User login
CDC specifies risk-reduction strategies for HIV-discordant conception
When HIV-discordant couples wish to conceive, several strategies can effectively prevent men from infecting their female partners, according to an analysis in Morbidity and Mortality Weekly Report.
Using sperm from an HIV-negative donor remains the safest choice, but current methods of sperm washing make in vitro fertilization (IVF) or intrauterine insemination (IUI) a low-risk alternative, especially when used together with highly active antiretroviral therapy (HAART) and preexposure prophylaxis (PrEP), wrote Jennifer F. Kawwass, MD, and her associates in the division of reproductive health at the Centers for Disease Control and Prevention, Atlanta (MMWR Morb Mortal Wkly Rep. 2017;66:554-7).
Previously, the CDC recommended against insemination with semen from HIV-infected men. The new report reverses that position, citing epidemiologic and laboratory evidence that conception can occur safely when HIV-discordant couples use combined strategies including HAART, PrEP, and either condomless intercourse limited to the time around ovulation, or sperm washing prior to IVF or IUI.![]()
Sperm washing has been around for about 2 decades, but current techniques appear to significantly cut the risk of HIV transmission, based on epidemiologic data. Among 11,500 IVF or IUI cycles in which uninfected females used washed sperm from their HIV-infected male partners, there have been no cases of HIV transmission to women or their children, the CDC authors emphasized. The use of HAART and PrEP can add an extra layer of protection, they added.
Serodiscordant couples may also consider or prefer natural conception. An HIV-infected male on HAART whose plasma viral load is undetectable can expect to infect his female partner only about 0.16 times for every 10,000 exposures during condomless intercourse, the report noted. Limiting condomless intercourse to the time of ovulation and using PrEP by the uninfected female can further minimize the risk of sexual transmission, the authors added. Experienced medical providers can help HIV-discordant couples further evaluate the “unique risk profile” of each approach to conception, they concluded.
Dr. Kawwass and her associates had no conflicts of interest.
When HIV-discordant couples wish to conceive, several strategies can effectively prevent men from infecting their female partners, according to an analysis in Morbidity and Mortality Weekly Report.
Using sperm from an HIV-negative donor remains the safest choice, but current methods of sperm washing make in vitro fertilization (IVF) or intrauterine insemination (IUI) a low-risk alternative, especially when used together with highly active antiretroviral therapy (HAART) and preexposure prophylaxis (PrEP), wrote Jennifer F. Kawwass, MD, and her associates in the division of reproductive health at the Centers for Disease Control and Prevention, Atlanta (MMWR Morb Mortal Wkly Rep. 2017;66:554-7).
Previously, the CDC recommended against insemination with semen from HIV-infected men. The new report reverses that position, citing epidemiologic and laboratory evidence that conception can occur safely when HIV-discordant couples use combined strategies including HAART, PrEP, and either condomless intercourse limited to the time around ovulation, or sperm washing prior to IVF or IUI.![]()
Sperm washing has been around for about 2 decades, but current techniques appear to significantly cut the risk of HIV transmission, based on epidemiologic data. Among 11,500 IVF or IUI cycles in which uninfected females used washed sperm from their HIV-infected male partners, there have been no cases of HIV transmission to women or their children, the CDC authors emphasized. The use of HAART and PrEP can add an extra layer of protection, they added.
Serodiscordant couples may also consider or prefer natural conception. An HIV-infected male on HAART whose plasma viral load is undetectable can expect to infect his female partner only about 0.16 times for every 10,000 exposures during condomless intercourse, the report noted. Limiting condomless intercourse to the time of ovulation and using PrEP by the uninfected female can further minimize the risk of sexual transmission, the authors added. Experienced medical providers can help HIV-discordant couples further evaluate the “unique risk profile” of each approach to conception, they concluded.
Dr. Kawwass and her associates had no conflicts of interest.
When HIV-discordant couples wish to conceive, several strategies can effectively prevent men from infecting their female partners, according to an analysis in Morbidity and Mortality Weekly Report.
Using sperm from an HIV-negative donor remains the safest choice, but current methods of sperm washing make in vitro fertilization (IVF) or intrauterine insemination (IUI) a low-risk alternative, especially when used together with highly active antiretroviral therapy (HAART) and preexposure prophylaxis (PrEP), wrote Jennifer F. Kawwass, MD, and her associates in the division of reproductive health at the Centers for Disease Control and Prevention, Atlanta (MMWR Morb Mortal Wkly Rep. 2017;66:554-7).
Previously, the CDC recommended against insemination with semen from HIV-infected men. The new report reverses that position, citing epidemiologic and laboratory evidence that conception can occur safely when HIV-discordant couples use combined strategies including HAART, PrEP, and either condomless intercourse limited to the time around ovulation, or sperm washing prior to IVF or IUI.![]()
Sperm washing has been around for about 2 decades, but current techniques appear to significantly cut the risk of HIV transmission, based on epidemiologic data. Among 11,500 IVF or IUI cycles in which uninfected females used washed sperm from their HIV-infected male partners, there have been no cases of HIV transmission to women or their children, the CDC authors emphasized. The use of HAART and PrEP can add an extra layer of protection, they added.
Serodiscordant couples may also consider or prefer natural conception. An HIV-infected male on HAART whose plasma viral load is undetectable can expect to infect his female partner only about 0.16 times for every 10,000 exposures during condomless intercourse, the report noted. Limiting condomless intercourse to the time of ovulation and using PrEP by the uninfected female can further minimize the risk of sexual transmission, the authors added. Experienced medical providers can help HIV-discordant couples further evaluate the “unique risk profile” of each approach to conception, they concluded.
Dr. Kawwass and her associates had no conflicts of interest.
FROM MMWR
U.S. malaria cases dipped slightly in 2014
The number of confirmed malaria cases reported in the United States in 2014 is the fourth highest annual total since 1973, according to the Centers for Disease Control and Prevention, but the 2014 number of 1,724 cases is down slightly from 1,741 – the previous year’s number of confirmed cases.
The CDC monitors malaria cases in part to identify any instances of local, rather than imported, transmission. For 2014, no cases of local transmission were reported.
Of the imported transmission cases for which the region of acquisition was known, 1,383 (82.1%) came from Africa and 160 (9.5%) from Asia, making up all but 62 of the imported cases. The four leading countries of origin in Africa were Nigeria, Ghana, Sierra Leone, and Liberia (346, 153, 133, and 125 cases, respectively). Most of the cases from Asia came from India, which accounted for 100 of the 160 cases.
Sierra Leone, Liberia, and Guinea were the countries primarily affected by the Ebola virus disease outbreak in 2014 and into 2015. The study authors, Kimberly E. Mace, PhD, and Paul M. Arguin, MD, noted in the May 26 Morbidity and Mortality Weekly Report that “Ebola negatively impacted the delivery of malaria care and prevention services in the Ebola-affected countries, which could have increased malaria morbidity and mortality” (MMWR Surveill Summ. 2017;66[12]:1-24).
“Despite progress in reducing global prevalence of malaria, the disease remains endemic in many regions and use of appropriate prevention measures by travelers is still inadequate,” they added.
Among all cases, 17% were classified as severe illness, including five deaths (a decrease from 10 deaths in 2013). All five patients who died reported not taking chemoprophylaxis during their travel. More than half (57.5%) of the patients reported that the purpose of their travel was to visit friends and relatives.
“Health care providers should talk to their patients, especially those who would travel to countries where malaria is endemic to visit friends and relatives, about upcoming travel plans and offer education and medicines to prevent malaria,” the authors wrote.
The number of confirmed malaria cases reported in the United States in 2014 is the fourth highest annual total since 1973, according to the Centers for Disease Control and Prevention, but the 2014 number of 1,724 cases is down slightly from 1,741 – the previous year’s number of confirmed cases.
The CDC monitors malaria cases in part to identify any instances of local, rather than imported, transmission. For 2014, no cases of local transmission were reported.
Of the imported transmission cases for which the region of acquisition was known, 1,383 (82.1%) came from Africa and 160 (9.5%) from Asia, making up all but 62 of the imported cases. The four leading countries of origin in Africa were Nigeria, Ghana, Sierra Leone, and Liberia (346, 153, 133, and 125 cases, respectively). Most of the cases from Asia came from India, which accounted for 100 of the 160 cases.
Sierra Leone, Liberia, and Guinea were the countries primarily affected by the Ebola virus disease outbreak in 2014 and into 2015. The study authors, Kimberly E. Mace, PhD, and Paul M. Arguin, MD, noted in the May 26 Morbidity and Mortality Weekly Report that “Ebola negatively impacted the delivery of malaria care and prevention services in the Ebola-affected countries, which could have increased malaria morbidity and mortality” (MMWR Surveill Summ. 2017;66[12]:1-24).
“Despite progress in reducing global prevalence of malaria, the disease remains endemic in many regions and use of appropriate prevention measures by travelers is still inadequate,” they added.
Among all cases, 17% were classified as severe illness, including five deaths (a decrease from 10 deaths in 2013). All five patients who died reported not taking chemoprophylaxis during their travel. More than half (57.5%) of the patients reported that the purpose of their travel was to visit friends and relatives.
“Health care providers should talk to their patients, especially those who would travel to countries where malaria is endemic to visit friends and relatives, about upcoming travel plans and offer education and medicines to prevent malaria,” the authors wrote.
The number of confirmed malaria cases reported in the United States in 2014 is the fourth highest annual total since 1973, according to the Centers for Disease Control and Prevention, but the 2014 number of 1,724 cases is down slightly from 1,741 – the previous year’s number of confirmed cases.
The CDC monitors malaria cases in part to identify any instances of local, rather than imported, transmission. For 2014, no cases of local transmission were reported.
Of the imported transmission cases for which the region of acquisition was known, 1,383 (82.1%) came from Africa and 160 (9.5%) from Asia, making up all but 62 of the imported cases. The four leading countries of origin in Africa were Nigeria, Ghana, Sierra Leone, and Liberia (346, 153, 133, and 125 cases, respectively). Most of the cases from Asia came from India, which accounted for 100 of the 160 cases.
Sierra Leone, Liberia, and Guinea were the countries primarily affected by the Ebola virus disease outbreak in 2014 and into 2015. The study authors, Kimberly E. Mace, PhD, and Paul M. Arguin, MD, noted in the May 26 Morbidity and Mortality Weekly Report that “Ebola negatively impacted the delivery of malaria care and prevention services in the Ebola-affected countries, which could have increased malaria morbidity and mortality” (MMWR Surveill Summ. 2017;66[12]:1-24).
“Despite progress in reducing global prevalence of malaria, the disease remains endemic in many regions and use of appropriate prevention measures by travelers is still inadequate,” they added.
Among all cases, 17% were classified as severe illness, including five deaths (a decrease from 10 deaths in 2013). All five patients who died reported not taking chemoprophylaxis during their travel. More than half (57.5%) of the patients reported that the purpose of their travel was to visit friends and relatives.
“Health care providers should talk to their patients, especially those who would travel to countries where malaria is endemic to visit friends and relatives, about upcoming travel plans and offer education and medicines to prevent malaria,” the authors wrote.
FROM MMWR
FDA approves Rebinyn for hemophilia B treatment
The Food and Drug Administration has approved Rebinyn (nonacog beta pegol, N9-GP) for the treatment of hemophilia B, according to a statement from Novo Nordisk.
Caused by deficient blood clotting factor IX activity, hemophilia B affects about 5,000 people in the United States. The disease is chronic and inherited and causes prolonged and spontaneous bleeding, particularly into muscles, joints, or internal organs. Rebinyn is intended to control and treat bleeding episodes, as well as provide perioperative management of bleeding.![]()
Find the full statement about Rebinyn’s approval on the Novo Nordisk website.
The Food and Drug Administration has approved Rebinyn (nonacog beta pegol, N9-GP) for the treatment of hemophilia B, according to a statement from Novo Nordisk.
Caused by deficient blood clotting factor IX activity, hemophilia B affects about 5,000 people in the United States. The disease is chronic and inherited and causes prolonged and spontaneous bleeding, particularly into muscles, joints, or internal organs. Rebinyn is intended to control and treat bleeding episodes, as well as provide perioperative management of bleeding.![]()
Find the full statement about Rebinyn’s approval on the Novo Nordisk website.
The Food and Drug Administration has approved Rebinyn (nonacog beta pegol, N9-GP) for the treatment of hemophilia B, according to a statement from Novo Nordisk.
Caused by deficient blood clotting factor IX activity, hemophilia B affects about 5,000 people in the United States. The disease is chronic and inherited and causes prolonged and spontaneous bleeding, particularly into muscles, joints, or internal organs. Rebinyn is intended to control and treat bleeding episodes, as well as provide perioperative management of bleeding.![]()
Find the full statement about Rebinyn’s approval on the Novo Nordisk website.
FDA advisory committee supports neratinib approval
of adults with early-stage HER2-positive breast cancer who received prior adjuvant trastuzumab-based therapy.
Members voting during the May 24 meeting expressed concern about the broadness of the indication proposed by the drug’s maker, Puma Biotechnology, but the majority said the risk-benefit profile of the drug is sufficient to support approval, particularly given the unmet need for such an agent. Those voting against approval argued that more data are needed to identify subpopulations of patients who would be most likely to benefit from treatment in order to narrow the indication.
“There are some unknowns that concern me. ... I don’t think I would treat as broadly as the indication describes,” he said, adding that it would be very difficult to decide which patients to treat, and it would be nice to have more data that provide predictive biomarkers to help determine who should be treated. “But at the end of the day I think it’s useful to have this as an option for treating patients.”
Lori M. Minasian, MD, also a temporary voting member from the National Institutes of Health, agreed.
“I think the option should be available,” she said, adding that the analysis was thorough. However, she, too, questioned the proposed indication.
“I remain concerned that the indication is far too broad, and ... I think we need greater understanding of which subsets of patients would be most responsive to this therapy,” she said.
Heidi D. Klepin, MD, of Wake Forest University in Winston-Salem, N.C. said she voted “yes” for similar reasons, despite concerns about the lack of data to help narrow the indication.
“I particularly felt it was important to support this indication because I think this is an unmet need, and I think the primary outcome is an important and relevant outcome for our patients, even though what we’re seeing effect-wise may be modest,” she said.
Patricia A. Spears, a breast cancer survivor and patient representative from Raleigh, N.C., however, voted against approval.
“I think it is important to get drugs out to patients, and I think this will benefit a certain subset of patients. I’m just not sure we know which ones yet. What we do is tend to put a lot of patients at risk to benefit just a few. We do that a lot,” she said.
She expressed concern that the treatment would be “tacked on to the end of trastuzumab” in too many cases without concern for whether the patient is likely to benefit.
Another member who voted no, Courtney J. Preusse, who was a consumer representative on the committee, said she struggled with the decision but ultimately decided that the benefit this drug adds beyond what already exists is “just not compelling.”
Neratinib is a kinase inhibitor that irreversibly binds to epidermal growth factor receptors, HER2, and HER4, and results of the phase III ExteNET trial presented to the committee by representatives of the applicant showed a statistically significant invasive disease-free survival benefit with treatment.
The invasive disease-free survival at 2 years in 1,420 patients with early stage HER2-positive breast cancer after adjuvant treatment with trastuzumab who were randomized to receive neratinib was 94.2%, compared with 91.9% in 1,420 who received placebo (stratified hazard ratio 0.66). Follow-up data from patients who reconsented to participate showed similar outcomes from 2 to 5 years post-randomization.
An exploratory subgroup analysis suggested a possible difference in the magnitude of benefit based on hormone receptor status, with a 51% reduction in recurrence risk among HR-positive patients, compared with a 7% reduction in HR-negative patients (hazard ratios, 0.49 and 0.93, respectively).
Some concern was raised regarding multiple amendments to the protocol during the study, but committee members who spoke about this expressed satisfaction with the way these issues were handled in terms of sensitivity analysis.
As for safety, diarrhea was the most frequently reported adverse reaction, occurring in 95% of treated patients. Grade 3 diarrhea occurred in 40% of treated patients. Additional data were presented showing that antidiarrheal prophylaxis was effective. Several patients who spoke during the public hearing portion of the meeting–some of whom had travel expenses paid by the applicant, said they either had no problem with diarrhea or that it was manageable. Nearly all of those who spoke during the open public hearing, including patients and family members, shared emotional stories about their personal battles with breast cancer and urged the committee to support approval of neratinib to expand the options available to patients.
The FDA will now consider the new drug application for neratinib, and although it is not bound by the advisory committee’s recommendation, it usually follows such recommendations.
The advisory committee members reported having no relevant conflicts of interest.
of adults with early-stage HER2-positive breast cancer who received prior adjuvant trastuzumab-based therapy.
Members voting during the May 24 meeting expressed concern about the broadness of the indication proposed by the drug’s maker, Puma Biotechnology, but the majority said the risk-benefit profile of the drug is sufficient to support approval, particularly given the unmet need for such an agent. Those voting against approval argued that more data are needed to identify subpopulations of patients who would be most likely to benefit from treatment in order to narrow the indication.
“There are some unknowns that concern me. ... I don’t think I would treat as broadly as the indication describes,” he said, adding that it would be very difficult to decide which patients to treat, and it would be nice to have more data that provide predictive biomarkers to help determine who should be treated. “But at the end of the day I think it’s useful to have this as an option for treating patients.”
Lori M. Minasian, MD, also a temporary voting member from the National Institutes of Health, agreed.
“I think the option should be available,” she said, adding that the analysis was thorough. However, she, too, questioned the proposed indication.
“I remain concerned that the indication is far too broad, and ... I think we need greater understanding of which subsets of patients would be most responsive to this therapy,” she said.
Heidi D. Klepin, MD, of Wake Forest University in Winston-Salem, N.C. said she voted “yes” for similar reasons, despite concerns about the lack of data to help narrow the indication.
“I particularly felt it was important to support this indication because I think this is an unmet need, and I think the primary outcome is an important and relevant outcome for our patients, even though what we’re seeing effect-wise may be modest,” she said.
Patricia A. Spears, a breast cancer survivor and patient representative from Raleigh, N.C., however, voted against approval.
“I think it is important to get drugs out to patients, and I think this will benefit a certain subset of patients. I’m just not sure we know which ones yet. What we do is tend to put a lot of patients at risk to benefit just a few. We do that a lot,” she said.
She expressed concern that the treatment would be “tacked on to the end of trastuzumab” in too many cases without concern for whether the patient is likely to benefit.
Another member who voted no, Courtney J. Preusse, who was a consumer representative on the committee, said she struggled with the decision but ultimately decided that the benefit this drug adds beyond what already exists is “just not compelling.”
Neratinib is a kinase inhibitor that irreversibly binds to epidermal growth factor receptors, HER2, and HER4, and results of the phase III ExteNET trial presented to the committee by representatives of the applicant showed a statistically significant invasive disease-free survival benefit with treatment.
The invasive disease-free survival at 2 years in 1,420 patients with early stage HER2-positive breast cancer after adjuvant treatment with trastuzumab who were randomized to receive neratinib was 94.2%, compared with 91.9% in 1,420 who received placebo (stratified hazard ratio 0.66). Follow-up data from patients who reconsented to participate showed similar outcomes from 2 to 5 years post-randomization.
An exploratory subgroup analysis suggested a possible difference in the magnitude of benefit based on hormone receptor status, with a 51% reduction in recurrence risk among HR-positive patients, compared with a 7% reduction in HR-negative patients (hazard ratios, 0.49 and 0.93, respectively).
Some concern was raised regarding multiple amendments to the protocol during the study, but committee members who spoke about this expressed satisfaction with the way these issues were handled in terms of sensitivity analysis.
As for safety, diarrhea was the most frequently reported adverse reaction, occurring in 95% of treated patients. Grade 3 diarrhea occurred in 40% of treated patients. Additional data were presented showing that antidiarrheal prophylaxis was effective. Several patients who spoke during the public hearing portion of the meeting–some of whom had travel expenses paid by the applicant, said they either had no problem with diarrhea or that it was manageable. Nearly all of those who spoke during the open public hearing, including patients and family members, shared emotional stories about their personal battles with breast cancer and urged the committee to support approval of neratinib to expand the options available to patients.
The FDA will now consider the new drug application for neratinib, and although it is not bound by the advisory committee’s recommendation, it usually follows such recommendations.
The advisory committee members reported having no relevant conflicts of interest.
of adults with early-stage HER2-positive breast cancer who received prior adjuvant trastuzumab-based therapy.
Members voting during the May 24 meeting expressed concern about the broadness of the indication proposed by the drug’s maker, Puma Biotechnology, but the majority said the risk-benefit profile of the drug is sufficient to support approval, particularly given the unmet need for such an agent. Those voting against approval argued that more data are needed to identify subpopulations of patients who would be most likely to benefit from treatment in order to narrow the indication.
“There are some unknowns that concern me. ... I don’t think I would treat as broadly as the indication describes,” he said, adding that it would be very difficult to decide which patients to treat, and it would be nice to have more data that provide predictive biomarkers to help determine who should be treated. “But at the end of the day I think it’s useful to have this as an option for treating patients.”
Lori M. Minasian, MD, also a temporary voting member from the National Institutes of Health, agreed.
“I think the option should be available,” she said, adding that the analysis was thorough. However, she, too, questioned the proposed indication.
“I remain concerned that the indication is far too broad, and ... I think we need greater understanding of which subsets of patients would be most responsive to this therapy,” she said.
Heidi D. Klepin, MD, of Wake Forest University in Winston-Salem, N.C. said she voted “yes” for similar reasons, despite concerns about the lack of data to help narrow the indication.
“I particularly felt it was important to support this indication because I think this is an unmet need, and I think the primary outcome is an important and relevant outcome for our patients, even though what we’re seeing effect-wise may be modest,” she said.
Patricia A. Spears, a breast cancer survivor and patient representative from Raleigh, N.C., however, voted against approval.
“I think it is important to get drugs out to patients, and I think this will benefit a certain subset of patients. I’m just not sure we know which ones yet. What we do is tend to put a lot of patients at risk to benefit just a few. We do that a lot,” she said.
She expressed concern that the treatment would be “tacked on to the end of trastuzumab” in too many cases without concern for whether the patient is likely to benefit.
Another member who voted no, Courtney J. Preusse, who was a consumer representative on the committee, said she struggled with the decision but ultimately decided that the benefit this drug adds beyond what already exists is “just not compelling.”
Neratinib is a kinase inhibitor that irreversibly binds to epidermal growth factor receptors, HER2, and HER4, and results of the phase III ExteNET trial presented to the committee by representatives of the applicant showed a statistically significant invasive disease-free survival benefit with treatment.
The invasive disease-free survival at 2 years in 1,420 patients with early stage HER2-positive breast cancer after adjuvant treatment with trastuzumab who were randomized to receive neratinib was 94.2%, compared with 91.9% in 1,420 who received placebo (stratified hazard ratio 0.66). Follow-up data from patients who reconsented to participate showed similar outcomes from 2 to 5 years post-randomization.
An exploratory subgroup analysis suggested a possible difference in the magnitude of benefit based on hormone receptor status, with a 51% reduction in recurrence risk among HR-positive patients, compared with a 7% reduction in HR-negative patients (hazard ratios, 0.49 and 0.93, respectively).
Some concern was raised regarding multiple amendments to the protocol during the study, but committee members who spoke about this expressed satisfaction with the way these issues were handled in terms of sensitivity analysis.
As for safety, diarrhea was the most frequently reported adverse reaction, occurring in 95% of treated patients. Grade 3 diarrhea occurred in 40% of treated patients. Additional data were presented showing that antidiarrheal prophylaxis was effective. Several patients who spoke during the public hearing portion of the meeting–some of whom had travel expenses paid by the applicant, said they either had no problem with diarrhea or that it was manageable. Nearly all of those who spoke during the open public hearing, including patients and family members, shared emotional stories about their personal battles with breast cancer and urged the committee to support approval of neratinib to expand the options available to patients.
The FDA will now consider the new drug application for neratinib, and although it is not bound by the advisory committee’s recommendation, it usually follows such recommendations.
The advisory committee members reported having no relevant conflicts of interest.
FDA approves generic Strattera for pediatric, adult ADHD patients
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
FDA panel backs licensure for epoetin alfa biosimilar
A Food and Drug Administration advisory committee voted overwhelmingly in support of licensure for Epoetin Hospira as a biosimilar product to epoetin alfa (Epogen/Procrit) for all approved Epogen/Procrit indications.
Most committee members who voted “yes” said they were hesitant to do so, however, because of safety concerns, particularly in HIV and oncology patients.
“I voted yes. I came in with concerns about the HIV and oncology patients. However, I do believe ... after hearing the justification, that it meets the [FDA regulatory guideline],” said temporary voting member and patient representative Karen E. Arscott, DO, of the Commonwealth Medical College, Scranton, Penn. “I would like to see extensive follow-up in these two population groups,” she said.
The concerns about HIV and oncology patients are related to the potential for immunogenicity-related events. Some members noted that the study populations were likely too small to detect these rare events.
Scott A. Waldman, MD, of Thomas Jefferson University, Philadelphia, also a temporary member, said he voted “yes” because there was “no substantial difference analytically, biologically, or clinically in what was tested.”
“I think the residual uncertainty of immunogenicity and hypersensitivity and the extrapolation across different patient populations will emerge in postmarketing surveillance. I think that’s when we’ll get the clearest picture of whether there really is any uncertainty in how these drugs perform,” he said.
For one member, however, the concerns were enough for a “no” vote.
“The analytical, clinical, and preclinical data support biosimilarity, and I strongly support approval for indications 1 and 4 based on the clinical data ... but I have residual concerns about lack of data, immunogenicity, basic safety data in patients with HIV and cancer and, for that reason, voted no for the broader indication,” said Thomas S. Uldrick, MD, of the HIV & AIDS Malignancy Branch at the Center for Cancer Research, National Cancer Institute, Bethesda, Md.
Gregory J. Riely, MD, of Memorial Sloan-Kettering Cancer Center in New York expressed similar concerns but said he found the data compelling.
“I understand the concerns around immunogenicity for HIV and cancer patients. I was somewhat reassured by the nonclinical data showing an absence of increased immunogenicity for this biosimilar,” he said.
To support its BLA for Epoetin Hospira, Hospira submitted data from four studies comparing it with the U.S.-licensed Epogen/Procrit and presented an analytical biosimilarity assessment and a nonclinical, clinical pharmacology and clinical biosimilarity assessment. The FDA analysis of the data considered chemistry, manufacturing, and controls, as well as pharmacology/toxicology, immunogenicity, clinical pharmacology, and clinical efficacy and safety.
Sumant Ramachandra, MD, of Pfizer Essential Health noted that the company is not currently seeking an interchangeability designation and that, while this is the first application for a biosimilar product to Amgen’s Epogen/Procrit product, which was approved in 1989, a “highly related epoetin product” from Pfizer (Retacrit) has been available in Europe for 9 years, with more than 363,000 patient years of treatment administered.
The demonstration of biosimilarity in the Epoetin Hospira data presented to the FDA, coupled with well-characterized nature of the reference product, “together support extrapolation across all conditions of use for the reference product,” he said, noting that the data demonstrate that the mechanism of action for both the biosimilar and reference product is the same, and that the immunogenicity profiles are consistent.
The FDA assessment of the data led to a similar conclusion that no clinically significant differences were found between the biosimilar and reference product.
Among other concerns expressed by advisory committee members and/or the public were in regard to the extrapolation of data from patients with chronic kidney disease on hemodialysis to other indications (although this is considered acceptable, according to FDA regulations) and to populations that weren’t studied and about the possibility that Epoetin Hospira would be forced on patients inappropriately, despite the fact that Hospira is not seeking an interchangeability designation. Patients and others speaking on behalf of various patient groups and advocacy organizations called for safeguards against such inappropriate substitution.
Acting committee chair, Brian I. Rini, MD, of the Cleveland Clinic Taussig Cancer Clinic, Ohio, said he voted “yes” because the product met all regulatory requirements but agreed that the “need for vigilance is exceedingly important not only for this drug but for all drugs in this circumstance.”
The FDA, which usually follows the recommendations of its advisory committees, will now consider the BLA for Epoetin Hospira.
In a statement released after the vote, Diem Nguyen, Pfizer Essential Health global president, Americas, said the committee’s recommendation for approval “reinforces the potential value of biosimilars in expanding access to additional high-quality treatment options for the patients in the U.S. who need them.”
The advisory committee members were screened and found to have no relevant conflicts of interest.
A Food and Drug Administration advisory committee voted overwhelmingly in support of licensure for Epoetin Hospira as a biosimilar product to epoetin alfa (Epogen/Procrit) for all approved Epogen/Procrit indications.
Most committee members who voted “yes” said they were hesitant to do so, however, because of safety concerns, particularly in HIV and oncology patients.
“I voted yes. I came in with concerns about the HIV and oncology patients. However, I do believe ... after hearing the justification, that it meets the [FDA regulatory guideline],” said temporary voting member and patient representative Karen E. Arscott, DO, of the Commonwealth Medical College, Scranton, Penn. “I would like to see extensive follow-up in these two population groups,” she said.
The concerns about HIV and oncology patients are related to the potential for immunogenicity-related events. Some members noted that the study populations were likely too small to detect these rare events.
Scott A. Waldman, MD, of Thomas Jefferson University, Philadelphia, also a temporary member, said he voted “yes” because there was “no substantial difference analytically, biologically, or clinically in what was tested.”
“I think the residual uncertainty of immunogenicity and hypersensitivity and the extrapolation across different patient populations will emerge in postmarketing surveillance. I think that’s when we’ll get the clearest picture of whether there really is any uncertainty in how these drugs perform,” he said.
For one member, however, the concerns were enough for a “no” vote.
“The analytical, clinical, and preclinical data support biosimilarity, and I strongly support approval for indications 1 and 4 based on the clinical data ... but I have residual concerns about lack of data, immunogenicity, basic safety data in patients with HIV and cancer and, for that reason, voted no for the broader indication,” said Thomas S. Uldrick, MD, of the HIV & AIDS Malignancy Branch at the Center for Cancer Research, National Cancer Institute, Bethesda, Md.
Gregory J. Riely, MD, of Memorial Sloan-Kettering Cancer Center in New York expressed similar concerns but said he found the data compelling.
“I understand the concerns around immunogenicity for HIV and cancer patients. I was somewhat reassured by the nonclinical data showing an absence of increased immunogenicity for this biosimilar,” he said.
To support its BLA for Epoetin Hospira, Hospira submitted data from four studies comparing it with the U.S.-licensed Epogen/Procrit and presented an analytical biosimilarity assessment and a nonclinical, clinical pharmacology and clinical biosimilarity assessment. The FDA analysis of the data considered chemistry, manufacturing, and controls, as well as pharmacology/toxicology, immunogenicity, clinical pharmacology, and clinical efficacy and safety.
Sumant Ramachandra, MD, of Pfizer Essential Health noted that the company is not currently seeking an interchangeability designation and that, while this is the first application for a biosimilar product to Amgen’s Epogen/Procrit product, which was approved in 1989, a “highly related epoetin product” from Pfizer (Retacrit) has been available in Europe for 9 years, with more than 363,000 patient years of treatment administered.
The demonstration of biosimilarity in the Epoetin Hospira data presented to the FDA, coupled with well-characterized nature of the reference product, “together support extrapolation across all conditions of use for the reference product,” he said, noting that the data demonstrate that the mechanism of action for both the biosimilar and reference product is the same, and that the immunogenicity profiles are consistent.
The FDA assessment of the data led to a similar conclusion that no clinically significant differences were found between the biosimilar and reference product.
Among other concerns expressed by advisory committee members and/or the public were in regard to the extrapolation of data from patients with chronic kidney disease on hemodialysis to other indications (although this is considered acceptable, according to FDA regulations) and to populations that weren’t studied and about the possibility that Epoetin Hospira would be forced on patients inappropriately, despite the fact that Hospira is not seeking an interchangeability designation. Patients and others speaking on behalf of various patient groups and advocacy organizations called for safeguards against such inappropriate substitution.
Acting committee chair, Brian I. Rini, MD, of the Cleveland Clinic Taussig Cancer Clinic, Ohio, said he voted “yes” because the product met all regulatory requirements but agreed that the “need for vigilance is exceedingly important not only for this drug but for all drugs in this circumstance.”
The FDA, which usually follows the recommendations of its advisory committees, will now consider the BLA for Epoetin Hospira.
In a statement released after the vote, Diem Nguyen, Pfizer Essential Health global president, Americas, said the committee’s recommendation for approval “reinforces the potential value of biosimilars in expanding access to additional high-quality treatment options for the patients in the U.S. who need them.”
The advisory committee members were screened and found to have no relevant conflicts of interest.
A Food and Drug Administration advisory committee voted overwhelmingly in support of licensure for Epoetin Hospira as a biosimilar product to epoetin alfa (Epogen/Procrit) for all approved Epogen/Procrit indications.
Most committee members who voted “yes” said they were hesitant to do so, however, because of safety concerns, particularly in HIV and oncology patients.
“I voted yes. I came in with concerns about the HIV and oncology patients. However, I do believe ... after hearing the justification, that it meets the [FDA regulatory guideline],” said temporary voting member and patient representative Karen E. Arscott, DO, of the Commonwealth Medical College, Scranton, Penn. “I would like to see extensive follow-up in these two population groups,” she said.
The concerns about HIV and oncology patients are related to the potential for immunogenicity-related events. Some members noted that the study populations were likely too small to detect these rare events.
Scott A. Waldman, MD, of Thomas Jefferson University, Philadelphia, also a temporary member, said he voted “yes” because there was “no substantial difference analytically, biologically, or clinically in what was tested.”
“I think the residual uncertainty of immunogenicity and hypersensitivity and the extrapolation across different patient populations will emerge in postmarketing surveillance. I think that’s when we’ll get the clearest picture of whether there really is any uncertainty in how these drugs perform,” he said.
For one member, however, the concerns were enough for a “no” vote.
“The analytical, clinical, and preclinical data support biosimilarity, and I strongly support approval for indications 1 and 4 based on the clinical data ... but I have residual concerns about lack of data, immunogenicity, basic safety data in patients with HIV and cancer and, for that reason, voted no for the broader indication,” said Thomas S. Uldrick, MD, of the HIV & AIDS Malignancy Branch at the Center for Cancer Research, National Cancer Institute, Bethesda, Md.
Gregory J. Riely, MD, of Memorial Sloan-Kettering Cancer Center in New York expressed similar concerns but said he found the data compelling.
“I understand the concerns around immunogenicity for HIV and cancer patients. I was somewhat reassured by the nonclinical data showing an absence of increased immunogenicity for this biosimilar,” he said.
To support its BLA for Epoetin Hospira, Hospira submitted data from four studies comparing it with the U.S.-licensed Epogen/Procrit and presented an analytical biosimilarity assessment and a nonclinical, clinical pharmacology and clinical biosimilarity assessment. The FDA analysis of the data considered chemistry, manufacturing, and controls, as well as pharmacology/toxicology, immunogenicity, clinical pharmacology, and clinical efficacy and safety.
Sumant Ramachandra, MD, of Pfizer Essential Health noted that the company is not currently seeking an interchangeability designation and that, while this is the first application for a biosimilar product to Amgen’s Epogen/Procrit product, which was approved in 1989, a “highly related epoetin product” from Pfizer (Retacrit) has been available in Europe for 9 years, with more than 363,000 patient years of treatment administered.
The demonstration of biosimilarity in the Epoetin Hospira data presented to the FDA, coupled with well-characterized nature of the reference product, “together support extrapolation across all conditions of use for the reference product,” he said, noting that the data demonstrate that the mechanism of action for both the biosimilar and reference product is the same, and that the immunogenicity profiles are consistent.
The FDA assessment of the data led to a similar conclusion that no clinically significant differences were found between the biosimilar and reference product.
Among other concerns expressed by advisory committee members and/or the public were in regard to the extrapolation of data from patients with chronic kidney disease on hemodialysis to other indications (although this is considered acceptable, according to FDA regulations) and to populations that weren’t studied and about the possibility that Epoetin Hospira would be forced on patients inappropriately, despite the fact that Hospira is not seeking an interchangeability designation. Patients and others speaking on behalf of various patient groups and advocacy organizations called for safeguards against such inappropriate substitution.
Acting committee chair, Brian I. Rini, MD, of the Cleveland Clinic Taussig Cancer Clinic, Ohio, said he voted “yes” because the product met all regulatory requirements but agreed that the “need for vigilance is exceedingly important not only for this drug but for all drugs in this circumstance.”
The FDA, which usually follows the recommendations of its advisory committees, will now consider the BLA for Epoetin Hospira.
In a statement released after the vote, Diem Nguyen, Pfizer Essential Health global president, Americas, said the committee’s recommendation for approval “reinforces the potential value of biosimilars in expanding access to additional high-quality treatment options for the patients in the U.S. who need them.”
The advisory committee members were screened and found to have no relevant conflicts of interest.
Key clinical point:
Major finding: The data demonstrate that the mechanism of action for both the biosimilar and reference product is the same, and that the immunogenicity profiles are consistent.
Data source: An FDA advisory committee review of epoetin alfa biosimilar Epoetin Hospira.
Disclosures: The advisory committee members were screened and found to have no relevant conflicts of interest.
Alzheimer’s mortality in U.S. grew from 1999 to 2014
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.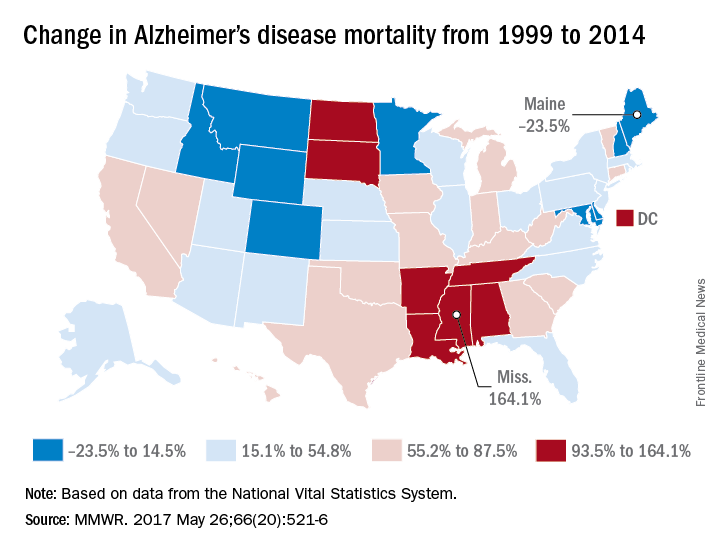
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
FROM MMWR
FDA advisory committee supports L-glutamine for SCD
The available data on the use of L-glutamine powder for treating sickle cell disease is favorable in terms of the agent’s overall benefit-risk profile, a majority of the Food and Drug Administration’s Oncologic Drugs Advisory Committee agreed during a meeting May 24.
L-glutamine powder, which is used as an oral solution for treating sickle cell disease, showed moderate benefit in a phase III study and a smaller phase II study, and if approved by the FDA – which usually follows the recommendations of its advisory committees – would be only the second treatment approved for the debilitating and sometimes deadly disease. The first, hydroxyurea, was approved for use in adults in 1998.
The committee voted 10-3 in favor of L-glutamate, after hearing from representatives of the new drug marketing applicant, Emmaus Medical, about the efficacy and safety data, as well as from FDA representatives who analyzed the data, physicians who treat sickle cell patients, patient advocates, and patients and their family members who gave emotional testimony in favor of approving this treatment.
“This is clearly a bad disease. It’s worse than cancer in many ways. I think probably mostly from a stigma standpoint it has a desperate need [for treatments],” said acting committee chair Brian I. Rini, MD, who voted in favor of the agent.
While there were some concerns about the data, including questions about methodologies, differential dropout rates between study arms, baseline characteristics that may have affected outcomes, and discrepancies between the Emmaus Medical data and the FDA’s analyses of the data, “all seemed to come down in favor of the agent,” Dr. Rini said, citing its “modest but consistent benefit” and low risk.
“I think one thing that’s strikingly clear is that even any sort of modest benefit in this community, given the sequelae of crises, is significant; it doesn’t take much to produce a clinical impact, and that should be motivation to study more drugs in this disease,” he said.
Another focus among those who voted “yes” was on the overwhelming need for treatment for sickle cell patients, who spend “a hugely disproportionate part of their life in the health care system, and who have had a tremendous burden imposed on them by their disease,” according to Harold J. Burstein, MD, PhD, of Dana-Farber Cancer Institute in Boston, who said he was swayed by the low risk of toxicity and the corroborating evidence in the phase II and III trials.
“What I took away was that one fewer hospital visit per year was a clinically compelling benefit for any individual or family or hospital that might be caring for patients with sickle cell disease,” he said, referencing a finding that treated patients had three visits, compared with four visits among patients in the placebo group, in the phase III study.
The data presented to the committee during the meeting by Yutaka Niihara, MD, of Emmaus Medical, included findings from a phase III randomized, placebo-controlled multicenter study (GLUSCC09-01) involving patients aged 5 years and older with sickle cell disease or beta-0 thalassemia who had at least two episodes of painful crises within the 12 months prior to screening. A total of 152 patients were randomized to receive oral L-glutamine at a dose of 0.3 mg/kg per day for 48 weeks followed by a 3-week tapering period, and 78 received placebo.
The Emmaus Medical analysis demonstrated a significant decrease in crisis events (median of 3 vs. 4) among treatment vs. placebo group patients, and the time to second crisis was delayed by 79 days in the treatment group (hazard ratio 0.68). The analysis, however, was complicated by the differential dropout rates (36% vs. 24% in the treatment and placebo arms, respectively), necessitating the use of imputation methods. Various methods were used to handle the missing data, and the findings with each of them favored L-glutamine, but each had important limitations, and while the FDA’s analysis of the various approaches showed that each favored L-glutamine over placebo with a range of reduction in the rates of crises from 0.4 to 0.9, this contributed to the decision by some panel member to vote against the agent.
The phase II study (Study 10478), which had a similar design, failed to meet its specified significance level for primary efficacy analysis, but showed a trend in favor of L-glutamine vs. placebo, Dr. Niihara said.
As for safety, Dr. Niihara reported that a safety population of 187 patients treated with L-glutamine and 111 treated with placebo in the phase II and III studies showed that most patients experienced a treatment-emergent adverse event – most often sickle cell anemia with crisis (66% and 72% in the groups, respectively) and acute chest syndrome (7% and 19%, respectively). Treatment-emergent adverse events led to withdrawal in 2.7% and 0.9% of patients, respectively. The most common adverse reactions were constipation, nausea, headache, cough, pain in the extremities, back pain, chest pain, and abdominal pain.
Bernard F. Cole, PhD, who was among the “no” votes, said he had concerns about “the limitations resulting from differential dropout” rates, which may have artificially shifted the risk profile.
“As a result of those limitations, it’s not clear whether patients at higher risk of a [sickle cell crisis] event might have disproportionately dropped out of the L-glutamine arm,” he explained. “My hope is that the sponsors can more thoroughly address the limitations of this pivotal trial with the FDA,” said Dr. Cole, a professor in the department of mathematics and statistics at the University of Vermont, Burlington.
Dr. Rini, despite his “yes” vote, agreed with the need for more data, noting that additional data analysis could help when it comes to clinical application of L-glutamine.
Specifically, more data regarding duration of therapy and quality data collection that “borrows from the cancer world ... in terms of rigor or data collection,” is needed, he said.
The committee members had no relevant conflicts of interests to disclose.
The available data on the use of L-glutamine powder for treating sickle cell disease is favorable in terms of the agent’s overall benefit-risk profile, a majority of the Food and Drug Administration’s Oncologic Drugs Advisory Committee agreed during a meeting May 24.
L-glutamine powder, which is used as an oral solution for treating sickle cell disease, showed moderate benefit in a phase III study and a smaller phase II study, and if approved by the FDA – which usually follows the recommendations of its advisory committees – would be only the second treatment approved for the debilitating and sometimes deadly disease. The first, hydroxyurea, was approved for use in adults in 1998.
The committee voted 10-3 in favor of L-glutamate, after hearing from representatives of the new drug marketing applicant, Emmaus Medical, about the efficacy and safety data, as well as from FDA representatives who analyzed the data, physicians who treat sickle cell patients, patient advocates, and patients and their family members who gave emotional testimony in favor of approving this treatment.
“This is clearly a bad disease. It’s worse than cancer in many ways. I think probably mostly from a stigma standpoint it has a desperate need [for treatments],” said acting committee chair Brian I. Rini, MD, who voted in favor of the agent.
While there were some concerns about the data, including questions about methodologies, differential dropout rates between study arms, baseline characteristics that may have affected outcomes, and discrepancies between the Emmaus Medical data and the FDA’s analyses of the data, “all seemed to come down in favor of the agent,” Dr. Rini said, citing its “modest but consistent benefit” and low risk.
“I think one thing that’s strikingly clear is that even any sort of modest benefit in this community, given the sequelae of crises, is significant; it doesn’t take much to produce a clinical impact, and that should be motivation to study more drugs in this disease,” he said.
Another focus among those who voted “yes” was on the overwhelming need for treatment for sickle cell patients, who spend “a hugely disproportionate part of their life in the health care system, and who have had a tremendous burden imposed on them by their disease,” according to Harold J. Burstein, MD, PhD, of Dana-Farber Cancer Institute in Boston, who said he was swayed by the low risk of toxicity and the corroborating evidence in the phase II and III trials.
“What I took away was that one fewer hospital visit per year was a clinically compelling benefit for any individual or family or hospital that might be caring for patients with sickle cell disease,” he said, referencing a finding that treated patients had three visits, compared with four visits among patients in the placebo group, in the phase III study.
The data presented to the committee during the meeting by Yutaka Niihara, MD, of Emmaus Medical, included findings from a phase III randomized, placebo-controlled multicenter study (GLUSCC09-01) involving patients aged 5 years and older with sickle cell disease or beta-0 thalassemia who had at least two episodes of painful crises within the 12 months prior to screening. A total of 152 patients were randomized to receive oral L-glutamine at a dose of 0.3 mg/kg per day for 48 weeks followed by a 3-week tapering period, and 78 received placebo.
The Emmaus Medical analysis demonstrated a significant decrease in crisis events (median of 3 vs. 4) among treatment vs. placebo group patients, and the time to second crisis was delayed by 79 days in the treatment group (hazard ratio 0.68). The analysis, however, was complicated by the differential dropout rates (36% vs. 24% in the treatment and placebo arms, respectively), necessitating the use of imputation methods. Various methods were used to handle the missing data, and the findings with each of them favored L-glutamine, but each had important limitations, and while the FDA’s analysis of the various approaches showed that each favored L-glutamine over placebo with a range of reduction in the rates of crises from 0.4 to 0.9, this contributed to the decision by some panel member to vote against the agent.
The phase II study (Study 10478), which had a similar design, failed to meet its specified significance level for primary efficacy analysis, but showed a trend in favor of L-glutamine vs. placebo, Dr. Niihara said.
As for safety, Dr. Niihara reported that a safety population of 187 patients treated with L-glutamine and 111 treated with placebo in the phase II and III studies showed that most patients experienced a treatment-emergent adverse event – most often sickle cell anemia with crisis (66% and 72% in the groups, respectively) and acute chest syndrome (7% and 19%, respectively). Treatment-emergent adverse events led to withdrawal in 2.7% and 0.9% of patients, respectively. The most common adverse reactions were constipation, nausea, headache, cough, pain in the extremities, back pain, chest pain, and abdominal pain.
Bernard F. Cole, PhD, who was among the “no” votes, said he had concerns about “the limitations resulting from differential dropout” rates, which may have artificially shifted the risk profile.
“As a result of those limitations, it’s not clear whether patients at higher risk of a [sickle cell crisis] event might have disproportionately dropped out of the L-glutamine arm,” he explained. “My hope is that the sponsors can more thoroughly address the limitations of this pivotal trial with the FDA,” said Dr. Cole, a professor in the department of mathematics and statistics at the University of Vermont, Burlington.
Dr. Rini, despite his “yes” vote, agreed with the need for more data, noting that additional data analysis could help when it comes to clinical application of L-glutamine.
Specifically, more data regarding duration of therapy and quality data collection that “borrows from the cancer world ... in terms of rigor or data collection,” is needed, he said.
The committee members had no relevant conflicts of interests to disclose.
The available data on the use of L-glutamine powder for treating sickle cell disease is favorable in terms of the agent’s overall benefit-risk profile, a majority of the Food and Drug Administration’s Oncologic Drugs Advisory Committee agreed during a meeting May 24.
L-glutamine powder, which is used as an oral solution for treating sickle cell disease, showed moderate benefit in a phase III study and a smaller phase II study, and if approved by the FDA – which usually follows the recommendations of its advisory committees – would be only the second treatment approved for the debilitating and sometimes deadly disease. The first, hydroxyurea, was approved for use in adults in 1998.
The committee voted 10-3 in favor of L-glutamate, after hearing from representatives of the new drug marketing applicant, Emmaus Medical, about the efficacy and safety data, as well as from FDA representatives who analyzed the data, physicians who treat sickle cell patients, patient advocates, and patients and their family members who gave emotional testimony in favor of approving this treatment.
“This is clearly a bad disease. It’s worse than cancer in many ways. I think probably mostly from a stigma standpoint it has a desperate need [for treatments],” said acting committee chair Brian I. Rini, MD, who voted in favor of the agent.
While there were some concerns about the data, including questions about methodologies, differential dropout rates between study arms, baseline characteristics that may have affected outcomes, and discrepancies between the Emmaus Medical data and the FDA’s analyses of the data, “all seemed to come down in favor of the agent,” Dr. Rini said, citing its “modest but consistent benefit” and low risk.
“I think one thing that’s strikingly clear is that even any sort of modest benefit in this community, given the sequelae of crises, is significant; it doesn’t take much to produce a clinical impact, and that should be motivation to study more drugs in this disease,” he said.
Another focus among those who voted “yes” was on the overwhelming need for treatment for sickle cell patients, who spend “a hugely disproportionate part of their life in the health care system, and who have had a tremendous burden imposed on them by their disease,” according to Harold J. Burstein, MD, PhD, of Dana-Farber Cancer Institute in Boston, who said he was swayed by the low risk of toxicity and the corroborating evidence in the phase II and III trials.
“What I took away was that one fewer hospital visit per year was a clinically compelling benefit for any individual or family or hospital that might be caring for patients with sickle cell disease,” he said, referencing a finding that treated patients had three visits, compared with four visits among patients in the placebo group, in the phase III study.
The data presented to the committee during the meeting by Yutaka Niihara, MD, of Emmaus Medical, included findings from a phase III randomized, placebo-controlled multicenter study (GLUSCC09-01) involving patients aged 5 years and older with sickle cell disease or beta-0 thalassemia who had at least two episodes of painful crises within the 12 months prior to screening. A total of 152 patients were randomized to receive oral L-glutamine at a dose of 0.3 mg/kg per day for 48 weeks followed by a 3-week tapering period, and 78 received placebo.
The Emmaus Medical analysis demonstrated a significant decrease in crisis events (median of 3 vs. 4) among treatment vs. placebo group patients, and the time to second crisis was delayed by 79 days in the treatment group (hazard ratio 0.68). The analysis, however, was complicated by the differential dropout rates (36% vs. 24% in the treatment and placebo arms, respectively), necessitating the use of imputation methods. Various methods were used to handle the missing data, and the findings with each of them favored L-glutamine, but each had important limitations, and while the FDA’s analysis of the various approaches showed that each favored L-glutamine over placebo with a range of reduction in the rates of crises from 0.4 to 0.9, this contributed to the decision by some panel member to vote against the agent.
The phase II study (Study 10478), which had a similar design, failed to meet its specified significance level for primary efficacy analysis, but showed a trend in favor of L-glutamine vs. placebo, Dr. Niihara said.
As for safety, Dr. Niihara reported that a safety population of 187 patients treated with L-glutamine and 111 treated with placebo in the phase II and III studies showed that most patients experienced a treatment-emergent adverse event – most often sickle cell anemia with crisis (66% and 72% in the groups, respectively) and acute chest syndrome (7% and 19%, respectively). Treatment-emergent adverse events led to withdrawal in 2.7% and 0.9% of patients, respectively. The most common adverse reactions were constipation, nausea, headache, cough, pain in the extremities, back pain, chest pain, and abdominal pain.
Bernard F. Cole, PhD, who was among the “no” votes, said he had concerns about “the limitations resulting from differential dropout” rates, which may have artificially shifted the risk profile.
“As a result of those limitations, it’s not clear whether patients at higher risk of a [sickle cell crisis] event might have disproportionately dropped out of the L-glutamine arm,” he explained. “My hope is that the sponsors can more thoroughly address the limitations of this pivotal trial with the FDA,” said Dr. Cole, a professor in the department of mathematics and statistics at the University of Vermont, Burlington.
Dr. Rini, despite his “yes” vote, agreed with the need for more data, noting that additional data analysis could help when it comes to clinical application of L-glutamine.
Specifically, more data regarding duration of therapy and quality data collection that “borrows from the cancer world ... in terms of rigor or data collection,” is needed, he said.
The committee members had no relevant conflicts of interests to disclose.
Key clinical point:
Major finding: In a phase III study, there was a significant decrease in crisis events (median of 3 vs. 4) among treatment vs. placebo group patients.
Data source: A total of 152 patients were randomized to receive oral L-glutamine at a dose of 0.3 mg/kg per day for 48 weeks followed by a 3-week tapering period, and 78 received placebo.
Disclosures: The committee members had no relevant conflicts of interests to disclose.
FDA okays pembrolizumab for certain solid tumors with common biomarker
In an accelerated approval process, the Food and Drug Administration has approved the use of the monoclonal antibody pembrolizumab for treatment of certain solid tumors that have a common biomarker, a first for the agency.
Pembrolizumab (Keytruda) was approved to treat patients whose tumors are metastatic or unresectable, and which have the biomarker microsatellite instability-high (MSI-H), also known as mismatch repair deficient (dMMR). This biomarker is found in many kinds of solid tumors, especially in colorectal and other gastrointestinal cancers, as well as endometrial cancer, and may make tumors more susceptible to host immune system activity. “Approximately 5% of patients with metastatic colorectal cancer have MSI-H or dMMR tumors,” according to the FDA statement announcing the approval.
Patients who are eligible for pembrolizumab under this approval are those whose solid tumors have progressed despite earlier treatment and who lack other treatment options; pembrolizumab was also approved for colorectal cancer patients whose cancers have progressed after treatment with some chemotherapy drugs.
Pembrolizumab received priority review from the FDA and was approved on the basis of five uncontrolled single-arm clinical trials. There were a total of 149 patients enrolled in the trials, and 15 different cancer types were represented. Colorectal and other gastrointestinal cancers and endometrial cancers were the most common types in the studies. Of the 149 patients, 39.6% had a complete or partial response, and of these responders, 78% had a response lasting at least 6 months.
Fatigue is a common side effect of pembrolizumab; anorexia, peripheral edema, rash, pruritis, hyperlipidemia, and electrolyte disturbances are also common. Because of the drug’s mechanism of action, it can cause immune-mediated side effects, such as pneumonitis, hepatitis, nephritis, and endocrinopathies.
Pembrolizumab is given as an intravenous infusion, usually once every 3 weeks. It targets the programmed death-1/programmed death-ligand 1 (PD-1/PDL-1) pathway to boost the immune system’s ability to target and kill cancer cells.
“This is an important first for the cancer community,” said Dr. Pazdur, who is also acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. The clinical trials were sponsored by Merck & Co., which markets Keytruda.
koakes@frontlinemedcom.com
On Twitter @karioakes
In an accelerated approval process, the Food and Drug Administration has approved the use of the monoclonal antibody pembrolizumab for treatment of certain solid tumors that have a common biomarker, a first for the agency.
Pembrolizumab (Keytruda) was approved to treat patients whose tumors are metastatic or unresectable, and which have the biomarker microsatellite instability-high (MSI-H), also known as mismatch repair deficient (dMMR). This biomarker is found in many kinds of solid tumors, especially in colorectal and other gastrointestinal cancers, as well as endometrial cancer, and may make tumors more susceptible to host immune system activity. “Approximately 5% of patients with metastatic colorectal cancer have MSI-H or dMMR tumors,” according to the FDA statement announcing the approval.
Patients who are eligible for pembrolizumab under this approval are those whose solid tumors have progressed despite earlier treatment and who lack other treatment options; pembrolizumab was also approved for colorectal cancer patients whose cancers have progressed after treatment with some chemotherapy drugs.
Pembrolizumab received priority review from the FDA and was approved on the basis of five uncontrolled single-arm clinical trials. There were a total of 149 patients enrolled in the trials, and 15 different cancer types were represented. Colorectal and other gastrointestinal cancers and endometrial cancers were the most common types in the studies. Of the 149 patients, 39.6% had a complete or partial response, and of these responders, 78% had a response lasting at least 6 months.
Fatigue is a common side effect of pembrolizumab; anorexia, peripheral edema, rash, pruritis, hyperlipidemia, and electrolyte disturbances are also common. Because of the drug’s mechanism of action, it can cause immune-mediated side effects, such as pneumonitis, hepatitis, nephritis, and endocrinopathies.
Pembrolizumab is given as an intravenous infusion, usually once every 3 weeks. It targets the programmed death-1/programmed death-ligand 1 (PD-1/PDL-1) pathway to boost the immune system’s ability to target and kill cancer cells.
“This is an important first for the cancer community,” said Dr. Pazdur, who is also acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. The clinical trials were sponsored by Merck & Co., which markets Keytruda.
koakes@frontlinemedcom.com
On Twitter @karioakes
In an accelerated approval process, the Food and Drug Administration has approved the use of the monoclonal antibody pembrolizumab for treatment of certain solid tumors that have a common biomarker, a first for the agency.
Pembrolizumab (Keytruda) was approved to treat patients whose tumors are metastatic or unresectable, and which have the biomarker microsatellite instability-high (MSI-H), also known as mismatch repair deficient (dMMR). This biomarker is found in many kinds of solid tumors, especially in colorectal and other gastrointestinal cancers, as well as endometrial cancer, and may make tumors more susceptible to host immune system activity. “Approximately 5% of patients with metastatic colorectal cancer have MSI-H or dMMR tumors,” according to the FDA statement announcing the approval.
Patients who are eligible for pembrolizumab under this approval are those whose solid tumors have progressed despite earlier treatment and who lack other treatment options; pembrolizumab was also approved for colorectal cancer patients whose cancers have progressed after treatment with some chemotherapy drugs.
Pembrolizumab received priority review from the FDA and was approved on the basis of five uncontrolled single-arm clinical trials. There were a total of 149 patients enrolled in the trials, and 15 different cancer types were represented. Colorectal and other gastrointestinal cancers and endometrial cancers were the most common types in the studies. Of the 149 patients, 39.6% had a complete or partial response, and of these responders, 78% had a response lasting at least 6 months.
Fatigue is a common side effect of pembrolizumab; anorexia, peripheral edema, rash, pruritis, hyperlipidemia, and electrolyte disturbances are also common. Because of the drug’s mechanism of action, it can cause immune-mediated side effects, such as pneumonitis, hepatitis, nephritis, and endocrinopathies.
Pembrolizumab is given as an intravenous infusion, usually once every 3 weeks. It targets the programmed death-1/programmed death-ligand 1 (PD-1/PDL-1) pathway to boost the immune system’s ability to target and kill cancer cells.
“This is an important first for the cancer community,” said Dr. Pazdur, who is also acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. The clinical trials were sponsored by Merck & Co., which markets Keytruda.
koakes@frontlinemedcom.com
On Twitter @karioakes
FDA approves sarilumab for DMARD-intolerant RA patients
The Food and Drug Administration has approved sarilumab (Kevzara), a human monoclonal antibody against the interleukin-6 receptor, for the treatment of adult patients with rheumatoid arthritis (RA), the manufacturer Regeneron Pharmaceuticals announced May 22.
Interleukin-6 is an important factor in RA, as excess amounts of IL-6 build up in the body and contribute to RA-associated inflammation. Sarilumab has been shown to bind to and reduce IL-6R signaling, and is intended for people who have shown inadequate response to or are intolerant to conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
“Not all currently available treatments work in all patients, and some patients may spend years cycling through different treatments without achieving their treatment goals. Sarilumab works differently from the most commonly used biologics, such as those in the anti-TNF [tumor necrosis factor] class, and is a welcome new option for patients and their physicians,” Alan Kivitz, MD, an investigator in sarilumab clinical trials and a rheumatologist in group practice in Duncansville, Pa., said in Regeneron’s announcement.
The recommended dosage of sarilumab is 200 mg once every 2 weeks given as a subcutaneous injection, which can be self-administered. The dosage can be reduced from 200 mg to 150 mg once every 2 weeks, as needed, to help manage certain laboratory abnormalities (neutropenia, thrombocytopenia, and liver enzyme elevations). Significant adverse events associated with sarilumab include weakening of the immune system, changes in certain laboratory tests, perforation in the stomach or intestines, increased risk of cancer, and serious allergic reaction.
The Food and Drug Administration has approved sarilumab (Kevzara), a human monoclonal antibody against the interleukin-6 receptor, for the treatment of adult patients with rheumatoid arthritis (RA), the manufacturer Regeneron Pharmaceuticals announced May 22.
Interleukin-6 is an important factor in RA, as excess amounts of IL-6 build up in the body and contribute to RA-associated inflammation. Sarilumab has been shown to bind to and reduce IL-6R signaling, and is intended for people who have shown inadequate response to or are intolerant to conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
“Not all currently available treatments work in all patients, and some patients may spend years cycling through different treatments without achieving their treatment goals. Sarilumab works differently from the most commonly used biologics, such as those in the anti-TNF [tumor necrosis factor] class, and is a welcome new option for patients and their physicians,” Alan Kivitz, MD, an investigator in sarilumab clinical trials and a rheumatologist in group practice in Duncansville, Pa., said in Regeneron’s announcement.
The recommended dosage of sarilumab is 200 mg once every 2 weeks given as a subcutaneous injection, which can be self-administered. The dosage can be reduced from 200 mg to 150 mg once every 2 weeks, as needed, to help manage certain laboratory abnormalities (neutropenia, thrombocytopenia, and liver enzyme elevations). Significant adverse events associated with sarilumab include weakening of the immune system, changes in certain laboratory tests, perforation in the stomach or intestines, increased risk of cancer, and serious allergic reaction.
The Food and Drug Administration has approved sarilumab (Kevzara), a human monoclonal antibody against the interleukin-6 receptor, for the treatment of adult patients with rheumatoid arthritis (RA), the manufacturer Regeneron Pharmaceuticals announced May 22.
Interleukin-6 is an important factor in RA, as excess amounts of IL-6 build up in the body and contribute to RA-associated inflammation. Sarilumab has been shown to bind to and reduce IL-6R signaling, and is intended for people who have shown inadequate response to or are intolerant to conventional synthetic disease-modifying antirheumatic drugs (DMARDs).
“Not all currently available treatments work in all patients, and some patients may spend years cycling through different treatments without achieving their treatment goals. Sarilumab works differently from the most commonly used biologics, such as those in the anti-TNF [tumor necrosis factor] class, and is a welcome new option for patients and their physicians,” Alan Kivitz, MD, an investigator in sarilumab clinical trials and a rheumatologist in group practice in Duncansville, Pa., said in Regeneron’s announcement.
The recommended dosage of sarilumab is 200 mg once every 2 weeks given as a subcutaneous injection, which can be self-administered. The dosage can be reduced from 200 mg to 150 mg once every 2 weeks, as needed, to help manage certain laboratory abnormalities (neutropenia, thrombocytopenia, and liver enzyme elevations). Significant adverse events associated with sarilumab include weakening of the immune system, changes in certain laboratory tests, perforation in the stomach or intestines, increased risk of cancer, and serious allergic reaction.