User login
Unintentional weight loss
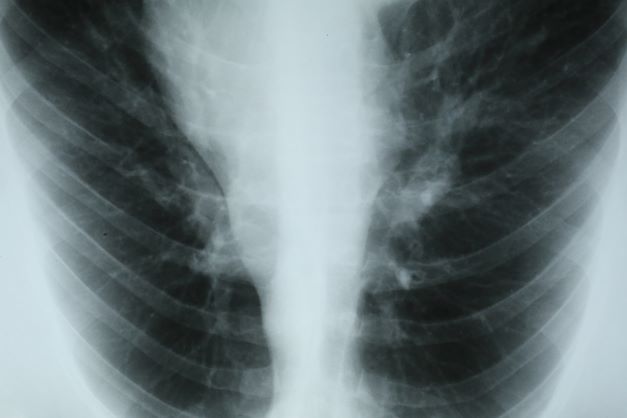
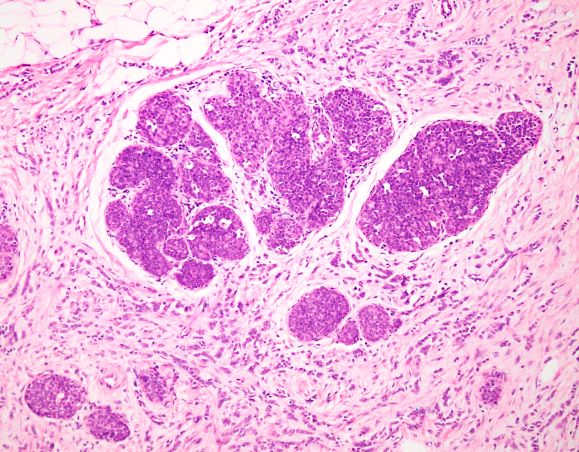
This patient's clinical presentation is consistent with a diagnosis of metastatic invasive lobular carcinoma, with nodal involvement.
Breast cancer is one of the most frequently diagnosed cancers worldwide. In Western countries, 1 in 8 women will be diagnosed with breast cancer at some point in their lives. Various histologic subtypes with specific clinical characteristics exist. Invasive lobular carcinoma (ILC) is the second most common subtype, accounting for an estimated 10%-15% of breast cancers. Over the past two decades, a significant increase has been observed in the incidence of ILC, particularly among postmenopausal women. Improved diagnostic techniques and the use of hormone replacement therapy may account for this increased incidence. White women have the highest incidence of ILC; however, compared with White women and women of other races, Black women experience the worst 5-year overall survival from ILC.
ILC arises in the mammary ducts (lobules) of the breast. Women with ILC are typically slightly older than women with invasive breast cancer of no special type at diagnosis (mean age 63.4 vs 59.5 years, respectively). Risk factors for ILC may include early menarche, use of progesterone-based hormone replacement therapy, late age at first live birth, and alcohol consumption.
In most cases, ILC does not form a discrete palpable mass until it has reached an advanced stage, making it more difficult to detect through physical examination or imaging. Patients often present with a large tumor and with nodal involvement. A slight thickening of the nipple, an exudative scab on the skin, or other changes in the skin, such as flushing or swelling, may be seen in patients presenting with advanced disease. Additionally, ILC tumors are often bilateral and multifocal.
ILC is predominantly a histopathologic diagnosis based on standard hematoxylin and eosin staining. Histologically, ILC is characterized by a proliferation of small cells that lack cohesion. These cells are often dispersed individually through a fibrous connective tissue; alternatively, they may be organized in single-file linear cords invading the stroma. A concentric pattern around normal ducts is often seen in the infiltrating cords. There is usually little host reaction of the background architecture. Round or notched ovoid nuclei are seen in the neoplastic cells, along with a thin rim of cytoplasm. Occasionally, an intracytoplasmic lumen is present and may harbor a central mucoid inclusion. Very few or no mitoses are seen.
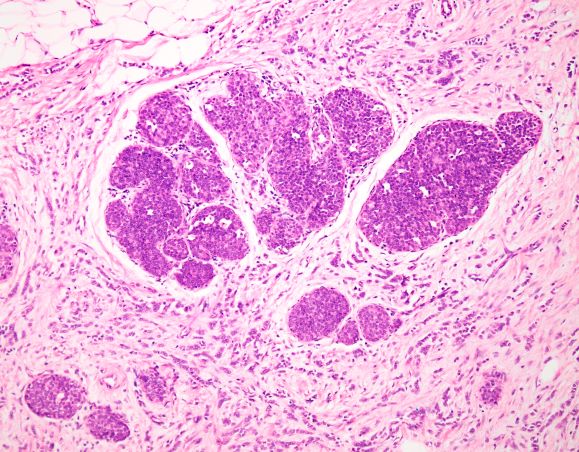
Several variants of ILC exist, all of which lack cell-to-cell cohesion. These include:
• Solid type
• Pleomorphic lobular carcinoma
• Tubulo-lobular variant
• Alveolar variant
• Mixed type
Complete loss of E-cadherin expression occurs in most ILCs, which can help to differentiate it from invasive ductal cancers or ductal carcinomas in situ. Diffuse cortical thickening without hilar mass effect is often seen in nodal metastases associated with ILC.
Most classic ILCs are estrogen receptor– and progesterone receptor–positive. Conversely, HER2 overexpression and amplification rarely occurs in ILC.
Late relapses more than 10 years after remission may occur. In addition to frequent bone and liver metastasis, ILC is associated with metastatic spread to unusual sites, including the peritoneum, gastrointestinal tract, urinary tract, leptomeninges, skin, orbit, and ovaries.
Mastectomy is often indicated in ILC. In the neoadjuvant setting, ILC is associated with low pathologic complete response rates. Endocrine therapy in the neoadjuvant setting is an emerging approach for some patients with ILC. According to 2022 National Comprehensive Cancer Network guidelines, adjuvant chemotherapy followed by endocrine therapy or endocrine therapy alone should be considered for pre- and postmenopausal patients with ILC.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation is consistent with a diagnosis of metastatic invasive lobular carcinoma, with nodal involvement.
Breast cancer is one of the most frequently diagnosed cancers worldwide. In Western countries, 1 in 8 women will be diagnosed with breast cancer at some point in their lives. Various histologic subtypes with specific clinical characteristics exist. Invasive lobular carcinoma (ILC) is the second most common subtype, accounting for an estimated 10%-15% of breast cancers. Over the past two decades, a significant increase has been observed in the incidence of ILC, particularly among postmenopausal women. Improved diagnostic techniques and the use of hormone replacement therapy may account for this increased incidence. White women have the highest incidence of ILC; however, compared with White women and women of other races, Black women experience the worst 5-year overall survival from ILC.
ILC arises in the mammary ducts (lobules) of the breast. Women with ILC are typically slightly older than women with invasive breast cancer of no special type at diagnosis (mean age 63.4 vs 59.5 years, respectively). Risk factors for ILC may include early menarche, use of progesterone-based hormone replacement therapy, late age at first live birth, and alcohol consumption.
In most cases, ILC does not form a discrete palpable mass until it has reached an advanced stage, making it more difficult to detect through physical examination or imaging. Patients often present with a large tumor and with nodal involvement. A slight thickening of the nipple, an exudative scab on the skin, or other changes in the skin, such as flushing or swelling, may be seen in patients presenting with advanced disease. Additionally, ILC tumors are often bilateral and multifocal.
ILC is predominantly a histopathologic diagnosis based on standard hematoxylin and eosin staining. Histologically, ILC is characterized by a proliferation of small cells that lack cohesion. These cells are often dispersed individually through a fibrous connective tissue; alternatively, they may be organized in single-file linear cords invading the stroma. A concentric pattern around normal ducts is often seen in the infiltrating cords. There is usually little host reaction of the background architecture. Round or notched ovoid nuclei are seen in the neoplastic cells, along with a thin rim of cytoplasm. Occasionally, an intracytoplasmic lumen is present and may harbor a central mucoid inclusion. Very few or no mitoses are seen.
Several variants of ILC exist, all of which lack cell-to-cell cohesion. These include:
• Solid type
• Pleomorphic lobular carcinoma
• Tubulo-lobular variant
• Alveolar variant
• Mixed type
Complete loss of E-cadherin expression occurs in most ILCs, which can help to differentiate it from invasive ductal cancers or ductal carcinomas in situ. Diffuse cortical thickening without hilar mass effect is often seen in nodal metastases associated with ILC.
Most classic ILCs are estrogen receptor– and progesterone receptor–positive. Conversely, HER2 overexpression and amplification rarely occurs in ILC.
Late relapses more than 10 years after remission may occur. In addition to frequent bone and liver metastasis, ILC is associated with metastatic spread to unusual sites, including the peritoneum, gastrointestinal tract, urinary tract, leptomeninges, skin, orbit, and ovaries.
Mastectomy is often indicated in ILC. In the neoadjuvant setting, ILC is associated with low pathologic complete response rates. Endocrine therapy in the neoadjuvant setting is an emerging approach for some patients with ILC. According to 2022 National Comprehensive Cancer Network guidelines, adjuvant chemotherapy followed by endocrine therapy or endocrine therapy alone should be considered for pre- and postmenopausal patients with ILC.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation is consistent with a diagnosis of metastatic invasive lobular carcinoma, with nodal involvement.
Breast cancer is one of the most frequently diagnosed cancers worldwide. In Western countries, 1 in 8 women will be diagnosed with breast cancer at some point in their lives. Various histologic subtypes with specific clinical characteristics exist. Invasive lobular carcinoma (ILC) is the second most common subtype, accounting for an estimated 10%-15% of breast cancers. Over the past two decades, a significant increase has been observed in the incidence of ILC, particularly among postmenopausal women. Improved diagnostic techniques and the use of hormone replacement therapy may account for this increased incidence. White women have the highest incidence of ILC; however, compared with White women and women of other races, Black women experience the worst 5-year overall survival from ILC.
ILC arises in the mammary ducts (lobules) of the breast. Women with ILC are typically slightly older than women with invasive breast cancer of no special type at diagnosis (mean age 63.4 vs 59.5 years, respectively). Risk factors for ILC may include early menarche, use of progesterone-based hormone replacement therapy, late age at first live birth, and alcohol consumption.
In most cases, ILC does not form a discrete palpable mass until it has reached an advanced stage, making it more difficult to detect through physical examination or imaging. Patients often present with a large tumor and with nodal involvement. A slight thickening of the nipple, an exudative scab on the skin, or other changes in the skin, such as flushing or swelling, may be seen in patients presenting with advanced disease. Additionally, ILC tumors are often bilateral and multifocal.
ILC is predominantly a histopathologic diagnosis based on standard hematoxylin and eosin staining. Histologically, ILC is characterized by a proliferation of small cells that lack cohesion. These cells are often dispersed individually through a fibrous connective tissue; alternatively, they may be organized in single-file linear cords invading the stroma. A concentric pattern around normal ducts is often seen in the infiltrating cords. There is usually little host reaction of the background architecture. Round or notched ovoid nuclei are seen in the neoplastic cells, along with a thin rim of cytoplasm. Occasionally, an intracytoplasmic lumen is present and may harbor a central mucoid inclusion. Very few or no mitoses are seen.
Several variants of ILC exist, all of which lack cell-to-cell cohesion. These include:
• Solid type
• Pleomorphic lobular carcinoma
• Tubulo-lobular variant
• Alveolar variant
• Mixed type
Complete loss of E-cadherin expression occurs in most ILCs, which can help to differentiate it from invasive ductal cancers or ductal carcinomas in situ. Diffuse cortical thickening without hilar mass effect is often seen in nodal metastases associated with ILC.
Most classic ILCs are estrogen receptor– and progesterone receptor–positive. Conversely, HER2 overexpression and amplification rarely occurs in ILC.
Late relapses more than 10 years after remission may occur. In addition to frequent bone and liver metastasis, ILC is associated with metastatic spread to unusual sites, including the peritoneum, gastrointestinal tract, urinary tract, leptomeninges, skin, orbit, and ovaries.
Mastectomy is often indicated in ILC. In the neoadjuvant setting, ILC is associated with low pathologic complete response rates. Endocrine therapy in the neoadjuvant setting is an emerging approach for some patients with ILC. According to 2022 National Comprehensive Cancer Network guidelines, adjuvant chemotherapy followed by endocrine therapy or endocrine therapy alone should be considered for pre- and postmenopausal patients with ILC.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
A 47-year-old woman presented for her annual gynecologic examination. Her current height and weight were 5 ft 4 in and 133 lb. This reflected a 9-lb weight loss since the previous visit. At completion of the height and weight intake by a nurse, the patient reported being surprised by this unintentional weight loss. Her previous medical history was unremarkable except for an advanced maternal age pregnancy 5 years earlier and dental implant surgery approximately 1 month earlier. The patient believed that her weight loss was related to her diminished appetite and transient difficulty chewing following her dental surgery. Laboratory findings were all within normal ranges except for a hemoglobin level of 9.4 g/dL. Physical examination revealed a palpable mass in the right upper outer quadrant of the right breast with slight thickening of the nipple and a right axillary mass. The patient's last bilateral screening mammogram 3 months earlier did not reveal any suspicious masses or lesions.
An ultrasound-guided biopsy of the right breast and axillary lymph node was performed. Histopathologic findings included small tumor cells without cohesion arranged in single files, loss of the long arm of chromosome 16, and a complete loss of E-cadherin expression on immunohistochemistry. Additionally, the tumor was estrogen receptor–positive/progesterone receptor–positive and human epidermal growth factor receptor 2–negative (ER+/PR+/HER2-).
Mild shortness of breath
This patient's clinical presentation of weight gain and associated symptoms are most closely related to a diagnosis of obesity. In addition, her laboratory findings are consistent with common obesity complications, including prediabetes and dyslipidemia, and her blood pressure is borderline high.
Obesity is a chronic, multifactorial disease with a complex pathogenesis comprising of genetic, biological, psychosocial, socioeconomic, and environmental factors. It is a heterogeneous disease characterized by a dysfunction of the normal pathways and mechanisms that are involved in body fat regulation (often referred to as weight regulation), which may lead to variable presentation and complications. According to the US Centers for Disease Control and Prevention, the highest age-adjusted prevalence of obesity is seen in non-Hispanic Black adults (49.9%), followed by Hispanic adults (45.6%), non-Hispanic White adults (41.4%), and non-Hispanic Asian adults (16.1%).
Epidemiologic studies have defined obesity as a BMI > 30, which is then subclassified into class 1 (BMI of 30-34.9), class 2 (BMI of 35-39.9), or class 3 (BMI ≥ 40) obesity. Though BMI is widely used to evaluate and classify obesity, it mainly represents general adiposity and can be confounded by excessive muscle mass or frailty. Guidelines from the American Diabetes Association state that in addition to weight and BMI, clinicians should consider weight distribution (including predisposition for central/visceral adipose deposition) and weight gain pattern and trajectory because these can help guide risk stratification and treatment options.
Increasingly, evidence supports visceral adiposity, or abdominal obesity, as a marker of cardiovascular risk. Abdominal obesity has been shown to be a strong independent predictor of mortality. On its own, BMI is an insufficient biomarker of abdominal obesity. Not all individuals with obesity have a central distribution of their weight; some individuals may have central obesity without meeting the criteria for the BMI definition of obesity. This can lead to misclassification and underdiagnosis of health risks in clinical practice. Consequently, numerous organizations and expert panels have recommended that waist circumference be measured along with BMI, specifically when the BMI < 35. Measurement of both BMI and waist circumference provides valuable opportunities to counsel patients regarding their risk for cardiovascular disease and other complications of obesity. Waist-to-hip ratio has also been shown to be a stronger predictor for mortality compared with BMI; however, it is rarely measured in clinical practice.
Although rarely performed outside of research settings, measurement of epicardial and pericardial fat via CT is also emerging as a potentially useful approach for informing predictive and precision medicine strategies. Recently, the Jackson Heart Study showed pericardial and visceral fat volumes were associated with incident heart failure, particularly heart failure with preserved ejection fraction, and all-cause mortality among Black participants even after adjusting for age, sex, education, and smoking status. Another recent study showed an increased risk of heart failure, particularly heart failure with preserved ejection fraction, among men and women with high pericardial fat volume. The Multi-Ethnic Study of Atherosclerosis showed that pericardial fat was associated with a higher risk of all-cause cardiovascular disease, hard atherosclerotic cardiovascular disease, and heart failure. Epicardial fat is directly correlated with BMI, visceral adiposity, and waist circumference.
Best practices for the management of obesity begin with recognizing and treating it as a complex chronic disease rather than the result of an individual's lifestyle choices. According to a 2020 joint international consensus statement for ending the stigma of obesity, the assumption that choosing to eat less and/or exercise more can entirely prevent or reverse obesity is contradicted by a definitive body of biological and clinical evidence that shows obesity results primarily from a complex combination of genetic, epigenetic, and environmental factors. When diagnosing patients with obesity, it may be helpful for clinicians to acknowledge that the term obesity is often perceived as an undesirable term because it has been associated with stigma but that it is in fact a clinical diagnosis, not a judgement. Many patients prefer the neutral term unhealthy weight over obesity.
As with other chronic diseases, individualized treatment and long-term support along with shared decision-making are essential for optimizing outcomes. Key components of obesity management include diet, exercise, and behavioral modification. In addition, an increasing array of pharmacologic therapies are also showing unprecedented efficacy for weight management, including several drugs that are also approved for the management of type 2 diabetes. In particular, the glucagonlike peptide 1 (GLP-1) agonists, semaglutide and liraglutide, and the novel glucose-dependent insulinotropic polypeptide (GIP)–GLP-1 receptor agonist, tirzepatide have been associated with significant weight loss. Semaglutide and liraglutide have been US Food and Drug Administration (FDA)–approved for chronic weight management and tirzepatide was granted fast track designation for the treatment of obesity by the FDA in October 2022. These drugs may also help to prevent the progression of prediabetes to diabetes. For individuals with severe obesity, metabolic and bariatric surgery is an effective treatment option that is associated with clinically significant and relatively sustained weight reduction in addition to significant amelioration of related complications.
W. Scott Butsch, MD, MSc, Director of Obesity Medicine, Bariatric and Metabolic Institute, Cleveland Clinic, Cleveland, Ohio.
Dr. Butsch has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Novo Nordisk, Inc.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation of weight gain and associated symptoms are most closely related to a diagnosis of obesity. In addition, her laboratory findings are consistent with common obesity complications, including prediabetes and dyslipidemia, and her blood pressure is borderline high.
Obesity is a chronic, multifactorial disease with a complex pathogenesis comprising of genetic, biological, psychosocial, socioeconomic, and environmental factors. It is a heterogeneous disease characterized by a dysfunction of the normal pathways and mechanisms that are involved in body fat regulation (often referred to as weight regulation), which may lead to variable presentation and complications. According to the US Centers for Disease Control and Prevention, the highest age-adjusted prevalence of obesity is seen in non-Hispanic Black adults (49.9%), followed by Hispanic adults (45.6%), non-Hispanic White adults (41.4%), and non-Hispanic Asian adults (16.1%).
Epidemiologic studies have defined obesity as a BMI > 30, which is then subclassified into class 1 (BMI of 30-34.9), class 2 (BMI of 35-39.9), or class 3 (BMI ≥ 40) obesity. Though BMI is widely used to evaluate and classify obesity, it mainly represents general adiposity and can be confounded by excessive muscle mass or frailty. Guidelines from the American Diabetes Association state that in addition to weight and BMI, clinicians should consider weight distribution (including predisposition for central/visceral adipose deposition) and weight gain pattern and trajectory because these can help guide risk stratification and treatment options.
Increasingly, evidence supports visceral adiposity, or abdominal obesity, as a marker of cardiovascular risk. Abdominal obesity has been shown to be a strong independent predictor of mortality. On its own, BMI is an insufficient biomarker of abdominal obesity. Not all individuals with obesity have a central distribution of their weight; some individuals may have central obesity without meeting the criteria for the BMI definition of obesity. This can lead to misclassification and underdiagnosis of health risks in clinical practice. Consequently, numerous organizations and expert panels have recommended that waist circumference be measured along with BMI, specifically when the BMI < 35. Measurement of both BMI and waist circumference provides valuable opportunities to counsel patients regarding their risk for cardiovascular disease and other complications of obesity. Waist-to-hip ratio has also been shown to be a stronger predictor for mortality compared with BMI; however, it is rarely measured in clinical practice.
Although rarely performed outside of research settings, measurement of epicardial and pericardial fat via CT is also emerging as a potentially useful approach for informing predictive and precision medicine strategies. Recently, the Jackson Heart Study showed pericardial and visceral fat volumes were associated with incident heart failure, particularly heart failure with preserved ejection fraction, and all-cause mortality among Black participants even after adjusting for age, sex, education, and smoking status. Another recent study showed an increased risk of heart failure, particularly heart failure with preserved ejection fraction, among men and women with high pericardial fat volume. The Multi-Ethnic Study of Atherosclerosis showed that pericardial fat was associated with a higher risk of all-cause cardiovascular disease, hard atherosclerotic cardiovascular disease, and heart failure. Epicardial fat is directly correlated with BMI, visceral adiposity, and waist circumference.
Best practices for the management of obesity begin with recognizing and treating it as a complex chronic disease rather than the result of an individual's lifestyle choices. According to a 2020 joint international consensus statement for ending the stigma of obesity, the assumption that choosing to eat less and/or exercise more can entirely prevent or reverse obesity is contradicted by a definitive body of biological and clinical evidence that shows obesity results primarily from a complex combination of genetic, epigenetic, and environmental factors. When diagnosing patients with obesity, it may be helpful for clinicians to acknowledge that the term obesity is often perceived as an undesirable term because it has been associated with stigma but that it is in fact a clinical diagnosis, not a judgement. Many patients prefer the neutral term unhealthy weight over obesity.
As with other chronic diseases, individualized treatment and long-term support along with shared decision-making are essential for optimizing outcomes. Key components of obesity management include diet, exercise, and behavioral modification. In addition, an increasing array of pharmacologic therapies are also showing unprecedented efficacy for weight management, including several drugs that are also approved for the management of type 2 diabetes. In particular, the glucagonlike peptide 1 (GLP-1) agonists, semaglutide and liraglutide, and the novel glucose-dependent insulinotropic polypeptide (GIP)–GLP-1 receptor agonist, tirzepatide have been associated with significant weight loss. Semaglutide and liraglutide have been US Food and Drug Administration (FDA)–approved for chronic weight management and tirzepatide was granted fast track designation for the treatment of obesity by the FDA in October 2022. These drugs may also help to prevent the progression of prediabetes to diabetes. For individuals with severe obesity, metabolic and bariatric surgery is an effective treatment option that is associated with clinically significant and relatively sustained weight reduction in addition to significant amelioration of related complications.
W. Scott Butsch, MD, MSc, Director of Obesity Medicine, Bariatric and Metabolic Institute, Cleveland Clinic, Cleveland, Ohio.
Dr. Butsch has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Novo Nordisk, Inc.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation of weight gain and associated symptoms are most closely related to a diagnosis of obesity. In addition, her laboratory findings are consistent with common obesity complications, including prediabetes and dyslipidemia, and her blood pressure is borderline high.
Obesity is a chronic, multifactorial disease with a complex pathogenesis comprising of genetic, biological, psychosocial, socioeconomic, and environmental factors. It is a heterogeneous disease characterized by a dysfunction of the normal pathways and mechanisms that are involved in body fat regulation (often referred to as weight regulation), which may lead to variable presentation and complications. According to the US Centers for Disease Control and Prevention, the highest age-adjusted prevalence of obesity is seen in non-Hispanic Black adults (49.9%), followed by Hispanic adults (45.6%), non-Hispanic White adults (41.4%), and non-Hispanic Asian adults (16.1%).
Epidemiologic studies have defined obesity as a BMI > 30, which is then subclassified into class 1 (BMI of 30-34.9), class 2 (BMI of 35-39.9), or class 3 (BMI ≥ 40) obesity. Though BMI is widely used to evaluate and classify obesity, it mainly represents general adiposity and can be confounded by excessive muscle mass or frailty. Guidelines from the American Diabetes Association state that in addition to weight and BMI, clinicians should consider weight distribution (including predisposition for central/visceral adipose deposition) and weight gain pattern and trajectory because these can help guide risk stratification and treatment options.
Increasingly, evidence supports visceral adiposity, or abdominal obesity, as a marker of cardiovascular risk. Abdominal obesity has been shown to be a strong independent predictor of mortality. On its own, BMI is an insufficient biomarker of abdominal obesity. Not all individuals with obesity have a central distribution of their weight; some individuals may have central obesity without meeting the criteria for the BMI definition of obesity. This can lead to misclassification and underdiagnosis of health risks in clinical practice. Consequently, numerous organizations and expert panels have recommended that waist circumference be measured along with BMI, specifically when the BMI < 35. Measurement of both BMI and waist circumference provides valuable opportunities to counsel patients regarding their risk for cardiovascular disease and other complications of obesity. Waist-to-hip ratio has also been shown to be a stronger predictor for mortality compared with BMI; however, it is rarely measured in clinical practice.
Although rarely performed outside of research settings, measurement of epicardial and pericardial fat via CT is also emerging as a potentially useful approach for informing predictive and precision medicine strategies. Recently, the Jackson Heart Study showed pericardial and visceral fat volumes were associated with incident heart failure, particularly heart failure with preserved ejection fraction, and all-cause mortality among Black participants even after adjusting for age, sex, education, and smoking status. Another recent study showed an increased risk of heart failure, particularly heart failure with preserved ejection fraction, among men and women with high pericardial fat volume. The Multi-Ethnic Study of Atherosclerosis showed that pericardial fat was associated with a higher risk of all-cause cardiovascular disease, hard atherosclerotic cardiovascular disease, and heart failure. Epicardial fat is directly correlated with BMI, visceral adiposity, and waist circumference.
Best practices for the management of obesity begin with recognizing and treating it as a complex chronic disease rather than the result of an individual's lifestyle choices. According to a 2020 joint international consensus statement for ending the stigma of obesity, the assumption that choosing to eat less and/or exercise more can entirely prevent or reverse obesity is contradicted by a definitive body of biological and clinical evidence that shows obesity results primarily from a complex combination of genetic, epigenetic, and environmental factors. When diagnosing patients with obesity, it may be helpful for clinicians to acknowledge that the term obesity is often perceived as an undesirable term because it has been associated with stigma but that it is in fact a clinical diagnosis, not a judgement. Many patients prefer the neutral term unhealthy weight over obesity.
As with other chronic diseases, individualized treatment and long-term support along with shared decision-making are essential for optimizing outcomes. Key components of obesity management include diet, exercise, and behavioral modification. In addition, an increasing array of pharmacologic therapies are also showing unprecedented efficacy for weight management, including several drugs that are also approved for the management of type 2 diabetes. In particular, the glucagonlike peptide 1 (GLP-1) agonists, semaglutide and liraglutide, and the novel glucose-dependent insulinotropic polypeptide (GIP)–GLP-1 receptor agonist, tirzepatide have been associated with significant weight loss. Semaglutide and liraglutide have been US Food and Drug Administration (FDA)–approved for chronic weight management and tirzepatide was granted fast track designation for the treatment of obesity by the FDA in October 2022. These drugs may also help to prevent the progression of prediabetes to diabetes. For individuals with severe obesity, metabolic and bariatric surgery is an effective treatment option that is associated with clinically significant and relatively sustained weight reduction in addition to significant amelioration of related complications.
W. Scott Butsch, MD, MSc, Director of Obesity Medicine, Bariatric and Metabolic Institute, Cleveland Clinic, Cleveland, Ohio.
Dr. Butsch has disclosed the following relevant financial relationships:
Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Novo Nordisk, Inc.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
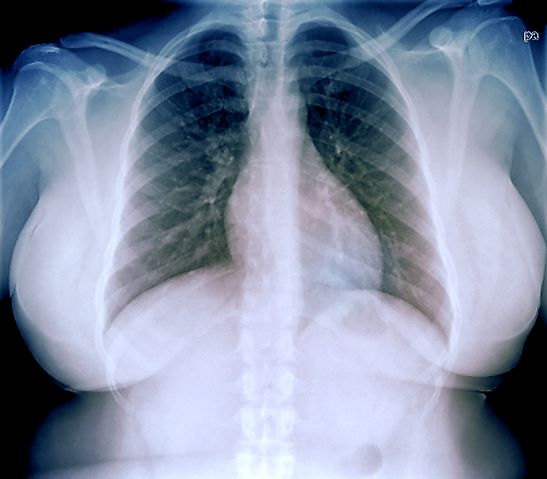
A 33-year-old African American woman presents for an initial consultation. The patient states that it has been several years since she received regular medical care because she did not have health insurance. She recently started a new job as an IT professional that has healthcare benefits. She does not currently take any medications. She reports mild shortness of breath upon exertion, which has worsened in the last year. She denies dizziness, chest pain, wheezing, cough, fever, or other associated symptoms. There is no history of any cardiac or pulmonary diseases as a child. The patient does not smoke or engage in recreational drug use. She is conscious of her diet and avoids red meat as well as sugary and processed foods. Although she was active in the past, she notes that she has been less intentional with her physical activity and has been living a more sedentary lifestyle recently. She has gained more than 40 lb over the past 3 years.
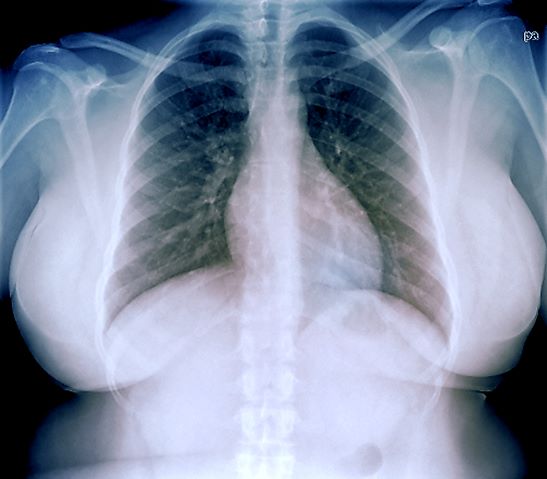
The patient is 5 ft 8 in, her weight is 266 lb (BMI 40.4), and her blood pressure is 140/90 mm Hg. Her pulse oximeter is 97%; however, this result should be interpreted with caution and in consideration of the patient's other signs and symptoms because numerous studies have shown inaccuracies in pulse oximeter readings among people with darker skin. Her physical exam is unremarkable except for a waist circumference of 49 in; breathing sounds are normal and no dermatologic abnormalities are noted.
An ECG is performed and is normal. A chest radiograph shows normal heart and blood vessel structures and airways of the lungs. Pertinent laboratory findings include A1c of 6.4%, HDL cholesterol of 37 mg/dL, LDL cholesterol of 185 mg/dL, serum creatinine of 1.1 mg/dL; AST of 27 U/L; ALT of 35 IU/L; and TSH of 4.2 mIU/L.
Abdominal pain and constipation
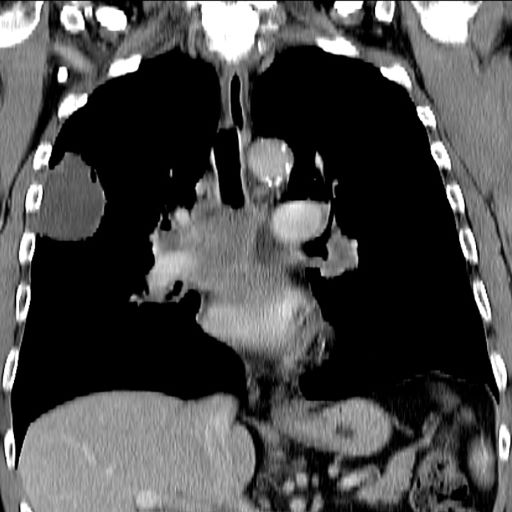
This patient's clinical presentation and endoscopy findings are consistent with a diagnosis of recurrent MCL presenting as a colonic mass.
MCL is an aggressive type of non-Hodgkin lymphoma that accounts for approximately 5%-7% of all lymphomas. Nearly 80% of patients have extranodal involvement at initial presentation, occurring in sites such as the bone marrow, spleen, Waldeyer ring, and the gastrointestinal (GI) tract. Secondary GI involvement in MCL (involving nodal and/or other extranodal tissue) is common and may be detected at diagnosis and/or relapse. In several retrospective studies, the prevalence of secondary GI involvement in MCL ranged from 15% to 30%. However, in later studies, routine endoscopies in patients with untreated MCL showed GI involvement in up to 90% of patients, despite most patients not reporting GI symptoms.
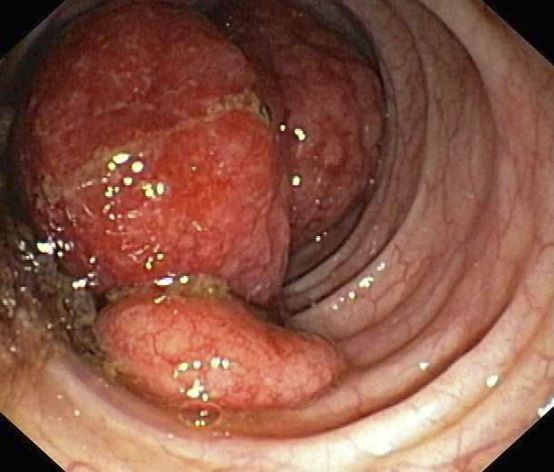
The colon is the most commonly involved GI site; however, both the upper and lower GI tract from the stomach to the colon can be involved. Lymphomatous polyposis is the most common endoscopic presentation of MCL, but polyp, mass, or even normal-appearing mucosa may also be seen.
New and emerging treatment options are helping to improve survival in patients with relapsed/refractory MCL. According to National Comprehensive Cancer Network guidelines, the preferred second-line and subsequent regimens are:
• Bruton tyrosine kinase (BTK) inhibitors:
o Acalabrutinib
o Ibrutinib ± rituximab
o Zanubrutinib
• Lenalidomide + rituximab (if BTK inhibitor is contraindicated)
Other regimens that may be useful in certain circumstances are:
• Bendamustine + rituximab (if not previously given)
• Bendamustine + rituximab + cytarabine (RBAC500) (if not previously given)
• Bortezomib ± rituximab
• RDHA (rituximab, dexamethasone, cytarabine) + platinum (carboplatin, cisplatin, or oxaliplatin) (if not previously given)
• GemOx (gemcitabine, oxaliplatin) + rituximab
• Ibrutinib, lenalidomide, rituximab (category 2B)
• Ibrutinib + venetoclax
• Venetoclax, lenalidomide, rituximab (category 2B)
• Venetoclax ± rituximab
Brexucabtagene autoleucel is suggested as third-line therapy, after chemoimmunotherapy and treatment with a BTK inhibitor.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and endoscopy findings are consistent with a diagnosis of recurrent MCL presenting as a colonic mass.
MCL is an aggressive type of non-Hodgkin lymphoma that accounts for approximately 5%-7% of all lymphomas. Nearly 80% of patients have extranodal involvement at initial presentation, occurring in sites such as the bone marrow, spleen, Waldeyer ring, and the gastrointestinal (GI) tract. Secondary GI involvement in MCL (involving nodal and/or other extranodal tissue) is common and may be detected at diagnosis and/or relapse. In several retrospective studies, the prevalence of secondary GI involvement in MCL ranged from 15% to 30%. However, in later studies, routine endoscopies in patients with untreated MCL showed GI involvement in up to 90% of patients, despite most patients not reporting GI symptoms.
The colon is the most commonly involved GI site; however, both the upper and lower GI tract from the stomach to the colon can be involved. Lymphomatous polyposis is the most common endoscopic presentation of MCL, but polyp, mass, or even normal-appearing mucosa may also be seen.
New and emerging treatment options are helping to improve survival in patients with relapsed/refractory MCL. According to National Comprehensive Cancer Network guidelines, the preferred second-line and subsequent regimens are:
• Bruton tyrosine kinase (BTK) inhibitors:
o Acalabrutinib
o Ibrutinib ± rituximab
o Zanubrutinib
• Lenalidomide + rituximab (if BTK inhibitor is contraindicated)
Other regimens that may be useful in certain circumstances are:
• Bendamustine + rituximab (if not previously given)
• Bendamustine + rituximab + cytarabine (RBAC500) (if not previously given)
• Bortezomib ± rituximab
• RDHA (rituximab, dexamethasone, cytarabine) + platinum (carboplatin, cisplatin, or oxaliplatin) (if not previously given)
• GemOx (gemcitabine, oxaliplatin) + rituximab
• Ibrutinib, lenalidomide, rituximab (category 2B)
• Ibrutinib + venetoclax
• Venetoclax, lenalidomide, rituximab (category 2B)
• Venetoclax ± rituximab
Brexucabtagene autoleucel is suggested as third-line therapy, after chemoimmunotherapy and treatment with a BTK inhibitor.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and endoscopy findings are consistent with a diagnosis of recurrent MCL presenting as a colonic mass.
MCL is an aggressive type of non-Hodgkin lymphoma that accounts for approximately 5%-7% of all lymphomas. Nearly 80% of patients have extranodal involvement at initial presentation, occurring in sites such as the bone marrow, spleen, Waldeyer ring, and the gastrointestinal (GI) tract. Secondary GI involvement in MCL (involving nodal and/or other extranodal tissue) is common and may be detected at diagnosis and/or relapse. In several retrospective studies, the prevalence of secondary GI involvement in MCL ranged from 15% to 30%. However, in later studies, routine endoscopies in patients with untreated MCL showed GI involvement in up to 90% of patients, despite most patients not reporting GI symptoms.
The colon is the most commonly involved GI site; however, both the upper and lower GI tract from the stomach to the colon can be involved. Lymphomatous polyposis is the most common endoscopic presentation of MCL, but polyp, mass, or even normal-appearing mucosa may also be seen.
New and emerging treatment options are helping to improve survival in patients with relapsed/refractory MCL. According to National Comprehensive Cancer Network guidelines, the preferred second-line and subsequent regimens are:
• Bruton tyrosine kinase (BTK) inhibitors:
o Acalabrutinib
o Ibrutinib ± rituximab
o Zanubrutinib
• Lenalidomide + rituximab (if BTK inhibitor is contraindicated)
Other regimens that may be useful in certain circumstances are:
• Bendamustine + rituximab (if not previously given)
• Bendamustine + rituximab + cytarabine (RBAC500) (if not previously given)
• Bortezomib ± rituximab
• RDHA (rituximab, dexamethasone, cytarabine) + platinum (carboplatin, cisplatin, or oxaliplatin) (if not previously given)
• GemOx (gemcitabine, oxaliplatin) + rituximab
• Ibrutinib, lenalidomide, rituximab (category 2B)
• Ibrutinib + venetoclax
• Venetoclax, lenalidomide, rituximab (category 2B)
• Venetoclax ± rituximab
Brexucabtagene autoleucel is suggested as third-line therapy, after chemoimmunotherapy and treatment with a BTK inhibitor.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
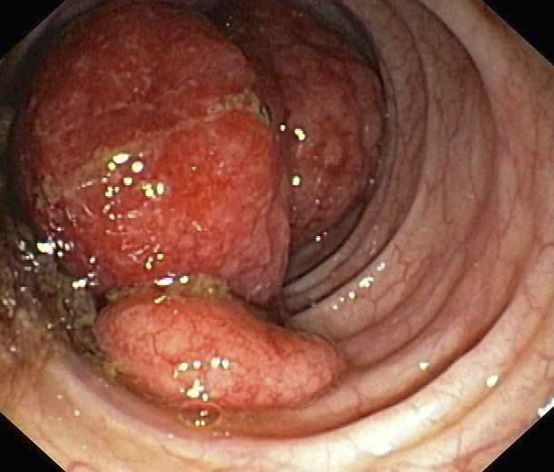
A 55-year-old White woman presents with complaints of left-sided abdominal pain and constipation of 10-day duration. The patient's prior medical history is notable for mantle cell lymphoma (MCL) treated 2 years earlier with RDHA (rituximab, dexamethasone, cytarabine) + platinum (carboplatin, cisplatin, or oxaliplatin) followed by autologous stem cell transplantation. No lymphadenopathy is noted on physical examination. Abdominal examination reveals abdominal distension, normal bowel sounds, and left lower quadrant tenderness to palpation without guarding, rigidity, or hepatosplenomegaly. Laboratory test results including CBC are within normal range. Endoscopy reveals a growth in the colon, as shown in the image.
Increasing fatigue and dry cough
This patient's clinical presentation is consistent with a diagnosis of superior vena cava syndrome (SVCS), secondary to SCLC.
SCLC is an aggressive, poorly differentiated, high-grade neuroendocrine carcinoma that accounts for approximately 13%-15% of all new lung cancer cases in the United States. SCLC has a propensity for early dissemination; as such, 80%-85% of patients are diagnosed with extensive disease (ES-SCLC). This is common in heavy smokers. Most SCLC tumors are found in hilar or perihilar areas; <5% present in peripheral locations. In many cases, invasion into the peribronchial tissue and lymph node can be clearly identified, with a typical circumferential spread along the submucosa of the bronchi.
Up to 10% of patients with SCLC develop SVCS, which comprises an array of signs and symptoms that result from the obstruction of blood flow through the thin-walled superior vena cava. Clinical symptoms may include cough, dyspnea, and orthopnea; facial edema and plethora, upper extremity swelling, and venous distension of the chest wall and neck are the most commonly encountered signs. Most cases of SVCS occur in patients with mediastinal tumors, although noncancerous causes (eg, thrombosis and fibrosing mediastinitis) can also give rise to it. The diagnosis of SVCS is usually made clinically and then confirmed with imaging (chest radiography, contrast-enhanced CT, duplex ultrasound, conventional venography, and/or magnetic resonance venography).
Though it was traditionally considered a virtual emergency, patients seldom experience life-threatening complications from SVCS. The goals of treatment are to alleviate the symptoms of SVC obstruction and treat the underlying disease process. Treatment approaches include radiation therapy, chemotherapy, open surgery, and endovenous recanalization; however, patients with clinical SVCS often achieve significant improvement in symptoms from conservative treatment approaches, including elevation of the head of the bed and supplemental oxygen. Systemic chemotherapy can effectively relieve the symptoms of SVCS obstruction, typically within 1-2 weeks of treatment initiation. Up to 80% of patients with SCLC and non-Hodgkin lymphoma may experience complete relief of SVCS symptoms with chemotherapy treatment.
Radiation therapy was once considered the standard approach to the management of SVCS in patients with cancer; however, endovenous recanalization can alleviate symptoms faster than radiation therapy — usually within 72 hours, whereas radiation therapy can take up to 2 weeks to provide relief. Endovascular therapy is also associated with higher efficacy rates than is radiation therapy.
Open surgery plays a limited role in the management of SVC obstruction, although it may be the best approach in select cases.
In cases involving brain edema, decreased cardiac output, or upper airway edema, emergency treatment is indicated.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation is consistent with a diagnosis of superior vena cava syndrome (SVCS), secondary to SCLC.
SCLC is an aggressive, poorly differentiated, high-grade neuroendocrine carcinoma that accounts for approximately 13%-15% of all new lung cancer cases in the United States. SCLC has a propensity for early dissemination; as such, 80%-85% of patients are diagnosed with extensive disease (ES-SCLC). This is common in heavy smokers. Most SCLC tumors are found in hilar or perihilar areas; <5% present in peripheral locations. In many cases, invasion into the peribronchial tissue and lymph node can be clearly identified, with a typical circumferential spread along the submucosa of the bronchi.
Up to 10% of patients with SCLC develop SVCS, which comprises an array of signs and symptoms that result from the obstruction of blood flow through the thin-walled superior vena cava. Clinical symptoms may include cough, dyspnea, and orthopnea; facial edema and plethora, upper extremity swelling, and venous distension of the chest wall and neck are the most commonly encountered signs. Most cases of SVCS occur in patients with mediastinal tumors, although noncancerous causes (eg, thrombosis and fibrosing mediastinitis) can also give rise to it. The diagnosis of SVCS is usually made clinically and then confirmed with imaging (chest radiography, contrast-enhanced CT, duplex ultrasound, conventional venography, and/or magnetic resonance venography).
Though it was traditionally considered a virtual emergency, patients seldom experience life-threatening complications from SVCS. The goals of treatment are to alleviate the symptoms of SVC obstruction and treat the underlying disease process. Treatment approaches include radiation therapy, chemotherapy, open surgery, and endovenous recanalization; however, patients with clinical SVCS often achieve significant improvement in symptoms from conservative treatment approaches, including elevation of the head of the bed and supplemental oxygen. Systemic chemotherapy can effectively relieve the symptoms of SVCS obstruction, typically within 1-2 weeks of treatment initiation. Up to 80% of patients with SCLC and non-Hodgkin lymphoma may experience complete relief of SVCS symptoms with chemotherapy treatment.
Radiation therapy was once considered the standard approach to the management of SVCS in patients with cancer; however, endovenous recanalization can alleviate symptoms faster than radiation therapy — usually within 72 hours, whereas radiation therapy can take up to 2 weeks to provide relief. Endovascular therapy is also associated with higher efficacy rates than is radiation therapy.
Open surgery plays a limited role in the management of SVC obstruction, although it may be the best approach in select cases.
In cases involving brain edema, decreased cardiac output, or upper airway edema, emergency treatment is indicated.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation is consistent with a diagnosis of superior vena cava syndrome (SVCS), secondary to SCLC.
SCLC is an aggressive, poorly differentiated, high-grade neuroendocrine carcinoma that accounts for approximately 13%-15% of all new lung cancer cases in the United States. SCLC has a propensity for early dissemination; as such, 80%-85% of patients are diagnosed with extensive disease (ES-SCLC). This is common in heavy smokers. Most SCLC tumors are found in hilar or perihilar areas; <5% present in peripheral locations. In many cases, invasion into the peribronchial tissue and lymph node can be clearly identified, with a typical circumferential spread along the submucosa of the bronchi.
Up to 10% of patients with SCLC develop SVCS, which comprises an array of signs and symptoms that result from the obstruction of blood flow through the thin-walled superior vena cava. Clinical symptoms may include cough, dyspnea, and orthopnea; facial edema and plethora, upper extremity swelling, and venous distension of the chest wall and neck are the most commonly encountered signs. Most cases of SVCS occur in patients with mediastinal tumors, although noncancerous causes (eg, thrombosis and fibrosing mediastinitis) can also give rise to it. The diagnosis of SVCS is usually made clinically and then confirmed with imaging (chest radiography, contrast-enhanced CT, duplex ultrasound, conventional venography, and/or magnetic resonance venography).
Though it was traditionally considered a virtual emergency, patients seldom experience life-threatening complications from SVCS. The goals of treatment are to alleviate the symptoms of SVC obstruction and treat the underlying disease process. Treatment approaches include radiation therapy, chemotherapy, open surgery, and endovenous recanalization; however, patients with clinical SVCS often achieve significant improvement in symptoms from conservative treatment approaches, including elevation of the head of the bed and supplemental oxygen. Systemic chemotherapy can effectively relieve the symptoms of SVCS obstruction, typically within 1-2 weeks of treatment initiation. Up to 80% of patients with SCLC and non-Hodgkin lymphoma may experience complete relief of SVCS symptoms with chemotherapy treatment.
Radiation therapy was once considered the standard approach to the management of SVCS in patients with cancer; however, endovenous recanalization can alleviate symptoms faster than radiation therapy — usually within 72 hours, whereas radiation therapy can take up to 2 weeks to provide relief. Endovascular therapy is also associated with higher efficacy rates than is radiation therapy.
Open surgery plays a limited role in the management of SVC obstruction, although it may be the best approach in select cases.
In cases involving brain edema, decreased cardiac output, or upper airway edema, emergency treatment is indicated.
Karl J. D'Silva, MD, Clinical Assistant Professor, Department of Medicine, Tufts University School of Medicine, Boston; Medical Director, Department of Oncology and Hematology, Lahey Hospital and Medical Center, Peabody, Massachusetts.
Karl J. D'Silva, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
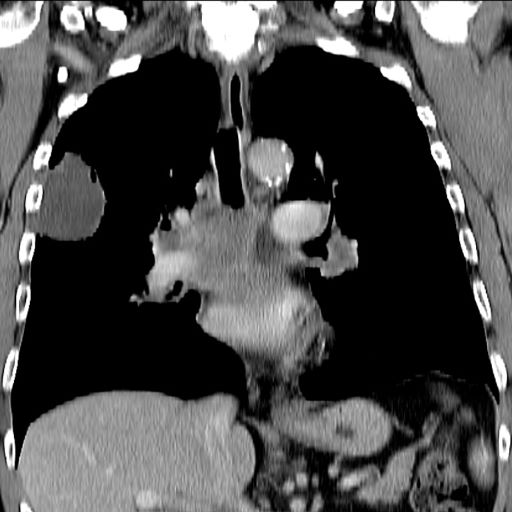
A 66-year-old African American man was diagnosed with small cell lung cancer (SCLC) after the discovery of an endobronchial tumor on bronchoscopy. A biopsy of the tumor was positive for SCLC and CT revealed multiple pulmonary nodules and extensive mediastinal nodal metastases. The patient completed his first cycle of carboplatin-based chemotherapy about 1 month ago. At today's visit, he presents with complaints of worsening symptoms over the past week or so; specifically, he reports increasing fatigue and shortness of breath, a dry cough, light-headedness, difficulty swallowing, and facial swelling. Physical examination reveals facial edema and venous distension of the neck and chest wall; blood pressure is 140/70 mm Hg, respiratory rate is 19 breaths/min, and pulse is 84 beats/min. The patient has a 45-pack-year smoking history and reports having two or three alcoholic drinks per day. His previous medical history is positive for hypertension, which is treated with enalapril 20 mg/day and metoprolol 200 mg/day. Complete blood cell count findings are all within normal range.
Lesions on upper arms
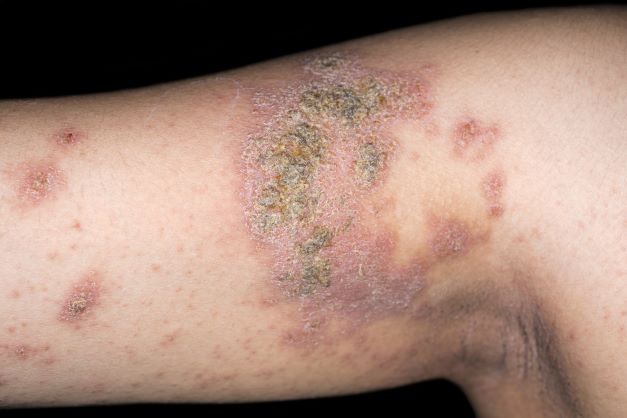
The patient is diagnosed with atopic dermatitis (AD) complicated by skin infection.
AD is the most common chronic pruritic inflammatory skin disorder that affects both children and adults. In the United States, up to 18% of children and 7% of adults are affected. Atopic dermatitis is associated with diminished quality of life, including disruption in activities of daily living, sleep disturbance, depression, and anxiety. Moreover, patients with AD have an increased risk for infections. A significantly higher prevalence of cutaneous and systemic infections is seen in patients with AD compared with individuals without AD.
Bacterial infections are common in AD and are usually caused by Staphylococcus aureus. Examples include impetigo, which typically presents with oozing serum that dries, resulting in a honey-crusted appearance surrounded by an erythematous base. Fluid-filled blisters (bullous impetigo) may also be present, which can be mistaken for eczema herpeticum (EH).
Nonpurulent skin and soft tissue infections (SSTIs) include erysipelas and cellulitis. In most cases, these infections begin in a focal skin area but spread rapidly across the affected sites such as the arms, legs, trunk, or face. Signs typically include focal erythema, swelling, warmth, and tenderness; fever and bacteremia may also be present.
Purulent SSTIs present as skin abscesses, involving fluctuant or nonfluctuant nodules or pustules surrounded by an erythematous swelling; the lesions may also be tender and warm. Methicillin-resistant S aureus (MRSA) is a common cause of purulent SSTIs.
Systemic complications of SSTI in AD may include bacteremia, osteomyelitis, septic arthritis, or bursitis; less often, endocarditis and staphylococcal scalded skin syndrome may occur. Clinicians should maintain a high index of suspicion for these complications in patients who present with an ill-looking appearance, lethargy, focal point tenderness of the bone, joint swelling, heart murmur, and widespread desquamation. Persistent elevated inflammatory markers (eg, C-reactive protein or erythrocyte sedimentation rate) should increase the level of suspicion.
Nonbacterial infections can occur concurrently with bacterial skin infections and the two can be difficult to distinguish. For example, EH results from the local spread of herpes simplex virus, which has a predilection for AD lesions. Early during EH, skin lesions appear as superficial clusters of dome‐shaped vesicles and/or small, round, punched‐out erosions. With progression, the lesions may become superficially infected with S aureus and may develop the characteristic honey-colored scale of impetigo.
Factors that contribute to the increased prevalence of infections in AD include skin barrier defects, suppression of cutaneous innate immunity by type 2 inflammation, S aureus colonization, allergen sensitivity, filaggrin loss-of-function mutation, and cutaneous dysbiosis.
Daily skin hydration and moisturization is a fundamental component of treatment for any patient with AD, both to treat the AD and prevent infection. Patients with AD should bathe daily, followed by gentle drying and application of a moisturizer or a prescribed topical medication. Standard topical anti-inflammatory medications, including topical corticosteroids and topical calcineurin inhibitors, improve skin barrier functions and have been reported to decrease S aureus colonization in AD lesions. Similarly, the monoclonal antibody dupilumab has been shown to decrease S aureus colonization and increase microbial diversity.
In the presence of an uncomplicated, nonpurulent skin infection, a beta-lactam antibiotic that covers both S aureus and beta-hemolytic streptococci (eg, cefazolin or cephalexin) may be sufficient, depending on clinical response or culture and in consideration of local epidemiology and resistance patterns. For patients with AD who present with a skin abscess, history of MRSA colonization, close contacts with a history of skin infections, or recent hospitalization, coverage for MRSA should be considered. Acceptable oral options for MRSA skin infections include clindamycin, doxycycline, trimethoprim-sulfamethoxazole, and linezolid, assuming that the isolate is susceptible in vitro. Topical mupirocin ointment can be used for patients with minor, localized skin infections (eg, impetigo).
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia.
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The patient is diagnosed with atopic dermatitis (AD) complicated by skin infection.
AD is the most common chronic pruritic inflammatory skin disorder that affects both children and adults. In the United States, up to 18% of children and 7% of adults are affected. Atopic dermatitis is associated with diminished quality of life, including disruption in activities of daily living, sleep disturbance, depression, and anxiety. Moreover, patients with AD have an increased risk for infections. A significantly higher prevalence of cutaneous and systemic infections is seen in patients with AD compared with individuals without AD.
Bacterial infections are common in AD and are usually caused by Staphylococcus aureus. Examples include impetigo, which typically presents with oozing serum that dries, resulting in a honey-crusted appearance surrounded by an erythematous base. Fluid-filled blisters (bullous impetigo) may also be present, which can be mistaken for eczema herpeticum (EH).
Nonpurulent skin and soft tissue infections (SSTIs) include erysipelas and cellulitis. In most cases, these infections begin in a focal skin area but spread rapidly across the affected sites such as the arms, legs, trunk, or face. Signs typically include focal erythema, swelling, warmth, and tenderness; fever and bacteremia may also be present.
Purulent SSTIs present as skin abscesses, involving fluctuant or nonfluctuant nodules or pustules surrounded by an erythematous swelling; the lesions may also be tender and warm. Methicillin-resistant S aureus (MRSA) is a common cause of purulent SSTIs.
Systemic complications of SSTI in AD may include bacteremia, osteomyelitis, septic arthritis, or bursitis; less often, endocarditis and staphylococcal scalded skin syndrome may occur. Clinicians should maintain a high index of suspicion for these complications in patients who present with an ill-looking appearance, lethargy, focal point tenderness of the bone, joint swelling, heart murmur, and widespread desquamation. Persistent elevated inflammatory markers (eg, C-reactive protein or erythrocyte sedimentation rate) should increase the level of suspicion.
Nonbacterial infections can occur concurrently with bacterial skin infections and the two can be difficult to distinguish. For example, EH results from the local spread of herpes simplex virus, which has a predilection for AD lesions. Early during EH, skin lesions appear as superficial clusters of dome‐shaped vesicles and/or small, round, punched‐out erosions. With progression, the lesions may become superficially infected with S aureus and may develop the characteristic honey-colored scale of impetigo.
Factors that contribute to the increased prevalence of infections in AD include skin barrier defects, suppression of cutaneous innate immunity by type 2 inflammation, S aureus colonization, allergen sensitivity, filaggrin loss-of-function mutation, and cutaneous dysbiosis.
Daily skin hydration and moisturization is a fundamental component of treatment for any patient with AD, both to treat the AD and prevent infection. Patients with AD should bathe daily, followed by gentle drying and application of a moisturizer or a prescribed topical medication. Standard topical anti-inflammatory medications, including topical corticosteroids and topical calcineurin inhibitors, improve skin barrier functions and have been reported to decrease S aureus colonization in AD lesions. Similarly, the monoclonal antibody dupilumab has been shown to decrease S aureus colonization and increase microbial diversity.
In the presence of an uncomplicated, nonpurulent skin infection, a beta-lactam antibiotic that covers both S aureus and beta-hemolytic streptococci (eg, cefazolin or cephalexin) may be sufficient, depending on clinical response or culture and in consideration of local epidemiology and resistance patterns. For patients with AD who present with a skin abscess, history of MRSA colonization, close contacts with a history of skin infections, or recent hospitalization, coverage for MRSA should be considered. Acceptable oral options for MRSA skin infections include clindamycin, doxycycline, trimethoprim-sulfamethoxazole, and linezolid, assuming that the isolate is susceptible in vitro. Topical mupirocin ointment can be used for patients with minor, localized skin infections (eg, impetigo).
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia.
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The patient is diagnosed with atopic dermatitis (AD) complicated by skin infection.
AD is the most common chronic pruritic inflammatory skin disorder that affects both children and adults. In the United States, up to 18% of children and 7% of adults are affected. Atopic dermatitis is associated with diminished quality of life, including disruption in activities of daily living, sleep disturbance, depression, and anxiety. Moreover, patients with AD have an increased risk for infections. A significantly higher prevalence of cutaneous and systemic infections is seen in patients with AD compared with individuals without AD.
Bacterial infections are common in AD and are usually caused by Staphylococcus aureus. Examples include impetigo, which typically presents with oozing serum that dries, resulting in a honey-crusted appearance surrounded by an erythematous base. Fluid-filled blisters (bullous impetigo) may also be present, which can be mistaken for eczema herpeticum (EH).
Nonpurulent skin and soft tissue infections (SSTIs) include erysipelas and cellulitis. In most cases, these infections begin in a focal skin area but spread rapidly across the affected sites such as the arms, legs, trunk, or face. Signs typically include focal erythema, swelling, warmth, and tenderness; fever and bacteremia may also be present.
Purulent SSTIs present as skin abscesses, involving fluctuant or nonfluctuant nodules or pustules surrounded by an erythematous swelling; the lesions may also be tender and warm. Methicillin-resistant S aureus (MRSA) is a common cause of purulent SSTIs.
Systemic complications of SSTI in AD may include bacteremia, osteomyelitis, septic arthritis, or bursitis; less often, endocarditis and staphylococcal scalded skin syndrome may occur. Clinicians should maintain a high index of suspicion for these complications in patients who present with an ill-looking appearance, lethargy, focal point tenderness of the bone, joint swelling, heart murmur, and widespread desquamation. Persistent elevated inflammatory markers (eg, C-reactive protein or erythrocyte sedimentation rate) should increase the level of suspicion.
Nonbacterial infections can occur concurrently with bacterial skin infections and the two can be difficult to distinguish. For example, EH results from the local spread of herpes simplex virus, which has a predilection for AD lesions. Early during EH, skin lesions appear as superficial clusters of dome‐shaped vesicles and/or small, round, punched‐out erosions. With progression, the lesions may become superficially infected with S aureus and may develop the characteristic honey-colored scale of impetigo.
Factors that contribute to the increased prevalence of infections in AD include skin barrier defects, suppression of cutaneous innate immunity by type 2 inflammation, S aureus colonization, allergen sensitivity, filaggrin loss-of-function mutation, and cutaneous dysbiosis.
Daily skin hydration and moisturization is a fundamental component of treatment for any patient with AD, both to treat the AD and prevent infection. Patients with AD should bathe daily, followed by gentle drying and application of a moisturizer or a prescribed topical medication. Standard topical anti-inflammatory medications, including topical corticosteroids and topical calcineurin inhibitors, improve skin barrier functions and have been reported to decrease S aureus colonization in AD lesions. Similarly, the monoclonal antibody dupilumab has been shown to decrease S aureus colonization and increase microbial diversity.
In the presence of an uncomplicated, nonpurulent skin infection, a beta-lactam antibiotic that covers both S aureus and beta-hemolytic streptococci (eg, cefazolin or cephalexin) may be sufficient, depending on clinical response or culture and in consideration of local epidemiology and resistance patterns. For patients with AD who present with a skin abscess, history of MRSA colonization, close contacts with a history of skin infections, or recent hospitalization, coverage for MRSA should be considered. Acceptable oral options for MRSA skin infections include clindamycin, doxycycline, trimethoprim-sulfamethoxazole, and linezolid, assuming that the isolate is susceptible in vitro. Topical mupirocin ointment can be used for patients with minor, localized skin infections (eg, impetigo).
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia.
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.

An 8-year-old girl presents with pruritic lesions on her upper arms. As an infant, the patient was treated for widespread dermatitis with topical steroids and emollients; recently, after a long symptom-free period, she has had multiple bouts of dermatitis on her face, knees, ankles, and elbows. According to the patient's mother, the patient bathes every 2-3 days to not dry out her skin. At the current visit, physical examination reveals scaly patches and plaques with a honey-colored crust surrounded by an erythematous base. No other family members are experiencing symptoms. There is a positive family history for atopy and asthma.
Fatigue and sporadic fever
This patient's findings are consistent with a diagnosis of malignant mantle cell lymphoma (MCL).
MCL is a rare and aggressive form of non-Hodgkin lymphoma that accounts for approximately 5%-7% of all lymphomas. MCL has a characteristic immunophenotype (ie, CD5+, CD10−, Bcl-2+, Bcl-6−, CD20+), with the t(11;14)(q13;q32) chromosomal translocation, and expression of cyclin D1. The median age at diagnosis is between 60 and 70 years. Approximately 70% of all cases occur in men.
The clinical presentation of MCL can vary. Patients may have asymptomatic monoclonal MCL type lymphocytosis or nonbulky nodal/extra nodal disease with minimal symptoms, or they may present with significant symptoms, progressive generalized lymphadenopathy, cytopenia, splenomegaly, and extranodal disease, including gastrointestinal involvement (lymphomatous polyposis), kidney involvement, involvement of other organs, or, rarely, central nervous system involvement. Disease involving multiple lymph nodes and other sites of the body is seen in most patients. Approximately 70% of patients present with stage IV disease requiring systemic treatment.
According to 2022 guidelines from the National Comprehensive Cancer Network (NCCN), essential components in the workup for MCL include:
• Physical examination, with attention to node-bearing areas, including Waldeyer ring, and to size of liver and spleen
• Assessment of performance status and B symptoms (ie, fever > 100.4°F [may be sporadic], drenching night sweats, unintentional weight loss of > 10% of body weight over 6 months or less)
• CBC with differential
• Comprehensive metabolic panel
• Serum lactate dehydrogenase (LDH) level (an important prognostic marker)
• PET/CT scan (including neck)
• Hepatitis B testing if treatment with rituximab is being contemplated
• Echocardiogram or multigated acquisition (MUGA) scan if anthracycline or anthracenedione-based regimen is indicated
• Pregnancy testing in women of childbearing age (if chemotherapy or radiation therapy is planned)
Additional testing may be indicated in specific circumstances, such as colonoscopy/endoscopy.
MCL remains challenging to treat. While 50%-90% of patients with MCL respond to combination chemotherapy, only 30% achieve a complete response. Median time to treatment failure is < 18 months.
When selecting systemic treatment for patients with MCL, clinicians should consider the availability of clinical trials for subsets of patients, eligibility for stem cell transplant (SCT), high-risk status (ie, blastoid MCL, high Ki-67% > 30%, or central nervous system involvement), age, and performance status. The addition of radiation to chemotherapy may be beneficial for patients with limited-stage, nonbulky disease, although this has not been confirmed in large, randomized studies. Outside of clinical trials, the usual approach for frontline treatment of MCL is chemoimmunotherapy with/without autologous SCT and with/without maintenance therapy.
Available options for primary MCL therapy in patients who require systemic therapy include:
• Single alkylating agents
• CVP (cyclophosphamide, vincristine, prednisone)
• CHOP (cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine [Oncovin], prednisone)
• Hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone) with or without rituximab
• R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone)
• Lenalidomide plus rituximab
• Hyper-CVAD with autologous SCT
Options for relapsed or refractory MCL include:
• R-hyper-CVAD
• Hyper-CVAD with or without rituximab followed by autologous SCT
• Nucleoside analogues and combinations
• Salvage chemotherapy combinations followed by autologous SCT
• Bortezomib
• Lenalidomide
• Ibrutinib
• Radioimmunotherapy
• Rituximab
• Rituximab and thalidomide combination
• Acalabrutinib
• High-dose chemotherapy with autologous bone marrow or SCT
• Brexucabtagene autoleucel
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's findings are consistent with a diagnosis of malignant mantle cell lymphoma (MCL).
MCL is a rare and aggressive form of non-Hodgkin lymphoma that accounts for approximately 5%-7% of all lymphomas. MCL has a characteristic immunophenotype (ie, CD5+, CD10−, Bcl-2+, Bcl-6−, CD20+), with the t(11;14)(q13;q32) chromosomal translocation, and expression of cyclin D1. The median age at diagnosis is between 60 and 70 years. Approximately 70% of all cases occur in men.
The clinical presentation of MCL can vary. Patients may have asymptomatic monoclonal MCL type lymphocytosis or nonbulky nodal/extra nodal disease with minimal symptoms, or they may present with significant symptoms, progressive generalized lymphadenopathy, cytopenia, splenomegaly, and extranodal disease, including gastrointestinal involvement (lymphomatous polyposis), kidney involvement, involvement of other organs, or, rarely, central nervous system involvement. Disease involving multiple lymph nodes and other sites of the body is seen in most patients. Approximately 70% of patients present with stage IV disease requiring systemic treatment.
According to 2022 guidelines from the National Comprehensive Cancer Network (NCCN), essential components in the workup for MCL include:
• Physical examination, with attention to node-bearing areas, including Waldeyer ring, and to size of liver and spleen
• Assessment of performance status and B symptoms (ie, fever > 100.4°F [may be sporadic], drenching night sweats, unintentional weight loss of > 10% of body weight over 6 months or less)
• CBC with differential
• Comprehensive metabolic panel
• Serum lactate dehydrogenase (LDH) level (an important prognostic marker)
• PET/CT scan (including neck)
• Hepatitis B testing if treatment with rituximab is being contemplated
• Echocardiogram or multigated acquisition (MUGA) scan if anthracycline or anthracenedione-based regimen is indicated
• Pregnancy testing in women of childbearing age (if chemotherapy or radiation therapy is planned)
Additional testing may be indicated in specific circumstances, such as colonoscopy/endoscopy.
MCL remains challenging to treat. While 50%-90% of patients with MCL respond to combination chemotherapy, only 30% achieve a complete response. Median time to treatment failure is < 18 months.
When selecting systemic treatment for patients with MCL, clinicians should consider the availability of clinical trials for subsets of patients, eligibility for stem cell transplant (SCT), high-risk status (ie, blastoid MCL, high Ki-67% > 30%, or central nervous system involvement), age, and performance status. The addition of radiation to chemotherapy may be beneficial for patients with limited-stage, nonbulky disease, although this has not been confirmed in large, randomized studies. Outside of clinical trials, the usual approach for frontline treatment of MCL is chemoimmunotherapy with/without autologous SCT and with/without maintenance therapy.
Available options for primary MCL therapy in patients who require systemic therapy include:
• Single alkylating agents
• CVP (cyclophosphamide, vincristine, prednisone)
• CHOP (cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine [Oncovin], prednisone)
• Hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone) with or without rituximab
• R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone)
• Lenalidomide plus rituximab
• Hyper-CVAD with autologous SCT
Options for relapsed or refractory MCL include:
• R-hyper-CVAD
• Hyper-CVAD with or without rituximab followed by autologous SCT
• Nucleoside analogues and combinations
• Salvage chemotherapy combinations followed by autologous SCT
• Bortezomib
• Lenalidomide
• Ibrutinib
• Radioimmunotherapy
• Rituximab
• Rituximab and thalidomide combination
• Acalabrutinib
• High-dose chemotherapy with autologous bone marrow or SCT
• Brexucabtagene autoleucel
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's findings are consistent with a diagnosis of malignant mantle cell lymphoma (MCL).
MCL is a rare and aggressive form of non-Hodgkin lymphoma that accounts for approximately 5%-7% of all lymphomas. MCL has a characteristic immunophenotype (ie, CD5+, CD10−, Bcl-2+, Bcl-6−, CD20+), with the t(11;14)(q13;q32) chromosomal translocation, and expression of cyclin D1. The median age at diagnosis is between 60 and 70 years. Approximately 70% of all cases occur in men.
The clinical presentation of MCL can vary. Patients may have asymptomatic monoclonal MCL type lymphocytosis or nonbulky nodal/extra nodal disease with minimal symptoms, or they may present with significant symptoms, progressive generalized lymphadenopathy, cytopenia, splenomegaly, and extranodal disease, including gastrointestinal involvement (lymphomatous polyposis), kidney involvement, involvement of other organs, or, rarely, central nervous system involvement. Disease involving multiple lymph nodes and other sites of the body is seen in most patients. Approximately 70% of patients present with stage IV disease requiring systemic treatment.
According to 2022 guidelines from the National Comprehensive Cancer Network (NCCN), essential components in the workup for MCL include:
• Physical examination, with attention to node-bearing areas, including Waldeyer ring, and to size of liver and spleen
• Assessment of performance status and B symptoms (ie, fever > 100.4°F [may be sporadic], drenching night sweats, unintentional weight loss of > 10% of body weight over 6 months or less)
• CBC with differential
• Comprehensive metabolic panel
• Serum lactate dehydrogenase (LDH) level (an important prognostic marker)
• PET/CT scan (including neck)
• Hepatitis B testing if treatment with rituximab is being contemplated
• Echocardiogram or multigated acquisition (MUGA) scan if anthracycline or anthracenedione-based regimen is indicated
• Pregnancy testing in women of childbearing age (if chemotherapy or radiation therapy is planned)
Additional testing may be indicated in specific circumstances, such as colonoscopy/endoscopy.
MCL remains challenging to treat. While 50%-90% of patients with MCL respond to combination chemotherapy, only 30% achieve a complete response. Median time to treatment failure is < 18 months.
When selecting systemic treatment for patients with MCL, clinicians should consider the availability of clinical trials for subsets of patients, eligibility for stem cell transplant (SCT), high-risk status (ie, blastoid MCL, high Ki-67% > 30%, or central nervous system involvement), age, and performance status. The addition of radiation to chemotherapy may be beneficial for patients with limited-stage, nonbulky disease, although this has not been confirmed in large, randomized studies. Outside of clinical trials, the usual approach for frontline treatment of MCL is chemoimmunotherapy with/without autologous SCT and with/without maintenance therapy.
Available options for primary MCL therapy in patients who require systemic therapy include:
• Single alkylating agents
• CVP (cyclophosphamide, vincristine, prednisone)
• CHOP (cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine [Oncovin], prednisone)
• Hyper-CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone) with or without rituximab
• R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone)
• Lenalidomide plus rituximab
• Hyper-CVAD with autologous SCT
Options for relapsed or refractory MCL include:
• R-hyper-CVAD
• Hyper-CVAD with or without rituximab followed by autologous SCT
• Nucleoside analogues and combinations
• Salvage chemotherapy combinations followed by autologous SCT
• Bortezomib
• Lenalidomide
• Ibrutinib
• Radioimmunotherapy
• Rituximab
• Rituximab and thalidomide combination
• Acalabrutinib
• High-dose chemotherapy with autologous bone marrow or SCT
• Brexucabtagene autoleucel
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
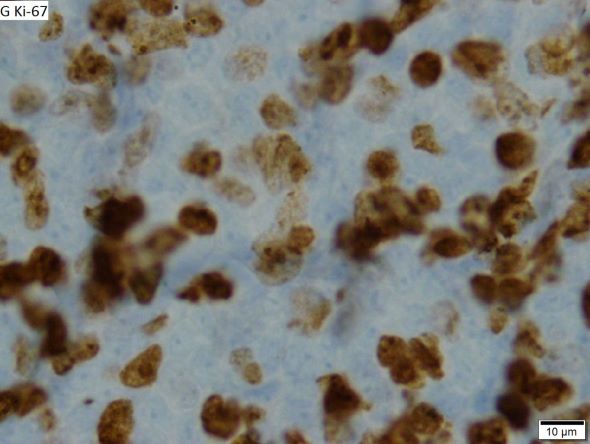
A 64-year-old Black man with a history of hypertension and hyperlipidemia presents with complaints of fatigue, sporadic fever > 100.4° F, and mild abdominal pain. The patient has lost 12 lb since he was last seen 9 months earlier. When questioned, he states that he simply doesn't have the appetite he once had. Physical examination reveals pallor; abdominal distension; lymphadenopathy in the anterior cervical, inguinal, and axillary regions; and palpable spleen and liver. CBC findings include RBC 4.4 x 106/µL; WBC 2400/μL; PLT 148,000/dL; MCV 57.8 fL; hematocrit 38%; and ALC 4200/µL. Immunophenotyping by flow cytometry and immunohistochemistry was positive for CD5 and CD19, with no expression of CD10 or CD23. Cyclin D1 was overexpressed.
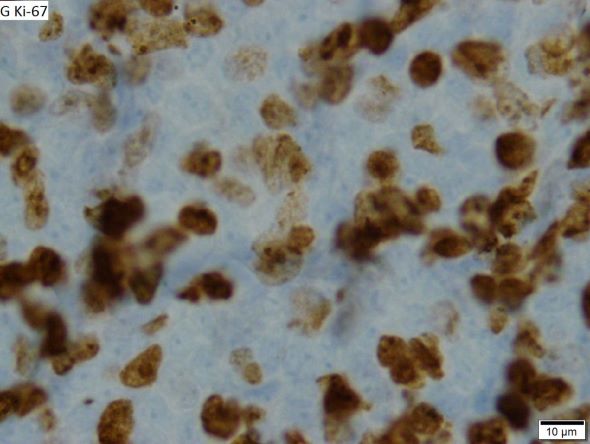
Red swollen eyelids
This patient's symptoms are consistent with a diagnosis of blepharitis.
Blepharitis is an inflammatory disorder of the eyelids that is frequently associated with bacterial colonization of the eyelid. Anatomically, it can be categorized as anterior blepharitis or posterior blepharitis. Anterior blepharitis refers to inflammation primarily positioned around the skin, eyelashes, and lash follicles and is usually further divided into staphylococcal and seborrheic variants. Posterior blepharitis involves the meibomian gland orifices, meibomian glands, tarsal plate, and blepharo-conjunctival junction.
Blepharitis can be acute or chronic. It is frequently associated with systemic diseases, such as rosacea, atopy, and seborrheic dermatitis, as well as ocular diseases, such as dry eye syndromes, chalazion, trichiasis, ectropion and entropion, infectious or other inflammatory conjunctivitis, and keratitis. Moreover, high rates of blepharitis have been reported in patients treated with dupilumab for atopic dermatitis.
Eye irritation, itching, erythema of the lids, flaking of the lid margins, and/or changes in the eyelashes are common presenting symptoms in patients with blepharitis. Other symptoms may include:
• Burning
• Watering
• Foreign-body sensation
• Crusting and mattering of the lashes and medial canthus
• Red lids
• Red eyes
• Photophobia
• Pain
• Decreased vision
• Visual fluctuations
• Heat, cold, alcohol, and spicy-food intolerance
The differential diagnosis for blepharitis includes bacterial keratitis, which is a serious ocular disorder that can lead to vision loss if not properly treated. Bacterial keratitis progresses rapidly and can result in corneal destruction within 24-48 hours with some particularly virulent bacteria. Patients with bacterial keratitis typically report rapid onset of pain, photophobia, and decreased vision.
Ocular rosacea should also be considered in the differential diagnosis of blepharitis, and the two conditions can co-occur. Patients with ocular rosacea may experience facial symptoms (eg, recurrent flushing episodes, persistent and/or recurrent midfacial erythema, papular and pustular lesions) in addition to ocular symptoms, which can range from minor irritation, foreign-body sensation, and blurry vision to severe ocular surface disruption and inflammatory keratitis.
Bacterial conjunctivitis involves inflammation of the bulbar and/or palpebral conjunctiva, whereas blepharitis involves inflammation of the eyelids only. Other conditions to consider in the diagnosis of blepharitis can be found here.
Given the unprecedented efficacy seen in clinical trials, dupilumab is emerging as a first-line therapeutic for moderate to severe atopic dermatitis. However, clinicians should be alert to ocular complications among their patients with atopic dermatitis who are being treated with dupilumab. In some patients, this may be because of preexisting meibomian gland disease and ocular surface disease. After a diagnosis of ocular complications, the continued use of dupilumab should be jointly evaluated by the ophthalmologist and dermatologist or allergist on the basis of the ocular risk vs systemic benefit. Treatment for blepharitis typically includes strict eyelid hygiene and topical antibiotic ointment; oral antibiotics can be beneficial for refractory disease.
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia.
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's symptoms are consistent with a diagnosis of blepharitis.
Blepharitis is an inflammatory disorder of the eyelids that is frequently associated with bacterial colonization of the eyelid. Anatomically, it can be categorized as anterior blepharitis or posterior blepharitis. Anterior blepharitis refers to inflammation primarily positioned around the skin, eyelashes, and lash follicles and is usually further divided into staphylococcal and seborrheic variants. Posterior blepharitis involves the meibomian gland orifices, meibomian glands, tarsal plate, and blepharo-conjunctival junction.
Blepharitis can be acute or chronic. It is frequently associated with systemic diseases, such as rosacea, atopy, and seborrheic dermatitis, as well as ocular diseases, such as dry eye syndromes, chalazion, trichiasis, ectropion and entropion, infectious or other inflammatory conjunctivitis, and keratitis. Moreover, high rates of blepharitis have been reported in patients treated with dupilumab for atopic dermatitis.
Eye irritation, itching, erythema of the lids, flaking of the lid margins, and/or changes in the eyelashes are common presenting symptoms in patients with blepharitis. Other symptoms may include:
• Burning
• Watering
• Foreign-body sensation
• Crusting and mattering of the lashes and medial canthus
• Red lids
• Red eyes
• Photophobia
• Pain
• Decreased vision
• Visual fluctuations
• Heat, cold, alcohol, and spicy-food intolerance
The differential diagnosis for blepharitis includes bacterial keratitis, which is a serious ocular disorder that can lead to vision loss if not properly treated. Bacterial keratitis progresses rapidly and can result in corneal destruction within 24-48 hours with some particularly virulent bacteria. Patients with bacterial keratitis typically report rapid onset of pain, photophobia, and decreased vision.
Ocular rosacea should also be considered in the differential diagnosis of blepharitis, and the two conditions can co-occur. Patients with ocular rosacea may experience facial symptoms (eg, recurrent flushing episodes, persistent and/or recurrent midfacial erythema, papular and pustular lesions) in addition to ocular symptoms, which can range from minor irritation, foreign-body sensation, and blurry vision to severe ocular surface disruption and inflammatory keratitis.
Bacterial conjunctivitis involves inflammation of the bulbar and/or palpebral conjunctiva, whereas blepharitis involves inflammation of the eyelids only. Other conditions to consider in the diagnosis of blepharitis can be found here.
Given the unprecedented efficacy seen in clinical trials, dupilumab is emerging as a first-line therapeutic for moderate to severe atopic dermatitis. However, clinicians should be alert to ocular complications among their patients with atopic dermatitis who are being treated with dupilumab. In some patients, this may be because of preexisting meibomian gland disease and ocular surface disease. After a diagnosis of ocular complications, the continued use of dupilumab should be jointly evaluated by the ophthalmologist and dermatologist or allergist on the basis of the ocular risk vs systemic benefit. Treatment for blepharitis typically includes strict eyelid hygiene and topical antibiotic ointment; oral antibiotics can be beneficial for refractory disease.
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia.
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's symptoms are consistent with a diagnosis of blepharitis.
Blepharitis is an inflammatory disorder of the eyelids that is frequently associated with bacterial colonization of the eyelid. Anatomically, it can be categorized as anterior blepharitis or posterior blepharitis. Anterior blepharitis refers to inflammation primarily positioned around the skin, eyelashes, and lash follicles and is usually further divided into staphylococcal and seborrheic variants. Posterior blepharitis involves the meibomian gland orifices, meibomian glands, tarsal plate, and blepharo-conjunctival junction.
Blepharitis can be acute or chronic. It is frequently associated with systemic diseases, such as rosacea, atopy, and seborrheic dermatitis, as well as ocular diseases, such as dry eye syndromes, chalazion, trichiasis, ectropion and entropion, infectious or other inflammatory conjunctivitis, and keratitis. Moreover, high rates of blepharitis have been reported in patients treated with dupilumab for atopic dermatitis.
Eye irritation, itching, erythema of the lids, flaking of the lid margins, and/or changes in the eyelashes are common presenting symptoms in patients with blepharitis. Other symptoms may include:
• Burning
• Watering
• Foreign-body sensation
• Crusting and mattering of the lashes and medial canthus
• Red lids
• Red eyes
• Photophobia
• Pain
• Decreased vision
• Visual fluctuations
• Heat, cold, alcohol, and spicy-food intolerance
The differential diagnosis for blepharitis includes bacterial keratitis, which is a serious ocular disorder that can lead to vision loss if not properly treated. Bacterial keratitis progresses rapidly and can result in corneal destruction within 24-48 hours with some particularly virulent bacteria. Patients with bacterial keratitis typically report rapid onset of pain, photophobia, and decreased vision.
Ocular rosacea should also be considered in the differential diagnosis of blepharitis, and the two conditions can co-occur. Patients with ocular rosacea may experience facial symptoms (eg, recurrent flushing episodes, persistent and/or recurrent midfacial erythema, papular and pustular lesions) in addition to ocular symptoms, which can range from minor irritation, foreign-body sensation, and blurry vision to severe ocular surface disruption and inflammatory keratitis.
Bacterial conjunctivitis involves inflammation of the bulbar and/or palpebral conjunctiva, whereas blepharitis involves inflammation of the eyelids only. Other conditions to consider in the diagnosis of blepharitis can be found here.
Given the unprecedented efficacy seen in clinical trials, dupilumab is emerging as a first-line therapeutic for moderate to severe atopic dermatitis. However, clinicians should be alert to ocular complications among their patients with atopic dermatitis who are being treated with dupilumab. In some patients, this may be because of preexisting meibomian gland disease and ocular surface disease. After a diagnosis of ocular complications, the continued use of dupilumab should be jointly evaluated by the ophthalmologist and dermatologist or allergist on the basis of the ocular risk vs systemic benefit. Treatment for blepharitis typically includes strict eyelid hygiene and topical antibiotic ointment; oral antibiotics can be beneficial for refractory disease.
William D. James, MD, Professor, Department of Dermatology, University of Pennsylvania, Philadelphia.
Disclosure: William D. James, MD, has disclosed the following relevant financial relationships:
Received income in an amount equal to or greater than $250 from: Elsevier.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
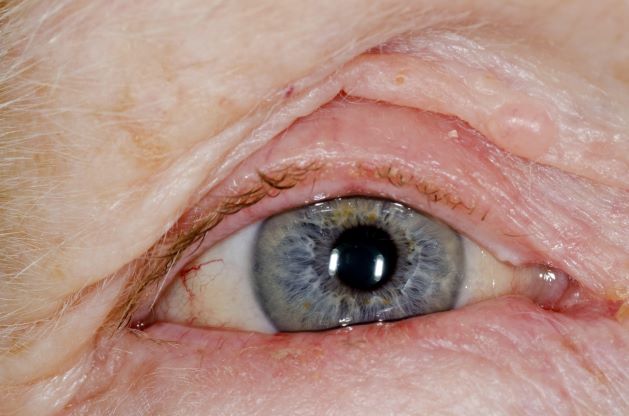
A 71-year-old woman was referred for an ophthalmologic examination by her dermatologist. The patient reports recent onset of red, swollen eyelids; ocular itching; and a burning sensation. Prior medical history includes severe atopic dermatitis, type 2 diabetes, and osteoarthritis. Current medications include metformin 1000 mg/d, celecoxib 200 mg/d, and clobetasol propionate 0.05% cream twice daily. The patient began receiving subcutaneous dupilumab 300 mg/once every 2 weeks about 6 weeks earlier.
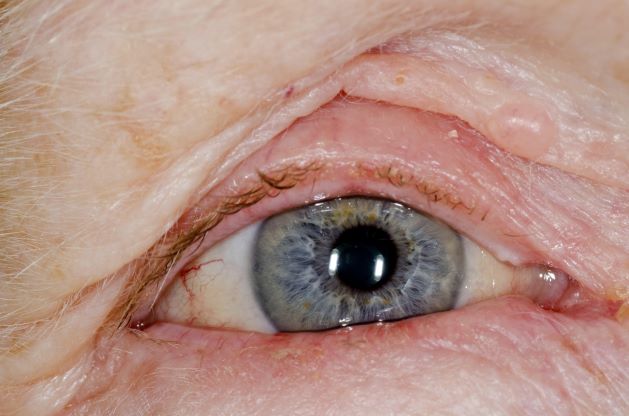
Right ankle pain and swelling
This patient's findings are consistent with a diagnosis of psoriatic enthesitis.
Enthesitis is a hallmark manifestation of psoriatic arthritis (PsA). Approximately 30% of patients with psoriasis are estimated to be affected by PsA, which belongs to the spondyloarthritis (SpA) family of inflammatory rheumatic diseases.
An enthesis is an attachment site of ligaments, tendons, and joint capsules to bone and is a key inflammatory target in SpA. It is a complex structure that dissipates biomechanical stress to preserve homeostasis. Entheses are anatomically and functionally integrated with bursa, fibrocartilage, and synovium in a synovial entheseal complex; biomechanical stress in this area may trigger inflammation. Enthesitis is an early manifestation of PsA that has been associated with radiographic peripheral/axial joint damage and severe disease, as well as reduced quality of life.
Enthesitis can be difficult to diagnose in clinical practice. Symptoms include tenderness, soreness, and pain at entheses on palpation, often without overt clinical evidence of inflammation. In contrast, dactylitis, another hallmark manifestation of PsA, can be recognized by swelling of an entire digit that is different from adjacent digits. Fibromyalgia frequently coexists with enthesitis, and it can be difficult to distinguish the two given the anatomic overlap between the tender points of fibromyalgia and many entheseal sites. Long-lasting morning stiffness and a sustained response to a course of steroids is more suggestive of enthesitis, whereas a higher number of somatoform symptoms is more suggestive of fibromyalgia.
Enthesitis is included in the Classification Criteria for Psoriatic Arthritis (CASPAR) as a hallmark of PsA. While it can be diagnosed clinically, imaging studies may be required, particularly in patients in whom symptoms may be difficult to discern. Evidence of enthesitis by conventional radiography includes bone cortex irregularities, erosions, entheseal soft tissue calcifications, and new bone formation; however, entheseal bone changes detected with conventional radiography appear relatively late in the disease process. Ultrasound is highly sensitive for assessing inflammation and can detect various features of enthesitis, such as increased thickness of tendon insertion, hypoechogenicity, erosions, enthesophytes, and subclinical enthesitis in people with PsA. MRI has the advantage of identifying perientheseal inflammation with adjacent bone marrow edema. Fat-suppressed MRI with or without gadolinium enhancement is a highly sensitive method for visualizing active enthesitis and can identify perientheseal inflammation with adjacent bone marrow edema.
Delayed treatment of PsA can result in irreversible joint damage and reduced quality of life; thus, patients with psoriasis should be closely monitored for early signs of its development, such as enthesitis. A thorough evaluation of the key clinical features of PsA (psoriasis, arthritis, enthesitis, dactylitis, and spondylitis), including evaluation of severity of each feature and impact on physical function and quality of life, is encouraged at each clinical encounter. Because patients may not understand the link between psoriasis and joint pain, specific probing questions can be helpful. Screening questionnaires to detect early signs and symptoms of PsA are available, such as the Psoriasis Epidemiology Screening Tool (PEST), Psoriatic Arthritis Screening and Evaluation (PASE) questionnaire, and Toronto Psoriatic Arthritis Screening (ToPAS) questionnaire. These and many others may be used to help dermatologists detect early signs and symptoms of PsA. Although these questionnaires all have limitations in sensitivity and specificity for the diagnosis of PsA, their use can still improve early diagnosis.
The treatment of PsA focuses on achieving the least amount of disease activity and inflammation possible; optimizing functional status, quality of life, and well-being; and preventing structural damage. Treatment decisions are based on the specific domains affected. Nonsteroidal anti-inflammatory drugs and corticosteroid injections are first-line treatments for enthesitis. Early use of tumor necrosis factor inhibitors (TNF) (adalimumab, certolizumab pegol, etanercept, infliximab, and golimumab) is recommended. Alternative biologic disease-modifying agents are indicated when these TNF inhibitors provide an inadequate response. They include ustekinumab (dual interleukin [IL]-12 and IL-23 inhibitor), secukinumab (IL-17A inhibitor), and apremilast (phosphodiesterase-4 inhibitor) and may be considered for patients with predominantly entheseal manifestations of PsA or dactylitis. Biological disease-modifying agents approved for PsA that have shown efficacy for enthesitis include ixekizumab (which targets IL-17A), abatacept (a T-cell inhibitor), guselkumab (monoclonal antibody), and ustekinumab (monoclonal antibody). Tofacitinib and upadacitinib, both oral Janus kinase inhibitors, may also be considered.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's findings are consistent with a diagnosis of psoriatic enthesitis.
Enthesitis is a hallmark manifestation of psoriatic arthritis (PsA). Approximately 30% of patients with psoriasis are estimated to be affected by PsA, which belongs to the spondyloarthritis (SpA) family of inflammatory rheumatic diseases.
An enthesis is an attachment site of ligaments, tendons, and joint capsules to bone and is a key inflammatory target in SpA. It is a complex structure that dissipates biomechanical stress to preserve homeostasis. Entheses are anatomically and functionally integrated with bursa, fibrocartilage, and synovium in a synovial entheseal complex; biomechanical stress in this area may trigger inflammation. Enthesitis is an early manifestation of PsA that has been associated with radiographic peripheral/axial joint damage and severe disease, as well as reduced quality of life.
Enthesitis can be difficult to diagnose in clinical practice. Symptoms include tenderness, soreness, and pain at entheses on palpation, often without overt clinical evidence of inflammation. In contrast, dactylitis, another hallmark manifestation of PsA, can be recognized by swelling of an entire digit that is different from adjacent digits. Fibromyalgia frequently coexists with enthesitis, and it can be difficult to distinguish the two given the anatomic overlap between the tender points of fibromyalgia and many entheseal sites. Long-lasting morning stiffness and a sustained response to a course of steroids is more suggestive of enthesitis, whereas a higher number of somatoform symptoms is more suggestive of fibromyalgia.
Enthesitis is included in the Classification Criteria for Psoriatic Arthritis (CASPAR) as a hallmark of PsA. While it can be diagnosed clinically, imaging studies may be required, particularly in patients in whom symptoms may be difficult to discern. Evidence of enthesitis by conventional radiography includes bone cortex irregularities, erosions, entheseal soft tissue calcifications, and new bone formation; however, entheseal bone changes detected with conventional radiography appear relatively late in the disease process. Ultrasound is highly sensitive for assessing inflammation and can detect various features of enthesitis, such as increased thickness of tendon insertion, hypoechogenicity, erosions, enthesophytes, and subclinical enthesitis in people with PsA. MRI has the advantage of identifying perientheseal inflammation with adjacent bone marrow edema. Fat-suppressed MRI with or without gadolinium enhancement is a highly sensitive method for visualizing active enthesitis and can identify perientheseal inflammation with adjacent bone marrow edema.
Delayed treatment of PsA can result in irreversible joint damage and reduced quality of life; thus, patients with psoriasis should be closely monitored for early signs of its development, such as enthesitis. A thorough evaluation of the key clinical features of PsA (psoriasis, arthritis, enthesitis, dactylitis, and spondylitis), including evaluation of severity of each feature and impact on physical function and quality of life, is encouraged at each clinical encounter. Because patients may not understand the link between psoriasis and joint pain, specific probing questions can be helpful. Screening questionnaires to detect early signs and symptoms of PsA are available, such as the Psoriasis Epidemiology Screening Tool (PEST), Psoriatic Arthritis Screening and Evaluation (PASE) questionnaire, and Toronto Psoriatic Arthritis Screening (ToPAS) questionnaire. These and many others may be used to help dermatologists detect early signs and symptoms of PsA. Although these questionnaires all have limitations in sensitivity and specificity for the diagnosis of PsA, their use can still improve early diagnosis.
The treatment of PsA focuses on achieving the least amount of disease activity and inflammation possible; optimizing functional status, quality of life, and well-being; and preventing structural damage. Treatment decisions are based on the specific domains affected. Nonsteroidal anti-inflammatory drugs and corticosteroid injections are first-line treatments for enthesitis. Early use of tumor necrosis factor inhibitors (TNF) (adalimumab, certolizumab pegol, etanercept, infliximab, and golimumab) is recommended. Alternative biologic disease-modifying agents are indicated when these TNF inhibitors provide an inadequate response. They include ustekinumab (dual interleukin [IL]-12 and IL-23 inhibitor), secukinumab (IL-17A inhibitor), and apremilast (phosphodiesterase-4 inhibitor) and may be considered for patients with predominantly entheseal manifestations of PsA or dactylitis. Biological disease-modifying agents approved for PsA that have shown efficacy for enthesitis include ixekizumab (which targets IL-17A), abatacept (a T-cell inhibitor), guselkumab (monoclonal antibody), and ustekinumab (monoclonal antibody). Tofacitinib and upadacitinib, both oral Janus kinase inhibitors, may also be considered.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's findings are consistent with a diagnosis of psoriatic enthesitis.
Enthesitis is a hallmark manifestation of psoriatic arthritis (PsA). Approximately 30% of patients with psoriasis are estimated to be affected by PsA, which belongs to the spondyloarthritis (SpA) family of inflammatory rheumatic diseases.
An enthesis is an attachment site of ligaments, tendons, and joint capsules to bone and is a key inflammatory target in SpA. It is a complex structure that dissipates biomechanical stress to preserve homeostasis. Entheses are anatomically and functionally integrated with bursa, fibrocartilage, and synovium in a synovial entheseal complex; biomechanical stress in this area may trigger inflammation. Enthesitis is an early manifestation of PsA that has been associated with radiographic peripheral/axial joint damage and severe disease, as well as reduced quality of life.
Enthesitis can be difficult to diagnose in clinical practice. Symptoms include tenderness, soreness, and pain at entheses on palpation, often without overt clinical evidence of inflammation. In contrast, dactylitis, another hallmark manifestation of PsA, can be recognized by swelling of an entire digit that is different from adjacent digits. Fibromyalgia frequently coexists with enthesitis, and it can be difficult to distinguish the two given the anatomic overlap between the tender points of fibromyalgia and many entheseal sites. Long-lasting morning stiffness and a sustained response to a course of steroids is more suggestive of enthesitis, whereas a higher number of somatoform symptoms is more suggestive of fibromyalgia.
Enthesitis is included in the Classification Criteria for Psoriatic Arthritis (CASPAR) as a hallmark of PsA. While it can be diagnosed clinically, imaging studies may be required, particularly in patients in whom symptoms may be difficult to discern. Evidence of enthesitis by conventional radiography includes bone cortex irregularities, erosions, entheseal soft tissue calcifications, and new bone formation; however, entheseal bone changes detected with conventional radiography appear relatively late in the disease process. Ultrasound is highly sensitive for assessing inflammation and can detect various features of enthesitis, such as increased thickness of tendon insertion, hypoechogenicity, erosions, enthesophytes, and subclinical enthesitis in people with PsA. MRI has the advantage of identifying perientheseal inflammation with adjacent bone marrow edema. Fat-suppressed MRI with or without gadolinium enhancement is a highly sensitive method for visualizing active enthesitis and can identify perientheseal inflammation with adjacent bone marrow edema.
Delayed treatment of PsA can result in irreversible joint damage and reduced quality of life; thus, patients with psoriasis should be closely monitored for early signs of its development, such as enthesitis. A thorough evaluation of the key clinical features of PsA (psoriasis, arthritis, enthesitis, dactylitis, and spondylitis), including evaluation of severity of each feature and impact on physical function and quality of life, is encouraged at each clinical encounter. Because patients may not understand the link between psoriasis and joint pain, specific probing questions can be helpful. Screening questionnaires to detect early signs and symptoms of PsA are available, such as the Psoriasis Epidemiology Screening Tool (PEST), Psoriatic Arthritis Screening and Evaluation (PASE) questionnaire, and Toronto Psoriatic Arthritis Screening (ToPAS) questionnaire. These and many others may be used to help dermatologists detect early signs and symptoms of PsA. Although these questionnaires all have limitations in sensitivity and specificity for the diagnosis of PsA, their use can still improve early diagnosis.
The treatment of PsA focuses on achieving the least amount of disease activity and inflammation possible; optimizing functional status, quality of life, and well-being; and preventing structural damage. Treatment decisions are based on the specific domains affected. Nonsteroidal anti-inflammatory drugs and corticosteroid injections are first-line treatments for enthesitis. Early use of tumor necrosis factor inhibitors (TNF) (adalimumab, certolizumab pegol, etanercept, infliximab, and golimumab) is recommended. Alternative biologic disease-modifying agents are indicated when these TNF inhibitors provide an inadequate response. They include ustekinumab (dual interleukin [IL]-12 and IL-23 inhibitor), secukinumab (IL-17A inhibitor), and apremilast (phosphodiesterase-4 inhibitor) and may be considered for patients with predominantly entheseal manifestations of PsA or dactylitis. Biological disease-modifying agents approved for PsA that have shown efficacy for enthesitis include ixekizumab (which targets IL-17A), abatacept (a T-cell inhibitor), guselkumab (monoclonal antibody), and ustekinumab (monoclonal antibody). Tofacitinib and upadacitinib, both oral Janus kinase inhibitors, may also be considered.
Herbert S. Diamond, MD, Professor of Medicine (retired), Temple University School of Medicine, University of Pittsburgh; Chairman, Department of Medicine Emeritus, Western Pennsylvania Hospital, Pittsburgh, PA.
Herbert S. Diamond, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
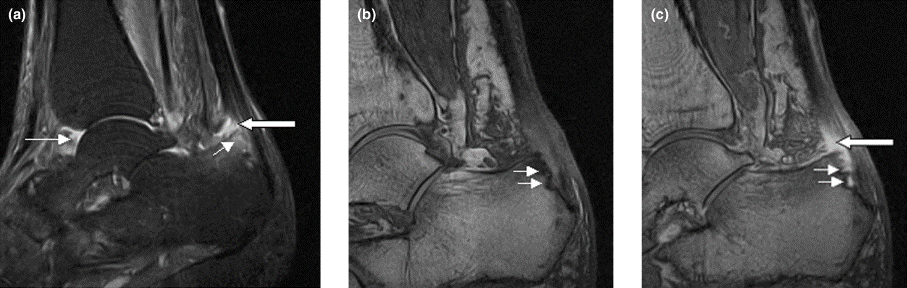
A 42-year-old woman with a 20-year history of plaque psoriasis presents with complaints of a 3-month history of pain, tenderness, and swelling in her right ankle and foot, of unknown origin. Physical examination reveals active psoriasis, with a Psoriasis Area and Severity Index (PASI) score of 6.7 and psoriatic nail dystrophy, including onycholysis, pitting, and hyperkeratosis. Tenderness and swelling are noted at the back of the heel. The patient denies any other complaints. Laboratory tests are normal, including negative rheumatoid factor and antinuclear factor. MRI reveals soft tissue and bone marrow edema below the Achilles insertion.
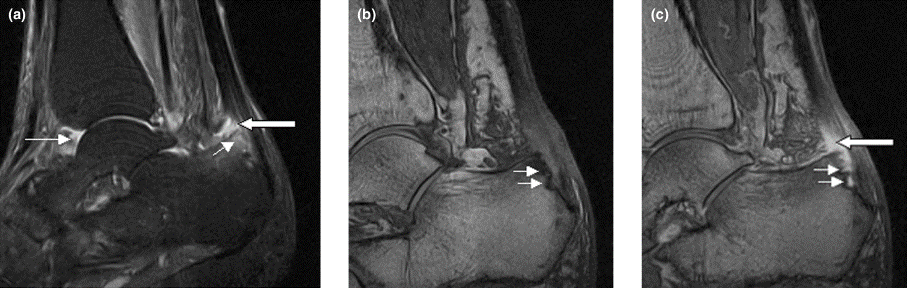
Decline in ambulatory function
Based on this patient's history and presentation, the likely diagnosis is primary progressive multiple sclerosis (PPMS). PPMS represents around 10% of MS cases and tends to develop about a decade later than relapsing MS. Unlike other forms of MS, this phenotype progresses steadily instead of in an episodic fashion like relapsing forms of MS. Most patients with PPMS present with gait difficulty because lesions often develop on the spinal cord. While relapsing-remitting MS (RRMS) is much more common among women than men, men with MS are more likely to have the progressive form.
Although this patient's MRI ultimately points to multiple sclerosis, his functional deficits may initially suggest other conditions in the differential diagnosis. Brainstem gliomas typically manifest in unsteady gait, weakness, double vision, difficulty swallowing, dysarthria, headache, drowsiness, nausea, and vomiting. Transverse myelitis often presents with rapid-onset weakness, sensory deficits, and bowel/bladder dysfunction. Musculoskeletal and neurologic symptoms are common in Lyme disease. B12 deficiency can present with worsening weakness and a sensory ataxia that can present as balance difficulties, but it would not cause focal lesions on the MRI, nor would it present with bladder symptoms. In addition, the patient's steady decline in function rules out RRMS.
PPMS is diagnosed with confirmation of gradual change in functional ability (often ambulation) over time without remission or relapse. These criteria include 1 full year of worsening neurologic function without asymptomatic periods as well as two of these signs of disease: brain lesion, two or more spinal cord lesions, and oligoclonal bands or elevated Immunoglobulin G index. These timing-specific criteria can delay diagnosis, as seen here.
Ocrelizumab is the only FDA-approved disease-modifying therapy (DMT) proven to alter disease progression in ambulatory patients with PPMS. American Academy of Neurology guidelines recommend ocrelizumab for patients with PPMS who are likely to benefit from this therapy. While it is thought that DMTs are more effective at targeting inflammation in RRMS than nerve degeneration in PPMS, these agents may show benefit for patients with active PPMS (relapse and/or evidence of new MRI activity) rather than inactive disease. A recent PPMS study concluded that among patients with relapse or disease activity, DMTs were associated with a significant reduction of long-term disability risk. Together with immunomodulatory therapy, rehabilitation can help manage symptoms.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ.
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
Based on this patient's history and presentation, the likely diagnosis is primary progressive multiple sclerosis (PPMS). PPMS represents around 10% of MS cases and tends to develop about a decade later than relapsing MS. Unlike other forms of MS, this phenotype progresses steadily instead of in an episodic fashion like relapsing forms of MS. Most patients with PPMS present with gait difficulty because lesions often develop on the spinal cord. While relapsing-remitting MS (RRMS) is much more common among women than men, men with MS are more likely to have the progressive form.
Although this patient's MRI ultimately points to multiple sclerosis, his functional deficits may initially suggest other conditions in the differential diagnosis. Brainstem gliomas typically manifest in unsteady gait, weakness, double vision, difficulty swallowing, dysarthria, headache, drowsiness, nausea, and vomiting. Transverse myelitis often presents with rapid-onset weakness, sensory deficits, and bowel/bladder dysfunction. Musculoskeletal and neurologic symptoms are common in Lyme disease. B12 deficiency can present with worsening weakness and a sensory ataxia that can present as balance difficulties, but it would not cause focal lesions on the MRI, nor would it present with bladder symptoms. In addition, the patient's steady decline in function rules out RRMS.
PPMS is diagnosed with confirmation of gradual change in functional ability (often ambulation) over time without remission or relapse. These criteria include 1 full year of worsening neurologic function without asymptomatic periods as well as two of these signs of disease: brain lesion, two or more spinal cord lesions, and oligoclonal bands or elevated Immunoglobulin G index. These timing-specific criteria can delay diagnosis, as seen here.
Ocrelizumab is the only FDA-approved disease-modifying therapy (DMT) proven to alter disease progression in ambulatory patients with PPMS. American Academy of Neurology guidelines recommend ocrelizumab for patients with PPMS who are likely to benefit from this therapy. While it is thought that DMTs are more effective at targeting inflammation in RRMS than nerve degeneration in PPMS, these agents may show benefit for patients with active PPMS (relapse and/or evidence of new MRI activity) rather than inactive disease. A recent PPMS study concluded that among patients with relapse or disease activity, DMTs were associated with a significant reduction of long-term disability risk. Together with immunomodulatory therapy, rehabilitation can help manage symptoms.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ.
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
Based on this patient's history and presentation, the likely diagnosis is primary progressive multiple sclerosis (PPMS). PPMS represents around 10% of MS cases and tends to develop about a decade later than relapsing MS. Unlike other forms of MS, this phenotype progresses steadily instead of in an episodic fashion like relapsing forms of MS. Most patients with PPMS present with gait difficulty because lesions often develop on the spinal cord. While relapsing-remitting MS (RRMS) is much more common among women than men, men with MS are more likely to have the progressive form.
Although this patient's MRI ultimately points to multiple sclerosis, his functional deficits may initially suggest other conditions in the differential diagnosis. Brainstem gliomas typically manifest in unsteady gait, weakness, double vision, difficulty swallowing, dysarthria, headache, drowsiness, nausea, and vomiting. Transverse myelitis often presents with rapid-onset weakness, sensory deficits, and bowel/bladder dysfunction. Musculoskeletal and neurologic symptoms are common in Lyme disease. B12 deficiency can present with worsening weakness and a sensory ataxia that can present as balance difficulties, but it would not cause focal lesions on the MRI, nor would it present with bladder symptoms. In addition, the patient's steady decline in function rules out RRMS.
PPMS is diagnosed with confirmation of gradual change in functional ability (often ambulation) over time without remission or relapse. These criteria include 1 full year of worsening neurologic function without asymptomatic periods as well as two of these signs of disease: brain lesion, two or more spinal cord lesions, and oligoclonal bands or elevated Immunoglobulin G index. These timing-specific criteria can delay diagnosis, as seen here.
Ocrelizumab is the only FDA-approved disease-modifying therapy (DMT) proven to alter disease progression in ambulatory patients with PPMS. American Academy of Neurology guidelines recommend ocrelizumab for patients with PPMS who are likely to benefit from this therapy. While it is thought that DMTs are more effective at targeting inflammation in RRMS than nerve degeneration in PPMS, these agents may show benefit for patients with active PPMS (relapse and/or evidence of new MRI activity) rather than inactive disease. A recent PPMS study concluded that among patients with relapse or disease activity, DMTs were associated with a significant reduction of long-term disability risk. Together with immunomodulatory therapy, rehabilitation can help manage symptoms.
Krupa Pandey, MD, Director, Multiple Sclerosis Center, Department of Neurology & Neuroscience Institute, Hackensack University Medical Center; Neurologist, Department of Neurology, Hackensack Meridian Health, Hackensack, NJ.
Krupa Pandey, MD, has serve(d) as a speaker or a member of a speakers bureau for: Bristol-Myers Squibb; Biogen; Alexion; Genentech; Sanofi-Genzyme.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
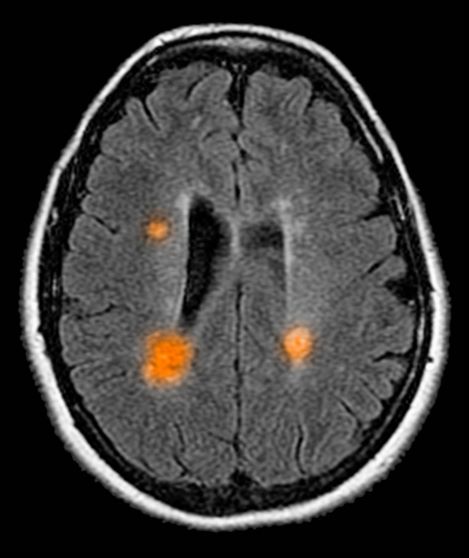
A 59-year-old man presents with worsening decline in ambulatory function and worsening bladder function. He reports "difficulty getting around" for the past year and a half, which he theorized might be because of arthritis, aging, or many years of biking. He presented to his primary care physician 2 months ago and was referred to rheumatology. His height is 5 ft 11 in and his weight is 166 lb (BMI 23.1). The patient subsequently reported a decreased attention span to the rheumatologist. He has no other significant medical or surgical history, though his brother has psoriatic arthritis. MRI shows multiple brain lesions without gadolinium enhancement and multiple spinal cord lesions.
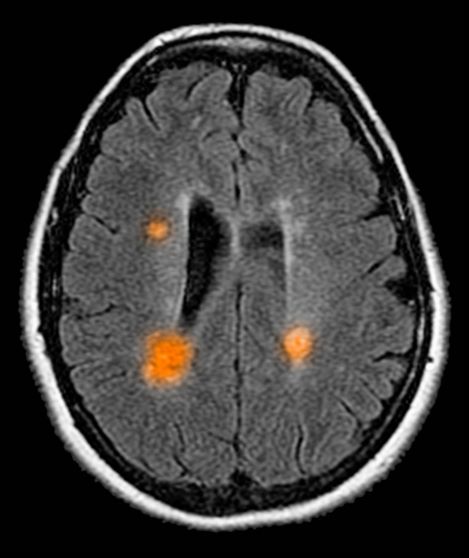
Chest tightness and wheezing
This patient's physical examination and imaging findings are consistent with a diagnosis of acute severe asthma. Agitation, breathlessness during rest, and a respiratory rate > 30 breaths/min are some manifestations of an acute severe episode. During severe episodes, accessory muscles of respiration are usually used, and suprasternal retractions are often present. The heart rate is > 120 beats/min and the respiratory rate is > 30 breaths/min. Loud biphasic (expiratory and inspiratory) wheezing can be heard, and pulsus paradoxus is often present (20-40 mm Hg). Oxyhemoglobin saturation with room air is < 91%. As the severity increases, the patient increasingly assumes a hunched-over sitting position with the hands supporting the torso, termed the tripod position.
Asthma is a chronic, heterogenous inflammatory airway disorder characterized by variable expiratory flow; airway wall thickening; respiratory symptoms; and exacerbations, which sometimes require hospitalization. According to the World Health Organization, asthma affected an estimated 262 million people in 2019. The presence of airway hyperresponsiveness or bronchial hyperreactivity in asthma is an exaggerated response to various exogenous and endogenous stimuli. Mechanisms implicated in the development of asthma include direct stimulation of airway smooth muscle and indirect stimulation by pharmacologically active substances from mediator-secreting cells, such as mast cells or nonmyelinated sensory neurons. The degree of airway hyperresponsiveness is associated with the clinical severity of asthma.
Acute severe asthma is a life-threatening emergency characterized by severe airflow limitation that is unresponsive to the typical appropriate bronchodilator therapy. As a result of pathophysiologic changes, airflow is severely restricted in severe asthma, leading to premature closure of the airway on expiration; impaired gas exchange; and dynamic hyperinflation, or air-trapping. In such cases, urgent action is essential to thwart serious outcomes, including mechanical ventilation and death.
Asthma severity is defined by the level of treatment required to control a patient's symptoms and exacerbations. According to the 2022 Global Initiative for Asthma (GINA) guidelines, a severe asthma exacerbation describes a patient who talks in words (rather than sentences); leans forward; is agitated; uses accessory respiratory muscles; and has a respiratory rate > 30 breaths/min, heart rate > 120 beats/min, oxygen saturation on air < 90%, and peak expiratory flow ≤ 50% of their best or of predicted value. Given the heterogeneity of asthma, patients with acute severe asthma may present with a variety of signs and symptoms, including dyspnea, chest tightness, cough and wheezing, agitation, drowsiness or signs of confusion, and significant breathlessness at rest.
Exposure to external agents, such as indoor and outdoor allergens, air pollutants, and respiratory tract infections (primarily viral), are the most common causes of asthma exacerbations, which vary in severity. Numerous other factors can trigger an asthma exacerbation, including exercise, weather changes, certain foods, additives, drugs, extreme emotional expressions, rhinitis, sinusitis, polyposis, gastroesophageal reflux, menstruation, and pregnancy. Importantly, a patient with known asthma of any level of severity can experience an asthma exacerbation, including patients with mild or well-controlled asthma.
Patients with a history of poorly controlled asthma or a recent exacerbation are at risk for an acute asthma exacerbation. Other risk factors include poor perception of airflow limitation, regular or overuse of short-acting beta agonists, incorrect inhaler technique, and suboptimal adherence to therapy. Comorbidities associated with risk for an acute asthma exacerbation include obesity, chronic rhinosinusitis, inducible laryngeal obstruction (vocal cord dysfunction), gastroesophageal reflux disease, chronic obstructive pulmonary disease, obstructive sleep apnea, bronchiectasis, cardiac disease, and kyphosis due to osteoporosis (followed by corticosteroid overuse). The lack of a written asthma action plan and socioeconomic factors are also associated with increased risk for a severe exacerbation.
In the emergency department setting, pharmacologic therapy of acute severe asthma should consist of a short-acting beta agonist, ipratropium bromide, systemic corticosteroids (oral or intravenous), and controlled oxygen therapy. Clinicians may also consider intravenous magnesium sulfate and high-dose inhaled corticosteroids. Once stable, patients should be treated with optimal asthma-controlling therapy, as outlined in GINA guidelines. Optimizing patients' inhaler technique and adherence to therapy are imperative, and comorbidities should be appropriately managed. Nonpharmacologic interventions, such as smoking cessation, pulmonary rehabilitation, exercise, weight loss, and influenza/COVID-19 vaccination, are also recommended as indicated.
Zab Mosenifar, MD, Medical Director, Women's Lung Institute; Executive Vice Chairman, Department of Medicine, Cedars Sinai Medical Center, Los Angeles, California.
Zab Mosenifar, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's physical examination and imaging findings are consistent with a diagnosis of acute severe asthma. Agitation, breathlessness during rest, and a respiratory rate > 30 breaths/min are some manifestations of an acute severe episode. During severe episodes, accessory muscles of respiration are usually used, and suprasternal retractions are often present. The heart rate is > 120 beats/min and the respiratory rate is > 30 breaths/min. Loud biphasic (expiratory and inspiratory) wheezing can be heard, and pulsus paradoxus is often present (20-40 mm Hg). Oxyhemoglobin saturation with room air is < 91%. As the severity increases, the patient increasingly assumes a hunched-over sitting position with the hands supporting the torso, termed the tripod position.
Asthma is a chronic, heterogenous inflammatory airway disorder characterized by variable expiratory flow; airway wall thickening; respiratory symptoms; and exacerbations, which sometimes require hospitalization. According to the World Health Organization, asthma affected an estimated 262 million people in 2019. The presence of airway hyperresponsiveness or bronchial hyperreactivity in asthma is an exaggerated response to various exogenous and endogenous stimuli. Mechanisms implicated in the development of asthma include direct stimulation of airway smooth muscle and indirect stimulation by pharmacologically active substances from mediator-secreting cells, such as mast cells or nonmyelinated sensory neurons. The degree of airway hyperresponsiveness is associated with the clinical severity of asthma.
Acute severe asthma is a life-threatening emergency characterized by severe airflow limitation that is unresponsive to the typical appropriate bronchodilator therapy. As a result of pathophysiologic changes, airflow is severely restricted in severe asthma, leading to premature closure of the airway on expiration; impaired gas exchange; and dynamic hyperinflation, or air-trapping. In such cases, urgent action is essential to thwart serious outcomes, including mechanical ventilation and death.
Asthma severity is defined by the level of treatment required to control a patient's symptoms and exacerbations. According to the 2022 Global Initiative for Asthma (GINA) guidelines, a severe asthma exacerbation describes a patient who talks in words (rather than sentences); leans forward; is agitated; uses accessory respiratory muscles; and has a respiratory rate > 30 breaths/min, heart rate > 120 beats/min, oxygen saturation on air < 90%, and peak expiratory flow ≤ 50% of their best or of predicted value. Given the heterogeneity of asthma, patients with acute severe asthma may present with a variety of signs and symptoms, including dyspnea, chest tightness, cough and wheezing, agitation, drowsiness or signs of confusion, and significant breathlessness at rest.
Exposure to external agents, such as indoor and outdoor allergens, air pollutants, and respiratory tract infections (primarily viral), are the most common causes of asthma exacerbations, which vary in severity. Numerous other factors can trigger an asthma exacerbation, including exercise, weather changes, certain foods, additives, drugs, extreme emotional expressions, rhinitis, sinusitis, polyposis, gastroesophageal reflux, menstruation, and pregnancy. Importantly, a patient with known asthma of any level of severity can experience an asthma exacerbation, including patients with mild or well-controlled asthma.
Patients with a history of poorly controlled asthma or a recent exacerbation are at risk for an acute asthma exacerbation. Other risk factors include poor perception of airflow limitation, regular or overuse of short-acting beta agonists, incorrect inhaler technique, and suboptimal adherence to therapy. Comorbidities associated with risk for an acute asthma exacerbation include obesity, chronic rhinosinusitis, inducible laryngeal obstruction (vocal cord dysfunction), gastroesophageal reflux disease, chronic obstructive pulmonary disease, obstructive sleep apnea, bronchiectasis, cardiac disease, and kyphosis due to osteoporosis (followed by corticosteroid overuse). The lack of a written asthma action plan and socioeconomic factors are also associated with increased risk for a severe exacerbation.
In the emergency department setting, pharmacologic therapy of acute severe asthma should consist of a short-acting beta agonist, ipratropium bromide, systemic corticosteroids (oral or intravenous), and controlled oxygen therapy. Clinicians may also consider intravenous magnesium sulfate and high-dose inhaled corticosteroids. Once stable, patients should be treated with optimal asthma-controlling therapy, as outlined in GINA guidelines. Optimizing patients' inhaler technique and adherence to therapy are imperative, and comorbidities should be appropriately managed. Nonpharmacologic interventions, such as smoking cessation, pulmonary rehabilitation, exercise, weight loss, and influenza/COVID-19 vaccination, are also recommended as indicated.
Zab Mosenifar, MD, Medical Director, Women's Lung Institute; Executive Vice Chairman, Department of Medicine, Cedars Sinai Medical Center, Los Angeles, California.
Zab Mosenifar, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's physical examination and imaging findings are consistent with a diagnosis of acute severe asthma. Agitation, breathlessness during rest, and a respiratory rate > 30 breaths/min are some manifestations of an acute severe episode. During severe episodes, accessory muscles of respiration are usually used, and suprasternal retractions are often present. The heart rate is > 120 beats/min and the respiratory rate is > 30 breaths/min. Loud biphasic (expiratory and inspiratory) wheezing can be heard, and pulsus paradoxus is often present (20-40 mm Hg). Oxyhemoglobin saturation with room air is < 91%. As the severity increases, the patient increasingly assumes a hunched-over sitting position with the hands supporting the torso, termed the tripod position.
Asthma is a chronic, heterogenous inflammatory airway disorder characterized by variable expiratory flow; airway wall thickening; respiratory symptoms; and exacerbations, which sometimes require hospitalization. According to the World Health Organization, asthma affected an estimated 262 million people in 2019. The presence of airway hyperresponsiveness or bronchial hyperreactivity in asthma is an exaggerated response to various exogenous and endogenous stimuli. Mechanisms implicated in the development of asthma include direct stimulation of airway smooth muscle and indirect stimulation by pharmacologically active substances from mediator-secreting cells, such as mast cells or nonmyelinated sensory neurons. The degree of airway hyperresponsiveness is associated with the clinical severity of asthma.
Acute severe asthma is a life-threatening emergency characterized by severe airflow limitation that is unresponsive to the typical appropriate bronchodilator therapy. As a result of pathophysiologic changes, airflow is severely restricted in severe asthma, leading to premature closure of the airway on expiration; impaired gas exchange; and dynamic hyperinflation, or air-trapping. In such cases, urgent action is essential to thwart serious outcomes, including mechanical ventilation and death.
Asthma severity is defined by the level of treatment required to control a patient's symptoms and exacerbations. According to the 2022 Global Initiative for Asthma (GINA) guidelines, a severe asthma exacerbation describes a patient who talks in words (rather than sentences); leans forward; is agitated; uses accessory respiratory muscles; and has a respiratory rate > 30 breaths/min, heart rate > 120 beats/min, oxygen saturation on air < 90%, and peak expiratory flow ≤ 50% of their best or of predicted value. Given the heterogeneity of asthma, patients with acute severe asthma may present with a variety of signs and symptoms, including dyspnea, chest tightness, cough and wheezing, agitation, drowsiness or signs of confusion, and significant breathlessness at rest.
Exposure to external agents, such as indoor and outdoor allergens, air pollutants, and respiratory tract infections (primarily viral), are the most common causes of asthma exacerbations, which vary in severity. Numerous other factors can trigger an asthma exacerbation, including exercise, weather changes, certain foods, additives, drugs, extreme emotional expressions, rhinitis, sinusitis, polyposis, gastroesophageal reflux, menstruation, and pregnancy. Importantly, a patient with known asthma of any level of severity can experience an asthma exacerbation, including patients with mild or well-controlled asthma.
Patients with a history of poorly controlled asthma or a recent exacerbation are at risk for an acute asthma exacerbation. Other risk factors include poor perception of airflow limitation, regular or overuse of short-acting beta agonists, incorrect inhaler technique, and suboptimal adherence to therapy. Comorbidities associated with risk for an acute asthma exacerbation include obesity, chronic rhinosinusitis, inducible laryngeal obstruction (vocal cord dysfunction), gastroesophageal reflux disease, chronic obstructive pulmonary disease, obstructive sleep apnea, bronchiectasis, cardiac disease, and kyphosis due to osteoporosis (followed by corticosteroid overuse). The lack of a written asthma action plan and socioeconomic factors are also associated with increased risk for a severe exacerbation.
In the emergency department setting, pharmacologic therapy of acute severe asthma should consist of a short-acting beta agonist, ipratropium bromide, systemic corticosteroids (oral or intravenous), and controlled oxygen therapy. Clinicians may also consider intravenous magnesium sulfate and high-dose inhaled corticosteroids. Once stable, patients should be treated with optimal asthma-controlling therapy, as outlined in GINA guidelines. Optimizing patients' inhaler technique and adherence to therapy are imperative, and comorbidities should be appropriately managed. Nonpharmacologic interventions, such as smoking cessation, pulmonary rehabilitation, exercise, weight loss, and influenza/COVID-19 vaccination, are also recommended as indicated.
Zab Mosenifar, MD, Medical Director, Women's Lung Institute; Executive Vice Chairman, Department of Medicine, Cedars Sinai Medical Center, Los Angeles, California.
Zab Mosenifar, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
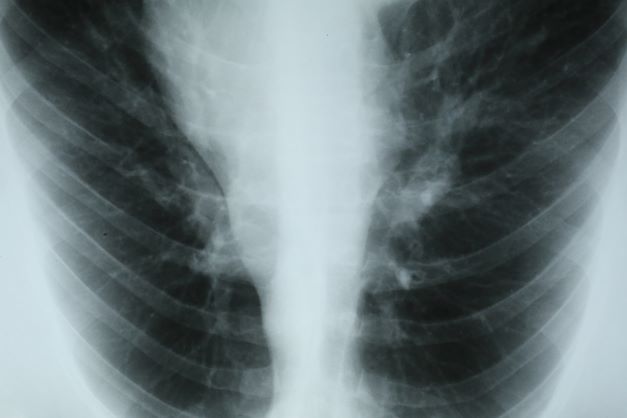
A 32-year-old Black man presents to the emergency department with severe dyspnea, chest tightness, and wheezing. The patient is sitting forward in the tripod position and appears agitated and confused. Use of accessory respiratory muscles and suprasternal retractions are noted. He reports an approximate 2-week history of rhinorrhea, cough, and mild fever, for which he has been taking an over-the-counter nonsteroidal anti-inflammatory agent and cough suppressant. His prior medical history is notable for obesity, type 2 diabetes, allergic rhinitis, mild asthma, and hypercholesterolemia. The patient is a current smoker (17 pack-years). Pertinent physical examination reveals a respiratory rate of 48 breaths/min, heart rate of 135 beats/min, 87% oxygen saturation, and peak expiratory flow of 300 L/min. Low biphasic wheezing can be heard. Rapid antigen and PCR tests for SARS-CoV-2 detected by nasopharyngeal swabs both come back negative. Chest radiography is ordered and shows pulmonary hyperinflation with bronchial wall thickening.