User login
FDA expands use of Toujeo to childhood type 1 diabetes
The Food and Drug Administration has expanded the indication for Toujeo (insulin glargine 300 units/mL injection; Sanofi) to include children as young as 6 years of age with type 1 diabetes.
The FDA first approved Toujeo in 2015 for adults with type 1 and type 2 diabetes, designed as a more potent follow-up to Sanofi’s top-selling insulin glargine (Lantus).
Last month, Sanofi reported positive results from the phase 3 EDITION JUNIOR trial of Toujeo in children and adolescents with type 1 diabetes. These were presented at the International Society for Pediatric and Adolescent Diabetes 45th Annual Conference in Boston.
In the trial, 463 children and adolescents (aged 6-17 years) treated for type 1 diabetes for at least 1 year and with A1c between 7.5% and 11.0% at screening were randomized to Toujeo or insulin glargine 100 units/mL (Gla-100); participants continued to take their existing mealtime insulin.
The primary endpoint was noninferior reduction in A1c after 26 weeks.
The study met its primary endpoint, confirming a noninferior reduction in A1c with Toujeo versus Gla-100 after 26 weeks (mean reduction, 0.4% vs. 0.4%; difference, 0.004%; 95% confidence interval, –0.17 to 0.18; upper bound was below the prespecified noninferiority margin of 0.3%).
Over 26 weeks, a comparable number of patients in each group experienced one or more hypoglycemic events documented at anytime over 24 hours. Numerically fewer patients taking Toujeo experienced severe hypoglycemia or experienced one or more episodes of hyperglycemia with ketosis compared with those taking Gla-100.
No unexpected safety concerns were reported based on the established profiles of both products, the company said.
In October 2019, the European Medicines Agency Committee for Medicinal Products for Human Use recommended approval of Toujeo for children age 6 years and older with diabetes.
For more diabetes and endocrinology news, follow us on Twitter and Facebook.
This story first appeared on Medscape.com.
The Food and Drug Administration has expanded the indication for Toujeo (insulin glargine 300 units/mL injection; Sanofi) to include children as young as 6 years of age with type 1 diabetes.
The FDA first approved Toujeo in 2015 for adults with type 1 and type 2 diabetes, designed as a more potent follow-up to Sanofi’s top-selling insulin glargine (Lantus).
Last month, Sanofi reported positive results from the phase 3 EDITION JUNIOR trial of Toujeo in children and adolescents with type 1 diabetes. These were presented at the International Society for Pediatric and Adolescent Diabetes 45th Annual Conference in Boston.
In the trial, 463 children and adolescents (aged 6-17 years) treated for type 1 diabetes for at least 1 year and with A1c between 7.5% and 11.0% at screening were randomized to Toujeo or insulin glargine 100 units/mL (Gla-100); participants continued to take their existing mealtime insulin.
The primary endpoint was noninferior reduction in A1c after 26 weeks.
The study met its primary endpoint, confirming a noninferior reduction in A1c with Toujeo versus Gla-100 after 26 weeks (mean reduction, 0.4% vs. 0.4%; difference, 0.004%; 95% confidence interval, –0.17 to 0.18; upper bound was below the prespecified noninferiority margin of 0.3%).
Over 26 weeks, a comparable number of patients in each group experienced one or more hypoglycemic events documented at anytime over 24 hours. Numerically fewer patients taking Toujeo experienced severe hypoglycemia or experienced one or more episodes of hyperglycemia with ketosis compared with those taking Gla-100.
No unexpected safety concerns were reported based on the established profiles of both products, the company said.
In October 2019, the European Medicines Agency Committee for Medicinal Products for Human Use recommended approval of Toujeo for children age 6 years and older with diabetes.
For more diabetes and endocrinology news, follow us on Twitter and Facebook.
This story first appeared on Medscape.com.
The Food and Drug Administration has expanded the indication for Toujeo (insulin glargine 300 units/mL injection; Sanofi) to include children as young as 6 years of age with type 1 diabetes.
The FDA first approved Toujeo in 2015 for adults with type 1 and type 2 diabetes, designed as a more potent follow-up to Sanofi’s top-selling insulin glargine (Lantus).
Last month, Sanofi reported positive results from the phase 3 EDITION JUNIOR trial of Toujeo in children and adolescents with type 1 diabetes. These were presented at the International Society for Pediatric and Adolescent Diabetes 45th Annual Conference in Boston.
In the trial, 463 children and adolescents (aged 6-17 years) treated for type 1 diabetes for at least 1 year and with A1c between 7.5% and 11.0% at screening were randomized to Toujeo or insulin glargine 100 units/mL (Gla-100); participants continued to take their existing mealtime insulin.
The primary endpoint was noninferior reduction in A1c after 26 weeks.
The study met its primary endpoint, confirming a noninferior reduction in A1c with Toujeo versus Gla-100 after 26 weeks (mean reduction, 0.4% vs. 0.4%; difference, 0.004%; 95% confidence interval, –0.17 to 0.18; upper bound was below the prespecified noninferiority margin of 0.3%).
Over 26 weeks, a comparable number of patients in each group experienced one or more hypoglycemic events documented at anytime over 24 hours. Numerically fewer patients taking Toujeo experienced severe hypoglycemia or experienced one or more episodes of hyperglycemia with ketosis compared with those taking Gla-100.
No unexpected safety concerns were reported based on the established profiles of both products, the company said.
In October 2019, the European Medicines Agency Committee for Medicinal Products for Human Use recommended approval of Toujeo for children age 6 years and older with diabetes.
For more diabetes and endocrinology news, follow us on Twitter and Facebook.
This story first appeared on Medscape.com.
CDC finds that efforts to reduce new HIV infections have stalled
, according to a Vital Signs report published by the Centers for Disease Control and Prevention based upon a simultaneous MMWR Early Release. The report indicates that many Americans with HIV are not aware of their status or are not receiving effective treatment. Furthermore, the data suggest that few Americans who could benefit from preexposure prophylaxis (PrEP), a daily pill that prevents HIV, are receiving it.
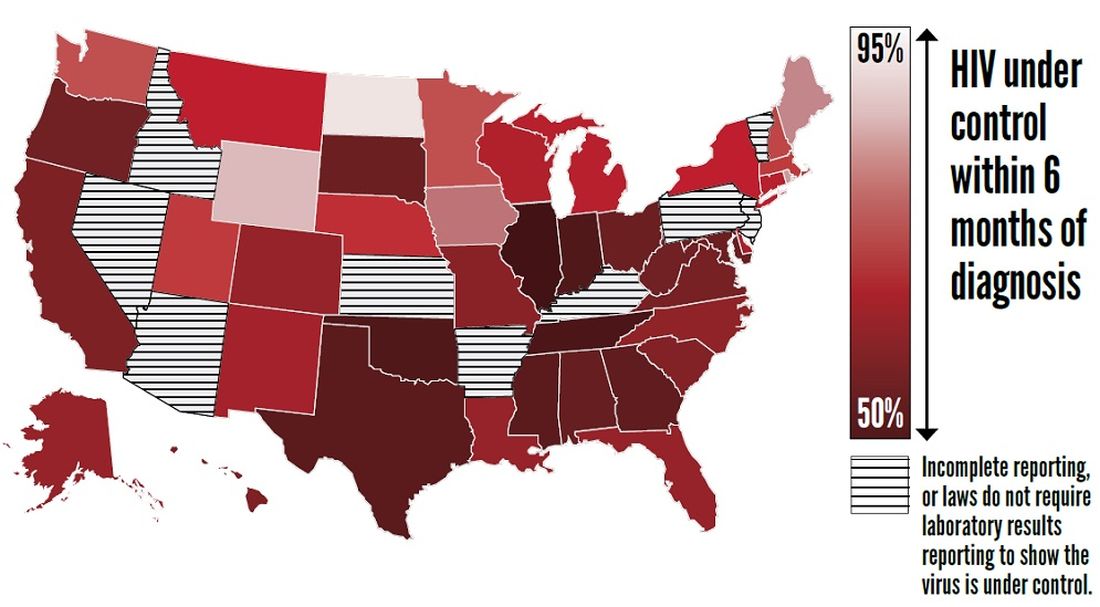
The report “shows that HIV testing, treatment, and prevention have not reached enough Americans, and it emphasizes the continued urgent need to increase these interventions,” said Jay C. Butler, MD, deputy director for infectious diseases at the CDC, at a press conference. “We made a lot of progress in the late ’90s and into the early part of the 21st century in reducing the number of new cases of HIV. But HIV prevention progress has stalled in America since 2013. This stalling underscores the need to increase resources, deploy new technologies, and build expertise, particularly in areas where they’re needed most.”
To achieve these objectives, the CDC has proposed a federal initiative called Ending the HIV Epidemic: A Plan for America. The goal of the initiative is to reduce new HIV infections by 90% by 2030, in part by expanding access to PrEP medications.
Data suggest shortcomings in diagnosis, treatment, and prevention
In its review of data on HIV testing and treatment in 2017, the Vital Signs report found that approximately 154,000 people with HIV (that is, 14% of the total population with HIV) were unaware that they had the virus. These patients consequently could not take advantage of HIV treatment to maintain health, control the virus, and prevent HIV transmission. Young people aged 13-24 years were less likely to know their HIV status than did those aged 25 years and older, according to the report.
Furthermore, approximately two-thirds (63%) of patients who knew that they had HIV had the virus under control through effective treatment. Young people and African Americans were least likely to have the virus under control, according to the report.
The report also examines data about treatment with PrEP in 2018. About 1.2 million Americans could benefit from PrEP, but only 219,700 (18%) of them had received a prescription for the drug. The eligible groups with the lowest rates of coverage were young people, African Americans, and Latinos.
The report presents a conservative estimate of PrEP coverage, however. Researchers examined data from 92% of prescriptions from retail pharmacies in the United States but did not include prescriptions from closed health care systems such as managed care organizations and military health plans. PrEP coverage in 2018 likely was higher than these estimates indicate, according to the CDC.
“There has been a rapid increase in the number of people taking PrEP over the past 3 years, but there is no doubt that PrEP uptake is too low,” said Eugene McCray, MD, director of CDC’s division of HIV/AIDS prevention. “We are working hard to increase access to PrEP, especially among gay and bisexual men, women, transgender people, young people, African Americans, and Latinos.”
The rate of new HIV infections has not decreased, but remained stable, according to the report. The CDC estimates that there were about 38,000 new infections per year from 2013 to 2017.
Proposed initiative focuses on areas of greatest need
The proposed Ending the HIV Epidemic initiative, if it is funded, will target the locations of greatest need throughout the country. Its initial focus will be on 50 areas that account for more than half of new HIV diagnoses, including 48 counties; San Juan, Puerto Rico; and Washington, D.C. It also will direct resources to seven states with high rates of infection in rural areas. In a second phase, the initiative will expand nationwide, provided that additional resources are made available.
The proposed initiative relies on four science-based strategies. First, it will aim to diagnose all Americans with HIV (at least 95% of HIV infections) as early as possible. Second, the initiative will enable people with HIV to receive treatment rapidly and effectively. The CDC’s target is to achieve viral suppression in at least 95% of people with diagnosed HIV. Third, the initiative will use proven interventions such as PrEP and syringe services programs to prevent new HIV transmissions. One related goal is for at least 50% of people who could benefit from PrEP to receive a prescription. Finally, the initiative is intended to respond quickly to potential HIV outbreaks and provide prevention and treatment to those who need them.
The U.S. Department of Health & Human Services already has taken steps to enable the initiative to be implemented quickly if it is funded in 2020. The department has provided funding to Baltimore City, Md.; DeKalb County, Ga.; and East Baton Rouge Parish, La. to begin pursuing parts of the initiative. These communities are encouraged to share the lessons of their experiences with other communities. HHS also has supported local efforts to develop plans under the initiative in all priority geographic areas. These plans draw upon recommendations from the community, HIV-planning bodies, and health care providers.
“Ending the HIV epidemic would be one of the greatest public health triumphs in our nation’s history,” said Dr. McCray.
SOURCES: Centers for Disease Control and Prevention. CDC Vital Signs. 2019 Dec 3. and Harris NS et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 3.
, according to a Vital Signs report published by the Centers for Disease Control and Prevention based upon a simultaneous MMWR Early Release. The report indicates that many Americans with HIV are not aware of their status or are not receiving effective treatment. Furthermore, the data suggest that few Americans who could benefit from preexposure prophylaxis (PrEP), a daily pill that prevents HIV, are receiving it.
The report “shows that HIV testing, treatment, and prevention have not reached enough Americans, and it emphasizes the continued urgent need to increase these interventions,” said Jay C. Butler, MD, deputy director for infectious diseases at the CDC, at a press conference. “We made a lot of progress in the late ’90s and into the early part of the 21st century in reducing the number of new cases of HIV. But HIV prevention progress has stalled in America since 2013. This stalling underscores the need to increase resources, deploy new technologies, and build expertise, particularly in areas where they’re needed most.”
To achieve these objectives, the CDC has proposed a federal initiative called Ending the HIV Epidemic: A Plan for America. The goal of the initiative is to reduce new HIV infections by 90% by 2030, in part by expanding access to PrEP medications.
Data suggest shortcomings in diagnosis, treatment, and prevention
In its review of data on HIV testing and treatment in 2017, the Vital Signs report found that approximately 154,000 people with HIV (that is, 14% of the total population with HIV) were unaware that they had the virus. These patients consequently could not take advantage of HIV treatment to maintain health, control the virus, and prevent HIV transmission. Young people aged 13-24 years were less likely to know their HIV status than did those aged 25 years and older, according to the report.
Furthermore, approximately two-thirds (63%) of patients who knew that they had HIV had the virus under control through effective treatment. Young people and African Americans were least likely to have the virus under control, according to the report.
The report also examines data about treatment with PrEP in 2018. About 1.2 million Americans could benefit from PrEP, but only 219,700 (18%) of them had received a prescription for the drug. The eligible groups with the lowest rates of coverage were young people, African Americans, and Latinos.
The report presents a conservative estimate of PrEP coverage, however. Researchers examined data from 92% of prescriptions from retail pharmacies in the United States but did not include prescriptions from closed health care systems such as managed care organizations and military health plans. PrEP coverage in 2018 likely was higher than these estimates indicate, according to the CDC.
“There has been a rapid increase in the number of people taking PrEP over the past 3 years, but there is no doubt that PrEP uptake is too low,” said Eugene McCray, MD, director of CDC’s division of HIV/AIDS prevention. “We are working hard to increase access to PrEP, especially among gay and bisexual men, women, transgender people, young people, African Americans, and Latinos.”
The rate of new HIV infections has not decreased, but remained stable, according to the report. The CDC estimates that there were about 38,000 new infections per year from 2013 to 2017.
Proposed initiative focuses on areas of greatest need
The proposed Ending the HIV Epidemic initiative, if it is funded, will target the locations of greatest need throughout the country. Its initial focus will be on 50 areas that account for more than half of new HIV diagnoses, including 48 counties; San Juan, Puerto Rico; and Washington, D.C. It also will direct resources to seven states with high rates of infection in rural areas. In a second phase, the initiative will expand nationwide, provided that additional resources are made available.
The proposed initiative relies on four science-based strategies. First, it will aim to diagnose all Americans with HIV (at least 95% of HIV infections) as early as possible. Second, the initiative will enable people with HIV to receive treatment rapidly and effectively. The CDC’s target is to achieve viral suppression in at least 95% of people with diagnosed HIV. Third, the initiative will use proven interventions such as PrEP and syringe services programs to prevent new HIV transmissions. One related goal is for at least 50% of people who could benefit from PrEP to receive a prescription. Finally, the initiative is intended to respond quickly to potential HIV outbreaks and provide prevention and treatment to those who need them.
The U.S. Department of Health & Human Services already has taken steps to enable the initiative to be implemented quickly if it is funded in 2020. The department has provided funding to Baltimore City, Md.; DeKalb County, Ga.; and East Baton Rouge Parish, La. to begin pursuing parts of the initiative. These communities are encouraged to share the lessons of their experiences with other communities. HHS also has supported local efforts to develop plans under the initiative in all priority geographic areas. These plans draw upon recommendations from the community, HIV-planning bodies, and health care providers.
“Ending the HIV epidemic would be one of the greatest public health triumphs in our nation’s history,” said Dr. McCray.
SOURCES: Centers for Disease Control and Prevention. CDC Vital Signs. 2019 Dec 3. and Harris NS et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 3.
, according to a Vital Signs report published by the Centers for Disease Control and Prevention based upon a simultaneous MMWR Early Release. The report indicates that many Americans with HIV are not aware of their status or are not receiving effective treatment. Furthermore, the data suggest that few Americans who could benefit from preexposure prophylaxis (PrEP), a daily pill that prevents HIV, are receiving it.
The report “shows that HIV testing, treatment, and prevention have not reached enough Americans, and it emphasizes the continued urgent need to increase these interventions,” said Jay C. Butler, MD, deputy director for infectious diseases at the CDC, at a press conference. “We made a lot of progress in the late ’90s and into the early part of the 21st century in reducing the number of new cases of HIV. But HIV prevention progress has stalled in America since 2013. This stalling underscores the need to increase resources, deploy new technologies, and build expertise, particularly in areas where they’re needed most.”
To achieve these objectives, the CDC has proposed a federal initiative called Ending the HIV Epidemic: A Plan for America. The goal of the initiative is to reduce new HIV infections by 90% by 2030, in part by expanding access to PrEP medications.
Data suggest shortcomings in diagnosis, treatment, and prevention
In its review of data on HIV testing and treatment in 2017, the Vital Signs report found that approximately 154,000 people with HIV (that is, 14% of the total population with HIV) were unaware that they had the virus. These patients consequently could not take advantage of HIV treatment to maintain health, control the virus, and prevent HIV transmission. Young people aged 13-24 years were less likely to know their HIV status than did those aged 25 years and older, according to the report.
Furthermore, approximately two-thirds (63%) of patients who knew that they had HIV had the virus under control through effective treatment. Young people and African Americans were least likely to have the virus under control, according to the report.
The report also examines data about treatment with PrEP in 2018. About 1.2 million Americans could benefit from PrEP, but only 219,700 (18%) of them had received a prescription for the drug. The eligible groups with the lowest rates of coverage were young people, African Americans, and Latinos.
The report presents a conservative estimate of PrEP coverage, however. Researchers examined data from 92% of prescriptions from retail pharmacies in the United States but did not include prescriptions from closed health care systems such as managed care organizations and military health plans. PrEP coverage in 2018 likely was higher than these estimates indicate, according to the CDC.
“There has been a rapid increase in the number of people taking PrEP over the past 3 years, but there is no doubt that PrEP uptake is too low,” said Eugene McCray, MD, director of CDC’s division of HIV/AIDS prevention. “We are working hard to increase access to PrEP, especially among gay and bisexual men, women, transgender people, young people, African Americans, and Latinos.”
The rate of new HIV infections has not decreased, but remained stable, according to the report. The CDC estimates that there were about 38,000 new infections per year from 2013 to 2017.
Proposed initiative focuses on areas of greatest need
The proposed Ending the HIV Epidemic initiative, if it is funded, will target the locations of greatest need throughout the country. Its initial focus will be on 50 areas that account for more than half of new HIV diagnoses, including 48 counties; San Juan, Puerto Rico; and Washington, D.C. It also will direct resources to seven states with high rates of infection in rural areas. In a second phase, the initiative will expand nationwide, provided that additional resources are made available.
The proposed initiative relies on four science-based strategies. First, it will aim to diagnose all Americans with HIV (at least 95% of HIV infections) as early as possible. Second, the initiative will enable people with HIV to receive treatment rapidly and effectively. The CDC’s target is to achieve viral suppression in at least 95% of people with diagnosed HIV. Third, the initiative will use proven interventions such as PrEP and syringe services programs to prevent new HIV transmissions. One related goal is for at least 50% of people who could benefit from PrEP to receive a prescription. Finally, the initiative is intended to respond quickly to potential HIV outbreaks and provide prevention and treatment to those who need them.
The U.S. Department of Health & Human Services already has taken steps to enable the initiative to be implemented quickly if it is funded in 2020. The department has provided funding to Baltimore City, Md.; DeKalb County, Ga.; and East Baton Rouge Parish, La. to begin pursuing parts of the initiative. These communities are encouraged to share the lessons of their experiences with other communities. HHS also has supported local efforts to develop plans under the initiative in all priority geographic areas. These plans draw upon recommendations from the community, HIV-planning bodies, and health care providers.
“Ending the HIV epidemic would be one of the greatest public health triumphs in our nation’s history,” said Dr. McCray.
SOURCES: Centers for Disease Control and Prevention. CDC Vital Signs. 2019 Dec 3. and Harris NS et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 3.
FROM THE CDC
FDA approves Tula system for recurrent pediatric ear infections
The Food and Drug Administration has approved the Tubes Under Local Anesthesia (Tula) System for treatment of recurrent ear infections (otitis media) via tympanostomy in young children, according to a release from the agency.

Consisting of Tymbion anesthetic, tympanostomy tubes developed by Tusker Medical, and several devices that deliver them into the ear drum,
“This approval has the potential to expand patient access to a treatment that can be administered in a physician’s office with local anesthesia and minimal discomfort,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health.
The approval was based on data from 222 children treated with the device, with a procedural success rate of 86% in children under 5 years and 89% in children aged 5-12 years. The most common adverse event was insufficient anesthetic.
The system should not be used in children with allergies to some local anesthetics or those younger than 6 months. It also is not intended for patients with preexisting issues with their eardrums, such as perforated ear drums, according to the press release.
The Tula system was granted a Breakthrough Device designation, which means the FDA provided intensive engagement and guidance during its development. The full release can be found on the FDA website.
The Food and Drug Administration has approved the Tubes Under Local Anesthesia (Tula) System for treatment of recurrent ear infections (otitis media) via tympanostomy in young children, according to a release from the agency.
Consisting of Tymbion anesthetic, tympanostomy tubes developed by Tusker Medical, and several devices that deliver them into the ear drum,
“This approval has the potential to expand patient access to a treatment that can be administered in a physician’s office with local anesthesia and minimal discomfort,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health.
The approval was based on data from 222 children treated with the device, with a procedural success rate of 86% in children under 5 years and 89% in children aged 5-12 years. The most common adverse event was insufficient anesthetic.
The system should not be used in children with allergies to some local anesthetics or those younger than 6 months. It also is not intended for patients with preexisting issues with their eardrums, such as perforated ear drums, according to the press release.
The Tula system was granted a Breakthrough Device designation, which means the FDA provided intensive engagement and guidance during its development. The full release can be found on the FDA website.
The Food and Drug Administration has approved the Tubes Under Local Anesthesia (Tula) System for treatment of recurrent ear infections (otitis media) via tympanostomy in young children, according to a release from the agency.
Consisting of Tymbion anesthetic, tympanostomy tubes developed by Tusker Medical, and several devices that deliver them into the ear drum,
“This approval has the potential to expand patient access to a treatment that can be administered in a physician’s office with local anesthesia and minimal discomfort,” said Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health.
The approval was based on data from 222 children treated with the device, with a procedural success rate of 86% in children under 5 years and 89% in children aged 5-12 years. The most common adverse event was insufficient anesthetic.
The system should not be used in children with allergies to some local anesthetics or those younger than 6 months. It also is not intended for patients with preexisting issues with their eardrums, such as perforated ear drums, according to the press release.
The Tula system was granted a Breakthrough Device designation, which means the FDA provided intensive engagement and guidance during its development. The full release can be found on the FDA website.
Obesity dropping in kids aged 2-4 years in WIC program

During 2010-2016, the prevalence of obesity among children in the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC) program significantly decreased in 73% of 56 states and territories, Liping Pan, MD, and colleagues reported in the Morbidity and Mortality Weekly Report.
The obesity prevalence decreases varied, but exceeded 3% in Guam, New Jersey, New Mexico, Northern Mariana Islands, Puerto Rico, Utah, and Virginia. Puerto Rico experienced the greatest benefit, with an 8% decrease in obesity among children aged 2-4 years enrolled in the WIC program, wrote Dr. Pan, an epidemiologist at the Centers for Disease Control and Prevention and coauthors.
Although the changes were small, the positive trend gains more import when viewed in light of the country’s long-term obesity trends, Dr. Pan and his team wrote. After a short-lived dip during 2007-2012, obesity has been on the rise among these children, jumping from 8% in 2012 to 14% in 2016. “Thus, even these small decreases indicate progress for this vulnerable WIC population,” the team said.
WIC extends nutritional assistance to families whose income is 185% or less of the federal poverty guideline or are eligible for other programs, as well as being deemed at nutritional risk.
The current study looked at obesity trends during 2010-2016 among 12,403,629 WIC recipients aged 2-4 years in all 50 U.S. states and five territories.
In 2010, obesity prevalence ranged from a low of 10% in Colorado to a high of 22% in Virginia. In Alaska, Puerto Rico, and Virginia, it was 20% or higher. Only in Colorado and Hawaii was obesity prevalence 10% or less among these children.
By 2016, obesity prevalence among children aged 2-4 years ranged from 8% in the Northern Mariana Islands to 19.8% in Alaska. It was less than 20% in all the states and territories, and less than 10% in Colorado, Guam, Hawaii, Northern Mariana Islands, Utah, and Wyoming.
It increased during 2010-2016, however, in Alabama (0.5%), North Carolina (0.6%), and West Virginia (2.2%).
The changes reflect the 2009 program revisions made to adhere to the 2005 Dietary Guidelines for Americans and the infant food and feeding practice guidelines of the American Academy of Pediatrics, Dr. Pan and associates wrote.
“The revised food packages include a broader range of healthy food options; promote fruit, vegetable, and whole wheat product purchases; support breastfeeding; and give WIC state and territory agencies more flexibility to accommodate cultural food preferences,” the authors noted.
In response to the changes, Dr. Pan and associates noted, authorized WIC stores began carrying healthier offerings. Tracking showed that children in the program consumed more fruits, vegetables, and whole grain products and less juice, white bread, and whole milk after the revisions than they did previously.
Despite the good news, childhood obesity rates still are too high and much remains to be done, they noted.
“Multiple approaches are needed to address and eliminate childhood obesity. The National Academy of Medicine and other groups have recommended a comprehensive and integrated approach that calls for positive changes in physical activity and food and beverage environments in multiple settings including home, early care and education [such as nutrition standards for food served], and community [such as neighborhood designs that encourage walking and biking] to promote healthy eating and physical activity for young children. Further implementation of these positive changes across the United States could further the decreases in child-hood obesity,” Dr. Pan and coauthors concluded.
Dr. Pan and coauthors had no financial disclosures.
SOURCE: Pan L et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 22;68(46):1057-61.
During 2010-2016, the prevalence of obesity among children in the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC) program significantly decreased in 73% of 56 states and territories, Liping Pan, MD, and colleagues reported in the Morbidity and Mortality Weekly Report.
The obesity prevalence decreases varied, but exceeded 3% in Guam, New Jersey, New Mexico, Northern Mariana Islands, Puerto Rico, Utah, and Virginia. Puerto Rico experienced the greatest benefit, with an 8% decrease in obesity among children aged 2-4 years enrolled in the WIC program, wrote Dr. Pan, an epidemiologist at the Centers for Disease Control and Prevention and coauthors.
Although the changes were small, the positive trend gains more import when viewed in light of the country’s long-term obesity trends, Dr. Pan and his team wrote. After a short-lived dip during 2007-2012, obesity has been on the rise among these children, jumping from 8% in 2012 to 14% in 2016. “Thus, even these small decreases indicate progress for this vulnerable WIC population,” the team said.
WIC extends nutritional assistance to families whose income is 185% or less of the federal poverty guideline or are eligible for other programs, as well as being deemed at nutritional risk.
The current study looked at obesity trends during 2010-2016 among 12,403,629 WIC recipients aged 2-4 years in all 50 U.S. states and five territories.
In 2010, obesity prevalence ranged from a low of 10% in Colorado to a high of 22% in Virginia. In Alaska, Puerto Rico, and Virginia, it was 20% or higher. Only in Colorado and Hawaii was obesity prevalence 10% or less among these children.
By 2016, obesity prevalence among children aged 2-4 years ranged from 8% in the Northern Mariana Islands to 19.8% in Alaska. It was less than 20% in all the states and territories, and less than 10% in Colorado, Guam, Hawaii, Northern Mariana Islands, Utah, and Wyoming.
It increased during 2010-2016, however, in Alabama (0.5%), North Carolina (0.6%), and West Virginia (2.2%).
The changes reflect the 2009 program revisions made to adhere to the 2005 Dietary Guidelines for Americans and the infant food and feeding practice guidelines of the American Academy of Pediatrics, Dr. Pan and associates wrote.
“The revised food packages include a broader range of healthy food options; promote fruit, vegetable, and whole wheat product purchases; support breastfeeding; and give WIC state and territory agencies more flexibility to accommodate cultural food preferences,” the authors noted.
In response to the changes, Dr. Pan and associates noted, authorized WIC stores began carrying healthier offerings. Tracking showed that children in the program consumed more fruits, vegetables, and whole grain products and less juice, white bread, and whole milk after the revisions than they did previously.
Despite the good news, childhood obesity rates still are too high and much remains to be done, they noted.
“Multiple approaches are needed to address and eliminate childhood obesity. The National Academy of Medicine and other groups have recommended a comprehensive and integrated approach that calls for positive changes in physical activity and food and beverage environments in multiple settings including home, early care and education [such as nutrition standards for food served], and community [such as neighborhood designs that encourage walking and biking] to promote healthy eating and physical activity for young children. Further implementation of these positive changes across the United States could further the decreases in child-hood obesity,” Dr. Pan and coauthors concluded.
Dr. Pan and coauthors had no financial disclosures.
SOURCE: Pan L et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 22;68(46):1057-61.
During 2010-2016, the prevalence of obesity among children in the Special Supplemental Nutrition Program for Women, Infants, and Children (WIC) program significantly decreased in 73% of 56 states and territories, Liping Pan, MD, and colleagues reported in the Morbidity and Mortality Weekly Report.
The obesity prevalence decreases varied, but exceeded 3% in Guam, New Jersey, New Mexico, Northern Mariana Islands, Puerto Rico, Utah, and Virginia. Puerto Rico experienced the greatest benefit, with an 8% decrease in obesity among children aged 2-4 years enrolled in the WIC program, wrote Dr. Pan, an epidemiologist at the Centers for Disease Control and Prevention and coauthors.
Although the changes were small, the positive trend gains more import when viewed in light of the country’s long-term obesity trends, Dr. Pan and his team wrote. After a short-lived dip during 2007-2012, obesity has been on the rise among these children, jumping from 8% in 2012 to 14% in 2016. “Thus, even these small decreases indicate progress for this vulnerable WIC population,” the team said.
WIC extends nutritional assistance to families whose income is 185% or less of the federal poverty guideline or are eligible for other programs, as well as being deemed at nutritional risk.
The current study looked at obesity trends during 2010-2016 among 12,403,629 WIC recipients aged 2-4 years in all 50 U.S. states and five territories.
In 2010, obesity prevalence ranged from a low of 10% in Colorado to a high of 22% in Virginia. In Alaska, Puerto Rico, and Virginia, it was 20% or higher. Only in Colorado and Hawaii was obesity prevalence 10% or less among these children.
By 2016, obesity prevalence among children aged 2-4 years ranged from 8% in the Northern Mariana Islands to 19.8% in Alaska. It was less than 20% in all the states and territories, and less than 10% in Colorado, Guam, Hawaii, Northern Mariana Islands, Utah, and Wyoming.
It increased during 2010-2016, however, in Alabama (0.5%), North Carolina (0.6%), and West Virginia (2.2%).
The changes reflect the 2009 program revisions made to adhere to the 2005 Dietary Guidelines for Americans and the infant food and feeding practice guidelines of the American Academy of Pediatrics, Dr. Pan and associates wrote.
“The revised food packages include a broader range of healthy food options; promote fruit, vegetable, and whole wheat product purchases; support breastfeeding; and give WIC state and territory agencies more flexibility to accommodate cultural food preferences,” the authors noted.
In response to the changes, Dr. Pan and associates noted, authorized WIC stores began carrying healthier offerings. Tracking showed that children in the program consumed more fruits, vegetables, and whole grain products and less juice, white bread, and whole milk after the revisions than they did previously.
Despite the good news, childhood obesity rates still are too high and much remains to be done, they noted.
“Multiple approaches are needed to address and eliminate childhood obesity. The National Academy of Medicine and other groups have recommended a comprehensive and integrated approach that calls for positive changes in physical activity and food and beverage environments in multiple settings including home, early care and education [such as nutrition standards for food served], and community [such as neighborhood designs that encourage walking and biking] to promote healthy eating and physical activity for young children. Further implementation of these positive changes across the United States could further the decreases in child-hood obesity,” Dr. Pan and coauthors concluded.
Dr. Pan and coauthors had no financial disclosures.
SOURCE: Pan L et al. MMWR Morb Mortal Wkly Rep. 2019 Nov 22;68(46):1057-61.
FROM THE MORBIDITY AND MORTALITY WEEKLY REPORT

FDA approves Oxbryta for sickle cell disease treatment
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
The Food and Drug Administration has approved voxelotor (Oxbryta) for adults and pediatric patients aged 12 years and older with sickle cell disease.
Approval was based on results from HOPE, a randomized, double-blind, placebo-controlled, multicenter trial of 274 patients with sickle cell disease (median age, 24 years) with a baseline hemoglobin level between 5.5 and 10.5 g/dL. Just over half of patients (51.1%) who received voxelotor at 1,500 mg had a hemoglobin increase of at least 1 g/dL over the 24-week study period, compared with 6.5% of patients who received placebo.
Patients in the 1,500-mg group also had reduced indirect bilirubin and percent reticulocyte count at –29.1% and –19.9%, respectively, compared with placebo, where the change was –3.2% and 4.5%, respectively.
The most common adverse events associated with voxelotor are headache, diarrhea, abdominal pain, nausea, rash, fatigue and pyrexia. The recommended voxelotor dose is 1,500 mg orally once daily with or without food, according to the FDA.
Alkermes submits NDA for new schizophrenia, bipolar I treatment
Alkermes has announced that it has submitted a New Drug Application to the Food and Drug Administration for the approval of ALKS 3831 (olanzapine/samidorphan) for the treatment of schizophrenia and bipolar I disorder.
Included in the application for the investigational, novel, once-daily, oral atypical antipsychotic drug candidate is data from the ENLIGHTEN-1 study, which evaluated the antipsychotic efficacy of ALKS 3831, compared with a placebo, over a 4-week period, as well as data from ENLIGHTEN-2, which compared weight gain with ALKS 3831 and olanzapine alone over a 6-month period.
“Antipsychotic medications are an important part of the treatment paradigm for both schizophrenia and bipolar I disorder, yet there remains a persistent unmet need for new treatments,” Craig Hopkinson, MD, chief medical officer and senior vice president of medicines development and medical affairs at Alkermes, said in a press release.
As a combination of olanzapine and samidorphan, Samidorphan, an opioid receptor antagonist, is structurally related to naltrexone.
Alkermes is seeking an indication for the treatment of schizophrenia and an indication for the treatment of manic or mixed episodes associated with bipolar I disorder as monotherapy or as an adjunct to lithium or valproate, as well as for maintenance treatment of bipolar I. Dosage strength would be 10 mg of samidorphan with 5, 10, 15, or 20 mg of olanzapine.
Find the full press release on the Alkermes website.
Alkermes has announced that it has submitted a New Drug Application to the Food and Drug Administration for the approval of ALKS 3831 (olanzapine/samidorphan) for the treatment of schizophrenia and bipolar I disorder.
Included in the application for the investigational, novel, once-daily, oral atypical antipsychotic drug candidate is data from the ENLIGHTEN-1 study, which evaluated the antipsychotic efficacy of ALKS 3831, compared with a placebo, over a 4-week period, as well as data from ENLIGHTEN-2, which compared weight gain with ALKS 3831 and olanzapine alone over a 6-month period.
“Antipsychotic medications are an important part of the treatment paradigm for both schizophrenia and bipolar I disorder, yet there remains a persistent unmet need for new treatments,” Craig Hopkinson, MD, chief medical officer and senior vice president of medicines development and medical affairs at Alkermes, said in a press release.
As a combination of olanzapine and samidorphan, Samidorphan, an opioid receptor antagonist, is structurally related to naltrexone.
Alkermes is seeking an indication for the treatment of schizophrenia and an indication for the treatment of manic or mixed episodes associated with bipolar I disorder as monotherapy or as an adjunct to lithium or valproate, as well as for maintenance treatment of bipolar I. Dosage strength would be 10 mg of samidorphan with 5, 10, 15, or 20 mg of olanzapine.
Find the full press release on the Alkermes website.
Alkermes has announced that it has submitted a New Drug Application to the Food and Drug Administration for the approval of ALKS 3831 (olanzapine/samidorphan) for the treatment of schizophrenia and bipolar I disorder.
Included in the application for the investigational, novel, once-daily, oral atypical antipsychotic drug candidate is data from the ENLIGHTEN-1 study, which evaluated the antipsychotic efficacy of ALKS 3831, compared with a placebo, over a 4-week period, as well as data from ENLIGHTEN-2, which compared weight gain with ALKS 3831 and olanzapine alone over a 6-month period.
“Antipsychotic medications are an important part of the treatment paradigm for both schizophrenia and bipolar I disorder, yet there remains a persistent unmet need for new treatments,” Craig Hopkinson, MD, chief medical officer and senior vice president of medicines development and medical affairs at Alkermes, said in a press release.
As a combination of olanzapine and samidorphan, Samidorphan, an opioid receptor antagonist, is structurally related to naltrexone.
Alkermes is seeking an indication for the treatment of schizophrenia and an indication for the treatment of manic or mixed episodes associated with bipolar I disorder as monotherapy or as an adjunct to lithium or valproate, as well as for maintenance treatment of bipolar I. Dosage strength would be 10 mg of samidorphan with 5, 10, 15, or 20 mg of olanzapine.
Find the full press release on the Alkermes website.
FDA okays cenobamate (Xcopri) for focal epilepsy
The Food and Drug Administration has approved cenobamate (Xcopri) for the treatment of partial-onset seizures in adult patients with epilepsy.

The approval on Nov. 21 was based on results from two randomized controlled trials that included more than 600 patients, the agency said in a press release.
Together, the trials showed that the study drug at doses of 100, 200, and 400 mg significantly reduced the percentage of seizures, compared with placebo.
The FDA notes that, although the recommended maintenance dose of the drug is 200 mg/day after titration, some patients may need to be titrated up to 400 mg/day.
“Xcopri is a new option to treat adults with partial-onset seizures, which is an often difficult-to-control condition that can have a significant impact on patient quality of life,” Billy Dunn, MD, director of the Office of Neuroscience in the Center for Drug Evaluation and Research at the FDA, said in a statement.
Adverse events
As reported by Medscape Medical News, results from one of the studies upon which this approval was based were published online last week in Lancet Neurology. The findings showed that both primary endpoints were met.
Although most treatment-emergent adverse events (AEs) were reported to be mild to moderate in severity, one of these participants receiving the 200 mg dose had a drug reaction with eosinophilia and systemic symptoms (DRESS).
The FDA noted that one patient in the other trial also died when the active drug was titrated rapidly.
In an open-label safety trial of 1,339 participants that was also reviewed by the FDA, there were no cases of DRESS when patients started cenobamate at 12.5 mg/day and the dose was adjusted every 2 weeks.
Because more patients who took the drug than those taking placebo had a shortening of the QT interval greater than 20 milliseconds, cenobamate shouldn’t be used in those with hypersensitivity to the drug “or any of the inactive ingredients in Xcopri or Familial Short QT syndrome,” the agency wrote, adding that QT shortening can be associated with ventricular fibrillation, a serious heart rhythm problem.
The FDA also noted that any patient taking an antiepileptic drug should be monitored for the emergence or worsening of depressive symptoms, suicidal thoughts or behaviors, or any other changes in mood.
The most common AEs reported in the trials were somnolence, dizziness, fatigue, and diplopia (double vision).
‘Welcome option’
“The approval of Xcopri will provide clinicians with an effective medication for our patients who are continuing to have focal [partial-onset] seizures,” Michael Sperling, MD, professor of neurology and director of the Jefferson Comprehensive Epilepsy Center, Philadelphia, and an investigator in the drug’s clinical development program, said in a press release from SK Life Science.
“It is very encouraging to see that patients receiving Xcopri saw significant reductions in frequency of seizures, with some even achieving zero seizures,” Dr. Sperling added.
“There is an urgent need to advance research and introduce new treatment options. The FDA approval of Xcopri for the treatment of partial-onset seizures is a welcome option for the epilepsy community,” Beth Lewin Dean, chief executive officer of Citizens United for Research in Epilepsy, said in the same release.
SK Life Science noted in a statement that the drug is expected to be available in the United States in the second quarter of 2020 “following scheduling review” by the Drug Enforcement Administration.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved cenobamate (Xcopri) for the treatment of partial-onset seizures in adult patients with epilepsy.
The approval on Nov. 21 was based on results from two randomized controlled trials that included more than 600 patients, the agency said in a press release.
Together, the trials showed that the study drug at doses of 100, 200, and 400 mg significantly reduced the percentage of seizures, compared with placebo.
The FDA notes that, although the recommended maintenance dose of the drug is 200 mg/day after titration, some patients may need to be titrated up to 400 mg/day.
“Xcopri is a new option to treat adults with partial-onset seizures, which is an often difficult-to-control condition that can have a significant impact on patient quality of life,” Billy Dunn, MD, director of the Office of Neuroscience in the Center for Drug Evaluation and Research at the FDA, said in a statement.
Adverse events
As reported by Medscape Medical News, results from one of the studies upon which this approval was based were published online last week in Lancet Neurology. The findings showed that both primary endpoints were met.
Although most treatment-emergent adverse events (AEs) were reported to be mild to moderate in severity, one of these participants receiving the 200 mg dose had a drug reaction with eosinophilia and systemic symptoms (DRESS).
The FDA noted that one patient in the other trial also died when the active drug was titrated rapidly.
In an open-label safety trial of 1,339 participants that was also reviewed by the FDA, there were no cases of DRESS when patients started cenobamate at 12.5 mg/day and the dose was adjusted every 2 weeks.
Because more patients who took the drug than those taking placebo had a shortening of the QT interval greater than 20 milliseconds, cenobamate shouldn’t be used in those with hypersensitivity to the drug “or any of the inactive ingredients in Xcopri or Familial Short QT syndrome,” the agency wrote, adding that QT shortening can be associated with ventricular fibrillation, a serious heart rhythm problem.
The FDA also noted that any patient taking an antiepileptic drug should be monitored for the emergence or worsening of depressive symptoms, suicidal thoughts or behaviors, or any other changes in mood.
The most common AEs reported in the trials were somnolence, dizziness, fatigue, and diplopia (double vision).
‘Welcome option’
“The approval of Xcopri will provide clinicians with an effective medication for our patients who are continuing to have focal [partial-onset] seizures,” Michael Sperling, MD, professor of neurology and director of the Jefferson Comprehensive Epilepsy Center, Philadelphia, and an investigator in the drug’s clinical development program, said in a press release from SK Life Science.
“It is very encouraging to see that patients receiving Xcopri saw significant reductions in frequency of seizures, with some even achieving zero seizures,” Dr. Sperling added.
“There is an urgent need to advance research and introduce new treatment options. The FDA approval of Xcopri for the treatment of partial-onset seizures is a welcome option for the epilepsy community,” Beth Lewin Dean, chief executive officer of Citizens United for Research in Epilepsy, said in the same release.
SK Life Science noted in a statement that the drug is expected to be available in the United States in the second quarter of 2020 “following scheduling review” by the Drug Enforcement Administration.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved cenobamate (Xcopri) for the treatment of partial-onset seizures in adult patients with epilepsy.
The approval on Nov. 21 was based on results from two randomized controlled trials that included more than 600 patients, the agency said in a press release.
Together, the trials showed that the study drug at doses of 100, 200, and 400 mg significantly reduced the percentage of seizures, compared with placebo.
The FDA notes that, although the recommended maintenance dose of the drug is 200 mg/day after titration, some patients may need to be titrated up to 400 mg/day.
“Xcopri is a new option to treat adults with partial-onset seizures, which is an often difficult-to-control condition that can have a significant impact on patient quality of life,” Billy Dunn, MD, director of the Office of Neuroscience in the Center for Drug Evaluation and Research at the FDA, said in a statement.
Adverse events
As reported by Medscape Medical News, results from one of the studies upon which this approval was based were published online last week in Lancet Neurology. The findings showed that both primary endpoints were met.
Although most treatment-emergent adverse events (AEs) were reported to be mild to moderate in severity, one of these participants receiving the 200 mg dose had a drug reaction with eosinophilia and systemic symptoms (DRESS).
The FDA noted that one patient in the other trial also died when the active drug was titrated rapidly.
In an open-label safety trial of 1,339 participants that was also reviewed by the FDA, there were no cases of DRESS when patients started cenobamate at 12.5 mg/day and the dose was adjusted every 2 weeks.
Because more patients who took the drug than those taking placebo had a shortening of the QT interval greater than 20 milliseconds, cenobamate shouldn’t be used in those with hypersensitivity to the drug “or any of the inactive ingredients in Xcopri or Familial Short QT syndrome,” the agency wrote, adding that QT shortening can be associated with ventricular fibrillation, a serious heart rhythm problem.
The FDA also noted that any patient taking an antiepileptic drug should be monitored for the emergence or worsening of depressive symptoms, suicidal thoughts or behaviors, or any other changes in mood.
The most common AEs reported in the trials were somnolence, dizziness, fatigue, and diplopia (double vision).
‘Welcome option’
“The approval of Xcopri will provide clinicians with an effective medication for our patients who are continuing to have focal [partial-onset] seizures,” Michael Sperling, MD, professor of neurology and director of the Jefferson Comprehensive Epilepsy Center, Philadelphia, and an investigator in the drug’s clinical development program, said in a press release from SK Life Science.
“It is very encouraging to see that patients receiving Xcopri saw significant reductions in frequency of seizures, with some even achieving zero seizures,” Dr. Sperling added.
“There is an urgent need to advance research and introduce new treatment options. The FDA approval of Xcopri for the treatment of partial-onset seizures is a welcome option for the epilepsy community,” Beth Lewin Dean, chief executive officer of Citizens United for Research in Epilepsy, said in the same release.
SK Life Science noted in a statement that the drug is expected to be available in the United States in the second quarter of 2020 “following scheduling review” by the Drug Enforcement Administration.
This story first appeared on Medscape.com.
FDA approves acalabrutinib for CLL, SLL treatment
The Food and Drug Administration has approved acalabrutinib (Calquence) as initial or subsequent treatment for adults with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The approval came as part of Project Orbis, a collaboration among the FDA, the Australian Therapeutic Goods Administration, and Health Canada. The program allows for the concurrent submission of review of oncology drug applications among the various agencies.
Acalabrutinib, a bruton tyrosin kinase inhibitor, is already approved in the United States for the treatment of adults with mantle cell lymphoma who have received at least one prior therapy. The FDA granted breakthrough therapy designation to acalabrutinib as a monotherapy for adults with CLL in August 2019, allowing for an expedited review.
The approval in CLL/SLL was based on results from two randomized clinical trials comparing acalabrutinib with other standard treatments. In the first trial, patients with previously untreated CLL who received acalabrutinib had a longer progression-free survival time, compared with patients who received standard treatment. A similar result was seen in the second trial among patients with previously treated CLL.
The most common adverse events associated with acalabrutinib include anemia, neutropenia, upper respiratory tract infection, thrombocytopenia, headache, diarrhea, and musculoskeletal pain. Patients receiving the drug should be monitored for symptoms of arrhythmia, serious infection, bleeding, and low blood count. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved acalabrutinib (Calquence) as initial or subsequent treatment for adults with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The approval came as part of Project Orbis, a collaboration among the FDA, the Australian Therapeutic Goods Administration, and Health Canada. The program allows for the concurrent submission of review of oncology drug applications among the various agencies.
Acalabrutinib, a bruton tyrosin kinase inhibitor, is already approved in the United States for the treatment of adults with mantle cell lymphoma who have received at least one prior therapy. The FDA granted breakthrough therapy designation to acalabrutinib as a monotherapy for adults with CLL in August 2019, allowing for an expedited review.
The approval in CLL/SLL was based on results from two randomized clinical trials comparing acalabrutinib with other standard treatments. In the first trial, patients with previously untreated CLL who received acalabrutinib had a longer progression-free survival time, compared with patients who received standard treatment. A similar result was seen in the second trial among patients with previously treated CLL.
The most common adverse events associated with acalabrutinib include anemia, neutropenia, upper respiratory tract infection, thrombocytopenia, headache, diarrhea, and musculoskeletal pain. Patients receiving the drug should be monitored for symptoms of arrhythmia, serious infection, bleeding, and low blood count. Full prescribing information can be found on the FDA website.
The Food and Drug Administration has approved acalabrutinib (Calquence) as initial or subsequent treatment for adults with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL).
The approval came as part of Project Orbis, a collaboration among the FDA, the Australian Therapeutic Goods Administration, and Health Canada. The program allows for the concurrent submission of review of oncology drug applications among the various agencies.
Acalabrutinib, a bruton tyrosin kinase inhibitor, is already approved in the United States for the treatment of adults with mantle cell lymphoma who have received at least one prior therapy. The FDA granted breakthrough therapy designation to acalabrutinib as a monotherapy for adults with CLL in August 2019, allowing for an expedited review.
The approval in CLL/SLL was based on results from two randomized clinical trials comparing acalabrutinib with other standard treatments. In the first trial, patients with previously untreated CLL who received acalabrutinib had a longer progression-free survival time, compared with patients who received standard treatment. A similar result was seen in the second trial among patients with previously treated CLL.
The most common adverse events associated with acalabrutinib include anemia, neutropenia, upper respiratory tract infection, thrombocytopenia, headache, diarrhea, and musculoskeletal pain. Patients receiving the drug should be monitored for symptoms of arrhythmia, serious infection, bleeding, and low blood count. Full prescribing information can be found on the FDA website.
FDA approves Givlaari for treatment of acute hepatic porphyria
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.

“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.
“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The Food and Drug Administration has approved givosiran (Givlaari) for the treatment of adult patients with acute hepatic porphyria, a genetic disorder that causes buildup of porphyrin molecules.
“This buildup can cause acute attacks, known as porphyria attacks, which can lead to severe pain and paralysis, respiratory failure, seizures, and mental status changes. These attacks occur suddenly and can produce permanent neurological damage and death. Prior to today’s approval, treatment options have only provided partial relief from the intense unremitting pain that characterizes these attacks,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
Approval for givosiran is based on results from a clinical trial of 94 patients with acute hepatic porphyria. Patients who received givosiran experienced 70% fewer porphyria attacks that required hospitalization, urgent health care visits, or home intravenous hemin injections compared with patients who received a placebo.
The most common adverse events associated with givosiran were nausea and injection site reactions. Patients receiving the medication should be monitored for anaphylactic reaction and renal function, and liver function should be tested before and periodically during treatment.
“The drug approved today can treat this disease by helping to reduce the number of attacks that disrupt the lives of patients,” said Dr. Pazdur, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
AASLD debrief: Five drugs show promise in NAFLD (and two do not)
BOSTON – For treatment of nonalcoholic fatty liver disease, cotadutide, licogliflozin, tropifexor, saroglitazar, and PF-05221304 are just a few of the drugs with promising data, Kathleen E. Corey, MD, MPH, said at the annual meeting of the American Association for the Study of Liver Diseases.

By contrast, selonsertib and emricasan did not achieve their endpoints in studies described here at the meeting, “but we have a lot to learn from them,” said Dr. Corey, director of the Mass General Fatty Liver Clinic and assistant professor at Harvard Medical School, Boston.
“This is an exciting time,” Dr. Corey said in a special debriefing oral session held on the final day of the conference. “There are many novel mechanisms of action out there, as well as some known mechanisms of action, with a considerable amount of promise.”
Cotadutide (MEDI0382)
Narha and coauthors (Abstract 35) described the effects of cotadutide, a GLP-1/glucagon receptor dual agonist on biomarkers of nonalcoholic steatohepatitis (NASH) at 26 weeks in overweight or obese patients with type 2 diabetes mellitus. In the randomized, phase 2b study, cotadutide produced superior reductions versus liraglutide, the GLP-1 receptor agonist, in alanine aminotransferase (ALT) and aspartate aminotransferase (AST), and body weight, which investigators said supported prospective trials of the drug for a potential indication in NASH.
“The adverse events were fairly typical for what we see with the GLP-1s – GI side effects that usually over 8 weeks improve,” Dr. Corey told attendees at the debrief session.
Licogliflozin (LIK066)
Interim analysis of a 12-week, randomized, placebo-controlled, phase 2a study showed that this SGLT1/2 inhibitor produced “robust” decreases in ALT and improvements in markers of hepatic and metabolic health in patients with NASH, according to Zhang and coauthors (Abstract L07).
Some 67% of those who received licogliflozin had at least a 30% decrease in their liver fat, while decreases in weight and hemoglobin A1c were also reported, according to Dr. Corey. “It was associated with diarrhea in about 97%, but this was considered mild, and certainly, we’re seeing good metabolic effects overall,” she said.
Tropifexor
Treatment for 12 weeks with this potent FXR agonist resulted in robust, dose-dependent reductions in hepatic fat and serum ALT in patients with fibrotic NASH, according to investigators in a phase 2 randomized, placebo-controlled trial known as FLIGHT-FXR (Abstract L04).
A total of 65% of patients achieved a 30% or greater reduction in liver fat, and decreases in weight and insulin resistance were reported. “Similar to other FXRs, they did have this concerning although potentially manageable increase in low-density lipoprotein (LDL)-cholesterol, and the adverse event of pruritis,” said Dr. Corey.
Saroglitazar
Gawrieh and coauthors presented results from EVIDENCES IV, a phase 2, double-blind, randomized, placebo-controlled study of saroglitazar, a novel dual peroxisome proliferator activated receptor (PPAR) alpha/gamma agonist, in patients with NAFLD or NASH (Abstract LO10).
The investigators found that 41% of patients achieved a 30% or greater relative reduction in liver fat, as well as reductions in hemoglobin A1c and lipids, but the treatment was “weight neutral,” Dr. Corey said, adding that no serious adverse events were reported.
PF-05221304
This liver-targeted acetyl-CoA carboxylase inhibitor (ACCI) demonstrated robust reduction in liver fat and ALT in a 16-week phase 2a, dose-ranging study in adults with NAFLD, according to Amin and coinvestigators (Abstract 31).
There was a “dramatic” decrease in liver fat in this study, said Dr. Corey, with 90% of treated patients experiencing a 30% or greater decrease. Side effects included a “significant” increase in triglycerides, she added, as well as transient increases in ALT and AST.
Selonsertib and emricasan
One agent not meeting study endpoints was selonsertib, an apoptosis signal-regulating kinase 1 (ASK1) inhibitor. While safe and well tolerated, the drug was nevertheless not effective as monotherapy in phase 3 double-blind, placebo-controlled trials including patients with advanced fibrosis due to NASH, investigators said (Abstract 64). Currently, the agent is being evaluated in combination with firsocostat – an ACCI – in a phase 2 study called ATLAS, according to the authors.
Emricasan, an oral pan-caspase inhibitor that suppresses apoptosis, did not improve fibrosis or resolve NASH in a multicenter, double-blind, placebo-controlled randomized trial, and may have even worsened histology, according to Dr. Corey. Investigators said further evaluation of the mechanisms underlying findings could provide insights into the role of necro-apoptosis in NASH pathophysiology (Abstract 61).
Dr. Corey provided disclosures related to BMS, Novo Nordisk, Boehringer Ingelheim, and Gilead.
BOSTON – For treatment of nonalcoholic fatty liver disease, cotadutide, licogliflozin, tropifexor, saroglitazar, and PF-05221304 are just a few of the drugs with promising data, Kathleen E. Corey, MD, MPH, said at the annual meeting of the American Association for the Study of Liver Diseases.
By contrast, selonsertib and emricasan did not achieve their endpoints in studies described here at the meeting, “but we have a lot to learn from them,” said Dr. Corey, director of the Mass General Fatty Liver Clinic and assistant professor at Harvard Medical School, Boston.
“This is an exciting time,” Dr. Corey said in a special debriefing oral session held on the final day of the conference. “There are many novel mechanisms of action out there, as well as some known mechanisms of action, with a considerable amount of promise.”
Cotadutide (MEDI0382)
Narha and coauthors (Abstract 35) described the effects of cotadutide, a GLP-1/glucagon receptor dual agonist on biomarkers of nonalcoholic steatohepatitis (NASH) at 26 weeks in overweight or obese patients with type 2 diabetes mellitus. In the randomized, phase 2b study, cotadutide produced superior reductions versus liraglutide, the GLP-1 receptor agonist, in alanine aminotransferase (ALT) and aspartate aminotransferase (AST), and body weight, which investigators said supported prospective trials of the drug for a potential indication in NASH.
“The adverse events were fairly typical for what we see with the GLP-1s – GI side effects that usually over 8 weeks improve,” Dr. Corey told attendees at the debrief session.
Licogliflozin (LIK066)
Interim analysis of a 12-week, randomized, placebo-controlled, phase 2a study showed that this SGLT1/2 inhibitor produced “robust” decreases in ALT and improvements in markers of hepatic and metabolic health in patients with NASH, according to Zhang and coauthors (Abstract L07).
Some 67% of those who received licogliflozin had at least a 30% decrease in their liver fat, while decreases in weight and hemoglobin A1c were also reported, according to Dr. Corey. “It was associated with diarrhea in about 97%, but this was considered mild, and certainly, we’re seeing good metabolic effects overall,” she said.
Tropifexor
Treatment for 12 weeks with this potent FXR agonist resulted in robust, dose-dependent reductions in hepatic fat and serum ALT in patients with fibrotic NASH, according to investigators in a phase 2 randomized, placebo-controlled trial known as FLIGHT-FXR (Abstract L04).
A total of 65% of patients achieved a 30% or greater reduction in liver fat, and decreases in weight and insulin resistance were reported. “Similar to other FXRs, they did have this concerning although potentially manageable increase in low-density lipoprotein (LDL)-cholesterol, and the adverse event of pruritis,” said Dr. Corey.
Saroglitazar
Gawrieh and coauthors presented results from EVIDENCES IV, a phase 2, double-blind, randomized, placebo-controlled study of saroglitazar, a novel dual peroxisome proliferator activated receptor (PPAR) alpha/gamma agonist, in patients with NAFLD or NASH (Abstract LO10).
The investigators found that 41% of patients achieved a 30% or greater relative reduction in liver fat, as well as reductions in hemoglobin A1c and lipids, but the treatment was “weight neutral,” Dr. Corey said, adding that no serious adverse events were reported.
PF-05221304
This liver-targeted acetyl-CoA carboxylase inhibitor (ACCI) demonstrated robust reduction in liver fat and ALT in a 16-week phase 2a, dose-ranging study in adults with NAFLD, according to Amin and coinvestigators (Abstract 31).
There was a “dramatic” decrease in liver fat in this study, said Dr. Corey, with 90% of treated patients experiencing a 30% or greater decrease. Side effects included a “significant” increase in triglycerides, she added, as well as transient increases in ALT and AST.
Selonsertib and emricasan
One agent not meeting study endpoints was selonsertib, an apoptosis signal-regulating kinase 1 (ASK1) inhibitor. While safe and well tolerated, the drug was nevertheless not effective as monotherapy in phase 3 double-blind, placebo-controlled trials including patients with advanced fibrosis due to NASH, investigators said (Abstract 64). Currently, the agent is being evaluated in combination with firsocostat – an ACCI – in a phase 2 study called ATLAS, according to the authors.
Emricasan, an oral pan-caspase inhibitor that suppresses apoptosis, did not improve fibrosis or resolve NASH in a multicenter, double-blind, placebo-controlled randomized trial, and may have even worsened histology, according to Dr. Corey. Investigators said further evaluation of the mechanisms underlying findings could provide insights into the role of necro-apoptosis in NASH pathophysiology (Abstract 61).
Dr. Corey provided disclosures related to BMS, Novo Nordisk, Boehringer Ingelheim, and Gilead.
BOSTON – For treatment of nonalcoholic fatty liver disease, cotadutide, licogliflozin, tropifexor, saroglitazar, and PF-05221304 are just a few of the drugs with promising data, Kathleen E. Corey, MD, MPH, said at the annual meeting of the American Association for the Study of Liver Diseases.
By contrast, selonsertib and emricasan did not achieve their endpoints in studies described here at the meeting, “but we have a lot to learn from them,” said Dr. Corey, director of the Mass General Fatty Liver Clinic and assistant professor at Harvard Medical School, Boston.
“This is an exciting time,” Dr. Corey said in a special debriefing oral session held on the final day of the conference. “There are many novel mechanisms of action out there, as well as some known mechanisms of action, with a considerable amount of promise.”
Cotadutide (MEDI0382)
Narha and coauthors (Abstract 35) described the effects of cotadutide, a GLP-1/glucagon receptor dual agonist on biomarkers of nonalcoholic steatohepatitis (NASH) at 26 weeks in overweight or obese patients with type 2 diabetes mellitus. In the randomized, phase 2b study, cotadutide produced superior reductions versus liraglutide, the GLP-1 receptor agonist, in alanine aminotransferase (ALT) and aspartate aminotransferase (AST), and body weight, which investigators said supported prospective trials of the drug for a potential indication in NASH.
“The adverse events were fairly typical for what we see with the GLP-1s – GI side effects that usually over 8 weeks improve,” Dr. Corey told attendees at the debrief session.
Licogliflozin (LIK066)
Interim analysis of a 12-week, randomized, placebo-controlled, phase 2a study showed that this SGLT1/2 inhibitor produced “robust” decreases in ALT and improvements in markers of hepatic and metabolic health in patients with NASH, according to Zhang and coauthors (Abstract L07).
Some 67% of those who received licogliflozin had at least a 30% decrease in their liver fat, while decreases in weight and hemoglobin A1c were also reported, according to Dr. Corey. “It was associated with diarrhea in about 97%, but this was considered mild, and certainly, we’re seeing good metabolic effects overall,” she said.
Tropifexor
Treatment for 12 weeks with this potent FXR agonist resulted in robust, dose-dependent reductions in hepatic fat and serum ALT in patients with fibrotic NASH, according to investigators in a phase 2 randomized, placebo-controlled trial known as FLIGHT-FXR (Abstract L04).
A total of 65% of patients achieved a 30% or greater reduction in liver fat, and decreases in weight and insulin resistance were reported. “Similar to other FXRs, they did have this concerning although potentially manageable increase in low-density lipoprotein (LDL)-cholesterol, and the adverse event of pruritis,” said Dr. Corey.
Saroglitazar
Gawrieh and coauthors presented results from EVIDENCES IV, a phase 2, double-blind, randomized, placebo-controlled study of saroglitazar, a novel dual peroxisome proliferator activated receptor (PPAR) alpha/gamma agonist, in patients with NAFLD or NASH (Abstract LO10).
The investigators found that 41% of patients achieved a 30% or greater relative reduction in liver fat, as well as reductions in hemoglobin A1c and lipids, but the treatment was “weight neutral,” Dr. Corey said, adding that no serious adverse events were reported.
PF-05221304
This liver-targeted acetyl-CoA carboxylase inhibitor (ACCI) demonstrated robust reduction in liver fat and ALT in a 16-week phase 2a, dose-ranging study in adults with NAFLD, according to Amin and coinvestigators (Abstract 31).
There was a “dramatic” decrease in liver fat in this study, said Dr. Corey, with 90% of treated patients experiencing a 30% or greater decrease. Side effects included a “significant” increase in triglycerides, she added, as well as transient increases in ALT and AST.
Selonsertib and emricasan
One agent not meeting study endpoints was selonsertib, an apoptosis signal-regulating kinase 1 (ASK1) inhibitor. While safe and well tolerated, the drug was nevertheless not effective as monotherapy in phase 3 double-blind, placebo-controlled trials including patients with advanced fibrosis due to NASH, investigators said (Abstract 64). Currently, the agent is being evaluated in combination with firsocostat – an ACCI – in a phase 2 study called ATLAS, according to the authors.
Emricasan, an oral pan-caspase inhibitor that suppresses apoptosis, did not improve fibrosis or resolve NASH in a multicenter, double-blind, placebo-controlled randomized trial, and may have even worsened histology, according to Dr. Corey. Investigators said further evaluation of the mechanisms underlying findings could provide insights into the role of necro-apoptosis in NASH pathophysiology (Abstract 61).
Dr. Corey provided disclosures related to BMS, Novo Nordisk, Boehringer Ingelheim, and Gilead.
REPORTING FROM THE LIVER MEETING 2019