User login
Diarrhea and weight loss
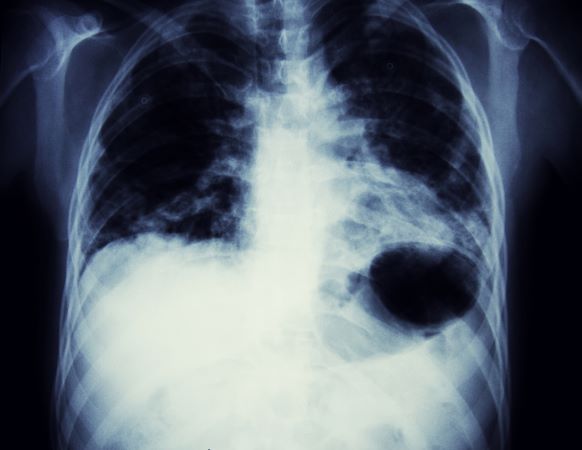
On the basis of the patient's presentation and history, this is probably a case of Crohn disease. Considering that the age of onset of Crohn disease has a bimodal distribution, this case is representative of late-onset disease. Among patients diagnosed with Crohn disease, the first peak is seen between 15 and 30 years of age, whereas the second peak, occurring in up to 15% of diagnoses, is observed mainly in women between 60 and 70 years of age. A significant proportion of Crohn disease cases are heritable. Patients of Ashkenazi Jewish descent are at higher risk of developing the condition than any other ethnic group.
According to American Gastroenterological Association guidelines, a diagnosis of inflammatory bowel disease (IBD) should be considered in older patients who present with diarrhea, rectal bleeding, urgency, abdominal pain, or weight loss. Fecal calprotectin or lactoferrin measurement may help identify patients who warrant further endoscopic evaluation. Colonoscopy is indicated for patients presenting with chronic diarrhea or hematochezia due to suspected IBD, microscopic colitis, or colorectal neoplasia.
Upon further workup for IBD, signs that suggest Crohn disease rather than ulcerative colitis (UC) are sparing of the rectum; discontinuous involvement with skip areas, deep, linear, or serpiginous ulcers of the colon; strictures; fistulas; or granulomatous inflammation. Antiglycan antibodies are more prevalent in Crohn disease than in ulcerative colitis, but they are not sensitive. Weight loss, perineal disease, fistulae, and obstruction are common in Crohn disease but uncommon in UC.
In treating Crohn disease among older adults, systemic corticosteroids are not indicated for maintenance therapy, though they may be used for induction therapy. When possible, nonsystemic corticosteroids should be used, or, if the phenotype prevents their use, early biological therapy. The decision to treat a patient with immunosuppressive drugs should be based on age, functional status, and comorbidities. Immunomodulatory treatments with lower risks for infection and cancer may be safer for patients with late-onset disease. For maintenance of remission, thiopurine monotherapy may be used, with consideration given to its risk for nonmelanoma skin cancers and lymphoma in older patients.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation and history, this is probably a case of Crohn disease. Considering that the age of onset of Crohn disease has a bimodal distribution, this case is representative of late-onset disease. Among patients diagnosed with Crohn disease, the first peak is seen between 15 and 30 years of age, whereas the second peak, occurring in up to 15% of diagnoses, is observed mainly in women between 60 and 70 years of age. A significant proportion of Crohn disease cases are heritable. Patients of Ashkenazi Jewish descent are at higher risk of developing the condition than any other ethnic group.
According to American Gastroenterological Association guidelines, a diagnosis of inflammatory bowel disease (IBD) should be considered in older patients who present with diarrhea, rectal bleeding, urgency, abdominal pain, or weight loss. Fecal calprotectin or lactoferrin measurement may help identify patients who warrant further endoscopic evaluation. Colonoscopy is indicated for patients presenting with chronic diarrhea or hematochezia due to suspected IBD, microscopic colitis, or colorectal neoplasia.
Upon further workup for IBD, signs that suggest Crohn disease rather than ulcerative colitis (UC) are sparing of the rectum; discontinuous involvement with skip areas, deep, linear, or serpiginous ulcers of the colon; strictures; fistulas; or granulomatous inflammation. Antiglycan antibodies are more prevalent in Crohn disease than in ulcerative colitis, but they are not sensitive. Weight loss, perineal disease, fistulae, and obstruction are common in Crohn disease but uncommon in UC.
In treating Crohn disease among older adults, systemic corticosteroids are not indicated for maintenance therapy, though they may be used for induction therapy. When possible, nonsystemic corticosteroids should be used, or, if the phenotype prevents their use, early biological therapy. The decision to treat a patient with immunosuppressive drugs should be based on age, functional status, and comorbidities. Immunomodulatory treatments with lower risks for infection and cancer may be safer for patients with late-onset disease. For maintenance of remission, thiopurine monotherapy may be used, with consideration given to its risk for nonmelanoma skin cancers and lymphoma in older patients.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation and history, this is probably a case of Crohn disease. Considering that the age of onset of Crohn disease has a bimodal distribution, this case is representative of late-onset disease. Among patients diagnosed with Crohn disease, the first peak is seen between 15 and 30 years of age, whereas the second peak, occurring in up to 15% of diagnoses, is observed mainly in women between 60 and 70 years of age. A significant proportion of Crohn disease cases are heritable. Patients of Ashkenazi Jewish descent are at higher risk of developing the condition than any other ethnic group.
According to American Gastroenterological Association guidelines, a diagnosis of inflammatory bowel disease (IBD) should be considered in older patients who present with diarrhea, rectal bleeding, urgency, abdominal pain, or weight loss. Fecal calprotectin or lactoferrin measurement may help identify patients who warrant further endoscopic evaluation. Colonoscopy is indicated for patients presenting with chronic diarrhea or hematochezia due to suspected IBD, microscopic colitis, or colorectal neoplasia.
Upon further workup for IBD, signs that suggest Crohn disease rather than ulcerative colitis (UC) are sparing of the rectum; discontinuous involvement with skip areas, deep, linear, or serpiginous ulcers of the colon; strictures; fistulas; or granulomatous inflammation. Antiglycan antibodies are more prevalent in Crohn disease than in ulcerative colitis, but they are not sensitive. Weight loss, perineal disease, fistulae, and obstruction are common in Crohn disease but uncommon in UC.
In treating Crohn disease among older adults, systemic corticosteroids are not indicated for maintenance therapy, though they may be used for induction therapy. When possible, nonsystemic corticosteroids should be used, or, if the phenotype prevents their use, early biological therapy. The decision to treat a patient with immunosuppressive drugs should be based on age, functional status, and comorbidities. Immunomodulatory treatments with lower risks for infection and cancer may be safer for patients with late-onset disease. For maintenance of remission, thiopurine monotherapy may be used, with consideration given to its risk for nonmelanoma skin cancers and lymphoma in older patients.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
A 65-year-old woman presents with diarrhea which began several months ago, abdominal pain, and a 10-lb weight loss. Height is 5 ft 3 in and weight is 120 lb (BMI 21.3). The patient notes that she typically does not have a sensitive stomach and is concerned by the onset of symptoms. Current medications are levothyroxine, alendronic acid, and hydrochlorothiazide. Family history is notable for pancreatic cancer on her mother's side; her daughter has celiac disease. She is of Ashkenazi Jewish descent. Body temperature is 100.2 °F and hemoglobin level is 12.9 g/dL. Colonoscopy shows ileitis with skip areas. Lab analysis is remarkable for antiglycan antibodies.
Severe ipsilateral headache
On the basis of the patient's presentation, family history, and personal history of headache, she seems to be presenting with hemiplegic migraine, an uncommon migraine subtype characterized by recurrent headaches associated with temporary unilateral hemiparesis or hemiplegia. The hemiparesis may resolve before the headache, as seen in the present case, or it may persist for days to weeks. These episodes are sometimes accompanied by ipsilateral numbness, tingling, or paresthesia, with or without a speech disturbance. Visual defects (ie, scintillating scotoma and hemianopia) and aphasia may occur.
Hemiplegic migraine can be sporadic or familial. Familial hemiplegic migraine is the only migraine subtype for which an autosomal dominant mode of inheritance has been identified. The onset is generally in adolescence between 12 and 17 years of age, with an estimated prevalence of 0.01%. Female patients are more likely to have these types of migraines.
Diagnosis of hemiplegic migraine is centered on exclusion of other possible causes of headache with motor weakness. When a patient presents with motor deficit, these symptoms can also be the result of a secondary headache rather than a primary headache disorder. Because of this neurologic aspect of presentation, the differential diagnosis is broad and should span other migraine subtypes, inflammatory or metabolic disorders, and mitochondrial diseases, as well as any condition that shows neurologic deficits without radiologic alterations. Pediatric patients with hemiplegic migraine are often misdiagnosed with epilepsy. Compared with hemiplegic migraine, seizures are much more brief, and any associated hemiparesis is usually characterized by limb jerking, head turning, and loss of consciousness. Of note, up to 7% of patients with familial hemiplegic migraine do eventually develop epilepsy. Although there are no telltale pathognomonic clinical, laboratory, or radiologic findings of hemiplegic migraine, electroencephalography may show asymmetric slow-wave activity contralateral to the hemiparesis.
In hemiplegic migraine, acute treatment options include antiemetics, NSAIDs, and nonnarcotic pain relievers; triptans and ergotamine preparations are contraindicated in this setting because of their potential vasoconstrictive effects. Even if episode frequency is low, the American Headache Society advises that prophylactic treatment should also be considered in the management of uncommon migraine subtypes such as this one.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation, family history, and personal history of headache, she seems to be presenting with hemiplegic migraine, an uncommon migraine subtype characterized by recurrent headaches associated with temporary unilateral hemiparesis or hemiplegia. The hemiparesis may resolve before the headache, as seen in the present case, or it may persist for days to weeks. These episodes are sometimes accompanied by ipsilateral numbness, tingling, or paresthesia, with or without a speech disturbance. Visual defects (ie, scintillating scotoma and hemianopia) and aphasia may occur.
Hemiplegic migraine can be sporadic or familial. Familial hemiplegic migraine is the only migraine subtype for which an autosomal dominant mode of inheritance has been identified. The onset is generally in adolescence between 12 and 17 years of age, with an estimated prevalence of 0.01%. Female patients are more likely to have these types of migraines.
Diagnosis of hemiplegic migraine is centered on exclusion of other possible causes of headache with motor weakness. When a patient presents with motor deficit, these symptoms can also be the result of a secondary headache rather than a primary headache disorder. Because of this neurologic aspect of presentation, the differential diagnosis is broad and should span other migraine subtypes, inflammatory or metabolic disorders, and mitochondrial diseases, as well as any condition that shows neurologic deficits without radiologic alterations. Pediatric patients with hemiplegic migraine are often misdiagnosed with epilepsy. Compared with hemiplegic migraine, seizures are much more brief, and any associated hemiparesis is usually characterized by limb jerking, head turning, and loss of consciousness. Of note, up to 7% of patients with familial hemiplegic migraine do eventually develop epilepsy. Although there are no telltale pathognomonic clinical, laboratory, or radiologic findings of hemiplegic migraine, electroencephalography may show asymmetric slow-wave activity contralateral to the hemiparesis.
In hemiplegic migraine, acute treatment options include antiemetics, NSAIDs, and nonnarcotic pain relievers; triptans and ergotamine preparations are contraindicated in this setting because of their potential vasoconstrictive effects. Even if episode frequency is low, the American Headache Society advises that prophylactic treatment should also be considered in the management of uncommon migraine subtypes such as this one.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation, family history, and personal history of headache, she seems to be presenting with hemiplegic migraine, an uncommon migraine subtype characterized by recurrent headaches associated with temporary unilateral hemiparesis or hemiplegia. The hemiparesis may resolve before the headache, as seen in the present case, or it may persist for days to weeks. These episodes are sometimes accompanied by ipsilateral numbness, tingling, or paresthesia, with or without a speech disturbance. Visual defects (ie, scintillating scotoma and hemianopia) and aphasia may occur.
Hemiplegic migraine can be sporadic or familial. Familial hemiplegic migraine is the only migraine subtype for which an autosomal dominant mode of inheritance has been identified. The onset is generally in adolescence between 12 and 17 years of age, with an estimated prevalence of 0.01%. Female patients are more likely to have these types of migraines.
Diagnosis of hemiplegic migraine is centered on exclusion of other possible causes of headache with motor weakness. When a patient presents with motor deficit, these symptoms can also be the result of a secondary headache rather than a primary headache disorder. Because of this neurologic aspect of presentation, the differential diagnosis is broad and should span other migraine subtypes, inflammatory or metabolic disorders, and mitochondrial diseases, as well as any condition that shows neurologic deficits without radiologic alterations. Pediatric patients with hemiplegic migraine are often misdiagnosed with epilepsy. Compared with hemiplegic migraine, seizures are much more brief, and any associated hemiparesis is usually characterized by limb jerking, head turning, and loss of consciousness. Of note, up to 7% of patients with familial hemiplegic migraine do eventually develop epilepsy. Although there are no telltale pathognomonic clinical, laboratory, or radiologic findings of hemiplegic migraine, electroencephalography may show asymmetric slow-wave activity contralateral to the hemiparesis.
In hemiplegic migraine, acute treatment options include antiemetics, NSAIDs, and nonnarcotic pain relievers; triptans and ergotamine preparations are contraindicated in this setting because of their potential vasoconstrictive effects. Even if episode frequency is low, the American Headache Society advises that prophylactic treatment should also be considered in the management of uncommon migraine subtypes such as this one.
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
A 16-year-old female patient presents with a severe ipsilateral headache. She describes that before the onset of head pain, she felt like she could not control her facial muscles on one side, and she was unable to speak in full sentences. She reports that these symptoms probably lasted an hour or so, and she was worried that she was experiencing an allergic reaction, though she reports no known allergies. In terms of family history, the patient explains that she does not have a close relationship with her father, but she recalls that he experienced similar episodes. She notes a history of frequent and recurrent headaches, varying in severity, for which she usually takes a high dose of nonsteroidal anti-inflammatory drugs (NSAIDs).
Pruritus and pitting edema
The 2020 Kidney Disease Improving Global Outcomes (KDIGO) diabetes management in CKD guideline states that most patients with diabetic nephropathy and an eGFR ≥ 30 mL/min/1.73 m2 benefit from treatment with both metformin and a sodium-glucose cotransporter 2 (SGLT2) inhibitor, which have been demonstrated to offer substantial benefits in reducing the risks for diabetic nephropathy and cardiovascular disease.
In patients who do not reach individualized targets with metformin and an SGLT2 inhibitor, or who are unable to use these medications, a long-acting glucagon-like peptide 1 (GLP-1) receptor antagonist may be used.
Metformin should be administered with caution to patients with CKD because it may increase the risk for lactic acidosis. It is contraindicated in patients with an eGFR < 30, but this patient's eGFR is adequate. Many clinicians might use a lower metformin dosage (1500 mg) as a precaution. Given how high his A1c is, adding a GLP-1 receptor antagonist is probably going to be needed because an SGLT2 inhibitor is only intermediate in terms of glucose reduction.
For control of his hypertension, the American Diabetes Association recommends either an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin receptor blocker (ARB) as first-line treatment. However, one agent alone is unlikely to control this patient's hypertension. At his level of eGFR, a thiazide diuretic is unlikely to be very effective. Therefore, a loop diuretic should be initiated with the ACE inhibitor or ARB, especially because he has edema.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The 2020 Kidney Disease Improving Global Outcomes (KDIGO) diabetes management in CKD guideline states that most patients with diabetic nephropathy and an eGFR ≥ 30 mL/min/1.73 m2 benefit from treatment with both metformin and a sodium-glucose cotransporter 2 (SGLT2) inhibitor, which have been demonstrated to offer substantial benefits in reducing the risks for diabetic nephropathy and cardiovascular disease.
In patients who do not reach individualized targets with metformin and an SGLT2 inhibitor, or who are unable to use these medications, a long-acting glucagon-like peptide 1 (GLP-1) receptor antagonist may be used.
Metformin should be administered with caution to patients with CKD because it may increase the risk for lactic acidosis. It is contraindicated in patients with an eGFR < 30, but this patient's eGFR is adequate. Many clinicians might use a lower metformin dosage (1500 mg) as a precaution. Given how high his A1c is, adding a GLP-1 receptor antagonist is probably going to be needed because an SGLT2 inhibitor is only intermediate in terms of glucose reduction.
For control of his hypertension, the American Diabetes Association recommends either an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin receptor blocker (ARB) as first-line treatment. However, one agent alone is unlikely to control this patient's hypertension. At his level of eGFR, a thiazide diuretic is unlikely to be very effective. Therefore, a loop diuretic should be initiated with the ACE inhibitor or ARB, especially because he has edema.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The 2020 Kidney Disease Improving Global Outcomes (KDIGO) diabetes management in CKD guideline states that most patients with diabetic nephropathy and an eGFR ≥ 30 mL/min/1.73 m2 benefit from treatment with both metformin and a sodium-glucose cotransporter 2 (SGLT2) inhibitor, which have been demonstrated to offer substantial benefits in reducing the risks for diabetic nephropathy and cardiovascular disease.
In patients who do not reach individualized targets with metformin and an SGLT2 inhibitor, or who are unable to use these medications, a long-acting glucagon-like peptide 1 (GLP-1) receptor antagonist may be used.
Metformin should be administered with caution to patients with CKD because it may increase the risk for lactic acidosis. It is contraindicated in patients with an eGFR < 30, but this patient's eGFR is adequate. Many clinicians might use a lower metformin dosage (1500 mg) as a precaution. Given how high his A1c is, adding a GLP-1 receptor antagonist is probably going to be needed because an SGLT2 inhibitor is only intermediate in terms of glucose reduction.
For control of his hypertension, the American Diabetes Association recommends either an angiotensin-converting enzyme (ACE) inhibitor or an angiotensin receptor blocker (ARB) as first-line treatment. However, one agent alone is unlikely to control this patient's hypertension. At his level of eGFR, a thiazide diuretic is unlikely to be very effective. Therefore, a loop diuretic should be initiated with the ACE inhibitor or ARB, especially because he has edema.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
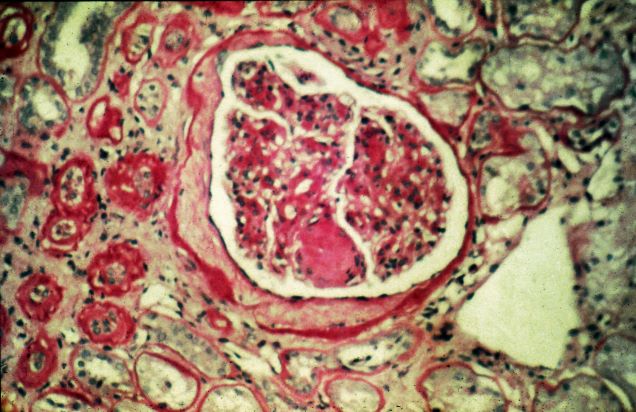
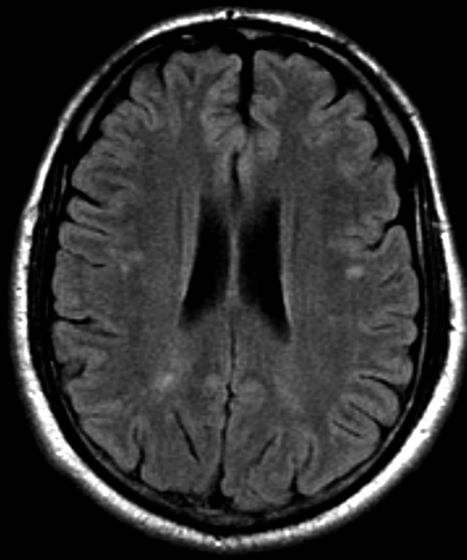
A 47-year-old Black man presents with shortness of breath, pruritus, and pitting edema of the bilateral extremities, which have been present for 6 weeks. He has a 7-year history of type 2 diabetes, hypertension, and hyperlipidemia, as well as a 30–pack-year history of smoking. His blood pressure is 160/95 mm Hg, heart rate is 97 beats/min (regular rate and rhythm), and respiration is 26 breaths/min. He also has proliferative retinopathy. He is 5 ft 10 in and weighs 220 lb (BMI 31.6). He is taking metformin 2550 mg/d. Other medications include simvastatin 20 mg, amlodipine 10 mg, and hydrochlorothiazide 25 mg. He admits to being nonadherent to his medication regimen. A year ago, his estimated glomerular filtration rate (eGFR) was 66 mL/min/1.73 m2 and he had 1+ proteinuria.
Laboratory tests reveal hemoglobin of 8.7 g/dL, creatinine of 3.4 g/dL, eGFR of 32 mL/min/1.73 m2, serum albumin of 3.3 g/dL, A1c of 8.8%, low-density lipoprotein of 143 mg/dL, high-density lipoprotein of 43 mg/dL, random glucose of 186 mg/dL, albumin-creatinine ratio of 3250 mg/g, calcium of 8.7 mg/dL, phosphorus of 4.2 mg/dL, plasma parathyroid hormone of 77 pg/mL, and C-reactive protein of 12.
In summary, this patient has normal albumin levels and increased proteinuria with decreased eGFR. His glucose level and A1c are not controlled. In addition, he has anemia, a low serum albumin level, and edema.
This patient has diabetic nephropathy and is at risk for a cardiovascular event because of his eGFR and long history of diabetes, hypertension, tobacco use, and hyperlipidemia. Intervention to control these risk factors should start immediately to prevent progression to chronic kidney disease (CKD).
Urinating multiple times per night
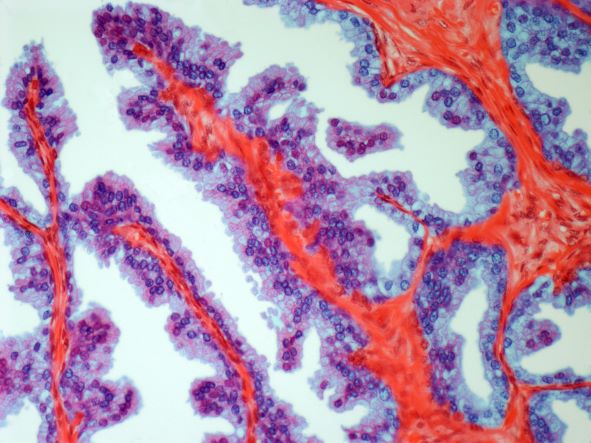
On the basis of the patient's history and presentation, this is likely a case of adenocarcinoma of the prostate. Although most patients with prostate cancer are diagnosed on screening, when localized symptoms do occur, they may include urinary frequency, decreased urine stream, urinary urgency, and hematuria. In some cases, these signs and symptoms may well be related to age-associated prostate enlargement or other conditions; benign prostatic hyperplasia, for example, can manifest in urinary symptoms and even elevate PSA (but because this patient does not report pain, nonbacterial prostatitis is unlikely). Symptomatic patients older than 50 years, such as the one in this case, should be screened for prostate cancer. Those with a PSA > 10 ng/mL are more than 50% likely to have prostate cancer.
National Comprehensive Cancer Network guidelines advise that needle biopsy of the prostate is indicated for tissue diagnosis in those with elevated PSA levels, preferably via a transrectal ultrasound. MRI can be used to assess lesions that are concerning for prostate cancer prior to biopsy. Lesions are then assigned Prostate Imaging Reporting and Data System (PI-RADS) scores depending on their location within the prostatic zones. A pathologic evaluation of the biopsy specimen will determine the patient's Gleason score. PSA density and PSA doubling time should be collected as well. The clinician should ask about high-risk germline mutations and estimate life expectancy because course of treatment is largely based on risk assessment.
Standard treatments for clinically localized prostate cancer include watchful waiting, active surveillance, radical prostatectomy, and radiation therapy. Active surveillance is often recommended for those who have very-low-risk disease because of the slow growth of certain types of prostate cancer. Radical prostatectomy is a viable option for any patient with localized disease that can be completely excised surgically, provided the patient has a life expectancy of 10 or more years and no serious comorbidities. In some patients, radical prostatectomy may be followed by radiation with or without a short course of hormone treatment, depending on risk factors for recurrence. Radiation therapy is also potentially curative in localized prostate cancer and may be delivered in the form of external-beam radiation therapy or brachytherapy. For asymptomatic patients who are older and/or have other serious underlying conditions, observation may be recommended.
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: CVICO Medical Solutions.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's history and presentation, this is likely a case of adenocarcinoma of the prostate. Although most patients with prostate cancer are diagnosed on screening, when localized symptoms do occur, they may include urinary frequency, decreased urine stream, urinary urgency, and hematuria. In some cases, these signs and symptoms may well be related to age-associated prostate enlargement or other conditions; benign prostatic hyperplasia, for example, can manifest in urinary symptoms and even elevate PSA (but because this patient does not report pain, nonbacterial prostatitis is unlikely). Symptomatic patients older than 50 years, such as the one in this case, should be screened for prostate cancer. Those with a PSA > 10 ng/mL are more than 50% likely to have prostate cancer.
National Comprehensive Cancer Network guidelines advise that needle biopsy of the prostate is indicated for tissue diagnosis in those with elevated PSA levels, preferably via a transrectal ultrasound. MRI can be used to assess lesions that are concerning for prostate cancer prior to biopsy. Lesions are then assigned Prostate Imaging Reporting and Data System (PI-RADS) scores depending on their location within the prostatic zones. A pathologic evaluation of the biopsy specimen will determine the patient's Gleason score. PSA density and PSA doubling time should be collected as well. The clinician should ask about high-risk germline mutations and estimate life expectancy because course of treatment is largely based on risk assessment.
Standard treatments for clinically localized prostate cancer include watchful waiting, active surveillance, radical prostatectomy, and radiation therapy. Active surveillance is often recommended for those who have very-low-risk disease because of the slow growth of certain types of prostate cancer. Radical prostatectomy is a viable option for any patient with localized disease that can be completely excised surgically, provided the patient has a life expectancy of 10 or more years and no serious comorbidities. In some patients, radical prostatectomy may be followed by radiation with or without a short course of hormone treatment, depending on risk factors for recurrence. Radiation therapy is also potentially curative in localized prostate cancer and may be delivered in the form of external-beam radiation therapy or brachytherapy. For asymptomatic patients who are older and/or have other serious underlying conditions, observation may be recommended.
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: CVICO Medical Solutions.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's history and presentation, this is likely a case of adenocarcinoma of the prostate. Although most patients with prostate cancer are diagnosed on screening, when localized symptoms do occur, they may include urinary frequency, decreased urine stream, urinary urgency, and hematuria. In some cases, these signs and symptoms may well be related to age-associated prostate enlargement or other conditions; benign prostatic hyperplasia, for example, can manifest in urinary symptoms and even elevate PSA (but because this patient does not report pain, nonbacterial prostatitis is unlikely). Symptomatic patients older than 50 years, such as the one in this case, should be screened for prostate cancer. Those with a PSA > 10 ng/mL are more than 50% likely to have prostate cancer.
National Comprehensive Cancer Network guidelines advise that needle biopsy of the prostate is indicated for tissue diagnosis in those with elevated PSA levels, preferably via a transrectal ultrasound. MRI can be used to assess lesions that are concerning for prostate cancer prior to biopsy. Lesions are then assigned Prostate Imaging Reporting and Data System (PI-RADS) scores depending on their location within the prostatic zones. A pathologic evaluation of the biopsy specimen will determine the patient's Gleason score. PSA density and PSA doubling time should be collected as well. The clinician should ask about high-risk germline mutations and estimate life expectancy because course of treatment is largely based on risk assessment.
Standard treatments for clinically localized prostate cancer include watchful waiting, active surveillance, radical prostatectomy, and radiation therapy. Active surveillance is often recommended for those who have very-low-risk disease because of the slow growth of certain types of prostate cancer. Radical prostatectomy is a viable option for any patient with localized disease that can be completely excised surgically, provided the patient has a life expectancy of 10 or more years and no serious comorbidities. In some patients, radical prostatectomy may be followed by radiation with or without a short course of hormone treatment, depending on risk factors for recurrence. Radiation therapy is also potentially curative in localized prostate cancer and may be delivered in the form of external-beam radiation therapy or brachytherapy. For asymptomatic patients who are older and/or have other serious underlying conditions, observation may be recommended.
Chad R. Tracy, MD, Professor; Director, Minimally Invasive Surgery, Department of Urology, University of Iowa Hospitals and Clinics, Iowa City, Iowa
Chad R. Tracy, MD, has disclosed the following relevant financial relationships:
Serve(d) as a consultant for: CVICO Medical Solutions.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
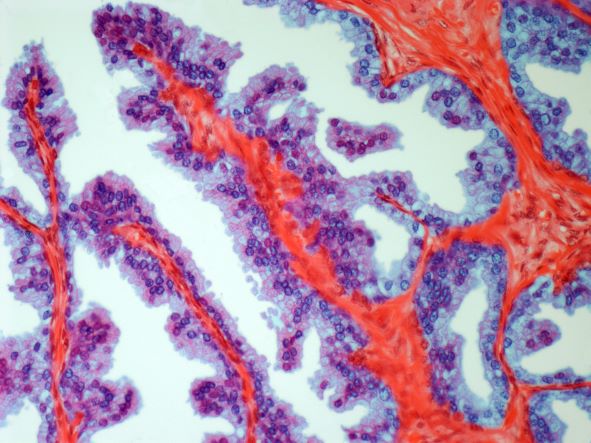
A 62-year-old man presents for routine prostate cancer screening. He notes that he has not been sleeping well as a result of getting up to urinate multiple times per night for the past few months. The patient underwent a prostate cancer screening about 26 months ago, and results were normal. On examination, digital rectal examination is normal, but prostate-specific antigen (PSA) levels are elevated at 10.2 ng/mL.
Persistent dry cough
On the basis of the patient's presentation, history, and imaging results, the likely diagnosis is metastatic small cell lung cancer (SCLC). Most patients with SCLC present with hematogenous metastases; only about one third present with limited disease confined to the chest that is amenable to multimodal therapy. Patients with SCLC often present with symptoms of widespread metastases, including weight loss, bone pain, and neurologic compromise. It is uncommon for patients to present with a solitary peripheral nodule. In earlier stages, the differential diagnosis of SCLC spans other neuroendocrine lung tumors and NSCLC, in particular, basaloid carcinoma, extrapulmonary small cell tumors, and lymphoma.
Because the concentration of circulating tumor cells in SCLC is among the highest of any solid tumor, SCLC is characterized by a rapid doubling time, high growth fraction, and early development of widespread metastases. It is likely for this reason that CT screening does not seem effective in detecting early-stage SCLC. Common sites of SCLC metastasis are the contralateral lung, the brain, liver, adrenal glands, and bone. Most cases of SCLC are caused by smoking.
Metastatic spread is often evident on radiologic exam, sometimes showing pleural and pericardial effusions. In general, workup for SCLC includes imaging (contrast-enhanced CT or F-FDG PET–CT of the chest, abdomen, and pelvis and brain MRI with contrast), blood tests (cell count, liver and kidney function, and lactate dehydrogenase), and ECG. Biopsies are generally procured by bronchoscopy with or without endobronchial ultrasonography; if accessible, a biopsy of a distal metastatic site may be obtained. Diagnosis of SCLC is confirmed by histopathologic examination via cytology.
Patients with extensive-stage SCLC are typically treated with systemic chemotherapy with or without immunotherapy. In the early stages, SCLC is very responsive to cytotoxic therapies, with response rates over 60% even in patients with metastatic disease. Until recently, the only second-line therapy for recurrent metastatic SCLC was the topoisomerase I inhibitor topotecan. However, lurbinectedin was granted accelerated approval for second-line therapy after demonstrating a 35% response rate in a single-arm phase 2 study of 105 patients. In addition, the anti–programmed cell death protein 1 monoclonal antibodies nivolumab and pembrolizumab were granted accelerated approval for third-line use. Finally, the National Comprehensive Cancer Network guidelines note that participation in clinical trials should be strongly encouraged for all patients with SCLC.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation, history, and imaging results, the likely diagnosis is metastatic small cell lung cancer (SCLC). Most patients with SCLC present with hematogenous metastases; only about one third present with limited disease confined to the chest that is amenable to multimodal therapy. Patients with SCLC often present with symptoms of widespread metastases, including weight loss, bone pain, and neurologic compromise. It is uncommon for patients to present with a solitary peripheral nodule. In earlier stages, the differential diagnosis of SCLC spans other neuroendocrine lung tumors and NSCLC, in particular, basaloid carcinoma, extrapulmonary small cell tumors, and lymphoma.
Because the concentration of circulating tumor cells in SCLC is among the highest of any solid tumor, SCLC is characterized by a rapid doubling time, high growth fraction, and early development of widespread metastases. It is likely for this reason that CT screening does not seem effective in detecting early-stage SCLC. Common sites of SCLC metastasis are the contralateral lung, the brain, liver, adrenal glands, and bone. Most cases of SCLC are caused by smoking.
Metastatic spread is often evident on radiologic exam, sometimes showing pleural and pericardial effusions. In general, workup for SCLC includes imaging (contrast-enhanced CT or F-FDG PET–CT of the chest, abdomen, and pelvis and brain MRI with contrast), blood tests (cell count, liver and kidney function, and lactate dehydrogenase), and ECG. Biopsies are generally procured by bronchoscopy with or without endobronchial ultrasonography; if accessible, a biopsy of a distal metastatic site may be obtained. Diagnosis of SCLC is confirmed by histopathologic examination via cytology.
Patients with extensive-stage SCLC are typically treated with systemic chemotherapy with or without immunotherapy. In the early stages, SCLC is very responsive to cytotoxic therapies, with response rates over 60% even in patients with metastatic disease. Until recently, the only second-line therapy for recurrent metastatic SCLC was the topoisomerase I inhibitor topotecan. However, lurbinectedin was granted accelerated approval for second-line therapy after demonstrating a 35% response rate in a single-arm phase 2 study of 105 patients. In addition, the anti–programmed cell death protein 1 monoclonal antibodies nivolumab and pembrolizumab were granted accelerated approval for third-line use. Finally, the National Comprehensive Cancer Network guidelines note that participation in clinical trials should be strongly encouraged for all patients with SCLC.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of the patient's presentation, history, and imaging results, the likely diagnosis is metastatic small cell lung cancer (SCLC). Most patients with SCLC present with hematogenous metastases; only about one third present with limited disease confined to the chest that is amenable to multimodal therapy. Patients with SCLC often present with symptoms of widespread metastases, including weight loss, bone pain, and neurologic compromise. It is uncommon for patients to present with a solitary peripheral nodule. In earlier stages, the differential diagnosis of SCLC spans other neuroendocrine lung tumors and NSCLC, in particular, basaloid carcinoma, extrapulmonary small cell tumors, and lymphoma.
Because the concentration of circulating tumor cells in SCLC is among the highest of any solid tumor, SCLC is characterized by a rapid doubling time, high growth fraction, and early development of widespread metastases. It is likely for this reason that CT screening does not seem effective in detecting early-stage SCLC. Common sites of SCLC metastasis are the contralateral lung, the brain, liver, adrenal glands, and bone. Most cases of SCLC are caused by smoking.
Metastatic spread is often evident on radiologic exam, sometimes showing pleural and pericardial effusions. In general, workup for SCLC includes imaging (contrast-enhanced CT or F-FDG PET–CT of the chest, abdomen, and pelvis and brain MRI with contrast), blood tests (cell count, liver and kidney function, and lactate dehydrogenase), and ECG. Biopsies are generally procured by bronchoscopy with or without endobronchial ultrasonography; if accessible, a biopsy of a distal metastatic site may be obtained. Diagnosis of SCLC is confirmed by histopathologic examination via cytology.
Patients with extensive-stage SCLC are typically treated with systemic chemotherapy with or without immunotherapy. In the early stages, SCLC is very responsive to cytotoxic therapies, with response rates over 60% even in patients with metastatic disease. Until recently, the only second-line therapy for recurrent metastatic SCLC was the topoisomerase I inhibitor topotecan. However, lurbinectedin was granted accelerated approval for second-line therapy after demonstrating a 35% response rate in a single-arm phase 2 study of 105 patients. In addition, the anti–programmed cell death protein 1 monoclonal antibodies nivolumab and pembrolizumab were granted accelerated approval for third-line use. Finally, the National Comprehensive Cancer Network guidelines note that participation in clinical trials should be strongly encouraged for all patients with SCLC.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
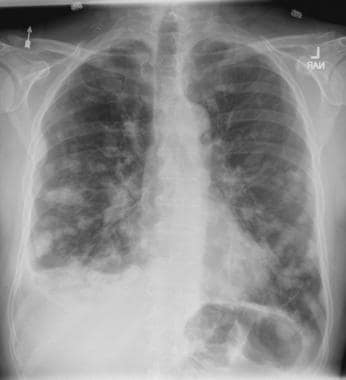
A 58-year-old man presents with a persistent dry cough that has developed over the past 8 weeks. He has lost about 8-10 lb in under 3 months. Height is 5 ft 10 in and weight is 172 lb (BMI 24.7). Although he quit smoking about 15 years ago, his wife still smokes. He has been screened twice for non–small cell lung cancer (NSCLC), most recently a year and a half ago. Chest radiograph shows multiple pulmonary nodules of varying sizes and a small right basal effusion.
Man with distal flexion deformities
On the basis of history and presentation, this patient's psoriatic disease has probably evolved to psoriatic arthritis mutilans (PAM). PAM is considered the most severe form of psoriatic arthritis (PsA), causing joint destruction and functional disability. It is estimated to affect about 5% of patients with PsA, with an equal sex distribution. Psoriatic nail dystrophy, a hallmark of PsA, appears to be a clinical biomarker of PAM development. Patients with PAM are generally younger at diagnosis than those with less severe forms of disease. Disease-modifying antirheumatic drugs and anti-TNF therapy do not appear to prevent the development of PAM, as evidenced by the present case.
In general, clinical presentation of PsA is heterogeneous and can be similar to that of other rheumatic diseases such as rheumatoid arthritis or osteoarthritis, complicating the differential diagnosis. The Classification Criteria for Psoriatic Arthritis (CASPAR) are considered the most sensitive diagnostic criteria, encompassing evidence of psoriasis; nail dystrophy; lab findings of typical autoantibodies (negative rheumatoid factor); and phenomena that are characteristic of PsA, like dactylitis.
Workup for PAM often includes radiography, ultrasound, and MRI or CT. With no established consensus, classification systems for the condition vary clinically and radiographically. Radiographic features suggestive of PAM include osteolysis or extended bone resorption; pencil-in-cup changes; joint subluxation; and, less often, ankylosis. Osteolysis has been defined as bone resorption with more than 50% loss of joint surface on both sides of the joint. Clinically, dissolution of the joint causes redundant, overlying skin with a telescoping motion of the digit. Other clinical features of PAM include digital shortening and flail joints. Of note, involvement of one small joint in the hands or feet is diagnostic of PAM.
In the setting of PsA, multiple genetic factors have been described, including presence of HLA-B27 and HLA-DRB1, but none are considered defining factors for the disease. A recent population-based study shows that presence of HLA-B27 was significantly increased among patients with PAM (45%) compared with patients with less severe PsA (13%) and healthy controls (13%).
According to the American College of Rheumatology guidelines, first-line therapy in adult patients who have active PsA and are treatment-naive is a TNFi biologic agent. For the patient in this case, who has active PsA despite treatment with TNFi biologic monotherapy, switching to a different TNFi biologic may be appropriate; however, switching to an interleukin-17 inhibitor may also be considered because this patient has severe disease. Data on the comparative efficacy of different biological agents for treatment of PAM are not yet available.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of history and presentation, this patient's psoriatic disease has probably evolved to psoriatic arthritis mutilans (PAM). PAM is considered the most severe form of psoriatic arthritis (PsA), causing joint destruction and functional disability. It is estimated to affect about 5% of patients with PsA, with an equal sex distribution. Psoriatic nail dystrophy, a hallmark of PsA, appears to be a clinical biomarker of PAM development. Patients with PAM are generally younger at diagnosis than those with less severe forms of disease. Disease-modifying antirheumatic drugs and anti-TNF therapy do not appear to prevent the development of PAM, as evidenced by the present case.
In general, clinical presentation of PsA is heterogeneous and can be similar to that of other rheumatic diseases such as rheumatoid arthritis or osteoarthritis, complicating the differential diagnosis. The Classification Criteria for Psoriatic Arthritis (CASPAR) are considered the most sensitive diagnostic criteria, encompassing evidence of psoriasis; nail dystrophy; lab findings of typical autoantibodies (negative rheumatoid factor); and phenomena that are characteristic of PsA, like dactylitis.
Workup for PAM often includes radiography, ultrasound, and MRI or CT. With no established consensus, classification systems for the condition vary clinically and radiographically. Radiographic features suggestive of PAM include osteolysis or extended bone resorption; pencil-in-cup changes; joint subluxation; and, less often, ankylosis. Osteolysis has been defined as bone resorption with more than 50% loss of joint surface on both sides of the joint. Clinically, dissolution of the joint causes redundant, overlying skin with a telescoping motion of the digit. Other clinical features of PAM include digital shortening and flail joints. Of note, involvement of one small joint in the hands or feet is diagnostic of PAM.
In the setting of PsA, multiple genetic factors have been described, including presence of HLA-B27 and HLA-DRB1, but none are considered defining factors for the disease. A recent population-based study shows that presence of HLA-B27 was significantly increased among patients with PAM (45%) compared with patients with less severe PsA (13%) and healthy controls (13%).
According to the American College of Rheumatology guidelines, first-line therapy in adult patients who have active PsA and are treatment-naive is a TNFi biologic agent. For the patient in this case, who has active PsA despite treatment with TNFi biologic monotherapy, switching to a different TNFi biologic may be appropriate; however, switching to an interleukin-17 inhibitor may also be considered because this patient has severe disease. Data on the comparative efficacy of different biological agents for treatment of PAM are not yet available.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
On the basis of history and presentation, this patient's psoriatic disease has probably evolved to psoriatic arthritis mutilans (PAM). PAM is considered the most severe form of psoriatic arthritis (PsA), causing joint destruction and functional disability. It is estimated to affect about 5% of patients with PsA, with an equal sex distribution. Psoriatic nail dystrophy, a hallmark of PsA, appears to be a clinical biomarker of PAM development. Patients with PAM are generally younger at diagnosis than those with less severe forms of disease. Disease-modifying antirheumatic drugs and anti-TNF therapy do not appear to prevent the development of PAM, as evidenced by the present case.
In general, clinical presentation of PsA is heterogeneous and can be similar to that of other rheumatic diseases such as rheumatoid arthritis or osteoarthritis, complicating the differential diagnosis. The Classification Criteria for Psoriatic Arthritis (CASPAR) are considered the most sensitive diagnostic criteria, encompassing evidence of psoriasis; nail dystrophy; lab findings of typical autoantibodies (negative rheumatoid factor); and phenomena that are characteristic of PsA, like dactylitis.
Workup for PAM often includes radiography, ultrasound, and MRI or CT. With no established consensus, classification systems for the condition vary clinically and radiographically. Radiographic features suggestive of PAM include osteolysis or extended bone resorption; pencil-in-cup changes; joint subluxation; and, less often, ankylosis. Osteolysis has been defined as bone resorption with more than 50% loss of joint surface on both sides of the joint. Clinically, dissolution of the joint causes redundant, overlying skin with a telescoping motion of the digit. Other clinical features of PAM include digital shortening and flail joints. Of note, involvement of one small joint in the hands or feet is diagnostic of PAM.
In the setting of PsA, multiple genetic factors have been described, including presence of HLA-B27 and HLA-DRB1, but none are considered defining factors for the disease. A recent population-based study shows that presence of HLA-B27 was significantly increased among patients with PAM (45%) compared with patients with less severe PsA (13%) and healthy controls (13%).
According to the American College of Rheumatology guidelines, first-line therapy in adult patients who have active PsA and are treatment-naive is a TNFi biologic agent. For the patient in this case, who has active PsA despite treatment with TNFi biologic monotherapy, switching to a different TNFi biologic may be appropriate; however, switching to an interleukin-17 inhibitor may also be considered because this patient has severe disease. Data on the comparative efficacy of different biological agents for treatment of PAM are not yet available.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
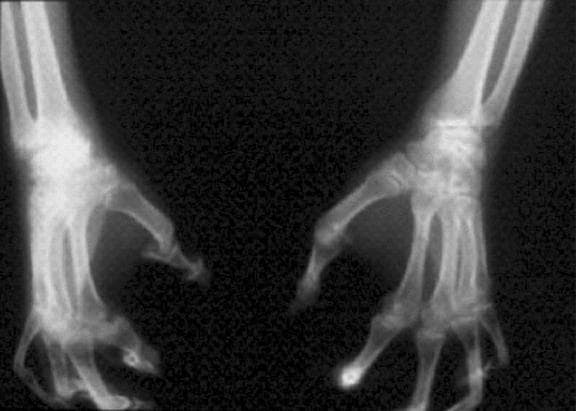
A 43-year-old man presents with distal flexion deformities and telescoping of the digits. The patient was diagnosed with psoriasis at age 31 and he has several immediate family members who previously received the same diagnosis. He has been treated intermittently with tumor necrosis factor inhibitor (TNFi) biologic monotherapy but admits to nonadherence when disease activity seems to quiet down. Radiography shows osteolysis and dissolution of the joint.

Abdominal cramping and diarrhea
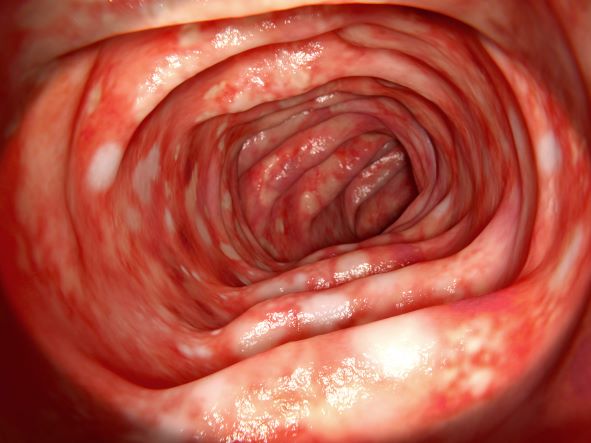
On the basis of the patient's history and presentation, the likely diagnosis is extensive UC. Extensive colitis is defined by the presence of disease activity proximal to the splenic flexure. Because disease activity in UC is dynamic, up to half of patients who present with proctitis and 70% of those who present with left-sided colitis go on to develop extensive colitis on follow-up.
On the basis of the workup, it appears that this patient's UC has transitioned from left-sided to extensive disease, given the loss of treatment response. Endoscopic evaluation of patients with loss of treatment response may reveal patchiness of the histologic activity, as seen in this case.
Extensive colitis is a poor prognostic factor in UC, as is systemic steroid requirement, young age at diagnosis, and an elevated C-reactive protein level or erythrocyte sedimentation rate, all of which are associated with higher rates of colectomy. Over time, patients living with extensive ulcerative colitis develop an increased risk for colorectal cancer. Routine colonoscopic screening and surveillance are recommended for these high-risk patients.
UC most often presents as a continuously inflamed segment involving the distal rectum and extending proximally. Endoscopic features of inflammation include loss of vascular markings; granularity and friability of the mucosa; erosions; and, in the setting of severe inflammation, ulcerations and spontaneous bleeding. The diagnosis of UC involves both a lower gastrointestinal endoscopic examination and histologic confirmation. In general, a complete colonoscopy including examination of the terminal ileum should be performed, allowing clinicians to assess the full extent of the disease while ruling out distal ileal involvement, which is characteristic of Crohn's disease.
Evaluation of UC during relapses should include assessment of symptom severity and potential triggers, including enteric infections, use of nonsteroidal anti-inflammatory drugs, and recent smoking cessation. Nonadherence to therapy is common in patients with UC and may lead to relapse.
To treat a patient like the one represented here, the American College of Gastroenterology guidelines recommend oral 5-ASA at a dose of at least 2 g/d to induce remission. However, because this patient lost response to this treatment, the next step in the guidelines are appropriate oral systemic corticosteroids.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships
On the basis of the patient's history and presentation, the likely diagnosis is extensive UC. Extensive colitis is defined by the presence of disease activity proximal to the splenic flexure. Because disease activity in UC is dynamic, up to half of patients who present with proctitis and 70% of those who present with left-sided colitis go on to develop extensive colitis on follow-up.
On the basis of the workup, it appears that this patient's UC has transitioned from left-sided to extensive disease, given the loss of treatment response. Endoscopic evaluation of patients with loss of treatment response may reveal patchiness of the histologic activity, as seen in this case.
Extensive colitis is a poor prognostic factor in UC, as is systemic steroid requirement, young age at diagnosis, and an elevated C-reactive protein level or erythrocyte sedimentation rate, all of which are associated with higher rates of colectomy. Over time, patients living with extensive ulcerative colitis develop an increased risk for colorectal cancer. Routine colonoscopic screening and surveillance are recommended for these high-risk patients.
UC most often presents as a continuously inflamed segment involving the distal rectum and extending proximally. Endoscopic features of inflammation include loss of vascular markings; granularity and friability of the mucosa; erosions; and, in the setting of severe inflammation, ulcerations and spontaneous bleeding. The diagnosis of UC involves both a lower gastrointestinal endoscopic examination and histologic confirmation. In general, a complete colonoscopy including examination of the terminal ileum should be performed, allowing clinicians to assess the full extent of the disease while ruling out distal ileal involvement, which is characteristic of Crohn's disease.
Evaluation of UC during relapses should include assessment of symptom severity and potential triggers, including enteric infections, use of nonsteroidal anti-inflammatory drugs, and recent smoking cessation. Nonadherence to therapy is common in patients with UC and may lead to relapse.
To treat a patient like the one represented here, the American College of Gastroenterology guidelines recommend oral 5-ASA at a dose of at least 2 g/d to induce remission. However, because this patient lost response to this treatment, the next step in the guidelines are appropriate oral systemic corticosteroids.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships
On the basis of the patient's history and presentation, the likely diagnosis is extensive UC. Extensive colitis is defined by the presence of disease activity proximal to the splenic flexure. Because disease activity in UC is dynamic, up to half of patients who present with proctitis and 70% of those who present with left-sided colitis go on to develop extensive colitis on follow-up.
On the basis of the workup, it appears that this patient's UC has transitioned from left-sided to extensive disease, given the loss of treatment response. Endoscopic evaluation of patients with loss of treatment response may reveal patchiness of the histologic activity, as seen in this case.
Extensive colitis is a poor prognostic factor in UC, as is systemic steroid requirement, young age at diagnosis, and an elevated C-reactive protein level or erythrocyte sedimentation rate, all of which are associated with higher rates of colectomy. Over time, patients living with extensive ulcerative colitis develop an increased risk for colorectal cancer. Routine colonoscopic screening and surveillance are recommended for these high-risk patients.
UC most often presents as a continuously inflamed segment involving the distal rectum and extending proximally. Endoscopic features of inflammation include loss of vascular markings; granularity and friability of the mucosa; erosions; and, in the setting of severe inflammation, ulcerations and spontaneous bleeding. The diagnosis of UC involves both a lower gastrointestinal endoscopic examination and histologic confirmation. In general, a complete colonoscopy including examination of the terminal ileum should be performed, allowing clinicians to assess the full extent of the disease while ruling out distal ileal involvement, which is characteristic of Crohn's disease.
Evaluation of UC during relapses should include assessment of symptom severity and potential triggers, including enteric infections, use of nonsteroidal anti-inflammatory drugs, and recent smoking cessation. Nonadherence to therapy is common in patients with UC and may lead to relapse.
To treat a patient like the one represented here, the American College of Gastroenterology guidelines recommend oral 5-ASA at a dose of at least 2 g/d to induce remission. However, because this patient lost response to this treatment, the next step in the guidelines are appropriate oral systemic corticosteroids.
Bhupinder S. Anand, MD, Professor, Department of Medicine, Baylor College of Medicine, Houston, TX
Bhupinder S. Anand, MD, has disclosed no relevant financial relationships
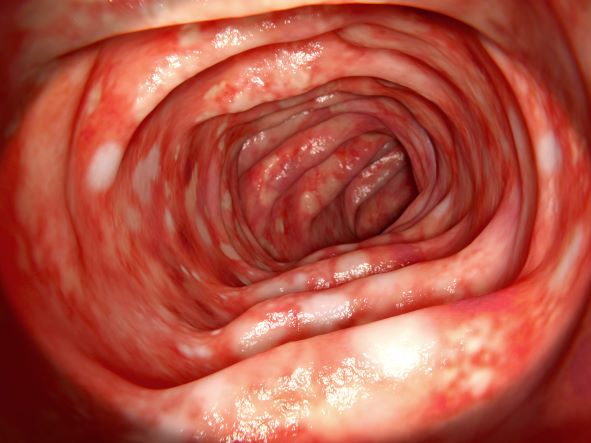
A 46-year-old man presents with abdominal cramping and diarrhea and reports about five bowel movements per day for the past 2 weeks. Height is 5 ft 9 in and weight is 157 lb (BMI, 23.2). History is significant for ulcerative colitis (UC), diagnosed about 20 years ago with proctitis and having progressed about 8 years ago to left-sided disease. He smoked "lightly" through his 20s. Until about a month ago, the patient had been able to maintain remission with oral 5-aminosalicylic acid (ASA) therapy (2 g/d). Endoscopy shows granularity and friability of the mucosa with the inflamed segment extending proximal to the splenic flexure, though there is patchiness of the histologic activity. Colonoscopy rules out distal ileal involvement. Stool culture is negative.
Recent unintended weight loss
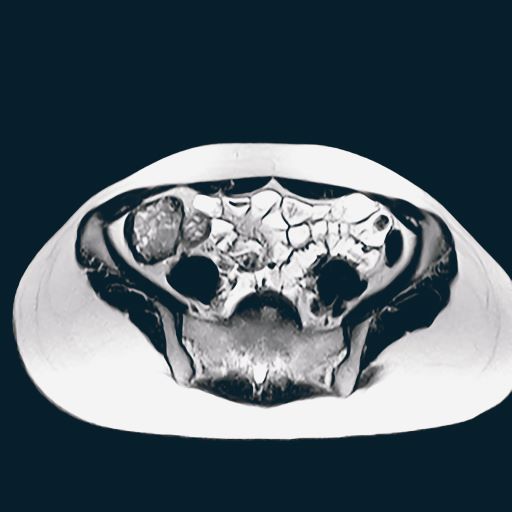
PCOS is most often defined according to the Rotterdam criteria, which stipulate that at least two of the following be present: irregular ovulation, biochemical/clinical hyperandrogenism, and polycystic ovaries (seen in the MRI scan above). Insulin resistance is part of the pathogenesis of PCOS, and insulin resistance is associated with T2D in PCOS.
In fact, PCOS is an independent risk factor for T2D, even after adjustment for BMI and obesity. Even normal-weight women with PCOS have an increased risk for T2D. More than half of women with PCOS develop T2D by age 40.
Even though family history and obesity are major contributors in the development of diabetes in patients with PCOS, diabetes can still occur in lean patients with PCOS who have no family history, mainly secondary to insulin resistance.
The Endocrine Society recommends that all individuals with PCOS undergo an oral glucose tolerance test every 3-5 years, with more frequent screening for those who develop symptoms of T2D, significant weight gain, or central adiposity. In guidelines published in 2015 by the American Association of Clinical Endocrinologists, the American College of Endocrinology, and the Androgen Excess and PCOS Society, an annual oral glucose tolerance test is recommended for patients with PCOS and impaired glucose tolerance, whereas those with a family history of T2D or a BMI above 30 should be screened every 1-2 years.
Management of T2D with PCOS is similar to that of T2D without PCOS. Accordingly, metformin and lifestyle changes are the treatments of choice; any antidiabetic agent may be added in patients who do not achieve glycemic targets despite treatment with metformin.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships
PCOS is most often defined according to the Rotterdam criteria, which stipulate that at least two of the following be present: irregular ovulation, biochemical/clinical hyperandrogenism, and polycystic ovaries (seen in the MRI scan above). Insulin resistance is part of the pathogenesis of PCOS, and insulin resistance is associated with T2D in PCOS.
In fact, PCOS is an independent risk factor for T2D, even after adjustment for BMI and obesity. Even normal-weight women with PCOS have an increased risk for T2D. More than half of women with PCOS develop T2D by age 40.
Even though family history and obesity are major contributors in the development of diabetes in patients with PCOS, diabetes can still occur in lean patients with PCOS who have no family history, mainly secondary to insulin resistance.
The Endocrine Society recommends that all individuals with PCOS undergo an oral glucose tolerance test every 3-5 years, with more frequent screening for those who develop symptoms of T2D, significant weight gain, or central adiposity. In guidelines published in 2015 by the American Association of Clinical Endocrinologists, the American College of Endocrinology, and the Androgen Excess and PCOS Society, an annual oral glucose tolerance test is recommended for patients with PCOS and impaired glucose tolerance, whereas those with a family history of T2D or a BMI above 30 should be screened every 1-2 years.
Management of T2D with PCOS is similar to that of T2D without PCOS. Accordingly, metformin and lifestyle changes are the treatments of choice; any antidiabetic agent may be added in patients who do not achieve glycemic targets despite treatment with metformin.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships
PCOS is most often defined according to the Rotterdam criteria, which stipulate that at least two of the following be present: irregular ovulation, biochemical/clinical hyperandrogenism, and polycystic ovaries (seen in the MRI scan above). Insulin resistance is part of the pathogenesis of PCOS, and insulin resistance is associated with T2D in PCOS.
In fact, PCOS is an independent risk factor for T2D, even after adjustment for BMI and obesity. Even normal-weight women with PCOS have an increased risk for T2D. More than half of women with PCOS develop T2D by age 40.
Even though family history and obesity are major contributors in the development of diabetes in patients with PCOS, diabetes can still occur in lean patients with PCOS who have no family history, mainly secondary to insulin resistance.
The Endocrine Society recommends that all individuals with PCOS undergo an oral glucose tolerance test every 3-5 years, with more frequent screening for those who develop symptoms of T2D, significant weight gain, or central adiposity. In guidelines published in 2015 by the American Association of Clinical Endocrinologists, the American College of Endocrinology, and the Androgen Excess and PCOS Society, an annual oral glucose tolerance test is recommended for patients with PCOS and impaired glucose tolerance, whereas those with a family history of T2D or a BMI above 30 should be screened every 1-2 years.
Management of T2D with PCOS is similar to that of T2D without PCOS. Accordingly, metformin and lifestyle changes are the treatments of choice; any antidiabetic agent may be added in patients who do not achieve glycemic targets despite treatment with metformin.
Romesh K. Khardori, MD, PhD, Professor, Department of Internal Medicine, Division of Diabetes, Endocrine, and Metabolic Disorders, Eastern Virginia Medical School; EVMS Medical Group, Norfolk, Virginia
Romesh K. Khardori, MD, PhD, has disclosed no relevant financial relationships
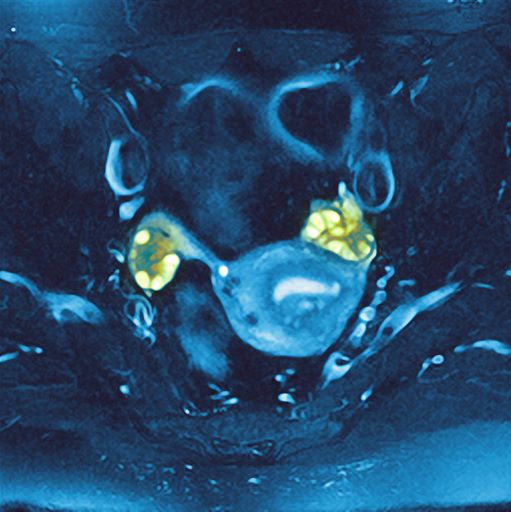
A 36-year-old woman presents with recent unintended weight loss of 12 lb in 2 months. Currently, she weighs 153 lb (BMI 24.7). She complains of increased thirst, increased urination, lack of energy, and fatigue.
Metabolic workup reveals that A1c is 7.1%, fasting blood glucose level is 131 mg/dL, oral glucose tolerance test level is 210 mg/dL, and random blood glucose level is 215 mg/dL, all of which are diagnostic for type 2 diabetes (T2D). She has no family history of diabetes.
A lipid panel shows a low-density lipoprotein cholesterol of 140 mg/dL, high-density lipoprotein cholesterol 38 mg/dL, and triglycerides 210 mg/dL. Blood pressure is 150/95 mm Hg.
The patient had been diagnosed with polycystic ovary syndrome (PCOS) at age 33, during a workup for infertility. At the time of her PCOS diagnosis, she weighed 190 lb (BMI, 30.7). She gave birth to an 8-lb son 14 months ago.

Retro-orbital headache and nausea
On the basis of his presentation, this patient is probably experiencing migraine with visual aura. Migraine is a condition in children and adolescents whose prevalence increases with age: 1%-3% between age 3 and 7 years, 4%-11% between age 7 and 11 years, and 8%-23% by age 15 years. Although migraine without aura is relatively uncommon in the pediatric population, visual aura is a hallmark sign of migraine headache and excludes the other headache types in the differential diagnosis. Basilar migraine is unlikely because the patient has not experienced symptoms that suggest occipital or brainstem area dysfunction post-aura.
The diagnosis of migraine is largely clinical, but the American Academy of Neurology (AAN) guidelines for the acute treatment of pediatric migraine recommend that when assessing children and adolescents with headache, clinicians should diagnose a specific headache type: primary, secondary, or other headache syndrome. Migraine in pediatric patients is often related to triggering factors such as infection, physical or psychological stress, or dietary choices, but on the basis of the patient's history, this headache appears to be primary in nature.
Most pediatric patients can achieve control of their migraines with acute treatments and benefit from nonprescription oral analgesics, including acetaminophen, ibuprofen, and naproxen. Clinicians should prescribe ibuprofen orally (10 mg/kg) as an initial treatment for children and adolescents with migraine. The US Food and Drug Administration has only approved certain triptans for pediatric patients: almotriptan, sumatriptan-naproxen, and zolmitriptan nasal spray for patients aged 12 years or older and rizatriptan for patients aged 6-17 years.
The AAN guidelines for the pharmacologic treatment of pediatric migraine prevention report that for those who experience migraine with aura, taking a triptan during the aura is safe, though it may be more effective when taken at the onset of head pain, as is the case with other acute treatments.
In pediatric patients, avoidance of known headache triggers is generally sufficient for migraine prevention. This includes managing anxiety, depression, attention-deficit/hyperactivity disorder, and other psychiatric comorbidities that can exacerbate headache. Lifestyle management also includes ensuring adequate sleep, exercise, hydration, and stress management.
The guidelines conclude that although the majority of randomized controlled trials exploring the efficacy of preventive medications in the pediatric population fail to demonstrate superiority to placebo, migraine prophylaxis should be considered when headaches occur with high frequency and severity and cause migraine-related disability based on the Pediatric Migraine Disability Assessment (PedMIDAS).
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships
On the basis of his presentation, this patient is probably experiencing migraine with visual aura. Migraine is a condition in children and adolescents whose prevalence increases with age: 1%-3% between age 3 and 7 years, 4%-11% between age 7 and 11 years, and 8%-23% by age 15 years. Although migraine without aura is relatively uncommon in the pediatric population, visual aura is a hallmark sign of migraine headache and excludes the other headache types in the differential diagnosis. Basilar migraine is unlikely because the patient has not experienced symptoms that suggest occipital or brainstem area dysfunction post-aura.
The diagnosis of migraine is largely clinical, but the American Academy of Neurology (AAN) guidelines for the acute treatment of pediatric migraine recommend that when assessing children and adolescents with headache, clinicians should diagnose a specific headache type: primary, secondary, or other headache syndrome. Migraine in pediatric patients is often related to triggering factors such as infection, physical or psychological stress, or dietary choices, but on the basis of the patient's history, this headache appears to be primary in nature.
Most pediatric patients can achieve control of their migraines with acute treatments and benefit from nonprescription oral analgesics, including acetaminophen, ibuprofen, and naproxen. Clinicians should prescribe ibuprofen orally (10 mg/kg) as an initial treatment for children and adolescents with migraine. The US Food and Drug Administration has only approved certain triptans for pediatric patients: almotriptan, sumatriptan-naproxen, and zolmitriptan nasal spray for patients aged 12 years or older and rizatriptan for patients aged 6-17 years.
The AAN guidelines for the pharmacologic treatment of pediatric migraine prevention report that for those who experience migraine with aura, taking a triptan during the aura is safe, though it may be more effective when taken at the onset of head pain, as is the case with other acute treatments.
In pediatric patients, avoidance of known headache triggers is generally sufficient for migraine prevention. This includes managing anxiety, depression, attention-deficit/hyperactivity disorder, and other psychiatric comorbidities that can exacerbate headache. Lifestyle management also includes ensuring adequate sleep, exercise, hydration, and stress management.
The guidelines conclude that although the majority of randomized controlled trials exploring the efficacy of preventive medications in the pediatric population fail to demonstrate superiority to placebo, migraine prophylaxis should be considered when headaches occur with high frequency and severity and cause migraine-related disability based on the Pediatric Migraine Disability Assessment (PedMIDAS).
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships
On the basis of his presentation, this patient is probably experiencing migraine with visual aura. Migraine is a condition in children and adolescents whose prevalence increases with age: 1%-3% between age 3 and 7 years, 4%-11% between age 7 and 11 years, and 8%-23% by age 15 years. Although migraine without aura is relatively uncommon in the pediatric population, visual aura is a hallmark sign of migraine headache and excludes the other headache types in the differential diagnosis. Basilar migraine is unlikely because the patient has not experienced symptoms that suggest occipital or brainstem area dysfunction post-aura.
The diagnosis of migraine is largely clinical, but the American Academy of Neurology (AAN) guidelines for the acute treatment of pediatric migraine recommend that when assessing children and adolescents with headache, clinicians should diagnose a specific headache type: primary, secondary, or other headache syndrome. Migraine in pediatric patients is often related to triggering factors such as infection, physical or psychological stress, or dietary choices, but on the basis of the patient's history, this headache appears to be primary in nature.
Most pediatric patients can achieve control of their migraines with acute treatments and benefit from nonprescription oral analgesics, including acetaminophen, ibuprofen, and naproxen. Clinicians should prescribe ibuprofen orally (10 mg/kg) as an initial treatment for children and adolescents with migraine. The US Food and Drug Administration has only approved certain triptans for pediatric patients: almotriptan, sumatriptan-naproxen, and zolmitriptan nasal spray for patients aged 12 years or older and rizatriptan for patients aged 6-17 years.
The AAN guidelines for the pharmacologic treatment of pediatric migraine prevention report that for those who experience migraine with aura, taking a triptan during the aura is safe, though it may be more effective when taken at the onset of head pain, as is the case with other acute treatments.
In pediatric patients, avoidance of known headache triggers is generally sufficient for migraine prevention. This includes managing anxiety, depression, attention-deficit/hyperactivity disorder, and other psychiatric comorbidities that can exacerbate headache. Lifestyle management also includes ensuring adequate sleep, exercise, hydration, and stress management.
The guidelines conclude that although the majority of randomized controlled trials exploring the efficacy of preventive medications in the pediatric population fail to demonstrate superiority to placebo, migraine prophylaxis should be considered when headaches occur with high frequency and severity and cause migraine-related disability based on the Pediatric Migraine Disability Assessment (PedMIDAS).
Jasmin Harpe, MD, MPH, Headache Fellow, Department of Neurology, Harvard University, John R. Graham Headache Center, Mass General Brigham, Boston, MA
Jasmin Harpe, MD, MPH, has disclosed no relevant financial relationships

A 7-year-old boy presents with a retro-orbital headache, nausea, and photophobia. Height is 4 ft 2 in and weight is 70 lb (BMI 19.7). The patient's mother reports that he described seeing "rainbow shapes" in his line of vision about 20 minutes before the onset of head pain and notes that she herself has a history of headache. The patient is nonfebrile but sweating and drowsy. Physical examination is unrevealing. The patient has no known allergies and is not currently on medication.

Cough and moderate hoarseness
Based on the patient's presentation, history, and imaging results, the likely diagnosis is non–small cell lung cancer (NSCLC) of an adenocarcinoma subtype. NSCLC accounts for about 80% of all lung cancer cases. Adenocarcinoma, in particular, is the most common type of lung cancer in the United States, accounting for about 40% of cases. This subtype is also the most common histology among nonsmokers. Still, individuals aged 55 to 77 years with a smoking history of 30 pack-years or more are considered to be the highest-risk group for lung cancer; those who quit less than 15 years ago — like the patient in the present case — are still considered to be in this risk group. Most cases of lung cancer are diagnosed at a late stage when symptoms have already begun to manifest. However, it should be noted that women are more likely to develop adenocarcinoma, are generally younger when they present with symptoms, and are more likely to present with localized disease. It remains to be proven whether the use of HRT affects the risk for lung cancer in women. Deaths from lung cancer, and in particular NSCLC, were shown to be higher among patients undergoing HRT, though no increase in lung cancer death was reported in women receiving estrogen alone.
In addition to the imaging described in this case, workup for NSCLC should include immunohistochemical (IHC) analyses to identify tumor type and lineage (adenocarcinoma, squamous cell carcinoma, metastatic malignancy, or primary pleural mesothelioma). Separate IHC analyses are then used to guide treatment decisions, identifying whether anaplastic lymphoma kinase inhibitor therapy or programmed death-ligand 1 inhibitor therapy would be appropriate.
Tissue should also be conserved for molecular testing. Management of NSCLC is primarily informed by the presence of targetable mutations. Among adenocarcinoma cases, the most common mutations are in the EGFR and KRAS genes. KRAS mutations, unlike EGFR mutations, are associated with a history of smoking and are considered prognostic biomarkers. Because overlapping targetable alterations are uncommon, patients who are confirmed to be harboring KRAS mutations will likely not benefit from additional molecular testing. Presence of the KRAS mutation suggests a poor response to EGFR tyrosine kinase inhibitors, though it does not appear to impact chemotherapeutic efficacy. Although no targeted therapies are yet available for this population, immune checkpoint inhibitors appear to be beneficial. National Comprehensive Cancer Network guidelines advise that all patients with adenocarcinoma be tested for EGFR mutations and that DNA mutational analysis is the preferred method.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Based on the patient's presentation, history, and imaging results, the likely diagnosis is non–small cell lung cancer (NSCLC) of an adenocarcinoma subtype. NSCLC accounts for about 80% of all lung cancer cases. Adenocarcinoma, in particular, is the most common type of lung cancer in the United States, accounting for about 40% of cases. This subtype is also the most common histology among nonsmokers. Still, individuals aged 55 to 77 years with a smoking history of 30 pack-years or more are considered to be the highest-risk group for lung cancer; those who quit less than 15 years ago — like the patient in the present case — are still considered to be in this risk group. Most cases of lung cancer are diagnosed at a late stage when symptoms have already begun to manifest. However, it should be noted that women are more likely to develop adenocarcinoma, are generally younger when they present with symptoms, and are more likely to present with localized disease. It remains to be proven whether the use of HRT affects the risk for lung cancer in women. Deaths from lung cancer, and in particular NSCLC, were shown to be higher among patients undergoing HRT, though no increase in lung cancer death was reported in women receiving estrogen alone.
In addition to the imaging described in this case, workup for NSCLC should include immunohistochemical (IHC) analyses to identify tumor type and lineage (adenocarcinoma, squamous cell carcinoma, metastatic malignancy, or primary pleural mesothelioma). Separate IHC analyses are then used to guide treatment decisions, identifying whether anaplastic lymphoma kinase inhibitor therapy or programmed death-ligand 1 inhibitor therapy would be appropriate.
Tissue should also be conserved for molecular testing. Management of NSCLC is primarily informed by the presence of targetable mutations. Among adenocarcinoma cases, the most common mutations are in the EGFR and KRAS genes. KRAS mutations, unlike EGFR mutations, are associated with a history of smoking and are considered prognostic biomarkers. Because overlapping targetable alterations are uncommon, patients who are confirmed to be harboring KRAS mutations will likely not benefit from additional molecular testing. Presence of the KRAS mutation suggests a poor response to EGFR tyrosine kinase inhibitors, though it does not appear to impact chemotherapeutic efficacy. Although no targeted therapies are yet available for this population, immune checkpoint inhibitors appear to be beneficial. National Comprehensive Cancer Network guidelines advise that all patients with adenocarcinoma be tested for EGFR mutations and that DNA mutational analysis is the preferred method.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Based on the patient's presentation, history, and imaging results, the likely diagnosis is non–small cell lung cancer (NSCLC) of an adenocarcinoma subtype. NSCLC accounts for about 80% of all lung cancer cases. Adenocarcinoma, in particular, is the most common type of lung cancer in the United States, accounting for about 40% of cases. This subtype is also the most common histology among nonsmokers. Still, individuals aged 55 to 77 years with a smoking history of 30 pack-years or more are considered to be the highest-risk group for lung cancer; those who quit less than 15 years ago — like the patient in the present case — are still considered to be in this risk group. Most cases of lung cancer are diagnosed at a late stage when symptoms have already begun to manifest. However, it should be noted that women are more likely to develop adenocarcinoma, are generally younger when they present with symptoms, and are more likely to present with localized disease. It remains to be proven whether the use of HRT affects the risk for lung cancer in women. Deaths from lung cancer, and in particular NSCLC, were shown to be higher among patients undergoing HRT, though no increase in lung cancer death was reported in women receiving estrogen alone.
In addition to the imaging described in this case, workup for NSCLC should include immunohistochemical (IHC) analyses to identify tumor type and lineage (adenocarcinoma, squamous cell carcinoma, metastatic malignancy, or primary pleural mesothelioma). Separate IHC analyses are then used to guide treatment decisions, identifying whether anaplastic lymphoma kinase inhibitor therapy or programmed death-ligand 1 inhibitor therapy would be appropriate.
Tissue should also be conserved for molecular testing. Management of NSCLC is primarily informed by the presence of targetable mutations. Among adenocarcinoma cases, the most common mutations are in the EGFR and KRAS genes. KRAS mutations, unlike EGFR mutations, are associated with a history of smoking and are considered prognostic biomarkers. Because overlapping targetable alterations are uncommon, patients who are confirmed to be harboring KRAS mutations will likely not benefit from additional molecular testing. Presence of the KRAS mutation suggests a poor response to EGFR tyrosine kinase inhibitors, though it does not appear to impact chemotherapeutic efficacy. Although no targeted therapies are yet available for this population, immune checkpoint inhibitors appear to be beneficial. National Comprehensive Cancer Network guidelines advise that all patients with adenocarcinoma be tested for EGFR mutations and that DNA mutational analysis is the preferred method.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
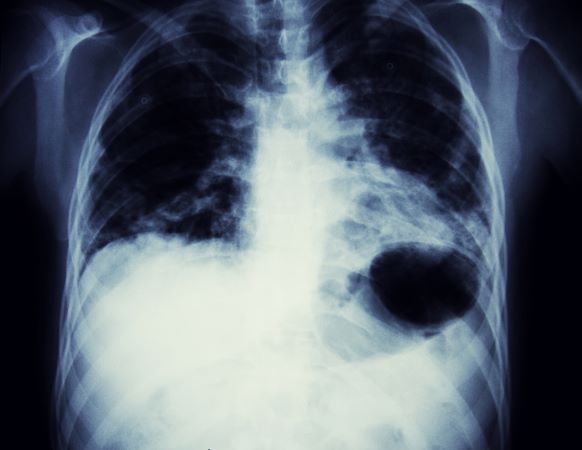
A 56-year-old woman presents with dyspnea, a persistent cough, and moderate hoarseness. She has no significant medical history other than thyroiditis. Her current medications include hormone replacement therapy (HRT). Although the patient reports a 20–pack-year history of smoking tobacco, she notes that she quit 11 years ago and has not been previously screened for lung cancer. A chest radiograph is ordered, which demonstrates a mass in the upper lobe of the right lung.