User login
Cancer Data Trends 2025
The annual issue of Cancer Data Trends, produced in collaboration with the Association of VA Hematology/Oncology (AVAHO), highlights the latest research in some of the top cancers impacting US veterans.

In this issue:
- Access, Race, and "Colon Age": Improving CRC Screening
- Lung Cancer: Mortality Trends in Veterans and New Treatments
- Racial Disparities, Germline Testing, and Improved Overall Survival in Prostate Cancer
- Breast and Uterine Cancer: Screening Guidelines, Genetic Testing, and Mortality Trends
- HCC Updates: Quality Care Framework and Risk Stratification Data
- Rising Kidney Cancer Cases and Emerging Treatments for Veterans
- Advances in Blood Cancer Care for Veterans
- AI-Based Risk Stratification for Oropharyngeal Carcinomas: AIROC
- Brain Cancer: Epidemiology, TBI, and New Treatments
The annual issue of Cancer Data Trends, produced in collaboration with the Association of VA Hematology/Oncology (AVAHO), highlights the latest research in some of the top cancers impacting US veterans.
In this issue:
- Access, Race, and "Colon Age": Improving CRC Screening
- Lung Cancer: Mortality Trends in Veterans and New Treatments
- Racial Disparities, Germline Testing, and Improved Overall Survival in Prostate Cancer
- Breast and Uterine Cancer: Screening Guidelines, Genetic Testing, and Mortality Trends
- HCC Updates: Quality Care Framework and Risk Stratification Data
- Rising Kidney Cancer Cases and Emerging Treatments for Veterans
- Advances in Blood Cancer Care for Veterans
- AI-Based Risk Stratification for Oropharyngeal Carcinomas: AIROC
- Brain Cancer: Epidemiology, TBI, and New Treatments
The annual issue of Cancer Data Trends, produced in collaboration with the Association of VA Hematology/Oncology (AVAHO), highlights the latest research in some of the top cancers impacting US veterans.
In this issue:
- Access, Race, and "Colon Age": Improving CRC Screening
- Lung Cancer: Mortality Trends in Veterans and New Treatments
- Racial Disparities, Germline Testing, and Improved Overall Survival in Prostate Cancer
- Breast and Uterine Cancer: Screening Guidelines, Genetic Testing, and Mortality Trends
- HCC Updates: Quality Care Framework and Risk Stratification Data
- Rising Kidney Cancer Cases and Emerging Treatments for Veterans
- Advances in Blood Cancer Care for Veterans
- AI-Based Risk Stratification for Oropharyngeal Carcinomas: AIROC
- Brain Cancer: Epidemiology, TBI, and New Treatments

Rare Blood Diseases the Focus of AVAHO Virtual Session
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
The Association of Veterans Affairs Hematology/Oncology (AVAHO) will provide more than education during a special virtual session on July 18 devoted to 4 rare and ultra-rare disorders in classical hematology. The program includes 2.25 free continuing education credits and the organization hopes it will spur hematology pathways within the US Department of Veterans Affairs (VA) National Oncology Program, according to AVAHO President Nicholas Burwick, MD.
“Guidance on hematology disorders lag far behind oncologic disorders at the national level,” said Burwick, who specializes in hematology and bone marrow transplant at the Veterans Affairs Puget Sound Health Care System in Seattle.
“Providers are facing real diagnostic and treatment decisions, and there are more and more treatment options available, some with significant cost implications,” Burwick said.
Burwick He is optimistic the session will have a lasting impact. He expanded on the 4 disorders slated for discussion at the meeting: aplastic anemia, thrombotic thrombocytopenic purpura (TTP), hereditary hemorrhagic telangiectasia (HHT), and acquired hemophilia A in a discussion with Federal Practitioner. The interview transcript has been edited for length and clarity.
What is aplastic anemia?
Aplastic anemia is a true bone marrow failure. The marrow stops producing cells. At a referral center like Seattle, we might see a few [cases] a year because we also do bone marrow transplant. Most facilities are probably seeing 1 or 2 a year at most.
Different mechanisms are behind it: immune-mediated causes, genetic predispositions, and acquired toxic injury. In the military population, aplastic anemia due to toxic exposure is a significant concern.
Who tends to develop aplastic anemia?
The age spectrum is broader than people think. We do see it in patients in their 40s and 50s, not just seniors. The general rule has been, if a patient is under 40, pursue transplant; over 40, pursue immunosuppressive therapy. However, that cutoff is somewhat arbitrary.
What are some things clinicians should understand about this disease?
Clinicians need to know the diagnostic criteria, what tests to run, how to put in transplant referral requests, and how to get a case on the radar at a transplant center like Nashville or Seattle.
Patients need referral quickly. They often don’t respond to standard treatments like growth factors, so they end up requiring a lot of transfusions. You don’t want to sit around.
Even if the transplant happens outside the VA, it usually runs through a transplant center for review first. There’s also the question of whether a condition qualifies as a service-connected disability or if the diagnosis is a presumptive condition for certain exposures.
How often do you see TTP, which produces small blood clots in blood vessels?
Some clinicians may see 1 case every 3 to 4 years, or possibly 1 case in an entire career. There is an inherited form and an acquired form. We’re primarily focused on the acquired autoimmune form, which can present in young adulthood or later.
What should clinicians know?
These patients come in needing urgent treatment. Recognizing the diagnosis quickly, ordering the right tests, and acting fast are all critical. This can be life-threatening within days without treatment.
Treatment involves plasma exchange, which not every facility is equipped to perform. There are also medications: steroids are standard, but there’s also caplacizumab, which is highly specific to TTP and unlikely to be stocked in a VA pharmacy because of how rarely it’s needed. Pharmacies often have to procure it on demand once the diagnosis is made, which can delay care. A key part of managing these cases is knowing who to reach out to and when to transfer a patient to an academic or community partner that has plasma exchange capability.
Are VA clinicians likely to see HHT, an inherited disorder that causes bleeding due to malformed blood vessels?
There’s a misconception that veterans don’t have inherited bleeding disorders, the assumption being that they wouldn’t have gotten into the military (had they had them). But many of these conditions don’t get diagnosed until later in life: symptoms can be mild initially or not present until young adulthood or later.
Patients might come in with recurrent nosebleeds or unexplained [gastrointestinal] bleeding. We probably all have patients with HHT in our practices without knowing it because we’re not always doing appropriate diagnostic testing.
How are treatments evolving?
Recent developments include both local options like laser treatment and systemic medications such as pomalidomide, which was approved recently, and bevacizumab. [The virtual session] speaker has been involved in clinical trials for pomalidomide and will speak to when to use these medications and how to choose between them now that there are options, both of which are expensive.
What is acquired hemophilia A?
It occurs when someone without a genetic predisposition loses their factor VIII activity due to an autoimmune process. It presents quickly, often with bleeding or bruising, and patients frequently show up in the [emergency department].
The treatment challenge is distinct from inherited hemophilia. Standard factor VIII replacement doesn’t work here because the autoantibody breaks it down. Treatment requires bypassing factor VIII—options include factor VIIa and emicizumab.
Emicizumab is FDA-approved for inherited hemophilia A and used off-label for the acquired form. Procuring it in a VA facility can be difficult, and that is exactly where a VA clinical pathway would help.
The VA doesn’t currently have strong hemophilia expertise internally. So, engaging with hemophilia treatment centers is important, as is developing subject matter experts within VA hematology who can serve as go-to resources for less-resourced facilities.
What unites these 4 rare hematology conditions?
There are common threads: rare presentations requiring urgent decision-making, diagnostic criteria that aren’t always familiar, and treatments that may be hard to procure quickly. VA-specific resources—pathways, referral contacts, teleoncology consults—can make the difference in patient outcomes.
A centralized virtual hematology hub, where providers could reach a knowledgeable hematologist for consultation, would go a long way. That’s what we’re ultimately trying to build toward.
The AVAHO Virtual Session on Rare and Ultra-Rare Hematologic Disorders will be held on July 18, 2026, from 12-2:30 p.m. EST.
The program is available to any health care professional who wants to learn more about the diagnosis and management of these hematologic disorders; 2.25 free continuing education credits are available.
The speakers are:
• Aplastic anemia: Emma Groarke, MB, BCh, BAO, MD, National Institutes of Health
• Thrombotic thrombocytopenic purpura: Yazan Abou-Ismail, MD, University of Utah
• Hereditary hemorrhagic telangiectasia: Hanny Al-Samkari, MD, Massachusetts General Hospital/Harvard Medical School; and
• Acquired hemophilia A, Aaron Boothby, MD, University of Washington/Fred Hutchinson Cancer Center.
The Fastest Way to Better Anticoagulants May Be a Land Snail
The Fastest Way to Better Anticoagulants May Be a Land Snail
The fastest way to a new anticoagulation therapy that prevents blood clots without prolonging bleeding or healing time may be a land snail.
A recent preclinical study in ACS Central Science investigating bioactive molecules derived from the land snail Camaena cicatricosa identified a compound that significantly reduced clot formation without prolonging bleeding time and wound healing in rodent models, directly challenging the assumption that effective anticoagulant therapy must inherently disrupt physiologic repair processes.
“I was quite excited,” said Lisha Lin, PhD, lead author on the study and research assistant at the State Key Laboratory of Phytochemistry and Natural Medicines at the Kunming Institute of Botany, Chinese Academy of Sciences, in Kunming, China. “A novel polysaccharide was identified, and more importantly, it showed anticoagulant activity by inhibiting a novel target, with different action mechanism from heparins.”
What Led to the Land Snail?
The choice of C cicatricosa reflects a shift in thinking. The team screened a range of mollusk-derived biomolecules in search of safer anticoagulation strategies, ultimately isolating a novel galactosylated glycosaminoglycan (CCG) from this terrestrial species.
“We compared the anticoagulant activity of glycosaminoglycans from three snail species,” said Lin. They initially studied polysaccharides from C cicatricosa, Achatina fulica, and Helix lucorum — all sulfated glycosaminoglycans with similar structures. “However, we only found CCG from [C cicatricosa] showed anticoagulant activity but not other snail polysaccharides.”
This unexpected selectivity proved crucial. “The result indicated that the galactose branches and special sulfate substitution were important,” she explained, helping to explain why this particular species stood out among structurally similar compounds.
While CCG shares some similarities with heparin-like molecules, it notably lacks the specific pentasaccharide sequence required for antithrombin binding. Researchers hypothesized that this absence could reduce bleeding risk while maintaining antithrombotic activity.
Rather than broadly inhibiting coagulation, the compound selectively disrupts the intrinsic tenase complex, a pathway more closely associated with pathologic thrombosis than with physiologic hemostasis. This mechanism’s selectivity is central to the study’s findings and helps explain why normal wound healing remained intact in preclinical models.
The isolated compound demonstrated a rare combination in anticoagulant research: potent inhibition of pathologic thrombosis with no significant increase in bleeding time and intact wound healing across multiple experimental models. The compound did not act as a broad-spectrum anticoagulant, instead selectively targeting pathways more relevant to disease-associated clot formation.
The Hope of Lowering Bleeding Risk
For decades, anticoagulation therapy has been built on the assumption that inhibiting clot formation inevitably increases bleeding risk. For physicians treating patients with deep vein thrombosis or atrial fibrillation or those requiring postsurgical attention, the balancing act is constant. Prevent clot formation aggressively enough and the bleeding risk rises. Reduce intensity and thrombosis risk returns.
Current therapies, including heparin and direct oral anticoagulants (DOACs), function by broadly targeting the coagulation cascade. This lack of specialty is precisely what limits them. Even when carefully dosed, the treatments interfere with both pathologic clot formation and physiologic hemostasis.
Physicians managing patients on heparin and DOACs frequently encounter recurrent epistaxis, gastrointestinal bleeding ranging from occult to clinically significant, and urogenital bleeding. A degree of mild bleeding after surgery is often expected and usually resolves on its own. However, it’s crucial to evaluate in the context of ongoing anticoagulation to rule out early signs of clinically significant complications.
“There is definitely a lot of interest in the concept of ‘uncoupling’ thrombosis from hemostasis,” said Yazan Abou-Ismail, MD, hematologist and associate professor of medicine at the University of Utah Health in Salt Lake City, who was not involved in the research. “This concept highlights the differences in pathways essential for normal hemostasis at sites of vessel injury, in contrast with those needed for clot propagation and blood vessel lumen occlusion.”
Abou-Ismail noted that this approach has been explored with Factor XI (FXI) inhibitors currently in clinical trials. However, he raised an important mechanistic concern.
“This mechanism may not necessarily accomplish that goal of uncoupling thrombosis from hemostasis, although it might at narrow therapeutic windows,” Abou-Ismail said. “The tenase complex is a central component of coagulation whose deficiency underlies hemophilia A and B, diseases that cause significant bleeding in humans, and it is more essential to hemostasis than [FXI inhibitors]. Tenase inhibition seems like it may pose a higher bleeding risk in humans.”
He explained that FXI inhibitors have a strong mechanistic rationale because they target the feedback loop that amplifies clot propagation, which is less essential for hemostasis. “FXI inhibitors have clinical trial data demonstrating that FXI inhibition is in fact associated with less bleeding compared to current established anticoagulants,” he said. “However, CCG disrupts the FIXa/FVIIIa intrinsic tenase complex itself, which is considered essential for hemostasis.”
Surprises and Confirmations
The researchers were not anticipating such a clear separation between antithrombotic activity and bleeding risk. The preservation of normal wound healing was equally surprising, directly challenging the belief that interfering with clot formation inevitably disrupts tissue repair.
However, the path to these conclusions was not straightforward. “The structural definition of complex macromolecules like sulfated polysaccharides is a common challenge in the research field,” Lin said. “We spent a lot of time to analyze the structure of CCG.”
Even after identifying the candidate compound, the team had to rigorously confirm that its effects were truly anticoagulant in nature, rather than secondary to anti-inflammatory or vascular remodeling properties. Mechanistic studies were essential in demonstrating its targeted disruption of the intrinsic tenase complex, helping to explain how thrombosis could be reduced without broadly impairing coagulation.
Lin is excited but knows patience and skepticism are needed. “Our research is still at the basic stage, but based on the available data, we may provide a potential anticoagulant option with low bleeding risk,” she said. “In the discussion section of our paper, we also stated that ‘the data suggest a wide therapeutic window of CCG, which may offer therapeutic advantages for patients with bleeding contraindication, such as elderly patients and those with renal failure, as well as for safer long-term anticoagulation.’”
A New Direction for Heparin Alternatives?
Despite these concerns, Abou-Ismail acknowledged that the research has genuinely noteworthy aspects. “A future anticoagulant with a novel mechanism of action may be useful in patients who have experienced anticoagulant failure or breakthrough thrombosis from currently established anticoagulants,” he said. “Having another option might be useful when all other options have failed or are not feasible.”
However, he added a note of caution: “If a therapeutic window exists where partial tenase disruption has antithrombotic effect that does not impair hemostasis, then that would definitely be a promising future finding, but it is too early to arrive at that conclusion with this study.”
The search for safer heparin alternatives has been ongoing for decades, but most candidates still operate within the same fundamental paradigm of broad coagulation inhibition. This snail can’t move fast enough.
Abou-Ismail reported having no relevant conflicts. Disclosure information for study authors is available in the original study publication.
A version of this article first appeared on Medscape.com.
The fastest way to a new anticoagulation therapy that prevents blood clots without prolonging bleeding or healing time may be a land snail.
A recent preclinical study in ACS Central Science investigating bioactive molecules derived from the land snail Camaena cicatricosa identified a compound that significantly reduced clot formation without prolonging bleeding time and wound healing in rodent models, directly challenging the assumption that effective anticoagulant therapy must inherently disrupt physiologic repair processes.
“I was quite excited,” said Lisha Lin, PhD, lead author on the study and research assistant at the State Key Laboratory of Phytochemistry and Natural Medicines at the Kunming Institute of Botany, Chinese Academy of Sciences, in Kunming, China. “A novel polysaccharide was identified, and more importantly, it showed anticoagulant activity by inhibiting a novel target, with different action mechanism from heparins.”
What Led to the Land Snail?
The choice of C cicatricosa reflects a shift in thinking. The team screened a range of mollusk-derived biomolecules in search of safer anticoagulation strategies, ultimately isolating a novel galactosylated glycosaminoglycan (CCG) from this terrestrial species.
“We compared the anticoagulant activity of glycosaminoglycans from three snail species,” said Lin. They initially studied polysaccharides from C cicatricosa, Achatina fulica, and Helix lucorum — all sulfated glycosaminoglycans with similar structures. “However, we only found CCG from [C cicatricosa] showed anticoagulant activity but not other snail polysaccharides.”
This unexpected selectivity proved crucial. “The result indicated that the galactose branches and special sulfate substitution were important,” she explained, helping to explain why this particular species stood out among structurally similar compounds.
While CCG shares some similarities with heparin-like molecules, it notably lacks the specific pentasaccharide sequence required for antithrombin binding. Researchers hypothesized that this absence could reduce bleeding risk while maintaining antithrombotic activity.
Rather than broadly inhibiting coagulation, the compound selectively disrupts the intrinsic tenase complex, a pathway more closely associated with pathologic thrombosis than with physiologic hemostasis. This mechanism’s selectivity is central to the study’s findings and helps explain why normal wound healing remained intact in preclinical models.
The isolated compound demonstrated a rare combination in anticoagulant research: potent inhibition of pathologic thrombosis with no significant increase in bleeding time and intact wound healing across multiple experimental models. The compound did not act as a broad-spectrum anticoagulant, instead selectively targeting pathways more relevant to disease-associated clot formation.
The Hope of Lowering Bleeding Risk
For decades, anticoagulation therapy has been built on the assumption that inhibiting clot formation inevitably increases bleeding risk. For physicians treating patients with deep vein thrombosis or atrial fibrillation or those requiring postsurgical attention, the balancing act is constant. Prevent clot formation aggressively enough and the bleeding risk rises. Reduce intensity and thrombosis risk returns.
Current therapies, including heparin and direct oral anticoagulants (DOACs), function by broadly targeting the coagulation cascade. This lack of specialty is precisely what limits them. Even when carefully dosed, the treatments interfere with both pathologic clot formation and physiologic hemostasis.
Physicians managing patients on heparin and DOACs frequently encounter recurrent epistaxis, gastrointestinal bleeding ranging from occult to clinically significant, and urogenital bleeding. A degree of mild bleeding after surgery is often expected and usually resolves on its own. However, it’s crucial to evaluate in the context of ongoing anticoagulation to rule out early signs of clinically significant complications.
“There is definitely a lot of interest in the concept of ‘uncoupling’ thrombosis from hemostasis,” said Yazan Abou-Ismail, MD, hematologist and associate professor of medicine at the University of Utah Health in Salt Lake City, who was not involved in the research. “This concept highlights the differences in pathways essential for normal hemostasis at sites of vessel injury, in contrast with those needed for clot propagation and blood vessel lumen occlusion.”
Abou-Ismail noted that this approach has been explored with Factor XI (FXI) inhibitors currently in clinical trials. However, he raised an important mechanistic concern.
“This mechanism may not necessarily accomplish that goal of uncoupling thrombosis from hemostasis, although it might at narrow therapeutic windows,” Abou-Ismail said. “The tenase complex is a central component of coagulation whose deficiency underlies hemophilia A and B, diseases that cause significant bleeding in humans, and it is more essential to hemostasis than [FXI inhibitors]. Tenase inhibition seems like it may pose a higher bleeding risk in humans.”
He explained that FXI inhibitors have a strong mechanistic rationale because they target the feedback loop that amplifies clot propagation, which is less essential for hemostasis. “FXI inhibitors have clinical trial data demonstrating that FXI inhibition is in fact associated with less bleeding compared to current established anticoagulants,” he said. “However, CCG disrupts the FIXa/FVIIIa intrinsic tenase complex itself, which is considered essential for hemostasis.”
Surprises and Confirmations
The researchers were not anticipating such a clear separation between antithrombotic activity and bleeding risk. The preservation of normal wound healing was equally surprising, directly challenging the belief that interfering with clot formation inevitably disrupts tissue repair.
However, the path to these conclusions was not straightforward. “The structural definition of complex macromolecules like sulfated polysaccharides is a common challenge in the research field,” Lin said. “We spent a lot of time to analyze the structure of CCG.”
Even after identifying the candidate compound, the team had to rigorously confirm that its effects were truly anticoagulant in nature, rather than secondary to anti-inflammatory or vascular remodeling properties. Mechanistic studies were essential in demonstrating its targeted disruption of the intrinsic tenase complex, helping to explain how thrombosis could be reduced without broadly impairing coagulation.
Lin is excited but knows patience and skepticism are needed. “Our research is still at the basic stage, but based on the available data, we may provide a potential anticoagulant option with low bleeding risk,” she said. “In the discussion section of our paper, we also stated that ‘the data suggest a wide therapeutic window of CCG, which may offer therapeutic advantages for patients with bleeding contraindication, such as elderly patients and those with renal failure, as well as for safer long-term anticoagulation.’”
A New Direction for Heparin Alternatives?
Despite these concerns, Abou-Ismail acknowledged that the research has genuinely noteworthy aspects. “A future anticoagulant with a novel mechanism of action may be useful in patients who have experienced anticoagulant failure or breakthrough thrombosis from currently established anticoagulants,” he said. “Having another option might be useful when all other options have failed or are not feasible.”
However, he added a note of caution: “If a therapeutic window exists where partial tenase disruption has antithrombotic effect that does not impair hemostasis, then that would definitely be a promising future finding, but it is too early to arrive at that conclusion with this study.”
The search for safer heparin alternatives has been ongoing for decades, but most candidates still operate within the same fundamental paradigm of broad coagulation inhibition. This snail can’t move fast enough.
Abou-Ismail reported having no relevant conflicts. Disclosure information for study authors is available in the original study publication.
A version of this article first appeared on Medscape.com.
The fastest way to a new anticoagulation therapy that prevents blood clots without prolonging bleeding or healing time may be a land snail.
A recent preclinical study in ACS Central Science investigating bioactive molecules derived from the land snail Camaena cicatricosa identified a compound that significantly reduced clot formation without prolonging bleeding time and wound healing in rodent models, directly challenging the assumption that effective anticoagulant therapy must inherently disrupt physiologic repair processes.
“I was quite excited,” said Lisha Lin, PhD, lead author on the study and research assistant at the State Key Laboratory of Phytochemistry and Natural Medicines at the Kunming Institute of Botany, Chinese Academy of Sciences, in Kunming, China. “A novel polysaccharide was identified, and more importantly, it showed anticoagulant activity by inhibiting a novel target, with different action mechanism from heparins.”
What Led to the Land Snail?
The choice of C cicatricosa reflects a shift in thinking. The team screened a range of mollusk-derived biomolecules in search of safer anticoagulation strategies, ultimately isolating a novel galactosylated glycosaminoglycan (CCG) from this terrestrial species.
“We compared the anticoagulant activity of glycosaminoglycans from three snail species,” said Lin. They initially studied polysaccharides from C cicatricosa, Achatina fulica, and Helix lucorum — all sulfated glycosaminoglycans with similar structures. “However, we only found CCG from [C cicatricosa] showed anticoagulant activity but not other snail polysaccharides.”
This unexpected selectivity proved crucial. “The result indicated that the galactose branches and special sulfate substitution were important,” she explained, helping to explain why this particular species stood out among structurally similar compounds.
While CCG shares some similarities with heparin-like molecules, it notably lacks the specific pentasaccharide sequence required for antithrombin binding. Researchers hypothesized that this absence could reduce bleeding risk while maintaining antithrombotic activity.
Rather than broadly inhibiting coagulation, the compound selectively disrupts the intrinsic tenase complex, a pathway more closely associated with pathologic thrombosis than with physiologic hemostasis. This mechanism’s selectivity is central to the study’s findings and helps explain why normal wound healing remained intact in preclinical models.
The isolated compound demonstrated a rare combination in anticoagulant research: potent inhibition of pathologic thrombosis with no significant increase in bleeding time and intact wound healing across multiple experimental models. The compound did not act as a broad-spectrum anticoagulant, instead selectively targeting pathways more relevant to disease-associated clot formation.
The Hope of Lowering Bleeding Risk
For decades, anticoagulation therapy has been built on the assumption that inhibiting clot formation inevitably increases bleeding risk. For physicians treating patients with deep vein thrombosis or atrial fibrillation or those requiring postsurgical attention, the balancing act is constant. Prevent clot formation aggressively enough and the bleeding risk rises. Reduce intensity and thrombosis risk returns.
Current therapies, including heparin and direct oral anticoagulants (DOACs), function by broadly targeting the coagulation cascade. This lack of specialty is precisely what limits them. Even when carefully dosed, the treatments interfere with both pathologic clot formation and physiologic hemostasis.
Physicians managing patients on heparin and DOACs frequently encounter recurrent epistaxis, gastrointestinal bleeding ranging from occult to clinically significant, and urogenital bleeding. A degree of mild bleeding after surgery is often expected and usually resolves on its own. However, it’s crucial to evaluate in the context of ongoing anticoagulation to rule out early signs of clinically significant complications.
“There is definitely a lot of interest in the concept of ‘uncoupling’ thrombosis from hemostasis,” said Yazan Abou-Ismail, MD, hematologist and associate professor of medicine at the University of Utah Health in Salt Lake City, who was not involved in the research. “This concept highlights the differences in pathways essential for normal hemostasis at sites of vessel injury, in contrast with those needed for clot propagation and blood vessel lumen occlusion.”
Abou-Ismail noted that this approach has been explored with Factor XI (FXI) inhibitors currently in clinical trials. However, he raised an important mechanistic concern.
“This mechanism may not necessarily accomplish that goal of uncoupling thrombosis from hemostasis, although it might at narrow therapeutic windows,” Abou-Ismail said. “The tenase complex is a central component of coagulation whose deficiency underlies hemophilia A and B, diseases that cause significant bleeding in humans, and it is more essential to hemostasis than [FXI inhibitors]. Tenase inhibition seems like it may pose a higher bleeding risk in humans.”
He explained that FXI inhibitors have a strong mechanistic rationale because they target the feedback loop that amplifies clot propagation, which is less essential for hemostasis. “FXI inhibitors have clinical trial data demonstrating that FXI inhibition is in fact associated with less bleeding compared to current established anticoagulants,” he said. “However, CCG disrupts the FIXa/FVIIIa intrinsic tenase complex itself, which is considered essential for hemostasis.”
Surprises and Confirmations
The researchers were not anticipating such a clear separation between antithrombotic activity and bleeding risk. The preservation of normal wound healing was equally surprising, directly challenging the belief that interfering with clot formation inevitably disrupts tissue repair.
However, the path to these conclusions was not straightforward. “The structural definition of complex macromolecules like sulfated polysaccharides is a common challenge in the research field,” Lin said. “We spent a lot of time to analyze the structure of CCG.”
Even after identifying the candidate compound, the team had to rigorously confirm that its effects were truly anticoagulant in nature, rather than secondary to anti-inflammatory or vascular remodeling properties. Mechanistic studies were essential in demonstrating its targeted disruption of the intrinsic tenase complex, helping to explain how thrombosis could be reduced without broadly impairing coagulation.
Lin is excited but knows patience and skepticism are needed. “Our research is still at the basic stage, but based on the available data, we may provide a potential anticoagulant option with low bleeding risk,” she said. “In the discussion section of our paper, we also stated that ‘the data suggest a wide therapeutic window of CCG, which may offer therapeutic advantages for patients with bleeding contraindication, such as elderly patients and those with renal failure, as well as for safer long-term anticoagulation.’”
A New Direction for Heparin Alternatives?
Despite these concerns, Abou-Ismail acknowledged that the research has genuinely noteworthy aspects. “A future anticoagulant with a novel mechanism of action may be useful in patients who have experienced anticoagulant failure or breakthrough thrombosis from currently established anticoagulants,” he said. “Having another option might be useful when all other options have failed or are not feasible.”
However, he added a note of caution: “If a therapeutic window exists where partial tenase disruption has antithrombotic effect that does not impair hemostasis, then that would definitely be a promising future finding, but it is too early to arrive at that conclusion with this study.”
The search for safer heparin alternatives has been ongoing for decades, but most candidates still operate within the same fundamental paradigm of broad coagulation inhibition. This snail can’t move fast enough.
Abou-Ismail reported having no relevant conflicts. Disclosure information for study authors is available in the original study publication.
A version of this article first appeared on Medscape.com.
The Fastest Way to Better Anticoagulants May Be a Land Snail
The Fastest Way to Better Anticoagulants May Be a Land Snail
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
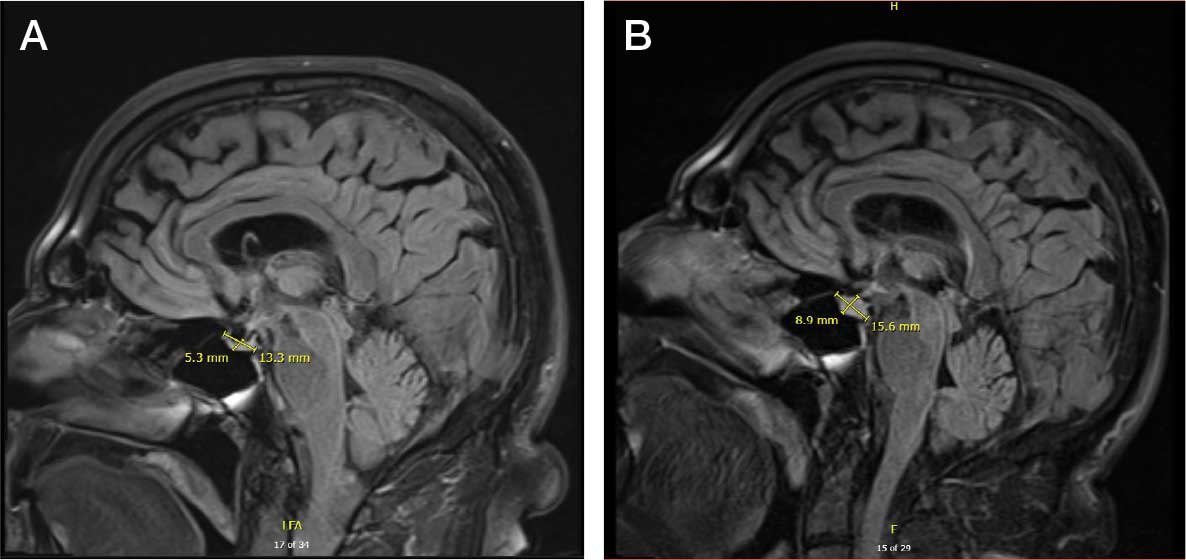
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.

No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
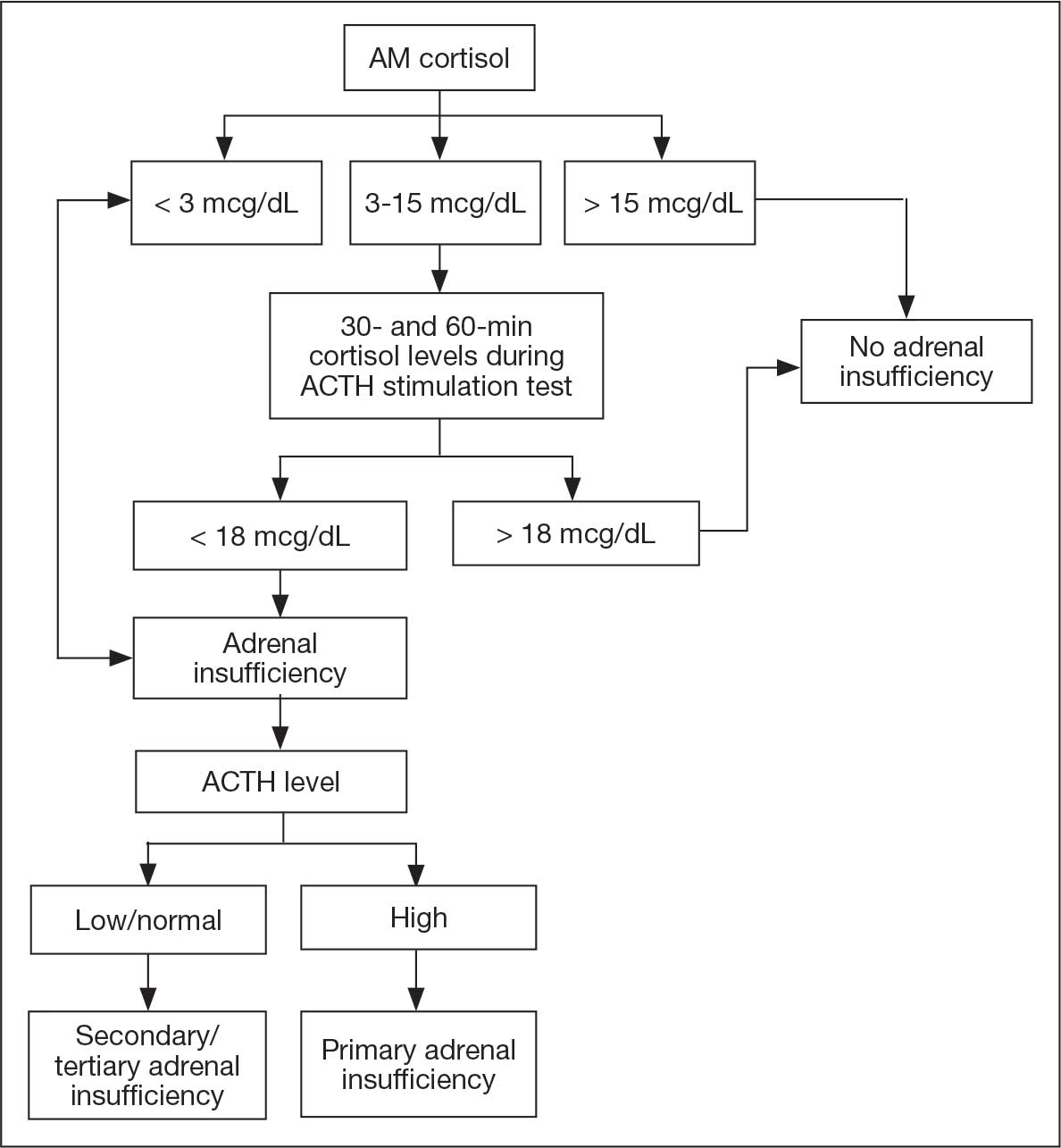
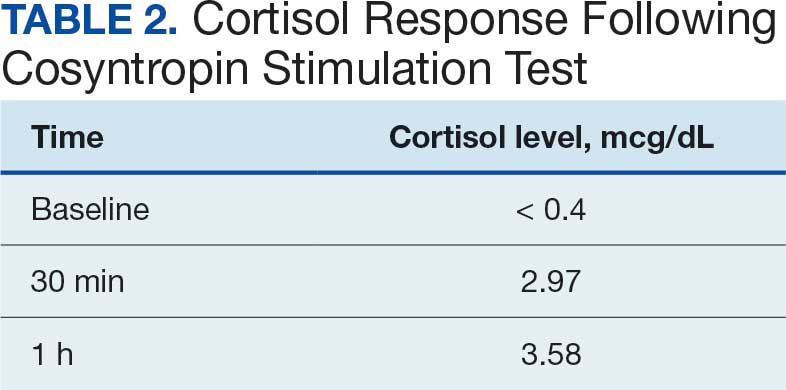
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.
No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
Immune checkpoint inhibitors (ICIs) have become important in oncology and represent an evolving area of therapeutics. Since their approval by the US Food and Drug Administration (FDA) in 2011, ICIs have been increasingly used as modalities in neoadjuvant and adjuvant treatment for resectable solid malignancies and in unresectable disease, such as advanced melanoma, and are associated with improved survival.1
Immune checkpoints are present on the cell surface of activated T cells as well as other immune cells like B cells and natural killer cells. By regulating the length and amplitude of the body’s innate immune response, they maintain immune homeostasis and prevent its overactivation. Immune checkpoints are often thought of as the brakes on the immune system.2
Two glycoproteins that act as immune checkpoints and are targeted by ICIs are cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1). CTLA-4 is upregulated on activated T cells. PD-1 is also expressed on activated T cells, as well as macrophages, B cells, and dendritic cells. Cancer cells can evade immune surveillance by exploiting immune checkpoint pathways. Inhibition of these checkpoints with ICIs reactivates T cells and enables the immune system to recognize and attack cancer cells more effectively. Ipilimumab blocks the activity of CTLA-4 on T cells. Nivolumab and pembrolizumab block the interaction between PD-1 on T cells and its ligand PD-L1 on cancer cells.3,4
Inhibition of these checkpoints is often effective in cancer treatment but can result in the loss of immunologic tolerance with resultant immune-related adverse events (irAEs) and potentially permanent autoimmune disorders. Autoreactive T cells can damage host cell tissues including the colon, lungs, liver, pituitary gland, thyroid, and skin. Severe irAEs include type 1 diabetes mellitus, myositis, nephritis, colitis, pneumonitis, hepatitis, uveitis, hypophysitis, and adrenalitis.4
Hypophysitis is inflammation of the pituitary gland, often with thickening of the pituitary stalk, resulting in dysfunction and hormone deficiencies. While primary hypophysitis is idiopathic, secondary hypophysitis is the result of an underlying condition such as exposure to an ICI. Immune-mediated inflammation of the pituitary gland in hypophysitis may disrupt corticotroph function, leading to adrenocorticotropic hormone (ACTH) deficiency. Early warning features are often vague and nonspecific, such as headache, fatigue, and weakness, which makes diagnosis challenging.3,5
CASE PRESENTATION
A 73-year-old male veteran with a history of metastatic melanoma on ipilimumab 3 mg/kg and nivolumab 1 mg/kg every 3 weeks (a standard combination regimen for advanced melanoma) presented to the emergency department (ED) with 2 weeks of cough, nausea, and severe headache 3 weeks after cycle 2 of combination ICI therapy. The patient had undergone excision of multiple sites of melanoma in situ with recurrence and disease progression after 5 cycles of pembrolizumab. He was subsequently started on combination ICI therapy.
On ED arrival, the patient was afebrile and saturating well on room air. He was normotensive but found to have orthostatic blood pressure. Physical examination was remarkable for dry oral mucosa and decreased skin turgor. Initial laboratory results were significant for hyponatremia of 123 mmol/L (reference range, 136-145 mmol/L), low-normal free thyroxine (T4) level of 0.5 ng/dL (reference range, 0.6-1.2 ng/dL), a low total triiodothyronine level of 32.14 ng/dL (reference range, 85-178 ng/dL), and a low thyrotropin level of 0.19 mIU/L (reference range, 0.35-5.50 mIU/L). Serum osmolarity was low at 259 mOsm/kg (reference range, 285-315 mOsm/kg), urine sodium was high at 168 mEq/L (reference, 20 mEq/L), and urine osmolarity was inappropriately concentrated at 726 mOsm/kg (reference range, 250-1000 mOsm/kg). The patient was admitted for additional testing. His morning cortisol level was within normal limits at 15 mcg/dL (reference range, 6.7-22.5 mcg/dL).
Computed tomography (CT) of the patient’s head revealed no acute findings. Chest CT revealed posterior right lower lobe mild ground-glass opacities, with possible ICI-induced pneumonitis. The patient received fluid resuscitation. Given concern for syndrome of inappropriate antidiuretic hormone secretion, the patient was started on 3 g salt tablets 3 times a day and urea 30 g powder daily. The etiology of the abnormal thyroid levels was unclear to endocrinology at that time. The differential diagnosis included a nonthyroidal illness or central hypothyroidism.
The patient started levothyroxine 75 mcg due to abnormal thyroid levels and persistent fatigue and fludrocortisone 0.1 mg daily to manage orthostatic hypotension. His sodium levels improved to 132 mmol/L over 6 days and he was discharged with levothyroxine 75 mcg daily, fludrocortisone 0.1 mg daily, 3 g salt tabs 3 times a day, urea 30 g powder daily, as well as oral cefpodoxime 500 mg twice daily for 3 days and azithromycin 500 mg once daily for 2 days (for a total of 10 days of antibiotic therapy) to treat potential occult pneumonia.
The patient returned to the ED 3 days after discharge following an outpatient oncology appointment with ongoing severe headaches and persistent nausea. There was concern for recurrent hyponatremia. His sodium level was within normal limits at 133 mmol/L. Repeat morning cortisol was low-normal at 9 mcg/dL. Magnetic resonance imaging (MRI) of the brain was negative for metastatic disease, but showed a slight interval increase in size of the pituitary gland compared with an MRI from 6 months prior, with mild fullness and a slightly convex superior margin near homogeneous enhancement, raising concern for infection or hypophysitis (Figure 1).
The patient was readmitted to the general medicine service and was given intravenous hydrocortisone 100 mg every 8 hours because of concern for central adrenal insufficiency due to grade 3 hypophysitis in the setting of MRI imaging and severe headaches (Table 1). He was not hypotensive at the time of hydrocortisone initiation and other vital signs were stable. A cosyntropin stimulation test—a standard diagnostic test for central adrenal insufficiency—was not performed because the patient had already started high-dose hydrocortisone. The patient’s free T4 on this admission remained low at 0.6 ng/dL.
No adjustments were made to his levothyroxine dose given that he recently began the medication and levels may lag after initiation. After a 4-day hospitalization, the decision was made to continue with the steroid taper and follow up with outpatient endocrinology to obtain a cosyntropin stimulation test to complete a full assessment of his pituitary axis (Figure 2). Repeat thyroid function testing for levothyroxine titration was arranged. The levothyroxine dosage was later increased to 88 mcg daily, but the patient discontinued the medication and remained euthyroid. Endocrinology attributed a nonthyroidal illness as the etiology of his hypothyroidism, likely euthyroid sick syndrome in the setting of illness. His hydrocortisone was tapered during outpatient care and fludrocortisone was discontinued due to hypertension.
One month after his second discharge, the patient presented to the ED with 2 weeks of dizziness, associated lightheadedness, and blurred vision when standing from a sitting position. Upon assessment, symptoms were attributed to poor oral intake. The patient’s vital signs were again positive for orthostatic hypotension, though refractory to adequate fluid replacement. Laboratory testing was significant for a low ACTH level of 3.0 pg/mL (reference range, 7.2-63.3 pg/mL). Given that the patient had not received steroids for 1 week, he underwent a cosyntropin stimulation test, which revealed a blunted response supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis (Table 2).
The patient was again readmitted to the general medicine service. A brain MRI showed interval shrinkage of the pituitary gland compared to imaging one month prior, which was attributed to hydrocortisone treatment during this month. CT of the patient’s abdomen demonstrated normal-sized adrenal glands. Positron emission tomography (PET)/CT showed no evidence of pituitary or adrenal metastases. Endocrinology recommended reinitiating oral hydrocortisone 50 mg in the morning and 50 mg around 3 pm daily with fludrocortisone 0.2 mg once daily, which resulted in near resolution of the patient’s symptoms. He was discharged after a 14-day hospitalization with home physical therapy services and endocrinology, nephrology, and oncology follow-up appointments.
The patient was readmitted twice to the general medicine service over the next 6 months for complications from hydrocortisone and fludrocortisone treatment including hypokalemia. He followed up with outpatient clinicians until his death 14 months later. He did not restart ICI therapy, and eventually joined a clinical trial for other advanced melanoma treatments at another institution. The patient’s family consented to the publication of this case report with the accompanying images.
DISCUSSION
The combination of ipilimumab (anti-CTLA-4 monoclonal antibody) and nivolumab (anti-PD-1 monoclonal antibody) is FDA-approved for treatment of advanced melanoma with the goal of harnessing complementary and synergistic mechanisms of dual therapy.6-8 Combination therapy, however, can increase the incidence of irAEs, which are often endocrine-related and more common in patients treated with dual immunotherapy than with monotherapy.9 Hypophysitis has the lowest reported fatality rate among ICI-related irAEs (< 1%), compared with higher mortality rates seen in myocarditis (25%-50%) and pneumonitis (10%-20%).4,10
The patient initially presented with ICI-related hypothyroidism, later identified as secondary (central) hypothyroidism. He was treated with levothyroxine until central hypothyroidism was confirmed. Subsequently, the patient developed headache, poor appetite, and lightheadedness, with MRI findings suggestive of hypophysitis, for which he was started on hydrocortisone. A component of primary adrenal insufficiency was initially considered, given the low ACTH level and blunted response to cosyntropin stimulation following prior high-dose steroid therapy. However, CT imaging demonstrated normal adrenal morphology without atrophy, supporting a diagnosis of central adrenal insufficiency secondary to ICI-induced hypophysitis.
The estimated incidence of ICI-induced hypophysitis is 1.5% to 13.3% with anti-CTLA-4 agents, 0.3% to 3.0% with anti-PD-1 agents, and can be as high as 12.8% with combination therapy.1 ICI-induced hypophysitis is believed to arise from the direct binding of ICI antibodies to their targets on anterior pituitary cells, such as corticotrophs, thyrotrophs, and gonadotrophs, triggering an immune response. One theory for targeting these cells is high CTLA-4 expression in the anterior pituitary gland.11 PD-1 therapies tend to manifest as either hypothyroidism, hyperthyroidism, Graves’ disease, diabetes, or adrenal insufficiency.10
A concern in patients with advanced melanoma is metastasis. Melanoma has a high propensity for brain metastasis.12 There was moderate suspicion for pituitary gland metastasis in this case, though pituitary metastasis more often manifests with symptoms of posterior pituitary gland deficiency, such as polyuria and polydipsia.13 The adrenal gland is the fourth-most common site for melanoma metastases, after the lung, liver, and bone.14 This patient had no evidence of pituitary or adrenal metastases on PET/CT. Therefore, his symptoms were most likely due to ICI therapy. Cases of ≥ 1 endocrine dysfunction have been reported as an ICI therapy irAE.15 In these situations, diagnosing primary and central adrenal insufficiency in the same patient is complex because hormone profiles are intertwined.
Many patients who develop hypophysitis from ICI therapy will require permanent replacement therapy. It is unclear whether low-dose replacement steroids have a significant effect on the efficacy of ICIs. Given that ICI treatment works by enhancing the immune system, medications that suppress the body’s immune system, such as steroids, could interfere with treatment efficacy. However, there are speculations that the development of irAEs is an indicator of effective treatment. In a phase 1 trial of a CTLA-4 blocker in patients with metastatic melanoma, there was a correlation between reduced CTLA-4 expression as well as low rates of melanoma recurrence and a higher incidence of irAEs.16
When assessing patients on ICI treatment, clinicians must remain vigilant for all potential irAEs, especially in patients receiving combination therapy. ICI-induced irAEs can present with vague and nonspecific symptoms. Concurrent endocrine irAEs, such as hypophysitis with thyroiditis or adrenalitis, are not uncommon in combination therapy and can complicate interpretation of hormone profiles. It is prudent for clinicians to review known risk factors. Hypophysitis is typically associated with older adult male patients.17,18
The irAEs of ICI therapy deeply affected the quality of life of the patient in this case, as he was often experiencing many of the clinical symptoms of his hormone insufficiencies as well as the treatment modalities, thus requiring repeated hospital admissions. The risks and benefits of continuing ICI therapy should be an ongoing discussion between the physician and patient and should take into account the acuity and severity of irAEs and oncological disease burden, among other variables. Given the severity of his AEs, the patient stopped ICI therapy and instead opted to enroll in a clinical trial at another institution for continued alternative treatments.
CONCLUSIONS
This case offers a lesson in the diagnostic challenges of vague symptoms in patients with cancer who are receiving ICI therapy. ICI therapy is widely used in the treatment of solid malignancies, and as its use increases, it is expected that clinicians will likely see more cases of irAEs in hospitalized patients. The vague presentation of irAEs can often lead to treatment delays, especially when > 1 irAE presents concurrently. There are ongoing studies researching potential ways to predict the likelihood of developing these irAEs. It is imperative that clinicians are aware of these ICI-related complications and that more research be conducted to understand patient quality of life and treatment guidance based on irAE severity and disease burden.
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
- Villani A, Potestio L, Fabbrocini G, et al. The treatment of advanced melanoma: therapeutic update. Int J Mol Sci. 2022;23:6388. doi:10.3390/ijms23126388
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252-264. doi:10.1038/nrc3239
- Chang LS, Barroso-Sousa R, Tolaney SM, et al. Endocrine toxicity of cancer immunotherapy targeting immune checkpoints. Endocr Rev. 2019;40:17-65. doi:10.1210/er.2018-00006
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med. 2017;23:540-547. doi:10.1038/nm.4321
- Jessel S, Weiss SA, Austin M, et al. Immune checkpoint inhibitor-induced hypophysitis and patterns of loss of pituitary function. Front Oncol. 2022;12:836859. doi:10.3389/fonc.2022.836859
- Betof AS, Nipp RD, Giobbie-Hurder A, et al. Impact of age on outcomes with immunotherapy for patients with melanoma. Oncologist. 2017;22:963-971. doi:10.1634/theoncologist.2016-0450
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133. doi:10.1056/NEJMoa1302369
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723. doi:10.1056/NEJMoa1003466
- Benhima N, Belbaraka R, Langouo Fontsa MD. Single agent vs combination immunotherapy in advanced melanoma: a review of the evidence. Curr Opin Oncol. 2024;36:69-73. doi:10.1097/CCO.0000000000001014
- Tong J, Kartolo A, Yeung C, et al. Long-term toxicities of immune checkpoint inhibitor (ICI) in melanoma patients. Curr Oncol. 2022;29:7953-7963. doi:10.3390/curroncol29100629
- Grouthier V, Lebrun-Vignes B, Moey M, et al. Immune checkpoint inhibitor-associated primary adrenal insufficiency: WHO VigiBase report analysis. Oncologist. 2020;25:696-701. doi:10.1634/theoncologist.2019-0555
- Park BC, Jung S, Wright JJ, et al. Recurrence of hypophysitis after immune checkpoint inhibitor rechallenge. Oncologist. 2022;27:e967-e969. doi:10.1093/oncolo/oyac220
- Zhang D, Wang Z, Shang D, et al. Incidence and prognosis of brain metastases in cutaneous melanoma patients: a population-based study. Melanoma Res. 2019;29:77-84. doi:10.1097/CMR.0000000000000538
- Barnabei A, Carpano S, Chiefari A, et al. Case report: ipilimumab-induced panhypophysitis: an infrequent occurrence and literature review. Front Oncol. 2020;10:582394. doi:10.3389/fonc.2020.582394
- Shortreed H, Burute N, Aseyev O. Management of undifferentiated adrenal gland metastases from malignant melanoma: case report. Front Oncol. 2024;14:1419827. doi:10.3389/fonc.2024.1419827
- Rossi S, Silvetti F, Bordoni M, et al. Pembrolizumab-induced thyroiditis, hypophysitis and adrenalitis: a case of triple endocrine dysfunction. JCEM Case Rep. 2024;2:luae200. doi:10.1210/jcemcr/luae200
- Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741-750. doi:10.1200/JCO.2005.01.128
- de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm Metab Res. 2019;51:145-156. doi:10.1055/a-0843-3366
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Diagnostic Challenge of Immune Checkpoint Inhibitor-Induced Hypophysitis in a Patient With Advanced Melanoma
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review
Cannabis has a long history of use for medicinal and recreational purposes. Research illustrates the potential benefits and increased prevalence of cannabis use in patients with cancer.1 Cannabis products have been shown to possess antineoplastic and palliative activity, improving nociceptive and neuropathic pain in addition to chemotherapy-related nausea and vomiting.2-5 Despite these developments and changing social attitudes toward cannabis, there remains a lack of comprehensive data on patient perspectives regarding its use, especially in regions where cannabis remains illegal. This knowledge gap is notable among veterans undergoing cancer treatment in states where cannabis is prohibited. Up to 57% of veterans report lifetime marijuana use, making it crucial to understand this population’s cannabis use patterns and potential interactions with cancer treatments.6
This observational study sought to determine the prevalence of cannabis use among patients undergoing cancer treatment at the US Department of Veterans Affairs (VA) Memphis Healthcare System and evaluate the potential risks associated with combining cannabis products with anticancer therapies.
METHODS
This prospective observational study identified cannabis use among veterans receiving antineoplastic therapy at the Lt. Col. Luke Weathers Jr. VA Medical Center (WJVAMC) and analyzed potential interactions between cannabis products and their cancer treatments. Participants included adults aged > 18 years undergoing antineoplastic therapy at WJVAMC who consented to the study. Data collection involved a written survey approved by the WJVAMC Institutional Review Board and verbal consent from participants. The survey asked participants about their cannabis use in the previous 90 days, including details on quantity, frequency, and method of consumption (eg, inhalation, oral, topical). No incentives were offered for participation.
Surveys from 50 patients who used cannabis were analyzed and their electronic health records were reviewed for sex, age, diagnosis, and antineoplastic regimen. This information was securely stored. A literature review was conducted using PubMed and the Cochrane Library to explore potential interactions between cannabis and the antineoplastic agents that were prescribed to patients in the study, focusing on toxicity, efficacy, or synergistic effects.
Patients were categorized into 4 groups based on treatment: cytotoxic chemotherapy, immunotherapy, endocrine therapy, and targeted therapy. Patients undergoing multiple types of therapies were included in each applicable category.
RESULTS
A total of 132 patients agreed to participate. Fifty patients (38%) acknowledged using cannabis products within 90 days. The patients that used cannabis products within 90 days of the survey reported the following malignancies: 8 patients (16%) had prostate cancer, 3 patients (6%) had hepatocellular carcinoma, 7 patients (14%) had pancreatic carcinoma, 5 patients (10%) had multiple myeloma, 3 patients (6%) had chronic lymphocytic leukemia, 9 patients (18%) had non-small cell lung cancer, 3 patients (6%) had breast cancer, 3 (6%) patients had bladder cancer, 2 patients (4%) had renal cell carcinoma, 1 (2%) patient had chronic myeloid leukemia, 1 (2%) patient had renal amyloid, 1 patient (2%) had supraglottic squamous cell carcinoma, 1 patient (2%) had esophageal carcinoma, 1 (2%) patient had small cell lung cancer, 1 (2%) patient had gastric cancer, and 1 patient (2%) had follicular lymphoma.
Five (10%) of the cannabis users were female, and 45 (90%) were male. Twenty-nine patients (58%) were aged 66 to 75 years, 16 (32%) were aged 56 to 65 years, 3 (6%) were aged 46 to 55 years, and 2 (4%) were aged 76 to 85 years.
Thirty-five patients (70%) inhaled cannabis as opposed to using it via other formulations or a combination (eg, inhalation and topical). Thirty-eight percent of patients used cannabis once daily, 24% used < 1 daily, and 28% used it ≥ 2 times daily. Five patients (10%) did not report the frequency of their cannabis use. Among the patients who reported cannabis use, 21 (42%) were undergoing cytotoxic chemotherapy, 19 (38%) were undergoing immunotherapy, 12 (24%) were undergoing targeted therapy, and 10 (20%) were undergoing endocrine therapy. Some patients were treated with multiple types of antineoplastic agents and were counted in multiple categories (Table 1).
Following a literature review of cannabis and antineoplastic agents, patients were evaluated for the potential effects of cannabis on their treatment. The literature review revealed that 31% of cytotoxic chemotherapy agents received by patients in this study might have increased toxicity, and 19% could have reduced efficacy when combined with cannabis. Among immunotherapy agents received by patients in this study, 70% might have decreased efficacy when combined with cannabis use. For targeted therapies, 35% could have increased toxicity, and 70% of endocrine agents could potentially have decreased efficacy (Table 2).
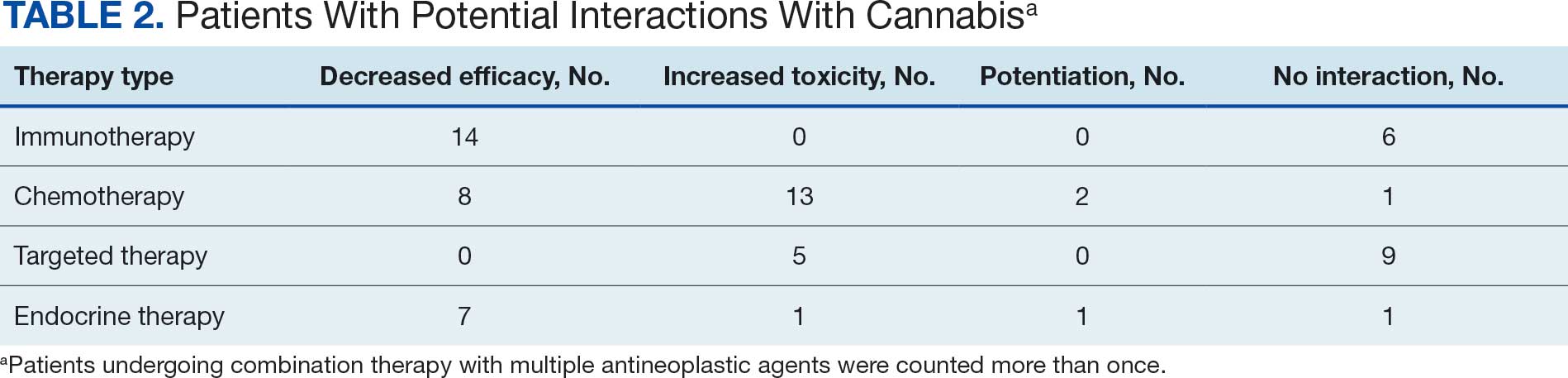
DISCUSSION
This prospective study corroborates previous research by demonstrating that more than one-third of patients receiving oncology care at WJVAMC use cannabis, most often inhaled. Cannabis use was observed among patients undergoing various cancer therapies, including cytotoxic chemotherapy, immunotherapy, targeted therapy, and endocrine therapy. The most common malignancies among cannabis users at WJVAMC include patients with lung cancer, prostate cancer, pancreatic cancer, and multiple myeloma. Cannabis use in patients with pancreatic cancer and multiple myeloma was significantly out of proportion to their prevalence at WJVAMC. This could potentially be due to their drastic effect on quality of life.
Cannabis use increased the risk of toxicity in patients treated with cytotoxic chemotherapy and targeted therapy. Cannabis use potentially decreased efficacy for patients treated with cytotoxic chemotherapy and/or immunotherapy. Cannabis use did not increase the risk of toxicity or efficacy in patients treated with endocrine therapy.
Antineoplastics/Cannabis Interactions
The potential interactions between cannabis and antineoplastic therapies administered at WJVAMC are worth exploring. While this review aims to shed light on possible interactions, it is important to acknowledge that much of the data is preliminary and derived from in vitro studies. The interactions should be interpreted as potential risks rather than established facts. Additional research is needed to confirm these interactions and effectively guide clinical practices. Understanding these dynamics is essential to optimize patient care and manage the complex interplay between cannabis use and cancer treatment.
Originating from Central Asia, the cannabis plant contains > 400 medicinally relevant compounds, of which about 100 are cannabinoids (CBs). Key CBs are cannabidiol (CBD), a nonpsychoactive compound, and ?-9-tetrahydrocannabinol (THC), a psychoactive compound. THC can make up 20% to 30% of the dry weight of female cannabis flowers.7
CBs act through the endocannabinoid system, involving CB1 and CB2 receptors, endogenous CBs like anandamide (AEA) and 2-arachidonoylglycerol, and various enzymes. These endogenous CBs, derived from arachidonic acid, play roles in cell growth and proliferation.8 In some studies, AEA has induced apoptosis in neuroblastoma cells and inhibited proliferation in breast cancer cells. However, other research suggests AEA may block apoptosis under certain conditions.9
CB receptors are transmembrane proteins that interact with CBs differently depending on tissue type and CB structure. Synthetic CBs are designed to target specific receptors, while natural CBs may act as both agonists and antagonists.10
Cytochrome P450 Metabolism
The human cytochrome P450 (CYP) 3A subfamily affects the metabolism of many therapeutic drugs, including cancer therapeutics.11 The various compositions of cannabis are primarily metabolized by the CYP450 pathway, the same as many cancer-directed pharmacologic treatments. CBs act as both CYP inducers and inhibitors. THC, for example, is a CYP inducer whereas CBD is a CYP inhibitor; both are found in the various compounds available for consumption.12,13 Pharmacology research has suggested potential interactions and effects on established adverse symptoms, but clinical data are lacking, and current research revealing interactions are only recognized in vitro.14
The Antineoplastic Activity of Cannabis
CBs can affect various cancer-related pathways such as PKB, AMPK, CAMKK-ß, mTOR, PDHK, HIF-1 a, and PPAR-γ. Δ-9-THC can selectively induce apoptosis in tumor cells without harming normal cells, though the exact mechanism remains unclear. Promising results from early mouse studies led to a 2006 human study where intracranial Δ-9-THC in patients with recurrent glioma yielded a median survival of 24 weeks, with 2 patients surviving > 1 year.15
In a 2022 review article, Cherkasova et al highlighted potential clinical benefits of cannabis across various cancers. They found that upregulated CB1 receptors in colon cancer might enhance the effect of 5-fluorouracil. However, many studies are preliminary and therefore not definitive.10
Additional research is needed to refine these findings. Challenges include variability in cannabis formulations, the complex tumor microenvironment, and the legal and psychoactive issues surrounding cannabis use. These factors complicate the design of multicenter randomized studies and may deter patients from disclosing cannabis use, thereby hindering efforts to fully understand its therapeutic potential.
Cannabis/Cytotoxic Chemotherapy Interactions
The chemotherapy agents used in this study included carboplatin, paclitaxel, 5-fluorouracil, etoposide, irinotecan, oxaliplatin, pemetrexed, docetaxel, cabazitaxel, T-DM1, gemcitabine, and cyclophosphamide. There is a paucity of research regarding the interactions between cytotoxic chemotherapy and cannabis. Most studies focused on CBD due to its inhibition of the CYP450 pathway, which is used for metabolizing cytotoxic chemotherapies. Through this mechanism, CBD could potentially increase the concentrations of chemotherapeutic agents, enhancing their toxicity.
When combined with irinotecan, cannabis can pose risks. Δ-9-THC undergoes first-pass metabolism in the liver, mediated by the CYP450 system and CYP3A4. The glucuronidation of irinotecan is mediated by uridine diphosphate glycosyltransferase, leading to its recirculation within the hepatic system and potentially increased toxicity due to prolonged drug presence. Cannabis may also compete with drug binding to albumin, altering the plasma concentrations of irinotecan and its conversion to the metabolite SN38.16
Cannabis products can affect chemotherapy levels by interacting with cellular transporters. The MRP1 transporter family, encoded by the ABCC gene family, is expressed mainly in the lung, kidney, skeletal muscle, and hematopoietic stem cells. A 2018 study investigating the effects of THC, CBD, and CBN on MRP1 transporters found that the presence of a cannabis component increased the concentration of vincristine 3-fold. Additional studies suggest the interaction with the CB1 receptor may lead to changes in the expression of MRP1 transporters.17
CBD inhibits the BCRP transporter, which functions as an efflux pump for methotrexate. Consequently, CBD can increase methotrexate levels, potentially enhancing efficacy but also worsening adverse effects.18
In pancreatic cancer, CBD specifically interacts with gemcitabine. CB1 and CB2 receptors are upregulated, and CBD inhibits the GPR55 receptor. These interactions may enhance the antineoplastic effect of gemcitabine, reducing cell cycle progression and growth.19
CBD also interacts with temozolomide (TMZ) by affecting extracellular vesicles used by cells for pro-oncogenic signaling and immune system evasion. Experiments on patient-derived glioblastoma cells, both chemotherapy-resistant and chemotherapy-sensitive, found that CBD increases the formation of extracellular vesicles with reduced levels of miR21 (pro-oncogenic) and elevated levels of miR126 (antioncogenic).20 CBD has also been found to decrease prohibitin levels, a protein associated with TMZ resistance.
In patients with glioblastoma, CBD combined with chemotherapeutic agents like TMZ, carmustine, doxorubicin, and cisplatin has shown increased sensitivity and improved tumor response. CBD is also known to inhibit NF-kB, a pathway that sustains tumor viability despite chemotherapy.21 Additionally, CBD inhibits the P-glycoprotein system, affecting chemotherapy efflux from neoplastic cells.14 In vitro studies have found that CBD is synergistic with bortezomib in inhibiting cancer cell viability. In another glioblastoma model, CBD enhanced the antiproliferative effects of both TMZ and carmustine.14
Different cannabis formulations may vary in how they interact with various cytotoxic chemotherapeutic agents. Some may potentiate the effects of chemotherapy and act synergistically to inhibit tumor growth, while others may lead to increased toxicity.10 More research is needed to determine which formulations, in combination with specific agents and doses, may have significant interactions that warrant adjustments in chemotherapy dosing.
Cannabis/Immunotherapy Interactions
Cannabis is an immunosuppressant. Data suggest the use of cannabis during immunotherapy worsens treatment outcomes in patients with cancer.22 Exogenous (THC) and endogenous (AEA) CBs negatively affect antitumor immunity by impairing the function of tumor-specific T cells via CB2 and by inhibiting the Jak1-STATs signaling in T cells through CNR2. Xiong et al found that THC reduces the therapeutic effect of anti-PD-1 therapy.22
In a prospective observational clinical study, Bar-Sela et al analyzed 102 patients with advanced cancer—of which 68 were cannabis users—that were started on immune checkpoint inhibitor therapy. The study found that cannabis users on anti-PD-1 (nivolumab, pembrolizumab), anti-CTLA-4 (ipilimumab), and anti-PD-L1 (durvalumab, atezolizumab) had a significant decrease in time to treatment progression and overall survival vs cannabis non-users.23 However, a 2023 study by Waissengrin et al found that concomitant use of medical cannabis with pembrolizumab had no harmful effect in advanced non-small cell lung cancer.24 Time to treatment progression of cannabis users did not differ from cannabis nonusers.25
Cannabis/Endocrine Therapy Interactions
In addition to having direct antineoplastic activity on tumor cells, data exist that show how cannabis affects the endocrine system. In animal models, cannabis has been found to suppress the whole hypothalamic-pituitary-adrenal axis as well as other hormones like thyroid, prolactin, and growth hormone. In breast cancer, cannabis competes with estrogen for the estrogen receptor and suppresses growth.26
The endocrine agents used by patients with cancer in this study were antiandrogens like abiraterone, enzalutamide, tamoxifen and anastrozole. Abiraterone is metabolized by CYP450 isoenzymes and uridine diphosphate glycosyltransferases. Cannabis inhibits both processes and therefore may lead to increased toxicities.27 Conversely, enzalutamide is a strong CYP3A inducer, and cannabis use during enzalutamide therapy may significantly increase the toxic effects of cannabis.
There is evidence that molecular pathways involving CB receptors and estrogens overlap, which may lead to interactions when antiestrogens are used in cannabis users with hormone receptor-positive breast cancer.26 In preclinical studies, tamoxifen has been shown to act as an inverse agonist on CB1 and CB2 receptors, though the significance of this finding is unclear. There is no research evaluating the effects of CBs on tamoxifen treatment. However, CBD has been found to potentiate the effectiveness of anastrozole or exemestane in breast cancer cell lines.28 Dobovišek et al demonstrated no inhibitory effect of CBD on the activity of tamoxifen, fulvestrant, or palbociclib in breast cancer cell lines.29 The interactions between hormone receptor-positive breast cancer and cannabinoids are complex, and the clinical significance of these interactions remains difficult to identify.
Cannabis/Targeted Therapy Interactions
The targeted therapies used by patients in this study included zanubrutinib, ibrutinib, sorafenib, acalabrutinib, dabrafenib, trametinib, trastuzumab, bevacizumab, daratumumab, and imatinib. Compared to other classes of cancer treatments, most studies have not demonstrated decreased efficacy or increased toxicity of targeted anticancer drugs when used concomitantly with CBD.29
Trastuzumab is a recombinant humanized monoclonal antibody that targets the proto-oncogene HER2/neu. It is used to treat select patients with metastatic breast cancer. Studies have shown that cannabis use does not attenuate the effectiveness of trastuzumab in HER2-positive and triple-negative breast cancer subtypes.29 One study found that CBD, in combination with chemotherapeutics and Bruton tyrosine kinase inhibitors, such as ibrutinib and zanubrutinib, has synergistic potential for treating diffuse large B-cell lymphoma and mantle cell lymphoma cell lines. This synergy is attributed to the CB1 antagonist activity of cannabis against diffuse large B-cell lymphoma and mantle cell lymphoma cell lines.30,31
Moreover, combining cannabinoids with bevacizumab (a monoclonal anti-VEGF antibody) has been shown to decrease tumor growth and intratumoral hypoxia in clinically relevant human glioblastoma models. This effect is mediated through the downregulation of HIF-1α.32 Long-term studies evaluating the potential harmful or synergistic potential of CBD on targeted anticancer therapy are needed.
CONCLUSIONS
This exploratory study of patients receiving cancer therapy at WJVAMC found a significant prevalence of concurrent cannabis use among patients undergoing antineoplastic treatments. Given that many antineoplastic agents are metabolized by the CYP450 enzyme system, the findings of this study suggest that concurrent cannabis use may pose risks of suboptimal therapeutic outcomes due to potential interactions affecting drug metabolism. These interactions could impact the efficacy and toxicity of the antineoplastic therapies, potentially leading to diminished therapeutic effects or exacerbated adverse reactions.
Patients should be informed regarding the potential decreased efficacy of immunotherapy with concurrent use of cannabis products. They should also be aware of the possibility of increased toxicity with other treatment modalities, though the exact impact on efficacy remains unclear. This highlights the necessity of caution when combining cannabis with prescribed cancer treatments.
While this study identified possible interactions, its data are preliminary and highlight the need for more rigorous research. Future studies should include larger, well-designed cohorts to compare outcomes between cannabis users and nonusers. Such research is essential to fully elucidate the clinical implications of cannabis use during cancer treatment, address the high prevalence of cannabis use among patients with cancer, and mitigate potential risks associated with combining cannabis products with antineoplastic therapies. This will ensure that treatment strategies are optimized for safety and efficacy in this complex patient population.
- Steele G, Arneson T, Zylla D. A comprehensive review of cannabis in patients with cancer: availability in the USA, general efficacy, and safety. Curr Oncol Rep. 2019;21:1-10. doi:10.1007/s11912-019-0757-7
- Brown D, Watson M, Schloss J. Pharmacological evidence of medicinal cannabis in oncology: a systematic review. Support Care Cancer. 2019;27:3195-320. doi:10.1007/s00520-019-04774-5
- Abrams DI. Integrating cannabis into clinical cancer care. Curr Oncol. 2016;23:S8-S14. doi:10.37.47/co.23.3099
- Serafimovska T, Darkovska-Serafimovska M, Stefkov G, Arsova-Sarafinovska Z, Balkanov T. Pharmacotherapeutic considerations for use of cannabinoids to relieve symptoms of nausea and vomiting induced by chemotherapy. Folia Medica (Plovdiv). 2020;62:668-678. doi:10.3897/folmed.62e51478
- Bar-Sela G, Zalman D, Semenysty V, Ballan E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: pilot study. Integr Cancer Ther. 2019;18:1534735419881498. doi:10.1177/1534735419881498
- Pederson ER, Villarosa-Hurlocker MC, Prince MA. Use of protective behavioral strategies among young adult veteran marijuana users. Cannabis. 2018;1:14-27.
- Schilling S, Melzer R, McCabe PF. Cannabis sativa. Curr Biol. 2020;30:R8-R9. doi:10.1016/j.cub.2019.10.039
- McDougle DR, Kambalyal A, Meling DD, Das A. Endocannabinoids anandamide and 2-arachidonoylglycerol are substrates for human CYP2J2 epoxygenase. J Pharmacol Exp Ther. 2014;351:616-627. doi:10.1124/jpet.114216598
- Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11:1121-1132. doi:10.1038/sj.cdd.4401442
- Cherkasova V, Wang B, Gerasymchuk M, Fiselier A, Kovalchuk O, Kovalchuk I. Use of cannabis and cannabinoids for treatment of cancer. Cancers (Basel). 2022;14:5142. doi:10.3390/cancers14205142
- Engels FK, Ten Tije AJ, Baker SD, et al. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin Pharmacol Ther. 2004;75:448-454. doi:10.1016/j.clpt.2004.01.001
- Alsherbiny MA, Li CG. Medicinal cannabis-potential drug interactions. Medicines (Basel). 2018;6:3. doi:10.3390/medicines6010003
- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46:86-95. doi:10.3109/03602532.2013.849268
- Opitz BJ, Ostroff ML, Whitman AC. The potential clinical implications and importance of drug interactions between anticancer agents and cannabidiol in patients with cancer. J Pharm Pract. 2020;33:506-512. doi:10.1177/0897190019828920
- Guzmán M, Duarte MJ, Blázquez C, et al. A pilot clinical study of D9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197-203. doi:10.1038/sj.bjc.6603236
- Kopjar N, Fuchs N, Brcic Karaconji I, et al. High doses of ?9-tetrahydrocannabinol might impair irinotecan chemotherapy: a review of potentially harmful interactions. Clin Drug Investig. 2020;40:775-787. doi:10.1007/s40261-020-00954-y
- Bouquié R, Deslandes G, Mazaré H, et al. Cannabis and anticancer drugs: societal usage and expected pharmacological interactions - a review. Fundam Clin Pharmacol. 2018;32:462-484. doi:10.1111/fcp.12373
- Buchtova T, Lukac D, Skrott Z, Chroma K, Bartek J, Mistrik M. Drug-drug interactions of cannabidiol with standard-of-care chemotherapeutics. Int J Mol Sci. 2023;24:2885. doi:10.3390/ijms24032885
- Sharafi G, He H, Nikfarjam M. Potential use of cannabinoids for the treatment of pancreatic cancer. J Pancreat Cancer. 2019;5:1-7. doi:10.1089/pancan.2018.0019
- Kosgodage US, Uysal-Onganer P, MacLatchy A, et al. Cannabidiol affects extracellular vesicle release, miR21 and miR126, and reduces prohibitin protein in glioblastoma multiforme cells. Transl Oncol. 2019;12:513-522. doi:10.1016/j.tranon.2018.12.004
- Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of cannabidiol in breast cancer. Mol Oncol. 2015;9:906-919. doi:10.1016/j.molonc.2014.12.010
- Xiong X, Chen S, Shen J, et al. Cannabis suppresses anti-tumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct Target Ther. 2022;7:99. doi:10.1038/s41392-022-00918-y
- Bar-Sela G, Cohen I, Campisi-Pinto S, et al. Cannabis consumption used by cancer patients during immunotherapy correlates with poor clinical outcome. Cancers (Basel). 2020;12:2447. doi:10.3390/cancers12092447
- Waissengrin B, Leshem Y, Taya M, et al. The use of medical cannabis concomitantly with immune checkpoint inhibitors in non-small cell lung cancer: a sigh of relief? Eur J Cancer. 2023;180:52-61. doi:10.1016/j.ejca.2022.11.022
- Sarsembayeva A, Schicho R. Cannabinoids and the endocannabinoid system in immunotherapy: helpful or harmful? Front Oncol. 2023;13:1296906. doi:10.3389/fonc.2023.1296906
- Kisková T, Mungenast F, Suváková M, Jäger W, Thalhammer T. Future aspects for cannabinoids in breast cancer therapy. Int J Mol Sci. 2019;20:1673. doi:10.3390/ijms20071673
- Woerdenbag HJ, Olinga P, Kok EA, et al. Potential, limitations and risks of cannabis-derived products in cancer treatment. Cancers (Basel). 2023;15:2119. doi:10.3390/cancers15072119
- Almeida CF, Teixeira N, Valente MJ, Vinggaard AM, Correia-da-Silva G, Amaral C. Cannabidiol as a promising adjuvant therapy for estrogen receptor-positive breast tumors: unveiling its benefits with aromatase inhibitors. Cancers (Basel). 2023;15:2517. doi:10.3390/cancers15092517
- Dobovišek L, Novak M, Krstanovic F, Borštnar S, Turnšek TL, Debeljak N. Effect of combining CBD with standard breast cancer therapeutics. Adv Cancer Biol Metastasis. 2022;4:100038. doi:10.1016/j.adcanc.2022.100038
- Strong T, Rauvolfova J, Jackson E, Pham LV, Bryant J. Synergistic effect of cannabidiol with conventional chemotherapy treatment. Blood. 2018;132:5382. doi:10.1182/blood-2018-99-116749
- Maggi F, Morelli MB, Tomassoni D, et al. The effects of cannabidiol via TRPV2 channel in chronic myeloid leukemia cells and its combination with imatinib. Cancer Sci. 2022;113:1235-1249. doi:10.1111/cas.15257
- Obad N, Janji B, Prestegarden L, et al. ATPS-59 improving efficacy of bevacizumab treatment in glioblastoma by targeting hif1 alpha. Neuro Oncol. 2015;17:v31. doi:10.1093/neuonc/nov204.59
Cannabis has a long history of use for medicinal and recreational purposes. Research illustrates the potential benefits and increased prevalence of cannabis use in patients with cancer.1 Cannabis products have been shown to possess antineoplastic and palliative activity, improving nociceptive and neuropathic pain in addition to chemotherapy-related nausea and vomiting.2-5 Despite these developments and changing social attitudes toward cannabis, there remains a lack of comprehensive data on patient perspectives regarding its use, especially in regions where cannabis remains illegal. This knowledge gap is notable among veterans undergoing cancer treatment in states where cannabis is prohibited. Up to 57% of veterans report lifetime marijuana use, making it crucial to understand this population’s cannabis use patterns and potential interactions with cancer treatments.6
This observational study sought to determine the prevalence of cannabis use among patients undergoing cancer treatment at the US Department of Veterans Affairs (VA) Memphis Healthcare System and evaluate the potential risks associated with combining cannabis products with anticancer therapies.
METHODS
This prospective observational study identified cannabis use among veterans receiving antineoplastic therapy at the Lt. Col. Luke Weathers Jr. VA Medical Center (WJVAMC) and analyzed potential interactions between cannabis products and their cancer treatments. Participants included adults aged > 18 years undergoing antineoplastic therapy at WJVAMC who consented to the study. Data collection involved a written survey approved by the WJVAMC Institutional Review Board and verbal consent from participants. The survey asked participants about their cannabis use in the previous 90 days, including details on quantity, frequency, and method of consumption (eg, inhalation, oral, topical). No incentives were offered for participation.
Surveys from 50 patients who used cannabis were analyzed and their electronic health records were reviewed for sex, age, diagnosis, and antineoplastic regimen. This information was securely stored. A literature review was conducted using PubMed and the Cochrane Library to explore potential interactions between cannabis and the antineoplastic agents that were prescribed to patients in the study, focusing on toxicity, efficacy, or synergistic effects.
Patients were categorized into 4 groups based on treatment: cytotoxic chemotherapy, immunotherapy, endocrine therapy, and targeted therapy. Patients undergoing multiple types of therapies were included in each applicable category.
RESULTS
A total of 132 patients agreed to participate. Fifty patients (38%) acknowledged using cannabis products within 90 days. The patients that used cannabis products within 90 days of the survey reported the following malignancies: 8 patients (16%) had prostate cancer, 3 patients (6%) had hepatocellular carcinoma, 7 patients (14%) had pancreatic carcinoma, 5 patients (10%) had multiple myeloma, 3 patients (6%) had chronic lymphocytic leukemia, 9 patients (18%) had non-small cell lung cancer, 3 patients (6%) had breast cancer, 3 (6%) patients had bladder cancer, 2 patients (4%) had renal cell carcinoma, 1 (2%) patient had chronic myeloid leukemia, 1 (2%) patient had renal amyloid, 1 patient (2%) had supraglottic squamous cell carcinoma, 1 patient (2%) had esophageal carcinoma, 1 (2%) patient had small cell lung cancer, 1 (2%) patient had gastric cancer, and 1 patient (2%) had follicular lymphoma.
Five (10%) of the cannabis users were female, and 45 (90%) were male. Twenty-nine patients (58%) were aged 66 to 75 years, 16 (32%) were aged 56 to 65 years, 3 (6%) were aged 46 to 55 years, and 2 (4%) were aged 76 to 85 years.
Thirty-five patients (70%) inhaled cannabis as opposed to using it via other formulations or a combination (eg, inhalation and topical). Thirty-eight percent of patients used cannabis once daily, 24% used < 1 daily, and 28% used it ≥ 2 times daily. Five patients (10%) did not report the frequency of their cannabis use. Among the patients who reported cannabis use, 21 (42%) were undergoing cytotoxic chemotherapy, 19 (38%) were undergoing immunotherapy, 12 (24%) were undergoing targeted therapy, and 10 (20%) were undergoing endocrine therapy. Some patients were treated with multiple types of antineoplastic agents and were counted in multiple categories (Table 1).
Following a literature review of cannabis and antineoplastic agents, patients were evaluated for the potential effects of cannabis on their treatment. The literature review revealed that 31% of cytotoxic chemotherapy agents received by patients in this study might have increased toxicity, and 19% could have reduced efficacy when combined with cannabis. Among immunotherapy agents received by patients in this study, 70% might have decreased efficacy when combined with cannabis use. For targeted therapies, 35% could have increased toxicity, and 70% of endocrine agents could potentially have decreased efficacy (Table 2).
DISCUSSION
This prospective study corroborates previous research by demonstrating that more than one-third of patients receiving oncology care at WJVAMC use cannabis, most often inhaled. Cannabis use was observed among patients undergoing various cancer therapies, including cytotoxic chemotherapy, immunotherapy, targeted therapy, and endocrine therapy. The most common malignancies among cannabis users at WJVAMC include patients with lung cancer, prostate cancer, pancreatic cancer, and multiple myeloma. Cannabis use in patients with pancreatic cancer and multiple myeloma was significantly out of proportion to their prevalence at WJVAMC. This could potentially be due to their drastic effect on quality of life.
Cannabis use increased the risk of toxicity in patients treated with cytotoxic chemotherapy and targeted therapy. Cannabis use potentially decreased efficacy for patients treated with cytotoxic chemotherapy and/or immunotherapy. Cannabis use did not increase the risk of toxicity or efficacy in patients treated with endocrine therapy.
Antineoplastics/Cannabis Interactions
The potential interactions between cannabis and antineoplastic therapies administered at WJVAMC are worth exploring. While this review aims to shed light on possible interactions, it is important to acknowledge that much of the data is preliminary and derived from in vitro studies. The interactions should be interpreted as potential risks rather than established facts. Additional research is needed to confirm these interactions and effectively guide clinical practices. Understanding these dynamics is essential to optimize patient care and manage the complex interplay between cannabis use and cancer treatment.
Originating from Central Asia, the cannabis plant contains > 400 medicinally relevant compounds, of which about 100 are cannabinoids (CBs). Key CBs are cannabidiol (CBD), a nonpsychoactive compound, and ?-9-tetrahydrocannabinol (THC), a psychoactive compound. THC can make up 20% to 30% of the dry weight of female cannabis flowers.7
CBs act through the endocannabinoid system, involving CB1 and CB2 receptors, endogenous CBs like anandamide (AEA) and 2-arachidonoylglycerol, and various enzymes. These endogenous CBs, derived from arachidonic acid, play roles in cell growth and proliferation.8 In some studies, AEA has induced apoptosis in neuroblastoma cells and inhibited proliferation in breast cancer cells. However, other research suggests AEA may block apoptosis under certain conditions.9
CB receptors are transmembrane proteins that interact with CBs differently depending on tissue type and CB structure. Synthetic CBs are designed to target specific receptors, while natural CBs may act as both agonists and antagonists.10
Cytochrome P450 Metabolism
The human cytochrome P450 (CYP) 3A subfamily affects the metabolism of many therapeutic drugs, including cancer therapeutics.11 The various compositions of cannabis are primarily metabolized by the CYP450 pathway, the same as many cancer-directed pharmacologic treatments. CBs act as both CYP inducers and inhibitors. THC, for example, is a CYP inducer whereas CBD is a CYP inhibitor; both are found in the various compounds available for consumption.12,13 Pharmacology research has suggested potential interactions and effects on established adverse symptoms, but clinical data are lacking, and current research revealing interactions are only recognized in vitro.14
The Antineoplastic Activity of Cannabis
CBs can affect various cancer-related pathways such as PKB, AMPK, CAMKK-ß, mTOR, PDHK, HIF-1 a, and PPAR-γ. Δ-9-THC can selectively induce apoptosis in tumor cells without harming normal cells, though the exact mechanism remains unclear. Promising results from early mouse studies led to a 2006 human study where intracranial Δ-9-THC in patients with recurrent glioma yielded a median survival of 24 weeks, with 2 patients surviving > 1 year.15
In a 2022 review article, Cherkasova et al highlighted potential clinical benefits of cannabis across various cancers. They found that upregulated CB1 receptors in colon cancer might enhance the effect of 5-fluorouracil. However, many studies are preliminary and therefore not definitive.10
Additional research is needed to refine these findings. Challenges include variability in cannabis formulations, the complex tumor microenvironment, and the legal and psychoactive issues surrounding cannabis use. These factors complicate the design of multicenter randomized studies and may deter patients from disclosing cannabis use, thereby hindering efforts to fully understand its therapeutic potential.
Cannabis/Cytotoxic Chemotherapy Interactions
The chemotherapy agents used in this study included carboplatin, paclitaxel, 5-fluorouracil, etoposide, irinotecan, oxaliplatin, pemetrexed, docetaxel, cabazitaxel, T-DM1, gemcitabine, and cyclophosphamide. There is a paucity of research regarding the interactions between cytotoxic chemotherapy and cannabis. Most studies focused on CBD due to its inhibition of the CYP450 pathway, which is used for metabolizing cytotoxic chemotherapies. Through this mechanism, CBD could potentially increase the concentrations of chemotherapeutic agents, enhancing their toxicity.
When combined with irinotecan, cannabis can pose risks. Δ-9-THC undergoes first-pass metabolism in the liver, mediated by the CYP450 system and CYP3A4. The glucuronidation of irinotecan is mediated by uridine diphosphate glycosyltransferase, leading to its recirculation within the hepatic system and potentially increased toxicity due to prolonged drug presence. Cannabis may also compete with drug binding to albumin, altering the plasma concentrations of irinotecan and its conversion to the metabolite SN38.16
Cannabis products can affect chemotherapy levels by interacting with cellular transporters. The MRP1 transporter family, encoded by the ABCC gene family, is expressed mainly in the lung, kidney, skeletal muscle, and hematopoietic stem cells. A 2018 study investigating the effects of THC, CBD, and CBN on MRP1 transporters found that the presence of a cannabis component increased the concentration of vincristine 3-fold. Additional studies suggest the interaction with the CB1 receptor may lead to changes in the expression of MRP1 transporters.17
CBD inhibits the BCRP transporter, which functions as an efflux pump for methotrexate. Consequently, CBD can increase methotrexate levels, potentially enhancing efficacy but also worsening adverse effects.18
In pancreatic cancer, CBD specifically interacts with gemcitabine. CB1 and CB2 receptors are upregulated, and CBD inhibits the GPR55 receptor. These interactions may enhance the antineoplastic effect of gemcitabine, reducing cell cycle progression and growth.19
CBD also interacts with temozolomide (TMZ) by affecting extracellular vesicles used by cells for pro-oncogenic signaling and immune system evasion. Experiments on patient-derived glioblastoma cells, both chemotherapy-resistant and chemotherapy-sensitive, found that CBD increases the formation of extracellular vesicles with reduced levels of miR21 (pro-oncogenic) and elevated levels of miR126 (antioncogenic).20 CBD has also been found to decrease prohibitin levels, a protein associated with TMZ resistance.
In patients with glioblastoma, CBD combined with chemotherapeutic agents like TMZ, carmustine, doxorubicin, and cisplatin has shown increased sensitivity and improved tumor response. CBD is also known to inhibit NF-kB, a pathway that sustains tumor viability despite chemotherapy.21 Additionally, CBD inhibits the P-glycoprotein system, affecting chemotherapy efflux from neoplastic cells.14 In vitro studies have found that CBD is synergistic with bortezomib in inhibiting cancer cell viability. In another glioblastoma model, CBD enhanced the antiproliferative effects of both TMZ and carmustine.14
Different cannabis formulations may vary in how they interact with various cytotoxic chemotherapeutic agents. Some may potentiate the effects of chemotherapy and act synergistically to inhibit tumor growth, while others may lead to increased toxicity.10 More research is needed to determine which formulations, in combination with specific agents and doses, may have significant interactions that warrant adjustments in chemotherapy dosing.
Cannabis/Immunotherapy Interactions
Cannabis is an immunosuppressant. Data suggest the use of cannabis during immunotherapy worsens treatment outcomes in patients with cancer.22 Exogenous (THC) and endogenous (AEA) CBs negatively affect antitumor immunity by impairing the function of tumor-specific T cells via CB2 and by inhibiting the Jak1-STATs signaling in T cells through CNR2. Xiong et al found that THC reduces the therapeutic effect of anti-PD-1 therapy.22
In a prospective observational clinical study, Bar-Sela et al analyzed 102 patients with advanced cancer—of which 68 were cannabis users—that were started on immune checkpoint inhibitor therapy. The study found that cannabis users on anti-PD-1 (nivolumab, pembrolizumab), anti-CTLA-4 (ipilimumab), and anti-PD-L1 (durvalumab, atezolizumab) had a significant decrease in time to treatment progression and overall survival vs cannabis non-users.23 However, a 2023 study by Waissengrin et al found that concomitant use of medical cannabis with pembrolizumab had no harmful effect in advanced non-small cell lung cancer.24 Time to treatment progression of cannabis users did not differ from cannabis nonusers.25
Cannabis/Endocrine Therapy Interactions
In addition to having direct antineoplastic activity on tumor cells, data exist that show how cannabis affects the endocrine system. In animal models, cannabis has been found to suppress the whole hypothalamic-pituitary-adrenal axis as well as other hormones like thyroid, prolactin, and growth hormone. In breast cancer, cannabis competes with estrogen for the estrogen receptor and suppresses growth.26
The endocrine agents used by patients with cancer in this study were antiandrogens like abiraterone, enzalutamide, tamoxifen and anastrozole. Abiraterone is metabolized by CYP450 isoenzymes and uridine diphosphate glycosyltransferases. Cannabis inhibits both processes and therefore may lead to increased toxicities.27 Conversely, enzalutamide is a strong CYP3A inducer, and cannabis use during enzalutamide therapy may significantly increase the toxic effects of cannabis.
There is evidence that molecular pathways involving CB receptors and estrogens overlap, which may lead to interactions when antiestrogens are used in cannabis users with hormone receptor-positive breast cancer.26 In preclinical studies, tamoxifen has been shown to act as an inverse agonist on CB1 and CB2 receptors, though the significance of this finding is unclear. There is no research evaluating the effects of CBs on tamoxifen treatment. However, CBD has been found to potentiate the effectiveness of anastrozole or exemestane in breast cancer cell lines.28 Dobovišek et al demonstrated no inhibitory effect of CBD on the activity of tamoxifen, fulvestrant, or palbociclib in breast cancer cell lines.29 The interactions between hormone receptor-positive breast cancer and cannabinoids are complex, and the clinical significance of these interactions remains difficult to identify.
Cannabis/Targeted Therapy Interactions
The targeted therapies used by patients in this study included zanubrutinib, ibrutinib, sorafenib, acalabrutinib, dabrafenib, trametinib, trastuzumab, bevacizumab, daratumumab, and imatinib. Compared to other classes of cancer treatments, most studies have not demonstrated decreased efficacy or increased toxicity of targeted anticancer drugs when used concomitantly with CBD.29
Trastuzumab is a recombinant humanized monoclonal antibody that targets the proto-oncogene HER2/neu. It is used to treat select patients with metastatic breast cancer. Studies have shown that cannabis use does not attenuate the effectiveness of trastuzumab in HER2-positive and triple-negative breast cancer subtypes.29 One study found that CBD, in combination with chemotherapeutics and Bruton tyrosine kinase inhibitors, such as ibrutinib and zanubrutinib, has synergistic potential for treating diffuse large B-cell lymphoma and mantle cell lymphoma cell lines. This synergy is attributed to the CB1 antagonist activity of cannabis against diffuse large B-cell lymphoma and mantle cell lymphoma cell lines.30,31
Moreover, combining cannabinoids with bevacizumab (a monoclonal anti-VEGF antibody) has been shown to decrease tumor growth and intratumoral hypoxia in clinically relevant human glioblastoma models. This effect is mediated through the downregulation of HIF-1α.32 Long-term studies evaluating the potential harmful or synergistic potential of CBD on targeted anticancer therapy are needed.
CONCLUSIONS
This exploratory study of patients receiving cancer therapy at WJVAMC found a significant prevalence of concurrent cannabis use among patients undergoing antineoplastic treatments. Given that many antineoplastic agents are metabolized by the CYP450 enzyme system, the findings of this study suggest that concurrent cannabis use may pose risks of suboptimal therapeutic outcomes due to potential interactions affecting drug metabolism. These interactions could impact the efficacy and toxicity of the antineoplastic therapies, potentially leading to diminished therapeutic effects or exacerbated adverse reactions.
Patients should be informed regarding the potential decreased efficacy of immunotherapy with concurrent use of cannabis products. They should also be aware of the possibility of increased toxicity with other treatment modalities, though the exact impact on efficacy remains unclear. This highlights the necessity of caution when combining cannabis with prescribed cancer treatments.
While this study identified possible interactions, its data are preliminary and highlight the need for more rigorous research. Future studies should include larger, well-designed cohorts to compare outcomes between cannabis users and nonusers. Such research is essential to fully elucidate the clinical implications of cannabis use during cancer treatment, address the high prevalence of cannabis use among patients with cancer, and mitigate potential risks associated with combining cannabis products with antineoplastic therapies. This will ensure that treatment strategies are optimized for safety and efficacy in this complex patient population.
Cannabis has a long history of use for medicinal and recreational purposes. Research illustrates the potential benefits and increased prevalence of cannabis use in patients with cancer.1 Cannabis products have been shown to possess antineoplastic and palliative activity, improving nociceptive and neuropathic pain in addition to chemotherapy-related nausea and vomiting.2-5 Despite these developments and changing social attitudes toward cannabis, there remains a lack of comprehensive data on patient perspectives regarding its use, especially in regions where cannabis remains illegal. This knowledge gap is notable among veterans undergoing cancer treatment in states where cannabis is prohibited. Up to 57% of veterans report lifetime marijuana use, making it crucial to understand this population’s cannabis use patterns and potential interactions with cancer treatments.6
This observational study sought to determine the prevalence of cannabis use among patients undergoing cancer treatment at the US Department of Veterans Affairs (VA) Memphis Healthcare System and evaluate the potential risks associated with combining cannabis products with anticancer therapies.
METHODS
This prospective observational study identified cannabis use among veterans receiving antineoplastic therapy at the Lt. Col. Luke Weathers Jr. VA Medical Center (WJVAMC) and analyzed potential interactions between cannabis products and their cancer treatments. Participants included adults aged > 18 years undergoing antineoplastic therapy at WJVAMC who consented to the study. Data collection involved a written survey approved by the WJVAMC Institutional Review Board and verbal consent from participants. The survey asked participants about their cannabis use in the previous 90 days, including details on quantity, frequency, and method of consumption (eg, inhalation, oral, topical). No incentives were offered for participation.
Surveys from 50 patients who used cannabis were analyzed and their electronic health records were reviewed for sex, age, diagnosis, and antineoplastic regimen. This information was securely stored. A literature review was conducted using PubMed and the Cochrane Library to explore potential interactions between cannabis and the antineoplastic agents that were prescribed to patients in the study, focusing on toxicity, efficacy, or synergistic effects.
Patients were categorized into 4 groups based on treatment: cytotoxic chemotherapy, immunotherapy, endocrine therapy, and targeted therapy. Patients undergoing multiple types of therapies were included in each applicable category.
RESULTS
A total of 132 patients agreed to participate. Fifty patients (38%) acknowledged using cannabis products within 90 days. The patients that used cannabis products within 90 days of the survey reported the following malignancies: 8 patients (16%) had prostate cancer, 3 patients (6%) had hepatocellular carcinoma, 7 patients (14%) had pancreatic carcinoma, 5 patients (10%) had multiple myeloma, 3 patients (6%) had chronic lymphocytic leukemia, 9 patients (18%) had non-small cell lung cancer, 3 patients (6%) had breast cancer, 3 (6%) patients had bladder cancer, 2 patients (4%) had renal cell carcinoma, 1 (2%) patient had chronic myeloid leukemia, 1 (2%) patient had renal amyloid, 1 patient (2%) had supraglottic squamous cell carcinoma, 1 patient (2%) had esophageal carcinoma, 1 (2%) patient had small cell lung cancer, 1 (2%) patient had gastric cancer, and 1 patient (2%) had follicular lymphoma.
Five (10%) of the cannabis users were female, and 45 (90%) were male. Twenty-nine patients (58%) were aged 66 to 75 years, 16 (32%) were aged 56 to 65 years, 3 (6%) were aged 46 to 55 years, and 2 (4%) were aged 76 to 85 years.
Thirty-five patients (70%) inhaled cannabis as opposed to using it via other formulations or a combination (eg, inhalation and topical). Thirty-eight percent of patients used cannabis once daily, 24% used < 1 daily, and 28% used it ≥ 2 times daily. Five patients (10%) did not report the frequency of their cannabis use. Among the patients who reported cannabis use, 21 (42%) were undergoing cytotoxic chemotherapy, 19 (38%) were undergoing immunotherapy, 12 (24%) were undergoing targeted therapy, and 10 (20%) were undergoing endocrine therapy. Some patients were treated with multiple types of antineoplastic agents and were counted in multiple categories (Table 1).
Following a literature review of cannabis and antineoplastic agents, patients were evaluated for the potential effects of cannabis on their treatment. The literature review revealed that 31% of cytotoxic chemotherapy agents received by patients in this study might have increased toxicity, and 19% could have reduced efficacy when combined with cannabis. Among immunotherapy agents received by patients in this study, 70% might have decreased efficacy when combined with cannabis use. For targeted therapies, 35% could have increased toxicity, and 70% of endocrine agents could potentially have decreased efficacy (Table 2).
DISCUSSION
This prospective study corroborates previous research by demonstrating that more than one-third of patients receiving oncology care at WJVAMC use cannabis, most often inhaled. Cannabis use was observed among patients undergoing various cancer therapies, including cytotoxic chemotherapy, immunotherapy, targeted therapy, and endocrine therapy. The most common malignancies among cannabis users at WJVAMC include patients with lung cancer, prostate cancer, pancreatic cancer, and multiple myeloma. Cannabis use in patients with pancreatic cancer and multiple myeloma was significantly out of proportion to their prevalence at WJVAMC. This could potentially be due to their drastic effect on quality of life.
Cannabis use increased the risk of toxicity in patients treated with cytotoxic chemotherapy and targeted therapy. Cannabis use potentially decreased efficacy for patients treated with cytotoxic chemotherapy and/or immunotherapy. Cannabis use did not increase the risk of toxicity or efficacy in patients treated with endocrine therapy.
Antineoplastics/Cannabis Interactions
The potential interactions between cannabis and antineoplastic therapies administered at WJVAMC are worth exploring. While this review aims to shed light on possible interactions, it is important to acknowledge that much of the data is preliminary and derived from in vitro studies. The interactions should be interpreted as potential risks rather than established facts. Additional research is needed to confirm these interactions and effectively guide clinical practices. Understanding these dynamics is essential to optimize patient care and manage the complex interplay between cannabis use and cancer treatment.
Originating from Central Asia, the cannabis plant contains > 400 medicinally relevant compounds, of which about 100 are cannabinoids (CBs). Key CBs are cannabidiol (CBD), a nonpsychoactive compound, and ?-9-tetrahydrocannabinol (THC), a psychoactive compound. THC can make up 20% to 30% of the dry weight of female cannabis flowers.7
CBs act through the endocannabinoid system, involving CB1 and CB2 receptors, endogenous CBs like anandamide (AEA) and 2-arachidonoylglycerol, and various enzymes. These endogenous CBs, derived from arachidonic acid, play roles in cell growth and proliferation.8 In some studies, AEA has induced apoptosis in neuroblastoma cells and inhibited proliferation in breast cancer cells. However, other research suggests AEA may block apoptosis under certain conditions.9
CB receptors are transmembrane proteins that interact with CBs differently depending on tissue type and CB structure. Synthetic CBs are designed to target specific receptors, while natural CBs may act as both agonists and antagonists.10
Cytochrome P450 Metabolism
The human cytochrome P450 (CYP) 3A subfamily affects the metabolism of many therapeutic drugs, including cancer therapeutics.11 The various compositions of cannabis are primarily metabolized by the CYP450 pathway, the same as many cancer-directed pharmacologic treatments. CBs act as both CYP inducers and inhibitors. THC, for example, is a CYP inducer whereas CBD is a CYP inhibitor; both are found in the various compounds available for consumption.12,13 Pharmacology research has suggested potential interactions and effects on established adverse symptoms, but clinical data are lacking, and current research revealing interactions are only recognized in vitro.14
The Antineoplastic Activity of Cannabis
CBs can affect various cancer-related pathways such as PKB, AMPK, CAMKK-ß, mTOR, PDHK, HIF-1 a, and PPAR-γ. Δ-9-THC can selectively induce apoptosis in tumor cells without harming normal cells, though the exact mechanism remains unclear. Promising results from early mouse studies led to a 2006 human study where intracranial Δ-9-THC in patients with recurrent glioma yielded a median survival of 24 weeks, with 2 patients surviving > 1 year.15
In a 2022 review article, Cherkasova et al highlighted potential clinical benefits of cannabis across various cancers. They found that upregulated CB1 receptors in colon cancer might enhance the effect of 5-fluorouracil. However, many studies are preliminary and therefore not definitive.10
Additional research is needed to refine these findings. Challenges include variability in cannabis formulations, the complex tumor microenvironment, and the legal and psychoactive issues surrounding cannabis use. These factors complicate the design of multicenter randomized studies and may deter patients from disclosing cannabis use, thereby hindering efforts to fully understand its therapeutic potential.
Cannabis/Cytotoxic Chemotherapy Interactions
The chemotherapy agents used in this study included carboplatin, paclitaxel, 5-fluorouracil, etoposide, irinotecan, oxaliplatin, pemetrexed, docetaxel, cabazitaxel, T-DM1, gemcitabine, and cyclophosphamide. There is a paucity of research regarding the interactions between cytotoxic chemotherapy and cannabis. Most studies focused on CBD due to its inhibition of the CYP450 pathway, which is used for metabolizing cytotoxic chemotherapies. Through this mechanism, CBD could potentially increase the concentrations of chemotherapeutic agents, enhancing their toxicity.
When combined with irinotecan, cannabis can pose risks. Δ-9-THC undergoes first-pass metabolism in the liver, mediated by the CYP450 system and CYP3A4. The glucuronidation of irinotecan is mediated by uridine diphosphate glycosyltransferase, leading to its recirculation within the hepatic system and potentially increased toxicity due to prolonged drug presence. Cannabis may also compete with drug binding to albumin, altering the plasma concentrations of irinotecan and its conversion to the metabolite SN38.16
Cannabis products can affect chemotherapy levels by interacting with cellular transporters. The MRP1 transporter family, encoded by the ABCC gene family, is expressed mainly in the lung, kidney, skeletal muscle, and hematopoietic stem cells. A 2018 study investigating the effects of THC, CBD, and CBN on MRP1 transporters found that the presence of a cannabis component increased the concentration of vincristine 3-fold. Additional studies suggest the interaction with the CB1 receptor may lead to changes in the expression of MRP1 transporters.17
CBD inhibits the BCRP transporter, which functions as an efflux pump for methotrexate. Consequently, CBD can increase methotrexate levels, potentially enhancing efficacy but also worsening adverse effects.18
In pancreatic cancer, CBD specifically interacts with gemcitabine. CB1 and CB2 receptors are upregulated, and CBD inhibits the GPR55 receptor. These interactions may enhance the antineoplastic effect of gemcitabine, reducing cell cycle progression and growth.19
CBD also interacts with temozolomide (TMZ) by affecting extracellular vesicles used by cells for pro-oncogenic signaling and immune system evasion. Experiments on patient-derived glioblastoma cells, both chemotherapy-resistant and chemotherapy-sensitive, found that CBD increases the formation of extracellular vesicles with reduced levels of miR21 (pro-oncogenic) and elevated levels of miR126 (antioncogenic).20 CBD has also been found to decrease prohibitin levels, a protein associated with TMZ resistance.
In patients with glioblastoma, CBD combined with chemotherapeutic agents like TMZ, carmustine, doxorubicin, and cisplatin has shown increased sensitivity and improved tumor response. CBD is also known to inhibit NF-kB, a pathway that sustains tumor viability despite chemotherapy.21 Additionally, CBD inhibits the P-glycoprotein system, affecting chemotherapy efflux from neoplastic cells.14 In vitro studies have found that CBD is synergistic with bortezomib in inhibiting cancer cell viability. In another glioblastoma model, CBD enhanced the antiproliferative effects of both TMZ and carmustine.14
Different cannabis formulations may vary in how they interact with various cytotoxic chemotherapeutic agents. Some may potentiate the effects of chemotherapy and act synergistically to inhibit tumor growth, while others may lead to increased toxicity.10 More research is needed to determine which formulations, in combination with specific agents and doses, may have significant interactions that warrant adjustments in chemotherapy dosing.
Cannabis/Immunotherapy Interactions
Cannabis is an immunosuppressant. Data suggest the use of cannabis during immunotherapy worsens treatment outcomes in patients with cancer.22 Exogenous (THC) and endogenous (AEA) CBs negatively affect antitumor immunity by impairing the function of tumor-specific T cells via CB2 and by inhibiting the Jak1-STATs signaling in T cells through CNR2. Xiong et al found that THC reduces the therapeutic effect of anti-PD-1 therapy.22
In a prospective observational clinical study, Bar-Sela et al analyzed 102 patients with advanced cancer—of which 68 were cannabis users—that were started on immune checkpoint inhibitor therapy. The study found that cannabis users on anti-PD-1 (nivolumab, pembrolizumab), anti-CTLA-4 (ipilimumab), and anti-PD-L1 (durvalumab, atezolizumab) had a significant decrease in time to treatment progression and overall survival vs cannabis non-users.23 However, a 2023 study by Waissengrin et al found that concomitant use of medical cannabis with pembrolizumab had no harmful effect in advanced non-small cell lung cancer.24 Time to treatment progression of cannabis users did not differ from cannabis nonusers.25
Cannabis/Endocrine Therapy Interactions
In addition to having direct antineoplastic activity on tumor cells, data exist that show how cannabis affects the endocrine system. In animal models, cannabis has been found to suppress the whole hypothalamic-pituitary-adrenal axis as well as other hormones like thyroid, prolactin, and growth hormone. In breast cancer, cannabis competes with estrogen for the estrogen receptor and suppresses growth.26
The endocrine agents used by patients with cancer in this study were antiandrogens like abiraterone, enzalutamide, tamoxifen and anastrozole. Abiraterone is metabolized by CYP450 isoenzymes and uridine diphosphate glycosyltransferases. Cannabis inhibits both processes and therefore may lead to increased toxicities.27 Conversely, enzalutamide is a strong CYP3A inducer, and cannabis use during enzalutamide therapy may significantly increase the toxic effects of cannabis.
There is evidence that molecular pathways involving CB receptors and estrogens overlap, which may lead to interactions when antiestrogens are used in cannabis users with hormone receptor-positive breast cancer.26 In preclinical studies, tamoxifen has been shown to act as an inverse agonist on CB1 and CB2 receptors, though the significance of this finding is unclear. There is no research evaluating the effects of CBs on tamoxifen treatment. However, CBD has been found to potentiate the effectiveness of anastrozole or exemestane in breast cancer cell lines.28 Dobovišek et al demonstrated no inhibitory effect of CBD on the activity of tamoxifen, fulvestrant, or palbociclib in breast cancer cell lines.29 The interactions between hormone receptor-positive breast cancer and cannabinoids are complex, and the clinical significance of these interactions remains difficult to identify.
Cannabis/Targeted Therapy Interactions
The targeted therapies used by patients in this study included zanubrutinib, ibrutinib, sorafenib, acalabrutinib, dabrafenib, trametinib, trastuzumab, bevacizumab, daratumumab, and imatinib. Compared to other classes of cancer treatments, most studies have not demonstrated decreased efficacy or increased toxicity of targeted anticancer drugs when used concomitantly with CBD.29
Trastuzumab is a recombinant humanized monoclonal antibody that targets the proto-oncogene HER2/neu. It is used to treat select patients with metastatic breast cancer. Studies have shown that cannabis use does not attenuate the effectiveness of trastuzumab in HER2-positive and triple-negative breast cancer subtypes.29 One study found that CBD, in combination with chemotherapeutics and Bruton tyrosine kinase inhibitors, such as ibrutinib and zanubrutinib, has synergistic potential for treating diffuse large B-cell lymphoma and mantle cell lymphoma cell lines. This synergy is attributed to the CB1 antagonist activity of cannabis against diffuse large B-cell lymphoma and mantle cell lymphoma cell lines.30,31
Moreover, combining cannabinoids with bevacizumab (a monoclonal anti-VEGF antibody) has been shown to decrease tumor growth and intratumoral hypoxia in clinically relevant human glioblastoma models. This effect is mediated through the downregulation of HIF-1α.32 Long-term studies evaluating the potential harmful or synergistic potential of CBD on targeted anticancer therapy are needed.
CONCLUSIONS
This exploratory study of patients receiving cancer therapy at WJVAMC found a significant prevalence of concurrent cannabis use among patients undergoing antineoplastic treatments. Given that many antineoplastic agents are metabolized by the CYP450 enzyme system, the findings of this study suggest that concurrent cannabis use may pose risks of suboptimal therapeutic outcomes due to potential interactions affecting drug metabolism. These interactions could impact the efficacy and toxicity of the antineoplastic therapies, potentially leading to diminished therapeutic effects or exacerbated adverse reactions.
Patients should be informed regarding the potential decreased efficacy of immunotherapy with concurrent use of cannabis products. They should also be aware of the possibility of increased toxicity with other treatment modalities, though the exact impact on efficacy remains unclear. This highlights the necessity of caution when combining cannabis with prescribed cancer treatments.
While this study identified possible interactions, its data are preliminary and highlight the need for more rigorous research. Future studies should include larger, well-designed cohorts to compare outcomes between cannabis users and nonusers. Such research is essential to fully elucidate the clinical implications of cannabis use during cancer treatment, address the high prevalence of cannabis use among patients with cancer, and mitigate potential risks associated with combining cannabis products with antineoplastic therapies. This will ensure that treatment strategies are optimized for safety and efficacy in this complex patient population.
- Steele G, Arneson T, Zylla D. A comprehensive review of cannabis in patients with cancer: availability in the USA, general efficacy, and safety. Curr Oncol Rep. 2019;21:1-10. doi:10.1007/s11912-019-0757-7
- Brown D, Watson M, Schloss J. Pharmacological evidence of medicinal cannabis in oncology: a systematic review. Support Care Cancer. 2019;27:3195-320. doi:10.1007/s00520-019-04774-5
- Abrams DI. Integrating cannabis into clinical cancer care. Curr Oncol. 2016;23:S8-S14. doi:10.37.47/co.23.3099
- Serafimovska T, Darkovska-Serafimovska M, Stefkov G, Arsova-Sarafinovska Z, Balkanov T. Pharmacotherapeutic considerations for use of cannabinoids to relieve symptoms of nausea and vomiting induced by chemotherapy. Folia Medica (Plovdiv). 2020;62:668-678. doi:10.3897/folmed.62e51478
- Bar-Sela G, Zalman D, Semenysty V, Ballan E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: pilot study. Integr Cancer Ther. 2019;18:1534735419881498. doi:10.1177/1534735419881498
- Pederson ER, Villarosa-Hurlocker MC, Prince MA. Use of protective behavioral strategies among young adult veteran marijuana users. Cannabis. 2018;1:14-27.
- Schilling S, Melzer R, McCabe PF. Cannabis sativa. Curr Biol. 2020;30:R8-R9. doi:10.1016/j.cub.2019.10.039
- McDougle DR, Kambalyal A, Meling DD, Das A. Endocannabinoids anandamide and 2-arachidonoylglycerol are substrates for human CYP2J2 epoxygenase. J Pharmacol Exp Ther. 2014;351:616-627. doi:10.1124/jpet.114216598
- Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11:1121-1132. doi:10.1038/sj.cdd.4401442
- Cherkasova V, Wang B, Gerasymchuk M, Fiselier A, Kovalchuk O, Kovalchuk I. Use of cannabis and cannabinoids for treatment of cancer. Cancers (Basel). 2022;14:5142. doi:10.3390/cancers14205142
- Engels FK, Ten Tije AJ, Baker SD, et al. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin Pharmacol Ther. 2004;75:448-454. doi:10.1016/j.clpt.2004.01.001
- Alsherbiny MA, Li CG. Medicinal cannabis-potential drug interactions. Medicines (Basel). 2018;6:3. doi:10.3390/medicines6010003
- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46:86-95. doi:10.3109/03602532.2013.849268
- Opitz BJ, Ostroff ML, Whitman AC. The potential clinical implications and importance of drug interactions between anticancer agents and cannabidiol in patients with cancer. J Pharm Pract. 2020;33:506-512. doi:10.1177/0897190019828920
- Guzmán M, Duarte MJ, Blázquez C, et al. A pilot clinical study of D9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197-203. doi:10.1038/sj.bjc.6603236
- Kopjar N, Fuchs N, Brcic Karaconji I, et al. High doses of ?9-tetrahydrocannabinol might impair irinotecan chemotherapy: a review of potentially harmful interactions. Clin Drug Investig. 2020;40:775-787. doi:10.1007/s40261-020-00954-y
- Bouquié R, Deslandes G, Mazaré H, et al. Cannabis and anticancer drugs: societal usage and expected pharmacological interactions - a review. Fundam Clin Pharmacol. 2018;32:462-484. doi:10.1111/fcp.12373
- Buchtova T, Lukac D, Skrott Z, Chroma K, Bartek J, Mistrik M. Drug-drug interactions of cannabidiol with standard-of-care chemotherapeutics. Int J Mol Sci. 2023;24:2885. doi:10.3390/ijms24032885
- Sharafi G, He H, Nikfarjam M. Potential use of cannabinoids for the treatment of pancreatic cancer. J Pancreat Cancer. 2019;5:1-7. doi:10.1089/pancan.2018.0019
- Kosgodage US, Uysal-Onganer P, MacLatchy A, et al. Cannabidiol affects extracellular vesicle release, miR21 and miR126, and reduces prohibitin protein in glioblastoma multiforme cells. Transl Oncol. 2019;12:513-522. doi:10.1016/j.tranon.2018.12.004
- Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of cannabidiol in breast cancer. Mol Oncol. 2015;9:906-919. doi:10.1016/j.molonc.2014.12.010
- Xiong X, Chen S, Shen J, et al. Cannabis suppresses anti-tumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct Target Ther. 2022;7:99. doi:10.1038/s41392-022-00918-y
- Bar-Sela G, Cohen I, Campisi-Pinto S, et al. Cannabis consumption used by cancer patients during immunotherapy correlates with poor clinical outcome. Cancers (Basel). 2020;12:2447. doi:10.3390/cancers12092447
- Waissengrin B, Leshem Y, Taya M, et al. The use of medical cannabis concomitantly with immune checkpoint inhibitors in non-small cell lung cancer: a sigh of relief? Eur J Cancer. 2023;180:52-61. doi:10.1016/j.ejca.2022.11.022
- Sarsembayeva A, Schicho R. Cannabinoids and the endocannabinoid system in immunotherapy: helpful or harmful? Front Oncol. 2023;13:1296906. doi:10.3389/fonc.2023.1296906
- Kisková T, Mungenast F, Suváková M, Jäger W, Thalhammer T. Future aspects for cannabinoids in breast cancer therapy. Int J Mol Sci. 2019;20:1673. doi:10.3390/ijms20071673
- Woerdenbag HJ, Olinga P, Kok EA, et al. Potential, limitations and risks of cannabis-derived products in cancer treatment. Cancers (Basel). 2023;15:2119. doi:10.3390/cancers15072119
- Almeida CF, Teixeira N, Valente MJ, Vinggaard AM, Correia-da-Silva G, Amaral C. Cannabidiol as a promising adjuvant therapy for estrogen receptor-positive breast tumors: unveiling its benefits with aromatase inhibitors. Cancers (Basel). 2023;15:2517. doi:10.3390/cancers15092517
- Dobovišek L, Novak M, Krstanovic F, Borštnar S, Turnšek TL, Debeljak N. Effect of combining CBD with standard breast cancer therapeutics. Adv Cancer Biol Metastasis. 2022;4:100038. doi:10.1016/j.adcanc.2022.100038
- Strong T, Rauvolfova J, Jackson E, Pham LV, Bryant J. Synergistic effect of cannabidiol with conventional chemotherapy treatment. Blood. 2018;132:5382. doi:10.1182/blood-2018-99-116749
- Maggi F, Morelli MB, Tomassoni D, et al. The effects of cannabidiol via TRPV2 channel in chronic myeloid leukemia cells and its combination with imatinib. Cancer Sci. 2022;113:1235-1249. doi:10.1111/cas.15257
- Obad N, Janji B, Prestegarden L, et al. ATPS-59 improving efficacy of bevacizumab treatment in glioblastoma by targeting hif1 alpha. Neuro Oncol. 2015;17:v31. doi:10.1093/neuonc/nov204.59
- Steele G, Arneson T, Zylla D. A comprehensive review of cannabis in patients with cancer: availability in the USA, general efficacy, and safety. Curr Oncol Rep. 2019;21:1-10. doi:10.1007/s11912-019-0757-7
- Brown D, Watson M, Schloss J. Pharmacological evidence of medicinal cannabis in oncology: a systematic review. Support Care Cancer. 2019;27:3195-320. doi:10.1007/s00520-019-04774-5
- Abrams DI. Integrating cannabis into clinical cancer care. Curr Oncol. 2016;23:S8-S14. doi:10.37.47/co.23.3099
- Serafimovska T, Darkovska-Serafimovska M, Stefkov G, Arsova-Sarafinovska Z, Balkanov T. Pharmacotherapeutic considerations for use of cannabinoids to relieve symptoms of nausea and vomiting induced by chemotherapy. Folia Medica (Plovdiv). 2020;62:668-678. doi:10.3897/folmed.62e51478
- Bar-Sela G, Zalman D, Semenysty V, Ballan E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: pilot study. Integr Cancer Ther. 2019;18:1534735419881498. doi:10.1177/1534735419881498
- Pederson ER, Villarosa-Hurlocker MC, Prince MA. Use of protective behavioral strategies among young adult veteran marijuana users. Cannabis. 2018;1:14-27.
- Schilling S, Melzer R, McCabe PF. Cannabis sativa. Curr Biol. 2020;30:R8-R9. doi:10.1016/j.cub.2019.10.039
- McDougle DR, Kambalyal A, Meling DD, Das A. Endocannabinoids anandamide and 2-arachidonoylglycerol are substrates for human CYP2J2 epoxygenase. J Pharmacol Exp Ther. 2014;351:616-627. doi:10.1124/jpet.114216598
- Movsesyan VA, Stoica BA, Yakovlev AG, et al. Anandamide-induced cell death in primary neuronal cultures: role of calpain and caspase pathways. Cell Death Differ. 2004;11:1121-1132. doi:10.1038/sj.cdd.4401442
- Cherkasova V, Wang B, Gerasymchuk M, Fiselier A, Kovalchuk O, Kovalchuk I. Use of cannabis and cannabinoids for treatment of cancer. Cancers (Basel). 2022;14:5142. doi:10.3390/cancers14205142
- Engels FK, Ten Tije AJ, Baker SD, et al. Effect of cytochrome P450 3A4 inhibition on the pharmacokinetics of docetaxel. Clin Pharmacol Ther. 2004;75:448-454. doi:10.1016/j.clpt.2004.01.001
- Alsherbiny MA, Li CG. Medicinal cannabis-potential drug interactions. Medicines (Basel). 2018;6:3. doi:10.3390/medicines6010003
- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46:86-95. doi:10.3109/03602532.2013.849268
- Opitz BJ, Ostroff ML, Whitman AC. The potential clinical implications and importance of drug interactions between anticancer agents and cannabidiol in patients with cancer. J Pharm Pract. 2020;33:506-512. doi:10.1177/0897190019828920
- Guzmán M, Duarte MJ, Blázquez C, et al. A pilot clinical study of D9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br J Cancer. 2006;95:197-203. doi:10.1038/sj.bjc.6603236
- Kopjar N, Fuchs N, Brcic Karaconji I, et al. High doses of ?9-tetrahydrocannabinol might impair irinotecan chemotherapy: a review of potentially harmful interactions. Clin Drug Investig. 2020;40:775-787. doi:10.1007/s40261-020-00954-y
- Bouquié R, Deslandes G, Mazaré H, et al. Cannabis and anticancer drugs: societal usage and expected pharmacological interactions - a review. Fundam Clin Pharmacol. 2018;32:462-484. doi:10.1111/fcp.12373
- Buchtova T, Lukac D, Skrott Z, Chroma K, Bartek J, Mistrik M. Drug-drug interactions of cannabidiol with standard-of-care chemotherapeutics. Int J Mol Sci. 2023;24:2885. doi:10.3390/ijms24032885
- Sharafi G, He H, Nikfarjam M. Potential use of cannabinoids for the treatment of pancreatic cancer. J Pancreat Cancer. 2019;5:1-7. doi:10.1089/pancan.2018.0019
- Kosgodage US, Uysal-Onganer P, MacLatchy A, et al. Cannabidiol affects extracellular vesicle release, miR21 and miR126, and reduces prohibitin protein in glioblastoma multiforme cells. Transl Oncol. 2019;12:513-522. doi:10.1016/j.tranon.2018.12.004
- Elbaz M, Nasser MW, Ravi J, et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: novel anti-tumor mechanisms of cannabidiol in breast cancer. Mol Oncol. 2015;9:906-919. doi:10.1016/j.molonc.2014.12.010
- Xiong X, Chen S, Shen J, et al. Cannabis suppresses anti-tumor immunity by inhibiting JAK/STAT signaling in T cells through CNR2. Signal Transduct Target Ther. 2022;7:99. doi:10.1038/s41392-022-00918-y
- Bar-Sela G, Cohen I, Campisi-Pinto S, et al. Cannabis consumption used by cancer patients during immunotherapy correlates with poor clinical outcome. Cancers (Basel). 2020;12:2447. doi:10.3390/cancers12092447
- Waissengrin B, Leshem Y, Taya M, et al. The use of medical cannabis concomitantly with immune checkpoint inhibitors in non-small cell lung cancer: a sigh of relief? Eur J Cancer. 2023;180:52-61. doi:10.1016/j.ejca.2022.11.022
- Sarsembayeva A, Schicho R. Cannabinoids and the endocannabinoid system in immunotherapy: helpful or harmful? Front Oncol. 2023;13:1296906. doi:10.3389/fonc.2023.1296906
- Kisková T, Mungenast F, Suváková M, Jäger W, Thalhammer T. Future aspects for cannabinoids in breast cancer therapy. Int J Mol Sci. 2019;20:1673. doi:10.3390/ijms20071673
- Woerdenbag HJ, Olinga P, Kok EA, et al. Potential, limitations and risks of cannabis-derived products in cancer treatment. Cancers (Basel). 2023;15:2119. doi:10.3390/cancers15072119
- Almeida CF, Teixeira N, Valente MJ, Vinggaard AM, Correia-da-Silva G, Amaral C. Cannabidiol as a promising adjuvant therapy for estrogen receptor-positive breast tumors: unveiling its benefits with aromatase inhibitors. Cancers (Basel). 2023;15:2517. doi:10.3390/cancers15092517
- Dobovišek L, Novak M, Krstanovic F, Borštnar S, Turnšek TL, Debeljak N. Effect of combining CBD with standard breast cancer therapeutics. Adv Cancer Biol Metastasis. 2022;4:100038. doi:10.1016/j.adcanc.2022.100038
- Strong T, Rauvolfova J, Jackson E, Pham LV, Bryant J. Synergistic effect of cannabidiol with conventional chemotherapy treatment. Blood. 2018;132:5382. doi:10.1182/blood-2018-99-116749
- Maggi F, Morelli MB, Tomassoni D, et al. The effects of cannabidiol via TRPV2 channel in chronic myeloid leukemia cells and its combination with imatinib. Cancer Sci. 2022;113:1235-1249. doi:10.1111/cas.15257
- Obad N, Janji B, Prestegarden L, et al. ATPS-59 improving efficacy of bevacizumab treatment in glioblastoma by targeting hif1 alpha. Neuro Oncol. 2015;17:v31. doi:10.1093/neuonc/nov204.59
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review
Cannabis Use by Veterans and Potential Interactions With Antineoplastic Agents: Analysis and Literature Review

VA Performs Its First ‘Bloodless’ Stem Cell Transplant
PHOENIX ‑ A US Department of Veterans Affairs (VA) hospital in Tennessee has performed the first “bloodless” autologous stem cell transplant within the Veterans Health Administration, treating a 61-year-old Jehovah’s Witness patient with multiple myeloma who traveled from California for the procedure.
The case, presented at the annual meeting of the Association of VA Hematology/Oncology, stated that “we should not withhold any therapies for patients who are Jehovah’s Witnesses out of fear of them bleeding out or having complications from anemia,” said Bhagirathbhai Dholaria, MBBS, an associate professor of medicine at Vanderbilt University Medical Center who worked with the VA Tennessee Valley Healthcare System in Nashville.
While Jehovah’s Witnesses accept medical treatment, their faith forbids blood transfusions, including of preoperative autologous blood, due to its interpretation of the Bible. The faith allows individuals to decide whether to accept stem cells collected from their blood or someone else’s “provided that blood components are not intentionally collected, stored, and reinfused along with the stem cells.”
There are an estimated 1.2 million Jehovah’s Witnesses in the US.
Traditional Stem Cell Transplants Require Blood Support
In conventional autologous stem cell transplants for multiple myeloma, high-dose chemotherapy temporarily wipes out the patient’s bone marrow for about 2 to 3 weeks, Dholaria explained. During this period, patients typically receive 2 units of packed red blood cells and platelet transfusions to prevent severe complications from anemia and low platelet counts.
“Because of this reason, Jehovah’s Witnesses have been traditionally denied these therapies,” Dholaria said.
However, bloodless autologous transplants have been performed for about 2 decades, and Vanderbilt University has been offering the procedures for about 3 years, according to Dholaria.
For the first bloodless procedure in the VA, the patient–who had an aggressive, newly diagnosed IgG kappa multiple myeloma–was evaluated.
“He had been treated by local doctors in California. Otherwise, he was actually in really good shape. Physically, he didn’t have any major issues,” Dholaria said. “So, he met the criteria for our bloodless protocol, and we decided to offer him the procedure.”
The team consulted ethics and legal departments and noted the patient’s blood product preferences in his electronic health record. The patient then underwent a preoptimization protocol that included erythropoiesis-stimulating agents, intravenous iron, and vitamin B12 supplementation to boost blood counts before the transplant, according to the case presentation.
Special Protocol Required in ‘Bloodless’ Procedures
After stem cell collection and chemotherapy, patients undergoing bloodless procedures receive aggressive growth factor support to minimize the duration and severity of cytopenia, Dholaria said. As part of the protocol, the care team uses pediatric tubes for blood draws to minimize blood loss and monitors patients closely on cardiac monitors, he added. In addition, blood draws are only performed every 3 days.
“We watch for any cardiac decompensation because these patients have severe anemia for a brief period of time. We make sure they don’t [have a] heart attack or arrhythmias,” Dholaria said. “Or if the platelets are too low, and they start oozing blood from the nose, gums, or gut, that needs to be dealt with accordingly.”
For bleeding complications, the team uses clotting factors and intravenous and oral medications to support remaining platelet function rather than platelet transfusions.
The patient in this case tolerated the transplant “exceptionally well with minimal complications,” according to the case presentation. He achieved full engraftment on day 14 after transplant and was discharged from inpatient care with continued monitoring through day 30.
“The patient was very compliant,” said Salyka Sengsayadeth, MD, medical director of the VA Tennessee Valley Healthcare System Stem Cell Transplant and Cellular Therapy Program and associate professor of medicine at Vanderbilt.
“He tolerated everything that we needed to do,” she said. “He called us when he needed to call us and did everything that we asked and recommended for him.”
The patient’s roughly 30-day hospital stay matched that of typical transplant patients, Sengsayadeth noted. His myeloma responded to treatment, and he returned to California, Dholaria said.
‘Bloodless’ Procedures Not for All Stem Cell Transplants
The case highlights the availability of stem cell transplants in the VA–they are only performed in Seattle and Nashville–and opportunities for patients who wish to avoid blood transfusions. Sengsayadeth said the bloodless protocol is available for patients without religious objections who simply prefer to avoid blood products.
Dholaria cautioned that bloodless protocol applies specifically to autologous transplants, where patients receive their own stem cells. The team does not plan to offer bloodless allogeneic transplants, which use donor stem cells for conditions like leukemia, due to higher risks. In addition, most Jehovah’s Witnesses decline allogeneic transplants because they do not accept stem cells from another person, Dholaria said.
Beyond multiple myeloma, the Tennessee Valley Healthcare System offers bloodless autologous transplants for various blood cancers, including non-Hodgkin lymphomas such as large B-cell lymphoma, follicular lymphoma, and mantle cell lymphoma, as well as lymphomas affecting the brain, Dholaria said.
Clinicians “should start thinking about this early on, as soon as the cancer diagnosis is made, to make the referral and get the patient on our radar,” Dholaria said.
Sengsayadeth said physicians within the VA typically know how to refer appropriate patients to her team. “They just send us an email or give us a call or a message to say ‘I have this patient. Do you think they’re someone I should send to you?’ We usually answer right back, and then we can proceed with the full evaluation if we think that’s a reasonable thing to do.”
‘Treated Like Family’
The patient, a Marine Corps veteran named Keith Cody, spoke about the procedure in a video interview. Cody said he was reluctant at first to undergo the procedure because he didn’t understand what it would accomplish.
“As I was doing the massive chemo every week, and then suffering with the side effects, I decided to ask again about this procedure and how it improves my quality of life,” he said.
At the time of the taping of the video, Cody was getting ready to go home to California. “They’ve told me that I’ll still need more time to get my energy back, but I do feel much better already,” he said.
He also praised the staff. “Everybody that we came across, I enjoyed the interactions. It’s actually sad to leave people behind that you really felt treated you like family.”
Dholaria discloses relationships with Janssen, Angiocrine, Pfizer, Poseida, MEI, Orcabio, Wugen, Allovir, Adicet, BMS, Molecular Templates, Atara, MJH, Arvinas, Janssen, ADC, Gilead, GSK, Caribou, F. Hoffmann-La Roche AG, Autolus, and Pierre Fabre.
Sengsayadeth has no disclosures.
PHOENIX ‑ A US Department of Veterans Affairs (VA) hospital in Tennessee has performed the first “bloodless” autologous stem cell transplant within the Veterans Health Administration, treating a 61-year-old Jehovah’s Witness patient with multiple myeloma who traveled from California for the procedure.
The case, presented at the annual meeting of the Association of VA Hematology/Oncology, stated that “we should not withhold any therapies for patients who are Jehovah’s Witnesses out of fear of them bleeding out or having complications from anemia,” said Bhagirathbhai Dholaria, MBBS, an associate professor of medicine at Vanderbilt University Medical Center who worked with the VA Tennessee Valley Healthcare System in Nashville.
While Jehovah’s Witnesses accept medical treatment, their faith forbids blood transfusions, including of preoperative autologous blood, due to its interpretation of the Bible. The faith allows individuals to decide whether to accept stem cells collected from their blood or someone else’s “provided that blood components are not intentionally collected, stored, and reinfused along with the stem cells.”
There are an estimated 1.2 million Jehovah’s Witnesses in the US.
Traditional Stem Cell Transplants Require Blood Support
In conventional autologous stem cell transplants for multiple myeloma, high-dose chemotherapy temporarily wipes out the patient’s bone marrow for about 2 to 3 weeks, Dholaria explained. During this period, patients typically receive 2 units of packed red blood cells and platelet transfusions to prevent severe complications from anemia and low platelet counts.
“Because of this reason, Jehovah’s Witnesses have been traditionally denied these therapies,” Dholaria said.
However, bloodless autologous transplants have been performed for about 2 decades, and Vanderbilt University has been offering the procedures for about 3 years, according to Dholaria.
For the first bloodless procedure in the VA, the patient–who had an aggressive, newly diagnosed IgG kappa multiple myeloma–was evaluated.
“He had been treated by local doctors in California. Otherwise, he was actually in really good shape. Physically, he didn’t have any major issues,” Dholaria said. “So, he met the criteria for our bloodless protocol, and we decided to offer him the procedure.”
The team consulted ethics and legal departments and noted the patient’s blood product preferences in his electronic health record. The patient then underwent a preoptimization protocol that included erythropoiesis-stimulating agents, intravenous iron, and vitamin B12 supplementation to boost blood counts before the transplant, according to the case presentation.
Special Protocol Required in ‘Bloodless’ Procedures
After stem cell collection and chemotherapy, patients undergoing bloodless procedures receive aggressive growth factor support to minimize the duration and severity of cytopenia, Dholaria said. As part of the protocol, the care team uses pediatric tubes for blood draws to minimize blood loss and monitors patients closely on cardiac monitors, he added. In addition, blood draws are only performed every 3 days.
“We watch for any cardiac decompensation because these patients have severe anemia for a brief period of time. We make sure they don’t [have a] heart attack or arrhythmias,” Dholaria said. “Or if the platelets are too low, and they start oozing blood from the nose, gums, or gut, that needs to be dealt with accordingly.”
For bleeding complications, the team uses clotting factors and intravenous and oral medications to support remaining platelet function rather than platelet transfusions.
The patient in this case tolerated the transplant “exceptionally well with minimal complications,” according to the case presentation. He achieved full engraftment on day 14 after transplant and was discharged from inpatient care with continued monitoring through day 30.
“The patient was very compliant,” said Salyka Sengsayadeth, MD, medical director of the VA Tennessee Valley Healthcare System Stem Cell Transplant and Cellular Therapy Program and associate professor of medicine at Vanderbilt.
“He tolerated everything that we needed to do,” she said. “He called us when he needed to call us and did everything that we asked and recommended for him.”
The patient’s roughly 30-day hospital stay matched that of typical transplant patients, Sengsayadeth noted. His myeloma responded to treatment, and he returned to California, Dholaria said.
‘Bloodless’ Procedures Not for All Stem Cell Transplants
The case highlights the availability of stem cell transplants in the VA–they are only performed in Seattle and Nashville–and opportunities for patients who wish to avoid blood transfusions. Sengsayadeth said the bloodless protocol is available for patients without religious objections who simply prefer to avoid blood products.
Dholaria cautioned that bloodless protocol applies specifically to autologous transplants, where patients receive their own stem cells. The team does not plan to offer bloodless allogeneic transplants, which use donor stem cells for conditions like leukemia, due to higher risks. In addition, most Jehovah’s Witnesses decline allogeneic transplants because they do not accept stem cells from another person, Dholaria said.
Beyond multiple myeloma, the Tennessee Valley Healthcare System offers bloodless autologous transplants for various blood cancers, including non-Hodgkin lymphomas such as large B-cell lymphoma, follicular lymphoma, and mantle cell lymphoma, as well as lymphomas affecting the brain, Dholaria said.
Clinicians “should start thinking about this early on, as soon as the cancer diagnosis is made, to make the referral and get the patient on our radar,” Dholaria said.
Sengsayadeth said physicians within the VA typically know how to refer appropriate patients to her team. “They just send us an email or give us a call or a message to say ‘I have this patient. Do you think they’re someone I should send to you?’ We usually answer right back, and then we can proceed with the full evaluation if we think that’s a reasonable thing to do.”
‘Treated Like Family’
The patient, a Marine Corps veteran named Keith Cody, spoke about the procedure in a video interview. Cody said he was reluctant at first to undergo the procedure because he didn’t understand what it would accomplish.
“As I was doing the massive chemo every week, and then suffering with the side effects, I decided to ask again about this procedure and how it improves my quality of life,” he said.
At the time of the taping of the video, Cody was getting ready to go home to California. “They’ve told me that I’ll still need more time to get my energy back, but I do feel much better already,” he said.
He also praised the staff. “Everybody that we came across, I enjoyed the interactions. It’s actually sad to leave people behind that you really felt treated you like family.”
Dholaria discloses relationships with Janssen, Angiocrine, Pfizer, Poseida, MEI, Orcabio, Wugen, Allovir, Adicet, BMS, Molecular Templates, Atara, MJH, Arvinas, Janssen, ADC, Gilead, GSK, Caribou, F. Hoffmann-La Roche AG, Autolus, and Pierre Fabre.
Sengsayadeth has no disclosures.
PHOENIX ‑ A US Department of Veterans Affairs (VA) hospital in Tennessee has performed the first “bloodless” autologous stem cell transplant within the Veterans Health Administration, treating a 61-year-old Jehovah’s Witness patient with multiple myeloma who traveled from California for the procedure.
The case, presented at the annual meeting of the Association of VA Hematology/Oncology, stated that “we should not withhold any therapies for patients who are Jehovah’s Witnesses out of fear of them bleeding out or having complications from anemia,” said Bhagirathbhai Dholaria, MBBS, an associate professor of medicine at Vanderbilt University Medical Center who worked with the VA Tennessee Valley Healthcare System in Nashville.
While Jehovah’s Witnesses accept medical treatment, their faith forbids blood transfusions, including of preoperative autologous blood, due to its interpretation of the Bible. The faith allows individuals to decide whether to accept stem cells collected from their blood or someone else’s “provided that blood components are not intentionally collected, stored, and reinfused along with the stem cells.”
There are an estimated 1.2 million Jehovah’s Witnesses in the US.
Traditional Stem Cell Transplants Require Blood Support
In conventional autologous stem cell transplants for multiple myeloma, high-dose chemotherapy temporarily wipes out the patient’s bone marrow for about 2 to 3 weeks, Dholaria explained. During this period, patients typically receive 2 units of packed red blood cells and platelet transfusions to prevent severe complications from anemia and low platelet counts.
“Because of this reason, Jehovah’s Witnesses have been traditionally denied these therapies,” Dholaria said.
However, bloodless autologous transplants have been performed for about 2 decades, and Vanderbilt University has been offering the procedures for about 3 years, according to Dholaria.
For the first bloodless procedure in the VA, the patient–who had an aggressive, newly diagnosed IgG kappa multiple myeloma–was evaluated.
“He had been treated by local doctors in California. Otherwise, he was actually in really good shape. Physically, he didn’t have any major issues,” Dholaria said. “So, he met the criteria for our bloodless protocol, and we decided to offer him the procedure.”
The team consulted ethics and legal departments and noted the patient’s blood product preferences in his electronic health record. The patient then underwent a preoptimization protocol that included erythropoiesis-stimulating agents, intravenous iron, and vitamin B12 supplementation to boost blood counts before the transplant, according to the case presentation.
Special Protocol Required in ‘Bloodless’ Procedures
After stem cell collection and chemotherapy, patients undergoing bloodless procedures receive aggressive growth factor support to minimize the duration and severity of cytopenia, Dholaria said. As part of the protocol, the care team uses pediatric tubes for blood draws to minimize blood loss and monitors patients closely on cardiac monitors, he added. In addition, blood draws are only performed every 3 days.
“We watch for any cardiac decompensation because these patients have severe anemia for a brief period of time. We make sure they don’t [have a] heart attack or arrhythmias,” Dholaria said. “Or if the platelets are too low, and they start oozing blood from the nose, gums, or gut, that needs to be dealt with accordingly.”
For bleeding complications, the team uses clotting factors and intravenous and oral medications to support remaining platelet function rather than platelet transfusions.
The patient in this case tolerated the transplant “exceptionally well with minimal complications,” according to the case presentation. He achieved full engraftment on day 14 after transplant and was discharged from inpatient care with continued monitoring through day 30.
“The patient was very compliant,” said Salyka Sengsayadeth, MD, medical director of the VA Tennessee Valley Healthcare System Stem Cell Transplant and Cellular Therapy Program and associate professor of medicine at Vanderbilt.
“He tolerated everything that we needed to do,” she said. “He called us when he needed to call us and did everything that we asked and recommended for him.”
The patient’s roughly 30-day hospital stay matched that of typical transplant patients, Sengsayadeth noted. His myeloma responded to treatment, and he returned to California, Dholaria said.
‘Bloodless’ Procedures Not for All Stem Cell Transplants
The case highlights the availability of stem cell transplants in the VA–they are only performed in Seattle and Nashville–and opportunities for patients who wish to avoid blood transfusions. Sengsayadeth said the bloodless protocol is available for patients without religious objections who simply prefer to avoid blood products.
Dholaria cautioned that bloodless protocol applies specifically to autologous transplants, where patients receive their own stem cells. The team does not plan to offer bloodless allogeneic transplants, which use donor stem cells for conditions like leukemia, due to higher risks. In addition, most Jehovah’s Witnesses decline allogeneic transplants because they do not accept stem cells from another person, Dholaria said.
Beyond multiple myeloma, the Tennessee Valley Healthcare System offers bloodless autologous transplants for various blood cancers, including non-Hodgkin lymphomas such as large B-cell lymphoma, follicular lymphoma, and mantle cell lymphoma, as well as lymphomas affecting the brain, Dholaria said.
Clinicians “should start thinking about this early on, as soon as the cancer diagnosis is made, to make the referral and get the patient on our radar,” Dholaria said.
Sengsayadeth said physicians within the VA typically know how to refer appropriate patients to her team. “They just send us an email or give us a call or a message to say ‘I have this patient. Do you think they’re someone I should send to you?’ We usually answer right back, and then we can proceed with the full evaluation if we think that’s a reasonable thing to do.”
‘Treated Like Family’
The patient, a Marine Corps veteran named Keith Cody, spoke about the procedure in a video interview. Cody said he was reluctant at first to undergo the procedure because he didn’t understand what it would accomplish.
“As I was doing the massive chemo every week, and then suffering with the side effects, I decided to ask again about this procedure and how it improves my quality of life,” he said.
At the time of the taping of the video, Cody was getting ready to go home to California. “They’ve told me that I’ll still need more time to get my energy back, but I do feel much better already,” he said.
He also praised the staff. “Everybody that we came across, I enjoyed the interactions. It’s actually sad to leave people behind that you really felt treated you like family.”
Dholaria discloses relationships with Janssen, Angiocrine, Pfizer, Poseida, MEI, Orcabio, Wugen, Allovir, Adicet, BMS, Molecular Templates, Atara, MJH, Arvinas, Janssen, ADC, Gilead, GSK, Caribou, F. Hoffmann-La Roche AG, Autolus, and Pierre Fabre.
Sengsayadeth has no disclosures.
Unique Presentation of Postpartum Hypereosinophilic Syndrome With Atypical Features and Therapeutic Challenges
Unique Presentation of Postpartum Hypereosinophilic Syndrome With Atypical Features and Therapeutic Challenges
Hypereosinophilic syndrome (HES) is defined by marked, persistent absolute eosinophil count (AEC) > 1500 cells/μL on ≥ 2 peripheral smears separated by ≥ 1 month with evidence of accompanied end-organ damage, in the absence of other causes of eosinophilia such as malignancy, atopy, or parasitic infections.1-5 Hypereosinophilic infiltration can impact almost every organ system; however, the most profound complications in patients with HES are related to leukemias and cardiac manifestations of the disease.3,4 Although rare, the associated morbidity and mortality of HES are considerable, making prompt recognition and treatment essential. Management involves targeted therapy based on pathologic classification of HES and on decreasing associated inflammation, fibrosis, and end-organ damage.3,5-7
The patient in this case report met the diagnostic criteria for HES. However, this patient had several clinical and laboratory features that made it difficult to characterize a specific HES variant. Moreover, she had additional immunomodulating factors in the setting of pregnancy. This is the first documented case of HES of undetermined etiology diagnosed postpartum and managed in the setting of a new pregnancy.2,8
CASE PRESENTATION
A 32-year-old female active-duty military service member with allergic rhinitis and a history of childhood eczema was referred to allergy/immunology for evaluation of a new, progressive pruritic rash. Symptoms started 3 months after the birth of her first child, with a new diffuse erythematous skin rash sparing her palms, soles, and mucosal surfaces. Given her history of atopy, the rash was initially treated as severe atopic dermatitis with appropriate topical medications. The rash gradually worsened, with the development of intermittent facial swelling, night sweats, dyspnea, recurrent epigastric abdominal pain, and nausea with vomiting, resulting in decreased oral intake and weight loss.
The patient was hospitalized and received an expedited multidisciplinary evaluation by dermatology, hematology/oncology, and gastroenterology. Her AEC of 4787 cells/μL peaked on admission and was markedly elevated from the 1070 cells/μL reported in the third trimester of her pregnancy. She was found to have mature eosinophilia on skin biopsy (Figure 1), endoscopic duodenal biopsy (Figure 2), peripheral blood smear (Figure 3), and bone marrow biopsy (Figure 4).
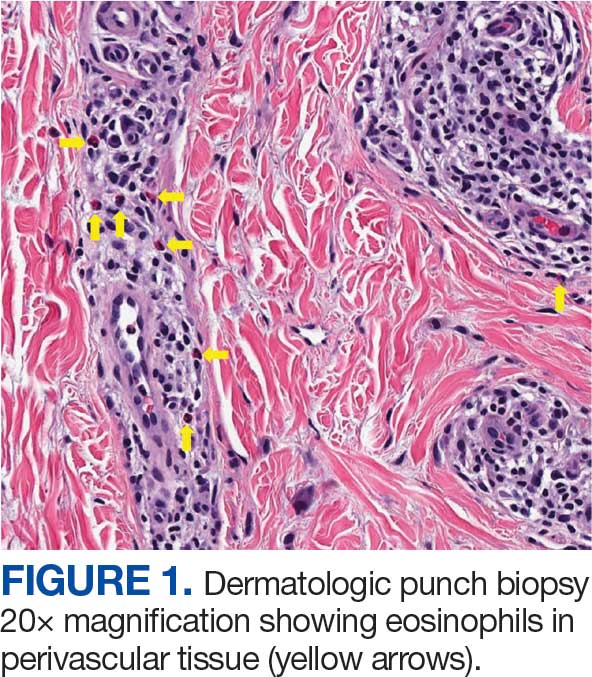
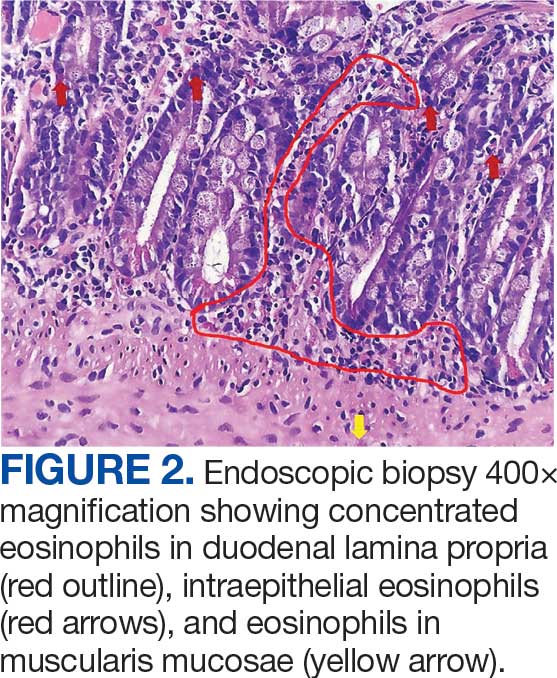
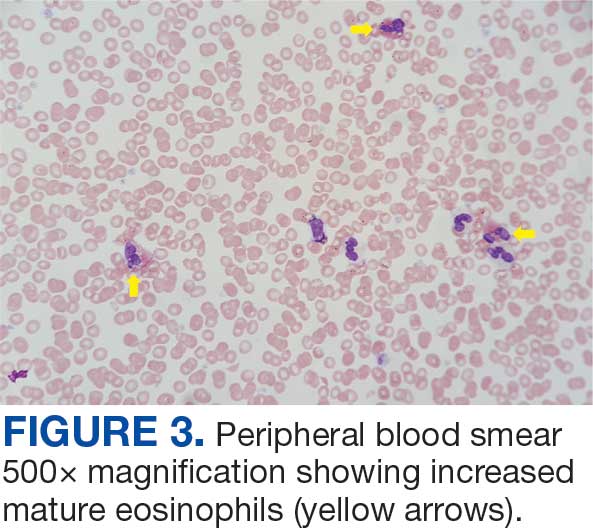
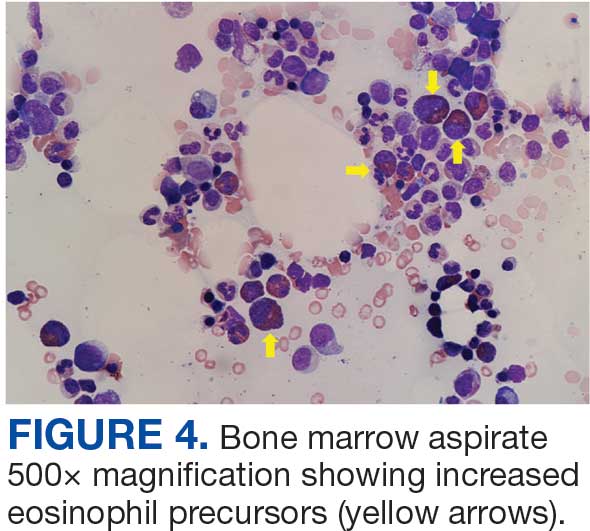
Radiographic imaging of the chest, abdomen, and pelvis revealed hepatomegaly without detectable neoplasm. There was no clinical evidence of cardiac involvement, and evaluation with electrocardiography and echocardiography did not indicate myocarditis. Extensive laboratory testing revealed no genetic mutations indicative of familial, myeloproliferative, or lymphocytic variants of HES.
The patient received topical emollients, omeprazole 40 mg daily, and ondansetron 8 mg 3 times daily as needed for symptom management, and was started on oral prednisone 40 mg daily with improvement in dyspnea, night sweats, and gastrointestinal complaints. During the patient's 6-day hospitalization and treatment, her AECs gradually decreased to 2110 cells/μL, and decreased to 1600 cells/μL over the course of a month, remaining in the hypereosinophilic range. The patient was discovered to be pregnant while symptoms were improving, resulting in stepwise discontinuation of oral steroids, but she reported continued improvement in symptoms.
DISCUSSION
Peripheral eosinophilia has a broad differential diagnoses, including HES, parasitic infections, atopic hypersensitivity diseases, eosinophilic lung diseases, eosinophilic gastrointestinal diseases, vasculitides such as eosinophilic granulomatosis with polyangiitis, genetic syndromes predisposing to eosinophilia, episodic angioedema with eosinophilia, and chronic metabolic disease with adrenal insufficiency.1-5 HES, although rare, is a disease process with potentially devastating associated morbidity and mortality if not promptly recognized and treated. HES is further delineated by hypereosinophilia with associated eosinophil-mediated organ damage or dysfunction.3-5
Clinical manifestations of HES can differ greatly depending on the HES variant and degree of organ involvement at the time of diagnosis and throughout the disease course. Patients with HES, as well as those with asymptomatic eosinophilia or hypereosinophilia, should be closely monitored for disease progression. In addition to trending peripheral AECs, clinicians should screen for symptoms of organ involvement and perform targeted evaluation of the suspected organs to promptly identify early signs of organ involvement and initiate treatment.1-4 Recommendations regarding screening intervals vary widely from monthly to annually, depending on a patient’s specific clinical picture.
HES has been subdivided into clinically relevant variants, including myeloproliferative (M-HES), T lymphocytic (L-HES), organ-restricted (or overlap) HES, familial HES, idiopathic HES, and specific syndromes with associated hypereosinophilia.3-5,9 Patients with M-HES have elevated circulating leukocyte precursors and clinical manifestations, including but not limited to hepatosplenomegaly, anemia, and thrombocytopenia. The most commonly associated genetic mutations include the FIP1L1-PDGFR-α fusion, BCR-ABL1, PDGFRA/B, JAK2, KIT, and FGFR1.3-6 L-HES usually has predominant skin and soft tissue involvement secondary to immunoglobulin E-mediated actions with clonal expansion of T cells (most commonly CD3-4+ or CD3+CD4-CD8-).3,5,6 Familial HES, a rare variant, follows an autosomal dominant inheritance pattern and is usually present at birth. It involves chromosome 5, which contains genes coding for cytokines that drive eosinophilic proliferation, including interleukin (IL)-3, IL-5, and granulocyte-macrophage colony-stimulating factor.5,9 Hypereosinophilia in the setting of end-organ damage restricted to a single organ is considered organ-restricted HES. There can be significant hepatic and gastrointestinal dysfunction, with or without malabsorption.
HES can also manifest with hematologic malignancy, restrictive obliterative cardiomyopathies, renal injury manifested by hematuria and electrolyte derangements, and neurologic complications including hemiparesis, dysarthria, and even coma.6 Endothelial damage due to eosinophil-driven inflammation can result in thrombus formation and increased risk of thromboembolic events in various organs.3 Idiopathic HES, otherwise known as HES of unknown etiology or significance, is a diagnosis of exclusion and constitutes a cohort of patients who do not fit into the other delineated categories.3-5 These patients often have multisystem involvement, making classification and treatment a challenge.5
The patient described in this case met the diagnostic criteria for HES, but her complicated clinical and laboratory features were challenging to characterize into a specific variant of HES. Organ-restricted HES was ruled out due to skin, marrow, and duodenal infiltration. She also had the potential for lung involvement based on her clinical symptoms, however no biopsy was obtained. Laboratory testing revealed no deletions or mutations indicative of familial, myeloproliferative, or lymphocytic variants. Her multisystem involvement without an underlying associated syndrome suggests idiopathic HES or HES of undetermined significance.1-5
Most patients with HES are diagnosed between the ages of 20 and 50 years.10 While HES has its peak incidence in the fourth decade of life, acute onset of new symptoms 3 months postpartum makes this an unusual presentation. In this unique case, it is important to highlight the role of the physiologic changes of pregnancy in inflammatory mediation. The physiologic changes that occur in pregnancy to ensure fetal tolerance can have profound implications for leukocyte count, AEC, and subsequent inflammatory responses. The phenomenon of inflammatory amelioration during pregnancy is well-documented, but there has only been 1 known published case report discussing decreasing HES symptoms during pregnancy with prepregnancy and postpartum hypereosinophilia.8 It is suggested that this amelioration is secondary to cortisol and progesterone shifts that occur in pregnancy. Physiologic increases in adrenocorticotropic hormone in pregnancy leads to subsequent secretion of endogenous steroids by the adrenal cortex. In turn, pregnancy can lead to leukocytosis and eosinopenia.8 Overall, pregnancy can have beneficial immunomodulating properties in the spectrum of hypereosinophilic syndromes. Even so, this patient with HES diagnosed postpartum remains at risk for the sequelae of hypereosinophilia, regardless of potential for AEC reduction during pregnancy. Therefore, treatment considerations need to be made with the safety of the maternal-fetal dyad as a priority.
Treatment
The treatment of symptomatic HES without acute life-threatening features or associated malignancy is generally determined by clinical variant.2-4 There is insufficient data to support initiation of treatment solely based on persistently elevated AEC. Patients with peripheral eosinophilia and hypereosinophilia should be monitored periodically with appropriate subspecialist evaluation for occult end-organ involvement, and targeted therapies should be deferred until an HES diagnosis.1-4 First-line therapy in most HES variants is systemic glucocorticoids.2,3,7 Since the disease course for this patient did not precisely match an HES variant, it was challenging to ascertain the optimal personalized treatment regimen. The approach to therapy was further complicated by newly identified pregnancy necessitating cessation of systemic glucocorticoids. In addition to glucocorticoids, hydroxyurea and interferon-α are among treatments historically used for HES, with tyrosine kinase inhibitors and monoclonal antibodies targeting IL-5 becoming more common.1-4 Although this patient may ultimately benefit from an IL-5 targeting biologic medication such as mepolizumab, safety in pregnancy is not well-studied and may require close clinical monitoring with treatment deferred until after delivery if possible.3,7,8,11
Military service members with frequent geographic relocation have additional barriers to timely diagnosis with often-limited access to subspecialty care depending on the duty station. While the patient was able to receive care at a large military medical center with many subspecialists, prompt recognition and timely referral to specialists would be even more critical at a smaller treatment facility. Depending on the severity and variant of HES, patients may warrant evaluation and treatment by hematology/oncology, cardiology, pulmonology, and immunology. Although HES can present in young children and older adults, this condition is most often diagnosed during the third and fourth decades of life, putting clinicians on the front line of hypereosinophilia identification and evaluation.10 Military physicians have the additional duty to not only think ahead in their diverse clinical settings to ensure proper care for patients, but also maintain a broad differential inclusive of more rare disease processes such as HES.
CONCLUSIONS
This case emphasizes how uncontrolled or untreated HES can lead to significant end-organ damage involving multiple systems and high morbidity. Prompt recognition of hypereosinophilia with potential HES can help expedite coordination of multidisciplinary care across multiple specialties to minimize delays in diagnosis and treatment. Doing so may minimize associated morbidity and mortality, especially in individuals located at more remote duty stations or deployed to austere environments.
- Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol. 2012;5:275-290. doi: 10.1586/ehm.12.14
- Klion A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology Am Soc Hematol Educ Program. 2018;2018:326-331. doi:10.1182/asheducation-2018.1.326
- Shomali W, Gotlib J. World health organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:129-148. doi:10.1002/ajh.26352
- Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev. 2021;49:100809. doi:10.1016/j.blre.2021.100809
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130:607-612.e9. doi:10.1016/j.jaci.2012.02.019
- Roufosse FE, Goldman M, Cogan E. Hypereosinophilic syndromes. Orphanet J Rare Dis. 2007;2:37. doi:10.1186/1750-1172-2-37
- Pitlick MM, Li JT, Pongdee T. Current and emerging biologic therapies targeting eosinophilic disorders. World Allergy Organ J. 2022;15:100676. doi:10.1016/j.waojou.2022.10067
- Ault P, Cortes J, Lynn A, Keating M, Verstovsek S. Pregnancy in a patient with hypereosinophilic syndrome. Leuk Res. 2009;33:186-187. doi:10.1016/j.leukres.2008.05.013
- Rioux JD, Stone VA, Daly MJ, et al. Familial eosinophilia maps to the cytokine gene cluster on human chromosomal region 5q31-q33. Am J Hum Genet. 1998;63:1086-1094. doi:10.1086/302053
- Williams KW, Ware J, Abiodun A, et al. Hypereosinophilia in children and adults: a retrospective comparison. J Allergy Clin Immunol Pract. 2016;4:941-947.e1. doi:10.1016/j.jaip.2016.03.020
- Pane F, Lefevre G, Kwon N, et al. Characterization of disease flares and impact of mepolizumab in patients with hypereosinophilic syndrome. Front Immunol. 2022;13:935996. doi:10.3389/fimmu.2022.935996
Hypereosinophilic syndrome (HES) is defined by marked, persistent absolute eosinophil count (AEC) > 1500 cells/μL on ≥ 2 peripheral smears separated by ≥ 1 month with evidence of accompanied end-organ damage, in the absence of other causes of eosinophilia such as malignancy, atopy, or parasitic infections.1-5 Hypereosinophilic infiltration can impact almost every organ system; however, the most profound complications in patients with HES are related to leukemias and cardiac manifestations of the disease.3,4 Although rare, the associated morbidity and mortality of HES are considerable, making prompt recognition and treatment essential. Management involves targeted therapy based on pathologic classification of HES and on decreasing associated inflammation, fibrosis, and end-organ damage.3,5-7
The patient in this case report met the diagnostic criteria for HES. However, this patient had several clinical and laboratory features that made it difficult to characterize a specific HES variant. Moreover, she had additional immunomodulating factors in the setting of pregnancy. This is the first documented case of HES of undetermined etiology diagnosed postpartum and managed in the setting of a new pregnancy.2,8
CASE PRESENTATION
A 32-year-old female active-duty military service member with allergic rhinitis and a history of childhood eczema was referred to allergy/immunology for evaluation of a new, progressive pruritic rash. Symptoms started 3 months after the birth of her first child, with a new diffuse erythematous skin rash sparing her palms, soles, and mucosal surfaces. Given her history of atopy, the rash was initially treated as severe atopic dermatitis with appropriate topical medications. The rash gradually worsened, with the development of intermittent facial swelling, night sweats, dyspnea, recurrent epigastric abdominal pain, and nausea with vomiting, resulting in decreased oral intake and weight loss.
The patient was hospitalized and received an expedited multidisciplinary evaluation by dermatology, hematology/oncology, and gastroenterology. Her AEC of 4787 cells/μL peaked on admission and was markedly elevated from the 1070 cells/μL reported in the third trimester of her pregnancy. She was found to have mature eosinophilia on skin biopsy (Figure 1), endoscopic duodenal biopsy (Figure 2), peripheral blood smear (Figure 3), and bone marrow biopsy (Figure 4).
Radiographic imaging of the chest, abdomen, and pelvis revealed hepatomegaly without detectable neoplasm. There was no clinical evidence of cardiac involvement, and evaluation with electrocardiography and echocardiography did not indicate myocarditis. Extensive laboratory testing revealed no genetic mutations indicative of familial, myeloproliferative, or lymphocytic variants of HES.
The patient received topical emollients, omeprazole 40 mg daily, and ondansetron 8 mg 3 times daily as needed for symptom management, and was started on oral prednisone 40 mg daily with improvement in dyspnea, night sweats, and gastrointestinal complaints. During the patient's 6-day hospitalization and treatment, her AECs gradually decreased to 2110 cells/μL, and decreased to 1600 cells/μL over the course of a month, remaining in the hypereosinophilic range. The patient was discovered to be pregnant while symptoms were improving, resulting in stepwise discontinuation of oral steroids, but she reported continued improvement in symptoms.
DISCUSSION
Peripheral eosinophilia has a broad differential diagnoses, including HES, parasitic infections, atopic hypersensitivity diseases, eosinophilic lung diseases, eosinophilic gastrointestinal diseases, vasculitides such as eosinophilic granulomatosis with polyangiitis, genetic syndromes predisposing to eosinophilia, episodic angioedema with eosinophilia, and chronic metabolic disease with adrenal insufficiency.1-5 HES, although rare, is a disease process with potentially devastating associated morbidity and mortality if not promptly recognized and treated. HES is further delineated by hypereosinophilia with associated eosinophil-mediated organ damage or dysfunction.3-5
Clinical manifestations of HES can differ greatly depending on the HES variant and degree of organ involvement at the time of diagnosis and throughout the disease course. Patients with HES, as well as those with asymptomatic eosinophilia or hypereosinophilia, should be closely monitored for disease progression. In addition to trending peripheral AECs, clinicians should screen for symptoms of organ involvement and perform targeted evaluation of the suspected organs to promptly identify early signs of organ involvement and initiate treatment.1-4 Recommendations regarding screening intervals vary widely from monthly to annually, depending on a patient’s specific clinical picture.
HES has been subdivided into clinically relevant variants, including myeloproliferative (M-HES), T lymphocytic (L-HES), organ-restricted (or overlap) HES, familial HES, idiopathic HES, and specific syndromes with associated hypereosinophilia.3-5,9 Patients with M-HES have elevated circulating leukocyte precursors and clinical manifestations, including but not limited to hepatosplenomegaly, anemia, and thrombocytopenia. The most commonly associated genetic mutations include the FIP1L1-PDGFR-α fusion, BCR-ABL1, PDGFRA/B, JAK2, KIT, and FGFR1.3-6 L-HES usually has predominant skin and soft tissue involvement secondary to immunoglobulin E-mediated actions with clonal expansion of T cells (most commonly CD3-4+ or CD3+CD4-CD8-).3,5,6 Familial HES, a rare variant, follows an autosomal dominant inheritance pattern and is usually present at birth. It involves chromosome 5, which contains genes coding for cytokines that drive eosinophilic proliferation, including interleukin (IL)-3, IL-5, and granulocyte-macrophage colony-stimulating factor.5,9 Hypereosinophilia in the setting of end-organ damage restricted to a single organ is considered organ-restricted HES. There can be significant hepatic and gastrointestinal dysfunction, with or without malabsorption.
HES can also manifest with hematologic malignancy, restrictive obliterative cardiomyopathies, renal injury manifested by hematuria and electrolyte derangements, and neurologic complications including hemiparesis, dysarthria, and even coma.6 Endothelial damage due to eosinophil-driven inflammation can result in thrombus formation and increased risk of thromboembolic events in various organs.3 Idiopathic HES, otherwise known as HES of unknown etiology or significance, is a diagnosis of exclusion and constitutes a cohort of patients who do not fit into the other delineated categories.3-5 These patients often have multisystem involvement, making classification and treatment a challenge.5
The patient described in this case met the diagnostic criteria for HES, but her complicated clinical and laboratory features were challenging to characterize into a specific variant of HES. Organ-restricted HES was ruled out due to skin, marrow, and duodenal infiltration. She also had the potential for lung involvement based on her clinical symptoms, however no biopsy was obtained. Laboratory testing revealed no deletions or mutations indicative of familial, myeloproliferative, or lymphocytic variants. Her multisystem involvement without an underlying associated syndrome suggests idiopathic HES or HES of undetermined significance.1-5
Most patients with HES are diagnosed between the ages of 20 and 50 years.10 While HES has its peak incidence in the fourth decade of life, acute onset of new symptoms 3 months postpartum makes this an unusual presentation. In this unique case, it is important to highlight the role of the physiologic changes of pregnancy in inflammatory mediation. The physiologic changes that occur in pregnancy to ensure fetal tolerance can have profound implications for leukocyte count, AEC, and subsequent inflammatory responses. The phenomenon of inflammatory amelioration during pregnancy is well-documented, but there has only been 1 known published case report discussing decreasing HES symptoms during pregnancy with prepregnancy and postpartum hypereosinophilia.8 It is suggested that this amelioration is secondary to cortisol and progesterone shifts that occur in pregnancy. Physiologic increases in adrenocorticotropic hormone in pregnancy leads to subsequent secretion of endogenous steroids by the adrenal cortex. In turn, pregnancy can lead to leukocytosis and eosinopenia.8 Overall, pregnancy can have beneficial immunomodulating properties in the spectrum of hypereosinophilic syndromes. Even so, this patient with HES diagnosed postpartum remains at risk for the sequelae of hypereosinophilia, regardless of potential for AEC reduction during pregnancy. Therefore, treatment considerations need to be made with the safety of the maternal-fetal dyad as a priority.
Treatment
The treatment of symptomatic HES without acute life-threatening features or associated malignancy is generally determined by clinical variant.2-4 There is insufficient data to support initiation of treatment solely based on persistently elevated AEC. Patients with peripheral eosinophilia and hypereosinophilia should be monitored periodically with appropriate subspecialist evaluation for occult end-organ involvement, and targeted therapies should be deferred until an HES diagnosis.1-4 First-line therapy in most HES variants is systemic glucocorticoids.2,3,7 Since the disease course for this patient did not precisely match an HES variant, it was challenging to ascertain the optimal personalized treatment regimen. The approach to therapy was further complicated by newly identified pregnancy necessitating cessation of systemic glucocorticoids. In addition to glucocorticoids, hydroxyurea and interferon-α are among treatments historically used for HES, with tyrosine kinase inhibitors and monoclonal antibodies targeting IL-5 becoming more common.1-4 Although this patient may ultimately benefit from an IL-5 targeting biologic medication such as mepolizumab, safety in pregnancy is not well-studied and may require close clinical monitoring with treatment deferred until after delivery if possible.3,7,8,11
Military service members with frequent geographic relocation have additional barriers to timely diagnosis with often-limited access to subspecialty care depending on the duty station. While the patient was able to receive care at a large military medical center with many subspecialists, prompt recognition and timely referral to specialists would be even more critical at a smaller treatment facility. Depending on the severity and variant of HES, patients may warrant evaluation and treatment by hematology/oncology, cardiology, pulmonology, and immunology. Although HES can present in young children and older adults, this condition is most often diagnosed during the third and fourth decades of life, putting clinicians on the front line of hypereosinophilia identification and evaluation.10 Military physicians have the additional duty to not only think ahead in their diverse clinical settings to ensure proper care for patients, but also maintain a broad differential inclusive of more rare disease processes such as HES.
CONCLUSIONS
This case emphasizes how uncontrolled or untreated HES can lead to significant end-organ damage involving multiple systems and high morbidity. Prompt recognition of hypereosinophilia with potential HES can help expedite coordination of multidisciplinary care across multiple specialties to minimize delays in diagnosis and treatment. Doing so may minimize associated morbidity and mortality, especially in individuals located at more remote duty stations or deployed to austere environments.
Hypereosinophilic syndrome (HES) is defined by marked, persistent absolute eosinophil count (AEC) > 1500 cells/μL on ≥ 2 peripheral smears separated by ≥ 1 month with evidence of accompanied end-organ damage, in the absence of other causes of eosinophilia such as malignancy, atopy, or parasitic infections.1-5 Hypereosinophilic infiltration can impact almost every organ system; however, the most profound complications in patients with HES are related to leukemias and cardiac manifestations of the disease.3,4 Although rare, the associated morbidity and mortality of HES are considerable, making prompt recognition and treatment essential. Management involves targeted therapy based on pathologic classification of HES and on decreasing associated inflammation, fibrosis, and end-organ damage.3,5-7
The patient in this case report met the diagnostic criteria for HES. However, this patient had several clinical and laboratory features that made it difficult to characterize a specific HES variant. Moreover, she had additional immunomodulating factors in the setting of pregnancy. This is the first documented case of HES of undetermined etiology diagnosed postpartum and managed in the setting of a new pregnancy.2,8
CASE PRESENTATION
A 32-year-old female active-duty military service member with allergic rhinitis and a history of childhood eczema was referred to allergy/immunology for evaluation of a new, progressive pruritic rash. Symptoms started 3 months after the birth of her first child, with a new diffuse erythematous skin rash sparing her palms, soles, and mucosal surfaces. Given her history of atopy, the rash was initially treated as severe atopic dermatitis with appropriate topical medications. The rash gradually worsened, with the development of intermittent facial swelling, night sweats, dyspnea, recurrent epigastric abdominal pain, and nausea with vomiting, resulting in decreased oral intake and weight loss.
The patient was hospitalized and received an expedited multidisciplinary evaluation by dermatology, hematology/oncology, and gastroenterology. Her AEC of 4787 cells/μL peaked on admission and was markedly elevated from the 1070 cells/μL reported in the third trimester of her pregnancy. She was found to have mature eosinophilia on skin biopsy (Figure 1), endoscopic duodenal biopsy (Figure 2), peripheral blood smear (Figure 3), and bone marrow biopsy (Figure 4).
Radiographic imaging of the chest, abdomen, and pelvis revealed hepatomegaly without detectable neoplasm. There was no clinical evidence of cardiac involvement, and evaluation with electrocardiography and echocardiography did not indicate myocarditis. Extensive laboratory testing revealed no genetic mutations indicative of familial, myeloproliferative, or lymphocytic variants of HES.
The patient received topical emollients, omeprazole 40 mg daily, and ondansetron 8 mg 3 times daily as needed for symptom management, and was started on oral prednisone 40 mg daily with improvement in dyspnea, night sweats, and gastrointestinal complaints. During the patient's 6-day hospitalization and treatment, her AECs gradually decreased to 2110 cells/μL, and decreased to 1600 cells/μL over the course of a month, remaining in the hypereosinophilic range. The patient was discovered to be pregnant while symptoms were improving, resulting in stepwise discontinuation of oral steroids, but she reported continued improvement in symptoms.
DISCUSSION
Peripheral eosinophilia has a broad differential diagnoses, including HES, parasitic infections, atopic hypersensitivity diseases, eosinophilic lung diseases, eosinophilic gastrointestinal diseases, vasculitides such as eosinophilic granulomatosis with polyangiitis, genetic syndromes predisposing to eosinophilia, episodic angioedema with eosinophilia, and chronic metabolic disease with adrenal insufficiency.1-5 HES, although rare, is a disease process with potentially devastating associated morbidity and mortality if not promptly recognized and treated. HES is further delineated by hypereosinophilia with associated eosinophil-mediated organ damage or dysfunction.3-5
Clinical manifestations of HES can differ greatly depending on the HES variant and degree of organ involvement at the time of diagnosis and throughout the disease course. Patients with HES, as well as those with asymptomatic eosinophilia or hypereosinophilia, should be closely monitored for disease progression. In addition to trending peripheral AECs, clinicians should screen for symptoms of organ involvement and perform targeted evaluation of the suspected organs to promptly identify early signs of organ involvement and initiate treatment.1-4 Recommendations regarding screening intervals vary widely from monthly to annually, depending on a patient’s specific clinical picture.
HES has been subdivided into clinically relevant variants, including myeloproliferative (M-HES), T lymphocytic (L-HES), organ-restricted (or overlap) HES, familial HES, idiopathic HES, and specific syndromes with associated hypereosinophilia.3-5,9 Patients with M-HES have elevated circulating leukocyte precursors and clinical manifestations, including but not limited to hepatosplenomegaly, anemia, and thrombocytopenia. The most commonly associated genetic mutations include the FIP1L1-PDGFR-α fusion, BCR-ABL1, PDGFRA/B, JAK2, KIT, and FGFR1.3-6 L-HES usually has predominant skin and soft tissue involvement secondary to immunoglobulin E-mediated actions with clonal expansion of T cells (most commonly CD3-4+ or CD3+CD4-CD8-).3,5,6 Familial HES, a rare variant, follows an autosomal dominant inheritance pattern and is usually present at birth. It involves chromosome 5, which contains genes coding for cytokines that drive eosinophilic proliferation, including interleukin (IL)-3, IL-5, and granulocyte-macrophage colony-stimulating factor.5,9 Hypereosinophilia in the setting of end-organ damage restricted to a single organ is considered organ-restricted HES. There can be significant hepatic and gastrointestinal dysfunction, with or without malabsorption.
HES can also manifest with hematologic malignancy, restrictive obliterative cardiomyopathies, renal injury manifested by hematuria and electrolyte derangements, and neurologic complications including hemiparesis, dysarthria, and even coma.6 Endothelial damage due to eosinophil-driven inflammation can result in thrombus formation and increased risk of thromboembolic events in various organs.3 Idiopathic HES, otherwise known as HES of unknown etiology or significance, is a diagnosis of exclusion and constitutes a cohort of patients who do not fit into the other delineated categories.3-5 These patients often have multisystem involvement, making classification and treatment a challenge.5
The patient described in this case met the diagnostic criteria for HES, but her complicated clinical and laboratory features were challenging to characterize into a specific variant of HES. Organ-restricted HES was ruled out due to skin, marrow, and duodenal infiltration. She also had the potential for lung involvement based on her clinical symptoms, however no biopsy was obtained. Laboratory testing revealed no deletions or mutations indicative of familial, myeloproliferative, or lymphocytic variants. Her multisystem involvement without an underlying associated syndrome suggests idiopathic HES or HES of undetermined significance.1-5
Most patients with HES are diagnosed between the ages of 20 and 50 years.10 While HES has its peak incidence in the fourth decade of life, acute onset of new symptoms 3 months postpartum makes this an unusual presentation. In this unique case, it is important to highlight the role of the physiologic changes of pregnancy in inflammatory mediation. The physiologic changes that occur in pregnancy to ensure fetal tolerance can have profound implications for leukocyte count, AEC, and subsequent inflammatory responses. The phenomenon of inflammatory amelioration during pregnancy is well-documented, but there has only been 1 known published case report discussing decreasing HES symptoms during pregnancy with prepregnancy and postpartum hypereosinophilia.8 It is suggested that this amelioration is secondary to cortisol and progesterone shifts that occur in pregnancy. Physiologic increases in adrenocorticotropic hormone in pregnancy leads to subsequent secretion of endogenous steroids by the adrenal cortex. In turn, pregnancy can lead to leukocytosis and eosinopenia.8 Overall, pregnancy can have beneficial immunomodulating properties in the spectrum of hypereosinophilic syndromes. Even so, this patient with HES diagnosed postpartum remains at risk for the sequelae of hypereosinophilia, regardless of potential for AEC reduction during pregnancy. Therefore, treatment considerations need to be made with the safety of the maternal-fetal dyad as a priority.
Treatment
The treatment of symptomatic HES without acute life-threatening features or associated malignancy is generally determined by clinical variant.2-4 There is insufficient data to support initiation of treatment solely based on persistently elevated AEC. Patients with peripheral eosinophilia and hypereosinophilia should be monitored periodically with appropriate subspecialist evaluation for occult end-organ involvement, and targeted therapies should be deferred until an HES diagnosis.1-4 First-line therapy in most HES variants is systemic glucocorticoids.2,3,7 Since the disease course for this patient did not precisely match an HES variant, it was challenging to ascertain the optimal personalized treatment regimen. The approach to therapy was further complicated by newly identified pregnancy necessitating cessation of systemic glucocorticoids. In addition to glucocorticoids, hydroxyurea and interferon-α are among treatments historically used for HES, with tyrosine kinase inhibitors and monoclonal antibodies targeting IL-5 becoming more common.1-4 Although this patient may ultimately benefit from an IL-5 targeting biologic medication such as mepolizumab, safety in pregnancy is not well-studied and may require close clinical monitoring with treatment deferred until after delivery if possible.3,7,8,11
Military service members with frequent geographic relocation have additional barriers to timely diagnosis with often-limited access to subspecialty care depending on the duty station. While the patient was able to receive care at a large military medical center with many subspecialists, prompt recognition and timely referral to specialists would be even more critical at a smaller treatment facility. Depending on the severity and variant of HES, patients may warrant evaluation and treatment by hematology/oncology, cardiology, pulmonology, and immunology. Although HES can present in young children and older adults, this condition is most often diagnosed during the third and fourth decades of life, putting clinicians on the front line of hypereosinophilia identification and evaluation.10 Military physicians have the additional duty to not only think ahead in their diverse clinical settings to ensure proper care for patients, but also maintain a broad differential inclusive of more rare disease processes such as HES.
CONCLUSIONS
This case emphasizes how uncontrolled or untreated HES can lead to significant end-organ damage involving multiple systems and high morbidity. Prompt recognition of hypereosinophilia with potential HES can help expedite coordination of multidisciplinary care across multiple specialties to minimize delays in diagnosis and treatment. Doing so may minimize associated morbidity and mortality, especially in individuals located at more remote duty stations or deployed to austere environments.
- Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol. 2012;5:275-290. doi: 10.1586/ehm.12.14
- Klion A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology Am Soc Hematol Educ Program. 2018;2018:326-331. doi:10.1182/asheducation-2018.1.326
- Shomali W, Gotlib J. World health organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:129-148. doi:10.1002/ajh.26352
- Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev. 2021;49:100809. doi:10.1016/j.blre.2021.100809
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130:607-612.e9. doi:10.1016/j.jaci.2012.02.019
- Roufosse FE, Goldman M, Cogan E. Hypereosinophilic syndromes. Orphanet J Rare Dis. 2007;2:37. doi:10.1186/1750-1172-2-37
- Pitlick MM, Li JT, Pongdee T. Current and emerging biologic therapies targeting eosinophilic disorders. World Allergy Organ J. 2022;15:100676. doi:10.1016/j.waojou.2022.10067
- Ault P, Cortes J, Lynn A, Keating M, Verstovsek S. Pregnancy in a patient with hypereosinophilic syndrome. Leuk Res. 2009;33:186-187. doi:10.1016/j.leukres.2008.05.013
- Rioux JD, Stone VA, Daly MJ, et al. Familial eosinophilia maps to the cytokine gene cluster on human chromosomal region 5q31-q33. Am J Hum Genet. 1998;63:1086-1094. doi:10.1086/302053
- Williams KW, Ware J, Abiodun A, et al. Hypereosinophilia in children and adults: a retrospective comparison. J Allergy Clin Immunol Pract. 2016;4:941-947.e1. doi:10.1016/j.jaip.2016.03.020
- Pane F, Lefevre G, Kwon N, et al. Characterization of disease flares and impact of mepolizumab in patients with hypereosinophilic syndrome. Front Immunol. 2022;13:935996. doi:10.3389/fimmu.2022.935996
- Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol. 2012;5:275-290. doi: 10.1586/ehm.12.14
- Klion A. Hypereosinophilic syndrome: approach to treatment in the era of precision medicine. Hematology Am Soc Hematol Educ Program. 2018;2018:326-331. doi:10.1182/asheducation-2018.1.326
- Shomali W, Gotlib J. World health organization-defined eosinophilic disorders: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:129-148. doi:10.1002/ajh.26352
- Helbig G, Klion AD. Hypereosinophilic syndromes - an enigmatic group of disorders with an intriguing clinical spectrum and challenging treatment. Blood Rev. 2021;49:100809. doi:10.1016/j.blre.2021.100809
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130:607-612.e9. doi:10.1016/j.jaci.2012.02.019
- Roufosse FE, Goldman M, Cogan E. Hypereosinophilic syndromes. Orphanet J Rare Dis. 2007;2:37. doi:10.1186/1750-1172-2-37
- Pitlick MM, Li JT, Pongdee T. Current and emerging biologic therapies targeting eosinophilic disorders. World Allergy Organ J. 2022;15:100676. doi:10.1016/j.waojou.2022.10067
- Ault P, Cortes J, Lynn A, Keating M, Verstovsek S. Pregnancy in a patient with hypereosinophilic syndrome. Leuk Res. 2009;33:186-187. doi:10.1016/j.leukres.2008.05.013
- Rioux JD, Stone VA, Daly MJ, et al. Familial eosinophilia maps to the cytokine gene cluster on human chromosomal region 5q31-q33. Am J Hum Genet. 1998;63:1086-1094. doi:10.1086/302053
- Williams KW, Ware J, Abiodun A, et al. Hypereosinophilia in children and adults: a retrospective comparison. J Allergy Clin Immunol Pract. 2016;4:941-947.e1. doi:10.1016/j.jaip.2016.03.020
- Pane F, Lefevre G, Kwon N, et al. Characterization of disease flares and impact of mepolizumab in patients with hypereosinophilic syndrome. Front Immunol. 2022;13:935996. doi:10.3389/fimmu.2022.935996
Unique Presentation of Postpartum Hypereosinophilic Syndrome With Atypical Features and Therapeutic Challenges
Unique Presentation of Postpartum Hypereosinophilic Syndrome With Atypical Features and Therapeutic Challenges
Hematology and Oncology Staffing Levels for Fiscal Years 19–24
Background
Department of Veterans Affairs (VA) faces a landscape of increasingly complex practice, especially in Hematology/Oncology (H/O), and a nationwide shortage of healthcare providers, while serving more Veterans than ever before. To understand current and future staffing needs, the VA National Oncology Program performed an assessment of H/O staffing, including attending physicians, residents/ fellows, licensed independent practitioners (LIPs) (nurse practitioners/physician assistants), and nurses for fiscal years (FY) 19–24.
Methods
Using VA Corporate Data Warehouse, we identified H/O visits in VA from 10/01/2018 through 09/30/2024 using stop codes. No-show (< 0.00001%) and National TeleOncology appointments (1%) were removed. We retrieved all notes associated with resulting visits and used area-ofspecialization and provider-type data to identify all attending physicians, trainees, LIPs, and nurses who authored or cosigned these notes. We identified H/O staff as 1. those associated with H/O clinic locations, 2. physicians who consistently cosigned H/O notes authored by fellows and LIPs associated with H/O locations, 3. fellows and LIPs authoring notes that were then cosigned by H/O physicians, and 4. nurses authoring notes associated with H/O visits.
Analysis
For each FY, we obtained total numbers of visits, unique patients, and care-providing staff by type. For validation, collaborating providers at several sites reviewed visit information, and a colleague also performed an independent, parallel data extraction. We adjusted FY totals to account for the growing patient population by dividing unique staff count by number of unique patients and multiplying by 200,000 (the approximate number of unique patients in FY19).
Results
From FY19 through FY24, VA Hematology/ Oncology saw a 14.6% rise in unique patients (from 232,084 to 265,926) and a 15.4% rise in visits (from 923,175 to 1,065,186). The absolute number of attendings rose by 4 (0.6%); of LIPs, by 138 (14.4%); and of nurses, by 142 (4.9%); trainees fell by 102 (4.3%). Adjusted to 200,000 patients, the number of attendings fell by 76 (12.3%); LIPs, by 1 (0.1%); trainees, by 335 (16.5%); and nurses, by 211 (8.4%).
Conclusions
Adjusted to number of Veterans, there are 10.4% fewer staff in Hematology/Oncology in FY24 compared to FY19.
Background
Department of Veterans Affairs (VA) faces a landscape of increasingly complex practice, especially in Hematology/Oncology (H/O), and a nationwide shortage of healthcare providers, while serving more Veterans than ever before. To understand current and future staffing needs, the VA National Oncology Program performed an assessment of H/O staffing, including attending physicians, residents/ fellows, licensed independent practitioners (LIPs) (nurse practitioners/physician assistants), and nurses for fiscal years (FY) 19–24.
Methods
Using VA Corporate Data Warehouse, we identified H/O visits in VA from 10/01/2018 through 09/30/2024 using stop codes. No-show (< 0.00001%) and National TeleOncology appointments (1%) were removed. We retrieved all notes associated with resulting visits and used area-ofspecialization and provider-type data to identify all attending physicians, trainees, LIPs, and nurses who authored or cosigned these notes. We identified H/O staff as 1. those associated with H/O clinic locations, 2. physicians who consistently cosigned H/O notes authored by fellows and LIPs associated with H/O locations, 3. fellows and LIPs authoring notes that were then cosigned by H/O physicians, and 4. nurses authoring notes associated with H/O visits.
Analysis
For each FY, we obtained total numbers of visits, unique patients, and care-providing staff by type. For validation, collaborating providers at several sites reviewed visit information, and a colleague also performed an independent, parallel data extraction. We adjusted FY totals to account for the growing patient population by dividing unique staff count by number of unique patients and multiplying by 200,000 (the approximate number of unique patients in FY19).
Results
From FY19 through FY24, VA Hematology/ Oncology saw a 14.6% rise in unique patients (from 232,084 to 265,926) and a 15.4% rise in visits (from 923,175 to 1,065,186). The absolute number of attendings rose by 4 (0.6%); of LIPs, by 138 (14.4%); and of nurses, by 142 (4.9%); trainees fell by 102 (4.3%). Adjusted to 200,000 patients, the number of attendings fell by 76 (12.3%); LIPs, by 1 (0.1%); trainees, by 335 (16.5%); and nurses, by 211 (8.4%).
Conclusions
Adjusted to number of Veterans, there are 10.4% fewer staff in Hematology/Oncology in FY24 compared to FY19.
Background
Department of Veterans Affairs (VA) faces a landscape of increasingly complex practice, especially in Hematology/Oncology (H/O), and a nationwide shortage of healthcare providers, while serving more Veterans than ever before. To understand current and future staffing needs, the VA National Oncology Program performed an assessment of H/O staffing, including attending physicians, residents/ fellows, licensed independent practitioners (LIPs) (nurse practitioners/physician assistants), and nurses for fiscal years (FY) 19–24.
Methods
Using VA Corporate Data Warehouse, we identified H/O visits in VA from 10/01/2018 through 09/30/2024 using stop codes. No-show (< 0.00001%) and National TeleOncology appointments (1%) were removed. We retrieved all notes associated with resulting visits and used area-ofspecialization and provider-type data to identify all attending physicians, trainees, LIPs, and nurses who authored or cosigned these notes. We identified H/O staff as 1. those associated with H/O clinic locations, 2. physicians who consistently cosigned H/O notes authored by fellows and LIPs associated with H/O locations, 3. fellows and LIPs authoring notes that were then cosigned by H/O physicians, and 4. nurses authoring notes associated with H/O visits.
Analysis
For each FY, we obtained total numbers of visits, unique patients, and care-providing staff by type. For validation, collaborating providers at several sites reviewed visit information, and a colleague also performed an independent, parallel data extraction. We adjusted FY totals to account for the growing patient population by dividing unique staff count by number of unique patients and multiplying by 200,000 (the approximate number of unique patients in FY19).
Results
From FY19 through FY24, VA Hematology/ Oncology saw a 14.6% rise in unique patients (from 232,084 to 265,926) and a 15.4% rise in visits (from 923,175 to 1,065,186). The absolute number of attendings rose by 4 (0.6%); of LIPs, by 138 (14.4%); and of nurses, by 142 (4.9%); trainees fell by 102 (4.3%). Adjusted to 200,000 patients, the number of attendings fell by 76 (12.3%); LIPs, by 1 (0.1%); trainees, by 335 (16.5%); and nurses, by 211 (8.4%).
Conclusions
Adjusted to number of Veterans, there are 10.4% fewer staff in Hematology/Oncology in FY24 compared to FY19.
Enhancing Coding Accuracy at the Hematology/Oncology Clinic: Is It Time to Hire a Dedicated Coder?
Background
Accurate clinical coding that reflects all diagnoses and problems addressed during a patient encounter is essential for the cancer program’s data quality, research initiatives, and securing VERA (Veterans Equitable Resource Allocation) funding. However, providers often face barriers such as limited time during patient visits and difficulty navigating Electronic health record (EHR) systems. These challenges lead to inaccurate coding, which undermines downstream data integrity. This quality improvement (QI) study aimed to identify these barriers and implement an intervention to improve coding accuracy, while also assessing the financial implications of improved documentation.
Methods
This QI study was conducted at the Albany Stratton VA Medical Center, focusing on hematology/ oncology outpatient encounters. A baseline chart audit of diagnosis codes from June 2023 revealed an accuracy rate of 69.8%. To address this, an intervention was implemented in which dedicated coders were assigned to support attending physicians in coding for over a two-week period. These coders reviewed and corrected diagnosis codes in real-time. A follow-up audit conducted after the intervention showed an improved coding accuracy of 82%.
Discussion/Implications
Coding remains a timeconsuming task for providers, made more difficult by EHR systems that are not user-friendly. This study demonstrated that involving dedicated coders significantly improves documentation accuracy—from 69% to 82%. In addition to data quality, the financial benefits are notable. A projected annual return on investment of $216,094 was calculated, based on an internal analysis showing that in a sample of 124 patients, 10% could have qualified for higher VERA funding based on accurate coding, generating an estimated $17,427 in additional reimbursement per patient. This cost-benefit ratio supports the recommendation to staff dedicated coders. Other interventions were also utilised, such as updating the national encounter form and auto-populating documentation in Dragon software, but had limited impact and did not directly address diagnosis accuracy respectively.
Conclusions
Targeted interventions improved coding accuracy, but sustainability remains a challenge due to time and system limitations. Future efforts should focus on hiring full-time coders. These steps can further enhance coding quality and potentially increase hospital revenue.
Background
Accurate clinical coding that reflects all diagnoses and problems addressed during a patient encounter is essential for the cancer program’s data quality, research initiatives, and securing VERA (Veterans Equitable Resource Allocation) funding. However, providers often face barriers such as limited time during patient visits and difficulty navigating Electronic health record (EHR) systems. These challenges lead to inaccurate coding, which undermines downstream data integrity. This quality improvement (QI) study aimed to identify these barriers and implement an intervention to improve coding accuracy, while also assessing the financial implications of improved documentation.
Methods
This QI study was conducted at the Albany Stratton VA Medical Center, focusing on hematology/ oncology outpatient encounters. A baseline chart audit of diagnosis codes from June 2023 revealed an accuracy rate of 69.8%. To address this, an intervention was implemented in which dedicated coders were assigned to support attending physicians in coding for over a two-week period. These coders reviewed and corrected diagnosis codes in real-time. A follow-up audit conducted after the intervention showed an improved coding accuracy of 82%.
Discussion/Implications
Coding remains a timeconsuming task for providers, made more difficult by EHR systems that are not user-friendly. This study demonstrated that involving dedicated coders significantly improves documentation accuracy—from 69% to 82%. In addition to data quality, the financial benefits are notable. A projected annual return on investment of $216,094 was calculated, based on an internal analysis showing that in a sample of 124 patients, 10% could have qualified for higher VERA funding based on accurate coding, generating an estimated $17,427 in additional reimbursement per patient. This cost-benefit ratio supports the recommendation to staff dedicated coders. Other interventions were also utilised, such as updating the national encounter form and auto-populating documentation in Dragon software, but had limited impact and did not directly address diagnosis accuracy respectively.
Conclusions
Targeted interventions improved coding accuracy, but sustainability remains a challenge due to time and system limitations. Future efforts should focus on hiring full-time coders. These steps can further enhance coding quality and potentially increase hospital revenue.
Background
Accurate clinical coding that reflects all diagnoses and problems addressed during a patient encounter is essential for the cancer program’s data quality, research initiatives, and securing VERA (Veterans Equitable Resource Allocation) funding. However, providers often face barriers such as limited time during patient visits and difficulty navigating Electronic health record (EHR) systems. These challenges lead to inaccurate coding, which undermines downstream data integrity. This quality improvement (QI) study aimed to identify these barriers and implement an intervention to improve coding accuracy, while also assessing the financial implications of improved documentation.
Methods
This QI study was conducted at the Albany Stratton VA Medical Center, focusing on hematology/ oncology outpatient encounters. A baseline chart audit of diagnosis codes from June 2023 revealed an accuracy rate of 69.8%. To address this, an intervention was implemented in which dedicated coders were assigned to support attending physicians in coding for over a two-week period. These coders reviewed and corrected diagnosis codes in real-time. A follow-up audit conducted after the intervention showed an improved coding accuracy of 82%.
Discussion/Implications
Coding remains a timeconsuming task for providers, made more difficult by EHR systems that are not user-friendly. This study demonstrated that involving dedicated coders significantly improves documentation accuracy—from 69% to 82%. In addition to data quality, the financial benefits are notable. A projected annual return on investment of $216,094 was calculated, based on an internal analysis showing that in a sample of 124 patients, 10% could have qualified for higher VERA funding based on accurate coding, generating an estimated $17,427 in additional reimbursement per patient. This cost-benefit ratio supports the recommendation to staff dedicated coders. Other interventions were also utilised, such as updating the national encounter form and auto-populating documentation in Dragon software, but had limited impact and did not directly address diagnosis accuracy respectively.
Conclusions
Targeted interventions improved coding accuracy, but sustainability remains a challenge due to time and system limitations. Future efforts should focus on hiring full-time coders. These steps can further enhance coding quality and potentially increase hospital revenue.
Evaluating the Implementation of 60-minute Iron Dextran Infusions at a Rural Health Center
Background
Due to risk for infusion-related reactions (IRR), administration of iron dextran requires an initial test dose with an extended monitoring period and subsequent doses given as a slow infusion over 2-3 hours. Safe use of a 60-minute iron dextran infusion protocol has been demonstrated previously at fully staffed academic teaching institutions. This study sought to determine the impact on patient safety and infusion clinic efficiency after implementing a 60-minute iron dextran administration protocol at a small, rural facility utilizing a decentralized clinical model.
Methods
This single-site, prospective, interventional study was conducted at a rural level 1C Veterans Affairs secondary care facility. The Hematology/Oncology clinic staffing includes one onsite clinical pharmacy practitioner (CPP) and advanced practice nurse. Remote providers complete patient encounters through video and telehealth modalities. A 60-minute iron dextran infusion service line agreement was designed by the Hematology/Oncology CPP and approved by the facility prior to data collection. The protocol included administration of a test dose and 15-minute monitoring period for treatment naïve patients. Pre-medications were allowed at the discretion of the ordering providers. All patients who received iron dextran between May 31, 2024 and April 14, 2025 per protocol were included in data analysis and results were stratified by treatment naïve and pre-treated patients. Outcomes included the proportion of patients experiencing any grade of IRR based on the Common Criteria for Adverse Events Version 5.0, and the average duration of administration. Descriptive statistics were used for safety and efficiency outcomes.
Results
Eighty patients received 103 iron dextran infusions and were included for analysis. Pre-medications were administered for 16 of the 103 (15.5%) included infusions. Two patients experienced grade 1 IRR (nausea) on 4 occasions (3.8%) which quickly resolved with intravenous ondansetron, and full iron dextran doses were received. The mean infusion time was 94 minutes in the treatment naïve cohort vs 71 minutes in the pre-treated cohort.
Conclusions
This study suggests a Hematology/ Oncology CPP developed iron dextran 60-minute infusion protocol may be safely and efficiently administered for qualifying patients in a decentralized, rural healthcare setting.
Background
Due to risk for infusion-related reactions (IRR), administration of iron dextran requires an initial test dose with an extended monitoring period and subsequent doses given as a slow infusion over 2-3 hours. Safe use of a 60-minute iron dextran infusion protocol has been demonstrated previously at fully staffed academic teaching institutions. This study sought to determine the impact on patient safety and infusion clinic efficiency after implementing a 60-minute iron dextran administration protocol at a small, rural facility utilizing a decentralized clinical model.
Methods
This single-site, prospective, interventional study was conducted at a rural level 1C Veterans Affairs secondary care facility. The Hematology/Oncology clinic staffing includes one onsite clinical pharmacy practitioner (CPP) and advanced practice nurse. Remote providers complete patient encounters through video and telehealth modalities. A 60-minute iron dextran infusion service line agreement was designed by the Hematology/Oncology CPP and approved by the facility prior to data collection. The protocol included administration of a test dose and 15-minute monitoring period for treatment naïve patients. Pre-medications were allowed at the discretion of the ordering providers. All patients who received iron dextran between May 31, 2024 and April 14, 2025 per protocol were included in data analysis and results were stratified by treatment naïve and pre-treated patients. Outcomes included the proportion of patients experiencing any grade of IRR based on the Common Criteria for Adverse Events Version 5.0, and the average duration of administration. Descriptive statistics were used for safety and efficiency outcomes.
Results
Eighty patients received 103 iron dextran infusions and were included for analysis. Pre-medications were administered for 16 of the 103 (15.5%) included infusions. Two patients experienced grade 1 IRR (nausea) on 4 occasions (3.8%) which quickly resolved with intravenous ondansetron, and full iron dextran doses were received. The mean infusion time was 94 minutes in the treatment naïve cohort vs 71 minutes in the pre-treated cohort.
Conclusions
This study suggests a Hematology/ Oncology CPP developed iron dextran 60-minute infusion protocol may be safely and efficiently administered for qualifying patients in a decentralized, rural healthcare setting.
Background
Due to risk for infusion-related reactions (IRR), administration of iron dextran requires an initial test dose with an extended monitoring period and subsequent doses given as a slow infusion over 2-3 hours. Safe use of a 60-minute iron dextran infusion protocol has been demonstrated previously at fully staffed academic teaching institutions. This study sought to determine the impact on patient safety and infusion clinic efficiency after implementing a 60-minute iron dextran administration protocol at a small, rural facility utilizing a decentralized clinical model.
Methods
This single-site, prospective, interventional study was conducted at a rural level 1C Veterans Affairs secondary care facility. The Hematology/Oncology clinic staffing includes one onsite clinical pharmacy practitioner (CPP) and advanced practice nurse. Remote providers complete patient encounters through video and telehealth modalities. A 60-minute iron dextran infusion service line agreement was designed by the Hematology/Oncology CPP and approved by the facility prior to data collection. The protocol included administration of a test dose and 15-minute monitoring period for treatment naïve patients. Pre-medications were allowed at the discretion of the ordering providers. All patients who received iron dextran between May 31, 2024 and April 14, 2025 per protocol were included in data analysis and results were stratified by treatment naïve and pre-treated patients. Outcomes included the proportion of patients experiencing any grade of IRR based on the Common Criteria for Adverse Events Version 5.0, and the average duration of administration. Descriptive statistics were used for safety and efficiency outcomes.
Results
Eighty patients received 103 iron dextran infusions and were included for analysis. Pre-medications were administered for 16 of the 103 (15.5%) included infusions. Two patients experienced grade 1 IRR (nausea) on 4 occasions (3.8%) which quickly resolved with intravenous ondansetron, and full iron dextran doses were received. The mean infusion time was 94 minutes in the treatment naïve cohort vs 71 minutes in the pre-treated cohort.
Conclusions
This study suggests a Hematology/ Oncology CPP developed iron dextran 60-minute infusion protocol may be safely and efficiently administered for qualifying patients in a decentralized, rural healthcare setting.