User login
FDA clears first fully disposable duodenoscope
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
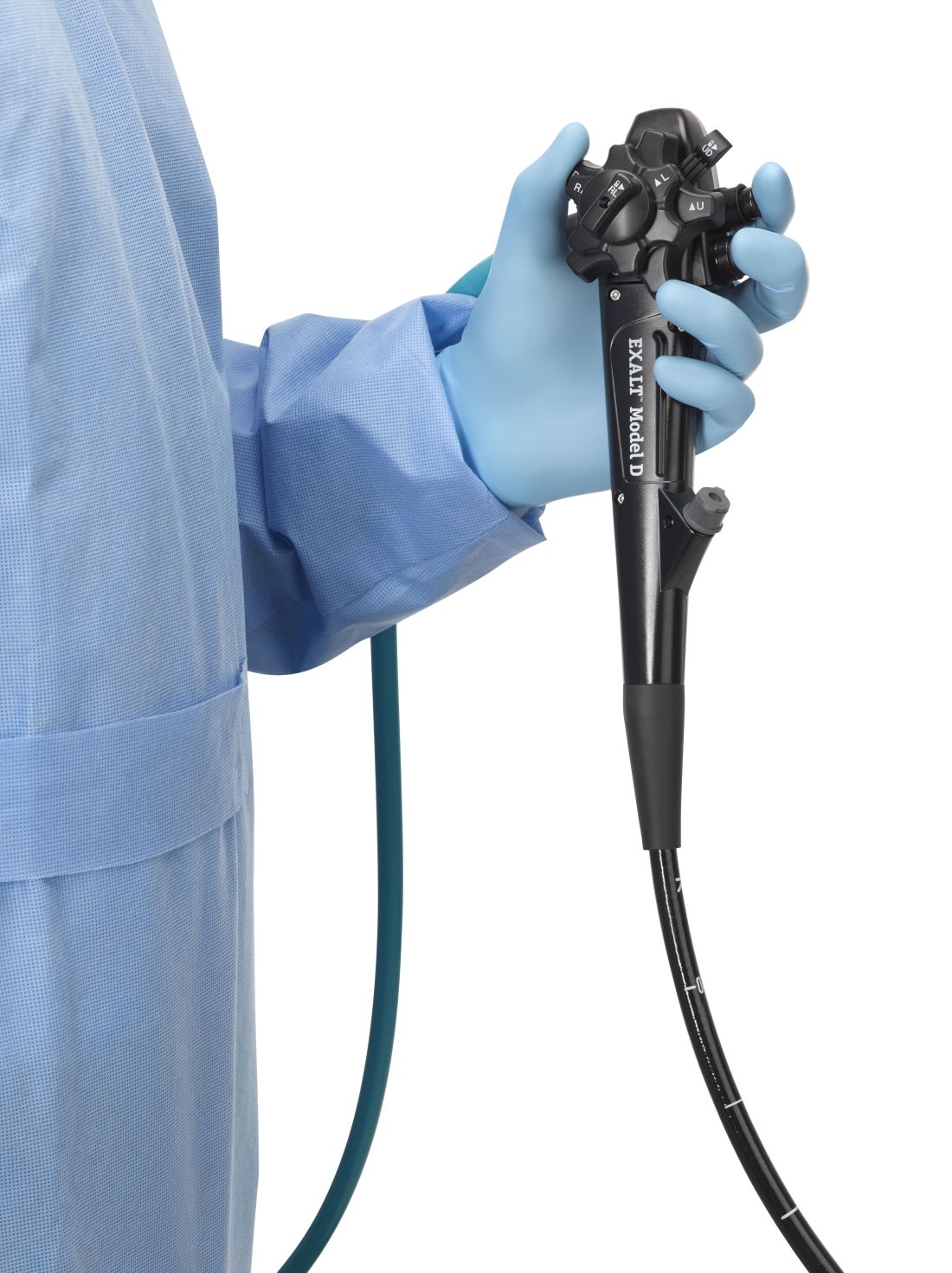
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test postive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
“The feel is a little different,” said Dr. Ketwaroo, who’s tried the new device, but “it’s pretty functional and probably okay to use in almost all endoscopic procedures that require ERCP.”
In a study funded by Boston Scientific, endoscopists reported a median overall satisfaction score of 9 out of 10 with the new scope (Clin Gastroenterol Hepatol. 2019 Nov 6. doi: 10.1016/j.cgh.2019.10.052).
As for using it at Baylor, Dr. Ketwaroo said, “we’re not sure yet; we are still evaluating it” and want to see if any problems emerge once it’s on the market. It’s also not clear if infection risks would be lower than with the disposable elevator model from Pentax, he added.
The Exalt Model D was granted breakthrough status by the FDA, and the agency worked closely with Boston Scientific to bring it to market.
“The availability of a fully disposable duodenoscope represents another major step forward for improving the safety of these devices, which are used in more than 500,000 procedures in the United States each year. The FDA continues to encourage innovative ways to improve the safety and effectiveness of these devices,” Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test postive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
“The feel is a little different,” said Dr. Ketwaroo, who’s tried the new device, but “it’s pretty functional and probably okay to use in almost all endoscopic procedures that require ERCP.”
In a study funded by Boston Scientific, endoscopists reported a median overall satisfaction score of 9 out of 10 with the new scope (Clin Gastroenterol Hepatol. 2019 Nov 6. doi: 10.1016/j.cgh.2019.10.052).
As for using it at Baylor, Dr. Ketwaroo said, “we’re not sure yet; we are still evaluating it” and want to see if any problems emerge once it’s on the market. It’s also not clear if infection risks would be lower than with the disposable elevator model from Pentax, he added.
The Exalt Model D was granted breakthrough status by the FDA, and the agency worked closely with Boston Scientific to bring it to market.
“The availability of a fully disposable duodenoscope represents another major step forward for improving the safety of these devices, which are used in more than 500,000 procedures in the United States each year. The FDA continues to encourage innovative ways to improve the safety and effectiveness of these devices,” Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test postive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
“The feel is a little different,” said Dr. Ketwaroo, who’s tried the new device, but “it’s pretty functional and probably okay to use in almost all endoscopic procedures that require ERCP.”
In a study funded by Boston Scientific, endoscopists reported a median overall satisfaction score of 9 out of 10 with the new scope (Clin Gastroenterol Hepatol. 2019 Nov 6. doi: 10.1016/j.cgh.2019.10.052).
As for using it at Baylor, Dr. Ketwaroo said, “we’re not sure yet; we are still evaluating it” and want to see if any problems emerge once it’s on the market. It’s also not clear if infection risks would be lower than with the disposable elevator model from Pentax, he added.
The Exalt Model D was granted breakthrough status by the FDA, and the agency worked closely with Boston Scientific to bring it to market.
“The availability of a fully disposable duodenoscope represents another major step forward for improving the safety of these devices, which are used in more than 500,000 procedures in the United States each year. The FDA continues to encourage innovative ways to improve the safety and effectiveness of these devices,” Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
Dr. Ketwaroo had no relevant financial disclosures.
FROM THE FDA
Icosapent ethyl approved for cardiovascular risk reduction
Icosapent ethyl (Vascepa) has gained an indication from the Food and Drug Administration for reduction of cardiovascular events in patients with high triglycerides who are at high risk for cardiovascular events. 
as an add-on to maximally tolerated statin therapy,” the agency said in an announcement.
The decision, announced on Dec. 13, was based primarily on results of REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial), which tested icosapent ethyl in 8,179 patients with either established cardiovascular disease or diabetes and at least one additional cardiovascular disease risk factor. It showed that patients who received icosapent ethyl had a statistically significant 25% relative risk reduction in the trial’s primary, composite endpoint (N Engl J Med. 2019 Jan 3;380[1]:11-22).
In a November meeting, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted unanimously for approval.
The agency notes that, in clinical trials, icosapent ethyl was linked to an increased risk of atrial fibrillation or atrial flutter requiring hospitalization, especially in patients with a history of either condition. The highly purified form of the ethyl ester of eicosapentaenoic acid was also associated with an increased risk of bleeding events, particularly in those taking blood-thinning drugs that increase the risk of bleeding, such as aspirin, clopidogrel, or warfarin.
The most common side effects reported in the clinical trials for icosapent ethyl were musculoskeletal pain, peripheral edema, atrial fibrillation, and arthralgia.
The complete indication is “as an adjunct to maximally tolerated statin therapy to reduce the risk of myocardial infarction, stroke, coronary revascularization, and unstable angina requiring hospitalization in adult patients with elevated triglyceride levels (at least 150 mg/dL) and established cardiovascular disease or diabetes mellitus and two or more additional risk factors for cardiovascular disease,” according to a statement from Amalin, which markets Vascepa.
The drug was approved in 2012 for the indication of cutting triglyceride levels once they reached at least 500 mg/dL.
Icosapent ethyl (Vascepa) has gained an indication from the Food and Drug Administration for reduction of cardiovascular events in patients with high triglycerides who are at high risk for cardiovascular events.
as an add-on to maximally tolerated statin therapy,” the agency said in an announcement.
The decision, announced on Dec. 13, was based primarily on results of REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial), which tested icosapent ethyl in 8,179 patients with either established cardiovascular disease or diabetes and at least one additional cardiovascular disease risk factor. It showed that patients who received icosapent ethyl had a statistically significant 25% relative risk reduction in the trial’s primary, composite endpoint (N Engl J Med. 2019 Jan 3;380[1]:11-22).
In a November meeting, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted unanimously for approval.
The agency notes that, in clinical trials, icosapent ethyl was linked to an increased risk of atrial fibrillation or atrial flutter requiring hospitalization, especially in patients with a history of either condition. The highly purified form of the ethyl ester of eicosapentaenoic acid was also associated with an increased risk of bleeding events, particularly in those taking blood-thinning drugs that increase the risk of bleeding, such as aspirin, clopidogrel, or warfarin.
The most common side effects reported in the clinical trials for icosapent ethyl were musculoskeletal pain, peripheral edema, atrial fibrillation, and arthralgia.
The complete indication is “as an adjunct to maximally tolerated statin therapy to reduce the risk of myocardial infarction, stroke, coronary revascularization, and unstable angina requiring hospitalization in adult patients with elevated triglyceride levels (at least 150 mg/dL) and established cardiovascular disease or diabetes mellitus and two or more additional risk factors for cardiovascular disease,” according to a statement from Amalin, which markets Vascepa.
The drug was approved in 2012 for the indication of cutting triglyceride levels once they reached at least 500 mg/dL.
Icosapent ethyl (Vascepa) has gained an indication from the Food and Drug Administration for reduction of cardiovascular events in patients with high triglycerides who are at high risk for cardiovascular events.
as an add-on to maximally tolerated statin therapy,” the agency said in an announcement.
The decision, announced on Dec. 13, was based primarily on results of REDUCE-IT (Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial), which tested icosapent ethyl in 8,179 patients with either established cardiovascular disease or diabetes and at least one additional cardiovascular disease risk factor. It showed that patients who received icosapent ethyl had a statistically significant 25% relative risk reduction in the trial’s primary, composite endpoint (N Engl J Med. 2019 Jan 3;380[1]:11-22).
In a November meeting, the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee voted unanimously for approval.
The agency notes that, in clinical trials, icosapent ethyl was linked to an increased risk of atrial fibrillation or atrial flutter requiring hospitalization, especially in patients with a history of either condition. The highly purified form of the ethyl ester of eicosapentaenoic acid was also associated with an increased risk of bleeding events, particularly in those taking blood-thinning drugs that increase the risk of bleeding, such as aspirin, clopidogrel, or warfarin.
The most common side effects reported in the clinical trials for icosapent ethyl were musculoskeletal pain, peripheral edema, atrial fibrillation, and arthralgia.
The complete indication is “as an adjunct to maximally tolerated statin therapy to reduce the risk of myocardial infarction, stroke, coronary revascularization, and unstable angina requiring hospitalization in adult patients with elevated triglyceride levels (at least 150 mg/dL) and established cardiovascular disease or diabetes mellitus and two or more additional risk factors for cardiovascular disease,” according to a statement from Amalin, which markets Vascepa.
The drug was approved in 2012 for the indication of cutting triglyceride levels once they reached at least 500 mg/dL.
FDA authorizes customizable automated glycemic controller
The Food and Drug Administration has authorized marketing of the Tandem Diabetes Care Control-IQ Technology, an interoperable automated glycemic controller, for use in a customizable glucose control system, according to a release from the agency.

The move also paves the way for the review and authorization of similar devices in the future.
The Control-IQ Technology controller coordinates with an alternate controller-enabled insulin pump and an integrated continuous glucose monitor, which can be made by other manufacturers as long they are compatible with this modular technology.
The agency reviewed data from a clinical study of 168 patients with type 1 diabetes who were randomized to use either the Control-IQ Technology controller installed on a Tandem t:slim X2 insulin pump, or a continuous glucose monitor and insulin pump without the Control-IQ controller. The findings showed that,
However, the agency noted that, although the system has been assessed for reliability, delays in insulin delivery remain possible and care should be taken when using it.
This authorization comes along with establishment of criteria and regulatory requirements that create a new regulatory classification for this type of device, whereby future devices of the same type and with the same purpose can go through the FDA’s 510(k) premarket process. Such a process would mean that, going forward, similar devices can “obtain marketing authorization by demonstrating substantial equivalence to a predicate device.”
More information can be found in the full release, available on the FDA website.
The Food and Drug Administration has authorized marketing of the Tandem Diabetes Care Control-IQ Technology, an interoperable automated glycemic controller, for use in a customizable glucose control system, according to a release from the agency.
The move also paves the way for the review and authorization of similar devices in the future.
The Control-IQ Technology controller coordinates with an alternate controller-enabled insulin pump and an integrated continuous glucose monitor, which can be made by other manufacturers as long they are compatible with this modular technology.
The agency reviewed data from a clinical study of 168 patients with type 1 diabetes who were randomized to use either the Control-IQ Technology controller installed on a Tandem t:slim X2 insulin pump, or a continuous glucose monitor and insulin pump without the Control-IQ controller. The findings showed that,
However, the agency noted that, although the system has been assessed for reliability, delays in insulin delivery remain possible and care should be taken when using it.
This authorization comes along with establishment of criteria and regulatory requirements that create a new regulatory classification for this type of device, whereby future devices of the same type and with the same purpose can go through the FDA’s 510(k) premarket process. Such a process would mean that, going forward, similar devices can “obtain marketing authorization by demonstrating substantial equivalence to a predicate device.”
More information can be found in the full release, available on the FDA website.
The Food and Drug Administration has authorized marketing of the Tandem Diabetes Care Control-IQ Technology, an interoperable automated glycemic controller, for use in a customizable glucose control system, according to a release from the agency.
The move also paves the way for the review and authorization of similar devices in the future.
The Control-IQ Technology controller coordinates with an alternate controller-enabled insulin pump and an integrated continuous glucose monitor, which can be made by other manufacturers as long they are compatible with this modular technology.
The agency reviewed data from a clinical study of 168 patients with type 1 diabetes who were randomized to use either the Control-IQ Technology controller installed on a Tandem t:slim X2 insulin pump, or a continuous glucose monitor and insulin pump without the Control-IQ controller. The findings showed that,
However, the agency noted that, although the system has been assessed for reliability, delays in insulin delivery remain possible and care should be taken when using it.
This authorization comes along with establishment of criteria and regulatory requirements that create a new regulatory classification for this type of device, whereby future devices of the same type and with the same purpose can go through the FDA’s 510(k) premarket process. Such a process would mean that, going forward, similar devices can “obtain marketing authorization by demonstrating substantial equivalence to a predicate device.”
More information can be found in the full release, available on the FDA website.
FDA panel rejects vernakalant bid for AFib cardioversion indication
for cardioversion of recent-onset atrial fibrillation (AFib).

It was the second time before an FDA advisory panel for vernakalant (Brinavess, Correvio International Sàrl), which the agency had declined to approve in 2008 due to safety concerns. That time, however, its advisors had given the agency a decidedly positive recommendation.
Since then, registry data collected for the drug’s resubmission seemed only to raise further safety issues, especially evidence that a single infusion may cause severe hypotension and suppress left ventricular function.
Some members of the Cardiovascular and Renal Drugs Advisory Committee (CRDAC), including a number who voted against approval, expressed hopes for further research aimed at identifying specific AFib patient groups who might safely benefit from vernakalant.
Of note, the drug has long been available for AFib cardioversion in Europe, where there are a number of other pharmacologic options, and was recently approved in Canada.
“We do recognize there’s a significant clinical need here,” observed Paul M. Ridker, MD, MPH, of Harvard Medical School and Brigham and Women’s Hospital Boston, a CRDAC panelist.
The results of the safety study that Correvio presented to the panel were “pretty marginal,” Dr. Ridker said. Given the negative safety signals and the available cardioversion alternatives, he questioned whether vernakalant represented a “substantial advance versus just another option. Right now, I’m not convinced it’s a substantial advance.”
FDA representatives were skeptical about vernakalant when they walked into the meeting room, as noted in briefing documents they had circulated beforehand. The drug’s safety experience under consideration included one case of ventricular arrhythmia and cardiogenic shock in a treated patient without apparent structural heart disease, who subsequently died. That case was much discussed throughout the meeting.
In its resubmission of vernakalant to regulators, Correvio also pointed to a significant unmet need for AFib cardioversion options in the United States, given the few alternatives.
For example, ibutilide is FDA-approved for recent-onset AFib or atrial flutter; but as the company and panelists noted, the drug isn’t often used for that indication. Patients with recent-onset AFib are often put on rate-control meds without cardioversion. Or clinicians may resort to electrical cardioversion, which can be logistically cumbersome and require anesthesia and generally a hospital stay.
Oral or intravenous amiodarone and oral dofetilide, flecainide, and propafenone are guideline-recommended but not actually FDA-approved for recent-onset AFib, the company noted.
Correvio made its “pre-infusion checklist” a core feature of its case. It was designed to guide selection of patients for vernakalant cardioversion based on contraindications such as a systolic blood pressure under 100 mm Hg, severe heart failure, aortic stenosis, severe bradycardia or heart block, or a prolonged QT interval.
In his presentation to the panel, FDA medical officer Preston Dunnmon, MD, said the safety results from the SPECTRUM registry, another main pillar of support for the vernakalant resubmission, “are not reassuring.”
As reasons, Dr. Dunnmon cited likely patient-selection bias and its high proportion of patients who were not prospectively enrolled; 21% were retrospectively entered from records.
Moreover, “the proposed preinfusion checklist will not reliably predict which subjects will experience serious cardiovascular adverse events with vernakalant,” he said.
“Vernakalant has induced harm that cannot be reliably predicted, prevented, or in some cases, treated. In contrast to vernakalant, electrical cardioversion and ibutilide pharmacologic cardioversion can cause adverse events, but these are transient and treatable,” he said.
Many on the panel agreed. “I thought the totality of evidence supported the hypothesis that this drug has a potential for a fatal side effect in a disease that you can live with, potentially, and that there are other treatments for,” said Julia B. Lewis, MD, Vanderbilt Medical Center, Nashville, Tenn., who chaired the CRDAC panel.
“The drug clearly converts atrial fibrillation, although it’s only transient,” observed John H. Alexander, MD, MHSc, Duke University, Durham, N.C., one of the two panelists who voted to recommend approval of vernakalant.
“And, there clearly is a serious safety signal in some populations of patients,” he agreed. “However, I was more reassured by the SPECTRUM data.” There is likely to be a low-risk group of patients for whom vernakalant could represent an important option that “outweighs the relatively low risk of serious complications,” Dr. Alexander said.
“So more work needs to be done to clarify who are the low risk patients where it would be favorable.”
Panelist Matthew Needleman, MD, Walter Reed National Military Medical Center, Bethesda, Md., also voted in favor of approval.
“We’ve all known patients with normal ejection fractions who keep coming in with symptomatic atrial fib, want to get out of it quickly, and get back to their lives. So having an option like this I think would be good for a very select group of patients,” Dr. Needleman said.
But the preinfusion checklist and other potential ways to select low-risk patients for vernakalant could potentially backfire, warned John M. Mandrola, MD, Baptist Medical Associates, Louisville, Ky., from the panel.
The FDA representatives had presented evidence that the drug can seriously depress ventricular function, and that the lower cardiac output is what leads to hypotension, he elaborated in an interview after the meeting.
If the checklist is used to exclude hemodynamically unstable patients from receiving vernakalant, he said, “Then you’re really giving this drug to relatively healthy patients for convenience, to decrease hospitalization or the hospital stay.”
The signal for substantial harm, Dr. Mandrola said, has to be balanced against that modest benefit.
Moreover, those in whom the drug doesn’t work may be left in a worse situation, he proposed. Only about half of patients are successfully converted on vernakalant, the company and FDA data suggested. The other half of patients who don’t achieve sinus rhythm on the drug still must face the significant hazards of depressed ejection fraction and hypotension, a high price to pay for an unsuccessful treatment.
Dr. Mandrola is Chief Cardiology Correspondent for theheart.org | Medscape Cardiology; his disclosure statement states no relevant financial relationships.
This article first appeared on Medscape.com.
for cardioversion of recent-onset atrial fibrillation (AFib).
It was the second time before an FDA advisory panel for vernakalant (Brinavess, Correvio International Sàrl), which the agency had declined to approve in 2008 due to safety concerns. That time, however, its advisors had given the agency a decidedly positive recommendation.
Since then, registry data collected for the drug’s resubmission seemed only to raise further safety issues, especially evidence that a single infusion may cause severe hypotension and suppress left ventricular function.
Some members of the Cardiovascular and Renal Drugs Advisory Committee (CRDAC), including a number who voted against approval, expressed hopes for further research aimed at identifying specific AFib patient groups who might safely benefit from vernakalant.
Of note, the drug has long been available for AFib cardioversion in Europe, where there are a number of other pharmacologic options, and was recently approved in Canada.
“We do recognize there’s a significant clinical need here,” observed Paul M. Ridker, MD, MPH, of Harvard Medical School and Brigham and Women’s Hospital Boston, a CRDAC panelist.
The results of the safety study that Correvio presented to the panel were “pretty marginal,” Dr. Ridker said. Given the negative safety signals and the available cardioversion alternatives, he questioned whether vernakalant represented a “substantial advance versus just another option. Right now, I’m not convinced it’s a substantial advance.”
FDA representatives were skeptical about vernakalant when they walked into the meeting room, as noted in briefing documents they had circulated beforehand. The drug’s safety experience under consideration included one case of ventricular arrhythmia and cardiogenic shock in a treated patient without apparent structural heart disease, who subsequently died. That case was much discussed throughout the meeting.
In its resubmission of vernakalant to regulators, Correvio also pointed to a significant unmet need for AFib cardioversion options in the United States, given the few alternatives.
For example, ibutilide is FDA-approved for recent-onset AFib or atrial flutter; but as the company and panelists noted, the drug isn’t often used for that indication. Patients with recent-onset AFib are often put on rate-control meds without cardioversion. Or clinicians may resort to electrical cardioversion, which can be logistically cumbersome and require anesthesia and generally a hospital stay.
Oral or intravenous amiodarone and oral dofetilide, flecainide, and propafenone are guideline-recommended but not actually FDA-approved for recent-onset AFib, the company noted.
Correvio made its “pre-infusion checklist” a core feature of its case. It was designed to guide selection of patients for vernakalant cardioversion based on contraindications such as a systolic blood pressure under 100 mm Hg, severe heart failure, aortic stenosis, severe bradycardia or heart block, or a prolonged QT interval.
In his presentation to the panel, FDA medical officer Preston Dunnmon, MD, said the safety results from the SPECTRUM registry, another main pillar of support for the vernakalant resubmission, “are not reassuring.”
As reasons, Dr. Dunnmon cited likely patient-selection bias and its high proportion of patients who were not prospectively enrolled; 21% were retrospectively entered from records.
Moreover, “the proposed preinfusion checklist will not reliably predict which subjects will experience serious cardiovascular adverse events with vernakalant,” he said.
“Vernakalant has induced harm that cannot be reliably predicted, prevented, or in some cases, treated. In contrast to vernakalant, electrical cardioversion and ibutilide pharmacologic cardioversion can cause adverse events, but these are transient and treatable,” he said.
Many on the panel agreed. “I thought the totality of evidence supported the hypothesis that this drug has a potential for a fatal side effect in a disease that you can live with, potentially, and that there are other treatments for,” said Julia B. Lewis, MD, Vanderbilt Medical Center, Nashville, Tenn., who chaired the CRDAC panel.
“The drug clearly converts atrial fibrillation, although it’s only transient,” observed John H. Alexander, MD, MHSc, Duke University, Durham, N.C., one of the two panelists who voted to recommend approval of vernakalant.
“And, there clearly is a serious safety signal in some populations of patients,” he agreed. “However, I was more reassured by the SPECTRUM data.” There is likely to be a low-risk group of patients for whom vernakalant could represent an important option that “outweighs the relatively low risk of serious complications,” Dr. Alexander said.
“So more work needs to be done to clarify who are the low risk patients where it would be favorable.”
Panelist Matthew Needleman, MD, Walter Reed National Military Medical Center, Bethesda, Md., also voted in favor of approval.
“We’ve all known patients with normal ejection fractions who keep coming in with symptomatic atrial fib, want to get out of it quickly, and get back to their lives. So having an option like this I think would be good for a very select group of patients,” Dr. Needleman said.
But the preinfusion checklist and other potential ways to select low-risk patients for vernakalant could potentially backfire, warned John M. Mandrola, MD, Baptist Medical Associates, Louisville, Ky., from the panel.
The FDA representatives had presented evidence that the drug can seriously depress ventricular function, and that the lower cardiac output is what leads to hypotension, he elaborated in an interview after the meeting.
If the checklist is used to exclude hemodynamically unstable patients from receiving vernakalant, he said, “Then you’re really giving this drug to relatively healthy patients for convenience, to decrease hospitalization or the hospital stay.”
The signal for substantial harm, Dr. Mandrola said, has to be balanced against that modest benefit.
Moreover, those in whom the drug doesn’t work may be left in a worse situation, he proposed. Only about half of patients are successfully converted on vernakalant, the company and FDA data suggested. The other half of patients who don’t achieve sinus rhythm on the drug still must face the significant hazards of depressed ejection fraction and hypotension, a high price to pay for an unsuccessful treatment.
Dr. Mandrola is Chief Cardiology Correspondent for theheart.org | Medscape Cardiology; his disclosure statement states no relevant financial relationships.
This article first appeared on Medscape.com.
for cardioversion of recent-onset atrial fibrillation (AFib).
It was the second time before an FDA advisory panel for vernakalant (Brinavess, Correvio International Sàrl), which the agency had declined to approve in 2008 due to safety concerns. That time, however, its advisors had given the agency a decidedly positive recommendation.
Since then, registry data collected for the drug’s resubmission seemed only to raise further safety issues, especially evidence that a single infusion may cause severe hypotension and suppress left ventricular function.
Some members of the Cardiovascular and Renal Drugs Advisory Committee (CRDAC), including a number who voted against approval, expressed hopes for further research aimed at identifying specific AFib patient groups who might safely benefit from vernakalant.
Of note, the drug has long been available for AFib cardioversion in Europe, where there are a number of other pharmacologic options, and was recently approved in Canada.
“We do recognize there’s a significant clinical need here,” observed Paul M. Ridker, MD, MPH, of Harvard Medical School and Brigham and Women’s Hospital Boston, a CRDAC panelist.
The results of the safety study that Correvio presented to the panel were “pretty marginal,” Dr. Ridker said. Given the negative safety signals and the available cardioversion alternatives, he questioned whether vernakalant represented a “substantial advance versus just another option. Right now, I’m not convinced it’s a substantial advance.”
FDA representatives were skeptical about vernakalant when they walked into the meeting room, as noted in briefing documents they had circulated beforehand. The drug’s safety experience under consideration included one case of ventricular arrhythmia and cardiogenic shock in a treated patient without apparent structural heart disease, who subsequently died. That case was much discussed throughout the meeting.
In its resubmission of vernakalant to regulators, Correvio also pointed to a significant unmet need for AFib cardioversion options in the United States, given the few alternatives.
For example, ibutilide is FDA-approved for recent-onset AFib or atrial flutter; but as the company and panelists noted, the drug isn’t often used for that indication. Patients with recent-onset AFib are often put on rate-control meds without cardioversion. Or clinicians may resort to electrical cardioversion, which can be logistically cumbersome and require anesthesia and generally a hospital stay.
Oral or intravenous amiodarone and oral dofetilide, flecainide, and propafenone are guideline-recommended but not actually FDA-approved for recent-onset AFib, the company noted.
Correvio made its “pre-infusion checklist” a core feature of its case. It was designed to guide selection of patients for vernakalant cardioversion based on contraindications such as a systolic blood pressure under 100 mm Hg, severe heart failure, aortic stenosis, severe bradycardia or heart block, or a prolonged QT interval.
In his presentation to the panel, FDA medical officer Preston Dunnmon, MD, said the safety results from the SPECTRUM registry, another main pillar of support for the vernakalant resubmission, “are not reassuring.”
As reasons, Dr. Dunnmon cited likely patient-selection bias and its high proportion of patients who were not prospectively enrolled; 21% were retrospectively entered from records.
Moreover, “the proposed preinfusion checklist will not reliably predict which subjects will experience serious cardiovascular adverse events with vernakalant,” he said.
“Vernakalant has induced harm that cannot be reliably predicted, prevented, or in some cases, treated. In contrast to vernakalant, electrical cardioversion and ibutilide pharmacologic cardioversion can cause adverse events, but these are transient and treatable,” he said.
Many on the panel agreed. “I thought the totality of evidence supported the hypothesis that this drug has a potential for a fatal side effect in a disease that you can live with, potentially, and that there are other treatments for,” said Julia B. Lewis, MD, Vanderbilt Medical Center, Nashville, Tenn., who chaired the CRDAC panel.
“The drug clearly converts atrial fibrillation, although it’s only transient,” observed John H. Alexander, MD, MHSc, Duke University, Durham, N.C., one of the two panelists who voted to recommend approval of vernakalant.
“And, there clearly is a serious safety signal in some populations of patients,” he agreed. “However, I was more reassured by the SPECTRUM data.” There is likely to be a low-risk group of patients for whom vernakalant could represent an important option that “outweighs the relatively low risk of serious complications,” Dr. Alexander said.
“So more work needs to be done to clarify who are the low risk patients where it would be favorable.”
Panelist Matthew Needleman, MD, Walter Reed National Military Medical Center, Bethesda, Md., also voted in favor of approval.
“We’ve all known patients with normal ejection fractions who keep coming in with symptomatic atrial fib, want to get out of it quickly, and get back to their lives. So having an option like this I think would be good for a very select group of patients,” Dr. Needleman said.
But the preinfusion checklist and other potential ways to select low-risk patients for vernakalant could potentially backfire, warned John M. Mandrola, MD, Baptist Medical Associates, Louisville, Ky., from the panel.
The FDA representatives had presented evidence that the drug can seriously depress ventricular function, and that the lower cardiac output is what leads to hypotension, he elaborated in an interview after the meeting.
If the checklist is used to exclude hemodynamically unstable patients from receiving vernakalant, he said, “Then you’re really giving this drug to relatively healthy patients for convenience, to decrease hospitalization or the hospital stay.”
The signal for substantial harm, Dr. Mandrola said, has to be balanced against that modest benefit.
Moreover, those in whom the drug doesn’t work may be left in a worse situation, he proposed. Only about half of patients are successfully converted on vernakalant, the company and FDA data suggested. The other half of patients who don’t achieve sinus rhythm on the drug still must face the significant hazards of depressed ejection fraction and hypotension, a high price to pay for an unsuccessful treatment.
Dr. Mandrola is Chief Cardiology Correspondent for theheart.org | Medscape Cardiology; his disclosure statement states no relevant financial relationships.
This article first appeared on Medscape.com.
FDA investigates NDMA contamination in metformin

This follows reports of low-level NDMA contamination of metformin in other countries and of a few regulatory agencies issuing recalls for the drug, according to a statement from Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research.
“There are no metformin recalls affecting the U.S. market at this time,” the agency emphasized in the statement. It said NDMA levels in affected medication have been low, at or even below the acceptable intake limit, and there is currently no evidence indicating that metformin drugs within the United States or European Union have been contaminated.
The FDA advised that patients should continue taking metformin alone or in combination with other drugs to control their diabetes and that it would be dangerous for them to stop taking the medication without first discussing it with their providers. It also recommended that providers continue to use metformin when “clinically appropriate” while the investigation is underway as there are no alternative therapies to treat the disease in the same way.
NDMA is a common contaminant that is found in water and some foods and has probable carcinogenic effects when exposure is too high. The acceptable daily intake for NDMA in the United States is 96 ng/day, according to the statement, though people who take in that amount or less every day for 70 years are not expected to have an increased risk of cancer.
Both the FDA and its counterpart, the European Medicines Agency, have recently investigated the presence of NDMA impurities in ranitidine, a drug used to reduce production of stomach acid, which led to several manufacturers issuing recalls for it.
The agencies have also investigated angiotensin II receptor blockers, which are used to treat hypertension, heart failure, and high blood pressure.
The presence of NDMA “can be related to the drug’s manufacturing process or its chemical structure or even the conditions in which they are stored or packaged. As food and drugs are processed in the body, nitrosamines, including NDMA, can be formed,” Dr. Woodcock noted in the statement.
“We are monitoring this issue closely to assess any potential impact on patients with diabetes,” said Robert W. Lash, MD, chief professional and clinical affairs officer of the Endocrine Society. “We have members around the world and are concerned about the possibility of carcinogenic impurities in medications, both in the United States and elsewhere.”
This follows reports of low-level NDMA contamination of metformin in other countries and of a few regulatory agencies issuing recalls for the drug, according to a statement from Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research.
“There are no metformin recalls affecting the U.S. market at this time,” the agency emphasized in the statement. It said NDMA levels in affected medication have been low, at or even below the acceptable intake limit, and there is currently no evidence indicating that metformin drugs within the United States or European Union have been contaminated.
The FDA advised that patients should continue taking metformin alone or in combination with other drugs to control their diabetes and that it would be dangerous for them to stop taking the medication without first discussing it with their providers. It also recommended that providers continue to use metformin when “clinically appropriate” while the investigation is underway as there are no alternative therapies to treat the disease in the same way.
NDMA is a common contaminant that is found in water and some foods and has probable carcinogenic effects when exposure is too high. The acceptable daily intake for NDMA in the United States is 96 ng/day, according to the statement, though people who take in that amount or less every day for 70 years are not expected to have an increased risk of cancer.
Both the FDA and its counterpart, the European Medicines Agency, have recently investigated the presence of NDMA impurities in ranitidine, a drug used to reduce production of stomach acid, which led to several manufacturers issuing recalls for it.
The agencies have also investigated angiotensin II receptor blockers, which are used to treat hypertension, heart failure, and high blood pressure.
The presence of NDMA “can be related to the drug’s manufacturing process or its chemical structure or even the conditions in which they are stored or packaged. As food and drugs are processed in the body, nitrosamines, including NDMA, can be formed,” Dr. Woodcock noted in the statement.
“We are monitoring this issue closely to assess any potential impact on patients with diabetes,” said Robert W. Lash, MD, chief professional and clinical affairs officer of the Endocrine Society. “We have members around the world and are concerned about the possibility of carcinogenic impurities in medications, both in the United States and elsewhere.”
This follows reports of low-level NDMA contamination of metformin in other countries and of a few regulatory agencies issuing recalls for the drug, according to a statement from Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research.
“There are no metformin recalls affecting the U.S. market at this time,” the agency emphasized in the statement. It said NDMA levels in affected medication have been low, at or even below the acceptable intake limit, and there is currently no evidence indicating that metformin drugs within the United States or European Union have been contaminated.
The FDA advised that patients should continue taking metformin alone or in combination with other drugs to control their diabetes and that it would be dangerous for them to stop taking the medication without first discussing it with their providers. It also recommended that providers continue to use metformin when “clinically appropriate” while the investigation is underway as there are no alternative therapies to treat the disease in the same way.
NDMA is a common contaminant that is found in water and some foods and has probable carcinogenic effects when exposure is too high. The acceptable daily intake for NDMA in the United States is 96 ng/day, according to the statement, though people who take in that amount or less every day for 70 years are not expected to have an increased risk of cancer.
Both the FDA and its counterpart, the European Medicines Agency, have recently investigated the presence of NDMA impurities in ranitidine, a drug used to reduce production of stomach acid, which led to several manufacturers issuing recalls for it.
The agencies have also investigated angiotensin II receptor blockers, which are used to treat hypertension, heart failure, and high blood pressure.
The presence of NDMA “can be related to the drug’s manufacturing process or its chemical structure or even the conditions in which they are stored or packaged. As food and drugs are processed in the body, nitrosamines, including NDMA, can be formed,” Dr. Woodcock noted in the statement.
“We are monitoring this issue closely to assess any potential impact on patients with diabetes,” said Robert W. Lash, MD, chief professional and clinical affairs officer of the Endocrine Society. “We have members around the world and are concerned about the possibility of carcinogenic impurities in medications, both in the United States and elsewhere.”
E-cigarette use, interest in flavors remains high among youth
, according to new findings from the Centers for Disease Control and Prevention.

Just over half of high school students and about a quarter of middle school students have ever tried a tobacco product, and more than a third of students have ever tried an e-cigarette, according to results from the 2019 National Youth Tobacco Survey. These results were published in the Morbidity and Mortality Weekly Report on Dec. 6.
Adolescent cigarette smoking rates have continued their decline, hitting their lowest rate ever in 2019, but e-cigarette use, or “vaping,” has continued to increase. E-cigarette use surpassed that of all other tobacco products in 2014 and has remained the most common—as well as the least likely to be perceived as harmful, researchers reported.
“Although most current youth tobacco product users are not daily users, estimates of frequent e-cigarette use among high school students were comparable to those observed for cigarette and smokeless tobacco product users in 2019,” wrote Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, and associates at the CDC and Food and Drug Administration. “Youth use of tobacco products in any form is unsafe, regardless of whether the products are smoked, smokeless, or electronic.”
The high prevalence of e-cigarette use was no surprise to Karen Wilson, MD, chief of the division of general pediatrics at the Icahn School of Medicine at Mount Sinai and Mount Sinai Kravis Children’s Hospital, New York, and chair of the American Academy of Pediatrics’ Tobacco Consortium.
“It also fits with what we’re seeing anecdotally,” Dr. Wilson said in an interview. “We hear the statistic that 30% of high school students are using them, but high school students will say it’s much more than that.”
It’s therefore important for physicians to be proactive in talking to youth about these products. “They should absolutely be screening for vaping and know all about the different products,” including JUUL, Suorin, nicotine toothpicks, and candies and other products, Dr. Wilson said. “Pediatricians need to be asking their teenagers open-ended questions about what are kids using now.”
The American Academy of Pediatrics has resources available to help pediatricians and families of youth using e-cigarettes and vaping devices, she added.
Main findings
The researchers reported data from the annual, cross-sectional National Youth Tobacco Survey, administered to U.S. students in public and private schools in all 50 states and the District of Columbia. The results were divided into middle school (grades 6-8) and high school (grades 9-12) from 251 participating schools between February 2019 and May 2019.
The survey has been done using pencil and paper questionnaires since it began in 1999, but this year’s surveys were digital for the first time. Among the 19,018 questionnaires completed (student response rate 85.3%), 8,837 were middle school and 10,097 were high school. The weighted analysis of results represents 27 million students: 11.9 million in middle school and 15 million in high school.
More than half (53.3%) of high school students reported ever having tried a tobacco product, and 31.2% reported having used one in the past 30 days. In middle school, 24.3% of students reported ever using a tobacco product, and 12.5% have used one in the past month.
Tobacco products include cigarettes (traditional/combusted), electronic cigarettes, cigars, smokeless tobacco, hookahs, pipe tobacco, and bidis, which are small brown cigarettes wrapped in leaves. Among the electronic tobacco products mentioned in the survey were NJOY, Blu, Vuse, MarkTen, Logic, Vapin Plus, eGo and Halo.
The most common product for youth to try was e-cigarettes, which 35% of middle and high school students had ever tried. Just under a quarter of students (23%) had used a tobacco product in the past month, and e-cigarettes were again the most commonly used overall by that group, cited by 20% of recent users. Cigars (5.3%), cigarettes (4.3%), smokeless tobacco (3.5%), hookahs (2.6%) and pipes (under 1%) were used much less frequently.
Frequent use, defined as at least 20 of the previous 30 days, was most common among youth using smokeless tobacco (34.1% of current users) and e-cigarettes (30.4%) and least common among cigar smokers (16.8%). Among those currently using any tobacco product, 24.7% said they had cravings for a product within the past month, and 13.7% wanted to use it within a half hour of waking up.
More than half of those who currently used any tobacco products (57.8%) were seriously considering quitting, and a similar proportion (57.5%) had stopped using all tobacco products for at least 1 day in an attempt to quit.
“Many [adolescents] will tell you they will use it until they don’t have the availability of getting it,” Dr. Wilson said. “The problem is that they’re becoming so addicted to the high-nicotine products that they’re going farther and farther out of their way to try to get these products so that they can satisfy their addictions.”
Policies restricting access, such as increasing the age for sales to 21 and increasing taxes on products, can reduce tobacco use among youth, Dr. Wilson said.
“It will encourage teenagers to get help for their addiction by using FDA-approved devices or nicotine replacement therapy and behavioral interventions rather than relying on an unproven and potentially dangerous product,” she said.
Reasons for use, flavor, and harm perception
The most common flavored tobacco product used among youth was e-cigarettes, reported by 68.8% of current e-cigarette users, followed by smokeless tobacco (48%), cigarettes (46.7%, only menthol), cigars, pipe tobacco, and hookahs.
The top reasons youth cited for trying e-cigarettes were curiosity (55.3%), a friend or family member’s use (30.8%), and their availability in a wide range of flavors (22.4%). Almost as popular as flavor availability was e-cigarette users’ interest in doing “tricks” with the product (21.2%).
The cross-sectional questionnaire method of the study precluded the ability to draw conclusions about why students might perceive a particular tobacco product as more or less harmful. However, public health officials have expressed concern that flavors reduce the perceived harm that can come from the products. Dr. Wilson said the attraction to e-cigarette flavors is “huge.”
“If electronic cigarettes were only available in tobacco flavor, I do not believe that many teenagers at all would try them,” Dr. Wilson said. “They think because they’re sweet and flavored that they actually aren’t harmful. It makes the kids think these are safe products.”
More than one in four students (28.2%) perceived intermittent e-cigarette use as causing little to no harm, and only 16.4% similarly saw little or no harm from intermittent hookah use, compared with 11.5% for smokeless tobacco and 9.5% for cigarettes. Less than a third of respondents (32.3%) saw intermittent e-cigarette use as causing a lot of harm, compared with much higher percentages for cigarettes (54.9%) and smokeless tobacco (52.5%).
Part of the problem with harm perception is the narrative promoted by e-cigarette companies, Dr. Wilson said.
“From the very beginning, they started with a campaign that called this harmless water vapor, which it is absolutely not,” she said. “It’s an aerosol of toxic chemicals and nicotine, which is addictive. We know that nicotine that can impact scores of cognitive tests and impulsivity. We have no idea what these really high levels [of nicotine] will do.”
Further, potential long-term harm is still an open question, she pointed out.
“We also know that these are particulates and toxins that are being inhaled into the lungs,” Dr. Wilson said. “We know they have some impact on asthma, and we don’t know what the impact is for using for 10 or 20 years.”
Curiosity about e-cigarettes and about traditional cigarettes were prevalent in similar proportions among youth who had never tried a tobacco product: 39.1% of never-users were curious about e-cigarettes, and 37% about traditional cigarettes. In addition to curiosity, researchers assess susceptibility among those who have never tried a tobacco product and found nearly identical susceptibility to e-cigarettes (45%) and traditional cigarettes (45.9%).
The survey also asked students about their exposure to tobacco advertising or promotions from a wide range of sources: convenience stores, supermarkets, gas stations, the Internet, television, video streaming, cinemas, and newspapers or magazines. Among the students who reported going to these sources, 69.3% had seen e-cigarette marketing, and 81.7% had seen marketing for other tobacco products, including cigarettes.
SOURCE: Wang TW et al. MMWR Surveill Summ. 2019 Nov 6;68(12):1-22. doi: 10.15585/mmwr.ss6812a1.
, according to new findings from the Centers for Disease Control and Prevention.
Just over half of high school students and about a quarter of middle school students have ever tried a tobacco product, and more than a third of students have ever tried an e-cigarette, according to results from the 2019 National Youth Tobacco Survey. These results were published in the Morbidity and Mortality Weekly Report on Dec. 6.
Adolescent cigarette smoking rates have continued their decline, hitting their lowest rate ever in 2019, but e-cigarette use, or “vaping,” has continued to increase. E-cigarette use surpassed that of all other tobacco products in 2014 and has remained the most common—as well as the least likely to be perceived as harmful, researchers reported.
“Although most current youth tobacco product users are not daily users, estimates of frequent e-cigarette use among high school students were comparable to those observed for cigarette and smokeless tobacco product users in 2019,” wrote Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, and associates at the CDC and Food and Drug Administration. “Youth use of tobacco products in any form is unsafe, regardless of whether the products are smoked, smokeless, or electronic.”
The high prevalence of e-cigarette use was no surprise to Karen Wilson, MD, chief of the division of general pediatrics at the Icahn School of Medicine at Mount Sinai and Mount Sinai Kravis Children’s Hospital, New York, and chair of the American Academy of Pediatrics’ Tobacco Consortium.
“It also fits with what we’re seeing anecdotally,” Dr. Wilson said in an interview. “We hear the statistic that 30% of high school students are using them, but high school students will say it’s much more than that.”
It’s therefore important for physicians to be proactive in talking to youth about these products. “They should absolutely be screening for vaping and know all about the different products,” including JUUL, Suorin, nicotine toothpicks, and candies and other products, Dr. Wilson said. “Pediatricians need to be asking their teenagers open-ended questions about what are kids using now.”
The American Academy of Pediatrics has resources available to help pediatricians and families of youth using e-cigarettes and vaping devices, she added.
Main findings
The researchers reported data from the annual, cross-sectional National Youth Tobacco Survey, administered to U.S. students in public and private schools in all 50 states and the District of Columbia. The results were divided into middle school (grades 6-8) and high school (grades 9-12) from 251 participating schools between February 2019 and May 2019.
The survey has been done using pencil and paper questionnaires since it began in 1999, but this year’s surveys were digital for the first time. Among the 19,018 questionnaires completed (student response rate 85.3%), 8,837 were middle school and 10,097 were high school. The weighted analysis of results represents 27 million students: 11.9 million in middle school and 15 million in high school.
More than half (53.3%) of high school students reported ever having tried a tobacco product, and 31.2% reported having used one in the past 30 days. In middle school, 24.3% of students reported ever using a tobacco product, and 12.5% have used one in the past month.
Tobacco products include cigarettes (traditional/combusted), electronic cigarettes, cigars, smokeless tobacco, hookahs, pipe tobacco, and bidis, which are small brown cigarettes wrapped in leaves. Among the electronic tobacco products mentioned in the survey were NJOY, Blu, Vuse, MarkTen, Logic, Vapin Plus, eGo and Halo.
The most common product for youth to try was e-cigarettes, which 35% of middle and high school students had ever tried. Just under a quarter of students (23%) had used a tobacco product in the past month, and e-cigarettes were again the most commonly used overall by that group, cited by 20% of recent users. Cigars (5.3%), cigarettes (4.3%), smokeless tobacco (3.5%), hookahs (2.6%) and pipes (under 1%) were used much less frequently.
Frequent use, defined as at least 20 of the previous 30 days, was most common among youth using smokeless tobacco (34.1% of current users) and e-cigarettes (30.4%) and least common among cigar smokers (16.8%). Among those currently using any tobacco product, 24.7% said they had cravings for a product within the past month, and 13.7% wanted to use it within a half hour of waking up.
More than half of those who currently used any tobacco products (57.8%) were seriously considering quitting, and a similar proportion (57.5%) had stopped using all tobacco products for at least 1 day in an attempt to quit.
“Many [adolescents] will tell you they will use it until they don’t have the availability of getting it,” Dr. Wilson said. “The problem is that they’re becoming so addicted to the high-nicotine products that they’re going farther and farther out of their way to try to get these products so that they can satisfy their addictions.”
Policies restricting access, such as increasing the age for sales to 21 and increasing taxes on products, can reduce tobacco use among youth, Dr. Wilson said.
“It will encourage teenagers to get help for their addiction by using FDA-approved devices or nicotine replacement therapy and behavioral interventions rather than relying on an unproven and potentially dangerous product,” she said.
Reasons for use, flavor, and harm perception
The most common flavored tobacco product used among youth was e-cigarettes, reported by 68.8% of current e-cigarette users, followed by smokeless tobacco (48%), cigarettes (46.7%, only menthol), cigars, pipe tobacco, and hookahs.
The top reasons youth cited for trying e-cigarettes were curiosity (55.3%), a friend or family member’s use (30.8%), and their availability in a wide range of flavors (22.4%). Almost as popular as flavor availability was e-cigarette users’ interest in doing “tricks” with the product (21.2%).
The cross-sectional questionnaire method of the study precluded the ability to draw conclusions about why students might perceive a particular tobacco product as more or less harmful. However, public health officials have expressed concern that flavors reduce the perceived harm that can come from the products. Dr. Wilson said the attraction to e-cigarette flavors is “huge.”
“If electronic cigarettes were only available in tobacco flavor, I do not believe that many teenagers at all would try them,” Dr. Wilson said. “They think because they’re sweet and flavored that they actually aren’t harmful. It makes the kids think these are safe products.”
More than one in four students (28.2%) perceived intermittent e-cigarette use as causing little to no harm, and only 16.4% similarly saw little or no harm from intermittent hookah use, compared with 11.5% for smokeless tobacco and 9.5% for cigarettes. Less than a third of respondents (32.3%) saw intermittent e-cigarette use as causing a lot of harm, compared with much higher percentages for cigarettes (54.9%) and smokeless tobacco (52.5%).
Part of the problem with harm perception is the narrative promoted by e-cigarette companies, Dr. Wilson said.
“From the very beginning, they started with a campaign that called this harmless water vapor, which it is absolutely not,” she said. “It’s an aerosol of toxic chemicals and nicotine, which is addictive. We know that nicotine that can impact scores of cognitive tests and impulsivity. We have no idea what these really high levels [of nicotine] will do.”
Further, potential long-term harm is still an open question, she pointed out.
“We also know that these are particulates and toxins that are being inhaled into the lungs,” Dr. Wilson said. “We know they have some impact on asthma, and we don’t know what the impact is for using for 10 or 20 years.”
Curiosity about e-cigarettes and about traditional cigarettes were prevalent in similar proportions among youth who had never tried a tobacco product: 39.1% of never-users were curious about e-cigarettes, and 37% about traditional cigarettes. In addition to curiosity, researchers assess susceptibility among those who have never tried a tobacco product and found nearly identical susceptibility to e-cigarettes (45%) and traditional cigarettes (45.9%).
The survey also asked students about their exposure to tobacco advertising or promotions from a wide range of sources: convenience stores, supermarkets, gas stations, the Internet, television, video streaming, cinemas, and newspapers or magazines. Among the students who reported going to these sources, 69.3% had seen e-cigarette marketing, and 81.7% had seen marketing for other tobacco products, including cigarettes.
SOURCE: Wang TW et al. MMWR Surveill Summ. 2019 Nov 6;68(12):1-22. doi: 10.15585/mmwr.ss6812a1.
, according to new findings from the Centers for Disease Control and Prevention.
Just over half of high school students and about a quarter of middle school students have ever tried a tobacco product, and more than a third of students have ever tried an e-cigarette, according to results from the 2019 National Youth Tobacco Survey. These results were published in the Morbidity and Mortality Weekly Report on Dec. 6.
Adolescent cigarette smoking rates have continued their decline, hitting their lowest rate ever in 2019, but e-cigarette use, or “vaping,” has continued to increase. E-cigarette use surpassed that of all other tobacco products in 2014 and has remained the most common—as well as the least likely to be perceived as harmful, researchers reported.
“Although most current youth tobacco product users are not daily users, estimates of frequent e-cigarette use among high school students were comparable to those observed for cigarette and smokeless tobacco product users in 2019,” wrote Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, and associates at the CDC and Food and Drug Administration. “Youth use of tobacco products in any form is unsafe, regardless of whether the products are smoked, smokeless, or electronic.”
The high prevalence of e-cigarette use was no surprise to Karen Wilson, MD, chief of the division of general pediatrics at the Icahn School of Medicine at Mount Sinai and Mount Sinai Kravis Children’s Hospital, New York, and chair of the American Academy of Pediatrics’ Tobacco Consortium.
“It also fits with what we’re seeing anecdotally,” Dr. Wilson said in an interview. “We hear the statistic that 30% of high school students are using them, but high school students will say it’s much more than that.”
It’s therefore important for physicians to be proactive in talking to youth about these products. “They should absolutely be screening for vaping and know all about the different products,” including JUUL, Suorin, nicotine toothpicks, and candies and other products, Dr. Wilson said. “Pediatricians need to be asking their teenagers open-ended questions about what are kids using now.”
The American Academy of Pediatrics has resources available to help pediatricians and families of youth using e-cigarettes and vaping devices, she added.
Main findings
The researchers reported data from the annual, cross-sectional National Youth Tobacco Survey, administered to U.S. students in public and private schools in all 50 states and the District of Columbia. The results were divided into middle school (grades 6-8) and high school (grades 9-12) from 251 participating schools between February 2019 and May 2019.
The survey has been done using pencil and paper questionnaires since it began in 1999, but this year’s surveys were digital for the first time. Among the 19,018 questionnaires completed (student response rate 85.3%), 8,837 were middle school and 10,097 were high school. The weighted analysis of results represents 27 million students: 11.9 million in middle school and 15 million in high school.
More than half (53.3%) of high school students reported ever having tried a tobacco product, and 31.2% reported having used one in the past 30 days. In middle school, 24.3% of students reported ever using a tobacco product, and 12.5% have used one in the past month.
Tobacco products include cigarettes (traditional/combusted), electronic cigarettes, cigars, smokeless tobacco, hookahs, pipe tobacco, and bidis, which are small brown cigarettes wrapped in leaves. Among the electronic tobacco products mentioned in the survey were NJOY, Blu, Vuse, MarkTen, Logic, Vapin Plus, eGo and Halo.
The most common product for youth to try was e-cigarettes, which 35% of middle and high school students had ever tried. Just under a quarter of students (23%) had used a tobacco product in the past month, and e-cigarettes were again the most commonly used overall by that group, cited by 20% of recent users. Cigars (5.3%), cigarettes (4.3%), smokeless tobacco (3.5%), hookahs (2.6%) and pipes (under 1%) were used much less frequently.
Frequent use, defined as at least 20 of the previous 30 days, was most common among youth using smokeless tobacco (34.1% of current users) and e-cigarettes (30.4%) and least common among cigar smokers (16.8%). Among those currently using any tobacco product, 24.7% said they had cravings for a product within the past month, and 13.7% wanted to use it within a half hour of waking up.
More than half of those who currently used any tobacco products (57.8%) were seriously considering quitting, and a similar proportion (57.5%) had stopped using all tobacco products for at least 1 day in an attempt to quit.
“Many [adolescents] will tell you they will use it until they don’t have the availability of getting it,” Dr. Wilson said. “The problem is that they’re becoming so addicted to the high-nicotine products that they’re going farther and farther out of their way to try to get these products so that they can satisfy their addictions.”
Policies restricting access, such as increasing the age for sales to 21 and increasing taxes on products, can reduce tobacco use among youth, Dr. Wilson said.
“It will encourage teenagers to get help for their addiction by using FDA-approved devices or nicotine replacement therapy and behavioral interventions rather than relying on an unproven and potentially dangerous product,” she said.
Reasons for use, flavor, and harm perception
The most common flavored tobacco product used among youth was e-cigarettes, reported by 68.8% of current e-cigarette users, followed by smokeless tobacco (48%), cigarettes (46.7%, only menthol), cigars, pipe tobacco, and hookahs.
The top reasons youth cited for trying e-cigarettes were curiosity (55.3%), a friend or family member’s use (30.8%), and their availability in a wide range of flavors (22.4%). Almost as popular as flavor availability was e-cigarette users’ interest in doing “tricks” with the product (21.2%).
The cross-sectional questionnaire method of the study precluded the ability to draw conclusions about why students might perceive a particular tobacco product as more or less harmful. However, public health officials have expressed concern that flavors reduce the perceived harm that can come from the products. Dr. Wilson said the attraction to e-cigarette flavors is “huge.”
“If electronic cigarettes were only available in tobacco flavor, I do not believe that many teenagers at all would try them,” Dr. Wilson said. “They think because they’re sweet and flavored that they actually aren’t harmful. It makes the kids think these are safe products.”
More than one in four students (28.2%) perceived intermittent e-cigarette use as causing little to no harm, and only 16.4% similarly saw little or no harm from intermittent hookah use, compared with 11.5% for smokeless tobacco and 9.5% for cigarettes. Less than a third of respondents (32.3%) saw intermittent e-cigarette use as causing a lot of harm, compared with much higher percentages for cigarettes (54.9%) and smokeless tobacco (52.5%).
Part of the problem with harm perception is the narrative promoted by e-cigarette companies, Dr. Wilson said.
“From the very beginning, they started with a campaign that called this harmless water vapor, which it is absolutely not,” she said. “It’s an aerosol of toxic chemicals and nicotine, which is addictive. We know that nicotine that can impact scores of cognitive tests and impulsivity. We have no idea what these really high levels [of nicotine] will do.”
Further, potential long-term harm is still an open question, she pointed out.
“We also know that these are particulates and toxins that are being inhaled into the lungs,” Dr. Wilson said. “We know they have some impact on asthma, and we don’t know what the impact is for using for 10 or 20 years.”
Curiosity about e-cigarettes and about traditional cigarettes were prevalent in similar proportions among youth who had never tried a tobacco product: 39.1% of never-users were curious about e-cigarettes, and 37% about traditional cigarettes. In addition to curiosity, researchers assess susceptibility among those who have never tried a tobacco product and found nearly identical susceptibility to e-cigarettes (45%) and traditional cigarettes (45.9%).
The survey also asked students about their exposure to tobacco advertising or promotions from a wide range of sources: convenience stores, supermarkets, gas stations, the Internet, television, video streaming, cinemas, and newspapers or magazines. Among the students who reported going to these sources, 69.3% had seen e-cigarette marketing, and 81.7% had seen marketing for other tobacco products, including cigarettes.
SOURCE: Wang TW et al. MMWR Surveill Summ. 2019 Nov 6;68(12):1-22. doi: 10.15585/mmwr.ss6812a1.
FROM THE MMWR

First generics for Gilenya approved by FDA
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generics of fingolimod (Gilenya) for the treatment of relapsing forms of multiple sclerosis.
The three generic fingolimod applications came from HEC Pharm, Biocon, and Sun Pharmaceutical Industries.
Fingolimod is a widely used, orally administered treatment option for relapsing forms of multiple sclerosis in adults. The most common adverse events associated with fingolimod in clinical trials include headache, elevation of liver enzymes, diarrhea, cough, influenza, sinusitis, back pain, abdominal pain, and pain in the extremities.
The drug must be dispensed with a medication guide that contains important information on its usage and risk, the FDA noted. Serious risks associated with fingolimod include slowing of the heart rate, vision problems, posterior reversible encephalopathy syndrome, respiratory problems, liver injury, increased blood pressure, skin cancer, and risk of serious infection including a rare and often deadly brain infection called progressive multifocal leukoencephalopathy. Fingolimod can also cause harm to a developing fetus.
Find the full press release on the FDA website.
FDA approves infliximab-axxq for numerous indications
The Food and Drug Administration has approved the biosimilar infliximab-axxq (Avsola) for various indications, making it the fourth biosimilar of infliximab (Remicade) to be cleared for marketing by the agency.
The tumor necrosis factor inhibitor is indicated for patients with Crohn’s disease or ulcerative colitis who are aged 6 years and older, RA in combination with methotrexate, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis. The approval is based on numerous trials. The most common adverse reactions are infections, infusion-related reactions, headache, and abdominal pain.
Full prescribing information can be found on the FDA website, as can more information about biosimilars.
The Food and Drug Administration has approved the biosimilar infliximab-axxq (Avsola) for various indications, making it the fourth biosimilar of infliximab (Remicade) to be cleared for marketing by the agency.
The tumor necrosis factor inhibitor is indicated for patients with Crohn’s disease or ulcerative colitis who are aged 6 years and older, RA in combination with methotrexate, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis. The approval is based on numerous trials. The most common adverse reactions are infections, infusion-related reactions, headache, and abdominal pain.
Full prescribing information can be found on the FDA website, as can more information about biosimilars.
The Food and Drug Administration has approved the biosimilar infliximab-axxq (Avsola) for various indications, making it the fourth biosimilar of infliximab (Remicade) to be cleared for marketing by the agency.
The tumor necrosis factor inhibitor is indicated for patients with Crohn’s disease or ulcerative colitis who are aged 6 years and older, RA in combination with methotrexate, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis. The approval is based on numerous trials. The most common adverse reactions are infections, infusion-related reactions, headache, and abdominal pain.
Full prescribing information can be found on the FDA website, as can more information about biosimilars.
FDA fast-tracks psilocybin for major depressive disorder
Psilocybin, a short-acting compound that is the psychoactive ingredient in “magic mushrooms,” has received a Breakthrough Therapy designation from the Food and Drug Administration for the treatment of adults with major depressive disorder.
The designation was given to the Usona Institute, a nonprofit medical research organization, and comes in the wake of Usona’s launch of a phase 2 clinical trial that will include about 80 participants at seven study sites across the United States, according to a press release. Two sites are currently recruiting patients, and the others are expected to begin recruiting in 2020.
Breakthrough Therapy designation as defined by the FDA means that, based on preliminary research, “the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint.” In this case, Usona is working with the University of Wisconsin’s University Hospital in Madison, and other collaborators, according to a presentation by Malynn Utzinger, MD, director of integrative medicine and cofounder of the organization.
More information on the Usona Institute and Usona’s clinical trials is available at https://usonaclinicaltrials.org/.
Psilocybin, a short-acting compound that is the psychoactive ingredient in “magic mushrooms,” has received a Breakthrough Therapy designation from the Food and Drug Administration for the treatment of adults with major depressive disorder.
The designation was given to the Usona Institute, a nonprofit medical research organization, and comes in the wake of Usona’s launch of a phase 2 clinical trial that will include about 80 participants at seven study sites across the United States, according to a press release. Two sites are currently recruiting patients, and the others are expected to begin recruiting in 2020.
Breakthrough Therapy designation as defined by the FDA means that, based on preliminary research, “the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint.” In this case, Usona is working with the University of Wisconsin’s University Hospital in Madison, and other collaborators, according to a presentation by Malynn Utzinger, MD, director of integrative medicine and cofounder of the organization.
More information on the Usona Institute and Usona’s clinical trials is available at https://usonaclinicaltrials.org/.
Psilocybin, a short-acting compound that is the psychoactive ingredient in “magic mushrooms,” has received a Breakthrough Therapy designation from the Food and Drug Administration for the treatment of adults with major depressive disorder.
The designation was given to the Usona Institute, a nonprofit medical research organization, and comes in the wake of Usona’s launch of a phase 2 clinical trial that will include about 80 participants at seven study sites across the United States, according to a press release. Two sites are currently recruiting patients, and the others are expected to begin recruiting in 2020.
Breakthrough Therapy designation as defined by the FDA means that, based on preliminary research, “the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint.” In this case, Usona is working with the University of Wisconsin’s University Hospital in Madison, and other collaborators, according to a presentation by Malynn Utzinger, MD, director of integrative medicine and cofounder of the organization.
More information on the Usona Institute and Usona’s clinical trials is available at https://usonaclinicaltrials.org/.
FDA approves atezolizumab combo as first line for advanced NSCLC
chemotherapy for first-line treatment of adults with metastatic, nonsquamous non–small cell lung cancer (NSCLC) with no EGFR or ALK genomic tumor aberrations.
Atezolizumab has been previously approved in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of adults with metastatic NSCLC with no EGFR or ALK genomic tumor aberrations. The monoclonal antibody is also approved to treat adults with metastatic NSCLC who have disease progression during or following chemotherapy, and for those with extensive-stage SCLC.
The current approval was based on a demonstrated improvement in overall survival in the phase 3 IMpower130 trial (NCT02367781). Median overall survival for advanced NSCLC patients who received atezolizumab in combination with chemotherapy was 18.6 months, compared with 13.9 months for patients who received chemotherapy alone (hazard ratio, 0.80; 95% confidence interval, 0.64-0.99; P = .0384) in the intention-to-treat wild-type population of 681 patients.
Grade 3-4 treatment-related adverse events were reported in 73.2% of people receiving atezolizumab plus chemotherapy, compared with 60.3% of people receiving chemotherapy alone, according to the company press release.
chemotherapy for first-line treatment of adults with metastatic, nonsquamous non–small cell lung cancer (NSCLC) with no EGFR or ALK genomic tumor aberrations.
Atezolizumab has been previously approved in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of adults with metastatic NSCLC with no EGFR or ALK genomic tumor aberrations. The monoclonal antibody is also approved to treat adults with metastatic NSCLC who have disease progression during or following chemotherapy, and for those with extensive-stage SCLC.
The current approval was based on a demonstrated improvement in overall survival in the phase 3 IMpower130 trial (NCT02367781). Median overall survival for advanced NSCLC patients who received atezolizumab in combination with chemotherapy was 18.6 months, compared with 13.9 months for patients who received chemotherapy alone (hazard ratio, 0.80; 95% confidence interval, 0.64-0.99; P = .0384) in the intention-to-treat wild-type population of 681 patients.
Grade 3-4 treatment-related adverse events were reported in 73.2% of people receiving atezolizumab plus chemotherapy, compared with 60.3% of people receiving chemotherapy alone, according to the company press release.
chemotherapy for first-line treatment of adults with metastatic, nonsquamous non–small cell lung cancer (NSCLC) with no EGFR or ALK genomic tumor aberrations.
Atezolizumab has been previously approved in combination with bevacizumab, paclitaxel, and carboplatin for the first-line treatment of adults with metastatic NSCLC with no EGFR or ALK genomic tumor aberrations. The monoclonal antibody is also approved to treat adults with metastatic NSCLC who have disease progression during or following chemotherapy, and for those with extensive-stage SCLC.
The current approval was based on a demonstrated improvement in overall survival in the phase 3 IMpower130 trial (NCT02367781). Median overall survival for advanced NSCLC patients who received atezolizumab in combination with chemotherapy was 18.6 months, compared with 13.9 months for patients who received chemotherapy alone (hazard ratio, 0.80; 95% confidence interval, 0.64-0.99; P = .0384) in the intention-to-treat wild-type population of 681 patients.
Grade 3-4 treatment-related adverse events were reported in 73.2% of people receiving atezolizumab plus chemotherapy, compared with 60.3% of people receiving chemotherapy alone, according to the company press release.