individuals who had this variant were more likely to develop symptoms earlier and to manifest them in atypical skin sites, including the scalp, neck, torso, and antecubital fossae. Additionally, even though their symptoms weren’t more severe than those without the variant, patients with the variant were more likely to require treatment with biologic agents.
Studies move genetics in HS forward
Writing in an accompanying editorial, Atlas Khan, PhD, and Lynn Petukhova, PhD, from Columbia University, New York, noted that both of these HS genetic studies “set a solid foundation for future studies aimed at understanding the biological and clinical relevance of new HS genetic evidence.”
“Each study suggests a series of experiments that will allow us to gain new knowledge about HS,” they wrote, including coming closer to providing patients with a genetic diagnosis.
In addition, evidence from the GWAS paper suggests that with “larger HS GWASs we will be able to better prioritize drug-repurposing studies,” concluded the editorialists. “Both of these goals will help to improve health outcomes for patients with HS and their family members.”
Dr. Sayed reported grants and/or personal fees from AbbVie, Novartis, UCB, Incyte, InflaRx, Alumis, and ChemoCentryx outside the submitted work; and serving as a volunteer member of the board of the HS Foundation and member of the European HS Foundation. The study in Malta was funded by the Government of Malta’s Tertiary Education Scholarship Scheme and Institutional Funds from the University of Malta. Dr. Mintoff reported grants from the Government of Malta Ministry for Education, Sport, Youth, Research and Innovation during the conduct of the study. Dr. Khan reported receiving grants from the National Institute of Diabetes and Digestive Kidney Diseases. Dr. Petukhova reported receiving grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Columbia University’s Precision Medicine Initiative, Herbert Irving Comprehensive Cancer Center, and Data Science Institute.
FROM JAMA DERMATOLOGY
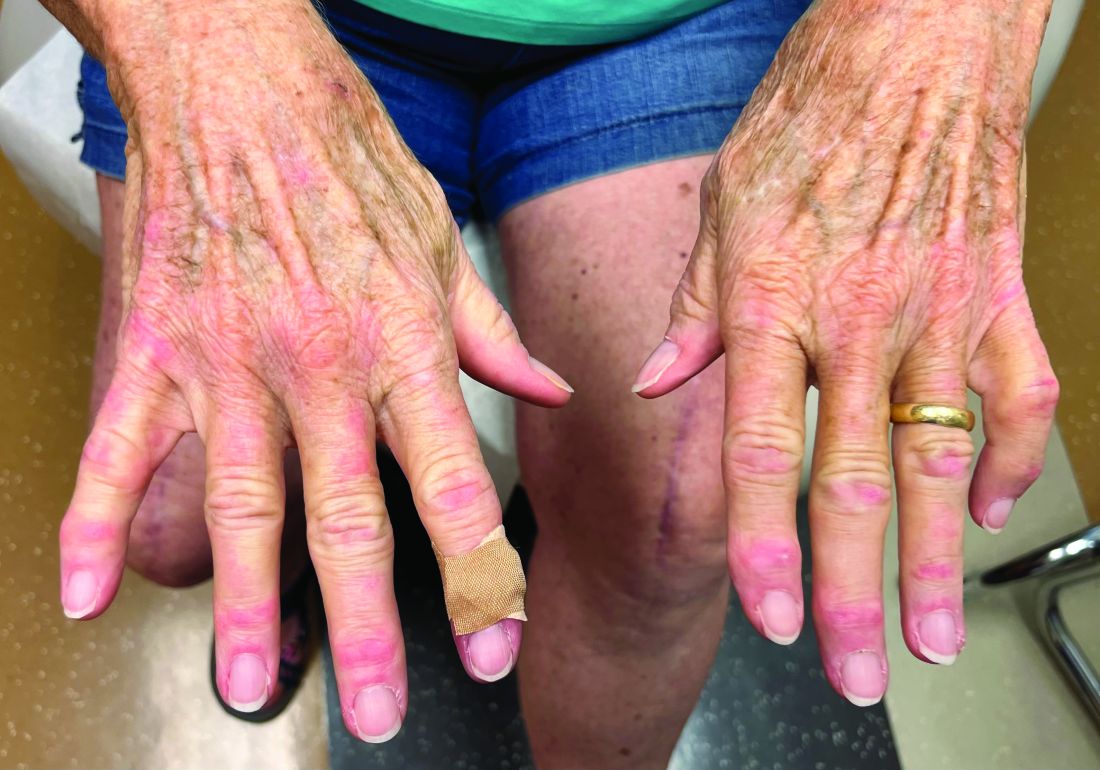
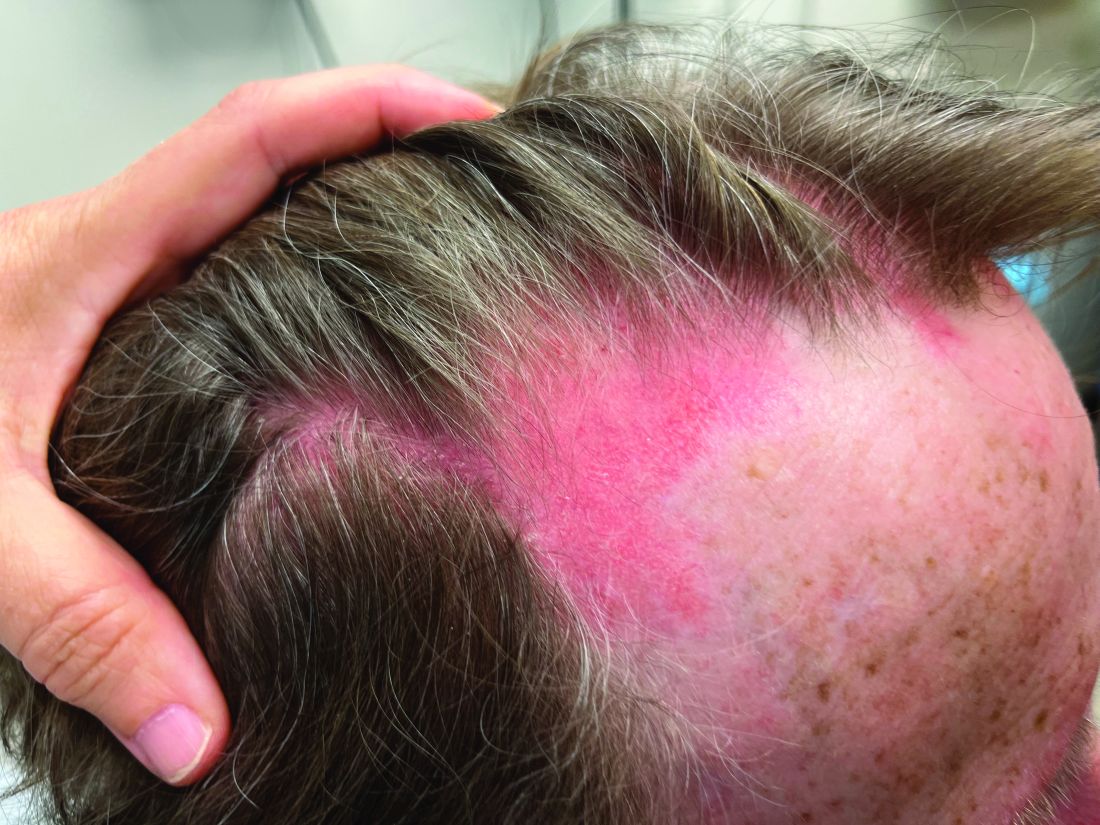
A 75-year-old White woman presented with diffuse erythema, scale, and pruritus on her scalp
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.

Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
Scalp cooling for chemo hair loss strikes out with patients
TOPLINE:
, compared with those who opted to forgo scalp cooling.
METHODOLOGY:
- Although studies have demonstrated the effectiveness of scalp cooling to reduce hair loss during breast cancer chemotherapy, most were in the setting of single-agent regimens instead of much more commonly used combined chemotherapy, and few studies assessed patients’ subjective experience.
- To get a real-world sense of the treatment, investigators compared outcomes in 75 women who opted to use the Orbis Paxman cooling cap during taxane/anthracycline-based chemotherapy sessions with 38 women with breast cancer patients who declined to use the cooling cap.
- The women were surveyed for hair loss perception, functional health, and body image at baseline, midchemotherapy, and at their last chemotherapy cycle, as well as at 3 months and 6-9 months following chemotherapy.
- The women were treated at the Medical University of Innsbruck, Austria, for various stages of breast cancer; about half were premenopausal.
TAKEAWAY:
- There was no significant difference between the scalp-cooling and control groups in patient-reported hair loss (P = .831), overall quality of life (P = .627), emotional functioning (P = .737), social functioning (P = .635), and body image (P = .463).
- On average, women stayed on treatment with the cooling cap for about 40% of the duration of their chemotherapy.
- Overall, 53 of 75 women (70.7%) stopped scalp cooling early, with most (73.9%) citing alopecia as the primary reason; only 30% completed treatment.
IN PRACTICE:
“The efficacy and tolerability of [scalp cooling] applied in a clinical routine setting ... appeared to be limited,” the authors concluded. “The further determination and up-front definition of criteria prognostic for effectiveness of [scalp cooling] may be helpful to identify patient subgroups that may experience a treatment benefit.”
SOURCE:
The work, led by Christine Brunner, Medical University of Innsbruck, Austria, was published in Breast Cancer: Targets and Therapy.
LIMITATIONS:
- Shorter intervals between surveys might have given a more granular understanding of patients’ experiences with scalp cooling.
- There were no biomarker assessments to help identify patients more likely to benefit.
DISCLOSURES:
The work was supported by the Medical University of Innsbruck. Dr. Brunner disclosed a grant from Paxman UK, maker of the cooling cap used in the study. Another investigator disclosed personal fees from AstraZeneca, Daiichi Sankyo, Gilead, Lilly, Novartis, and Sirius.
A version of this article first appeared on Medscape.com.
TOPLINE:
, compared with those who opted to forgo scalp cooling.
METHODOLOGY:
- Although studies have demonstrated the effectiveness of scalp cooling to reduce hair loss during breast cancer chemotherapy, most were in the setting of single-agent regimens instead of much more commonly used combined chemotherapy, and few studies assessed patients’ subjective experience.
- To get a real-world sense of the treatment, investigators compared outcomes in 75 women who opted to use the Orbis Paxman cooling cap during taxane/anthracycline-based chemotherapy sessions with 38 women with breast cancer patients who declined to use the cooling cap.
- The women were surveyed for hair loss perception, functional health, and body image at baseline, midchemotherapy, and at their last chemotherapy cycle, as well as at 3 months and 6-9 months following chemotherapy.
- The women were treated at the Medical University of Innsbruck, Austria, for various stages of breast cancer; about half were premenopausal.
TAKEAWAY:
- There was no significant difference between the scalp-cooling and control groups in patient-reported hair loss (P = .831), overall quality of life (P = .627), emotional functioning (P = .737), social functioning (P = .635), and body image (P = .463).
- On average, women stayed on treatment with the cooling cap for about 40% of the duration of their chemotherapy.
- Overall, 53 of 75 women (70.7%) stopped scalp cooling early, with most (73.9%) citing alopecia as the primary reason; only 30% completed treatment.
IN PRACTICE:
“The efficacy and tolerability of [scalp cooling] applied in a clinical routine setting ... appeared to be limited,” the authors concluded. “The further determination and up-front definition of criteria prognostic for effectiveness of [scalp cooling] may be helpful to identify patient subgroups that may experience a treatment benefit.”
SOURCE:
The work, led by Christine Brunner, Medical University of Innsbruck, Austria, was published in Breast Cancer: Targets and Therapy.
LIMITATIONS:
- Shorter intervals between surveys might have given a more granular understanding of patients’ experiences with scalp cooling.
- There were no biomarker assessments to help identify patients more likely to benefit.
DISCLOSURES:
The work was supported by the Medical University of Innsbruck. Dr. Brunner disclosed a grant from Paxman UK, maker of the cooling cap used in the study. Another investigator disclosed personal fees from AstraZeneca, Daiichi Sankyo, Gilead, Lilly, Novartis, and Sirius.
A version of this article first appeared on Medscape.com.
TOPLINE:
, compared with those who opted to forgo scalp cooling.
METHODOLOGY:
- Although studies have demonstrated the effectiveness of scalp cooling to reduce hair loss during breast cancer chemotherapy, most were in the setting of single-agent regimens instead of much more commonly used combined chemotherapy, and few studies assessed patients’ subjective experience.
- To get a real-world sense of the treatment, investigators compared outcomes in 75 women who opted to use the Orbis Paxman cooling cap during taxane/anthracycline-based chemotherapy sessions with 38 women with breast cancer patients who declined to use the cooling cap.
- The women were surveyed for hair loss perception, functional health, and body image at baseline, midchemotherapy, and at their last chemotherapy cycle, as well as at 3 months and 6-9 months following chemotherapy.
- The women were treated at the Medical University of Innsbruck, Austria, for various stages of breast cancer; about half were premenopausal.
TAKEAWAY:
- There was no significant difference between the scalp-cooling and control groups in patient-reported hair loss (P = .831), overall quality of life (P = .627), emotional functioning (P = .737), social functioning (P = .635), and body image (P = .463).
- On average, women stayed on treatment with the cooling cap for about 40% of the duration of their chemotherapy.
- Overall, 53 of 75 women (70.7%) stopped scalp cooling early, with most (73.9%) citing alopecia as the primary reason; only 30% completed treatment.
IN PRACTICE:
“The efficacy and tolerability of [scalp cooling] applied in a clinical routine setting ... appeared to be limited,” the authors concluded. “The further determination and up-front definition of criteria prognostic for effectiveness of [scalp cooling] may be helpful to identify patient subgroups that may experience a treatment benefit.”
SOURCE:
The work, led by Christine Brunner, Medical University of Innsbruck, Austria, was published in Breast Cancer: Targets and Therapy.
LIMITATIONS:
- Shorter intervals between surveys might have given a more granular understanding of patients’ experiences with scalp cooling.
- There were no biomarker assessments to help identify patients more likely to benefit.
DISCLOSURES:
The work was supported by the Medical University of Innsbruck. Dr. Brunner disclosed a grant from Paxman UK, maker of the cooling cap used in the study. Another investigator disclosed personal fees from AstraZeneca, Daiichi Sankyo, Gilead, Lilly, Novartis, and Sirius.
A version of this article first appeared on Medscape.com.
BREAST CANCER: TARGETS AND THERAPY
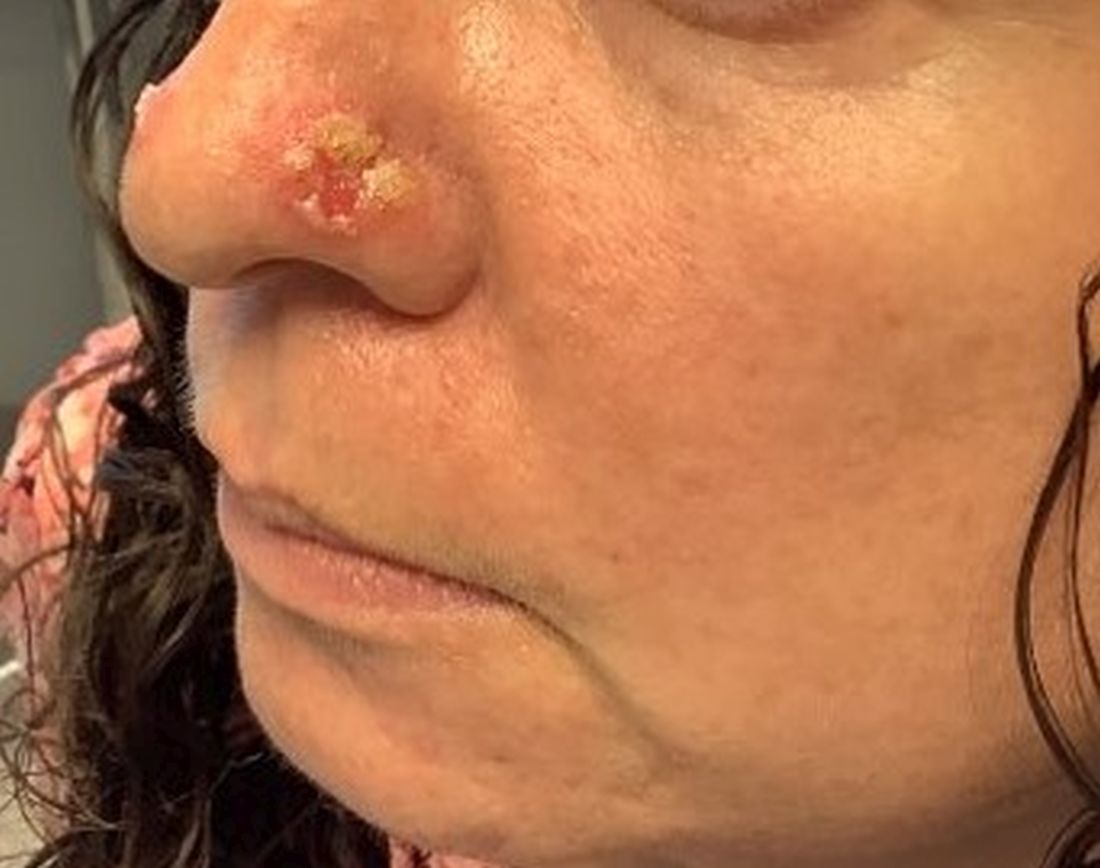
A 45-year-old White woman with no significant medical history presented with a 1-month history of lesions on the nose and right cheek
Cultures for bacteria, varicella zoster virus, herpes simplex virus, and mpox virus were all negative. A biopsy revealed suprabasilar acantholysis with follicular involvement in association with blister formation and inflammation. Direct immunofluorescence was positive for suprabasilar IgG and C3 deposition, consistent with pemphigus vulgaris (PV).
. There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, flaccid blistering lesions are present that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions may involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
Treatment is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid-sparing agent such as mycophenolate mofetil. Other steroid-sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
This case and the photos are from Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Cultures for bacteria, varicella zoster virus, herpes simplex virus, and mpox virus were all negative. A biopsy revealed suprabasilar acantholysis with follicular involvement in association with blister formation and inflammation. Direct immunofluorescence was positive for suprabasilar IgG and C3 deposition, consistent with pemphigus vulgaris (PV).
. There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, flaccid blistering lesions are present that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions may involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
Treatment is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid-sparing agent such as mycophenolate mofetil. Other steroid-sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
This case and the photos are from Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Cultures for bacteria, varicella zoster virus, herpes simplex virus, and mpox virus were all negative. A biopsy revealed suprabasilar acantholysis with follicular involvement in association with blister formation and inflammation. Direct immunofluorescence was positive for suprabasilar IgG and C3 deposition, consistent with pemphigus vulgaris (PV).
. There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, flaccid blistering lesions are present that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions may involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
Treatment is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid-sparing agent such as mycophenolate mofetil. Other steroid-sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
This case and the photos are from Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
FDA approves cantharidin for molluscum contagiosum
On July 21, 2023, .
The product is a drug-device combination that contains a formulation of cantharidin solution (0.7%), delivered topically via a single-use applicator, which allows for precise dosing and targeted administration. According to a press release from Verrica Pharmaceuticals, cantharidin is expected to be available by September 2023 and should be administered only by a trained health care professional; it is not for use in the home.
The approval of the product, also known as VP-102, is based on results from two identical multicenter phase 3 randomized, double-blind, placebo-controlled trials that evaluated the drug’s safety and efficacy in patients 2 years of age and older diagnosed with molluscum: Cantharidin Application in Molluscum Patients-1 (CAMP-1) and CAMP-2. Patients in both trials met the primary endpoint of complete clearance of all treatable molluscum lesions. Specifically, 46% of CAMP-1 participants treated with VP-102 achieved complete clearance of molluscum lesions compared with 18% of participants in the vehicle group (P < .0001), while 54% of CAMP-2 participants treated with VP-102 achieved complete clearance of molluscum lesions compared with 13% of participants in the vehicle group (P < .0001).
A post hoc analysis of both trials found that complete clearance of all lesions was significantly higher in the VP-102 group than vehicle across all body regions. It also found that there were no serious adverse reactions reported in the trials. Adverse reactions were mostly mild to moderate and included application site vesicles, erythema, pain, dryness, scab, discoloration, pruritus, and edema.
The product will be marketed as Ycanth.
In March of 2023, the FDA accepted a new drug application for another treatment for molluscum contagiosum, berdazimer gel 10.3%. That product is being developed by Novan.
On July 21, 2023, .
The product is a drug-device combination that contains a formulation of cantharidin solution (0.7%), delivered topically via a single-use applicator, which allows for precise dosing and targeted administration. According to a press release from Verrica Pharmaceuticals, cantharidin is expected to be available by September 2023 and should be administered only by a trained health care professional; it is not for use in the home.
The approval of the product, also known as VP-102, is based on results from two identical multicenter phase 3 randomized, double-blind, placebo-controlled trials that evaluated the drug’s safety and efficacy in patients 2 years of age and older diagnosed with molluscum: Cantharidin Application in Molluscum Patients-1 (CAMP-1) and CAMP-2. Patients in both trials met the primary endpoint of complete clearance of all treatable molluscum lesions. Specifically, 46% of CAMP-1 participants treated with VP-102 achieved complete clearance of molluscum lesions compared with 18% of participants in the vehicle group (P < .0001), while 54% of CAMP-2 participants treated with VP-102 achieved complete clearance of molluscum lesions compared with 13% of participants in the vehicle group (P < .0001).
A post hoc analysis of both trials found that complete clearance of all lesions was significantly higher in the VP-102 group than vehicle across all body regions. It also found that there were no serious adverse reactions reported in the trials. Adverse reactions were mostly mild to moderate and included application site vesicles, erythema, pain, dryness, scab, discoloration, pruritus, and edema.
The product will be marketed as Ycanth.
In March of 2023, the FDA accepted a new drug application for another treatment for molluscum contagiosum, berdazimer gel 10.3%. That product is being developed by Novan.
On July 21, 2023, .
The product is a drug-device combination that contains a formulation of cantharidin solution (0.7%), delivered topically via a single-use applicator, which allows for precise dosing and targeted administration. According to a press release from Verrica Pharmaceuticals, cantharidin is expected to be available by September 2023 and should be administered only by a trained health care professional; it is not for use in the home.
The approval of the product, also known as VP-102, is based on results from two identical multicenter phase 3 randomized, double-blind, placebo-controlled trials that evaluated the drug’s safety and efficacy in patients 2 years of age and older diagnosed with molluscum: Cantharidin Application in Molluscum Patients-1 (CAMP-1) and CAMP-2. Patients in both trials met the primary endpoint of complete clearance of all treatable molluscum lesions. Specifically, 46% of CAMP-1 participants treated with VP-102 achieved complete clearance of molluscum lesions compared with 18% of participants in the vehicle group (P < .0001), while 54% of CAMP-2 participants treated with VP-102 achieved complete clearance of molluscum lesions compared with 13% of participants in the vehicle group (P < .0001).
A post hoc analysis of both trials found that complete clearance of all lesions was significantly higher in the VP-102 group than vehicle across all body regions. It also found that there were no serious adverse reactions reported in the trials. Adverse reactions were mostly mild to moderate and included application site vesicles, erythema, pain, dryness, scab, discoloration, pruritus, and edema.
The product will be marketed as Ycanth.
In March of 2023, the FDA accepted a new drug application for another treatment for molluscum contagiosum, berdazimer gel 10.3%. That product is being developed by Novan.
Study examines pediatric skin biopsy trends at a tertiary care center
.
In addition, fewer biopsies were performed in the first 3 years of the global COVID-19 pandemic than in the previous 3 years.
These findings from a retrospective analysis were presented during a poster session at the annual meeting of the Society for Pediatric Dermatology. The analysis set out to evaluate which patients required biopsy, which skin conditions were sampled, and if practice patterns changed following the start of the COVID-19 pandemic.
“The work is important because very few pediatric patients, relative to adult patients seen in dermatology clinics, have a biopsy done,” Kelly M. Cordoro, MD, one of the study authors, told this news organization.
“Approximately 1%-4% of pediatric patients visiting a dermatology clinic will have a biopsy done as compared to 30%-50% of adult patients. Understanding what is being biopsied in children sheds light on the medical decision-making required to decide when a biopsy is necessary,” said Dr. Cordoro, chief of pediatric dermatology at UCSF.
For the study, the researchers retrospectively reviewed 1,196 biopsy specimens from 1,080 unique patients that were performed by pediatric dermatologists at UCSF from 2017 to 2022. Half of the patients were female, their mean age was 11.5 years, and they ranged in age from 1 day to 61 years. Nearly half of biopsies (47%) were performed in patients aged 12-18 years and one-quarter (25.6%) were performed in those aged 6-11 years. In the remaining biopsies, 6.6% came from patients younger than 1 year, 5.8% of those aged 1-2 years, 7.3% from those aged 3-5 years, and 3.9% each in those aged 19-21 years and in those older than 21 years.
The five most common biopsy results were compound nevus (99 biopsies), pyogenic granuloma (96), spongiotic dermatitis (57), intradermal nevus (53), and pilomatricoma (40).
The researchers identified 30 malignant diagnoses in 28 unique patients, most commonly mycosis fungoides (in 16 patients with a median age of 12.5 years), basal cell carcinoma (in 5 patients with a median age of 9 years), and dermatofibrosarcoma protuberans (in 4 patients with a median age of 2 years).
There was no significant sex-based difference in the number of biopsies performed at a given age (P = .47), but Dr. Cordoro and colleagues noted a statistically significant decrease in the number of biopsies during the pandemic compared with the 3 years prior to the pandemic (P = .04).
“There was a slight uptick in 2022, although it remains to be seen whether this trend will continue,” they wrote in their abstract. “While the most common diagnoses in the years leading up to – versus following the start of the pandemic – were similar, there was one clear outlier. The histopathologic diagnosis of pernio spiked in 2020, reflecting the ‘COVID toes’ phenomenon”.
In an interview, Dr. Cordoro said that growths and rashes in children of all ages can, and should, be biopsied, but special considerations are necessary depending on the patient’s age and context.
“Our data showed that neoplastic conditions were biopsied more often than inflammatory conditions, with an emphasis on lesions that required removal (such as pyogenic granuloma), raised concerns for atypia (nevi), or had implications for systemic management (such as Langerhans cell histiocytosis and graft-versus-host disease). Importantly, cutaneous malignancies in children are rare but do occur, and a high index of suspicion is required when approaching any child with a complex neoplasm or rash.”
Dr. Cordoro characterized the medical decision making and rationale for biopsying skin lesions and rashes in children as “a complex process that involves weighing the risks of the biopsy itself against the benefit of the information it will provide; shared decision-making with the caregivers, the patient (if age-appropriate), and other members of the health care team; age of the child and clinical context; and whether the biopsy can be done at the bedside or requires sedation.”
Based on the study results, Dr. Cordoro said, the rationale to proceed with a biopsy boils down to three main goals: To make or confirm a diagnosis, to make decisions about management, and/or the biopsy itself is therapeutic.
UCSF dermatopathology fellow Suzanne W. Birmingham, MD, performed the study in collaboration with Dr. Cordoro and UCSF dermatopathologist Thaddeus W. Mully, MD. Additional analyses of this data set are in progress. The researchers reported having no relevant financial disclosures.
.
In addition, fewer biopsies were performed in the first 3 years of the global COVID-19 pandemic than in the previous 3 years.
These findings from a retrospective analysis were presented during a poster session at the annual meeting of the Society for Pediatric Dermatology. The analysis set out to evaluate which patients required biopsy, which skin conditions were sampled, and if practice patterns changed following the start of the COVID-19 pandemic.
“The work is important because very few pediatric patients, relative to adult patients seen in dermatology clinics, have a biopsy done,” Kelly M. Cordoro, MD, one of the study authors, told this news organization.
“Approximately 1%-4% of pediatric patients visiting a dermatology clinic will have a biopsy done as compared to 30%-50% of adult patients. Understanding what is being biopsied in children sheds light on the medical decision-making required to decide when a biopsy is necessary,” said Dr. Cordoro, chief of pediatric dermatology at UCSF.
For the study, the researchers retrospectively reviewed 1,196 biopsy specimens from 1,080 unique patients that were performed by pediatric dermatologists at UCSF from 2017 to 2022. Half of the patients were female, their mean age was 11.5 years, and they ranged in age from 1 day to 61 years. Nearly half of biopsies (47%) were performed in patients aged 12-18 years and one-quarter (25.6%) were performed in those aged 6-11 years. In the remaining biopsies, 6.6% came from patients younger than 1 year, 5.8% of those aged 1-2 years, 7.3% from those aged 3-5 years, and 3.9% each in those aged 19-21 years and in those older than 21 years.
The five most common biopsy results were compound nevus (99 biopsies), pyogenic granuloma (96), spongiotic dermatitis (57), intradermal nevus (53), and pilomatricoma (40).
The researchers identified 30 malignant diagnoses in 28 unique patients, most commonly mycosis fungoides (in 16 patients with a median age of 12.5 years), basal cell carcinoma (in 5 patients with a median age of 9 years), and dermatofibrosarcoma protuberans (in 4 patients with a median age of 2 years).
There was no significant sex-based difference in the number of biopsies performed at a given age (P = .47), but Dr. Cordoro and colleagues noted a statistically significant decrease in the number of biopsies during the pandemic compared with the 3 years prior to the pandemic (P = .04).
“There was a slight uptick in 2022, although it remains to be seen whether this trend will continue,” they wrote in their abstract. “While the most common diagnoses in the years leading up to – versus following the start of the pandemic – were similar, there was one clear outlier. The histopathologic diagnosis of pernio spiked in 2020, reflecting the ‘COVID toes’ phenomenon”.
In an interview, Dr. Cordoro said that growths and rashes in children of all ages can, and should, be biopsied, but special considerations are necessary depending on the patient’s age and context.
“Our data showed that neoplastic conditions were biopsied more often than inflammatory conditions, with an emphasis on lesions that required removal (such as pyogenic granuloma), raised concerns for atypia (nevi), or had implications for systemic management (such as Langerhans cell histiocytosis and graft-versus-host disease). Importantly, cutaneous malignancies in children are rare but do occur, and a high index of suspicion is required when approaching any child with a complex neoplasm or rash.”
Dr. Cordoro characterized the medical decision making and rationale for biopsying skin lesions and rashes in children as “a complex process that involves weighing the risks of the biopsy itself against the benefit of the information it will provide; shared decision-making with the caregivers, the patient (if age-appropriate), and other members of the health care team; age of the child and clinical context; and whether the biopsy can be done at the bedside or requires sedation.”
Based on the study results, Dr. Cordoro said, the rationale to proceed with a biopsy boils down to three main goals: To make or confirm a diagnosis, to make decisions about management, and/or the biopsy itself is therapeutic.
UCSF dermatopathology fellow Suzanne W. Birmingham, MD, performed the study in collaboration with Dr. Cordoro and UCSF dermatopathologist Thaddeus W. Mully, MD. Additional analyses of this data set are in progress. The researchers reported having no relevant financial disclosures.
.
In addition, fewer biopsies were performed in the first 3 years of the global COVID-19 pandemic than in the previous 3 years.
These findings from a retrospective analysis were presented during a poster session at the annual meeting of the Society for Pediatric Dermatology. The analysis set out to evaluate which patients required biopsy, which skin conditions were sampled, and if practice patterns changed following the start of the COVID-19 pandemic.
“The work is important because very few pediatric patients, relative to adult patients seen in dermatology clinics, have a biopsy done,” Kelly M. Cordoro, MD, one of the study authors, told this news organization.
“Approximately 1%-4% of pediatric patients visiting a dermatology clinic will have a biopsy done as compared to 30%-50% of adult patients. Understanding what is being biopsied in children sheds light on the medical decision-making required to decide when a biopsy is necessary,” said Dr. Cordoro, chief of pediatric dermatology at UCSF.
For the study, the researchers retrospectively reviewed 1,196 biopsy specimens from 1,080 unique patients that were performed by pediatric dermatologists at UCSF from 2017 to 2022. Half of the patients were female, their mean age was 11.5 years, and they ranged in age from 1 day to 61 years. Nearly half of biopsies (47%) were performed in patients aged 12-18 years and one-quarter (25.6%) were performed in those aged 6-11 years. In the remaining biopsies, 6.6% came from patients younger than 1 year, 5.8% of those aged 1-2 years, 7.3% from those aged 3-5 years, and 3.9% each in those aged 19-21 years and in those older than 21 years.
The five most common biopsy results were compound nevus (99 biopsies), pyogenic granuloma (96), spongiotic dermatitis (57), intradermal nevus (53), and pilomatricoma (40).
The researchers identified 30 malignant diagnoses in 28 unique patients, most commonly mycosis fungoides (in 16 patients with a median age of 12.5 years), basal cell carcinoma (in 5 patients with a median age of 9 years), and dermatofibrosarcoma protuberans (in 4 patients with a median age of 2 years).
There was no significant sex-based difference in the number of biopsies performed at a given age (P = .47), but Dr. Cordoro and colleagues noted a statistically significant decrease in the number of biopsies during the pandemic compared with the 3 years prior to the pandemic (P = .04).
“There was a slight uptick in 2022, although it remains to be seen whether this trend will continue,” they wrote in their abstract. “While the most common diagnoses in the years leading up to – versus following the start of the pandemic – were similar, there was one clear outlier. The histopathologic diagnosis of pernio spiked in 2020, reflecting the ‘COVID toes’ phenomenon”.
In an interview, Dr. Cordoro said that growths and rashes in children of all ages can, and should, be biopsied, but special considerations are necessary depending on the patient’s age and context.
“Our data showed that neoplastic conditions were biopsied more often than inflammatory conditions, with an emphasis on lesions that required removal (such as pyogenic granuloma), raised concerns for atypia (nevi), or had implications for systemic management (such as Langerhans cell histiocytosis and graft-versus-host disease). Importantly, cutaneous malignancies in children are rare but do occur, and a high index of suspicion is required when approaching any child with a complex neoplasm or rash.”
Dr. Cordoro characterized the medical decision making and rationale for biopsying skin lesions and rashes in children as “a complex process that involves weighing the risks of the biopsy itself against the benefit of the information it will provide; shared decision-making with the caregivers, the patient (if age-appropriate), and other members of the health care team; age of the child and clinical context; and whether the biopsy can be done at the bedside or requires sedation.”
Based on the study results, Dr. Cordoro said, the rationale to proceed with a biopsy boils down to three main goals: To make or confirm a diagnosis, to make decisions about management, and/or the biopsy itself is therapeutic.
UCSF dermatopathology fellow Suzanne W. Birmingham, MD, performed the study in collaboration with Dr. Cordoro and UCSF dermatopathologist Thaddeus W. Mully, MD. Additional analyses of this data set are in progress. The researchers reported having no relevant financial disclosures.
FROM SPD 2023
Indian Health Service dermatologist: ‘I saw a real need to be of service’
After completing his dermatology residency at Johns Hopkins Hospital in 2010, Christopher Bengson, MD, MHS, then a Lieutenant Commander in the U.S. Public Health Service, accepted an offer to become a full-time dermatologist at Phoenix Indian Medical Center (PIMC) in Arizona, fulfilling a long desire to provide care for underserved individuals. Thirteen years later, .
As one of the largest hospitals in the IHS system, PIMC provides direct health care services to a population of more than 156,000, including tribal members from The Fort McDowell Yavapai Nation, the Salt River Pima-Maricopa Indian Community, and the San Lucy District of the Tohono O’odham Nation, the Tonto Apache Tribe, the Yavapai-Apache Indian Tribe, and the Yavapai-Prescott Indian Tribe. Dr. Bengson also cares for tribal members who travel to PIMC from as far away as Washington State and Hawaii to receive dermatologic care.

“There is a disproportionate number of Native American patients that come in with severe psoriasis, hidradenitis suppurativa, and dissecting cellulitis of the scalp compared to the general U.S. population, and I’ve been surprised by how many have nonmelanoma skin cancers and autoimmune connective tissue diseases like lupus, as the prevailing sentiment among his patients is that Native people do not get skin cancer,” he said in an interview. “Those who travel great distances are those who come see me for the surgical removal of skin cancers.”
Interesting cases he’s seen in his nearly 13 years on the job include Epstein-Barr virus-induced NK/T-cell lymphoma, anaplastic large cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma, and necrobiotic xanthogranuloma, “tumors that have generally gone to tertiary care facilities for treatment, but we’ve been able to manage here.”
In 2017, Dr. Bengson was appointed as the IHS’s first chief clinical consultant for dermatology, a post that provides him the opportunity to interface with Native people and IHS-affiliated clinicians nationwide regarding skin-related questions and concerns. As the only full-time dermatologist employed by the IHS, he also views his role as providing an opportunity to change the perception that some Native Americans may still hold about federally delivered health care, “where there may be a cultural distrust of government health care in indigenous communities, driven by generational historical traumas that have come out of boarding schools, population relocation to desolate and isolated areas of the country, and contracts that were simply not honored,” he explained.
“While none of these issues are new, what has been great for me is that I’m going on 13 years of being at the same facility, and I’ve treated family members, their kids, and even their grandkids. In some ways the primary barrier of continuity of care – at least at PIMC – has been eliminated by me just being here for a long period of time.”
In Dr. Bengson’s opinion, efforts to improve access to attract more Native Americans to dermatology are laudable, including the American Academy of Dermatology’s Pathways Program, which aims to increase the number of dermatology residents from Black, Latino, and indigenous communities from approximately 100 residents to 250 residents by 2027, or by over 150%, through community-based engagement strategies that begin in high school.
“To have an objective benchmark is encouraging,” he said. However, he encourages dermatology residency program directors to rethink how they recruit Native Americans, many of whom hail from rural areas. “If you’re recruiting primarily from urban settings, you’re very unlikely to include Native Americans as a larger group of minorities,” he said. “When you look at the number of department chairs who are Native American, it’s on the order of 0.1%, [so] it’s no surprise that dermatologists coming out of a residency program don’t want to go to reservations to provide dermatologic care. We pay a lot of lip service to mentorship programs and things like that, but you need a mentor who follows you through the process – and it’s a long process.”
He believes that residency program directors should reconsider the metrics used to select dermatology residents and should consider the degree of adversity that a Native American applicant may have had to overcome to make it to the residency selection committees.
Despite obstacles to attracting young Native Americans to a career in medicine, Dr. Bengson sees encouraging signs ahead. Some of his Native American patients and family members of patients have enrolled in medical school and have asked to rotate with him at PIMC at the premedical and medical student level. “Some have moved on, not necessarily to dermatology, but to other specialties and careers in health care,” he said. “When you have such high rates of obesity, diabetes, hypertension, coronary artery disease, and stroke in Native American communities, nodulocystic acne and other skin conditions that are not threats to life and limb become less of a priority. We need to get more people in the pipeline to deliver medical services even if it may not be in dermatology, as the need for dedicated health care professionals is so great across all disciplines.”
After completing his dermatology residency at Johns Hopkins Hospital in 2010, Christopher Bengson, MD, MHS, then a Lieutenant Commander in the U.S. Public Health Service, accepted an offer to become a full-time dermatologist at Phoenix Indian Medical Center (PIMC) in Arizona, fulfilling a long desire to provide care for underserved individuals. Thirteen years later, .
As one of the largest hospitals in the IHS system, PIMC provides direct health care services to a population of more than 156,000, including tribal members from The Fort McDowell Yavapai Nation, the Salt River Pima-Maricopa Indian Community, and the San Lucy District of the Tohono O’odham Nation, the Tonto Apache Tribe, the Yavapai-Apache Indian Tribe, and the Yavapai-Prescott Indian Tribe. Dr. Bengson also cares for tribal members who travel to PIMC from as far away as Washington State and Hawaii to receive dermatologic care.
“There is a disproportionate number of Native American patients that come in with severe psoriasis, hidradenitis suppurativa, and dissecting cellulitis of the scalp compared to the general U.S. population, and I’ve been surprised by how many have nonmelanoma skin cancers and autoimmune connective tissue diseases like lupus, as the prevailing sentiment among his patients is that Native people do not get skin cancer,” he said in an interview. “Those who travel great distances are those who come see me for the surgical removal of skin cancers.”
Interesting cases he’s seen in his nearly 13 years on the job include Epstein-Barr virus-induced NK/T-cell lymphoma, anaplastic large cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma, and necrobiotic xanthogranuloma, “tumors that have generally gone to tertiary care facilities for treatment, but we’ve been able to manage here.”
In 2017, Dr. Bengson was appointed as the IHS’s first chief clinical consultant for dermatology, a post that provides him the opportunity to interface with Native people and IHS-affiliated clinicians nationwide regarding skin-related questions and concerns. As the only full-time dermatologist employed by the IHS, he also views his role as providing an opportunity to change the perception that some Native Americans may still hold about federally delivered health care, “where there may be a cultural distrust of government health care in indigenous communities, driven by generational historical traumas that have come out of boarding schools, population relocation to desolate and isolated areas of the country, and contracts that were simply not honored,” he explained.
“While none of these issues are new, what has been great for me is that I’m going on 13 years of being at the same facility, and I’ve treated family members, their kids, and even their grandkids. In some ways the primary barrier of continuity of care – at least at PIMC – has been eliminated by me just being here for a long period of time.”
In Dr. Bengson’s opinion, efforts to improve access to attract more Native Americans to dermatology are laudable, including the American Academy of Dermatology’s Pathways Program, which aims to increase the number of dermatology residents from Black, Latino, and indigenous communities from approximately 100 residents to 250 residents by 2027, or by over 150%, through community-based engagement strategies that begin in high school.
“To have an objective benchmark is encouraging,” he said. However, he encourages dermatology residency program directors to rethink how they recruit Native Americans, many of whom hail from rural areas. “If you’re recruiting primarily from urban settings, you’re very unlikely to include Native Americans as a larger group of minorities,” he said. “When you look at the number of department chairs who are Native American, it’s on the order of 0.1%, [so] it’s no surprise that dermatologists coming out of a residency program don’t want to go to reservations to provide dermatologic care. We pay a lot of lip service to mentorship programs and things like that, but you need a mentor who follows you through the process – and it’s a long process.”
He believes that residency program directors should reconsider the metrics used to select dermatology residents and should consider the degree of adversity that a Native American applicant may have had to overcome to make it to the residency selection committees.
Despite obstacles to attracting young Native Americans to a career in medicine, Dr. Bengson sees encouraging signs ahead. Some of his Native American patients and family members of patients have enrolled in medical school and have asked to rotate with him at PIMC at the premedical and medical student level. “Some have moved on, not necessarily to dermatology, but to other specialties and careers in health care,” he said. “When you have such high rates of obesity, diabetes, hypertension, coronary artery disease, and stroke in Native American communities, nodulocystic acne and other skin conditions that are not threats to life and limb become less of a priority. We need to get more people in the pipeline to deliver medical services even if it may not be in dermatology, as the need for dedicated health care professionals is so great across all disciplines.”
After completing his dermatology residency at Johns Hopkins Hospital in 2010, Christopher Bengson, MD, MHS, then a Lieutenant Commander in the U.S. Public Health Service, accepted an offer to become a full-time dermatologist at Phoenix Indian Medical Center (PIMC) in Arizona, fulfilling a long desire to provide care for underserved individuals. Thirteen years later, .
As one of the largest hospitals in the IHS system, PIMC provides direct health care services to a population of more than 156,000, including tribal members from The Fort McDowell Yavapai Nation, the Salt River Pima-Maricopa Indian Community, and the San Lucy District of the Tohono O’odham Nation, the Tonto Apache Tribe, the Yavapai-Apache Indian Tribe, and the Yavapai-Prescott Indian Tribe. Dr. Bengson also cares for tribal members who travel to PIMC from as far away as Washington State and Hawaii to receive dermatologic care.
“There is a disproportionate number of Native American patients that come in with severe psoriasis, hidradenitis suppurativa, and dissecting cellulitis of the scalp compared to the general U.S. population, and I’ve been surprised by how many have nonmelanoma skin cancers and autoimmune connective tissue diseases like lupus, as the prevailing sentiment among his patients is that Native people do not get skin cancer,” he said in an interview. “Those who travel great distances are those who come see me for the surgical removal of skin cancers.”
Interesting cases he’s seen in his nearly 13 years on the job include Epstein-Barr virus-induced NK/T-cell lymphoma, anaplastic large cell lymphoma, subcutaneous panniculitis-like T-cell lymphoma, and necrobiotic xanthogranuloma, “tumors that have generally gone to tertiary care facilities for treatment, but we’ve been able to manage here.”
In 2017, Dr. Bengson was appointed as the IHS’s first chief clinical consultant for dermatology, a post that provides him the opportunity to interface with Native people and IHS-affiliated clinicians nationwide regarding skin-related questions and concerns. As the only full-time dermatologist employed by the IHS, he also views his role as providing an opportunity to change the perception that some Native Americans may still hold about federally delivered health care, “where there may be a cultural distrust of government health care in indigenous communities, driven by generational historical traumas that have come out of boarding schools, population relocation to desolate and isolated areas of the country, and contracts that were simply not honored,” he explained.
“While none of these issues are new, what has been great for me is that I’m going on 13 years of being at the same facility, and I’ve treated family members, their kids, and even their grandkids. In some ways the primary barrier of continuity of care – at least at PIMC – has been eliminated by me just being here for a long period of time.”
In Dr. Bengson’s opinion, efforts to improve access to attract more Native Americans to dermatology are laudable, including the American Academy of Dermatology’s Pathways Program, which aims to increase the number of dermatology residents from Black, Latino, and indigenous communities from approximately 100 residents to 250 residents by 2027, or by over 150%, through community-based engagement strategies that begin in high school.
“To have an objective benchmark is encouraging,” he said. However, he encourages dermatology residency program directors to rethink how they recruit Native Americans, many of whom hail from rural areas. “If you’re recruiting primarily from urban settings, you’re very unlikely to include Native Americans as a larger group of minorities,” he said. “When you look at the number of department chairs who are Native American, it’s on the order of 0.1%, [so] it’s no surprise that dermatologists coming out of a residency program don’t want to go to reservations to provide dermatologic care. We pay a lot of lip service to mentorship programs and things like that, but you need a mentor who follows you through the process – and it’s a long process.”
He believes that residency program directors should reconsider the metrics used to select dermatology residents and should consider the degree of adversity that a Native American applicant may have had to overcome to make it to the residency selection committees.
Despite obstacles to attracting young Native Americans to a career in medicine, Dr. Bengson sees encouraging signs ahead. Some of his Native American patients and family members of patients have enrolled in medical school and have asked to rotate with him at PIMC at the premedical and medical student level. “Some have moved on, not necessarily to dermatology, but to other specialties and careers in health care,” he said. “When you have such high rates of obesity, diabetes, hypertension, coronary artery disease, and stroke in Native American communities, nodulocystic acne and other skin conditions that are not threats to life and limb become less of a priority. We need to get more people in the pipeline to deliver medical services even if it may not be in dermatology, as the need for dedicated health care professionals is so great across all disciplines.”