User login
Farxiga granted Priority Review for treatment of adults with HFrEF
The Food and Drug Administration has accepted a supplemental New Drug Application and granted Priority Review for dapagliflozin (Farxiga) for the reduction of risk of cardiovascular death or worsening of heart failure in adult patients with heart failure with reduced ejection fraction (HFrEF).

The application was based on results from the landmark, phase 3 DAPA-HF trial, published in September 2019 in the New England Journal of Medicine. The study showed that dapagliflozin plus standard care reduced the incidence of cardiovascular death and worsening of heart failure versus placebo in patients with HFrEF.
Dapagliflozin was granted Fast Track designation for heart failure by the FDA in September 2019. In August 2019, the FDA also granted Fast Track designation to dapagliflozin for the delayed progression of renal failure and prevention of cardiovascular and renal death in patients with chronic kidney disease.
The drug is currently indicated for the improvement of glycemic control in adults with type 2 diabetes as either monotherapy or in combination. The FDA approved dapagliflozin in October 2019 for the reduction of heart failure hospitalization risk in patients with type 2 diabetes and cardiovascular risk factors.
“Farxiga is well established in the treatment of type 2 diabetes and this Priority Review shows its potential to also impact millions of patients with heart failure. If approved, Farxiga will be the first and only medicine of its kind indicated to treat patients with heart failure,” said Mene Pangalos, executive vice president of biopharmaceutical research and development at AstraZeneca.
Find the full press release on the AstraZeneca website.
The Food and Drug Administration has accepted a supplemental New Drug Application and granted Priority Review for dapagliflozin (Farxiga) for the reduction of risk of cardiovascular death or worsening of heart failure in adult patients with heart failure with reduced ejection fraction (HFrEF).
The application was based on results from the landmark, phase 3 DAPA-HF trial, published in September 2019 in the New England Journal of Medicine. The study showed that dapagliflozin plus standard care reduced the incidence of cardiovascular death and worsening of heart failure versus placebo in patients with HFrEF.
Dapagliflozin was granted Fast Track designation for heart failure by the FDA in September 2019. In August 2019, the FDA also granted Fast Track designation to dapagliflozin for the delayed progression of renal failure and prevention of cardiovascular and renal death in patients with chronic kidney disease.
The drug is currently indicated for the improvement of glycemic control in adults with type 2 diabetes as either monotherapy or in combination. The FDA approved dapagliflozin in October 2019 for the reduction of heart failure hospitalization risk in patients with type 2 diabetes and cardiovascular risk factors.
“Farxiga is well established in the treatment of type 2 diabetes and this Priority Review shows its potential to also impact millions of patients with heart failure. If approved, Farxiga will be the first and only medicine of its kind indicated to treat patients with heart failure,” said Mene Pangalos, executive vice president of biopharmaceutical research and development at AstraZeneca.
Find the full press release on the AstraZeneca website.
The Food and Drug Administration has accepted a supplemental New Drug Application and granted Priority Review for dapagliflozin (Farxiga) for the reduction of risk of cardiovascular death or worsening of heart failure in adult patients with heart failure with reduced ejection fraction (HFrEF).
The application was based on results from the landmark, phase 3 DAPA-HF trial, published in September 2019 in the New England Journal of Medicine. The study showed that dapagliflozin plus standard care reduced the incidence of cardiovascular death and worsening of heart failure versus placebo in patients with HFrEF.
Dapagliflozin was granted Fast Track designation for heart failure by the FDA in September 2019. In August 2019, the FDA also granted Fast Track designation to dapagliflozin for the delayed progression of renal failure and prevention of cardiovascular and renal death in patients with chronic kidney disease.
The drug is currently indicated for the improvement of glycemic control in adults with type 2 diabetes as either monotherapy or in combination. The FDA approved dapagliflozin in October 2019 for the reduction of heart failure hospitalization risk in patients with type 2 diabetes and cardiovascular risk factors.
“Farxiga is well established in the treatment of type 2 diabetes and this Priority Review shows its potential to also impact millions of patients with heart failure. If approved, Farxiga will be the first and only medicine of its kind indicated to treat patients with heart failure,” said Mene Pangalos, executive vice president of biopharmaceutical research and development at AstraZeneca.
Find the full press release on the AstraZeneca website.
FDA okays first generics for Eliquis
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.

The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
FDA okays ubrogepant for acute migraine treatment
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.

Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.
Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.
Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
FDA approves Caplyta to treat schizophrenia in adults
The Food and Drug Administration has approved lumateperone for the treatment of schizophrenia in adults.
Lumateperone, an atypical antipsychotic, will be marketed by Intra-Cellular Therapies as Caplyta, according to a Dec. 23 statement from the company.
In the first, 335 patients with schizophrenia were randomized to two doses of lumateperone, an active comparator, or placebo. Those randomized to the approved 42-mg dose of lumateperone showed a statistically significant reduction in total score on the Positive and Negative Syndrome Scale (PANSS), compared with patients in the other groups. The median age in this study was 42 years; 17% of patients were female, 19% were white, and 78% were black.
In the second study, 450 patients diagnosed with schizophrenia were randomized in a double-blind fashion to one of two doses of lumateperone or placebo. Patients taking the approved dose showed a statistically significant reduction from baseline to day 28 in PANSS total score. In this study, patients’ median age was 44 years; 23% were female, 26% were white, and 66% were black.
Treatment with lumateperone appears to feature a more favorable cardiometabolic profile than that of other approved antipsychotic agents.
Patients treated with lumateperone for at least a year showed significant reductions in LDL cholesterol, total cholesterol, serum prolactin, and body weight, compared with baseline values recorded when participants were on various standard-of-care antipsychotics prior to switching, Suresh Durgam, MD, reported at the annual congress of the European College of Neuropsychopharmacology. Other cardiometabolic parameters, including fasting blood glucose, insulin, triglycerides, and HDL cholesterol, showed only negligible change over the course of study, according to Dr. Durgam, a psychiatrist and senior vice president for late-stage clinical development and medical affairs at Intra-Cellular Therapies.
The most common adverse events with lumateperone were somnolence (24% vs. 10% on placebo) and dry mouth (6% vs. 2%).
Approval of lumateperone hit a snag last summer when the FDA canceled the Psychopharmacologic Drugs Advisory Committee meeting it previously had called for to review the new drug application for lumateperone. The agency said the meeting was canceled because of “new information regarding the application.”
At the time, Intra-Cellular Therapies noted that it had provided additional information to the FDA to meet agency requests. This information was related to nonclinical studies.
“The FDA canceled the advisory committee meeting to allow sufficient time to review this new and any forthcoming information as they continue” to review the new drug application for lumateperone, Intra-Cellular said a statement.
The company plans to launch Caplyta late in the first quarter of 2020.
Bruce Jancin and Kerry Dooley Young contributed to this report.
The Food and Drug Administration has approved lumateperone for the treatment of schizophrenia in adults.
Lumateperone, an atypical antipsychotic, will be marketed by Intra-Cellular Therapies as Caplyta, according to a Dec. 23 statement from the company.
In the first, 335 patients with schizophrenia were randomized to two doses of lumateperone, an active comparator, or placebo. Those randomized to the approved 42-mg dose of lumateperone showed a statistically significant reduction in total score on the Positive and Negative Syndrome Scale (PANSS), compared with patients in the other groups. The median age in this study was 42 years; 17% of patients were female, 19% were white, and 78% were black.
In the second study, 450 patients diagnosed with schizophrenia were randomized in a double-blind fashion to one of two doses of lumateperone or placebo. Patients taking the approved dose showed a statistically significant reduction from baseline to day 28 in PANSS total score. In this study, patients’ median age was 44 years; 23% were female, 26% were white, and 66% were black.
Treatment with lumateperone appears to feature a more favorable cardiometabolic profile than that of other approved antipsychotic agents.
Patients treated with lumateperone for at least a year showed significant reductions in LDL cholesterol, total cholesterol, serum prolactin, and body weight, compared with baseline values recorded when participants were on various standard-of-care antipsychotics prior to switching, Suresh Durgam, MD, reported at the annual congress of the European College of Neuropsychopharmacology. Other cardiometabolic parameters, including fasting blood glucose, insulin, triglycerides, and HDL cholesterol, showed only negligible change over the course of study, according to Dr. Durgam, a psychiatrist and senior vice president for late-stage clinical development and medical affairs at Intra-Cellular Therapies.
The most common adverse events with lumateperone were somnolence (24% vs. 10% on placebo) and dry mouth (6% vs. 2%).
Approval of lumateperone hit a snag last summer when the FDA canceled the Psychopharmacologic Drugs Advisory Committee meeting it previously had called for to review the new drug application for lumateperone. The agency said the meeting was canceled because of “new information regarding the application.”
At the time, Intra-Cellular Therapies noted that it had provided additional information to the FDA to meet agency requests. This information was related to nonclinical studies.
“The FDA canceled the advisory committee meeting to allow sufficient time to review this new and any forthcoming information as they continue” to review the new drug application for lumateperone, Intra-Cellular said a statement.
The company plans to launch Caplyta late in the first quarter of 2020.
Bruce Jancin and Kerry Dooley Young contributed to this report.
The Food and Drug Administration has approved lumateperone for the treatment of schizophrenia in adults.
Lumateperone, an atypical antipsychotic, will be marketed by Intra-Cellular Therapies as Caplyta, according to a Dec. 23 statement from the company.
In the first, 335 patients with schizophrenia were randomized to two doses of lumateperone, an active comparator, or placebo. Those randomized to the approved 42-mg dose of lumateperone showed a statistically significant reduction in total score on the Positive and Negative Syndrome Scale (PANSS), compared with patients in the other groups. The median age in this study was 42 years; 17% of patients were female, 19% were white, and 78% were black.
In the second study, 450 patients diagnosed with schizophrenia were randomized in a double-blind fashion to one of two doses of lumateperone or placebo. Patients taking the approved dose showed a statistically significant reduction from baseline to day 28 in PANSS total score. In this study, patients’ median age was 44 years; 23% were female, 26% were white, and 66% were black.
Treatment with lumateperone appears to feature a more favorable cardiometabolic profile than that of other approved antipsychotic agents.
Patients treated with lumateperone for at least a year showed significant reductions in LDL cholesterol, total cholesterol, serum prolactin, and body weight, compared with baseline values recorded when participants were on various standard-of-care antipsychotics prior to switching, Suresh Durgam, MD, reported at the annual congress of the European College of Neuropsychopharmacology. Other cardiometabolic parameters, including fasting blood glucose, insulin, triglycerides, and HDL cholesterol, showed only negligible change over the course of study, according to Dr. Durgam, a psychiatrist and senior vice president for late-stage clinical development and medical affairs at Intra-Cellular Therapies.
The most common adverse events with lumateperone were somnolence (24% vs. 10% on placebo) and dry mouth (6% vs. 2%).
Approval of lumateperone hit a snag last summer when the FDA canceled the Psychopharmacologic Drugs Advisory Committee meeting it previously had called for to review the new drug application for lumateperone. The agency said the meeting was canceled because of “new information regarding the application.”
At the time, Intra-Cellular Therapies noted that it had provided additional information to the FDA to meet agency requests. This information was related to nonclinical studies.
“The FDA canceled the advisory committee meeting to allow sufficient time to review this new and any forthcoming information as they continue” to review the new drug application for lumateperone, Intra-Cellular said a statement.
The company plans to launch Caplyta late in the first quarter of 2020.
Bruce Jancin and Kerry Dooley Young contributed to this report.
FDA panel okays teprotumumab for thyroid eye disease
TED is a rare autoimmune disease that causes the eyes to bulge (proptosis) and can lead to blindness. It is also known as thyroid-associated ophthalmopathy, Graves ophthalmopathy, and Graves orbitopathy. Current treatment is aimed at relief of symptoms and includes corticosteroids and orbital decompression.
“TED can affect patients both physically and emotionally, limiting their ability to perform everyday activities like driving, working, reading, sleeping, and participating in social activities,” Jeff Todd, president and chief executive officer, Prevent Blindness, said in a news release.
“As an organization dedicated to helping patients with vision impairment and those who are at significant risk, we are extremely encouraged by today’s vote and hopeful this will change the future of TED treatment by giving patients an option that has been shown to improve the painful and vision-threatening aspects of the disease,” Mr. Todd said.
Teprotumumab was granted fast-track status in April 2015 and breakthrough therapy designation in July 2016. It received orphan drug designation on June 19, 2019. If approved, it would be the first approved treatment for this indication.
All 12 members of the Dermatologic and Ophthalmic Drugs Advisory Committee of the FDA voted to recommend approval of teprotumumab.
“It’s clearly a pleasure to participate in seeing a drug being designed and moving forward in a clinical trial for a disease that really has not been treatable for us in the past,” said voting committee member Timothy Murray, MD, MBA, of the Bascom Palmer Eye Institute, Miami.
Efficacy demonstrated
The FDA advisory committee considered data from two randomized, double-masked, placebo-controlled, parallel-group studies that were similar in design and that demonstrated efficacy.
The studies included a total of 171 patients, fewer than 90 of whom were treated with teprotumumab, the agency explained in a briefing document. “This is a considerably smaller database than the common safety database of greater than 300 patients treated with a course of therapy,” it observed.
The study required that patients have proptosis but did not require that they have “progressive forward motion of the globe,” it noted.
The primary objective – a reduction in proptosis of 2 or more mm – was met in 82% of patients who received teprotumumab, compared with 16% of those who received placebo.
Review study No. 1 (TED01RV) was a phase 2 study, and review study No. 2 (OPTIC) was a phase 3 study. Both trials included a 24-week treatment period during which participants received teprotumumab or placebo intravenously every 3 weeks for a total of eight doses. Patients in both studies underwent a follow-up period during which they received no further treatment.
Some patients experienced a reduction in proptosis as early as 6 weeks after they received the first infusion. In an extension of TED01RV, that reduction extended for at least 4 weeks after the last infusion. For about 60% of responders, no relapse had occurred by week 72. The extension of OPTIC and an open-label treatment period for those with no response to placebo or teprotumumab are ongoing.
“I welcome the addition of this drug to our armamentarium to treat this horrible, horrible disease,” said temporary voting committee member John F. Stamler, MD, PhD, clinical instructor, ophthalmology and visual sciences, University of Iowa, Iowa City.
Adverse events
Teprotumumab inhibits the insulinlike growth factor–1 receptor, which can interfere with the body’s ability to regulate glucose, particularly in patients with diabetes. In the study, some patients with diabetes required additional insulin for glycemic control.
In three study participants whose baseline fasting blood glucose levels were normal, blood glucose levels were found to be elevated at one or more visits during the treatment period. None had a history of diabetes mellitus, but for two, baseline hemoglobin A1c levels were elevated.
Five or more patients reported loss of hearing (hypoacusis), and others reported tinnitus. One of them experienced a spontaneous return of hearing the day after the hypoacusis developed, whereas for others, hearing did not return until after teprotumumab treatment was completed. The mechanism of action for hearing loss is unclear.
Panel members felt the potential for hearing loss is important and that patients should receive some type of monitoring, but they did not all agree on when that testing should occur or who should be responsible for getting it done.
“It strikes me that, if this drug were approved, there would be centers that would be interested in undertaking independent studies of hearing loss in treated patients, and that that could be done outside the sponsor’s responsibility and probably would be of interest to independent investigators,” noted committee chairperson James Chodosh, MD, MPH, of Massachusetts Eye and Ear and Harvard Medical School, Boston.
Benefits outweigh risks
More than one-third of patients (36%) experienced gastrointestinal complaints such as nausea and diarrhea (12% each) and abdominal pain (5%). None of the cases caused any patient to discontinue the study drug.
The overall incidence of muscle spasms was more than three times higher in the teprotumumab group (32%) than in the placebo group (9.5%).
One patient stopped taking teprotumumab after being hospitalized for Escherichia coli sepsis and dehydration, and another participant stopped after experiencing an episode of inflammatory bowel disease. It is not clear whether there is a causal association between teprotumumab and inflammatory bowel disease.
No study participants died.
Panel members expressed concern about safety in the longer term or in patients who receive multiple courses of teprotumumab, but they felt the potential benefits from teprotumumab outweigh the risks. The committee also wanted more information about the effects of teprotumumab with respect to glucose control, hearing loss, and other outcomes that are very important to patients, such as alopecia.
The panel favored including diarrhea in the list of adverse events in the label and felt that there should be a warning about inflammatory bowel disease.
“We’re finally going to be able to get a lot of people some help,” concluded voting committee member Sidney Gicheru, MD, LaserCare Eye Center, Irving, Texas.
A version of this story originally appeared on Medscape.com.
TED is a rare autoimmune disease that causes the eyes to bulge (proptosis) and can lead to blindness. It is also known as thyroid-associated ophthalmopathy, Graves ophthalmopathy, and Graves orbitopathy. Current treatment is aimed at relief of symptoms and includes corticosteroids and orbital decompression.
“TED can affect patients both physically and emotionally, limiting their ability to perform everyday activities like driving, working, reading, sleeping, and participating in social activities,” Jeff Todd, president and chief executive officer, Prevent Blindness, said in a news release.
“As an organization dedicated to helping patients with vision impairment and those who are at significant risk, we are extremely encouraged by today’s vote and hopeful this will change the future of TED treatment by giving patients an option that has been shown to improve the painful and vision-threatening aspects of the disease,” Mr. Todd said.
Teprotumumab was granted fast-track status in April 2015 and breakthrough therapy designation in July 2016. It received orphan drug designation on June 19, 2019. If approved, it would be the first approved treatment for this indication.
All 12 members of the Dermatologic and Ophthalmic Drugs Advisory Committee of the FDA voted to recommend approval of teprotumumab.
“It’s clearly a pleasure to participate in seeing a drug being designed and moving forward in a clinical trial for a disease that really has not been treatable for us in the past,” said voting committee member Timothy Murray, MD, MBA, of the Bascom Palmer Eye Institute, Miami.
Efficacy demonstrated
The FDA advisory committee considered data from two randomized, double-masked, placebo-controlled, parallel-group studies that were similar in design and that demonstrated efficacy.
The studies included a total of 171 patients, fewer than 90 of whom were treated with teprotumumab, the agency explained in a briefing document. “This is a considerably smaller database than the common safety database of greater than 300 patients treated with a course of therapy,” it observed.
The study required that patients have proptosis but did not require that they have “progressive forward motion of the globe,” it noted.
The primary objective – a reduction in proptosis of 2 or more mm – was met in 82% of patients who received teprotumumab, compared with 16% of those who received placebo.
Review study No. 1 (TED01RV) was a phase 2 study, and review study No. 2 (OPTIC) was a phase 3 study. Both trials included a 24-week treatment period during which participants received teprotumumab or placebo intravenously every 3 weeks for a total of eight doses. Patients in both studies underwent a follow-up period during which they received no further treatment.
Some patients experienced a reduction in proptosis as early as 6 weeks after they received the first infusion. In an extension of TED01RV, that reduction extended for at least 4 weeks after the last infusion. For about 60% of responders, no relapse had occurred by week 72. The extension of OPTIC and an open-label treatment period for those with no response to placebo or teprotumumab are ongoing.
“I welcome the addition of this drug to our armamentarium to treat this horrible, horrible disease,” said temporary voting committee member John F. Stamler, MD, PhD, clinical instructor, ophthalmology and visual sciences, University of Iowa, Iowa City.
Adverse events
Teprotumumab inhibits the insulinlike growth factor–1 receptor, which can interfere with the body’s ability to regulate glucose, particularly in patients with diabetes. In the study, some patients with diabetes required additional insulin for glycemic control.
In three study participants whose baseline fasting blood glucose levels were normal, blood glucose levels were found to be elevated at one or more visits during the treatment period. None had a history of diabetes mellitus, but for two, baseline hemoglobin A1c levels were elevated.
Five or more patients reported loss of hearing (hypoacusis), and others reported tinnitus. One of them experienced a spontaneous return of hearing the day after the hypoacusis developed, whereas for others, hearing did not return until after teprotumumab treatment was completed. The mechanism of action for hearing loss is unclear.
Panel members felt the potential for hearing loss is important and that patients should receive some type of monitoring, but they did not all agree on when that testing should occur or who should be responsible for getting it done.
“It strikes me that, if this drug were approved, there would be centers that would be interested in undertaking independent studies of hearing loss in treated patients, and that that could be done outside the sponsor’s responsibility and probably would be of interest to independent investigators,” noted committee chairperson James Chodosh, MD, MPH, of Massachusetts Eye and Ear and Harvard Medical School, Boston.
Benefits outweigh risks
More than one-third of patients (36%) experienced gastrointestinal complaints such as nausea and diarrhea (12% each) and abdominal pain (5%). None of the cases caused any patient to discontinue the study drug.
The overall incidence of muscle spasms was more than three times higher in the teprotumumab group (32%) than in the placebo group (9.5%).
One patient stopped taking teprotumumab after being hospitalized for Escherichia coli sepsis and dehydration, and another participant stopped after experiencing an episode of inflammatory bowel disease. It is not clear whether there is a causal association between teprotumumab and inflammatory bowel disease.
No study participants died.
Panel members expressed concern about safety in the longer term or in patients who receive multiple courses of teprotumumab, but they felt the potential benefits from teprotumumab outweigh the risks. The committee also wanted more information about the effects of teprotumumab with respect to glucose control, hearing loss, and other outcomes that are very important to patients, such as alopecia.
The panel favored including diarrhea in the list of adverse events in the label and felt that there should be a warning about inflammatory bowel disease.
“We’re finally going to be able to get a lot of people some help,” concluded voting committee member Sidney Gicheru, MD, LaserCare Eye Center, Irving, Texas.
A version of this story originally appeared on Medscape.com.
TED is a rare autoimmune disease that causes the eyes to bulge (proptosis) and can lead to blindness. It is also known as thyroid-associated ophthalmopathy, Graves ophthalmopathy, and Graves orbitopathy. Current treatment is aimed at relief of symptoms and includes corticosteroids and orbital decompression.
“TED can affect patients both physically and emotionally, limiting their ability to perform everyday activities like driving, working, reading, sleeping, and participating in social activities,” Jeff Todd, president and chief executive officer, Prevent Blindness, said in a news release.
“As an organization dedicated to helping patients with vision impairment and those who are at significant risk, we are extremely encouraged by today’s vote and hopeful this will change the future of TED treatment by giving patients an option that has been shown to improve the painful and vision-threatening aspects of the disease,” Mr. Todd said.
Teprotumumab was granted fast-track status in April 2015 and breakthrough therapy designation in July 2016. It received orphan drug designation on June 19, 2019. If approved, it would be the first approved treatment for this indication.
All 12 members of the Dermatologic and Ophthalmic Drugs Advisory Committee of the FDA voted to recommend approval of teprotumumab.
“It’s clearly a pleasure to participate in seeing a drug being designed and moving forward in a clinical trial for a disease that really has not been treatable for us in the past,” said voting committee member Timothy Murray, MD, MBA, of the Bascom Palmer Eye Institute, Miami.
Efficacy demonstrated
The FDA advisory committee considered data from two randomized, double-masked, placebo-controlled, parallel-group studies that were similar in design and that demonstrated efficacy.
The studies included a total of 171 patients, fewer than 90 of whom were treated with teprotumumab, the agency explained in a briefing document. “This is a considerably smaller database than the common safety database of greater than 300 patients treated with a course of therapy,” it observed.
The study required that patients have proptosis but did not require that they have “progressive forward motion of the globe,” it noted.
The primary objective – a reduction in proptosis of 2 or more mm – was met in 82% of patients who received teprotumumab, compared with 16% of those who received placebo.
Review study No. 1 (TED01RV) was a phase 2 study, and review study No. 2 (OPTIC) was a phase 3 study. Both trials included a 24-week treatment period during which participants received teprotumumab or placebo intravenously every 3 weeks for a total of eight doses. Patients in both studies underwent a follow-up period during which they received no further treatment.
Some patients experienced a reduction in proptosis as early as 6 weeks after they received the first infusion. In an extension of TED01RV, that reduction extended for at least 4 weeks after the last infusion. For about 60% of responders, no relapse had occurred by week 72. The extension of OPTIC and an open-label treatment period for those with no response to placebo or teprotumumab are ongoing.
“I welcome the addition of this drug to our armamentarium to treat this horrible, horrible disease,” said temporary voting committee member John F. Stamler, MD, PhD, clinical instructor, ophthalmology and visual sciences, University of Iowa, Iowa City.
Adverse events
Teprotumumab inhibits the insulinlike growth factor–1 receptor, which can interfere with the body’s ability to regulate glucose, particularly in patients with diabetes. In the study, some patients with diabetes required additional insulin for glycemic control.
In three study participants whose baseline fasting blood glucose levels were normal, blood glucose levels were found to be elevated at one or more visits during the treatment period. None had a history of diabetes mellitus, but for two, baseline hemoglobin A1c levels were elevated.
Five or more patients reported loss of hearing (hypoacusis), and others reported tinnitus. One of them experienced a spontaneous return of hearing the day after the hypoacusis developed, whereas for others, hearing did not return until after teprotumumab treatment was completed. The mechanism of action for hearing loss is unclear.
Panel members felt the potential for hearing loss is important and that patients should receive some type of monitoring, but they did not all agree on when that testing should occur or who should be responsible for getting it done.
“It strikes me that, if this drug were approved, there would be centers that would be interested in undertaking independent studies of hearing loss in treated patients, and that that could be done outside the sponsor’s responsibility and probably would be of interest to independent investigators,” noted committee chairperson James Chodosh, MD, MPH, of Massachusetts Eye and Ear and Harvard Medical School, Boston.
Benefits outweigh risks
More than one-third of patients (36%) experienced gastrointestinal complaints such as nausea and diarrhea (12% each) and abdominal pain (5%). None of the cases caused any patient to discontinue the study drug.
The overall incidence of muscle spasms was more than three times higher in the teprotumumab group (32%) than in the placebo group (9.5%).
One patient stopped taking teprotumumab after being hospitalized for Escherichia coli sepsis and dehydration, and another participant stopped after experiencing an episode of inflammatory bowel disease. It is not clear whether there is a causal association between teprotumumab and inflammatory bowel disease.
No study participants died.
Panel members expressed concern about safety in the longer term or in patients who receive multiple courses of teprotumumab, but they felt the potential benefits from teprotumumab outweigh the risks. The committee also wanted more information about the effects of teprotumumab with respect to glucose control, hearing loss, and other outcomes that are very important to patients, such as alopecia.
The panel favored including diarrhea in the list of adverse events in the label and felt that there should be a warning about inflammatory bowel disease.
“We’re finally going to be able to get a lot of people some help,” concluded voting committee member Sidney Gicheru, MD, LaserCare Eye Center, Irving, Texas.
A version of this story originally appeared on Medscape.com.
Influenza activity continues to be unusually high
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
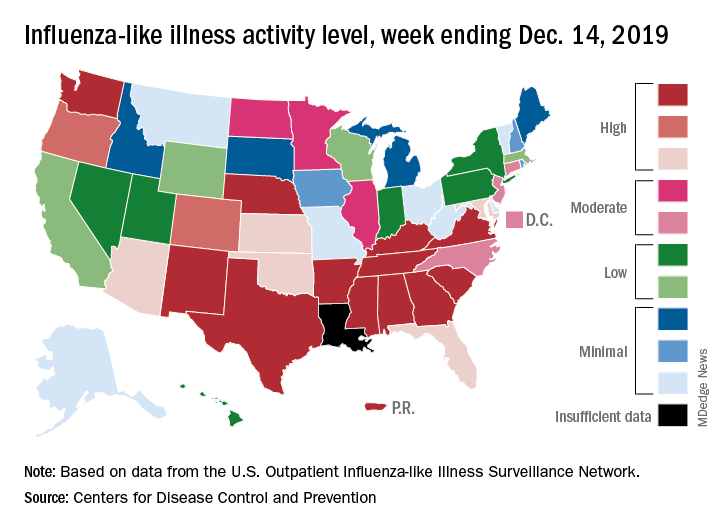
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
FDA approves antibody-drug conjugate for advanced urothelial cancer
The Food and Drug Administration has granted accelerated approval to enfortumab vedotin-ejfv (Padcev) for the treatment of adult patients with locally advanced or metastatic urothelial cancer that has previously been treated with a programmed cell death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.
The conjugate was approved based on overall response rate in a trial of 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy, the FDA said in a press statement.
The overall response rate was 44%, with 12% having a complete response and 32% having a partial response. The median duration of response was 7.6 months.
The most common side effects for patients were fatigue, peripheral neuropathy, decreased appetite, rash, alopecia, nausea, altered taste, diarrhea, dry eye, pruritis, and dry skin. Patients may experience hyperglycemia, and blood sugar levels should be monitored closely in patients receiving enfortumab vedotin-ejfv, the FDA said.
Patients may experience eye disorders, and health care professionals may consider prophylactic artificial tears for dry eyes and referral to an ophthalmologist for any new symptoms related to the eye, the agency said. The FDA also advises telling patients of reproductive age to use effective contraception during treatment, and for a period of time thereafter. Women who are pregnant or breastfeeding should not take the antibody-drug conjugate because it may cause harm to a developing fetus or newborn baby or cause delivery complications.
The Food and Drug Administration has granted accelerated approval to enfortumab vedotin-ejfv (Padcev) for the treatment of adult patients with locally advanced or metastatic urothelial cancer that has previously been treated with a programmed cell death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.
The conjugate was approved based on overall response rate in a trial of 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy, the FDA said in a press statement.
The overall response rate was 44%, with 12% having a complete response and 32% having a partial response. The median duration of response was 7.6 months.
The most common side effects for patients were fatigue, peripheral neuropathy, decreased appetite, rash, alopecia, nausea, altered taste, diarrhea, dry eye, pruritis, and dry skin. Patients may experience hyperglycemia, and blood sugar levels should be monitored closely in patients receiving enfortumab vedotin-ejfv, the FDA said.
Patients may experience eye disorders, and health care professionals may consider prophylactic artificial tears for dry eyes and referral to an ophthalmologist for any new symptoms related to the eye, the agency said. The FDA also advises telling patients of reproductive age to use effective contraception during treatment, and for a period of time thereafter. Women who are pregnant or breastfeeding should not take the antibody-drug conjugate because it may cause harm to a developing fetus or newborn baby or cause delivery complications.
The Food and Drug Administration has granted accelerated approval to enfortumab vedotin-ejfv (Padcev) for the treatment of adult patients with locally advanced or metastatic urothelial cancer that has previously been treated with a programmed cell death protein 1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.
The conjugate was approved based on overall response rate in a trial of 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy, the FDA said in a press statement.
The overall response rate was 44%, with 12% having a complete response and 32% having a partial response. The median duration of response was 7.6 months.
The most common side effects for patients were fatigue, peripheral neuropathy, decreased appetite, rash, alopecia, nausea, altered taste, diarrhea, dry eye, pruritis, and dry skin. Patients may experience hyperglycemia, and blood sugar levels should be monitored closely in patients receiving enfortumab vedotin-ejfv, the FDA said.
Patients may experience eye disorders, and health care professionals may consider prophylactic artificial tears for dry eyes and referral to an ophthalmologist for any new symptoms related to the eye, the agency said. The FDA also advises telling patients of reproductive age to use effective contraception during treatment, and for a period of time thereafter. Women who are pregnant or breastfeeding should not take the antibody-drug conjugate because it may cause harm to a developing fetus or newborn baby or cause delivery complications.
HHS drug importation proposals aim to address high costs
The Department of Health & Human Services is taking the first steps in allowing drugs to be imported into the United States.

HHS proposes to offer two different pathways for importation: One allowing states to design programs to import certain drugs directly from Canada and another allowing manufacturers to obtain a new National Drug Code (NDC) number to import their own Food and Drug Administration–approved products manufactured outside of the United States.
“The importation proposals we are rolling out ... are a historic step forward in efforts to bring down drug prices and out-of-pocket costs,” HHS Secretary Alex Azar said during a Dec. 17, 2019, press conference. “New pathways for importation can move us toward a more open and competitive marketplace that supplies American patients with safe, effective, affordable prescription drugs.”
The proposals were made public on Dec. 18, the day the House Rules committee was scheduled to vote on impeaching President Trump.
He emphasized that these proposals “are both important steps in advancing the FDA’s safe-importation action plan, [which] aims to insure that importation is done in a way that prioritizes safety and includes elements to help insure importation does not put patients or the U.S. drug supply chain at risk.”
The pathway for states to import drugs from Canada will be proposed through the federal regulatory process. The notice of proposed rulemaking, which implements authority for FDA regulation of importation granted in the Medicare Modernization Act of 2003, will outline a process by which states, potentially working with wholesalers and/or pharmacies, will submit proposals for FDA review and approval on how they would implement an importation program.
Only certain drugs would be eligible for importation from Canada under this proposal. The drugs would need to be approved in Canada and, except for Canadian labeling, need to meet the conditions of an FDA-approved new drug application or abbreviated new drug application.
Controlled substances, biologics, intravenously injected drugs, drugs with a risk evaluation and management strategy, and drugs injected into the spinal column or eye would be excluded from importation.
Drugs coming in from Canada would be relabeled with U.S.-approved labels and would be subject to testing to ensure they are authentic, not degraded, and compliant with U.S. standards.
States would be required to show that importing drugs poses no additional risk in public health and safety and it would result in the reduction of costs, according to information provided by HHS.
Many of the most expensive drugs, as well as insulins, would not be eligible for importation under this pathway, Mr. Azar acknowledged, adding that “I would envision that as we demonstrate the safety as well as the cost savings from this pathway, [this could serve as] a pilot and a proof of concept that Congress could then look to and potentially take up for more complex molecules that involve cold-chain storage and more complex distribution channels.”
The proposed regulations do not offer any estimates on how much savings could be achieved. He said that there is no way to estimate which states might develop importation plans and how those plans might work.
The second proposed pathway would involve FDA guidance to manufacturers allowing them to import their own FDA-approved products manufactured abroad. Under this proposal, there would be no restriction on which type or kind of FDA-approved product to be imported.
“The FDA has become aware that manufacturers of some brand-name drugs want to offer their drugs at lower costs in the U.S. market but, due to certain challenges in the private market, are not readily [able] to do so without obtaining a different national drug code for their drugs,” Adm. Brett Giroir, MD, HHS assistant secretary for health, said during the press conference.
Obtaining a separate NDC for imported drugs could address the challenges, particularly those posed by the incentives to raise list prices and offer higher rebates to pharmacy benefit managers, Mr. Azar said.
The draft guidance outlines procedures manufacturers could follow to get that NDC for those products and how manufacturers can demonstrate that these products meet U.S. regulatory standards. Products imported in this pathway could be made available to patients in hospitals, physician offices, and pharmacies. Generic drugs are not part of this guidance, but the proposed guidance asked for feedback on whether a similar approach is needed for generic products.
“This would potentially allow for the sale of these drugs at lower prices than currently offered to American consumers, giving drugmakers new flexibility to reduce list prices,” Mr. Azar said.
The proposed regulation on state-level importation will have a 75-day comment period from the day it is published in the Federal Register, and Mr. Azar said that the FDA is committing resources to getting the comments analyzed and reflected in the final rule.
“We will be moving as quickly as we possibly can,” Mr. Azar said, adding that the FDA guidance to manufacturers may move more quickly through its approval process because it is not a formal rule.
The Department of Health & Human Services is taking the first steps in allowing drugs to be imported into the United States.
HHS proposes to offer two different pathways for importation: One allowing states to design programs to import certain drugs directly from Canada and another allowing manufacturers to obtain a new National Drug Code (NDC) number to import their own Food and Drug Administration–approved products manufactured outside of the United States.
“The importation proposals we are rolling out ... are a historic step forward in efforts to bring down drug prices and out-of-pocket costs,” HHS Secretary Alex Azar said during a Dec. 17, 2019, press conference. “New pathways for importation can move us toward a more open and competitive marketplace that supplies American patients with safe, effective, affordable prescription drugs.”
The proposals were made public on Dec. 18, the day the House Rules committee was scheduled to vote on impeaching President Trump.
He emphasized that these proposals “are both important steps in advancing the FDA’s safe-importation action plan, [which] aims to insure that importation is done in a way that prioritizes safety and includes elements to help insure importation does not put patients or the U.S. drug supply chain at risk.”
The pathway for states to import drugs from Canada will be proposed through the federal regulatory process. The notice of proposed rulemaking, which implements authority for FDA regulation of importation granted in the Medicare Modernization Act of 2003, will outline a process by which states, potentially working with wholesalers and/or pharmacies, will submit proposals for FDA review and approval on how they would implement an importation program.
Only certain drugs would be eligible for importation from Canada under this proposal. The drugs would need to be approved in Canada and, except for Canadian labeling, need to meet the conditions of an FDA-approved new drug application or abbreviated new drug application.
Controlled substances, biologics, intravenously injected drugs, drugs with a risk evaluation and management strategy, and drugs injected into the spinal column or eye would be excluded from importation.
Drugs coming in from Canada would be relabeled with U.S.-approved labels and would be subject to testing to ensure they are authentic, not degraded, and compliant with U.S. standards.
States would be required to show that importing drugs poses no additional risk in public health and safety and it would result in the reduction of costs, according to information provided by HHS.
Many of the most expensive drugs, as well as insulins, would not be eligible for importation under this pathway, Mr. Azar acknowledged, adding that “I would envision that as we demonstrate the safety as well as the cost savings from this pathway, [this could serve as] a pilot and a proof of concept that Congress could then look to and potentially take up for more complex molecules that involve cold-chain storage and more complex distribution channels.”
The proposed regulations do not offer any estimates on how much savings could be achieved. He said that there is no way to estimate which states might develop importation plans and how those plans might work.
The second proposed pathway would involve FDA guidance to manufacturers allowing them to import their own FDA-approved products manufactured abroad. Under this proposal, there would be no restriction on which type or kind of FDA-approved product to be imported.
“The FDA has become aware that manufacturers of some brand-name drugs want to offer their drugs at lower costs in the U.S. market but, due to certain challenges in the private market, are not readily [able] to do so without obtaining a different national drug code for their drugs,” Adm. Brett Giroir, MD, HHS assistant secretary for health, said during the press conference.
Obtaining a separate NDC for imported drugs could address the challenges, particularly those posed by the incentives to raise list prices and offer higher rebates to pharmacy benefit managers, Mr. Azar said.
The draft guidance outlines procedures manufacturers could follow to get that NDC for those products and how manufacturers can demonstrate that these products meet U.S. regulatory standards. Products imported in this pathway could be made available to patients in hospitals, physician offices, and pharmacies. Generic drugs are not part of this guidance, but the proposed guidance asked for feedback on whether a similar approach is needed for generic products.
“This would potentially allow for the sale of these drugs at lower prices than currently offered to American consumers, giving drugmakers new flexibility to reduce list prices,” Mr. Azar said.
The proposed regulation on state-level importation will have a 75-day comment period from the day it is published in the Federal Register, and Mr. Azar said that the FDA is committing resources to getting the comments analyzed and reflected in the final rule.
“We will be moving as quickly as we possibly can,” Mr. Azar said, adding that the FDA guidance to manufacturers may move more quickly through its approval process because it is not a formal rule.
The Department of Health & Human Services is taking the first steps in allowing drugs to be imported into the United States.
HHS proposes to offer two different pathways for importation: One allowing states to design programs to import certain drugs directly from Canada and another allowing manufacturers to obtain a new National Drug Code (NDC) number to import their own Food and Drug Administration–approved products manufactured outside of the United States.
“The importation proposals we are rolling out ... are a historic step forward in efforts to bring down drug prices and out-of-pocket costs,” HHS Secretary Alex Azar said during a Dec. 17, 2019, press conference. “New pathways for importation can move us toward a more open and competitive marketplace that supplies American patients with safe, effective, affordable prescription drugs.”
The proposals were made public on Dec. 18, the day the House Rules committee was scheduled to vote on impeaching President Trump.
He emphasized that these proposals “are both important steps in advancing the FDA’s safe-importation action plan, [which] aims to insure that importation is done in a way that prioritizes safety and includes elements to help insure importation does not put patients or the U.S. drug supply chain at risk.”
The pathway for states to import drugs from Canada will be proposed through the federal regulatory process. The notice of proposed rulemaking, which implements authority for FDA regulation of importation granted in the Medicare Modernization Act of 2003, will outline a process by which states, potentially working with wholesalers and/or pharmacies, will submit proposals for FDA review and approval on how they would implement an importation program.
Only certain drugs would be eligible for importation from Canada under this proposal. The drugs would need to be approved in Canada and, except for Canadian labeling, need to meet the conditions of an FDA-approved new drug application or abbreviated new drug application.
Controlled substances, biologics, intravenously injected drugs, drugs with a risk evaluation and management strategy, and drugs injected into the spinal column or eye would be excluded from importation.
Drugs coming in from Canada would be relabeled with U.S.-approved labels and would be subject to testing to ensure they are authentic, not degraded, and compliant with U.S. standards.
States would be required to show that importing drugs poses no additional risk in public health and safety and it would result in the reduction of costs, according to information provided by HHS.
Many of the most expensive drugs, as well as insulins, would not be eligible for importation under this pathway, Mr. Azar acknowledged, adding that “I would envision that as we demonstrate the safety as well as the cost savings from this pathway, [this could serve as] a pilot and a proof of concept that Congress could then look to and potentially take up for more complex molecules that involve cold-chain storage and more complex distribution channels.”
The proposed regulations do not offer any estimates on how much savings could be achieved. He said that there is no way to estimate which states might develop importation plans and how those plans might work.
The second proposed pathway would involve FDA guidance to manufacturers allowing them to import their own FDA-approved products manufactured abroad. Under this proposal, there would be no restriction on which type or kind of FDA-approved product to be imported.
“The FDA has become aware that manufacturers of some brand-name drugs want to offer their drugs at lower costs in the U.S. market but, due to certain challenges in the private market, are not readily [able] to do so without obtaining a different national drug code for their drugs,” Adm. Brett Giroir, MD, HHS assistant secretary for health, said during the press conference.
Obtaining a separate NDC for imported drugs could address the challenges, particularly those posed by the incentives to raise list prices and offer higher rebates to pharmacy benefit managers, Mr. Azar said.
The draft guidance outlines procedures manufacturers could follow to get that NDC for those products and how manufacturers can demonstrate that these products meet U.S. regulatory standards. Products imported in this pathway could be made available to patients in hospitals, physician offices, and pharmacies. Generic drugs are not part of this guidance, but the proposed guidance asked for feedback on whether a similar approach is needed for generic products.
“This would potentially allow for the sale of these drugs at lower prices than currently offered to American consumers, giving drugmakers new flexibility to reduce list prices,” Mr. Azar said.
The proposed regulation on state-level importation will have a 75-day comment period from the day it is published in the Federal Register, and Mr. Azar said that the FDA is committing resources to getting the comments analyzed and reflected in the final rule.
“We will be moving as quickly as we possibly can,” Mr. Azar said, adding that the FDA guidance to manufacturers may move more quickly through its approval process because it is not a formal rule.
FDA expands Xtandi approval to mCSPC
The Food and Drug Administration has approved enzalutamide (Xtandi) for patients with metastatic castration-sensitive prostate cancer (mCSPC).
The drug was previously approved for patients with castration-resistant prostate cancer.
Approval was based on radiographic progression-free survival (rPFS) improvement in ARCHES, a trial of 1,150 patients with mCSPC randomized to receive either enzalutamide or placebo daily. All patients received a gonadotropin-releasing hormone analogue or had a prior bilateral orchiectomy.
Median rPFS was not reached in the enzalutamide, arm compared with 19.4 months (95% confidence interval, 16.6 to not reached) in the placebo arm (HR 0.39; 95% CI, 0.30-0.50; P less than .0001), the FDA said in a statement.
The most common adverse reactions in enzalutamide-treated patients in ARCHES were hot flush, asthenia/fatigue, hypertension, fractures, and musculoskeletal pain.
The recommended dose is 160 mg (four 40 mg capsules) administered orally once daily with or without food, the FDA said.
lnikolaides@mdedge.com
The Food and Drug Administration has approved enzalutamide (Xtandi) for patients with metastatic castration-sensitive prostate cancer (mCSPC).
The drug was previously approved for patients with castration-resistant prostate cancer.
Approval was based on radiographic progression-free survival (rPFS) improvement in ARCHES, a trial of 1,150 patients with mCSPC randomized to receive either enzalutamide or placebo daily. All patients received a gonadotropin-releasing hormone analogue or had a prior bilateral orchiectomy.
Median rPFS was not reached in the enzalutamide, arm compared with 19.4 months (95% confidence interval, 16.6 to not reached) in the placebo arm (HR 0.39; 95% CI, 0.30-0.50; P less than .0001), the FDA said in a statement.
The most common adverse reactions in enzalutamide-treated patients in ARCHES were hot flush, asthenia/fatigue, hypertension, fractures, and musculoskeletal pain.
The recommended dose is 160 mg (four 40 mg capsules) administered orally once daily with or without food, the FDA said.
lnikolaides@mdedge.com
The Food and Drug Administration has approved enzalutamide (Xtandi) for patients with metastatic castration-sensitive prostate cancer (mCSPC).
The drug was previously approved for patients with castration-resistant prostate cancer.
Approval was based on radiographic progression-free survival (rPFS) improvement in ARCHES, a trial of 1,150 patients with mCSPC randomized to receive either enzalutamide or placebo daily. All patients received a gonadotropin-releasing hormone analogue or had a prior bilateral orchiectomy.
Median rPFS was not reached in the enzalutamide, arm compared with 19.4 months (95% confidence interval, 16.6 to not reached) in the placebo arm (HR 0.39; 95% CI, 0.30-0.50; P less than .0001), the FDA said in a statement.
The most common adverse reactions in enzalutamide-treated patients in ARCHES were hot flush, asthenia/fatigue, hypertension, fractures, and musculoskeletal pain.
The recommended dose is 160 mg (four 40 mg capsules) administered orally once daily with or without food, the FDA said.
lnikolaides@mdedge.com
FDA approves Vyondys 53 for Duchenne muscular dystrophy subtype
The Food and Drug Administration has granted accelerated approval to Vyondys 53 (golodirsen) to treat patients with Duchenne muscular dystrophy (DMD) who have a mutation of the dystrophin gene that is amenable to exon 53 skipping. About 8% of patients with DMD have this type of mutation. Further research is required to establish whether the antisense oligonucleotide provides clinical benefit, the agency said.
Separately, the agency approved the first newborn screening test for DMD.
DMD is a “rare and devastating disease,” said Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research.
“Patients ... who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping will now have available the first treatment targeted specifically for this disease subtype,” Dr. Dunn said in a news release. “Use of the accelerated approval pathway will make Vyondys 53 available to patients based on initial data, and we look forward to learning more about the drug’s clinical benefit from the ongoing confirmatory clinical trial.”
A surrogate endpoint
The FDA approved Vyondys 53 based on the surrogate endpoint of increased dystrophin production in the skeletal muscle in some patients treated with the drug. Sarepta Therapeutics, the developer of Vyondys 53, evaluated the treatment in a two-part clinical study. In the first part, eight patients with DMD received Vyondys 53, and four received placebo. In the second part, 25 patients, including the 12 patients from the first part, received open-label treatment. Dystrophin levels increased from 0.10% of normal at baseline to 1.02% of normal after at least 48 weeks of treatment.
A placebo-controlled, confirmatory trial is expected to conclude by 2024, the company said. If the trial does not confirm clinical benefit, the FDA could withdraw approval of the drug.
The most common side effects in patients who received Vyondys 53 include headache, fever, fall, cough, vomiting, abdominal pain, cold symptoms, and nausea. Some patients had hypersensitivity reactions. Renal toxicity occurred in animal studies of golodirsen, but not in the clinical studies. Renal toxicity, however, has occurred after treatment with other antisense oligonucleotides, the FDA noted.
Sarepta said Vyondys 53, an injection, would be available immediately. The drug is the company’s second RNA exon-skipping treatment for DMD. The FDA approved the first treatment, Exondys 51 (eteplirsen), in 2016. Together, the two drugs can treat about 20% of patients with DMD, the company said.
Newborn screening
On the same day, Dec. 12, 2019, the FDA authorized marketing of the first test to aid in newborn screening for DMD. Although authorization for the GSP Neonatal Creatine Kinase–MM kit enables laboratories to add this test to their newborn screening panel, it “does not signal a recommendation for DMD to be added ... as a condition for which newborn screening is recommended,” the agency said. In addition, the FDA noted that the kit is not meant to diagnose DMD or to screen for other muscular dystrophies.
The GSP Neonatal Creatine Kinase–MM kit measures the concentration of CK-MM, a type of protein that increases when there is muscle damage. The test measures CK-MM in dried blood samples collected from a newborn’s heel 24-48 hours after birth. Elevated levels may indicate DMD, but physicians must confirm the diagnosis using other methods, such as muscle biopsies, genetic testing, and other laboratory tests.
DMD primarily affects boys, and patients often do not have a family history of the condition. About 1 in 3,600 male live-born infants worldwide have DMD. Symptom onset usually occurs between the ages of 3 and 5 years.
The FDA reviewed the kit through the de novo premarket review pathway for low to moderate risk devices. In a clinical study of 3,041 newborns, the kit identified the four screened newborns who had DMD-causing genetic mutations. In addition, the test correctly identified 30 samples from newborns with clinically confirmed cases of DMD.
PerkinElmer developed the GSP Neonatal Creatine Kinase–MM kit.
The Food and Drug Administration has granted accelerated approval to Vyondys 53 (golodirsen) to treat patients with Duchenne muscular dystrophy (DMD) who have a mutation of the dystrophin gene that is amenable to exon 53 skipping. About 8% of patients with DMD have this type of mutation. Further research is required to establish whether the antisense oligonucleotide provides clinical benefit, the agency said.
Separately, the agency approved the first newborn screening test for DMD.
DMD is a “rare and devastating disease,” said Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research.
“Patients ... who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping will now have available the first treatment targeted specifically for this disease subtype,” Dr. Dunn said in a news release. “Use of the accelerated approval pathway will make Vyondys 53 available to patients based on initial data, and we look forward to learning more about the drug’s clinical benefit from the ongoing confirmatory clinical trial.”
A surrogate endpoint
The FDA approved Vyondys 53 based on the surrogate endpoint of increased dystrophin production in the skeletal muscle in some patients treated with the drug. Sarepta Therapeutics, the developer of Vyondys 53, evaluated the treatment in a two-part clinical study. In the first part, eight patients with DMD received Vyondys 53, and four received placebo. In the second part, 25 patients, including the 12 patients from the first part, received open-label treatment. Dystrophin levels increased from 0.10% of normal at baseline to 1.02% of normal after at least 48 weeks of treatment.
A placebo-controlled, confirmatory trial is expected to conclude by 2024, the company said. If the trial does not confirm clinical benefit, the FDA could withdraw approval of the drug.
The most common side effects in patients who received Vyondys 53 include headache, fever, fall, cough, vomiting, abdominal pain, cold symptoms, and nausea. Some patients had hypersensitivity reactions. Renal toxicity occurred in animal studies of golodirsen, but not in the clinical studies. Renal toxicity, however, has occurred after treatment with other antisense oligonucleotides, the FDA noted.
Sarepta said Vyondys 53, an injection, would be available immediately. The drug is the company’s second RNA exon-skipping treatment for DMD. The FDA approved the first treatment, Exondys 51 (eteplirsen), in 2016. Together, the two drugs can treat about 20% of patients with DMD, the company said.
Newborn screening
On the same day, Dec. 12, 2019, the FDA authorized marketing of the first test to aid in newborn screening for DMD. Although authorization for the GSP Neonatal Creatine Kinase–MM kit enables laboratories to add this test to their newborn screening panel, it “does not signal a recommendation for DMD to be added ... as a condition for which newborn screening is recommended,” the agency said. In addition, the FDA noted that the kit is not meant to diagnose DMD or to screen for other muscular dystrophies.
The GSP Neonatal Creatine Kinase–MM kit measures the concentration of CK-MM, a type of protein that increases when there is muscle damage. The test measures CK-MM in dried blood samples collected from a newborn’s heel 24-48 hours after birth. Elevated levels may indicate DMD, but physicians must confirm the diagnosis using other methods, such as muscle biopsies, genetic testing, and other laboratory tests.
DMD primarily affects boys, and patients often do not have a family history of the condition. About 1 in 3,600 male live-born infants worldwide have DMD. Symptom onset usually occurs between the ages of 3 and 5 years.
The FDA reviewed the kit through the de novo premarket review pathway for low to moderate risk devices. In a clinical study of 3,041 newborns, the kit identified the four screened newborns who had DMD-causing genetic mutations. In addition, the test correctly identified 30 samples from newborns with clinically confirmed cases of DMD.
PerkinElmer developed the GSP Neonatal Creatine Kinase–MM kit.
The Food and Drug Administration has granted accelerated approval to Vyondys 53 (golodirsen) to treat patients with Duchenne muscular dystrophy (DMD) who have a mutation of the dystrophin gene that is amenable to exon 53 skipping. About 8% of patients with DMD have this type of mutation. Further research is required to establish whether the antisense oligonucleotide provides clinical benefit, the agency said.
Separately, the agency approved the first newborn screening test for DMD.
DMD is a “rare and devastating disease,” said Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research.
“Patients ... who have a confirmed mutation of the dystrophin gene amenable to exon 53 skipping will now have available the first treatment targeted specifically for this disease subtype,” Dr. Dunn said in a news release. “Use of the accelerated approval pathway will make Vyondys 53 available to patients based on initial data, and we look forward to learning more about the drug’s clinical benefit from the ongoing confirmatory clinical trial.”
A surrogate endpoint
The FDA approved Vyondys 53 based on the surrogate endpoint of increased dystrophin production in the skeletal muscle in some patients treated with the drug. Sarepta Therapeutics, the developer of Vyondys 53, evaluated the treatment in a two-part clinical study. In the first part, eight patients with DMD received Vyondys 53, and four received placebo. In the second part, 25 patients, including the 12 patients from the first part, received open-label treatment. Dystrophin levels increased from 0.10% of normal at baseline to 1.02% of normal after at least 48 weeks of treatment.
A placebo-controlled, confirmatory trial is expected to conclude by 2024, the company said. If the trial does not confirm clinical benefit, the FDA could withdraw approval of the drug.
The most common side effects in patients who received Vyondys 53 include headache, fever, fall, cough, vomiting, abdominal pain, cold symptoms, and nausea. Some patients had hypersensitivity reactions. Renal toxicity occurred in animal studies of golodirsen, but not in the clinical studies. Renal toxicity, however, has occurred after treatment with other antisense oligonucleotides, the FDA noted.
Sarepta said Vyondys 53, an injection, would be available immediately. The drug is the company’s second RNA exon-skipping treatment for DMD. The FDA approved the first treatment, Exondys 51 (eteplirsen), in 2016. Together, the two drugs can treat about 20% of patients with DMD, the company said.
Newborn screening
On the same day, Dec. 12, 2019, the FDA authorized marketing of the first test to aid in newborn screening for DMD. Although authorization for the GSP Neonatal Creatine Kinase–MM kit enables laboratories to add this test to their newborn screening panel, it “does not signal a recommendation for DMD to be added ... as a condition for which newborn screening is recommended,” the agency said. In addition, the FDA noted that the kit is not meant to diagnose DMD or to screen for other muscular dystrophies.
The GSP Neonatal Creatine Kinase–MM kit measures the concentration of CK-MM, a type of protein that increases when there is muscle damage. The test measures CK-MM in dried blood samples collected from a newborn’s heel 24-48 hours after birth. Elevated levels may indicate DMD, but physicians must confirm the diagnosis using other methods, such as muscle biopsies, genetic testing, and other laboratory tests.
DMD primarily affects boys, and patients often do not have a family history of the condition. About 1 in 3,600 male live-born infants worldwide have DMD. Symptom onset usually occurs between the ages of 3 and 5 years.
The FDA reviewed the kit through the de novo premarket review pathway for low to moderate risk devices. In a clinical study of 3,041 newborns, the kit identified the four screened newborns who had DMD-causing genetic mutations. In addition, the test correctly identified 30 samples from newborns with clinically confirmed cases of DMD.
PerkinElmer developed the GSP Neonatal Creatine Kinase–MM kit.