User login
FDA approves Qwo for treatment of cellulite
. The drug is the first injectable treatment for cellulite to receive regulatory approval.

In cellulite, fibrous septae are a primary contributing factor. The septae make up the fibrous connective tissue that connects the skin perpendicularly to the fascia below and tether the skin, drawing it downward and leading to a mattress-like appearance, commonly referred to as “dimpling.” When injected into the treatment area, Qwo is thought to release the fibrous septae enzymatically by specifically targeting types 1 and 3 collagen, which may result in smoothing of the skin and an improved appearance of cellulite.
The most common side effects of Qwo include injection site bruising, pain, areas of hardness, itching, redness, discoloration, swelling, and warmth in the treatment area.
Qwo is expected to be available throughout the United States at aesthetic health care practitioner’s offices starting in Spring 2021.
“Qwo could be a game-changer for many women with cellulite,” Anne Chapas, MD, a board-certified dermatologist at Union Square Laser Dermatology in New York, said in a press release. “I am thrilled there will now be an FDA-approved injectable treatment option proven to address a root cause of cellulite. What is exciting about Qwo is that it is a cutting-edge cellulite treatment, without the cutting,” Dr. Chapas, said.
. The drug is the first injectable treatment for cellulite to receive regulatory approval.
In cellulite, fibrous septae are a primary contributing factor. The septae make up the fibrous connective tissue that connects the skin perpendicularly to the fascia below and tether the skin, drawing it downward and leading to a mattress-like appearance, commonly referred to as “dimpling.” When injected into the treatment area, Qwo is thought to release the fibrous septae enzymatically by specifically targeting types 1 and 3 collagen, which may result in smoothing of the skin and an improved appearance of cellulite.
The most common side effects of Qwo include injection site bruising, pain, areas of hardness, itching, redness, discoloration, swelling, and warmth in the treatment area.
Qwo is expected to be available throughout the United States at aesthetic health care practitioner’s offices starting in Spring 2021.
“Qwo could be a game-changer for many women with cellulite,” Anne Chapas, MD, a board-certified dermatologist at Union Square Laser Dermatology in New York, said in a press release. “I am thrilled there will now be an FDA-approved injectable treatment option proven to address a root cause of cellulite. What is exciting about Qwo is that it is a cutting-edge cellulite treatment, without the cutting,” Dr. Chapas, said.
. The drug is the first injectable treatment for cellulite to receive regulatory approval.
In cellulite, fibrous septae are a primary contributing factor. The septae make up the fibrous connective tissue that connects the skin perpendicularly to the fascia below and tether the skin, drawing it downward and leading to a mattress-like appearance, commonly referred to as “dimpling.” When injected into the treatment area, Qwo is thought to release the fibrous septae enzymatically by specifically targeting types 1 and 3 collagen, which may result in smoothing of the skin and an improved appearance of cellulite.
The most common side effects of Qwo include injection site bruising, pain, areas of hardness, itching, redness, discoloration, swelling, and warmth in the treatment area.
Qwo is expected to be available throughout the United States at aesthetic health care practitioner’s offices starting in Spring 2021.
“Qwo could be a game-changer for many women with cellulite,” Anne Chapas, MD, a board-certified dermatologist at Union Square Laser Dermatology in New York, said in a press release. “I am thrilled there will now be an FDA-approved injectable treatment option proven to address a root cause of cellulite. What is exciting about Qwo is that it is a cutting-edge cellulite treatment, without the cutting,” Dr. Chapas, said.
FDA approves avelumab as maintenance for urothelial carcinoma
The Food and Administration has approved a new indication for the PD-L1 inhibitor avelumab.
Physicians can now prescribe avelumab (Bavencio) as maintenance treatment for patients with locally advanced or metastatic urothelial carcinoma (UC) that has not progressed after first-line platinum-containing chemotherapy.
The new indication adds to avelumab use in other patient populations, including people with locally advanced or metastatic UC who experience disease progression during or following platinum-containing chemotherapy. The FDA also previously approved avelumab for patients who experienced UC progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
The FDA first approved marketing of avelumab in 2017. Other uses include treatment of metastatic Merkel cell carcinoma and first-line treatment of advanced renal cell carcinoma in combination with axitinib.
The new maintenance therapy indication for avelumab is based on efficacy demonstrated in the JAVELIN Bladder 100 trial. Results from this trial were presented as part of the American Society of Clinical Oncology virtual scientific program.
Investigators randomly assigned 700 patients with unresectable, locally advanced or metastatic UC to intravenous avelumab and best supportive care or best supportive care alone. All participants had UC that had not progressed after first-line platinum-containing chemotherapy.
The median overall survival was 21.4 months in the avelumab arm and 14.3 months in the best supportive care–alone arm (hazard ratio, 0.69; 95% confidence interval, 0.56-0.86). This difference was statistically significant (P = .001).
Avelumab also was associated with significantly longer overall survival in the 51% of participants with PD-L1–positive tumors (hazard ratio, 0.56; 95% confidence interval, 0.40-0.79; P < .001).
Results from the JAVELIN Bladder 100 trial allowed the FDA to convert an initial accelerated approval of avelumab to a regular approval.
Fatigue, musculoskeletal pain, urinary tract infection, and rash were the most common adverse events reported in 20% or more of trial participants. In all, 28% of patients experienced serious adverse events, and one patient died from sepsis during the trial.
Recommended avelumab dosing is 800 mg administered as an intravenous infusion over 60 minutes every 2 weeks until disease progresses or toxicity becomes unacceptable.
See the full prescribing information for more details.
The Food and Administration has approved a new indication for the PD-L1 inhibitor avelumab.
Physicians can now prescribe avelumab (Bavencio) as maintenance treatment for patients with locally advanced or metastatic urothelial carcinoma (UC) that has not progressed after first-line platinum-containing chemotherapy.
The new indication adds to avelumab use in other patient populations, including people with locally advanced or metastatic UC who experience disease progression during or following platinum-containing chemotherapy. The FDA also previously approved avelumab for patients who experienced UC progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
The FDA first approved marketing of avelumab in 2017. Other uses include treatment of metastatic Merkel cell carcinoma and first-line treatment of advanced renal cell carcinoma in combination with axitinib.
The new maintenance therapy indication for avelumab is based on efficacy demonstrated in the JAVELIN Bladder 100 trial. Results from this trial were presented as part of the American Society of Clinical Oncology virtual scientific program.
Investigators randomly assigned 700 patients with unresectable, locally advanced or metastatic UC to intravenous avelumab and best supportive care or best supportive care alone. All participants had UC that had not progressed after first-line platinum-containing chemotherapy.
The median overall survival was 21.4 months in the avelumab arm and 14.3 months in the best supportive care–alone arm (hazard ratio, 0.69; 95% confidence interval, 0.56-0.86). This difference was statistically significant (P = .001).
Avelumab also was associated with significantly longer overall survival in the 51% of participants with PD-L1–positive tumors (hazard ratio, 0.56; 95% confidence interval, 0.40-0.79; P < .001).
Results from the JAVELIN Bladder 100 trial allowed the FDA to convert an initial accelerated approval of avelumab to a regular approval.
Fatigue, musculoskeletal pain, urinary tract infection, and rash were the most common adverse events reported in 20% or more of trial participants. In all, 28% of patients experienced serious adverse events, and one patient died from sepsis during the trial.
Recommended avelumab dosing is 800 mg administered as an intravenous infusion over 60 minutes every 2 weeks until disease progresses or toxicity becomes unacceptable.
See the full prescribing information for more details.
The Food and Administration has approved a new indication for the PD-L1 inhibitor avelumab.
Physicians can now prescribe avelumab (Bavencio) as maintenance treatment for patients with locally advanced or metastatic urothelial carcinoma (UC) that has not progressed after first-line platinum-containing chemotherapy.
The new indication adds to avelumab use in other patient populations, including people with locally advanced or metastatic UC who experience disease progression during or following platinum-containing chemotherapy. The FDA also previously approved avelumab for patients who experienced UC progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
The FDA first approved marketing of avelumab in 2017. Other uses include treatment of metastatic Merkel cell carcinoma and first-line treatment of advanced renal cell carcinoma in combination with axitinib.
The new maintenance therapy indication for avelumab is based on efficacy demonstrated in the JAVELIN Bladder 100 trial. Results from this trial were presented as part of the American Society of Clinical Oncology virtual scientific program.
Investigators randomly assigned 700 patients with unresectable, locally advanced or metastatic UC to intravenous avelumab and best supportive care or best supportive care alone. All participants had UC that had not progressed after first-line platinum-containing chemotherapy.
The median overall survival was 21.4 months in the avelumab arm and 14.3 months in the best supportive care–alone arm (hazard ratio, 0.69; 95% confidence interval, 0.56-0.86). This difference was statistically significant (P = .001).
Avelumab also was associated with significantly longer overall survival in the 51% of participants with PD-L1–positive tumors (hazard ratio, 0.56; 95% confidence interval, 0.40-0.79; P < .001).
Results from the JAVELIN Bladder 100 trial allowed the FDA to convert an initial accelerated approval of avelumab to a regular approval.
Fatigue, musculoskeletal pain, urinary tract infection, and rash were the most common adverse events reported in 20% or more of trial participants. In all, 28% of patients experienced serious adverse events, and one patient died from sepsis during the trial.
Recommended avelumab dosing is 800 mg administered as an intravenous infusion over 60 minutes every 2 weeks until disease progresses or toxicity becomes unacceptable.
See the full prescribing information for more details.
FDA approves new indications for pembrolizumab
The Food and Drug Administration recently announced two new types of cancer that can be treated by the anti–PD-1 antibody pembrolizumab.

The new indications expand the use of pembrolizumab (Keytruda) to include treatment of patients with unresectable or metastatic tumor mutational burden–high (TMB-H) solid tumors as well as patients with cutaneous squamous cell carcinoma (cSCC). The FDA announced the new indications just 8 days apart, on June 16 and June 24.
In addition, on June 29, the FDA approved a third new indication for pembrolizumab, this time as first-line treatment for patients with unresectable or metastatic microsatellite instability–high or mismatch repair–deficient colorectal cancer.
The new approvals add to a wide range of oncology indications for which pembrolizumab can be used.
Accelerated approval to treat solid tumors
The FDA granted accelerated approval for pembrolizumab to treat children and adults with unresectable or metastatic TMB-H solid tumors that progressed after previous treatment or in instances where there are no satisfactory alternative treatment options.
The tumor mutational burden must be confirmed by an FDA-approved test. To that end, the FDA approved the FoundationOneCDx assay, which is designed to help physicians determine which patients meet the threshold for TMB-H malignancies (10 or more mutations per megabase).
The efficacy of pembrolizumab in TMB-H solid tumors was investigated in 10 cohorts from the multicenter, open-label KEYNOTE-158 trial. Participants received 200 mg of pembrolizumab intravenously every 3 weeks until their disease progressed or they experienced unacceptable toxicity.
Within this population, 102 patients had tumors that met the TMB-H definition. In this group, the overall response rate was 29%, including a 25% partial response rate and a 4% complete response rate.
The median duration of response was not reached, but 57% of participants experienced a response lasting 12 months or longer, and 50% had a response lasting 24 months or longer.
The most common adverse events associated with pembrolizumab in this trial were fatigue, musculoskeletal pain, decreased appetite, pruritus, diarrhea, nausea, rash, pyrexia, cough, dyspnea, constipation, pain, and abdominal pain. Pembrolizumab is associated with immune-mediated side effects, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, and skin adverse reactions, the FDA noted.
Safety and efficacy of pembrolizumab in pediatric patients with TMB-H central nervous system cancers have not been established.
New option for recurrent or metastatic cSCC
Physicians treating patients with cSCC that is not curable by surgery or radiation now have pembrolizumab to consider as another treatment option.
The cSCC approval is based on results of the multicenter, open-label KEYNOTE-629 trial. The dosage regimen was 200 mg of pembrolizumab intravenously every 3 weeks until cancer progressed, unacceptable toxicity arose, or 24 months of treatment were completed.
The objective response rate was 34%, and the median duration of response was not reached.
Adverse events were similar to those occurring in patients who received pembrolizumab as a single agent in other clinical trials, the FDA noted.
The Food and Drug Administration recently announced two new types of cancer that can be treated by the anti–PD-1 antibody pembrolizumab.
The new indications expand the use of pembrolizumab (Keytruda) to include treatment of patients with unresectable or metastatic tumor mutational burden–high (TMB-H) solid tumors as well as patients with cutaneous squamous cell carcinoma (cSCC). The FDA announced the new indications just 8 days apart, on June 16 and June 24.
In addition, on June 29, the FDA approved a third new indication for pembrolizumab, this time as first-line treatment for patients with unresectable or metastatic microsatellite instability–high or mismatch repair–deficient colorectal cancer.
The new approvals add to a wide range of oncology indications for which pembrolizumab can be used.
Accelerated approval to treat solid tumors
The FDA granted accelerated approval for pembrolizumab to treat children and adults with unresectable or metastatic TMB-H solid tumors that progressed after previous treatment or in instances where there are no satisfactory alternative treatment options.
The tumor mutational burden must be confirmed by an FDA-approved test. To that end, the FDA approved the FoundationOneCDx assay, which is designed to help physicians determine which patients meet the threshold for TMB-H malignancies (10 or more mutations per megabase).
The efficacy of pembrolizumab in TMB-H solid tumors was investigated in 10 cohorts from the multicenter, open-label KEYNOTE-158 trial. Participants received 200 mg of pembrolizumab intravenously every 3 weeks until their disease progressed or they experienced unacceptable toxicity.
Within this population, 102 patients had tumors that met the TMB-H definition. In this group, the overall response rate was 29%, including a 25% partial response rate and a 4% complete response rate.
The median duration of response was not reached, but 57% of participants experienced a response lasting 12 months or longer, and 50% had a response lasting 24 months or longer.
The most common adverse events associated with pembrolizumab in this trial were fatigue, musculoskeletal pain, decreased appetite, pruritus, diarrhea, nausea, rash, pyrexia, cough, dyspnea, constipation, pain, and abdominal pain. Pembrolizumab is associated with immune-mediated side effects, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, and skin adverse reactions, the FDA noted.
Safety and efficacy of pembrolizumab in pediatric patients with TMB-H central nervous system cancers have not been established.
New option for recurrent or metastatic cSCC
Physicians treating patients with cSCC that is not curable by surgery or radiation now have pembrolizumab to consider as another treatment option.
The cSCC approval is based on results of the multicenter, open-label KEYNOTE-629 trial. The dosage regimen was 200 mg of pembrolizumab intravenously every 3 weeks until cancer progressed, unacceptable toxicity arose, or 24 months of treatment were completed.
The objective response rate was 34%, and the median duration of response was not reached.
Adverse events were similar to those occurring in patients who received pembrolizumab as a single agent in other clinical trials, the FDA noted.
The Food and Drug Administration recently announced two new types of cancer that can be treated by the anti–PD-1 antibody pembrolizumab.
The new indications expand the use of pembrolizumab (Keytruda) to include treatment of patients with unresectable or metastatic tumor mutational burden–high (TMB-H) solid tumors as well as patients with cutaneous squamous cell carcinoma (cSCC). The FDA announced the new indications just 8 days apart, on June 16 and June 24.
In addition, on June 29, the FDA approved a third new indication for pembrolizumab, this time as first-line treatment for patients with unresectable or metastatic microsatellite instability–high or mismatch repair–deficient colorectal cancer.
The new approvals add to a wide range of oncology indications for which pembrolizumab can be used.
Accelerated approval to treat solid tumors
The FDA granted accelerated approval for pembrolizumab to treat children and adults with unresectable or metastatic TMB-H solid tumors that progressed after previous treatment or in instances where there are no satisfactory alternative treatment options.
The tumor mutational burden must be confirmed by an FDA-approved test. To that end, the FDA approved the FoundationOneCDx assay, which is designed to help physicians determine which patients meet the threshold for TMB-H malignancies (10 or more mutations per megabase).
The efficacy of pembrolizumab in TMB-H solid tumors was investigated in 10 cohorts from the multicenter, open-label KEYNOTE-158 trial. Participants received 200 mg of pembrolizumab intravenously every 3 weeks until their disease progressed or they experienced unacceptable toxicity.
Within this population, 102 patients had tumors that met the TMB-H definition. In this group, the overall response rate was 29%, including a 25% partial response rate and a 4% complete response rate.
The median duration of response was not reached, but 57% of participants experienced a response lasting 12 months or longer, and 50% had a response lasting 24 months or longer.
The most common adverse events associated with pembrolizumab in this trial were fatigue, musculoskeletal pain, decreased appetite, pruritus, diarrhea, nausea, rash, pyrexia, cough, dyspnea, constipation, pain, and abdominal pain. Pembrolizumab is associated with immune-mediated side effects, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, and skin adverse reactions, the FDA noted.
Safety and efficacy of pembrolizumab in pediatric patients with TMB-H central nervous system cancers have not been established.
New option for recurrent or metastatic cSCC
Physicians treating patients with cSCC that is not curable by surgery or radiation now have pembrolizumab to consider as another treatment option.
The cSCC approval is based on results of the multicenter, open-label KEYNOTE-629 trial. The dosage regimen was 200 mg of pembrolizumab intravenously every 3 weeks until cancer progressed, unacceptable toxicity arose, or 24 months of treatment were completed.
The objective response rate was 34%, and the median duration of response was not reached.
Adverse events were similar to those occurring in patients who received pembrolizumab as a single agent in other clinical trials, the FDA noted.
Pembro approved for first-line use in MSI-H/dMMR colorectal cancer
This is the first time that an immunotherapy agent has been approved in this setting and as monotherapy, without added chemotherapy.
“Metastatic colorectal cancer is a serious and life-threatening disease with a poor prognosis,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Oncologic Diseases at the FDA Center for Drug Evaluation and Research, in a statement. “Available current therapy with chemotherapy combinations and other biologics are associated with substantial toxicity.”
“Having a nonchemotherapy option available for selected patients is a noteworthy paradigm shift in treatment,” he commented.
Pembrolizumab is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, and has demonstrated robust antitumor activity and a favorable safety profile in multiple tumor types.
The current approval is based on data from the multicenter, randomized KEYNOTE-177 trial, which found that pembrolizumab more than doubled median progression-free survival (PFS) compared with chemotherapy, the current standard of care. The study results were presented earlier this year at the American Society of Clinical Oncology (ASCO) virtual scientific program, as reported by Medscape Medical News. In a commentary about that study, David Kerr, MD, professor of cancer medicine at the Oxford Cancer Centre, UK, said: “We know that MSI-H/dMMR tumors occur in only 4% or 5% of all patients in the advanced setting. Nevertheless, for that small but important subgroup, we now have a well-tolerated new treatment.”
New standard of care
The KEYNOTE-177 trial involved 307 patients with previously untreated unresectable or metastatic MSI-H or dMMR colorectal cancer. They were randomized to receive either pembrolizumab at 200 mg every 3 weeks for up to 35 cycles (n = 153) or to the investigators’ choice of chemotherapy (n = 154).
Chemotherapy regimens were modified FOLFOX (5-fluorouracil, leucovorin, oxaliplatin) used alone or in combination with either bevacizumab or cetuximab, or FOLFIRI (leucovorin, fluorouracil, irinotecan) alone or in combination with either bevacizumab or cetuximab.
Crossover from the chemotherapy arm to immunotherapy was permitted for up to 35 cycles if the patient experienced disease progression confirmed by central review.
The primary endpoints were PFS and overall survival, and the trial would be considered successful if either primary endpoint was met.
Treatment with pembrolizumab monotherapy significantly reduced the risk of disease progression or death by 40% (HR, 0.60; 95% CI, 0.45 - 0.80; P = .0004), with a median PFS of 16.5 months versus 8.2 months for chemotherapy. At the time of the PFS analysis, overall survival data were not yet mature (66% of the required number of events for final analysis).
The overall response rate with pembrolizumab was 44%, with a complete response achieved in 11% of patients and a partial response rate of 33%. For the chemotherapy arm, the overall response rate was 33%, with a complete response rate of 4% and a partial response rate of 29%.
The median duration of response was not reached in the pembrolizumab arm versus 10.6 months with chemotherapy.
“In the past, no medical treatment has shown such difference in terms of improvement of PFS in metastatic colorectal cancer,” said study investigator Thierry André, MD, of Hôpital Saint Antoine in Paris, France, at a press briefing held prior to the presentation at the ASCO meeting.
At that time, Michael J. Overman, MD, of the University of Texas MD Anderson Cancer Center in Houston, told Medscape Medical News that “I think this is setting a new standard of care.”
Grade 3 or greater treatment-related adverse events occurred in 22% of patients on pembrolizumab and 66% on chemotherapy.
Immune-mediated adverse events and infusion reactions were more common with pembrolizumab than chemotherapy (31% and 13%, respectively). Adverse events that were common with chemotherapy included gastrointestinal events, fatigue, neutropenia, and peripheral sensory neuropathy.
This article first appeared on Medscape.com.
This is the first time that an immunotherapy agent has been approved in this setting and as monotherapy, without added chemotherapy.
“Metastatic colorectal cancer is a serious and life-threatening disease with a poor prognosis,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Oncologic Diseases at the FDA Center for Drug Evaluation and Research, in a statement. “Available current therapy with chemotherapy combinations and other biologics are associated with substantial toxicity.”
“Having a nonchemotherapy option available for selected patients is a noteworthy paradigm shift in treatment,” he commented.
Pembrolizumab is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, and has demonstrated robust antitumor activity and a favorable safety profile in multiple tumor types.
The current approval is based on data from the multicenter, randomized KEYNOTE-177 trial, which found that pembrolizumab more than doubled median progression-free survival (PFS) compared with chemotherapy, the current standard of care. The study results were presented earlier this year at the American Society of Clinical Oncology (ASCO) virtual scientific program, as reported by Medscape Medical News. In a commentary about that study, David Kerr, MD, professor of cancer medicine at the Oxford Cancer Centre, UK, said: “We know that MSI-H/dMMR tumors occur in only 4% or 5% of all patients in the advanced setting. Nevertheless, for that small but important subgroup, we now have a well-tolerated new treatment.”
New standard of care
The KEYNOTE-177 trial involved 307 patients with previously untreated unresectable or metastatic MSI-H or dMMR colorectal cancer. They were randomized to receive either pembrolizumab at 200 mg every 3 weeks for up to 35 cycles (n = 153) or to the investigators’ choice of chemotherapy (n = 154).
Chemotherapy regimens were modified FOLFOX (5-fluorouracil, leucovorin, oxaliplatin) used alone or in combination with either bevacizumab or cetuximab, or FOLFIRI (leucovorin, fluorouracil, irinotecan) alone or in combination with either bevacizumab or cetuximab.
Crossover from the chemotherapy arm to immunotherapy was permitted for up to 35 cycles if the patient experienced disease progression confirmed by central review.
The primary endpoints were PFS and overall survival, and the trial would be considered successful if either primary endpoint was met.
Treatment with pembrolizumab monotherapy significantly reduced the risk of disease progression or death by 40% (HR, 0.60; 95% CI, 0.45 - 0.80; P = .0004), with a median PFS of 16.5 months versus 8.2 months for chemotherapy. At the time of the PFS analysis, overall survival data were not yet mature (66% of the required number of events for final analysis).
The overall response rate with pembrolizumab was 44%, with a complete response achieved in 11% of patients and a partial response rate of 33%. For the chemotherapy arm, the overall response rate was 33%, with a complete response rate of 4% and a partial response rate of 29%.
The median duration of response was not reached in the pembrolizumab arm versus 10.6 months with chemotherapy.
“In the past, no medical treatment has shown such difference in terms of improvement of PFS in metastatic colorectal cancer,” said study investigator Thierry André, MD, of Hôpital Saint Antoine in Paris, France, at a press briefing held prior to the presentation at the ASCO meeting.
At that time, Michael J. Overman, MD, of the University of Texas MD Anderson Cancer Center in Houston, told Medscape Medical News that “I think this is setting a new standard of care.”
Grade 3 or greater treatment-related adverse events occurred in 22% of patients on pembrolizumab and 66% on chemotherapy.
Immune-mediated adverse events and infusion reactions were more common with pembrolizumab than chemotherapy (31% and 13%, respectively). Adverse events that were common with chemotherapy included gastrointestinal events, fatigue, neutropenia, and peripheral sensory neuropathy.
This article first appeared on Medscape.com.
This is the first time that an immunotherapy agent has been approved in this setting and as monotherapy, without added chemotherapy.
“Metastatic colorectal cancer is a serious and life-threatening disease with a poor prognosis,” said Richard Pazdur, MD, director of the FDA Oncology Center of Excellence and acting director of the Office of Oncologic Diseases at the FDA Center for Drug Evaluation and Research, in a statement. “Available current therapy with chemotherapy combinations and other biologics are associated with substantial toxicity.”
“Having a nonchemotherapy option available for selected patients is a noteworthy paradigm shift in treatment,” he commented.
Pembrolizumab is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, and has demonstrated robust antitumor activity and a favorable safety profile in multiple tumor types.
The current approval is based on data from the multicenter, randomized KEYNOTE-177 trial, which found that pembrolizumab more than doubled median progression-free survival (PFS) compared with chemotherapy, the current standard of care. The study results were presented earlier this year at the American Society of Clinical Oncology (ASCO) virtual scientific program, as reported by Medscape Medical News. In a commentary about that study, David Kerr, MD, professor of cancer medicine at the Oxford Cancer Centre, UK, said: “We know that MSI-H/dMMR tumors occur in only 4% or 5% of all patients in the advanced setting. Nevertheless, for that small but important subgroup, we now have a well-tolerated new treatment.”
New standard of care
The KEYNOTE-177 trial involved 307 patients with previously untreated unresectable or metastatic MSI-H or dMMR colorectal cancer. They were randomized to receive either pembrolizumab at 200 mg every 3 weeks for up to 35 cycles (n = 153) or to the investigators’ choice of chemotherapy (n = 154).
Chemotherapy regimens were modified FOLFOX (5-fluorouracil, leucovorin, oxaliplatin) used alone or in combination with either bevacizumab or cetuximab, or FOLFIRI (leucovorin, fluorouracil, irinotecan) alone or in combination with either bevacizumab or cetuximab.
Crossover from the chemotherapy arm to immunotherapy was permitted for up to 35 cycles if the patient experienced disease progression confirmed by central review.
The primary endpoints were PFS and overall survival, and the trial would be considered successful if either primary endpoint was met.
Treatment with pembrolizumab monotherapy significantly reduced the risk of disease progression or death by 40% (HR, 0.60; 95% CI, 0.45 - 0.80; P = .0004), with a median PFS of 16.5 months versus 8.2 months for chemotherapy. At the time of the PFS analysis, overall survival data were not yet mature (66% of the required number of events for final analysis).
The overall response rate with pembrolizumab was 44%, with a complete response achieved in 11% of patients and a partial response rate of 33%. For the chemotherapy arm, the overall response rate was 33%, with a complete response rate of 4% and a partial response rate of 29%.
The median duration of response was not reached in the pembrolizumab arm versus 10.6 months with chemotherapy.
“In the past, no medical treatment has shown such difference in terms of improvement of PFS in metastatic colorectal cancer,” said study investigator Thierry André, MD, of Hôpital Saint Antoine in Paris, France, at a press briefing held prior to the presentation at the ASCO meeting.
At that time, Michael J. Overman, MD, of the University of Texas MD Anderson Cancer Center in Houston, told Medscape Medical News that “I think this is setting a new standard of care.”
Grade 3 or greater treatment-related adverse events occurred in 22% of patients on pembrolizumab and 66% on chemotherapy.
Immune-mediated adverse events and infusion reactions were more common with pembrolizumab than chemotherapy (31% and 13%, respectively). Adverse events that were common with chemotherapy included gastrointestinal events, fatigue, neutropenia, and peripheral sensory neuropathy.
This article first appeared on Medscape.com.
FDA approves in-home breast cancer treatment
Advantageous for infusion centers?
The Food and Drug Administration approved a combination of pertuzumab (Perjeta, Genentech/Roche), trastuzumab (Herceptin, Genentech/Roche) and hyaluronidase (Phesgo, Genentech/Roche) that is administered subcutaneously – rather than intravenously – for the treatment of early and metastatic HER2-positive breast cancers.
Phesgo is initially used in combination with chemotherapy at an infusion center but could continue to be administered in a patient’s home by a qualified health care professional once chemotherapy is complete, according to the FDA.
Administration takes approximately 8 minutes for the initial loading dose and approximately 5 minutes for maintenance doses, according to a Genentech press statement. This compares favorably with the 150 minutes needed for the combined loading dose of intravenous pertuzumab and trastuzumab, and the 60-150 minutes for intravenous maintenance infusions, the company said.
“Currently, most patients with HER2-positive breast cancer receive trastuzumab and pertuzumab at infusion centers. With a new administration route, Phesgo offers an outpatient option for patients to receive trastuzumab and pertuzumab,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in an agency press release.
“The fixed-dose combination of trastuzumab and pertuzumab offers a simpler, faster, and easier treatment experience for patients with HER2-positive breast cancer,” said Antoinette Tan, MD, MHSc, chief of breast medical oncology at Levine Cancer Institute, Charlotte, N.C., in the company statement.
Dr. Tan also said that home administration “can be advantageous for patients and infusion centers.”
However, in April, the Community Oncology Alliance strenuously objected to this type of treatment in a patient’s home, as reported by Medscape Medical News.
The group, which represents U.S. community-based practices, said it “fundamentally opposes home infusion of chemotherapy, cancer immunotherapy, and cancer treatment supportive drugs because of serious patient safety concerns.”
The FDA’s approval was based on the results of the pivotal phase 3 FeDeriCa trial, a noninferiority study in patients with HER2-positive early breast cancer, which demonstrated that the new product had comparable efficacy and safety as intravenous pertuzumab and intravenous trastuzumab.
In terms of efficacy, the subcutaneous product demonstrated noninferior plasma levels of pertuzumab, which was the primary endpoint, when compared with IV administration of pertuzumab.
Safety was comparable between the two approaches, with no new safety signals using the subcutaneous delivery method, including no “meaningful difference” in cardiac toxicity, according to Genentech. However, there were more administration-related reactions with the new product. The most common adverse events in both groups were alopecia, nausea, diarrhea, and anemia.
The new product uses a drug delivery technology (Enhanze, Halozyme Therapeutics) that employs a proprietary enzyme that temporarily degrades hyaluronan, a glycosaminoglycan or chain of natural sugars in the body, to facilitate the dispersion and absorption of injected therapeutic drugs, according to Genentech.
In May, at the European Society for Medical Oncology Breast Cancer Virtual Meeting 2020, investigators of the phase 2 PHranceSCa study reported that “more than 80%” of patients preferred subcutaneous to intravenous administration of pertuzumab and trastuzumab.
This article first appeared on Medscape.com.
Advantageous for infusion centers?
Advantageous for infusion centers?
The Food and Drug Administration approved a combination of pertuzumab (Perjeta, Genentech/Roche), trastuzumab (Herceptin, Genentech/Roche) and hyaluronidase (Phesgo, Genentech/Roche) that is administered subcutaneously – rather than intravenously – for the treatment of early and metastatic HER2-positive breast cancers.
Phesgo is initially used in combination with chemotherapy at an infusion center but could continue to be administered in a patient’s home by a qualified health care professional once chemotherapy is complete, according to the FDA.
Administration takes approximately 8 minutes for the initial loading dose and approximately 5 minutes for maintenance doses, according to a Genentech press statement. This compares favorably with the 150 minutes needed for the combined loading dose of intravenous pertuzumab and trastuzumab, and the 60-150 minutes for intravenous maintenance infusions, the company said.
“Currently, most patients with HER2-positive breast cancer receive trastuzumab and pertuzumab at infusion centers. With a new administration route, Phesgo offers an outpatient option for patients to receive trastuzumab and pertuzumab,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in an agency press release.
“The fixed-dose combination of trastuzumab and pertuzumab offers a simpler, faster, and easier treatment experience for patients with HER2-positive breast cancer,” said Antoinette Tan, MD, MHSc, chief of breast medical oncology at Levine Cancer Institute, Charlotte, N.C., in the company statement.
Dr. Tan also said that home administration “can be advantageous for patients and infusion centers.”
However, in April, the Community Oncology Alliance strenuously objected to this type of treatment in a patient’s home, as reported by Medscape Medical News.
The group, which represents U.S. community-based practices, said it “fundamentally opposes home infusion of chemotherapy, cancer immunotherapy, and cancer treatment supportive drugs because of serious patient safety concerns.”
The FDA’s approval was based on the results of the pivotal phase 3 FeDeriCa trial, a noninferiority study in patients with HER2-positive early breast cancer, which demonstrated that the new product had comparable efficacy and safety as intravenous pertuzumab and intravenous trastuzumab.
In terms of efficacy, the subcutaneous product demonstrated noninferior plasma levels of pertuzumab, which was the primary endpoint, when compared with IV administration of pertuzumab.
Safety was comparable between the two approaches, with no new safety signals using the subcutaneous delivery method, including no “meaningful difference” in cardiac toxicity, according to Genentech. However, there were more administration-related reactions with the new product. The most common adverse events in both groups were alopecia, nausea, diarrhea, and anemia.
The new product uses a drug delivery technology (Enhanze, Halozyme Therapeutics) that employs a proprietary enzyme that temporarily degrades hyaluronan, a glycosaminoglycan or chain of natural sugars in the body, to facilitate the dispersion and absorption of injected therapeutic drugs, according to Genentech.
In May, at the European Society for Medical Oncology Breast Cancer Virtual Meeting 2020, investigators of the phase 2 PHranceSCa study reported that “more than 80%” of patients preferred subcutaneous to intravenous administration of pertuzumab and trastuzumab.
This article first appeared on Medscape.com.
The Food and Drug Administration approved a combination of pertuzumab (Perjeta, Genentech/Roche), trastuzumab (Herceptin, Genentech/Roche) and hyaluronidase (Phesgo, Genentech/Roche) that is administered subcutaneously – rather than intravenously – for the treatment of early and metastatic HER2-positive breast cancers.
Phesgo is initially used in combination with chemotherapy at an infusion center but could continue to be administered in a patient’s home by a qualified health care professional once chemotherapy is complete, according to the FDA.
Administration takes approximately 8 minutes for the initial loading dose and approximately 5 minutes for maintenance doses, according to a Genentech press statement. This compares favorably with the 150 minutes needed for the combined loading dose of intravenous pertuzumab and trastuzumab, and the 60-150 minutes for intravenous maintenance infusions, the company said.
“Currently, most patients with HER2-positive breast cancer receive trastuzumab and pertuzumab at infusion centers. With a new administration route, Phesgo offers an outpatient option for patients to receive trastuzumab and pertuzumab,” said Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in an agency press release.
“The fixed-dose combination of trastuzumab and pertuzumab offers a simpler, faster, and easier treatment experience for patients with HER2-positive breast cancer,” said Antoinette Tan, MD, MHSc, chief of breast medical oncology at Levine Cancer Institute, Charlotte, N.C., in the company statement.
Dr. Tan also said that home administration “can be advantageous for patients and infusion centers.”
However, in April, the Community Oncology Alliance strenuously objected to this type of treatment in a patient’s home, as reported by Medscape Medical News.
The group, which represents U.S. community-based practices, said it “fundamentally opposes home infusion of chemotherapy, cancer immunotherapy, and cancer treatment supportive drugs because of serious patient safety concerns.”
The FDA’s approval was based on the results of the pivotal phase 3 FeDeriCa trial, a noninferiority study in patients with HER2-positive early breast cancer, which demonstrated that the new product had comparable efficacy and safety as intravenous pertuzumab and intravenous trastuzumab.
In terms of efficacy, the subcutaneous product demonstrated noninferior plasma levels of pertuzumab, which was the primary endpoint, when compared with IV administration of pertuzumab.
Safety was comparable between the two approaches, with no new safety signals using the subcutaneous delivery method, including no “meaningful difference” in cardiac toxicity, according to Genentech. However, there were more administration-related reactions with the new product. The most common adverse events in both groups were alopecia, nausea, diarrhea, and anemia.
The new product uses a drug delivery technology (Enhanze, Halozyme Therapeutics) that employs a proprietary enzyme that temporarily degrades hyaluronan, a glycosaminoglycan or chain of natural sugars in the body, to facilitate the dispersion and absorption of injected therapeutic drugs, according to Genentech.
In May, at the European Society for Medical Oncology Breast Cancer Virtual Meeting 2020, investigators of the phase 2 PHranceSCa study reported that “more than 80%” of patients preferred subcutaneous to intravenous administration of pertuzumab and trastuzumab.
This article first appeared on Medscape.com.
FDA approves first oral somatostatin analog for acromegaly
who previously responded to and tolerated octreotide or lanreotide injections.
“People living with acromegaly experience many challenges associated with injectable therapies and are in need of new treatment options,” Jill Sisco, president of Acromegaly Community, a patient support group, said in a Chiasma press release.
“The entire acromegaly community has long awaited oral therapeutic options and it is gratifying to see that the FDA has now approved the first oral somatostatin analog (SSA) therapy with the potential to make a significant impact in the lives of people with acromegaly and their caregivers,” she added.
Acromegaly, a rare, chronic disease usually caused by a benign pituitary tumor that leads to excess production of growth hormone and insulin-like growth factor-1 (IGF-1) hormone, can be cured through the successful surgical removal of the pituitary tumor. However, management of the disease remains a lifelong challenge for many who must rely on chronic injections.
The new oral formulation of octreotide is the first and only oral somatostatin analog approved by the FDA.
The approval was based on the results of the 9-month, phase 3 pivotal CHIASMA OPTIMAL clinical trial, involving 56 adults with acromegaly controlled by injectable SSAs.
The patients, who were randomized 1:1 to octreotide capsules or placebo, were dose-titrated from 40 mg/day up to a maximum of 80 mg/day, equaling two capsules in the morning and two in the evening.
The study met its primary endpoint. Overall, 58% of patients taking octreotide maintained IGF-1 response compared with 19% of those on placebo at the end of 9 months (P = .008), according to the average of the last two IGF-1 levels that were 1 times or less the upper limit of normal, assessed at weeks 34 and 36.
The trial also met its secondary endpoints, which included the proportion of patients who maintain growth hormone response at week 36 compared with screening; time to loss of response; and proportion of patients requiring reversion to prior treatment.
Safety data were favorable. Adverse reactions to the drug, detailed in the prescribing information, include cholelithiasis and associated complications; hyperglycemia and hypoglycemia; thyroid function abnormalities; cardiac function abnormalities; decreased vitamin B12 levels, and abnormal Schilling’s test results.
Results from the clinical trial “are encouraging for patients with acromegaly,” the study’s principal investigator, Susan Samson, MD, PhD, of Baylor College of Medicine, Houston, said in the Chiasma statement.
“Based on data from the CHIASMA OPTIMAL trial showing patients on therapy being able to maintain mean IGF-1 levels within the normal range at the end of treatment, I believe oral octreotide capsules hold meaningful promise for patients with this disease and will address a long-standing unmet treatment need,” she added.
Chiasma reports that it expects Mycapssa to be available in the fourth quarter of 2020, pending FDA approval of a planned manufacturing supplement to the approved new drug application.
The company further plans to provide patient support services including assistance with insurance providers and specialty pharmacies and support in incorporating treatment into patients’ daily routines.
Despite effective biochemical control of growth hormone, many patients with acromegaly continue to suffer symptoms, mainly because of comorbidities, so it is important that these are also adequately treated, a consensus group concluded earlier this year.
The CHIASMA OPTIMAL trial was funded by Chiasma.
A version of this article originally appeared on Medscape.com.
who previously responded to and tolerated octreotide or lanreotide injections.
“People living with acromegaly experience many challenges associated with injectable therapies and are in need of new treatment options,” Jill Sisco, president of Acromegaly Community, a patient support group, said in a Chiasma press release.
“The entire acromegaly community has long awaited oral therapeutic options and it is gratifying to see that the FDA has now approved the first oral somatostatin analog (SSA) therapy with the potential to make a significant impact in the lives of people with acromegaly and their caregivers,” she added.
Acromegaly, a rare, chronic disease usually caused by a benign pituitary tumor that leads to excess production of growth hormone and insulin-like growth factor-1 (IGF-1) hormone, can be cured through the successful surgical removal of the pituitary tumor. However, management of the disease remains a lifelong challenge for many who must rely on chronic injections.
The new oral formulation of octreotide is the first and only oral somatostatin analog approved by the FDA.
The approval was based on the results of the 9-month, phase 3 pivotal CHIASMA OPTIMAL clinical trial, involving 56 adults with acromegaly controlled by injectable SSAs.
The patients, who were randomized 1:1 to octreotide capsules or placebo, were dose-titrated from 40 mg/day up to a maximum of 80 mg/day, equaling two capsules in the morning and two in the evening.
The study met its primary endpoint. Overall, 58% of patients taking octreotide maintained IGF-1 response compared with 19% of those on placebo at the end of 9 months (P = .008), according to the average of the last two IGF-1 levels that were 1 times or less the upper limit of normal, assessed at weeks 34 and 36.
The trial also met its secondary endpoints, which included the proportion of patients who maintain growth hormone response at week 36 compared with screening; time to loss of response; and proportion of patients requiring reversion to prior treatment.
Safety data were favorable. Adverse reactions to the drug, detailed in the prescribing information, include cholelithiasis and associated complications; hyperglycemia and hypoglycemia; thyroid function abnormalities; cardiac function abnormalities; decreased vitamin B12 levels, and abnormal Schilling’s test results.
Results from the clinical trial “are encouraging for patients with acromegaly,” the study’s principal investigator, Susan Samson, MD, PhD, of Baylor College of Medicine, Houston, said in the Chiasma statement.
“Based on data from the CHIASMA OPTIMAL trial showing patients on therapy being able to maintain mean IGF-1 levels within the normal range at the end of treatment, I believe oral octreotide capsules hold meaningful promise for patients with this disease and will address a long-standing unmet treatment need,” she added.
Chiasma reports that it expects Mycapssa to be available in the fourth quarter of 2020, pending FDA approval of a planned manufacturing supplement to the approved new drug application.
The company further plans to provide patient support services including assistance with insurance providers and specialty pharmacies and support in incorporating treatment into patients’ daily routines.
Despite effective biochemical control of growth hormone, many patients with acromegaly continue to suffer symptoms, mainly because of comorbidities, so it is important that these are also adequately treated, a consensus group concluded earlier this year.
The CHIASMA OPTIMAL trial was funded by Chiasma.
A version of this article originally appeared on Medscape.com.
who previously responded to and tolerated octreotide or lanreotide injections.
“People living with acromegaly experience many challenges associated with injectable therapies and are in need of new treatment options,” Jill Sisco, president of Acromegaly Community, a patient support group, said in a Chiasma press release.
“The entire acromegaly community has long awaited oral therapeutic options and it is gratifying to see that the FDA has now approved the first oral somatostatin analog (SSA) therapy with the potential to make a significant impact in the lives of people with acromegaly and their caregivers,” she added.
Acromegaly, a rare, chronic disease usually caused by a benign pituitary tumor that leads to excess production of growth hormone and insulin-like growth factor-1 (IGF-1) hormone, can be cured through the successful surgical removal of the pituitary tumor. However, management of the disease remains a lifelong challenge for many who must rely on chronic injections.
The new oral formulation of octreotide is the first and only oral somatostatin analog approved by the FDA.
The approval was based on the results of the 9-month, phase 3 pivotal CHIASMA OPTIMAL clinical trial, involving 56 adults with acromegaly controlled by injectable SSAs.
The patients, who were randomized 1:1 to octreotide capsules or placebo, were dose-titrated from 40 mg/day up to a maximum of 80 mg/day, equaling two capsules in the morning and two in the evening.
The study met its primary endpoint. Overall, 58% of patients taking octreotide maintained IGF-1 response compared with 19% of those on placebo at the end of 9 months (P = .008), according to the average of the last two IGF-1 levels that were 1 times or less the upper limit of normal, assessed at weeks 34 and 36.
The trial also met its secondary endpoints, which included the proportion of patients who maintain growth hormone response at week 36 compared with screening; time to loss of response; and proportion of patients requiring reversion to prior treatment.
Safety data were favorable. Adverse reactions to the drug, detailed in the prescribing information, include cholelithiasis and associated complications; hyperglycemia and hypoglycemia; thyroid function abnormalities; cardiac function abnormalities; decreased vitamin B12 levels, and abnormal Schilling’s test results.
Results from the clinical trial “are encouraging for patients with acromegaly,” the study’s principal investigator, Susan Samson, MD, PhD, of Baylor College of Medicine, Houston, said in the Chiasma statement.
“Based on data from the CHIASMA OPTIMAL trial showing patients on therapy being able to maintain mean IGF-1 levels within the normal range at the end of treatment, I believe oral octreotide capsules hold meaningful promise for patients with this disease and will address a long-standing unmet treatment need,” she added.
Chiasma reports that it expects Mycapssa to be available in the fourth quarter of 2020, pending FDA approval of a planned manufacturing supplement to the approved new drug application.
The company further plans to provide patient support services including assistance with insurance providers and specialty pharmacies and support in incorporating treatment into patients’ daily routines.
Despite effective biochemical control of growth hormone, many patients with acromegaly continue to suffer symptoms, mainly because of comorbidities, so it is important that these are also adequately treated, a consensus group concluded earlier this year.
The CHIASMA OPTIMAL trial was funded by Chiasma.
A version of this article originally appeared on Medscape.com.
FDA approves new treatment for Dravet syndrome
Dravet syndrome is a rare childhood-onset epilepsy characterized by frequent, drug-resistant convulsive seizures that may contribute to intellectual disability and impairments in motor control, behavior, and cognition, as well as an increased risk of sudden unexpected death in epilepsy (SUDEP).
Dravet syndrome takes a “tremendous toll on both patients and their families. Fintepla offers an additional effective treatment option for the treatment of seizures associated with Dravet syndrome,” Billy Dunn, MD, director, Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The FDA approved fenfluramine for Dravet syndrome based on the results of two randomized, double-blind, placebo-controlled phase 3 trials involving children ages 2 to 18 years with Dravet syndrome.
In both studies, children treated with fenfluramine experienced significantly greater reductions in the frequency of convulsive seizures than did their peers who received placebo. These reductions occurred within 3 to 4 weeks, and remained generally consistent over the 14- to 15-week treatment periods, the FDA said.
“There remains a huge unmet need for the many Dravet syndrome patients who continue to experience frequent severe seizures even while taking one or more of the currently available antiseizure medications,” Joseph Sullivan, MD, who worked on the fenfluramine for Dravet syndrome studies, said in a news release.
Given the “profound reductions” in convulsive seizure frequency seen in the clinical trials, combined with the “ongoing, robust safety monitoring,” fenfluramine offers “an extremely important treatment option for Dravet syndrome patients,” said Dr. Sullivan, director of the Pediatric Epilepsy Center of Excellence at the University of California San Francisco (UCSF) Benioff Children’s Hospital.
Fenfluramine is an anorectic agent that was used to treat obesity until it was removed from the market in 1997 over reports of increased risk of valvular heart disease when prescribed in higher doses and most often when prescribed with phentermine. The combination of the two drugs was known as fen-phen.
In the clinical trials of Dravet syndrome, the most common adverse reactions were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; increased blood pressure; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; and status epilepticus.
The Fintepla label has a boxed warning stating that the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). Due to these risks, patients must undergo echocardiography before treatment, every 6 months during treatment, and once 3 to 6 months after treatment is stopped.
If signs of VHD, PAH, or other cardiac abnormalities are present, clinicians should weigh the benefits and risks of continuing treatment with Fintepla, the FDA said.
Fintepla is available only through a risk evaluation and mitigation strategy (REMS) program, which requires physicians who prescribe the drug and pharmacies that dispense it to be certified in the Fintepla REMS and that patients be enrolled in the program.
As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring to receive the drug.
Fintepla will be available to certified prescribers in the United States in July. Zogenix is launching Zogenix Central, a comprehensive support service that will provide ongoing product assistance to patients, caregivers, and their medical teams. Further information is available online.
This article first appeared on Medscape.com.
Dravet syndrome is a rare childhood-onset epilepsy characterized by frequent, drug-resistant convulsive seizures that may contribute to intellectual disability and impairments in motor control, behavior, and cognition, as well as an increased risk of sudden unexpected death in epilepsy (SUDEP).
Dravet syndrome takes a “tremendous toll on both patients and their families. Fintepla offers an additional effective treatment option for the treatment of seizures associated with Dravet syndrome,” Billy Dunn, MD, director, Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The FDA approved fenfluramine for Dravet syndrome based on the results of two randomized, double-blind, placebo-controlled phase 3 trials involving children ages 2 to 18 years with Dravet syndrome.
In both studies, children treated with fenfluramine experienced significantly greater reductions in the frequency of convulsive seizures than did their peers who received placebo. These reductions occurred within 3 to 4 weeks, and remained generally consistent over the 14- to 15-week treatment periods, the FDA said.
“There remains a huge unmet need for the many Dravet syndrome patients who continue to experience frequent severe seizures even while taking one or more of the currently available antiseizure medications,” Joseph Sullivan, MD, who worked on the fenfluramine for Dravet syndrome studies, said in a news release.
Given the “profound reductions” in convulsive seizure frequency seen in the clinical trials, combined with the “ongoing, robust safety monitoring,” fenfluramine offers “an extremely important treatment option for Dravet syndrome patients,” said Dr. Sullivan, director of the Pediatric Epilepsy Center of Excellence at the University of California San Francisco (UCSF) Benioff Children’s Hospital.
Fenfluramine is an anorectic agent that was used to treat obesity until it was removed from the market in 1997 over reports of increased risk of valvular heart disease when prescribed in higher doses and most often when prescribed with phentermine. The combination of the two drugs was known as fen-phen.
In the clinical trials of Dravet syndrome, the most common adverse reactions were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; increased blood pressure; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; and status epilepticus.
The Fintepla label has a boxed warning stating that the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). Due to these risks, patients must undergo echocardiography before treatment, every 6 months during treatment, and once 3 to 6 months after treatment is stopped.
If signs of VHD, PAH, or other cardiac abnormalities are present, clinicians should weigh the benefits and risks of continuing treatment with Fintepla, the FDA said.
Fintepla is available only through a risk evaluation and mitigation strategy (REMS) program, which requires physicians who prescribe the drug and pharmacies that dispense it to be certified in the Fintepla REMS and that patients be enrolled in the program.
As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring to receive the drug.
Fintepla will be available to certified prescribers in the United States in July. Zogenix is launching Zogenix Central, a comprehensive support service that will provide ongoing product assistance to patients, caregivers, and their medical teams. Further information is available online.
This article first appeared on Medscape.com.
Dravet syndrome is a rare childhood-onset epilepsy characterized by frequent, drug-resistant convulsive seizures that may contribute to intellectual disability and impairments in motor control, behavior, and cognition, as well as an increased risk of sudden unexpected death in epilepsy (SUDEP).
Dravet syndrome takes a “tremendous toll on both patients and their families. Fintepla offers an additional effective treatment option for the treatment of seizures associated with Dravet syndrome,” Billy Dunn, MD, director, Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research, said in a news release.
The FDA approved fenfluramine for Dravet syndrome based on the results of two randomized, double-blind, placebo-controlled phase 3 trials involving children ages 2 to 18 years with Dravet syndrome.
In both studies, children treated with fenfluramine experienced significantly greater reductions in the frequency of convulsive seizures than did their peers who received placebo. These reductions occurred within 3 to 4 weeks, and remained generally consistent over the 14- to 15-week treatment periods, the FDA said.
“There remains a huge unmet need for the many Dravet syndrome patients who continue to experience frequent severe seizures even while taking one or more of the currently available antiseizure medications,” Joseph Sullivan, MD, who worked on the fenfluramine for Dravet syndrome studies, said in a news release.
Given the “profound reductions” in convulsive seizure frequency seen in the clinical trials, combined with the “ongoing, robust safety monitoring,” fenfluramine offers “an extremely important treatment option for Dravet syndrome patients,” said Dr. Sullivan, director of the Pediatric Epilepsy Center of Excellence at the University of California San Francisco (UCSF) Benioff Children’s Hospital.
Fenfluramine is an anorectic agent that was used to treat obesity until it was removed from the market in 1997 over reports of increased risk of valvular heart disease when prescribed in higher doses and most often when prescribed with phentermine. The combination of the two drugs was known as fen-phen.
In the clinical trials of Dravet syndrome, the most common adverse reactions were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; increased blood pressure; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; and status epilepticus.
The Fintepla label has a boxed warning stating that the drug is associated with valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). Due to these risks, patients must undergo echocardiography before treatment, every 6 months during treatment, and once 3 to 6 months after treatment is stopped.
If signs of VHD, PAH, or other cardiac abnormalities are present, clinicians should weigh the benefits and risks of continuing treatment with Fintepla, the FDA said.
Fintepla is available only through a risk evaluation and mitigation strategy (REMS) program, which requires physicians who prescribe the drug and pharmacies that dispense it to be certified in the Fintepla REMS and that patients be enrolled in the program.
As part of the REMS requirements, prescribers and patients must adhere to the required cardiac monitoring to receive the drug.
Fintepla will be available to certified prescribers in the United States in July. Zogenix is launching Zogenix Central, a comprehensive support service that will provide ongoing product assistance to patients, caregivers, and their medical teams. Further information is available online.
This article first appeared on Medscape.com.
New quadrivalent meningococcal vaccine joins VFC arsenal
No changes to the current meningococcal vaccination recommendations were made. The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP) voted 14-0 to include MenACWY-TT as an option for vaccination against meningococcal serogroups A, C, W, and Y in the VFC program. The vote took place in a virtual meeting held on June 24.
The currently available MenACWY vaccines in the United States are MenACWY-D (Menactra), MenACWY-CRW (Menveo), and MenACWY-TT (MedQuadfi), with MenACWY-TT approved by the Food and Drug Administration in April 2020.
Meningococcal vaccination is currently recommended for adolescents, with one dose at age 11 or 12 years and a booster at age 16 years, as well as individuals aged 2 months and older at increased risk for meningococcal disease, according to Lucy McNamara, PhD, of the CDC’s National Center for Immunization and Respiratory Diseases.
Dr. McNamara presented considerations from the Meningococcal Work Group, which determined that the inclusion of MenACWY-TT “is of public health importance given recent vaccine licensure and to support security of vaccine supply.”
The Work Group reviewed 10 studies (phase 2 or 3) of MenACWY-TT that included data on short-term immune response, persistence of immune response, immune interference because of coadministration with other routine adolescent vaccines, and incidence of serious adverse events. Overall, the data showed noninferiority of MenACWY-TT, compared with other available products, in terms of response rates, as well as higher levels of immune response in some studies. Serious adverse events were similar, and none determined to be associated with the vaccines.
ACIP member Paul Hunter, MD, of the University of Milwaukee, Wisc., expressed some concerns about pain or side effects for the new vaccine and Tdap when given together. However, a study of coadministration of MedACWY-TT and Tdap, compared with Tdap alone, showed no impact on geometric mean titer ratios.
Overall, the Work Group concluded that “desirable effects outweigh undesirable effects” and that the data favor the inclusion of MenACWY-TT as an option for meningococcal vaccination.
The committee members and Dr. McNamara had no relevant financial conflicts to disclose.
No changes to the current meningococcal vaccination recommendations were made. The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP) voted 14-0 to include MenACWY-TT as an option for vaccination against meningococcal serogroups A, C, W, and Y in the VFC program. The vote took place in a virtual meeting held on June 24.
The currently available MenACWY vaccines in the United States are MenACWY-D (Menactra), MenACWY-CRW (Menveo), and MenACWY-TT (MedQuadfi), with MenACWY-TT approved by the Food and Drug Administration in April 2020.
Meningococcal vaccination is currently recommended for adolescents, with one dose at age 11 or 12 years and a booster at age 16 years, as well as individuals aged 2 months and older at increased risk for meningococcal disease, according to Lucy McNamara, PhD, of the CDC’s National Center for Immunization and Respiratory Diseases.
Dr. McNamara presented considerations from the Meningococcal Work Group, which determined that the inclusion of MenACWY-TT “is of public health importance given recent vaccine licensure and to support security of vaccine supply.”
The Work Group reviewed 10 studies (phase 2 or 3) of MenACWY-TT that included data on short-term immune response, persistence of immune response, immune interference because of coadministration with other routine adolescent vaccines, and incidence of serious adverse events. Overall, the data showed noninferiority of MenACWY-TT, compared with other available products, in terms of response rates, as well as higher levels of immune response in some studies. Serious adverse events were similar, and none determined to be associated with the vaccines.
ACIP member Paul Hunter, MD, of the University of Milwaukee, Wisc., expressed some concerns about pain or side effects for the new vaccine and Tdap when given together. However, a study of coadministration of MedACWY-TT and Tdap, compared with Tdap alone, showed no impact on geometric mean titer ratios.
Overall, the Work Group concluded that “desirable effects outweigh undesirable effects” and that the data favor the inclusion of MenACWY-TT as an option for meningococcal vaccination.
The committee members and Dr. McNamara had no relevant financial conflicts to disclose.
No changes to the current meningococcal vaccination recommendations were made. The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices (ACIP) voted 14-0 to include MenACWY-TT as an option for vaccination against meningococcal serogroups A, C, W, and Y in the VFC program. The vote took place in a virtual meeting held on June 24.
The currently available MenACWY vaccines in the United States are MenACWY-D (Menactra), MenACWY-CRW (Menveo), and MenACWY-TT (MedQuadfi), with MenACWY-TT approved by the Food and Drug Administration in April 2020.
Meningococcal vaccination is currently recommended for adolescents, with one dose at age 11 or 12 years and a booster at age 16 years, as well as individuals aged 2 months and older at increased risk for meningococcal disease, according to Lucy McNamara, PhD, of the CDC’s National Center for Immunization and Respiratory Diseases.
Dr. McNamara presented considerations from the Meningococcal Work Group, which determined that the inclusion of MenACWY-TT “is of public health importance given recent vaccine licensure and to support security of vaccine supply.”
The Work Group reviewed 10 studies (phase 2 or 3) of MenACWY-TT that included data on short-term immune response, persistence of immune response, immune interference because of coadministration with other routine adolescent vaccines, and incidence of serious adverse events. Overall, the data showed noninferiority of MenACWY-TT, compared with other available products, in terms of response rates, as well as higher levels of immune response in some studies. Serious adverse events were similar, and none determined to be associated with the vaccines.
ACIP member Paul Hunter, MD, of the University of Milwaukee, Wisc., expressed some concerns about pain or side effects for the new vaccine and Tdap when given together. However, a study of coadministration of MedACWY-TT and Tdap, compared with Tdap alone, showed no impact on geometric mean titer ratios.
Overall, the Work Group concluded that “desirable effects outweigh undesirable effects” and that the data favor the inclusion of MenACWY-TT as an option for meningococcal vaccination.
The committee members and Dr. McNamara had no relevant financial conflicts to disclose.
FDA approves selinexor for relapsed/refractory DLBCL
The Food and Drug Administration has granted accelerated approval of selinexor, a nuclear transport inhibitor, for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
Selinexor (marketed as XPOVIO by Karyopharm Therapeutics) is intended for adult patients with relapsed/refractory DLBCL, not otherwise specified, including DLBCL arising from follicular lymphoma, after at least two lines of systemic therapy, according to the FDA’s announcement.
The FDA granted selinexor accelerated approval for this indication based on the response rate seen in the SADAL trial. Continued approval for this indication “may be contingent upon verification and description of clinical benefit in confirmatory trials,” according to the FDA.
The SADAL trial (NCT02227251) was a phase 2, single-arm, open-label study of patients with DLBCL who had previously received two to five systemic regimens. The patients received selinexor at 60 mg orally on days 1 and 3 of each week.
Results in 134 patients showed an overall response rate of 29% (95% confidence interval: 22-38), with complete responses in 13% of patients. Of 39 patients who achieved a partial or complete response, 38% had a response duration of at least 6 months, and 15% had a response duration of at least 12 months, according to the FDA announcement.
The most common adverse reactions in this trial were fatigue, nausea, diarrhea, appetite decrease, weight decrease, constipation, vomiting, and pyrexia. Grade 3-4 laboratory abnormalities occurred in 15% or more of the patients, and comprised thrombocytopenia, lymphopenia, neutropenia, anemia, and hyponatremia.
Serious adverse reactions occurred in 46% of patients, most often from infection. Gastrointestinal toxicity occurred in 80% of patients, and any-grade hyponatremia occurred in 61%. Central neurological adverse reactions occurred in 25% of patients, including dizziness and mental status changes, according to the announcement.
Warnings and precautions for adverse events – including thrombocytopenia, neutropenia, gastrointestinal toxicity, hyponatremia, serious infection, neurological toxicity, and embryo-fetal toxicity – are provided in the prescribing information.
Selinexor acts through the inhibition of exportin-1 and leads to an accumulation of tumor suppressor proteins, a reduction in oncoproteins, and apoptosis of cancer cells. The drug was previously approved in 2019 for the treatment of relapsed/refractory multiple myeloma.
The SADAL trial was sponsored by Karyopharm Therapeutics.
SOURCE: U.S. Food and Drug Administration. 2020. Approval announcement.
The Food and Drug Administration has granted accelerated approval of selinexor, a nuclear transport inhibitor, for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
Selinexor (marketed as XPOVIO by Karyopharm Therapeutics) is intended for adult patients with relapsed/refractory DLBCL, not otherwise specified, including DLBCL arising from follicular lymphoma, after at least two lines of systemic therapy, according to the FDA’s announcement.
The FDA granted selinexor accelerated approval for this indication based on the response rate seen in the SADAL trial. Continued approval for this indication “may be contingent upon verification and description of clinical benefit in confirmatory trials,” according to the FDA.
The SADAL trial (NCT02227251) was a phase 2, single-arm, open-label study of patients with DLBCL who had previously received two to five systemic regimens. The patients received selinexor at 60 mg orally on days 1 and 3 of each week.
Results in 134 patients showed an overall response rate of 29% (95% confidence interval: 22-38), with complete responses in 13% of patients. Of 39 patients who achieved a partial or complete response, 38% had a response duration of at least 6 months, and 15% had a response duration of at least 12 months, according to the FDA announcement.
The most common adverse reactions in this trial were fatigue, nausea, diarrhea, appetite decrease, weight decrease, constipation, vomiting, and pyrexia. Grade 3-4 laboratory abnormalities occurred in 15% or more of the patients, and comprised thrombocytopenia, lymphopenia, neutropenia, anemia, and hyponatremia.
Serious adverse reactions occurred in 46% of patients, most often from infection. Gastrointestinal toxicity occurred in 80% of patients, and any-grade hyponatremia occurred in 61%. Central neurological adverse reactions occurred in 25% of patients, including dizziness and mental status changes, according to the announcement.
Warnings and precautions for adverse events – including thrombocytopenia, neutropenia, gastrointestinal toxicity, hyponatremia, serious infection, neurological toxicity, and embryo-fetal toxicity – are provided in the prescribing information.
Selinexor acts through the inhibition of exportin-1 and leads to an accumulation of tumor suppressor proteins, a reduction in oncoproteins, and apoptosis of cancer cells. The drug was previously approved in 2019 for the treatment of relapsed/refractory multiple myeloma.
The SADAL trial was sponsored by Karyopharm Therapeutics.
SOURCE: U.S. Food and Drug Administration. 2020. Approval announcement.
The Food and Drug Administration has granted accelerated approval of selinexor, a nuclear transport inhibitor, for the treatment of relapsed/refractory diffuse large B-cell lymphoma (DLBCL).
Selinexor (marketed as XPOVIO by Karyopharm Therapeutics) is intended for adult patients with relapsed/refractory DLBCL, not otherwise specified, including DLBCL arising from follicular lymphoma, after at least two lines of systemic therapy, according to the FDA’s announcement.
The FDA granted selinexor accelerated approval for this indication based on the response rate seen in the SADAL trial. Continued approval for this indication “may be contingent upon verification and description of clinical benefit in confirmatory trials,” according to the FDA.
The SADAL trial (NCT02227251) was a phase 2, single-arm, open-label study of patients with DLBCL who had previously received two to five systemic regimens. The patients received selinexor at 60 mg orally on days 1 and 3 of each week.
Results in 134 patients showed an overall response rate of 29% (95% confidence interval: 22-38), with complete responses in 13% of patients. Of 39 patients who achieved a partial or complete response, 38% had a response duration of at least 6 months, and 15% had a response duration of at least 12 months, according to the FDA announcement.
The most common adverse reactions in this trial were fatigue, nausea, diarrhea, appetite decrease, weight decrease, constipation, vomiting, and pyrexia. Grade 3-4 laboratory abnormalities occurred in 15% or more of the patients, and comprised thrombocytopenia, lymphopenia, neutropenia, anemia, and hyponatremia.
Serious adverse reactions occurred in 46% of patients, most often from infection. Gastrointestinal toxicity occurred in 80% of patients, and any-grade hyponatremia occurred in 61%. Central neurological adverse reactions occurred in 25% of patients, including dizziness and mental status changes, according to the announcement.
Warnings and precautions for adverse events – including thrombocytopenia, neutropenia, gastrointestinal toxicity, hyponatremia, serious infection, neurological toxicity, and embryo-fetal toxicity – are provided in the prescribing information.
Selinexor acts through the inhibition of exportin-1 and leads to an accumulation of tumor suppressor proteins, a reduction in oncoproteins, and apoptosis of cancer cells. The drug was previously approved in 2019 for the treatment of relapsed/refractory multiple myeloma.
The SADAL trial was sponsored by Karyopharm Therapeutics.
SOURCE: U.S. Food and Drug Administration. 2020. Approval announcement.
FROM THE FDA
ED visits for life-threatening conditions declined early in COVID-19 pandemic
ED visits for myocardial infarction, stroke, and hyperglycemic crisis dropped substantially in the 10 weeks after COVID-19 was declared a national emergency on March 13, according to the Centers for Disease Control and Prevention.
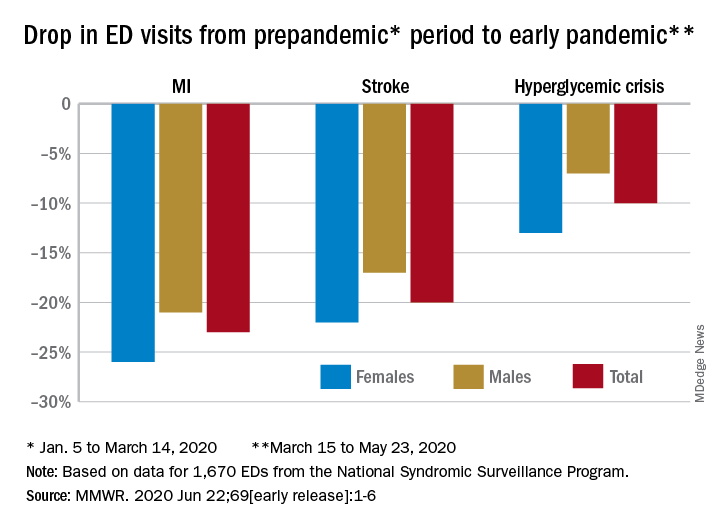
Compared with the 10-week period from Jan. 5 to March 14, ED visits were down by 23% for MI, 20% for stroke, and 10% for hyperglycemic crisis from March 15 to May 23, Samantha J. Lange, MPH, and associates at the CDC reported June 22 in the Morbidity and Mortality Weekly Report.
“A short-term decline of this magnitude … is biologically implausible for MI and stroke, especially for older adults, and unlikely for hyperglycemic crisis, and the finding suggests that patients with these conditions either could not access care or were delaying or avoiding seeking care during the early pandemic period,” they wrote.
The largest decreases in the actual number of visits for MI occurred among both men (down by 2,114, –24%) and women (down by 1,459, –25%) aged 65-74 years. For stroke, men aged 65-74 years had 1,406 (–19%) fewer visits to the ED and women 75-84 years had 1,642 (–23%) fewer visits, the CDC researchers said.
For hypoglycemic crisis, the largest declines during the early pandemic period occurred among younger adults: ED visits for men and women aged 18-44 years were down, respectively, by 419 (–8%) and 775 (–16%), they reported based on data from the National Syndromic Surveillance Program.
“Decreases in ED visits for hyperglycemic crisis might be less striking because patient recognition of this crisis is typically augmented by home glucose monitoring and not reliant upon symptoms alone, as is the case for MI and stroke,” Ms. Lange and her associates noted.
Charting weekly visit numbers showed that the drop for all three conditions actually started the week before the emergency was declared and reached its nadir the week after (March 22) for MI and 2 weeks later (March 29) for stroke and hypoglycemic crisis.
Visits for hypoglycemic crisis have largely returned to normal since those low points, but MI and stroke visits “remain below prepandemic levels” despite gradual increases through April and May, they said.
It has been reported that “deaths not associated with confirmed or probable COVID-19 might have been directly or indirectly attributed to the pandemic. The striking decline in ED visits for acute life-threatening conditions might partially explain observed excess mortality not associated with COVID-19,” the investigators wrote.
ED visits for myocardial infarction, stroke, and hyperglycemic crisis dropped substantially in the 10 weeks after COVID-19 was declared a national emergency on March 13, according to the Centers for Disease Control and Prevention.
Compared with the 10-week period from Jan. 5 to March 14, ED visits were down by 23% for MI, 20% for stroke, and 10% for hyperglycemic crisis from March 15 to May 23, Samantha J. Lange, MPH, and associates at the CDC reported June 22 in the Morbidity and Mortality Weekly Report.
“A short-term decline of this magnitude … is biologically implausible for MI and stroke, especially for older adults, and unlikely for hyperglycemic crisis, and the finding suggests that patients with these conditions either could not access care or were delaying or avoiding seeking care during the early pandemic period,” they wrote.
The largest decreases in the actual number of visits for MI occurred among both men (down by 2,114, –24%) and women (down by 1,459, –25%) aged 65-74 years. For stroke, men aged 65-74 years had 1,406 (–19%) fewer visits to the ED and women 75-84 years had 1,642 (–23%) fewer visits, the CDC researchers said.
For hypoglycemic crisis, the largest declines during the early pandemic period occurred among younger adults: ED visits for men and women aged 18-44 years were down, respectively, by 419 (–8%) and 775 (–16%), they reported based on data from the National Syndromic Surveillance Program.
“Decreases in ED visits for hyperglycemic crisis might be less striking because patient recognition of this crisis is typically augmented by home glucose monitoring and not reliant upon symptoms alone, as is the case for MI and stroke,” Ms. Lange and her associates noted.
Charting weekly visit numbers showed that the drop for all three conditions actually started the week before the emergency was declared and reached its nadir the week after (March 22) for MI and 2 weeks later (March 29) for stroke and hypoglycemic crisis.
Visits for hypoglycemic crisis have largely returned to normal since those low points, but MI and stroke visits “remain below prepandemic levels” despite gradual increases through April and May, they said.
It has been reported that “deaths not associated with confirmed or probable COVID-19 might have been directly or indirectly attributed to the pandemic. The striking decline in ED visits for acute life-threatening conditions might partially explain observed excess mortality not associated with COVID-19,” the investigators wrote.
ED visits for myocardial infarction, stroke, and hyperglycemic crisis dropped substantially in the 10 weeks after COVID-19 was declared a national emergency on March 13, according to the Centers for Disease Control and Prevention.
Compared with the 10-week period from Jan. 5 to March 14, ED visits were down by 23% for MI, 20% for stroke, and 10% for hyperglycemic crisis from March 15 to May 23, Samantha J. Lange, MPH, and associates at the CDC reported June 22 in the Morbidity and Mortality Weekly Report.
“A short-term decline of this magnitude … is biologically implausible for MI and stroke, especially for older adults, and unlikely for hyperglycemic crisis, and the finding suggests that patients with these conditions either could not access care or were delaying or avoiding seeking care during the early pandemic period,” they wrote.
The largest decreases in the actual number of visits for MI occurred among both men (down by 2,114, –24%) and women (down by 1,459, –25%) aged 65-74 years. For stroke, men aged 65-74 years had 1,406 (–19%) fewer visits to the ED and women 75-84 years had 1,642 (–23%) fewer visits, the CDC researchers said.
For hypoglycemic crisis, the largest declines during the early pandemic period occurred among younger adults: ED visits for men and women aged 18-44 years were down, respectively, by 419 (–8%) and 775 (–16%), they reported based on data from the National Syndromic Surveillance Program.
“Decreases in ED visits for hyperglycemic crisis might be less striking because patient recognition of this crisis is typically augmented by home glucose monitoring and not reliant upon symptoms alone, as is the case for MI and stroke,” Ms. Lange and her associates noted.
Charting weekly visit numbers showed that the drop for all three conditions actually started the week before the emergency was declared and reached its nadir the week after (March 22) for MI and 2 weeks later (March 29) for stroke and hypoglycemic crisis.
Visits for hypoglycemic crisis have largely returned to normal since those low points, but MI and stroke visits “remain below prepandemic levels” despite gradual increases through April and May, they said.
It has been reported that “deaths not associated with confirmed or probable COVID-19 might have been directly or indirectly attributed to the pandemic. The striking decline in ED visits for acute life-threatening conditions might partially explain observed excess mortality not associated with COVID-19,” the investigators wrote.
FROM MMWR