User login
FDA approves first specific treatment for giant cell arteritis
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.
FDA approves pembrolizumab for advanced urothelial carcinoma
The Food and Drug Administration has granted regular approval to pembrolizumab (Keytruda) for patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
Accelerated approval was granted for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
Approval of pembrolizumab for the second-line indication was based on an improvement in overall survival and objective response rate (ORR) in the KEYNOTE-045 trial. Median overall survival was 10.3 months for patients randomized to receive pembrolizumab (n = 270) every 3 weeks, compared with 7.4 months for patients randomized to receive the investigator’s choice of a chemotherapy regimen (paclitaxel [n = 84], docetaxel [n = 84], or vinflunine [n = 87]) every 3 weeks (hazard ratio, 0.73; 95% confidence interval: 0.59-0.91, P =.004). ORR was 21% for pembrolizumab and 11% for chemotherapy (P = .002). All patients had locally advanced or metastatic urothelial carcinoma with disease progression on or after platinum-containing chemotherapy. No statistically significant difference in progression-free survival between the two arms was observed, the FDA said in a statement.
The accelerated approval for the first-line indication was based on an ORR of 28.6% (95% CI, 24-34) in KEYNOTE-052, a single-arm, open-label trial in 370 patients with locally advanced or metastatic urothelial carcinoma who were deemed not eligible for cisplatin-containing chemotherapy. The median response duration was not reached (range, 1.4+-17.8+ months).
The most common adverse reactions reported for those receiving pembrolizumab in either of the two trials included fatigue, musculoskeletal pain, pruritus, decreased appetite, nausea, diarrhea, constipation, and rash. Discontinuation of pembrolizumab occurred in 8% of patients in KEYNOTE-045 and in 11% in KEYNOTE-052. Serious adverse reactions occurred in approximately 40% of pembrolizumab-treated patients, the FDA said.
The recommended pembrolizumab dose and schedule for the treatment of urothelial carcinoma is 200 mg as an intravenous infusion over 30 minutes every 3 weeks. Full prescribing information is available the FDA website.
The Food and Drug Administration has granted regular approval to pembrolizumab (Keytruda) for patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
Accelerated approval was granted for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
Approval of pembrolizumab for the second-line indication was based on an improvement in overall survival and objective response rate (ORR) in the KEYNOTE-045 trial. Median overall survival was 10.3 months for patients randomized to receive pembrolizumab (n = 270) every 3 weeks, compared with 7.4 months for patients randomized to receive the investigator’s choice of a chemotherapy regimen (paclitaxel [n = 84], docetaxel [n = 84], or vinflunine [n = 87]) every 3 weeks (hazard ratio, 0.73; 95% confidence interval: 0.59-0.91, P =.004). ORR was 21% for pembrolizumab and 11% for chemotherapy (P = .002). All patients had locally advanced or metastatic urothelial carcinoma with disease progression on or after platinum-containing chemotherapy. No statistically significant difference in progression-free survival between the two arms was observed, the FDA said in a statement.
The accelerated approval for the first-line indication was based on an ORR of 28.6% (95% CI, 24-34) in KEYNOTE-052, a single-arm, open-label trial in 370 patients with locally advanced or metastatic urothelial carcinoma who were deemed not eligible for cisplatin-containing chemotherapy. The median response duration was not reached (range, 1.4+-17.8+ months).
The most common adverse reactions reported for those receiving pembrolizumab in either of the two trials included fatigue, musculoskeletal pain, pruritus, decreased appetite, nausea, diarrhea, constipation, and rash. Discontinuation of pembrolizumab occurred in 8% of patients in KEYNOTE-045 and in 11% in KEYNOTE-052. Serious adverse reactions occurred in approximately 40% of pembrolizumab-treated patients, the FDA said.
The recommended pembrolizumab dose and schedule for the treatment of urothelial carcinoma is 200 mg as an intravenous infusion over 30 minutes every 3 weeks. Full prescribing information is available the FDA website.
The Food and Drug Administration has granted regular approval to pembrolizumab (Keytruda) for patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
Accelerated approval was granted for patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin-containing chemotherapy.
Approval of pembrolizumab for the second-line indication was based on an improvement in overall survival and objective response rate (ORR) in the KEYNOTE-045 trial. Median overall survival was 10.3 months for patients randomized to receive pembrolizumab (n = 270) every 3 weeks, compared with 7.4 months for patients randomized to receive the investigator’s choice of a chemotherapy regimen (paclitaxel [n = 84], docetaxel [n = 84], or vinflunine [n = 87]) every 3 weeks (hazard ratio, 0.73; 95% confidence interval: 0.59-0.91, P =.004). ORR was 21% for pembrolizumab and 11% for chemotherapy (P = .002). All patients had locally advanced or metastatic urothelial carcinoma with disease progression on or after platinum-containing chemotherapy. No statistically significant difference in progression-free survival between the two arms was observed, the FDA said in a statement.
The accelerated approval for the first-line indication was based on an ORR of 28.6% (95% CI, 24-34) in KEYNOTE-052, a single-arm, open-label trial in 370 patients with locally advanced or metastatic urothelial carcinoma who were deemed not eligible for cisplatin-containing chemotherapy. The median response duration was not reached (range, 1.4+-17.8+ months).
The most common adverse reactions reported for those receiving pembrolizumab in either of the two trials included fatigue, musculoskeletal pain, pruritus, decreased appetite, nausea, diarrhea, constipation, and rash. Discontinuation of pembrolizumab occurred in 8% of patients in KEYNOTE-045 and in 11% in KEYNOTE-052. Serious adverse reactions occurred in approximately 40% of pembrolizumab-treated patients, the FDA said.
The recommended pembrolizumab dose and schedule for the treatment of urothelial carcinoma is 200 mg as an intravenous infusion over 30 minutes every 3 weeks. Full prescribing information is available the FDA website.
FDA: Some blood lead tests have reported falsely low levels since 2014
The Food and Drug Administration is recommending repeat lead testing of young children and at-risk women who were tested via a venous blood sample. The recommendation was issued May 17 after the agency discovered a probable 3-year history of inaccurate tests by the nation’s largest lead test distributor.
The tests should be repeated with a capillary sample in children younger than 6 years of age as of May 17, and all pregnant or breastfeeding women who had a lead level of 10 mcg/dL or lower in a venous blood draw tested with any system made by Magellan Diagnostics, FDA representatives said during at a press briefing.
“We do have evidence of a problem with falsely lower lead reading with venous blood,” said Dr. Shuren, director of the FDA Center for Devices and Radiological Health. “Based on the information we have now, we don’t know how often we see this inaccuracy in samples, or how much lower it is. There is a wide variation and a small amount of data. We need to do further testing to see how big the problem is and the root cause. However, we do have enough data to be confident that we don’t have the problem in capillary blood.”
The warning includes all tests run on four of Magellan Diagnostics’ lead testing systems: LeadCare; LeadCare II; LeadCare Plus; and LeadCare Ultra. All LeadCare systems can be used with blood from a finger or heel stick, including the LeadCare II system – one found in many doctors’ offices and clinics. In addition, some laboratories offer other methods of lead testing, which are not now believed to be affected.
At this point, Magellan isn’t required to pay for any retesting. Tim Hill, acting director of the Center for Medicaid and CHIP Services, who was also on the call, confirmed that retesting will be covered for Medicaid and CHIP recipients. Patients with private insurance will have to contact their insurance companies to ascertain coverage, he said.
“Our first priority is to be sure folks get retested through our programs,” Mr. Hill said during the briefing. “We don’t want reimbursement to hold up the retesting. My understanding is that talks with Magellan with regard to their liability are ongoing.”
Regulators discovered the extent of the problem in March, after Magellan submitted a 510(k) premarket notification for a new iteration of its point-of-care test, FDA spokesperson Tara Goodin said in an interview. The new product was based on a kit approved in 2013, so all tests run on venous blood since then are in question.
During the data review, FDA discovered that customers began complaining to Magellan about inaccurate results on venous blood in 2014. Magellan issued three customer notifications letters (primarily to laboratories) alerting them to testing inaccuracies and recommending mitigations designed to address them. These customer notifications were issued on Nov. 24, 2014; Nov. 4, 2016; and April 28, 2017. In these, the company indicated that about 2.5% of patients whose tests were below the level of medical concern could actually have enough lead to warrant intervention. Magellan suggested that the problem could be solved by holding all samples for 24 hours before mixing them with the reagent – a step the company said would reduce risk of a misread to zero.
Regulators disagreed, Ms. Goodin said.
“Based on available information, the FDA believes that Magellan should have determined that the risk of an inaccurate test result and the number of people that could be adversely affected was much higher than they estimated and that their mitigation might not be adequate to address the increased risk. Instead, the company submitted a malfunction report in 2015 related to an observed increased frequency of falsely low test results in the LeadCare Ultra system that the firm indicated it first identified through the August 2014 complaint.”
In the malfunction report, the company characterized this issue as a Class III recall, which the FDA defines as a situation in which use of or exposure to a violative product is not likely to cause adverse health consequences.
The scope of the problem became apparent only after Magellan submitted its 510(k) paperwork in March, Ms. Goodin said.
“After reviewing initial data available from Magellan on these inaccuracies and their mitigations, the FDA was unable to identify the root cause of the inaccuracies, the frequency and extent of the inaccuracies, or to confirm that the mitigations are effective. While the FDA’s investigation is in its early stages, we did not want to delay warning health care professionals and laboratories about the risk of testing inaccuracies and encouraging parents and at-risk adults to follow the CDC’s recommendations.”
As soon as FDA identified the issue as a potential public health risk, it began working with the Centers for Medicare & Medicaid Services to issue recommendations for laboratories, health care professionals, and at-risk individuals.
“The FDA prioritized communicating to the public about this issue but is also aggressively investigating this issue to determine the cause of the inaccurate results and will provide updates as more is learned,” Ms. Goodin said. “This includes reviewing data provided by the company, requesting additional information from Magellan regarding the issue, and inspecting the company’s facility. The FDA has also requested an independent analysis of the test. We are aggressively investigating this issue and have already sent staff to inspect Magellan’s facility.”
Phone calls to Magellan for clarification on its mitigation procedures, the potential impact on customers, and the history of the LeadCare series’ approvals were not returned at press time, and the company had no prepared statement. A safety communication was posted to its website.
Since 2014, Magellan has run 8 million blood lead tests. Based on the company’s 2.5% estimate of misreads, 200,000 patients tested with the kit could have dangerously high blood lead levels. Currently, the FDA has no official estimate of how many tests were run on venous blood, how many of those returned inaccurate results, or even what caused the tests to read out with falsely low levels, Dr. Shuren said.
“We are investigating the cause, however when [Magellan’s prior test kits] came on the market, there was data supporting their accuracy. The root cause of this, we don’t know. It may not be specific to the test; it may have to do with the tubes, the reactions with chemicals, the way it’s processed. We are looking into all [of] these.”
On Twitter @Alz_gal
The Food and Drug Administration is recommending repeat lead testing of young children and at-risk women who were tested via a venous blood sample. The recommendation was issued May 17 after the agency discovered a probable 3-year history of inaccurate tests by the nation’s largest lead test distributor.
The tests should be repeated with a capillary sample in children younger than 6 years of age as of May 17, and all pregnant or breastfeeding women who had a lead level of 10 mcg/dL or lower in a venous blood draw tested with any system made by Magellan Diagnostics, FDA representatives said during at a press briefing.
“We do have evidence of a problem with falsely lower lead reading with venous blood,” said Dr. Shuren, director of the FDA Center for Devices and Radiological Health. “Based on the information we have now, we don’t know how often we see this inaccuracy in samples, or how much lower it is. There is a wide variation and a small amount of data. We need to do further testing to see how big the problem is and the root cause. However, we do have enough data to be confident that we don’t have the problem in capillary blood.”
The warning includes all tests run on four of Magellan Diagnostics’ lead testing systems: LeadCare; LeadCare II; LeadCare Plus; and LeadCare Ultra. All LeadCare systems can be used with blood from a finger or heel stick, including the LeadCare II system – one found in many doctors’ offices and clinics. In addition, some laboratories offer other methods of lead testing, which are not now believed to be affected.
At this point, Magellan isn’t required to pay for any retesting. Tim Hill, acting director of the Center for Medicaid and CHIP Services, who was also on the call, confirmed that retesting will be covered for Medicaid and CHIP recipients. Patients with private insurance will have to contact their insurance companies to ascertain coverage, he said.
“Our first priority is to be sure folks get retested through our programs,” Mr. Hill said during the briefing. “We don’t want reimbursement to hold up the retesting. My understanding is that talks with Magellan with regard to their liability are ongoing.”
Regulators discovered the extent of the problem in March, after Magellan submitted a 510(k) premarket notification for a new iteration of its point-of-care test, FDA spokesperson Tara Goodin said in an interview. The new product was based on a kit approved in 2013, so all tests run on venous blood since then are in question.
During the data review, FDA discovered that customers began complaining to Magellan about inaccurate results on venous blood in 2014. Magellan issued three customer notifications letters (primarily to laboratories) alerting them to testing inaccuracies and recommending mitigations designed to address them. These customer notifications were issued on Nov. 24, 2014; Nov. 4, 2016; and April 28, 2017. In these, the company indicated that about 2.5% of patients whose tests were below the level of medical concern could actually have enough lead to warrant intervention. Magellan suggested that the problem could be solved by holding all samples for 24 hours before mixing them with the reagent – a step the company said would reduce risk of a misread to zero.
Regulators disagreed, Ms. Goodin said.
“Based on available information, the FDA believes that Magellan should have determined that the risk of an inaccurate test result and the number of people that could be adversely affected was much higher than they estimated and that their mitigation might not be adequate to address the increased risk. Instead, the company submitted a malfunction report in 2015 related to an observed increased frequency of falsely low test results in the LeadCare Ultra system that the firm indicated it first identified through the August 2014 complaint.”
In the malfunction report, the company characterized this issue as a Class III recall, which the FDA defines as a situation in which use of or exposure to a violative product is not likely to cause adverse health consequences.
The scope of the problem became apparent only after Magellan submitted its 510(k) paperwork in March, Ms. Goodin said.
“After reviewing initial data available from Magellan on these inaccuracies and their mitigations, the FDA was unable to identify the root cause of the inaccuracies, the frequency and extent of the inaccuracies, or to confirm that the mitigations are effective. While the FDA’s investigation is in its early stages, we did not want to delay warning health care professionals and laboratories about the risk of testing inaccuracies and encouraging parents and at-risk adults to follow the CDC’s recommendations.”
As soon as FDA identified the issue as a potential public health risk, it began working with the Centers for Medicare & Medicaid Services to issue recommendations for laboratories, health care professionals, and at-risk individuals.
“The FDA prioritized communicating to the public about this issue but is also aggressively investigating this issue to determine the cause of the inaccurate results and will provide updates as more is learned,” Ms. Goodin said. “This includes reviewing data provided by the company, requesting additional information from Magellan regarding the issue, and inspecting the company’s facility. The FDA has also requested an independent analysis of the test. We are aggressively investigating this issue and have already sent staff to inspect Magellan’s facility.”
Phone calls to Magellan for clarification on its mitigation procedures, the potential impact on customers, and the history of the LeadCare series’ approvals were not returned at press time, and the company had no prepared statement. A safety communication was posted to its website.
Since 2014, Magellan has run 8 million blood lead tests. Based on the company’s 2.5% estimate of misreads, 200,000 patients tested with the kit could have dangerously high blood lead levels. Currently, the FDA has no official estimate of how many tests were run on venous blood, how many of those returned inaccurate results, or even what caused the tests to read out with falsely low levels, Dr. Shuren said.
“We are investigating the cause, however when [Magellan’s prior test kits] came on the market, there was data supporting their accuracy. The root cause of this, we don’t know. It may not be specific to the test; it may have to do with the tubes, the reactions with chemicals, the way it’s processed. We are looking into all [of] these.”
On Twitter @Alz_gal
The Food and Drug Administration is recommending repeat lead testing of young children and at-risk women who were tested via a venous blood sample. The recommendation was issued May 17 after the agency discovered a probable 3-year history of inaccurate tests by the nation’s largest lead test distributor.
The tests should be repeated with a capillary sample in children younger than 6 years of age as of May 17, and all pregnant or breastfeeding women who had a lead level of 10 mcg/dL or lower in a venous blood draw tested with any system made by Magellan Diagnostics, FDA representatives said during at a press briefing.
“We do have evidence of a problem with falsely lower lead reading with venous blood,” said Dr. Shuren, director of the FDA Center for Devices and Radiological Health. “Based on the information we have now, we don’t know how often we see this inaccuracy in samples, or how much lower it is. There is a wide variation and a small amount of data. We need to do further testing to see how big the problem is and the root cause. However, we do have enough data to be confident that we don’t have the problem in capillary blood.”
The warning includes all tests run on four of Magellan Diagnostics’ lead testing systems: LeadCare; LeadCare II; LeadCare Plus; and LeadCare Ultra. All LeadCare systems can be used with blood from a finger or heel stick, including the LeadCare II system – one found in many doctors’ offices and clinics. In addition, some laboratories offer other methods of lead testing, which are not now believed to be affected.
At this point, Magellan isn’t required to pay for any retesting. Tim Hill, acting director of the Center for Medicaid and CHIP Services, who was also on the call, confirmed that retesting will be covered for Medicaid and CHIP recipients. Patients with private insurance will have to contact their insurance companies to ascertain coverage, he said.
“Our first priority is to be sure folks get retested through our programs,” Mr. Hill said during the briefing. “We don’t want reimbursement to hold up the retesting. My understanding is that talks with Magellan with regard to their liability are ongoing.”
Regulators discovered the extent of the problem in March, after Magellan submitted a 510(k) premarket notification for a new iteration of its point-of-care test, FDA spokesperson Tara Goodin said in an interview. The new product was based on a kit approved in 2013, so all tests run on venous blood since then are in question.
During the data review, FDA discovered that customers began complaining to Magellan about inaccurate results on venous blood in 2014. Magellan issued three customer notifications letters (primarily to laboratories) alerting them to testing inaccuracies and recommending mitigations designed to address them. These customer notifications were issued on Nov. 24, 2014; Nov. 4, 2016; and April 28, 2017. In these, the company indicated that about 2.5% of patients whose tests were below the level of medical concern could actually have enough lead to warrant intervention. Magellan suggested that the problem could be solved by holding all samples for 24 hours before mixing them with the reagent – a step the company said would reduce risk of a misread to zero.
Regulators disagreed, Ms. Goodin said.
“Based on available information, the FDA believes that Magellan should have determined that the risk of an inaccurate test result and the number of people that could be adversely affected was much higher than they estimated and that their mitigation might not be adequate to address the increased risk. Instead, the company submitted a malfunction report in 2015 related to an observed increased frequency of falsely low test results in the LeadCare Ultra system that the firm indicated it first identified through the August 2014 complaint.”
In the malfunction report, the company characterized this issue as a Class III recall, which the FDA defines as a situation in which use of or exposure to a violative product is not likely to cause adverse health consequences.
The scope of the problem became apparent only after Magellan submitted its 510(k) paperwork in March, Ms. Goodin said.
“After reviewing initial data available from Magellan on these inaccuracies and their mitigations, the FDA was unable to identify the root cause of the inaccuracies, the frequency and extent of the inaccuracies, or to confirm that the mitigations are effective. While the FDA’s investigation is in its early stages, we did not want to delay warning health care professionals and laboratories about the risk of testing inaccuracies and encouraging parents and at-risk adults to follow the CDC’s recommendations.”
As soon as FDA identified the issue as a potential public health risk, it began working with the Centers for Medicare & Medicaid Services to issue recommendations for laboratories, health care professionals, and at-risk individuals.
“The FDA prioritized communicating to the public about this issue but is also aggressively investigating this issue to determine the cause of the inaccurate results and will provide updates as more is learned,” Ms. Goodin said. “This includes reviewing data provided by the company, requesting additional information from Magellan regarding the issue, and inspecting the company’s facility. The FDA has also requested an independent analysis of the test. We are aggressively investigating this issue and have already sent staff to inspect Magellan’s facility.”
Phone calls to Magellan for clarification on its mitigation procedures, the potential impact on customers, and the history of the LeadCare series’ approvals were not returned at press time, and the company had no prepared statement. A safety communication was posted to its website.
Since 2014, Magellan has run 8 million blood lead tests. Based on the company’s 2.5% estimate of misreads, 200,000 patients tested with the kit could have dangerously high blood lead levels. Currently, the FDA has no official estimate of how many tests were run on venous blood, how many of those returned inaccurate results, or even what caused the tests to read out with falsely low levels, Dr. Shuren said.
“We are investigating the cause, however when [Magellan’s prior test kits] came on the market, there was data supporting their accuracy. The root cause of this, we don’t know. It may not be specific to the test; it may have to do with the tubes, the reactions with chemicals, the way it’s processed. We are looking into all [of] these.”
On Twitter @Alz_gal
Few states fully back HCV prevention, treatment
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”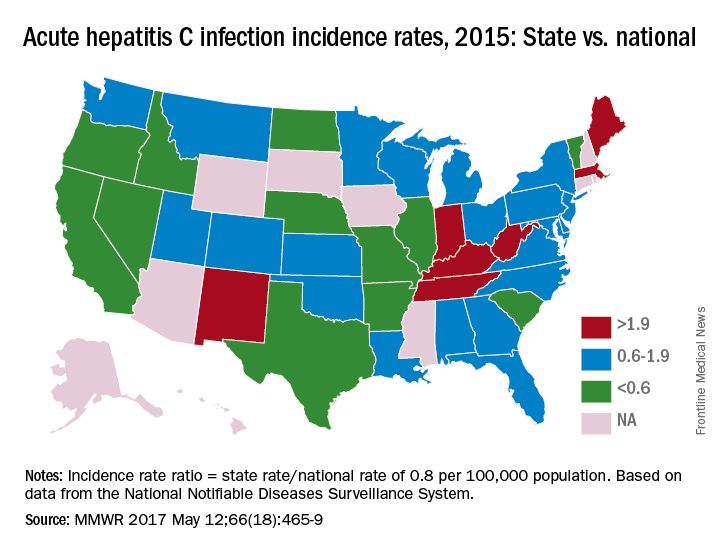
The “most comprehensive” laws on prevention through clean needle access as of 2016 were found in Maine, Nevada, and Utah, with laws in 12 other states categorized as “more comprehensive” and 18 states falling into the “least comprehensive” category. On the Medicaid side of the equation, 16 states had permissive policies that did not require sobriety or required only screening and counseling before treatment, 24 states had restrictive policies that requited sobriety, and 10 states had no policy available, the report showed (MMWR. 2017 May 12:66[18]:465-9).
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”
The “most comprehensive” laws on prevention through clean needle access as of 2016 were found in Maine, Nevada, and Utah, with laws in 12 other states categorized as “more comprehensive” and 18 states falling into the “least comprehensive” category. On the Medicaid side of the equation, 16 states had permissive policies that did not require sobriety or required only screening and counseling before treatment, 24 states had restrictive policies that requited sobriety, and 10 states had no policy available, the report showed (MMWR. 2017 May 12:66[18]:465-9).
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”
The “most comprehensive” laws on prevention through clean needle access as of 2016 were found in Maine, Nevada, and Utah, with laws in 12 other states categorized as “more comprehensive” and 18 states falling into the “least comprehensive” category. On the Medicaid side of the equation, 16 states had permissive policies that did not require sobriety or required only screening and counseling before treatment, 24 states had restrictive policies that requited sobriety, and 10 states had no policy available, the report showed (MMWR. 2017 May 12:66[18]:465-9).
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
FROM MMWR
FDA approves pembrolizumab for first-line advanced NSCLC
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor pembrolizumab in combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic nonsquamous non–small cell lung cancer (NSCLC).
The immunotherapy pembrolizumab was approved as a second-line treatment for metastatic NSCLC in 2015.
First-line approval was based on an improved overall response rate (ORR) and progression-free survival (PFS) in a cohort of 123 patients within an open-label, multicohort study (KEYNOTE-21). Enrollees in cohort G1 had locally advanced or metastatic NSCLC and no prior systemic treatment for metastatic disease. They were randomized to receive either pembrolizumab, in combination with pemetrexed and carboplatin (PC) for four cycles followed by pembrolizumab for a maximum of 24 months (n = 60) or PC alone (n = 63). Randomization was stratified by PD-L1 tumor expression (tumor proportion score [TPS] less than 1% vs. TPS greater than or equal to 1%).![]()
The hazard ratio for PFS was 0.53 (95% CI: 0.31, 0.91, P = .0205). The median PFS was 13.0 months for the pembrolizumab plus PC arm and 8.9 months for the PC-alone arm. In the TPS less than 1% subgroup, the ORR was 57% and 13% in the pembrolizumab-plus-PC and in the PC-alone arms, respectively. In the TPS greater-than-or-equal-to-1% subgroup, the ORR was 54% in the pembrolizumab-plus-PC arm and 38% in the pembrolizumab-plus-PC arm, the FDA said.
There were serious adverse events in 41% of the patients in the pembrolizumab-plus-PC arm compared with 28% in the PC-alone arm. Pembrolizumab was discontinued for adverse reactions in 10% of patients, most commonly due to acute kidney injury. The most common grade 3-4 adverse reactions were fatigue, dyspnea, nausea, vomiting, diarrhea, and rash.
The FDA cautioned that immune-mediated adverse reactions can occur with pembrolizumab including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis. Based on the severity of the adverse reaction, pembrolizumab should be withheld or discontinued and corticosteroids administered when appropriate. The recommended dose and schedule for NSCLC is 200 mg as an intravenous infusion every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor pembrolizumab in combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic nonsquamous non–small cell lung cancer (NSCLC).
The immunotherapy pembrolizumab was approved as a second-line treatment for metastatic NSCLC in 2015.
First-line approval was based on an improved overall response rate (ORR) and progression-free survival (PFS) in a cohort of 123 patients within an open-label, multicohort study (KEYNOTE-21). Enrollees in cohort G1 had locally advanced or metastatic NSCLC and no prior systemic treatment for metastatic disease. They were randomized to receive either pembrolizumab, in combination with pemetrexed and carboplatin (PC) for four cycles followed by pembrolizumab for a maximum of 24 months (n = 60) or PC alone (n = 63). Randomization was stratified by PD-L1 tumor expression (tumor proportion score [TPS] less than 1% vs. TPS greater than or equal to 1%).![]()
The hazard ratio for PFS was 0.53 (95% CI: 0.31, 0.91, P = .0205). The median PFS was 13.0 months for the pembrolizumab plus PC arm and 8.9 months for the PC-alone arm. In the TPS less than 1% subgroup, the ORR was 57% and 13% in the pembrolizumab-plus-PC and in the PC-alone arms, respectively. In the TPS greater-than-or-equal-to-1% subgroup, the ORR was 54% in the pembrolizumab-plus-PC arm and 38% in the pembrolizumab-plus-PC arm, the FDA said.
There were serious adverse events in 41% of the patients in the pembrolizumab-plus-PC arm compared with 28% in the PC-alone arm. Pembrolizumab was discontinued for adverse reactions in 10% of patients, most commonly due to acute kidney injury. The most common grade 3-4 adverse reactions were fatigue, dyspnea, nausea, vomiting, diarrhea, and rash.
The FDA cautioned that immune-mediated adverse reactions can occur with pembrolizumab including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis. Based on the severity of the adverse reaction, pembrolizumab should be withheld or discontinued and corticosteroids administered when appropriate. The recommended dose and schedule for NSCLC is 200 mg as an intravenous infusion every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor pembrolizumab in combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic nonsquamous non–small cell lung cancer (NSCLC).
The immunotherapy pembrolizumab was approved as a second-line treatment for metastatic NSCLC in 2015.
First-line approval was based on an improved overall response rate (ORR) and progression-free survival (PFS) in a cohort of 123 patients within an open-label, multicohort study (KEYNOTE-21). Enrollees in cohort G1 had locally advanced or metastatic NSCLC and no prior systemic treatment for metastatic disease. They were randomized to receive either pembrolizumab, in combination with pemetrexed and carboplatin (PC) for four cycles followed by pembrolizumab for a maximum of 24 months (n = 60) or PC alone (n = 63). Randomization was stratified by PD-L1 tumor expression (tumor proportion score [TPS] less than 1% vs. TPS greater than or equal to 1%).![]()
The hazard ratio for PFS was 0.53 (95% CI: 0.31, 0.91, P = .0205). The median PFS was 13.0 months for the pembrolizumab plus PC arm and 8.9 months for the PC-alone arm. In the TPS less than 1% subgroup, the ORR was 57% and 13% in the pembrolizumab-plus-PC and in the PC-alone arms, respectively. In the TPS greater-than-or-equal-to-1% subgroup, the ORR was 54% in the pembrolizumab-plus-PC arm and 38% in the pembrolizumab-plus-PC arm, the FDA said.
There were serious adverse events in 41% of the patients in the pembrolizumab-plus-PC arm compared with 28% in the PC-alone arm. Pembrolizumab was discontinued for adverse reactions in 10% of patients, most commonly due to acute kidney injury. The most common grade 3-4 adverse reactions were fatigue, dyspnea, nausea, vomiting, diarrhea, and rash.
The FDA cautioned that immune-mediated adverse reactions can occur with pembrolizumab including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis. Based on the severity of the adverse reaction, pembrolizumab should be withheld or discontinued and corticosteroids administered when appropriate. The recommended dose and schedule for NSCLC is 200 mg as an intravenous infusion every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression.
Pembrolizumab is marketed as Keytruda by Merck.
FDA approves avelumab for advanced urothelial carcinoma
The Food and Drug Administration has granted accelerated approval to avelumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
Approval was based on a confirmed overall response rate (ORR) of 16.1% in 26 patients who had been followed for at least 6 months in a single-arm, multicenter study that enrolled 242 patients with locally advanced or metastatic urothelial carcinoma whose disease progressed following platinum-containing neoadjuvant or adjuvant chemotherapy. The ORR was 13.3% among 30 patients who had been followed for at least 13 weeks. Median time to response was 2.0 months (range 1.3-11.0). The median response duration had not been reached in patients followed for at least 13 weeks or at least 6 months, but ranged from 1.4+ to 17.4+ months in both groups, the FDA said in a statement.![]()
Serious adverse reactions, including urinary tract infection/urosepsis, abdominal pain, musculoskeletal pain, creatinine increased/renal failure, dehydration, hematuria/urinary tract hemorrhage, intestinal obstruction/small intestinal obstruction, and pyrexia, were reported in 41% of patients, causing death in 6% of patients. The most common adverse reactions were infusion-related reaction, musculoskeletal pain, nausea, decreased appetite, and urinary tract infection.
The FDA recommended an intravenous infusion of 10 mg/kg over 60 minutes every 2 weeks, and premedication with an antihistamine and acetaminophen prior to the first four infusions of avelumab.
Full prescribing information is available here.
The drug is being marketed as Bavencio by EMD Serono.
The Food and Drug Administration has granted accelerated approval to avelumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
Approval was based on a confirmed overall response rate (ORR) of 16.1% in 26 patients who had been followed for at least 6 months in a single-arm, multicenter study that enrolled 242 patients with locally advanced or metastatic urothelial carcinoma whose disease progressed following platinum-containing neoadjuvant or adjuvant chemotherapy. The ORR was 13.3% among 30 patients who had been followed for at least 13 weeks. Median time to response was 2.0 months (range 1.3-11.0). The median response duration had not been reached in patients followed for at least 13 weeks or at least 6 months, but ranged from 1.4+ to 17.4+ months in both groups, the FDA said in a statement.![]()
Serious adverse reactions, including urinary tract infection/urosepsis, abdominal pain, musculoskeletal pain, creatinine increased/renal failure, dehydration, hematuria/urinary tract hemorrhage, intestinal obstruction/small intestinal obstruction, and pyrexia, were reported in 41% of patients, causing death in 6% of patients. The most common adverse reactions were infusion-related reaction, musculoskeletal pain, nausea, decreased appetite, and urinary tract infection.
The FDA recommended an intravenous infusion of 10 mg/kg over 60 minutes every 2 weeks, and premedication with an antihistamine and acetaminophen prior to the first four infusions of avelumab.
Full prescribing information is available here.
The drug is being marketed as Bavencio by EMD Serono.
The Food and Drug Administration has granted accelerated approval to avelumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant platinum-containing chemotherapy.
Approval was based on a confirmed overall response rate (ORR) of 16.1% in 26 patients who had been followed for at least 6 months in a single-arm, multicenter study that enrolled 242 patients with locally advanced or metastatic urothelial carcinoma whose disease progressed following platinum-containing neoadjuvant or adjuvant chemotherapy. The ORR was 13.3% among 30 patients who had been followed for at least 13 weeks. Median time to response was 2.0 months (range 1.3-11.0). The median response duration had not been reached in patients followed for at least 13 weeks or at least 6 months, but ranged from 1.4+ to 17.4+ months in both groups, the FDA said in a statement.![]()
Serious adverse reactions, including urinary tract infection/urosepsis, abdominal pain, musculoskeletal pain, creatinine increased/renal failure, dehydration, hematuria/urinary tract hemorrhage, intestinal obstruction/small intestinal obstruction, and pyrexia, were reported in 41% of patients, causing death in 6% of patients. The most common adverse reactions were infusion-related reaction, musculoskeletal pain, nausea, decreased appetite, and urinary tract infection.
The FDA recommended an intravenous infusion of 10 mg/kg over 60 minutes every 2 weeks, and premedication with an antihistamine and acetaminophen prior to the first four infusions of avelumab.
Full prescribing information is available here.
The drug is being marketed as Bavencio by EMD Serono.
FDA approves first new drug for ALS in decades
The Food and Drug Administration approved the antioxidant drug edaravone on May 5 for the treatment of amyotrophic lateral sclerosis, making it only the second drug ever to be approved by the agency for the motor neuron disease.
The FDA granted approval for edaravone, to be marketed by Mitsubishi Tanabe Pharma America under the brand name Radicava, through its orphan drug pathway, which is meant for drugs used to treat rare diseases or conditions. The Centers for Disease Control and Prevention estimates that amyotrophic lateral sclerosis (ALS) affects 12,000-15,000 Americans.
Mitsubishi Tanabe Pharma America demonstrated the efficacy of edaravone in a 6-month trial of 137 Japanese ALS patients. At 24 weeks, individuals who received edaravone had less decline on a clinical assessment of daily functioning, the ALS Functional Rating Scale-Revised (ALSFRS-R), compared with those who received a placebo. The difference in decline between the two groups was 33%, or a total of 2.49 points, on the ALSFRS-R. Most of the patients in the study also received the only other drug approved for ALS, riluzole (Rilutek).
Edaravone is thought to confer neuroprotection in part through its free radical–scavenging activity.
The adverse events most often reported by clinical trial participants who took edaravone included bruising and gait disturbance. The FDA also warned that edaravone is associated with hives, swelling, or shortness of breath, and allergic reactions to an ingredient in the drug, sodium bisulfite, which may cause anaphylactic symptoms that can be life-threatening in people with sulfite sensitivity.
The drug is administered via intravenous infusion with an initial treatment cycle of daily dosing for 14 days, followed by a 14-day drug-free period. Subsequent treatment cycles consist of dosing on 10 of 14 days, followed by 14 days drug-free.
Mitsubishi Tanabe Pharma America said in a statement that it has created a patient access program called Searchlight Support for people with ALS who are prescribed the drug. The program provides personal case management, reimbursement support, and 24/7 clinical support.
In 2015, edaravone was approved for use as a treatment for ALS in Japan and South Korea.
The Food and Drug Administration approved the antioxidant drug edaravone on May 5 for the treatment of amyotrophic lateral sclerosis, making it only the second drug ever to be approved by the agency for the motor neuron disease.
The FDA granted approval for edaravone, to be marketed by Mitsubishi Tanabe Pharma America under the brand name Radicava, through its orphan drug pathway, which is meant for drugs used to treat rare diseases or conditions. The Centers for Disease Control and Prevention estimates that amyotrophic lateral sclerosis (ALS) affects 12,000-15,000 Americans.
Mitsubishi Tanabe Pharma America demonstrated the efficacy of edaravone in a 6-month trial of 137 Japanese ALS patients. At 24 weeks, individuals who received edaravone had less decline on a clinical assessment of daily functioning, the ALS Functional Rating Scale-Revised (ALSFRS-R), compared with those who received a placebo. The difference in decline between the two groups was 33%, or a total of 2.49 points, on the ALSFRS-R. Most of the patients in the study also received the only other drug approved for ALS, riluzole (Rilutek).
Edaravone is thought to confer neuroprotection in part through its free radical–scavenging activity.
The adverse events most often reported by clinical trial participants who took edaravone included bruising and gait disturbance. The FDA also warned that edaravone is associated with hives, swelling, or shortness of breath, and allergic reactions to an ingredient in the drug, sodium bisulfite, which may cause anaphylactic symptoms that can be life-threatening in people with sulfite sensitivity.
The drug is administered via intravenous infusion with an initial treatment cycle of daily dosing for 14 days, followed by a 14-day drug-free period. Subsequent treatment cycles consist of dosing on 10 of 14 days, followed by 14 days drug-free.
Mitsubishi Tanabe Pharma America said in a statement that it has created a patient access program called Searchlight Support for people with ALS who are prescribed the drug. The program provides personal case management, reimbursement support, and 24/7 clinical support.
In 2015, edaravone was approved for use as a treatment for ALS in Japan and South Korea.
The Food and Drug Administration approved the antioxidant drug edaravone on May 5 for the treatment of amyotrophic lateral sclerosis, making it only the second drug ever to be approved by the agency for the motor neuron disease.
The FDA granted approval for edaravone, to be marketed by Mitsubishi Tanabe Pharma America under the brand name Radicava, through its orphan drug pathway, which is meant for drugs used to treat rare diseases or conditions. The Centers for Disease Control and Prevention estimates that amyotrophic lateral sclerosis (ALS) affects 12,000-15,000 Americans.
Mitsubishi Tanabe Pharma America demonstrated the efficacy of edaravone in a 6-month trial of 137 Japanese ALS patients. At 24 weeks, individuals who received edaravone had less decline on a clinical assessment of daily functioning, the ALS Functional Rating Scale-Revised (ALSFRS-R), compared with those who received a placebo. The difference in decline between the two groups was 33%, or a total of 2.49 points, on the ALSFRS-R. Most of the patients in the study also received the only other drug approved for ALS, riluzole (Rilutek).
Edaravone is thought to confer neuroprotection in part through its free radical–scavenging activity.
The adverse events most often reported by clinical trial participants who took edaravone included bruising and gait disturbance. The FDA also warned that edaravone is associated with hives, swelling, or shortness of breath, and allergic reactions to an ingredient in the drug, sodium bisulfite, which may cause anaphylactic symptoms that can be life-threatening in people with sulfite sensitivity.
The drug is administered via intravenous infusion with an initial treatment cycle of daily dosing for 14 days, followed by a 14-day drug-free period. Subsequent treatment cycles consist of dosing on 10 of 14 days, followed by 14 days drug-free.
Mitsubishi Tanabe Pharma America said in a statement that it has created a patient access program called Searchlight Support for people with ALS who are prescribed the drug. The program provides personal case management, reimbursement support, and 24/7 clinical support.
In 2015, edaravone was approved for use as a treatment for ALS in Japan and South Korea.
FDA approves abaloparatide for postmenopausal osteoporosis
The Food and Drug Administration has approved abaloparatide (Tymlos) for postmenopausal women with osteoporosis at high risk for fracture.
Abaloparatide was approved based on 18-month results from the ACTIVE trial and 6-month results from the ACTIVExtend trial. Patients in the ACTIVE trial showed an relative risk reduction of 86% for new vertebral fractures and 43% for nonvertebral fractures, compared with placebo, according to a statement from manufacturer Radius Health. Results were similar regardless of age, years since menopause, presence or absence of prior fracture (vertebral or nonvertebral), and bone mineral density at baseline.
Find the full statement on the Radius Health website.
The Food and Drug Administration has approved abaloparatide (Tymlos) for postmenopausal women with osteoporosis at high risk for fracture.
Abaloparatide was approved based on 18-month results from the ACTIVE trial and 6-month results from the ACTIVExtend trial. Patients in the ACTIVE trial showed an relative risk reduction of 86% for new vertebral fractures and 43% for nonvertebral fractures, compared with placebo, according to a statement from manufacturer Radius Health. Results were similar regardless of age, years since menopause, presence or absence of prior fracture (vertebral or nonvertebral), and bone mineral density at baseline.
Find the full statement on the Radius Health website.
The Food and Drug Administration has approved abaloparatide (Tymlos) for postmenopausal women with osteoporosis at high risk for fracture.
Abaloparatide was approved based on 18-month results from the ACTIVE trial and 6-month results from the ACTIVExtend trial. Patients in the ACTIVE trial showed an relative risk reduction of 86% for new vertebral fractures and 43% for nonvertebral fractures, compared with placebo, according to a statement from manufacturer Radius Health. Results were similar regardless of age, years since menopause, presence or absence of prior fracture (vertebral or nonvertebral), and bone mineral density at baseline.
Find the full statement on the Radius Health website.
Durvalumab approved for advanced urothelial carcinoma
for patients with locally advanced or metastatic urothelial carcinoma who have disease progression after prior treatment with a platinum-containing chemotherapy.
The agency also approved a complementary diagnostic for the assessment of the PD-L1 protein the tumor tissue.
The most common adverse reactions were fatigue, musculoskeletal pain, constipation, decreased appetite, nausea, peripheral edema, and urinary tract infection. Pneumonitis, hepatitis, colitis, thyroid disease, adrenal insufficiency, and diabetes also occurred in patients taking durvalumab.
The recommended dose of durvalumab is 10 mg/kg IV over a period of 60 minutes, every 2 weeks, until disease progression or unacceptable toxicity occurs. Full prescribing information is available here.
Durvalumab is marketed as Imfinzi by AstraZeneca. The complementary diagnostic is the Ventana PD-L1 (SP263) Assay from Ventana Medical Systems.
for patients with locally advanced or metastatic urothelial carcinoma who have disease progression after prior treatment with a platinum-containing chemotherapy.
The agency also approved a complementary diagnostic for the assessment of the PD-L1 protein the tumor tissue.
The most common adverse reactions were fatigue, musculoskeletal pain, constipation, decreased appetite, nausea, peripheral edema, and urinary tract infection. Pneumonitis, hepatitis, colitis, thyroid disease, adrenal insufficiency, and diabetes also occurred in patients taking durvalumab.
The recommended dose of durvalumab is 10 mg/kg IV over a period of 60 minutes, every 2 weeks, until disease progression or unacceptable toxicity occurs. Full prescribing information is available here.
Durvalumab is marketed as Imfinzi by AstraZeneca. The complementary diagnostic is the Ventana PD-L1 (SP263) Assay from Ventana Medical Systems.
for patients with locally advanced or metastatic urothelial carcinoma who have disease progression after prior treatment with a platinum-containing chemotherapy.
The agency also approved a complementary diagnostic for the assessment of the PD-L1 protein the tumor tissue.
The most common adverse reactions were fatigue, musculoskeletal pain, constipation, decreased appetite, nausea, peripheral edema, and urinary tract infection. Pneumonitis, hepatitis, colitis, thyroid disease, adrenal insufficiency, and diabetes also occurred in patients taking durvalumab.
The recommended dose of durvalumab is 10 mg/kg IV over a period of 60 minutes, every 2 weeks, until disease progression or unacceptable toxicity occurs. Full prescribing information is available here.
Durvalumab is marketed as Imfinzi by AstraZeneca. The complementary diagnostic is the Ventana PD-L1 (SP263) Assay from Ventana Medical Systems.
FDA approves brigatinib for second-line advanced ALK-positive NSCLC
The Food and Drug Administration has granted accelerated approval to brigatinib for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)–positive non–small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib.
Approval was based on a meaningful and durable overall response rate in a two-arm, open-label, multicenter trial of 222 patients with locally advanced or metastatic ALK-positive NSCLC who had progressed on crizotinib. Patients were randomized to brigatinib orally either 90 mg once daily (112 patients) or 180 mg once daily following a 7-day lead-in at 90 mg once daily (110 patients).
The median duration of response was 13.8 months in both arms.
The most common adverse reactions in 219 patients who received at least one dose were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and interstitial lung disease/pneumonitis. The rate of fatal adverse reactions was 3.7%; they were pneumonia in two patients and sudden death, dyspnea, respiratory failure, pulmonary embolism, bacterial meningitis, and urosepsis in one patient each. Visual disturbances also occurred in patients receiving brigatinib. The FDA cautions that patients receiving brigatinib should be monitored for new or worsening respiratory symptoms; hypertension; bradycardia; visual symptoms; and elevations in amylase, lipase, blood glucose, and creatine phosphokinase.
The recommended dosing of brigatinib, marketed as Alunbrig by Takeda Pharmaceutical, is 90 mg orally once daily for the first 7 days then, if tolerated, increase to 180 mg orally once daily.
Full prescribing information is available here.
The Food and Drug Administration has granted accelerated approval to brigatinib for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)–positive non–small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib.
Approval was based on a meaningful and durable overall response rate in a two-arm, open-label, multicenter trial of 222 patients with locally advanced or metastatic ALK-positive NSCLC who had progressed on crizotinib. Patients were randomized to brigatinib orally either 90 mg once daily (112 patients) or 180 mg once daily following a 7-day lead-in at 90 mg once daily (110 patients).
The median duration of response was 13.8 months in both arms.
The most common adverse reactions in 219 patients who received at least one dose were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and interstitial lung disease/pneumonitis. The rate of fatal adverse reactions was 3.7%; they were pneumonia in two patients and sudden death, dyspnea, respiratory failure, pulmonary embolism, bacterial meningitis, and urosepsis in one patient each. Visual disturbances also occurred in patients receiving brigatinib. The FDA cautions that patients receiving brigatinib should be monitored for new or worsening respiratory symptoms; hypertension; bradycardia; visual symptoms; and elevations in amylase, lipase, blood glucose, and creatine phosphokinase.
The recommended dosing of brigatinib, marketed as Alunbrig by Takeda Pharmaceutical, is 90 mg orally once daily for the first 7 days then, if tolerated, increase to 180 mg orally once daily.
Full prescribing information is available here.
The Food and Drug Administration has granted accelerated approval to brigatinib for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)–positive non–small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib.
Approval was based on a meaningful and durable overall response rate in a two-arm, open-label, multicenter trial of 222 patients with locally advanced or metastatic ALK-positive NSCLC who had progressed on crizotinib. Patients were randomized to brigatinib orally either 90 mg once daily (112 patients) or 180 mg once daily following a 7-day lead-in at 90 mg once daily (110 patients).
The median duration of response was 13.8 months in both arms.
The most common adverse reactions in 219 patients who received at least one dose were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and interstitial lung disease/pneumonitis. The rate of fatal adverse reactions was 3.7%; they were pneumonia in two patients and sudden death, dyspnea, respiratory failure, pulmonary embolism, bacterial meningitis, and urosepsis in one patient each. Visual disturbances also occurred in patients receiving brigatinib. The FDA cautions that patients receiving brigatinib should be monitored for new or worsening respiratory symptoms; hypertension; bradycardia; visual symptoms; and elevations in amylase, lipase, blood glucose, and creatine phosphokinase.
The recommended dosing of brigatinib, marketed as Alunbrig by Takeda Pharmaceutical, is 90 mg orally once daily for the first 7 days then, if tolerated, increase to 180 mg orally once daily.
Full prescribing information is available here.